
code
stringlengths 501
5.19M
| package
stringlengths 2
81
| path
stringlengths 9
304
| filename
stringlengths 4
145
|
|---|---|---|---|
from alphacoders_downloader.util.utils import limit_tasks, clear_line, print_error
from alphacoders_downloader.util.arguments_builder import ArgumentsBuilder
from alphacoders_downloader.util.utils import create_folder_recursively
from alphacoders_downloader.exceptions import WallpapersNotFounds
from alphacoders_downloader.util.cursor import HiddenCursor, show
from alphacoders_downloader.util.progress_bar import ProgressBar
from alphacoders_downloader import __version__, __license__
from alphacoders_downloader.util.spinner import Spinner
from bs4 import BeautifulSoup
from typing import Union
import setproctitle
import aiofiles
import asyncio
import aiohttp
import shutil
import sys
import os
class AlphacodersDownloader:
def __init__(
self, url: str, path: str, size: int, client_session: aiohttp.ClientSession
):
self.url = url
self.path = path if path[-1] == os.sep else path + os.sep
self.size = size
self.client_session = client_session
self.temp_path = self.path + "temp" + os.sep
self.page_char = ""
self.progress_bar: Union[ProgressBar, None] = None
self.spinner: Spinner = Spinner()
self.temp_images_list = []
self.images_list = []
self.links_len = 0
self.links_got = 0
self.total_size_to_download = 0
self.total_size_downloaded = 0
async def fetch_images(self, image_url: str):
images_link_list_split = image_url.split("/")
images_name_file_thumb = images_link_list_split[len(images_link_list_split) - 1]
images_name_file = images_name_file_thumb.split("-")[
len(images_name_file_thumb.split("-")) - 1
]
if os.path.isfile(os.path.join(self.path, images_name_file)) is False:
image_url = image_url.replace(images_name_file_thumb, "") + images_name_file
async with self.client_session.head(image_url) as request:
file_size = int(request.headers["Content-Length"])
self.total_size_to_download += file_size
self.images_list.append([image_url, images_name_file, file_size])
async def parse_url(self, url: str):
async with self.client_session.get(
url, cookies={"AlphaCodersView": "paged"}
) as request:
page = BeautifulSoup(await request.text(), "html.parser")
find_images_urls = page.find("div", {"id": "page_container"}).find_all(
"div", "thumb-container-big"
)
if find_images_urls is None:
raise WallpapersNotFounds(url)
for a_element in find_images_urls:
href = str(a_element.find("img").get("src"))
if (
href.startswith("https://images") or href.startswith("https://mfiles")
) and href not in self.temp_images_list:
self.temp_images_list.append(href)
self.links_got += 1
a_elements = page.find("div", {"class": "pagination-simple center"}).find_all(
"a"
)
changed_element = None
if a_elements is not None and a_elements:
for a_element in a_elements:
if a_element.text.strip() == "Next >>":
changed_element = a_element
break
if changed_element is not None:
url = changed_element.get("href")
try:
url_spliced = url.split("&page=")[1]
except IndexError:
url_spliced = url.split("?page=")[1]
await self.parse_url(f"{self.url}{self.page_char}page=" + url_spliced)
async def download(self, element: list):
path = os.path.join(self.path, element[1])
if os.path.isfile(path) is False:
temp_path = os.path.join(self.temp_path, element[1])
headers = {}
temp_file_exist = os.path.isfile(temp_path)
if temp_file_exist:
file_size = os.stat(temp_path).st_size
headers["Range"] = f"bytes={file_size}-"
file_downloaded = 0
async with self.client_session.get(element[0], headers=headers) as request:
try:
write_mode = "ab" if temp_file_exist else "wb"
async with aiofiles.open(temp_path, write_mode) as file:
try:
async for data in request.content.iter_chunked(
int(self.size / 8)
):
await file.write(data)
file_downloaded += len(data)
self.progress_bar.progress(len(data))
except asyncio.TimeoutError:
self.progress_bar.progress(
element[2] - file_downloaded, True
)
return self.progress_bar.print_error(
f"Download of file: {element[0]} has been timeout."
)
except aiohttp.ClientPayloadError:
self.progress_bar.progress(element[2] - file_downloaded, True)
return self.progress_bar.print_error(
f"Download of file: {element[0]} raise ClientPayloadError."
)
if os.path.isfile(temp_path):
shutil.move(temp_path, path)
else:
self.progress_bar.progress(element[2], True)
async def start(self):
self.spinner.set_text("Recovery of the URLS of all the pages...")
await self.spinner.start()
create_folder_recursively(self.path)
create_folder_recursively(self.temp_path)
self.spinner.set_text("Recovery of the URLS of all the wallpapers...")
self.page_char = (
"&" if "https://mobile.alphacoders.com/" not in self.url else "?"
)
await self.parse_url(f"{self.url}{self.page_char}page=1")
self.spinner.set_text("Recovery of the informations about wallpapers...")
await limit_tasks(
10, *[self.fetch_images(element) for element in self.temp_images_list]
)
self.temp_images_list.clear()
self.spinner.stop()
self.progress_bar = ProgressBar(
self.total_size_to_download, "Downloading wallpapers", speed=True
)
await limit_tasks(10, *[self.download(element) for element in self.images_list])
shutil.rmtree(self.temp_path)
print("\033[1mCompleted!\033[0m")
class CommandsHandler:
@staticmethod
async def download(command_return: dict):
wallpapers_url = command_return["args"][command_return["args"].index("-S") + 1]
if "https://" not in wallpapers_url and "alphacoders.com" not in wallpapers_url:
print_error("This URL isn't correct.")
sys.exit()
path_to_download = command_return["args"][
command_return["args"].index("-P") + 1
]
if os.access(os.path.dirname(path_to_download), os.W_OK) is False:
print_error("This path isn't correct.")
sys.exit()
size = 2048
if "-D" in command_return["args"]:
download_index = command_return["args"].index("-D") + 1
if download_index < len(command_return["args"]):
converted_size = int(command_return["args"][download_index])
if (
converted_size % 8 == 0
and (converted_size / 8) > 0
and ((converted_size / 8) % 8) == 0
):
size = converted_size
async with aiohttp.ClientSession() as client_session:
await AlphacodersDownloader(
wallpapers_url, path_to_download, size, client_session
).start()
@staticmethod
def get_version(_):
version_text = f"\033[1mAlphacodersDownloader {__version__}\033[0m\n"
version_text += (
"Created by \033[1mAsthowen\033[0m - \033[[email protected]\033[0m\n"
)
version_text += f"License: \033[1m{__license__}\033[0m"
print(version_text)
async def main():
setproctitle.setproctitle("AlphacodersDownloader")
if len(sys.argv) <= 1:
url = ""
while "https://" not in url and "alphacoders.com" not in url:
url = input(
"Please enter the download url (e.g. "
"https://wall.alphacoders.com/search.php?search=sword+art+online). > "
).replace(" ", "")
clear_line()
path = ""
while os.access(os.path.dirname(path), os.W_OK) is False:
path = input(
"Please enter the folder where the images are saved (e.g. ~/downloads/wallpapers/). > "
)
clear_line()
size = None
change_size = False
while size is None:
if change_size is False:
change_size_input = input(
"Do you want to change the default download limit of 2Mo/s (y/n)? > "
)
clear_line()
if change_size_input.lower() in ("y", "yes") or change_size:
change_size = True
new_size_input = input(
"Enter the new speed limit (must be in Ko, and be a multiple of 8) > "
)
clear_line()
if new_size_input.isdigit():
converted = int(new_size_input)
if (
converted % 8 == 0
and (converted / 8) > 0
and ((converted / 8) % 8) == 0
):
size = int(new_size_input)
else:
size = 2048
with HiddenCursor() as _:
async with aiohttp.ClientSession() as client_session:
await AlphacodersDownloader(url, path, size, client_session).start()
else:
parser = ArgumentsBuilder(
"A script for download wallpapers on https://alphacoders.com/.",
"alphacoders-downloader",
)
parser.add_argument(
"-S",
action=CommandsHandler().download,
description="Download wallpapers.",
command_usage="-S wallpapers_url -P path -D 1024",
)
parser.add_argument(
"-V",
action=CommandsHandler.get_version,
description="Get version infos.",
command_usage="-V",
)
with HiddenCursor() as _:
await parser.build()
def start():
# pylint: disable=W0703
try:
os.get_terminal_size(0)
asyncio.get_event_loop().run_until_complete(main())
except OSError:
print_error(
"Your terminal does not support all the features needed for AlphacodersDownloader, please use "
"another one."
)
show()
except KeyboardInterrupt:
clear_line()
print("Stop the script...")
show()
except Exception as exception:
print_error(str(exception))
show()
|
AlphacodersDownloader
|
/AlphacodersDownloader-0.1.4.3-py3-none-any.whl/alphacoders_downloader/alphacoders_downloader.py
|
alphacoders_downloader.py
|
from alphacoders_downloader.util.utils import print_error
import asyncio
import sys
class ArgumentsBuilder:
def __init__(self, description: str, command_base: str, args: list = None):
if args is None:
args = sys.argv
self.description = description
self.command_base = command_base
self.args = args
self.arguments = {}
self.__help_content = None
def add_argument(
self,
argument_name: str,
action=None,
description: str = None,
command_usage: str = None,
):
self.arguments[argument_name.lower()] = {
"action": action,
"description": description,
"command_usage": command_usage,
}
def build_help(self):
if self.__help_content is None:
self.__help_content = (
f"\n\033[1m{self.description}\033[0m\n\nCommand list:\n"
)
for _, command_json in self.arguments.items():
self.__help_content += (
f'・\033[1m{self.command_base} {command_json["command_usage"]}\033['
f'0m | {command_json["description"]}\n'
)
print(self.__help_content)
async def build(self):
if len(self.args) == 1 or "--help" in self.args or "-h" in self.args:
return self.build_help()
has_been_found = False
for argument in self.args:
argument = argument.lower()
if argument in self.arguments:
has_been_found = True
self.arguments[argument]["args"] = self.args
if asyncio.iscoroutinefunction(self.arguments[argument]["action"]):
await self.arguments[argument]["action"](self.arguments[argument])
else:
self.arguments[argument]["action"](self.arguments[argument])
if has_been_found is False:
print_error(
f"\033[1mThis command doesn't exist. Please check the command: {self.command_base} -H.\033[0m"
)
|
AlphacodersDownloader
|
/AlphacodersDownloader-0.1.4.3-py3-none-any.whl/alphacoders_downloader/util/arguments_builder.py
|
arguments_builder.py
|
from alphacoders_downloader.util.utils import clear_line
import time
import math
import os
class ProgressBar:
def __init__(self, total: float, prefix: str, speed: bool = False):
self.total = total
self.prefix = prefix
self.speed = speed
self.iteration: float = 0
self.iteration_eta: float = 0
self.__speed_value = ""
self.__speed_latest_value = 0
self.__speed_latest_time = 0
self.__speed_eta = ""
self.__can_use_data = 0
self.is_started = False
self.__progress_bar_chars = ("▏", "▎", "▍", "▌", "▋", "▊", "▉")
def set_total(self, total: float):
self.total = total
def set_prefix(self, prefix: str):
self.prefix = prefix
def set_iteration(self, iteration: float, ignore_speed=False):
self.iteration = iteration
if ignore_speed is False:
self.iteration_eta = iteration
def append_iteration(self, iteration: float, ignore_speed=False):
self.iteration += iteration
if ignore_speed is False:
self.iteration_eta += iteration
def set_progress_at_0(self):
self.progress(0)
self.__update_progress_bar()
def set_progress_bar_parameters(
self,
total: float = None,
prefix: str = None,
iteration: float = None,
progress_at_0: bool = False,
):
if total is not None:
self.set_total(total)
if prefix is not None:
self.set_prefix(prefix)
if iteration is not None:
self.set_iteration(iteration)
if progress_at_0:
self.set_progress_at_0()
def progress(self, iteration: float, ignore_speed=False):
if 0 < iteration <= self.total:
if self.speed and time.time() - self.__speed_latest_time >= 1:
if self.__can_use_data >= 2:
current_eta = self.iteration_eta - self.__speed_latest_value
self.__speed_value = " - " + self.__parse_size(current_eta) + " "
self.__speed_eta = "| " + self.__parse_duration(
math.trunc(
(
(self.total - (self.iteration - self.iteration_eta))
- self.iteration_eta
)
/ current_eta
)
)
else:
self.__can_use_data += 1
self.__speed_latest_time = time.time()
self.__speed_latest_value = self.iteration_eta
self.append_iteration(iteration, ignore_speed)
self.__update_progress_bar()
self.is_started = True
def print_error(self, text: str):
if self.is_started:
print()
clear_line()
print("\033[91m" + text + "\033[0m")
def __update_progress_bar(self):
terminal_size = os.get_terminal_size(0).columns
place_to_print = terminal_size - len(self.prefix) - 8 - 14 - 14
percentage = 100 * (self.iteration / float(self.total))
filled_length = int(place_to_print * self.iteration // self.total)
additional_progress = self.__progress_bar_chars[
int(((place_to_print * self.iteration / self.total) % 1) / (1 / 7))
]
progress_chars = (
"█" * filled_length
+ additional_progress
+ " " * (place_to_print - filled_length - 1)
)
to_print = f"{self.prefix} [{progress_chars}] {percentage:.2f}%{self.__speed_value}{self.__speed_eta}"
print(f"{to_print}{(terminal_size - len(to_print)) * ' '}", end="\r")
if self.iteration == self.total:
print()
clear_line()
self.is_started = False
@staticmethod
def __parse_size(num) -> str:
for unit in ("B", "KB", "MB", "GB", "TB", "PB", "EB", "ZB"):
if abs(num) < 1024.0:
return f"{num:3.1f}{unit}/s"
num /= 1024.0
return "0B/s"
@staticmethod
def __parse_duration(duration: int) -> str:
minutes, seconds = divmod(duration, 60)
hours, minutes = divmod(minutes, 60)
days, hours = divmod(hours, 24)
return f"{f'{days}d' if days > 0 else ''}{f'{hours}h' if hours > 0 else ''}{f'{minutes}m' if minutes > 0 else ''}{f'{seconds}s' if seconds > 0 else ''} "
|
AlphacodersDownloader
|
/AlphacodersDownloader-0.1.4.3-py3-none-any.whl/alphacoders_downloader/util/progress_bar.py
|
progress_bar.py
|
# This file is part of alquimia.
# alquimia is free software: you can redistribute it and/or modify
# it under the terms of the GNU Lesser General Public License as published by
# the Free Software Foundation, either version 3 of the License, or
# (at your option) any later version.
# This program is distributed in the hope that it will be useful,
# but WITHOUT ANY WARRANTY; without even the implied warranty of
# MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the
# GNU General Public License for more details.
# You should have received a copy of the GNU General Public License
# along with this program. If not, see <http://www.gnu.org/licenses/>.
import jsonschema
import logging
import copy
from sqlalchemy.orm import relationship
from sqlalchemy import Column, ForeignKey, Table
from alquimia import SCHEMA, DATA_TYPES
from alquimia.utils import log
from alquimia.models_attrs_reflect import ModelsAtrrsReflect
class AmbiguousRelationshipsError(Exception):
def __init__(self, model_name, rel_name, logger=logging):
message = "%s.%s and %s.%s relationships is ambiguous!" \
% (model_name, rel_name, rel_name, model_name)
log(logger, 'critical', message)
Exception.__init__(self, message)
class ModelsAttributes(ModelsAtrrsReflect):
def __init__(self, dict_, metadata, data_types=DATA_TYPES, logger=logging):
dict_ = copy.deepcopy(dict_)
jsonschema.validate(dict_, SCHEMA)
self._data_types = data_types
ModelsAtrrsReflect.__init__(self, metadata, logger, *[dict_])
def _build_columns(self, model_name, model):
new_model = {}
for k, v in model.iteritems():
new_model[k] = type(v)(v)
model = new_model
model.pop('relationships', None)
model['id'] = {'type': 'integer', 'primary_key': True}
for col_name, column in model.iteritems():
if not isinstance(column, dict):
type_ = column
column = {'args': [col_name, type_]}
else:
column['args'] = [col_name, column.pop('type')]
self._build_column_instance(column, col_name, model_name)
def _build_rel_attr_dict(self, new_rels, rel):
if not isinstance(rel, dict):
new_rels[rel] = {}
elif not isinstance(rel.values()[0], dict):
new_rels[rel.keys()[0]] = {rel.values()[0]: True}
else:
new_rels[rel.keys()[0]] = rel.values()[0].copy()
def _build_relationships_dict(self, rels):
new_rels = {}
if not isinstance(rels, dict):
if isinstance(rels, str):
rels = [rels]
for rel in rels:
self._build_rel_attr_dict(new_rels, rel)
else:
for k, v in rels.iteritems():
self._build_rel_attr_dict(new_rels, {k: v})
return new_rels
def _build_relationship_column(self, rel_name, model_name, primary_key, oto):
foreign_key = {
'args': [rel_name+'.id'],
'onupdate': 'CASCADE',
'ondelete': 'CASCADE'
}
rel_col_name = rel_name+'_id'
column = {'args': [rel_col_name, 'integer', foreign_key],
'autoincrement': False}
if primary_key:
column['primary_key'] = True
if oto:
column['unique'] = True
self._build_column_instance(column, rel_col_name, model_name)
def _build_relationships(self, model_name, rels_dict):
rels = {}
for rel_name, rel in rels_dict.iteritems():
if rel.pop('many-to-many', False):
mtm_table_name = '%s_%s_association' % \
(model_name, rel_name)
self._build_many_to_many_table(model_name, rel_name,
mtm_table_name)
self._build_many_to_many_rel(rel_name,
model_name, mtm_table_name)
else:
is_oto = rel.pop('one-to-one', False) or rel_name == model_name
self._build_relationship_column(rel_name, model_name,
rel.pop('primary_key', False), is_oto)
if is_oto:
id_column = self[model_name]['id'] \
if rel_name == model_name else None
self._build_one_to_one_rel(rel_name, model_name, id_column)
else:
self._build_many_to_one_rel(rel_name, model_name)
return rels
def _build_column_instance(self, column, col_name, model_name):
if len(column['args']) == 3:
fk = column['args'][2]
column['args'][2] = ForeignKey(*fk.pop('args'), **fk)
column['args'][1] = self._data_types[column['args'][1]]
self[model_name][col_name] = Column(*column.pop('args'), **column)
def _build_many_to_many_table(self, rel1_name, rel2_name, table_name):
col1 = Column(rel2_name+'_id', self._data_types['integer'],
ForeignKey(rel2_name+'.id', onupdate='CASCADE', ondelete='CASCADE'),
primary_key=True, autoincrement=False)
col2 = Column(rel1_name+'_id', self._data_types['integer'],
ForeignKey(rel1_name+'.id', onupdate='CASCADE', ondelete='CASCADE'),
primary_key=True, autoincrement=False)
Table(table_name, self._metadata, col1, col2)
def _check_rels(self, models_rels):
new_mr = {m: r.copy() for m, r in models_rels.iteritems()}
for mdl_name, rels in models_rels.iteritems():
for rel_name, rel in rels.iteritems():
if mdl_name in new_mr[rel_name] and mdl_name != rel_name:
rel2 = models_rels[rel_name][mdl_name]
rel_mtm = rel.get('many-to-many', False)
rel2_mtm = rel2.get('many-to-many', False)
rel_oto = rel.get('one-to-one', False)
rel2_oto = rel2.get('one-to-one', False)
if (not rel_mtm or not rel2_mtm) and \
(not rel_oto or not rel2_oto):
raise AmbiguousRelationshipsError(mdl_name, rel_name)
message = 'Removed relationship %s.%s duplicated from '\
'%s.%s' % (mdl_name, rel_name, rel_name, mdl_name)
log(self._logger, 'warning', message)
new_mr[mdl_name].pop(rel_name)
models_rels.clear()
models_rels.update(new_mr)
def _build(self, dict_):
models_rels = {}
for model_name, model in dict_.iteritems():
rels = model.get('relationships', {})
models_rels[model_name] = self._build_relationships_dict(rels)
self._init_attrs(model_name)
self._check_rels(models_rels)
for model_name, model in dict_.iteritems():
self._build_columns(model_name, model)
self._build_relationships(model_name, models_rels[model_name])
self[model_name]['__tablename__'] = model_name
|
Alquimia
|
/alquimia-0.7.2.tar.gz/alquimia-0.7.2/alquimia/models_attrs.py
|
models_attrs.py
|
# This file is part of alquimia.
# alquimia is free software: you can redistribute it and/or modify
# it under the terms of the GNU Lesser General Public License as published by
# the Free Software Foundation, either version 3 of the License, or
# (at your option) any later version.
# This program is distributed in the hope that it will be useful,
# but WITHOUT ANY WARRANTY; without even the implied warranty of
# MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the
# GNU General Public License for more details.
# You should have received a copy of the GNU General Public License
# along with this program. If not, see <http://www.gnu.org/licenses/>.
from sqlalchemy.ext.declarative.api import DeclarativeMeta
from sqlalchemy.orm.attributes import InstrumentedAttribute
from sqlalchemy.orm.exc import NoResultFound
from sqlalchemy import or_
from alquimia import utils
class AlquimiaModelMeta(DeclarativeMeta):
def __init__(cls, classname, bases, dict_):
DeclarativeMeta.__init__(cls, classname, bases, dict_)
attrs = {k:v for k,v in cls.__dict__.items() \
if isinstance(v, InstrumentedAttribute)}
cls.__attrs__ = cls.__attributes__ = attrs
cls._current_pos = 0
def __getitem__(cls, attr_name):
try:
return cls.__attributes__[attr_name]
except KeyError, e:
raise KeyError(e.message)
def __iter__(cls):
return cls
def __contains__(cls, item):
return item in cls.__attrs__
def next(cls):
cls._current_pos += 1
if cls._current_pos >= len(cls.keys()):
cls._current_pos = 0
raise StopIteration
else:
return cls.keys()[cls._current_pos - 1]
@property
def keys(cls):
return cls.__attrs__.keys
@property
def values(cls):
return cls.__attrs__.values
@property
def items(cls):
return cls.__attrs__.items
@property
def iteritems(cls):
return cls.__attrs__.iteritems
def _build_objs(cls, obj):
if not isinstance(obj, list):
obj = [obj]
objs = []
for each in obj:
if not isinstance(each, cls):
each = cls(**each)
objs.append(each)
return objs
def _update_rec(cls, new_values, obj):
for prop_name, new_value in new_values.iteritems():
if isinstance(new_value, dict):
cls._update_rec(new_value, obj[prop_name])
elif isinstance(new_value, list):
new_list = []
model = type(obj)[prop_name].model
for each_value in new_value:
if 'id' in each_value:
each_obj = cls._get_obj_by_id(model, each_value['id'])
cls._update_rec(each_value, each_obj)
else:
each_obj = model(**each_value)
new_list.append(each_obj)
obj[prop_name] = new_list
else:
obj[prop_name] = new_value
def _get_obj_by_id(cls, model, id_):
try:
return cls._session.query(model).filter(model.id == id_).one()
except NoResultFound:
raise TypeError("invalid id '%s'" % id_)
def insert(cls, objs):
objs_ = cls._build_objs(objs)
cls._session.commit()
if not isinstance(objs, list):
objs_ = objs_[0]
return objs_
def update(cls, new_values):
objs = []
if not isinstance(new_values, list):
new_values = [new_values]
for new_value in new_values:
try:
id_ = new_value.pop('id')
except KeyError:
raise KeyError('values must have id property!')
obj = cls._get_obj_by_id(cls, id_)
cls._update_rec(new_value, obj)
objs.append(obj)
cls._session.commit()
objs = objs[0] if len(objs) == 1 else objs
return objs
def delete(cls, ids):
session = cls._session
if not isinstance(ids, list):
ids = [ids]
for id_ in ids:
try:
int(id_)
except ValueError:
session.rollback()
raise TypeError('%s.delete just receive ids (integer)!' \
' No delete operation was done.' % cls)
session.query(cls).filter(cls.id == id_).delete()
session.commit()
def _parse_filters(cls, query_dict, obj, filters):
for prop_name, prop in query_dict.iteritems():
if prop_name == '_or':
filters_ = []
[cls._parse_filters(subfilter, obj, filters_) for subfilter in prop]
filters.append(or_(*filters_))
else:
if hasattr(obj, 'model'):
obj = obj.model
if isinstance(prop, dict):
cls._parse_filters(prop, obj[prop_name], filters)
elif isinstance(prop, list):
cls._parse_filters({'_or': prop}, obj[prop_name], filters)
else:
filter_ = (obj[prop_name] == prop) if not isinstance(prop, str) \
else obj[prop_name].like(prop)
filters.append(filter_)
return filters
def query(cls, filters={}):
filters = cls._parse_filters(filters, cls, [])
return cls._session.query(cls).filter(*filters)
|
Alquimia
|
/alquimia-0.7.2.tar.gz/alquimia-0.7.2/alquimia/modelmeta.py
|
modelmeta.py
|
# This file is part of alquimia.
# alquimia is free software: you can redistribute it and/or modify
# it under the terms of the GNU Lesser General Public License as published by
# the Free Software Foundation, either version 3 of the License, or
# (at your option) any later version.
# This program is distributed in the hope that it will be useful,
# but WITHOUT ANY WARRANTY; without even the implied warranty of
# MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the
# GNU General Public License for more details.
# You should have received a copy of the GNU General Public License
# along with this program. If not, see <http://www.gnu.org/licenses/>.
import logging
from sqlalchemy.orm import relationship
from alquimia.utils import log
class OneToOneManyToManyError(Exception):
def __init__(self, model_name, rel_name, logger=logging):
message = '%s.%s is a one-to-one relationship but ' \
'was mapped as many-to-many!' % (model_name, rel_name)
log(logger, 'critical', message)
Exception.__init__(self, message)
class ModelsAtrrsReflect(dict):
def __init__(self, metadata, logger=logging, *args):
self._logger = logger
self._metadata = metadata
self._rels = {}
self._build(*args)
def _build_rel_instance(self, rel_name, table_name, update_kargs={}):
kwargs = {'cascade': 'all'}
kwargs.update(update_kargs)
self[table_name][rel_name] = relationship(rel_name, **kwargs)
def _add_rel(self, rel_type, rel_name, table_name, args={}):
self[table_name][rel_type].append(rel_name)
self[table_name]['relationships'].append(rel_name)
self._build_rel_instance(rel_name, table_name, args)
def _build_many_to_many_rel(self, rel_name, table_name, mtm_table):
if rel_name == table_name:
raise OneToOneManyToManyError(table_name, rel_name)
args = {'secondary': mtm_table}
self._add_rel('mtm', rel_name, table_name, args)
self._add_rel('mtm', table_name, rel_name, args)
def _build_many_to_one_rel(self, rel_name, table_name):
self._add_rel('mto', rel_name, table_name)
args = {'cascade': 'all,delete-orphan'}
self._add_rel('otm', table_name, rel_name, args)
def _build_one_to_one_rel(self, rel_name, table_name, id_column=None):
args = {'uselist': False, 'single_parent': True,
'cascade': 'all,delete-orphan'}
if id_column is not None:
args['remote_side'] = [id_column]
self._add_rel('oto', rel_name, table_name, args)
if not rel_name == table_name:
self._add_rel('oto', table_name, rel_name, args)
def _build_relationships(self, table):
for fk in table.foreign_keys:
rel_name = fk.column.table.name
id_column = table.c['id'] if rel_name == table.name else None
if id_column is not None or fk.parent.unique:
self._build_one_to_one_rel(rel_name, table.name, id_column)
else:
self._build_many_to_one_rel(rel_name, table.name)
mtm_tables = list(self._mtm_tables.values())
for mtm_table in mtm_tables:
mtm_rels = list(mtm_table.columns.keys())
table_rel = table.name+'_id'
if table_rel in mtm_rels:
mtm_rels.remove(table_rel)
rel_name = mtm_rels[0][:-3]
self._build_many_to_many_rel(rel_name, table.name,
mtm_table.name)
self._mtm_tables.pop(mtm_table.name)
def _keep_mtm_tables(self):
self._mtm_tables = {}
for table in self._metadata.tables.values():
fks = table.foreign_keys
if len(fks) == len(table.c) == 2:
is_pks = True
for fk in fks:
if not fk.column.primary_key:
is_pks = False
break
if not is_pks:
continue
self._mtm_tables[table.name] = table
def _init_attrs(self, table_name):
self[table_name] = {
'mtm': [],
'mto': [],
'oto': [],
'otm': [],
'relationships': [],
'columns': [],
'session': None
}
def _build(self, *args):
self._metadata.reflect()
self._keep_mtm_tables()
attrs = {}
tables = [table for table in self._metadata.tables.values() \
if table.name not in self._mtm_tables]
for table in tables:
self._init_attrs(str(table.name))
for table in tables:
if table.name not in self._mtm_tables:
self._build_relationships(table)
self[table.name]['__table__'] = table
|
Alquimia
|
/alquimia-0.7.2.tar.gz/alquimia-0.7.2/alquimia/models_attrs_reflect.py
|
models_attrs_reflect.py
|
# This file is part of alquimia.
# alquimia is free software: you can redistribute it and/or modify
# it under the terms of the GNU Lesser General Public License as published by
# the Free Software Foundation, either version 3 of the License, or
# (at your option) any later version.
# This program is distributed in the hope that it will be useful,
# but WITHOUT ANY WARRANTY; without even the implied warranty of
# MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the
# GNU General Public License for more details.
# You should have received a copy of the GNU General Public License
# along with this program. If not, see <http://www.gnu.org/licenses/>.
from sqlalchemy.orm.exc import DetachedInstanceError
class AlquimiaModel(object):
def __init__(self, **kwargs):
if 'id' in kwargs:
raise Exception("Can't add objects with id!")
for prop_name, prop in kwargs.iteritems():
if isinstance(prop, dict):
self[prop_name] = type(self)[prop_name].model(**prop)
elif isinstance(prop, list):
self[prop_name] = [type(self)[prop_name].model(**prop_) \
if isinstance(prop_, dict) else prop_ for prop_ in prop]
else:
self[prop_name] = prop
self._session.add(self)
self._current_pos = 0
def __setitem__(self, item, value):
self._check_attr(item)
setattr(self, item, value)
def __getitem__(self, item):
self._check_attr(item)
try:
return getattr(self, item)
except DetachedInstanceError:
self._session.add(self)
return getattr(self, item)
def __repr__(self):
return repr(self.todict())
def __iter__(self):
return self
def next(self):
self._current_pos += 1
if self._current_pos >= len(self.keys()):
self._current_pos = 0
raise StopIteration
else:
return self.keys()[self._current_pos - 1]
def _check_attr(self, attr_name):
if not attr_name in type(self):
raise TypeError("'%s' is not a valid %s attribute!" %
(attr_name, type(self).__name__))
def todict(self, rec_stack=None):
if rec_stack is None:
rec_stack = []
if self in rec_stack:
return None
dict_ = {}
rec_stack.append(self)
for prop_name, prop in self.items():
if not prop_name.endswith('_id'):
if isinstance(prop, list):
propl = []
for each in prop:
each = each.todict(rec_stack)
if each is not None:
propl.append(each)
prop = propl if propl else None
elif isinstance(prop, AlquimiaModel):
prop = prop.todict(rec_stack)
if prop is not None:
dict_[prop_name] = prop
return dict_
def has_key(self, key):
self._check_attr(key)
return hasattr(self, key)
def keys(self):
return type(self).keys()
def items(self):
return [(k, self[k]) for k in self.keys()]
def remove(self):
self._session.delete(self)
self._session.commit()
def save(self):
self._session.commit()
|
Alquimia
|
/alquimia-0.7.2.tar.gz/alquimia-0.7.2/alquimia/model.py
|
model.py
|
# This file is part of alquimia.
# alquimia is free software: you can redistribute it and/or modify
# it under the terms of the GNU Lesser General Public License as published by
# the Free Software Foundation, either version 3 of the License, or
# (at your option) any later version.
# This program is distributed in the hope that it will be useful,
# but WITHOUT ANY WARRANTY; without even the implied warranty of
# MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the
# GNU General Public License for more details.
# You should have received a copy of the GNU General Public License
# along with this program. If not, see <http://www.gnu.org/licenses/>.
import logging
from sqlalchemy import create_engine
from sqlalchemy.ext.declarative import declarative_base
from sqlalchemy.orm import sessionmaker
from sqlalchemy.orm.relationships import RelationshipProperty
from sqlalchemy.orm import ColumnProperty
from alquimia.model import AlquimiaModel
from alquimia.modelmeta import AlquimiaModelMeta
from alquimia.models_attrs import ModelsAttributes
from alquimia.models_attrs_reflect import ModelsAtrrsReflect
from alquimia import DATA_TYPES
class AlquimiaModels(dict):
def __init__(self, db_url, dict_=None, data_types=DATA_TYPES,
create=False, logger=logging):
engine = create_engine(db_url)
base_model = declarative_base(engine, metaclass=AlquimiaModelMeta,
cls=AlquimiaModel, constructor=AlquimiaModel.__init__)
self._session_class = sessionmaker(engine)
self._session = self._session_class()
self.metadata = base_model.metadata
if dict_ is not None:
attrs = ModelsAttributes(dict_, self.metadata, data_types, logger)
else:
attrs = ModelsAtrrsReflect(self.metadata, logger)
self._build(base_model, attrs)
if create:
self.metadata.create_all()
def _build(self, base_model, models_attrs):
models = {}
for model_name, attrs in models_attrs.iteritems():
attrs.update({'_session': self._session})
model = type(model_name, (base_model,), attrs)
models[model_name] = model
for model in models.values():
model.__mapper__.relationships
for attr_name, attr in model.iteritems():
if isinstance(attr.prop, RelationshipProperty):
setattr(attr, 'model', models[attr_name])
else:
model.columns.append(attr_name)
self.update(models)
def clean(self):
self._session.expunge_all()
|
Alquimia
|
/alquimia-0.7.2.tar.gz/alquimia-0.7.2/alquimia/models.py
|
models.py
|
AltAnalyze
=======================
AltAnalyze is an extremely user-friendly and open-source toolkit that can be used for a broad range of genomics analyses. These analyses include the direct processing of raw RNASeq or microarray data files, advanced methods for single-cell population discovery with ICGS, differential expression analyses, analysis of alternative splicing/promoter/polyadenylation and advanced isoform function prediction analysis (protein, domain and microRNA targeting). Multiple advanced visualization tools and à la carte analysis methods are supported in AltAnalyze (e.g., network, pathway, splicing graph). AltAnalyze is compatible with various data inputs for RNASeq data (FASTQ, BAM, BED), microarray platforms (Gene 1.0, Exon 1.0, junction and 3' arrays). This software requires no advanced knowledge of bioinformatics programs or scripting or advanced computer hardware. User friendly `videos <https://www.google.com/search?q=altanalyze&tbm=vid&cad=h>`_, `online tutorials <https://github.com/nsalomonis/altanalyze/wiki/Tutorials>`_ and `blog posts <http://altanalyze.blogspot.com/>`_ are also available.
AltAnalyze documentation can be found on `Read The Docs <http://altanalyze.readthedocs.io/en/latest/>`_ and stand-alone archives are provided at `sourceforge <https://sourceforge.net/projects/altanalyze/files/>`_ as well as at `GitHub <https://github.com/nsalomonis/altanalyze>`_.
.. image:: https://raw.githubusercontent.com/wiki/nsalomonis/altanalyze/images/AltAnalyzeOverview.gif
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/README.rst
|
README.rst
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"""This module contains fairly generic methods for reading different GO-Elite gene relationship
files, user input and denominator files, and summarizing quantitative and gene annotation data."""
import sys, string
import os.path
import unique
import math
from stats_scripts import statistics
try: from import_scripts import OBO_import
except Exception: pass
import export
import re
###### File Import Functions ######
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv" or ".owl" in entry or ".gpml" in entry or ".xml" in entry or ".gmt" in entry or ".tab" in entry: dir_list2.append(entry)
return dir_list2
###### Classes ######
class GrabFiles:
def setdirectory(self,value):
self.data = value
def display(self):
print self.data
def searchdirectory(self,search_term):
#self is an instance while self.data is the value of the instance
all_matching,file_dir,file = getDirectoryFiles(self.data,str(search_term))
#if len(file)<1: print self.data,search_term,'not found', filepath(self.data)
return file_dir,file
def getAllFiles(self,search_term):
#self is an instance while self.data is the value of the instance
all_matching,file_dir,file = getDirectoryFiles(self.data,str(search_term))
#if len(file)<1: print search_term,'not found'
return all_matching
def getMatchingFolders(self,search_term):
dir_list = unique.read_directory(self.data); matching = ''
root_dir = filepath('')
for folder in dir_list:
if search_term in folder and '.' not in folder:
matching = filepath(self.data[1:]+'/'+folder)
if root_dir not in matching:
matching = filepath(root_dir +'/'+ matching) ### python bug????
return matching
def getDirectoryFiles(import_dir, search_term):
exact_file = ''; exact_file_dir=''; all_matching=[]
if '.txt' in import_dir:
import_dir = export.findParentDir(import_dir)
dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
for data in dir_list: #loop through each file in the directory to output results
present = verifyFile(import_dir+'/'+data) ### Check to see if the file is present in formatting the full file_dir
if present == True:
affy_data_dir = import_dir+'/'+data
else: affy_data_dir = import_dir[1:]+'/'+data
if search_term in affy_data_dir and '._' not in affy_data_dir:
if 'version.txt' not in affy_data_dir: exact_file_dir = affy_data_dir; exact_file = data; all_matching.append(exact_file_dir)
return all_matching, exact_file_dir,exact_file
def verifyFile(filename):
present = False
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): present = True; break
except Exception: present = False
return present
class GeneRelationships:
def __init__(self,uid,gene,terms,uid_system,gene_system):
self._uid = uid; self._gene = gene; self._terms = terms
self._uid_system = uid_system; self._gene_system = gene_system
def UniqueID(self): return self._uid
def GeneID(self): return self._gene
def GOIDs(self): return self._terms
def GOIDInts(self):
goid_list = self._terms; goid_list_int = []
for goid in goid_list: goid_list_int.append(int(goid))
return goid_list_int
def GOIDStrings(self):
goid_list = self._terms; goid_list_str = []
for goid in goid_list:
if ':' not in goid: goid_list_str.append('GO:'+goid)
return goid_list_str
def MAPPs(self):
mapp_list = self._terms
return mapp_list
def UIDSystem(self): return self.uid_system
def GeneIDSystem(self): return self._gene_system
def setUIDValues(self,uid_values):
self._uid_values = uid_values
def UIDValues(self): return self._uid_values
def Report(self):
output = self.UniqueID()
return output
def __repr__(self): return self.Report()
class GeneAnnotations:
def __init__(self,gene,symbol,name,gene_system):
self._gene = gene; self._symbol = symbol; self._name = name;
self._gene_system = gene_system
def GeneID(self): return self._gene
def Symbol(self): return self._symbol
def SymbolLower(self): return string.lower(self._symbol)
def Description(self): return self._name
def GeneIDSystem(self): return self._gene_system
def Report(self):
output = self.GeneID()+'|'+self.Symbol()
return output
def __repr__(self): return self.Report()
class RedundantRelationships:
def __init__(self,redundant_names,inverse_names):
self._redundant_names = redundant_names; self._inverse_names = inverse_names
def RedundantNames(self):
redundant_with_terms = string.join(self._redundant_names,'|')
return redundant_with_terms
def InverseNames(self):
redundant_with_terms = string.join(self._inverse_names,'|')
return redundant_with_terms
def Report(self):
output = self.RedundantNames()+'|'+self.InverseNames()
return output
def __repr__(self): return self.Report()
def cleanUpLine(line):
data = string.replace(line,'\n','')
data = string.replace(data,'\c','')
data = string.replace(data,'\r','')
data = string.replace(data,'"','')
return data
def importGeneric(filename):
fn=filepath(filename); key_db = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
key_db[t[0]] = t[1:]
return key_db
def importGenericDB(filename):
try:
key_db=collections.OrderedDict() ### Retain order if possible
except Exception:
key_db={}
fn=filepath(filename); x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#': x=x
elif x==0:
x=1
headers = t
else:
try: key_db[t[0]].append(t[1:])
except Exception: key_db[t[0]] = [t[1:]]
return key_db, headers
###### Begin Gene-Relationship Parsing ######
def importGeneData(species_code,mod):
if 'export' in mod: export_status,mod = mod
else: export_status = 'no'
###Pass in species_code, mod
program_type,database_dir = unique.whatProgramIsThis()
gene_import_dir = '/'+database_dir+'/'+species_code+'/gene'
g = GrabFiles(); g.setdirectory(gene_import_dir)
filedir,filename = g.searchdirectory(mod) ###Identify gene files corresponding to a particular MOD
system_type = string.replace(filename,'.txt','')
fn=filepath(filedir); gene_annotations={}; x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x == 0: x = 1
else:
try:
gene = t[0]; symbol = t[1]; name = t[2]
except:
gene = t[0]; symbol = t[0]; name = t[0]
else:
s = GeneAnnotations(gene, symbol, name, system_type)
gene_annotations[gene] = s
if export_status == 'export':
export_dir = database_dir+'/'+species_code+'/uid-gene/'+mod+'-Symbol'+'.txt'
data = export.ExportFile(export_dir)
data.write('GeneID\tSymbol\n')
for gene in gene_annotations:
s = gene_annotations[gene]
if len(s.Symbol())>0:
data.write(gene+'\t'+s.Symbol()+'\n')
data.close()
else:
return gene_annotations
def buildNestedAssociations(species_code):
print "Nested gene association, not yet build for",species_code
print "Proceeding to build nested associations for this species\n"
export_databases = 'no'; genmapp_mod = 'Ensembl' ### default
import GO_Elite; system_codes,source_types,mod_types = GO_Elite.getSourceData()
OBO_import.buildNestedOntologyAssociations(species_code,export_databases,mod_types,genmapp_mod)
def importGeneToOntologyData(species_code,mod,gotype,ontology_type):
program_type,database_dir = unique.whatProgramIsThis()
if gotype == 'nested':
geneGO_import_dir = '/'+database_dir+'/'+species_code+'/nested'
if ontology_type == 'GeneOntology': ontology_type = 'GO.'
mod += '_to_Nested'
elif gotype == 'null':
geneGO_import_dir = '/'+database_dir+'/'+species_code+'/gene-go'
gg = GrabFiles(); gg.setdirectory(geneGO_import_dir)
try: filedir,file = gg.searchdirectory(mod+'-'+ontology_type) ###Identify gene files corresponding to a particular MOD
except Exception:
if gotype == 'nested':
buildNestedAssociations(species_code)
filedir,file = gg.searchdirectory(mod+'-'+ontology_type)
global gene_to_go; x=0
fn=filepath(filedir); gene_to_go={}
for line in open(fn,'rU').xreadlines():
if gotype == 'nested' and x==0: x = 1
else:
if 'Ontology' not in line and 'ID' not in line: ### Header included in input from AP or BioMart
#data = cleanUpLine(line)
data = line.strip()
t = string.split(data,'\t')
if len(t)>1:
try: gene = t[0]; goid = t[1]
except IndexError: print [line],t, 'Ontology-gene relationship not valid...please clean up',filedir;sys.exit()
goid = string.replace(goid,'GO:','') ### Included in input from AP
### If supplied without proceeding zeros
if len(goid)<7 and len(goid)>0:
extended_goid=goid
while len(extended_goid)< 7: extended_goid = '0'+extended_goid
goid = extended_goid
if len(gene)>0 and len(goid)>0:
if ':' not in goid:
goid = 'GO:'+goid
try: gene_to_go[gene].append(goid)
except KeyError: gene_to_go[gene]= [goid]
return gene_to_go
def importGeneMAPPData(species_code,mod):
program_type,database_dir = unique.whatProgramIsThis()
geneMAPP_import_dir = '/'+database_dir+'/'+species_code+'/gene-mapp'
gm = GrabFiles(); gm.setdirectory(geneMAPP_import_dir)
filedir,file = gm.searchdirectory(mod) ### Identify gene files corresponding to a particular MOD
global gene_to_mapp; x = True
fn=filepath(filedir); gene_to_mapp={}
for line in open(fn,'rU').xreadlines():
#data = cleanUpLine(line)
data = line.strip()
if x: x=False
else:
t = string.split(data,'\t')
gene = t[0]; mapp = t[2]
try: gene_to_mapp[gene].append(mapp)
except KeyError: gene_to_mapp[gene]= [mapp]
return gene_to_mapp
def exportCustomPathwayMappings(gene_to_custom,mod,system_codes,custom_sets_folder):
if '.txt' in custom_sets_folder:
export_dir = custom_sets_folder
else:
export_dir = custom_sets_folder+'/CustomGeneSets/custom_gene_set.txt'
#print 'Exporting:',export_dir
try: data = export.ExportFile(export_dir)
except Exception: data = export.ExportFile(export_dir[1:])
for system_code in system_codes:
if system_codes[system_code] == mod: mod_code = system_code
data.write('GeneID\t'+mod+'\tPathway\n'); relationships=0
gene_to_custom = eliminate_redundant_dict_values(gene_to_custom)
for gene in gene_to_custom:
for pathway in gene_to_custom[gene]:
values = string.join([gene,mod_code,pathway],'\t')+'\n'
data.write(values); relationships+=1
data.close()
#print relationships,'Custom pathway-to-ID relationships exported...'
def exportNodeInteractions(pathway_db,mod,custom_sets_folder):
import GO_Elite
system_codes,source_types,mod_types = GO_Elite.getSourceData()
export_dir = custom_sets_folder+'/Interactomes/interactions.txt'
print 'Exporting:',export_dir
try: data = export.ExportFile(export_dir)
except Exception: data = export.ExportFile(export_dir[1:])
for system_code in system_codes:
if system_codes[system_code] == mod: mod_code = system_code
try: gene_to_symbol_db = getGeneToUid(species_code,('hide',mod+'-Symbol.txt')); #print mod_source, 'relationships imported.'
except Exception: gene_to_symbol_db={}
data.write('Symbol1\tInteractionType\tSymbol2\tGeneID1\tGeneID2\tPathway\n'); relationships=0
for pathway_id in pathway_db:
wpd = pathway_db[pathway_id]
for itd in wpd.Interactions():
gi1 = itd.GeneObject1()
gi2 = itd.GeneObject2()
if len(gene_to_symbol_db)>0:
try:
if gi1.ModID()[0] in gene_to_symbol_db:
symbol = gene_to_symbol_db[gi1.ModID()[0]][0]
if len(symbol)>0:
gi1.setLabel(symbol) ### Replace the WikiPathways user annnotated symbol with a MOD symbol
except Exception:
None
try:
if gi2.ModID()[0] in gene_to_symbol_db:
symbol = gene_to_symbol_db[gi2.ModID()[0]][0]
if len(symbol)>0:
gi2.setLabel(symbol) ### Replace the WikiPathways user annnotated symbol with a MOD symbol
except Exception:
None
try:
values = string.join([gi1.Label(),itd.InteractionType(),gi2.Label(),gi1.ModID()[0],gi2.ModID()[0],wpd.Pathway()],'\t')
relationships+=1
try:
values = cleanUpLine(values)+'\n' ### get rid of any end-of lines introduced by the labels
data.write(values)
except Exception:
try: ### Occurs when 'ascii' codec can't encode characters due to UnicodeEncodeError
values = string.join(['',itd.InteractionType(),gi2.Label(),gi1.ModID()[0],gi2.ModID()[0],wpd.Pathway()],'\t')
values = cleanUpLine(values)+'\n' ### get rid of any end-of lines introduced by the labels
data.write(values)
except Exception:
values = string.join([gi1.Label(),itd.InteractionType(),'',gi1.ModID()[0],gi2.ModID()[0],wpd.Pathway()],'\t')
values = cleanUpLine(values)+'\n' ### get rid of any end-of lines introduced by the labels
try: data.write(values)
except Exception: None ### Occurs due to illegal characters
except AttributeError,e:
#print e
null=[] ### Occurs if a MODID is not present for one of the nodes
data.close()
print relationships,'Interactions exported...'
def importGeneCustomData(species_code,system_codes,custom_sets_folder,mod):
print 'Importing custom pathway relationships...'
#print 'Trying to import text data'
gene_to_custom = importTextCustomData(species_code,system_codes,custom_sets_folder,mod)
#print len(gene_to_custom)
#print 'Trying to import gmt data'
gmt_data = parseGMT(custom_sets_folder)
#print 'Trying to import gpml data'
gpml_data,pathway_db = parseGPML(custom_sets_folder)
#print 'Trying to import biopax data'
biopax_data = parseBioPax(custom_sets_folder)
#print 'Unifying gene systems for biopax'
gene_to_BioPax = unifyGeneSystems(biopax_data,species_code,mod); #print len(gene_to_BioPax)
#print 'Unifying gene systems for WP'
gene_to_WP = unifyGeneSystems(gpml_data,species_code,mod); #print len(gene_to_WP)
#print 'Unifying gene systems for gmt'
gene_to_GMT = unifyGeneSystems(gmt_data,species_code,mod); #print len(gene_to_WP)
#print 'Combine WP-biopax'
gene_to_xml = combineDBs(gene_to_WP,gene_to_BioPax)
#print 'Combine xml-text'
gene_to_custom = combineDBs(gene_to_xml,gene_to_custom)
#print 'Combine gmt-other'
gene_to_custom = combineDBs(gene_to_GMT,gene_to_custom)
exportCustomPathwayMappings(gene_to_custom,mod,system_codes,custom_sets_folder)
if len(gene_to_WP)>0: ### Export all pathway interactions
try: exportNodeInteractions(pathway_db,mod,custom_sets_folder)
except Exception: null=[]
"""
### Combine WikiPathway associations with the custom
try: gene_to_mapp = importGeneMAPPData(species_code,mod)
except Exception: gene_to_mapp = {}
for gene in gene_to_mapp:
for mapp in gene_to_mapp[gene]:
try: gene_to_custom[gene].append(mapp)
except KeyError: gene_to_custom[gene]= [mapp]"""
return gene_to_custom
def importTextCustomData(species_code,system_codes,custom_sets_folder,mod):
program_type,database_dir = unique.whatProgramIsThis()
gm = GrabFiles(); gm.setdirectory(custom_sets_folder); system = None
filedirs = gm.getAllFiles('.txt')
global gene_to_custom
gene_to_custom={}
file_gene_to_custom={}
for filedir in filedirs:
try:
file = string.split(filedir,'/')[-1]
print "Reading custom gene set",filedir
fn=filepath(filedir); x = 1
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if x==0: x=1
else:
x+=1
t = string.split(data,'\t')
try:
gene = t[0]; mapp = t[2]; system_code = t[1]
if system_code in system_codes: system = system_codes[system_code]
else:
if x == 3: print system_code, "is not a recognized system code. Skipping import of",file; break
except Exception:
if len(t)>0:
gene = t[0]
if len(t)==1: ### Hence, no system code is provided and only one gene-set is indicated per file
source_data = predictIDSource(t[0],system_codes)
if len(source_data)>0: system = source_data; mapp = file
else:
if x == 3: print file, 'is not propperly formatted (skipping import of relationships)'; break
elif len(t)==2:
if t[1] in system_codes: system = system_codes[t[1]]; mapp = file ### Hence, system code is provided by only one gene-set is indicated per file
else:
source_data = predictIDSource(t[0],system_codes)
if len(source_data)>0: system = source_data; mapp = t[1]
else:
if x == 3: print file, 'is not propperly formatted (skipping import of relationships)'; break
else: continue ### Skip line
try: file_gene_to_custom[gene].append(mapp)
except KeyError: file_gene_to_custom[gene]= [mapp]
#print [system, mod, len(file_gene_to_custom)]
### If the system code is not the MOD - Convert to the MOD
if (system != mod) and (system != None):
mod_source = 'hide',mod+'-'+system+'.txt'
try: gene_to_source_id = getGeneToUid(species_code,mod_source)
except Exception: print mod_source,'relationships not found. Skipping import of',file; break
source_to_gene = OBO_import.swapKeyValues(gene_to_source_id)
if system == 'Symbol': source_to_gene = lowerAllIDs(source_to_gene)
for source_id in file_gene_to_custom:
original_source_id = source_id ### necessary when Symbol
if system == 'Symbol': source_id = string.lower(source_id)
if source_id in source_to_gene:
for gene in source_to_gene[source_id]:
try: gene_to_custom[gene] += file_gene_to_custom[original_source_id]
except Exception: gene_to_custom[gene] = file_gene_to_custom[original_source_id]
else:
for gene in file_gene_to_custom:
try: gene_to_custom[gene] += file_gene_to_custom[gene]
except Exception: gene_to_custom[gene] = file_gene_to_custom[gene]
except Exception:
print file, 'not formatted propperly!'
file_gene_to_custom={} ### Clear this file specific object
return gene_to_custom
def grabFileRelationships(filename):
filename = string.replace(filename,'.txt','')
system1,system2 = string.split(filename,'-')
return system1,system2
def importUidGeneSimple(species_code,mod_source):
program_type,database_dir = unique.whatProgramIsThis()
geneUID_import_dir = '/'+database_dir+'/'+species_code+'/uid-gene'
ug = GrabFiles(); ug.setdirectory(geneUID_import_dir)
filedir,file = ug.searchdirectory(mod_source) ### Identify gene files corresponding to a particular MOD
gene_to_uid={}; x = 0
fn=filepath(filedir)
for line in open(fn,'rU').xreadlines():
#data = cleanUpLine(line)
data = line.strip()
if x==0: x=1
else:
t = string.split(data,'\t')
gene = t[0]; uid = t[1]
try: gene_to_uid[gene].append(uid)
except KeyError: gene_to_uid[gene]= [uid]
return gene_to_uid
def augmentEnsemblGO(species_code):
ontology_type = 'GeneOntology'
entrez_ens = importUidGeneSimple(species_code,'EntrezGene-Ensembl')
try: ens_go=importGeneToOntologyData(species_code,'Ensembl','null',ontology_type)
except Exception: ens_go = {}
try: entrez_go=importGeneToOntologyData(species_code,'Entrez','null',ontology_type)
except Exception: entrez_go = {}
ens_go_translated = {}
for entrez in entrez_go:
if entrez in entrez_ens:
if len(entrez_ens[entrez])<3: ### Limit bad associations
#print entrez,entrez_ens[entrez];kill
for ens in entrez_ens[entrez]: ens_go_translated[ens] = entrez_go[entrez]
### Add these relationships to the original
for ens in ens_go_translated:
try: ens_go[ens] = unique.unique(ens_go[ens] + ens_go_translated[ens])
except Exception: ens_go[ens] = ens_go_translated[ens]
program_type,database_dir = unique.whatProgramIsThis()
export_dir = database_dir+'/'+species_code+'/gene-go/Ensembl-GeneOntology.txt'
print 'Exporting augmented:',export_dir
try: data = export.ExportFile(export_dir)
except Exception: data = export.ExportFile(export_dir[1:])
data.write('GeneID'+'\tGO ID\n')
for gene in ens_go:
for goid in ens_go[gene]:
values = string.join([gene,goid],'\t')+'\n'
data.write(values)
data.close()
### Build Nested
export_databases = 'no'; genmapp_mod = 'Ensembl'
full_path_db,path_id_to_goid,null = OBO_import.buildNestedOntologyAssociations(species_code,export_databases,['Ensembl'],genmapp_mod)
def importOntologyUIDGeneData(species_code,mod_source,gene_to_go,denominator_source_ids):
program_type,database_dir = unique.whatProgramIsThis()
geneUID_import_dir = '/'+database_dir+'/'+species_code+'/uid-gene'
ug = GrabFiles(); ug.setdirectory(geneUID_import_dir)
#print "begining to parse",mod_source, 'from',geneGO_import_dir
filedir,filename = ug.searchdirectory(mod_source) ###Identify gene files corresponding to a particular MOD
fn=filepath(filedir); uid_to_go={}; count=0; x=0
uid_system,gene_system = grabFileRelationships(filename)
for line in open(fn,'rU').xreadlines(): count+=1
original_increment = int(count/10); increment = original_increment
for line in open(fn,'rU').xreadlines():
#data = cleanUpLine(line); x+=1
data = line.strip(); x+=1
if program_type == 'GO-Elite':
if x == increment: increment+=original_increment; print '*',
t = string.split(data,'\t')
uid = t[1]; gene = t[0]
try:
if len(denominator_source_ids)>0:
null=denominator_source_ids[uid] ### requires that the source ID be in the list of analyzed denominators
goid = gene_to_go[gene]
y = GeneRelationships(uid,gene,goid,uid_system,gene_system)
try: uid_to_go[uid].append(y)
except KeyError: uid_to_go[uid] = [y]
except Exception: null=[]
try:
if len(denominator_source_ids)>0:
null=denominator_source_ids[uid] ### requires that the source ID be in the list of analyzed denominators
except Exception: null=[]
return uid_to_go,uid_system
def importGeneSetUIDGeneData(species_code,mod_source,gene_to_mapp,denominator_source_ids):
program_type,database_dir = unique.whatProgramIsThis()
geneUID_import_dir = '/'+database_dir+'/'+species_code+'/uid-gene'
ug = GrabFiles(); ug.setdirectory(geneUID_import_dir)
#print "begining to parse",mod_source, 'from',geneGO_import_dir
filedir,filename = ug.searchdirectory(mod_source) ###Identify gene files corresponding to a particular MOD
fn=filepath(filedir); uid_to_mapp={}; count=0; x=0
uid_system,gene_system = grabFileRelationships(filename)
for line in open(fn,'rU').xreadlines(): count+=1
original_increment = int(count/10); increment = original_increment
for line in open(fn,'rU').xreadlines():
#data = cleanUpLine(line); x+=1
data = line.strip(); x+=1
if program_type == 'GO-Elite':
if x == increment: increment+=original_increment; print '*',
t = string.split(data,'\t')
uid = t[1]; gene = t[0]
try:
if len(denominator_source_ids)>0:
null=denominator_source_ids[uid] ### requires that the source ID be in the list of analyzed denominators
mapp_name = gene_to_mapp[gene]
y = GeneRelationships(uid,gene,mapp_name,uid_system,gene_system)
try: uid_to_mapp[uid].append(y)
except KeyError: uid_to_mapp[uid] = [y]
except Exception: null=[]
return uid_to_mapp,uid_system
def eliminate_redundant_dict_values(database):
db1={}
for key in database: list = unique.unique(database[key]); list.sort(); db1[key] = list
return db1
def getGeneToUid(species_code,mod_source,display=True):
if 'hide' in mod_source: show_progress, mod_source = mod_source
elif display==False: show_progress = 'no'
else: show_progress = 'yes'
program_type,database_dir = unique.whatProgramIsThis()
import_dir = '/'+database_dir+'/'+species_code+'/uid-gene'
ug = GrabFiles(); ug.setdirectory(import_dir)
filedir,filename = ug.searchdirectory(mod_source) ###Identify gene files corresponding to a particular MOD
fn=filepath(filedir); gene_to_uid={}; count = 0; x=0
uid_system,gene_system = grabFileRelationships(filename)
for line in open(fn,'r').xreadlines(): count+=1
original_increment = int(count/10); increment = original_increment
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line); x+=1
if x == increment and show_progress == 'yes':
increment+=original_increment; print '*',
t = string.split(data,'\t')
uid = t[1]; gene = t[0]
try: gene_to_uid[gene].append(uid)
except KeyError: gene_to_uid[gene] = [uid]
gene_to_uid = eliminate_redundant_dict_values(gene_to_uid)
return gene_to_uid
def getGeneToUidNoExon(species_code,mod_source):
gene_to_uid = getGeneToUid(species_code,mod_source)
try:
probeset_db = simpleExonImporter(species_code); filtered_db={}
for gene in gene_to_uid:
for uid in gene_to_uid[gene]:
try: probeset_db[uid]
except KeyError: ### Only inlcude probesets not in the exon database
try: filtered_db[gene].append(uid)
except KeyError: filtered_db[gene] = [uid]
return filtered_db
except Exception: return gene_to_uid
def simpleExonImporter(species_code):
filename = 'AltDatabase/'+species_code+'/exon/'+species_code+'_Ensembl_probesets.txt'
fn=filepath(filename); probeset_db={}
for line in open(fn,'rU').xreadlines():
#data = cleanUpLine(line)
data = line.strip()
data = string.split(data,'\t')
probeset_db[data[0]]=[]
return probeset_db
def predictIDSource(id,system_codes):
au = ['A','B','C','D','E','F','G','H','I','J','K','L','M','N','O','P','Q','R','S','T','U','V','W','X','Y','Z']
nm = ['1','2','3','4','5','6','7','8','9']
affy_suffix = '_at'; ensembl_prefix = 'ENS'; source_data = ''; id_type = 'Symbol'; id_types={}
if len(id)>3:
if affy_suffix == id[-3:]: id_type = 'Affymetrix'
elif ensembl_prefix == id[:3]: id_type = 'Ensembl'
elif id[2] == '.': id_type = 'Unigene'
elif (id[0] in au and id[1] in nm) or '_' in id: id_type = 'UniProt'
else:
try:
int_val = int(id)
if len(id) == 7: id_type = 'Affymetrix' ###All newer Affymetrix transcript_cluster_ids and probesets (can be mistaken for EntrezGene IDs)
else: id_type = 'EntrezGene'
except ValueError: null = []
try: id_types[id_type]+=1
except Exception: id_types[id_type]=1
###If the user changes the names of the above id_types, we need to verify that the id_type is in the database
if len(id_type)>0 and len(id_types)>0:
id_type_count=[]
for i in id_types:
id_type_count.append((id_types[i],i))
id_type_count.sort()
#print id_type_count
id_type = id_type_count[-1][-1]
for code in system_codes:
if system_codes[code] == id_type: source_data = id_type
return source_data
def predictIDSourceSimple(id):
au = ['A','B','C','D','E','F','G','H','I','J','K','L','M','N','O','P','Q','R','S','T','U','V','W','X','Y','Z']
nm = ['1','2','3','4','5','6','7','8','9']
affy_suffix = '_at'; ensembl_prefix = 'ENS'; source_data = ''; id_type = 'Sy'; id_types={}
if len(id)>3:
if affy_suffix == id[-3:]: id_type = 'X'
elif ensembl_prefix == id[:3]:
if ' ' in id:
id_type = 'En:Sy'
else:
id_type = 'En'
elif id[2] == '.': id_type = 'Ug'
#elif (id[0] in au and id[1] in nm) or '_' in id: id_type = 'S'
else:
try:
int_val = int(id)
if len(id) == 7: id_type = 'X' ###All newer Affymetrix transcript_cluster_ids and probesets (can be mistaken for EntrezGene IDs)
else: id_type = 'L'
except ValueError: null = []
if id_type != 'En' and ensembl_prefix in id and ':' in id:
prefix = string.split(id,':')[0]
if ensembl_prefix not in prefix and ' ' in id:
id_type = '$En:Sy'
else:
id_type = 'Ae'
return id_type
def addNewCustomSystem(filedir,system,save_option,species_code):
print 'Adding new custom system (be patient)' ### Print statement here forces the status window to appear quicker otherwise stalls
gene_annotations={}
if 'update' in save_option:
print "Importing existing",species_code,system,"relationships."
gene_annotations = importGeneData(species_code,mod)
fn=filepath(filedir); length3=0; lengthnot3=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if len(t[0])>0:
try:
### Allow the user to only include one column here
gene = t[0]
try: symbol = t[1]
except Exception: symbol = gene
try: name = ''
except Exception: name = ''
s = GeneAnnotations(gene,symbol,name,'')
gene_annotations[gene] = s
except Exception:
print 'Unexpected error in the following line'; print t, filedir;kill
if len(gene_annotations)>0:
print 'Writing new annotation file:',species_code,system
export_dir = 'Databases/'+species_code+'/gene/'+system+'.txt'
data = export.ExportFile(export_dir)
data.write('UID\tSymbol\tDescription\n')
for gene in gene_annotations:
s = gene_annotations[gene]
data.write(string.join([gene,s.Symbol(),s.Description()],'\t')+'\n')
data.close()
return 'exported'
else: return 'not-exported'
def importGeneSetsIntoDatabase(source_file,species_code,mod):
source_filename = export.findFilename(source_file)
export.deleteFolder('BuildDBs/temp') ### Delete any previous data
destination_dir = filepath('BuildDBs/temp/'+source_filename)
export.customFileCopy(source_file,destination_dir)
#print destination_dir
custom_sets_folder = export.findParentDir(destination_dir)
import GO_Elite; system_codes,source_types,mod_types = GO_Elite.getSourceData()
gene_to_custom = importGeneCustomData(species_code,system_codes,custom_sets_folder,mod)
return gene_to_custom
def addNewCustomRelationships(filedir,relationship_file,save_option,species_code):
print 'Adding new custom relationships (be patient)' ### Print statement here forces the status window to appear quicker otherwise stalls
relationship_filename = export.findFilename(relationship_file)
mod,data_type = string.split(relationship_filename,'-')
mod_id_to_related={}
if 'update' in save_option or '.txt' not in filedir:
if 'gene-mapp' in relationship_file:
if '.txt' not in filedir: ### Then these are probably gmt, gpml or owl
mod_id_to_related = importGeneSetsIntoDatabase(filedir,species_code,mod)
else:
mod_id_to_related = importGeneMAPPData(species_code,mod)
elif 'gene-go' in relationship_file:
mod_id_to_related=importGeneToOntologyData(species_code,mod,'null',data_type)
else: mod_id_to_related=importUidGeneSimple(species_code,mod+'-'+data_type+'.txt')
fn=filepath(filedir); length2=0; lengthnot2=0
if '.txt' in fn:
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
data_length = len(t)
### Check the length of the input file
if data_length==2 or data_length==3: #This can occur if users load a custom gene set file created by GO-Elite
length2+=1
if len(t[0])>0 and len(t[-1])>0:
if ',' in t[0]: keys = string.split(t[0],',') ### These can be present in the MGI phenotype ontology gene association files
else: keys = [t[0]]
for key in keys:
try: mod_id_to_related[key].append(t[-1])
except KeyError: mod_id_to_related[key] = [t[-1]]
else: lengthnot2+=1
else: length2+=1
if length2>lengthnot2:
###Ensure that the input file is almost completely 2 columns
print 'Writing new relationship file:',species_code,relationship_file
if 'Databases' in relationship_file:
export_dir = relationship_file ### Sometimes we just include the entire path for clarification
if 'mapp' in export_dir: data_type = 'MAPP'
else: data_type = 'Ontology'
elif data_type == 'MAPP': export_dir = 'Databases/'+species_code+'/gene-mapp/'+relationship_file+'.txt'
elif data_type == 'Ontology':
export_dir = 'Databases/'+species_code+'/gene-go/'+relationship_file+'.txt'
else: export_dir = 'Databases/'+species_code+'/uid-gene/'+relationship_file+'.txt'
if data_type == 'Ontology':
### To trigger a rebuild of the ontology nested, must deleted the existing nested for this ontology
nested_path = filepath(string.replace(relationship_file,'gene-go','nested'))
nested_path = string.replace(nested_path,'GeneOntology','GO')
obo=string.split(nested_path,'-')[-1]
nested_path = string.replace(nested_path,'-'+obo,'_to_Nested-'+obo) ### Will break if there is another '-' in the name
try: os.remove(nested_path)
except Exception: null=[]
data = export.ExportFile(export_dir)
data.write('UID\t\tRelated ID\n')
for mod_id in mod_id_to_related:
for related_id in mod_id_to_related[mod_id]:
if data_type == 'MAPP': ###Pathway files have 3 rather than 2 columns
data.write(string.join([mod_id,'',related_id],'\t')+'\n')
else: data.write(string.join([mod_id,related_id],'\t')+'\n')
data.close()
return 'exported'
else: return 'not-exported'
def importUIDsForMAPPFinderQuery(filedir,system_codes,return_uid_values):
fn=filepath(filedir); x=0; uid_list = {}; uid_value_db = {}; source_data = ''; value_len_list = []
system_code_found = 'no'; first_uid=''; first_line = 'no lines in file'; underscore = False
source_data_db={}; error=''; bad_alphanumerics=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
tnull = string.join(t,''); ### Indicates whether the line is blank - even if tabs are present
alphanumeric = string.join(re.findall(r"\w",data))
if x==0:
x=1; value_headers = t[1:]
else:
if len(tnull)>0:
if len(alphanumeric)==0: bad_alphanumerics+=1
first_uid = t[0]; first_line = t
x+=1; source_data_read = ''
uid = t[0]
uid = string.replace(uid,'---','') ###occurs for Affymetrix gene ID input
uids = string.split(uid,' /// ') ###occurs for Affymetrix gene IDs where there are multiple associations
for uid in uids:
if len(uid) > 0 and uid != ' ':
if uid[0]==' ': uid = uid[1:]
if uid[-1]==' ': uid = uid[:-1]
all_alphanumeric = re.findall(r"\w",uid)
if len(all_alphanumeric)>0: uid_list[uid]=[]
if len(t)>1: ###Therefore there is either gene values or system data associated with imported primary IDs
try:
system = t[1]
system = string.replace(system,' ','')
if system in system_codes:
#if len(source_data)<1: source_data = system_codes[t[1]]; system_code_found = 'yes'
source_data = system_codes[system]; system_code_found = 'yes'; source_data_read = source_data
if len(t)==2: values = [] ###Therefore, there is system code data but no associated values
else: values = t[2:]
else: values = t[1:]
value_len_list.append(len(values))
if len(values)>0:
if len(uid) > 0 and uid != ' ':
all_alphanumeric = re.findall(r"\w",uid)
if len(all_alphanumeric)>0: uid_value_db[uid] = values
if '_' in uid: underscore=True
except Exception:
source_data_read =''
if len(source_data_read)<1:
for uid in uids:
source_data_read = predictIDSource(uid,system_codes)
if len(source_data_read)>0: source_data = source_data_read
try: source_data_db[source_data_read]+=1
except Exception: source_data_db[source_data_read]=1
first_uid = string.replace(first_uid,'Worksheet','!!!!')
filenames = string.split(filedir,'/'); filename = filenames[-1]
if x==1:
error = 'No results in input file:'+filename
print error
elif '!!!!' in first_uid:
error = 'WARNING!!! There appears to be a formatting file issue with the file:\n"'+filename+'"\nPlease correct and re-run (should be tab-delimited text).'
elif len(source_data_db)>1:
#error = 'WARNING!!! There is more than one gene system (e.g., Ensembl and EntrezGene) in the file:\n"'+filename+'"\nPlease correct and re-run.'
sources = []
for s in source_data_db:
sources.append([source_data_db[s],s])
sources.sort(); source_data = sources[-1][1]
#print 'Using the system code:', source_data, "(multiple systems present)"
elif source_data == '':
error = 'WARNING!!! No System Code identified in:\n"'+filename+'"\nPlease provide in input text file and re-run.\n If System Code column present, the file format may be incorrect.'
try: error +='Possible system code: '+system+' not recognized.'
except Exception: None
elif x>0 and bad_alphanumerics>1:
error = 'WARNING!!! Invalid text file encoding found (invalid alphanumeric values). Please resave text file in a standard tab-delimited format'
if underscore and system == 'S':
error += '\nSwissProt IDs of the type P53_HUMAN (P04637) are not recognized as propper UniProt IDs. Consider using alternative compatible IDs (e.g., P04637).'
if return_uid_values == 'yes' and len(value_len_list)>0:
value_len_list.sort(); longest_value_list = value_len_list[-1]
for uid in uid_value_db:
values = uid_value_db[uid]
if len(values) != longest_value_list:
while len(values) != longest_value_list: values.append('')
if system_code_found == 'yes': value_headers = value_headers[1:]###Therfore, column #2 has system code data
return uid_list,uid_value_db,value_headers,error
elif return_uid_values == 'yes': return uid_list,uid_value_db,value_headers,error
else: return uid_list,source_data,error
def getAllDenominators(denom_search_dir,system_codes):
import mappfinder; denom_all_db={}
m = GrabFiles(); m.setdirectory(denom_search_dir)
if len(denom_search_dir)>0: denom_dir_list = mappfinder.readDirText(denom_search_dir)
else: denom_dir_list = []
for denominator_file in denom_dir_list:
gene_file_dir, gene_file = m.searchdirectory(denominator_file)
denom_gene_list,source_data_denom,error_message = importUIDsForMAPPFinderQuery(denom_search_dir+'/'+gene_file,system_codes,'no')
for i in denom_gene_list: denom_all_db[i]=[]
return denom_all_db
def grabNestedGeneToOntologyAssociations(species_code,mod,source_data,system_codes,denom_search_dir,ontology_type):
program_type,database_dir = unique.whatProgramIsThis()
global printout; printout = 'yes'
gene_annotations = importGeneData(species_code,mod)
### Filter source IDs for those in all user denominator files
denominator_source_ids = getAllDenominators(denom_search_dir,system_codes)
try: gene_to_go = importGeneToOntologyData(species_code,mod,'nested',ontology_type)
except Exception:
print "Warning...the MOD you have selected:",mod,ontology_type,"is missing the appropriate relationship files",
print "necessary to run GO-Elite. Either replace the missing files ("+database_dir+'/'+species_code+') or select a different MOD at runtime.'
print 'Exiting program.'; forceExit
if source_data != mod:
mod_source = mod+'-'+source_data+'.txt'
uid_to_go,uid_system = importOntologyUIDGeneData(species_code,mod_source,gene_to_go,denominator_source_ids)
gene_to_go = convertGeneDBtoGeneObjects(gene_to_go,mod)
for gene in gene_to_go: uid_to_go[gene] = gene_to_go[gene]
return uid_to_go, uid_system, gene_annotations
else:
gene_to_go = convertGeneDBtoGeneObjects(gene_to_go,mod)
return gene_to_go, mod, gene_annotations
def grabNestedGeneToPathwayAssociations(species_code,mod,source_data,system_codes,custom_sets_folder,denom_search_dir,ontology_type):
program_type,database_dir = unique.whatProgramIsThis()
global printout; printout = 'yes'
gene_annotations = importGeneData(species_code,mod)
### Filter source IDs for those in all user denominator files
denominator_source_ids = getAllDenominators(denom_search_dir,system_codes)
try:
if ontology_type == 'WikiPathways': ontology_type = 'MAPP'
gene_to_mapp = importGeneMAPPData(species_code,mod+'-'+ontology_type)
except Exception:
### If a custom collection, shouldn't be found in the gene-mapp directory
try: gene_to_mapp = importGeneCustomData(species_code,system_codes,custom_sets_folder,mod)
except Exception: gene_to_mapp = {}
if source_data != mod:
mod_source = mod+'-'+source_data+'.txt'
uid_to_mapp,uid_system = importGeneSetUIDGeneData(species_code,mod_source,gene_to_mapp,denominator_source_ids)
gene_to_mapp = convertGeneDBtoGeneObjects(gene_to_mapp,mod)
### Combine the gene and UID data in order to later filter out gene's linked to an arrayid that are not linked to the pathway (common for some pathways)
for gene in gene_to_mapp: uid_to_mapp[gene] = gene_to_mapp[gene]
return uid_to_mapp, uid_system, gene_annotations
else:
gene_to_mapp = convertGeneDBtoGeneObjects(gene_to_mapp,mod)
return gene_to_mapp, mod, gene_annotations
def convertGeneDBtoGeneObjects(gene_to_pathway,mod):
gene_to_pathway_objects={}
for gene in gene_to_pathway:
pathways = gene_to_pathway[gene]
y = GeneRelationships(gene,gene,pathways,mod,mod) ###Note: in the analagous function for unique_ids, pathways is called pathway but is still a list (correct this at some point)
try: gene_to_pathway_objects[gene].append(y)
except KeyError: gene_to_pathway_objects[gene] = [y]
return gene_to_pathway_objects
def matchInputIDsToGOEliteTerms(gene_file_dir,go_elite_output_dir,system_codes,mappfinder_file,nested_collapsed_go_tree,uid_to_go,gene_annotations,full_go_name_db,uid_system,prev_combined_associations,prev_combined_gene_ranking):
global combined_associations ###This variable exists as a global under GO-Elite main as well, but needs to be re-initialized here as well
global combined_gene_ranking; global value_headers
combined_associations = prev_combined_associations
combined_gene_ranking = prev_combined_gene_ranking
try: uid_list,uid_value_db,value_headers,error = importUIDsForMAPPFinderQuery(gene_file_dir,system_codes,'yes')
except Exception: uid_list,uid_value_db,value_headers,error = importUIDsForMAPPFinderQuery(gene_file_dir[1:],system_codes,'yes')
filename_list = string.split(mappfinder_file,"/")
filename = filename_list[-1]
related_goid_db={}
for uid in uid_list:
try:
for y in uid_to_go[uid]: ### Different instances for each gene associated with the uid (e.g. multiple probeset to gene associations)
try: related_goids = y.GOIDs()
except Exception: print uid, y;kill
###If there are numerical values associated with the primary unique ID, store these with the GeneRelationships instance data
if uid in uid_value_db:
uid_values = uid_value_db[uid]
y.setUIDValues(uid_values)
else: y.setUIDValues([])
for goid in related_goids:
if goid in nested_collapsed_go_tree:
try: related_goid_db[goid].append(y)
except KeyError: related_goid_db[goid] = [y] ###store probesets linked to each goid
except Exception: null=[]
###Reduce related_goid_db to unique GO to gene relationships (rather than unique ids)
for goid in related_goid_db:
gene_db={}
for y in related_goid_db[goid]:
if y.GeneID() in uid_to_go: ### Ensures that genes not in the GO term, but linked to an array id are excluded
try: gene_db[y.GeneID()].append(y)
except KeyError: gene_db[y.GeneID()]=[y]
related_goid_db[goid] = gene_db
go_elite_gene_associations={}; gene_uid_data={}
for parent_goid in nested_collapsed_go_tree:
go_associated = [] ###Add all genes belonging to the parent
if parent_goid in related_goid_db:
gene_db = related_goid_db[parent_goid]
for gene in gene_db: ###Too complicated to store this information in the next list: Create a database a call later
gene_uid_data[gene] = gene_db[gene]
go_associated.append(gene)
go_associated = unique.unique(go_associated)
go_elite_gene_associations[parent_goid] = go_associated
return exportGeneGOAssociations(filename,go_elite_output_dir,go_elite_gene_associations,gene_uid_data,gene_annotations,full_go_name_db,uid_system)
def matchInputIDsToMAPPEliteTerms(gene_file_dir,go_elite_output_dir,system_codes,mappfinder_file,uid_to_mapp,zscore_mapp,gene_annotations,uid_system,prev_combined_associations,prev_combined_gene_ranking):
global combined_associations ###This variable exists as a global under GO-Elite main as well, but needs to be re-initialized here as well
global combined_gene_ranking; global value_headers
combined_associations = prev_combined_associations
combined_gene_ranking = prev_combined_gene_ranking
try: uid_list,uid_value_db,value_headers,error = importUIDsForMAPPFinderQuery(gene_file_dir,system_codes,'yes')
except Exception: uid_list,uid_value_db,value_headers,error = importUIDsForMAPPFinderQuery(gene_file_dir[1:],system_codes,'yes')
filename_list = string.split(mappfinder_file,"/")
filename = filename_list[-1]
related_mapp_db={}
for uid in uid_list:
if uid in uid_to_mapp: ###Find probesets in input list with MAPP annotataions
for y in uid_to_mapp[uid]:
related_mapps = y.MAPPs()
###If there are numerical values associated with the primary unique ID, store these with the GeneRelationships instance data
if uid in uid_value_db:
uid_values = uid_value_db[uid]
y.setUIDValues(uid_values)
else: y.setUIDValues([])
for mapp in related_mapps:
if mapp in zscore_mapp: ###Thus this MAPP is a GO-elite Local term
try: related_mapp_db[mapp].append(y)
except KeyError: related_mapp_db[mapp] = [y] ###store probesets linked to each MAPP
###Reduce related_mapp_db to unique GO to gene relationships (rather than unique ids)
for mapp in related_mapp_db:
gene_db={}
for y in related_mapp_db[mapp]:
if y.GeneID() in uid_to_mapp: ### Ensures that genes not on the pathway, but linked to an array id are excluded
try: gene_db[y.GeneID()].append(y)
except KeyError: gene_db[y.GeneID()]=[y]
related_mapp_db[mapp] = gene_db
go_elite_gene_associations={}; gene_uid_data={}
for mapp in zscore_mapp: ###Redundant with above, but preserving the structure of the analagous GO processing function
mapp_associated = []
if mapp in related_mapp_db:
gene_db = related_mapp_db[mapp]
for gene in gene_db: ###Too complicated to store this information in the next list: Create a database a call later
gene_uid_data[gene] = gene_db[gene]
mapp_associated.append(gene)
mapp_associated = unique.unique(mapp_associated)
go_elite_gene_associations[mapp] = mapp_associated
full_mapp_name_db={}
return exportGeneGOAssociations(filename,go_elite_output_dir,go_elite_gene_associations,gene_uid_data,gene_annotations,full_mapp_name_db,uid_system)
def simpleGenePathwayImport(species,geneset_type,pathway,OntologyID,directory):
###Import a gene-set and only store geneIDs for that pathway
associated_IDs={}
if geneset_type == 'WikiPathways': geneset_type = 'MAPP'
filename = 'AltDatabase/goelite/'+species+'/'+directory+'/'+'Ensembl-'+geneset_type+'.txt'
if directory == 'nested':
if geneset_type == 'GeneOntology': geneset_type = 'GO'
if len(OntologyID)>1: pathway = OntologyID
else: pathway = lookupOntologyID(geneset_type,pathway) ### Translates from name to ID
filename = 'AltDatabase/goelite/'+species+'/'+directory+'/'+'Ensembl_to_Nested-'+geneset_type+'.txt'
fn=filepath(filename)
### Imports a geneset category and stores pathway-level names
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
#print [t[-1]],[pathway,OntologyID]
if t[-1] == pathway:
associated_IDs[t[0]] = None
return associated_IDs
def filterGeneToUID(species_code,mod,source,filter_db):
mod_source = mod+'-'+source
if 'hide' in mod_source: show_progress, mod_source = mod_source
else: show_progress = 'yes'
program_type,database_dir = unique.whatProgramIsThis()
import_dir = '/'+database_dir+'/'+species_code+'/uid-gene'
ug = GrabFiles(); ug.setdirectory(import_dir)
filedir,filename = ug.searchdirectory(mod_source) ###Identify gene files corresponding to a particular MOD
fn=filepath(filedir); gene_to_uid={}; count = 0; x=0
uid_system,gene_system = grabFileRelationships(filename)
for line in open(fn,'rU').xreadlines():
#data = cleanUpLine(line)
data = line.strip()
if x==0: x=1
else:
t = string.split(data,'\t')
if t[0] in filter_db or t[1] in filter_db:
uid = t[1]; gene = t[0]
try: gene_to_uid[uid].append(gene)
except KeyError: gene_to_uid[uid] = [gene]
elif len(filter_db)==0:
uid = t[1]; gene = t[0]
try: gene_to_uid[uid].append(gene)
except KeyError: gene_to_uid[uid] = [gene]
gene_to_uid = eliminate_redundant_dict_values(gene_to_uid)
return gene_to_uid
def lookupOntologyID(geneset_type,ontology_name,type='name'):
if geneset_type == 'GeneOntology': geneset_type = 'go'
filename = 'AltDatabase/goelite/OBO/builds/'+geneset_type+'_annotations.txt'
fn=filepath(filename)
### Imports a geneset category and stores pathway-level names
i=0
for line in open(fn,'rU').xreadlines():
if i==0: i=1 ### Skip the header
else:
#data = cleanUpLine(line)
data = line.strip()
t = string.split(data,'\t')
geneset_category = t[1]
ontologyID = t[0]
if type=='ID':
if ontologyID == ontology_name:
ontology_id = geneset_category; break
elif geneset_category == ontology_name:
ontology_id = t[0]; break
return ontology_id
def swapKeyValues(db):
swapped={}
for key in db:
values = list(db[key]) ###If the value is not a list, make a list
for value in values:
try: swapped[value].append(key)
except KeyError: swapped[value] = [key]
swapped = eliminate_redundant_dict_values(swapped)
return swapped
def identifyRedundantPathways(go_elite_gene_associations):
for goid_or_mapp1 in go_elite_gene_associations:
for goid_or_mapp2 in go_elite_gene_associations:
if goid_or_mapp1 != goid_or_mapp2:
genes1 = go_elite_gene_associations[goid_or_mapp1]; total_genes1 = len(genes1)
genes2 = go_elite_gene_associations[goid_or_mapp2]; total_genes1 = len(genes2)
common_genes1 = []; common_genes2 = []
for gene in genes1:
if gene in genes2: common_genes1.append(gene)
for gene in genes2:
if gene in gene1: common_genes2.append(gene)
###Can get recipricol hits (unique_common_genes1 = unique_common_genes2), or hits in just one
def exportGeneGOAssociations(filename,go_elite_output_dir,go_elite_gene_associations,gene_uid_data,gene_annotations,full_go_name_db,uid_system):
gene_annotation_filename = filename[0:-4]+ '_' +uid_system+"-gene-associations.txt"
gene_ranking_filename = filename[0:-4]+ '_' +uid_system+"-Gene-Rankings.txt"
original_gene_annotation_filename = gene_annotation_filename
original_gene_ranking_filename = gene_ranking_filename
if len(go_elite_output_dir) == 0:
gene_annotation_filename = "output/gene_associations/" + gene_annotation_filename
gene_ranking_filename = "output/gene_associations/" + gene_ranking_filename
new_dir = 'output/gene_associations'
else:
gene_annotation_filename = go_elite_output_dir+'/gene_associations/'+gene_annotation_filename
gene_ranking_filename = go_elite_output_dir+'/gene_associations/'+gene_ranking_filename
new_dir = go_elite_output_dir
go_gene_annotation_db={}; go_values_db={}
data = export.ExportFile(gene_annotation_filename)
title = ['GeneID','Symbol','Description','UID-str','parent GOID','GO Name']+value_headers
title = string.join(title,'\t')
combined_associations[original_gene_annotation_filename]=[''] ###create a new key (filename) and append all entries to it
combined_associations[original_gene_annotation_filename].append(title)
data.write(title+'\n'); gene_centric_results={}; gene_to_goid={}
for parent_goid in go_elite_gene_associations:
try: go_name = full_go_name_db[parent_goid]
except KeyError: go_name = parent_goid
for gene in go_elite_gene_associations[parent_goid]:
uid_data = gene_uid_data[gene]
uid_list=[]; uid_value_db={}; indexes=[]; uid_value_list = []; uid_mean_value_list = []
for y in uid_data:
uid_list.append(y.UniqueID())
index = 0
for value in y.UIDValues():
try: uid_value_db[index].append(value)
except KeyError: uid_value_db[index] = [value]
indexes.append(index); index+=1
indexes = unique.unique(indexes); indexes.sort()
for index in indexes:
value_str = string.join(uid_value_db[index],'|')
try: value_mean = statistics.avg(uid_value_db[index])
except ValueError: value_mean = 0
uid_value_list.append(value_str); uid_mean_value_list.append(value_mean)
uid_str = string.join(uid_list,'|')
try:
s = gene_annotations[gene]
info = [s.GeneID(),s.Symbol(),s.Description(),uid_str,str(parent_goid),go_name]+uid_value_list
except KeyError:
s = GeneAnnotations(gene,'','','')
info = [gene,'','',uid_str,str(parent_goid),go_name]
###Record these associations to include in the main GO-elite output results file
try: go_gene_annotation_db[parent_goid].append(s)
except KeyError: go_gene_annotation_db[parent_goid] = [s]
try: go_values_db[parent_goid].append(uid_mean_value_list)
except KeyError: go_values_db[parent_goid] = [uid_mean_value_list]
try: gene_centric_results[gene,uid_str].append(go_name)
except KeyError: gene_centric_results[gene,uid_str] = [go_name]
try: gene_to_goid[gene].append(parent_goid)
except KeyError: gene_to_goid[gene] = [parent_goid]
try: info = string.join(info,'\t')
except TypeError: print info;kill
combined_associations[original_gene_annotation_filename].append(info)
data.write(info+'\n')
data.close()
#print 'Nested gene associations (re-derived) written for GO-Elite results to:\n',gene_annotation_filename
data = export.ExportFile(gene_ranking_filename); sorted_gene_ranking=[]
title = ['GeneID','Symbol','Description','#GO-Terms Associated','Percent GO-Terms Associated','UID-str','GO Names']
title = string.join(title,'\t')
combined_gene_ranking[original_gene_ranking_filename]=['']
combined_gene_ranking[original_gene_ranking_filename].append(title)
data.write(title+'\n')
for (gene,uid_str) in gene_centric_results:
go_terms = gene_centric_results[(gene,uid_str)]
num_go_terms = len(go_terms); total_go_elite_terms = len(go_elite_gene_associations); percent = float(num_go_terms)/total_go_elite_terms
go_name_str = string.join(go_terms,' // ')
try:
s = gene_annotations[gene]
info = [s.GeneID(),s.Symbol(),s.Description(),str(num_go_terms),str(percent),uid_str,go_name_str]
except KeyError:
info = [gene,'','',str(num_go_terms),str(percent),uid_str,go_name_str]
info = string.join(info,'\t')
sorted_gene_ranking.append((num_go_terms,info))
sorted_gene_ranking.sort(); sorted_gene_ranking.reverse()
for (rank,info) in sorted_gene_ranking:
combined_gene_ranking[original_gene_ranking_filename].append(info)
data.write(info+'\n')
data.close()
###Additionaly identify redundant GO-terms based solely on gene content
goids_with_redundant_genes={}
for parent_goid in go_elite_gene_associations:
go_count_db={}
for gene in go_elite_gene_associations[parent_goid]:
associated_goids = gene_to_goid[gene]
for goid in associated_goids:
###Count the number of genes associated with each goid associated with all genes (linked to a parent_goid)
try: go_count_db[goid] += 1
except KeyError: go_count_db[goid] = 1
##### MAJOR SOURCE OF KEYERRORS DUE TO MISMATCHING DATABASES OR ARRAY FILES - RAISE EXCEPTION IN GO-ELITE.PY
del go_count_db[parent_goid] ###this goterm would otherwise be redundant
for goid in go_count_db:
overlap_gene_count = go_count_db[goid] ###number of overlaping genes between the parent_goid and current goid
total_gene_count = len(go_elite_gene_associations[goid])
if overlap_gene_count == total_gene_count:
try: parent_go_name = full_go_name_db[parent_goid]
except KeyError: parent_go_name = parent_goid
try: go_name = full_go_name_db[goid]
except KeyError: go_name = goid
try: goids_with_redundant_genes[goid].append((parent_go_name,parent_goid))
except KeyError: goids_with_redundant_genes[goid] = [(parent_go_name,parent_goid)]
inverse_goids_with_redundant_genes={}
###Now, select the inverse of redundant GOIDs (report parent redundancies for each redundant term
all_redundant={}
for child_goid in goids_with_redundant_genes:
try: child_go_name = full_go_name_db[child_goid]
except KeyError: child_go_name = child_goid
parent_go_names = []
for (parent_go_name,parent_goid) in goids_with_redundant_genes[child_goid]:
parent_go_names.append(parent_go_name) ###we just want to report the name, but we needed the goid for this function
try: inverse_goids_with_redundant_genes[parent_goid].append(child_go_name)
except KeyError: inverse_goids_with_redundant_genes[parent_goid] = [child_go_name]
all_redundant[parent_goid] = []
goids_with_redundant_genes[child_goid] = parent_go_names ###replace the existing entry with a list of names only
all_redundant[child_goid] = []
goids_with_redundant_genes = eliminate_redundant_dict_values(goids_with_redundant_genes)
inverse_goids_with_redundant_genes = eliminate_redundant_dict_values(inverse_goids_with_redundant_genes)
goids_with_redundant_genes2={} ###Combine the redundant and inverse data and store as instances of RedundantRelationships
for goid in all_redundant:
redundant_go_names=[' ']; inverse_go_names=[' ']
if goid in goids_with_redundant_genes: redundant_go_names = goids_with_redundant_genes[goid]
if goid in inverse_goids_with_redundant_genes: inverse_go_names = inverse_goids_with_redundant_genes[goid]
rr = RedundantRelationships(redundant_go_names,inverse_go_names)
goids_with_redundant_genes2[goid] = rr
goids_with_redundant_genes = goids_with_redundant_genes2
#for go_name in goids_with_redundant_genes: print go_name,goids_with_redundant_genes[go_name]
###For each column of values, summarize the numerical values (take the mean) for each GO/MAPP term
for goid in go_values_db:
index_db={}; index_ls=[]
for vals in go_values_db[goid]:
index = 0
for val in vals:
if val != 0: ###only occurs when the value was non-numeric, otherwise it's a float
try: index_db[index].append(val)
except KeyError: index_db[index] = [val]
index_ls.append(index) ###for sorting through
index+=1
index_ls = unique.unique(index_ls); index_ls.sort()
summary_values = []; summary_stdev_values = []
for index in index_ls:
try:
try: avg_val = statistics.avg(index_db[index]); summary_values.append(str(avg_val))
except KeyError: summary_values.append('')
try: stdev_val = statistics.stdev(index_db[index]); summary_stdev_values.append(str(stdev_val))
except KeyError: summary_stdev_values.append('')
except ValueError: summary_values.append(''); summary_stdev_values.append('')
go_values_db[goid] = summary_values, summary_stdev_values
#print 'Gene counts (re-derived) for GO-Elite results writen to:\n',gene_ranking_filename
return combined_associations,combined_gene_ranking,go_gene_annotation_db,go_values_db,value_headers,goids_with_redundant_genes,len(gene_to_goid)
def exportCombinedAssociations(combined_associations,go_elite_output_folder,file_suffix):
new_filename = 'pruned-'+file_suffix+'.txt'
if len(go_elite_output_folder)==0: output_results = 'output/gene_associations/' + new_filename
else: output_results = go_elite_output_folder +'/gene_associations/'+ new_filename
data = export.ExportFile(output_results)
for file in combined_associations:
for line in combined_associations[file]:
info = file +'\t'+ line +'\n'
data.write(info)
data.close()
#print 'Combined gene associations (re-derived) written for GO-Elite results to:\n',output_results
def swapAndExportSystems(species_code,system1,system2):
program_type,database_dir = unique.whatProgramIsThis()
geneUID_import_dir = '/'+database_dir+'/'+species_code+'/uid-gene'
ug = GrabFiles(); ug.setdirectory(geneUID_import_dir)
filedir,file = ug.searchdirectory(system1+'-'+system2) ### Identify gene files corresponding to a particular MOD
gene_to_uid={}; x = 0
fn=filepath(filedir)
for line in open(fn,'rU').xreadlines():
#data = cleanUpLine(line)
data = line.strip()
if x==0: x=1
else:
t = string.split(data,'\t')
id1 = t[0]; id2 = t[1]
try: gene_to_uid[id2].append(id1)
except KeyError: gene_to_uid[id2]= [id1]
export_dir = geneUID_import_dir[1:]+'/'+system2+'-'+system1+'.txt'
try: data = export.ExportFile(export_dir)
except Exception: data = export.ExportFile(export_dir[1:])
data.write(system2+'\t'+system1+'\n')
for id2 in gene_to_uid:
for id1 in gene_to_uid[id2]:
values = string.join([id2,id1],'\t')+'\n'
data.write(values)
data.close()
class GeneIDInfo:
def __init__(self,system_name,geneID,pathway):
self.system_name = system_name; self.geneID = geneID; self.pathway = pathway
self.geneID = string.replace(geneID,'...','')
#print self.geneID, system_name
try:
if ' ' == self.geneID[-1]: self.geneID = self.geneID[:-1]
except Exception: null=[]
def GeneID(self): return str(self.geneID)
def System(self): return str(self.system_name)
def Pathway(self):
try: return str(self.pathway)
except Exception: return 'invalid'
def setGraphID(self,graphID): self.graphID = graphID
def setGroupID(self,groupid): self.groupid = groupid
def GraphID(self): return self.graphID
def GroupID(self): return self.groupid
def setLabel(self,label): self.label = label
def Label(self): return self.label
def setModID(self,mod_list): self.mod_list = mod_list
def ModID(self): return self.mod_list
def Report(self):
try: output = self.GeneID()+'|'+self.System()
except Exception: print self.Label()
return output
def __repr__(self): return self.Report()
def parseGMT(custom_sets_folder):
### File format from the Broad Institutes MSigDB ()
gm = GrabFiles(); gm.setdirectory(custom_sets_folder); system = None
filedirs = gm.getAllFiles('.gmt') ### Identify gene files corresponding to a particular MOD
gene_data=[]
for gmt in filedirs:
gmt=filepath(gmt)
for line in open(gmt,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
pathway_name = t[0]; url = t[1]; geneids = t[2:]
try: null=int(geneids[0]); system_name = 'Entrez Gene'
except Exception: system_name = 'Symbol'
for id in geneids:
gi = GeneIDInfo(system_name,id,pathway_name)
gene_data.append(gi)
return gene_data
def exportWikiPathwayData(species_name,pathway_db,type):
export_dir = 'BuildDBs/wikipathways/wikipathways_'+type+'_data_'+species_name+'.tab'
export_data = export.ExportFile(export_dir)
title = ['Pathway Name', 'Organism', 'Gene Ontology', 'Url to WikiPathways', 'Last Changed', 'Last Revision', 'Author', 'Count', 'Entrez Gene', 'Ensembl', 'Uniprot/TrEMBL', 'UniGene', 'RefSeq', 'MOD', 'PubChem', 'CAS', 'ChEBI']
title = string.join(title,'\t')+'\n'
export_data.write(title)
for wpid in pathway_db:
wpd = pathway_db[wpid]
values = [wpd.Pathway(), wpd.Organism(), '', wpd.URL(), '', wpd.Revision(), '', wpd.OriginalCount(), wpd.EntrezGene(), wpd.Ensembl()]
values +=[wpd.Uniprot(), wpd.Unigene(), wpd.Refseq(), wpd.MOD(), wpd.Pubchem(), wpd.CAS(), wpd.Chebi()]
values = string.join(values,'\t')
values = string.replace(values,'\n','') ###Included by mistake
try: export_data.write(values+'\n')
except Exception: pass
export_data.close()
#print 'WikiPathways data exported to:',export_dir
def clusterPrimaryPathwayGeneSystems(species_code,pathway_db):
st = systemTranslation()
for wpid in pathway_db:
wpd = pathway_db[wpid]
xml_data = wpd.PathwayGeneData()
ensembl=[]; entrez=[]; refseq=[]; unigene=[]; uniprot=[]; cas=[]; chebi=[]; pubchem=[]; mod=[]
### If the MOD gene IDs are in the pathway then add these
for gi in xml_data:
if len(gi.GeneID())>0:
source_data = gi.System()
if 'Ensembl' in source_data: source_data = 'Ensembl'
if source_data in st: source_data = st[source_data] ### convert the name to the GO-Elite compatible name
if source_data == '' and gi.GeneID()[:2]=='WP': source_data = 'WikiPathways'
if source_data == 'Ensembl': ensembl.append(gi.GeneID())
elif source_data == 'EntrezGene': entrez.append(gi.GeneID())
elif string.lower(source_data) == 'refseq': refseq.append(gi.GeneID())
elif string.lower(source_data) == 'unigene': unigene.append(gi.GeneID())
elif 'uniprot' in string.lower(source_data): uniprot.append(gi.GeneID())
elif string.lower(source_data) == 'swissprot': uniprot.append(gi.GeneID())
elif 'trembl' in string.lower(source_data): uniprot.append(gi.GeneID())
elif string.lower(source_data) == 'cas': cas.append(gi.GeneID())
elif string.lower(source_data) == 'chebi': chebi.append(gi.GeneID())
elif string.lower(source_data) == 'pubchem': pubchem.append(gi.GeneID())
else: mod.append(gi.GeneID()+'('+source_data+')')
wpd.setGeneData(ensembl,uniprot,refseq,unigene,entrez,mod,pubchem,cas,chebi)
wpd.setOriginalCount(wpd.Count()) ### Since the mapped count will include all infered, only reference the original count
def convertAllGPML(specific_species,all_species):
import update; import UI
global species_code
try: species_names = UI.remoteSpeciesInfo('yes') ### GO-Elite version
except Exception: species_names = UI.remoteSpeciesAlt() ### AltAnalyze version
for species_code in species_names:
if all_species == 'yes' or species_code in specific_species:
species_name = species_names[species_code].SpeciesName()
species = string.replace(species_name,' ','_')
### Clear and create the output dir
try: export.deleteFolder('GPML')
except Exception: null=[]
try: os.mkdir(filepath('GPML'))
except Exception: null=[]
### Download all species GPML from .zip
#url = 'http://wikipathways.org//wpi/cache/wikipathways_'+species+'_Curation-AnalysisCollection__gpml.zip'
url = 'http://data.wikipathways.org/20190410/gpml/wikipathways-20190410-gpml-'+species+'.zip'
print url
fln,status = update.download(url,'GPML/','')
if 'Internet' not in status:
if len(specific_species) == 1:
print 'Including the latest WikiPathways associations'
gene_data,pathway_db = parseGPML('/GPML') ### Get pathway associations (gene and annotation)
clusterPrimaryPathwayGeneSystems(species_code,pathway_db)
exportWikiPathwayData(species_name,pathway_db,'native')
### Create a "MAPPED" version (mapped will contain BAD MAPPINGS provide by the source database)!!!
ensembl_to_WP = unifyGeneSystems(gene_data,species_code,'Ensembl') ### convert primary systems to MOD IDs
hmdb_to_WP = unifyGeneSystems(gene_data,species_code,'HMDB') ### convert primary systems to MOD IDs
convertBetweenSystems(species_code,pathway_db,ensembl_to_WP,hmdb_to_WP) ### convert MOD IDs to all related primary systems (opposite as last method)
exportWikiPathwayData(species_name,pathway_db,'mapped')
if len(pathway_db)>0: ### Export all pathway interactions
try: exportNodeInteractions(pathway_db,'Ensembl','GPML')
except Exception: null=[]
def convertBetweenSystems(species_code,pathway_db,ensembl_to_WP,hmdb_to_WP):
WP_to_ensembl = OBO_import.swapKeyValues(ensembl_to_WP)
WP_to_hmdb = OBO_import.swapKeyValues(hmdb_to_WP)
ens_uniprot = getRelated(species_code,'Ensembl-'+'Uniprot')
ens_refseq = getRelated(species_code,'Ensembl-'+'RefSeq')
ens_unigene = getRelated(species_code,'Ensembl-'+'UniGene')
ens_entrez = getRelated(species_code,'Ensembl-'+'EntrezGene')
hmdb_pubchem = getRelated(species_code,'HMDB-'+'PubChem')
hmdb_cas = getRelated(species_code,'HMDB-'+'CAS')
hmdb_chebi = getRelated(species_code,'HMDB-'+'ChEBI')
for wpid in pathway_db:
wpd = pathway_db[wpid]
try: ens_ids = WP_to_ensembl[wpd.Pathway()]
except Exception: ens_ids=[]; #print wpid,len(WP_to_ensembl);sys.exit()
try: hmdb_ids = WP_to_hmdb[wpd.Pathway()]
except Exception: hmdb_ids=[]
#print wpid,wpid.Pathway(),hmdb_ids;sys.exit()
ensembl,uniprot,refseq,unigene,entrez,mod = wpd.GeneDataSystems()
pubchem,cas,chebi = wpd.ChemSystems()
uniprot = getConverted(ens_uniprot,ens_ids) +uniprot
refseq = getConverted(ens_refseq,ens_ids) +refseq
unigene = getConverted(ens_unigene,ens_ids) +unigene
entrez = getConverted(ens_entrez,ens_ids) +entrez
ensembl = ens_ids + ensembl
pubchem = getConverted(hmdb_pubchem,hmdb_ids) +pubchem
cas = getConverted(hmdb_cas,hmdb_ids) +cas
chebi = getConverted(hmdb_chebi,hmdb_ids) +chebi
### Reset these
wpd.setGeneData(ensembl,uniprot,refseq,unigene,entrez,mod,pubchem,cas,chebi)
def getConverted(gene_to_source_id,mod_ids):
source_ids=[]
for id in mod_ids:
try:
for i in gene_to_source_id[id]: source_ids.append(i)
except Exception: null=[]
return source_ids
def getRelated(species_code,mod_source):
try:
gene_to_source_id = getGeneToUid(species_code,('hide',mod_source)); #print mod_source, 'relationships imported.'
except Exception: gene_to_source_id={}
return gene_to_source_id
class WikiPathwaysData:
def __init__(self,pathway,wpid,revision,organism,gi):
self.pathway = pathway; self.wpid = wpid; self.revision = revision; self.gi = gi
self.organism = organism
def Pathway(self): return self.pathway
def WPID(self): return self.wpid
def URL(self): return 'http://www.wikipathways.org/index.php/Pathway:'+self.wpid
def Organism(self): return self.organism
def Revision(self): return self.revision
def PathwayGeneData(self): return self.gi
def Report(self):
output = self.WPID()
def setGeneData(self,ensembl,uniprot,refseq,unigene,entrez,mod,pubchem,cas,chebi):
self.ensembl=ensembl;self.uniprot=uniprot;self.refseq=refseq;self.unigene=unigene
self.entrez=entrez;self.mod=mod;self.pubchem=pubchem;self.cas=cas;self.chebi=chebi
combined = ensembl+uniprot+refseq+unigene+entrez+mod+pubchem+cas+chebi
self.count = len(unique.unique(combined))
def setOriginalCount(self,original_count): self.original_count = original_count
def OriginalCount(self): return self.original_count
def setInteractions(self,interactions): self.interactions = interactions
def Interactions(self): return self.interactions
def GeneDataSystems(self): return self.ensembl,self.uniprot,self.refseq,self.unigene,self.entrez,self.mod
def ChemSystems(self): return self.pubchem,self.cas,self.chebi
def Ensembl(self): return self.Join(self.ensembl)
def Uniprot(self): return self.Join(self.uniprot)
def Refseq(self): return self.Join(self.refseq)
def Unigene(self): return self.Join(self.unigene)
def EntrezGene(self): return self.Join(self.entrez)
def MOD(self): return self.Join(self.mod)
def Pubchem(self): return self.Join(self.pubchem)
def CAS(self): return self.Join(self.cas)
def Chebi(self): return self.Join(self.chebi)
def Join(self,ls): return string.join(unique.unique(ls),',')
def Count(self): return str(self.count)
def __repr__(self): return self.Report()
class InteractionData:
def __init__(self, gene1,gene2,int_type):
self.gene1 = gene1; self.gene2 = gene2; self.int_type = int_type
def GeneObject1(self): return self.gene1
def GeneObject2(self): return self.gene2
def InteractionType(self): return self.int_type
class EdgeData:
def __init__(self, graphid1,graphid2,int_type):
self.graphid1 = graphid1; self.graphid2 = graphid2; self.int_type = int_type
def GraphID1(self): return self.graphid1
def GraphID2(self): return self.graphid2
def InteractionType(self): return self.int_type
def parseGPML(custom_sets_folder):
import xml.dom.minidom
from xml.dom.minidom import Node
from xml.dom.minidom import parse, parseString
gm = GrabFiles(); gm.setdirectory(custom_sets_folder); system = None
filedirs = gm.getAllFiles('.gpml') ### Identify gene files corresponding to a particular MOD
gene_data=[]; pathway_db={}
for xml in filedirs:
pathway_gene_data=[]
complexes_data={}
edge_data=[]
#graph_node_data=[]
pathway_type = 'GPML'
xml=filepath(xml)
filename = string.split(xml,'/')[-1]
try: wpid = string.split(filename,'_')[-2]
except Exception: wpid = filename[:-5]
revision = string.split(filename,'_')[-1][:-5]
try: dom = parse(xml)
except Except: continue
tags = dom.getElementsByTagName('Xref')
data_node_tags = dom.getElementsByTagName('DataNode')
groups = dom.getElementsByTagName('Group') ### complexes
edges = dom.getElementsByTagName('Point') ### interacting nodes/complexes
pathway_tag = dom.getElementsByTagName('Pathway')
comment_tag = dom.getElementsByTagName('Comment')
#print comment_tag.nodeValue; sys.exit()
for pn in pathway_tag:
pathway_name = pn.getAttribute("Name")
organism = pn.getAttribute("Organism")
data_source = pn.getAttribute("Data-Source")
for ed in edges:
### Store internal graph data for pathway edges to build gene interaction networks later
graphid = ed.getAttribute("GraphRef")
edge_type = ed.getAttribute("ArrowHead")
if edge_type == '': edge_pair = [graphid] ### either just a graphical line or the begining of a node-node edge
else:
try:
edge_pair.append(graphid)
edd = EdgeData(str(edge_pair[0]),str(edge_pair[1]),str(edge_type))
edge_data.append(edd)
except Exception:
None ### Can happen with some pathways
for gd in groups:
### Specific to groups and complexes
groupID = gd.getAttribute("GroupId") ### Group node ID (same as "GroupRef")
graphID = gd.getAttribute("GraphId") ### GPML node ID
complexes_data[str(groupID)] = str(graphID)
for cm in comment_tag:
pathway_type = cm.getAttribute("Source")
### This is an issue with KEGG pathways who's names are truncated
try:
extended_comment_text = cm.childNodes[0].nodeValue
if 'truncated' in extended_comment_text:
pathway_name = string.split(extended_comment_text,': ')[-1]
except IndexError: null=[]
if 'Kegg' in pathway_type:
if '?' in data_source:
pathway_id = string.split(data_source,'?')[-1]
pathway_name += ':KEGG-'+pathway_id
if 'WP' not in wpid:
wpid = pathway_id
for i in data_node_tags:
#print i.toxml()
id = ''
system_name = ''
for x in i.childNodes: ### DataNode is the parent attribute with children GraphId, TextLabel, Xref
if x.nodeName == 'Xref': ### Since the attributes we want are children of these nodes, must find the parents first
system_name = x.getAttribute("Database") ### System Code
id = x.getAttribute("ID") ### Gene or metabolite ID
label = i.getAttribute("TextLabel") ### Gene or metabolite label
type = i.getAttribute('Type') #E.g.', GeneProduct, Metabolite
graphID = i.getAttribute("GraphId") ### WikiPathways graph ID
groupID = i.getAttribute('GroupRef')### Group node ID
#graph_node_data.append([graphID,groupID,label,type])
try:
gi = GeneIDInfo(str(system_name),str(id),pathway_name)
gi.setGroupID(str(groupID)) ### Include internal graph IDs for determining edges
gi.setGraphID(graphID)
gi.setLabel(label)
if len(id)>0 or 'Tissue' in pathway_name: ### Applies to the Lineage Profiler pathway which doesn't have IDs
gene_data.append(gi)
pathway_gene_data.append(gi)
except:
#Can occur as - UnicodeEncodeError: 'ascii' codec can't encode character u'\xa0' in position 15: ordinal not in range(128)
pass
wpd=WikiPathwaysData(pathway_name,wpid,revision,organism,pathway_gene_data)
pathway_db[wpid]=wpd
interaction_data = getInteractions(complexes_data,edge_data,wpd)
wpd.setInteractions(interaction_data)
return gene_data,pathway_db
def getInteractions(complexes_data,edge_data,wpd):
### Annotate the interactions between nodes and between groups of nodes from WikiPathways
interaction_data=[]
wpid = wpd.WPID()
graphID_db={}
for gi in wpd.PathwayGeneData():
graphID_db[gi.GraphID()] = [gi] ### graphID for the node
if gi.GroupID() in complexes_data:
graph_id = complexes_data[gi.GroupID()]
try: graphID_db[graph_id].append(gi) ### graphID for the group of nodes with putative edges
except Exception: graphID_db[graph_id] = [gi]
for eed in edge_data:
if eed.GraphID1() != '' and eed.GraphID2() != '':
try:
gi_list1 = graphID_db[eed.GraphID1()]
gi_list2 = graphID_db[eed.GraphID2()]
for gi1 in gi_list1:
for gi2 in gi_list2:
intd = InteractionData(gi1,gi2,eed.InteractionType())
interaction_data.append(intd)
except KeyError: null=[] ### Typically occurs for interactions with Labels and similar objects
return interaction_data
def getGPMLGraphData(custom_sets_folder,species_code,mod):
""" Calls methods to import GPML data and retrieve MOD IDs (Ensembl/Entrez) for GPML graphIDs """
gpml_data,pathway_db = parseGPML(custom_sets_folder)
gene_to_WP = unifyGeneSystems(gpml_data,species_code,mod)
return pathway_db ### This object contains all pathways and all ID associations
def parseGPML2():
import urllib
from xml.dom import minidom
gpml = 'http://wikipathways.org//wpi/wpi.php?action=downloadFile&type=gpml&pwTitle=Pathway:WP201'
#pathway = string.split(gpml,'/')[-1]; pathway = string.split(pathway,'.')[0]
dom = minidom.parse(urllib.urlopen(gpml))
tags = dom.getElementsByTagName('Xref')
pathway_tag = dom.getElementsByTagName('Pathway')
for pn in pathway_tag:
pathway_name = pn.getAttribute("Name")
organism = pn.getAttribute("Organism")
print pathway_name,organism;kill
gene_data=[]
for i in tags:
system_name = i.getAttribute("Database")
id = i.getAttribute("ID")
gi = GeneIDInfo(system_name,id,pathway_name)
gene_data.append(gi)
return gene_data
def parseBioPax3(custom_sets_folder):
import xml.dom.minidom
from xml.dom.minidom import Node
from xml.dom.minidom import parse, parseString
gm = GrabFiles(); gm.setdirectory(custom_sets_folder); system = None
filedirs = gm.getAllFiles('.owl') ### Identify gene files corresponding to a particular MOD
gene_data=[]
for xml in filedirs:
xml=filepath(xml)
for line in open(xml,'rU').xreadlines():
data = cleanUpLine(line)
if 'pathway rdf:ID=' in data: ##<pathway rdf:ID="EGFR1_pathway_Human">
pathway_name = string.split(data,'pathway rdf:ID')[-1][:-1]
#test
def processXMLline(data_type,line):
data_types = ['NAME','pathway']
for data_type in data_types:
full_str='<bp:'+data_type+' rdf:datatype="xsd:string">'
if full_str in line:
value = string.replace(line,full_str,'')
def parseBioPax(custom_sets_folder):
#import urllib
from xml.dom import minidom
from xml.dom.minidom import Node
import xml.dom.minidom
from xml.dom.minidom import Node
from xml.dom.minidom import parse, parseString
gm = GrabFiles(); gm.setdirectory(custom_sets_folder); system = None
filedirs = gm.getAllFiles('.owl') ### Identify gene files corresponding to a particular MOD
gene_data=[]
for xml in filedirs:
#print xml
#xml = 'http://www.netpath.org/data/biopax/NetPath_4.owl'
#pathway = string.split(xml,'/')[-1]; pathway = string.split(pathway,'.')[0]
#dom = minidom.parse(urllib.urlopen(xml))
xml=filepath(xml)
dom = parse(xml); pathway_name=[]
tags = dom.getElementsByTagName('unificationXref')
pathway_tag = dom.getElementsByTagName('pathway')
for pathway_name in pathway_tag: pathway_name = pathway_name.getAttribute("rdf:ID")
if len(pathway_name)==0:
pathway_tag = dom.getElementsByTagName('bp:NAME')
for pathway_name in pathway_tag: pathway_name = pathway_name.getAttribute("rdf:datatype='xsd:string'")
#print pathway_name
for node in tags:
id_object = node.getElementsByTagName("ID")
db_object = node.getElementsByTagName("DB")
for node2 in id_object:
id = ''
for node3 in node2.childNodes:
if node3.nodeType == Node.TEXT_NODE: id+= node3.data
for node2 in db_object:
system_name = ''
for node3 in node2.childNodes:
if node3.nodeType == Node.TEXT_NODE: system_name+= node3.data
gi = GeneIDInfo(system_name,id,pathway_name)
gene_data.append(gi)
#print gene_data
return gene_data
def parseBioPax2(filename):
import urllib
from xml.dom import minidom
from xml.dom.minidom import Node
xml = 'http://www.netpath.org/data/biopax/NetPath_4.owl'
#pathway = string.split(xml,'/')[-1]; pathway = string.split(pathway,'.')[0]
dom = minidom.parse(urllib.urlopen(xml))
tags = dom.getElementsByTagName('unificationXref')
pathway_tag = dom.getElementsByTagName('pathway')
for pathway_name in pathway_tag: pathway_name = pathway_name.getAttribute("rdf:ID")
gene_data=[]
for node in tags:
id_object = node.getElementsByTagName("ID")
db_object = node.getElementsByTagName("DB")
for node2 in id_object:
id = ''
for node3 in node2.childNodes:
if node3.nodeType == Node.TEXT_NODE: id+= node3.data
for node2 in db_object:
system_name = ''
for node3 in node2.childNodes:
if node3.nodeType == Node.TEXT_NODE: system_name+= node3.data
gi = GeneIDInfo(system_name,id,pathway_name)
gene_data.append(gi)
return gene_data
def systemTranslation():
st={}
st['Entrez Gene']='EntrezGene'
st['refseq']='RefSeq'
st['uniprot']='Uniprot'
st['TubercuList']='Ensembl'
st['Kegg Compound']='KeggCompound'
st['Uniprot/TrEMBL']='Uniprot'
st['SwissProt']='Uniprot'
return st
def unifyGeneSystems(xml_data,species_code,mod):
systems_db={}
for gi in xml_data:
try:
try: systems_db[gi.System()]+=1
except KeyError: systems_db[gi.System()]=1
except Exception: print gi;kill
#for i in systems_db: print i
### Import and combine all secondary systems
system_ids={}
if 'Symbol' not in systems_db: systems_db['Symbol']=1
st = systemTranslation();
for source_data in systems_db:
original_source = source_data
if 'Ensembl' in source_data: source_data = 'Ensembl'
if source_data in st: source_data = st[source_data] ### convert the name to the GO-Elite compatible name
if source_data != mod:
mod_source = mod+'-'+source_data+'.txt'
try: gene_to_source_id = getGeneToUid(species_code,('hide',mod_source)); #print mod_source, 'relationships imported.'
except Exception: gene_to_source_id={}
#print len(gene_to_source_id),mod_source,source_data
source_to_gene = OBO_import.swapKeyValues(gene_to_source_id)
system_ids[original_source]=source_to_gene
for system in system_ids:
if system == 'Symbol':
source_to_gene = system_ids[system]
source_to_gene = lowerAllIDs(source_to_gene)
system_ids[system] = source_to_gene
mod_pathway={}
### Convert source IDs to MOD IDs
mapped=0
for gi in xml_data:
if gi.System() in system_ids:
source_to_gene = system_ids[gi.System()]
geneID = gi.GeneID()
if gi.System() == 'Symbol':
geneID = string.lower(geneID)
if geneID in source_to_gene:
for mod_id in source_to_gene[geneID]:
mapped+=1
try: mod_pathway[mod_id].append(gi.Pathway())
except Exception: mod_pathway[mod_id] = [gi.Pathway()]
gi.setModID(source_to_gene[geneID]) ### Update this object to include associated MOD IDs (e.g., Ensembl or Entrez)
else:
source_to_gene = system_ids['Symbol'] ### Assume the missing ID is a symbol
geneID = string.lower(geneID)
if geneID in source_to_gene:
for mod_id in source_to_gene[geneID]:
mapped+=1
try: mod_pathway[mod_id].append(gi.Pathway())
except Exception: mod_pathway[mod_id] = [gi.Pathway()]
gi.setModID(source_to_gene[geneID]) ### Update this object to include associated MOD IDs (e.g., Ensembl or Entrez)
#print mapped;sys.exit()
### If the MOD gene IDs are in the pathway then add these
for gi in xml_data:
source_data = gi.System()
if 'Ensembl' in source_data: source_data = 'Ensembl'
if source_data in st: source_data = st[source_data] ### convert the name to the GO-Elite compatible name
if source_data == mod:
try: mod_pathway[gi.GeneID()].append(gi.Pathway())
except Exception: mod_pathway[gi.GeneID()] = [gi.Pathway()]
gi.setModID([gi.GeneID()])
#print len(system_ids),len(mod_pathway),len(mod_pathway)
return mod_pathway
def lowerAllIDs(source_to_gene):
source_to_gene2={}
for source in source_to_gene:
source_to_gene2[string.lower(source)] = source_to_gene[source]
return source_to_gene2
def combineDBs(db1,db2):
for id in db1:
if id in db2: db2[id]+=db1[id]
else: db2[id]=db1[id]
return db2
def IDconverter(filename,species_code,input_system_name, output_system_name,analysis=None):
""" This is a function built to convert the IDs in an input file from one system to another while preserving the original members """
if 'HMDB' in input_system_name or 'HMDB' in output_system_name: mod = 'HMDB'
elif 'ChEBI' in input_system_name or 'ChEBI' in output_system_name: mod = 'HMDB'
elif 'KeggCompound' in input_system_name or 'KeggCompound' in output_system_name: mod = 'HMDB'
elif 'CAS' in input_system_name or 'CAS' in output_system_name: mod = 'HMDB'
elif 'PubChem' in input_system_name or 'PubChem' in output_system_name: mod = 'HMDB'
else: mod = 'Ensembl'
print 'Attempting to convert IDs from ', export.findFilename(filename)
input_data_db, headers = importGenericDB(filename)
if input_system_name == mod: ### This is or MOD
source1_to_gene={}
for id in input_data_db:
source1_to_gene[id] = [id] ### make the primary ensembl ID = ensembl ID
else:
gene_to_source1 = getGeneToUid(species_code,mod+'-'+input_system_name)
source1_to_gene = OBO_import.swapKeyValues(gene_to_source1)
if output_system_name == mod: ### This is or MOD
gene_to_source2={}
gene_annotations = importGeneData(species_code,mod)
for gene in gene_annotations:
if 'LRG_' not in gene: ### Bad gene IDs from Ensembl
gene_to_source2[gene] = [gene] ###import and use all availalable Ensembl IDs
else:
gene_to_source2 = getGeneToUid(species_code,mod+'-'+output_system_name)
converted=0
converted_ids={}
for id in input_data_db:
secondary_ids = ''
genes = ''
if id in source1_to_gene:
genes = source1_to_gene[id]
for gene in genes:
if gene in gene_to_source2:
secondary_ids = gene_to_source2[gene]
secondary_ids = string.join(secondary_ids,'|')
genes = string.join(genes,'|')
converted+=1
converted_ids[id] = secondary_ids, genes
if analysis != 'signature':
if '.txt' in filename:
filename = string.replace(filename,'.txt','-'+output_system_name+'.txt')
else:
filename = filename[:-4]+'-'+output_system_name+'.txt'
export_data = export.ExportFile(filename)
headers = string.join([output_system_name,mod+' IDs']+headers,'\t')+'\n'
export_data.write(headers)
for id in input_data_db:
secondary_ids, genes = converted_ids[id]
for t in input_data_db[id]:
export_values = string.join([secondary_ids,genes]+[id]+t,'\t')+'\n'
export_data.write(export_values)
export_data.close()
print ''
print converted, 'input',input_system_name,'IDs converted to',output_system_name,'out of',len(input_data_db)
filename = export.findFilename(filename)
return filename
else:
#print len(input_data_db), len(converted_ids)
return converted_ids, input_data_db
if __name__ == '__main__':
species_code = 'Hs'; mod = 'Ensembl'; gotype='nested'
filedir = 'C:/Documents and Settings/Nathan Salomonis/My Documents/GO-Elite_120beta/Databases/EnsMart56Plus/Ce/gene/EntrezGene.txt'
system = 'Macaroni'
gene_annotations = importGeneData('Hs','EntrezGene')
for i in gene_annotations:
print i, gene_annotations[i].Symbol(); break
print len(gene_annotations)
sys.exit()
import GO_Elite
system_codes,source_types,mod_types = GO_Elite.getSourceData()
#custom_sets_folder = '/test'
#importGeneCustomData(species_code,system_codes,custom_sets_folder,mod); sys.exit()
### Test interactions export
custom_sets_folder = 'GPML'
species_code = 'Hs'; mod = 'Ensembl'
gene_to_symbol_db = getGeneToUid(species_code,('hide',mod+'-Symbol.txt')); #print mod_source, 'relationships imported.'
gpml_data,pathway_db = parseGPML(custom_sets_folder)
gene_to_WP = unifyGeneSystems(gpml_data,species_code,mod)
exportNodeInteractions(pathway_db,mod,custom_sets_folder)
sys.exit()
biopax_data = parseBioPax('/test'); sys.exit()
gene_data,pathway_db = parseGPML('/test')
print len(gpml_data)
mod = 'Ensembl'
gene_to_BioPax = unifyGeneSystems(biopax_data,species_code,mod)
#gene_to_WP = unifyGeneSystems(gpml_data,species_code,mod)
#gene_to_BioPax = combineDBs(gene_to_WP,gene_to_BioPax)
for i in gene_to_BioPax:
print i, gene_to_BioPax[i]
print len(gene_to_BioPax); sys.exit()
addNewCustomSystem(filedir,system,'yes','Ms'); kill
addNewCustomRelationships('test.txt','Ensembl-MAPP','update','Mm');kill
importGeneToOntologyData(species_code,mod,gotype,ontology_type);kill
species_name = 'Homo Sapiens'; species_code = 'Hs'; source_data = 'EntrezGene'; mod = 'EntrezGene'
system_codes={}; system_codes['X'] = 'Affymetrix'
import_dir = '/input/GenesToQuery/'+species_code
m = GrabFiles(); m.setdirectory(import_dir)
dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
for mappfinder_input in dir_list: #loop through each file in the directory
permuted_z_scores={}; original_go_z_score_data={}; original_mapp_z_score_data={}
gene_file_dir, gene_file = m.searchdirectory(mappfinder_input)
###Import Input gene/source-id lists
input_gene_list,source_data_input,error = importUIDsForMAPPFinderQuery('input/GenesToQuery/'+species_code+'/'+gene_file,system_codes,'no'); input_count = len(input_gene_list)
#!/usr/bin/python
###########################
#Program: GO-elite.py
#Author: Nathan Salomonis
#Date: 12/12/06
#Website: http://www.genmapp.org
#Email: [email protected]
###########################
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/gene_associations.py
|
gene_associations.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"""This module contains methods for performing over-representation analysis (ORA) on input gene
lists provided by the user relative to denominator gene lists for nested Gene Ontology relationships
and WikiPathway biological pathways. These methods include a permutation based analysis and multiple
hypthesis correction."""
import sys, string
import os.path, platform
import unique
import math
import time
import gene_associations; reload(gene_associations)
try: from import_scripts import OBO_import
except Exception: pass
import GO_Elite
from stats_scripts import statistics
import random
import UI
import export; reload(export)
import re
from stats_scripts import fishers_exact_test
import traceback
import warnings
try:
from scipy import stats
except Exception:
pass ### scipy is not required but is used as a faster implementation of Fisher Exact Test when present
################# Parse directory files
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
try:
dir_list = unique.read_directory(sub_dir)
except Exception:
dir_list=[] ### Directory does not exist
dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv": dir_list2.append(entry)
return dir_list2
def readDirText(sub_dir):
if sub_dir == None or sub_dir == '':
dir_list2 = [] ### For automatically assigned denominators
elif '.txt' in sub_dir:
dir_list2 = [export.findFilename(sub_dir)] ### for pooled analyses - analyze only the specific file direction provided
else:
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" and '._' not in entry: dir_list2.append(entry)
return dir_list2
###### Classes ######
class GrabFiles:
def setdirectory(self,value):
if '.txt' in value:
value = export.findParentDir(value) ### For pooled analyses where only one file is submitted
self.data = value
def display(self):
print self.data
def directory(self):
return self.data
def searchdirectory(self,search_term):
#self is an instance while self.data is the value of the instance
all_matching,file_dir,file = gene_associations.getDirectoryFiles(self.data,str(search_term))
#if len(file)<1: print search_term,'not found'
return file_dir,file
def getAllFiles(self,search_term):
#self is an instance while self.data is the value of the instance
all_matching,file_dir,file = gene_associations.getDirectoryFiles(self.data,str(search_term))
#if len(file)<1: print search_term,'not found'
return all_matching
################# Import and Annotate Data
def eliminate_redundant_dict_values(database):
db1={}
for key in database: list = unique.unique(database[key]); list.sort(); db1[key] = list
return db1
def swapKeyValues(db):
swapped={}
for key in db:
values = db[key]
for value in values:
try: swapped[value].append(key)
except KeyError: swapped[value] = [key]
return swapped
def identifyGeneFiles(import_dir,gene_file):
split_name = string.split(gene_file,'.')
e = GrabFiles(); e.setdirectory(import_dir)
dir_files = read_directory(import_dir)
if len(split_name)>2:
prefix_id = split_name[0]+'.'
denominator_file_dir,denominator_file = e.searchdirectory(prefix_id)
else: denominator_file_dir =''
if len(dir_files)==1 or denominator_file_dir=='':
try: denominator_file_dir,denominator_file = e.searchdirectory(dir_files[0])
except IndexError:
print_out = "WARNING: No denominator file included in\nthe GeneQuery/DenominatorGenes directory.\nTo proceed, place all denominator\nIDs in a file in that directory."
if PoolVar: q.put([print_out]); return None
ForceCriticalError(print_out)
return denominator_file_dir
def associateInputSourceWithGene(source_to_gene,source_id_list):
gene_db={}; count_null = 0
for source_id in source_id_list:
try:
gene_ids = source_to_gene[source_id]
for gene_id in gene_ids:
try: gene_db[gene_id].append(source_id)
except KeyError: gene_db[gene_id] = [source_id]
except KeyError: count_null+=1
#print count_null, 'source IDs not imported'
return gene_db
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def importVersionData(dir):
global OBO_date
program_type,database_dir = unique.whatProgramIsThis(); parent_dir = ''
if program_type == 'AltAnalyze': parent_dir = 'AltDatabase/goelite/'
dir = parent_dir+dir
filename = dir+'version.txt'; fn=filepath(filename)
for line in open(fn,'r').readlines():
data = cleanUpLine(line)
OBO_version, OBO_date = string.split(data,'\t')
return OBO_date
def checkDenominatorMatchesInput(input_gene_list,denominator_gene_list,gene_file):
for id in input_gene_list:
try: null = denominator_gene_list[id] ###this object was changed from a list to a dictionary for efficiency
except KeyError: ###Only occurs if an input ID is NOT found in the denominator
all_alphanumeric = string.join(re.findall(r"\w",id))
print_out = 'Identifier: '+'"'+id+'"'+' not found in Denominator set '+str(len(id))+' '+all_alphanumeric+' '+str(len(input_gene_list))+' '+str(len(denominator_gene_list))+'\n'
print_out = 'WARNING!!! Job stopped... Denominator gene list\ndoes not match the input gene list for\n%s' % gene_file
if PoolVar: q.put([print_out]); return None
ForceCriticalError(print_out)
def formatTime(start_time,end_time):
intgr,decim = string.split(str(end_time-start_time),'.')
### Alternatively, use - round(end_time-start_time,1)
return intgr+'.'+decim[0]
def importGeneData():
### Simple dictionary for verifying ID sets (important when wrong species selected)
program_type,database_dir = unique.whatProgramIsThis()
gene_import_dir = database_dir+'/'+species_code+'/gene/'+mod+'.txt'
fn=filepath(gene_import_dir); gene_annotations={}; x=0
for line in open(fn,'rU').xreadlines():
data = line.strip()
t = string.split(data,'\t')
if x == 0: x = 1
else:
gene_annotations[t[0]] = t[0]
return gene_annotations
def checkCorrectSystemAndSpecies(input_gene_list):
### Verify that the input IDs are the correct system and species
gene_annotations = importGeneData()
missing=True
while missing:
for i in input_gene_list:
try:
null=gene_annotations[i]
missing=False
except Exception: pass
break
return missing
def generateMAPPFinderScores(species_title,species_id,source,mod_db,system_Codes,permute,resources_to_analyze,file_dirs,parent_root,poolVar=False,Q=None,Multi=None):
global mappfinder_output_dir; global custom_sets_folder; global root; root = parent_root
global mapp_to_mod_genes; global ontology_to_mod_genes; global system_codes; system_codes = system_Codes
global q; q=Q; global mlp; mlp = Multi; global display; poolVar = True
criterion_input_folder, criterion_denom_folder, output_dir, custom_sets_folder = file_dirs
previous_denominator_file_dir = ''
ontology_to_mod_genes={}; mapp_to_mod_genes={}; global test; test = 'no'; global PoolVar; PoolVar=poolVar
program_type,database_dir = unique.whatProgramIsThis()
if resources_to_analyze == 'Gene Ontology': resources_to_analyze = 'GeneOntology'
if len(output_dir) == 0: mappfinder_output_dir = 'input/MAPPFinder'
else: mappfinder_output_dir = output_dir + '/GO-Elite_results/CompleteResults/ORA'
denom_folder = criterion_denom_folder
global source_data; source_data = source; global mod; mod = mod_db
global species_code; species_code = species_id
global species_name; species_name = species_title; global gene_to_mapp
global permutations; permutations = permute
global eliminate_redundant_genes; eliminate_redundant_genes = 'yes'
global permuted_z_scores; global ontology_annotations
global original_ontology_z_score_data; global original_mapp_z_score_data
global input_gene_list; global denominator_gene_list
global gene_file; global denom_file_status
global input_count; global denom_count; global gene_annotations
global source_to_gene; global use_FET
if permutations == "FisherExactTest":
use_FET = 'yes' ### Use Fisher's Exact test instead of permutation-based p-values
permutations = 0
else:
use_FET = 'no'
if poolVar: display = False
else: display = True
start_time = time.time()
gene_annotations = gene_associations.importGeneData(species_code,mod)
OBO_date = importVersionData('OBO/')
if len(criterion_input_folder) == 0: import_dir = '/input/GenesToQuery/'+species_code
else: import_dir = criterion_input_folder
m = GrabFiles(); m.setdirectory(import_dir)
import_dir_alt = m.directory()
try: dir_list = readDirText(import_dir) #send a sub_directory to a function to identify all files in a directory
except Exception:
print_out = 'Warning! Input directory location is not a valid folder. Exiting GO-Elite.'
try:
if PoolVar: q.put([print_out]); return None
except Exception: pass
ForceCriticalError(print_out)
try: denom_dir_list = readDirText(criterion_denom_folder)
except Exception:
print_out = 'Warning! Denominator directory location is not a valid folder. Exiting GO-Elite.'
try:
if PoolVar: q.put([print_out]); return None
except Exception: pass
ForceCriticalError(print_out)
if len(dir_list)==0:
error_message = 'No files with the extension ".txt" found in the input directory.'
try:
if PoolVar: q.put([print_out]); return None
except Exception: pass
ForceCriticalError(error_message)
if len(denom_dir_list)==0:
skipDenomImport=True #### No denominator supplied by the user
#error_message = 'No files with the extension ".txt" found in the denominator directory.'
#if PoolVar: q.put([print_out]); return None
#ForceCriticalError(error_message)
else:
skipDenomImport=False
inputs_analyzed=0
for mappfinder_input in dir_list: #loop through each file in the directory
permuted_z_scores={}; original_ontology_z_score_data={}; original_mapp_z_score_data={}
if PoolVar==False:
print 'Performing over-representation analysis (ORA) on',mappfinder_input
gene_file_dir, gene_file = m.searchdirectory(mappfinder_input)
###Import Input gene/source-id lists
input_gene_list,source_data_input,error_message = gene_associations.importUIDsForMAPPFinderQuery(import_dir_alt+'/'+gene_file,system_codes,'no'); input_count = len(input_gene_list)
if 'No results' in error_message:
continue
if 'WARNING!!!' in error_message: ### Warn the user about SwissProt issues when importing the denominator
try:
if PoolVar: q.put([print_out]); return None
except Exception: pass
ForceCriticalError(error_message)
continue
if skipDenomImport==False:
denominator_file_dir = identifyGeneFiles(denom_folder,gene_file) ###input is in input\Genes, denominator in
try:
denominator_file_dir = identifyGeneFiles(denom_folder,gene_file) ###input is in input\Genes, denominator in
denominator_file = string.split(denominator_file_dir,'/')[-1]
if PoolVar==False:
print 'Using:', denominator_file,'for the denominator.'
except Exception:
print_out = "WARNING: No denominator file included in\nthe Denominator directory.\nTo proceed, place all denominator\nIDs in a file in that directory."
try:
if PoolVar: q.put([print_out]); return None
except Exception: pass
ForceCriticalError(print_out)
else:
denominator_file_dir = None
if denominator_file_dir == previous_denominator_file_dir: denom_file_status = 'old'
else: denom_file_status = 'new'
if denom_file_status == 'new':
if skipDenomImport==False:
previous_denominator_file_dir = denominator_file_dir
denominator_gene_list,source_data_denom,error_message = gene_associations.importUIDsForMAPPFinderQuery(denominator_file_dir,system_codes,'no')
denom_count = len(denominator_gene_list)
if 'SwissProt' in error_message and 'WARNING!!!' not in error_message:
if len(input_gene_list)==0:
error_message+='\nNo valid input IDs found. Exiting GO-Elite.'
if PoolVar: q.put([print_out]); return None
ForceCriticalError(error_message)
else:
if PoolVar: q.put([print_out]); return None
ForceCriticalError(error_message)
elif len(error_message)>0:
if PoolVar: q.put([print_out]); return None
ForceCriticalError(error_message)
if len(denominator_gene_list) == len(input_gene_list):
print_out = 'Input and Denominator lists have identical counts.\nPlease load a propper denominator set (containing\nthe input list with all assayed gene IDs) before proceeding.'
if PoolVar: q.put([print_out]); return None
ForceCriticalError(print_out)
original_denominator_gene_list=[]
for id in denominator_gene_list: original_denominator_gene_list.append(id) ###need this to be a valid list not dictionary for permutation analysis
else:
### Pull in all MOD IDs as a surrogate denominator
denominator_gene_list = importGeneData()
original_denominator_gene_list=[]
for id in denominator_gene_list: original_denominator_gene_list.append(id)
denom_count = len(denominator_gene_list)
if len(source_data_input)>0: source_data = source_data_input ###over-ride source_data if a source was identified from the input file
if source_data != mod:
if denom_file_status == 'new':
mod_source = mod+'-'+source_data+'.txt'
try:
gene_to_source_id = gene_associations.getGeneToUid(species_code,mod_source,display=display)
if PoolVar==False:
print mod_source, 'imported'
except Exception:
try:
if mod=='EntrezGene': mod = 'Ensembl'
else: mod = 'EntrezGene'
if PoolVar==False:
print 'The primary system (MOD) has been switched from',mod_db,'to',mod,'\n('+mod_db,'not supported for the %s ID system).' % source_data
mod_source = mod+'-'+source_data+'.txt'
gene_to_source_id = gene_associations.getGeneToUid(species_code,mod_source,display=display)
except Exception:
print_out = "WARNING: The primary gene ID system '"+mod+"'\ndoes not support relationships with '"+ source_data +"'.\nRe-run using a supported primary ID system."
try:
if PoolVar: q.put([print_out]); return None
except Exception: pass
ForceCriticalError(print_out)
source_to_gene = OBO_import.swapKeyValues(gene_to_source_id)
if skipDenomImport==False:
denominator_gene_list = associateInputSourceWithGene(source_to_gene,denominator_gene_list)
### Introduced the below method in version 1.2.1 to improve permutation speed (no longer need to search all source IDs)
### Only includes source ID to gene relationships represented in the denominator file (needed for Affymetrix)
source_to_gene = OBO_import.swapKeyValues(denominator_gene_list)
###Replace input lists with corresponding MOD IDs
input_gene_list = associateInputSourceWithGene(source_to_gene,input_gene_list)
else:
if len(input_gene_list)>3:
missing = checkCorrectSystemAndSpecies(input_gene_list)
if missing:
print_out = "None of the input IDs match the selected mod (%s) or species (%s) for %s." % (mod,species_name,gene_file)
print_out += '\nVerify the correct ID system is indicated in the input file and the correct species seleced.'
try:
if PoolVar: q.put([print_out]); return None
except Exception: pass
ForceCriticalError(print_out)
if skipDenomImport==False:
checkDenominatorMatchesInput(input_gene_list,denominator_gene_list,gene_file) ###This is for only the associated MOD IDs
gd = GrabFiles(); gd.setdirectory('/'+database_dir+'/'+species_code+'/gene-mapp')
available_genesets = reorganizeResourceList(gd.getAllFiles(mod))
od = GrabFiles(); od.setdirectory('/'+database_dir+'/'+species_code+'/gene-go')
available_ontologies = reorganizeResourceList(od.getAllFiles(mod))
input_gene_count = len(input_gene_list) ###Count number of genes associated with source input IDs
if len(input_gene_list)==0 or len(denominator_gene_list)==0:
if len(input_gene_list)==0:
try:
print_out = 'WARNING!!!! None of the input IDs provided map to genes for '+mappfinder_input+'. Check to make sure the selected species is correct.'
print_out += '\nSelected species: '+species_name
print_out += '\nInput ID system: '+str(source_data_input)
print_out += '\nPrimary ID system (MOD): '+str(mod)
if PoolVar: q.put([print_out]); return None
except Exception:
pass
ForceCriticalError(print_out)
if len(denominator_gene_list)==0:
try:
print_out = 'WARNING!!!! None of the denominator IDs provided map to genes for '+denominator_file_dir+'. Check to make sure the selected species is correct.'
print_out += '\nSelected species: '+species_name
print_out += '\nDenominator ID system: '+str(source)
print_out += '\nPrimary ID system (MOD):'+str(mod)
if PoolVar: q.put([print_out]); return None
ForceCriticalError(print_out)
except Exception:
pass
elif len(available_ontologies) == 0 and len(available_genesets) == 0:
print_out = 'WARNING!!!! No Ontology or GeneSets appear to be available for this species. Please supply and re-analyze.'
try:
if PoolVar: q.put([print_out]); return None
except Exception: pass
ForceCriticalError(print_out)
else:
""" Perform permutation analysis and ORA on available GeneSets or Ontologies"""
inputs_analyzed+=1
global permute_inputs; permute_inputs=[]
if permutations != 0 or use_FET == 'no':
buildPermutationDatabase(original_denominator_gene_list,input_count)
run_status = 0
### Analyzed ontologies
if len(available_ontologies)>0:
if PoolVar==False:
print ' Analyzing input ID list with available ontologies'
for ontology_dir in available_ontologies:
ontology_type = getResourceType(ontology_dir)
permuted_z_scores={}; original_ontology_z_score_data={}
#print ontology_type, resources_to_analyze
if (resources_to_analyze == ontology_type) or (resources_to_analyze == 'all') or (resources_to_analyze == 'both' and ontology_type == 'GeneOntology'):
ontology_annotations = importOntologyAnnotations(species_code,ontology_type)
if ontology_annotations!=None: ### Occurs when the files are named or formatted correctly
status, ontology_to_mod_genes = performOntologyORA(ontology_dir)
run_status += status
### Analyzed gene-sets
if len(available_genesets)>0:
if PoolVar==False:
print ' Analyzing input ID list with available gene-sets'
""" The resources_to_analyze can be one GO-Elite category (e.g., BioMarkers) or multiple (WikiPathways, PathwayCommons).
The below code checks for an exact match or then a partial match if not specific match."""
for geneset_dir in available_genesets:
geneset_type = getResourceType(geneset_dir)
permuted_z_scores={}; original_mapp_z_score_data={}
if geneset_type == resources_to_analyze:
status, mapp_to_mod_genes = performGeneSetORA(geneset_dir)
run_status += status
elif resources_to_analyze == 'all' or (resources_to_analyze == 'both' and geneset_type == 'Pathways'):
status, mapp_to_mod_genes = performGeneSetORA(geneset_dir)
run_status += status
if len(custom_sets_folder)>0:
### Hence - Analyze User Supplied GeneSets
permuted_z_scores={}; original_mapp_z_score_data={}
run_status += performGeneSetORA('UserSuppliedAssociations')[0]
permute_inputs=[]; permute_mapp_inputs=[]
ontology_input_gene_count=[]; mapp_input_gene_count=[]
if run_status == 0:
### Returns the number of successfully analyzed gene-set databases
program_type,database_dir = unique.whatProgramIsThis()
print_out = "Warning!!! Either the MOD you have selected: "+mod+"\nis missing the appropriate relationship files necessary to run GO-Elite\nor you have selected an invalid resource to analyze. Either replace\nthe missing MOD files in "+database_dir+'/'+species_code+' sub-directories or\nselect a different MOD at run-time.'
if PoolVar: q.put([print_out]); return None
ForceCriticalError(print_out)
end_time = time.time()
time_diff = formatTime(start_time,end_time)
if PoolVar==False:
print 'ORA analyses finished in %s seconds' % time_diff
try:
q.put([ontology_to_mod_genes, mapp_to_mod_genes,time_diff,mappfinder_input,resources_to_analyze])
except Exception:
q = [ontology_to_mod_genes, mapp_to_mod_genes,time_diff,mappfinder_input,resources_to_analyze]
return q ###Return the MOD genes associated with each GO term and MAPP
def importOntologyAnnotations(species_code,ontology_type):
try:
system_codes,source_types,mod_types = GO_Elite.getSourceData()
verified_nested = OBO_import.verifyNestedFileCreation(species_code,mod_types,ontology_type)
if verified_nested == 'no': force_error
ontology_annotations = OBO_import.importPreviousOntologyAnnotations(ontology_type)
except Exception:
try:
### Occurs when the annotation file isn't built yet - if so try to build
OBO_import.buildNestedOntologyAssociations(species_code,mod_types,ontology_type,display=display)
ontology_annotations = OBO_import.importPreviousOntologyAnnotations(ontology_type)
except Exception:
### Occurs for non-ontologies
#print 'error 2',traceback.format_exc()
ontology_annotations=None
return ontology_annotations
def getResourceType(pathway_dir):
pathway_type = string.split(pathway_dir,'-')[-1][:-4]
if pathway_type == 'MAPP':
pathway_type = 'Pathways'
return pathway_type
def reorganizeResourceList(pathway_list):
### Make sure that WikiPathways and GO are analyzed last, so that gene results are also reported last to GO_Elite.py
add_pathway=[]
pathway_list_reorganized=[]
for pathway in pathway_list:
if '-MAPP.txt' in pathway: add_pathway.append(pathway)
elif '-GeneOntology.txt' in pathway: add_pathway.append(pathway)
else: pathway_list_reorganized.append(pathway)
pathway_list_reorganized+=add_pathway
return pathway_list_reorganized
def ForceCriticalError(print_out):
print '\n'+print_out+'\n'
"""
if len(sys.argv[1:])<2: ### Don't create a Tkinter window if command-line options supplied
try: UI.WarningWindow(print_out,'Error Encountered!'); root.destroy(); GO_Elite.importGOEliteParameters('yes'); #sys.exit()
except Exception: None
else:
#sys.exit()
pass
"""
def buildPermutationDatabase(original_denominator_gene_list,input_count):
if PoolVar==False:
print "Building %d permuted ID sets" % permutations,
global k; k=0; x=0
try: original_increment = int(permutations/10); increment = original_increment
except Exception: null=None
if PoolVar==False:
if permutations!=0: print '*',
start_time = time.time() ### Build Permutation Identifier Database
while x<permutations:
if x == increment and PoolVar==False:
increment+=original_increment; print '*',
try: permute_input_list = random.sample(original_denominator_gene_list,input_count); x+=1
except ValueError:
print_out = 'Input count>Denominator '+str(len(original_denominator_gene_list))+' '+str(input_count)+'\n terminating'
if PoolVar: q.put([print_out]); return None
ForceCriticalError(print_out)
#permute_input_list = random.sample(denominator_gene_list,len(input_gene_list)); x+=1
#permute_input_list = random.shuffle(original_denominator_gene_list); x+=1; permute_input_list = permute_input_list[:input_count]
if source_data!=mod: ###Store the randomly choosen input lists for GenMAPP MAPP Permutation analysis
permute_input_list = associateInputSourceWithGene(source_to_gene,permute_input_list)
if len(permute_input_list)>len(input_gene_list): k+=1
permute_inputs.append(permute_input_list)
end_time = time.time()
time_diff = formatTime(start_time,end_time)
if PoolVar==False:
print 'completed in %s seconds' % time_diff
def swapKeyValues(db):
rev_db = {}
for i, k_list in db.iteritems():
for k in k_list:
try: rev_db[k].append(i)
except Exception: rev_db[k] = [i]
return rev_db
def performGeneSetORA(geneset_dir):
""" Perform over-representation analysis (ORA) on any provided Gene Set """
start_time = time.time()
geneset_type = getResourceType(geneset_dir)
#permuted_z_scores={}; original_mapp_z_score_data={}
if geneset_type == 'Pathways': geneset_type = 'WikiPathways'
### Since MAPP tables can be provided by the user, allow the file to be missing
if geneset_dir == 'UserSuppliedAssociations':
gene_to_mapp = gene_associations.importGeneCustomData(species_code,system_codes,custom_sets_folder,mod)
geneset_type = geneset_dir
else:
try: gene_to_mapp = gene_associations.importGeneMAPPData(species_code,geneset_dir)
except Exception: gene_to_mapp = {}
mapp_to_gene = swapKeyValues(gene_to_mapp)
if len(gene_to_mapp)==0:
return 0, None
else:
###Calculate primary z-scores for GeneSets
#print len(input_gene_list), len(gene_to_mapp)
mapp_to_mod_genes = getGenesInPathway(input_gene_list,gene_to_mapp) ### For summary reporting
mapp_input_gene_count,Rm,input_linked_mapp = countGenesInPathway(input_gene_list,gene_to_mapp,'yes')
mapp_denominator_gene_count,Nm,denom_linked_mapp = countGenesInPathway(denominator_gene_list,gene_to_mapp,'yes')
#print Nm,"unique genes, linked to GeneSets and in dataset and", Rm, "unique GeneSets\n linked genes matching criterion."
#zstart_time = time.time()
try:
#exhaustiveMultiZScores(mapp_input_gene_count,mapp_denominator_gene_count,Nm,Rm,mapp_to_gene,'MAPP')
if PoolVar==False:
try: multiZScores(mapp_input_gene_count,mapp_denominator_gene_count,Nm,Rm,mapp_to_gene,'MAPP')
except Exception: calculateZScores(mapp_input_gene_count,mapp_denominator_gene_count,Nm,Rm,mapp_to_gene,'MAPP')
else:
calculateZScores(mapp_input_gene_count,mapp_denominator_gene_count,Nm,Rm,mapp_to_gene,'MAPP')
except Exception:
calculateZScores(mapp_input_gene_count,mapp_denominator_gene_count,Nm,Rm,mapp_to_gene,'MAPP')
#print len(permuted_z_scores), len(original_mapp_z_score_data)
#print time.time()-zstart_time, 'seconds...';sys.exit()
if use_FET == 'no':
permute_mapp_inputs=[]
###Begin GeneSets Permutation Analysis
try: original_increment = int(permutations/10); increment = original_increment
except Exception: null=None
x=0
if PoolVar==False:
if permutations!=0: print '*',
for permute_input_list in permute_inputs:
if PoolVar==False:
if x == increment: increment+=original_increment; print '*',
x+=1
permute_mapp_input_gene_count,null,null = countGenesInPathway(permute_input_list,gene_to_mapp,'no')
permute_mapp_inputs.append(permute_mapp_input_gene_count)
calculatePermuteZScores(permute_mapp_inputs,mapp_denominator_gene_count,Nm,Rm)
calculatePermuteStats(original_mapp_z_score_data)
adjustPermuteStats(original_mapp_z_score_data)
mapp_headers = formatHeaders(gene_file,input_count,input_linked_mapp,denom_count,denom_linked_mapp,Rm,Nm,'MAPP',OBO_date)
exportPathwayData(original_mapp_z_score_data,gene_file,mapp_headers,geneset_type,'local')
### Export all gene associations (added in version 1.21)
exportPathwayToGeneAssociations(mapp_to_mod_genes,mod,gene_file,gene_annotations,geneset_type,'local')
end_time = time.time()
time_diff = formatTime(start_time,end_time)
if PoolVar==False:
print "Initial results for %s calculated in %s seconds" % (geneset_type,time_diff)
permute_mapp_inputs=[]
return 1, mapp_to_mod_genes
def performOntologyORA(ontology_dir):
""" Perform over-representation analysis (ORA) on any provided Ontology """
start_time = time.time()
ontology_type = getResourceType(ontology_dir)
######### Import Gene-to-Nested-Ontology #########
gene_to_ontology = gene_associations.importGeneToOntologyData(species_code,mod,'nested',ontology_type)
ontology_to_gene = OBO_import.swapKeyValues(gene_to_ontology)
if len(gene_to_ontology)==0:
return 0, None
else:
######### Calculate primary z-scores for GO terms
#a = time.time()
ontology_to_mod_genes = getGenesInPathway(input_gene_list,gene_to_ontology) ### For summary gene reporting
#b = time.time(); print 'a',b-a
ontology_input_gene_count,Rg,input_linked_ontology = countGenesInPathway(input_gene_list,gene_to_ontology,'yes')
#c = time.time(); print 'b',c-b
ontology_denominator_gene_count,Ng,denom_linked_ontology = countGenesInPathway(denominator_gene_list,gene_to_ontology,'yes')
#d = time.time(); print 'c',d-c
#print Ng,"unique genes, linked to GO and in dataset and", Rg, "unique GO linked genes matching criterion."
try:
if PoolVar==False:
multiZScores(ontology_input_gene_count,ontology_denominator_gene_count,Ng,Rg,ontology_to_gene,'Ontology')
else:
calculateZScores(ontology_input_gene_count,ontology_denominator_gene_count,Ng,Rg,ontology_to_gene,'Ontology')
except Exception: calculateZScores(ontology_input_gene_count,ontology_denominator_gene_count,Ng,Rg,ontology_to_gene,'Ontology')
#e = time.time(); print 'd',e-d; sys.exit()
if use_FET == 'no':
###Begining Ontology Permutation Analysis
try: original_increment = int(permutations/10); increment = original_increment
except Exception: null=None
x=0
permute_ontology_inputs=[]
if PoolVar==False:
if permutations!=0: print '*',
for permute_input_list in permute_inputs:
### http://docs.python.org/library/multiprocessing.html
if PoolVar==False:
if x == increment: increment+=original_increment; print '*',
x+=1
permute_ontology_input_gene_count,null,null = countGenesInPathway(permute_input_list,gene_to_ontology,'no'); permute_input_list=[]
permute_ontology_inputs.append(permute_ontology_input_gene_count)
if PoolVar==False:
if permutations !=0: print 'Ontology finished'
calculatePermuteZScores(permute_ontology_inputs,ontology_denominator_gene_count,Ng,Rg)
calculatePermuteStats(original_ontology_z_score_data)
adjustPermuteStats(original_ontology_z_score_data)
go_headers = formatHeaders(gene_file,input_count,input_linked_ontology,denom_count,denom_linked_ontology,Rg,Ng,'Ontology',OBO_date)
exportPathwayData(original_ontology_z_score_data,gene_file,go_headers,ontology_type,'Ontology')
### Export all gene associations (added in version 1.21)
exportPathwayToGeneAssociations(ontology_to_mod_genes,mod,gene_file,gene_annotations,ontology_type,'Ontology')
end_time = time.time()
time_diff = formatTime(start_time,end_time)
if PoolVar==False:
print "Initial results for %s calculated in %s seconds" % (ontology_type,time_diff)
permute_ontology_inputs=[]
return 1, ontology_to_mod_genes
def exportPathwayToGeneAssociations(pathway_to_mod_genes,mod,gene_file,gene_annotations,resource_name,pathway_type):
headers = string.join([mod,'symbol',resource_name],'\t')+'\n'
if resource_name == 'GeneOntology': resource_name = 'GO' ### Makes the output filename compatible with GenMAPP-CS plugin filenames
if resource_name == 'WikiPathways': resource_name = 'local' ### Makes the output filename compatible with GenMAPP-CS plugin filenames
new_file = mappfinder_output_dir+'/'+gene_file[:-4]+'-'+resource_name+'-associations.tab'
data = export.ExportFile(new_file); data.write(headers)
for pathway in pathway_to_mod_genes:
for gene in pathway_to_mod_genes[pathway]:
try: symbol = gene_annotations[gene].Symbol()
except Exception: symbol = ''
if pathway_type == 'Ontology' and ':' not in pathway: pathway = 'GO:'+ pathway
values = string.join([gene,symbol,pathway],'\t')+'\n'
data.write(values)
data.close()
def formatHeaders(gene_file,input_count,input_linked,denom_count,denom_linked,R,N,pathway_type,OBO_date):
headers = []
headers.append('GO-Elite ORA Results')
headers.append('File:')
headers.append('Table:')
if pathway_type == 'Ontology':
headers.append('Database: Based on OBO-Database version: '+OBO_date)
headers.append('colors:')
t = time.localtime(); dt = str(t[1])+'/'+str(t[2])+'/'+str(t[0])
headers.append(dt)
headers.append(species_name)
headers.append('Pvalues = true')
headers.append('Calculation Summary:')
headers.append(str(input_count)+' '+source_data+' source identifiers supplied in the input file:'+gene_file)
headers.append(str(input_linked)+' source identifiers meeting the filter linked to a '+mod+' ID.')
headers.append(str(R)+' genes meeting the criterion linked to a term.')
headers.append(str(denom_count)+' source identifiers in this dataset.')
headers.append(str(denom_linked)+' source identifiers linked to a '+mod+' ID.')
headers.append(str(N)+' Genes linked to a term.')
headers.append('The z score is based on an N of '+str(N)+' and a R of '+str(R)+' distinct genes in all terms.\n')
if use_FET == 'yes': prob = "FisherExactP"
else: prob = "PermuteP"
if pathway_type == 'Ontology':
title = ['Ontology-ID','Ontology Name','Ontology Type','Number Changed','Number Measured','Number in Ontology','Percent Changed','Percent Present','Z Score',prob,'AdjustedP']
title = string.join(title,'\t'); headers.append(title)
else:
title = ['Gene-Set Name','Number Changed','Number Measured','Number in Gene-Set','Percent Changed','Percent Present','Z Score',prob,'AdjustedP']
title = string.join(title,'\t'); headers.append(title)
header_str = string.join(headers,'\n')
return header_str+'\n'
def exportPathwayData(original_pathway_z_score_data,gene_file,headers,resource_name,pathway_type,altOutputDir=None):
if resource_name == 'GeneOntology': resource_name = 'GO' ### Makes the output filename compatible with GenMAPP-CS plugin filenames
if resource_name == 'WikiPathways': resource_name = 'local' ### Makes the output filename compatible with GenMAPP-CS plugin filenames
new_file = mappfinder_output_dir+'/'+gene_file[:-4]+'-'+resource_name+'.txt'
global sort_results
data = export.ExportFile(new_file); data.write(headers); sort_results=[]
#print "Results for",len(original_pathway_z_score_data),"pathways exported to",new_file
for pathway in original_pathway_z_score_data:
zsd=original_pathway_z_score_data[pathway]
try: results = [zsd.Changed(), zsd.Measured(), zsd.InPathway(), zsd.PercentChanged(), zsd.PercentPresent(), zsd.ZScore(), zsd.PermuteP(), zsd.AdjP()]
except AttributeError:
return traceback.format_exc()
#print pathway,len(permuted_z_scores[pathway]);kill
try: ###This is unnecessary, unless using the non-nested GO associations (which can have out of sync GOIDs)
if pathway_type == 'Ontology':
s = ontology_annotations[pathway]
annotations = [s.OntologyID(),s.OntologyTerm(),s.OntologyType()]; results = annotations + results
else:
results = [pathway] + results
results = string.join(results,'\t') + '\n'
sort_results.append([float(zsd.ZScore()),-1/float(zsd.Measured()),results])
except KeyError: null = []
sort_results.sort(); sort_results.reverse()
for values in sort_results:
results = values[2]
data.write(results)
data.close()
def swapKeyValuesTuple(db):
swapped={}
for key in db:
values = tuple(db[key]) ###If the value is not a list, make a list
swapped[values] = [key]
swapped = eliminate_redundant_dict_values(swapped)
return swapped
class ZScoreData:
def __init__(self,pathway,changed,measured,zscore,null_z,in_pathway):
self._pathway = pathway; self._changed = changed; self._measured = measured
self._zscore = zscore; self._null_z = null_z; self._in_pathway = in_pathway
def PathwayID(self): return self._pathway
def Changed(self): return str(int(self._changed))
def Measured(self): return str(int(self._measured))
def InPathway(self): return str(self._in_pathway)
def ZScore(self): return str(self._zscore)
def SetP(self,p): self._permute_p = p
def PermuteP(self): return str(self._permute_p)
def SetAdjP(self,adjp): self._adj_p = adjp
def AdjP(self): return str(self._adj_p)
def setAssociatedIDs(self,ids): self.ids = ids
def AssociatedIDs(self): return self.ids
def PercentChanged(self):
try: pc = float(self.Changed())/float(self.Measured())*100
except Exception: pc = 0
return str(pc)
def PercentPresent(self):
try: pp = float(self.Measured())/float(self.InPathway())*100
except Exception: pp = 0
return str(pp)
def NullZ(self): return self._null_z
def Report(self):
output = self.PathwayID()
return output
def __repr__(self): return self.Report()
def workerExhaustive(queue,pathway,genes_in_pathway,pathway_input_gene_count,pathway_denominator_gene_count,N,R,pathway_type):
permuted_z_scores_instance={}; original_mapp_z_score_data_instance={}; original_ontology_z_score_data_instance={}
""" Exhaustive multiprocessing execution """
try:
n = pathway_denominator_gene_count[pathway]
try: r = pathway_input_gene_count[pathway]
except Exception: r = 0.0000
except Exception: n = 0.0000; r = 0.0000
if n != 0:
try: z = Zscore(r,n,N,R)
except ZeroDivisionError: z = 0.0000
try: null_z = Zscore(0,n,N,R)
except ZeroDivisionError: null_z = 0.000
zsd = ZScoreData(pathway,r,n,z,null_z,genes_in_pathway)
if pathway_type == 'Ontology': original_ontology_z_score_data_instance[pathway] = zsd
else: original_mapp_z_score_data_instance[pathway] = zsd
permuted_z_scores_instance[pathway] = [z]
#if '06878' in pathway: print pathway, z, null_z, r,n, N, R;kill
if use_FET == 'yes':
### Alternatively calculate p using the Fisher's Exact Test
p = FishersExactTest(r,n,R,N)
zsd.SetP(p)
queue.put((permuted_z_scores_instance,original_mapp_z_score_data_instance,original_ontology_z_score_data_instance))
def exhaustiveMultiZScores(pathway_input_gene_count,pathway_denominator_gene_count,N,R,pathway_db,pathway_type):
""" Exhaustive multiprocessing - create a process for each entry in the dictionary for Z-score calcualtion """
procs=list()
queue = mlp.Queue()
for pathway in pathway_db:
genes_in_pathway = len(pathway_db[pathway])
p = mlp.Process(target=workerExhaustive, args=(queue,pathway,genes_in_pathway,pathway_input_gene_count,pathway_denominator_gene_count,N,R,pathway_type))
procs.append(p)
p.start()
permuted_z_scores_list=[]; original_mapp_z_score_data_list=[]; original_ontology_z_score_data_list=[]
for _ in procs:
val = queue.get()
permuted_z_scores_list.append(val[0]); original_mapp_z_score_data_list.append(val[1])
original_ontology_z_score_data_list.append(val[2])
for p in procs:
p.join()
for i in permuted_z_scores_list: permuted_z_scores.update(i)
for i in original_mapp_z_score_data_list: original_mapp_z_score_data.update(i)
for i in original_ontology_z_score_data_list: original_ontology_z_score_data.update(i)
def multiZScores(pathway_input_gene_count,pathway_denominator_gene_count,N,R,pathway_db,pathway_type):
""" Create a finate pool of processes (4) for Z-score calculation """
if mlp.cpu_count() < 3:
processors = mlp.cpu_count()
else: processors = 4
pool = mlp.Pool(processes=processors)
si = (len(pathway_db)/processors)
s = si; b=0
db_ls=[]
if len(pathway_db)<10: forceError ### will si to be zero and an infanite loop
while s<len(pathway_db):
db_ls.append(dict(pathway_db.items()[b:s]))
b+=si; s+=si
db_ls.append(dict(pathway_db.items()[b:s]))
### Create an instance of MultiZscoreWorker (store the variables to save memory)
workerMulti = MultiZscoreWorker(pathway_input_gene_count,pathway_denominator_gene_count,N,R,pathway_type,use_FET)
results = pool.map(workerMulti,db_ls)
pool.close(); pool.join(); pool = None
permuted_z_scores_list=[]; original_mapp_z_score_data_list=[]; original_ontology_z_score_data_list=[]
for (a,b,c) in results:
permuted_z_scores_list.append(a); original_mapp_z_score_data_list.append(b)
original_ontology_z_score_data_list.append(c)
for i in permuted_z_scores_list: permuted_z_scores.update(i)
for i in original_mapp_z_score_data_list: original_mapp_z_score_data.update(i)
for i in original_ontology_z_score_data_list: original_ontology_z_score_data.update(i)
class MultiZscoreWorker:
def __init__(self,pathway_input_gene_count,pathway_denominator_gene_count,N,R,pathway_type,use_FET):
self.pathway_input_gene_count = pathway_input_gene_count
self.pathway_denominator_gene_count = pathway_denominator_gene_count
self.N = N
self.R = R
self.pathway_type = pathway_type
self.use_FET = use_FET
def __call__(self,pathway_db):
N = self.N; R = self.R; use_FET = self.use_FET
zt=0; nzt=0; zsdt=0; rt=0; ft=0
permuted_z_scores_instance={}; original_mapp_z_score_data_instance={}; original_ontology_z_score_data_instance={}
for pathway in pathway_db:
genes_in_pathway = len(pathway_db[pathway])
try:
n = self.pathway_denominator_gene_count[pathway]
#r1 = time.time()
try: r = self.pathway_input_gene_count[pathway]
except Exception: r = 0.0000
#rt += time.time() - r1
except Exception: n = 0.0000; r = 0.0000
if n != 0:
z1 = time.time()
try: z = Zscore(r,n,N,R)
except ZeroDivisionError: z = 0.0000
#zt += time.time() - z1
#nz1 = time.time()
try: null_z = Zscore(0,n,N,R)
except ZeroDivisionError: null_z = 0.000
#nzt+= time.time() - nz1
#zsd1 = time.time()
zsd = ZScoreData(pathway,r,n,z,null_z,genes_in_pathway)
#zsdt+= time.time() - zsd1
if self.pathway_type == 'Ontology': original_ontology_z_score_data_instance[pathway] = zsd
else: original_mapp_z_score_data_instance[pathway] = zsd
permuted_z_scores_instance[pathway] = [z]
#if '06878' in pathway: print pathway, z, null_z, r,n, N, R;kill
if use_FET == 'yes':
### Alternatively calculate p using the Fisher's Exact Test
#ft1 = time.time()
p = FishersExactTest(r,n,R,N)
#ft+= time.time() - ft1
zsd.SetP(p)
#print zt,nzt,zsdt,rt,ft ### Used for efficiency evaluation
return permuted_z_scores_instance,original_mapp_z_score_data_instance, original_ontology_z_score_data_instance
def calculateZScores(pathway_input_gene_count,pathway_denominator_gene_count,N,R,pathway_db,pathway_type):
"""where N is the total number of genes measured:
R is the total number of genes meeting the criterion:
n is the total number of genes in this specific MAPP:
r is the number of genes meeting the criterion in this MAPP: """
for pathway in pathway_db:
try:
n = pathway_denominator_gene_count[pathway]
try: r = pathway_input_gene_count[pathway]
except Exception: r = 0.0000
except Exception: n = 0.0000; r = 0.0000
if n != 0:
try: z = Zscore(r,n,N,R)
except ZeroDivisionError: z = 0.0000
try: null_z = Zscore(0,n,N,R)
except ZeroDivisionError: null_z = 0.000
genes_in_pathway = len(pathway_db[pathway])
zsd = ZScoreData(pathway,r,n,z,null_z,genes_in_pathway)
if pathway_type == 'Ontology': original_ontology_z_score_data[pathway] = zsd
else: original_mapp_z_score_data[pathway] = zsd
permuted_z_scores[pathway] = [z]
#if '06878' in pathway: print pathway, z, null_z, r,n, N, R;kill
if use_FET == 'yes':
### Alternatively calculate p using the Fisher's Exact Test
p = FishersExactTest(r,n,R,N)
zsd.SetP(p)
def Zscore(r,n,N,R):
N=float(N) ### This bring all other values into float space
z = (r - n*(R/N))/math.sqrt(n*(R/N)*(1-(R/N))*(1-((n-1)/(N-1))))
return z
def calculatePermuteZScores(permute_pathway_inputs,pathway_denominator_gene_count,N,R):
for pathway_input_gene_count in permute_pathway_inputs:
for pathway in pathway_input_gene_count:
r = pathway_input_gene_count[pathway]
n = pathway_denominator_gene_count[pathway]
try: z = statistics.zscore(r,n,N,R)
except ZeroDivisionError: z = 0
permuted_z_scores[pathway].append(abs(z))
#if pathway == '0005488':
#a.append(r)
def calculatePermuteStats(original_pathway_z_score_data):
for pathway in original_pathway_z_score_data:
zsd = original_pathway_z_score_data[pathway]
z = abs(permuted_z_scores[pathway][0])
permute_scores = permuted_z_scores[pathway][1:] ###Exclude the true value
nullz = zsd.NullZ()
if abs(nullz) == z: ###Only add the nullz values if they can count towards the p-value (if equal to the original z)
null_z_to_add = permutations - len(permute_scores)
permute_scores+=[abs(nullz)]*null_z_to_add ###Add null_z's in proportion to the amount of times there were not genes found for that pathway
if len(permute_scores)>0: p = permute_p(permute_scores,z)
else: p = 0
#if p>1: p=1
zsd.SetP(p)
def adjustPermuteStats(original_pathway_z_score_data):
#1. Sort ascending the original input p value vector. Call this spval. Keep the original indecies so you can sort back.
#2. Define a new vector called tmp. tmp= spval. tmp will contain the BH p values.
#3. m is the length of tmp (also spval)
#4. i=m-1
#5 tmp[ i ]=min(tmp[i+1], min((m/i)*spval[ i ],1)) - second to last, last, last/second to last
#6. i=m-2
#7 tmp[ i ]=min(tmp[i+1], min((m/i)*spval[ i ],1))
#8 repeat step 7 for m-3, m-4,... until i=1
#9. sort tmp back to the original order of the input p values.
global spval; spval=[]; adj_p_list=[]
for pathway in original_pathway_z_score_data:
zsd = original_pathway_z_score_data[pathway]
p = float(zsd.PermuteP())
spval.append([p,pathway])
spval.sort(); tmp = spval; m = len(spval); i=m-2; x=0 ###Step 1-4
l=0
while i > -1:
adjp = min(tmp[i+1][0], min((float(m)/(i+1))*spval[i][0],1))
tmp[i]=adjp,tmp[i][1]; i -= 1
if adjp !=0: adj_p_list.append(adjp) ### get the minimum adjp
for (adjp,pathway) in tmp:
try:
if adjp == 0: adjp = min(adj_p_list)
except Exception: null=[]
zsd = original_pathway_z_score_data[pathway]
zsd.SetAdjP(adjp)
def adjustPermuteStatsTemp(pval_db):
global spval; spval=[]
for element in pval_db:
zsd = pval_db[element]
try:
try: p = float(zsd.PermuteP())
except AttributeError: p = float(zsd[0]) ### When values are indeces rather than objects
except Exception: p = 1
spval.append([p,element])
spval.sort(); tmp = spval; m = len(spval); i=m-2; x=0 ###Step 1-4
#spval.sort(); tmp = spval; m = len(spval)-1; i=m-1; x=0 ###Step 1-4
while i > -1:
tmp[i]=min(tmp[i+1][0], min((float(m)/(i+1))*spval[i][0],1)),tmp[i][1]; i -= 1
for (adjp,element) in tmp:
zsd = pval_db[element]
try: zsd.SetAdjP(adjp)
except AttributeError: zsd[1] = adjp ### When values are indeces rather than objects
def permute_p(null_list,true_value):
y = 0; z = 0; x = permutations
for value in null_list:
if value >= true_value: y += 1
#if true_value > 8: global a; a = null_list; print true_value,y,x;kill
return (float(y)/float(x)) ###Multiply probabilty x2?
def FishersExactTest(r,n,R,N):
"""
N is the total number of genes measured (Ensembl linked from denom) (total number of ) (number of exons evaluated)
R is the total number of genes meeting the criterion (Ensembl linked from input) (number of exonic/intronic regions overlaping with any CLIP peeks)
n is the total number of genes in this specific MAPP (Ensembl denom in MAPP) (number of exonic/intronic regions associated with the SF)
r is the number of genes meeting the criterion in this MAPP (Ensembl input in MAPP) (number of exonic/intronic regions with peeks overlapping with the SF)
With these values, we must create a 2x2 contingency table for a Fisher's Exact Test
that reports:
+---+---+ a is the # of IDs in the term regulated
| a | b | b is the # of IDs in the term not-regulated
+---+---+ c is the # of IDs not-in-term and regulated
| c | d | d is the # of IDs not-in-term and not-regulated
+---+---+
If we know r=20, R=80, n=437 and N=14480
+----+-----+
| 20 | 417 | 437
+----+-----+
| 65 |13978| 14043
+----+-----+
85 14395 14480
"""
a = r; b = n-r; c=R-r; d=N-R-b
table = [[int(a),int(b)], [int(c),int(d)]]
"""
print a,b; print c,d
from stats_scripts import fishers_exact_test; table = [[a,b], [c,d]]
ft = fishers_exact_test.FishersExactTest(table)
print ft.probability_of_table(table); print ft.two_tail_p()
print ft.right_tail_p(); print ft.left_tail_p()
"""
try: ### Scipy version - cuts down rutime by ~1/3rd the time
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
oddsratio, pvalue = stats.fisher_exact(table)
return pvalue
except Exception:
ft = fishers_exact_test.FishersExactTest(table)
return ft.two_tail_p()
def getGenesInPathway(gene_list,gene_to_pathway):
###This function is similar to countGenesInPathway, but is used to return the genes associated with a pathway
### Can be used to improve downstream annotation speed when this file is present rather than re-derive
pathway_to_gene={}
for gene in gene_list:
if gene in gene_to_pathway:
pathways = gene_to_pathway[gene]
for pathway in pathways:
try: pathway_to_gene[pathway].append(gene)
except KeyError: pathway_to_gene[pathway] = [gene]
return pathway_to_gene
def countGenesInPathway(gene_list,gene_to_pathway,count_linked_source):
pathway_count={}; associated_genes={}; linked_source={}
### Add genes to a dictionary of pathways to get unique counts (could count directly, but biased by redundant source-id associations with MOD)
for gene in gene_list:
if source_data != mod and eliminate_redundant_genes == 'yes':
gene_id = tuple(gene_list[gene]) ### switches gene with list of source_ids (if made unique, decreased redundant association)
if count_linked_source == 'yes':
for id in gene_id: linked_source[id] = []
else: gene_id = gene; linked_source[gene_id] = []
try:
pathways = gene_to_pathway[gene]
associated_genes[gene_id] = []
for pathway in pathways:
try: pathway_count[pathway][gene_id]=None ### more efficent storage for length determination below
except: pathway_count[pathway] = {gene_id:None}
except Exception: pass
### Count unique gene or source set associations per pathway
unique_associated_gene_count = len(associated_genes)
linked_count = len(linked_source)
for pathway in pathway_count:
pathway_count[pathway] = len(pathway_count[pathway])
return pathway_count, unique_associated_gene_count, linked_count
if __name__ == '__main__':
#r=20; R=85; n=437; N=14480
species_name = 'Galus galus'; species_code = 'Gg'; source_data = 'EnsTranscript'; mod = 'Ensembl'
species_name = 'Mus musculus'; species_code = 'Mm'; source_data = 'EntrezGene'; mod = 'EntrezGene'
species_name = 'Homo sapiens'; species_code = 'Hs'; source_data = 'Ensembl'; mod = 'Ensembl'
system_codes={}; system_codes['L'] = 'EntrezGene'; system_codes['En'] = 'Ensembl'; system_codes['X'] = 'Affymetrix'
file_dirs = 'C:/Documents and Settings/Nathan/Desktop/GenMAPP/Mm_sample/input_list_small','C:/Documents and Settings/Nathan/Desktop/GenMAPP/Mm_sample/denominator','C:/Documents and Settings/Nathan/Desktop/GenMAPP/Mm_sample'
file_dirs = '/Users/nsalomonis/Desktop/GOElite-test/input','/Users/nsalomonis/Desktop/GOElite-test/denom','/Users/nsalomonis/Desktop/GOElite-test','/Users/nsalomonis/Desktop/GOElite-test/miR'
permute = 20000
permute = 'FisherExactTest'
generateMAPPFinderScores(species_name,species_code,source_data,mod,system_codes,permute,'all',file_dirs,'')
#!/usr/bin/python
###########################
#Program: GO-elite.py
#Author: Nathan Salomonis
#Date: 12/12/06
#Website: http://www.genmapp.org
#Email: [email protected]
###########################
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/mappfinder.py
|
mappfinder.py
|
import matplotlib
import matplotlib.pyplot as pylab
from matplotlib.path import Path
import matplotlib.patches as patches
import numpy
import string
import time
import random
import math
import sys, os
import sqlite3
import export
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def filepath(filename):
try:
import unique ### local to AltAnalyze
fn = unique.filepath(filename)
except Exception:
### Should work fine when run as a script with this (AltAnalyze code is specific for packaging with AltAnalyze)
dir=os.path.dirname(dirfile.__file__)
try: dir_list = os.listdir(filename); fn = filename ### test to see if the path can be found (then it is the full path)
except Exception: fn=os.path.join(dir,filename)
return fn
##### SQLite Database Access ######
def createSchemaTextFile():
schema_filename = 'relational_databases/AltAnalyze_schema.sql'
export_data = export.ExportFile(schema_filename)
schema_text ='''-- Schema for species specific AltAnalyze transcript data.
-- Genes store general information on each Ensembl gene ID
create table genes (
id text primary key,
name text,
description text,
chr text,
strand text
);
-- Exon_regions store coordinates and annotations for AltAnalyze defined unique exon regions
create table exons (
id text,
start integer,
end integer,
gene text not null references genes(id)
);
-- Junctions store coordinates (5' donor and 3' acceptor splice site) and annotations
create table junctions (
id text,
start integer,
end integer,
gene text not null references genes(id)
);
-- Transcripts store basic transcript data
create table transcripts (
id text primary key,
gene text not null references genes(id)
);
-- CDS store basic transcript data
create table cds (
id text primary key references transcripts(id),
start integer,
end integer
);
-- Transcript_exons store coordinates for Ensembl and UCSC defined transcript exons
create table transcript_exons (
start integer,
end integer,
transcript text not null references transcripts(id)
);
-- Proteins store basic protein info
create table proteins (
id text primary key,
genome_start integer,
genome_end integer,
transcript text not null references transcripts(id)
);
-- Protein_features store InterPro and UniProt protein feature info
create table protein_feature (
id text,
name text,
genome_start integer,
genome_end integer,
protein text not null references proteins(id)
);
'''
### We will need to augment the database with protein feature annotations for
export_data.write(schema_text)
export_data.close()
def populateSQLite(species,platform):
global conn
""" Since we wish to work with only one gene at a time which can be associated with a lot of data
it would be more memory efficient transfer this data to a propper relational database for each query """
db_filename = filepath('AltDatabase/'+species+'/'+platform+'/AltAnalyze.db') ### store in user directory
schema_filename = filepath('AltDatabase/'+species+'/'+platform+'/AltAnalyze_schema.sql')
### Check to see if the database exists already and if not creat it
db_is_new = not os.path.exists(db_filename)
with sqlite3.connect(db_filename) as conn:
if db_is_new:
createSchemaTextFile()
print 'Creating schema'
with open(schema_filename, 'rt') as f:
schema = f.read()
conn.executescript(schema)
print 'Inserting initial data'
species = 'Hs'
importAllTranscriptData(species)
else:
print 'Database exists, assume schema does too.'
retreiveDatabaseFields()
sys.exit()
#return conn
def retreiveDatabaseFields():
""" Retreive data from specific fields from the database """
cursor = conn.cursor()
id = 'ENSG00000114127'
query = "select id, name, description, chr, strand from genes where id = ?"
cursor.execute(query,(id,)) ### In this way, don't have to use %s and specify type
#if only one entry applicable
#id, name, description, chr, strand = cursor.fetchone()
#print '%s %s %s %s %s' % (id, name, description, chr, strand);sys.exit()
for row in cursor.fetchall():
id, name, description, chr, strand = row
print '%s %s %s %s %s' % (id, name, description, chr, strand)
def bulkLoading():
import csv
import sqlite3
import sys
db_filename = 'todo.db'
data_filename = sys.argv[1]
SQL = """insert into task (details, priority, status, deadline, project)
values (:details, :priority, 'active', :deadline, :project)
"""
with open(data_filename, 'rt') as csv_file:
csv_reader = csv.DictReader(csv_file)
with sqlite3.connect(db_filename) as conn:
cursor = conn.cursor()
cursor.executemany(SQL, csv_reader)
def verifyFile(filename):
fn=filepath(filename)
try:
for line in open(fn,'rU').xreadlines(): found = True; break
except Exception: found = False
return found
def isoformViewer():
"""
Make a "broken" horizontal bar plot, ie one with gaps
"""
fig = pylab.figure()
ax = fig.add_subplot(111)
ax.broken_barh([ (110, 30), (150, 10) ] , (10, 5), facecolors=('gray','blue')) # (position, length) - top row
ax.broken_barh([ (10, 50), (100, 20), (130, 10)] , (20, 5),
facecolors=('red', 'yellow', 'green')) # (position, length) - next row down
### Straight line
pylab.plot((140,150),(12.5,12.5),lw=2,color = 'red') ### x coordinates of the line, y-coordinates of the line, line-thickness - iterate through a list of coordinates to do this
### Curved line
verts = [
(140, 15), # P0
(145, 20), # (x coordinate, half distance to this y coordinate)
(150, 15), # P2
]
codes = [Path.MOVETO,
Path.CURVE4,
Path.CURVE4,
]
path = Path(verts, codes)
patch = patches.PathPatch(path, facecolor='none', lw=2, edgecolor = 'green')
ax.add_patch(patch)
#midpt = cubic_bezier(pts, .5)
ax.text(142, 17.7, '25 reads')
### End-curved line
ax.set_ylim(5,35)
ax.set_xlim(0,200)
ax.set_xlabel('Transcript Exons')
ax.set_yticks([15,25])
ax.set_yticklabels(['isoform A', 'isoform B'])
ax.grid(True)
"""
ax.annotate('alternative splice site', (61, 25),
xytext=(0.8, 0.9), textcoords='axes fraction',
arrowprops=dict(facecolor='black', shrink=0.05),
fontsize=16,
horizontalalignment='right', verticalalignment='top')
"""
pylab.show()
def countReadsGenomicInterval():
### Could use an interval tree implementation - http://informatics.malariagen.net/2011/07/07/using-interval-trees-to-query-genome-annotations-by-position/
""" other options -
http://www.biostars.org/post/show/99/fast-interval-intersection-methodologies/
PMID: 17061921
http://pypi.python.org/pypi/fastinterval
"""
a = numpy.array([0,1,2,3,4,5,6,7,8,9]) ### could be genomic start of reads from a chromosome
count = ((25 < a) & (a < 100)).sum() ### gives the total count
class FeatureData:
def __init__(self,start,end,annotation):
self.start = start; self.end = end; self.annotation = annotation
def Start(self): return self.start
def End(self): return self.end
def Annotation(self): return self.annotation
class GeneData:
def __init__(self,chr,strand):
self.chr = chr; self.strand = strand
def setAnnotations(self,symbol,description):
self.symbol = symbol; self.description = description
def Chr(self): return self.chr
def Strand(self): return self.strand
def Symbol(self): return self.symbol
def Description(self): return self.description
class TranscriptData:
def __init__(self,strand,start,stop):
self.start = start; self.stop = stop; self.strand = strand
def Strand(self): return self.strand
def Start(self): return self.start
def Stop(self): return self.stop
def importExonAnnotations(species,type):
start_time = time.time()
if 'exons' in type:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt'
else:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_junction.txt'
fn=filepath(filename); x=0; exon_annotation_db={}; gene_annotation_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: x=1
else:
gene, exonid, chr, strand, start, end, constitutive_call, ens_exon_ids, splice_events, splice_junctions = t; proceed = 'yes'
if proceed == 'yes': #if gene == 'ENSG00000111671':
if type == 'junctions':
exon1_start,exon1_end = string.split(start,'|')
exon2_start,exon2_end = string.split(end,'|')
if strand == '-':
exon1_end,exon1_start = exon1_start,exon1_end
exon2_end,exon2_start = exon2_start,exon2_end
start = int(exon1_end); end = int(exon2_start)
else:
start = int(exon1_end); end = int(exon2_start)
else:
start = int(start); end = int(end)
if gene not in gene_annotation_db:
gd = GeneData(chr,strand)
gene_annotation_db[gene]=gd
### Store this data in the SQL database
command = """insert into %s (id, start, end, gene)
values ('%s', %d, %d, '%s')""" % (type,exonid,start,end,gene)
conn.execute(command)
#gi = FeatureData(start,end,exonid)
#try: exon_annotation_db[gene].append(gi)
#except KeyError: exon_annotation_db[gene]=[gi]
time_diff = str(round(time.time()-start_time,1))
print 'Dataset import in %s seconds' % time_diff
if type == 'exons':
return gene_annotation_db
def importGeneAnnotations(species,gene_annotation_db):
start_time = time.time()
gene_annotation_file = "AltDatabase/ensembl/"+species+"/"+species+"_Ensembl-annotations_simple.txt"
fn=filepath(gene_annotation_file)
count = 0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if count == 0: count = 1
else:
gene, description, symbol = string.split(data,'\t')
description = string.replace(description,"'","") ### single ' will cause problems
#gene_annotation_db[gene].setAnnotations(symbol, description) ### don't need to store this
chr = gene_annotation_db[gene].Chr()
strand = gene_annotation_db[gene].Strand()
### Store this data in the SQL database
command = """insert into genes (id, name, description, chr, strand)
values ('%s', '%s', '%s', '%s', '%s')""" % (gene,symbol,description,chr,strand)
try: conn.execute(command)
except Exception:
print [command];sys.exit()
del gene_annotation_db
time_diff = str(round(time.time()-start_time,1))
#print 'Dataset import in %s seconds' % time_diff
def importProcessedSpliceData(filename):
start_time = time.time()
fn=filepath(filename)
count = 0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if count == 0: count = 1
else:
gene, symbol, description = string.split(data,'\t')
gene_annotation_db[gene].setAnnotations(symbol, description)
time_diff = str(round(time.time()-start_time,1))
print 'Dataset import in %s seconds' % time_diff
def importEnsExonStructureData(filename,option):
start_time = time.time()
fn=filepath(filename); count=0; last_transcript = ''
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if count==0: count=1
else:
gene, chr, strand, start, end, ens_exonid, constitutive_exon, transcript = t
end = int(end); start = int(start)
if option == 'SQL':
### Store this data in the SQL database
command = """insert into transcripts (id, gene)
values ('%s', '%s')""" % (transcript, gene)
try: conn.execute(command)
except Exception: None ### Occurs when transcript in database already
command = """insert into transcript_exons (start, end, transcript)
values ('%d', '%d', '%s')""" % (start, end, transcript)
conn.execute(command)
elif option == 'transcript':
### Need to separate the two types for in silico translation
if 'Ensembl' in filename:
type = 'Ensembl'
else:
type = 'GenBank'
try: gene_transcript_db[gene].append((transcript,type))
except Exception: gene_transcript_db[gene] = [(transcript,type)]
elif option == 'exon':
if transcript in cds_location_db and transcript not in cds_genomic_db: # and strand == '-1'
cds_start, cds_stop = cds_location_db[transcript]
cds_start, cds_stop = int(cds_start), int(cds_stop)
if transcript != last_transcript:
cumulative = 0
last_transcript = transcript
if strand == '-1': start_correction = -3; end_correction = 2
else: start_correction = 0; end_correction = 0
diff1 = cumulative-cds_start
diff2 = ((end-start+cumulative) - cds_start)+2
diff3 = cumulative-cds_stop
diff4 = ((end-start+cumulative) - cds_stop)+2
if diff1 <= 0 and diff2 > 0: ### CDS start is in the first exon
exon_length = abs(end-start)+1
coding_bp_in_exon = exon_length - cds_start
if strand == '-1':
cds_genomic_start = start + coding_bp_in_exon + start_correction
else:
cds_genomic_start = end + coding_bp_in_exon
if diff3 < 0 and diff4 >= 0: ### CDS start is in the first exon
coding_bp_in_exon = cds_stop
if strand == '-1':
cds_genomic_stop = end - coding_bp_in_exon + start_correction
else:
cds_genomic_stop = start + coding_bp_in_exon
try:
cds_genomic_db[transcript] = cds_genomic_start,cds_genomic_stop
except Exception:
print cds_start, cds_stop, transcript, cds_genomic_stop; sys.exit()
if transcript == 'ENST00000326513':
print chr+':'+str(cds_genomic_stop)+'-'+str(cds_genomic_start)
del cds_genomic_stop
del cds_genomic_start
if transcript == 'ENST00000326513':
print diff1,diff2,diff3,diff4
print end,start,cumulative,cds_start,cds_stop
cumulative += (end-start)
"""
cumulative = 1
last_transcript = transcript
diff1 = cumulative-cds_start
diff2 = ((end-start+cumulative) - cds_start)+1
diff3 = cumulative-cds_stop
diff4 = ((end-start+cumulative) - cds_stop)+1
if diff1 <= 0 and diff2 > 0: ### CDS start is in the first exon
if strand == '1':
cds_genomic_start = end - diff2 + 1
else:
cds_genomic_start = start + diff2
if diff3 < 0 and diff4 >= 0: ### CDS start is in the first exon
if strand == '1':
cds_genomic_stop = end - diff4
else:
cds_genomic_stop = start + diff4 - 1
try:
cds_genomic_db[transcript] = cds_genomic_start,cds_genomic_stop
except Exception:
print cds_start, cds_stop, transcript, cds_genomic_stop; sys.exit()
if transcript == 'ENST00000436739':
print chr+':'+str(cds_genomic_stop)+'-'+str(cds_genomic_start);sys.exit()
del cds_genomic_stop
del cds_genomic_start
if transcript == 'ENST00000436739':
print diff1,diff2,diff3,diff4
print end,start,cumulative,cds_stop
cumulative += (end-start)
"""
#sys.exit()
time_diff = str(round(time.time()-start_time,1))
print 'Dataset import in %s seconds' % time_diff
def importProteinFeatureData(species,cds_location_db):
filename = 'AltDatabase/ensembl/'+species+'/ProteinFeatureIsoform_complete.txt'
db={}
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
transcript, protein, residue_start, residue_stop, feature_annotation, feature_id = t
if transcript in cds_location_db:
cds_start,cds_end = cds_location_db[transcript]
cds_feature_start = cds_start+(int(residue_start)*3)
cds_feature_end = cds_start+(int(residue_stop)*3)
#print residue_start, residue_stop
#print cds_start,cds_end
#print transcript, protein, feature_annotation, feature_id, cds_feature_start, cds_feature_end;sys.exit()
if 'blah' in transcript:
command = """insert into transcripts (id, gene)
values ('%s', '%s')""" % (transcript, gene)
try: conn.execute(command)
except Exception: None ### Occurs when transcript in database already
### There are often many features that overlap within a transcript, so consistently pick just one
if transcript in transcript_feature_db:
db = transcript_feature_db[transcript]
db[cds_feature_start, cds_feature_end].append([feature_annotation, feature_id])
else:
db={}
db[cds_feature_start, cds_feature_end]=[[feature_annotation, feature_id]]
transcript_feature_db[transcript] = db
#for transcript in transcript_feature_db:
print len(db)
def importAllTranscriptData(species):
"""
gene_annotation_db = importExonAnnotations(species,'exons') ### stores gene annotations and adds exon data to SQL database
importExonAnnotations(species,'junctions') ### adds junction data to SQL database
importGeneAnnotations(species,gene_annotation_db) ### adds gene annotations to SQL database
option = 'SQL'
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_transcript-annotations.txt'
importEnsExonStructureData(filename,option)
filename = 'AltDatabase/ucsc/'+species+'/'+species+'_UCSC_transcript_structure_mrna.txt'
try: importEnsExonStructureData(filename,option)
except Exception: None ### Not available for all species - needs to be built prior to transcript model creation
"""
cds_location_db = importCDSsimple(species) ### Import all CDS coordinates
importProteinFeatureData(species,cds_location_db)
processed_splice_file = '/Users/nsalomonis/Desktop/AltExonViewer/ProcessedSpliceData/Hs_RNASeq_H9_ES_vs_BJ1_Fibroblast.ExpCutoff-2.0_average-splicing-index-ProcessedSpliceData.txt'
def importCDSsimple(species):
""" Reads in the combined mRNA CDS coordinates compiled in the IdentifyAltIsoforms.importCDScoordinates() """
cds_location_db={}
filename = 'AltDatabase/ensembl/'+species +'/AllTranscriptCDSPositions.txt'
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
line_data = cleanUpLine(line) #remove endline
mRNA_AC,start,end = string.split(line_data,'\t')
start,end = int(start),int(end)
#command = """insert into cds (id, start, end)
#values ('%s', '%s', '%s')""" % (transcript, start, end)
#try: conn.execute(command)
#except Exception: None ### Occurs when transcript in database already
cds_location_db[mRNA_AC] = start, end
return cds_location_db
def alignAllDomainsToTranscripts(species,platform):
""" This function is only run during the database build process to create files available for subsequent download.
This recapitulates several functions executed during the database build process but does so explicitely for each
isoform with the goal of obtained genomic coordinates of each protein feature post de novo sequence alignment.
This includes all Ensembl proteins, UCSC mRNAs and in silico translated RNAs """
### Import all transcript to gene associations for Ensembl and UCSC transcripts
global gene_transcript_db
gene_transcript_db={}
option = 'transcript'
print 'Importing transcript data into memory'
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_transcript-annotations.txt'
importEnsExonStructureData(filename,option)
filename = 'AltDatabase/ucsc/'+species+'/'+species+'_UCSC_transcript_structure_mrna.txt'
try: importEnsExonStructureData(filename,option)
except Exception: None ### Not available for all species - needs to be built prior to transcript model creation
from build_scripts import FeatureAlignment
ucsc_transcripts={}
gene_db = {}
gene_transcript_db = FeatureAlignment.eliminateRedundant(gene_transcript_db)
for gene in gene_transcript_db:
for (ac,type) in gene_transcript_db[gene]:
if type != 'Ensembl':
ucsc_transcripts[ac]=[] ### Store all the untranslated UCSC mRNAs
gene_db[gene] = [gene] ### mimics the necessary structure for FeatureAlignment
### Identify untranslated Ensembl transcripts
print 'Importing Ensembl transcript to protein'
ens_transcript_protein_db = importEnsemblTranscriptAssociations(species)
### Import protein ID and protein sequence into a dictionary
#global protein_sequence_db
#protein_sequence_db = FeatureAlignment.remoteEnsemblProtSeqImport(species) ### All Ensembl protein sequences
"""This code imports all protein sequences (NCBI, Ensembl, in silico translated) associated with optimal isoform pairs,
however, not all isoforms analyzed in the database are here, hence, this should be considered a subset of in silico
translated Ensembl mRNAs, UCSC ,RNAs, and known analyzed UCSC proteins"""
#ucsc_transcripts={}
#ucsc_transcripts['BC065499']=[]
#ucsc_transcripts['AK309510']=[] ### in silico translated
#ens_transcript_protein_db={}
### Download or translate ANY AND ALL mRNAs considered by AltAnalyze via in silico translation
from build_scripts import IdentifyAltIsoforms
analysis_type = 'fetch_new' # analysis_type = 'fetch' ???
#IdentifyAltIsoforms.remoteTranslateRNAs(species,ucsc_transcripts,ens_transcript_protein_db,analysis_type)
### Derive all protein ID, domain and genomic coordinate data from Ensembl and UniProt
""" This data is available for Ensembl and UniProt isoforms but we re-derive the associations based on sequence for completeness """
### Get the domain sequences and genomic coordinates
"""
# for testing
gt = {}; y=0
for gene in gene_db:
if y < 20:
gt[gene] = gene_db[gene]
else: break
y+=1
"""
protein_ft_db,domain_gene_counts = FeatureAlignment.grab_exon_level_feature_calls(species,platform,gene_db)
from build_scripts import ExonAnalyze_module
seq_files, mRNA_protein_seq_db = IdentifyAltIsoforms.importProteinSequences(species,'getSequence') ### Import all available protein sequences (downloaded or in silico)
coordinate_type = 'genomic'; #coordinate_type = 'protein'
ExonAnalyze_module.getFeatureIsoformGenomePositions(species,protein_ft_db,mRNA_protein_seq_db,gene_transcript_db,coordinate_type)
### We may need to augment the above domain coordinate to isoform information with the Ensembl and UniProt files (if seq alignment failed for some reason - see grab_exon_level_feature_calls)!
def importEnsemblTranscriptAssociations(species):
""" Import all protein ID-gene relationships used in AltAnalyze. Requires accessing multiple flat files """
ens_transcript_protein_db={}
### Import Ensembl protein IDs
import gene_associations
gene_import_dir = '/AltDatabase/ensembl/'+species
g = gene_associations.GrabFiles()
g.setdirectory(gene_import_dir)
filedir,filename = g.searchdirectory('Ensembl_Protein__')
fn=filepath(filedir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
gene,transcript,protein = string.split(data,'\t')
if len(protein)==0:
ens_transcript_protein_db[transcript] = transcript ### Infer protein sequence (in silico translation)
else:
ens_transcript_protein_db[transcript] = protein
return ens_transcript_protein_db
def getCodingGenomicCoordinates(species):
global cds_location_db
global cds_genomic_db
from build_scripts import IdentifyAltIsoforms
cds_location_db = IdentifyAltIsoforms.importCDScoordinates(species)
#print cds_location_db['ENST00000436739'];sys.exit()
cds_genomic_db={}
option = 'exon'
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_transcript-annotations.txt'
importEnsExonStructureData(filename,option)
filename = 'AltDatabase/ucsc/'+species+'/'+species+'_UCSC_transcript_structure_mrna.txt'
try: importEnsExonStructureData(filename,option)
except Exception: None ### Not available for all species - needs to be built prior to transcript model creation
def buildAltExonDatabases(species,platform):
alignAllDomainsToTranscripts(species,platform)
if __name__ == '__main__':
#isoformViewer();sys.exit()
species = 'Mm'; type = 'exon'; platform = 'RNASeq'
#importAllTranscriptData(species); sys.exit()
#getCodingGenomicCoordinates(species)
#importProteinFeatureData(species)
buildAltExonDatabases(species,platform)
sys.exit()
populateSQLite()
#sys.exit()
#importAllTranscriptData(species);sys.exit()
#test()
isoformViewer()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/AltExonViewer.py
|
AltExonViewer.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys, string
from stats_scripts import statistics
import math
import os.path
import unique
import copy
import time
import export
import traceback
import warnings
#from stats_scripts import salstat_stats; reload(salstat_stats)
try:
from scipy import stats
use_scipy = True
import numpy
except Exception:
use_scipy = False ### scipy is not required but is used as a faster implementation of Fisher Exact Test when present
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir)
return dir_list
def makeUnique(item):
db1={}; list1=[]; k=0
for i in item:
try: db1[i]=[]
except TypeError: db1[tuple(i)]=[]; k=1
for i in db1:
if k==0: list1.append(i)
else: list1.append(list(i))
list1.sort()
return list1
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def returnLargeGlobalVars():
### Prints all large global variables retained in memory (taking up space)
all = [var for var in globals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(globals()[var])>1:
print var, len(globals()[var])
except Exception: null=[]
def clearObjectsFromMemory(db_to_clear):
db_keys={}
for key in db_to_clear: db_keys[key]=[]
for key in db_keys:
try: del db_to_clear[key]
except Exception:
try:
for i in key: del i ### For lists of tuples
except Exception: del key ### For plain lists
######### Below code deals is specific to this module #########
def runLineageProfiler(species,array_type,exp_input,exp_output,codingtype,compendium_platform,customMarkers=False):
"""
print species
print array_type
print export.findFilename(exp_input)
print export.findFilename(exp_output)
print codingtype
print compendium_platform
print customMarkers
"""
global exp_output_file; exp_output_file = exp_output; global targetPlatform
global tissue_specific_db; global expession_subset; global tissues; global sample_headers
global analysis_type; global coding_type; coding_type = codingtype
global tissue_to_gene; tissue_to_gene = {}; global platform; global cutoff
global customMarkerFile; global keyed_by; global compendiumPlatform
customMarkerFile = customMarkers; compendiumPlatform = compendium_platform
global correlate_by_order; correlate_by_order = 'no'
global rho_threshold; rho_threshold = -1
global correlate_to_tissue_specific; correlate_to_tissue_specific = 'no'
platform = array_type
cutoff = 0.01
global value_type
global missingValuesPresent
if 'stats.' in exp_input:
value_type = 'calls'
else:
value_type = 'expression'
tissue_specific_db={}; expession_subset=[]; sample_headers=[]; tissues=[]
if len(array_type)==2:
### When a user-supplied expression is provided (no ExpressionOutput files provided - importGeneIDTranslations)
vendor, array_type = array_type
platform = array_type
else: vendor = 'Not needed'
if 'other:' in vendor:
vendor = string.replace(vendor,'other:','')
array_type = "3'array"
if 'RawSplice' in exp_input or 'FullDatasets' in exp_input or coding_type == 'AltExon':
analysis_type = 'AltExon'
if platform != compendium_platform: ### If the input IDs are not Affymetrix Exon 1.0 ST probesets, then translate to the appropriate system
translate_to_genearray = 'no'
targetPlatform = compendium_platform
translation_db = importExonIDTranslations(array_type,species,translate_to_genearray)
keyed_by = 'translation'
else: translation_db=[]; keyed_by = 'primaryID'; targetPlatform = compendium_platform
elif array_type == "3'array" or array_type == 'AltMouse':
### Get arrayID to Ensembl associations
if vendor != 'Not needed':
### When no ExpressionOutput files provided (user supplied matrix)
translation_db = importVendorToEnsemblTranslations(species,vendor,exp_input)
else:
try: translation_db = importGeneIDTranslations(exp_output)
except: translation_db = importVendorToEnsemblTranslations(species,'Symbol',exp_input)
keyed_by = 'translation'
targetPlatform = compendium_platform
analysis_type = 'geneLevel'
else:
translation_db=[]; keyed_by = 'primaryID'; targetPlatform = compendium_platform; analysis_type = 'geneLevel'
if compendium_platform == "3'array" and array_type != "3'array":
keyed_by = 'ensembl' ### ensembl is not indicated anywhere but avoides key by primaryID and translation -> works for RNASeq
targetPlatform = compendium_platform ### Overides above
""" Determine if a PSI file with missing values """
if vendor == 'PSI':
missingValuesPresent = True
else:
missingValuesPresent = importTissueSpecificProfiles(species,checkForMissingValues=True)
try: importTissueSpecificProfiles(species)
except Exception:
try:
try:
targetPlatform = 'exon'
importTissueSpecificProfiles(species)
except Exception:
try:
targetPlatform = 'gene'
importTissueSpecificProfiles(species)
except Exception:
targetPlatform = "3'array"
importTissueSpecificProfiles(species)
except Exception,e:
print traceback.format_exc()
print 'No compatible compendiums present...'
forceTissueSpecificProfileError
try: importGeneExpressionValues(exp_input,tissue_specific_db,translation_db,species=species)
except:
print "Changing platform to 3'array"
array_type = "3'array"
exp_input = string.replace(exp_input,'-steady-state.txt','.txt')
importGeneExpressionValues(exp_input,tissue_specific_db,translation_db,species=species)
### If the incorrect gene system was indicated re-run with generic parameters
if len(expession_subset)==0 and (array_type == "3'array" or array_type == 'AltMouse' or array_type == 'Other'):
translation_db=[]; keyed_by = 'primaryID'; targetPlatform = compendium_platform; analysis_type = 'geneLevel'
tissue_specific_db={}
try: importTissueSpecificProfiles(species)
except Exception:
try: targetPlatform = 'exon'; importTissueSpecificProfiles(species)
except Exception:
try: targetPlatform = 'gene'; importTissueSpecificProfiles(species)
except Exception: targetPlatform = "3'array"; importTissueSpecificProfiles(species)
importGeneExpressionValues(exp_input,tissue_specific_db,translation_db,species=species)
zscore_output_dir = analyzeTissueSpecificExpressionPatterns(expInput=exp_input)
return zscore_output_dir
def importVendorToEnsemblTranslations(species,vendor,exp_input):
translation_db={}
"""
### Faster method but possibly not as good
uid_db = simpleUIDImport(exp_input)
import gene_associations
### Use the same annotation method that is used to create the ExpressionOutput annotations
array_to_ens = gene_associations.filterGeneToUID(species,'Ensembl',vendor,associated_IDs)
for arrayid in array_to_ens:
ensembl_list = array_to_ens[arrayid]
try: translation_db[arrayid] = ensembl_list[0] ### This first Ensembl is ranked as the most likely valid based on various metrics in getArrayAnnotationsFromGOElite
except Exception: None
"""
translation_db={}
from import_scripts import BuildAffymetrixAssociations
### Use the same annotation method that is used to create the ExpressionOutput annotations
use_go = 'yes'
conventional_array_db={}
conventional_array_db = BuildAffymetrixAssociations.getUIDAnnotationsFromGOElite(conventional_array_db,species,vendor,use_go)
for arrayid in conventional_array_db:
ca = conventional_array_db[arrayid]
ens = ca.Ensembl()
try: translation_db[arrayid] = ens[0] ### This first Ensembl is ranked as the most likely valid based on various metrics in getArrayAnnotationsFromGOElite
except Exception: None
return translation_db
def importTissueSpecificProfiles(species,checkForMissingValues=False):
if analysis_type == 'AltExon':
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_'+targetPlatform +'_tissue-specific_AltExon_protein_coding.txt'
else:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_'+targetPlatform +'_tissue-specific_'+coding_type+'.txt'
if customMarkerFile != False and customMarkerFile != None:
if len(customMarkerFile)>0:
filename = customMarkerFile
#filename = 'AltDatabase/ensembl/'+species+'/random.txt'
#print 'Target platform used for analysis:',species, targetPlatform, coding_type
if value_type == 'calls':
filename = string.replace(filename,'.txt','_stats.txt')
fn=filepath(filename); x=0
tissue_index = 1
tissues_added={}
missing_values_present = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
print 'Importing the tissue compedium database:',export.findFilename(filename)
headers = t; x=1; index=0
for i in headers:
if 'UID' == i: ens_index = index; uid_index = index
if analysis_type == 'AltExon': ens_index = ens_index ### Assigned above when analyzing probesets
elif 'Ensembl' in i: ens_index = index
if 'marker-in' in i: tissue_index = index+1; marker_in = index
index+=1
try:
for i in t[tissue_index:]: tissues.append(i)
except Exception:
for i in t[1:]: tissues.append(i)
if keyed_by == 'primaryID':
try: ens_index = uid_index
except Exception: None
else:
try:
gene = t[0]
try: gene = string.split(gene,'|')[0] ### Only consider the first listed gene - this gene is the best option based on ExpressionBuilder rankings
except Exception: pass
tissue_exp = map(float, t[1:])
tissue_specific_db[gene]=x,tissue_exp ### Use this to only grab relevant gene expression profiles from the input dataset
except Exception:
try: gene = string.split(gene,'|')[0] ### Only consider the first listed gene - this gene is the best option based on ExpressionBuilder rankings
except Exception: pass
#if 'Pluripotent Stem Cells' in t[marker_in] or 'Heart' in t[marker_in]:
#if t[marker_in] not in tissues_added: ### Only add the first instance of a gene for that tissue - used more for testing to quickly run the analysis
tissue_exp = t[tissue_index:]
if '' in tissue_exp:
missing_values_present = True
### If missing values present (PSI values)
tissue_exp = ['0.000101' if i=='' else i for i in tissue_exp]
tissue_exp = map(float,tissue_exp)
if value_type == 'calls':
tissue_exp = produceDetectionCalls(tissue_exp,platform) ### 0 or 1 calls
tissue_specific_db[gene]=x,tissue_exp ### Use this to only grab relevant gene expression profiles from the input dataset
try: tissues_added[t[marker_in]]=[] ### Not needed currently
except Exception: pass
x+=1
print len(tissue_specific_db), 'genes in the tissue compendium database'
if correlate_to_tissue_specific == 'yes':
try: importTissueCorrelations(filename)
except Exception:
null=[]
#print '\nNo tissue-specific correlations file present. Skipping analysis.'; kill
if checkForMissingValues:
return missing_values_present
def importTissueCorrelations(filename):
filename = string.replace(filename,'specific','specific_correlations')
fn=filepath(filename); x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if x==0: x=1 ### Ignore header line
else:
uid,symbol,rho,tissue = string.split(data,'\t')
if float(rho)>rho_threshold: ### Variable used for testing different thresholds internally
try: tissue_to_gene[tissue].append(uid)
except Exception: tissue_to_gene[tissue] = [uid]
def simpleUIDImport(filename):
"""Import the UIDs in the gene expression file"""
uid_db={}
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
uid_db[string.split(data,'\t')[0]]=[]
return uid_db
def importGeneExpressionValues(filename,tissue_specific_db,translation_db,useLog=False,previouslyRun=False,species=None):
### Import gene-level expression raw values
fn=filepath(filename); x=0; genes_added={}; gene_expression_db={}
dataset_name = export.findFilename(filename)
max_val=0
print 'importing:',dataset_name
try:
import gene_associations, OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception: symbol_to_gene={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
if '#' not in data:
for i in t[1:]: sample_headers.append(i)
x=1
else:
gene = t[0]
try: gene = string.split(t[0],'|')[0]
except Exception: pass
#if '-' not in gene and ':E' in gene: print gene;sys.exit()
if analysis_type == 'AltExon':
try: ens_gene,exon = string.split(gene,'-')[:2]
except Exception: exon = gene
gene = exon
if keyed_by == 'translation': ### alternative value is 'primaryID'
"""if gene == 'ENSMUSG00000025915-E19.3':
for i in translation_db: print [i], len(translation_db); break
print gene, [translation_db[gene]];sys.exit()"""
try: gene = translation_db[gene] ### Ensembl annotations
except Exception: pass
try: gene = symbol_to_gene[gene][0] ### If RNASeq is the selected platform and Symbol is the uid
except Exception: pass
if gene in tissue_specific_db:
index,tissue_exp=tissue_specific_db[gene]
try: genes_added[gene]+=1
except Exception: genes_added[gene]=1
proceed=True
try:
exp_vals = t[1:]
if '' in exp_vals:
### If missing values present (PSI values)
exp_vals = ['0.000101' if i=='' else i for i in exp_vals]
useLog = False
exp_vals = map(float, exp_vals)
if platform == 'RNASeq':
if max(exp_vals)>max_val: max_val = max(exp_vals)
#if max(exp_vals)<3: proceed=False
if useLog==False:
exp_vals = map(lambda x: math.log(x+1,2),exp_vals)
if value_type == 'calls': ### Hence, this is a DABG or RNA-Seq expression
exp_vals = produceDetectionCalls(exp_vals,targetPlatform) ### 0 or 1 calls
if proceed:
gene_expression_db[gene] = [index,exp_vals]
except Exception:
print 'Non-numeric values detected:'
x = 5
print t[:x]
while x < t:
t[x:x+5]
x+=5
print 'Formatting error encountered in:',dataset_name; forceError
"""else:
for gene in tissue_specific_db:
if 'Ndufa9:ENSMUSG00000000399:I2.1-E3.1' in gene:
print gene, 'dog';sys.exit()
print gene;kill"""
print len(gene_expression_db), 'matching genes in the dataset and tissue compendium database'
for gene in genes_added:
if genes_added[gene]>1:
del gene_expression_db[gene] ### delete entries that are present in the input set multiple times (not trustworthy)
else: expession_subset.append(gene_expression_db[gene]) ### These contain the rank order and expression
#print len(expession_subset);sys.exit()
expession_subset.sort() ### This order now matches that of
gene_expression_db=[]
if max_val<20 and platform == 'RNASeq' and previouslyRun==False: ### Only allow to happen once
importGeneExpressionValues(filename,tissue_specific_db,translation_db,useLog=True,previouslyRun=True,species=species)
def produceDetectionCalls(values,Platform):
# Platform can be the compendium platform (targetPlatform) or analyzed data platform (platform or array_type)
new=[]
for value in values:
if Platform == 'RNASeq':
if value>1:
new.append(1) ### expressed
else:
new.append(0)
else:
if value<cutoff: new.append(1)
else: new.append(0)
return new
def importGeneIDTranslations(filename):
### Import ExpressionOutput/DATASET file to obtain Ensembl associations (typically for Affymetrix 3' arrays)
fn=filepath(filename); x=0; translation_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
headers = t; x=1; index=0
for i in headers:
if 'Ensembl' in i: ens_index = index; break
index+=1
else:
uid = t[0]
ens_geneids = t[ens_index]
ens_geneid = string.split(ens_geneids,'|')[0] ### In v.2.0.5, the first ID is the best protein coding candidate
if len(ens_geneid)>0:
translation_db[uid] = ens_geneid
return translation_db
def remoteImportExonIDTranslations(array_type,species,translate_to_genearray,targetplatform):
global targetPlatform; targetPlatform = targetplatform
translation_db = importExonIDTranslations(array_type,species,translate_to_genearray)
return translation_db
def importExonIDTranslations(array_type,species,translate_to_genearray):
gene_translation_db={}; gene_translation_db2={}
if targetPlatform == 'gene' and translate_to_genearray == 'no':
### Get gene array to exon array probeset associations
gene_translation_db = importExonIDTranslations('gene',species,'yes')
for geneid in gene_translation_db:
exonid = gene_translation_db[geneid]
gene_translation_db2[exonid] = geneid
#print exonid, geneid
translation_db = gene_translation_db2
else:
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_'+array_type+'-exon_probesets.txt'
### Import exon array to target platform translations (built for DomainGraph visualization)
fn=filepath(filename); x=0; translation_db={}
print 'Importing the translation file',export.findFilename(fn)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: x=1
else:
platform_id,exon_id = t
if targetPlatform == 'gene' and translate_to_genearray == 'no':
try:
translation_db[platform_id] = gene_translation_db[exon_id] ### return RNA-Seq to gene array probeset ID
#print platform_id, exon_id, gene_translation_db[exon_id];sys.exit()
except Exception: null=[]
else:
translation_db[platform_id] = exon_id
del gene_translation_db; del gene_translation_db2
return translation_db
def analyzeTissueSpecificExpressionPatterns(expInput=None):
tissue_specific_sorted = []; genes_present={}; tissue_exp_db={}; gene_order_db={}; gene_order=[]
for (index,vals) in expession_subset: genes_present[index]=[]
for gene in tissue_specific_db:
tissue_specific_sorted.append(tissue_specific_db[gene])
# tissue_specific_db[gene][1]
#print tissue_specific_db[gene][1].count(0.000101);sys.exit()
gene_order_db[tissue_specific_db[gene][0]] = gene ### index order (this index was created before filtering)
tissue_specific_sorted.sort()
new_index=0
for (index,tissue_exp) in tissue_specific_sorted:
try:
null=genes_present[index]
i=0
gene_order.append([new_index,gene_order_db[index]]); new_index+=1
for f in tissue_exp:
### The order of the tissue specific expression profiles is based on the import gene order
try: tissue_exp_db[tissues[i]].append(f)
except Exception: tissue_exp_db[tissues[i]] = [f]
i+=1
except Exception:
#print gene;sys.exit()
null=[] ### Gene is not present in the input dataset
### Organize sample expression, with the same gene order as the tissue expression set
sample_exp_db={}
for (index,exp_vals) in expession_subset:
i=0
for f in exp_vals:
### The order of the tissue specific expression profiles is based on the import gene order
try: sample_exp_db[sample_headers[i]].append(f)
except Exception: sample_exp_db[sample_headers[i]] = [f]
i+=1
if correlate_by_order == 'yes':
### Rather than correlate to the absolute expression order, correlate to the order of expression (lowest to highest)
sample_exp_db = replaceExpressionWithOrder(sample_exp_db)
tissue_exp_db = replaceExpressionWithOrder(tissue_exp_db)
global tissue_comparison_scores; tissue_comparison_scores={}
if correlate_to_tissue_specific == 'yes':
### Create a gene_index that reflects the current position of each gene
gene_index={}
for (i,gene) in gene_order: gene_index[gene] = i
### Create a tissue to gene-index from the gene_index
tissue_to_index={}
for tissue in tissue_to_gene:
for gene in tissue_to_gene[tissue]:
if gene in gene_index: ### Some are not in both tissue and sample datasets
index = gene_index[gene] ### Store by index, since the tissue and expression lists are sorted by index
try: tissue_to_index[tissue].append(index)
except Exception: tissue_to_index[tissue] = [index]
tissue_to_index[tissue].sort()
sample_exp_db,tissue_exp_db = returnTissueSpecificExpressionProfiles(sample_exp_db,tissue_exp_db,tissue_to_index)
PearsonCorrelationAnalysis(sample_exp_db,tissue_exp_db)
sample_exp_db=[]; tissue_exp_db=[]
zscore_output_dir = exportCorrelationResults(expInput)
return zscore_output_dir
def returnTissueSpecificExpressionProfiles(sample_exp_db,tissue_exp_db,tissue_to_index):
tissue_exp_db_abreviated={}
sample_exp_db_abreviated={} ### This db is designed differently than the non-tissue specific (keyed by known tissues)
### Build the tissue specific expression profiles
for tissue in tissue_exp_db:
tissue_exp_db_abreviated[tissue] = []
for index in tissue_to_index[tissue]:
tissue_exp_db_abreviated[tissue].append(tissue_exp_db[tissue][index]) ### populate with just marker expression profiles
### Build the sample specific expression profiles
for sample in sample_exp_db:
sample_tissue_exp_db={}
sample_exp_db[sample]
for tissue in tissue_to_index:
sample_tissue_exp_db[tissue] = []
for index in tissue_to_index[tissue]:
sample_tissue_exp_db[tissue].append(sample_exp_db[sample][index])
sample_exp_db_abreviated[sample] = sample_tissue_exp_db
return sample_exp_db_abreviated, tissue_exp_db_abreviated
def replaceExpressionWithOrder(sample_exp_db):
for sample in sample_exp_db:
sample_exp_sorted=[]; i=0
for exp_val in sample_exp_db[sample]: sample_exp_sorted.append([exp_val,i]); i+=1
sample_exp_sorted.sort(); sample_exp_resort = []; order = 0
for (exp_val,i) in sample_exp_sorted: sample_exp_resort.append([i,order]); order+=1
sample_exp_resort.sort(); sample_exp_sorted=[] ### Order lowest expression to highest
for (i,o) in sample_exp_resort: sample_exp_sorted.append(o) ### The expression order replaces the expression, in the original order
sample_exp_db[sample] = sample_exp_sorted ### Replace exp with order
return sample_exp_db
def PearsonCorrelationAnalysis(sample_exp_db,tissue_exp_db):
print "Beginning LineageProfiler analysis"; k=0
original_increment = int(len(tissue_exp_db)/15.00); increment = original_increment
p = 1 ### Default value if not calculated
for tissue in tissue_exp_db:
#print k,"of",len(tissue_exp_db),"classifier tissue/cell-types"
if k == increment: increment+=original_increment; print '*',
k+=1
tissue_expression_list = tissue_exp_db[tissue]
for sample in sample_exp_db:
if correlate_to_tissue_specific == 'yes':
### Keyed by tissue specific sample profiles
sample_expression_list = sample_exp_db[sample][tissue] ### dictionary as the value for sample_exp_db[sample]
#print tissue, sample_expression_list
#print tissue_expression_list; sys.exit()
else: sample_expression_list = sample_exp_db[sample]
try:
### p-value is likely useful to report (not supreemly accurate but likely sufficient)
if missingValuesPresent:
### For PSI values
tissue_expression_list = numpy.ma.masked_values(tissue_expression_list,0.000101)
#tissue_expression_list = numpy.ma.array([numpy.nan if i==0.000101 else i for i in tissue_expression_list])
sample_expression_list = numpy.ma.masked_values(sample_expression_list,0.000101)
#tissue_expression_list = numpy.ma.array([numpy.nan if i==0.000101 else i for i in tissue_expression_list])
updated_tissue_expression_list=[]
updated_sample_expression_list=[]
i=0
coefr=numpy.ma.corrcoef(tissue_expression_list,sample_expression_list)
rho = coefr[0][1]
"""
if sample == 'Cmp.21':
#print rho
#print tissue_expression_list[:10]
#print string.join(map(str,tissue_expression_list[:20]),'\t')
#print sample_expression_list[:10]
#print string.join(map(str,sample_expression_list[:20]),'\t')
#coefr=numpy.ma.corrcoef(numpy.array(tissue_expression_list[:10]),numpy.array(sample_expression_list[:10]))
print tissue, sample, rho, len(tissue_expression_list), len(sample_expression_list)
"""
else:
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
rho,p = stats.pearsonr(tissue_expression_list,sample_expression_list)
except Exception:
#print traceback.format_exc(); sys.exit()
### simple pure python implementation - no scipy required (not as fast though and no p-value)
rho = pearson(tissue_expression_list,sample_expression_list)
#tst = salstat_stats.TwoSampleTests(tissue_expression_list,sample_expression_list)
#pp,pr = tst.PearsonsCorrelation()
#sp,sr = tst.SpearmansCorrelation()
#print tissue, sample
#if rho>.5: print [rho, pr, sr],[pp,sp];sys.exit()
#if rho<.5: print [rho, pr, sr],[pp,sp];sys.exit()
try: tissue_comparison_scores[tissue].append([rho,p,sample])
except Exception: tissue_comparison_scores[tissue] = [[rho,p,sample]]
sample_exp_db=[]; tissue_exp_db=[]
print 'Correlation analysis finished'
def pearson(array1,array2):
item = 0; sum_a = 0; sum_b = 0; sum_c = 0
while item < len(array1):
a = (array1[item] - avg(array1))*(array2[item] - avg(array2))
b = math.pow((array1[item] - avg(array1)),2)
c = math.pow((array2[item] - avg(array2)),2)
sum_a = sum_a + a
sum_b = sum_b + b
sum_c = sum_c + c
item = item + 1
r = sum_a/math.sqrt(sum_b*sum_c)
return r
def avg(array):
return sum(array)/len(array)
def adjustPValues():
""" Can be applied to calculate an FDR p-value on the p-value reported by scipy.
Currently this method is not employed since the p-values are not sufficiently
stringent or appropriate for this type of analysis """
all_sample_data={}
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]:
all_sample_data[sample] = db = {} ### populate this dictionary and create sub-dictionaries
break
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]:
gs = statistics.GroupStats('','',p)
all_sample_data[sample][tissue] = gs
for sample in all_sample_data:
statistics.adjustPermuteStats(all_sample_data[sample])
for tissue in tissue_comparison_scores:
scores = []
for (r,p,sample) in tissue_comparison_scores[tissue]:
p = all_sample_data[sample][tissue].AdjP()
scores.append([r,p,sample])
tissue_comparison_scores[tissue] = scores
def replacePearsonPvalueWithZscore():
all_sample_data={}
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]:
all_sample_data[sample] = [] ### populate this dictionary and create sub-dictionaries
break
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]:
all_sample_data[sample].append(r)
sample_stats={}
all_dataset_rho_values=[]
### Get average and standard deviation for all sample rho's
for sample in all_sample_data:
all_dataset_rho_values+=all_sample_data[sample]
avg=statistics.avg(all_sample_data[sample])
stdev=statistics.stdev(all_sample_data[sample])
sample_stats[sample]=avg,stdev
global_rho_avg = statistics.avg(all_dataset_rho_values)
global_rho_stdev = statistics.stdev(all_dataset_rho_values)
### Replace the p-value for each rho
for tissue in tissue_comparison_scores:
scores = []
for (r,p,sample) in tissue_comparison_scores[tissue]:
#u,s=sample_stats[sample]
#z = (r-u)/s
z = (r-global_rho_avg)/global_rho_stdev ### Instead of doing this for the sample background, do it relative to all analyzed samples
scores.append([r,z,sample])
tissue_comparison_scores[tissue] = scores
def exportCorrelationResults(exp_input):
input_file = export.findFilename(exp_input)
if '.txt' in exp_output_file:
corr_output_file = string.replace(exp_output_file,'DATASET','LineageCorrelations')
else: ### Occurs when processing a non-standard AltAnalyze file
corr_output_file = exp_output_file+'/'+input_file
corr_output_file = string.replace(corr_output_file,'.txt','-'+coding_type+'-'+compendiumPlatform+'.txt')
if analysis_type == 'AltExon':
corr_output_file = string.replace(corr_output_file,coding_type,'AltExon')
filename = export.findFilename(corr_output_file)
score_data = export.ExportFile(corr_output_file)
if use_scipy:
zscore_output_dir = string.replace(corr_output_file,'.txt','-zscores.txt')
probability_data = export.ExportFile(zscore_output_dir)
#adjustPValues()
replacePearsonPvalueWithZscore()
### Make title row
headers=['Sample_name']
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]: headers.append(sample)
break
title_row = string.join(headers,'\t')+'\n'
score_data.write(title_row)
if use_scipy:
probability_data.write(title_row)
### Export correlation data
tissue_scores = {}; tissue_probabilities={}; tissue_score_list = [] ### store and rank tissues according to max(score)
for tissue in tissue_comparison_scores:
scores=[]
probabilities=[]
for (r,p,sample) in tissue_comparison_scores[tissue]:
scores.append(r)
probabilities.append(p)
tissue_score_list.append((max(scores),tissue))
tissue_scores[tissue] = string.join(map(str,[tissue]+scores),'\t')+'\n' ### export line
if use_scipy:
tissue_probabilities[tissue] = string.join(map(str,[tissue]+probabilities),'\t')+'\n'
tissue_score_list.sort()
tissue_score_list.reverse()
for (score,tissue) in tissue_score_list:
score_data.write(tissue_scores[tissue])
if use_scipy:
probability_data.write(tissue_probabilities[tissue])
score_data.close()
if use_scipy:
probability_data.close()
print filename,'exported...'
return zscore_output_dir
def visualizeLineageZscores(zscore_output_dir,grouped_lineage_zscore_dir,graphic_links):
from visualization_scripts import clustering
### Perform hierarchical clustering on the LineageProfiler Zscores
graphic_links = clustering.runHCOnly(zscore_output_dir,graphic_links)
return graphic_links
if __name__ == '__main__':
species = 'Hs'
array_type = "3'array"
vendor = 'Affymetrix'
vendor = 'other:Symbol'
vendor = 'other:Ensembl'
#vendor = 'RNASeq'
array_type = "exon"
#array_type = "3'array"
#array_type = "RNASeq"
compendium_platform = "3'array"
compendium_platform = "exon"
#compendium_platform = "gene"
#array_type = "junction"
codingtype = 'ncRNA'
codingtype = 'protein_coding'
#codingtype = 'AltExon'
array_type = vendor, array_type
exp_input = "/Users/saljh8/Documents/1-conferences/GE/LineageMarkerAnalysis/Synapse-ICGS-EB-Ensembl.txt"
exp_output = "/Users/saljh8/Documents/1-conferences/GE/LineageMarkerAnalysis/temp.txt"
#customMarkers = "/Users/nsalomonis/Desktop/dataAnalysis/qPCR/PAM50/AltAnalyze/ExpressionOutput/MarkerFinder/AVERAGE-training.txt"
customMarkers = False
runLineageProfiler(species,array_type,exp_input,exp_output,codingtype,compendium_platform,customMarkers)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/LineageProfiler.py
|
LineageProfiler.py
|
import os.path, sys, shutil
import os
import string, re
import subprocess
import numpy as np
import unique
import traceback
import wx
import wx.lib.scrolledpanel
import wx.grid as gridlib
try:
import warnings
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
import matplotlib
#try: matplotlib.use('TkAgg')
#except Exception: pass
#import matplotlib.pyplot as plt ### Backend conflict issue when called prior to the actual Wx window appearing
#matplotlib.use('WXAgg')
from matplotlib.backends.backend_wx import NavigationToolbar2Wx
from matplotlib.figure import Figure
from numpy import arange, sin, pi
except Exception: pass
if os.name == 'nt': bheight=20
else: bheight=10
rootDirectory = unique.filepath(str(os.getcwd()))
currentDirectory = unique.filepath(str(os.getcwd())) + "/" + "Config/" ### NS-91615 alternative to __file__
currentDirectory = string.replace(currentDirectory,'AltAnalyzeViewer.app/Contents/Resources','')
os.chdir(currentDirectory)
parentDirectory = str(os.getcwd()) ### NS-91615 gives the parent AltAnalyze directory
sys.path.insert(1,parentDirectory) ### NS-91615 adds the AltAnalyze modules to the system path to from visualization_scripts import clustering and others
import UI
#These classes set up the "tab" feature in the program, allowing you to switch the viewer to different modes.
class PageTwo(wx.Panel):
def __init__(self, parent):
wx.Panel.__init__(self, parent)
self.SetBackgroundColour("white")
myGrid = ""
class PageThree(wx.Panel):
def __init__(self, parent):
wx.Panel.__init__(self, parent)
self.SetBackgroundColour("white")
class PageFour(wx.Panel):
def __init__(self, parent):
wx.Panel.__init__(self, parent)
class PageFive(wx.Panel):
def __init__(self, parent):
wx.Panel.__init__(self, parent)
class Main(wx.Frame):
def __init__(self,parent,id):
wx.Frame.__init__(self, parent, id,'AltAnalyze Results Viewer', size=(900,610))
self.Show()
self.Maximize(True) #### This allows the frame to resize to the host machine's max size
self.heatmap_translation = {}
self.heatmap_run = {}
self.species = 'Hs'
self.platform = 'RNASeq'
self.geneset_type = 'WikiPathways'
self.supported_genesets = []
self.runPCA = False
self.SetBackgroundColour((230, 230, 230))
self.species=''
#PANELS & WIDGETS
#self.panel is one of the TOP PANELS. These are used for title display, the open project button, and sort & filter buttons.
self.panel = wx.Panel(self, id=2, pos=(200,0), size=(600,45), style=wx.RAISED_BORDER)
self.panel.SetBackgroundColour((110, 150, 250))
#Panel 2 is the main view panel.
self.panel2 = wx.Panel(self, id=3, pos=(200,50), size=(1400,605), style=wx.RAISED_BORDER)
self.panel2.SetBackgroundColour((218, 218, 218))
#Panel 3 contains the pseudo-directory tree.
self.panel3 = wx.Panel(self, id=4, pos=(0,50), size=(200,625), style=wx.RAISED_BORDER)
self.panel3.SetBackgroundColour("white")
self.panel4 = wx.Panel(self, id=5, pos=(200,650), size=(1400,150), style=wx.RAISED_BORDER)
self.panel4.SetBackgroundColour("black")
#These are the other top panels.
self.panel_left = wx.Panel(self, id=12, pos=(0,0), size=(200,45), style=wx.RAISED_BORDER)
self.panel_left.SetBackgroundColour((218, 218, 218))
self.panel_right = wx.Panel(self, id=11, pos=(1100,0), size=(200,45), style=wx.RAISED_BORDER)
self.panel_right.SetBackgroundColour((218, 218, 218))
self.panel_right2 = wx.Panel(self, id=13, pos=(1300,0), size=(300,45), style=wx.RAISED_BORDER)
self.panel_right2.SetBackgroundColour((218, 218, 218))
self.panel_right2.SetMaxSize([300, 45])
#Lines 81-93 set up the user input box for the "sort" function (used on the table).
self.sortbox = wx.TextCtrl(self.panel_right2, id=7, pos=(55,10), size=(40,25))
wx.Button(self.panel_right2, id=8, label="Sort", pos=(5, 12), size=(40, bheight))
self.Bind(wx.EVT_BUTTON, self.SortTablefromButton, id=8)
self.AscendingRadio = wx.RadioButton(self.panel_right2, id=17, label="Sort", pos=(100, 3), size=(12, 12))
self.DescendingRadio = wx.RadioButton(self.panel_right2, id=18, label="Sort", pos=(100, 23), size=(12, 12))
font = wx.Font(10, wx.SWISS, wx.NORMAL, wx.BOLD)
self.AscendingOpt = wx.StaticText(self.panel_right2, label="Ascending", pos=(115, 1))
self.AscendingOpt.SetFont(font)
self.DescendingOpt = wx.StaticText(self.panel_right2, label="Descending", pos=(115, 21))
self.DescendingOpt.SetFont(font)
#Lines 96-98 set up the user input box for the "filter" function (used on the table).
self.filterbox = wx.TextCtrl(self.panel_right, id=9, pos=(60,10), size=(125,25))
wx.Button(self.panel_right, id=10, label="Filter", pos=(0, 12), size=(50, bheight))
self.Bind(wx.EVT_BUTTON, self.FilterTablefromButton, id=10)
#Lines 101-103 set up the in-program log.
self.control = wx.TextCtrl(self.panel4, id=6, pos=(1,1), size=(1400,150), style=wx.TE_MULTILINE)
self.control.write("Welcome to AltAnalyze Results Viewer!" + "\n")
self.Show(True)
self.main_results_directory = ""
#self.browser is the "directory tree" where groups of files are instantiated in self.browser2.
self.browser = wx.TreeCtrl(self.panel3, id=2000, pos=(0,0), size=(200,325))
#self.browser2 is the "file group" where groups of files are accessed, respective to the directory selected in self.browser.
self.browser2 = wx.TreeCtrl(self.panel3, id=2001, pos=(0,325), size=(200,325))
self.tree = self.browser
#self.sortdict groups the table headers to integers---this works with sort function.
self.sortdict = {"A" : 0, "B" : 1, "C" : 2, "D" : 3, "E" : 4, "F" : 5, "G" : 6, "H" : 7, "I" : 8, "J" : 9, "K" : 10, "L" : 11, "M" : 12, "N" : 13, "O" : 14, "P" : 15, "Q" : 16, "R" : 17, "S" : 18, "T" : 19, "U" : 20, "V" : 21, "W" : 22, "X" : 23, "Y" : 24, "Z" : 25, "AA" : 26, "AB" : 27, "AC" : 28, "AD" : 29, "AE" : 30, "AF" : 31, "AG" : 32, "AH" : 33, "AI" : 34, "AJ" : 35, "AK" : 36, "AL" : 37, "AM" : 38, "AN" : 39, "AO" : 40, "AP" : 41, "AQ" : 42, "AR" : 43, "AS" : 44, "AT" : 45, "AU" : 46, "AV" : 47, "AW" : 48, "AX" : 49, "AY" : 50, "AZ" : 51}
#SIZER--main sizer for the program.
ver = wx.BoxSizer(wx.VERTICAL)
verpan2 = wx.BoxSizer(wx.VERTICAL)
hpan1 = wx.BoxSizer(wx.HORIZONTAL)
hpan2 = wx.BoxSizer(wx.HORIZONTAL)
hpan3 = wx.BoxSizer(wx.HORIZONTAL)
verpan2.Add(self.panel2, 8, wx.ALL|wx.EXPAND, 2)
hpan1.Add(self.panel_left, 5, wx.ALL|wx.EXPAND, 2)
hpan1.Add(self.panel, 24, wx.ALL|wx.EXPAND, 2)
hpan1.Add(self.panel_right, 3, wx.ALL|wx.EXPAND, 2)
hpan1.Add(self.panel_right2, 3, wx.ALL|wx.EXPAND, 2)
hpan2.Add(self.panel3, 1, wx.ALL|wx.EXPAND, 2)
hpan2.Add(verpan2, 7, wx.ALL|wx.EXPAND, 2)
hpan3.Add(self.panel4, 1, wx.ALL|wx.EXPAND, 2)
ver.Add(hpan1, 1, wx.EXPAND)
ver.Add(hpan2, 18, wx.EXPAND)
ver.Add(hpan3, 4, wx.EXPAND)
self.browser.SetSize(self.panel3.GetSize())
self.SetSizer(ver)
#TABS: lines 137-159 instantiate the tabs for the main viewing panel.
self.nb = wx.Notebook(self.panel2, id=7829, style = wx.NB_BOTTOM)
self.page1 = wx.ScrolledWindow(self.nb, wx.ID_ANY, wx.DefaultPosition, wx.DefaultSize, wx.HSCROLL|wx.VSCROLL )
self.page1.SetScrollRate( 5, 5 )
self.page2 = PageTwo(self.nb)
self.page3 = PageThree(self.nb)
self.page4 = PageFour(self.nb)
self.nb.AddPage(self.page2, "Table")
self.nb.AddPage(self.page1, "PNG")
self.nb.AddPage(self.page3, "Interactive")
self.page3.SetBackgroundColour((218, 218, 218))
sizer = wx.BoxSizer()
sizer.Add(self.nb, 1, wx.EXPAND)
self.panel2.SetSizer(sizer)
self.page1.SetBackgroundColour("white")
self.myGrid = gridlib.Grid(self.page2, id=1002)
#self.myGrid.CreateGrid(100, self.dataset_file_length) ### Sets this at 400 columns rather than 100 - Excel like
self.Bind(gridlib.EVT_GRID_CELL_RIGHT_CLICK, self.GridRightClick, id=1002)
self.Bind(gridlib.EVT_GRID_CELL_LEFT_DCLICK, self.GridRowColor, id=1002)
self.HighlightedCells = []
gridsizer = wx.BoxSizer(wx.VERTICAL)
gridsizer.Add(self.myGrid)
self.page2.SetSizer(gridsizer)
self.page2.Layout()
#In the event that the interactive tab is chosen, a function must immediately run.
self.Bind(wx.EVT_NOTEBOOK_PAGE_CHANGED, self.InteractiveTabChoose, id=7829)
#INTERACTIVE PANEL LAYOUT: lines 167-212
#Pca Setup
self.RunButton1 = wx.Button(self.page3, id=43, label="Run", pos=(275, 150), size=(120, bheight))
self.Bind(wx.EVT_BUTTON, self.InteractiveRun, id=43)
self.Divider1 = self.ln = wx.StaticLine(self.page3, pos=(5,100))
self.ln.SetSize((415,10))
IntTitleFont = wx.Font(15, wx.SWISS, wx.NORMAL, wx.BOLD)
self.InteractiveTitle = wx.StaticText(self.page3, label="Main Dataset Parameters", pos=(10, 15))
self.InteractiveDefaultMessage = wx.StaticText(self.page3, label="No interactive options available.", pos=(10, 45))
self.InteractiveTitle.SetFont(IntTitleFont)
self.IntFileTxt = wx.TextCtrl(self.page3, id=43, pos=(105,45), size=(375,20))
self.InteractiveFileLabel = wx.StaticText(self.page3, label="Selected File:", pos=(10, 45))
self.Yes1Label = wx.StaticText(self.page3, label="Yes", pos=(305, 80))
self.No1Label = wx.StaticText(self.page3, label="No", pos=(375, 80))
self.D_3DLabel = wx.StaticText(self.page3, label="3D", pos=(305, 120))
self.D_2DLabel = wx.StaticText(self.page3, label="2D", pos=(375, 120))
self.IncludeLabelsRadio = wx.RadioButton(self.page3, id=40, pos=(285, 83), size=(12, 12), style=wx.RB_GROUP)
self.No1Radio = wx.RadioButton(self.page3, id=41, pos=(355, 83), size=(12, 12))
self.IncludeLabelsRadio.SetValue(True)
#self.EnterPCAGenes = wx.TextCtrl(self.page3, id=48, pos=(105,45), size=(375,20))
self.D_3DRadio = wx.RadioButton(self.page3, id=46, pos=(285, 123), size=(12, 12), style=wx.RB_GROUP)
self.D_2DRadio = wx.RadioButton(self.page3, id=47, pos=(355, 123), size=(12, 12))
self.D_3DRadio.SetValue(True)
self.Opt1Desc = wx.StaticText(self.page3, label="Display sample labels next to each object", pos=(10, 80))
self.Opt2Desc = wx.StaticText(self.page3, label="Dimensions to display", pos=(10, 120))
self.IntFileTxt.Hide()
self.InteractiveFileLabel.Hide()
self.Yes1Label.Hide()
self.No1Label.Hide()
self.D_3DLabel.Hide()
self.D_2DLabel.Hide()
self.IncludeLabelsRadio.Hide()
self.No1Radio.Hide()
self.D_3DRadio.Hide()
self.D_2DRadio.Hide()
self.Opt1Desc.Hide()
self.Opt2Desc.Hide()
self.RunButton1.Hide()
self.Divider1.Hide()
#TERMINAL SETUP
TxtBox = wx.BoxSizer(wx.VERTICAL)
TxtBox.Add(self.control, 1, wx.EXPAND)
self.panel4.SetSizer(TxtBox)
self.panel4.Layout()
#SELECTION LIST
self.TopSelectList = []
self.SearchArray = []
self.SearchArrayFiltered = []
self.TopID = ""
self.ColoredCellList = []
#LOGO
self.png = wx.Image("logo.gif", wx.BITMAP_TYPE_ANY).ConvertToBitmap()
self.LOGO = wx.StaticBitmap(self.page1, -1, self.png, (0,0), (self.png.GetWidth(), self.png.GetHeight()), style=wx.ALIGN_CENTER)
imgsizer_v = wx.BoxSizer(wx.VERTICAL)
imgsizer_v.Add(self.LOGO, proportion=0, flag=wx.ALIGN_CENTER_HORIZONTAL|wx.ALIGN_CENTER_VERTICAL)
self.page1.SetSizer(imgsizer_v)
self.page1.Layout()
self.nb.SetSelection(1)
browspan = wx.BoxSizer(wx.VERTICAL)
browspan.Add(self.browser, 1, wx.EXPAND)
browspan.Add(self.browser2, 1, wx.EXPAND)
self.panel3.SetSizer(browspan)
self.PanelTitle = wx.StaticText(self.panel, label="", pos=(210, 15))
#Open Button
ButtonMan = wx.Button(self.panel_left, id=1001, label="Open Project", pos=(0,0), size=(100,100))
self.Bind(wx.EVT_BUTTON, self.OnOpen, id=1001)
OpenSizer = wx.BoxSizer(wx.HORIZONTAL)
OpenSizer.Add(ButtonMan, 1, wx.EXPAND)
self.panel_left.SetSizer(OpenSizer)
#STATUS BAR CREATE --- not all of these are currently functional. The "edit" menu still needs to be implemented.
status = self.CreateStatusBar()
menubar = wx.MenuBar()
file = wx.Menu()
edit = wx.Menu()
view = wx.Menu()
search = wx.Menu()
filter_table = wx.Menu()
help_menu = wx.Menu()
open_menu = wx.Menu()
open_menu.Append(120, 'Project')
open_menu.Append(121, 'File')
file.AppendMenu(101, '&Open\tCtrl+O', open_menu)
file.Append(102, '&Save\tCtrl+S', 'Save the document')
file.AppendSeparator()
file.Append(103, 'Options', '')
file.AppendSeparator()
quit = wx.MenuItem(file, 105, '&Quit\tCtrl+Q', 'Quit the Application')
file.AppendItem(quit)
edit.Append(109, 'Undo', '')
edit.Append(110, 'Redo', '')
edit.AppendSeparator()
edit.Append(106, '&Cut\tCtrl+X', '')
edit.Append(107, '&Copy\tCtrl+C', '')
edit.Append(108, '&Paste\tCtrl+V', '')
edit.AppendSeparator()
edit.Append(111, '&Select All\tCtrl+A', '')
view.Append(112, '&Clear Panel\tCtrl+.', '')
search.Append(113, 'Tree', '')
search.Append(114, 'Table', '')
filter_table.Append(116, 'Filter', '')
filter_table.Append(117, 'Sort', '')
help_menu.AppendSeparator()
help_menu.Append(139, 'Help', '')
help_menu.Append(140, 'About', '')
menubar.Append(file, "File")
menubar.Append(edit, "Edit")
menubar.Append(view, "View")
menubar.Append(search, "Search")
menubar.Append(filter_table, "Table")
menubar.Append(help_menu, "Help")
self.SetMenuBar(menubar)
#STATUS BAR BINDINGS
self.Bind(wx.EVT_MENU, self.OnOpen, id=120)
self.Bind(wx.EVT_MENU, self.OnOpenSingleFile, id=121)
self.Bind(wx.EVT_MENU, self.OnQuit, id=105)
self.Bind(wx.EVT_MENU, self.ClearVisualPanel, id=112)
self.Bind(wx.EVT_MENU, self.TreeSearch, id=113)
self.Bind(wx.EVT_MENU, self.GridSearch, id=114)
self.Bind(wx.EVT_MENU, self.FilterTable, id=116)
self.Bind(wx.EVT_MENU, self.SortTable, id=117)
self.Bind(wx.EVT_MENU, self.OnAbout, id=140)
self.Bind(wx.EVT_MENU, self.OnHelp, id=139)
self.Layout()
def OnQuit(self, event):
popup = wx.MessageDialog(None, "Are you sure you want to quit?", "Warning", wx.YES_NO)
popup_answer = popup.ShowModal()
#print popup_answer
if(popup_answer == 5103):
self.Close()
else:
return
def GridRowColor(self, event):
#This colors any row that has been selected and resets it accordingly: may be removed in future versions.
if len(self.HighlightedCells) > 0:
for i in self.HighlightedCells:
self.myGrid.SetCellBackgroundColour(i[0], i[1], (255, 255, 255))
self.HighlightedCells = []
self.GridRowEvent = event.GetRow()
for i in range(50):
self.myGrid.SetCellBackgroundColour(self.GridRowEvent, i, (235, 255, 255))
self.HighlightedCells.append((self.GridRowEvent, i))
def GridRightClick(self, event):
#Pop-up menu instantiation for a right click on the table.
self.GridRowEvent = event.GetRow()
# only do this part the first time so the events are only bound once
if not hasattr(self, "popupID3"):
self.popupID1 = wx.NewId()
self.popupID2 = wx.NewId()
if self.analyzeSplicing:
self.popupID3 = wx.NewId()
self.popupID4 = wx.NewId()
self.popupID5 = wx.NewId()
self.Bind(wx.EVT_MENU, self.GeneExpressionSummaryPlot, id=self.popupID1)
self.Bind(wx.EVT_MENU, self.PrintGraphVariables, id=self.popupID2)
if self.analyzeSplicing:
self.Bind(wx.EVT_MENU, self.AltExonViewInitiate, id=self.popupID3)
self.Bind(wx.EVT_MENU, self.IsoformViewInitiate, id=self.popupID4)
self.Bind(wx.EVT_MENU, self.SashimiPlotInitiate, id=self.popupID5)
# build the menu
menu = wx.Menu()
itemOne = menu.Append(self.popupID1, "Gene Plot")
#itemTwo = menu.Append(self.popupID2, "Print Variables")
if self.analyzeSplicing:
itemThree = menu.Append(self.popupID3, "Exon Plot")
itemFour = menu.Append(self.popupID4, "Isoform Plot")
itemFive = menu.Append(self.popupID5, "SashimiPlot")
# show the popup menu
self.PopupMenu(menu)
menu.Destroy()
def AltExonViewInitiate(self, event):
### Temporary option for exon visualization until the main tool is complete and database can be bundled with the program
i=0; values=[]
while i<1000:
try:
val = str(self.myGrid.GetCellValue(self.GridRowEvent, i))
values.append(val)
if ('G000' in val) and '->' not in val:
geneID_temp = string.split(val,":")[0]
if ('G000' in geneID_temp) and '->' not in geneID_temp:
geneID = geneID_temp
if ' ' in geneID:
geneID = string.split(geneID,' ')[0]
else:
geneID_temp = string.split(val,":")[1]
if ('G000' in geneID_temp):
geneID = geneID_temp
if ' ' in geneID:
geneID = string.split(geneID,' ')[0]
i+=1
except Exception: break
datasetDir = self.main_results_directory
#print datasetDir
self.control.write("Plotting... " + geneID + "\n")
data_type = 'raw expression'
show_introns = 'no'
analysisType = 'graph-plot'
exp_dir = unique.filepath(datasetDir+'/ExpressionInput')
#print exp_dir
exp_file = UI.getValidExpFile(exp_dir)
#print print exp_file
UI.altExonViewer(self.species,self.platform,exp_file,geneID,show_introns,analysisType,'')
def IsoformViewInitiate(self, event):
#print os.getcwd()
#This function is a part of the pop-up menu for the table: it plots a gene and protein level view.
os.chdir(parentDirectory)
t = os.getcwd()
#self.control.write(str(os.listdir(t)) + "\n")
gene = self.myGrid.GetCellValue(self.GridRowEvent, 0)
i=0; values=[]; spliced_junctions=[]
while i<1000:
try:
val = str(self.myGrid.GetCellValue(self.GridRowEvent, i))
values.append(val)
if ('G000' in val) and 'ENSP' not in val and 'ENST' not in val and '->' not in val:
geneID_temp = string.split(val,":")[0]
if ('G000' in geneID_temp) and '->' not in geneID_temp:
geneID = geneID_temp
if ' ' in geneID:
geneID = string.split(geneID,' ')[0]
elif '->' in geneID_temp: pass
else:
geneID_temp = string.split(val,":")[1]
if ('G000' in geneID_temp):
geneID = geneID_temp
if ' ' in geneID:
geneID = string.split(geneID,' ')[0]
i+=1
except Exception: break
#print [geneID]
self.control.write("Plotting... " + geneID + "\n")
from visualization_scripts import ExPlot
reload(ExPlot)
ExPlot.remoteGene(geneID,self.species,self.main_results_directory,self.CurrentFile)
#Q = subprocess.Popen(['python', 'ExPlot13.py', str(R)])
#os.chdir(currentDirectory)
def SashimiPlotInitiate(self, event):
#This function is a part of the pop-up menu for the table: it plots a SashimiPlot
datasetDir = str(self.main_results_directory)
geneID = None
#self.control.write(str(os.listdir(t)) + "\n")
i=0; values=[]; spliced_junctions=[]
while i<1000:
try:
val = str(self.myGrid.GetCellValue(self.GridRowEvent, i))
values.append(val)
if ('G000' in val) and ':E' in val:
#if 'ASPIRE' in self.DirFileTxt:
if ':ENS' in val:
val = 'ENS'+string.split(val,':ENS')[1]
val = string.replace(val,'|', ' ')
#Can also refer to MarkerFinder files
if ' ' in val:
if '.' not in string.split(val,' ')[1]:
val = string.split(val,' ')[0] ### get the gene
if 'Combined-junction' in self.DirFileTxt:
if '-' in val and '|' in val:
junctions = string.split(val,'|')[0]
val = 'ENS'+string.split(junctions,'-ENS')[-1]
spliced_junctions.append(val) ### exclusion junction
if 'index' in self.DirFileTxt: ### Splicing-index analysis
spliced_junctions.append(val)
elif '-' in val:
spliced_junctions.append(val) ### junction-level
if ('G000' in val) and geneID == None and '->' not in val:
geneID = string.split(val,":")[0]
if ' ' in geneID:
geneID = string.split(geneID,' ')[0]
i+=1
except Exception: break
if len(spliced_junctions)>0:
spliced_junctions = [spliced_junctions[-1]] ### Select the exclusion junction
else:
spliced_junctions = [geneID]
if 'DATASET' in self.DirFileTxt:
spliced_junctions = [geneID]
from visualization_scripts import SashimiPlot
reload(SashimiPlot)
self.control.write("Attempting to build SashimiPlots for " + str(spliced_junctions[0]) + "\n")
SashimiPlot.remoteSashimiPlot(self.species,datasetDir,datasetDir,None,events=spliced_junctions,show=True) ### assuming the bam files are in the root-dir
def GeneExpressionSummaryPlot(self, event):
#This function is a part of the pop-up menu for the table: it plots expression levels.
Wikipathway_Flag = 0
Protein_Flag = 0
VarGridSet = []
try:
for i in range(3000):
try:
p = self.myGrid.GetCellValue(0, i)
VarGridSet.append(p)
except Exception:
pass
for i in VarGridSet:
y = re.findall("WikiPathways", i)
if len(y) > 0:
Wikipathway_Flag = 1
break
if Wikipathway_Flag == 0:
for i in VarGridSet:
y = re.findall("Select Protein Classes", i)
if len(y) > 0:
Protein_Flag = 1
break
if Protein_Flag == 1:
VariableBox = []
for i in range(len(VarGridSet)):
y = re.findall("avg", VarGridSet[i])
if(len(y) > 0):
VariableBox.append(i)
if Wikipathway_Flag == 1:
VariableBox = []
for i in range(len(VarGridSet)):
y = re.findall("avg", VarGridSet[i])
if(len(y) > 0):
VariableBox.append(i)
q_barrel = []
for i in VariableBox:
q_box = []
q = i
for p in range(500):
if(q < 0):
break
q = q - 1
#Regular expression is needed to find the appropriate columns to match from.
FLAG_log_fold = re.findall("log_fold",VarGridSet[q])
FLAG_adjp = re.findall("adjp",VarGridSet[q])
FLAG_rawp = re.findall("rawp",VarGridSet[q])
FLAG_wiki = re.findall("Wiki",VarGridSet[q])
FLAG_pc = re.findall("Protein Classes",VarGridSet[q])
FLAG_avg = re.findall("avg",VarGridSet[q])
if(len(FLAG_log_fold) > 0 or len(FLAG_adjp) > 0 or len(FLAG_rawp) > 0 or len(FLAG_wiki) > 0 or len(FLAG_pc) > 0 or len(FLAG_avg) > 0):
break
q_box.append(q)
q_barrel.append((q_box))
Values_List = []
HeaderList = []
TitleList = self.myGrid.GetCellValue(self.GridRowEvent, 0)
for i in VariableBox:
HeaderList.append(self.myGrid.GetCellValue(0, i))
for box in q_barrel:
output_box = []
for value in box:
output_var = self.myGrid.GetCellValue(self.GridRowEvent, value)
output_box.append(float(output_var))
Values_List.append((output_box))
self.control.write("Plotting values from: " + str(self.myGrid.GetCellValue(self.GridRowEvent, 0)) + "\n")
Output_Values_List = []
Output_std_err = []
for box in Values_List:
T = 0
for item in box:
T = T + item
output_item = T / float(len(box))
Output_Values_List.append(output_item)
for box in Values_List:
box_std = np.std(box)
box_power = np.power((len(box)), 0.5)
std_err = box_std / float(box_power)
Output_std_err.append(std_err)
n_groups = len(Output_Values_List)
#PLOTTING STARTS --
means_men = Output_Values_List
import matplotlib.pyplot as plt
fig, ax = plt.subplots()
index = np.arange(n_groups)
bar_width = 0.35
pos = bar_width / float(2)
opacity = 0.4
error_config = {'ecolor': '0.3'}
with warnings.catch_warnings():
rects1 = plt.bar((index + pos), Output_Values_List, bar_width,
alpha=opacity,
color='b',
yerr=Output_std_err,
label="")
#plt.title(self.myGrid.GetCellValue(self.GridRowEvent, 2))
plt.title(TitleList)
plt.xticks(index + bar_width, HeaderList)
plt.legend()
plt.tight_layout()
plt.show()
#-- PLOTTING STOPS
except Exception:
self.control.write("Plot failed to output... only applicalbe for the file with prefix DATASET")
def PrintGraphVariables(self, event):
#This function is a part of the pop-up menu for the table: it prints the variables for the expression levels. Used for testing mainly.
Wikipathway_Flag = 0
Protein_Flag = 0
VarGridSet = []
for i in range(100):
p = self.myGrid.GetCellValue(0, i)
VarGridSet.append(p)
for i in VarGridSet:
y = re.findall("WikiPathways", i)
if len(y) > 0:
Wikipathway_Flag = 1
break
if Wikipathway_Flag == 0:
for i in VarGridSet:
y = re.findall("Select Protein Classes", i)
if len(y) > 0:
Protein_Flag = 1
break
if Protein_Flag == 1:
VariableBox = []
for i in range(len(VarGridSet)):
y = re.findall("avg", VarGridSet[i])
if(len(y) > 0):
VariableBox.append(i)
if Wikipathway_Flag == 1:
VariableBox = []
for i in range(len(VarGridSet)):
y = re.findall("avg", VarGridSet[i])
if(len(y) > 0):
VariableBox.append(i)
q_barrel = []
for i in VariableBox:
q_box = []
q = i
for p in range(500):
if(q < 0):
break
q = q - 1
FLAG_log_fold = re.findall("log_fold",VarGridSet[q])
FLAG_adjp = re.findall("adjp",VarGridSet[q])
FLAG_rawp = re.findall("rawp",VarGridSet[q])
FLAG_wiki = re.findall("Wiki",VarGridSet[q])
FLAG_pc = re.findall("Protein Classes",VarGridSet[q])
FLAG_avg = re.findall("avg",VarGridSet[q])
if(len(FLAG_log_fold) > 0 or len(FLAG_adjp) > 0 or len(FLAG_rawp) > 0 or len(FLAG_wiki) > 0 or len(FLAG_pc) > 0 or len(FLAG_avg) > 0):
break
q_box.append(q)
q_barrel.append((q_box))
self.control.write("Selected Row: " + str(self.myGrid.GetCellValue(self.GridRowEvent, 0)) + "\n")
self.control.write("Selected Columns: " + str(q_barrel) + "\n")
Values_List = []
HeaderList = []
for i in VariableBox:
HeaderList.append(self.myGrid.GetCellValue(0, i))
for box in q_barrel:
output_box = []
for value in box:
output_var = self.myGrid.GetCellValue(self.GridRowEvent, value)
output_box.append(float(output_var))
Values_List.append((output_box))
self.control.write("Selected Values: " + str(Values_List) + "\n")
def InteractiveTabChoose(self, event):
#If the interactive tab is chosen, a plot will immediately appear with the default variables.
try:
#The PCA and Heatmap flags are set; a different UI will appear for each of them.
PCA_RegEx = re.findall("PCA", self.DirFile)
Heatmap_RegEx = re.findall("hierarchical", self.DirFile)
if(self.nb.GetSelection() == 2):
if(len(PCA_RegEx) > 0 or len(Heatmap_RegEx) > 0):
self.InteractiveRun(event)
except:
pass
def getDatasetVariables(self):
for file in os.listdir(self.main_results_directory):
if 'AltAnalyze_report' in file and '.log' in file:
log_file = unique.filepath(self.main_results_directory+'/'+file)
log_contents = open(log_file, "rU")
species = ' species: '
platform = ' method: '
for line in log_contents:
line = line.rstrip()
if species in line:
self.species = string.split(line,species)[1]
if platform in line:
self.platform = string.split(line,platform)[1]
try:
self.supported_genesets = UI.listAllGeneSetCategories(self.species,'WikiPathways','gene-mapp')
self.geneset_type = 'WikiPathways'
except Exception:
try:
self.supported_genesets = UI.listAllGeneSetCategories(self.species,'GeneOntology','gene-mapp')
self.geneset_type = 'GeneOntology'
except Exception:
self.supported_genesets = []
self.geneset_type = 'None Selected'
#print 'Using',self.geneset_type, len(self.supported_genesets),'pathways'
break
try:
for file in os.listdir(self.main_results_directory+'/ExpressionOutput'):
if 'DATASET' in file:
dataset_file = unique.filepath(self.main_results_directory+'/ExpressionOutput/'+file)
for line in open(dataset_file,'rU').xreadlines():
self.dataset_file_length = len(string.split(line,'\t'))
break
except Exception:
pass
try:
if self.dataset_file_length<50:
self.dataset_file_length=50
except Exception:
self.dataset_file_length=50
self.myGrid.CreateGrid(100, self.dataset_file_length) ### Re-set the grid width based on the DATASET- file width
def OnOpen(self, event):
#Bound to the open tab from the menu and the "Open Project" button.
openFileDialog = wx.DirDialog(None, "Choose project", "", wx.DD_DEFAULT_STYLE | wx.DD_DIR_MUST_EXIST)
if openFileDialog.ShowModal() == wx.ID_CANCEL:
return
#self.input stream is the path of our project's main directory.
self.main_results_directory = openFileDialog.GetPath()
if (len(self.main_results_directory) > 0):
if self.species == '':
self.getDatasetVariables()
self.SearchArray = []
self.SearchArrayFiltered = []
self.control.write("Working..." + "\n")
#FLAG COLLECT
root = 'Data'
for (dirpath, dirnames, filenames) in os.walk(root):
for dirname in dirnames:
#fullpath = os.path.join(dirpath, dirname)
fullpath = currentDirectory+'/'+dirpath+'/'+dirname
for filename in sorted(filenames):
if filename == "location.txt":
#file_fullpath = unique.filepath(os.path.join(dirpath, filename))
file_fullpath = currentDirectory+'/'+dirpath+'/'+filename
file_location = open(file_fullpath, "r")
fl_array = []
for line in file_location:
line = line.rstrip(); line = string.replace(line,'"','')
line = line.split("\r")
if len(line) > 1:
fl_array.append(line[0])
fl_array.append(line[1])
else:
fl_array.append(line[0])
file_location.close()
#if dirname == 'ExonGraph': print fl_array
if(len(fl_array) == 3):
fl_array.append(dirpath)
self.SearchArray.append(fl_array)
self.control.write("Opening project at: " + self.main_results_directory + "\n")
self.browser2.DeleteAllItems()
#SEARCH USING FLAGS
count = 0
for FLAG in self.SearchArray:
if((FLAG[0][-1] != "/") and (FLAG[0][-1] != "\\")):
SearchingFlag = FLAG[0] + "/"
SearchingFlag = FLAG[0]
SearchingFlagPath = self.main_results_directory + "/" + SearchingFlag
try:
SFP_Contents = os.listdir(SearchingFlagPath)
for filename in SFP_Contents:
Search_base = FLAG[1]
Search_base = Search_base.split(":")
Search_base = Search_base[1]
Split_Extension = str(FLAG[2])
Split_Extension = Split_Extension.split(":")
S_E = str(Split_Extension[1]).split(",")
GOOD_FLAG = 0
if(Search_base != "*"):
for i in S_E:
if(filename[-4:] == i):
GOOD_FLAG = 1
if(Search_base != "*"):
candidate = re.findall(Search_base, filename)
if(Search_base == "*"):
candidate = "True"
GOOD_FLAG = 1
if (len(Search_base) == 0 or GOOD_FLAG == 0):
continue
if len(candidate) > 0:
self.SearchArrayFiltered.append(FLAG)
except:
continue
count = count + 1
#AVAILABLE DATA SET
try:
shutil.rmtree("AvailableData")
except:
pass
for i in self.SearchArrayFiltered:
AvailablePath = "Available" + i[3]
if '\\' in AvailablePath: ### Windows
AvailablePath = string.replace(AvailablePath,'/','\\')
if '/' in AvailablePath:
Path_List = AvailablePath.split("/")
else:
Path_List = AvailablePath.split("\\")
Created_Directory = ""
for directorynum in range(len(Path_List)):
if directorynum == 0:
Created_Directory = Created_Directory + Path_List[directorynum]
try:
os.mkdir(Created_Directory)
except:
continue
else:
Created_Directory = Created_Directory + "/" + Path_List[directorynum]
try:
os.mkdir(Created_Directory)
except:
continue
#TOP BROWSER SET
root = 'AvailableData'
color_root = [253, 253, 253]
self.tree.DeleteAllItems()
self.ids = {root : self.tree.AddRoot(root)}
self.analyzeSplicing=False
for (dirpath, dirnames, filenames) in os.walk(root):
#print 'x',[dirpath, dirnames, filenames]#;sys.exit()
for dirname in dirnames:
#print dirpath, dirname
if 'Splicing' in dirpath: self.analyzeSplicing=True
fullpath = os.path.join(dirpath, dirname)
#print currentDirectory+'/'+dirpath
self.ids[fullpath] = self.tree.AppendItem(self.ids[dirpath], dirname)
DisplayColor = [255, 255, 255]
DisplayColor[0] = color_root[0] - len(dirpath)
DisplayColor[1] = color_root[1] - len(dirpath)
DisplayColor[2] = color_root[2] - len(dirpath)
self.tree.SetItemBackgroundColour(self.ids[fullpath], DisplayColor)
for i in self.SearchArrayFiltered:
SearchRoot = "Available" + i[3]
if(SearchRoot == fullpath):
SearchSplit = i[1].split(":")
SearchSplit = SearchSplit[1]
SearchSplit = SearchSplit + ";" + i[0]
SearchSplit = SearchSplit + ";" + i[2]
DisplayColor = [130, 170, 250]
self.tree.SetItemData(self.ids[fullpath],wx.TreeItemData(SearchSplit))
self.tree.SetItemBackgroundColour(self.ids[fullpath], DisplayColor)
self.tree.SetItemBackgroundColour(self.ids[root], [100, 140, 240])
self.tree.Expand(self.ids[root])
try: self.Bind(wx.EVT_TREE_ITEM_ACTIVATED, self.SelectedTopTreeID, self.tree)
except Exception: pass
#OPENING DISPLAY
try:
self.LOGO.Destroy()
except:
pass
self.png = wx.Image(rootDirectory+"/Config/no-image-available.png", wx.BITMAP_TYPE_ANY).ConvertToBitmap()
self.LOGO = wx.StaticBitmap(self.page1, -1, self.png, (0,0), (self.png.GetWidth(), self.png.GetHeight()), style=wx.ALIGN_CENTER)
imgsizer_v = wx.BoxSizer(wx.VERTICAL)
imgsizer_v.Add(self.LOGO, proportion=0, flag=wx.ALIGN_CENTER_HORIZONTAL|wx.ALIGN_CENTER_VERTICAL)
self.page1.SetSizer(imgsizer_v)
self.page1.Layout()
self.control.write("Resetting grid..." + "\n")
self.control.write("Currently displaying: " + "SUMMARY" + "\n")
self.myGrid.ClearGrid()
if 'ExpressionInput' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'ExpressionInput')[0]
if 'AltResults' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'AltResults')[0]
if 'ExpressionOutput' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'ExpressionOutput')[0]
if 'GO-Elite' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'GO-Elite')[0]
if 'ICGS' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'ICGS')[0]
if 'DataPlots' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'DataPlots')[0]
if 'AltExpression' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'AltExpression')[0]
if 'AltDatabase' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'AltDatabase')[0]
if 'ExonPlots' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'ExonPlots')[0]
if 'SashimiPlots' in self.main_results_directory:
self.main_results_directory = string.split(self.main_results_directory,'SashimiPlots')[0]
opening_display_folder = self.main_results_directory + "/ExpressionOutput"
try:
list_contents = os.listdir(opening_display_folder)
target_file = ""
for file in list_contents:
candidate = re.findall("SUMMARY", file)
if len(candidate) > 0:
target_file = file
break
except Exception:
opening_display_folder = self.main_results_directory
list_contents = os.listdir(opening_display_folder)
for file in list_contents:
candidate = re.findall(".log", file)
if len(candidate) > 0:
target_file = file ### get the last log file
target_file = unique.filepath(opening_display_folder + "/" + target_file)
opened_target_file = open(target_file, "r")
opened_target_file_contents = []
for line in opened_target_file:
line = line.rstrip(); line = string.replace(line,'"','')
line = line.split("\t")
if len(line)==1: line += ['']*5
opened_target_file_contents.append((line))
self.table_length = len(opened_target_file_contents)
for cell in self.ColoredCellList:
try: self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
except Exception: pass
self.ColoredCellList = []
x_count = 0
for item_list in opened_target_file_contents:
y_count = 0
for item in item_list:
try:
self.myGrid.SetCellValue(x_count, y_count, item)
except Exception:
pass ### if the length of the row is 0
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
self.myGrid.AutoSize()
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
#This line always sets the opening display to the "Table" tab.
self.nb.SetSelection(0)
def OnOpenSingleFile(self, event):
#Opens only one file as opposed to the whole project; possibly unstable and needs further testing.
openFileDialog = wx.FileDialog(self, "", "", "", "", wx.FD_OPEN | wx.FD_FILE_MUST_EXIST)
if openFileDialog.ShowModal() == wx.ID_CANCEL:
return
single_input_stream = openFileDialog.GetPath()
self.control.write(str(single_input_stream) + "\n")
if single_input_stream[-4:] == ".txt":
self.myGrid.ClearGrid()
self.DirFileTxt = single_input_stream
self.DirFile = single_input_stream
table_file = open(self.DirFileTxt, "r")
table_file_contents = []
for line in table_file:
line = line.rstrip(); line = string.replace(line,'"','')
line = line.split("\t")
if(len(table_file_contents) >= 5000):
break
table_file_contents.append((line))
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
x_count = 0
for item_list in table_file_contents:
y_count = 0
for item in item_list:
self.myGrid.SetCellValue(x_count, y_count, item)
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
self.myGrid.AutoSize()
for i in range(50):
try:
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
except Exception: pass
self.page2.Layout()
if single_input_stream[-4:] == ".png":
self.myGrid.ClearGrid()
try:
self.LOGO.Destroy()
except:
pass
self.png = wx.Image(single_input_stream, wx.BITMAP_TYPE_ANY).ConvertToBitmap()
self.LOGO = wx.StaticBitmap(self.page1, -1, self.png, (0,0), (self.png.GetWidth(), self.png.GetHeight()), style=wx.ALIGN_CENTER)
imgsizer_v = wx.BoxSizer(wx.VERTICAL)
imgsizer_v.Add(self.LOGO, proportion=0, flag=wx.ALIGN_CENTER_HORIZONTAL|wx.ALIGN_CENTER_VERTICAL)
self.page1.SetSizer(imgsizer_v)
self.page1.Layout()
if single_input_stream[-4:] == ".pdf":
#http://wxpython.org/Phoenix/docs/html/lib.pdfviewer.html
pass
def OnSave(self, event):
#Save function is currently not implemented but is a priority for future updates.
saveFileDialog = wx.FileDialog(self, "", "", "", "", wx.FD_SAVE | wx.FD_OVERWRITE_PROMPT)
if saveFileDialog.ShowModal() == wx.ID_CANCEL:
return
def OnSearch(self, event):
#This handles the search prompt pop-up box when using "search -> table" from the status bar menu.
popup = wx.TextEntryDialog(None, "Enter filter for results.", "Search", "Enter search here.")
if popup.ShowModal()==wx.ID_OK:
answer=popup.GetValue()
popup.Destroy()
else:
popup.Destroy()
return
def TreeSearch(self, event):
#Search tree function: searches the tree for a given phrase and opens the tree to that object.
popup = wx.TextEntryDialog(None, "Search the browser tree for directories and files.", "Search", "Enter search here.")
if popup.ShowModal()==wx.ID_OK:
answer=popup.GetValue()
self.control.write("K" + str(answer) + "\n")
os.chdir(currentDirectory) ### NS-91615 alternative to __file__
rootman = "AvailableData"
search_box = []
found = ""
for (dirpath, dirnames, filenames) in os.walk(rootman):
for dirname in dirnames:
fullpath = dirpath + "/" + dirname
search_box.append(fullpath)
self.control.write("Searching..." + "\n")
for path in search_box:
path2 = path.split("/")
search_candidate = path2[-1]
self.control.write(search_candidate + " " + str(answer) + "\n")
if(str(answer) == search_candidate):
found = path
break
self.control.write(found + "\n")
tree_recreate = found.split("/")
treepath = ""
self.control.write(str(range(len(tree_recreate))) + "\n")
tree_length = len(tree_recreate)
last_tree_value = len(tree_recreate) - 1
for i in range(tree_length):
self.control.write(str(i) + "\n")
if(i == 0):
self.tree.Expand(self.ids[tree_recreate[i]])
treepath = treepath + tree_recreate[i]
self.control.write(treepath + "\n")
if(i > 0 and i < last_tree_value):
treepath = treepath + "/" + tree_recreate[i]
self.control.write(treepath + "\n")
self.tree.Expand(self.ids[treepath])
if(i == last_tree_value):
treepath = treepath + "/" + tree_recreate[i]
self.control.write(treepath + "\n")
self.tree.SelectItem(self.ids[treepath])
popup.Destroy()
else:
popup.Destroy()
return
def GridSearch(self, event):
#Search table function: this searchs the table and highlights the search query in the table; also zooms to the nearest match.
popup = wx.TextEntryDialog(None, "Search the table.", "Search", "Enter search here.")
if popup.ShowModal()==wx.ID_OK:
PageDownFound = "False"
match_count = 0
answer=popup.GetValue()
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
if(self.table_length > 5100):
y_range = range(5100)
y_range = range(self.table_length)
x_range = range(100)
y_count = 0
for number in y_range:
x_count = 0
for number in x_range:
cellvalue = self.myGrid.GetCellValue(y_count, x_count)
gridmatch = re.findall(answer, cellvalue)
if(len(gridmatch) > 0):
if(PageDownFound == "False"):
PageScrollY = y_count
PageScrollX = x_count
PageDownFound = "True"
match_count = match_count + 1
self.ColoredCellList.append((y_count, x_count))
self.myGrid.SetCellBackgroundColour(y_count, x_count, (255, 255, 125))
x_count = x_count + 1
y_count = y_count + 1
#"MakeCellVisible" zooms to the given coordinates.
self.myGrid.MakeCellVisible(PageScrollY, PageScrollX)
terminal_list = []
for cell in self.ColoredCellList:
newrow = cell[0] + 1
newcolumn = cell[1] + 1
terminal_list.append((newrow, newcolumn))
self.control.write(str(match_count) + " matches found for " + answer + "\n")
self.control.write("At positions (row, column): " + str(terminal_list) + "\n")
popup.Destroy()
self.nb.SetSelection(0)
else:
popup.Destroy()
return
def FilterTable(self, event):
#The filter function displays ONLY the rows that have matches for the given search. Does not delete the filtered out data---table data is still fully functional and usable.
popup = wx.TextEntryDialog(None, "Filter the table.", "Search", "Enter filter phrase.")
if popup.ShowModal()==wx.ID_OK:
self.myGrid.ClearGrid()
answer=popup.GetValue()
try:
table_file = open(self.DirFileTxt, "r")
table_file_contents = []
count = 0
for line in table_file:
line = line.rstrip(); line = string.replace(line,'"','')
regex_test = re.findall(answer.upper(), line.upper())
line = line.split("\t")
if(len(regex_test) > 0 or count == 0):
if(len(table_file_contents) >= 5100):
break
table_file_contents.append((line))
count = count + 1
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
x_count = 0
for item_list in table_file_contents:
y_count = 0
for item in item_list:
self.myGrid.SetCellValue(x_count, y_count, item)
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
self.myGrid.AutoSize()
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
self.control.write("Unable to open txt." + "\n")
self.nb.SetSelection(0)
def SortTable(self, event):
#The sort function re-writes the table sorting by either descending or ascending values in a given column.
popup = wx.TextEntryDialog(None, "Sort the table.", "Sort", "Which column to sort from?")
if popup.ShowModal()==wx.ID_OK:
self.myGrid.ClearGrid()
answer=popup.GetValue()
answer = answer.upper()
try:
table_file = open(self.DirFileTxt, "r")
table_file_contents = []
pre_sort2 = []
header = []
t_c = 0
column_clusters_flat = 0
for line in table_file:
line=string.replace(line,'Insufficient Expression','0')
try:
line = line.rstrip(); line = string.replace(line,'"','')
line = line.split("\t")
if(t_c == 0):
header.append((line))
t_c = t_c + 1
continue
if(line[0] == "column_clusters-flat"):
header.append((line))
column_clusters_flat = 1
continue
line_sort_select = line[self.sortdict[answer]]
pre_sort1 = []
count = 0
for i in line:
if(count == 0):
try:
pre_sort1.append(float(line_sort_select))
except:
pre_sort1.append(line_sort_select)
pre_sort1.append(i)
if(count == self.sortdict[answer]):
count = count + 1
continue
if(count != 0):
pre_sort1.append(i)
count = count + 1
pre_sort2.append((pre_sort1))
except:
continue
table_file_contents.append(header[0])
if(column_clusters_flat == 1):
table_file_contents.append(header[1])
pre_sort2 = sorted(pre_sort2, reverse = True)
for line in pre_sort2:
try:
final_count1 = 0
final_count2 = 1
send_list = []
for item in line:
if(final_count1 == 0):
send_list.append(line[final_count2])
if(final_count1 == self.sortdict[answer]):
send_list.append(str(line[0]))
if(final_count1 != 0 and final_count1 != self.sortdict[answer]):
if(final_count1 < self.sortdict[answer]):
send_list.append(line[final_count2])
if(final_count1 > self.sortdict[answer]):
send_list.append(line[final_count1])
final_count1 = final_count1 + 1
final_count2 = final_count2 + 1
if(len(table_file_contents) >= 5100):
break
table_file_contents.append((send_list))
except:
continue
n_table_file_contents = []
if(answer.upper() == "A"):
for i in range(len(table_file_contents)):
if(i == 0):
max_length = len(table_file_contents[i])
if(max_length < len(table_file_contents[i])):
n_l = table_file_contents[i][2:]
else:
n_l = table_file_contents[i]
n_table_file_contents.append((n_l))
table_file_contents = n_table_file_contents
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
x_count = 0
for item_list in table_file_contents:
y_count = 0
for item in item_list:
self.myGrid.SetCellValue(x_count, y_count, item)
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
self.myGrid.AutoSizeRows(True)
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
self.control.write("Unable to sort." + "\n")
self.nb.SetSelection(0)
def FilterTablefromButton(self, event):
#Same filter function as before, but this function is bound to the button in the top-right corner of the main GUI.
self.myGrid.ClearGrid()
#In single line text boxes, you must always set 0 to the GetLineText value; 0 represents the first and only line.
answer = self.filterbox.GetLineText(0)
try:
try:
self.myGrid.DeleteRows(100, self.AppendTotal, True)
except:
pass
table_file_contents = []
count = 0
for line in open(self.DirFileTxt,'rU').xreadlines():
line = line.rstrip(); line = string.replace(line,'"','')
regex_test = re.findall(answer.upper(), line.upper())
line = line.split("\t")
if(len(regex_test) > 0 or count == 0):
if(len(table_file_contents) >= 5100):
break
table_file_contents.append((line))
count = count + 1
self.table_length = len(table_file_contents)
self.control.write("Table Length: " + str(self.table_length) + "\n")
if(self.table_length > 100):
self.AppendTotal = self.table_length - 100
self.myGrid.AppendRows(self.AppendTotal, True)
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
x_count = 0
for item_list in table_file_contents:
y_count = 0
for item in item_list:
self.myGrid.SetCellValue(x_count, y_count, item)
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
self.myGrid.AutoSize()
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
self.control.write("Unable to open txt." + "\n")
self.nb.SetSelection(0)
def SortTablefromButton(self, event):
#Same sort function as before, but this function is bound to the button in the top-right corner of the main GUI.
answer = self.sortbox.GetLineText(0)
self.myGrid.ClearGrid()
answer = answer.upper()
try:
table_file = open(self.DirFileTxt, "r")
table_file_contents = []
pre_sort2 = []
header = []
t_c = 0
column_clusters_flat = 0
for line in table_file:
line=string.replace(line,'Insufficient Expression','0')
try:
line = line.rstrip(); line = string.replace(line,'"','')
line = line.split("\t")
if(t_c == 0):
header.append((line))
t_c = t_c + 1
continue
if(line[0] == "column_clusters-flat"):
header.append((line))
column_clusters_flat = 1
continue
line_sort_select = line[self.sortdict[answer]]
pre_sort1 = []
count = 0
for i in line:
if(count == 0):
try:
pre_sort1.append(float(line_sort_select))
except:
pre_sort1.append(line_sort_select)
pre_sort1.append(i)
if(count == self.sortdict[answer]):
count = count + 1
continue
if(count != 0):
pre_sort1.append(i)
count = count + 1
pre_sort2.append((pre_sort1))
except:
continue
table_file_contents.append(header[0])
if(column_clusters_flat == 1):
table_file_contents.append(header[1])
if(self.DescendingRadio.GetValue() == True):
pre_sort2 = sorted(pre_sort2, reverse = True)
if(self.AscendingRadio.GetValue() == True):
pre_sort2 = sorted(pre_sort2)
for line in pre_sort2:
try:
final_count1 = 0
final_count2 = 1
send_list = []
for item in line:
if(final_count1 == 0):
send_list.append(line[final_count2])
if(final_count1 == self.sortdict[answer]):
send_list.append(str(line[0]))
if(final_count1 != 0 and final_count1 != self.sortdict[answer]):
if(final_count1 < self.sortdict[answer]):
send_list.append(line[final_count2])
if(final_count1 > self.sortdict[answer]):
send_list.append(line[final_count1])
final_count1 = final_count1 + 1
final_count2 = final_count2 + 1
if(len(table_file_contents) >= 5100):
break
table_file_contents.append((send_list))
except:
continue
n_table_file_contents = []
if(answer.upper() == "A"):
for i in range(len(table_file_contents)):
if(i == 0):
max_length = len(table_file_contents[i])
if(max_length < len(table_file_contents[i])):
n_l = table_file_contents[i][2:]
else:
n_l = table_file_contents[i]
n_table_file_contents.append((n_l))
table_file_contents = n_table_file_contents
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
x_count = 0
for item_list in table_file_contents:
y_count = 0
for item in item_list:
self.myGrid.SetCellValue(x_count, y_count, item)
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
self.myGrid.AutoSizeRows(True)
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
self.control.write("Unable to sort." + "\n")
self.nb.SetSelection(0)
def SelectedTopTreeID(self, event):
item = event.GetItem()
try:
#This handles the selection of an item in the TOP tree browser.
item = event.GetItem()
itemObject = self.tree.GetItemData(item).GetData()
SearchObject = itemObject.split(";")
SearchSuffix = SearchObject[0]
SearchPath = SearchObject[1]
SearchExtension = SearchObject[2]
SearchExtension = SearchExtension.split(":")
SearchExtension = SearchExtension[1:]
SearchExtension = SearchExtension[0]
SearchExtension = SearchExtension.split(",")
#SELECTION IMPLEMENT
ID_Strings = []
self.TopSelectList = []
self.TopID = SearchSuffix
root = self.main_results_directory + "/" + SearchPath
root_display = self.main_results_directory + "/" + SearchPath
root_contents = os.listdir(root)
root_contents_display = os.listdir(root)
for obj in root_contents:
if(SearchSuffix != "*"):
FindList = re.findall(SearchSuffix, obj)
if(len(FindList) > 0):
self.TopSelectList.append(obj)
#print obj
self.browser2.DeleteAllItems()
for filename in root_contents:
if(SearchSuffix != "*"):
FindList2 = re.findall(SearchSuffix, filename)
if(len(FindList2) > 0):
display_name = filename[0:-4]
ID_Strings.append(display_name)
else:
if(filename[-4] == "."):
display_name = filename[0:-4]
if "AVERAGE-" not in display_name and "COUNTS-" not in display_name:
ID_Strings.append(display_name)
ID_Strings = list(set(ID_Strings))
change_path = currentDirectory + "/UseDir" ### NS-91615 alternative to __file__
shutil.rmtree("UseDir")
os.mkdir("UseDir")
#self.control.write(ID_Strings[0] + "\n")
os.chdir(change_path)
for marker in ID_Strings:
try:
os.mkdir(marker)
except:
pass
os.chdir(currentDirectory) ### NS-91615 alternative to __file__
root = "UseDir"
color_root2 = [223, 250, 223]
self.ids2 = {root : self.browser2.AddRoot(root)}
for (dirpath, dirnames, filenames) in os.walk(root):
color_root2[0] = color_root2[0] - 1
color_root2[1] = color_root2[1] - 0
color_root2[2] = color_root2[2] - 1
for dirname in dirnames:
#self.control.write(str(SearchExtension) + "\n")
Extensions = dirname + "|" + str(SearchExtension) + "|" + str(SearchPath)
fullpath = os.path.join(dirpath, dirname)
self.ids2[fullpath] = self.browser2.AppendItem(self.ids2[dirpath], dirname)
self.browser2.SetItemData(self.ids2[fullpath],wx.TreeItemData(Extensions))
T = re.findall("DATASET", fullpath)
if(len(T) > 0):
self.browser2.SetItemBackgroundColour(self.ids2[fullpath], [250, 100, 100])
else:
self.browser2.SetItemBackgroundColour(self.ids2[fullpath], [130, 170, 250])
self.browser2.SetItemBackgroundColour(self.ids2[root], [110, 150, 250])
self.Bind(wx.EVT_TREE_ITEM_ACTIVATED, self.SelectedBottomTreeID, self.browser2)
self.browser2.ExpandAll()
#OPENING DISPLAY
display_file_selected = ""
TXT_FLAG = 0
PNG_FLAG = 0
if(root_display[-1] != "/"):
root_display = root_display + "/"
for possible in root_contents_display:
total_filepath = unique.filepath(root_display + possible)
if(possible[-4:] == ".txt"):
self.control.write("Displaying File: " + str(total_filepath) + "\n")
display_file_selected = total_filepath
break
TXT_FLAG = 0
PNG_FLAG = 0
#self.control.write(str(os.listdir(root)) + "\n")
#self.control.write(str(SearchExtension) + "\n")
for i in SearchExtension:
if(i == ".txt"):
TXT_FLAG = 1
#self.control.write(str(i) + "\n")
if(i == ".png"):
PNG_FLAG = 1
#self.control.write(str(i) + "\n")
if(root_display[-1] != "/"):
root_display = root_display + "/"
Pitch = os.listdir(root)
PitchSelect = Pitch[0]
self.CurrentFile = PitchSelect
#self.control.write(str(PitchSelect) + " " + root_display + "\n")
self.DirFile = unique.filepath(root_display + PitchSelect)
self.IntFileTxt.Clear()
self.IntFileTxt.write(self.DirFile)
self.DirFileTxt = unique.filepath(root_display + PitchSelect + ".txt")
DirFilePng = unique.filepath(root_display + PitchSelect + ".png")
self.myGrid.ClearGrid()
title_name = PitchSelect
try:
self.LOGO.Destroy()
except:
pass
try:
self.PanelTitle.Destroy()
except:
pass
font = wx.Font(16, wx.SWISS, wx.NORMAL, wx.BOLD)
self.PanelTitle = wx.StaticText(self.panel, label=title_name, pos=(5, 7))
self.PanelTitle.SetFont(font)
if(TXT_FLAG == 1):
try:
self.myGrid.DeleteRows(100, self.AppendTotal, True)
except:
pass
try:
#First time the DATASET file is imported
#font = wx.Font(16, wx.DECORATIVE, wx.BOLD, wx.NORMAL)
#self.PanelTitle = wx.StaticText(self.panel, label=title_name, pos=(210, 15))
#self.PanelTitle.SetFont(font)
#table_file = open(self.DirFileTxt, "rU")
table_file_contents = []
column_lengths = []
count=0
for line in open(self.DirFileTxt,'rU').xreadlines():
line = line.rstrip(); line = string.replace(line,'"','')
line = line.split("\t")
column_lengths.append(len(line))
table_file_contents.append((line))
if count>2000: break
count+=1
self.max_column_length = max(column_lengths)
self.table_length = len(table_file_contents)
if(self.table_length > 100 and self.table_length < 5000):
self.AppendTotal = self.table_length - 100
self.myGrid.AppendRows(self.AppendTotal, True)
if(self.table_length >= 5000):
self.AppendTotal = 5000
self.myGrid.AppendRows(self.AppendTotal, True)
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
x_count = 0
try:
for item_list in table_file_contents:
y_count = 0
for item in item_list:
self.myGrid.SetCellValue(x_count, y_count, item)
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
except:
pass
self.myGrid.AutoSize()
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
TXT_FLAG = 0
self.control.write("Unable to open txt." + "\n")
try:
self.myGrid.AutoSize()
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
pass
if(PNG_FLAG == 1):
try:
open(DirFilePng, "r")
self.png = wx.Image(DirFilePng, wx.BITMAP_TYPE_ANY).ConvertToBitmap()
self.LOGO = wx.StaticBitmap(self.page1, -1, self.png, (0,0), (self.png.GetWidth(), self.png.GetHeight()), style=wx.ALIGN_CENTER)
imgsizer_v = wx.BoxSizer(wx.VERTICAL)
imgsizer_v.Add(self.LOGO, proportion=0, flag=wx.ALIGN_CENTER_HORIZONTAL|wx.ALIGN_CENTER_VERTICAL)
self.page1.SetSizer(imgsizer_v)
self.page1.Layout()
except:
PNG_FLAG = 0
self.control.write("Unable to open png." + "\n")
try:
self.root_widget_id = 500
self.root_widget_text = 550
for i in range(self.root_widget_id, self.root_widget_end):
self.heatmap_ids[i].Destroy()
for i in range(self.root_widget_text, self.rwtend):
self.heatmap_ids[i].Destroy()
self.RunButton2.Destroy()
except:
pass
self.InteractivePanelUpdate(event)
if(PNG_FLAG == 1 and TXT_FLAG == 0):
self.nb.SetSelection(1)
self.Layout()
self.page1.Layout()
if(PNG_FLAG == 0 and TXT_FLAG == 1):
self.nb.SetSelection(0)
if(PNG_FLAG == 1 and TXT_FLAG == 1):
self.nb.SetSelection(1)
self.Layout()
self.page1.Layout()
except Exception: pass
def SelectedBottomTreeID(self, event):
#This handles the selection of an item in the BOTTOM tree browser; represents a file most of the time.
item = event.GetItem()
itemObject = self.browser2.GetItemData(item).GetData()
Parameters = itemObject.split("|")
file_extension = Parameters[1][1:-1]
file_extension.replace("'", "")
file_extension = file_extension.split(",")
file_exts = []
TXT_FLAG = 0
PNG_FLAG = 0
for i in file_extension:
i = i.replace("'", "")
i = i.replace(" ", "")
file_exts.append(i)
for i in file_exts:
if(i == ".txt"):
TXT_FLAG = 1
if(i == ".png"):
PNG_FLAG = 1
DirPath = self.main_results_directory + "/" + Parameters[2]
if(DirPath[-1] != "/"):
DirPath = DirPath + "/"
DirFile = DirPath + Parameters[0]
self.CurrentFile = DirFile
self.control.write("Displaying file: " + DirFile + "\n")
title_name = DirFile.split("/")
title_name = title_name[-1]
self.DirFile = unique.filepath(DirFile)
self.IntFileTxt.Clear()
self.IntFileTxt.write(self.DirFile)
self.DirFileTxt = DirFile + ".txt"
DirFilePng = DirFile + ".png"
self.myGrid.ClearGrid()
try:
self.LOGO.Destroy()
except:
pass
try:
self.PanelTitle.Destroy()
except:
pass
font = wx.Font(16, wx.SWISS, wx.NORMAL, wx.BOLD)
self.PanelTitle = wx.StaticText(self.panel, label=title_name, pos=(5, 7))
self.PanelTitle.SetFont(font)
#PNG_FLAG and TXT_FLAG are flags that sense the presence of an image or text file.
if(PNG_FLAG == 1):
try:
open(DirFilePng, "r")
self.png = wx.Image(DirFilePng, wx.BITMAP_TYPE_ANY).ConvertToBitmap()
self.LOGO = wx.StaticBitmap(self.page1, -1, self.png, (0,0), (self.png.GetWidth(), self.png.GetHeight()), style=wx.ALIGN_CENTER)
imgsizer_v = wx.BoxSizer(wx.VERTICAL)
imgsizer_v.Add(self.LOGO, proportion=0, flag=wx.ALIGN_CENTER_HORIZONTAL|wx.ALIGN_CENTER_VERTICAL)
self.page1.SetSizer(imgsizer_v)
self.page1.Layout()
except:
PNG_FLAG = 0
self.control.write("Unable to open png." + "\n")
if(TXT_FLAG == 1):
try:
self.myGrid.DeleteRows(100, self.AppendTotal, True)
except:
pass
try:
count=0
#table_file = open(self.DirFileTxt, "r")
table_file_contents = []
column_lengths = []
for line in open(self.DirFileTxt,'rU').xreadlines():
line = line.rstrip(); line = string.replace(line,'"','')
line = line.split("\t")
column_lengths.append(len(line))
table_file_contents.append((line))
count+=1
if count>2000:break
self.max_column_length = max(column_lengths)
self.table_length = len(table_file_contents)
if(self.table_length > 100 and self.table_length < 5000):
self.AppendTotal = self.table_length - 100
self.myGrid.AppendRows(self.AppendTotal, True)
if(self.table_length >= 5000):
self.AppendTotal = 5000
self.myGrid.AppendRows(self.AppendTotal, True)
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
x_count = 0
for item_list in table_file_contents:
y_count = 0
for item in item_list:
try: self.myGrid.SetCellValue(x_count, y_count, item) ###Here
except Exception:
### Unclear why this is throwing an error
#print traceback.format_exc()
#print x_count, y_count, item;sys.exit()
pass
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
self.myGrid.AutoSize()
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
print traceback.format_exc()
TXT_FLAG = 0
self.control.write("Unable to open txt." + "\n")
DATASET_FIND_FLAG = re.findall("DATASET", self.DirFileTxt)
count=0
if(len(DATASET_FIND_FLAG) > 0):
try:
#table_file = open(self.DirFileTxt, "rU")
table_file_contents = []
pre_sort2 = []
header = []
t_c = 0
column_clusters_flat = 0
answer = "AC"
for line in open(self.DirFileTxt,'rU').xreadlines():
#for line in table_file:
count+=1
if count>2000:
break
try:
line = line.rstrip(); line = string.replace(line,'"','')
line = line.split("\t")
if(t_c == 0):
header.append((line))
t_c = t_c + 1
index=0
for i in line:
if 'ANOVA-rawp' in i: answer = index
index+=1
continue
if(line[0] == "column_clusters-flat"):
header.append((line))
column_clusters_flat = 1
continue
line_sort_select = line[answer]
pre_sort1 = []
count = 0
for i in line:
if(count == 0):
try:
pre_sort1.append(float(line_sort_select))
except:
pre_sort1.append(line_sort_select)
pre_sort1.append(i)
if(count == answer):
count = count + 1
continue
if(count != 0):
pre_sort1.append(i)
count = count + 1
pre_sort2.append((pre_sort1))
except:
continue
table_file_contents.append(header[0])
if(column_clusters_flat == 1):
table_file_contents.append(header[1])
pre_sort2 = sorted(pre_sort2)
for line in pre_sort2:
try:
final_count1 = 0
final_count2 = 1
send_list = []
for item in line:
if(final_count1 == 0):
send_list.append(line[final_count2])
if(final_count1 == answer):
send_list.append(str(line[0]))
if(final_count1 != 0 and final_count1 != answer):
if(final_count1 < answer):
send_list.append(line[final_count2])
if(final_count1 > answer):
send_list.append(line[final_count1])
final_count1 = final_count1 + 1
final_count2 = final_count2 + 1
if(len(table_file_contents) >= 5100):
break
table_file_contents.append((send_list))
except:
continue
for cell in self.ColoredCellList:
self.myGrid.SetCellBackgroundColour(cell[0], cell[1], wx.WHITE)
self.ColoredCellList = []
x_count = 0
try:
for item_list in table_file_contents:
y_count = 0
for item in item_list:
self.myGrid.SetCellValue(x_count, y_count, item)
if(x_count == 0):
self.myGrid.SetCellFont(x_count, y_count, wx.Font(12, wx.SWISS, wx.NORMAL, wx.BOLD))
y_count = y_count + 1
x_count = x_count + 1
except:
pass
self.myGrid.AutoSizeRows(True)
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
self.control.write("Unable to sort." + "\n")
self.nb.SetSelection(0)
self.InteractivePanelUpdate(event)
try:
self.myGrid.AutoSize()
for i in range(50):
colsize = self.myGrid.GetColSize(i)
if(colsize > 200):
self.myGrid.SetColSize(i, 200)
self.page2.Layout()
except:
pass
if(PNG_FLAG == 1 and TXT_FLAG == 0):
self.nb.SetSelection(1)
self.Layout()
self.page1.Layout()
if(PNG_FLAG == 0 and TXT_FLAG == 1):
self.nb.SetSelection(0)
if(PNG_FLAG == 1 and TXT_FLAG == 1):
self.nb.SetSelection(1)
self.Layout()
self.page1.Layout()
def InteractivePanelUpdate(self, event):
#Both the PCA UI and Heatmap UI share the same panel, so buttons and text boxes (as well as other GUI) will have to be destroyed/hidden
#whenever a new type of interactivity is selected.
self.IntFileTxt.Hide()
self.InteractiveFileLabel.Hide()
self.Yes1Label.Hide()
self.No1Label.Hide()
self.D_3DLabel.Hide()
self.D_2DLabel.Hide()
self.IncludeLabelsRadio.Hide()
self.No1Radio.Hide()
self.D_3DRadio.Hide()
self.D_2DRadio.Hide()
self.Opt1Desc.Hide()
self.Opt2Desc.Hide()
self.RunButton1.Hide()
self.Divider1.Hide()
self.InteractiveDefaultMessage.Hide()
try:
self.root_widget_id = 500
self.root_widget_text = 550
for i in range(self.root_widget_id, self.root_widget_end):
self.heatmap_ids[i].Destroy()
for i in range(self.root_widget_text, self.rwtend):
self.heatmap_ids[i].Destroy()
self.RunButton2.Destroy()
except:
pass
PCA_RegEx = re.findall("PCA", self.DirFile)
if(len(PCA_RegEx) > 0):
self.IntFileTxt.Show()
self.InteractiveFileLabel.Show()
self.Yes1Label.Show()
self.No1Label.Show()
self.D_3DLabel.Show()
self.D_2DLabel.Show()
self.IncludeLabelsRadio.Show()
self.No1Radio.Show()
self.D_3DRadio.Show()
self.D_2DRadio.Show()
self.Opt1Desc.Show()
self.Opt2Desc.Show()
self.RunButton1.Show()
self.Divider1.Show()
Heatmap_RegEx = re.findall("hierarchical", self.DirFile)
if(len(Heatmap_RegEx) > 0):
#Heatmap Setup
os.chdir(parentDirectory)
options_open = open(unique.filepath(currentDirectory+"/options.txt"), "rU")
heatmap_array = []
self.heatmap_ids = {}
self.heatmap_translation = {}
supported_geneset_types = UI.getSupportedGeneSetTypes(self.species,'gene-mapp')
supported_geneset_types += UI.getSupportedGeneSetTypes(self.species,'gene-go')
supported_geneset_types_alt = [self.geneset_type]
supported_genesets = self.supported_genesets
for line in options_open:
line = line.split("\t")
variable_name,displayed_title,display_object,group,notes,description,global_default,options = line[:8]
options = string.split(options,'|')
if(group == "heatmap"):
if(display_object == "file"):
continue
od = UI.OptionData(variable_name,displayed_title,display_object,notes,options,global_default)
od.setDefaultOption(global_default)
#"""
if variable_name == 'ClusterGOElite':
od.setArrayOptions(['None Selected','all']+supported_geneset_types)
elif variable_name == 'GeneSetSelection':
od.setArrayOptions(['None Selected']+supported_geneset_types_alt)
elif variable_name == 'PathwaySelection':
od.setArrayOptions(['None Selected']+supported_genesets)
elif od.DefaultOption() == '':
od.setDefaultOption(od.Options()[0])
if od.DefaultOption() == '---':
od.setDefaultOption('')#"""
heatmap_array.append(od)
#heatmap_array.append((line[1], line[2], line[7], line[6]))
os.chdir(currentDirectory)
root_widget_y_pos = 45
self.root_widget_id = 500
self.root_widget_text = 550
for od in heatmap_array:
#od.VariableName()
id = wx.NewId()
#print od.VariableName(),od.Options()
self.heatmap_translation[od.VariableName()] = self.root_widget_id
self.heatmap_ids[self.root_widget_text] = wx.StaticText(self.page3, self.root_widget_text, label=od.Display(), pos=(150, root_widget_y_pos))
if(od.DisplayObject() == "comboBox" or od.DisplayObject() == "multiple-comboBox"):
self.heatmap_ids[self.root_widget_id] = wx.ComboBox(self.page3, self.root_widget_id, od.DefaultOption(), (10, root_widget_y_pos), (120,25), od.Options(), wx.CB_DROPDOWN)
else:
self.heatmap_ids[self.root_widget_id] = wx.TextCtrl(self.page3, self.root_widget_id, od.DefaultOption(), (10, root_widget_y_pos), (120,25))
self.root_widget_id = self.root_widget_id + 1
self.root_widget_text = self.root_widget_text + 1
root_widget_y_pos = root_widget_y_pos + 25
self.rwtend = self.root_widget_text
self.root_widget_end = self.root_widget_id
self.RunButton2 = wx.Button(self.page3, id=599, label="Run", pos=(175, (self.root_widget_end + 10)), size=(120, bheight))
self.Bind(wx.EVT_BUTTON, self.InteractiveRun, id=599)
if(len(PCA_RegEx) == 0 and len(Heatmap_RegEx) == 0):
self.InteractiveDefaultMessage.Show()
def ClearVisualPanel(self, event):
#Deletes the current image on the viewing panel. Unstable and mostly broken; may be removed from future versions.
popup = wx.MessageDialog(None, "Are you sure you want to clear the visual panel?", "Warning", wx.YES_NO)
popup_answer = popup.ShowModal()
if(popup_answer == 5103):
try:
self.LOGO.Destroy()
self.panel2.Layout()
except:
pass
try:
self.myGrid.ClearGrid()
self.panel2.Layout()
except:
pass
popup.Destroy()
self.control.write("Visual panel cleared." + "\n")
else:
return
def InteractiveRun(self, event):
#This function is bound to the "Run" button on the interactive tab GUI. Generates an interactive plot.
#Currently updates on the panel are a priority and many changes may come with it.
RegExHeat = re.findall("hierarchical", self.DirFile)
if(len(RegExHeat) > 0):
for VariableName in self.heatmap_translation:
#self.control.write(str(self.heatmap_ids[self.heatmap_translation[VariableName]].GetValue()) + " " + str(VariableName) + " " + str(self.heatmap_ids[self.heatmap_translation[VariableName]]) + "\n")
try:
self.heatmap_translation[VariableName] = str(self.heatmap_ids[self.heatmap_translation[VariableName]].GetValue())
#print self.heatmap_translation[VariableName]
except Exception: pass
try:
#self.control.write(self.DirFile + "\n")
input_file_dir = self.DirFile + ".txt"
column_metric = self.heatmap_translation['column_metric']; #self.control.write(column_metric + "\n")
column_method = self.heatmap_translation['column_method']; #self.control.write(column_method + "\n")
row_metric = self.heatmap_translation['row_metric']; #self.control.write(row_metric + "\n")
row_method = self.heatmap_translation['row_method']; #self.control.write(row_method+ "\n")
color_gradient = self.heatmap_translation['color_selection']; #self.control.write(color_gradient + "\n")
cluster_rows = self.heatmap_translation['cluster_rows']; #self.control.write(cluster_rows + "\n")
cluster_columns = self.heatmap_translation['cluster_columns']; #self.control.write(cluster_columns + "\n")
normalization = self.heatmap_translation['normalization']; #self.control.write(normalization + "\n")
contrast = self.heatmap_translation['contrast']; #self.control.write(contrast + "\n")
transpose = self.heatmap_translation['transpose']; #self.control.write(transpose + "\n")
GeneSetSelection = self.heatmap_translation['GeneSetSelection']; #self.control.write(GeneSetSelection + "\n")
PathwaySelection = self.heatmap_translation['PathwaySelection']; #self.control.write(PathwaySelection + "\n")
OntologyID = self.heatmap_translation['OntologyID']; #self.control.write(OntologyID + "\n")
GeneSelection = self.heatmap_translation['GeneSelection']; #self.control.write(GeneSelection + "\n")
justShowTheseIDs = self.heatmap_translation['JustShowTheseIDs']; #self.control.write(JustShowTheseIDs + "\n")
HeatmapAdvanced = self.heatmap_translation['HeatmapAdvanced']; #self.control.write(HeatmapAdvanced + "\n")
clusterGOElite = self.heatmap_translation['ClusterGOElite']; #self.control.write(ClusterGOElite + "\n")
heatmapGeneSets = self.heatmap_translation['heatmapGeneSets']; #self.control.write(heatmapGeneSets + "\n")
if cluster_rows == 'no': row_method = None
if cluster_columns == 'no': column_method = None
HeatmapAdvanced = (HeatmapAdvanced,)
#print ['JustShowTheseIDs',justShowTheseIDs]
if self.DirFile not in self.heatmap_run:
self.heatmap_run[self.DirFile]=None
### occurs when automatically running the heatmap
column_method = None
row_method = None
color_gradient = 'yellow_black_blue'
normalization = 'median'
translate={'None Selected':'','Exclude Cell Cycle Effects':'excludeCellCycle','Top Correlated Only':'top','Positive Correlations Only':'positive','Perform Iterative Discovery':'driver', 'Intra-Correlated Only':'IntraCorrelatedOnly', 'Perform Monocle':'monocle'}
try:
if 'None Selected' in HeatmapAdvanced: ('None Selected')
except Exception: HeatmapAdvanced = ('None Selected')
if ('None Selected' in HeatmapAdvanced and len(HeatmapAdvanced)==1) or 'None Selected' == HeatmapAdvanced: pass
else:
#print HeatmapAdvanced,'kill'
try:
GeneSelection += ' '+string.join(list(HeatmapAdvanced),' ')
for name in translate:
GeneSelection = string.replace(GeneSelection,name,translate[name])
GeneSelection = string.replace(GeneSelection,' ',' ')
if 'top' in GeneSelection or 'driver' in GeneSelection or 'excludeCellCycle' in GeneSelection or 'positive' in GeneSelection or 'IntraCorrelatedOnly' in GeneSelection:
GeneSelection+=' amplify'
except Exception: pass
GeneSetSelection = string.replace(GeneSetSelection,'\n',' ')
GeneSetSelection = string.replace(GeneSetSelection,'\r',' ')
if justShowTheseIDs == '': justShowTheseIDs = 'None Selected'
if GeneSetSelection== '': GeneSetSelection = 'None Selected'
if PathwaySelection== '': PathwaySelection = 'None Selected'
try: rho = float(self.heatmap_translation['CorrelationCutoff'])
except Exception: rho=None
if transpose == 'yes': transpose = True
else: transpose = False
vendor = 'RNASeq'
color_gradient = string.replace(color_gradient,'-','_')
if GeneSetSelection != 'None Selected' or GeneSelection != '' or normalization != 'NA' or JustShowTheseIDs != '' or JustShowTheseIDs != 'None Selected':
gsp = UI.GeneSelectionParameters(self.species,self.platform,vendor)
if rho!=None:
try:
gsp.setRhoCutoff(rho)
GeneSelection = 'amplify '+GeneSelection
except Exception: print 'Must enter a valid Pearson correlation cutoff (float)',traceback.format_exc()
gsp.setGeneSet(GeneSetSelection)
gsp.setPathwaySelect(PathwaySelection)
gsp.setGeneSelection(GeneSelection)
gsp.setOntologyID(OntologyID)
gsp.setTranspose(transpose)
gsp.setNormalize(normalization)
gsp.setJustShowTheseIDs(justShowTheseIDs)
gsp.setClusterGOElite(clusterGOElite)
transpose = gsp ### this allows methods that don't transmit this object to also work
if row_method == 'no': row_method = None
if column_method == 'no': column_method = None
#print [GeneSetSelection, PathwaySelection,OntologyID]
remoteCallToAltAnalyze = False
#try: print [gsp.ClusterGOElite()]
#except Exception: print 'dog', traceback.format_exc()
except Exception:
print traceback.format_exc()
if remoteCallToAltAnalyze == False:
try: UI.createHeatMap(input_file_dir, row_method, row_metric, column_method, column_metric, color_gradient, transpose, contrast, None, display=True)
except Exception: print traceback.format_exc()
else:
try:
command = ['--image', 'hierarchical','--species', self.species,'--platform',self.platform,'--input',input_file_dir, '--display', 'True']
command += ['--column_method',str(column_method),'--column_metric',column_metric]
command += ['--row_method',str(row_method),'--row_metric',row_metric]
command += ['--normalization',normalization,'--transpose',str(transpose),'--contrast',contrast,'--color_gradient',color_gradient]
#print command
command_str = string.join(['']+command,' ')
#print command
package_path = unique.filepath('python')
mac_package_path = string.replace(package_path,'python','AltAnalyze.app/Contents/MacOS/AltAnalyze')
#os.system(mac_package_path+command_str);sys.exit()
import subprocess
#subprocess.call([mac_package_path, 'C:\\test.txt'])
usePopen = True
if os.name == 'nt':
command = [mac_package_path]+command
DETACHED_PROCESS = 0x00000008
pid = subprocess.Popen(command, creationflags=DETACHED_PROCESS).pid
else:
command = [mac_package_path]+command
if usePopen:
alt_command = ["start"]+command
alt_command = ["start",mac_package_path]
subprocess.call(command) #works but runs in back of the application, detatched
if usePopen==False:
### sampe issue as subprocess.Popen
pid = os.fork()
if pid ==0:
os.execv(mac_package_path,command) ### Kills the parent app
os._exit(0)
"""
retcode = subprocess.call([
apt_file, "-d", cdf_file, "--kill-list", kill_list_dir, "-a", algorithm, "-o", output_dir,
"--cel-files", cel_dir, "-a", "pm-mm,mas5-detect.calls=1.pairs=1"])"""
except Exception:
print traceback.format_exc()
else:
os.chdir(parentDirectory)
RegExMatch = re.findall("exp.", self.DirFile)
if(len(RegExMatch) == 0):
InputFile = self.DirFile.replace("-3D", "")
InputFile = InputFile.replace("-PCA", "")
InputFile = InputFile.replace("DataPlots/Clustering-", "ExpressionOutput/Clustering/")
input_file_dir= InputFile + ".txt"
else:
InputFile = self.DirFile.replace("-3D", "")
InputFile = InputFile.replace("-PCA", "")
InputFile = InputFile.replace("DataPlots/Clustering-", "ExpressionInput/")
input_file_dir= InputFile + ".txt"
if(self.IncludeLabelsRadio.GetValue() == True):
include_labels= 'yes'
else:
include_labels= 'no'
pca_algorithm = 'SVD'
transpose = False
if self.runPCA == False:
include_labels = 'no'
if(self.D_3DRadio.GetValue() == True):
plotType = '3D'
else:
plotType = '2D'
display = True
self.runPCA = True
count,columns = self.verifyFileLength(input_file_dir)
if columns == 3: plotType = '2D' ### only 2 components possible for 2 samples
if count>0:
UI.performPCA(input_file_dir, include_labels, pca_algorithm, transpose, None, plotType=plotType, display=display)
else:
self.control.write('PCA input file not present: '+input_file_dir+'\n')
os.chdir(currentDirectory)
self.InteractivePanelUpdate(event)
def verifyFileLength(self,filename):
count = 0; columns=0
try:
fn=unique.filepath(filename)
for line in open(fn,'rU').xreadlines():
t = string.split(line,'\t')
columns = len(t)
count+=1
if count>9: break
except Exception: null=[]
return count,columns
def OnAbout(self, event):
#Brings up the developer information. Non-functional currently but will be updated eventually.
dial = wx.MessageDialog(None, 'AltAnalyze Results Viewer\nVersion 0.5\n2015', 'About', wx.OK)
dial.ShowModal()
def OnHelp(self, event):
#Brings up the tutorial and dorumentation. Will be updated to a .pdf in the future.
os.chdir(parentDirectory)
ManualPath = rootDirectory + "/Documentation/ViewerManual.pdf"
subprocess.Popen(['open', ManualPath])
os.chdir(currentDirectory)
class ImageFrame(wx.Frame):
#Obsolete code, will be removed almost certainly.
title = "Image"
def __init__(self):
wx.Frame.__init__(self, None, title=self.title)
def remoteViewer(app):
fr = Main(parent=None,id=1)
fr.Show()
app.MainLoop()
if __name__ == "__main__":
app = wx.App(False)
fr = Main(parent=None,id=1)
fr.Show()
app.MainLoop()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/AltAnalyzeViewer.py
|
AltAnalyzeViewer.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys, string
import os.path
import bisect
import unique
from stats_scripts import statistics
import math
import reorder_arrays
try: from build_scripts import ExonArray
except: pass
import export
import copy
import time
import traceback
import UI
from import_scripts import BuildAffymetrixAssociations; reload(BuildAffymetrixAssociations)
try:
from scipy import average as Average
except Exception:
try: from statistics import avg as Average
except: pass
use_Tkinter = 'no'
try:
from Tkinter import *
use_Tkinter = 'yes'
except ImportError: use_Tkinter = 'yes'; print "\nPmw or Tkinter not found... Tkinter print out not available";
debug_mode = 'no'
### Method specific global variables most easily initialized here
cluster_id=0
cluster_name='clu_0'
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv": dir_list2.append(entry)
return dir_list2
def returnLargeGlobalVars():
### Prints all large global variables retained in memory (taking up space)
all = [var for var in globals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(globals()[var])>1:
print var, len(globals()[var])
except Exception: null=[]
def clearObjectsFromMemory(db_to_clear):
db_keys={}
for key in db_to_clear: db_keys[key]=[]
for key in db_keys: del db_to_clear[key]
################# Begin Analysis from parsing files
def checkArrayHeaders(expr_input_dir,expr_group_dir):
array_names, array_linker_db = getArrayHeaders(expr_input_dir)
expr_group_list,expr_group_db = importArrayGroups(expr_group_dir,array_linker_db)
def getArrayHeaders(expr_input_dir):
### This method is used to check to see if the array headers in the groups and expression files match
fn=filepath(expr_input_dir); x = 0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
headers = string.split(data,'\t')
if data[0] != '#':
### differentiate data from column headers
if x == 1: break ### Exit out of loop, since we only want the array names
if x == 0: ### only grab headers if it's the first row
array_names = []; array_linker_db = {}; d = 0
for entry in headers[1:]: entry = string.replace(entry,'"',''); array_names.append(entry)
for array in array_names: array = string.replace(array,'\r',''); array_linker_db[array] = d; d +=1
x = 1
return array_names, array_linker_db
def checkExpressionFileFormat(expFile,reportNegatives=False,filterIDs=False):
""" Determine if the data is log, non-log and increment value for log calculation """
firstLine=True; convert=False
inputMax=0; inputMin=10000; increment=0
expressed_values={}
startIndex = 1
for line in open(expFile,'rU').xreadlines():
line = cleanUpLine(line)
key = string.split(line,'\t')[0]
t = string.split(line,'\t')
if firstLine:
headers = line
if 'row_clusters-flat' == t[1]:
startIndex = 2
firstLine = False
else:
if 'column_clusters-flat' in t:
continue ### skip this row if analyzing a clustered heatmap file
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
if filterIDs!=False:
if uid not in filterIDs:
continue
if '' in t[1:]:
values = [0 if x=='' else x for x in t[startIndex:]]
elif 'NA' in t[1:]:
values = [0 if x=='NA' else x for x in t[startIndex:]]
else:
values = t[1:]
try: values = map(lambda x: float(x), values)
except Exception:
print values
print traceback.format_exc()
if max(values)>inputMax: inputMax = max(values)
if min(values)<inputMin: inputMin = min(values)
if inputMax>100: ### Thus, not log values
expressionDataFormat = 'non-log'
if inputMin<=1: #if inputMin<=1:
increment = inputMin+1
convert = True
else:
expressionDataFormat = "log"
#print expressionDataFormat,increment,convert
if reportNegatives == False:
return expressionDataFormat,increment,convert
else:
### Report if negative values are present
increment = inputMin
if convert: ### Should rarely be the case, as this would indicate that a non-log folds are present in the file
increment = increment+1
return expressionDataFormat,increment,convert
def calculate_expression_measures(expr_input_dir,expr_group_dir,experiment_name,comp_group_dir,probeset_db,annotate_db):
print "Processing the expression file:",expr_input_dir
try: expressionDataFormat,increment,convertNonLogToLog = checkExpressionFileFormat(expr_input_dir)
except Exception:
print traceback.format_exc()
expressionDataFormat = expression_data_format; increment = 0
if expressionDataFormat == 'non-log': convertNonLogToLog=True
else: convertNonLogToLog = False
#print convertNonLogToLog, expressionDataFormat, increment
global array_fold_headers; global summary_filtering_stats; global raw_data_comp_headers; global array_folds
fn1=filepath(expr_input_dir)
x = 0; y = 0; d = 0
blanksPresent=False
array_folds={}
for line in open(fn1,'rU').xreadlines():
data = cleanUpLine(line)
if data[0] != '#' and data[0] != '!':
fold_data = string.split(data,'\t')
try: arrayid = fold_data[0]
except Exception: arrayid = 'UID'
if len(arrayid)>0:
if arrayid[0]== ' ':
try: arrayid = arrayid[1:] ### Cufflinks issue
except Exception: arrayid = ' ' ### can be the first row UID column as blank
if 'ENSG' in arrayid and '.' in arrayid:
arrayid = string.split(arrayid,'.')[0]
else:
arrayid = 'UID'
#if 'counts.' in expr_input_dir: arrayid,coordinates = string.split(arrayid,'=') ### needed for exon-level analyses only
### differentiate data from column headers
if x == 1:
fold_data = fold_data[1:]; fold_data2=[]
for fold in fold_data:
fold = string.replace(fold,'"','')
try:
fold = float(fold); fold_data2.append(fold)
except Exception:
fold_data2.append('')
blanksPresent = True
"""
print_out = 'WARNING!!! The ID'+arrayid+ 'has an invalid expression value:'+[fold]+'\n. Correct and re-run'
try: UI.WarningWindow(print_out,'Critical Error - Exiting Program!!!'); sys.exit()
except NameError: print print_out; sys.exit()
"""
if expressionDataFormat == 'non-log' and (convertNonLogToLog or array_type == 'RNASeq'):
fold_data3=[] ###Convert numeric expression to log fold (previous to version 2.05 added 1)
for fold in fold_data2:
try:
log_fold = math.log((float(fold)+increment),2) ### changed from - log_fold = math.log((float(fold)+1),2) - version 2.05
fold_data3.append(log_fold)
except ValueError: ###Not an ideal situation: Value is negative - Convert to zero
if float(fold)<=0:
log_fold = math.log(1.01,2); fold_data3.append(log_fold)
else:
fold_data3.append('')
blanksPresent = True
"""
print_out = 'WARNING!!! The ID'+arrayid+ 'has an invalid expression value:'+fold+'\n. Correct and re-run'
try: UI.WarningWindow(print_out,'Critical Error - Exiting Program!!!'); sys.exit()
except NameError: print print_out; sys.exit()
"""
fold_data2 = fold_data3
if (array_type == "AltMouse"):
if arrayid in probeset_db: array_folds[arrayid] = fold_data2; y = y+1
else: array_folds[arrayid] = fold_data2; y = y+1
else: #only grab headers if it's the first row
array_names = []; array_linker_db = {}
for entry in fold_data[1:]:
entry = string.replace(entry,'"','')
if len(entry)>0: array_names.append(entry)
for array in array_names: #use this to have an orignal index order of arrays
array = string.replace(array,'\r','') ###This occured once... not sure why
array_linker_db[array] = d; d +=1
#add this aftwards since these will also be used as index values
x = 1
print len(array_folds),"IDs imported...beginning to calculate statistics for all group comparisons"
expr_group_list,expr_group_db,dataType = importArrayGroups(expr_group_dir,array_linker_db,checkInputType=True)
comp_group_list, comp_group_list2 = importComparisonGroups(comp_group_dir)
if dataType=='Kallisto':
return None, None
if 'RPKM' in norm and 'counts.' in expr_input_dir: normalization_method = 'RPKM-counts' ### process as counts if analyzing the counts file
else: normalization_method = norm
if expressionDataFormat == 'non-log': logvalues=False
else: logvalues=True
if convertNonLogToLog: logvalues = True
try:
array_folds, array_fold_headers, summary_filtering_stats,raw_data_comp_headers = reorder_arrays.reorder(array_folds,array_names,expr_group_list,
comp_group_list,probeset_db,include_raw_data,array_type,normalization_method,fl,logvalues=logvalues,blanksPresent=blanksPresent)
except Exception:
print traceback.format_exc(),'\n'
print_out = 'AltAnalyze encountered an error with the format of the expression file.\nIf the data was designated as log intensities and it is not, then re-run as non-log.'
try: UI.WarningWindow(print_out,'Critical Error - Exiting Program!!!'); root.destroy(); force_exit ### Forces the error log to pop-up
except NameError: print print_out; sys.exit()
### Integrate maximum counts for each gene for the purpose of filtering (RNASeq data only)
if array_type == 'RNASeq' and 'counts.' not in expr_input_dir: addMaxReadCounts(expr_input_dir)
### Export these results to a DATASET statistics and annotation results file
if 'counts.' not in expr_input_dir:
if array_type == 'RNASeq' and norm == 'RPKM':
filterRNASeq(count_statistics_db)
### Export count summary in GenMAPP format
if include_raw_data == 'yes': headers = removeRawCountData(array_fold_headers)
else: headers = array_fold_headers
exportDataForGenMAPP(headers,'counts')
exportAnalyzedData(comp_group_list2,expr_group_db)
### Export formatted results for input as an expression dataset into GenMAPP or PathVisio
if data_type == 'expression':
if include_raw_data == 'yes': headers = removeRawData(array_fold_headers)
else: headers = array_fold_headers
exportDataForGenMAPP(headers,'expression')
try: clearObjectsFromMemory(summary_filtering_stats); clearObjectsFromMemory(array_folds)
except Exception: null=[]
try: clearObjectsFromMemory(summary_filtering_stats); summary_filtering_stats=[]
except Exception: null=[]
else:
### When performing an RNASeq analysis on RPKM data, we first perform these analyses on the raw counts to remove fold changes for low expressing genes
"""count_statistics_db={}; count_statistics_headers=[]
for key in array_folds:
count_statistics_db[key] = array_folds[key]
for name in array_fold_headers: count_statistics_headers.append(name)"""
try: clearObjectsFromMemory(summary_filtering_stats)
except Exception: null=[]
try: clearObjectsFromMemory(summary_filtering_stats); summary_filtering_stats=[]
except Exception: null=[]
return array_folds, array_fold_headers
def filterRNASeq(counts_db):
### Parse through the raw count data summary statistics and annotate any comparisons considered NOT EXPRESSED by read count filtering as not expressed (on top of RPKM filtering)
reassigned = 0; re = 0
for gene in counts_db:
i=0 ### keep track of the index (same as RPKM index)
for val in counts_db[gene]:
if val =='Insufficient Expression':
#print val, i, array_folds[gene][i];kill
if array_folds[gene][i] != 'Insufficient Expression': reassigned = gene, array_folds[gene][i]
array_folds[gene][i] = 'Insufficient Expression' ### Re-assign the fold changes to this non-numeric value
re+=1
i+=1
#print reassigned, re
def addMaxReadCounts(filename):
import RNASeq
max_count_db,array_names = RNASeq.importGeneCounts(filename,'max')
for gene in summary_filtering_stats:
gs = summary_filtering_stats[gene]
gs.setMaxCount(max_count_db[gene]) ### Shouldn't cause an error, but we want to get an exception if it does (something is wrong with the analysis)
def simplerGroupImport(group_dir):
if 'exp.' in group_dir or 'filteredExp.' in group_dir:
group_dir = string.replace(group_dir,'exp.','groups.')
group_dir = string.replace(group_dir,'filteredExp.','groups.')
import collections
try: sample_group_db = collections.OrderedDict()
except Exception:
try:
import ordereddict
sample_group_db = ordereddict.OrderedDict()
except Exception:
sample_group_db={}
fn = filepath(group_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
try:
group_data = string.split(data,'\t')
sample_filename = group_data[0]
group_name = group_data[-1]
if len(group_data)>3:
forceError
except Exception:
#print 'Non-Standard Groups file or missing relationships'
print string.split(data,'\t')[:10], 'more than 3 columns present in groups file'
kill
sample_group_db[sample_filename] = group_name
return sample_group_db
def simpleGroupImport(group_dir,splitHeaders=False, ignoreComps=False, reverseOrder=False):
""" Used for calculating fold changes prior to clustering for individual samples (genomtric folds) """
import collections
try:
### OrderedDict used to return the keys in the orders added for markerFinder
group_sample_db=collections.OrderedDict()
group_name_db=collections.OrderedDict()
group_name_sample_db=collections.OrderedDict()
group_db=collections.OrderedDict()
except Exception:
try:
import ordereddict
group_sample_db = ordereddict.OrderedDict()
group_name_db=ordereddict.OrderedDict()
group_name_sample_db=ordereddict.OrderedDict()
group_db=ordereddict.OrderedDict()
except Exception:
group_sample_db={}
group_name_db={}
group_name_sample_db={}
group_db={}
sample_list=[]
group_dir = verifyExpressionFile(group_dir)
fn = filepath(group_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
try: sample_filename,group_number,group_name = string.split(data,'\t')
except Exception:
print traceback.format_exc()
print "\nWARNING!!! Impropper groups file format detected. Terminating AltAnalyze. The groups file must have only three columns (sampleName, groupNumber, groupName).\n"
forceGroupsError
if splitHeaders:
if '~' in sample_filename: sample_filename = string.split(sample_filename,'~')[-1]
group_sample_db[sample_filename] = group_name+':'+sample_filename
if reverseOrder==False:
try: group_name_sample_db[group_name].append(group_name+':'+sample_filename)
except Exception: group_name_sample_db[group_name] = [group_name+':'+sample_filename]
else:
try: group_name_sample_db[group_name].append(sample_filename)
except Exception: group_name_sample_db[group_name] = [sample_filename]
sample_list.append(sample_filename)
group_db[sample_filename] = group_name
group_name_db[group_number]=group_name ### used by simpleCompsImport
### Get the comparisons indicated by the user
if ignoreComps==False: ### Not required for some analyses
comps_name_db,comp_groups = simpleCompsImport(group_dir,group_name_db,reverseOrder=reverseOrder)
else:
comps_name_db={}; comp_groups=[]
return sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db
def simpleCompsImport(group_dir,group_name_db,reverseOrder=False):
""" Used for calculating fold changes prior to clustering for individual samples (genomtric folds) """
comps_dir = string.replace(group_dir,'groups.','comps.')
import collections
comps_name_db=collections.OrderedDict()
comp_groups=[]
comps_dir = verifyExpressionFile(comps_dir)
fn = filepath(comps_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
try:
exp_group_num,con_group_num = string.split(data,'\t')
if reverseOrder:
con_group_num, exp_group_num = exp_group_num,con_group_num
exp_group_name = group_name_db[exp_group_num]
con_group_name = group_name_db[con_group_num]
try: comps_name_db[con_group_name].append(exp_group_name)
except Exception:
#comps_name_db[con_group_name] = [exp_group_name] ### If we don't want to include the control samples
comps_name_db[con_group_name] = [con_group_name] ### Add the control group versus itself the first time
comps_name_db[con_group_name].append(exp_group_name)
### Keep track of the order of the groups for ordering the cluster inputs
if con_group_name not in comp_groups:
comp_groups.append(con_group_name)
if exp_group_name not in comp_groups:
comp_groups.append(exp_group_name)
except Exception: pass ### Occurs if there are dummy lines in the file (returns with no values)
return comps_name_db,comp_groups
def importArrayGroups(expr_group_dir,array_linker_db,checkInputType=False):
new_index_order = 0
import collections
updated_groups = collections.OrderedDict()
expr_group_list=[]
expr_group_db = {} ### use when writing out data
fn=filepath(expr_group_dir)
data_type='null'
try:
try:
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
length = string.join(t,'') ### Some lines can be blank
if len(length)>2:
array_header,group,group_name = t
group = int(group)
### compare new to original index order of arrays
try:
original_index_order = array_linker_db[array_header]
except:
if array_header+'.bed' in array_linker_db:
new_header = array_header+'.bed'
original_index_order = array_linker_db[new_header]
updated_groups[new_header]=group,group_name
elif array_header[:-4] in array_linker_db:
new_header = array_header[:-4]
original_index_order = array_linker_db[new_header]
updated_groups[new_header]=group,group_name
else:
print_out = 'WARNING!!! At least one sample-ID listed in the "groups." file (e.g.,'+array_header+')'+'\n is not in the sample "exp." file. See the new file "arrays." with all "exp." header names\nand correct "groups."'
try: UI.WarningWindow(print_out,'Critical Error - Exiting Program!!!')
except Exception: print print_out
exportArrayHeaders(expr_group_dir,array_linker_db)
try: root.destroy(); sys.exit()
except Exception: sys.exit()
entry = new_index_order, original_index_order, group, group_name
expr_group_list.append(entry)
new_index_order += 1 ### add this aftwards since these will also be used as index values
expr_group_db[str(group)] = group_name
expr_group_list.sort() ### sorting put's this in the original array order
except ValueError:
print_out = 'The group number "'+group+'" is not a valid integer. Correct before proceeding.'
try: UI.WarningWindow(print_out,'Critical Error - Exiting Program!!!'); root.destroy(); sys.exit()
except Exception: print print_out; sys.exit()
except Exception,e:
print traceback.format_exc(),'\n'
exportArrayHeaders(expr_group_dir,array_linker_db)
print_out = 'No groups or comps files found for'+expr_group_dir+'... exiting program.'
try: UI.WarningWindow(print_out,'Critical Error - Exiting Program!!!'); root.destroy(); sys.exit()
except Exception: print print_out; sys.exit()
if len(updated_groups)>0 and checkInputType:
import shutil
try: ### When a Kallisto TPM and a gene-RPKM file co-exist (prioritize differential analysis of Kallisto)
shutil.copy(expr_group_dir,string.replace(expr_group_dir,'.txt','-Kallisto.txt'))
scr_exp_dir = string.replace(expr_group_dir,'groups.','Kallisto_Results/exp.')
dst_exp_dir = string.replace(expr_group_dir,'groups.','exp.')
shutil.copy(scr_exp_dir,string.replace(dst_exp_dir,'.txt','-Kallisto.txt'))
src_comps_dir = string.replace(expr_group_dir,'groups.','comps.')
shutil.copy(src_comps_dir,string.replace(src_comps_dir,'.txt','-Kallisto.txt'))
except:
pass
data_type='Kallisto'
exportUpdatedGroups(expr_group_dir,updated_groups)
if checkInputType:
return expr_group_list,expr_group_db,data_type
else:
return expr_group_list,expr_group_db
def exportUpdatedGroups(expr_group_dir,updated_groups):
eo = export.ExportFile(expr_group_dir)
for sample in updated_groups:
eo.write(sample+'\t'+str(updated_groups[sample][0])+'\t'+updated_groups[sample][1]+'\n')
eo.close()
print 'The groups file has been updated with bed file sample names'
def exportArrayHeaders(expr_group_dir,array_linker_db):
new_file = string.replace(expr_group_dir,'groups.','arrays.')
new_file = string.replace(new_file,'exp.','arrays.')
new_file = string.replace(new_file,'counts.','arrays.')
if 'arrays.' not in new_file: new_file = 'arrays.' + new_file ### Can occur if the file does not have 'exp.' in it
fn=filepath(new_file); data = open(fn,'w')
for array in array_linker_db: data.write(array+'\n')
data.close()
def importComparisonGroups(comp_group_dir):
comp_group_list=[]; comp_group_list2=[]
try:
fn=filepath(comp_group_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
groups = string.split(data,'\t')
groups2 = groups[0],groups[1] #as a list these would be unhashable
comp_group_list.append(groups)
comp_group_list2.append(groups2)
except Exception: null=[] ### Occcurs when no file present
return comp_group_list, comp_group_list2
def importMicrornaAssociations(species,report):
filename = 'AltDatabase/Ensembl/'+species+'/'+species+'_microRNA-Ensembl.txt'
fn=filepath(filename); ensembl_microRNA_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
miR,ens_geneid,sources = string.split(data,'\t')
miR_annot = miR+'('+sources+')'
try: ensembl_microRNA_db[ens_geneid].append(miR_annot)
except KeyError: ensembl_microRNA_db[ens_geneid] = [miR_annot]
###Optionally filter out miRs with evidence from just one algorithm (options are 'any' and 'muliple'
for gene in ensembl_microRNA_db:
miRs = ensembl_microRNA_db[gene]; miRs.sort()
if report == 'multiple':
miRs2=[]
for mir in miRs:
if '|' in mir: miRs2.append(mir)
miRs=miRs2
miRs = string.join(miRs,', ')
ensembl_microRNA_db[gene] = miRs
return ensembl_microRNA_db
def importSystemCodes():
filename = 'Config/source_data.txt'
fn=filepath(filename); x=0; systems={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
system_name=t[0];system_code=t[1]
if x==0: x=1
else: systems[system_name] = system_code
return systems
def exportDataForGenMAPP(headers,input_type):
###Export summary columns for GenMAPP analysis
systems = importSystemCodes()
GenMAPP_file = expression_dataset_output_dir + 'GenMAPP-'+experiment_name+'.txt'
if 'counts' in input_type:
GenMAPP_file = string.replace(GenMAPP_file,'GenMAPP-','COUNTS-')
try: genmapp = export.createExportFile(GenMAPP_file,expression_dataset_output_dir[:-1])
except RuntimeError:
export.isFileOpen(GenMAPP_file,expression_dataset_output_dir[:-1])
genmapp = export.createExportFile(GenMAPP_file,expression_dataset_output_dir[:-1])
if array_type == "3'array" and 'Ensembl' not in vendor:
if vendor == 'Affymetrix': system_code = 'X'
elif vendor == 'Illumina': system_code = 'Il'
elif vendor == 'Agilent': system_code = 'Ag'
elif vendor == 'Codelink': system_code = 'Co'
else:
### This is another system selected by the user
system = string.replace(vendor,'other:','')
try: system_code = systems[system]
except Exception: system_code = 'Sy'
elif array_type != 'AltMouse': system_code = 'En'
else:
try: system_code = systems[vendor]
except Exception: system_code = 'X'
genmapp_title = ['GeneID','SystemCode'] + headers
genmapp_title = string.join(genmapp_title,'\t')+'\t'+'ANOVA-rawp'+'\t'+'ANOVA-adjp'+'\t'+'largest fold'+'\n'
genmapp.write(genmapp_title)
for probeset in array_folds:
if 'ENS' in probeset and (' ' in probeset or '_' in probeset or ':' in probeset or '-' in probeset) and len(probeset)>9:
system_code = 'En'
ensembl_gene = 'ENS'+string.split(probeset,'ENS')[1]
if ' ' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,' ')[0]
if '_' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,'_')[0]
if ':' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,':')[0]
if '-' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,'-')[0]
data_val = ensembl_gene+'\t'+system_code
elif ('ENS' in probeset or 'ENF' in probeset) and system_code == 'Sy' and len(probeset)>9:
system_code = 'En'
data_val = probeset+'\t'+system_code
else:
data_val = probeset+'\t'+system_code
for value in array_folds[probeset]: data_val += '\t'+ str(value)
gs = summary_filtering_stats[probeset]
data_val += '\t'+ str(gs.Pval()) +'\t'+ str(gs.AdjP()) +'\t'+ str(gs.LogFold()) +'\n'
genmapp.write(data_val)
genmapp.close()
exportGOEliteInput(headers,system_code)
print 'Exported GO-Elite input files...'
def buildCriterion(ge_fold_cutoffs, ge_pvalue_cutoffs, ge_ptype, main_output_folder, operation, UseDownRegulatedLabel=False, genesToExclude={}):
global array_folds; global m_cutoff; global p_cutoff; global expression_dataset_output_dir
global ptype_to_use; global use_downregulated_label; use_downregulated_label = UseDownRegulatedLabel
m_cutoff = math.log(float(ge_fold_cutoffs),2); p_cutoff = ge_pvalue_cutoffs; ptype_to_use = ge_ptype
expression_dataset_output_dir = string.replace(main_output_folder,'GO-Elite','ExpressionOutput/')
dir_list = read_directory(expression_dataset_output_dir[:-1])
if operation == 'summary': filetype = 'DATASET-'
else: filetype = 'GenMAPP-'
for filename in dir_list:
if filetype in filename:
fn=filepath(expression_dataset_output_dir+filename)
array_folds = {}; x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line); t = string.split(data,'\t')
if x==0: x=1; headers = t[1:-2]
else:
values = t[1:-2]; probeset = t[0]; system_code = t[1]
if probeset not in genesToExclude: ### E.g., sex-associated or pseudogenes
array_folds[probeset] = values
if operation == 'summary':
exportGeneRegulationSummary(filename,headers,system_code)
else:
input_files_exported = exportGOEliteInput(headers,system_code)
array_folds=[]
def excludeGenesImport(filename):
fn=filepath(filename)
exclude_genes = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
uid = string.split(data,'\t')[0]
exclude_genes[uid] = None
return exclude_genes
def importCountSummary():
### Copied code from buildCriterion
count_summary_db={}
indexed_headers={}
filetype = 'COUNTS-'
dir_list = read_directory(expression_dataset_output_dir[:-1])
for filename in dir_list:
if filetype in filename:
fn=filepath(expression_dataset_output_dir+filename)
count_summary_db = {}; x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line); t = string.split(data,'\t')
if x==0:
x=1; i=0
for header in t:
indexed_headers[header]=i
i+=1
else:
values = t[1:-2]; probeset = t[0]; system_code = t[1]
count_summary_db[probeset] = values
return count_summary_db, indexed_headers
def exportGOEliteInput(headers,system_code):
### Filter statistics based on user-defined thresholds as input for GO-Elite analysis
criterion_db={}; denominator_geneids={}; index = 0; ttest=[]
for column in headers:
if 'ANOVA' in ptype_to_use and ptype_to_use in column: ttest.append(index) ### Not currently implemented
elif ptype_to_use in column and 'ANOVA' not in column: ttest.append(index)
lfi = 2 ### relative logfold index position
if ptype_to_use == 'adjp': lfi = 3
index+=1
### Had to introduce the below code to see if any p-values for a criterion are < 1 (otherwise, include them for GO-Elite)
exclude_p1={}
for probeset in array_folds:
index = 0; af = array_folds[probeset]
for value in array_folds[probeset]:
if index in ttest:
criterion_name = headers[index][5:]
if criterion_name not in exclude_p1:
try: p_value = float(value)
except Exception: p_value = 1 ### Occurs when a p-value is annotated as 'Insufficient Expression'
if p_value < 1:
exclude_p1[criterion_name] = True # Hence, at least one gene has a p<1
index+=1
for probeset in array_folds:
index = 0; af = array_folds[probeset]
for value in array_folds[probeset]:
denominator_geneids[probeset]=[]
if index in ttest:
criterion_name = headers[index][5:]
if use_downregulated_label==False:
rcn = string.split(criterion_name,'_vs_'); rcn.reverse() ### re-label all downregulated as up (reverse the numerator/denominator)
reverse_criterion_names = string.join(rcn,'_vs_')
regulation_call = '-upregulated'
else:
reverse_criterion_names = criterion_name
regulation_call = '-downregulated'
try: log_fold = float(af[index-lfi])
except Exception: log_fold = 0 ### Occurs when a fold change is annotated as 'Insufficient Expression'
try: p_value = float(value)
except Exception: p_value = 1 ### Occurs when a p-value is annotated as 'Insufficient Expression'
try: excl_p1 = exclude_p1[criterion_name] ### You can have adjusted p-values that are equal to 1
except Exception: excl_p1 = False #Make True to exclude ALL non-adjp < sig value entries
#print [log_fold, m_cutoff, p_value, p_cutoff];sys.exit()
if abs(log_fold)>m_cutoff and (p_value<p_cutoff or (p_value==1 and excl_p1==False)):
#if p_value == 1: print log_fold, probeset,[value]; sys.exit()
try: criterion_db[criterion_name].append((probeset,log_fold,p_value))
except KeyError: criterion_db[criterion_name] = [(probeset,log_fold,p_value)]
if log_fold>0:
try: criterion_db[criterion_name+'-upregulated'].append((probeset,log_fold,p_value))
except KeyError: criterion_db[criterion_name+'-upregulated'] = [(probeset,log_fold,p_value)]
else:
if use_downregulated_label==False:
log_fold = abs(log_fold)
try: criterion_db[reverse_criterion_names+regulation_call].append((probeset,log_fold,p_value))
except KeyError: criterion_db[reverse_criterion_names+regulation_call] = [(probeset,log_fold,p_value)]
index += 1
### Format these statistical filtering parameters as a string to include in the file as a record
if m_cutoff<0: fold_cutoff = -1/math.pow(2,m_cutoff)
else: fold_cutoff = math.pow(2,m_cutoff)
stat_filters = ' (Regulation criterion: fold > '+str(fold_cutoff)+' and '+ptype_to_use+ ' p-value < '+str(p_cutoff)+')'
stat_filters_filename = '-fold'+str(fold_cutoff)+'_'+ptype_to_use+str(p_cutoff)
### Format these lists to export as tab-delimited text files
if len(criterion_db)>0:
### Export denominator gene IDs
input_files_exported = 'yes'
expression_dir = string.replace(expression_dataset_output_dir,'ExpressionOutput/','')
goelite_file = expression_dir +'GO-Elite/denominator/GE.denominator.txt'
goelite = export.createExportFile(goelite_file,expression_dir+'GO-Elite/denominator')
goelite_title = ['GeneID','SystemCode']
goelite_title = string.join(goelite_title,'\t')+'\n'; goelite.write(goelite_title)
for probeset in denominator_geneids:
try:
if 'ENS' in probeset and (' ' in probeset or '_' in probeset or ':' in probeset or '-' in probeset) and len(probeset)>9:
system_code = 'En'
ensembl_gene = 'ENS'+string.split(probeset,'ENS')[1]
if ' ' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,' ')[0]
if '_' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,'_')[0]
if ':' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,':')[0]
if '-' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,'-')[0]
probeset = ensembl_gene
elif ':' in probeset:
probeset = string.split(probeset,':')[0]
system_code = 'Sy'
except Exception:
pass
if ('ENS' in probeset or 'ENF' in probeset) and system_code == 'Sy' and len(probeset)>9:
system_code = 'En'
values = string.join([probeset,system_code],'\t')+'\n'; goelite.write(values)
goelite.close()
### Export criterion gene IDs and minimal data
for criterion_name in criterion_db:
if criterion_name[-1] == ' ': criterion_file_name = criterion_name[:-1]
else: criterion_file_name = criterion_name
if 'upregulated' in criterion_name: elitedir = 'upregulated'
elif 'downregulated' in criterion_name: elitedir = 'downregulated'
else: elitedir = 'regulated'
goelite_file = expression_dir + 'GO-Elite/'+elitedir+'/GE.'+criterion_file_name+stat_filters_filename+'.txt'
goelite = export.ExportFile(goelite_file)
goelite_title = ['GeneID'+stat_filters,'SystemCode',criterion_name+'-log_fold',criterion_name+'-p_value']
goelite_title = string.join(goelite_title,'\t')+'\n'; goelite.write(goelite_title)
for (probeset,log_fold,p_value) in criterion_db[criterion_name]:
try:
if 'ENS' in probeset and (' ' in probeset or '_' in probeset or ':' in probeset or '-' in probeset):
system_code = 'En'
ensembl_gene = 'ENS'+string.split(probeset,'ENS')[1]
if ' ' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,' ')[0]
if '_' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,'_')[0]
if ':' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,':')[0]
if '-' in ensembl_gene:
ensembl_gene = string.split(ensembl_gene,'-')[0]
probeset = ensembl_gene
elif ':' in probeset:
probeset = string.split(probeset,':')[0]
system_code = 'Sy'
except Exception:
pass
values = string.join([probeset,system_code,str(log_fold),str(p_value)],'\t')+'\n'
goelite.write(values)
goelite.close()
else: input_files_exported = 'no'
return input_files_exported
def exportGeneRegulationSummary(filename,headers,system_code):
"""
1) Exports summary results description - Performs a series of targetted queries to report the number
of coding and non-coding genes expressed along with various regulation and annotation parameters.
2) Exports a global regulated expression table - Values are log2 geometric folds relative to baseline
of the entire row (all samples) for any criterion met (see ptype_to_use, m_cutoff, p_cutoff). Optionally
cluster these results downstream and perform QC analyses."""
criterion_db={}; detected_exp_db={}; denominator_geneids={}; index = 0; ttest=[]; avg_columns=[]; all_criterion=[]; all_groups=[]
search_miR = 'miR-1('
coding_types = ['protein_coding','ncRNA']
for column in headers:
if 'ANOVA' in ptype_to_use and ptype_to_use in column: ttest.append(index) ### Not currently implemented
elif ptype_to_use in column and 'ANOVA' not in column: ttest.append(index)
lfi = 2 ### relative logfold index position
if ptype_to_use == 'adjp': lfi = 3
index+=1
if 'Protein Classes' in column: pc = index-1
if 'microRNA' in column: mi = index-1
if 'avg-' in column: avg_columns.append(index-1)
if 'Symbol' in column: sy = index-1
try: count_summary_db,indexed_headers = importCountSummary()
except Exception: count_summary_db={}
### Had to introduce the below code to see if any p-values for a criterion are < 1 (otherwise, include them for GO-Elite)
exclude_p1={}
for probeset in array_folds:
index = 0; af = array_folds[probeset]
for value in array_folds[probeset]:
if index in ttest:
criterion_name = headers[index][5:]
if criterion_name not in exclude_p1:
try: p_value = float(value)
except Exception: p_value = 1 ### Occurs when a p-value is annotated as 'Insufficient Expression'
if p_value < 1:
exclude_p1[criterion_name] = True # Hence, at least one gene has a p<1
index+=1
genes_to_import={}; probeset_symbol={}
for probeset in array_folds:
index = 0; af = array_folds[probeset]
probeset_symbol[probeset] = af[sy]
for value in array_folds[probeset]:
denominator_geneids[probeset]=[]
if index in avg_columns:
column_name = headers[index]
group_name = column_name[4:]
try: protein_class = af[pc]
except Exception: protein_class = 'NULL'
proceed = False
if array_type == 'RNASeq':
if norm == 'RPKM':
try: ### Counts file should be present but if not, still proceed
i2 = indexed_headers[column_name]
if float(af[index])>gene_rpkm_threshold and count_summary_db[probeset][i2]>gene_exp_threshold:
#if float(af[index])>5 and count_summary_db[probeset][i2]>50:
proceed = True
except Exception:
proceed = True
exp_info = probeset, af[index],count_summary_db[probeset][i2] ### keep track of the expression info
else:
if float(af[index])>expr_threshold:
proceed = True
exp_info = probeset, expr_threshold,expr_threshold
if proceed==True:
if group_name not in all_groups: all_groups.append(group_name)
if 'protein_coding' in protein_class:
try: detected_exp_db[group_name,'protein_coding'].append(exp_info)
except KeyError: detected_exp_db[group_name,'protein_coding']=[exp_info]
else:
try: detected_exp_db[group_name,'ncRNA'].append(exp_info)
except KeyError: detected_exp_db[group_name,'ncRNA']=[exp_info]
if index in ttest:
criterion_name = headers[index][5:]
try: log_fold = float(af[index-lfi])
except Exception: log_fold = 0 ### Occurs when a fold change is annotated as 'Insufficient Expression'
try: p_value = float(value)
except Exception: p_value = 1 ### Occurs when a p-value is annotated as 'Insufficient Expression'
try: excl_p1 = exclude_p1[criterion_name] ### You can have adjusted p-values that are equal to 1
except Exception: excl_p1 = False #Make True to exclude ALL non-adjp < sig value entries
try: protein_class = af[pc]
except Exception: protein_class = 'NULL'
if abs(log_fold)>m_cutoff and (p_value<p_cutoff or (p_value==1 and excl_p1==False)):
if criterion_name not in all_criterion: all_criterion.append(criterion_name)
try: criterion_db[criterion_name]+=1
except KeyError: criterion_db[criterion_name] = 1
genes_to_import[probeset]=[] ### All, regulated genes (any criterion)
if 'protein_coding' in protein_class:
if log_fold>0:
try: criterion_db[criterion_name,'upregulated','protein_coding']+=1
except KeyError: criterion_db[criterion_name,'upregulated','protein_coding'] = 1
try:
if 'miR-1(' in af[mi]:
try: criterion_db[criterion_name,'upregulated','protein_coding',search_miR[:-1]]+=1
except KeyError: criterion_db[criterion_name,'upregulated','protein_coding',search_miR[:-1]] = 1
except Exception: None ### occurs when mi not present
else:
try: criterion_db[criterion_name,'downregulated','protein_coding']+=1
except KeyError: criterion_db[criterion_name,'downregulated','protein_coding'] = 1
try:
if 'miR-1(' in af[mi]:
try: criterion_db[criterion_name,'downregulated','protein_coding',search_miR[:-1]]+=1
except KeyError: criterion_db[criterion_name,'downregulated','protein_coding',search_miR[:-1]] = 1
except Exception: None ### occurs when mi not present
else:
if protein_class == 'NULL':
class_name = 'unclassified'
else:
class_name = 'ncRNA'
if log_fold>0:
try: criterion_db[criterion_name,'upregulated',class_name]+=1
except KeyError: criterion_db[criterion_name,'upregulated',class_name] = 1
try:
if 'miR-1(' in af[mi]:
try: criterion_db[criterion_name,'upregulated',class_name,search_miR[:-1]]+=1
except KeyError: criterion_db[criterion_name,'upregulated',class_name,search_miR[:-1]] = 1
except Exception: None ### occurs when mi not present
else:
try: criterion_db[criterion_name,'downregulated',class_name]+=1
except KeyError: criterion_db[criterion_name,'downregulated',class_name] = 1
try:
if 'miR-1(' in af[mi]:
try: criterion_db[criterion_name,'downregulated',class_name,search_miR[:-1]]+=1
except KeyError: criterion_db[criterion_name,'downregulated',class_name,search_miR[:-1]] = 1
except Exception: None ### occurs when mi not present
index += 1
if len(criterion_db)>0:
try: exportGeometricFolds(expression_dataset_output_dir+filename,array_type,genes_to_import,probeset_symbol)
except Exception,e:
print 'Failed to export geometric folds due to:'
print e ### Don't exit the analysis just report the problem
print traceback.format_exc()
None
### Export lists of expressed genes
all_expressed={}
for (group_name,coding_type) in detected_exp_db:
eo = export.ExportFile(expression_dataset_output_dir+'/ExpressedGenes/'+group_name+'-'+coding_type+'.txt')
eo.write('GeneID\tRPKM\tCounts\n')
for (gene,rpkm,counts) in detected_exp_db[(group_name,coding_type)]:
eo.write(gene+'\t'+str(rpkm)+'\t'+str(counts)+'\n')
all_expressed[gene]=[]
try: eo.close()
except Exception: pass
filename = string.replace(filename,'DATASET-','SUMMARY-')
filename = string.replace(filename,'GenMAPP-','SUMMARY-')
summary_path = expression_dataset_output_dir +filename
export_data = export.ExportFile(summary_path)
print 'Export summary gene expression results to:',filename
### Output Number of Expressed Genes
title = ['Biological group']
for group_name in all_groups: title.append(group_name)
title = string.join(title,'\t')+'\n'; export_data.write(title)
if array_type == 'RNASeq':
### Only really informative for RNA-Seq data right now, since DABG gene-level stats are not calculated (too time-intensive for this one statistic)
for coding_type in coding_types:
if coding_type == 'protein_coding': values = ['Expressed protein-coding genes']
else: values = ['Expressed ncRNAs']
for group in all_groups:
for group_name in detected_exp_db:
if group in group_name and coding_type in group_name:
values.append(str(len(detected_exp_db[group_name])))
values = string.join(values,'\t')+'\n'; export_data.write(values)
export_data.write('\n')
if m_cutoff<0: fold_cutoff = -1/math.pow(2,m_cutoff)
else: fold_cutoff = math.pow(2,m_cutoff)
### Export criterion gene IDs and minimal data
export_data.write('Regulation criterion: fold > '+str(fold_cutoff)+' and '+ptype_to_use+ ' p-value < '+str(p_cutoff)+'\n\n')
for criterion in all_criterion:
title = [criterion,'up','down','up-'+search_miR[:-1],'down-'+search_miR[:-1]]
title = string.join(title,'\t')+'\n'; export_data.write(title)
for coding_type in coding_types:
values = ['Regulated '+coding_type+' genes']
for criterion_name in criterion_db:
if len(criterion_name)==3:
if criterion in criterion_name and ('upregulated',coding_type) == criterion_name[1:]:
values.append(str(criterion_db[criterion_name]))
if len(values)==1: values.append('0')
for criterion_name in criterion_db:
if len(criterion_name)==3:
if criterion in criterion_name and ('downregulated',coding_type) == criterion_name[1:]:
values.append(str(criterion_db[criterion_name]))
if len(values)==2: values.append('0')
for criterion_name in criterion_db:
if len(criterion_name)==4:
if criterion in criterion_name and ('upregulated',coding_type) == criterion_name[1:-1]:
values.append(str(criterion_db[criterion_name]))
if len(values)==3: values.append('0')
for criterion_name in criterion_db:
if len(criterion_name)==4:
if criterion in criterion_name and ('downregulated',coding_type) == criterion_name[1:-1]:
values.append(str(criterion_db[criterion_name]))
if len(values)==4: values.append('0')
#print values;sys.exit()
values = string.join(values,'\t')+'\n'; export_data.write(values)
export_data.write('\n')
export_data.close()
def exportGeometricFolds(filename,platform,genes_to_import,probeset_symbol,exportOutliers=True,exportRelative=True,customPath=None,convertNonLogToLog=False):
#print expression_data_format
#print platform
#print len(genes_to_import)
#print exportOutliers
#print exportRelative
#print customPath
""" Import sample and gene expression values from input file, filter, calculate geometric folds
and export. Use for clustering and QC."""
#print '\n',filename
filename = string.replace(filename,'///','/')
filename = string.replace(filename,'//','/')
status = 'yes'
convertGeneToSymbol = True
if 'ExpressionOutput' in filename:
filename = string.replace(filename,'-steady-state.txt','.txt')
export_path = string.replace(filename,'ExpressionOutput','ExpressionOutput/Clustering')
export_path = string.replace(export_path,'DATASET-','SampleLogFolds-') ### compared to all-sample mean
export_path2 = string.replace(export_path,'SampleLogFolds-','OutlierLogFolds-') ### compared to all-sample mean
export_path3 = string.replace(export_path,'SampleLogFolds-','RelativeSampleLogFolds-') ### compared to control groups
filename = string.replace(filename,'ExpressionOutput','ExpressionInput')
filename = string.replace(filename,'DATASET-','exp.')
groups_dir = string.replace(filename,'exp.','groups.')
if platform != "3'array" and platform != "AltMouse":
### This is the extension for gene-level results for exon sensitive platfomrs
filename1 = string.replace(filename,'.txt','-steady-state.txt')
status = verifyFile(filename1)
if status == 'yes':
filename = filename1
status = verifyFile(filename)
if status != 'yes':
filename = string.replace(filename,'exp.','')
filename = string.replace(filename,'ExpressionInput','')
status = verifyFile(filename)
if customPath!=None:
### If an alternative output path is desired
export_path = customPath
print len(genes_to_import), 'genes with data to export...'
try:
if expression_data_format == 'non-log' and platform != 'RNASeq': convertNonLogToLog = True
except Exception: pass
if status != 'yes':
print "Clustering expression file not exported due to missing file:"
print filename
if status == 'yes':
export_data = export.ExportFile(export_path)
if exportOutliers: export_outliers = export.ExportFile(export_path2)
if exportRelative: export_relative = export.ExportFile(export_path3)
print 'Export inputs for clustering to:',export_path
expressionDataFormat,increment,convertNonLogToLog = checkExpressionFileFormat(filename)
#print expressionDataFormat,increment,convertNonLogToLog
fn=filepath(filename); row_number=0; exp_db={}; relative_headers_exported = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#' and row_number==0: row_number = 0
elif row_number==0:
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = simpleGroupImport(groups_dir)
try: sample_index_list = map(lambda x: t[1:].index(x), sample_list) ### lookup index of each sample in the ordered group sample list
except Exception:
missing=[]
for x in sample_list:
if x not in t[1:]: missing.append(x)
print 'missing:',missing
print t
print sample_list
print filename, groups_dir
print 'Unknown Error!!! Skipping cluster input file build (check column and row formats for conflicts)'; forceExit
new_sample_list = map(lambda x: group_sample_db[x], sample_list) ### lookup index of each sample in the ordered group sample list
title = string.join([t[0]]+new_sample_list,'\t')+'\n' ### output the new sample order (group file order)
export_data.write(title)
if exportOutliers: export_outliers.write(title)
if exportRelative:
### Used for the relative fold calculation
group_index_db={}
for x in sample_list:
group_name = group_db[x]
sample_index = t[1:].index(x)
try: group_index_db[group_name].append(sample_index)
except Exception: group_index_db[group_name] = [sample_index] ### dictionary of group to input file sample indexes
row_number=1
else:
gene = t[0]
if platform == 'RNASeq':
### Convert to log2 RPKM values - or counts
try: values = map(lambda x: math.log(float(x)+increment,2), t[1:])
except Exception:
if convertNonLogToLog:
values = logTransformWithNAs(t[1:],increment)
else:
values = TransformWithNAs(t[1:])
else:
try:
if convertNonLogToLog:
values = map(lambda x: math.log(float(x)+increment,2), t[1:])
else:
values = map(float,t[1:])
except Exception:
if convertNonLogToLog:
values = logTransformWithNAs(t[1:],increment)
else:
values = TransformWithNAs(t[1:])
### Calculate log-fold values relative to the mean of all sample expression values
values = map(lambda x: values[x], sample_index_list) ### simple and fast way to reorganize the samples
try: avg = statistics.avg(values)
except Exception:
values2=[]
for v in values:
try: values2.append(float(v))
except Exception: pass
try: avg = statistics.avg(values2)
except Exception:
if len(values2)>0: avg = values2[0]
else: avg = 0
try: log_folds = map(lambda x: (x-avg), values)
except Exception:
log_folds=[]
for x in values:
try: log_folds.append(x-avg)
except Exception: log_folds.append('')
if gene in genes_to_import:
### Genes regulated in any user-indicated comparison according to the fold and pvalue cutoffs provided
log_folds = map(lambda x: str(x), log_folds)
try:
"""
if convertGeneToSymbol:
if gene == probeset_symbol[gene]:
gene2 = gene
convertGeneToSymbol = False
else:
gene2 = gene+' '+probeset_symbol[gene]
else:
gene2 = gene
"""
gene2 = gene+' '+probeset_symbol[gene]
except Exception: gene2 = gene
#print [gene2,gene];sys.exit()
if len(t[1:])!=len(log_folds):
log_folds = t[1:] ### If NAs - output the original values
export_data.write(string.join([gene2]+log_folds,'\t')+'\n')
if exportRelative:
### Calculate log-fold values relative to the mean of each valid group comparison
control_group_avg={}; comps_exp_db={}
for group_name in comps_name_db: ### control group names
con_group_values = map(lambda x: values[x], group_index_db[group_name]) ### simple and fast way to reorganize the samples
try: control_group_avg[group_name] = statistics.avg(con_group_values) ### store the mean value of each control group
except Exception:
con_group_values2=[]
for val in con_group_values:
try: con_group_values2.append(float(val))
except Exception: pass
try: control_group_avg[group_name] = statistics.avg(con_group_values)
except Exception:
if len(con_group_values)>0:
control_group_avg[group_name] = con_group_values[0]
else: control_group_avg[group_name] = 0.0
for exp_group in comps_name_db[group_name]:
try: comps_exp_db[exp_group].append(group_name) ### Create a reversed version of the comps_name_db, list experimental as the key
except Exception: comps_exp_db[exp_group] = [group_name]
relative_log_folds=[] ### append all new log folds to this list
relative_column_names=[]
for group_name in comp_groups:
if group_name in comps_exp_db: ### Hence, the group has a designated control (controls may not) - could have the control group be a control for the control samples
group_values = map(lambda x: values[x], group_index_db[group_name]) ### simple and fast way to reorganize the samples
for control_group_name in comps_exp_db[group_name]:
con_avg = control_group_avg[control_group_name]
try:
relative_log_folds += map(lambda x: str(x-con_avg), group_values) ### calculate log-folds and convert to strings
except Exception:
relative_log_folds=[]
for x in group_values:
try: relative_log_folds.append(str(x-con_avg))
except Exception: relative_log_folds.append('')
if relative_headers_exported == False:
exp_sample_names = group_name_sample_db[group_name]
relative_column_names += map(lambda x: (x+' vs '+control_group_name), exp_sample_names) ### add column names indicating the comparison
if relative_headers_exported == False:
title = string.join(['UID']+relative_column_names,'\t')+'\n' ### Export column headers for the relative fold changes
export_relative.write(title)
relative_headers_exported = True
if len(t[1:])!=len(relative_log_folds):
relative_log_folds = t[1:] ### If NAs - output the original values
export_relative.write(string.join([gene2]+relative_log_folds,'\t')+'\n')
elif exportOutliers:
### When a gene is regulated and not significant, export to the outlier set
try: gene2 = gene+' '+probeset_symbol[gene]
except Exception: gene2 = gene
### These are defaults we may allow the user to control later
log_folds = [0 if x=='' else x for x in log_folds] ### using list comprehension, replace '' with 0
if max([max(log_folds),abs(min(log_folds))])>1:
proceed = True
if platform == 'RNASeq':
if max(values)<0.1: proceed = False
if proceed == True:
log_folds = map(lambda x: str(x), log_folds)
if len(t[1:])!=len(log_folds):
log_folds = t[1:] ### If NAs - output the original values
export_outliers.write(string.join([gene2]+log_folds,'\t')+'\n')
row_number+=1 ### Keep track of the first gene as to write out column headers for the relative outputs
export_data.close()
if exportOutliers: export_outliers.close()
if exportRelative: export_relative.close()
def logTransformWithNAs(values,increment):
values2=[]
for x in values:
try: values2.append(math.log(float(x)+increment,2))
except Exception:
values2.append('')
return values2
def TransformWithNAs(values):
values2=[]
for x in values:
try: values2.append(float(x))
except Exception:
values2.append('')
return values2
def importAndOrganizeLineageOutputs(expr_input,filename,platform):
""" This function takes LineageProfiler z-scores and organizes the samples into groups
takes the mean results for each group and looks for changes in lineage associations """
groups_dir = string.replace(expr_input,'exp.','groups.')
groups_dir = string.replace(groups_dir,'-steady-state.txt','.txt') ### groups is for the non-steady-state file
export_path = string.replace(filename,'ExpressionOutput','ExpressionOutput/Clustering')
export_path = string.replace(export_path,'.txt','-groups.txt')
export_data = export.ExportFile(export_path)
export_pathF = string.replace(export_path,'.txt','_filtered.txt')
export_dataF = export.ExportFile(export_pathF)
print 'Export inputs for clustering to:',export_path
fn=filepath(filename); row_number=0; exp_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#': row_number = 0
elif row_number==0:
group_index_db={}
### use comps in the future to visualize group comparison changes
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = simpleGroupImport(groups_dir)
for x in sample_list:
group_name = group_db[x]
sample_index = t[1:].index(x)
try: group_index_db[group_name].append(sample_index)
except Exception: group_index_db[group_name] = [sample_index] ### dictionary of group to input file sample indexes
groups = map(str, group_index_db) ### store group names
new_sample_list = map(lambda x: group_db[x], sample_list) ### lookup index of each sample in the ordered group sample list
title = string.join([t[0]]+groups,'\t')+'\n' ### output the new sample order (group file order)
export_data.write(title)
export_dataF.write(title)
row_number=1
else:
tissue = t[0]
if platform == 'RNASeq' and 'LineageCorrelations' not in filename:
### Convert to log2 RPKM values - or counts
values = map(lambda x: math.log(float(x),2), t[1:])
else:
values = map(float,t[1:])
avg_z=[]; avg_z_float=[]
for group_name in group_index_db:
group_values = map(lambda x: values[x], group_index_db[group_name]) ### simple and fast way to reorganize the samples
avg = statistics.avg(group_values)
avg_z.append(str(avg))
avg_z_float.append(avg)
export_data.write(string.join([tissue]+avg_z,'\t')+'\n')
if max(avg_z_float)>1:
export_dataF.write(string.join([tissue]+avg_z,'\t')+'\n')
export_data.close(); export_dataF.close()
return export_path,export_pathF
def removeRawData(array_fold_headers):
### Prior to exporting data for GenMAPP, remove raw data columns
columns_with_stats=[]; i=0; stat_headers = ['avg', 'log_fold', 'fold', 'rawp', 'adjp']; filtered_headers=[]
for header in array_fold_headers:
broken_header = string.split(header,'-')
### Only keep those headers and indexes with recognized ExpressionBuilder inserted prefixes
if broken_header[0] in stat_headers: columns_with_stats.append(i); filtered_headers.append(header)
i+=1
for probeset in array_folds:
filtered_list=[]
for i in columns_with_stats: filtered_list.append(array_folds[probeset][i])
array_folds[probeset] = filtered_list ### Re-assign values of the db
return filtered_headers
def removeRawCountData(array_fold_headers):
### Prior to exporting data for GenMAPP, remove raw data columns
columns_with_stats=[]; i=0; stat_headers = ['avg', 'log_fold', 'fold', 'rawp', 'adjp']; filtered_headers=[]
for header in array_fold_headers:
broken_header = string.split(header,'-')
### Only keep those headers and indexes with recognized ExpressionBuilder inserted prefixes
if broken_header[0] in stat_headers: columns_with_stats.append(i); filtered_headers.append(header)
i+=1
for probeset in count_statistics_db:
filtered_list=[]
for i in columns_with_stats: filtered_list.append(count_statistics_db[probeset][i])
count_statistics_db[probeset] = filtered_list ### Re-assign values of the db
return filtered_headers
def exportAnalyzedData(comp_group_list2,expr_group_db):
report = 'multiple'; report = 'single'
try: ensembl_microRNA_db = importMicrornaAssociations(species,report)
except IOError: ensembl_microRNA_db={}
if array_type != "AltMouse" and array_type != "3'array":
try:
from build_scripts import EnsemblImport
gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,array_type,'key_by_array')
except Exception: gene_location_db={}
if data_type == 'expression':
new_file = expression_dataset_output_dir + 'DATASET-'+experiment_name+'.txt'
try: data = export.createExportFile(new_file,expression_dataset_output_dir[:-1])
except RuntimeError:
export.isFileOpen(new_file,expression_dataset_output_dir[:-1])
data = export.createExportFile(new_file,expression_dataset_output_dir[:-1])
try: custom_annotation_dbase = importCustomAnnotations()
except Exception: custom_annotation_dbase={}
x=0;y=0;z=0
for arrayid in array_folds:
if arrayid in annotate_db and arrayid in probeset_db: x = 1
if arrayid in annotate_db: y = 1
if arrayid in conventional_array_db: z = 1
break
Vendor = vendor ### Need to rename as re-assigning will cause a global conflict error
for arrayid in array_folds:
if 'ENS' in arrayid and Vendor == 'Symbol':
Vendor = 'Ensembl'
break
if array_type != "AltMouse" and (array_type != "3'array" or 'Ensembl' in Vendor):
#annotate_db[gene] = symbol, definition,rna_processing
#probeset_db[gene] = transcluster_string, exon_id_string
title = ['Ensembl_gene','Definition','Symbol','Transcript_cluster_ids','Constitutive_exons_used','Constitutive_IDs_used','Putative microRNA binding sites','Select Cellular Compartments','Select Protein Classes','Chromosome','Strand','Genomic Gene Corrdinates','GO-Biological Process','GO-Molecular Function','GO-Cellular Component','WikiPathways']
title = string.join(title,'\t')
elif arrayCode == 3: ### Code indicates this array probes only for small RNAs
title = ['Probeset ID','Sequence Type','Transcript ID','Species Scientific Name','Genomic Location']
title = string.join(title,'\t')
elif x == 1:
title = "Probesets" +'\t'+ 'Definition' +'\t'+ 'Symbol' +'\t'+ 'affygene' +'\t'+ 'exons' +'\t'+ 'probe_type_call' +'\t'+ 'ensembl'
elif y==1: title = "Probesets" +'\t'+ 'Symbol' +'\t'+ 'Definition'
elif array_type == "3'array":
title = ['Probesets','Symbol','Definition','Ensembl_id','Entrez_id','Unigene_id','GO-Process','GO-Function','GO-Component','Pathway_info','Putative microRNA binding sites','Select Cellular Compartments','Select Protein Classes']
title = string.join(title,'\t')
else: title = "Probesets"
for entry in array_fold_headers: title = title + '\t' + entry
title += '\t'+ 'ANOVA-rawp' +'\t'+ 'ANOVA-adjp' +'\t'+'largest fold'
if array_type == 'RNASeq': title += '\t'+ 'maximum sample read count'
data.write(title+'\n')
for arrayid in array_folds:
if arrayCode == 3:
ca = conventional_array_db[arrayid]
definition = ca.Description()
symbol = ca.Symbol()
data_val = [arrayid,ca.Description(),ca.Symbol(),ca.Species(),ca.Coordinates()]
data_val = string.join(data_val,'\t')
elif array_type != 'AltMouse' and (array_type != "3'array" or 'Ensembl' in Vendor):
try: definition = annotate_db[arrayid][0]; symbol = annotate_db[arrayid][1]; rna_processing = annotate_db[arrayid][2]
except Exception: definition=''; symbol=''; rna_processing=''
report = 'all'
try: miRs = ensembl_microRNA_db[arrayid]
except KeyError: miRs = ''
try:
trans_cluster = probeset_db[arrayid][0]
exon_ids = probeset_db[arrayid][1]
probesets = probeset_db[arrayid][2]
except Exception:
trans_cluster='';exon_ids='';probesets=''
try: compartment,custom_class = custom_annotation_dbase[arrayid]
except KeyError: compartment=''; custom_class=''
try: chr,strand,start,end = gene_location_db[arrayid]
except Exception: chr=''; strand=''; strand=''; start=''; end=''
try: pi = conventional_array_db[arrayid]; process = pi.Process(); function=pi.Function(); component=pi.Component(); pathway = pi.Pathway()
except Exception: process=''; function=''; component=''; pathway=''
data_val = [arrayid,symbol,definition,trans_cluster,exon_ids,probesets,miRs,compartment,custom_class,chr,strand,start+'-'+end,process,function,component,pathway]
data_val = string.join(data_val,'\t')
elif arrayid in annotate_db and arrayid in probeset_db: ### This is for the AltMouse array
symbol = annotate_db[arrayid][0]
definition = annotate_db[arrayid][1]
affygene = probeset_db[arrayid][0][0:-1] #probeset_db[probeset] = affygene,exons,probe_type_call,ensembl
exons = probeset_db[arrayid][1]
probe_type_call = probeset_db[arrayid][2]
ensembl = probeset_db[arrayid][3]
data_val = arrayid +'\t'+ definition +'\t'+ symbol +'\t'+ affygene +'\t'+ exons +'\t'+ probe_type_call +'\t'+ ensembl
elif arrayid in annotate_db:
definition = annotate_db[arrayid][0]
symbol = annotate_db[arrayid][1]
data_val = arrayid +'\t'+ definition +'\t'+ symbol
elif array_type == "3'array" and 'Ensembl' not in Vendor:
try:
ca = conventional_array_db[arrayid]
definition = ca.Description()
symbol = ca.Symbol()
ens = ca.EnsemblString()
entrez = ca.EntrezString()
unigene = ca.UnigeneString()
pathway_info = ca.PathwayInfo()
component = ca.GOComponentNames(); process = ca.GOProcessNames(); function = ca.GOFunctionNames()
compartment=''; custom_class=''; miRs=''
if len(ens)>0:
if ens[0]=='|': ens = ens[1:]
store=[]
for ens_gene in ca.Ensembl(): ### Add Custom Annotation layer
try: compartment,custom_class = custom_annotation_dbase[ens_gene]
except KeyError: null=[]
try: miRs = ensembl_microRNA_db[ens_gene]
except KeyError: null=[]
if 'protein_coding' in custom_class and len(store)==0: ### Use the first instance only
store = miRs,compartment,custom_class+'('+ens_gene+')'
if len(store)>0: ### pick the Ensembl with protein coding annotation to represent (as opposed to aligning annotated pseudo genes)
miRs,compartment,custom_class = store
except KeyError:
definition=''; symbol=''; ens=''; entrez=''; unigene=''; pathway_info=''
process=''; function=''; component=''; compartment='' ;custom_class=''; miRs=''
data_val = [arrayid,symbol,definition,ens,entrez,unigene,process,function,component,pathway_info,miRs,compartment,custom_class]
data_val = string.join(data_val,'\t')
else:
data_val = arrayid
for value in array_folds[arrayid]:
data_val = data_val + '\t' + str(value)
gs = summary_filtering_stats[arrayid]
#if arrayid == '1623863_a_at': print [gs.LogFold()]
data_val += '\t'+ str(gs.Pval()) +'\t'+ str(gs.AdjP()) +'\t'+ str(gs.LogFold())
if array_type == 'RNASeq': data_val+= '\t'+ gs.MaxCount()
data_val = string.replace(data_val,'\n','')
data.write(data_val+'\n')
data.close()
print "Full Dataset with statistics:",'DATASET-'+experiment_name+'.txt', 'written'
gene_location_db=[]
ensembl_microRNA_db=[]
custom_annotation_dbase=[]
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def eliminate_redundant_dict_values(database):
db1={}
for key in database:
list = unique.unique(database[key])
list.sort()
db1[key] = list
return db1
def convert_to_list(database):
db1=[]; db2=[]; temp_list=[]
for key in database:
list = database[key]
#print key,list,dog #32 [(2, 1.1480585565447154), (3, 0.72959188370731742), (0, 0.0), (1, -0.60729064216260165)]
list.sort()
temp_list=[]
temp_list.append(key)
for entry in list:
avg_fold = entry[1]
temp_list.append(avg_fold)
#print temp_list, dog #[32, 0.0, -0.60729064216260165, 1.1480585565447154, 0.72959188370731742]
db1.append(temp_list)
db1.sort()
return db1
def import_annotations(filename):
fn=filepath(filename)
annotation_dbase = {}
for line in open(fn,'rU').xreadlines():
try:
data = cleanUpLine(line)
try: probeset,definition,symbol,rna_processing = string.split(data,'\t')
except ValueError:
probeset,definition,symbol = string.split(data,'\t')
rna_processing = ''
annotation_dbase[probeset] = definition, symbol,rna_processing
except ValueError: continue
return annotation_dbase
def importCustomAnnotations():
### Combine non-coding Ensembl gene annotations with UniProt functional annotations
try: custom_annotation_dbase = importTranscriptBiotypeAnnotations(species)
except Exception: custom_annotation_dbase = {}
try: housekeeping_genes=BuildAffymetrixAssociations.getHousekeepingGenes(species)
except Exception: housekeeping_genes=[]
print len(custom_annotation_dbase),'Ensembl Biotypes and', len(housekeeping_genes),'housekeeping genes.'
for ens_gene in housekeeping_genes:
if ens_gene not in custom_annotation_dbase: custom_annotation_dbase[ens_gene] = '','housekeeping'
else:
compartment,custom_class = custom_annotation_dbase[ens_gene]
custom_class+='|housekeeping'
custom_annotation_dbase[ens_gene] = compartment,custom_class
filename = 'AltDatabase/uniprot/'+species+'/custom_annotations.txt'
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
ens_gene,compartment,custom_class = t[:3]
if ens_gene in custom_annotation_dbase:
biotype = custom_annotation_dbase[ens_gene][1]
if len(custom_class)>0: custom_class+='|'+biotype
else: custom_class=biotype
custom_annotation_dbase[ens_gene] = compartment,custom_class
return custom_annotation_dbase
def importTranscriptBiotypeAnnotations(species):
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_transcript-biotypes.txt'
fn=filepath(filename); biotype_db = {}; custom_annotation_dbase={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
gene,transcript,biotype = string.split(data,'\t')
### Determine if only one annotation is associated with each gene
try: biotype_db[gene][biotype]=[]
except Exception: biotype_db[gene] = db = {biotype:[]}
for gene in biotype_db:
db = biotype_db[gene]
if len(db)==1 and 'protein_coding' not in db:
for biotype in db: ### Non-coding gene annotation
custom_annotation_dbase[gene] = '',biotype
elif 'protein_coding' in db:
custom_annotation_dbase[gene] = '','protein_coding'
elif 'transcribed_unprocessed_pseudogene' in db:
custom_annotation_dbase[gene] = '','transcribed_unprocessed_pseudogene'
else:
ls=[] ### otherwise include all gene annotations
for i in db: ls.append(i)
ls = string.join(ls,'|')
custom_annotation_dbase[gene] = '',ls
return custom_annotation_dbase
def importAltMerge(import_type):
### Import Probeset annotations
try:
ensembl_db={}; fn=filepath('AltDatabase/Mm/AltMouse/AltMouse-Ensembl.txt')
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
affygene,ensembl = string.split(data,'\t')
ensembl_db[affygene]=ensembl
#print len(ensembl_db),'Ensembl-AltMouse relationships imported'
except TypeError: null=[]
### Import Probeset annotations
probeset_annotation_file = "AltDatabase/"+species+'/'+array_type+'/'+ "MASTER-probeset-transcript.txt"
probeset_db = {}; constitutive_db = {}; fn=filepath(probeset_annotation_file); replacements=0
for line in open(fn,'rU').xreadlines():
probeset_data = cleanUpLine(line)
probeset,affygene,exons,transcript_num,transcripts,probe_type_call,ensembl,block_exon_ids,block_structure,comparison_info = string.split(probeset_data,'\t')
if probeset == "Probeset": continue
else:
if affygene[:-1] in ensembl_db: ensembl = ensembl_db[affygene[:-1]]; replacements+=1
if import_type == 'full': ### Mimics the structure of ExonArrayEnsemblRules.reimportEnsemblProbesets() dictionary probe_association_db
probe_data = affygene,affygene,exons,'','core'
probeset_db[probeset] = probe_data
else: probeset_db[probeset] = affygene,exons,probe_type_call,ensembl
if probe_type_call == 'gene':
try: constitutive_db[affygene].append(probeset)
except KeyError: constitutive_db[affygene] = [probeset]
return probeset_db, constitutive_db
def parse_custom_annotations(filename):
custom_array_db = {}
x=0
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
array_data = data
array_id,probeset,other = string.split(array_data,'\t') #remove endline
custom_array_db[array_id] = probeset,other
print len(custom_array_db), "custom array entries process"
return custom_array_db
def remoteLineageProfiler(params,expr_input_dir,ArrayType,Species,Vendor,customMarkers=False,specificPlatform=False,visualizeNetworks=True):
global species
global array_type
global vendor
global remoteAnalysis
global fl
remoteAnalysis = True
species = Species
array_type = ArrayType
vendor = Vendor
fl = params
graphics_links = []
if 'ExpressionInput' in expr_input_dir:
output_dir = string.replace(expr_input_dir,'ExpressionInput', 'ExpressionOutput')
root_dir = export.findParentDir(output_dir)
else:
root_dir = export.findParentDir(expr_input_dir)+'ExpressionOutput/'
try:
### If this directory exists, create a global variable for it
dir_list = read_directory(root_dir[:-1])
global expression_dataset_output_dir
global experiment_name
experiment_name = string.replace(export.findFilename(expr_input_dir)[:-4],'exp.','')
expression_dataset_output_dir = root_dir
except Exception:
#print traceback.format_exc()
None
graphic_links = performLineageProfiler(expr_input_dir,graphics_links,customMarkers,specificPlatform=specificPlatform,visualizeNetworks=visualizeNetworks)
return graphic_links
def performLineageProfiler(expr_input_dir,graphic_links,customMarkers=False,specificPlatform=False,visualizeNetworks=True):
try:
from visualization_scripts import WikiPathways_webservice
import LineageProfiler; reload(LineageProfiler)
start_time = time.time()
try:
compendium_type = fl.CompendiumType()
compendium_platform = fl.CompendiumPlatform()
except Exception:
compendium_type = 'protein_coding'
compendium_platform = 'exon'
#print 'Compendium platform selected:',compendium_platform
print 'Biological data type to examine:',compendium_type
try: ### Works when expression_dataset_output_dir is defined
exp_output = expression_dataset_output_dir + 'DATASET-'+experiment_name+'.txt'
#try: array_type_data = vendor, array_type
array_type_data = array_type
except Exception: ### Otherwise, user directly supplied file is used
array_type_data = vendor, array_type
exp_output = export.findParentDir(expr_input_dir)+'/LineageCorrelations-'+export.findFilename(expr_input_dir)
if specificPlatform == False:
compendium_platform = 'exon'
status = False
compareToAll=False
"""
print species
print array_type_data
print expr_input_dir
print exp_output
print compendium_type
print compendium_platform
print customMarkers
"""
if 'steady-state.txt' in expr_input_dir:
status = verifyFile(expr_input_dir)
if status != 'yes':
expr_input_dir = string.replace(expr_input_dir,'-steady-state.txt','.txt')
array_type_data = "3'array"
try:
zscore_output_dir1 = LineageProfiler.runLineageProfiler(species,array_type_data,
expr_input_dir,exp_output,compendium_type,compendium_platform,customMarkers); status = True
#zscore_output_dir1 = None
except Exception:
print traceback.format_exc(),'\n'
zscore_output_dir1 = None
if compareToAll:
try:
zscore_output_dir2 = LineageProfiler.runLineageProfiler(species,array_type_data,expr_input_dir, exp_output,compendium_type,'gene',customMarkers); status = True
#zscore_output_dir2 = None
except Exception: zscore_output_dir2 = None
try:
zscore_output_dir3 = LineageProfiler.runLineageProfiler(species,array_type_data,expr_input_dir, exp_output,compendium_type,"3'array",customMarkers); status = True
#zscore_output_dir3 = None
except Exception: zscore_output_dir3 = None
zscore_output_dirs=[zscore_output_dir1,zscore_output_dir2,zscore_output_dir3]
else:
zscore_output_dirs=[zscore_output_dir1]
### Create a combined zscore_output_dir output using all predictions
zscore_output_dir = combineLPResultFiles(zscore_output_dirs)
if status == False:
#print traceback.format_exc(),'\n'
if species != 'Mm' and species != 'Hs':
print 'LineageProfiler analysis failed (possibly unsupported species).'
else:
time_diff = str(round(time.time()-start_time,1))
print 'LineageProfiler analysis completed in %s seconds' % time_diff
try: ### If the file a groups file exists in the expected directory structure -> export a groups z-score file
export_path, export_filtered_path = importAndOrganizeLineageOutputs(expr_input_dir,zscore_output_dir,array_type)
except Exception:
export_path = zscore_output_dir ### keeps the sample z-score file as input
export_filtered_path = None
### Output a heat map of the sample Z-score correlations
graphic_links = LineageProfiler.visualizeLineageZscores(zscore_output_dir,export_path,graphic_links)
if export_filtered_path != None:
try: LineageProfiler.visualizeLineageZscores(export_filtered_path,export_path,graphic_links) ### Just output a heatmap of filtered grouped terms
except Exception: pass
### Color the TissueMap from WikiPathways using their webservice
if customMarkers==False and visualizeNetworks:
print 'Coloring LineageMap profiles using WikiPathways webservice...'
graphic_links = WikiPathways_webservice.viewLineageProfilerResults(export_path,graphic_links)
except Exception:
print traceback.format_exc(),'\n'
### Analysis may not be supported for species or data is incompatible
try:
if remoteAnalysis:
if species != 'Mm' and species != 'Hs':
print 'LineageProfiler analysis failed (possibly unsupported species).'
#print traceback.format_exc(),'\n'
except Exception:
pass
return graphic_links
def combineLPResultFiles(input_files):
combined_sample_cell_db={}
celltypes=[]
for fn in input_files:
if fn != None:
firstLine=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t=string.split(data,'\t')
if firstLine:
headers = t[1:]
firstLine=False
else:
cell_type = t[0]
if cell_type not in celltypes:
celltypes.append(cell_type)
zscores = t[1:]
for sample in headers:
z = float(zscores[headers.index(sample)])
try:
cell_zscores = combined_sample_cell_db[sample]
try: cell_zscores[cell_type].append(z)
except Exception: cell_zscores[cell_type]=[z]
except Exception:
combined_sample_cell_db[sample] = {cell_type:[z]}
try:
headers.sort()
celltypes.sort()
for i in input_files:
if i!=None:
output_file = string.join(string.split(i,'-')[:-2],'-')+'-zscores.txt'
o = export.ExportFile(output_file)
o.write(string.join(['LineagePredictions']+headers,'\t')+'\n')
break
for cell_type in celltypes:
values = [cell_type]
for sample in headers:
cell_zscores = combined_sample_cell_db[sample]
#try: cell_zscores[cell_type].sort()
#except Exception: cell_zscores[cell_type] = [0]
selectedZ=str(cell_zscores[cell_type][0])
#if 'Breast' in sample and cell_type=='Breast': print cell_zscores['Breast'],sample, selectedZ;sys.exit()
#selectedZ=str(statistics.avg(cell_zscores[cell_type]))
values.append(selectedZ)
o.write(string.join(values,'\t')+'\n')
o.close()
except Exception: pass
try: returnRowHeaderForMaxEntry(output_file,10)
except Exception: pass
return output_file
def visualizeQCPlots(expr_input_dir):
original_expr_input_dir = expr_input_dir
expr_input_dir = string.replace(expr_input_dir,'-steady-state','') ### We want the full exon/probeset-level expression file
try:
from visualization_scripts import clustering
from visualization_scripts import QC
print 'Building quality control graphs...'
if array_type == 'RNASeq':
counts_input_dir = string.replace(expr_input_dir,'exp.','counts.')
graphic_links = QC.outputRNASeqQC(counts_input_dir)
else:
graphic_links = QC.outputArrayQC(expr_input_dir)
print 'Building hierarchical cluster graphs...'
paths = getSampleLogFoldFilenames(expr_input_dir)
graphic_links = clustering.outputClusters(paths,graphic_links, Normalize='median',Species=species)
try: graphic_links = clustering.runPCAonly(original_expr_input_dir,graphic_links,False,plotType='2D',display=False)
except Exception: pass
except Exception:
print 'Unable to generate QC plots:'
print traceback.format_exc()
try: graphic_links = graphic_links
except Exception: graphic_links=None ### Matplotlib likely not installed - or other unknown issue
return graphic_links
def getSampleLogFoldFilenames(filename):
if '/' in filename: delim = '/'
else: delim = '\\'
if 'ExpressionInput' in filename:
export_path = string.replace(filename,'ExpressionInput','ExpressionOutput/Clustering')
path1 = string.replace(export_path,'exp.','SampleLogFolds-')
path2 = string.replace(export_path,'exp.','OutlierLogFolds-')
path3 = string.replace(export_path,'exp.','RelativeSampleLogFolds-')
paths = [path1,path2,path3]
else:
paths = string.split(filename,delim)
path1 = string.join(paths[:-1],delim)+'/ExpressionOutput/Clustering/SampleLogFolds-'+paths[-1]
path2 = string.replace(path1,'SampleLogFolds-','OutlierLogFolds-')
path3 = string.replace(path1,'SampleLogFolds-','RelativeSampleLogFolds-')
paths = [path1,path2,path3]
return paths
def importGeneAnnotations(species):
### Used for internal testing
gene_annotation_file = "AltDatabase/ensembl/"+species+"/"+species+"_Ensembl-annotations_simple.txt"
fn=filepath(gene_annotation_file)
count = 0; annotate_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if count == 0: count = 1
else:
gene, description, symbol = string.split(data,'\t')
annotate_db[gene] = symbol,description,''
return annotate_db
def remoteExpressionBuilder(Species,Array_type,dabg_p,expression_threshold,
avg_all_for_ss,Expression_data_format,Vendor,
constitutive_source,data_source,Include_raw_data,
perform_alt_analysis,GE_fold_cutoffs,GE_pvalue_cutoffs,
GE_ptype,exp_file_location_db,Root):
start_time = time.time()
global root; root = Root
#def remoteExpressionBuilder():
global species; global array_type ; species = Species; array_type = Array_type; global altanalyze_files; global vendor; vendor = Vendor
global filter_by_dabg; filter_by_dabg = 'yes' ### shouldn't matter, since the program should just continue on without it
global expression_data_format; global expression_dataset_output_dir; global root_dir; global data_type
global conventional_array_db; global custom_array_db; global constitutive_db; global include_raw_data; global experiment_name
global annotate_db; global probeset_db; global process_custom; global m_cutoff; global p_cutoff; global ptype_to_use; global norm
global arrayCode; arrayCode = 0; global probability_statistic; global fl; global use_downregulated_label; use_downregulated_label = True
global count_statistics_db; global count_statistics_headers; count_statistics_db = {}
include_raw_data = Include_raw_data; expression_data_format = Expression_data_format
data_type = 'expression' ###Default, otherwise is 'dabg'
d = "core"; e = "extendend"; f = "full"; exons_to_grab = d ### Currently, not used by the program... intended as an option for ExonArrayAffymetrixRules full annotation (deprecated)
### Original options and defaults
"""
dabg_p = 0.75; data_type = 'expression' ###used for expression analysis when dealing with AltMouse arrays
a = "3'array"; b = "exon"; c = "AltMouse"; e = "custom"; array_type = c
l = 'log'; n = 'non-log'; expression_data_format = l
w = 'Agilent'; x = 'Affymetrix'; y = 'Ensembl'; z = 'default'; data_source = y; constitutive_source = z; vendor = x
hs = 'Hs'; mm = 'Mm'; dr = 'Dr'; rn = 'Rn'; species = mm
include_raw_data = 'yes'
expression_threshold = 70 ### Based on suggestion from BMC Genomics. 2006 Dec 27;7:325. PMID: 17192196, for hu-exon 1.0 st array
avg_all_for_ss = 'no' ###Default is 'no' since we don't want all probes averaged for the exon arrays
"""
ct = 'count'; avg = 'average'; filter_method = avg
filter_by_dabg = 'yes'
m_cutoff = m_cutoff = math.log(float(GE_fold_cutoffs),2); p_cutoff = float(GE_pvalue_cutoffs); ptype_to_use = GE_ptype
if array_type=="3'array":
platform_description = "gene-level"
else:
platform_description = array_type
print "Beginning to process the",species,platform_description,'dataset'
process_custom = 'no'
if array_type == "custom": ### Keep this code for now, even though not currently used
import_dir = '/AltDatabase/affymetrix/custom'
dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
for affy_data in dir_list: #loop through each file in the directory to output results
affy_data_dir = 'AltDatabase/affymetrix/custom/'+affy_data
custom_array_db = parse_custom_annotations(affy_data_dir)
array_type = a; process_custom = 'yes'
if array_type == "AltMouse":
print "Processing AltMouse splicing data"
original_probeset_db,constitutive_db = importAltMerge('basic')
probe_annotation_file = "AltDatabase/"+species+'/'+ array_type+'/'+array_type+"_annotations.txt"
original_annotate_db = import_annotations(probe_annotation_file)
conventional_array_db = []
elif array_type == "3'array" and 'Ensembl' not in vendor: ### If user supplied IDs are from Ensembl - doesn't matter the vendor
original_vendor = vendor
if 'other:' in vendor:
vendor = string.replace(vendor,'other:','')
process_go='yes';extract_go_names='yes';extract_pathway_names='yes'
probeset_db = []; annotate_db = []
constitutive_db = ""; conventional_array_db = {}
affy_data_dir = 'AltDatabase/affymetrix'
if vendor == 'Affymetrix':
try: conventional_array_db, arrayCode = BuildAffymetrixAssociations.importAffymetrixAnnotations(affy_data_dir,species,process_go,extract_go_names,extract_pathway_names)
except Exception: print 'Error in processing CSV data. Getting this data from GO-Elite annotations instead.'
if vendor == 'Affymetrix' and len(conventional_array_db)>0: use_go='no'
else: use_go = 'yes'
try:
print "Adding additional gene, GO and WikiPathways annotations"
conventional_array_db = BuildAffymetrixAssociations.getUIDAnnotationsFromGOElite(conventional_array_db,species,vendor,use_go)
except Exception: print "Additional annotation import failed"
print len(conventional_array_db), "Array IDs with annotations from",vendor,"annotation files imported."
vendor = original_vendor
elif array_type != "AltMouse":
probeset_db = []; annotate_db = []; constitutive_db = []; conventional_array_db = []
### The below function gathers GO annotations from the GO-Elite database (not Affymetrix as the module name implies)
conventional_array_db = BuildAffymetrixAssociations.getEnsemblAnnotationsFromGOElite(species)
if 'Ensembl' in vendor:
annotate_db = importGeneAnnotations(species) ### populate annotate_db - mimicking export structure of exon array
original_platform = array_type
global expr_threshold; global dabg_pval; global gene_exp_threshold; global gene_rpkm_threshold; dabg_pval = dabg_p
altanalyze_files = []; datasets_with_all_necessary_files=0
for dataset in exp_file_location_db:
experiment_name = string.replace(dataset,'exp.',''); experiment_name = string.replace(experiment_name,'.txt','')
fl = exp_file_location_db[dataset]
expr_input_dir = fl.ExpFile()
stats_input_dir = fl.StatsFile()
expr_group_dir = fl.GroupsFile()
comp_group_dir = fl.CompsFile()
try: batch_effects = fl.BatchEffectRemoval()
except Exception: batch_effects = 'NA'
try: norm = fl.FeatureNormalization()
except Exception: norm = 'NA'
try: probability_statistic = fl.ProbabilityStatistic()
except Exception: probability_statistic = 'unpaired t-test'
try: gene_exp_threshold = fl.GeneExpThreshold()
except Exception: gene_exp_threshold = 0
try: gene_rpkm_threshold = fl.RPKMThreshold()
except Exception: gene_rpkm_threshold = 0
if expression_data_format == 'log':
try: expr_threshold = math.log(float(expression_threshold),2)
except Exception: expr_threshold = 0 ### Applies to RNASeq datasets
else:
try: expr_threshold = float(expression_threshold)
except Exception: expr_threshold = 0
residuals_input_dir = string.replace(expr_input_dir,'exp.','residuals.')
root_dir = fl.RootDir()
datasets_with_all_necessary_files +=1
checkArrayHeaders(expr_input_dir,expr_group_dir)
expression_dataset_output_dir = root_dir+"ExpressionOutput/"
if batch_effects == 'yes':
try:
from stats_scripts import combat
combat.runPyCombat(fl)
except Exception:
print_out = 'Batch effect removal analysis (py-combat) failed due to an uknown error:'
print traceback.format_exc()
try: UI.WarningWindow(print_out,'Critical Error - Exiting Program!!!'); root.destroy(); sys.exit()
except Exception: print print_out; sys.exit()
if array_type != "3'array": #array_type != 'AltMouse' and
try: probeset_db,annotate_db,comparison_filename_list = ExonArray.getAnnotations(fl,array_type,dabg_p,expression_threshold,data_source,vendor,constitutive_source,species,avg_all_for_ss,filter_by_dabg,perform_alt_analysis,expression_data_format)
except Exception, e:
print traceback.format_exc()
print_out = 'Error ecountered for the '+species+', '+array_type+' dataset. Check to ensure that:\n(1) the correct platform and species were selected and\n(2) some expression values are present in ExpressionInput/exp.YourDataset.txt'
try: UI.WarningWindow(print_out,'Critical Error - Exiting Program!!!'); root.destroy(); sys.exit()
except Exception: print print_out; sys.exit()
if array_type != 'AltMouse': expr_input_dir = expr_input_dir[:-4]+'-steady-state.txt'
else: probeset_db = original_probeset_db; annotate_db = original_annotate_db
for file in comparison_filename_list: altanalyze_files.append(file)
residual_file_status = ExonArray.verifyFile(residuals_input_dir)
### Separate residual file into comparison files for AltAnalyze (if running FIRMA)
if residual_file_status == 'found':
ExonArray.processResiduals(fl,Array_type,Species,perform_alt_analysis)
"""
from build_scripts import ExonArrayEnsemblRules
source_biotype = array_type, root_dir
probeset_db,annotate_db,constitutive_gene_db,splicing_analysis_db = ExonArrayEnsemblRules.getAnnotations('no',constitutive_source,source_biotype,species)
expr_input_dir = expr_input_dir[:-4]+'-steady-state.txt'
"""
if norm == 'RPKM' and array_type == 'RNASeq':
### Separately analyze steady-state counts first, to replace fold changes
counts_expr_dir = string.replace(expr_input_dir,'exp.','counts.')
if 'counts.' not in counts_expr_dir: counts_expr_dir = 'counts.'+counts_expr_dir ### Occurs if 'exp.' not in the filename
count_statistics_db, count_statistics_headers = calculate_expression_measures(counts_expr_dir,expr_group_dir,experiment_name,comp_group_dir,probeset_db,annotate_db)
if count_statistics_headers==None:
### Indicates that the Kallisto expressio file should be used instead of the steady-state RPKM file
expr_input_dir = string.replace(expr_input_dir,'-steady-state.txt','-Kallisto.txt')
expr_group_dir = string.replace(expr_group_dir,'.txt','-Kallisto.txt')
array_type = "3'array"; arrayCode=0; vendor='Ensembl'
experiment_name += '-Kallisto'
fl.setKallistoFile(expr_input_dir)
annotate_db = importGeneAnnotations(species)
conventional_array_db = BuildAffymetrixAssociations.getEnsemblAnnotationsFromGOElite(species)
probeset_db={}
original_platform = 'RNASeq'
calculate_expression_measures(expr_input_dir,expr_group_dir,experiment_name,comp_group_dir,probeset_db,annotate_db)
buildCriterion(GE_fold_cutoffs, p_cutoff, ptype_to_use, root_dir+'/ExpressionOutput/','summary') ###Outputs a summary of the dataset and all comparisons to ExpressionOutput/summary.txt
#except Exception: null=[]
graphic_links = None
if fl.ProducePlots() == 'yes':
graphic_links = visualizeQCPlots(expr_input_dir)
if fl.PerformLineageProfiler() == 'yes':
if graphic_links==None: graphic_links = []
graphic_links = performLineageProfiler(expr_input_dir,graphic_links) ### Correlate gene-level expression values with known cells and tissues
if graphic_links != None:
fl.setGraphicLinks(graphic_links) ### Uses Matplotlib to export QC and clustering plots
annotate_db={}; probeset_db={}; constitutive_db={}; array_fold_db={}; raw_data_comps={}; conventional_array_db=[]
clearObjectsFromMemory(conventional_array_db); conventional_array_db=[]
try: clearObjectsFromMemory(summary_filtering_stats); summary_filtering_stats=[]
except Exception: null=[]
try: clearObjectsFromMemory(array_folds); array_folds=[]
except Exception: null=[]
try: clearObjectsFromMemory(count_statistics_db); count_statistics_db=[]
except Exception: null=[]
#print 'after deleted'; returnLargeGlobalVars()
### Generate the NI file if possible
try: calculateNormalizedIntensities(root_dir,species,array_type,avg_all_for_SS=avg_all_for_ss)
except Exception: pass
if datasets_with_all_necessary_files == 0:
###Thus no files were found with valid inputs for all file types
print 'WARNING....No propperly named datasets were found. ExpressionBuilder requires that there are at least 3 files with the prefixes "exp.", "groups." and "comps.", with the following dataset name being identical with all three files.'
print "...check these file names before running again."
inp = sys.stdin.readline(); sys.exit()
altanalyze_files = unique.unique(altanalyze_files) ###currently not used, since declaring altanalyze_files a global is problematic (not available from ExonArray... could add though)
if (array_type != "3'array" and perform_alt_analysis != 'expression') or original_platform == 'RNASeq':
from stats_scripts import FilterDabg; reload(FilterDabg)
altanalyze_output = FilterDabg.remoteRun(fl,species,original_platform,expression_threshold,filter_method,dabg_p,expression_data_format,altanalyze_files,avg_all_for_ss)
return 'continue',altanalyze_output
else:
end_time = time.time(); time_diff = int(end_time-start_time)
return 'stop'
def verifyFile(filename):
fn=filepath(filename)
try:
for line in open(fn,'rU').xreadlines(): found = 'yes'; break
except Exception: found = 'no'
return found
def verifyExpressionFile(filename):
""" Unlike the above, corrects the expression file path if not found """
fn=filepath(filename)
try:
for line in open(fn,'rU').xreadlines(): break
except Exception:
fn = string.replace(fn,'ExpressionInput/','') ### This file is in the parent path presumably (otherwise can't find it)
return fn
def exportSignatures(db,directory,species):
import gene_associations
export_file = export.findParentDir(directory[:-2]+'.txt')
if 'AltExon' in directory:
export_file+='signatures/AltExon-signatures.txt'
else:
export_file+='signatures/GeneExpression-signatures.txt'
export_object = export.ExportFile(export_file)
if 'AltExon' in directory:
export_object.write('symbol\tentrez\tensembl\tsource\turl\tname\tAltAnalyze-ExonID\tASPIRE|Splicing-Index LogFold\tGenomicLocation\n') ### Header line
else:
export_object.write('symbol\tentrez\tsource\turl\tname\tLogFold\tBH-adjp-value\n') ### Header line
url = 'http://code.google.com/p/altanalyze/wiki/PCBC_C4_compendium'
source = 'AltAnalyze'
sig = 0
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
for filename in gene_conversion_db:
db,input_data_db = gene_conversion_db[filename]
filename = string.replace(filename,'.txt','')
filename = string.replace(filename,'GE.','')
filename = string.replace(filename,'-upregulated','')
for ensembl in db:
sig+=1
try: symbol = gene_to_symbol[ensembl][0]
except Exception: continue
entrezgenes = db[ensembl][0]
entrezgenes = string.split(entrezgenes,'|')
statistics = input_data_db[ensembl][0][1:]
#print [statistics];sys.exit()
for entrez in entrezgenes:
### output format: symbol, entrez, source, url, name
values = string.join([symbol,entrez,ensembl,source,url,filename]+statistics,'\t')+'\n'
export_object.write(values)
export_object.close()
print 'Exported',sig,'signatures to:'
print export_file
def runGOElite(species,directory):
""" Separate pipeline for automating GO-Elite when re-generating criterion - Currently used outside of any pipelines """
mod = 'Ensembl'
pathway_permutations = 'FisherExactTest'
filter_method = 'z-score'
z_threshold = 1.96
p_val_threshold = 0.05
change_threshold = 2
resources_to_analyze = 'local'
returnPathways = 'yes'
root = None
import GO_Elite
directory = string.replace(directory,'ExpressionOutput','')
results_dir = directory
print '\nBeginning to run GO-Elite analysis on all results'
elite_input_dirs = ['regulated']#,'upregulated','downregulated','MarkerFinder'] ### 'AltExon' Run GO-Elite multiple times to ensure heatmaps are useful and to better organize results
for elite_dir in elite_input_dirs:
if elite_dir == 'AltExon': returnPathways = 'no'
else: returnPathways = 'yes'
file_dirs = results_dir+'GO-Elite/'+elite_dir,results_dir+'GO-Elite/denominator',results_dir+'GO-Elite/'+elite_dir
input_dir = results_dir+'GO-Elite/'+elite_dir
try: input_files = read_directory(input_dir) ### Are there any files to analyze?
except Exception: input_files = []
if len(input_files)>0:
variables = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,root
try: GO_Elite.remoteAnalysis(variables,'non-UI',Multi=mlp)
except Exception: pass
def filterDatasetFile(main_output_folder):
global array_folds; global expression_dataset_output_dir
expression_dataset_output_dir = string.replace(main_output_folder,'GO-Elite','ExpressionOutput/')
dir_list = read_directory(expression_dataset_output_dir[:-1])
for filename in dir_list:
if 'DATASET-' in filename:
dataset_fn=filepath(expression_dataset_output_dir+filename)
x=0
for line in open(dataset_fn,'rU').xreadlines():
data = cleanUpLine(line); t = string.split(data,'\t')
if x==0: headers = t; break
### Get a list of column header titles to include
fn=filepath('Config/DATASET-headers.txt')
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line); t = string.split(data,'\t')
columns_to_include = t
### Filter statistics based on user-defined thresholds as input for GO-Elite analysis
criterion_db={}; denominator_geneids={}; index = 0; indexes=[]; avg_indexes=[]
for column in headers:
if 'avg-' in column: avg_indexes.append(index)
elif 'log_fold-' in column: indexes.append(index)
elif 'fold-' in column: indexes.append(index)
elif 'rawp-' in column: indexes.append(index)
elif 'adjp-' in column: indexes.append(index)
elif 'ANOVA' in column: indexes.append(index)
elif column in columns_to_include: indexes.append(index)
index+=1
###Export out the filtered file
dataset_fn_filtered = string.replace(dataset_fn,'.txt','-abreviated.txt')
dataset_fn_filtered = string.replace(dataset_fn_filtered,'ExpressionOutput','ExpressionOutput/filtered')
data_filter = export.ExportFile(dataset_fn_filtered)
firstLine=True
for line in open(dataset_fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine=False
if 'maximum sample read count' in t: ### For RNA-Seq
h=[]
for l in t:
if 'avg-' in l: h.append(string.replace(l,'avg-','avg_RPKM-'))
else: h.append(l)
t=h
values = map(lambda x: t[x], indexes)
avg_values = map(lambda x: t[x], avg_indexes) ### Specifically grab only the average-expression values (not anything else)
values = string.join(values+avg_values,'\t')+'\n'
data_filter.write(values)
data_filter.close()
def importProbesetRegions(species,platform):
filename = 'AltDatabase/'+species+'/'+platform+'/'+species+'_Ensembl_probesets.txt'
fn=filepath(filename)
region_db = {}
firstRow=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow: firstRow = False
else:
probeset = t[0]
gene = t[2]
region = gene+':'+string.replace(t[12],'-','.')
region_db[region] = probeset
return region_db
def buildAltExonClusterInputs(input_folder,species,platform,dataType='AltExonConfirmed'):
alternative_exon_db={}
dir_list = read_directory(input_folder)
if platform == 'junction':
region_db = importProbesetRegions(species,platform)
for filename in dir_list:
if '.txt' in filename:
proceed = True
if platform == 'RNASeq':
if 'ASPIRE' in filename or 'egress' in filename: proceed = False ### Need to create a special analysis just for reciprocal junctions
if proceed: ### Don't include splicing-index results for RNA-Seq
fn=filepath(input_folder+filename)
x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: x=1
else:
if platform == 'junction' and 'GO-Elite' in input_folder:
altExon = t[2]
if altExon in region_db:
altExon = region_db[altExon]
elif 'splicing-index' in filename:
altExon = t[-1]
elif 'ASPIRE' in filename or 'egress' in filename:
altExon = t[-1]
altExon = string.split(altExon,' | ')
else:
altExon = t[2]
#if float(t[3])<0.001:
alternative_exon_db[altExon]=None
print len(alternative_exon_db), 'alternative exon IDs imported'
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
input_folder = string.split(input_folder,'GO-Elite')[0]+'AltResults/RawSpliceData/'+species+'/splicing-index/'
dir_list = read_directory(input_folder)
exported_IDs=0
added={}
for filename in dir_list:
if '.txt' in filename and ('_average' not in filename) or len(dir_list)==1: ### We only want the group-comparison SI file
export_dir = string.split(input_folder,'RawSpliceData')[0]+'Clustering/'+dataType+'-'+filename
export_data = export.ExportFile(export_dir)
fn=filepath(input_folder+filename)
x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
headers = t[2:] ### first two columsn are gene and ExonID
export_data.write(string.join(headers,'\t')+'\n') ### write header row
x=1
else:
if platform != 'RNASeq':
if 'ENS' in t[0]: #ENSG human
gene = t[0]; altExon = t[2]
else:
altExon = t[0]
else:
gene = t[0]; altExon = t[2]
if ';' in altExon:
altExon1, altExon2 = string.split(altExon,';')
altExons = [altExon1,gene+':'+altExon2]
else:
altExons = [altExon]
for altExon in altExons:
if altExon in alternative_exon_db and altExon not in added:
added[altExon]=[]
#values = map(lambda x: float(x)*-1, t[3:]) #reverse the fold for cluster visualization
values = map(lambda x: float(x), t[3:])
#print len(headers),len(values);sys.exit()
avg = statistics.avg(values)
log_folds = map(lambda x: str(x-avg), values) ### calculate log folds and make these strings
values = string.join([altExon]+log_folds,'\t')+'\n' ### [t[3]]+ before log_folds?
if gene in gene_to_symbol: symbol = gene_to_symbol[gene][0]+" "
else: symbol = ''
export_data.write(symbol+values)
exported_IDs+=1
print exported_IDs, 'exported ID values for clustering'
export_data.close()
return export_dir, exported_IDs
def exportHeatmap(filename,useHOPACH=True, color_gradient='red_black_sky',normalize=False,columnMethod='average',size=0,graphics=[]):
from visualization_scripts import clustering
row_method = 'weighted'; row_metric = 'cosine'; column_method = 'average'; column_metric = 'euclidean'; transpose = False
try:
if columnMethod !=None:
column_method = columnMethod
if size < 7000:
graphics = clustering.runHCexplicit(filename, graphics, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=normalize)
except Exception:
print 'Clustering failed for:',filename
return graphics
def meanCenterPSI(filename):
firstLine=True
output_file = filename[:-4]+'-cluster.txt'
export_obj = export.ExportFile(output_file)
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
export_obj.write(line)
firstLine = False
else:
try:
avg = averageWithNulls(t[1:])
values = map(lambda x: replaceNulls(x,avg), t[1:])
export_obj.write(string.join([t[0]]+values,'\t')+'\n')
except Exception: pass
return output_file
def filterJunctionExpression(filename,minPercentPresent=None):
output_file = filename[:-4]+'-filter.txt'
export_obj = export.ExportFile(output_file)
filtered_lines = []; filtered_size=0; filtered_size_stringent=0
firstLine = True
size=0; imported_num=0
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
export_obj.write(line)
firstLine = False
else:
imported_num+=1
if '_' in t[0] or '-ENS' in t[0] or '-' not in t[0]:
pass
else:
try: a,b = string.split(t[0],'|')
except Exception:
try: n,a,b = string.split(t[0],' ')
except Exception: continue
if '-' in a and '-' in b:
vals = map(lambda x: countNulls(x),t[1:])
percent_present = sum(vals)/(len(vals)*1.00)
if percent_present>0.1:
filtered_lines.append([percent_present,line])
size+=1
if percent_present>0.5:
filtered_size+=1
if percent_present>0.85:
filtered_size_stringent+=1
percent_imported = ((1.00*size)/imported_num)
if minPercentPresent!=None:
size=0
filtered_lines.sort(); filtered_lines.reverse()
for (percent_present,line) in filtered_lines:
if percent_present>minPercentPresent:
export_obj.write(line)
size+=1
elif size < 8000 and percent_imported<0.5: ### The filtered is at least 30% the size of the imported
for (percent_present,line) in filtered_lines:
export_obj.write(line)
else:
size=0
to_import = int(imported_num*0.3)
#print "Filtering down to", to_import, "entries."
filtered_lines.sort(); filtered_lines.reverse()
for (percent_present,line) in filtered_lines:
if filtered_size_stringent>8000:
if percent_present>0.95:
export_obj.write(line)
size+=1
elif filtered_size>8000:
if percent_present>0.85:
export_obj.write(line)
size+=1
else:
if percent_present>0.5:
export_obj.write(line)
size+=1
print 'Filtered RI-PSI entries to',size
export_obj.close()
return output_file,size
def countNulls(val):
if float(val) == 0:
return 0
else: return 1
def calculateNormalizedIntensities(root_dir, species, array_type, avg_all_for_SS = 'no', probeset_type = 'core', analysis_type = 'processed', expFile = False):
""" Since it is a time-consuming step that is needed for visualizing SI values for exons, it is faster
to efficiently calculate NI values in advance. """
if array_type == 'gene': platform = 'GeneArray'
elif array_type == 'exon': platform = 'ExonArray'
elif array_type == 'junction': platform = 'JunctionArray'
else: platform = array_type
#alt_exon_exp_dir = root_dir+'/AltExpression/FullDatasets/'+platform+'/'+species ### This file doesn't exist if only one pairwise comparison group
alt_exon_exp_dir = root_dir+'/ExpressionInput/' ### This file doesn't exist if only one pairwise comparison group
dir_list = read_directory(alt_exon_exp_dir)
for file in dir_list:
if '.txt' in file and 'exp.' in file and 'steady' not in file:
selected_file = file[4:]
fn=filepath(alt_exon_exp_dir+'/'+file)
#sample_group_db = simplerGroupImport(string.replace(fn,'exp.','groups.'))
if analysis_type == 'raw':
### Create a filtered exon and junction expression file outside the typical workflow
import RNASeq
exp_threshold=5; rpkm_threshold=5
expressed_uids_rpkm = RNASeq.getMaxCounts(expFile,rpkm_threshold)
expressed_uids_counts = RNASeq.getMaxCounts(string.replace(expFile,'exp.','counts.'),exp_threshold)
expressed_uids = expressed_uids_rpkm.viewkeys() & expressed_uids_counts.viewkeys() ### common
fn = root_dir+'/AltExpression/FilteredDataset/'+platform+'/'+species+'/'+ export.findFilename(expFile)### This file doesn't exist if only one pairwise comparison group
expressed_uids_rpkm = RNASeq.getMaxCounts(expFile,rpkm_threshold,filterExport=expressed_uids,filterExportDir=fn)
### Get the gene values
gene_db={};
if analysis_type == 'raw':
### Get these directly from the steady-state file
exp_dir=string.replace(expFile,'.txt','-steady-state.txt')
firstLine = True
low_diff_exp_genes={}
for line in open(exp_dir,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine = False
ge_header = t
else:
values = map(lambda x: math.log(float(x),2), t[1:])
gene_db[t[0]]=values
elif array_type == 'RNASeq':
firstLine=True
fn_steady = fn[:-4]+'-steady-state.txt'
for line in open(fn_steady,'rU').xreadlines():
if firstLine: firstLine = False
elif ':' in line[:50]: pass
else:
data = cleanUpLine(line)
t = string.split(data,'\t')
try:
values = map(lambda x: float(x), t[1:])
values = map(lambda x: math.log(x,2),values)
gene_db[t[0]]=values
except Exception: pass
else:
import AltAnalyze
exon_db, constitutive_probeset_db = AltAnalyze.importSplicingAnnotations(array_type,species,probeset_type,avg_all_for_SS,root_dir)
gene_db={}; firstLine=True
for line in open(fn,'rU').xreadlines():
if firstLine: firstLine = False
else:
data = cleanUpLine(line)
t = string.split(data,'\t')
if t[0] in constitutive_probeset_db:
gene = constitutive_probeset_db[t[0]]
values = map(lambda x: float(x), t[1:])
try: gene_db[gene].append(values) ### combine these
except Exception: gene_db[gene] = [values]
for gene in gene_db:
#print gene, gene_db[gene]
constitutive_values = zip(*gene_db[gene]) ### combine values from multiple lists into lists of lists
#print constitutive_values
values = map(lambda x: Average(x), constitutive_values)
#print values; sys.exit()
gene_db[gene] = values
if len(gene_db)>0:
alt_exon_output_dir = root_dir+'/AltResults/RawSpliceData/'+species+'/splicing-index/'+selected_file
if analysis_type == 'raw':
alt_exon_output_dir = string.replace(alt_exon_output_dir,'RawSpliceData','RawSpliceDataTemp')
export_obj = export.ExportFile(alt_exon_output_dir)
print 'Exporting exon-level Normalized Intensity file to:',alt_exon_output_dir
print len(gene_db),'genes with data imported'
firstLine=True
exon_entries=0; saved_entries=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine = False
values = string.join(['Gene','ExonID','probesetID']+t[1:],'\t')+'\n'
export_obj.write(values)
else:
if (':' in t[0]) or array_type != 'RNASeq':
feature_values = map(lambda x: float(x), t[1:])
if analysis_type == 'raw' or array_type == 'RNASeq':
feature_values = map(lambda x: math.log(x,2), feature_values)
if array_type == 'RNASeq':
gene = string.split(t[0],':')[0]
else:
try: gene = exon_db[t[0]].GeneID()
except Exception: gene = None
if gene != None:
exon_entries+=1
if gene in gene_db:
gene_values = gene_db[gene]
if ('-' not in t[0]) or analysis_type == 'raw':
NI_values = [logratio(value) for value in zip(*[feature_values,gene_values])]
NI_values = map(lambda x: str(x), NI_values)
NI_values = string.join([gene,'NA',t[0]]+NI_values,'\t')+'\n'
export_obj.write(NI_values)
saved_entries+=1
export_obj.close()
print exon_entries, 'found with',saved_entries,'entries normalized.'
return alt_exon_output_dir
def compareRawJunctionExpression(root_dir,platform,species,critical_exon_db,expFile,min_events=0,med_events=0):
expFile = exportSorted(expFile, 0) ### Sort the expression file
from scipy import stats
exported=[]
retained_introns=[]
features_examined={}
junction_locations = {}
critical_exon_gene_db={}
critical_junction_pair_db={}
psi_db={}
#min_events = 4; med_events = 13
#min_events = 0; med_events = 0
print 'Begining reciprocal junction/intron retention unbiased analyses (min_events=%d, med_events=%d)' % (min_events,med_events)
for altexon in critical_exon_db:
gene = string.split(altexon,':')[0]
inclusion_list,exclusion_list = critical_exon_db[altexon]
try:
critical_exon_gene_db[gene].append(critical_exon_db[altexon])
except Exception:
critical_exon_gene_db[gene] = [critical_exon_db[altexon]]
for i in inclusion_list:
for e in exclusion_list:
try: critical_junction_pair_db[i,e].append(altexon)
except Exception: critical_junction_pair_db[i,e] = [altexon]
export_dir = root_dir+'AltResults/AlternativeOutput/'+species+'_'+platform+'_top_alt_junctions-PSI.txt'
export_data = export.ExportFile(export_dir)
clust_export_dir = root_dir+'AltResults/AlternativeOutput/'+species+'_'+platform+'_top_alt_junctions-PSI-clust.txt'
clust_export_data = export.ExportFile(clust_export_dir)
### Included a new file with cluster and junction information
clusterID_export_dir=root_dir+'AltResults/AlternativeOutput/'+species+'_'+platform+'_top_alt_junctions-PSI-ClusterIDs.txt'
clusterID_export_data = export.ExportFile(clusterID_export_dir)
def ratio(values):
#print values
#return float(values[0]+1)/(values[1]+1)
try:return float(values[0])/(values[1])
except Exception: return 0.0
def dPSI(incl_exp,total_exp,indexes,min_events):
indexes.sort()
incl_filt = map(lambda x: incl_exp[x],indexes) ### Only consider expressed cells
total_filt = map(lambda x: total_exp[x],indexes)
dpsi_values = [ratio(value) for value in zip(*[incl_filt,total_filt])]
dpsi_values.sort()
if len(dpsi_values)==1:
return dpsi_values[0]
elif min_events == 0:
return dpsi_values[-1]-dpsi_values[0]
else: return dpsi_values[-1*min_events]-dpsi_values[min_events]
#return max(dpsi_values)-min(dpsi_values) ### percent change in isoform expression
def getIndexes(events,min_exp_thresh):
i=0
indexes=[]
for e in events:
if e>min_exp_thresh: indexes.append(i) ### minimum expression value (changed from 5 to 10 8/5/2016)
i+=1
return indexes
def diff(values,demon,num):
return values[num]-values[demon]
def diffCompare(incl_exp,excl_exp,incl,excl):
if max(incl_exp)>max(excl_exp): denom = 1;num = 0
else: denom = 0; num=1
diff_values = [diff(value,denom,num) for value in zip(*[incl_exp,excl_exp])]
if min(diff_values)==0 and ('-' not in incl or '-' not in excl): ### subtract out the overlap if all reads in the inclusion exon are confounded by the junction 1nt reads
if denom == 1:
feature_exp_db[incl] = diff_values
else:
feature_exp_db[excl] = diff_values
return True
else: return False
def junctionComparisonMethod(incl,excl):
useAllJunctionsForExcl=True
#min_exp = 4; med_exp = 9 #min_exp = 19; med_exp = 19
min_exp=9;med_exp=19
incl_exp = feature_exp_db[incl]
excl_exp = feature_exp_db[excl]
if useAllJunctionsForExcl:
gene = string.split(excl,':')[0]
try: gene_exp = gene_junction_denom[gene]
except Exception:
try:
gene = string.split(incl,':')[0]
gene_exp = gene_junction_denom[gene]
except Exception: gene_exp=[]
rho,p = stats.pearsonr(incl_exp,excl_exp)
### See if the data for one feature is confounded by another
### The example is exon-inclusion with 1nt overlap getting the associated reads from BedTools
status = diffCompare(incl_exp,excl_exp,incl,excl)
if status: ### update the difference generated values
excl_exp = feature_exp_db[excl]
rho,p = stats.pearsonr(incl_exp,excl_exp)
#if 'ENSMUSG00000009350:E14.2_87617106-E15.1' in incl: print feature_exp_db[incl],'a'
num_incl_events = sum(i > min_exp for i in incl_exp)
num_excl_events = sum(i > min_exp for i in excl_exp)
combined = [sum(value) for value in zip(*[incl_exp,excl_exp])]
total_number_junctions = sum(i > min_exp for i in combined)
### Calculate a delta PSI value for each expressed cell
combined = [sum(value) for value in zip(*[incl_exp,excl_exp])]
exp_indexes = getIndexes(combined,med_exp)
try: dpsi = dPSI(incl_exp,combined,exp_indexes,min_events)
except Exception: dpsi = 1
dpsi_values = [ratio(value) for value in zip(*[incl_exp,combined])]
dpsi_values = nullReplace(dpsi_values,combined,min_exp)
psi_db[incl,excl] = dpsi_values ### optionally output these at the end
#if incl == 'ENSMUSG00000019505:E3.2_62365772-E3.6_62366457' and excl == 'ENSMUSG00000029162:E7.2-E7.4':
#dpsi2 = dPSI(excl_exp,combined,exp_indexes)
#if 'ENSMUSG00000009350:E14.2_87617106-E15.1' in incl: print feature_exp_db[incl],'b'
if '-' in incl and '-' in excl:
try: max_ratio = expressionRatio(incl_exp,excl_exp,num_incl_events,num_excl_events) # Checks to see if the predominant isoform is expressed at significantly higher levels
except Exception: max_ratio = 0
try:
max_gene_ratio = max([ratio(value) for value in zip(*[incl_exp,gene_exp])])
except Exception: max_gene_ratio = 0
else: max_ratio = 1; max_gene_ratio = 0
#if 'ENSMUSG00000009350:E14.2_87617106-E15.1' in incl: print feature_exp_db[incl],'c'
#if num_incl_events > 15 and num_excl_events > 15 and total_number_junctions>30 and max_ratio>0.5: ### ensures high expression of the minor isoform
#if ((num_incl_events > 15 and num_excl_events > 7) or (num_incl_events > 7 and num_excl_events > 15)) and total_number_junctions>20 and max_ratio>0.3:
#if 'ENSG00000100650' in incl: print incl,excl,max_ratio,num_incl_events,num_excl_events,dpsi,rho
if ((num_incl_events > med_events and num_excl_events > min_events) or (num_incl_events > min_events and num_excl_events > med_events)) and total_number_junctions>(min_events*2) and max_ratio>0.1: ### ensures high expression of the minor isoform
#print rho
if dpsi > 0.15:
return max_ratio,num_incl_events,num_excl_events,dpsi,rho,max_gene_ratio,True
else:
return max_ratio,num_incl_events,num_excl_events,dpsi,rho,max_gene_ratio,False
else:
return max_ratio,num_incl_events,num_excl_events,dpsi,rho,max_gene_ratio,False
def intronComparisonMethod(query):
upstream_exon = string.replace(query,'I','E')
if '_' in query:
intronic_region = string.split(query,'_')[0] ### Unclear why this is needed, but sometimes only an _ intronic region exists for the intron
if intronic_region not in junction_locations: ### Ensures we are not using this instead of a real intron region (e.g., I2.1_1234 and I2.1)
upstream_exon = string.replace(intronic_region,'I','E')
try:
pos1,pos2 = junction_locations[upstream_exon][0]
upstream_exon_len = abs(pos2-pos1)
except Exception: upstream_exon_len = None
downstream_exon = getDownstreamExon(upstream_exon)
try:
pos1,pos2 = junction_locations[downstream_exon][0]
downstream_exon_len = abs(pos2-pos1)
except Exception: downstream_exon_len=None
pos1,pos2 = junction_locations[query][0]
intron_len = abs(pos2-pos1)
try:
up_rpk = max(feature_exp_db[upstream_exon])/float(upstream_exon_len)
if downstream_exon_len!=None:
down_rpk = max(feature_exp_db[downstream_exon])/float(downstream_exon_len)
adjacent_rpk = max([up_rpk,down_rpk]) ### get the most conservative estimate
adjacent_rpk = up_rpk
intron_rel_exp = (max(feature_exp_db[query])/float(intron_len))/adjacent_rpk
return intron_rel_exp
except Exception:
return 0
def compareJunctionExpression(gene):
regulated_junctions={}
inclusion_max_psi={}
try: symbol,description = gene_annotations[gene]
except Exception: symbol='';description=''
if gene in critical_exon_gene_db:
critical_exon_list = critical_exon_gene_db[gene]
critical_exon_list = unique.unique(critical_exon_list)
for (inclusion_list,exclusion_list) in critical_exon_list:
inclusion_list = unique.unique(inclusion_list)
exclusion_list = unique.unique(exclusion_list)
for incl in inclusion_list:
for excl in exclusion_list:
if excl != incl:
pair = [incl,excl]; pair.sort()
features_examined[incl]=[]
try:
max_ratio,num_incl_events,num_excl_events,dpsi,rho,max_all_psi,proceed = junctionComparisonMethod(incl,excl)
inclusion_max_psi[incl] = max_all_psi
#if 'ENSMUSG00000027680:E21.1-E22.1' in incl: print incl, excl,max_ratio,num_incl_events,num_excl_events,dpsi,rho,proceed
if proceed:
"""if max_ratio<0.1:
if num_excl_events > num_incl_events:
print max_ratio
print max(incl_exp)
print statistics.median(excl_exp)
print incl_exp
print excl_exp;sys.exit()"""
#if 'ENSMUSG00000009350:E14.2_87617106-E15.1' in incl: print feature_exp_db[incl]
try:
altexons = unique.unique(critical_junction_pair_db[incl,excl])
except:
altexons=[]
altexons = string.join(altexons,'|')
if num_excl_events > num_incl_events:
#print max_ratio, '\t',gene
regulated_junctions[incl]=rho,excl,num_incl_events,num_excl_events,'incl',dpsi,rho,max_ratio,altexons
else:
#print max_ratio, '\t',gene
regulated_junctions[excl]=rho,incl,num_excl_events,num_incl_events,'excl',dpsi,rho,max_ratio,altexons
#if 'ENSMUSG00000009350:E14.2_87617106-E15.1' in incl: print feature_exp_db[incl]
#if rho < -0.3:
#print incl, excl, rho
#print incl_exp
#print excl_exp
#print sum(i > 5 for i in incl_exp)
#print sum(i > 5 for i in excl_exp)
except Exception: pass
#lower_index = int(len(rpkms)*0.2)
#upper_index = len(rpkms)-lower_index
for reg_junction in regulated_junctions:
ref_junction = regulated_junctions[reg_junction][1]
altexons = regulated_junctions[reg_junction][-1]
max_ratio = regulated_junctions[reg_junction][-2]
rho = regulated_junctions[reg_junction][-3]
dpsi = regulated_junctions[reg_junction][-4]
#if 'ENSMUSG00000009350:E14.2_87617106-E15.1' in incl: print feature_exp_db[incl]
### Perform a sanity check for junction comparisons
reg_pos,reg_loc = junction_locations[reg_junction]
ref_pos,ref_loc = junction_locations[ref_junction]
positions = reg_pos+ref_pos
positions.sort()
values = map(lambda x: str(x+1),feature_exp_db[reg_junction])
try: values = psi_db[reg_junction,ref_junction]
except Exception: values = psi_db[ref_junction,reg_junction]; reg_junction,ref_junction = ref_junction,reg_junction
try: max_incl_psi = str(inclusion_max_psi[reg_junction])
except Exception: max_incl_psi = '0'
if reg_pos == positions[:2] or reg_pos == positions[-2:]:
#print 'Impropper junctions excluded',reg_junction,ref_junction,positions,reg_pos
pass
elif ('-' not in reg_junction or '-' not in ref_junction) and platform != 'junction': ### Possible retained intron
if '-' not in reg_junction: query = reg_junction
elif '-' not in ref_junction: query = ref_junction
if 'I' in query:
intron_rel_exp = intronComparisonMethod(query)
if intron_rel_exp>0.15:
"""
print upstream_exon, query, intron_len, upstream_exon_len,(max(feature_exp_db[query])/float(intron_len)),(max(feature_exp_db[upstream_exon])/float(upstream_exon_len))
print max(feature_exp_db[query])
print junction_locations[query]
print max(feature_exp_db[upstream_exon])
print junction_locations[upstream_exon]
print intron_rel_exp
"""
if platform == 'junction':
try: reg_junction = probeset_junction_db[reg_junction] + ' ' +reg_junction
except Exception: pass
try: ref_junction = probeset_junction_db[ref_junction] + ' ' +ref_junction
except Exception: pass
export_data.write(string.join([symbol,description,reg_junction,ref_junction,altexons,str(max_ratio),str(dpsi),str(rho),max_incl_psi,reg_loc+'|'+ref_loc,'intron-retained']+values,'\t')+'\n')
avg = averageWithNulls(values)
values = map(lambda x: replaceNulls(x,avg), values)
clust_export_data.write(string.join([symbol+':'+reg_junction+'|'+ref_junction]+values,'\t')+'\n')
retained_introns.append(reg_junction)
retained_introns.append(ref_junction)
else:
try: avg = averageWithNulls(values)
except Exception: continue
if platform == 'junction':
try: reg_junction = probeset_junction_db[reg_junction] + ' ' +reg_junction
except Exception: pass
try: ref_junction = probeset_junction_db[ref_junction] + ' ' +ref_junction
except Exception: pass
export_data.write(string.join([symbol,description,reg_junction,ref_junction,altexons,str(max_ratio),str(dpsi),str(rho),max_incl_psi,reg_loc+'|'+ref_loc,'junctions']+values,'\t')+'\n')
values = map(lambda x: replaceNulls(x,avg), values)
clust_export_data.write(string.join([symbol+':'+reg_junction+'|'+ref_junction]+values,'\t')+'\n')
exported.append(reg_junction)
exported.append(ref_junction)
### Predict novel undetected events from above
junctions=[]; other=[]; features=[]; feature_pos=[]; loc_db={}; coord_db={}; junctions_to_compare=[]; regulated_junctions={};inclusion_max_psi={}
#max_ratio,proceed = junctionComparisonMethod(incl,excl)
for feature in feature_exp_db:
### The below code is for overlapping junctions not found from the above analysis (could include exons and junctions)
if feature not in exported and feature not in retained_introns:
if '-' in feature and platform != 'junction':
junctions.append(feature)
else:
other.append(feature)
features.append(feature)
try:
pos1,pos2 = junction_locations[feature][0]
feature_pos.append(pos1); feature_pos.append(pos2)
try:loc_db[pos1].append(feature)
except Exception: loc_db[pos1] = [feature]
try:loc_db[pos2].append(feature)
except Exception: loc_db[pos2] = [feature]
try: coord_db[pos1,pos2].append(feature)
except Exception: coord_db[pos1,pos2] = [feature]
except Exception: pass ### occurs for junction arrays if the probeset ID is not in the database
feature_pos = unique.unique(feature_pos); feature_pos.sort() ### These are the unique positions sorted
overlapping_features=[]
additional_possible_retained_introns=[] ### catch some funky intron retention events
### Get initial list of overlapping features
for feature in features: ### e.g., some junction
try:
pos1,pos2 = junction_locations[feature][0] ### coordinates of that junction
i1 = feature_pos.index(pos1) ### index position of the junctions
i2 = feature_pos.index(pos2)
#print feature, i1, i2, pos1, pos2
if (i1-i2) != 1:
overlapping_features.append(feature)
if 'I' in feature and '_' in feature:
possible_intron = string.split(feature,'_')[0]
if possible_intron not in features:
additional_possible_retained_introns.append((i1,i2,feature))
except Exception:
pass
for feature in overlapping_features:
#if feature not in features_examined: ### Remove this to allow for other reasonable junction or junction intron pairs that were not significant above
### get overlapping feature pairs
pos1,pos2 = junction_locations[feature][0]
i1 = feature_pos.index(pos1)
i2 = feature_pos.index(pos2)
### Include a search for funky intron retention events (needed due to some weird intron retention issue)
for (in1,in2,f2) in additional_possible_retained_introns:
if i1<=in1 and i2>=in2 and feature!=f2:
junctions_to_compare.append([feature,f2])
for i in range(i1+1,i2):
overlapping = loc_db[feature_pos[i]]
for o in overlapping:
if o not in features_examined and '-' in o and '-' in feature and platform != 'junction':
#print feature, o
#print junction_locations[feature][0]
#print junction_locations[o][0]
junctions_to_compare.append([feature,o])
#print 'junctions_to_compare:',junctions_to_compare
### Since this is the same coordinates, should be finding intron retention pairs
for coord in coord_db:
features = unique.unique(coord_db[coord])
if len(features)>1:
for f in features:
for g in features:
if g!=f:
fs = [g,f]; fs.sort()
if g not in exported and f not in exported:
if g not in retained_introns and f not in retained_introns:
junctions_to_compare.append(fs)
unique.unique(junctions_to_compare)
for (incl,excl) in junctions_to_compare: #Not really incl, excl, just features
max_ratio,num_incl_events,num_excl_events,dpsi,rho,max_all_psi,proceed = junctionComparisonMethod(incl,excl)
inclusion_max_psi[incl] = max_all_psi
#if 'ENSG00000100650' in incl: print incl,excl, max_ratio, proceed, rho, num_incl_events, num_excl_events, 'k'
if proceed:
altexons = ''
if num_excl_events > num_incl_events:
#print max_ratio, '\t',gene
regulated_junctions[incl]=rho,excl,num_incl_events,num_excl_events,'incl',dpsi,rho,max_ratio,altexons
else:
#print max_ratio, '\t',gene
regulated_junctions[excl]=rho,incl,num_excl_events,num_incl_events,'excl',dpsi,rho,max_ratio,altexons
for reg_junction in regulated_junctions:
ref_junction = regulated_junctions[reg_junction][1]
altexons = regulated_junctions[reg_junction][-1]
max_ratio = regulated_junctions[reg_junction][-2]
rho = regulated_junctions[reg_junction][-3]
dpsi = regulated_junctions[reg_junction][-4]
### Perform a sanity check for junction comparisons
reg_pos,reg_loc = junction_locations[reg_junction]
ref_pos,ref_loc = junction_locations[ref_junction]
positions = reg_pos+ref_pos
positions.sort()
values = map(lambda x: str(x+1),feature_exp_db[reg_junction])
try: values = psi_db[reg_junction,ref_junction]
except Exception: values = psi_db[ref_junction,reg_junction]; reg_junction,ref_junction = ref_junction,reg_junction
try: max_incl_psi = str(inclusion_max_psi[reg_junction])
except Exception: max_incl_psi = '0'
if ('-' not in reg_junction or '-' not in ref_junction) and platform != 'junction': ### Possible retained intron
if '-' not in reg_junction: query = reg_junction
elif '-' not in ref_junction: query = ref_junction
intron_rel_exp = intronComparisonMethod(query)
if intron_rel_exp>0.15:
if platform == 'junction':
try: reg_junction = probeset_junction_db[reg_junction] + ' ' +reg_junction
except Exception: pass
try: ref_junction = probeset_junction_db[ref_junction] + ' ' +ref_junction
except Exception: pass
export_data.write(string.join([symbol,description,reg_junction,ref_junction,altexons,str(max_ratio),str(dpsi),str(rho),max_incl_psi,reg_loc+'|'+ref_loc,'exon-retained']+values,'\t')+'\n')
avg = averageWithNulls(values)
values = map(lambda x: replaceNulls(x,avg), values)
clust_export_data.write(string.join([symbol+':'+reg_junction+'|'+ref_junction]+values,'\t')+'\n')
retained_introns.append(reg_junction)
retained_introns.append(ref_junction)
#print query
else:
if platform == 'junction':
try: reg_junction = probeset_junction_db[reg_junction] + ' ' +reg_junction
except Exception: pass
try: ref_junction = probeset_junction_db[ref_junction] + ' ' +ref_junction
except Exception: pass
export_data.write(string.join([symbol,description,reg_junction,ref_junction,altexons,str(max_ratio),str(dpsi),str(rho),'0',reg_loc+'|'+ref_loc,'others']+values,'\t')+'\n')
avg = averageWithNulls(values)
values = map(lambda x: replaceNulls(x,avg), values)
clust_export_data.write(string.join([symbol+':'+reg_junction+'|'+ref_junction]+values,'\t')+'\n')
exported.append(reg_junction)
exported.append(ref_junction)
#print ref_junction,reg_junction
if platform == 'junction' or platform == 'AltMouse':
probeset_gene_db={}
from build_scripts import ExonArrayEnsemblRules
if platform == 'junction':
export_exon_filename = 'AltDatabase/'+species+'/'+platform+'/'+species+'_Ensembl_probesets.txt'
if platform == 'AltMouse':
export_exon_filename = 'AltDatabase/'+species+'/'+platform+'/'+species+'_Ensembl_probesets.txt'
ensembl_probeset_db = ExonArrayEnsemblRules.reimportEnsemblProbesetsForSeqExtraction(export_exon_filename,'only-junctions',{})
for gene in ensembl_probeset_db:
for uid,pos,location in ensembl_probeset_db[gene]:
junction_locations[uid] = pos,location
probeset_gene_db[uid]=gene
def filterByLocalJunctionExp(gene,features):
try: symbol,description = gene_annotations[gene]
except Exception: symbol='';description=''
global cluster_id
global cluster_name
begin_time = time.time()
count=0
junctions_to_compare={}
overlapping_junctions_exp={}
ovelapping_pos={}
existing=[]
overlapping_junctions_test={}
for feature in feature_exp_db:
feature_exp = feature_exp_db[feature]
if '-' in feature:
pos1,pos2 = junction_locations[feature][0]
for f2 in feature_exp_db:
flag=False
if '-' in f2:
if f2!=feature:
f2_exp = feature_exp_db[f2]
alt_pos1,alt_pos2 = junction_locations[f2][0]
positions = [pos1,pos2,alt_pos1,alt_pos2]
positions.sort()
diff = positions.index(pos2)-positions.index(pos1)
if diff!=1:
#try: junctions_to_compare[feature].append(f2)
#except Exception: junctions_to_compare[feature] = [f2]
try:
overlapping_junctions_exp[feature].append([f2_exp,f2])
except Exception:
overlapping_junctions_exp[feature] = [[f2_exp,f2]]
flag=True
else:
diff = positions.index(alt_pos2)-positions.index(alt_pos1)
if diff!=1:
try:
overlapping_junctions_exp[feature].append([f2_exp,f2])
except Exception:
overlapping_junctions_exp[feature] = [[f2_exp,f2]]
#overlapping_junctions_test[count]=[feature,]
flag=True
if flag==True:
if feature not in existing and f2 not in existing:
count=count+1
overlapping_junctions_test[count]=[feature,]
overlapping_junctions_test[count].append(f2)
existing.append(feature)
existing.append(f2)
if feature in existing and f2 not in existing:
for i in overlapping_junctions_test:
if feature in overlapping_junctions_test[i]:
overlapping_junctions_test[i].append(f2)
existing.append(f2)
if f2 in existing and feature not in existing:
for i in overlapping_junctions_test:
if f2 in overlapping_junctions_test[i]:
overlapping_junctions_test[i].append(feature)
existing.append(feature)
if feature in existing and f2 in existing:
for i in overlapping_junctions_test:
if feature in overlapping_junctions_test[i]:
loc1=i
if f2 in overlapping_junctions_test[i]:
loc2=i
if loc1!=loc2:
for jun in overlapping_junctions_test[loc2]:
if jun not in overlapping_junctions_test[loc1]:
overlapping_junctions_test[loc1].append(jun)
del overlapping_junctions_test[loc2]
cluster_junction={}
#Finding clusters and corresponding junctions
for count in overlapping_junctions_test:
for feature in overlapping_junctions_test[count]:
cluster_junction[feature]=cluster_name
clusterID_export_data.write(cluster_name+"\t"+feature+"\t"+junction_locations[feature][1]+"\n")
cluster_id+=1
cluster_name=string.join('clu_'+str(cluster_id))
cluster_name=cluster_name.replace(" ","")
#duration = time.time() - begin_time
#print duration, 'seconds'
expressed_junctions=[]
for feature in overlapping_junctions_exp:
counts = map(lambda x: x[0], overlapping_junctions_exp[feature])
combined = [sum(value) for value in zip(*counts)]
#if feature == 'ENSG00000002586:E1.5-E4.1':
#print feature
#print combined
#print overlapping_junctions_exp[feature][0];sys.exit()
#dpsi_values = [ratio(value) for value in zip(*[overlapping_junctions_exp[feature][0],combined])]
#print feature
#print overlapping_junctions[feature]
#print overlapping_junctions_exp[feature]
#print combined;sys.exit()
exclusion_id = feature+'|exclusion'
feature_exp_db[exclusion_id] = combined
#rho : replace with clusterid
max_ratio,num_incl_events,num_excl_events,dpsi,rho,max_all_psi,proceed = junctionComparisonMethod(feature,exclusion_id)
if proceed:
fe1,fe2 = string.split(feature,'-')
if '_' in fe1 and '_' in fe2: pass
else:
#"""
top_excl_junction=[]
for (exp_ls,f2) in overlapping_junctions_exp[feature]:
top_excl_junction.append([statistics.avg(exp_ls),f2])
top_excl_junction.sort()
#print top_excl_junction[-8:]
#print statistics.avg(feature_exp_db[feature])
top_excl_junction = top_excl_junction[-1][-1]
t1,t2 = string.split(top_excl_junction,'-')
altexons = []
if t1!=fe1: altexons.append(fe1)
if t2!=fe2: altexons.append(gene+':'+fe2)
altexons = string.join(altexons,'|')
reg_pos,reg_loc = junction_locations[feature]
ref_pos,ref_loc = junction_locations[top_excl_junction]
#print [feature, dpsi,rho]
#top_excl_junctions = map(lambda x: x[-1], top_excl_junction[-5:])
#print top_excl_junctions;sys.exit()
#for i in top_excl_junctions: max_ratio,num_incl_events,num_excl_events,dpsi,rho,max_all_psi,proceed = junctionComparisonMethod(feature,i); print i, dpsi,rho
values = psi_db[feature,exclusion_id]
max_incl_psi = str(getMax(values))
# adding cluster information to PSI file
combined_ID = symbol+':'+feature+"|"+top_excl_junction
export_data.write(string.join([symbol,description,feature,top_excl_junction,altexons,str(max_ratio),str(dpsi),cluster_junction[feature],combined_ID,reg_loc+'|'+ref_loc,'junctions']+values,'\t')+'\n')
avg = averageWithNulls(values)
values_imputed = map(lambda x: replaceNulls(x,avg), values)
clust_export_data.write(string.join([symbol+':'+feature+'|'+top_excl_junction]+values_imputed,'\t')+'\n')
exported.append(feature)
exported.append(top_excl_junction)
#sys.exit()
gene_annotations = getGeneAnnotations(species)
firstLine = True
feature_exp_db={}
gene_junction_denom={} ### Determine the max junction counts per gene per sample
regulated_junctions = {}
genes_examined=0; gene_increment=1000
prior_gene = None
gene = None
""" Import all exon-exon and exon-intron junction reads for a gene. Then look for
genomic overlapping coordinates to compute PSI models for each splicing event
and report exon-exon junctions clusters prior to annotating (EventAnnotation) """
for line in open(expFile,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine = False
ge_header = t
if len(t)<4:
### If only two samples reduce the below numbers to the minimum number of observations
min_events=0;med_events=0
additional_headers = string.join(['Symbol','Description','Examined-Junction','Background-Major-Junction','AltExons',"PME","dPSI",'ClusterID','UID','Coordinates','feature']+t[1:],'\t')+'\n'
export_data.write(additional_headers)
clust_export_data.write(line)
else:
uid = t[0] ### Junction identifier
if '=' in uid:
try: uid,location = string.split(uid,'=')
except Exception: print t[0];sys.exit()
pos1,pos2 = string.split(string.split(location,':')[-1],'-')
pos = [int(pos1),int(pos2)]
pos.sort()
junction_locations[uid] = pos,location ### use the to report position and verify compared junctions
if gene == 'ENSG00000100650': ### For testing
proceed = True
else: proceed = True
if platform == 'RNASeq':
gene = string.split(uid,':')[0]
else:
if uid in probeset_gene_db:
gene = probeset_gene_db[uid]
else: proceed = False
if proceed:
counts = map(lambda x: float(x), t[1:])
if platform == 'junction' or platform == 'AltMouse':
counts = map(lambda x: int(math.pow(2,x)), counts) #log transform these instead, to make like junction counts
if '-' in uid or uid in junction_locations:
#try: gene_junction_denom[gene].append(counts)
#except Exception: gene_junction_denom[gene] = [counts]
pass
if genes_examined==gene_increment:
gene_increment+=1000
print '*',
if gene != prior_gene and prior_gene !=None:
genes_examined+=1
#if len(gene_junction_denom)>0:
""" Now that all your exons have been imported into this sorted counts file,
determine overlapping junctions to compute and quantify alternative splicing events """
if prior_gene == '!ENSG00000198001': ### For testing
novel_junc_count = 0
for junc in feature_exp_db:
if "_" in junc: novel_junc_count+=1
if novel_junc_count>5000:
### Indicates genomic variation resulting in broad diversity
### Will prevent function from running in a reasonable amount of time
pass
else:
filterByLocalJunctionExp(prior_gene,feature_exp_db)
#try: gene_junction_denom[prior_gene] = [max(value) for value in zip(*gene_junction_denom[prior_gene])] # sum the junction counts for all junctions across the gene
#except Exception: pass
if platform == 'RNASeq':
filterByLocalJunctionExp(prior_gene,feature_exp_db)
else:
compareJunctionExpression(prior_gene)
feature_exp_db={}
gene_junction_denom={}
if max(counts)>4:
feature_exp_db[uid] = counts
prior_gene = gene
#compareJunctionExpression(gene)
export_data.close()
clust_export_data.close()
clusterID_export_data.close()
graphic_links=[]
if (len(exported)/2)<7000:
if (len(exported)/2)<4000:
graphic_links = exportHeatmap(clust_export_dir,useHOPACH=False,color_gradient='yellow_black_blue',normalize=True,columnMethod='hopach',size=len(exported)/2)
else:
clust_export_dir,size = filterJunctionExpression(clust_export_dir)
if size<4000:
try: graphic_links = exportHeatmap(clust_export_dir,useHOPACH=False,color_gradient='yellow_black_blue',normalize=True,columnMethod='hopach',size=len(exported)/2,filter=True)
except Exception: graphic_links=[]
print len(exported)/2,'junctions exported' #,len(retained_introns)/2, 'retained introns exported...'
return graphic_links, clust_export_dir
def getGeneAnnotations(species):
gene_annotations={}
fn = filepath('AltDatabase/ensembl/'+species+'/'+species+'_Ensembl-annotations_simple.txt')
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
ensembl,description,symbol = string.split(data,'\t')
gene_annotations[ensembl] = symbol,description
return gene_annotations
def getDownstreamExon(upstream_exon):
gene,exon = string.split(upstream_exon,':')
downstream_exon = gene+':E'+str(int(string.split(exon,'.')[0][1:])+1)+'.1'
return downstream_exon
def expressionRatio(incl_exp,excl_exp,num_incl_events,num_excl_events):
### Selects the minor isoform and looks at its relative expression
if num_incl_events>num_excl_events:
max_ratio = max(excl_exp)/max(incl_exp)
else:
max_ratio = max(incl_exp)/max(excl_exp)
return max_ratio
def unbiasedComparisonSpliceProfiles(root_dir,species,platform,expFile=None,min_events=-1,med_events=-1): # 4 9
""" This is prototype code to identify critical splicing events (SI-exon-level) from single cell data prior to group assignment """
begin_time = time.time()
if platform == 'RNASeq': avg_all_for_SS = 'yes'
else: avg_all_for_SS = 'no'
agglomerate_inclusion_probesets = 'no'
probeset_type = 'core'
try:from build_scripts import JunctionArray
except:
try: import JunctionArray
except: pass
try: import AltAnalyze
except: pass
buildFromDeNovoJunctionsOnly=True
if buildFromDeNovoJunctionsOnly and platform=='RNASeq':
alt_junction_db={}
else:
exon_db, constitutive_probeset_db = AltAnalyze.importSplicingAnnotations(platform,species,probeset_type,avg_all_for_SS,root_dir)
alt_junction_db,critical_exon_db,exon_dbase,exon_inclusion_db,exon_db = JunctionArray.getPutativeSpliceEvents(species,platform,exon_db,agglomerate_inclusion_probesets,root_dir)
#print 'Number of Genes with Examined Splice Events:',len(alt_junction_db)
if platform == 'junction':
global probeset_junction_db; probeset_junction_db={}
#alt_junction_db = {'ENSG00000100650':alt_junction_db['ENSG00000100650']}
critical_exon_db={}
for affygene in alt_junction_db:
for event in alt_junction_db[affygene]:
for critical_exon in event.CriticalExonList():
critical_exon = affygene+':'+critical_exon
try:
#print event.InclusionJunction(), event.ExclusionJunction();sys.exit()
inclusion_list,exclusion_list = critical_exon_db[critical_exon]
if '-' in event.InclusionProbeset() or (platform == 'junction' and '-' in event.InclusionJunction()):
inclusion_list.append(event.InclusionProbeset())
exclusion_list.append(event.ExclusionProbeset())
if platform == 'junction':
probeset_junction_db[event.InclusionProbeset()] = event.InclusionJunction()
probeset_junction_db[event.ExclusionProbeset()] = event.ExclusionJunction()
except Exception:
if '-' in event.InclusionProbeset() or (platform == 'junction' and '-' in event.InclusionJunction()):
inclusion_list = [event.InclusionProbeset()]
else: inclusion_list=[]
exclusion_list = [event.ExclusionProbeset()]
#inclusion_list.append(critical_exon)
inclusion_list = unique.unique(inclusion_list)
exclusion_list = unique.unique(exclusion_list)
if len(inclusion_list)>0 and len(exclusion_list)>0:
critical_exon_db[critical_exon] = inclusion_list,exclusion_list
elif 'I' in critical_exon and '_' not in critical_exon and '.1' in critical_exon:
critical_exon_db[critical_exon] = [critical_exon],exclusion_list
#if affygene == 'ENSMUSG00000004952':
#if '.1' not in critical_exon: print critical_exon,inclusion_list,exclusion_list
if expFile != None:
graphic_links, cluster_input = compareRawJunctionExpression(root_dir,platform,species,critical_exon_db,expFile,min_events=min_events,med_events=med_events)
print 'finished in',int(time.time()-begin_time), 'seconds'
return graphic_links, cluster_input
### Determine the location of the gene expression file
input_folder = root_dir+'AltResults/RawSpliceDataTemp/'+species+'/splicing-index/'
dir_list = read_directory(input_folder) ### get all of the RawSplice files
for filename in dir_list:
if '.txt' in filename and ('_average' not in filename):
dataset_name = filename
input_dir = input_folder + dataset_name
exportSorted(input_dir, 2)
for filename in dir_list:
if '.txt' in filename and ('_average' not in filename):
dataset_name = filename
input_dir = input_folder + dataset_name
import RNASeq
biological_categories = RNASeq.importBiologicalRelationships(species)
genes = biological_categories['protein_coding']
#genes = biological_categories['BioMarker']
genes.update(biological_categories['transcription regulator'])
genes.update(biological_categories['splicing regulator'])
genes.update(biological_categories['kinase'])
genes.update(biological_categories['GPCR'])
### Import gene expression summaries to exclude high differential genes
fn=filepath(root_dir+'/ExpressionInput/exp.'+dataset_name[:-4]+'-steady-state.txt')
firstLine = True
low_diff_exp_genes={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine = False
ge_header = t
else:
rpkms = map(lambda x: float(x), t[1:])
rpkms.sort()
lower_index = int(len(rpkms)*0.2); upper_index = len(rpkms)-lower_index
gene = t[0]
#if (max(rpkms)/min(rpkms))<5: ### Max allowed differential expression
#if (rpkms[upper_index]/rpkms[lower_index])<5:
#if gene == 'ENSMUSG00000078812': print statistics.avg(rpkms)
if gene in genes and statistics.avg(rpkms)>5:
low_diff_exp_genes[gene]=rpkms
#print low_diff_exp_genes['ENSMUSG00000078812']
print len(low_diff_exp_genes), 'genes with less than 5-fold differential expression'
import gene_associations; from scipy import stats
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
exported_IDs=0; ids_exmained=0; genes_examined=0; gene_increment=1000
added={}; prior_gene = False; gene_comparison_db={}
print 'Begining to quickly find alternative exons...'
for filename in dir_list:
if '.txt' in filename and ('_average' not in filename) or len(dir_list)==1: ### We only want the group-comparison SI file
export_dir = string.split(input_folder,'RawSpliceData')[0]+'Unbiased/'+filename
export_data = export.ExportFile(export_dir)
fn=filepath(input_folder+filename)
x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
headers = t[2:] ### first two columsn are gene and ExonID
#print headers
#print ge_header;sys.exit()
export_data.write(string.join(headers,'\t')+'\n') ### write header row
x=1
else:
if platform != 'RNASeq':
if 'ENS' in t[0]: #ENSG human
gene = t[0]; altExon = t[2]
else:
altExon = t[0]
else:
gene = t[0]; altExon = t[2]
if genes_examined==gene_increment:
gene_increment+=1000
print '*',
if gene != prior_gene:
genes_examined+=1
hits={}
for gene in gene_comparison_db:
feature_db = gene_comparison_db[gene]
if gene == 'ENSMUSG00000078812':
for i in feature_db: print i
for altexon in feature_db:
#print altexon
if altexon in critical_exon_db:
inclusion_list,exclusion_list = critical_exon_db[altexon]
#print inclusion_list, 'incl'
altexon_SI = feature_db[altexon]
for incl in inclusion_list:
if incl in feature_db:
incl_SI = feature_db[incl]
with warnings.catch_warnings():
warnings.filterwarnings("ignore") ### hides import warnings
try: rho,p = stats.pearsonr(altexon_SI,incl_SI)
except Exception: rho = 1
#print rho, altexon,incl
#print string.join(map(str,altexon_SI),'\t')
#print string.join(map(str,incl_SI),'\t');sys.exit()
if rho>0.4:
hits[altexon]=[]
if gene == 'ENSMUSG00000078812':print '***', incl
#print inclusion_list, 'excl'
for excl in inclusion_list:
if excl in feature_db:
excl_SI = feature_db[excl]
with warnings.catch_warnings():
warnings.filterwarnings("ignore") ### hides import warnings
rho,p = stats.pearsonr(altexon_SI,excl_SI)
if rho<-0.4:
hits[altexon]=[]
if gene == 'ENSMUSG00000078812': print '***', excl
if gene == 'ENSMUSG00000078812': print hits
for altExon in hits:
added[altExon]=[]
log_folds = feature_db[altExon]
log_folds = map(str, log_folds)
values = string.join([altExon]+log_folds,'\t')+'\n' ### [t[3]]+ before log_folds?
if gene in gene_to_symbol: symbol = gene_to_symbol[gene][0]+" "
else: symbol = ''
export_data.write(symbol+values)
exported_IDs+=1
gene_comparison_db={}
#if exported_IDs> 1: sys.exit()
prior_gene = gene
if ';' in altExon:
altExon1, altExon2 = string.split(altExon,';')
altExons = [altExon1,gene+':'+altExon2]
else:
altExons = [altExon]
for altExon in altExons:
if altExon not in added and gene in low_diff_exp_genes: #altExon in alternative_exon_db and
#if altExon == 'ENSMUSG00000022841:E7.2':
ids_exmained+=1
#values = map(lambda x: float(x)*-1, t[3:]) #reverse the fold for cluster visualization
values = map(lambda x: float(x), t[3:])
#print len(headers),len(values);sys.exit()
avg = statistics.avg(values)
log_folds = map(lambda x: x-avg, values) ### calculate log folds and make these strings
i=0; si_exp_list = [] ### store the pairs of SI and gene expression for each sample
rpkms = list(low_diff_exp_genes[gene])
for si in log_folds: si_exp_list.append([rpkms[i],si]); i+=1
rpkms.sort()
si_exp_list.sort() ### This object contains both gene expression and SI values
max_rpkm = rpkms[-1]
half_max_rpkm = max_rpkm/2 ### Only look at genes in which there is less than a 2 fold differnce
s = bisect.bisect_right(rpkms,half_max_rpkm)
si_highGeneExp = map(lambda (rpkm,si): si, si_exp_list[s:])
#print si_exp_list[s:]
#cv = statistics.stdev(si_highGeneExp)/statistics.avg(si_highGeneExp)
si_highGeneExp.sort()
try:
biggest_diff = si_highGeneExp[-2]-si_highGeneExp[1]
#print biggest_diff
#print cv
if gene == 'ENSG00000009413':
print altExon, biggest_diff
print si_highGeneExp
if biggest_diff>2 and len(si_highGeneExp)>20:
try:
feature_db = gene_comparison_db[gene]
feature_db[altExon] = log_folds
except Exception:
feature_db={}
feature_db[altExon] = log_folds
gene_comparison_db[gene] = feature_db
#added[altExon]=[]
#log_folds = map(str, log_folds)
#values = string.join([altExon]+log_folds,'\t')+'\n' ### [t[3]]+ before log_folds?
#if gene in gene_to_symbol: symbol = gene_to_symbol[gene][0]+" "
#else: symbol = ''
#export_data.write(symbol+values)
#exported_IDs+=1
except Exception: pass ### Occurs with less than 4 samples in the si_highGeneExp set
print exported_IDs, 'exported ID values for clustering out of',ids_exmained
export_data.close()
return export_dir, exported_IDs
def AllGroupsNIComparison(root_dir, species, array_type):
if array_type == 'RNASeq': avg_all_for_SS = 'yes'
else: avg_all_for_SS = 'no'
agglomerate_inclusion_probesets = 'no'
#calculateNormalizedIntensities(root_dir, species, array_type, avg_all_for_SS = avg_all_for_SS, probeset_type = 'core')
### This analysis is designed for datasets without known variables (e.g., single cell seq)
from build_scripts import JunctionArray
exon_db, constitutive_probeset_db = AltAnalyze.importSplicingAnnotations(array_type,species,probeset_type,avg_all_for_SS,root_dir)
alt_junction_db,critical_exon_db,exon_dbase,exon_inclusion_db,exon_db = JunctionArray.getPutativeSpliceEvents(species,array_type,exon_db,agglomerate_inclusion_probesets,root_dir)
print 'Number of Genes with Examined Splice Events:',len(alt_junction_db)
for affygene in alt_junction_db:
for event in alt_junction_db[affygene]:
event.InclusionProbeset()
event.ExclusionProbeset()
def createExpressionSQLdb(species,platform,expFile):
""" Store junction/exon RPKMs or probesets expression in a SQL database"""
start=time.time()
from import_scripts import SQLInterface
DBname = 'FeatureExpression'
schema_text ='''-- Schema for species specific AltAnalyze junction/exon expression data.
-- Genes store general information on each Ensembl gene ID
create table ExonExp (
uid text primary key,
gene text,
expression text
);
'''
conn = SQLInterface.populateSQLite(species,platform,DBname,schema_text=schema_text) ### conn is the database connnection interface
### Populate the database
print 'importing', expFile
fn=filepath(expFile)
for line in open(fn,'r').xreadlines():
data = line.strip()
t = string.split(data,'\t')
uid = t[0]; expression = string.join(t[1:],'\t')
try: gene = string.split(uid,':')[0]
except Exception: print 'not RNASeq - function not supported';kill
#print exonID,gene,sequence
### Store this data in the SQL database
command = """insert into ExonExp (uid, gene, expression)
values ('%s', '%s','%s')""" % (uid,gene,expression)
conn.execute(command)
conn.commit() ### Needed to commit changes
conn.close()
time_diff = str(round(time.time()-start,1))
print 'Exon/Junction Expression Data added to SQLite database in %s seconds' % time_diff
def logratio(list):
return list[0] - list[1]
def matchAndCorrelate(prime, secondary, output_dir, rho_cutoff):
### Take two files and correlate their IDs to any matching
export_object = export.ExportFile(output_dir[:-4]+'-'+str(rho_cutoff)+'.txt')
export_object.write('Feature1\tFeature2\trho\n')
firstLine = True; prime_db={}
for line in open(prime,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine = False
else:
prime_db[t[0]] = map(float,t[1:])
firstLine = True; secondary_db={}; key_db={}
for line in open(secondary,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine = False
else:
try: gene_id,probe_id = string.split(t[0],':')
except Exception: gene_id = t[0]; probe_id = t[0]
try: secondary_db[gene_id].append((map(float,t[1:]),probe_id))
except Exception: secondary_db[gene_id] = [(map(float,t[1:]),probe_id)]
from scipy import stats
top_correlated={}
for gene in prime_db:
prime_profile = prime_db[gene]
if gene in secondary_db:
for (secondary_db_profile, probe_id) in secondary_db[gene]:
rho,p = stats.pearsonr(prime_profile,secondary_db_profile)
if rho > rho_cutoff or rho < -1*rho_cutoff:
#print gene, '\t',probe_id, '\t',rho
export_object.write(gene+'\t'+probe_id+'\t'+str(rho)+'\n')
export_object.close()
def getMax(values):
values2=[]
for i in values:
try: values2.append(float(i))
except Exception: pass
return max(values2)
def replaceNulls(x,avg):
if x=='':
return '0'
else:
return str(float(x)-avg)
def nullReplace(dpsi_values,combined,min_exp): ### Don't count un-detected genes in later stats
null_replaced=[]
i=0
for v in combined:
if v<(min_exp): null_replaced.append('') #changed July3 2017(min_exp+1)-Meenakshi
else: null_replaced.append(str(dpsi_values[i]))
i+=1
return null_replaced
def averageWithNulls(values):
avg_vals=[]
for i in values:
try: avg_vals.append(float(i))
except Exception: pass
avg = statistics.avg(avg_vals)
return avg
def expressionSortImport(filename,filter_db=None):
firstLine = True; exp_db={}; lines=0; max_var = 3
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
headers = t[1:]
header_ind = map(lambda x: (x,headers.index(x)),headers) ### store header index values
header_ind.sort()
#print header_ind
headers_ind = map(lambda (x,i): i,header_ind)
firstLine = False
else:
try: exp_data = map(float,t[1:])
except Exception:
exp_data=[]
for value in t[1:]:
try: value = float(value)
except Exception: pass
exp_data.append(value)
exp_data = map(lambda i: exp_data[i],headers_ind)
if filter_db != None:
key = t[0]
if ':' in key:
key = string.split(key,':')[0]
max_var = 0
if key in filter_db:
if max(exp_data)>max_var:
exp_db[key] = exp_data
else:
exp_db[t[0]] = exp_data
lines+=1
print len(exp_db),'IDs imported from', export.findFilename(filename)
return exp_db
def featureCorrelate(species,query_dir,feature_dir,output_dir,feature_type):
#python ExpressionBuilder.py --species Hs --i /Volumes/SEQ-DATA/SingleCell-Churko/Filtered/Unsupervised-AllExons/AltResults/Unbiased/junctions-2-5/top_alt_junctions_clust-TTN_all_selected.txt --additional /Volumes/SEQ-DATA/SingleCell-Churko/Filtered/Unsupervised-AllExons/ExpressionInput/exp.CM-TTN-steady-state.txt --analysis featureCorrelate --var "splicing regulator"
### Correlate features in a file to feature-specific gene expression data (e.g., "splicing regulator")
try: export_object = export.ExportFile(output_dir[:-4]+'-'+feature_type+'.txt')
except Exception: export_object = export.ExportFile(output_dir[:-4]+'-None.txt')
export_object.write('UID\tFeature\trho\n')
import RNASeq; import ExpressionBuilder
biological_categories = RNASeq.importBiologicalRelationships(species)
gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
if feature_type != None:
filter_genes = biological_categories[feature_type]
else:
filter_genes=None
try: print len(filter_genes),feature_type,'genes imported for comparison...'
except Exception: pass
query_db = expressionSortImport(query_dir)
feature_db = expressionSortImport(feature_dir,filter_genes)
from scipy import stats
top_correlated={}
for uid in query_db:
query_exp_profile = query_db[uid]
for gene in feature_db:
feature_exp_profile = feature_db[gene]
try: rho,p = stats.pearsonr(query_exp_profile,feature_exp_profile)
except Exception:
### If missing values are present, only correlate to where the values are present
query_exp_profile2=[]
feature_exp_profile2=[]
i=0
for v in query_exp_profile:
if v!='':
query_exp_profile2.append(query_exp_profile[i])
feature_exp_profile2.append(feature_exp_profile[i])
i+=1
if len(feature_exp_profile2)>20:
rho,p = stats.pearsonr(query_exp_profile2,feature_exp_profile2)
else:
rho = 0
try: symbol = gene_to_symbol_db[gene]
except Exception: symbol = gene
try: top_correlated[uid].append([abs(rho),symbol[0],rho])
except Exception: top_correlated[uid]=[[abs(rho),symbol[0],rho]]
for uid in top_correlated:
res = top_correlated[uid]
res.sort()
feature = res[-1][1]
rho = res[-1][-1]
export_object.write(uid+'\t'+feature+'\t'+str(rho)+'\n')
export_object.close()
def lncRNANeighborCorrelationAnalysis(dataset_dir):
### dataset_dir is the ExpressionOuput DATASET file location
#Get all analyzed genes and coordinates
print 'Importing the DATASET file'
global gene_symbol_db
fn=filepath(dataset_dir); gene_coordinate_db={}; all_lncRNA_db={}; coord_gene_db={}; gene_symbol_db={}
chr_coord_list=[]; positive_strand=[]; negative_strand=[]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
geneID = t[0]
symbol = t[2]
protein_class = t[8]
chr = t[9]
strand = t[10]
coordinates = tuple(string.split(t[11],'-'))
coord_list = chr,coordinates,strand
gene_coordinate_db[geneID]=coord_list
coord_gene_db[coord_list] = geneID
gene_symbol_db[geneID] = symbol
chr_coord_list.append(coord_list)
if '+' in strand:
positive_strand.append(coord_list)
else:
negative_strand.append(coord_list)
if 'lincRNA' in protein_class or 'lncRNA' in protein_class:
all_lncRNA_db[geneID]=[]
chr_coord_list.sort(); positive_strand.sort(); negative_strand.sort()
useClusterFile = False
#Get all significantly differentially expressed genes
if useClusterFile:
cluster_file = string.replace(dataset_dir,'ExpressionOutput','ExpressionOutput/Clustering/')
cluster_file = string.replace(cluster_file,'DATASET-','SampleLogFolds-')
else:
cluster_file = string.replace(dataset_dir,'ExpressionOutput','ExpressionInput')
cluster_file = string.replace(cluster_file,'DATASET-','exp.')
cluster_file = string.replace(cluster_file,'.txt','-steady-state.txt')
print 'Importing the cluster file'
fn=filepath(cluster_file); differentially_exp_db={}; lncRNA_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
uid = string.split(data,'\t')[0]
if ' ' in uid:
uid = string.split(uid,' ')[0]
differentially_exp_db[uid]=[]
if uid in all_lncRNA_db:
lncRNA_db[uid]=[]
#import random
#lncRNA_db = random.sample(differentially_exp_db,len(lncRNA_db))
print 'Number of lncRNAs regulated in clusters:',len(lncRNA_db)
#Get the MarkerFinder cluster assignments of all analyzed genes
root_dir = string.split(dataset_dir,'ExpressionOutput')[0]
markerfinder = root_dir+'ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
print 'Importing the MarkerFinder file'
fn=filepath(markerfinder); cluster_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
geneID = t[0]
cluster = t[-1]
if geneID in differentially_exp_db:
cluster_db[geneID] = cluster
cluster_regulated_lncRNAs={}
for geneID in lncRNA_db:
try:
cluster_regulated_lncRNAs[cluster_db[geneID]]+=1
except Exception:
try: cluster_regulated_lncRNAs[cluster_db[geneID]]=1
except Exception: pass
for cluster in cluster_regulated_lncRNAs:
print cluster, cluster_regulated_lncRNAs[cluster]
print 'Searching for lncRNA positional correlations'
direction_list=['both','forward','reverse']
print '\tExamining both strands'
for direction in direction_list:
nmc = searchUpOrDownstreamGenes(lncRNA_db,gene_coordinate_db,cluster_db,coord_gene_db,chr_coord_list,direction)
#print len(nmc),len(chr_coord_list)
print '\tExamining the positive strand'
for direction in direction_list:
nmc = searchUpOrDownstreamGenes(lncRNA_db,gene_coordinate_db,cluster_db,coord_gene_db,positive_strand,direction)
#print len(nmc),len(positive_strand)
print '\tExamining the negative strand'
for direction in direction_list:
nmc = searchUpOrDownstreamGenes(lncRNA_db,gene_coordinate_db,cluster_db,coord_gene_db,negative_strand,direction)
#print len(nmc),len(negative_strand)
def searchUpOrDownstreamGenes(lncRNA_db,gene_coordinate_db,cluster_db,coord_gene_db,coord_list,direction):
neighbor_matching_cluster_db={}; multiple_lncRNAs=0; number_of_neighbors=0
for geneID in lncRNA_db:
coordinates = gene_coordinate_db[geneID]
if coordinates in coord_list: ### strand dependent
rank_index = coord_list.index(coordinates)
if geneID in cluster_db:
cluster = cluster_db[geneID]
if direction == 'forward':
search_pos = [4, 3,2,1]
search_pos = [1]
elif direction == 'reverse':
search_pos = [-4, -3,-2,-1]
search_pos = [-1]
else:
search_pos = [4,3,2,1,-3,-2,-1, -4]
search_pos = [1,-1]
for oi in search_pos:
i = coord_list[rank_index-oi]
neighbor_gene = coord_gene_db[i]
symbol = gene_symbol_db[neighbor_gene]
if neighbor_gene in cluster_db and neighbor_gene not in lncRNA_db and neighbor_gene != geneID and '.' not in symbol:
ncluster = cluster_db[neighbor_gene]
if cluster == ncluster:
if neighbor_gene in lncRNA_db:
multiple_lncRNAs+=1
try: neighbor_matching_cluster_db[geneID]+=1; number_of_neighbors+=1
except Exception: neighbor_matching_cluster_db[geneID]=1; number_of_neighbors+=1
print cluster,gene_symbol_db[geneID],gene_symbol_db[neighbor_gene]
#print 'multiple_lncRNAs:', multiple_lncRNAs, number_of_neighbors
return neighbor_matching_cluster_db
def getHighestExpressingGenes(input_file,output_dir,topReported):
### Sorts genes based on RPKM (ignore read counts)
bisectValues = False
if topReported<100:
bisectValues = True
firstLine = True
sampleExpression_db={}
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
if firstLine:
headers = values[1:]
for i in headers:
sampleExpression_db[i]=[]
firstLine = False
print len(values)
else:
gene = values[0]
i=0
for rpkm in values[1:]:
sampleExpression_db[headers[i]].append((float(rpkm),gene))
i+=1
for sample in sampleExpression_db:
Sample = string.replace(sample,'.bed','')
Sample = string.replace(Sample,'.cel','')
Sample = string.replace(Sample,'.CEL','')
Sample = string.replace(Sample,':','-')
export_object = export.ExportFile(output_dir+'/'+Sample+'-top_'+str(topReported)+'.txt')
export_object.write('Genes\tSystemCode\tChanged\n')
sampleExpression_db[sample].sort()
if bisectValues:
rpkms = map(lambda x: x[0], sampleExpression_db[sample])
print rpkms[-5:]
s = bisect.bisect_right(rpkms,float(topReported))
topExpGenes = map(lambda x: str(x[1]), sampleExpression_db[sample][-1*(len(rpkms)-s):])
print Sample,len(topExpGenes), s
else:
topExpGenes = map(lambda x: str(x[1]), sampleExpression_db[sample][-1*topReported:])
for gene in topExpGenes:
if 'ENS' in gene or 'ENF' in gene: system = 'En'
else: system = 'Sy'
export_object.write(gene+'\t'+system+'\t1\n')
export_object.close()
print 'The top',topReported,'expressing genes have been exported to',output_file
def returnRowHeaderForMaxEntry(filename,top):
### Used for enrichment analysis matrices to find the most significant term for each comparison/group/sample
output_file = filename[:-4]+'_top%d.txt' % top
export_object = export.ExportFile(output_file)
from visualization_scripts import clustering; import numpy
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(filename,reverseOrder=False)
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
x=0
for row in matrix:
comparison = row_header[x]
copied_row_values = list(row)
copied_row_values.sort()
max_vals = copied_row_values[-1*top:]
max_vals.reverse()
term = column_header[list(row).index(max_vals[0])]
term+= '('+str(max_vals[0])[:4]+')|'
if top>1:
term+= column_header[list(row).index(max_vals[1])]
term+= '('+str(max_vals[1])[:4]+')|'
if top>2:
term+= column_header[list(row).index(max_vals[2])]
term+= '('+str(max_vals[2])[:4]+')|'
if top>3:
term+= column_header[list(row).index(max_vals[3])]
term+= '('+str(max_vals[3])[:4]+')|'
if top>4:
term+= column_header[list(row).index(max_vals[4])]
term+= '('+str(max_vals[4])[:4]+')|'
if top>5:
term+= column_header[list(row).index(max_vals[5])]
term+= '('+str(max_vals[5])[:4]+')|'
if top>6:
term+= column_header[list(row).index(max_vals[6])]
term+= '('+str(max_vals[6])[:4]+')|'
if top>7:
term+= column_header[list(row).index(max_vals[7])]
term+= '('+str(max_vals[7])[:4]+')|'
if top>8:
term+= column_header[list(row).index(max_vals[8])]
term+= '('+str(max_vals[8])[:4]+')|'
if top>9:
term+= column_header[list(row).index(max_vals[9])]
term+= '('+str(max_vals[9])[:4]+')|'
if top>10:
term+= column_header[list(row).index(max_vals[10])]
term+= '('+str(max_vals[10])[:4]+')|'
#print comparison, term
export_object.write(comparison+'\t'+term+'\n')
x+=1
export_object.close()
def orderHeatmapByMarkerFinderOrder(clustered_file):
output_file = clustered_file[:-4]+'_MarkerFinderOrdered.txt'
export_object = export.ExportFile(output_file)
firstLine = True
geneOrder=[]
arrayOrder={}
for line in open(input_file,'rU').xreadlines():
data = line[:-1]
values = string.split(data,'\t')
if firstLine:
headers = values[1:]
for i in headers:
group,sample = string.split(i,':')
try: arrayOrder[group].append(i)
except Exception: arrayOrder[group] = [i]
firstLine = False
else:
gene = values[0]
def exportSorted(filename, sort_col, excludeHeader=True):
### efficient method to sort a big file without storing everything in memory
### http://stackoverflow.com/questions/7079473/sorting-large-text-data
ouput_file = filename[:-4]+'-sorted' ### temporary
index = []
f = open(filename)
firstLine = True
while True:
offset = f.tell()
line = f.readline()
if not line: break
length = len(line)
col = line.split('\t')[sort_col].strip()
if firstLine:
header = line
firstLine = False
if excludeHeader == False:
index.append((col, offset, length))
else:
index.append((col, offset, length))
f.close()
index.sort()
o = open(ouput_file,'w')
f = open(filename)
if excludeHeader:
o.write(header)
for col, offset, length in index:
#print col, offset, length
f.seek(offset)
o.write(f.read(length))
o.close()
try:
### Error occurs when the file can't be deleted due to system permissions
os.remove(filename)
os.rename(ouput_file,filename)
return filename
except Exception:
return ouput_file
def importJunctionPositions(species,array_type):
### Look up the junction coordinates for the region
if array_type == 'RNASeq':
probesets = 'junctions'
else:
probesets = 'probesets'
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_'+probesets+'.txt'
fn=filepath(filename)
region_db = {}
firstRow=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow: firstRow = False
else:
probeset = t[0]
gene = t[2]
chr = t[4]
if '|' in t[13]:
region1 = string.split(t[13],'|')[1]
region2 = string.split(t[14],'|')[0]
junction_coord = region1+'-'+region2
region_db[probeset] = junction_coord
region_db[junction_coord] = gene+':'+t[12],chr ### Junction region for this version of Ensembl
return region_db
def convertArrayReciprocalJunctionToCoordinates(species,array_type,dir_path,start_version,end_version):
""" Script for taking junction array defined ASPIRE or LinearRegression junction pairs, extracting the region coordinates
and exporting those coordinates with end_version EnsMart block and region IDs"""
UI.exportDBversion(start_version) ### Database EnsMart version
region_db = importJunctionPositions(species,array_type)
comparison_db={}
dir_list = UI.read_directory(dir_path)
for filename in dir_list:
if '.txt' in filename:
comparsion = string.split(filename,'.')[0]
proceed = False
if ('ASPIRE' in filename or 'egress' in filename) and ('GENE' not in filename and 'inclusion' in filename): proceed = True ### Need to create a special analysis just for reciprocal junctions
if proceed: ### Don't include splicing-index results for RNA-Seq
comparison_db[comparsion] = {}
fn=filepath(dir_path+'/'+filename)
x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
p1 = t.index('probeset1')
p2 = t.index('probeset2')
reg_call = t.index('regulation_call')
e1 = t.index('exons1')
x=1
else:
if '-' in t[e1]:
jc1 = region_db[t[p1]]
jc2 = region_db[t[p2]]
chr = region_db[jc1][1]
db = comparison_db[comparsion]
db[jc1,jc2] = t[reg_call],chr
UI.exportDBversion(end_version) ### Database EnsMart version
converted_comparison_db = {}
region_db2 = importJunctionPositions(species,'RNASeq')
eo = export.ExportFile(dir_path+'/converted_junction_events.txt')
succeed=0; fail=0
for comparison in comparison_db:
for (j1,j2) in comparison_db[comparison]:
reg_call,chr = comparison_db[comparison][(j1,j2)]
if j1 in region_db2 and j2 in region_db2:
junction1_id,chr = region_db2[j1]
junction2_id,chr = region_db2[j2]
#print junction1_id, junction2_id, j1,j2, comparison;sys.exit()
else:
junction1_id=''
junction2_id=''
j1=chr+':'+j1
j2=chr+':'+j2
values = string.join([comparison,j1,j2,junction1_id,junction2_id,reg_call],'\t')+'\n'
eo.write(values)
eo.close()
def convertPSIJunctionIDsToPositions(psi_file,regulated_file):
""" Links up PSI genomic positions with IDs in a significantly differentially regulated PSI results file """
fn=filepath(psi_file)
x=0
coord_db = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
symbol = t.index('Symbol')
minor = t.index('Examined-Junction')
major = t.index('Background-Major-Junction')
coord = t.index('Coordinates')
x=1
else:
uid = t[symbol]+':'+t[minor]+'|'+t[major]
coordinates = t[coord]
coord_db[uid] = coordinates
dir_path = export.findParentDir(regulated_file)
comparison = export.findFilename(regulated_file)
eo = export.ExportFile(dir_path+'/coordinate_PSI_events.txt')
fn=filepath(regulated_file)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
event = t[0]
event = string.replace(event,'@',':')
event = string.replace(event,'&',':')
event = string.replace(event,'__','|')
regulation = t[1]
if event in coord_db:
coordinates = coord_db[event]
values = string.join([comparison,event,coordinates,regulation],'\t')+'\n'
eo.write(values)
eo.close()
if __name__ == '__main__':
#predictSplicingEventTypes('ENSG00000123352:E15.4-E16.1','ENSG00000123352:E15.3-E16.1');sys.exit()
test=False
if test:
directory = '/Volumes/SEQ-DATA/AML_junction/AltResults/AlternativeOutput/'
dir_list = read_directory(directory)
for file in dir_list:
if 'PSI-clust' in file:
filename = meanCenterPSI(directory+'/'+file)
#filterJunctionExpression(filename,minPercentPresent=0.75)
#exportHeatmap('/Volumes/My Passport/AML-LAML/LAML1/AltResults/AlternativeOutput/Hs_RNASeq_top_alt_junctions-PSI-clust-filt.txt',color_gradient='yellow_black_blue',columnMethod='hopach')
#sys.exit()
#convertPSIJunctionIDsToPositions('/Volumes/SEQ-DATA/Grimeslab/TopHat/AltResults/AlternativeOutput/Mm_RNASeq_top_alt_junctions-PSI.txt','/Users/saljh8/Documents/1-dataAnalysis/SplicingFactors/Grimes-MarkerFinder-v2.txt.txt')
#convertArrayReciprocalJunctionToCoordinates('Hs','junction','/Volumes/Time Machine Backups/dataAnalysis/SplicingFactor/Hs/hglue/Marto/AltResults/AlternativeOutput','EnsMart65','EnsMart72')
#sys.exit()
fold = 2
pval = 0.05
ptype = 'rawp'
species = 'Hs'
analysis = 'goelite'
array_type = "3'array"
norm = 'RPKM'
graphic_links=[]
additional = None
use_downregulated_labels=True
excludeGenes = None
expression_data_format = 'non-log'
expression_data_format = 'log'
var = None
################ Comand-line arguments ################
import getopt
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Please designate a tab-delimited input expression file in the command-line"
print "Example: python ExpressionBuilder.py --i '/Users/me/GSEXXX/ExpressionOutput' --p 0.05 --f 1.5 --ptype rawp --analysis summary --direction up --platform RNASeq"
sys.exit()
#Building GO-Elite inputs and running GO-Elite in batch
#python ExpressionBuilder.py --i /Users/saljh8/Desktop/C4-hESC/ExpressionOutput --p 0.05 --f 2 --ptype adjp --analysis goelite --direction down --platform gene --species Hs --additional goelite
#Generating signatures
#python ExpressionBuilder.py --i /Users/saljh8/Desktop/C4-hESC/GO-Elite/upregulated/ --analysis signature --inputSource Ensembl --outputSource EntrezGene
#Filtering expression datasets
#python ExpressionBuilder.py --i /Users/saljh8/Desktop/C4-hESC/ExpressionOutput --analysis filter
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['i=','o=','f=','p=','a=','platform=',
'ptype=','analysis=','species=','direction=',
'inputSource=','outputSource=', 'additional=',
'excludeGenes=','var='])
#print sys.argv[1:]
for opt, arg in options:
if opt == '--i': directory=arg
elif opt == '--o': output_file=arg
elif opt == '--f': fold=float(arg)
elif opt == '--p': pval=float(arg)
elif opt == '--ptype': ptype=arg
elif opt == '--analysis' or opt == '--a': analysis=arg
elif opt == '--species': species=arg
elif opt == '--platform': array_type=arg
elif opt == '--inputSource': input_source=arg
elif opt == '--outputSource': output_source=arg
elif opt == '--additional': additional=arg
elif opt == '--excludeGenes': excludeGenes=arg ### File location for text file with genes to exclude
elif opt == '--var': var=arg
elif opt == '--direction':
if 'own' in arg:
use_downregulated_labels = True
else:
use_downregulated_labels = False
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
### Allow for import of genes to exclude (e.g., sex-associated or pseudogenes)
try: genesToExclude = excludeGenesImport(excludeGenes)
except Exception: genesToExclude = {}
print analysis
if array_type == 'RNASeq':
gene_exp_threshold = 50
gene_rpkm_threshold = 3
if analysis == 'matchAndCorrelate':
matchAndCorrelate(directory, var, output_source, additional)
if analysis == 'returnRowHeaderForMaxEntry':
### Used primarily for combining LineageProfiler z-scores to report the top categories across compendiums
try: returnRowHeaderForMaxEntry(directory,int(var))
except Exception: pass
if analysis == 'featureCorrelate':
try: output_file = output_file
except Exception: output_file=directory
featureCorrelate(species,directory,additional,output_file,var)
if analysis == 'MarkerFinderOrder':
### Used for combining the all gene MarkerFinder ordered results with already clustered results (e.g., significant gene)
### to return significantly differentially expressed genes (expressed sufficiently) and cluster samples within classes
### but order by MarkerFinder correlations and groups
orderHeatmapByMarkerFinderOrder(directory)
if analysis == 'unbiased':
#python ExpressionBuilder.py --species Hs --platform RNASeq --i "/Volumes/My Passport/salomonis2/SRP042161_GBM-single-cell/bams/" --a unbiased --additional "/Volumes/My Passport/salomonis2/SRP042161_GBM-single-cell/bams/ExpressionInput/counts.GBM_scRNA-Seq.txt"
import RNASeq
#export_dir = '/Volumes/SEQ-DATA/Grimes/14018_gmp-pro/Lattice/Full/AltResults/Unbiased/DataPlots/Clustering-myeloblast-hierarchical_euclidean_euclidean.txt'
#export_dir = '/Volumes/SEQ-DATA/SingleCell-Churko/AltResults/Unbiased/DataPlots/Clustering-CM-hierarchical_euclidean_euclidean.txt'
#calculateNormalizedIntensities(directory, species, array_type, analysis_type = 'raw', expFile = additional)
var = unbiasedComparisonSpliceProfiles(directory,species,array_type,expFile=additional,min_events=1,med_events=1)
#export_dir, exported_IDs = var
#print export_dir
#RNASeq.correlateClusteredGenes(export_dir)
if analysis == 'highest-expressing':
getHighestExpressingGenes(directory,output_file,float(var))
if analysis == 'lncRNA':
lncRNANeighborCorrelationAnalysis(directory)
if analysis == 'NI':
calculateNormalizedIntensities(directory,species,array_type)
if analysis == 'AltExonConfirmed':
### Grab the alternative exons in the AltExonConfirmed GO-Elite folder, combine them and filter the splicing-index raw table
input_dir = directory+'/AltExonConfirmed/'
cluster_file, rows_in_file = buildAltExonClusterInputs(input_dir,species,array_type,dataType='AltExonConfirmed')
if rows_in_file < 7000:
exportHeatmap(cluster_file,size=rows_in_file)
if analysis == 'goelite' or analysis == 'summary':
#python ExpressionBuilder.py --f 2 --p 0.05 --ptype adjp --analysis summary --i /inputs
buildCriterion(fold, pval, ptype, directory+'/',analysis,UseDownRegulatedLabel=use_downregulated_labels,genesToExclude=genesToExclude)
if additional == 'goelite':
import multiprocessing as mlp
runGOElite(species,directory)
if analysis == 'filter':
filterDatasetFile(directory+'/')
if analysis == 'signature':
import gene_associations
directory+='/'; gene_conversion_db={}
dir_list = read_directory(directory)
for file in dir_list:
filename = directory+'/'+file
db,input_data_db = gene_associations.IDconverter(filename, species, input_source, output_source,analysis=analysis)
gene_conversion_db[file] = db,input_data_db
exportSignatures(gene_conversion_db,directory,species)
if analysis == 'QC':
graphic_links = visualizeQCPlots(directory)
elif analysis == 'LineageProfiler':
graphic_links = performLineageProfiler(directory,graphic_links)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/ExpressionBuilder.py
|
ExpressionBuilder.py
|
###update
#Copyright 2005-2008 J. David Gladstone Institutes, San Francisco California
#Author Nathan Salomonis - [email protected]
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"""This module contains generic methods for downloading and decompressing files designated
online files and coordinating specific database build operations for all AltAnalyze supported
gene ID systems with other program modules. """
import os
import sys
import unique
import string
import export
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv": dir_list2.append(entry)
return dir_list2
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def zipDirectory(dir):
#http://www.testingreflections.com/node/view/8173
import zipfile
dir = filepath(dir); zip_file = dir+'.zip'
p = string.split(dir,'/'); top=p[-1]
zip = zipfile.ZipFile(zip_file, 'w', compression=zipfile.ZIP_DEFLATED)
root_len = len(os.path.abspath(dir))
for root, dirs, files in os.walk(dir):
archive_root = os.path.abspath(root)[root_len:]
for f in files:
fullpath = os.path.join(root, f)
archive_name = os.path.join(top+archive_root, f)
zip.write(fullpath, archive_name, zipfile.ZIP_DEFLATED)
zip.close()
return zip_file
def unzipFiles(filename,dir):
import zipfile
output_filepath = filepath(dir+filename)
try:
zfile = zipfile.ZipFile(output_filepath)
for name in zfile.namelist():
if name.endswith('/'):null=[] ### Don't need to export
else:
try: outfile = export.ExportFile(dir+name)
except Exception: outfile = export.ExportFile(dir+name[1:])
outfile.write(zfile.read(name)); outfile.close()
#print 'Zip extracted to:',output_filepath
status = 'completed'
except Exception, e:
print e
print 'WARNING!!!! The zip file',output_filepath,'does not appear to be a valid zip archive file or is currupt.'
status = 'failed'
return status
def download(url,dir,file_type):
try: dp = download_protocol(url,dir,file_type); gz_filepath, status = dp.getStatus()
except Exception:
gz_filepath='failed'; status = "Internet connection was not established. Re-establsih and try again."
if status == 'remove':
#print "\nRemoving zip file:",gz_filepath
try: os.remove(gz_filepath); status = 'removed'
except Exception: null=[] ### Not sure why this error occurs since the file is not open
return gz_filepath, status
class download_protocol:
def __init__(self,url,dir,file_type):
"""Copy the contents of a file from a given URL to a local file."""
filename = url.split('/')[-1]
if len(file_type) == 2: filename, file_type = file_type ### Added this feature for when a file has an invalid filename
output_filepath_object = export.createExportFile(dir+filename,dir[:-1])
output_filepath = filepath(dir+filename)
print "Downloading the following file:",filename,' ',
self.original_increment = 10
self.increment = 0
import urllib
from urllib import urlretrieve
try:
try: webfile, msg = urlretrieve(url,output_filepath,reporthook=self.reporthookFunction)
except IOError:
if 'Binary' in traceback.format_exc(): #IOError: [Errno ftp error] 200 Switching to Binary mode.
### https://bugs.python.org/issue1067702 - some machines the socket doesn't close and causes an error - reload to close the socket
reload(urllib)
webfile, msg = urlretrieve(url,output_filepath,reporthook=self.reporthookFunction)
reload(urllib)
except:
print 'Unknown URL error encountered...'; forceURLError
print ''
print "\nFile downloaded to:",output_filepath
if '.zip' in filename:
try: decompressZipStackOverflow(filename,dir); status = 'completed'
except Exception:
status = unzipFiles(filename,dir)
if status == 'failed': print 'Zip Extraction failed'
self.gz_filepath = filepath(output_filepath); self.status = 'remove'
print "zip file extracted..."
elif '.gz' in filename:
self.gz_filepath = output_filepath
if len(file_type)==0: extension = '.gz'
else: extension = 'gz'
decompressed_filepath = string.replace(self.gz_filepath,extension,file_type)
### Below code can be too memory intensive
#file_size = os.path.getsize(output_filepath)
#megabtyes = file_size/1000000.00
#if megabtyes>5000: force_error ### force_error is an undefined variable which causes an exception
import gzip; content = gzip.GzipFile(self.gz_filepath, 'rb')
data = open(decompressed_filepath,'wb')
#print "\nExtracting downloaded file:",self.gz_filepath
import shutil; shutil.copyfileobj(content,data)
self.status = 'remove'
else: self.gz_filepath = ''; self.status = 'NA'
def getStatus(self): return self.gz_filepath, self.status
def reporthookFunction(self, blocks_read, block_size, total_size):
if not blocks_read:
print 'Connection opened. Downloading (be patient)'
if total_size < 0:
# Unknown size
print 'Read %d blocks' % blocks_read
else:
amount_read = blocks_read * block_size
percent_read = ((amount_read)*1.00/total_size)*100
if percent_read>self.increment:
#print '%d%% downloaded' % self.increment
print '*',
self.increment += self.original_increment
#print 'Read %d blocks, or %d/%d' % (blocks_read, amount_read, (amount_read/total_size)*100.000)
def createExportFile(new_file,dir):
try:
fn=filepath(new_file); file_var = open(fn,'w')
except IOError:
#print "IOError", fn
fn = filepath(dir)
try:
os.mkdir(fn) ###Re-Create directory if deleted
#print fn, 'written'
except OSError: createExportDir(new_file,dir) ###Occurs if the parent directory is also missing
fn=filepath(new_file); file_var = open(fn,'w')
return file_var
def downloadCurrentVersionUI(filename,secondary_dir,file_type,root):
continue_analysis = downloadCurrentVersion(filename,secondary_dir,file_type)
if continue_analysis == 'no':
import UI
root.destroy(); UI.getUserParameters('no'); sys.exit()
root.destroy()
def downloadCurrentVersion(filename,secondary_dir,file_type):
import UI
file_location_defaults = UI.importDefaultFileLocations()
uds = file_location_defaults['url'] ### Get the location of the download site from Config/default-files.csv
for ud in uds: url_dir = ud.Location() ### Only one entry
dir = export.findParentDir(filename)
filename = export.findFilename(filename)
url = url_dir+secondary_dir+'/'+filename
file,status = download(url,dir,file_type); continue_analysis = 'yes'
if 'Internet' in status:
print_out = "File:\n"+url+"\ncould not be found on server or internet connection is unavailable."
try:
UI.WarningWindow(print_out,'WARNING!!!')
continue_analysis = 'no'
except Exception:
print url
print 'cannot be downloaded';die
elif status == 'remove':
try: os.remove(file) ### Not sure why this works now and not before
except Exception: status = status
return continue_analysis
def decompressZipStackOverflow(zip_file,dir):
zip_file = filepath(dir+zip_file)
###http://stackoverflow.com/questions/339053/how-do-you-unzip-very-large-files-in-python
import zipfile
import zlib
src = open(zip_file,"rb")
zf = zipfile.ZipFile(src)
for m in zf.infolist():
# Examine the header
#print m.filename, m.header_offset, m.compress_size, repr(m.extra), repr(m.comment)
src.seek(m.header_offset)
src.read(30) # Good to use struct to unpack this.
nm= src.read(len(m.filename))
if len(m.extra) > 0: ex= src.read(len(m.extra))
if len(m.comment) > 0: cm=src.read(len(m.comment))
# Build a decompression object
decomp= zlib.decompressobj(-15)
# This can be done with a loop reading blocks
out=open(filepath(dir+m.filename), "wb")
result=decomp.decompress(src.read(m.compress_size))
out.write(result); result=decomp.flush()
out.write(result); out.close()
zf.close()
src.close()
if __name__ == '__main__':
dp = download_protocol('http://may2009.archive.ensembl.org/biomart/martresults/136?file=martquery_1117221814_599.txt.gz','downloaded','')
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/download.py
|
download.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"""This module contains instructions for recalling operating system file and directory paths,
eliminating redundant list entries, removing unecessary file paths from py2app or py2exe
and reading the propper Ensembl database version to allow for version specific access."""
import sys, string
import os.path, platform
import traceback
from os.path import expanduser
userHomeDir = expanduser("~")+'/altanalyze/'
ignoreHome = False
py2app_adj = '/GO_Elite.app/Contents/Resources/Python/site-packages.zip'
py2app_adj1 = '/GO_Elite.app/Contents/Resources/lib/python2.4/site-packages.zip'
py2app_adj2 = '/GO_Elite.app/Contents/Resources/lib/python2.5/site-packages.zip'
py2app_adj3 = '/GO_Elite.app/Contents/Resources/lib/python2.6/site-packages.zip'
py2app_adj4 = '/GO_Elite.app/Contents/Resources/lib/python2.7/site-packages.zip'
py2exe_adj = '\\library.zip' ###py2exe
cx_Freeze_adj = '/library.zip'
pyinstaller_adj = '/GO_Elite.app/Contents/MacOS'
py2app_ge_dirs = [py2app_adj,py2exe_adj,py2app_adj1,py2app_adj2,py2app_adj3,py2app_adj4,cx_Freeze_adj,pyinstaller_adj]
py2app_adj = '/AltAnalyze.app/Contents/Resources/Python/site-packages.zip'
py2app_adj1 = '/AltAnalyze.app/Contents/Resources/lib/python2.4/site-packages.zip'
py2app_adj2 = '/AltAnalyze.app/Contents/Resources/lib/python2.5/site-packages.zip'
py2app_adj3 = '/AltAnalyze.app/Contents/Resources/lib/python2.6/site-packages.zip'
py2app_adj4 = '/AltAnalyze.app/Contents/Resources/lib/python2.7/site-packages.zip'
py2exe_adj = '\\library.zip' ###py2exe
cx_Freeze_adj = '/library.zip'
pyinstaller_adj = '/AltAnalyze.app/Contents/MacOS'
pyinstaller_adj2 = '/AltAnalyze.app/Contents/Resources'
py2app_aa_dirs = [py2app_adj,py2app_adj1,py2exe_adj,py2app_adj2,py2app_adj3,py2app_adj4,cx_Freeze_adj,pyinstaller_adj,pyinstaller_adj2]
py2app_dirs = py2app_ge_dirs + py2app_aa_dirs
for i in py2app_aa_dirs:
i = string.replace(i,'AltAnalyze.app','AltAnalyzeViewer.app')
py2app_dirs.append(i)
if ('linux' in sys.platform or 'posix' in sys.platform) and getattr(sys, 'frozen', False): ### For PyInstaller
application_path = os.path.dirname(sys.executable)
#application_path = sys._MEIPASS ### should be the same as the above
else:
if '..' in __file__:
""" Indicates the file callin unique.py is in a subdirectory """
try:
if '.py' in __file__:
import export
application_path = export.findParentDir(string.split(__file__,'..')[0][:-1])
else:
application_path = os.getcwd()
except Exception:
application_path = os.getcwd()
else:
application_path = os.path.dirname(__file__)
if len(application_path)==0:
application_path = os.getcwd()
if 'AltAnalyze?' in application_path:
application_path = string.replace(application_path,'//','/')
application_path = string.replace(application_path,'\\','/') ### If /// present
application_path = string.split(application_path,'AltAnalyze?')[0]
if 'GO_Elite?' in application_path:
application_path = string.replace(application_path,'//','/')
application_path = string.replace(application_path,'\\','/') ### If /// present
application_path = string.split(application_path,'GO_Elite?')[0]
for py2app_dir in py2app_dirs:
application_path = string.replace(application_path,py2app_dir,'')
def applicationPath():
return application_path
def filepath(filename,force=None):
altDatabaseCheck = True
#dir=os.path.dirname(dirfile.__file__) #directory file is input as a variable under the main
dir = application_path
"""
if os.path.isfile(filename):
fn = filename
return fn
elif os.path.isfile(dir+'/'+filename):
fn = filename
return fn
#"""
""" If a local file without the full path (e.g., Config/options.txt). Checks in the software directory."""
import export
parent_dir = export.findParentDir(filename)
actual_file = export.findFilename(filename)
try:
#if os.path.exists(dir+'/'+parent_dir):
dir_list = os.listdir(dir+'/'+parent_dir)
fn = dir+'/'+parent_dir+'/'+actual_file
if '.txt' in fn or '.log' in fn:
return fn
except:
pass
if filename== '': ### Windows will actually recognize '' as the AltAnalyze root in certain situations but not others
fn = dir
elif ':' in filename:
fn = filename
else:
try:
try:
dir_list = os.listdir(dir+'/'+filename)
fn = dir+'/'+filename
except:
dir_list = os.listdir(filename)
fn = filename ### test to see if the path can be found (then it is the full path)
except Exception:
fn=os.path.join(dir,filename)
fileExists = os.path.isfile(fn)
#print 'filename:',filename, fileExists
""""When AltAnalyze installed through pypi - AltDatabase and possibly Config in user-directory """
if 'Config' in fn:
if fileExists == False and force !='application-path' and ignoreHome==False:
fn=os.path.join(userHomeDir,filename)
if 'AltDatabase' in fn:
getCurrentGeneDatabaseVersion()
fn = correctGeneDatabaseDir(fn)
altanalyze_dir = string.split(fn,'AltDatabase')[0]+'AltDatabase'
### Check the AltDatabase dir not the fn, since the fn may not exist yet
fileExists = os.path.isfile(altanalyze_dir)
try:
dir_list = os.listdir(altanalyze_dir)
fileExists=True
except Exception: pass
#print 2, [fn],fileExists
if fileExists == False and ignoreHome==False:
fn=os.path.join(userHomeDir,filename)
fn = correctGeneDatabaseDir(fn)
altDatabaseCheck = False
if '/Volumes/' in filename and altDatabaseCheck:
filenames = string.split(filename,'/Volumes/'); fn = '/Volumes/'+filenames[-1]
for py2app_dir in py2app_dirs: fn = string.replace(fn,py2app_dir,'')
if (('Databases' in fn) or ('AltDatabase' in fn)) and altDatabaseCheck:
getCurrentGeneDatabaseVersion()
fn = correctGeneDatabaseDir(fn)
fn = string.replace(fn,'.txt.txt','.txt')
fn = string.replace(fn,'//','/')
fn = string.replace(fn,'//','/') ### If /// present
return fn
def read_directory(sub_dir):
dir=application_path
for py2app_dir in py2app_dirs: dir = string.replace(dir,py2app_dir,'')
if 'Databases' in sub_dir or 'AltDatabase' in sub_dir:
getCurrentGeneDatabaseVersion()
sub_dir = correctGeneDatabaseDir(sub_dir)
try: dir_list = os.listdir(dir+sub_dir)
except Exception:
try: dir_list = os.listdir(sub_dir) ### For linux
except Exception:
dir = userHomeDir ### When AltAnalyze installed through pypi - AltDatabase in user-directory
dir_list = os.listdir(dir+sub_dir)
try: dir_list.remove('.DS_Store') ### This is needed on a mac
except Exception: null=[]
#print dir, sub_dir
return dir_list
def returnDirectories(sub_dir):
dir=application_path
if 'Databases' in sub_dir or 'AltDatabase' in sub_dir:
getCurrentGeneDatabaseVersion()
sub_dir = correctGeneDatabaseDir(sub_dir)
for py2app_dir in py2app_dirs:
dir = string.replace(dir,py2app_dir,'')
try: dir_list = os.listdir(dir + sub_dir)
except Exception:
try: dir_list = os.listdir(sub_dir) ### For linux
except Exception: print dir, sub_dir; bad_exit
return dir_list
def returnDirectoriesNoReplace(sub_dir,search=None):
dir=application_path
for py2app_dir in py2app_dirs:
dir = string.replace(dir,py2app_dir,'')
try: dir_list = os.listdir(dir + sub_dir)
except Exception:
try: dir_list = os.listdir(sub_dir) ### For linux
except Exception:
try: dir_list = os.listdir(sub_dir[1:]) ### For linux
except: dir_list = os.listdir(userHomeDir + sub_dir)
if search!=None:
for file in dir_list:
if search in file:
file = string.replace(file,'.lnk','')
dir_list = file
return dir_list
def refDir():
reference_dir=application_path #directory file is input as a variable under the main
for py2app_dir in py2app_dirs:
reference_dir = string.replace(reference_dir,py2app_adj,'')
return reference_dir
def whatProgramIsThis():
reference_dir = refDir()
if 'AltAnalyze' in reference_dir: type = 'AltAnalyze'; database_dir = 'AltDatabase/goelite/'
elif 'GO-Elite' in reference_dir: type = 'GO-Elite'; database_dir = 'Databases/'
else: database_dir = 'AltDatabase/goelite/'; type = 'AltAnalyze'
return type,database_dir
def correctGeneDatabaseDir(fn):
try:
proceed = 'no'
alt_version = 'AltDatabase/'+gene_database_dir
elite_version = 'Databases/'+gene_database_dir
fn=string.replace(fn,'//','/'); fn=string.replace(fn,'\\','/')
if (alt_version not in fn) and (elite_version not in fn): proceed = 'yes' ### If the user creates that contains EnsMart
if gene_database_dir not in fn: proceed = 'yes'
if 'EnsMart' in fn: proceed = 'no'
if proceed == 'yes':
fn = string.replace(fn,'Databases','Databases/'+gene_database_dir)
if 'AltDatabase/affymetrix' not in fn and 'NoVersion' not in fn and 'AltDatabase/primer3' not in fn \
and 'AltDatabase/TreeView' not in fn and 'AltDatabase/kallisto' not in fn and 'AltDatabase/tools' not in fn \
and 'AltDatabase/subreads' not in fn:
if 'AltDatabase' in fn:
fn = string.replace(fn,'AltDatabase','AltDatabase/'+gene_database_dir)
fn = string.replace(fn,'NoVersion','') ### When the text 'NoVersion' is in a filepath, is tells the program to ignore it for adding the database version
except Exception: null = ''
return fn
def getCurrentGeneDatabaseVersion():
global gene_database_dir
try:
filename = 'Config/version.txt'
fn=filepath(filename)
for line in open(fn,'r').readlines():
gene_database_dir, previous_date = string.split(line,'\t')
except Exception:
import UI
gene_database_dir=''
try:
for db_version in db_versions:
if 'EnsMart' in db_version:
gene_database_dir = db_version; UI.exportDBversion(db_version)
break
except Exception:
pass
return gene_database_dir
def unique(s):
#we need to remove duplicates from a list, unsuccessfully tried many different methods
#so I found the below function at: http://aspn.activestate.com/ASPN/Cookbook/Python/Recipe/52560
n = len(s)
if n == 0: return []
u = {}
try:
for x in s: u[x] = 1
except TypeError: del u # move on to the next method
else: return u.keys()
try: t = list(s); t.sort()
except TypeError: del t # move on to the next method
else:
assert n > 0
last = t[0]; lasti = i = 1
while i < n:
if t[i] != last: t[lasti] = last = t[i]; lasti += 1
i += 1
return t[:lasti]
u = []
for x in s:
if x not in u: u.append(x)
return u
def dictionary(s):
d={}
for i in s:
try: d[i]=[]
except TypeError: d[tuple(i)]=[]
return d
def unique_db(s):
d={}; t=[]
for i in s:
try: d[i]=[]
except TypeError: d[tuple(i)]=[]
for i in d: t.append(i)
return t
def list(d):
t=[]
for i in d: t.append(i)
return t
def exportVersionData(version,version_date,dir,force='application-path'):
new_file = dir+'version.txt'
new_file_default = filepath(new_file,force=force) ### can use user directory local or application local
print new_file_default;sys.exit()
try:
data.write(str(version)+'\t'+str(version_date)+'\n'); data.close()
except:
data = export.ExportFile(new_file)
data.write(str(version)+'\t'+str(version_date)+'\n'); data.close()
if __name__ == '__main__':
#db = returnDirectoriesNoReplace('/AltDatabase',search='EnsMart')
exportVersionData('EnsMart72','12/17/19','Config/')
path = filepath('Config/',force=None)
print path
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/unique.py
|
unique.py
|
import sys, string
import os.path
import unique
import export
import gene_associations
import traceback
import time
################# Parse directory files
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir)
#add in code to prevent folder names from being included
dir_list2 = []
for file in dir_list:
lf = string.lower(file)
if '.txt' in lf or '.sif' in lf or '.tab' in lf: dir_list2.append(file)
return dir_list2
################# Begin Analysis from parsing files
def getEnsemblGeneData(filename):
fn=filepath(filename)
global ensembl_symbol_db; ensembl_symbol_db={}; global symbol_ensembl_db; symbol_ensembl_db={}
for line in open(fn,'rU').xreadlines():
data,null = string.split(line,'\n')
t = string.split(data,'\t')
ensembl=t[0];symbol=t[1]
### Have to do this in order to get the WEIRD chromosomal assocaitions and the normal to the same genes
try: symbol_ensembl_db[symbol].append(ensembl)
except Exception: symbol_ensembl_db[symbol] = [ensembl]
try: symbol_ensembl_db[string.lower(symbol)].append(ensembl)
except Exception: symbol_ensembl_db[string.lower(symbol)] = [ensembl]
try: symbol_ensembl_db[symbol.title()].append(ensembl)
except Exception: symbol_ensembl_db[symbol.title()] = [ensembl]
ensembl_symbol_db[ensembl] = symbol
def getHMDBData(species):
program_type,database_dir = unique.whatProgramIsThis()
filename = database_dir+'/'+species+'/gene/HMDB.txt'
x=0
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if x==0: x=1
else:
t = string.split(data,'\t')
try: hmdb_id,symbol,description,secondary_id,iupac,cas_number,chebi_id,pubchem_compound_id,Pathways,ProteinNames = t
except Exception:
### Bad Tab introduced from HMDB
hmdb_id = t[0]; symbol = t[1]; ProteinNames = t[-1]
symbol_hmdb_db[symbol]=hmdb_id
hmdb_symbol_db[hmdb_id] = symbol
ProteinNames=string.split(ProteinNames,',')
### Add gene-metabolite interactions to databases
for protein_name in ProteinNames:
try:
for ensembl in symbol_ensembl_db[protein_name]:
z = InteractionInformation(hmdb_id,ensembl,'HMDB','Metabolic')
interaction_annotation_dbase[ensembl,hmdb_id] = z ### This is the interaction direction that is appropriate
try: interaction_db[hmdb_id][ensembl]=1
except KeyError: db = {ensembl:1}; interaction_db[hmdb_id] = db ###weight of 1 (weights currently not-supported)
try: interaction_db[ensembl][hmdb_id]=1
except KeyError: db = {hmdb_id:1}; interaction_db[ensembl] = db ###weight of 1 (weights currently not-supported)
except Exception: None
def verifyFile(filename):
status = 'not found'
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = 'found';break
except Exception: status = 'not found'
return status
def importInteractionDatabases(interactionDirs):
""" Import multiple interaction format file types (designated by the user) """
exclude=[]
for file in interactionDirs:
status = verifyFile(file)
if status == 'not found':
exclude.append(file)
for i in exclude:
interactionDirs.remove(i)
for fn in interactionDirs: #loop through each file in the directory to output results
x=0; imported=0; stored=0
file = export.findFilename(fn)
count=0
print "Parsing interactions from:",file
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
count+=1
if x==0: x=1
#elif 'PAZAR' in data or 'Amadeus' in data:x+=0
else:
obligatory = False
imported+=1
proceed = True
source=''
interaction_type = 'interaction'
try:
symbol1,interaction_type, symbol2, ensembl1,ensembl2,source = t
ens_ls1=[ensembl1]; ens_ls2=[ensembl2]
if 'HMDB' in ensembl1:
ensembl1 = string.replace(ensembl1,' ','') ### HMDB ID sometimes proceeded by ' '
symbol_hmdb_db[symbol1]=ensembl1
hmdb_symbol_db[ensembl1] = symbol1
interaction_type = 'Metabolic'
if 'HMDB' in ensembl2:
ensembl2 = string.replace(ensembl2,' ','') ### HMDB ID sometimes proceeded by ' '
symbol_hmdb_db[symbol2]=ensembl2
hmdb_symbol_db[ensembl2] = symbol2
interaction_type = 'Metabolic'
except Exception:
try:
ensembl1,ensembl2,symbol1,symbol2,interaction_type=t
if ensembl1 == '':
try:
ens_ls1 = symbol_ensembl_db[symbol1]
ens_ls2 = symbol_ensembl_db[symbol2]
except Exception: None
except Exception:
proceed = False
if proceed: ### If the interaction data conformed to one of the two above types (typically two valid interacting gene IDs)
if (len(ens_ls1)>0 and len(ens_ls2)>0):
secondary_proceed = True
stored+=1
for ensembl1 in ens_ls1:
for ensembl2 in ens_ls2:
"""
if (ensembl1,ensembl2) == ('ENSG00000111704','ENSG00000152284'):
print t;sys.exit()
if (ensembl1,ensembl2) == ('ENSG00000152284','ENSG00000111704'):
print t;sys.exit()
"""
if 'WikiPathways' in file or 'KEGG' in file:
if ensembl2 != ensembl1:
if (ensembl2,ensembl1) in interaction_annotation_dbase:
del interaction_annotation_dbase[(ensembl2,ensembl1)]
### Exclude redundant entries with fewer interaction details (e.g., arrow direction BIOGRID) - overwrite with the opposite gene arrangement below
if (ensembl1,ensembl2) in interaction_annotation_dbase:
if interaction_annotation_dbase[(ensembl1,ensembl2)].InteractionType() !='physical':
secondary_proceed = False ### Don't overwrite a more informative annotation like transcriptional regulation or microRNA targeting
if 'DrugBank' in fn:
source = 'DrugBank'
interaction_type = 'drugInteraction'
obligatory=True
ensembl1, ensembl2 = ensembl2, ensembl1 ### switch the order of these (drugs reported as first ID and gene as the second)
if secondary_proceed:
z = InteractionInformation(ensembl1,ensembl2,source,interaction_type)
interaction_annotation_dbase[ensembl1,ensembl2] = z
#z = InteractionInformation(ensembl2,ensembl1,source,interaction_type)
#interaction_annotation_dbase[ensembl2,ensembl1] = z
try: interaction_db[ensembl1][ensembl2]=1
except KeyError: db = {ensembl2:1}; interaction_db[ensembl1] = db ###weight of 1 (weights currently not-supported)
try: interaction_db[ensembl2][ensembl1]=1
except KeyError: db = {ensembl1:1}; interaction_db[ensembl2] = db ###weight of 1 (weights currently not-supported)
if obligatory and source in obligatoryList: ### Include these in the final pathway if linked to any input node (e.g., miRNAs, drugs)
try: obligatory_interactions[ensembl1][ensembl2]=1
except KeyError: db = {ensembl2:1}; obligatory_interactions[ensembl1] = db ###weight of 1 (weights currentlynot-supported)
elif source in secondDegreeObligatoryCategories:
try: second_degree_obligatory[ensembl1][ensembl2]=1
except KeyError: db = {ensembl2:1}; second_degree_obligatory[ensembl1] = db ###weight of 1 (weights currently not-supported)
else:
proceed = False
try:
ID1, null, ID2 = t
proceed = True
except Exception:
try:
ID1, ID2 = t
proceed = True
except Exception:
None
if proceed:
if 'microRNATargets' in fn:
if 'mir' in ID2: prefix = 'MIR'
else: prefix = 'LET'
ID2='MIR'+string.split(ID2,'-')[2] ### Ensembl naming convention
source = 'microRNATargets'
interaction_type = 'microRNAInteraction'
obligatory=True
try: ID_ls1 = symbol_ensembl_db[ID1]
except Exception: ID_ls1 = [ID1]
try: ID_ls2 = symbol_ensembl_db[ID2]
except Exception: ID_ls2 = [ID2]
"""if 'microRNATargets' in fn:
if '*' not in ID2: print ID_ls2;sys.exit()"""
addInteractions = True
for ID1 in ID_ls1:
for ID2 in ID_ls2:
z = InteractionInformation(ID2,ID1,source,interaction_type)
interaction_annotation_dbase[ID2,ID1] = z ### This is the interaction direction that is appropriate
try: interaction_db[ID1][ID2]=1
except KeyError: db = {ID2:1}; interaction_db[ID1] = db ###weight of 1 (weights currently supported)
try: interaction_db[ID2][ID1]=1
except KeyError: db = {ID1:1}; interaction_db[ID2] = db ###weight of 1 (weights currently supported)
if source in secondDegreeObligatoryCategories:
try: second_degree_obligatory[ID1][ID2]=1
except KeyError: db = {ID2:1}; second_degree_obligatory[ID1] = db ###weight of 1 (weights currently supported)
elif obligatory and source in obligatoryList: ### Include these in the final pathway if linked to any input node (e.g., miRNAs, drugs)
try: obligatory_interactions[ID1][ID2]=1
except KeyError: db = {ID2:1}; obligatory_interactions[ID1] = db ###weight of 1 (weights currently supported)
### Evaluate the most promiscous interactors (e.g., UBC)
remove_list=[]
for ID in interaction_db:
if len(interaction_db[ID])>20000:
remove_list.append(ID)
#print len(interaction_db[ID]),ensembl_symbol_db[ID]
for ID in remove_list:
#print 'removing', ID
del interaction_db[ID]
blackList[ID] = []
print 'Imported interactions:',len(interaction_annotation_dbase)
class InteractionInformation:
def __init__(self, ensembl1, ensembl2, source, interaction_type):
self._ensembl1 = ensembl1; self._ensembl2 = ensembl2; self._source = source
self._interaction_type = interaction_type
def Ensembl1(self): return self._ensembl1
def Ensembl2(self): return self._ensembl2
def Source(self): return self._source
def InteractionType(self): return self._interaction_type
def Report(self):
output = self.Ensembl1()+'|'+self.Ensembl2()
return output
def __repr__(self): return self.Report()
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def importqueryResults(species,dir_file,id_db):
global query_db; query_db = {}
query_interactions={} ### This is the final list of shown interactions
if dir_file == None:
fileRead = dir_file
elif '.' in dir_file:
fn=filepath(dir_file)
fileRead = open(fn,'rU').xreadlines()
else:
fileRead = dir_file ### This is a list of IDs passed to this function rather than in a file
if len(id_db)==0: ### Otherwise, already provided gene IDs to query
translated=0
count=0
try:
x=0
for line in fileRead:
count+=1
try:
data = cleanUpLine(line)
t = string.split(data,'\t')
except Exception:
t = line
if x==1: x = 1 ### no longer statement since the first row may be a valid ID(s)
else:
id = t[0]
ensembl_ls1=[]
if id in ensembl_symbol_db:
symbol = ensembl_symbol_db[id]
query_db[id] = symbol
ensembl_ls1 = [id]
translated+=1
elif id in symbol_ensembl_db:
ensembl_ls1 = symbol_ensembl_db[id]
translated+=1
for ensembl in ensembl_ls1:
query_db[ensembl] = id
elif id in symbol_hmdb_db:
hmdb = symbol_hmdb_db[id]
query_db[hmdb] = id
elif id in hmdb_symbol_db:
symbol = hmdb_symbol_db[id]
query_db[id] = symbol
else:
query_db[id] = id ### Currently not dealt with
ensembl_ls1 = [id]
### If a SIF file add genes and interactions
if len(t)>1 and 'SIF' in inputDataType: ### Potentially SIF format
interaction_type = t[1]
try: id2 = t[2]
except Exception: id2 = t[1]; interaction_type = 'undetermined'
ensembl_ls2=[]
if id2 in ensembl_symbol_db:
symbol = ensembl_symbol_db[id2]
query_db[id2] = symbol
ensembl_ls2 = [id2]
elif id2 in symbol_ensembl_db:
ensembl_ls2 = symbol_ensembl_db[id2]
for ensembl in ensembl_ls2:
query_db[ensembl] = id2
elif id2 in symbol_hmdb_db:
hmdb = symbol_hmdb_db[id2]
query_db[hmdb] = id2
elif id2 in hmdb_symbol_db:
symbol = hmdb_symbol_db[id2]
query_db[id2] = symbol
else:
query_db[id2] = id2
for ensembl1 in ensembl_ls1:
for ensembl2 in ensembl_ls2:
try: query_interactions[ensembl1].append(ensembl2)
except Exception: query_interactions[ensembl1] = [ensembl2]
z = InteractionInformation(ensembl1,ensembl2,'custom',interaction_type)
interaction_annotation_dbase[ensembl1,ensembl2] = z
except Exception:
print traceback.format_exc()
print 'No valid directories or IDs provided. Exiting.'; kill
if translated==0:
from visualization_scripts import WikiPathways_webservice
try: query_db = WikiPathways_webservice.importDataSimple(dir_file,None,MOD='Ensembl',Species=species)[0]
except Exception: ### If metabolomics
query_db = WikiPathways_webservice.importDataSimple(dir_file,None,MOD='HMDB',Species=species)[0]
### Translate the Ensembl IDs to symbols (where possible)
for id in query_db:
if id in ensembl_symbol_db:
symbol = ensembl_symbol_db[id]
else:
symbol=id
query_db[id] = symbol
else:
for id in id_db:
if id_db[id]==None:
try: id_db[id] = ensembl_symbol_db[id] ### Save symbol (done for imported pathway genes)
except Exception: id_db[id]=id
query_db = id_db ### Input gene IDs (not in a file)
print 'Number of IDs from', dir_file, 'is', len(query_db)
return query_db,query_interactions,dir_file
def associateQueryGenesWithInteractions(query_db,query_interactions,dir_file):
suffix=''
if dir_file!=None:
if len(dir_file)!=0:
suffix='-'+intNameShort+'_'+export.findFilename(dir_file)[:-4]
if len(suffix)==0:
try: suffix = '_'+FileName
except Exception: None
file_name = 'AltAnalyze-network'+suffix
query_interactions_unique={}
interacting_genes={}
connections = 1
primary=0
secondary=0
terciary=0
for ensemblGene in query_db:
if ensemblGene in interaction_db:
for interacting_ensembl in interaction_db[ensemblGene]:
if interacting_ensembl not in blackList:
###Only allow direct interactions found in query
if interacting_ensembl in query_db:
try: query_interactions[ensemblGene].append(interacting_ensembl)
except KeyError: query_interactions[ensemblGene] = [interacting_ensembl]
try: query_interactions[interacting_ensembl].append(ensemblGene)
except KeyError: query_interactions[interacting_ensembl] = [ensemblGene]
primary+=1
if degrees == 2 or degrees == 'indirect':
try: interacting_genes[interacting_ensembl].append(ensemblGene)
except KeyError: interacting_genes[interacting_ensembl] = [ensemblGene]
elif degrees == 'allInteracting' or degrees == 'all possible':
try: query_interactions[ensemblGene].append(interacting_ensembl)
except KeyError: query_interactions[ensemblGene] = [interacting_ensembl]
if interacting_ensembl in secondaryQueryIDs: ### IDs in the expression file
secondary+=1 ### When indirect degrees selected, no additional power added by this (only for direct or shortest path)
try: query_interactions[ensemblGene].append(interacting_ensembl)
except KeyError: query_interactions[ensemblGene] = [interacting_ensembl]
if ensemblGene in second_degree_obligatory:
for interacting_ensembl in second_degree_obligatory[ensemblGene]:
try: interacting_genes[interacting_ensembl].append(ensemblGene)
except KeyError: interacting_genes[interacting_ensembl] = [ensemblGene]
### Include indirect interactions to secondaryQueryIDs from the expression file
if degrees == 2 or degrees == 'indirect':
for ensemblGene in secondaryQueryIDs:
if ensemblGene in interaction_db:
for interacting_ensembl in interaction_db[ensemblGene]:
if interacting_ensembl not in blackList:
try:
interacting_genes[interacting_ensembl].append(ensemblGene)
terciary+=1#; print interacting_ensembl
except KeyError: None ### Only increase the interacting_genes count if the interacting partner is present from the primary query list
#print primary,secondary,terciary
### Report the number of unique interacting genes
for interacting_ensembl in interacting_genes:
if len(interacting_genes[interacting_ensembl])==1:
interacting_genes[interacting_ensembl] = 1
else:
unique_interactions = unique.unique(interacting_genes[interacting_ensembl])
interacting_genes[interacting_ensembl] = len(unique_interactions)
query_indirect_interactions={}; indirect_interacting_gene_list=[]; interacting_gene_list=[]; added=[]
if degrees=='shortestPath' or degrees=='shortest path': ### Typically identifying the single smallest path(s) between two nodes.
query_indirect_interactions, indirect_interacting_gene_list, interacting_gene_list = evaluateShortestPath(query_db,interaction_db,10)
else:
if degrees==2 or degrees=='indirect' or len(secondDegreeObligatoryCategories)>0:
for ensembl in interacting_genes:
if interacting_genes[ensembl] > connections:
if ensembl in interaction_db: ### Only nodes removed due to promiscuity will not be found
for interacting_ensembl in interaction_db[ensembl]:
if interacting_ensembl in query_db or interacting_ensembl in secondaryQueryIDs:
try: query_indirect_interactions[interacting_ensembl].append(ensembl)
except KeyError: query_indirect_interactions[interacting_ensembl] = [ensembl]
###Record the highest linked nodes
indirect_interacting_gene_list.append((interacting_genes[ensembl],ensembl))
if len(obligatory_interactions)>0: ### Include always
all_reported_genes = combineDBs(query_interactions,query_indirect_interactions) ### combinesDBs and returns a unique list of genes
for ensemblGene in all_reported_genes: ###This only includes genes in the original input list
if ensemblGene in obligatory_interactions:
for interacting_ensembl in obligatory_interactions[ensemblGene]:
#symbol = ensembl_symbol_db[ensemblGene]
try: query_interactions[ensemblGene].append(interacting_ensembl)
except KeyError: query_interactions[ensemblGene] = [interacting_ensembl]
z = dict(query_interactions.items() + query_indirect_interactions.items())
interaction_restricted_db={}
for ensembl in z:
interacting_nodes = z[ensembl]
for node in interacting_nodes:
if ensembl in interaction_restricted_db:
db = interaction_restricted_db[ensembl]
db[node] = 1
else: interaction_restricted_db[ensembl] = {node:1}
if node in interaction_restricted_db:
db = interaction_restricted_db[node]
db[ensembl] = 1
else: interaction_restricted_db[node] = {ensembl:1}
if degrees==2 or degrees=='indirect': ### get rid of non-specific interactions
query_indirect_interactions, indirect_interacting_gene_list, interacting_gene_list = evaluateShortestPath(query_db,interaction_restricted_db,4)
###Record the highest linked nodes
for ensembl in query_interactions:
linked_nodes = len(unique.unique(query_interactions[ensembl]))
interacting_gene_list.append((linked_nodes,ensembl))
interacting_gene_list.sort(); interacting_gene_list.reverse()
indirect_interacting_gene_list.sort(); indirect_interacting_gene_list.reverse()
print "Length of query_interactions:",len(query_interactions)
query_interactions_unique=[]
for gene1 in query_interactions:
for gene2 in query_interactions[gene1]:
temp = []; temp.append(gene2); temp.append(gene1)#; temp.sort()
if gene1 == gene2: interaction_type = 'self'
else: interaction_type = 'distinct'
temp.append(interaction_type); temp.reverse()
query_interactions_unique.append(temp)
for gene1 in query_indirect_interactions:
for gene2 in query_indirect_interactions[gene1]:
temp = []; temp.append(gene2); temp.append(gene1)#; temp.sort()
if gene1 == gene2: interaction_type = 'self'
else: interaction_type = 'indirect'
temp.append(interaction_type); temp.reverse()
query_interactions_unique.append(temp)
query_interactions_unique = unique.unique(query_interactions_unique)
query_interactions_unique.sort()
###Write out nodes linked to many other nodes
new_file = outputDir+'/networks/'+file_name+ '-interactions_'+str(degrees)+'_degrees_summary.txt'
data = export.ExportFile(new_file)
for (linked_nodes,ensembl) in interacting_gene_list:
try: symbol = query_db[ensembl]
except KeyError: symbol = ensembl_symbol_db[ensembl]
data.write(str(linked_nodes)+'\t'+ensembl+'\t'+symbol+'\t'+'direct'+'\n')
for (linked_nodes,ensembl) in indirect_interacting_gene_list:
try: symbol = query_db[ensembl]
except KeyError:
try: symbol = ensembl_symbol_db[ensembl]
except KeyError: symbol = ensembl
if 'HMDB' in symbol:
try: symbol = hmdb_symbol_db[ensembl]
except Exception: pass
data.write(str(linked_nodes)+'\t'+ensembl+'\t'+symbol+'\t'+'indirect'+'\n')
data.close()
regulated_gene_db = query_db
sif_export,symbol_pair_unique = exportInteractionData(file_name,query_interactions_unique,regulated_gene_db)
return sif_export,symbol_pair_unique
def combineDBs(db1,db2):
### combinesDBs and returns a unique list of genes
new_db={}
for i in db1:
new_db[i]=[]
for k in db1[i]:
new_db[k]=[]
for i in db2:
new_db[i]=[]
for k in db2[i]:
new_db[k]=[]
return new_db
def evaluateShortestPath(query_db,interaction_restricted_db,depth):
interactions_found=0
start_time = time.time()
query_indirect_interactions={}; indirect_interacting_gene_list=[]; interacting_gene_list=[]; added=[]
print 'Performing shortest path analysis on %s IDs...' % len(query_db),
for gene1 in query_db:
for gene2 in query_db:
if (gene1,gene2) not in added and (gene2,gene1) not in added:
if gene1 != gene2 and gene1 in interaction_restricted_db and gene2 in interaction_restricted_db:
try:
path = shortest_path(interaction_restricted_db,gene1,gene2,depth)
added.append((gene1,gene2))
i=1
while i<len(path): ### Add the relationship pairs
try: query_indirect_interactions[path[i-1]].append(path[i])
except Exception: query_indirect_interactions[path[i-1]]=[path[i]]
interactions_found+=1
i+=1
except Exception:
#tb = traceback.format_exc()
pass
if len(query_indirect_interactions)==0:
print 'None of the query genes interacting in the selected interaction databases...'; queryGeneError
print interactions_found, 'interactions found in', time.time()-start_time, 'seconds'
return query_indirect_interactions, indirect_interacting_gene_list, interacting_gene_list
def shortest_path(G, start, end, depth):
#http://code.activestate.com/recipes/119466-dijkstras-algorithm-for-shortest-paths/
import heapq
def flatten(L): # Flatten linked list of form [0,[1,[2,[]]]]
while len(L) > 0:
yield L[0]
L = L[1]
q = [(0, start, ())] # Heap of (cost, path_head, path_rest).
visited = set() # Visited vertices.
while True:
(cost, v1, path) = heapq.heappop(q)
if v1 not in visited and v1 in G:
visited.add(v1)
if v1 == end:
final_path = list(flatten(path))[::-1] + [v1]
if len(final_path)<depth:
return final_path
else:
return None
path = (v1, path)
for (v2, cost2) in G[v1].iteritems():
if v2 not in visited:
heapq.heappush(q, (cost + cost2, v2, path))
def exportInteractionData(file_name,query_interactions_unique,regulated_gene_db):
file_name = string.replace(file_name,':','-')
new_file = outputDir+'/networks/'+file_name + '-interactions_'+str(degrees)+'.txt'
sif_export = outputDir+'/networks/'+file_name + '-interactions_'+str(degrees)+'.sif'
fn=filepath(new_file); fn2=filepath(sif_export)
data = open(fn,'w'); data2 = open(fn2,'w')
added = {} ### Don't add the same entry twice
symbol_added={}; symbol_pair_unique={}
for (interaction_type,gene1,gene2) in query_interactions_unique:
try: symbol1 = query_db[gene1]
except KeyError:
try: symbol1 = ensembl_symbol_db[gene1]
except KeyError: symbol1 = gene1
if 'HMDB' in symbol1:
symbol1 = hmdb_symbol_db[gene1]
try: symbol2 = query_db[gene2]
except KeyError:
try: symbol2 = ensembl_symbol_db[gene2]
except KeyError: symbol2 = gene2
if 'HMDB' in symbol2:
symbol2 = hmdb_symbol_db[gene2]
gene_pair = ''; symbol_pair=''; direction = 'interactsWith'
if (gene1,gene2) in interaction_annotation_dbase: gene_pair = gene1,gene2; symbol_pair = symbol1,symbol2
elif (gene2,gene1) in interaction_annotation_dbase: gene_pair = gene2,gene1; symbol_pair = symbol2,symbol1
else: print gene1, gene2, symbol1, symbol2; kill
if len(gene_pair)>0:
y = interaction_annotation_dbase[gene_pair]
gene1,gene2 = gene_pair ### This is the proper order of the interaction
symbol1,symbol2 = symbol_pair
interaction_type = y.InteractionType()
if interaction_type == 'drugInteraction':
### Switch their order
gene1, gene2, symbol1, symbol2 = gene2, gene1, symbol2, symbol1
direction = interaction_type
if (gene_pair,direction) not in added:
added[(gene_pair,direction)]=[]
data.write(gene1+'\t'+gene2+'\t'+symbol1+'\t'+symbol2+'\t'+interaction_type+'\n')
if len(symbol1)>1 and len(symbol2)>1 and (symbol_pair,direction) not in symbol_added:
if symbol1 != symbol2:
data2.write(symbol1+'\t'+direction+'\t'+symbol2+'\n')
symbol_added[(symbol_pair,direction)]=[]
symbol_pair_unique[symbol_pair]=[]
data.close(); data2.close()
print "Interaction data exported"
return sif_export,symbol_pair_unique
def eliminate_redundant_dict_values(database):
db1={}
for key in database:
list = unique.unique(database[key])
list.sort()
db1[key] = list
return db1
def importInteractionData(interactionDirs):
global interaction_db; interaction_db = {}
global interaction_annotation_dbase; interaction_annotation_dbase = {}
global obligatory_interactions; obligatory_interactions={}
global second_degree_obligatory; second_degree_obligatory={}
global blackList; blackList = {}
###Collect both Human and Mouse interactions (Mouse directly sorted in interaction_db
importInteractionDatabases(interactionDirs)
def interactionPermuteTest(species,Degrees,inputType,inputDir,outputdir,interactionDirs,Genes=None,
geneSetType=None,PathwayFilter=None,OntologyID=None,directory=None,expressionFile=None,
obligatorySet=None,secondarySet=None,IncludeExpIDs=False):
global degrees
global outputDir
global inputDataType
global obligatoryList ### Add these if connected to anything
global secondaryQueryIDs
global secondDegreeObligatoryCategories ### Add if common to anything in the input - Indicates systems to apply this to
global symbol_hmdb_db; symbol_hmdb_db={}; global hmdb_symbol_db; hmdb_symbol_db={} ### Create an annotation database for HMDB IDs
global FileName
secondaryQueryIDs = {}
degrees = Degrees
outputDir = outputdir
inputDataType = inputType
obligatoryList = obligatorySet
secondDegreeObligatoryCategories=[]
if obligatoryList == None:
obligatoryList=[]
if expressionFile == None:
expressionFile = inputDir ### If it doesn't contain expression values, view as yellow nodes
if secondarySet!= None and (degrees==1 or degrees=='direct'): ### If degrees == 2, this is redundant
### This currently adds alot of predictions - either make more stringent or currently exclude
secondDegreeObligatoryCategories = secondarySet
if PathwayFilter != None: FileName = PathwayFilter
elif OntologyID != None: FileName = OntologyID
elif Genes != None: FileName = Genes
### Import Ensembl-Symbol annotations
getEnsemblGeneData('AltDatabase/ensembl/'+species+'/'+species+'_Ensembl-annotations.txt')
### Import interaction databases indicated in interactionDirs
importInteractionData(interactionDirs)
getHMDBData(species) ### overwrite the symbol annotation from any HMDB that comes from a WikiPathway or KEGG pathway that we also include (for consistent official annotation)
input_IDs = getGeneIDs(Genes)
try: input_IDs = gene_associations.simpleGenePathwayImport(species,geneSetType,PathwayFilter,OntologyID,directory)
except Exception: None
permutations = 10000; p = 0
secondaryQueryIDs = importqueryResults(species,expressionFile,{})[0]
input_IDs,query_interactions,dir_file = importqueryResults(species,inputDir,input_IDs) ### Get the number of unique genes
sif_file, original_symbol_pair_unique = associateQueryGenesWithInteractions(input_IDs,query_interactions,dir_file)
#print len(original_symbol_pair_unique)
ensembl_unique = map(lambda x: x, ensembl_symbol_db)
interaction_lengths = []
import random
while p < permutations:
random_inputs = random.sample(ensembl_unique,len(input_IDs))
random_input_db={}
#print len(random_inputs), len(input_IDs); sys.exit()
for i in random_inputs: random_input_db[i]=i
secondaryQueryIDs = importqueryResults(species,random_inputs,{})[0]
input_IDs,query_interactions,dir_file = importqueryResults(species,inputDir,input_IDs)
sif_file, symbol_pair_unique = associateQueryGenesWithInteractions(input_IDs,query_interactions,inputDir)
#print len(symbol_pair_unique);sys.exit()
interaction_lengths.append(len(symbol_pair_unique))
p+=1
interaction_lengths.sort(); interaction_lengths.reverse()
y = len(original_symbol_pair_unique)
print 'permuted length distribution:',interaction_lengths
print 'original length:',y
k=0
for i in interaction_lengths:
if i>=y: k+=1
print 'p-value:',float(k)/float(permutations)
def buildInteractions(species,Degrees,inputType,inputDir,outputdir,interactionDirs,Genes=None,
geneSetType=None,PathwayFilter=None,OntologyID=None,directory=None,expressionFile=None,
obligatorySet=None,secondarySet=None,IncludeExpIDs=False):
global degrees
global outputDir
global inputDataType
global obligatoryList ### Add these if connected to anything
global secondaryQueryIDs
global secondDegreeObligatoryCategories ### Add if common to anything in the input - Indicates systems to apply this to
global symbol_hmdb_db; symbol_hmdb_db={}; global hmdb_symbol_db; hmdb_symbol_db={} ### Create an annotation database for HMDB IDs
global FileName
global intNameShort
secondaryQueryIDs = {}
degrees = Degrees
outputDir = outputdir
inputDataType = inputType
obligatoryList = obligatorySet
secondDegreeObligatoryCategories=[]
intNameShort=''
if obligatoryList == None:
obligatoryList=[]
if expressionFile == None:
expressionFile = inputDir ### If it doesn't contain expression values, view as yellow nodes
if secondarySet!= None and (degrees==1 or degrees=='direct'): ### If degrees == 2, this is redundant
### This currently adds alot of predictions - either make more stringent or currently exclude
secondDegreeObligatoryCategories = secondarySet
if PathwayFilter != None:
if len(PathwayFilter)==1:
FileName = PathwayFilter[0]
if isinstance(PathwayFilter, tuple) or isinstance(PathwayFilter, list):
FileName = string.join(list(PathwayFilter),' ')
FileName = string.replace(FileName,':','-')
else:
FileName = PathwayFilter
if len(FileName)>40:
FileName = FileName[:40]
elif OntologyID != None: FileName = OntologyID
elif Genes != None: FileName = Genes
### Import Ensembl-Symbol annotations
getEnsemblGeneData('AltDatabase/ensembl/'+species+'/'+species+'_Ensembl-annotations.txt')
if len(interactionDirs[0]) == 1: interactionDirs = [interactionDirs]
### Import interaction databases indicated in interactionDirs
for i in interactionDirs:
print i
i = export.findFilename(i)
i=string.split(i,'-')[1]
intNameShort+=i[0]
importInteractionData(interactionDirs)
getHMDBData(species) ### overwrite the symbol annotation from any HMDB that comes from a WikiPathway or KEGG pathway that we also include (for consistent official annotation)
input_IDs = getGeneIDs(Genes)
try:
if isinstance(PathwayFilter, tuple):
for pathway in PathwayFilter:
IDs = gene_associations.simpleGenePathwayImport(species,geneSetType,pathway,OntologyID,directory)
for id in IDs:input_IDs[id]=None
else:
input_IDs = gene_associations.simpleGenePathwayImport(species,geneSetType,PathwayFilter,OntologyID,directory)
except Exception: None
if expressionFile == None or len(expressionFile)==0:
expressionFile = exportSelectedIDs(input_IDs) ### create an expression file
elif IncludeExpIDs: ### Prioritize selection of IDs for interactions WITH the primary query set (not among expression input IDs)
secondaryQueryIDs = importqueryResults(species,expressionFile,{})[0]
input_IDs,query_interactions,dir_file = importqueryResults(species,inputDir,input_IDs)
sif_file,symbol_pair_unique = associateQueryGenesWithInteractions(input_IDs,query_interactions,dir_file)
output_filename = exportGraphImage(species,sif_file,expressionFile)
return output_filename
def exportSelectedIDs(input_IDs):
expressionFile = outputDir+'/networks/IDList.txt'
data = export.ExportFile(expressionFile)
data.write('UID\tSystemCode\n')
for id in input_IDs:
if 'HMDB' in id:
id = hmdb_symbol_db[id]
data.write(id+'\tEn\n')
data.close()
return expressionFile
def exportGraphImage(species,sif_file,expressionFile):
from visualization_scripts import clustering
output_filename = clustering.buildGraphFromSIF('Ensembl',species,sif_file,expressionFile)
return output_filename
def getGeneIDs(Genes):
input_IDs={}
if Genes == None: None
elif len(Genes)>0:
### Get IDs from list of gene IDs
Genes=string.replace(Genes,'|',',')
Genes=string.replace(Genes,' ',',')
if ',' in Genes: Genes = string.split(Genes,',')
else: Genes = [Genes]
for i in Genes:
if len(i)>0:
if i in symbol_ensembl_db:
for ensembl in symbol_ensembl_db[i]:
input_IDs[ensembl]=i ### Translate to Ensembl
elif i in symbol_hmdb_db:
hmdb=symbol_hmdb_db[i]
symbol = hmdb_symbol_db[hmdb] ### Get the official symbol
input_IDs[hmdb]=symbol ### Translate to HMDB
else:
try: input_IDs[i] = ensembl_symbol_db[i] ### If an input Ensembl ID
except Exception: input_IDs[i] = i ### Currently not dealt with
return input_IDs
def remoteBuildNetworks(species, outputDir, interactions=['WikiPathways','KEGG','TFTargets'],degrees='direct'):
""" Attempts to output regulatory/interaction networks from a directory of input files """
print degrees
directory = 'gene-mapp'
interactionDirs=[]
obligatorySet=[] ### Always include interactions from these if associated with any input ID period
secondarySet=[]
inputType = 'IDs'
for i in interactions:
fn = filepath('AltDatabase/goelite/'+species+'/gene-interactions/Ensembl-'+i+'.txt')
interactionDirs.append(fn)
pdfs=[]
dir_list = read_directory(outputDir)
for file in dir_list:
if 'GE.' in file:
input_file_dir = outputDir+'/'+file
try:
output_filename = buildInteractions(species,degrees,inputType,input_file_dir,outputDir,interactionDirs,
directory=outputDir,expressionFile=input_file_dir, IncludeExpIDs=True)
try: pdfs.append(output_filename[:-4]+'.pdf')
except: pass
except: pass
return pdfs
if __name__ == '__main__':
import getopt
species = 'Hs'
degrees = 'direct'
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Insufficient options provided";sys.exit()
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['i=','species=', 'degrees='])
for opt, arg in options:
if opt == '--i': output_dir=arg
elif opt == '--species': species=arg
elif opt == '--degrees': degrees = arg
remoteBuildNetworks(species,output_dir,degrees=degrees); sys.exit()
remoteBuildNetworks('Mm', '/Users/saljh8/Desktop/DemoData/cellHarmony/Mouse_BoneMarrow/inputFile/cellHarmony/DifferentialExpression_Fold_2.0_adjp_0.05')
sys.exit()
Species = 'Hs'
Degrees = 2
inputType = 'IDs'
inputDir=''
inputDir='/Users/nsalomonis/Desktop/dataAnalysis/Sarwal/Urine-AR-increased/met/networks/AltAnalyze-network_Met.inceased_AR_1.5fold_metabolite-interactions_shortest path.sif'
inputDir='/Users/saljh8/Documents/1-dataAnalysis/PaulTang/ARVC_genes.txt'
obligatorySet = []#['drugInteraction']#'microRNAInteraction'
Genes = 'POU5F1,NANOG,TCF7L1,WNT1,CTNNB1,SOX2,TCF4,GSK3B'
Genes = 'Glucose'; Degrees = 'shortestPath'; Degrees = 'indirect'; Degrees = 'all possible'
Genes = ''; Degrees='indirect'
interactionDirs = []
Genes=''
outputdir = filepath('AltAnalyze/test')
outputdir = '/Users/saljh8/Desktop/Archived/Documents/1-manuscripts/Salomonis/SIDS-WikiPathways/Interactomics/'
interaction_root = 'AltDatabase/goelite/'+Species+'/gene-interactions'
files = read_directory('AltDatabase/goelite/'+Species+'/gene-interactions')
rooot = '/Users/nsalomonis/Desktop/dataAnalysis/Sarwal/CTOTC/AltAnalyze Based/GO-Elite/MarkerFinder/'
expressionFile=None
expressionFile = '/Users/nsalomonis/Desktop/dataAnalysis/Sarwal/Urine-AR-increased/UrinProteomics_Kidney-All/GO-Elite/input/GE.AR_vs_STA-fold1.5_rawp0.05.txt'
expressionFile = '/Users/nsalomonis/Desktop/dataAnalysis/Sarwal/BKVN infection/GO-Elite/input/AR_vs_norm_adjp05.txt'
expressionFile = '/Users/nsalomonis/Desktop/dataAnalysis/Sarwal/Blood AR-BK/AR-STA/Batches/overlap/AR_vs_STA_p0.05_fold1_common.txt'
expressionFile=None
#files2 = read_directory(rooot)
#inputType = 'SIF'
for file in files:
if 'micro' not in file and 'all-Drug' not in file and 'GRID' not in file and 'Drug' not in file and 'TF' not in file: # and 'TF' not in file and 'KEGG' not in file:
interactionDirs.append(filepath(interaction_root+'/'+file))
#"""
inputDir='/Users/saljh8/Desktop/Archived/Documents/1-manuscripts/Salomonis/SIDS-WikiPathways/Interactomics/CoreGeneSet67/core_SIDS.txt'
expressionFile = '/Users/saljh8/Desktop/Archived/Documents/1-manuscripts/Salomonis/SIDS-WikiPathways/Interactomics/Proteomics/proteomics_kinney.txt'
interactionPermuteTest(Species,Degrees,inputType,inputDir,outputdir,interactionDirs,Genes=Genes,obligatorySet=obligatorySet,expressionFile=expressionFile, IncludeExpIDs=True)
sys.exit()
buildInteractions(Species,Degrees,inputType,inputDir,outputdir,interactionDirs,Genes=Genes,obligatorySet=obligatorySet,expressionFile=expressionFile, IncludeExpIDs=True)
sys.exit()
#"""
#canonical Wnt signaling: GO:0060070
# BioMarkers 'Pluripotent Stem Cells' 'gene-mapp'
#inputDir = '/Users/nsalomonis/Desktop/dataAnalysis/Sarwal/Diabetes-Blood/ACR/log2/MergedFiles-Symbol_ACR.txt'
#inputDir = '/Users/nsalomonis/Desktop/dataAnalysis/SplicingFactors/RBM20_splicing_network.txt'; inputType = 'SIF'
#inputDir = '/Users/nsalomonis/Documents/1-manuscripts/Salomonis/SIDS-WikiPathways/67_SIDS-genes.txt'
#Genes=None
#exportGraphImage(Species,'/Users/nsalomonis/Desktop/AltAnalyze/AltAnalyze/test/networks/AltAnalyze-network-interactions_1degrees.sif',inputDir);sys.exit()
#buildInteractions(Species,Degrees,inputType,inputDir,outputdir,interactionDirs,Genes=None,obligatorySet=obligatorySet,geneSetType='BioMarkers',PathwayFilter='Pluripotent Stem Cells',directory='gene-mapp')
buildInteractions(Species,Degrees,inputType,inputDir,outputdir,interactionDirs,Genes=Genes,obligatorySet=obligatorySet,expressionFile=expressionFile)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/InteractionBuilder.py
|
InteractionBuilder.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"""This module contains generic methods for creating export paths that include new
multiple nested folders and deleting all files within a directory."""
import os
import sys
import string
import unique
import shutil
import UI
if sys.platform == "win32":
mode = 'wb' ### writes as in a binary mode versus text mode which introduces extra cariage returns in Windows
else:
mode = 'w' ### writes text mode which can introduce extra carriage return characters (e.g., /r)
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir)
return dir_list
def getParentDir(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
return string.split(filename,'/')[-2]
def findParentDir(filename):
### :: reverses string
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'//','/') ### If /// present
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1 ### get just the parent directory
return filename[:x]
def findFilename(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'//','/') ### If /// present
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1 ### get just the parent directory
return filename[x:]
def ExportFile(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
dir = findParentDir(filename)
try: file_var = createExportFile(filename,dir)
except RuntimeError:
isFileOpen(filename,dir)
file_var = createExportFile(filename,dir)
return file_var
def customFileMove(old_fn,new_fn):
old_fn = filepath(old_fn)
new_fn = filepath(new_fn)
raw = ExportFile(new_fn)
for line in open(old_fn,'rU').xreadlines():
if line[0]!='#': ### Applies to Affymetrix APT data (screws up combat)
raw.write(line)
raw.close()
os.remove(old_fn)
def customFileCopy(old_fn,new_fn):
old_fn = filepath(old_fn)
new_fn = filepath(new_fn)
raw = ExportFile(new_fn)
for line in open(old_fn,'rU').xreadlines():
if line[0]!='#': ### Applies to Affymetrix APT data (screws up combat)
raw.write(line)
raw.close()
def isFileOpen(new_file,dir):
try:
file_open = 'yes'
dir_list = read_directory(dir)
if len(dir_list)>0:
while file_open == 'yes':
file_open = 'no'
for file in dir_list:
if file in new_file: ###Thus the file is open
try:
fn=filepath(new_file);
file_var = open(fn,mode)
"""
except Exception:
try: os.chmod(fn,0777) ### It's rare, but this can be a write issue
except Exception:
print "This user account does not have write priveledges to change the file:"
print fn,"\nPlease login as an administrator or re-install the software as a non-admin.";sys.exit()
file_var = open(fn,'w')"""
file_open = 'no'
except IOError:
print_out = 'Results file: '+fn+ '\nis open...can not re-write.\nPlease close file and select "OK".'
try: UI.WarningWindow(print_out,' OK ');
except Exception:
print print_out; print 'Please correct (hit return to continue)'
inp = sys.stdin.readline()
file_open = 'yes'
except OSError: null = []
def createExportFile(new_file,dir):
try:
#isFileOpen(new_file,dir) ###Creates problems on Mac - not clear why
fn=filepath(new_file)
file_var = open(fn,mode)
"""except Exception:
try: os.chmod(fn,0777) ### It's rare, but this can be a write issue
except Exception:
print "This user account does not have write priveledges to change the file:"
print fn,"\nPlease login as an administrator or re-install the software as a non-admin.";sys.exit()
file_var = open(fn,'w')"""
except Exception:
createExportDir(new_file,dir) ###Occurs if the parent directory is also missing
fn=filepath(new_file)
file_var = open(fn,mode)
"""except Exception:
try: os.chmod(fn,0777) ### It's rare, but this can be a write issue
except Exception:
print "This user account does not have write priveledges to change the file:"
print fn,"\nPlease login as an administrator or re-install the software as a non-admin.";sys.exit()
file_var = open(fn,'w')"""
return file_var
def createExportDirAlt(new_file,dir):
### Original method for creating a directory path that is not present
### Works by going backwards (not ideal)
dir = string.replace(dir,'//','/')
dir = string.replace(dir,'\\','/')
dir = string.replace(dir,'\\','/')
dir_ls = string.split(dir,'/')
if len(dir_ls) != 1:
index = 1
while index < (len(dir_ls)+1):
parent_dir = string.join(dir_ls[:index],'/')
index+=1
try:pfn = filepath(parent_dir); os.mkdir(pfn)
except OSError: continue
createExportFile(new_file,dir)
else: print "Parent directory not found locally for", dir_ls
def createExportDir(new_file,dir):
### New method for creating a directory path that is not present
### Works by going forward (check if a base path is present and then go up)
dir = string.replace(dir,'//','/')
dir = string.replace(dir,'\\','/')
dir = string.replace(dir,'\\','/')
dir = filepath(dir)
dir_ls = string.split(dir,'/')
i = 1; paths_added = 'no'
while i <= len(dir_ls):
new_dir = string.join(dir_ls[:i],'/')
status = verifyDirectory(new_dir)
if status == 'no':
try: os.mkdir(new_dir); paths_added = 'yes'
except Exception: paths_added = 'yes'
i+=1
if paths_added == 'yes':
try:
fn=filepath(new_file)
file_var = open(fn,mode)
except Exception:
print "Parent directory not found locally for", [dir,new_file]
#else: print "Parent directory not found locally for", [dir,new_file]; sys.exit()
def createDirPath(dir):
### New method for creating a directory path that is not present
### Works by going forward (check if a base path is present and then go up)
dir = string.replace(dir,'//','/')
dir = string.replace(dir,'\\','/')
dir = string.replace(dir,'\\','/')
dir = filepath(dir)
dir_ls = string.split(dir,'/')
i = 1; paths_added = 'no'
while i <= len(dir_ls):
new_dir = string.join(dir_ls[:i],'/')
status = verifyDirectory(new_dir)
if status == 'no':
try: os.mkdir(new_dir); paths_added = 'yes'
except Exception: paths_added = 'yes'
i+=1
def verifyDirectory(dir):
try: dir_list = read_directory(dir); verified = 'yes'
except Exception: verified = 'no'
#print 'verify',[dir],verified
if verified == 'no': ### Can occur for the AltDatabase dir, since EnsMart62 will be added an not found
try: dir_list = os.listdir(dir); verified = 'yes'
except Exception: verified = 'no'
return verified
def createExportFolder(dir):
dir = string.replace(dir,'//','/')
dir = string.replace(dir,'\\','/')
dir = string.replace(dir,'\\','/')
dir_ls = string.split(dir,'/')
if len(dir_ls) != 1:
index = 1
while index < (len(dir_ls)+1):
parent_dir = string.join(dir_ls[:index],'/')
index+=1
#print "Trying to create the directory:",parent_dir
try:
pfn = filepath(parent_dir)
#print pfn
os.mkdir(pfn)
#print parent_dir, 'written' #, index-1, len(dir_ls)
except OSError:
#print "Can not write this dir"
#break
continue
else: print "Parent directory not found locally for", dir_ls
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def cleanFile(source_file,removeExtra=None):
### Some files have extra odd encoding that results in blank new lines in the extracted file
### For succeptible directories copy all files line by line, removing existing end of lines
file = findFilename(source_file); temp_file = 'tempdir/'+file
data = ExportFile(temp_file)
fn=filepath(source_file)
for line in open(fn,'rU').xreadlines():
line = cleanUpLine(line)
writeFile=True
if removeExtra!=None:
if line[0]==removeExtra: writeFile=False
if len(line)>0 and writeFile: data.write(line+'\n')
data.close()
### Replace old file with new file
copyFile(temp_file,source_file)
os.remove(temp_file)
print 'copied',file
def cleanAndFilterAllFiles(source_dir,output_dir,filter_file):
fn=filepath(filter_file)
filter_ids={} ### gene, protein and transcript IDs
for line in open(fn,'rU').xreadlines():
line = cleanUpLine(line)
filter_ids[line]=[]
#source_file = '/Users/saljh8/Desktop/testX/AltAnalyze/AltXDatabase/EnsMart72/goelite/Hs/gene-interactions/Ensembl-BioGRID.txt'
#destination_file = '/Users/saljh8/Desktop/testX/AltAnalyze/AltXDatabase/EnsMart72_filtered/goelite/Hs/gene-interactions/Ensembl-BioGRID.txt'
#cleanAndFilterFile(source_file,destination_file,filter_ids);sys.exit()
exclude_names = ['.DS','EntrezGene', 'HMDB', 'DrugBank']
for root, subFolders, files in os.walk(source_dir):
for f in files:
exclude = False
for i in exclude_names:
if i in f: exclude = True
if exclude==False:
source_file = root+'/'+f
destination_file = string.replace(source_file,source_dir,output_dir)
cleanAndFilterFile(source_file,destination_file,filter_ids)
def cleanAndFilterFile(source_file,destination_file,filter_ids):
data = ExportFile(destination_file)
count=0
firstLine=True
for line in open(source_file,'rU').xreadlines():
if firstLine:
data.write(line)
firstLine=False
else:
writeOut=False
line = cleanUpLine(line)
t = string.split(line,'\t')
if t[0] in filter_ids:
writeOut=True
elif ':' in t[0]:
uid = string.split(t[0],':')[0]
if uid in filter_ids:
writeOut=True
else:
if len(t)>1:
if t[1] in filter_ids:
writeOut=True
elif ':' in t[1]:
uid = string.split(t[1],':')[0]
if uid in filter_ids:
writeOut=True
if len(t)>2:
if t[2] in filter_ids:
writeOut=True
if len(t)>3:
if t[3] in filter_ids:
writeOut=True
if len(t)>4:
if t[4] in filter_ids:
writeOut=True
if len(t)>5:
if t[5] in filter_ids:
writeOut=True
if writeOut:
data.write(line+'\n')
count+=1
data.close()
print 'copied',source_file
if count==0: ### There are no relevant entries in the file, so copy over the entire file
print '*****',source_file
copyFile(source_file,destination_file)
def copyFile(source_file,destination_file):
dir = findParentDir(destination_file)
try: createExportFolder(dir)
except Exception: null=[] ### already exists
shutil.copyfile(source_file,destination_file)
#print '\nFile copied to:',destination_file
def deleteFolder(dir):
try:
dir = filepath(dir); dir_list = read_directory(dir) ### Get all files in directory
#try: print 'deleting dir:',dir
#except Exception: null=None ### Occurs due to a Tkinter issue sometimes
for file in dir_list:
fn = filepath(dir+'/'+file)
try:
if '.' in file: os.remove(fn)
else: deleteFolder(fn) ### Remove subdirectories
except Exception: None
os.removedirs(dir)
#print dir
return 'success'
except OSError: return 'failed'
if __name__ == '__main__':
import getopt
source_dir=None
filter_file=None
output_dir=None
print 'Filtering AltAnalyze Database based on supplied Ensembl gene, protein and transcript IDs'
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print 'please provide sufficient arguments (--source, --destination, --filter)',sys.exit()
#Filtering samples in a datasets
#python SampleSelect.py --i /Users/saljh8/Desktop/C4-hESC/ExpressionInput/exp.C4.txt --f /Users/saljh8/Desktop/C4-hESC/ExpressionInput/groups.C4.txt
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['source=','destination=','filter='])
#print sys.argv[1:]
for opt, arg in options:
if opt == '--source': source_dir=arg
elif opt == '--destination': output_dir=arg
elif opt == '--filter': filter_rows=True
output_file = input_file[:-4]+'-filtered.txt'
if filter_file != None and source_dir != None and output_dir != None:
cleanAndFilterAllFiles(source_dir,output_dir,filter_file)
sys.exit()
customFileCopy('/Volumes/SEQ-DATA/IlluminaBodyMap/pooled/pooled/Thyroid-ERR030872__exons.bed','/Volumes/SEQ-DATA/IlluminaBodyMap/pooled/Thyroid-ERR030872__exons.bed');sys.exit()
createExportFolder('Databases/null/null'); sys.exit()
createExportDir('C:/Users/Nathan Salomonis/Desktop/Gladstone/1-datasets/RNASeq/hESC-NP/TopHat-hESC_differentiation/AltExpression/pre-filtered/counts/a.txt','C:/Users/Nathan Salomonis/Desktop/Gladstone/1-datasets/RNASeq/hESC-NP/TopHat-hESC_differentiation/AltExpression/pre-filtered/counts'); sys.exit()
deleteFolder('BuildDBs/Entrez/Gene2GO');kill
fn = '/Users/nsalomonis/Desktop/GSE13297_RAW//AltExpression/ExonArray/Hs/Hs_Exon_CS_vs_hESC.p5_average.txt'
createExportFile(fn,'/Users/nsalomonis/Desktop/GSE13297_RAW//AltExpression/ExonArray/Hs')
ExportFile(fn)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/export.py
|
export.py
|
# AltAnalyze #
An automated cross-platform workflow for RNA-Seq gene, splicing and pathway analysis
AltAnalyze is an extremely user-friendly and open-source analysis tool that can be used for a broad range of genomics analyses. These analyses include the direct processing of raw [RNASeq](https://github.com/nsalomonis/altanalyze/wiki/RNASeq) or microarray data files, advanced methods for [single-cell population discovery](http://altanalyze.blogspot.com/2016/08/introducing-powerful-and-easy-to-use.html), differential expression analyses, analysis of alternative splicing/promoter/polyadenylation and advanced isoform function prediction analysis (protein, domain and microRNA targeting). Multiple advanced visualization tools and [à la carte analysis methods](https://github.com/nsalomonis/altanalyze/wiki/Tutorials) are supported in AltAnalyze (e.g., network, pathway, splicing graph). AltAnalyze is compatible with various data inputs for [RNASeq](https://github.com/nsalomonis/altanalyze/wiki/RNASeq) data ([FASTQ](http://altanalyze.blogspot.com/2016/08/using-ultrafast-sequence.html), [BAM](http://altanalyze.blogspot.com/2016/08/bye-bye-bed-files-welcome-bam.html), BED), microarray platforms ([Gene 1.0](https://github.com/nsalomonis/altanalyze/wiki/AffyGeneArray), [Exon 1.0](https://github.com/nsalomonis/altanalyze/wiki/AffyExonArray), [junction](https://github.com/nsalomonis/altanalyze/wiki/JAY) and [3' arrays](https://github.com/nsalomonis/altanalyze/wiki/CompatibleArrays)) for automated gene expression and splicing analysis. This software requires no advanced knowledge of bioinformatics programs or scripting or advanced computer hardware. User friendly [videos](https://www.google.com/#q=altanalyze&tbm=vid), [online tutorials](https://github.com/nsalomonis/altanalyze/wiki/Tutorials) and [blog posts](http://altanalyze.blogspot.com/) are also available.
# Dependencies #
If installed from PyPI (pip install AltAnalyze), the below dependencies should be included in the installed package. When running from source code you will need to install the following libraries.
* Required: Python 2.7, numpy, scipy, matplotlib, sklearn (scikit-learn)
* Recommended: umap-learn, nimfa, numba, python-louvain, annoy, networkx, R 3+, fastcluster, pillow, pysam, requests, pandas, patsy, lxml, python-igraph, cairo
[AltAnalyze documentation](http://altanalyze.readthedocs.io/), stand-alone archives are provided at [sourceforge](https://sourceforge.net/projects/altanalyze/files/) as well as at [github](https://github.com/nsalomonis/altanalyze). For questions not addressed here, please [contact us](https://github.com/nsalomonis/altanalyze/wiki/ContactUs).
**News Update** [12/16/18](https://github.com/nsalomonis/altanalyze/wiki/News)
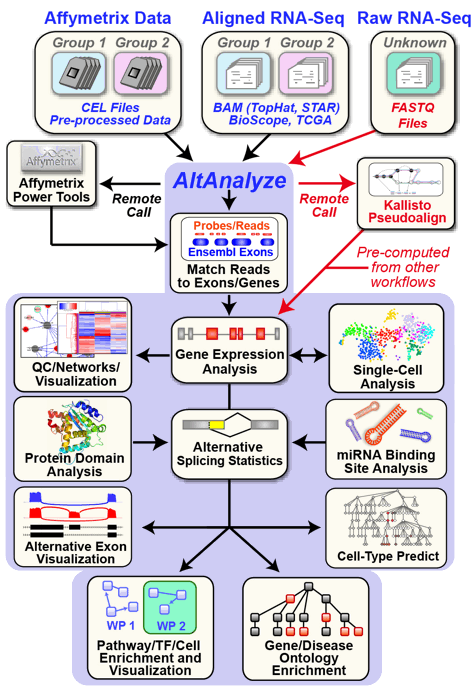
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/README.md
|
README.md
|
###update
#Copyright 2005-2008 J. David Gladstone Institutes, San Francisco California
#Author Nathan Salomonis - [email protected]
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"""This module contains generic methods for downloading and decompressing files designated
online files and coordinating specific database build operations for all AltAnalyze supported
gene ID systems with other program modules. """
import os
import sys
import unique
import string
import export
import traceback
try: from build_scripts import ExonArray
except Exception: null=[]
import traceback
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv": dir_list2.append(entry)
return dir_list2
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def zipDirectory(dir):
#http://www.testingreflections.com/node/view/8173
import zipfile
dir = filepath(dir); zip_file = dir+'.zip'
p = string.split(dir,'/'); top=p[-1]
zip = zipfile.ZipFile(zip_file, 'w', compression=zipfile.ZIP_DEFLATED)
root_len = len(os.path.abspath(dir))
for root, dirs, files in os.walk(dir):
archive_root = os.path.abspath(root)[root_len:]
for f in files:
fullpath = os.path.join(root, f)
archive_name = os.path.join(top+archive_root, f)
zip.write(fullpath, archive_name, zipfile.ZIP_DEFLATED)
zip.close()
return zip_file
def unzipFiles(filename,dir):
import zipfile
output_filepath = filepath(dir+'/'+filename)
try:
zfile = zipfile.ZipFile(output_filepath)
for name in zfile.namelist():
if name.endswith('/'):null=[] ### Don't need to export
else:
if 'EnsMart' in name and 'EnsMart' in dir:
dir = export.findParentDir(dir[:-1]) ### Remove EnsMart suffix directory
try: outfile = export.ExportFile(filepath(dir+name))
except Exception: outfile = export.ExportFile(filepath(dir+name[1:]))
outfile.write(zfile.read(name)); outfile.close()
#print 'Zip extracted to:',output_filepath
status = 'completed'
except Exception, e:
try:
### Use the operating system's unzip if all else fails
extracted_path = string.replace(output_filepath,'.zip','')
try: os.remove(extracted_path) ### This is necessary, otherwise the empty file created above will require user authorization to delete
except Exception: null=[]
subprocessUnzip(dir,output_filepath)
status = 'completed'
except IOError:
print e
print 'WARNING!!!! The zip file',output_filepath,'does not appear to be a valid zip archive file or is currupt.'
status = 'failed'
return status
def subprocessUnzip(dir,output_filepath):
import subprocess
dir = filepath(dir)
subprocess.Popen(["unzip", "-d", dir, output_filepath]).wait()
############## Update Databases ##############
def buildJunctionExonAnnotations(species,array_type,specific_array_type,force,genomic_build):
### Get UCSC associations (download databases if necessary)
mRNA_Type = 'mrna'; run_from_scratch = 'yes'; force='no'
export_all_associations = 'no' ### YES only for protein prediction analysis
#buildUCSCAnnoationFiles(species,mRNA_Type,export_all_associations,run_from_scratch,force)
### Get genomic locations and initial annotations for exon sequences (exon pobesets and junctions)
from build_scripts import JunctionArray
from build_scripts import JunctionArrayEnsemblRules
""" The following functions:
1) Extract transcript cluster-to-gene annotations
2) Extract exon sequences for junctions and exon probesets from the Affymetrix annotation file (version 2.0),
3) Map these sequences to Ensembl gene sequences (build specific) plus and minus 2KB, upstream and downstream
4) Obtain AltAnalyze exon region annotations and obtain full-length exon sequences for each exon probeset
5) Consoladate these into an Ensembl_probeset.txt file (rather than Ensembl_junction_probeset.txt) with junctions
having a single probeset identifier.
6) Determine which junctions and junction-exons represent recipricol junctions using:
a) AltAnalyze identified recipricol junctions from Ensembl and UCSC and
b) Affymetrix suggested recipricol junctions based on common exon cluster annotations, creating
Mm_junction_comps_updated.txt.
c) De novo comparison of all exon-junction region IDs for all junctions using the EnsemblImport method compareJunctions().
"""
### Steps 1-3
JunctionArray.getJunctionExonLocations(species,array_type,specific_array_type)
### Step 4
JunctionArrayEnsemblRules.getAnnotations(species,array_type,'yes',force)
### Step 5-6
JunctionArray.identifyJunctionComps(species,array_type,specific_array_type)
def buildAltMouseExonAnnotations(species,array_type,force,genomic_build):
"""Code required to:
1) Extract out Affymetrix provided exon sequence (probeset sequence extracted from "probeset_sequence_reversed.txt", derived
directly from the Affymetrix AltMouse probe annotation file), from the "SEQUENCE-transcript-dbase.txt" (built using
dump-chip1 .gff sequence and AltMerge-Peptide Informatics script "sequence_analysis_AltMouse_refseq.py").
2) Once exported, grab full length exon sequences using exon/intron coordinates matches to full-length gene sequences with 2kb
flanking sequence to efficiently predict microRNA binding site exclusion (reAnnotateCriticalExonSequences) and later for
coordinate mapping to get exons aligning with UCSC annotated splicing annotations and exons. This sequence data replaced
the previous file (don't need to re-run this - see rederive_exonseq == 'yes' below for reference).
3) Match the updated exon sequences to the most recent genomic coordinates and build the exact equivalent of the exon array
Mm_Ensembl_probeset.txt database (same structure and ExonArrayEnsemblRules.py code). This involves running EnsemblImport.
This code should be run before the exon array location build code since the "Mm_Ensembl_probeset.txt" is created and then re-
written as "Mm_AltMouse_Ensembl_probeset.txt".
"""
from build_scripts import JunctionArray
from build_scripts import JunctionArrayEnsemblRules
rederive_exonseq = 'no'
### Only needs to be run once, to export exon sequence for AltMouse array the original (1 and 2 above)
if rederive_exonseq == 'yes':
import AltAnalyze
from import_scripts import ExonAnnotate_module
from build_scripts import ExonAnalyze_module
agglomerate_inclusion_probesets = 'no'; onlyAnalyzeJunctions='no'
probeset_annotations_file = "AltDatabase/"+species+"/"+array_type+"/"+"MASTER-probeset-transcript.txt"
verifyFile(probeset_annotations_file,array_type) ### Will force download if missing
exon_db={}; filtered_arrayids={};filter_status='no'
constituitive_probeset_db,exon_db,genes_being_analyzed = AltAnalyze.importSplicingAnnotationDatabase(probeset_annotations_file,array_type,filtered_arrayids,filter_status)
alt_junction_db,critical_exon_db,exon_dbase,exon_inclusion_db,exon_db = ExonAnnotate_module.identifyPutativeSpliceEvents(exon_db,constituitive_probeset_db,{},agglomerate_inclusion_probesets,onlyAnalyzeJunctions)
ExonAnnotate_module.exportJunctionComparisons(alt_junction_db,critical_exon_db,exon_dbase)
print "Finished exporting junctions used in AltMouse array comparisons."
ExonAnalyze_module.exportAltMouseExonSequence()
JunctionArray.reAnnotateCriticalExonSequences(species,array_type)
### Get UCSC associations (download databases if necessary)
mRNA_Type = 'mrna'; run_from_scratch = 'yes'
export_all_associations = 'no' ### YES only for protein prediction analysis
buildUCSCAnnoationFiles(species,mRNA_Type,export_all_associations,run_from_scratch,force)
reannotate_exon_seq = 'yes'
print 'genomic_build', genomic_build
if genomic_build == 'new':
### Need to run with every new genomic build (match up new coordinates
print "Begining to derive exon sequence from new genomic build"
JunctionArray.identifyCriticalExonLocations(species,array_type)
reannotate_exon_seq = 'yes'
JunctionArrayEnsemblRules.getAnnotations(species,array_type,reannotate_exon_seq,force)
### Download files required during AltAnalyze analysis but not during the database build process
filename = "AltDatabase/"+species+"/"+array_type+"/"+"MASTER-probeset-transcript.txt"
verifyFile(filename,array_type) ### Will force download if missing
filename = "AltDatabase/"+species+'/'+ array_type+'/'+array_type+"_annotations.txt"
verifyFile(filename,array_type) ### Will force download if missing
def buildExonArrayExonAnnotations(species, array_type, force):
### Get UCSC associations (download databases if necessary)
mRNA_Type = 'mrna'; run_from_scratch = 'yes'
export_all_associations = 'no' ### YES only for protein prediction analysis
buildUCSCAnnoationFiles(species,mRNA_Type,export_all_associations,run_from_scratch,force)
from build_scripts import ExonArrayEnsemblRules; reload(ExonArrayEnsemblRules)
process_from_scratch='yes'
constitutive_source='default'
### Build the databases and return the variables (not used here)
source_biotype = 'mRNA'
if array_type == 'gene': source_biotype = 'gene'
probeset_db,annotate_db,constitutive_gene_db,splicing_analysis_db = ExonArrayEnsemblRules.getAnnotations(process_from_scratch,constitutive_source,source_biotype,species)
def getFTPData(ftp_server,subdir,filename_search):
### This is a more generic function for downloading FTP files based on input directories and a search term
from ftplib import FTP
print 'Connecting to',ftp_server
ftp = FTP(ftp_server); ftp.login()
ftp.cwd(subdir)
ftpfilenames = []; ftp.dir(ftpfilenames.append); ftp.quit()
matching=[]
for line in ftpfilenames:
line = string.split(line,' '); file_dir = line[-1]
dir = 'ftp://'+ftp_server+subdir+'/'+file_dir
#print dir
if filename_search in dir and '.md5' not in dir: matching.append(dir)
if len(matching)==1:
return matching[0]
elif len(matching)==0:
print filename_search, 'not found at',ftp_server+subdir
return string.replace(filename_search,'.gz','')
else:
return matching
def verifyFile(filename,server_folder):
fn=filepath(filename); counts=0
try:
for line in open(fn,'rU').xreadlines():
if len(line)>2: counts+=1 ### Needed for connection error files
if counts>10: break
except Exception:
counts=0
if server_folder == 'counts': ### Used if the file cannot be downloaded from http://www.altanalyze.org
return counts
elif counts == 0:
if server_folder == None: server_folder = 'AltMouse'
continue_analysis = downloadCurrentVersion(filename,server_folder,'')
if continue_analysis == 'no' and 'nnot' not in filename:
print 'The file:\n',filename, '\nis missing and cannot be found online. Please save to the designated directory or contact AltAnalyze support.'
else:
return counts
def getFileLocations(species_code,search_term):
### The three supplied variables determine the type of data to obtain from default-files.csv
import UI
file_location_defaults = UI.importDefaultFileLocations()
sfl = file_location_defaults[search_term]
for sf in sfl:
if species_code in sf.Species(): filename = sf.Location()
return filename
def buildUniProtFunctAnnotations(species,force):
import UI
file_location_defaults = UI.importDefaultFileLocations()
"""Identify the appropriate download location for the UniProt database for the selected species"""
uis = file_location_defaults['UniProt']
trembl_filename_url=''
for ui in uis:
if species in ui.Species(): uniprot_filename_url = ui.Location()
species_codes = importSpeciesInfo(); species_full = species_codes[species].SpeciesName()
from build_scripts import ExtractUniProtFunctAnnot; reload(ExtractUniProtFunctAnnot)
ExtractUniProtFunctAnnot.runExtractUniProt(species,species_full,uniprot_filename_url,trembl_filename_url,force)
class SpeciesData:
def __init__(self, abrev, species):
self._abrev = abrev; self._species = species
def SpeciesCode(self): return self._abrev
def SpeciesName(self): return self._species
def __repr__(self): return self.Report()
def importSpeciesInfo():
filename = 'Config/species_all.txt'; x=0
fn=filepath(filename); species_codes={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
abrev = t[0]; species = t[1]
if x==0: x=1
else:
sd = SpeciesData(abrev,species)
species_codes[abrev] = sd
return species_codes
def downloadSuppressPrintOuts(url,dir,file_type):
global Suppress_Printouts
Suppress_Printouts = 'yes'
return download(url,dir,file_type)
def download(url,dir,file_type):
global suppress_printouts
try: suppress_printouts = Suppress_Printouts
except Exception: suppress_printouts = 'no'
try: dp = download_protocol(url,dir,file_type); output_filepath, status = dp.getStatus(); fp = output_filepath
except Exception:
#print traceback.format_exc()
try:
dir = unique.filepath(dir) ### Can result in the wrong filepath exported for AltDatabase RNA-Seq zip files (don't include by default)
dp = download_protocol(url,dir,file_type); output_filepath, status = dp.getStatus(); fp = output_filepath
except Exception:
output_filepath='failed'; status = "Internet connection not established. Re-establish and try again."
fp = filepath(dir+url.split('/')[-1]) ### Remove this empty object if saved
if 'Internet' not in status:
if '.zip' in fp or '.gz' in fp or '.tar' in fp:
#print "\nRemoving zip file:",fp
try: os.remove(fp); status = 'removed'
except Exception: null=[] ### Not sure why this error occurs since the file is not open
#print "\nRemoving zip file:",string.replace(fp,'.gz','')
if '.tar' in fp:
try: os.remove(string.replace(fp,'.gz',''))
except Exception: null=[]
return output_filepath, status
class download_protocol:
def __init__(self,url,dir,file_type):
try: self.suppress = suppress_printouts
except Exception: self.suppress = 'no'
"""Copy the contents of a file from a given URL to a local file."""
filename = url.split('/')[-1]; self.status = ''
#print [url, dir, file_type]
#dir = unique.filepath(dir) ### Can screw up directory structures
if file_type == None: file_type =''
if len(file_type) == 2: filename, file_type = file_type ### Added this feature for when a file has an invalid filename
output_filepath = unique.filepath(dir+filename, force='application-path')
dir = export.findParentDir(output_filepath)
output_filepath_object = export.createExportFile(output_filepath,dir[:-1])
self.output_filepath = output_filepath
if self.suppress == 'no':
print "Downloading the following file:",filename,' ',
self.original_increment = 5
self.increment = 0
import urllib
from urllib import urlretrieve
#if 'gene.txt.gz' in url: print [self.reporthookFunction];sys.exit()
try:
try: webfile, msg = urlretrieve(url,output_filepath,reporthook=self.reporthookFunction)
except IOError:
if 'Binary' in traceback.format_exc(): #IOError: [Errno ftp error] 200 Switching to Binary mode.
### https://bugs.python.org/issue1067702 - some machines the socket doesn't close and causes an error - reload to close the socket
reload(urllib)
webfile, msg = urlretrieve(url,output_filepath,reporthook=self.reporthookFunction)
reload(urllib)
except:
print 'Unknown URL error encountered...'; forceURLError
if self.suppress == 'no': print ''
self.testFile()
if self.suppress == 'no': print self.status
if 'Internet' not in self.status:
if '.zip' in filename:
if self.suppress == 'no': print "Extracting zip file...",
try: decompressZipStackOverflow(filename,dir); status = 'completed'
except Exception:
#print 'Native unzip not present...trying python unzip methods...'
status = unzipFiles(filename,dir)
if status == 'failed': print 'zip extraction failed!'
self.gz_filepath = filepath(output_filepath); self.status = 'remove'
if self.suppress == 'no': print "zip file extracted"
elif '.gz' in filename:
self.gz_filepath = output_filepath
if len(file_type)==0: extension = '.gz'
else: extension = 'gz'
decompressed_filepath = string.replace(self.gz_filepath,extension,file_type)
### Below code can be too memory intensive
#file_size = os.path.getsize(output_filepath)
#megabtyes = file_size/1000000.00
#if megabtyes>5000: force_error ### force_error is an undefined variable which causes an exception
import gzip; content = gzip.GzipFile(self.gz_filepath, 'rb')
data = open(decompressed_filepath,'wb')
#print "\nExtracting downloaded file:",self.gz_filepath
import shutil; shutil.copyfileobj(content,data)
# http://pythonicprose.blogspot.com/2009/10/python-extract-or-unzip-tar-file.html
os.chdir(filepath(dir))
if '.tar' in decompressed_filepath:
import tarfile
tfile = tarfile.open(decompressed_filepath)
tfile.extractall()
tfile.close()
tar_dir = string.replace(decompressed_filepath,'.tar','')
self.status = 'remove'
else: self.gz_filepath = ''; self.status = 'remove'
def testFile(self):
fn=filepath(self.output_filepath)
try:
for line in open(fn,'rU').xreadlines():
if '!DOCTYPE' in line: self.status = "Internet connection not established. Re-establish and try again."
break
except Exception: null=[]
def getStatus(self): return self.output_filepath, self.status
def reporthookFunction(self, blocks_read, block_size, total_size):
if not blocks_read:
if self.suppress == 'no':
print 'Connection opened. Downloading (be patient)'
if total_size < 0:
# Unknown size
if self.suppress == 'no':
print 'Read %d blocks' % blocks_read
else:
amount_read = blocks_read * block_size
percent_read = ((amount_read)*1.00/total_size)*100
if percent_read>self.increment:
#print '%d%% downloaded' % self.increment
if self.suppress == 'no':
print '*',
self.increment += self.original_increment
#print 'Read %d blocks, or %d/%d' % (blocks_read, amount_read, (amount_read/total_size)*100.000)
def createExportFile(new_file,dir):
try:
fn=filepath(new_file); file_var = open(fn,'w')
except IOError:
#print "IOError", fn
fn = filepath(dir)
try:
os.mkdir(fn) ###Re-Create directory if deleted
#print fn, 'written'
except OSError: createExportDir(new_file,dir) ###Occurs if the parent directory is also missing
fn=filepath(new_file); file_var = open(fn,'w')
return file_var
def downloadCurrentVersionUI(filename,secondary_dir,file_type,root):
continue_analysis = downloadCurrentVersion(filename,secondary_dir,file_type)
if continue_analysis == 'no' and 'nnot' not in filename:
import UI
try: root.destroy(); UI.getUserParameters('no'); sys.exit()
except Exception: sys.exit()
try: root.destroy()
except Exception(): null=[] ### Occurs when running from command-line
def downloadCurrentVersion(filename,secondary_dir,file_type):
import UI
file_location_defaults = UI.importDefaultFileLocations()
ud = file_location_defaults['url'] ### Get the location of the download site from Config/default-files.csv
url_dir = ud.Location() ### Only one entry
dir = export.findParentDir(filename)
dir = string.replace(dir,'hGlue','') ### Used since the hGlue data is in a sub-directory
filename = export.findFilename(filename)
url = url_dir+secondary_dir+'/'+filename
file,status = download(url,dir,file_type); continue_analysis = 'yes'
if 'Internet' in status and 'nnot' not in filename: ### Exclude for Affymetrix annotation files
print_out = "File:\n"+url+"\ncould not be found on the server or an internet connection is unavailable."
if len(sys.argv)<2:
try:
UI.WarningWindow(print_out,'WARNING!!!')
continue_analysis = 'no'
except Exception:
print 'cannot be downloaded';force_error
else: print 'cannot be downloaded';force_error
elif status == 'remove' and ('.zip' in file or '.tar' in file or '.gz' in file):
try: os.remove(file) ### Not sure why this works now and not before
except Exception: status = status
return continue_analysis
def decompressZipStackOverflow(original_zip_file,dir):
zip_file = filepath(dir+original_zip_file)
#print 'Using OS native unzip software to extract'
###http://stackoverflow.com/questions/339053/how-do-you-unzip-very-large-files-in-python
import zipfile
import zlib
src = open(zip_file,"rb")
zf = zipfile.ZipFile(src)
for m in zf.infolist():
# Examine the header
#print m.filename, m.header_offset, m.compress_size, repr(m.extra), repr(m.comment)
src.seek(m.header_offset)
src.read(30) # Good to use struct to unpack this.
nm= src.read(len(m.filename))
if len(m.extra) > 0: ex= src.read(len(m.extra))
if len(m.comment) > 0: cm=src.read(len(m.comment))
# Build a decompression object
decomp= zlib.decompressobj(-15)
# This can be done with a loop reading blocks
out=open(filepath(dir+m.filename), "wb")
result=decomp.decompress(src.read(m.compress_size))
out.write(result); result=decomp.flush()
out.write(result); out.close()
zf.close()
src.close()
def buildUCSCAnnoationFiles(species,mRNA_Type,export_all_associations,run_from_scratch,force):
### Test whether files already exist and if not downloads/builds them
if export_all_associations == 'no': ### Applies to EnsemblImport.py analyses
filename = 'AltDatabase/ucsc/'+species+'/'+species+'_UCSC_transcript_structure_mrna.txt'
else: ### Applies to the file used for Domain-level analyses
filename = 'AltDatabase/ucsc/'+species+'/'+species+'_UCSC_transcript_structure_COMPLETE-mrna.txt'
counts = verifyFile(filename,'counts')
if counts<9:
from build_scripts import UCSCImport
try: UCSCImport.runUCSCEnsemblAssociations(species,mRNA_Type,export_all_associations,run_from_scratch,force)
except Exception: UCSCImport.exportNullDatabases(species) ### used for species not supported by UCSC
def executeParameters(species,array_type,force,genomic_build,update_uniprot,update_ensembl,update_probeset_to_ensembl,update_domain,update_miRs,update_all,update_miR_seq,ensembl_version):
if '|' in array_type: array_type, specific_array_type = string.split(array_type,'|') ### To destinguish between array sub-types, like the HJAY and hGlue
else: specific_array_type = array_type
if update_all == 'yes':
update_uniprot='yes'; update_ensembl='yes'; update_probeset_to_ensembl='yes'; update_domain='yes'; update_miRs = 'yes'
if update_ensembl == 'yes':
from build_scripts import EnsemblSQL; reload(EnsemblSQL)
""" Used to grab all essential Ensembl annotations previously obtained via BioMart"""
configType = 'Advanced'; analysisType = 'AltAnalyzeDBs'; externalDBName = ''
EnsemblSQL.buildEnsemblRelationalTablesFromSQL(species,configType,analysisType,externalDBName,ensembl_version,force)
""" Used to grab Ensembl-to-External gene associations"""
configType = 'Basic'; analysisType = 'ExternalOnly'; externalDBName = 'Uniprot/SWISSPROT'
EnsemblSQL.buildEnsemblRelationalTablesFromSQL(species,configType,analysisType,externalDBName,ensembl_version,force)
""" Used to grab Ensembl full gene sequence plus promoter and 3'UTRs """
if array_type == 'AltMouse' or array_type == 'junction' or array_type == 'RNASeq':
EnsemblSQL.getFullGeneSequences(ensembl_version,species)
if update_uniprot == 'yes':
###Might need to delete the existing versions of downloaded databases or force download
buildUniProtFunctAnnotations(species,force)
if update_probeset_to_ensembl == 'yes':
if species == 'Mm' and array_type == 'AltMouse':
buildAltMouseExonAnnotations(species,array_type,force,genomic_build)
elif array_type == 'junction':
buildJunctionExonAnnotations(species,array_type,specific_array_type,force,genomic_build)
elif array_type == 'RNASeq':
import RNASeq; test_status = 'no'; data_type = 'mRNA'
RNASeq.getEnsemblAssociations(species,data_type,test_status,force)
else: buildExonArrayExonAnnotations(species,array_type,force)
if update_domain == 'yes':
if array_type == 'RNASeq':
only_rely_on_coordinate_mapping = True ### This will provide more accurate results as many junctions have missing sequences
else:
only_rely_on_coordinate_mapping = False
from build_scripts import FeatureAlignment
from build_scripts import JunctionArray
from build_scripts import mRNASeqAlign
from build_scripts import IdentifyAltIsoforms
### Get UCSC associations for all Ensembl linked genes (download databases if necessary) if species == 'Mm' and array_type == 'AltMouse':
mRNA_Type = 'mrna'; run_from_scratch = 'yes'
export_all_associations = 'yes' ### YES only for protein prediction analysis
buildUCSCAnnoationFiles(species,mRNA_Type,export_all_associations,run_from_scratch,force)
if (species == 'Mm' and array_type == 'AltMouse'):
"""Imports and re-exports array-Ensembl annotations"""
null = JunctionArray.importArrayAnnotations(species,array_type); null={}
if (species == 'Mm' and array_type == 'AltMouse') or array_type == 'junction' or array_type == 'RNASeq':
if only_rely_on_coordinate_mapping == False:
"""Performs probeset sequence aligment to Ensembl and UCSC transcripts. To do: Need to setup download if files missing"""
analysis_type = 'reciprocal'
mRNASeqAlign.alignProbesetsToTranscripts(species,array_type,analysis_type,force)
run_seqcomp = 'no'
if only_rely_on_coordinate_mapping == False:
IdentifyAltIsoforms.runProgram(species,array_type,'null',force,run_seqcomp)
FeatureAlignment.findDomainsByGenomeCoordinates(species,array_type,'null')
if array_type == 'junction' or array_type == 'RNASeq':
if only_rely_on_coordinate_mapping == False:
### For junction probeset sequences from mRNASeqAlign(), find and assess alternative proteins - export to the folder 'junction'
mRNASeqAlign.alignProbesetsToTranscripts(species,array_type,'single',force)
IdentifyAltIsoforms.runProgram(species,array_type,'junction',force,run_seqcomp)
FeatureAlignment.findDomainsByGenomeCoordinates(species,array_type,'junction')
### For exon probesets (and junction exons) align and assess alternative proteins - export to the folder 'exon'
IdentifyAltIsoforms.runProgram(species,array_type,'exon',force,run_seqcomp)
FeatureAlignment.findDomainsByGenomeCoordinates(species,array_type,'exon') # not needed
""" Repeat above with CoordinateBasedMatching = True """
### Peform coordinate based junction mapping to transcripts (requires certain sequence files built in IdentifyAltIosofmrs)
analysis_type = 'reciprocal'
mRNASeqAlign.alignProbesetsToTranscripts(species,array_type,analysis_type,force,CoordinateBasedMatching = True)
IdentifyAltIsoforms.runProgram(species,array_type,'null',force,run_seqcomp)
FeatureAlignment.findDomainsByGenomeCoordinates(species,array_type,'null')
mRNASeqAlign.alignProbesetsToTranscripts(species,array_type,'single',force,CoordinateBasedMatching = True)
IdentifyAltIsoforms.runProgram(species,array_type,'junction',force,run_seqcomp)
FeatureAlignment.findDomainsByGenomeCoordinates(species,array_type,'junction')
IdentifyAltIsoforms.runProgram(species,array_type,'exon',force,run_seqcomp)
if array_type == 'RNASeq':
JunctionArray.combineExonJunctionAnnotations(species,array_type)
if update_miRs == 'yes':
if update_miR_seq == 'yes':
from build_scripts import MatchMiRTargetPredictions; only_add_sequence_to_previous_results = 'no'
MatchMiRTargetPredictions.runProgram(species,force,only_add_sequence_to_previous_results)
if array_type == 'exon' or array_type == 'gene':
from build_scripts import ExonSeqModule
stringency = 'strict'; process_microRNA_predictions = 'yes'; mir_source = 'multiple'
ExonSeqModule.runProgram(species,array_type,process_microRNA_predictions,mir_source,stringency)
stringency = 'lax'
ExonSeqModule.runProgram(species,array_type,process_microRNA_predictions,mir_source,stringency)
ExonArray.exportMetaProbesets(array_type,species) ### Export metaprobesets for this build
else:
from build_scripts import JunctionSeqModule
stringency = 'strict'; mir_source = 'multiple'
JunctionSeqModule.runProgram(species,array_type,mir_source,stringency,force)
stringency = 'lax'
JunctionSeqModule.runProgram(species,array_type,mir_source,stringency,force)
if array_type == 'junction':
try:
from build_scripts import JunctionArray; from build_scripts import JunctionArrayEnsemblRules
JunctionArray.filterForCriticalExons(species,array_type)
JunctionArray.overRideExonEntriesWithJunctions(species,array_type)
JunctionArrayEnsemblRules.annotateJunctionIDsAsExon(species,array_type)
ExonArray.exportMetaProbesets(array_type,species) ### Export metaprobesets for this build
except IOError: print 'No built junction files to analyze';sys.exit()
if array_type == 'RNASeq' and (species == 'Hs' or species == 'Mm' or species == 'Rn'):
from build_scripts import JunctionArray; from build_scripts import JunctionArrayEnsemblRules
try: JunctionArrayEnsemblRules.annotateJunctionIDsAsExon(species,array_type)
except IOError: print 'No Ensembl_exons.txt file to analyze';sys.exit()
try:
filename = 'AltDatabase/'+species+'/SequenceData/miRBS-combined_gene-targets.txt'; ef=filepath(filename)
er = string.replace(ef,species+'/SequenceData/miRBS-combined_gene-targets.txt','ensembl/'+species+'/'+species+'_microRNA-Ensembl.txt')
import shutil; shutil.copyfile(ef,er)
except Exception: null=[]
if array_type != 'RNASeq':
### Get the probeset-probe relationships from online - needed for FIRMA analysis
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_probeset-probes.txt'
if array_type == 'junction' and 'lue' in specific_array_type:
server_folder = 'junction/hGlue'
verifyFile(filename,server_folder) ### Will force download if missing
verifyFile('AltDatabase/'+species+'/'+array_type+'/platform.txt',server_folder) ### Will force download if missing
elif array_type != 'AltMouse': verifyFile(filename,array_type) ### Will force download if missing
if (array_type == 'exon' or array_type == 'AltMouse') and species != 'Rn':
try:
### Available for select exon-arrays and AltMouse
probeset_to_remove_file = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_probes_to_remove.txt'
verifyFile(probeset_to_remove_file,array_type)
except Exception: null=[]
def temp(array_type,species):
specific_array_type = 'hGlue'
ExonArray.exportMetaProbesets(array_type,species) ### Export metaprobesets for this build
if array_type != 'RNASeq':
### Get the probeset-probe relationships from online - needed for FIRMA analysis
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_probeset-probes.txt'
if array_type == 'junction' and 'lue' in specific_array_type:
server_folder = 'junction/hGlue'
verifyFile(filename,server_folder) ### Will force download if missing
verifyFile('AltDatabase/'+species+'/'+array_type+'/platform.txt',server_folder) ### Will force download if missing
elif array_type != 'AltMouse': verifyFile(filename,array_type) ### Will force download if missing
if (array_type == 'exon' or array_type == 'AltMouse') and species != 'Rn':
try:
### Available for select exon-arrays and AltMouse
probeset_to_remove_file = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_probes_to_remove.txt'
verifyFile(probeset_to_remove_file,array_type)
except Exception: null=[]
if __name__ == '__main__':
dir = '/home/nsalomonis/software/AltAnalyze_v.2.0.5-Py/AltDatabase/EnsMart65/miRBS/'; filename = 'UTR_Sequences.txt.zip'
array_type = 'gene'; species = 'Mm'
filename = "AltDatabase/"+species+'/'+ array_type+'/'+array_type+"_annotations.txt"
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_probeset-probes.txt'
#verifyFile(filename,array_type);sys.exit()
#temp(array_type,species); sys.exit()
#unzipFiles(filename,dir); kill
#zipDirectory('Rn/EnsMart49');kill
#unzipFiles('Rn.zip', 'AltDatabaseNoVersion/');kill
#filename = 'http://altanalyze.org/archiveDBs/LibraryFiles/Mouse430_2.zip'
#filename = 'AltDatabase/affymetrix/LibraryFiles/Mouse430_2.zip'
filename = 'AltDatabase/Mm_RNASeq.zip'; dir = 'AltDatabase/updated/EnsMart72'
#downloadCurrentVersionUI(filename,dir,'','')
import update
dp = update.download_protocol('ftp://ftp.ensembl.org/pub/release-72/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh37.72.cdna.all.fa.gz','AltDatabase/Hs/SequenceData/','');sys.exit()
#dp = update.download_protocol('ftp://ftp.ensembl.org/pub/release-72/mysql/macaca_mulatta_core_72_10/gene.txt.gz','AltDatabase/ensembl/Ma/EnsemblSQL/','');sys.exit()
#kill
#target_folder = 'Databases/Ci'; zipDirectory(target_folder)
filename = 'Mm_RNASeq.zip'; dir = 'AltDatabase/EnsMart72/'
unzipFiles(filename, dir);sys.exit()
#buildUniProtFunctAnnotations('Hs',force='no')
species = 'Hs'; array_type = 'junction'; force = 'yes'; run_seqcomp = 'no'
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_probeset-probes.txt'
filename = string.replace(filename,'junction','junction/hGlue') ### Due this also for the hGlue array
#verifyFile(filename,'junction/hGlue')
#print filename; sys.exit()
#from build_scripts import IdentifyAltIsoforms
#IdentifyAltIsoforms.runProgram(species,array_type,'exon',force,run_seqcomp); sys.exit()
import UI
#date = UI.TimeStamp(); file_type = ('wikipathways_'+date+'.tab','.txt')
url ='http://www.wikipathways.org/wpi/pathway_content_flatfile.php?output=tab'
output = 'BuildDBs/wikipathways/'
file_type = ''
url = 'http://www.genmapp.org/go_elite/Databases/EnsMart56/Cs.zip'
output = 'Databases/'
url = 'http://www.altanalyze.org/archiveDBs/Cytoscape/cytoscape.tar.gz'
output = ''
dp = download_protocol(url,output,file_type);sys.exit()
fln,status = download(url,output,file_type)
print status;sys.exit()
url='http://altanalyze.org/archiveDBs/LibraryFiles/MoEx-1_0-st-v1.r2.antigenomic.bgp.gz'
dir='AltDatabase/affymetrix/LibraryFiles/'
file_type = ''
download(url,dir,file_type)
species='Hs'; array_type = 'junction'
#"""
array_type = ['RNASeq']; species = ['Mm']; force = 'yes'; ensembl_version = '60'
update_uniprot='no'; update_ensembl='no'; update_probeset_to_ensembl='yes'; update_domain='yes'; update_all='no'; update_miRs='yes'
proceed = 'yes'; genomic_build = 'old'; update_miR_seq = 'yes'
for specific_species in species:
for platform_name in array_type:
if platform_name == 'AltMouse' and specific_species == 'Mm': proceed = 'yes'
elif platform_name == 'exon' or platform_name == 'gene' or platform_name == 'junction': proceed = 'yes'
else: proceed = 'no'
if proceed == 'yes':
print "Analyzing", specific_species, platform_name
executeParameters(specific_species,platform_name,force,genomic_build,update_uniprot,update_ensembl,update_probeset_to_ensembl,update_domain,update_miRs,update_all,update_miR_seq,ensembl_version)
#"""
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/update.py
|
update.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys, string, os
from stats_scripts import statistics
import math
import os.path
import unique
import update
import copy
import time
import export
from build_scripts import EnsemblImport; reload(EnsemblImport)
try: from build_scripts import JunctionArrayEnsemblRules
except Exception: pass ### occurs with circular imports
try: from build_scripts import JunctionArray; reload(JunctionArray)
except Exception: pass ### occurs with circular imports
try: from build_scripts import ExonArrayEnsemblRules
except Exception: pass ### occurs with circular imports
import multiprocessing
import logging
import traceback
import warnings
import bisect
import shutil
from visualization_scripts import clustering; reload(clustering)
try:
import scipy
import scipy.cluster.hierarchy as sch
import scipy.spatial.distance as dist
except Exception: pass
try: import numpy
except Exception: pass
LegacyMode = True
try:
from scipy import average as Average
from scipy import stats
except Exception:
try: from statistics import avg as Average
except Exception: pass ### occurs with circular imports
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list_clean=[]
dir_list = unique.read_directory(sub_dir)
for filepath in dir_list:
if 'log.txt' not in filepath and '.log' not in filepath:
dir_list_clean.append(filepath)
return dir_list_clean
def makeUnique(item):
db1={}; list1=[]; k=0
for i in item:
try: db1[i]=[]
except TypeError: db1[tuple(i)]=[]; k=1
for i in db1:
if k==0: list1.append(i)
else: list1.append(list(i))
list1.sort()
return list1
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
######### Below code deals with building the AltDatabase #########
def collapseNoveExonBoundaries(novel_exon_coordinates,dataset_dir):
""" Merge exon predictions based on junction measurments from TopHat. The predicted exons are
bound by the identified splice site and the consensus length of reads in that sample"""
dataset_dir = string.replace(dataset_dir,'exp.','ExpressionInput/novel.')
export_data,status = AppendOrWrite(dataset_dir) ### Export all novel exons
if status == 'not found':
export_data.write('GeneID\tStrand\tExonID\tCoordinates\n')
novel_gene_exon_db={}
for (chr,coord) in novel_exon_coordinates:
key = (chr,coord)
ji,side,coord2 = novel_exon_coordinates[(chr,coord)]
try:
if side == 'left': ### left corresponds to the position of coord
intron = string.split(string.split(ji.ExonRegionID(),'-')[1][:2],'.')[0]
else:
intron = string.split(string.split(ji.ExonRegionID(),'-'),'.')[0]
ls = [coord,coord2]
ls.sort() ### The order of this is variable
if ji.Strand() == '-':
coord2,coord = ls
else: coord,coord2 = ls
if 'I' in intron and ji.Novel() == 'side':
#if 'ENSG00000221983' == ji.GeneID():
try: novel_gene_exon_db[ji.GeneID(),ji.Strand(),intron].append((coord,coord2,ji,key,side))
except Exception: novel_gene_exon_db[ji.GeneID(),ji.Strand(),intron] = [(coord,coord2,ji,key,side)]
except Exception: pass
outdatedExons={} ### merging novel exons, delete one of the two original
for key in novel_gene_exon_db:
firstNovel=True ### First putative novel exon coordinates examined for that gene
novel_gene_exon_db[key].sort()
if key[1]=='-':
novel_gene_exon_db[key].reverse()
for (c1,c2,ji,k,s) in novel_gene_exon_db[key]:
if firstNovel==False:
#print [c1,l2] #abs(c1-l2);sys.exit()
### see if the difference between the start position of the second exon is less than 300 nt away from the end of the last
if abs(c2-l1) < 300 and os!=s: ### 80% of human exons are less than 200nt - PMID: 15217358
proceed = True
#if key[1]=='-':
if c2 in k:
novel_exon_coordinates[k] = ji,s,l1
outdatedExons[ok]=None ### merged out entry
elif l1 in ok:
novel_exon_coordinates[ok] = li,os,c2
outdatedExons[k]=None ### merged out entry
else:
proceed = False ### Hence, the two splice-site ends are pointing to two distinct versus one common exons
"""
if c2 == 18683670 or l1 == 18683670:
print key,abs(c2-l1), c1, c2, l1, l2, li.ExonRegionID(), ji.ExonRegionID();
print k,novel_exon_coordinates[k]
print ok,novel_exon_coordinates[ok]
"""
if proceed:
values = string.join([ji.GeneID(),ji.Strand(),key[2],ji.Chr()+':'+str(l1)+'-'+str(c2)],'\t')+'\n'
export_data.write(values)
### For negative strand genes, c1 is larger than c2 but is the 5' begining of the exon
l1,l2,li,ok,os = c1,c2,ji,k,s ### record the last entry
firstNovel=False
for key in outdatedExons: ### Delete the non-merged entry
del novel_exon_coordinates[key]
export_data.close()
return novel_exon_coordinates
def exportNovelExonToBedCoordinates(species,novel_exon_coordinates,chr_status,searchChr=None):
### Export the novel exon coordinates based on those in the junction BED file to examine the differential expression of the predicted novel exon
#bamToBed -i accepted_hits.bam -split| coverageBed -a stdin -b /home/databases/hESC_differentiation_exons.bed > day20_7B__exons-novel.bed
bed_export_path = filepath('AltDatabase/'+species+'/RNASeq/chr/'+species + '_Ensembl_exons'+searchChr+'.bed')
bed_data = open(bed_export_path,'w') ### Appends to existing file
for (chr,coord) in novel_exon_coordinates:
ji,side,coord2 = novel_exon_coordinates[(chr,coord)]
if side == 'left': start,stop = coord,coord2
if side == 'right': start,stop = coord2,coord
try: gene = ji.GeneID()
except Exception: gene = 'NA'
if gene == None: gene = 'NA'
if gene == None: gene = 'NA'
if gene != 'NA': ### Including these has no benefit for AltAnalyze (just slows down alignment and piles up memory)
if ji.Strand() == '-': stop,start=start,stop
if chr_status == False:
chr = string.replace(chr,'chr','') ### This will thus match up to the BAM files
a = [start,stop]; a.sort(); start,stop = a
bed_values = [chr,str(start),str(stop),gene,'0',str(ji.Strand())]
bed_values = cleanUpLine(string.join(bed_values,'\t'))+'\n'
bed_data.write(bed_values)
bed_data.close()
return bed_export_path
def moveBAMtoBEDFile(species,dataset_name,root_dir):
bed_export_path = filepath('AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_exons.bed')
dataset_name = string.replace(dataset_name,'exp.','')
new_fn = root_dir+'/BAMtoBED/'+species + '_'+dataset_name+'_exons.bed'
new_fn = string.replace(new_fn,'.txt','')
print 'Writing exon-level coordinates to BED file:'
print new_fn
catFiles(bed_export_path,'chr') ### concatenate the files ot the main AltDatabase directory then move
export.customFileMove(bed_export_path,new_fn)
return new_fn
def reformatExonFile(species,type,chr_status):
if type == 'exon':
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt'
export_path = 'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_exons.txt'
### Used by BEDTools to get counts per specific AltAnalyze exon region (should augment with de novo regions identified from junction analyses)
bed_export_path = 'AltDatabase/'+species+'/RNASeq/chr/'+species + '_Ensembl_exons.bed'
bed_data = export.ExportFile(bed_export_path)
else:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_junction.txt'
export_path = 'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_junctions.txt'
print 'Writing',export_path
try:
export_data = export.ExportFile(export_path)
fn=filepath(filename); x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
x+=1
export_title = ['AltAnalyzeID','exon_id','ensembl_gene_id','transcript_cluster_id','chromosome','strand','probeset_start','probeset_stop']
export_title +=['affy_class','constitutive_probeset','ens_exon_ids','ens_constitutive_status','exon_region','exon-region-start(s)','exon-region-stop(s)','splice_events','splice_junctions']
export_title = string.join(export_title,'\t')+'\n'; export_data.write(export_title)
else:
try: gene, exonid, chr, strand, start, stop, constitutive_call, ens_exon_ids, splice_events, splice_junctions = t
except Exception: print t;kill
if chr == 'chrM': chr = 'chrMT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if chr == 'M': chr = 'MT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention,
if constitutive_call == 'yes': ens_constitutive_status = '1'
else: ens_constitutive_status = '0'
export_values = [gene+':'+exonid, exonid, gene, '', chr, strand, start, stop, 'known', constitutive_call, ens_exon_ids, ens_constitutive_status]
export_values+= [exonid, start, stop, splice_events, splice_junctions]
export_values = string.join(export_values,'\t')+'\n'; export_data.write(export_values)
if type == 'exon':
if chr_status == False:
chr = string.replace(chr,'chr','') ### This will thus match up to the BAM files
bed_values = [chr,start,stop,gene+':'+exonid+'_'+ens_exon_ids,'0',strand]
bed_values = string.join(bed_values,'\t')+'\n'; bed_data.write(bed_values)
export_data.close()
except: pass ### occurs for machines with write permission errors to the AltAnalyze directory (fixed in 2.1.4)
try:
if type == 'exon': bed_data.close()
except: pass
def importExonAnnotations(species,type,search_chr):
if 'exon' in type:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt'
else:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_junction.txt'
fn=filepath(filename); x=0; exon_annotation_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: x=1
else:
gene, exonid, chr, strand, start, stop, constitutive_call, ens_exon_ids, splice_events, splice_junctions = t; proceed = 'yes'
if chr == 'chrM': chr = 'chrMT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if chr == 'M': chr = 'MT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if len(search_chr)>0:
if chr != search_chr: proceed = 'no'
if proceed == 'yes':
if type == 'exon': start = int(start); stop = int(stop)
ea = EnsemblImport.ExonAnnotationsSimple(chr, strand, start, stop, gene, ens_exon_ids, constitutive_call, exonid, splice_events, splice_junctions)
if type == 'junction_coordinates':
exon1_start,exon1_stop = string.split(start,'|')
exon2_start,exon2_stop = string.split(stop,'|')
if strand == '-':
exon1_stop,exon1_start = exon1_start,exon1_stop
exon2_stop,exon2_start = exon2_start,exon2_stop
#if gene == 'ENSMUSG00000027340': print chr,int(exon1_stop),int(exon2_start)
exon_annotation_db[chr,int(exon1_stop),int(exon2_start)]=ea
elif type == 'distal-exon':
exon_annotation_db[gene] = exonid
else:
try: exon_annotation_db[gene].append(ea)
except KeyError: exon_annotation_db[gene]=[ea]
return exon_annotation_db
def exportKnownJunctionComparisons(species):
gene_junction_db = JunctionArrayEnsemblRules.importEnsemblUCSCAltJunctions(species,'standard')
gene_intronjunction_db = JunctionArrayEnsemblRules.importEnsemblUCSCAltJunctions(species,'_intronic')
for i in gene_intronjunction_db: gene_junction_db[i]=[]
gene_junction_db2={}
for (gene,critical_exon,incl_junction,excl_junction) in gene_junction_db:
critical_exons = string.split(critical_exon,'|')
for critical_exon in critical_exons:
try: gene_junction_db2[gene,incl_junction,excl_junction].append(critical_exon)
except Exception: gene_junction_db2[gene,incl_junction,excl_junction] = [critical_exon]
gene_junction_db = gene_junction_db2; gene_junction_db2=[]
junction_export = 'AltDatabase/' + species + '/RNASeq/'+ species + '_junction_comps.txt'
fn=filepath(junction_export); data = open(fn,'w')
print "Exporting",junction_export
title = 'gene'+'\t'+'critical_exon'+'\t'+'exclusion_junction_region'+'\t'+'inclusion_junction_region'+'\t'+'exclusion_probeset'+'\t'+'inclusion_probeset'+'\t'+'data_source'+'\n'
data.write(title); temp_list=[]
for (gene,incl_junction,excl_junction) in gene_junction_db:
critical_exons = unique.unique(gene_junction_db[(gene,incl_junction,excl_junction)])
critical_exon = string.join(critical_exons,'|')
temp_list.append(string.join([gene,critical_exon,excl_junction,incl_junction,gene+':'+excl_junction,gene+':'+incl_junction,'AltAnalyze'],'\t')+'\n')
temp_list = unique.unique(temp_list)
for i in temp_list: data.write(i)
data.close()
def getExonAndJunctionSequences(species):
export_exon_filename = 'AltDatabase/'+species+'/RNASeq/'+species+'_Ensembl_exons.txt'
ensembl_exon_db = ExonArrayEnsemblRules.reimportEnsemblProbesetsForSeqExtraction(export_exon_filename,'null',{})
### Import just the probeset region for mRNA alignment analysis
analysis_type = ('region_only','get_sequence'); array_type = 'RNASeq'
dir = 'AltDatabase/'+species+'/SequenceData/chr/'+species; gene_seq_filename = dir+'_gene-seq-2000_flank.fa'
ensembl_exon_db = EnsemblImport.import_sequence_data(gene_seq_filename,ensembl_exon_db,species,analysis_type)
critical_exon_file = 'AltDatabase/'+species+'/'+ array_type + '/' + array_type+'_critical-exon-seq.txt'
getCriticalJunctionSequences(critical_exon_file,species,ensembl_exon_db)
"""
### Import the full Ensembl exon sequence (not just the probeset region) for miRNA binding site analysis
analysis_type = 'get_sequence'; array_type = 'RNASeq'
dir = 'AltDatabase/'+species+'/SequenceData/chr/'+species; gene_seq_filename = dir+'_gene-seq-2000_flank.fa'
ensembl_exon_db = EnsemblImport.import_sequence_data(gene_seq_filename,ensembl_exon_db,species,analysis_type)
"""
critical_exon_file = 'AltDatabase/'+species+'/'+ array_type + '/' + array_type+'_critical-exon-seq.txt'
updateCriticalExonSequences(critical_exon_file, ensembl_exon_db)
def updateCriticalExonSequences(filename,ensembl_exon_db):
exon_seq_db_filename = filename[:-4]+'_updated.txt'
exonseq_data = export.ExportFile(exon_seq_db_filename)
critical_exon_seq_db={}; null_count={}
for gene in ensembl_exon_db:
gene_exon_data={}
for probe_data in ensembl_exon_db[gene]:
exon_id,((probe_start,probe_stop,probeset_id,exon_class,transcript_clust),ed) = probe_data
try: gene_exon_data[probeset_id] = ed.ExonSeq()
except Exception: null_count[gene]=[] ### Occurs for non-chromosomal DNA (could also download this sequence though)
if len(gene_exon_data)>0: critical_exon_seq_db[gene] = gene_exon_data
print len(null_count),'genes not assigned sequenced (e.g.,non-chromosomal)'
ensembl_exon_db=[]
### Export exon sequences
for gene in critical_exon_seq_db:
gene_exon_data = critical_exon_seq_db[gene]
for probeset in gene_exon_data:
critical_exon_seq = gene_exon_data[probeset]
values = [probeset,'',critical_exon_seq]
values = string.join(values,'\t')+'\n'
exonseq_data.write(values)
exonseq_data.close()
print exon_seq_db_filename, 'exported....'
def getCriticalJunctionSequences(filename,species,ensembl_exon_db):
### Assemble and export junction sequences
junction_seq_db_filename = string.replace(filename,'exon-seq','junction-seq')
junctionseq_data = export.ExportFile(junction_seq_db_filename)
critical_exon_seq_db={}; null_count={}
for gene in ensembl_exon_db:
gene_exon_data={}
for probe_data in ensembl_exon_db[gene]:
exon_id,((probe_start,probe_stop,probeset_id,exon_class,transcript_clust),ed) = probe_data
try: gene_exon_data[probeset_id] = ed.ExonSeq()
except Exception: null_count[gene]=[] ### Occurs for non-chromosomal DNA (could also download this sequence though)
if len(gene_exon_data)>0: critical_exon_seq_db[gene] = gene_exon_data
print len(null_count),'genes not assigned sequenced (e.g.,non-chromosomal)'
ensembl_exon_db=[]
junction_annotation_db = importExonAnnotations(species,'junction',[])
for gene in junction_annotation_db:
if gene in critical_exon_seq_db:
gene_exon_data = critical_exon_seq_db[gene]
for jd in junction_annotation_db[gene]:
exon1,exon2=string.split(jd.ExonRegionIDs(),'-')
p1=gene+':'+exon1
p2=gene+':'+exon2
p1_seq=gene_exon_data[p1][-15:]
p2_seq=gene_exon_data[p2][:15]
junction_seq = p1_seq+'|'+p2_seq
junctionseq_data.write(gene+':'+jd.ExonRegionIDs()+'\t'+junction_seq+'\t\n')
junctionseq_data.close()
print junction_seq_db_filename, 'exported....'
def getEnsemblAssociations(species,data_type,test_status,force):
### Get UCSC associations (download databases if necessary)
from build_scripts import UCSCImport
mRNA_Type = 'mrna'; run_from_scratch = 'yes'
export_all_associations = 'no' ### YES only for protein prediction analysis
update.buildUCSCAnnoationFiles(species,mRNA_Type,export_all_associations,run_from_scratch,force)
null = EnsemblImport.getEnsemblAssociations(species,data_type,test_status); null=[]
reformatExonFile(species,'exon',True); reformatExonFile(species,'junction',True)
exportKnownJunctionComparisons(species)
getExonAndJunctionSequences(species)
######### Below code deals with user read alignment as opposed to building the AltDatabase #########
class ExonInfo:
def __init__(self,start,unique_id,annotation):
self.start = start; self.unique_id = unique_id; self.annotation = annotation
def ReadStart(self): return self.start
def UniqueID(self): return self.unique_id
def Annotation(self): return self.annotation
def setExonRegionData(self,rd): self.rd = rd
def ExonRegionData(self): return self.rd
def setExonRegionID(self,region_id): self.region_id = region_id
def ExonRegionID(self): return self.region_id
def setAlignmentRegion(self,region_type): self.region_type = region_type
def AlignmentRegion(self): return self.region_type
def __repr__(self): return "ExonData values"
class JunctionData:
def __init__(self,chr,strand,exon1_stop,exon2_start,junction_id,biotype):
self.chr = chr; self.strand = strand; self._chr = chr
self.exon1_stop = exon1_stop; self.exon2_start = exon2_start
self.junction_id = junction_id; self.biotype = biotype
#self.reads = reads; self.condition = condition
self.left_exon = None; self.right_exon = None; self.jd = None; self.gene_id = None
self.trans_splicing = None
self.splice_events=''
self.splice_junctions=''
self.seq_length=''
self.uid = None
def Chr(self): return self.chr
def Strand(self): return self.strand
def Exon1Stop(self): return self.exon1_stop
def Exon2Start(self): return self.exon2_start
def setExon1Stop(self,exon1_stop): self.exon1_stop = exon1_stop
def setExon2Start(self,exon2_start): self.exon2_start = exon2_start
def setSeqLength(self,seq_length): self.seq_length = seq_length
def SeqLength(self): return self.seq_length
def BioType(self): return self.biotype
def checkExonPosition(self,exon_pos):
if exon_pos == self.Exon1Stop(): return 'left'
else: return 'right'
### These are used to report novel exon boundaries
def setExon1Start(self,exon1_start): self.exon1_start = exon1_start
def setExon2Stop(self,exon2_stop): self.exon2_stop = exon2_stop
def Exon1Start(self): return self.exon1_start
def Exon2Stop(self): return self.exon2_stop
def Reads(self): return self.reads
def JunctionID(self): return self.junction_id
def Condition(self): return self.condition
def setExonAnnotations(self,jd):
self.jd = jd
self.splice_events = jd.AssociatedSplicingEvent()
self.splice_junctions = jd.AssociatedSplicingJunctions()
self.exon_region = jd.ExonRegionIDs()
self.exonid = jd.ExonID()
self.gene_id = jd.GeneID()
self.uid = jd.GeneID()+':'+jd.ExonRegionIDs()
def ExonAnnotations(self): return self.jd
def setLeftExonAnnotations(self,ld): self.gene_id,self.left_exon = ld
def LeftExonAnnotations(self): return self.left_exon
def setRightExonAnnotations(self,rd): self.secondary_geneid,self.right_exon = rd
def RightExonAnnotations(self): return self.right_exon
def setGeneID(self,geneid): self.gene_id = geneid
def GeneID(self): return self.gene_id
def setSecondaryGeneID(self,secondary_geneid): self.secondary_geneid = secondary_geneid
def SecondaryGeneID(self): return self.secondary_geneid
def setTransSplicing(self): self.trans_splicing = 'yes'
def TransSplicing(self): return self.trans_splicing
def SpliceSitesFound(self):
if self.jd != None: sites_found = 'both'
elif self.left_exon != None and self.right_exon != None: sites_found = 'both'
elif self.left_exon != None: sites_found = 'left'
elif self.right_exon != None: sites_found = 'right'
else: sites_found = None
return sites_found
def setConstitutive(self,constitutive): self.constitutive = constitutive
def Constitutive(self): return self.constitutive
def setAssociatedSplicingEvent(self,splice_events): self.splice_events = splice_events
def AssociatedSplicingEvent(self): return self.splice_events
def setAssociatedSplicingJunctions(self,splice_junctions): self.splice_junctions = splice_junctions
def AssociatedSplicingJunctions(self): return self.splice_junctions
def setExonID(self,exonid): self.exonid = exonid
def ExonID(self): return self.exonid
def setExonRegionID(self,exon_region): self.exon_region = exon_region
def ExonRegionID(self): return self.exon_region
def setUniqueID(self,uid): self.uid = uid
def UniqueID(self): return self.uid
def setLeftExonRegionData(self,li): self.li = li
def LeftExonRegionData(self): return self.li
def setRightExonRegionData(self,ri): self.ri = ri
def RightExonRegionData(self): return self.ri
def setNovel(self, side): self.side = side
def Novel(self): return self.side
def __repr__(self): return "JunctionData values"
def checkBEDFileFormat(bed_dir,root_dir):
""" This method checks to see if the BED files (junction or exon) have 'chr' proceeding the chr number.
It also checks to see if some files have two underscores and one has none or if double underscores are missing from all."""
dir_list = read_directory(bed_dir)
x=0
break_now = False
chr_present = False
condition_db={}
for filename in dir_list:
fn=filepath(bed_dir+filename)
#if ('.bed' in fn or '.BED' in fn): delim = 'r'
delim = 'rU'
if '.tab' in string.lower(filename) or '.bed' in string.lower(filename) or '.junction_quantification.txt' in string.lower(filename):
condition_db[filename]=[]
for line in open(fn,delim).xreadlines(): ### changed rU to r to remove \r effectively, rather than read as end-lines
if line[0] == '#': x=0 ### BioScope
elif x == 0: x=1 ###skip the first line
elif x < 10: ### Only check the first 10 lines
if 'chr' in line: ### Need to look at multiple input formats (chr could be in t[0] or t[1])
chr_present = True
x+=1
else:
break_now = True
break
if break_now == True:
break
### Check to see if exon.bed and junction.bed file names are propper or faulty (which will result in downstream errors)
double_underscores=[]
no_doubles=[]
for condition in condition_db:
if '__' in condition:
double_underscores.append(condition)
else:
no_doubles.append(condition)
exon_beds=[]
junctions_beds=[]
if len(double_underscores)>0 and len(no_doubles)>0:
### Hence, a problem is likely due to inconsistent naming
print 'The input files appear to have inconsistent naming. If both exon and junction sample data are present, make sure they are named propperly.'
print 'For example: cancer1__exon.bed, cancer1__junction.bed (double underscore required to match these samples up)!'
print 'Exiting AltAnalyze'; forceError
elif len(no_doubles)>0:
for condition in no_doubles:
condition = string.lower(condition)
if 'exon' in condition:
exon_beds.append(condition)
if 'junction' in condition:
junctions_beds.append(condition)
if len(exon_beds)>0 and len(junctions_beds)>0:
print 'The input files appear to have inconsistent naming. If both exon and junction sample data are present, make sure they are named propperly.'
print 'For example: cancer1__exon.bed, cancer1__junction.bed (double underscore required to match these samples up)!'
print 'Exiting AltAnalyze'; forceError
return chr_present
def getStrandMappingData(species):
splicesite_db={}
refExonCoordinateFile = unique.filepath('AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt')
firstLine=True
for line in open(refExonCoordinateFile,'rU').xreadlines():
if firstLine: firstLine=False
else:
line = line.rstrip('\n')
t = string.split(line,'\t'); #'gene', 'exon-id', 'chromosome', 'strand', 'exon-region-start(s)', 'exon-region-stop(s)', 'constitutive_call', 'ens_exon_ids', 'splice_events', 'splice_junctions'
geneID, exon, chr, strand, start, stop = t[:6]
splicesite_db[chr,int(start)]=strand
splicesite_db[chr,int(stop)]=strand
return splicesite_db
def importBEDFile(bed_dir,root_dir,species,normalize_feature_exp,getReads=False,searchChr=None,getBiotype=None,testImport=False,filteredJunctions=None):
dir_list = read_directory(bed_dir)
begin_time = time.time()
if 'chr' not in searchChr:
searchChr = 'chr'+searchChr
condition_count_db={}; neg_count=0; pos_count=0; junction_db={}; biotypes={}; algorithms={}; exon_len_db={}; splicesite_db={}
if testImport == 'yes': print "Reading user RNA-seq input data files"
for filename in dir_list:
count_db={}; rows=0
fn=filepath(bed_dir+filename)
condition = export.findFilename(fn)
if '__' in condition:
### Allow multiple junction files per sample to be combined (e.g. canonical and non-canonical junction alignments)
condition=string.split(condition,'__')[0]+filename[-4:]
if ('.bed' in fn or '.BED' in fn or '.tab' in fn or '.TAB' in fn or '.junction_quantification.txt' in fn) and '._' not in condition:
if ('.bed' in fn or '.BED' in fn): delim = 'r'
else: delim = 'rU'
### The below code removes .txt if still in the filename along with .tab or .bed
if '.tab' in fn: condition = string.replace(condition,'.txt','.tab')
elif '.bed' in fn: condition = string.replace(condition,'.txt','.bed')
if '.TAB' in fn: condition = string.replace(condition,'.txt','.TAB')
elif '.BED' in fn: condition = string.replace(condition,'.txt','.BED')
if testImport == 'yes': print "Reading the bed file", [fn], condition
### If the BED was manually created on a Mac, will neeed 'rU' - test this
for line in open(fn,delim).xreadlines(): break
if len(line)>500: delim = 'rU'
for line in open(fn,delim).xreadlines(): ### changed rU to r to remove \r effectively, rather than read as end-lines
data = cleanUpLine(line)
t = string.split(data,'\t')
rows+=1
if rows==1 or '#' == data[0]:
format_description = data
algorithm = 'Unknown'
if 'TopHat' in format_description: algorithm = 'TopHat'
elif 'HMMSplicer' in format_description: algorithm = 'HMMSplicer'
elif 'SpliceMap junctions' in format_description: algorithm = 'SpliceMap'
elif t[0] == 'E1': algorithm = 'BioScope-junction'
elif '# filterOrphanedMates=' in data or 'alignmentFilteringMode=' in data or '#number_of_mapped_reads=' in data:
algorithm = 'BioScope-exon'
elif '.junction_quantification.txt' in fn:
algorithm = 'TCGA format'
if 'barcode' in t: junction_position = 1
else: junction_position = 0
if 'Hybridization' in line:
rows = 0
elif '.tab' in fn and len(t)==9:
try: start = float(t[1]) ### expect this to be a numerical coordinate
except Exception: continue
algorithm = 'STAR'
strand = '-' ### If no strand exists
rows=2 ### allows this first row to be processed
if len(splicesite_db)==0: ### get strand to pos info
splicesite_db = getStrandMappingData(species)
if testImport == 'yes': print condition, algorithm
if rows>1:
try:
if ':' in t[0]:
chr = string.split(t[0],':')[0]
else: chr = t[0]
if 'chr' not in chr:
chr = 'chr'+chr
if searchChr == chr or ('BioScope' in algorithm and searchChr == t[1]): proceed = True
elif searchChr == 'chrMT' and ('BioScope' not in algorithm):
if 'M' in chr and len(chr)<6: proceed = True ### If you don't have the length, any random thing with an M will get included
else: proceed = False
else: proceed = False
except IndexError:
print 'The input file:\n',filename
print 'is not formated as expected (format='+algorithm+').'
print 'search chromosome:',searchChr
print t; force_bad_exit
if proceed:
proceed = False
if '.tab' in fn or '.TAB' in fn:
### Applies to non-BED format Junction and Exon inputs (BioScope)
if 'BioScope' in algorithm:
if algorithm == 'BioScope-exon': ### Not BED format
chr,source,data_type,start,end,reads,strand,null,gene_info=t[:9]
if 'chr' not in chr: chr = 'chr'+chr
if data_type == 'exon': ### Can also be CDS
gene_info,test,rpkm_info,null = string.split(gene_info,';')
symbol = string.split(gene_info,' ')[-1]
#refseq = string.split(transcript_info,' ')[-1]
rpkm = string.split(rpkm_info,' ')[-1]
#if normalize_feature_exp == 'RPKM': reads = rpkm ### The RPKM should be adjusted +1 counts, so don't use this
biotype = 'exon'; biotypes[biotype]=[]
exon1_stop,exon2_start = int(start),int(end); junction_id=''
### Adjust exon positions - not ideal but necessary. Needed as a result of exon regions overlapping by 1nt (due to build process)
exon1_stop+=1; exon2_start-=1
#if float(reads)>4 or getReads:
proceed = True ### Added in version 2.0.9 to remove rare novel isoforms
seq_length = abs(exon1_stop-exon2_start)
if algorithm == 'BioScope-junction':
chr = t[1]; strand = t[2]; exon1_stop = int(t[4]); exon2_start = int(t[8]); count_paired = t[17]; count_single = t[19]; score=t[21]
if 'chr' not in chr: chr = 'chr'+chr
try: exon1_start = int(t[3]); exon2_stop = int(t[9])
except Exception: pass ### If missing, these are not assigned
reads = str(int(float(count_paired))+int(float(count_single))) ### Users will either have paired or single read (this uses either)
biotype = 'junction'; biotypes[biotype]=[]; junction_id=''
if float(reads)>4 or getReads: proceed = True ### Added in version 2.0.9 to remove rare novel isoforms
seq_length = abs(float(exon1_stop-exon2_start))
if 'STAR' in algorithm:
chr = t[0]; exon1_stop = int(t[1])-1; exon2_start = int(t[2])+1; strand=''
if 'chr' not in chr: chr = 'chr'+chr
reads = str(int(t[7])+int(t[6]))
biotype = 'junction'; biotypes[biotype]=[]; junction_id=''
if float(reads)>4 or getReads: proceed = True ### Added in version 2.0.9 to remove rare novel isoforms
if (chr,exon1_stop) in splicesite_db:
strand = splicesite_db[chr,exon1_stop]
elif (chr,exon2_start) in splicesite_db:
strand = splicesite_db[chr,exon2_start]
#else: proceed = False
seq_length = abs(float(exon1_stop-exon2_start))
if strand == '-': ### switch the orientation of the positions
exon1_stop,exon2_start=exon2_start,exon1_stop
exon1_start = exon1_stop; exon2_stop = exon2_start
#if 9996685==exon1_stop and 10002682==exon2_stop:
#print chr, strand, reads, exon1_stop, exon2_start,proceed;sys.exit()
else:
try:
if algorithm == 'TCGA format':
coordinates = string.split(t[junction_position],',')
try: chr,pos1,strand = string.split(coordinates[0],':')
except Exception: print t;sys.exit()
chr,pos2,strand = string.split(coordinates[1],':')
if 'chr' not in chr: chr = 'chr'+chr
if abs(int(pos1)-int(pos2))<2:
pos2 = str(int(pos2)-1) ### This is the bed format conversion with exons of 0 length
exon1_start, exon2_stop = pos1, pos2
reads = t[junction_position+1]
junction_id = t[junction_position]
exon1_len=0; exon2_len=0
else:
### Applies to BED format Junction input
chr, exon1_start, exon2_stop, junction_id, reads, strand, null, null, null, null, lengths, null = t
if 'chr' not in chr: chr = 'chr'+chr
exon1_len,exon2_len=string.split(lengths,',')[:2]; exon1_len = int(exon1_len); exon2_len = int(exon2_len)
exon1_start = int(exon1_start); exon2_stop = int(exon2_stop)
biotype = 'junction'; biotypes[biotype]=[]
if strand == '-':
if (exon1_len+exon2_len)==0: ### Kallisto-Splice directly reports these coordinates
exon1_stop = exon1_start
exon2_start = exon2_stop
else:
exon1_stop = exon1_start+exon1_len; exon2_start=exon2_stop-exon2_len+1
### Exons have the opposite order
a = exon1_start,exon1_stop; b = exon2_start,exon2_stop
exon1_stop,exon1_start = b; exon2_stop,exon2_start = a
else:
if (exon1_len+exon2_len)==0: ### Kallisto-Splice directly reports these coordinates
exon1_stop = exon1_start
exon2_start= exon2_stop
else:
exon1_stop = exon1_start+exon1_len; exon2_start=exon2_stop-exon2_len+1
if float(reads)>4 or getReads: proceed = True
if algorithm == 'HMMSplicer':
if '|junc=' in junction_id: reads = string.split(junction_id,'|junc=')[-1]
else: proceed = False
if algorithm == 'SpliceMap':
if ')' in junction_id and len(junction_id)>1: reads = string.split(junction_id,')')[0][1:]
else: proceed = False
seq_length = abs(float(exon1_stop-exon2_start)) ### Junction distance
except Exception,e:
#print traceback.format_exc();sys.exit()
### Applies to BED format exon input (BEDTools export)
# bamToBed -i accepted_hits.bam -split| coverageBed -a stdin -b /home/nsalomonis/databases/Mm_Ensembl_exons.bed > day0_8B__exons.bed
try: chr, start, end, exon_id, null, strand, reads, bp_coverage, bp_total, percent_coverage = t
except Exception:
print 'The file',fn,'does not appear to be propperly formatted as input.'
print t; force_exception
if 'chr' not in chr: chr = 'chr'+chr
algorithm = 'TopHat-exon'; biotype = 'exon'; biotypes[biotype]=[]
exon1_stop,exon2_start = int(start),int(end); junction_id=exon_id; seq_length = float(bp_total)
if seq_length == 0:
seq_length = abs(float(exon1_stop-exon2_start))
### Adjust exon positions - not ideal but necessary. Needed as a result of exon regions overlapping by 1nt (due to build process)
exon1_stop+=1; exon2_start-=1
#if float(reads)>4 or getReads: ### Added in version 2.0.9 to remove rare novel isoforms
proceed = True
#else: proceed = False
if proceed:
if 'chr' not in chr:
chr = 'chr'+chr ### Add the chromosome prefix
if chr == 'chrM': chr = 'chrMT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if chr == 'M': chr = 'MT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if strand == '+': pos_count+=1
else: neg_count+=1
if getReads and seq_length>0:
if getBiotype == biotype:
if biotype == 'junction':
### We filtered for junctions>4 reads before, now we include all reads for expressed junctions
if (chr,exon1_stop,exon2_start) in filteredJunctions:
count_db[chr,exon1_stop,exon2_start] = reads
try: exon_len_db[chr,exon1_stop,exon2_start] = seq_length
except Exception: exon_len_db[chr,exon1_stop,exon2_start] = []
else:
count_db[chr,exon1_stop,exon2_start] = reads
try: exon_len_db[chr,exon1_stop,exon2_start] = seq_length
except Exception: exon_len_db[chr,exon1_stop,exon2_start] = []
elif seq_length>0:
if (chr,exon1_stop,exon2_start) not in junction_db:
ji = JunctionData(chr,strand,exon1_stop,exon2_start,junction_id,biotype)
junction_db[chr,exon1_stop,exon2_start] = ji
try: ji.setSeqLength(seq_length) ### If RPKM imported or calculated
except Exception: null=[]
try: ji.setExon1Start(exon1_start);ji.setExon2Stop(exon2_stop)
except Exception: null=[]
key = chr,exon1_stop,exon2_start
algorithms[algorithm]=[]
if getReads:
if condition in condition_count_db:
### combine the data from the different files for the same sample junction alignments
count_db1 = condition_count_db[condition]
for key in count_db:
if key not in count_db1: count_db1[key] = count_db[key]
else:
combined_counts = int(count_db1[key])+int(count_db[key])
count_db1[key] = str(combined_counts)
condition_count_db[condition]=count_db1
else:
try: condition_count_db[condition] = count_db
except Exception: null=[] ### Occurs for other text files in the directory that are not used for the analysis
end_time = time.time()
if testImport == 'yes': print 'Read coordinates imported in',int(end_time-begin_time),'seconds'
if getReads:
#print len(exon_len_db), getBiotype, 'read counts present for',algorithm
return condition_count_db,exon_len_db,biotypes,algorithms
else:
if testImport == 'yes':
if 'exon' not in biotypes and 'BioScope' not in algorithm:
print len(junction_db),'junctions present in',algorithm,'format BED files.' # ('+str(pos_count),str(neg_count)+' by strand).'
elif 'exon' in biotypes and 'BioScope' not in algorithm:
print len(junction_db),'sequence identifiers present in input files.'
else: print len(junction_db),'sequence identifiers present in BioScope input files.'
return junction_db,biotypes,algorithms
def importExonCoordinates(probeCoordinateFile,search_chr,getBiotype):
probe_coordinate_db={}
junction_db={}
biotypes={}
x=0
fn=filepath(probeCoordinateFile)
for line in open(fn,'rU').xreadlines(): ### changed rU to r to remove \r effectively, rather than read as end-lines
data = cleanUpLine(line)
if x==0: x=1
else:
t = string.split(data,'\t')
probe_id = t[0]; probeset_id=t[1]; chr=t[2]; strand=t[3]; start=t[4]; end=t[5]
exon1_stop,exon2_start = int(start),int(end)
seq_length = abs(float(exon1_stop-exon2_start))
if 'chr' not in chr:
chr = 'chr'+chr ### Add the chromosome prefix
if chr == 'chrM': chr = 'chrMT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if search_chr == chr or search_chr == None:
try: biotype = t[6]
except Exception:
if seq_length>25:biotype = 'junction'
else: biotype = 'exon'
if strand == '-':
exon1_stop,exon2_start = exon2_start, exon1_stop ### this is their actual 5' -> 3' orientation
if biotype == 'junction':
exon1_start,exon2_stop = exon1_stop,exon2_start
else:
exon1_stop+=1; exon2_start-=1
biotypes[biotype]=[]
if getBiotype == biotype or getBiotype == None:
ji = JunctionData(chr,strand,exon1_stop,exon2_start,probe_id,biotype)
junction_db[chr,exon1_stop,exon2_start] = ji
try: ji.setSeqLength(seq_length) ### If RPKM imported or calculated
except Exception: null=[]
try: ji.setExon1Start(exon1_start);ji.setExon2Stop(exon2_stop)
except Exception: null=[]
probe_coordinate_db[probe_id] = chr,exon1_stop,exon2_start ### Import the expression data for the correct chromosomes with these IDs
return probe_coordinate_db, junction_db, biotypes
def importExpressionMatrix(exp_dir,root_dir,species,fl,getReads,search_chr=None,getBiotype=None):
""" Non-RNA-Seq expression data (typically Affymetrix microarray) import and mapping to an external probe-coordinate database """
begin_time = time.time()
condition_count_db={}; neg_count=0; pos_count=0; algorithms={}; exon_len_db={}
probe_coordinate_db, junction_db, biotypes = importExonCoordinates(fl.ExonMapFile(),search_chr,getBiotype)
x=0
fn=filepath(exp_dir)[:-1]
condition = export.findFilename(fn)
### If the BED was manually created on a Mac, will neeed 'rU' - test this
for line in open(fn,'rU').xreadlines(): ### changed rU to r to remove \r effectively, rather than read as end-lines
data = cleanUpLine(line)
t = string.split(data,'\t')
if '#' == data[0]: None
elif x==0:
if 'block' in t:
start_index = 7
else:
start_index = 1
headers = t[start_index:]
x=1
else:
proceed = 'yes' ### restrict by chromosome with minimum line parsing (unless we want counts instead)
probe_id=t[0]
if probe_id in probe_coordinate_db:
key = probe_coordinate_db[probe_id]
if getReads == 'no':
pass
else:
expression_data = t[start_index:]
i=0
for sample in headers:
if sample in condition_count_db:
count_db = condition_count_db[sample]
count_db[key] = expression_data[i]
exon_len_db[key]=[]
else:
count_db={}
count_db[key] = expression_data[i]
condition_count_db[sample] = count_db
exon_len_db[key]=[]
i+=1
algorithms['ProbeData']=[]
end_time = time.time()
if testImport == 'yes': print 'Probe data imported in',int(end_time-begin_time),'seconds'
if getReads == 'yes':
return condition_count_db,exon_len_db,biotypes,algorithms
else:
return junction_db,biotypes,algorithms
def adjustCounts(condition_count_db,exon_len_db):
for key in exon_len_db:
try:
null=exon_len_db[key]
for condition in condition_count_db:
count_db = condition_count_db[condition]
try: read_count = float(count_db[key])+1 ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
except KeyError: read_count = 1 ###Was zero, but needs to be one for more realistic log2 fold calculations
count_db[key] = str(read_count) ### Replace original counts with adjusted counts
except Exception: null=[]
return condition_count_db
def calculateRPKM(condition_count_db,exon_len_db,biotype_to_examine):
"""Determines the total number of reads in a sample and then calculates RPMK relative to a pre-determined junction length (60).
60 was choosen, based on Illumina single-end read lengths of 35 (5 nt allowed overhand on either side of the junction)"""
### Get the total number of mapped reads
mapped_reads={}
for condition in condition_count_db:
mapped_reads[condition]=0
count_db = condition_count_db[condition]
for key in count_db:
read_count = count_db[key]
mapped_reads[condition]+=float(read_count)
### Use the average_total_reads when no counts reported such that 0 counts are comparable
average_total_reads = 0
for i in mapped_reads:
average_total_reads+=mapped_reads[i]
if testImport == 'yes':
print 'condition:',i,'total reads:',mapped_reads[i]
average_total_reads = average_total_reads/len(condition_count_db)
if testImport == 'yes':
print 'average_total_reads:',average_total_reads
k=0
c=math.pow(10.0,9.0)
for key in exon_len_db:
try:
for condition in condition_count_db:
total_mapped_reads = mapped_reads[condition]
try: read_count = float(condition_count_db[condition][key])+1 ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
except KeyError: read_count = 1 ###Was zero, but needs to be one for more realistic log2 fold calculations
if biotype_to_examine == 'junction': region_length = 60.0
else:
try: region_length = exon_len_db[key]
except Exception: continue ### This should only occur during testing (when restricting to one or few chromosomes)
if read_count == 1: ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
rpkm = c*(float(read_count)/(float(average_total_reads)*region_length))
try:
if region_length == 0:
region_length = abs(int(key[2]-key[1]))
rpkm = c*(read_count/(float(total_mapped_reads)*region_length))
except Exception:
print condition, key
print 'Error Encountered... Exon or Junction of zero length encoutered... RPKM failed... Exiting AltAnalyze.'
print 'This error may be due to inconsistent file naming. If both exon and junction sample data is present, make sure they are named propperly.'
print 'For example: cancer1__exon.bed, cancer1__junction.bed (double underscore required to match these samples up)!'
print [read_count,total_mapped_reads,region_length];k=1; forceError
condition_count_db[condition][key] = str(rpkm) ### Replace original counts with RPMK
except Exception:
if k == 1: kill
null=[]
return condition_count_db
def calculateGeneLevelStatistics(steady_state_export,species,expressed_gene_exon_db,normalize_feature_exp,array_names,fl,excludeLowExp=True,exportRPKMs=False):
global UserOptions; UserOptions = fl
exp_file = string.replace(steady_state_export,'-steady-state','')
if normalize_feature_exp == 'RPKM':
exp_dbase, all_exp_features, array_count = importRawCountData(exp_file,expressed_gene_exon_db,excludeLowExp=excludeLowExp)
steady_state_db = obtainGeneCounts(expressed_gene_exon_db,species,exp_dbase,array_count,normalize_feature_exp,excludeLowExp=excludeLowExp); exp_dbase=[]
exportGeneCounts(steady_state_export,array_names,steady_state_db)
steady_state_db = calculateGeneRPKM(steady_state_db)
if exportRPKMs:
exportGeneCounts(steady_state_export,array_names,steady_state_db,dataType='RPKMs')
else:
exp_dbase, all_exp_features, array_count = importNormalizedCountData(exp_file,expressed_gene_exon_db)
steady_state_db = obtainGeneCounts(expressed_gene_exon_db,species,exp_dbase,array_count,normalize_feature_exp); exp_dbase=[]
exportGeneCounts(steady_state_export,array_names,steady_state_db)
return steady_state_db, all_exp_features
def exportGeneCounts(steady_state_export,headers,gene_count_db,dataType='counts'):
### In addition to RPKM gene-level data, export gene level counts and lengths (should be able to calculate gene RPKMs from this file)
if dataType=='counts':
export_path = string.replace(steady_state_export,'exp.','counts.')
else:
export_path = steady_state_export
export_data = export.ExportFile(export_path)
title = string.join(['Ensembl']+headers,'\t')+'\n'
export_data.write(title)
for gene in gene_count_db:
sample_counts=[]
for count_data in gene_count_db[gene]:
try: read_count,region_length = count_data
except Exception: read_count = count_data
sample_counts.append(str(read_count))
sample_counts = string.join([gene]+sample_counts,'\t')+'\n'
export_data.write(sample_counts)
export_data.close()
def importGeneCounts(filename,import_type):
### Import non-normalized original counts and return the max value
counts_filename = string.replace(filename,'exp.','counts.')
status = verifyFile(counts_filename)
if status == 'not found': ### Occurs for non-normalized counts
counts_filename = filename
fn=filepath(counts_filename); x=0; count_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: array_names = t[1:]; x=1
else:
gene = t[0]
if import_type == 'max':
count_db[gene] = str(max(map(float,t[1:])))
else:
count_db[gene] = map(float,t[1:])
return count_db,array_names
def calculateGeneRPKM(gene_count_db):
"""Determines the total number of reads in a sample and then calculates RPMK relative to a pre-determined junction length (60).
60 was choosen, based on Illumina single-end read lengths of 35 (5 nt allowed overhand on either side of the junction)"""
### Get the total number of mapped reads (relative to all gene aligned rather than genome aligned exon reads)
mapped_reads={}
for gene in gene_count_db:
index=0
for (read_count,total_len) in gene_count_db[gene]:
try: mapped_reads[index]+=float(read_count)
except Exception: mapped_reads[index]=float(read_count)
index+=1
### Use the average_total_reads when no counts reported such that 0 counts are comparable
average_total_reads = 0
for i in mapped_reads: average_total_reads+=mapped_reads[i]
average_total_reads = average_total_reads/(index+1) ###
c=math.pow(10.0,9.0)
for gene in gene_count_db:
index=0; rpkms = []
for (read_count,region_length) in gene_count_db[gene]:
total_mapped_reads = mapped_reads[index]
#print [read_count],[region_length],[total_mapped_reads]
#if gene == 'ENSMUSG00000028186': print [read_count, index, total_mapped_reads,average_total_reads,region_length]
if read_count == 0: read_count=1; rpkm = c*(float(read_count)/(float(average_total_reads)*region_length)) ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
else:
try: rpkm = c*(float(read_count+1)/(float(total_mapped_reads)*region_length)) ### read count is incremented +1 (see next line)
except Exception: read_count=1; rpkm = c*(float(read_count)/(float(average_total_reads)*region_length)) ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
#if gene == 'ENSMUSG00000028186': print rpkm,read_count,index,total_mapped_reads,average_total_reads,region_length
#if gene == 'ENSMUSG00000026049': print gene_count_db[gene], mapped_reads[index], rpkm
rpkms.append(rpkm)
index+=1
gene_count_db[gene] = rpkms ### Replace original counts with RPMK
return gene_count_db
def deleteOldAnnotations(species,root_dir,dataset_name):
db_dir = root_dir+'AltDatabase/'+species
try:
status = export.deleteFolder(db_dir)
if status == 'success':
print "...Previous experiment database deleted"
except Exception: null=[]
count_dir = root_dir+'ExpressionInput/Counts'
try: status = export.deleteFolder(count_dir)
except Exception: pass
if 'exp.' not in dataset_name: dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name: dataset_name+='.txt'
export_path = root_dir+'ExpressionInput/'+dataset_name
try: os.remove(filepath(export_path))
except Exception: null=[]
try: os.remove(filepath(string.replace(export_path,'exp.','counts.')))
except Exception: null=[]
try: os.remove(filepath(string.replace(export_path,'exp.','novel.')))
except Exception: null=[]
from copy_reg import pickle
from types import MethodType
def _pickle_method(method):
func_name = method.im_func.__name__
obj = method.im_self
cls = method.im_class
return _unpickle_method, (func_name, obj, cls)
def _unpickle_method(func_name, obj, cls):
for cls in cls.mro():
try:
func = cls.__dict__[func_name]
except KeyError:
pass
else:
break
return func.__get__(obj, cls)
def call_it(instance, name, args=(), kwargs=None):
"indirect caller for instance methods and multiprocessing"
if kwargs is None:
kwargs = {}
return getattr(instance, name)(*args, **kwargs)
def alignExonsAndJunctionsToEnsembl(species,exp_file_location_db,dataset_name,Multi=None):
fl = exp_file_location_db[dataset_name]
try: multiThreading = fl.multiThreading()
except Exception: multiThreading = True
print 'multiThreading:',multiThreading
normalize_feature_exp = fl.FeatureNormalization()
testImport='no'
if 'demo_data' in fl.ExpFile():
### If the input files are in the AltAnalyze test directory, only analyze select chromosomes
print 'Running AltAnalyze in TEST MODE... restricting to select chromosomes only!!!!!'
testImport='yes'
rnaseq_begin_time = time.time()
p = AlignExonsAndJunctionsToEnsembl(species,exp_file_location_db,dataset_name,testImport)
chromosomes = p.getChromosomes()
### The following files need to be produced from chromosome specific sets later
countsFile = p.countsFile()
exonFile = p.exonFile()
junctionFile = p.junctionFile()
junctionCompFile = p.junctionCompFile()
novelJunctionAnnotations = p.novelJunctionAnnotations()
#chromosomes = ['chrMT']
#p('chrY'); p('chr1'); p('chr2')
#chromosomes = ['chr8','chr17']
multiprocessing_pipe = True
if 'exp.' not in dataset_name:
dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name:
dataset_name+='.txt'
try:
mlp=Multi
pool_size = mlp.cpu_count()
print 'Using %d processes' % pool_size
if multiprocessing_pipe and multiThreading:
### This is like pool, but less efficient (needed to get print outs)
s = pool_size; b=0
chr_blocks=[]
while s<len(chromosomes):
chr_blocks.append(chromosomes[b:s])
b+=pool_size; s+=pool_size
chr_blocks.append(chromosomes[b:s])
queue = mlp.Queue()
results=[]
#parent_conn, child_conn=multiprocessing.Pipe()
for chromosomes in chr_blocks:
procs=list()
#print 'Block size:',len(chromosomes)
for search_chr in chromosomes:
proc = mlp.Process(target=p, args=(queue,search_chr)) ### passing sys.stdout unfortunately doesn't work to pass the Tk string
procs.append(proc)
proc.start()
for _ in procs:
val = queue.get()
if p.AnalysisMode() == 'GUI': print '*',
results.append(val)
for proc in procs:
proc.join()
elif multiThreading:
pool = mlp.Pool(processes=pool_size)
chr_vars=[]
for search_chr in chromosomes:
chr_vars.append(([],search_chr)) ### As an alternative for the pipe version above, pass an empty list rather than queue
results = pool.map(p, chr_vars) ### worker jobs initiated in tandem
try:pool.close(); pool.join(); pool = None
except Exception: pass
else:
forceThreadingError
print 'Read exon and junction mapping complete'
except Exception,e:
#print e
print 'Proceeding with single-processor version align...'
try: proc.close; proc.join; proc = None
except Exception: pass
try: pool.close(); pool.join(); pool = None
except Exception: pass
results=[] ### For single-thread compatible versions of Python
for search_chr in chromosomes:
result = p([],search_chr)
results.append(result)
results_organized=[]
for result_set in results:
if len(result_set[0])>0: ### Sometimes chromsomes are missing
biotypes = result_set[0]
results_organized.append(list(result_set[1:]))
pooled_results = [sum(value) for value in zip(*results_organized)] # combine these counts
pooled_results = [biotypes]+pooled_results
p.setCountsOverview(pooled_results) # store as retreivable objects
catFiles(countsFile,'Counts')
catFiles(junctionFile,'junctions')
catFiles(exonFile,'exons')
catFiles(junctionCompFile,'comps')
catFiles(novelJunctionAnnotations,'denovo')
if normalize_feature_exp == 'RPKM':
fastRPKMCalculate(countsFile)
rnaseq_end_time = time.time()
print '...RNA-seq import completed in',int(rnaseq_end_time-rnaseq_begin_time),'seconds\n'
biotypes = p.outputResults()
return biotypes
def alignCoordinatesToGeneExternal(species,coordinates_to_annotate):
chr_strand_gene_dbs,location_gene_db,chromosomes,gene_location_db = getChromosomeStrandCoordinates(species,'no')
read_aligned_to_gene=0
for (chr,strand) in coordinates_to_annotate:
if (chr,strand) in chr_strand_gene_dbs:
chr_gene_locations = chr_strand_gene_dbs[chr,strand]
chr_reads = coordinates_to_annotate[chr,strand]
chr_gene_locations.sort(); chr_reads.sort()
### Set GeneID for each coordinate object (primary and seconardary GeneIDs)
read_aligned_to_gene=geneAlign(chr,chr_gene_locations,location_gene_db,chr_reads,'no',read_aligned_to_gene)
### Gene objects will be updated
def catFiles(outFileDir,folder):
""" Concatenate all the chromosomal files but retain only the first header """
root_dir = export.findParentDir(outFileDir)+folder+'/'
dir_list = read_directory(root_dir)
firstFile=True
with open(filepath(outFileDir), 'w') as outfile:
for fname in dir_list:
chr_file = root_dir+fname
header=True
with open(filepath(chr_file)) as infile:
for line in infile:
if header:
header=False
if firstFile:
outfile.write(line)
firstFile=False
else: outfile.write(line)
export.deleteFolder(root_dir)
def error(msg, *args):
return multiprocessing.get_logger().error(msg, *args)
class AlignExonsAndJunctionsToEnsembl:
def setCountsOverview(self, overview):
self.biotypes_store, self.known_count, self.novel_junction_count, self.trans_splicing_reads, self.junctions_without_exon_gene_alignments, self.exons_without_gene_alignment_count = overview
def getChromosomes(self):
chr_list=list()
for c in self.chromosomes:
### Sort chromosome by int number
ci=string.replace(c,'chr','')
try: ci = int(ci)
except Exception: pass
chr_list.append((ci,c))
chr_list.sort()
chr_list2=list()
for (i,c) in chr_list: chr_list2.append(c) ### sorted
return chr_list2
def countsFile(self):
return string.replace(self.expfile,'exp.','counts.')
def junctionFile(self):
junction_file = self.root_dir+'AltDatabase/'+self.species+'/RNASeq/'+self.species + '_Ensembl_junctions.txt'
return junction_file
def exonFile(self):
exon_file = self.root_dir+'AltDatabase/'+self.species+'/RNASeq/'+self.species + '_Ensembl_exons.txt'
return exon_file
def junctionCompFile(self):
junction_comp_file = self.root_dir+'AltDatabase/'+self.species+'/RNASeq/'+self.species + '_junction_comps_updated.txt'
return junction_comp_file
def novelJunctionAnnotations(self):
junction_annotation_file = self.root_dir+'AltDatabase/ensembl/'+self.species+'/'+self.species + '_alternative_junctions_de-novo.txt'
return junction_annotation_file
def AnalysisMode(self): return self.analysisMode
def __init__(self,species,exp_file_location_db,dataset_name,testImport):
self.species = species; self.dataset_name = dataset_name
self.testImport = testImport
fl = exp_file_location_db[dataset_name]
bed_dir=fl.BEDFileDir()
root_dir=fl.RootDir()
#self.stdout = fl.STDOUT()
try: platformType = fl.PlatformType()
except Exception: platformType = 'RNASeq'
try: analysisMode = fl.AnalysisMode()
except Exception: analysisMode = 'GUI'
### This occurs when run using the BAMtoBED pipeline in the GUI
if 'exp.' not in dataset_name:
dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name:
dataset_name+='.txt'
self.dataset_name = dataset_name
### Import experimentally identified junction splice-sites
normalize_feature_exp = fl.FeatureNormalization()
if platformType == 'RNASeq':
chr_status = checkBEDFileFormat(bed_dir,root_dir) ### If false, need to remove 'chr' from the search_chr
else:
chr_status = True
#self.fl = fl # Can not pass this object in pool or it breaks
self.platformType = platformType
self.analysisMode = analysisMode
self.root_dir = root_dir
self.normalize_feature_exp = normalize_feature_exp
self.bed_dir = bed_dir
self.chr_status = chr_status
self.exonBedBuildStatus = fl.ExonBedBuildStatus()
self.expfile = root_dir+'ExpressionInput/'+dataset_name
if testImport == 'yes':
print 'Chromosome annotation detected =',chr_status
#if self.exonBedBuildStatus == 'yes':
reformatExonFile(species,'exon',chr_status) ### exports BED format exons for exon expression extraction
"""
Strategies to reduce memory in RNASeq:
1) (done)Delete old AltDatabase-local version if it exists before starting
2) (done)Check to see if a file exists before writing it and if so append rather than create
3) (done)Get counts last and normalize last in for exons and junctions separately.
4) (done)Delete objects explicitly before importing any new data (define a new function that just does this).
5) (done)Get all chromosomes first then parse exon and junction coordinate data on a per known chromosome basis.
6) (done)Prior to deleting all junction/exon object info for each chromsome, save the coordinate(key)-to-annotation information for the read count export file."""
### Delete any existing annotation databases that currently exist (redundant with below)
deleteOldAnnotations(species,root_dir,dataset_name)
###Define variables to report once reads for all chromosomes have been aligned
#global self.known_count; global self.novel_junction_count; global self.one_found; global self.not_found; global self.both_found; global self.trans_splicing_reads
#global self.junctions_without_exon_gene_alignments; global self.exons_without_gene_alignment_count; global self.junction_simple_db; global self.chr_strand_gene_dbs
self.known_count=0; self.novel_junction_count=0; self.one_found=0; self.not_found=0; self.both_found=0; self.trans_splicing_reads=0
self.junctions_without_exon_gene_alignments=0; self.exons_without_gene_alignment_count=0; self.junction_simple_db={}
###Begin Chromosome specific read to exon alignments
self.chr_strand_gene_dbs,self.location_gene_db,chromosomes,self.gene_location_db = getChromosomeStrandCoordinates(species,testImport)
self.chromosomes = chromosomes
print "Processing exon/junction coordinates sequentially by chromosome"
print "Note: this step is time intensive (can be hours) and no print statements may post for a while"
def outputResults(self):
exportDatasetLinkedGenes(self.species,self.gene_location_db,self.root_dir) ### Include an entry for gene IDs to include constitutive expression for RPKM normalized data
chr_gene_locations=[]; self.location_gene_db=[]; self.chr_strand_gene_dbs=[]
#print 'user coordinates imported/processed'
#print 'Importing read counts from coordinate data...'
biotypes = self.biotypes_store
### Output summary statistics
if self.normalize_feature_exp != 'none':
print self.normalize_feature_exp, 'normalization complete'
if 'junction' in biotypes:
print 'Imported Junction Statistics:'
print ' ',self.known_count, 'junctions found in Ensembl/UCSC and',self.novel_junction_count,'are novel'
print ' ',self.trans_splicing_reads,'trans-splicing junctions found (two aligning Ensembl genes)'
print ' ',self.junctions_without_exon_gene_alignments, 'junctions where neither splice-site aligned to a gene'
if (float(self.known_count)*10)<float(self.novel_junction_count):
print '\nWARNING!!!!! Few junctions aligned to known exons. Ensure that the AltAnalyze Ensembl database\nversion matches the genome build aligned to!\n'
if 'exon' in biotypes:
print 'Imported Exon Statistics:'
print ' ',self.exons_without_gene_alignment_count, 'exons where neither aligned to a gene'
print 'User databases and read counts written to:', self.root_dir[:-1]+'ExpressionInput'
### END CHROMOSOME SPECIFIC ANALYSES
if self.exonBedBuildStatus == 'yes':
bedfile = moveBAMtoBEDFile(self.species,self.dataset_name,self.root_dir)
print 'Exon BED file updated with novel exon predictions from junction file'
return bedfile; sys.exit()
clearObjectsFromMemory(self.junction_simple_db); self.junction_simple_db=[]
return biotypes
def test(self, search_chr):
print search_chr
def __call__(self, queue, search_chr):
try:
#sys.stdout = self.stdout
platformType = self.platformType
testImport = self.testImport
species = self.species
dataset_name = self.dataset_name
platformType = self.platformType
analysisMode = self.analysisMode
root_dir = self.root_dir
normalize_feature_exp = self.normalize_feature_exp
bed_dir = self.bed_dir
chr_status = self.chr_status
junction_annotations={}
if chr_status == False:
searchchr = string.replace(search_chr,'chr','')
else:
searchchr = search_chr
if platformType == 'RNASeq':
junction_db,biotypes,algorithms = importBEDFile(bed_dir,root_dir,species,normalize_feature_exp,searchChr=searchchr,testImport=testImport)
else:
normalize_feature_exp = 'quantile'
junction_db,biotypes,algorithms = importExpressionMatrix(bed_dir,root_dir,species,fl,'no',search_chr=searchchr)
self.biotypes_store = biotypes
if len(junction_db)>0:
### Determine which kind of data is being imported, junctions, exons or both
unmapped_exon_db={}
if 'junction' in biotypes:
### Get all known junction splice-sites
ens_junction_coord_db = importExonAnnotations(species,'junction_coordinates',search_chr)
if testImport == 'yes':
print len(ens_junction_coord_db),'Ensembl/UCSC junctions imported'
### Identify known junctions sites found in the experimental dataset (perfect match)
novel_junction_db={}; novel_exon_db={}
for key in junction_db:
ji=junction_db[key]
if ji.BioType()=='junction':
if key in ens_junction_coord_db:
jd=ens_junction_coord_db[key]
ji.setExonAnnotations(jd)
self.known_count+=1
else:
novel_junction_db[key]=junction_db[key]; self.novel_junction_count+=1
#if 75953254 in key: print key; sys.exit()
else:
unmapped_exon_db[key]=junction_db[key]
ens_exon_db = importExonAnnotations(species,'exon',search_chr)
if 'junction' in biotypes:
if testImport == 'yes':
print self.known_count, 'junctions found in Ensembl/UCSC and',len(novel_junction_db),'are novel.'
### Separate each junction into a 5' and 3' splice site (exon1_coord_db and exon2_coord_db)
exon1_coord_db={}; exon2_coord_db={}
for (chr,exon1_stop,exon2_start) in ens_junction_coord_db:
jd = ens_junction_coord_db[(chr,exon1_stop,exon2_start)]
exon1_coord_db[chr,exon1_stop] = jd.GeneID(),string.split(jd.ExonRegionIDs(),'-')[0]
exon2_coord_db[chr,exon2_start] = jd.GeneID(),string.split(jd.ExonRegionIDs(),'-')[1]
clearObjectsFromMemory(ens_junction_coord_db); ens_junction_coord_db=[] ### Clear object from memory
### Get and re-format individual exon info
exon_region_db={}
#if 'exon' not in biotypes:
for gene in ens_exon_db:
for rd in ens_exon_db[gene]:
exon_region_db[gene,rd.ExonRegionIDs()]=rd
### Add the exon annotations from the known junctions to the exons to export dictionary
exons_to_export={}
for key in junction_db:
ji=junction_db[key]
if ji.ExonAnnotations() != None:
jd = ji.ExonAnnotations()
exon1, exon2 = string.split(jd.ExonRegionIDs(),'-')
key1 = jd.GeneID(),exon1; key2 = jd.GeneID(),exon2
exons_to_export[key1] = exon_region_db[key1]
exons_to_export[key2] = exon_region_db[key2]
### For novel experimental junctions, identify those with at least one matching known 5' or 3' site
exons_not_identified = {}; novel_exon_coordinates={}
for (chr,exon1_stop,exon2_start) in novel_junction_db:
ji = novel_junction_db[(chr,exon1_stop,exon2_start)]
coord = [exon1_stop,exon2_start]; coord.sort()
if (chr,exon1_stop) in exon1_coord_db and (chr,exon2_start) in exon2_coord_db:
### Assign exon annotations to junctions where both splice-sites are known in Ensembl/UCSC
### Store the exon objects, genes and regions (le is a tuple of gene and exon region ID)
### Do this later for the below un-assigned exons
le=exon1_coord_db[(chr,exon1_stop)]; ji.setLeftExonAnnotations(le); ji.setLeftExonRegionData(exon_region_db[le])
re=exon2_coord_db[(chr,exon2_start)]; ji.setRightExonAnnotations(re); ji.setRightExonRegionData(exon_region_db[re])
if le[0] != re[0]: ### Indicates Trans-splicing (e.g., chr7:52,677,568-52,711,750 mouse mm9)
ji.setTransSplicing(); #print exon1_stop,le,exon2_start,re,ji.Chr(),ji.Strand()
self.both_found+=1; #print 'five',(chr,exon1_stop,exon2_start),exon1_coord_db[(chr,exon1_stop)]
else:
if (chr,exon1_stop) in exon1_coord_db: ### hence, exon1_stop is known, so report the coordinates of exon2 as novel
le=exon1_coord_db[(chr,exon1_stop)]; ji.setLeftExonAnnotations(le)
self.one_found+=1; #print 'three',(chr,exon1_stop,exon2_start),exon1_coord_db[(chr,exon1_stop)]
novel_exon_coordinates[ji.Chr(),exon2_start] = ji,'left',ji.Exon2Stop() ### Employ this strategy to avoid duplicate exons with differing lengths (mainly an issue if analyzing only exons results)
ji.setNovel('side')
elif (chr,exon2_start) in exon2_coord_db: ### hence, exon2_start is known, so report the coordinates of exon1 as novel
re=exon2_coord_db[(chr,exon2_start)]; ji.setRightExonAnnotations(re) ### In very rare cases, a gene can be assigned here, even though the splice-site is on the opposite strand (not worthwhile filtering out)
self.one_found+=1; #print 'three',(chr,exon1_stop,exon2_start),exon1_coord_db[(chr,exon1_stop)]
novel_exon_coordinates[ji.Chr(),exon1_stop] = ji,'right',ji.Exon1Start()
ji.setNovel('side')
else:
self.not_found+=1; #if self.not_found < 10: print (chr,exon1_stop,exon2_start)
novel_exon_coordinates[ji.Chr(),exon1_stop] = ji,'right',ji.Exon1Start()
novel_exon_coordinates[ji.Chr(),exon2_start] = ji,'left',ji.Exon2Stop()
ji.setNovel('both')
### We examine reads where one splice-site aligns to a known but the other not, to determine if trans-splicing occurs
try: exons_not_identified[chr,ji.Strand()].append((coord,ji))
except KeyError: exons_not_identified[chr,ji.Strand()] = [(coord,ji)]
"""
if fl.ExonBedBuildStatus() == 'no':
exportNovelJunctions(species,novel_junction_db,condition_count_db,root_dir,dataset_name,'junction') ### Includes known exons
"""
#print self.both_found, ' where both and', self.one_found, 'where one splice-site are known out of',self.both_found+self.one_found+self.not_found
#print 'Novel junctions where both splice-sites are known:',self.both_found
#print 'Novel junctions where one splice-site is known:',self.one_found
#print 'Novel junctions where the splice-sites are not known:',self.not_found
clearObjectsFromMemory(exon_region_db); exon_region_db=[] ### Clear memory of this object
read_aligned_to_gene=0
for (chr,strand) in exons_not_identified:
if (chr,strand) in self.chr_strand_gene_dbs:
chr_gene_locations = self.chr_strand_gene_dbs[chr,strand]
chr_reads = exons_not_identified[chr,strand]
chr_gene_locations.sort(); chr_reads.sort()
### Set GeneID for each coordinate object (primary and seconardary GeneIDs)
read_aligned_to_gene=geneAlign(chr,chr_gene_locations,self.location_gene_db,chr_reads,'no',read_aligned_to_gene)
#print read_aligned_to_gene, 'novel junctions aligned to Ensembl genes out of',self.one_found+self.not_found
clearObjectsFromMemory(exons_not_identified); exons_not_identified=[] ## Clear memory of this object
for key in novel_junction_db:
(chr,exon1_stop,exon2_start) = key
ji=novel_junction_db[key]
if ji.GeneID() == None:
try:
if ji.SecondaryGeneID() != None:
### Occurs if mapping is to the 5'UTR of a gene for the left splice-site (novel alternative promoter)
ji.setGeneID(ji.SecondaryGeneID()); ji.setSecondaryGeneID(''); #print key, ji.GeneID(), ji.Strand(), ji.SecondaryGeneID()
except Exception: null=[]
if ji.GeneID() != None:
geneid = ji.GeneID()
proceed = 'no'
if ji.SpliceSitesFound() == None: proceed = 'yes'; coordinates = [exon1_stop,exon2_start]
elif ji.SpliceSitesFound() == 'left': proceed = 'yes'; coordinates = [exon1_stop,exon2_start]
elif ji.SpliceSitesFound() == 'right': proceed = 'yes'; coordinates = [exon1_stop,exon2_start]
if proceed == 'yes':
for coordinate in coordinates:
if ji.TransSplicing() == 'yes':
#print ji.Chr(),ji.GeneID(), ji.SecondaryGeneID(), ji.Exon1Stop(), ji.Exon2Start()
self.trans_splicing_reads+=1
if ji.checkExonPosition(coordinate) == 'right': geneid = ji.SecondaryGeneID()
if abs(exon2_start-exon1_stop)==1: eventType = 'novel-exon-intron' ### Indicates intron-exon boundary (intron retention)
else: eventType = 'novel'
exon_data = (coordinate,ji.Chr()+'-'+str(coordinate),eventType)
try: novel_exon_db[geneid].append(exon_data)
except KeyError: novel_exon_db[geneid] = [exon_data]
else:
### write these out
self.junctions_without_exon_gene_alignments+=1
### Remove redundant exon entries and store objects
for key in novel_exon_db:
exon_data_objects=[]
exon_data_list = unique.unique(novel_exon_db[key])
exon_data_list.sort()
for e in exon_data_list:
ed = ExonInfo(e[0],e[1],e[2])
exon_data_objects.append(ed)
novel_exon_db[key] = exon_data_objects
#print self.trans_splicing_reads,'trans-splicing junctions found (two aligning Ensembl genes).'
#print self.junctions_without_exon_gene_alignments, 'junctions where neither splice-site aligned to a gene'
#if 'X' in search_chr: print len(ens_exon_db),len(ens_exon_db['ENSMUSG00000044424'])
alignReadsToExons(novel_exon_db,ens_exon_db,testImport=testImport)
### Link exon annotations up with novel junctions
junction_region_db,exons_to_export = annotateNovelJunctions(novel_junction_db,novel_exon_db,exons_to_export)
### Add the exon region data from known Ensembl/UCSC matched junctions to junction_region_db for recipricol junction analysis
for key in junction_db:
ji=junction_db[key]; jd = ji.ExonAnnotations()
try:
uid = jd.GeneID()+':'+jd.ExonRegionIDs(); ji.setUniqueID(uid)
try: junction_region_db[jd.GeneID()].append((formatID(uid),jd.ExonRegionIDs()))
except KeyError: junction_region_db[jd.GeneID()] = [(formatID(uid),jd.ExonRegionIDs())]
except AttributeError: null=[] ### Occurs since not all entries in the dictionary are perfect junction matches
try: novel_exon_coordinates = collapseNoveExonBoundaries(novel_exon_coordinates,root_dir+dataset_name) ### Joins inferred novel exon-IDs (5' and 3' splice sites) from adjacent and close junction predictions
except Exception: pass ### No errors encountered before
#if self.exonBedBuildStatus == 'yes':
### Append to the exported BED format exon coordinate file
bedfile = exportNovelExonToBedCoordinates(species,novel_exon_coordinates,chr_status,searchChr=searchchr)
### Identify reciprocol junctions and retrieve splice-event annotations for exons and inclusion junctions
junction_annotations,critical_exon_annotations = JunctionArray.inferJunctionComps(species,('RNASeq',junction_region_db,root_dir),searchChr=searchchr)
clearObjectsFromMemory(junction_region_db); junction_region_db=[]
### Reformat these dictionaries to combine annotations from multiple reciprocol junctions
junction_annotations = combineExonAnnotations(junction_annotations)
critical_exon_annotations = combineExonAnnotations(critical_exon_annotations)
if 'exon' in biotypes:
if testImport == 'yes':
print len(unmapped_exon_db),'exon genomic locations imported.'
### Create a new dictionary keyed by chromosome and strand
exons_not_aligned={}
for (chr,exon1_stop,exon2_start) in unmapped_exon_db:
ji = unmapped_exon_db[(chr,exon1_stop,exon2_start)]
coord = [exon1_stop,exon2_start]; coord.sort()
try: exons_not_aligned[chr,ji.Strand()].append((coord,ji))
except KeyError: exons_not_aligned[chr,ji.Strand()] = [(coord,ji)]
read_aligned_to_gene=0
for (chr,strand) in exons_not_aligned:
if (chr,strand) in self.chr_strand_gene_dbs:
chr_gene_locations = self.chr_strand_gene_dbs[chr,strand]
chr_reads = exons_not_aligned[chr,strand]
chr_gene_locations.sort(); chr_reads.sort()
read_aligned_to_gene=geneAlign(chr,chr_gene_locations,self.location_gene_db,chr_reads,'no',read_aligned_to_gene)
#print read_aligned_to_gene, 'exons aligned to Ensembl genes out of',self.one_found+self.not_found
align_exon_db={}; exons_without_gene_alignments={}; multigene_exon=0
for key in unmapped_exon_db:
(chr,exon1_stop,exon2_start) = key
ji=unmapped_exon_db[key]
if ji.GeneID() == None:
try:
if ji.SecondaryGeneID() != None:
### Occurs if mapping outside known exon boundaries for one side of the exon
ji.setGeneID(ji.SecondaryGeneID()); ji.setSecondaryGeneID(''); #print key, ji.GeneID(), ji.Strand(), ji.SecondaryGeneID()
except Exception: null=[]
else:
if 'ENS' in ji.JunctionID():
if ji.GeneID() not in ji.JunctionID(): ### Hence, there were probably two overlapping Ensembl genes and the wrong was assigned based on the initial annotations
original_geneid = string.split(ji.JunctionID(),':')[0]
if original_geneid in ens_exon_db: ji.setGeneID(original_geneid) #check if in ens_exon_db (since chromosome specific)
if ji.GeneID() != None:
geneid = ji.GeneID()
coordinates = [exon1_stop,exon2_start]
for coordinate in coordinates:
if ji.TransSplicing() != 'yes': ### This shouldn't occur for exons
exon_data = (coordinate,ji.Chr()+'-'+str(coordinate),'novel')
try: align_exon_db[geneid].append(exon_data)
except KeyError: align_exon_db[geneid] = [exon_data]
else:
multigene_exon+=1 ### Shouldn't occur due to a fix in the gene-alignment method which will find the correct gene on the 2nd interation
else: exons_without_gene_alignments[key]=ji; self.exons_without_gene_alignment_count+=1
### Remove redundant exon entries and store objects (this step may be unnecessary)
for key in align_exon_db:
exon_data_objects=[]
exon_data_list = unique.unique(align_exon_db[key])
exon_data_list.sort()
for e in exon_data_list:
ed = ExonInfo(e[0],e[1],e[2])
exon_data_objects.append(ed)
align_exon_db[key] = exon_data_objects
#print self.exons_without_gene_alignment_count, 'exons where neither aligned to a gene'
#if self.exons_without_gene_alignment_count>3000: print 'NOTE: Poor mapping of these exons may be due to an older build of\nEnsembl than the current version. Update BAMtoBED mappings to correct.'
begin_time = time.time()
alignReadsToExons(align_exon_db,ens_exon_db)
end_time = time.time()
if testImport == 'yes':
print 'Exon sequences aligned to exon regions in',int(end_time-begin_time),'seconds'
### Combine the start and end region alignments into a single exon annotation entry
combineDetectedExons(unmapped_exon_db,align_exon_db,novel_exon_db)
clearObjectsFromMemory(unmapped_exon_db); clearObjectsFromMemory(align_exon_db); clearObjectsFromMemory(novel_exon_db)
unmapped_exon_db=[]; align_exon_db=[]; novel_exon_db=[]
"""
if fl.ExonBedBuildStatus() == 'no':
exportNovelJunctions(species,exons_without_gene_alignments,condition_count_db,root_dir,dataset_name,'exon') ### Includes known exons
"""
clearObjectsFromMemory(exons_without_gene_alignments); exons_without_gene_alignments=[]
### Export both exon and junction annotations
if 'junction' in biotypes:
### Export the novel user exon annotations
exportDatasetLinkedExons(species,exons_to_export,critical_exon_annotations,root_dir,testImport=testImport,searchChr=searchchr)
### Export the novel user exon-junction annotations (original junction_db objects updated by above processing)
exportDatasetLinkedJunctions(species,junction_db,junction_annotations,root_dir,testImport=testImport,searchChr=searchchr)
### Clear memory once results are exported (don't want to delete actively used objects)
if 'junction' in biotypes:
clearObjectsFromMemory(exons_to_export); clearObjectsFromMemory(critical_exon_annotations)
clearObjectsFromMemory(novel_junction_db); novel_junction_db=[]
clearObjectsFromMemory(novel_exon_coordinates); novel_exon_coordinates=[]
exons_to_export=[]; critical_exon_annotations=[]
clearObjectsFromMemory(exon1_coord_db); clearObjectsFromMemory(exon2_coord_db)
exon1_coord_db=[]; exon2_coord_db=[]
if 'exon' in biotypes:
clearObjectsFromMemory(exons_not_aligned); exons_not_aligned=[]
clearObjectsFromMemory(ens_exon_db); ens_exon_db=[]
### Add chromsome specific junction_db data to a simple whole genome dictionary
for key in junction_db:
ji = junction_db[key]
if ji.GeneID()!=None and ji.UniqueID()!=None: self.junction_simple_db[key]=ji.UniqueID()
#returnLargeGlobalVars()
clearObjectsFromMemory(junction_db); clearObjectsFromMemory(junction_annotations)
junction_db=[]; junction_annotations=[]; chr_reads=[]
for biotype in biotypes:
### Import Read Counts (do this last to conserve memory)
if platformType == 'RNASeq':
condition_count_db,exon_len_db,biotypes2,algorithms = importBEDFile(bed_dir,root_dir,species,normalize_feature_exp,getReads=True,searchChr=searchchr,getBiotype=biotype,testImport=testImport,filteredJunctions=self.junction_simple_db)
else:
condition_count_db,exon_len_db,biotypes2,algorithms = importExpressionMatrix(bed_dir,root_dir,species,fl,'yes',getBiotype=biotype)
###First export original counts, rather than quantile normalized or RPKM
self.exportJunctionCounts(species,self.junction_simple_db,exon_len_db,condition_count_db,root_dir,dataset_name,biotype,'counts',searchChr=searchchr)
clearObjectsFromMemory(condition_count_db); clearObjectsFromMemory(exon_len_db); condition_count_db=[]; exon_len_db=[]
if analysisMode == 'commandline':
print 'finished parsing data for chromosome:',search_chr ### Unix platforms are not displaying the progress in real-time
else:
pass #print "*",
try: queue.put([self.biotypes_store, self.known_count, self.novel_junction_count, self.trans_splicing_reads, self.junctions_without_exon_gene_alignments, self.exons_without_gene_alignment_count])
except Exception:
### If queue is not a multiprocessing object
queue = [self.biotypes_store, self.known_count, self.novel_junction_count, self.trans_splicing_reads, self.junctions_without_exon_gene_alignments, self.exons_without_gene_alignment_count]
return queue
except Exception:
print traceback.format_exc()
error(traceback.format_exc())
multiprocessing.log_to_stderr().setLevel(logging.DEBUG)
raise
def exportJunctionCounts(self,species,junction_simple_db,exon_len_db,condition_count_db,root_dir,dataset_name,biotype,count_type,searchChr=None):
if 'exp.' not in dataset_name: dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name: dataset_name+='.txt'
export_path = root_dir+'ExpressionInput/'+dataset_name
if count_type == 'counts':
export_path = string.replace(export_path,'exp.','counts.') ### separately export counts
if searchChr !=None:
export_path = string.replace(export_path,'ExpressionInput','ExpressionInput/Counts')
export_path = string.replace(export_path,'.txt','.'+searchChr+'.txt')
self.countsFile = export_path
if self.testImport == 'yes':
print 'Writing',export_path
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
title = ['AltAnalyze_ID']
for condition in condition_count_db: title.append(condition)
export_data.write(string.join(title,'\t')+'\n')
for key in self.junction_simple_db:
chr,exon1_stop,exon2_start = key
if biotype == 'junction':
coordinates = chr+':'+str(exon1_stop)+'-'+str(exon2_start)
elif biotype == 'exon':
coordinates = chr+':'+str(exon1_stop-1)+'-'+str(exon2_start+1)
try:
null=exon_len_db[key]
if count_type == 'counts': values = [self.junction_simple_db[key]+'='+coordinates]
else: values = [self.junction_simple_db[key]]
for condition in condition_count_db: ###Memory crash here
count_db = condition_count_db[condition]
try: read_count = count_db[key]
except KeyError: read_count = '0'
values.append(read_count)
export_data.write(string.join(values,'\t')+'\n')
except Exception: null=[]
export_data.close()
def countsDir(self):
return self.countsFile
def calculateRPKMsFromGeneCounts(filename,species,AdjustExpression):
""" Manual way of calculating gene RPKMs from gene counts only """
gene_lengths = getGeneExonLengths(species)
fastRPKMCalculate(filename,GeneLengths=gene_lengths,AdjustExpression=AdjustExpression)
def fastRPKMCalculate(counts_file,GeneLengths=None,AdjustExpression=True):
export_path = string.replace(counts_file,'counts.','exp.')
export_data = export.ExportFile(export_path) ### Write this new file
fn=filepath(counts_file); header=True
exon_sum_array=[]; junction_sum_array=[]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
samples = t[1:]
header=False
exon_sum_array=[0]*len(samples)
junction_sum_array=[0]*len(samples)
else:
try: values = map(float,t[1:])
except Exception:
print traceback.format_exc()
print t
badCountsLine
### get the total reads/sample
if '-' in string.split(t[0],'=')[0]:
junction_sum_array = [sum(value) for value in zip(*[junction_sum_array,values])]
else:
exon_sum_array = [sum(value) for value in zip(*[exon_sum_array,values])]
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides warnings associated with Scipy for n=1 sample comparisons
jatr=Average(junction_sum_array) # Average of the total maped reads
eatr=Average(exon_sum_array) # Average of the total maped reads
if AdjustExpression:
offset = 1
else:
offset = 0
header=True
c=math.pow(10.0,9.0)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
export_data.write(line) ### Write header
header=False
else:
try:
exon_id,coordinates = string.split(t[0],'=')
coordinates = string.split(coordinates,':')[1]
coordinates = string.split(coordinates,'-')
l=abs(int(coordinates[1])-int(coordinates[0])) ### read-length
except Exception: ### Manual way of calculating gene RPKMs from gene counts only
exon_id = t[0]
try: l = GeneLengths[exon_id]
except Exception: continue #Occurs when Ensembl genes supplied from an external analysis
try: read_counts = map(lambda x: int(x)+offset, t[1:])
except Exception: read_counts = map(lambda x: int(float(x))+offset, t[1:])
if '-' in exon_id:
count_stats = zip(read_counts,junction_sum_array)
atr = jatr
l=60
else:
count_stats = zip(read_counts,exon_sum_array)
atr = eatr
values=[]
#rpkm = map(lambda (r,t): c*(r/(t*l)), count_stats) ### Efficent way to convert to rpkm, but doesn't work for 0 counts
for (r,t) in count_stats:
if r == 1: ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
t = atr
try:
rpkm = str(c*(r/(t*l)))
#print c,r,t,l,exon_id,rpkm;sys.exit()
values.append(rpkm)
except Exception,e:
print e
print t[0]
print 'Error Encountered... Exon or Junction of zero length encoutered... RPKM failed... Exiting AltAnalyze.'
print 'This error may be due to inconsistent file naming. If both exon and junction sample data is present, make sure they are named propperly.'
print 'For example: cancer1__exon.bed, cancer1__junction.bed (double underscore required to match these samples up)!'
print [r,t,l];k=1; forceError
values = string.join([exon_id]+values,'\t')+'\n'
export_data.write(values)
export_data.close()
def mergeCountFiles(counts_file1,counts_file2):
### Used internally to merge count files that are very large and too time-consuming to recreate (regenerate them)
export_path = string.replace(counts_file2,'counts.','temp-counts.')
export_data = export.ExportFile(export_path) ### Write this new file
fn=filepath(counts_file1); header=True
count_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
samples = t[1:]
header=False
si = samples.index('H9.102.2.5.bed')+1
else:
try: value = t[si]
except Exception: print t; sys.exit()
### get the total reads/sample
count_db[t[0]] = value
fn=filepath(counts_file2); header=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
samples = t[1:]
header=False
si = samples.index('H9.102.2.5.bed')+1
export_data.write(line)
else:
try: t[si] = count_db[t[0]]
except Exception: pass ### keep the current value
export_data.write(string.join(t,'\t')+'\n')
export_data.close()
def getGeneExonLengths(species):
gene_lengths={}
filename = 'AltDatabase/'+species+'/RNASeq/'+species+'_Ensembl_exons.txt'
fn=filepath(filename)
firstLine=True
for line in open(fn,'rU').xreadlines():
line = line.rstrip('\n')
if firstLine:
firstLine=False
else:
t = string.split(line,'\t')
geneID = t[2]; start = int(t[6]); end = int(t[7]); exonID = t[1]
if 'E' in exonID:
try: gene_lengths[geneID]+=abs(end-start)
except Exception: gene_lengths[geneID]=abs(end-start)
return gene_lengths
def importRawCountData(filename,expressed_gene_exon_db,excludeLowExp=True):
""" Identifies exons or junctions to evaluate gene-level expression. This function, as it is currently written:
1) examines the RPKM and original read counts associated with all exons
2) removes exons/junctions that do not meet their respective RPKM AND read count cutoffs
3) returns ONLY those exons and genes deemed expressed, whether constitutive selected or all exons
"""
### Get expression values for exon/junctions to analyze
seq_ids_to_import={}
for gene in expressed_gene_exon_db:
for exonid in expressed_gene_exon_db[gene]: seq_ids_to_import[exonid]=[]
### Define thresholds
exon_exp_threshold = UserOptions.ExonExpThreshold()
junction_exp_threshold = UserOptions.JunctionExpThreshold()
exon_rpkm_threshold = UserOptions.ExonRPKMThreshold()
gene_rpkm_threshold = UserOptions.RPKMThreshold()
gene_exp_threshold = UserOptions.GeneExpThreshold()
### Import RPKM normalized expression values
fn=filepath(filename); x=0; rpkm_dbase={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: array_names = t[1:]; x=1
else:
exon_id=t[0]
max_count=max(map(float,t[1:]))
if max_count>=exon_rpkm_threshold or excludeLowExp==False: rpkm_dbase[exon_id]=[] ### Only retain exons/junctions meeting the RPKM threshold
### Import non-normalized original counts
counts_filename = string.replace(filename,'exp.','counts.')
fn=filepath(counts_filename); x=0; exp_dbase={}
all_exp_features={} ### Don't filter for only gene-expression reporting
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: array_names = t[1:]; x=1
else:
exon_id,coordinates = string.split(t[0],'=')
coordinates = string.split(coordinates,':')[1]
coordinates = string.split(coordinates,'-')
length=abs(int(coordinates[1])-int(coordinates[0]))
max_count=max(map(float,t[1:])); proceed = 'no'
if '-' in exon_id:
length = 60.0
if max_count>=junction_exp_threshold or excludeLowExp==False:
### Only considered when exon data is not present in the analysis
proceed = 'yes'
elif max_count>=exon_exp_threshold or excludeLowExp==False: proceed = 'yes'
if proceed == 'yes' and exon_id in rpkm_dbase: ### Ensures that the maximum sample (not group) user defined count threshold is achieved at the exon or junction-level
all_exp_features[exon_id]=None
if exon_id in seq_ids_to_import:### Forces an error if not in the steady-state pre-determined set (CS or all-exons) - INCLUDE HERE TO FILTER ALL FEATURES
exp_dbase[exon_id] = t[1:],length ### Include sequence length for normalization
for exon in exp_dbase: array_count = len(exp_dbase[exon][0]); break
try:null=array_count
except Exception:
print 'No exons or junctions considered expressed (based user thresholds). Exiting analysis.'; force_exit
return exp_dbase, all_exp_features, array_count
def importNormalizedCountData(filename,expressed_gene_exon_db):
### Get expression values for exon/junctions to analyze
seq_ids_to_import={}
for gene in expressed_gene_exon_db:
for exonid in expressed_gene_exon_db[gene]: seq_ids_to_import[exonid]=[]
### Define thresholds
exon_exp_threshold = UserOptions.ExonExpThreshold()
junction_exp_threshold = UserOptions.JunctionExpThreshold()
exon_rpkm_threshold = UserOptions.ExonRPKMThreshold()
gene_rpkm_threshold = UserOptions.RPKMThreshold()
gene_exp_threshold = UserOptions.GeneExpThreshold()
### Import non-normalized original counts
fn=filepath(filename); x=0; exp_dbase={}
all_exp_features={} ### Don't filter for only gene-expression reporting
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: array_names = t[1:]; x=1
else:
exon_id=t[0]; proceed = 'no'
max_count=max(map(float,t[1:]))
if '-' in exon_id:
if max_count>=junction_exp_threshold: proceed = 'yes'
elif max_count>=exon_exp_threshold: proceed = 'yes'
if proceed == 'yes': ### Ensures that the maximum sample (not group) user defined count threshold is achieved at the exon or junction-level
all_exp_features[exon_id]=None
if exon_id in seq_ids_to_import: ### If a "constitutive" or exon-level feature (filter missing prior to 2.0.8 - bug)
exp_dbase[exon_id] = t[1:],0 ### Add the zero just to comply with the raw count input format (indicates exon length)
for exon in exp_dbase: array_count = len(exp_dbase[exon][0]); break
return exp_dbase, all_exp_features, array_count
def obtainGeneCounts(expressed_gene_exon_db,species,exp_dbase,array_count,normalize_feature_exp,excludeLowExp=True):
###Calculate avg expression for each sample for each exon (using constitutive or all exon values)
if excludeLowExp == False:
gene_lengths = getGeneExonLengths(species)
steady_state_db={}
for gene in expressed_gene_exon_db:
x = 0; gene_sum=0
exon_list = expressed_gene_exon_db[gene]
while x < array_count:
exp_list=[]; len_list=[]
for exon in exon_list:
try:
exp_val = exp_dbase[exon][0][x]
if normalize_feature_exp == 'RPKM':
### Decided to include all exons, expressed or not to prevent including lowly expressed exons that are long, that can bias the expression call
#if float(exp_val) != 0: ### Here, we use the original raw count data, whereas above is the adjusted quantile or raw count data
exp_list.append(exp_val); len_list.append(exp_dbase[exon][1]) ### This is for RNASeq -> don't include undetected exons - made in v.204
else: exp_list.append(exp_val) #elif float(exp_val) != 1:
except KeyError: null =[] ###occurs if the expression exon list is missing some of these exons
try:
if len(exp_list)==0:
for exon in exon_list:
try:
exp_list.append(exp_dbase[exon][0][x]); len_list.append(exp_dbase[exon][1])
#kill
except KeyError: null=[] ### Gene entries will cause this error, since they are in the database but not in the count file
if normalize_feature_exp == 'RPKM':
sum_const_exp=sum(map(float,exp_list)); gene_sum+=sum_const_exp
sum_length=sum(len_list) ### can have different lengths for each sample, since only expressed exons are considered
if excludeLowExp == False:
sum_length = gene_lengths[gene] ### Uses the all annotated exon lengths
### Add only one avg-expression value for each array, this loop
try: steady_state_db[gene].append((sum_const_exp,sum_length))
except KeyError: steady_state_db[gene] = [(sum_const_exp,sum_length)]
else:
avg_const_exp=Average(exp_list)
if avg_const_exp != 1: gene_sum+=avg_const_exp
### Add only one avg-expression value for each array, this loop
try: steady_state_db[gene].append(avg_const_exp)
except KeyError: steady_state_db[gene] = [avg_const_exp]
except Exception: null=[] ### Occurs when processing a truncated dataset (for testing usually) - no values for the gene should be included
x += 1
if gene_sum==0:
try:
del steady_state_db[gene] ### Hence, no genes showed evidence of expression (most critical for RNA-Seq)
except Exception: null=[] ### Error occurs when a gene is added to the database from self.location_gene_db, but is not expressed
return steady_state_db
def returnLargeGlobalVars():
### Prints all large global variables retained in memory (taking up space)
all = [var for var in globals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(globals()[var])>1:
print var, len(globals()[var])
except Exception: null=[]
def clearObjectsFromMemory(db_to_clear):
db_keys={}
for key in db_to_clear: db_keys[key]=[]
for key in db_keys:
try: del db_to_clear[key]
except Exception:
try:
for i in key: del i ### For lists of tuples
except Exception: del key ### For plain lists
def verifyFile(filename):
status = 'not found'
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = 'found';break
except Exception: status = 'not found'
return status
def AppendOrWrite(export_path):
export_path = filepath(export_path)
status = verifyFile(export_path)
if status == 'not found':
export_data = export.ExportFile(export_path) ### Write this new file
else:
export_data = open(export_path,'a') ### Appends to existing file
return export_data, status
def quantileNormalizationSimple(condition_count_db):
### Basic quantile normalization method (average ranked expression values)
### Get all junction or exon entries
key_db={}
for condition in condition_count_db:
count_db = condition_count_db[condition]
for key in count_db: key_db[key]=[]
condition_unnormalized_db={}
for key in key_db:
### Only look at the specific biotype of interest for each normalization
for condition in condition_count_db:
count_db = condition_count_db[condition]
try:
count = float(count_db[key])+1 ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
count_db[key] = [] ### Set equal to null as a temporary measure to save memory
except KeyError: count = 1.00 ###Was zero, but needs to be one for more realistic log2 fold calculations
### store the minimal information to recover the original count and ID data prior to quantile normalization
try: condition_unnormalized_db[condition].append([count,key])
except Exception: condition_unnormalized_db[condition]=[[count,key]]
quantile_normalize_db={}; key_db={}
for condition in condition_unnormalized_db:
condition_unnormalized_db[condition].sort() ### Sort lists by count number
rank=0 ### thus, the ID is the rank order of counts
for (count,key) in condition_unnormalized_db[condition]:
try: quantile_normalize_db[rank].append(count)
except KeyError: quantile_normalize_db[rank] = [count]
rank+=1
### Get the average value for each index
for rank in quantile_normalize_db:
quantile_normalize_db[rank] = Average(quantile_normalize_db[rank])
for condition in condition_unnormalized_db:
rank=0
count_db = condition_count_db[condition]
for (count,key) in condition_unnormalized_db[condition]:
avg_count = quantile_normalize_db[rank]
rank+=1
count_db[key] = str(avg_count) ### re-set this value to the normalized value
try:
clearObjectsFromMemory(condition_unnormalized_db); condition_unnormalized_db = []
clearObjectsFromMemory(quantile_normalize_db); quantile_normalize_db = []
except Exception: None
return condition_count_db
def combineExonAnnotations(db):
for i in db:
list1=[]; list2=[]
for (junctions,splice_event) in db[i]:
list1.append(junctions); list2.append(splice_event)
junctions = EnsemblImport.combineAnnotations(list1)
splice_event = EnsemblImport.combineAnnotations(list2)
db[i] = junctions,splice_event
return db
def formatID(id):
### JunctionArray methods handle IDs with ":" different than those that lack this
return string.replace(id,':','@')
def filterChromosomes(chromosome_names):
### If transcriptome only aligned to Ensembl reference, many chromosomes are not real
updated_chromosomes=[]
chr_count=0
for chr in chromosome_names:
if 'chr' in chr and len(chr)<7:
chr_count+=1
updated_chromosomes.append(chr)
if chr_count>1:
return updated_chromosomes
else:
return chromosome_names
def getChromosomeStrandCoordinates(species,testImport):
### For novel junctions with no known-splice site, map to genes
gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,'RNASeq','key_by_array')
chr_strand_gene_db = {}; location_gene_db = {}; chromosome_names={}; all_chromosomes={}
for gene in gene_location_db:
chr,strand,start,end = gene_location_db[gene]
location_gene_db[chr,int(start),int(end)] = gene,strand
try: chr_strand_gene_db[chr,strand].append((int(start),int(end)))
except KeyError: chr_strand_gene_db[chr,strand] = [(int(start),int(end))]
if testImport == 'yes':
if chr=='chr1': chromosome_names[chr]=[]
#if chr=='chr19': chromosome_names[chr]=[] ### Gene rich chromosome
#if chr=='chrMT': chromosome_names[chr]=[] ### Gene rich chromosome
elif len(chr)<7: chromosome_names[chr]=[]
all_chromosomes[chr]=[]
#chromosome_names = filterChromosomes(chromosome_names)
### Some organisms aren't organized into classical chromosomes (why I don't know)
if len(chromosome_names)<10 and len(all_chromosomes)>9 and testImport=='no': chromosome_names = all_chromosomes
return chr_strand_gene_db,location_gene_db,chromosome_names,gene_location_db
def exportDatasetLinkedExons(species,exons_to_export,critical_exon_annotations,root_dir,testImport=None,searchChr=None):
export_path = root_dir+'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_exons.txt'
if searchChr != None:
export_path = string.replace(export_path,'RNASeq/'+species,'RNASeq/exons/'+species)
export_path = string.replace(export_path,'.txt','.'+searchChr+'.txt')
if testImport == 'yes': print 'Writing',export_path
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
export_title = ['AltAnalyzeID','exon_id','ensembl_gene_id','transcript_cluster_id','chromosome','strand','probeset_start','probeset_stop']
export_title +=['class','constitutive_probeset','ens_exon_ids','ens_constitutive_status','exon_region','exon-region-start(s)','exon-region-stop(s)','splice_events','splice_junctions']
export_title = string.join(export_title,'\t')+'\n'; export_data.write(export_title)
### We stored these in a dictionary to make sure each exon is written only once and so we can organize by gene
exons_to_export_list=[]
for key in exons_to_export:
ed = exons_to_export[key]
exons_to_export_list.append((key,ed))
exons_to_export_list.sort()
for (key,ed) in exons_to_export_list:
constitutive_call = 'no'; ens_constitutive_status = '0'
try:
red = ed.ExonRegionData()
exon_region = ed.ExonRegionID()
start = str(ed.ReadStart()); stop = start
if '-' not in exon_region and '_' not in exon_region: annotation = 'known'
else: annotation = 'novel'
except Exception:
red = ed ### For annotated exons, no difference in the annotations
exon_region = ed.ExonRegionIDs()
start = str(red.ExonStart()); stop = str(red.ExonStop())
constitutive_call = red.Constitutive()
if constitutive_call == 'yes': ens_constitutive_status = '1'
annotation = 'known'
uid = red.GeneID()+':'+exon_region
splice_events = red.AssociatedSplicingEvent(); splice_junctions = red.AssociatedSplicingJunctions()
if uid in critical_exon_annotations:
splice_junctions,splice_events = critical_exon_annotations[uid]
export_values = [uid, exon_region, red.GeneID(), '', red.Chr(), red.Strand(), start, stop, annotation, constitutive_call, red.ExonID(), ens_constitutive_status]
export_values+= [exon_region, str(red.ExonStart()), str(red.ExonStop()), splice_events, splice_junctions]
export_values = string.join(export_values,'\t')+'\n'; export_data.write(export_values)
export_data.close()
def exportNovelJunctions(species,novel_junction_db,condition_count_db,root_dir,dataset_name,biotype):
if 'exp.' not in dataset_name: dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name: dataset_name+='.txt'
dataset_name = string.replace(dataset_name,'exp','novel')
dataset_name = string.replace(dataset_name,'.txt','.'+biotype+'.txt')
export_path = root_dir+'ExpressionInput/'+dataset_name
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
title = ['chr','strand','start','stop','start Ensembl','end Ensembl','known start', 'known end']
for condition in condition_count_db: title.append(condition)
export_data.write(string.join(title,'\t')+'\n')
for key in novel_junction_db:
ji = novel_junction_db[key]
try: gene1 = str(ji.GeneID())
except Exception: gene1=''
try: gene2 = str(ji.SecondaryGeneID())
except Exception: gene2 = 'None'
try: le = str(ji.LeftExonAnnotations())
except Exception: le = ''
try: re = str(ji.RightExonAnnotations())
except Exception: re = ''
if biotype == 'junction':
values = [ji.Chr(), ji.Strand(), str(ji.Exon1Stop()), str(ji.Exon2Start())]
elif biotype == 'exon':
values = [ji.Chr(), ji.Strand(), str(ji.Exon1Stop()-1), str(ji.Exon2Start()+1)] ### correct for initial adjustment
values += [gene1,gene2,le,re]
for condition in condition_count_db:
count_db = condition_count_db[condition]
try: read_count = count_db[key]
except KeyError: read_count = '0'
values.append(read_count)
export_data.write(string.join(values,'\t')+'\n')
export_data.close()
def exportDatasetLinkedGenes(species,gene_location_db,root_dir):
"""Include an entry for gene IDs to include constitutive expression for RPKM normalized data"""
export_path = root_dir+'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_junctions.txt'
export_data,status = AppendOrWrite(export_path)
for gene in gene_location_db:
chr,strand,start,end = gene_location_db[gene]
export_values = [gene, 'E0.1',gene, '', chr, strand, str(start), str(end), 'known', 'yes', gene, '1']
export_values+= ['E0.1', str(start), str(end), '', '']
export_values = string.join(export_values,'\t')+'\n'; export_data.write(export_values)
export_data.close()
def exportDatasetLinkedJunctions(species,junction_db,junction_annotations,root_dir,testImport=False,searchChr=None):
export_path = root_dir+'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_junctions.txt'
if searchChr != None:
export_path = string.replace(export_path,'RNASeq/'+species,'RNASeq/junctions/'+species)
export_path = string.replace(export_path,'.txt','.'+searchChr+'.txt')
if testImport == 'yes': print 'Writing',export_path
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
export_title = ['AltAnalyzeID','exon_id','ensembl_gene_id','transcript_cluster_id','chromosome','strand','probeset_start','probeset_stop']
export_title +=['class','constitutive_probeset','ens_exon_ids','ens_constitutive_status','exon_region','exon-region-start(s)','exon-region-stop(s)','splice_events','splice_junctions']
export_title = string.join(export_title,'\t')+'\n'; export_data.write(export_title)
for key in junction_db:
(chr,exon1_stop,exon2_start) = key
ji=junction_db[key]
#print key, ji.UniqueID(), ji.GeneID()
if ji.GeneID()!=None and ji.UniqueID()!=None:
if ji.UniqueID() in junction_annotations: ### Obtained from JunctionArray.inferJunctionComps()
junctions,splice_events = junction_annotations[ji.UniqueID()]
if ji.TransSplicing() == 'yes':
if len(splice_events)>0: splice_events+= '|trans-splicing'
else: splice_events = 'trans-splicing'
ji.setAssociatedSplicingEvent(splice_events); ji.setAssociatedSplicingJunctions(junctions)
elif ji.TransSplicing() == 'yes':
ji.setAssociatedSplicingEvent('trans-splicing')
try:
try: constitutive_call = ji.Constitutive()
except Exception:
jd = ji.ExonAnnotations()
constitutive_call = jd.Constitutive()
if constitutive_call == 'yes': ens_constitutive_status = '1'
else: ens_constitutive_status = '0'
annotation = 'known'
except Exception:
constitutive_call = 'no'; ens_constitutive_status = '0'; annotation = 'novel'
if 'I' in ji.ExonRegionID() or 'U' in ji.ExonRegionID() or '_' in ji.ExonRegionID():
annotation = 'novel' ### Not previously indicated well (as I remember) for exon-level reads - so do this
export_values = [ji.UniqueID(), ji.ExonRegionID(), ji.GeneID(), '', ji.Chr(), ji.Strand(), str(ji.Exon1Stop()), str(ji.Exon2Start()), annotation, constitutive_call, ji.ExonID(), ens_constitutive_status]
export_values+= [ji.ExonRegionID(), str(ji.Exon1Stop()), str(ji.Exon2Start()), ji.AssociatedSplicingEvent(), ji.AssociatedSplicingJunctions()]
export_values = string.join(export_values,'\t')+'\n'; export_data.write(export_values)
export_data.close()
def combineDetectedExons(unmapped_exon_db,align_exon_db,novel_exon_db):
### Used for exon alignments (both start position and end position aligned to exon/intron/UTR regions)
### Reformat align_exon_db to easily lookup exon data
aligned_exon_lookup_db={}
for gene in align_exon_db:
for ed in align_exon_db[gene]:
aligned_exon_lookup_db[gene,ed.ReadStart()]=ed
#if gene == 'ENSMUSG00000064181': print ed.ReadStart(),ed.ExonRegionID()
### Reformat novel_exon_db to easily lookup exon data - created from junction analysis (rename above exons to match novel junctions)
novel_exon_lookup_db={}
for gene in novel_exon_db:
for ed in novel_exon_db[gene]:
try:
### Only store exons that are found in the novel exon file
null = aligned_exon_lookup_db[gene,ed.ReadStart()+1] ### offset introduced on import
novel_exon_lookup_db[gene,ed.ReadStart()+1]=ed
except Exception: null=[]
try:
### Only store exons that are found in the novel exon file
null = aligned_exon_lookup_db[gene,ed.ReadStart()-1] ### offset introduced on import
novel_exon_lookup_db[gene,ed.ReadStart()-1]=ed
except Exception: null=[]
### Lookup the propper exon region ID and gene ID to format the unique ID and export coordinates
x = 0
for key in unmapped_exon_db:
(chr,exon1_stop,exon2_start) = key
ji=unmapped_exon_db[key]
proceed = 'no'
if ji.GeneID() != None:
e1 = (ji.GeneID(),exon1_stop)
e2 = (ji.GeneID(),exon2_start)
exon_info=[]; override_annotation = None; found=[]
try: null = aligned_exon_lookup_db[e1]; found.append(1)
except Exception: null=[]
try: null = aligned_exon_lookup_db[e2]; found.append(2)
except Exception: null=[]
try: null = novel_exon_lookup_db[e1]; override_annotation = 1
except Exception:
try: null = novel_exon_lookup_db[e2]; override_annotation = 2
except Exception: null=[]
if len(found)>0:
### Below is not the simplist way to do this, but should be the fastest
if 1 in found: exon_info.append(aligned_exon_lookup_db[e1])
if 2 in found: exon_info.append(aligned_exon_lookup_db[e2])
if len(exon_info) == 2: ed1,ed2 = exon_info
else:
ed1 = exon_info[0]; ed2 = ed1; x+=1 ### if only one splice site aligned to a gene region (shouldn't occur)
if x == 2: null=[]; #print 'SOME EXONS FOUND WITH ONLY ONE ALIGNING POSITION...',key,ji.GeneID(),ed1.ExonRegionID(),e1,e2
try: red1 = ed1.ExonRegionData(); red2 = ed2.ExonRegionData()
except Exception:
"""
print [ji.GeneID(), ji.Chr(), key]
print e1, e2
try: print ed1.ExonRegionData()
except Exception: 'ed1 failed'
try: print ed2.ExonRegionData()
except Exception: 'ed2 failed'
"""
continue
region1 = ed1.ExonRegionID(); region2 = ed2.ExonRegionID()
#print region1,region2,ji.GeneID(),ji.Chr(),ji.Strand()
try: splice_junctions = EnsemblImport.combineAnnotations([red1.AssociatedSplicingJunctions(),red2.AssociatedSplicingJunctions()])
except Exception: print red1, red2;sys.exit()
splice_events = EnsemblImport.combineAnnotations([red1.AssociatedSplicingEvent(),red2.AssociatedSplicingEvent()])
ji.setAssociatedSplicingJunctions(splice_junctions)
ji.setAssociatedSplicingEvent(splice_events)
ens_exon_ids = EnsemblImport.combineAnnotations([red1.ExonID(),red2.ExonID()])
ji.setExonID(ens_exon_ids)
if red1.Constitutive() == 'yes' or red2.Constitutive() == 'yes': constitutive_call = 'yes'
else: constitutive_call = 'no'
ji.setConstitutive(constitutive_call)
report_both_regions = 'no'
try:
### If the annotations are from a BED file produced by AltAnalyze, novel alternative splice sites may be present
### if the below variable is not created, then this exon may over-ride the annotated exon region (e.g., E15.1 is over-written by E15.1_1234;E15.1_1256)
if 'ENS' in ji.JunctionID() and ':' not in ji.JunctionID(): report_both_regions = 'yes'
except Exception: null=[]
try:
### If the annotations are from a BED file produced by AltAnalyze, it is possible for to a known exon to share a splice-site coordinate
### with a novel junction exon. This will cause both to have the same override_annotation. Prevent this with the below 2nd override
if 'ENS' in ji.JunctionID() and ':' in ji.JunctionID(): override_annotation = None
except Exception: null=[]
if override_annotation != None:
if '_' in region1: region1 = string.split(region1,'_')[0]+'_'+str(int(string.split(region1,'_')[-1])-1)
if '_' in region2: region2 = string.split(region2,'_')[0]+'_'+str(int(string.split(region2,'_')[-1])+1)
if override_annotation == 1: region_id = region1 ### This forces a TopHat exon to be named for the splice-site position
else: region_id = region2
else:
if report_both_regions == 'no':
### Don't include specific start and end coordinates if inside a known exon
if ed1.AlignmentRegion() == 'exon': region1 = string.split(region1,'_')[0]
if ed2.AlignmentRegion() == 'exon': region2 = string.split(region2,'_')[0]
if ed1.AlignmentRegion() == 'full-intron' and ed2.AlignmentRegion() == 'full-intron':
region1 = string.split(region1,'_')[0]; region2 = string.split(region2,'_')[0]
### Below adjustmements need to compenstate for adjustments made upon import
if '_' in region1: region1 = string.split(region1,'_')[0]+'_'+str(int(string.split(region1,'_')[-1])-1)
if '_' in region2: region2 = string.split(region2,'_')[0]+'_'+str(int(string.split(region2,'_')[-1])+1)
ji.setExon1Stop(ji.Exon1Stop()-1); ji.setExon2Start(ji.Exon2Start()+1)
if override_annotation != None: null=[] ### It is already assigned above
elif region1 == region2: region_id = region1
elif ji.Strand() == '+': region_id = region1+';'+region2
else: region_id = region2+';'+region1 ### start and stop or genomically assigned
uid = ji.GeneID()+':'+region_id
#try: exon_region_db[ji.GeneID()].append((formatID(uid),region_id))
#except KeyError: exon_region_db[ji.GeneID()]=[(formatID(uid),region_id)]
ji.setExonRegionID(region_id)
ji.setUniqueID(uid) ### hgu133
### Export format for new exons to add to the existing critical exon database (those in exon_region_db are combined with analyzed junctions)
#exons_to_export[ji.GeneID(),region_id] = ji
else:
#print key, ji.GeneID(), ji.JunctionID(); sys.exit()
null=[] ### Occurs because two genes are overlapping
#return exons_to_export
def annotateNovelJunctions(novel_junction_db,novel_exon_db,exons_to_export):
### Reformat novel_exon_db to easily lookup exon data
novel_exon_lookup_db={}
for gene in novel_exon_db:
for ed in novel_exon_db[gene]:
novel_exon_lookup_db[gene,ed.ReadStart()]=ed
### Lookup the propper exon region ID and gene ID to format the unique ID and export coordinates
junction_region_db={}
unknown_gene_junctions={}
for key in novel_junction_db:
(chr,exon1_stop,exon2_start) = key
ji=novel_junction_db[key]
proceed = 'no'
if ji.GeneID() != None:
if ji.SpliceSitesFound() != 'both':
e1 = (ji.GeneID(),exon1_stop)
if ji.TransSplicing() == 'yes':
e2 = (ji.SecondaryGeneID(),exon2_start)
else: e2 = (ji.GeneID(),exon2_start)
if e1 in novel_exon_lookup_db and e2 in novel_exon_lookup_db:
proceed = 'yes'
try: ed1 = novel_exon_lookup_db[e1]; red1 = ed1.ExonRegionData(); gene1 = e1[0]
except Exception:
print chr, key, e1; kill
ed2 = novel_exon_lookup_db[e2]; red2 = ed2.ExonRegionData(); gene2 = e2[0]
### If the splice-site was a match to a known junciton splice site, use it instead of that identified by exon-region location overlapp
if ji.LeftExonAnnotations() != None: region1 = ji.LeftExonAnnotations()
else: region1 = ed1.ExonRegionID(); exons_to_export[gene1,region1] = ed1
if ji.RightExonAnnotations() != None: region2 = ji.RightExonAnnotations()
else: region2 = ed2.ExonRegionID(); exons_to_export[gene2,region2] = ed2
#print region1,region2,ji.GeneID(),ji.Chr(),ji.Strand(), ji.LeftExonAnnotations(), ji.RightExonAnnotations()
else:
proceed = 'yes'
region1 = ji.LeftExonAnnotations()
region2 = ji.RightExonAnnotations()
red1 = ji.LeftExonRegionData()
red2 = ji.RightExonRegionData()
### Store the individual exons for export
gene1 = ji.GeneID()
if ji.TransSplicing() == 'yes': gene2 = ji.SecondaryGeneID()
else: gene2 = ji.GeneID()
exons_to_export[gene1,region1] = red1
exons_to_export[gene2,region2] = red2
if proceed == 'yes':
try: splice_junctions = EnsemblImport.combineAnnotations([red1.AssociatedSplicingJunctions(),red2.AssociatedSplicingJunctions()])
except Exception: print red1, red2;sys.exit()
splice_events = EnsemblImport.combineAnnotations([red1.AssociatedSplicingEvent(),red2.AssociatedSplicingEvent()])
ji.setAssociatedSplicingJunctions(splice_junctions)
ji.setAssociatedSplicingEvent(splice_events)
ens_exon_ids = EnsemblImport.combineAnnotations([red1.ExonID(),red2.ExonID()])
ji.setExonID(ens_exon_ids)
if ji.TransSplicing() == 'yes':
uid = ji.GeneID()+':'+region1+'-'+ji.SecondaryGeneID()+':'+region2
region_id = uid
### When trans-splicing occurs, add the data twice to junction_region_db for the two different genes
### in JunctionArray.inferJunctionComps, establish two separate gene junctions with a unique ID for the non-gene exon
try: junction_region_db[ji.GeneID()].append((formatID(uid),region1+'-'+'U1000.1_'+str(ji.Exon2Start())))
except KeyError: junction_region_db[ji.GeneID()]=[(formatID(uid),region1+'-'+'U1000.1_'+str(ji.Exon2Start()))]
try: junction_region_db[ji.SecondaryGeneID()].append((formatID(uid),'U0.1_'+str(ji.Exon1Stop())+'-'+region2))
except KeyError: junction_region_db[ji.SecondaryGeneID()]=[(formatID(uid),'U0.1_'+str(ji.Exon1Stop())+'-'+region2)]
else:
uid = ji.GeneID()+':'+region1+'-'+region2
region_id = region1+'-'+region2
try: junction_region_db[ji.GeneID()].append((formatID(uid),region_id))
except KeyError: junction_region_db[ji.GeneID()]=[(formatID(uid),region_id)]
ji.setExonRegionID(region_id)
ji.setUniqueID(uid)
else:
unknown_gene_junctions[key]=[]
return junction_region_db,exons_to_export
def alignReadsToExons(novel_exon_db,ens_exon_db,testImport=False):
### Simple method for aligning a single coordinate to an exon/intron region of an already matched gene
examined_exons=0; aligned_exons=0
for gene in ens_exon_db: #novel_exon_db
try:
region_numbers=[]; region_starts=[]; region_stops=[]
for ed in novel_exon_db[gene]:
examined_exons+=1; aligned_status=0; index=-1
for rd in ens_exon_db[gene]:
index+=1 ### keep track of exon/intron we are in
region_numbers.append(int(string.split(rd.ExonRegionIDs()[1:],'.')[0]))
if rd.Strand() == '-': region_starts.append(rd.ExonStop()); region_stops.append(rd.ExonStart())
else: region_starts.append(rd.ExonStart()); region_stops.append(rd.ExonStop())
#print [rd.ExonStart(),rd.ExonStop(), rd.Strand()]
#print [ed.ReadStart(),rd.ExonStart(),rd.ExonStop()]
if ed.ReadStart()>=rd.ExonStart() and ed.ReadStart()<=rd.ExonStop():
ed.setAlignmentRegion('exon')
if 'I' in rd.ExonRegionIDs(): ### In an annotated intron
ed.setAlignmentRegion('intron')
ord = rd; updated = None
try: ### If the splice site is a novel 3' splice site then annotate as the 3' exon (less than 50nt away)
nrd = ens_exon_db[gene][index+1]
if (abs(ed.ReadStart()-nrd.ExonStart())<3) or (abs(ed.ReadStart()-nrd.ExonStop())<3):
ed.setAlignmentRegion('full-intron') ### this is the start/end of intron coordinates
elif (abs(ed.ReadStart()-nrd.ExonStart())<50) or (abs(ed.ReadStart()-nrd.ExonStop())<50): rd = nrd; updated = 1
except Exception: null=[]
try:
prd = ens_exon_db[gene][index-1]
if (abs(ed.ReadStart()-prd.ExonStart())<3) or (abs(ed.ReadStart()-prd.ExonStop())<3):
ed.setAlignmentRegion('full-intron')### this is the start/end of intron coordinates
elif (abs(ed.ReadStart()-prd.ExonStart())<50) or (abs(ed.ReadStart()-prd.ExonStop())<50):
if updated==1: rd = ord; ###Hence the intron is too small to descriminate between alt5' and alt3' exons
else: rd = prd
except Exception: null=[]
ed.setExonRegionData(rd); aligned_exons+=1; aligned_status=1
if rd.ExonStop()==ed.ReadStart():
ed.setExonRegionID(rd.ExonRegionIDs())
elif rd.ExonStart()==ed.ReadStart():
ed.setExonRegionID(rd.ExonRegionIDs())
elif 'exon-intron' in ed.Annotation(): ### intron retention
ed.setExonRegionID(rd.ExonRegionIDs()) ### Hence there is a 1nt difference between read
else:
ed.setExonRegionID(rd.ExonRegionIDs()+'_'+str(ed.ReadStart()))
break
if aligned_status == 0: ### non-exon/intron alinging sequences
region_numbers.sort(); region_starts.sort(); region_stops.sort()
if (rd.Strand() == '+' and ed.ReadStart()>=rd.ExonStop()) or (rd.Strand() == '-' and rd.ExonStop()>=ed.ReadStart()):
### Applicable to 3'UTR (or other trans-splicing) aligning
utr_id = 'U'+str(region_numbers[-1])+'.1_'+str(ed.ReadStart())
ud = EnsemblImport.ExonAnnotationsSimple(rd.Chr(),rd.Strand(),region_stops[-1],region_stops[-1],gene,'','no',utr_id,'','')
ed.setExonRegionID(utr_id)
else:
### Applicable to 5'UTR (or other trans-splicing) aligning
utr_id = 'U0.1'+'_'+str(ed.ReadStart())
ud = EnsemblImport.ExonAnnotationsSimple(rd.Chr(),rd.Strand(),region_starts[0],region_starts[0],gene,'','no',utr_id,'','')
ed.setExonRegionID(utr_id)
ed.setExonRegionData(ud)
ed.setAlignmentRegion('UTR')
except Exception: null=[]
if testImport == 'yes': print aligned_exons, 'splice sites aligned to exon region out of', examined_exons
def geneAlign(chr,chr_gene_locations,location_gene_db,chr_reads,switch_coord,read_aligned_to_gene):
""" This function aligns the start or end position for each feature (junction or exon) to a gene, in two
steps by calling this function twice. In the second interation, the coordinates are reversed """
index = 0 ### Don't examine genes already looked at
genes_assigned = 0; trans_splicing=[]
for (coord,ji) in chr_reads: ### junction coordinates or exon coordinates with gene object
if index >5: index -=5 ### It is possible for some genes to overlap, so set back the index of genomically ranked genes each time
gene_id_obtained = 'no'
if switch_coord == 'no': rs,re=coord ### reverse the coordinates for the second iteration
else: re,rs=coord ### first-interation coordinates (start and end)
while index < len(chr_gene_locations):
cs,ce = chr_gene_locations[index]
#print [re,rs,cs,ce, ji.Chromosome()];sys.exit()
### Determine if the first listed coordinate lies within the gene
if cs <= rs and ce >= rs:
### Yes, it does
gene,strand = location_gene_db[chr,cs,ce]
if switch_coord == 'yes': ### Only applies to coordinates, where the end-position didn't lie in the same gene as the start-position
if cs <= re and ce >= re:
### This occurs when the first iteration detects a partial overlap, but the gene containing both coordinates is downstream
### Hence, not trans-splicing
ji.setGeneID(gene)
break
first_geneid = ji.GeneID() ### see what gene was assigned in the first iteration (start position only)
#print ['trans',coord, first_geneid, gene] ### Note: in rare cases, an exon can overlap with two genes (bad Ensembl annotations?)
ji.setTransSplicing()
side = ji.checkExonPosition(rs)
if side == 'left':
ji.setGeneID(gene)
ji.setSecondaryGeneID(first_geneid)
else:
ji.setSecondaryGeneID(gene)
#if ji.GeneID() == None: print 'B',coord, ji.GeneID(), secondaryGeneID()
#print ji.GeneID(), ji.SecondaryGeneID();kill
genes_assigned+=1; gene_id_obtained = 'yes'
### Check to see if this gene represents a multi-gene spanning region (overlaps with multiple gene loci)
try:
### This code was used to check and see if the gene is multi-spanning. Appears that the < sign is wrong > anyways, never go to the next gene unless the next read has passed it
#cs2,ce2 = chr_gene_locations[index+1]
#if cs2 < ce: index+=1 ### Continue analysis (if above is correct, the gene will have already been assigned)
#else: break
break
except Exception: break
else:
### First iteration, store the identified gene ID (only looking at the start position)
ji.setGeneID(gene); gene_id_obtained = 'yes'
#print gene, rs, re, cs, ce
### Check the end position, to ensure it is also lies within the gene region
if cs <= re and ce >= re:
genes_assigned+=1
else:
### Hence, the end lies outside the gene region
trans_splicing.append((coord,ji))
### Check to see if this gene represents a multi-gene spanning region (overlaps with multiple gene loci)
try:
### This code was used to check and see if the gene is multi-spanning. Appears that the < sign is wrong > anyways, never go to the next gene unless the next read has passed it
#cs2,ce2 = chr_gene_locations[index+1]
#if cs2 < ce: index+=1 ### Continue analysis (if above is correct, the gene will have already been assigned)
#else: break
break
except Exception: break
else:
if rs < ce and re < ce: break
elif switch_coord == 'no' and cs <= re and ce >= re:
### This can occur if the left junction splice site is in an exon and the other is the UTR as opposed to another gene
gene,strand = location_gene_db[chr,cs,ce]
ji.setSecondaryGeneID(gene); gene_id_obtained = 'yes'
#print gene, coord, ji.Strand(), ji.GeneID()
index+=1
if gene_id_obtained == 'no':
### These often appear to be genes predicted by tBLASTn at UCSC but not by Ensembl (e.g., chr17:27,089,652-27,092,318 mouse mm9)
null=[]
#ji.setGeneID(None) ### This is not necessary, since if one exon does not align to a gene it is still a valid alignment
#print chr,coord
read_aligned_to_gene += genes_assigned
#print genes_assigned, chr, 'Gene IDs assigned out of', len(chr_reads)
#print len(trans_splicing),'reads with evidence of trans-splicing'
### For any coordinate-pair where the end-position doesn't lie within the same gene as the start, re-run for those to see which gene they are in
if switch_coord == 'no' and len(trans_splicing)>0:
read_aligned_to_gene = geneAlign(chr,chr_gene_locations,location_gene_db,trans_splicing,'yes',read_aligned_to_gene)
return read_aligned_to_gene
def getNovelExonCoordinates(species,root_dir):
""" Currently, any novel exon determined during initial RNA-Seq read annotation with defined start and end coordinates, only has
the exon-end coordinate, not start, in it's name. However, the start and stop are indicated in the counts.Experiment.txt file.
To get this, we parse that file and only store exons with an I or U in them and then correct for this in the matching function below """
exp_dir = root_dir+'/ExpressionInput/'
dir_list = read_directory(exp_dir)
counts_file = None
for file in dir_list:
if 'counts.' in file and 'steady' not in file:
counts_file = file
### Example
#ENSG00000137076:I17.1_35718353=chr9:35718353-35718403 (novel exon coordinates - just sorted, not necessarily in the correct order)
#ENSG00000137076:E17.1-I17.1_35718403=chr9:35718809-35718403 (5' supporting junction)
#ENSG00000137076:I17.1_35718353-E18.1=chr9:35718353-35717783 (3' supporting junction)
#here, once we see that I17.1_35718353 is the exon ID, we know we need to get the function with -I17.1_35718403 (always the second value)
if counts_file!=None:
fn=filepath(exp_dir+counts_file)
print 'Reading counts file'
novel_exon_db = parseCountFile(fn,'exons',{}) ### Get novel exons
print 'Reading counts file'
novel_exon_db = parseCountFile(fn,'junctions',novel_exon_db) ### Get novel exons
return novel_exon_db
def getMaxCounts(fn,cutoff,filterExport=False,filterExportDir=False):
firstLine=True
expressed_uids={}
if filterExport != False:
eo=export.ExportFile(filterExportDir)
for line in open(fn,'rU').xreadlines():
Line = cleanUpLine(line)
t = string.split(Line,'\t')
key = t[0]
if firstLine:
firstLine = False
if filterExport != False:
eo.write(line)
else:
if filterExport != False:
if key in filterExport:
eo.write(line)
else:
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
try: maxExp = max(map(lambda x: float(x), t[1:])); #print maxExp;sys.exit()
except Exception:
#print t[1:];sys.exit()
if 'NA' in t[1:]:
tn = [0 if x=='NA' else x for x in t[1:]] ### Replace NAs
maxExp = max(map(lambda x: float(x), tn))
elif '' in t[1:]:
tn = [0 if x=='' else x for x in t[1:]] ### Replace blanks
maxExp = max(map(lambda x: float(x), tn))
else:
maxExp=cutoff+1
#gene = string.split(uid,':')[0]
if maxExp > cutoff:
expressed_uids[uid] = []
return expressed_uids
def check_for_ADT(gene):
if '.ADT' in gene or '-ADT' in gene:
return True
elif len(gene)>17 and '-' in gene:
if len(string.split(gene,'-')[1])>11:
return True
else:
return False
else:
return False
def importBiologicalRelationships(species):
### Combine non-coding Ensembl gene annotations with UniProt functional annotations
import ExpressionBuilder
custom_annotation_dbase={}
try: coding_db = ExpressionBuilder.importTranscriptBiotypeAnnotations(species)
except Exception: coding_db = {}
try: gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
except Exception: gene_to_symbol_db = {}
for gene in coding_db:
#coding_type = string.split(coding_db[gene][-1],'|')
coding_type = coding_db[gene][-1]
if 'protein_coding' in coding_type:
coding_type = 'protein_coding'
else:
coding_type = 'ncRNA'
status = check_for_ADT(gene)
if gene in gene_to_symbol_db:
symbol = string.lower(gene_to_symbol_db[gene][0])
### The below genes cause issues with many single cell datasets in terms of being highly correlated
if 'rpl'==symbol[:3] or 'rps'==symbol[:3] or 'mt-'==symbol[:3] or '.' in symbol or 'gm'==symbol[:2]:
coding_type = 'ncRNA'
try: gene_db = custom_annotation_dbase[coding_type]; gene_db[gene]=[]
except Exception: custom_annotation_dbase[coding_type] = {gene:[]}
filename = 'AltDatabase/uniprot/'+species+'/custom_annotations.txt'
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
ens_gene,compartment,custom_class = t[:3]
if 'GPCR' in custom_class:
custom_class = ['GPCR']
else:
custom_class = string.split(custom_class,'|')
custom_class = string.split(compartment,'|')+custom_class
for cc in custom_class:
try: gene_db = custom_annotation_dbase[cc]; gene_db[ens_gene]=[]
except Exception: custom_annotation_dbase[cc] = {ens_gene:[]}
#custom_annotation_dbase={}
try:
filename = 'AltDatabase/goelite/'+species+'/gene-mapp/Ensembl-BioMarkers.txt'
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
gene,null,celltype = t[:3]
try: gene_db = custom_annotation_dbase['BioMarker']; gene_db[gene]=[]
except Exception: custom_annotation_dbase['BioMarker'] = {gene:[]}
#print len(custom_annotation_dbase), 'gene classes imported'
except Exception: pass
return custom_annotation_dbase
def importGeneSets(geneSetType,filterType=None,geneAnnotations=None,speciesName=None):
try: speciesName = species
except: pass
gene_db={}
if 'Ontology' in geneSetType:
filename = 'AltDatabase/goelite/'+speciesName+'/nested/Ensembl_to_Nested-GO.txt'
ontology=True
else:
filename = 'AltDatabase/goelite/'+speciesName+'/gene-mapp/Ensembl-'+geneSetType+'.txt'
ontology=False
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if ontology:
gene,category = t
else: gene,null,category = t[:3]
if filterType==None:
try: gene_db[gene].append(category)
except Exception: gene_db[gene] = [category]
elif filterType in category:
if gene in geneAnnotations:
gene = geneAnnotations[gene][0]
gene_db[gene]=[]
return gene_db
def singleCellRNASeqWorkflow(Species, platform, expFile, mlp, exp_threshold=0, rpkm_threshold=5, drivers=False, parameters = None, reportOnly=False):
global species
global rho_cutoff
species = Species
removeOutliers = False
if parameters != None:
rpkm_threshold = parameters.ExpressionCutoff()
exp_threshold = parameters.CountsCutoff()
rho_cutoff = parameters.RhoCutoff()
restrictBy = parameters.RestrictBy()
try: numGenesExp = parameters.NumGenesExp()-1
except: numGenesExp = 499
try: removeOutliers = parameters.RemoveOutliers()
except Exception: pass
if platform == 'exons' or platform == 'PSI':
rpkm_threshold=0
exp_threshold=0
else:
rho_cutoff = 0.4
restrictBy = 'protein_coding'
onlyIncludeDrivers=True
if platform != 'exons' and platform != 'PSI':
try: platform = checkExpressionFileFormat(expFile,platform)
except:
if '-steady-state' in expFile:
expFile = string.replace(expFile,'-steady-state','') ### Occurs for Kallisto processed
platform = checkExpressionFileFormat(expFile,platform)
if platform != 'RNASeq':
if rpkm_threshold>1.9999:
rpkm_threshold = math.log(rpkm_threshold,2) ### log2 transform
if removeOutliers:
### Remove samples with low relative number of genes expressed
try:
print '***Removing outlier samples***'
from import_scripts import sampleIndexSelection
reload(sampleIndexSelection)
output_file = expFile[:-4]+'-OutliersRemoved.txt'
sampleIndexSelection.statisticallyFilterFile(expFile,output_file,rpkm_threshold,minGeneCutoff=numGenesExp)
if 'exp.' in expFile:
### move the original groups and comps files
groups_file = string.replace(expFile,'exp.','groups.')
groups_file = string.replace(groups_file,'-steady-state','')
groups_filtered_file = groups_file[:-4]+'-OutliersRemoved.txt'
#comps_file = string.replace(groups_file,'groups.','comps.')
#comps_filtered_file = string.replace(groups_filtered_file,'groups.','comps.')
#counts_file = string.replace(expFile,'exp.','counts.')
#counts_filtered_file = string.replace(output_file,'exp.','counts.')
try: shutil.copyfile(groups_file,groups_filtered_file) ### if present copy over
except Exception: pass
try: shutil.copyfile(comps_file,comps_filtered_file) ### if present copy over
except Exception: pass
#try: shutil.copyfile(counts_file,counts_filtered_file) ### if present copy over
#except Exception: pass
expFile = output_file
print ''
except Exception:
print '***Filtering FAILED***'
print traceback.format_exc()
expressed_uids_rpkm = getMaxCounts(expFile,rpkm_threshold)
try: expressed_uids_counts = getMaxCounts(string.replace(expFile,'exp.','counts.'),exp_threshold)
except Exception: expressed_uids_counts=expressed_uids_rpkm
if len(expressed_uids_counts) > 0:
try: expressed_uids = expressed_uids_rpkm.viewkeys() & expressed_uids_counts.viewkeys() ### common
except Exception: expressed_uids = getOverlappingKeys(expressed_uids_rpkm,expressed_uids_counts)
else:
expressed_uids = expressed_uids_rpkm
if reportOnly:
print '.',
else:
print 'Genes filtered by counts:',len(expressed_uids_counts)
print 'Genes filtered by expression:',len(expressed_uids_rpkm),len(expressed_uids)
#expressed_uids = filterByProteinAnnotation(species,expressed_uids)
print len(expressed_uids), 'expressed genes by RPKM/TPM (%d) and counts (%d)' % (rpkm_threshold,exp_threshold)
from import_scripts import OBO_import; import ExpressionBuilder
gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
try: biological_categories = importBiologicalRelationships(species)
except Exception:
restrictBy = None
biological_categories={}
print 'Missing annotation file in:','AltDatabase/uniprot/'+species+'/custom_annotations.txt !!!!!'
if restrictBy !=None:
if reportOnly:
print '.',
else:
print 'Attempting to restrict analysis to protein coding genes only (flag --RestrictBy protein_coding)'
genes = biological_categories['protein_coding']
genes_temp=dict(genes)
for gene in genes_temp:
if gene in gene_to_symbol_db:
genes[gene_to_symbol_db[gene][0]]=[] ### add symbols
genes_temp={}
else:
genes = {}
for i in expressed_uids: genes[i]=[]
"""
genes.update(biological_categories['BioMarker'])
genes.update(biological_categories['transcription regulator'])
genes.update(biological_categories['splicing regulator'])
genes.update(biological_categories['kinase'])
genes.update(biological_categories['GPCR'])
"""
expressed_uids_db={}; guide_genes={}
for id in expressed_uids: expressed_uids_db[id]=[]
if platform == 'exons' or platform == 'PSI': ### For splicing-index value filtering
expressed_uids=[]
for uid in expressed_uids_db:
geneID = string.split(uid,':')[0]
geneID = string.split(geneID,' ')[-1]
if geneID in genes: expressed_uids.append(uid)
else:
expressed_uids2=[]
for gene in expressed_uids:
ADT_status = check_for_ADT(gene)
if ADT_status:
expressed_uids2.append(gene)
elif gene in genes:
expressed_uids2.append(gene)
expressed_uids = expressed_uids2
expressed_uids_db2={}
for id in expressed_uids: expressed_uids_db2[id]=[]
if drivers != False:
guide_genes = getDrivers(drivers)
if onlyIncludeDrivers:
try: expressed_uids = guide_genes.viewkeys() & expressed_uids_db2.viewkeys() ### common
except Exception: expressed_uids = getOverlappingKeys(guide_genes,expressed_uids_db2)
if len(expressed_uids)<100:
print '\nNOTE: The input IDs do not sufficiently map to annotated protein coding genes...',
print 'skipping protein coding annotation filtering.'
expressed_uids=[]
for uid in expressed_uids_db:
expressed_uids.append(uid)
if reportOnly:
print '.',
else:
print len(expressed_uids), 'expressed IDs being further analyzed'
print_out,n = findCommonExpressionProfiles(expFile,species,platform,expressed_uids,guide_genes,mlp,parameters=parameters,reportOnly=reportOnly)
return print_out,n
def getOverlappingKeys(db1,db2):
db3=[]
for key in db1:
if key in db2:
db3.append(key)
return db3
def getDrivers(filename):
fn = filepath(filename)
firstLine=True
drivers={}
for line in open(fn,'rU').xreadlines():
line = line.rstrip()
t = string.split(line,'\t')
if firstLine: firstLine = False
else:
gene = t[0]
drivers[gene]=[]
print 'Imported %d guide genes' % len(drivers)
return drivers
def filterByProteinAnnotation(species,expressed_uids):
import ExpressionBuilder
custom_annotation_dbase = ExpressionBuilder.importTranscriptBiotypeAnnotations(species)
expressed_uids_protein=[]
for gene in expressed_uids:
if gene in custom_annotation_dbase:
compartment,custom_class = custom_annotation_dbase[gene]
if 'protein_coding' in custom_class:
expressed_uids_protein.append(gene)
if len(expressed_uids_protein)>10:
return expressed_uids_protein
else:
return expressed_uids
def CoeffVar(expFile,platform,expressed_uids,fold=2,samplesDiffering=2,guideGenes=[]):
firstLine=True
expressed_values={}
expressed_values_filtered={}
cv_list=[]
for line in open(expFile,'rU').xreadlines():
key = string.split(line,'\t')[0]
t = string.split(line,'\t')
if firstLine:
headers = line
firstLine = False
else:
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
values = map(lambda x: float(x), t[1:])
#gene = string.split(uid,':')[0]
if uid in expressed_uids:
vs = list(values); vs.sort()
cv = statistics.stdev(values)/statistics.avg(values)
if samplesDiffering<1: samplesDiffering=1
if platform == 'RNASeq':
if (vs[-1*samplesDiffering]/vs[samplesDiffering])>fold: ### Ensures that atleast 4 samples are significantly different in the set
expressed_values[uid] = values
cv_list.append((cv,uid))
else:
if (vs[-1*samplesDiffering]-vs[samplesDiffering])>fold: ### Ensures that atleast 4 samples are significantly different in the set
expressed_values[uid] = values
cv_list.append((cv,uid))
if uid in guideGenes:
expressed_values[uid] = values
cv_list.append((10000,uid)) ### Very high CV
cv_list.sort()
cv_list.reverse()
x=0
for (cv,uid) in cv_list:
x+=1
"""
if uid == 'ENSMUSG00000003882':
print x, 'ilr7'
"""
for (cv,uid) in cv_list[:5000]:
expressed_values_filtered[uid] = expressed_values[uid]
return expressed_values_filtered, fold, samplesDiffering, headers
def determinePattern(vs):
max_vs = max(vs)
min_vs = min(vs)
lower_max = max_vs - (max_vs*0.01)
upper_min = abs(max_vs)*0.01
s = bisect.bisect_right(vs,upper_min) ### starting low 15% index position
e = bisect.bisect_left(vs,lower_max) ### ending upper 85% index position
#print vs
#print max_vs, min_vs
#print lower_max, upper_min
#print s, e
avg = statistics.avg(vs[s:e+1])
m = bisect.bisect_left(vs,avg)
ratio = vs[m]/vs[((e-s)/2)+s-2] ### If the ratio is close to 1, a sigmoidal or linear pattern likely exists
print ratio
#sys.exit()
return ratio
def checkExpressionFileFormat(expFile,platform):
firstLine=True
inputMax=0; inputMin=10000
expressed_values={}
rows=0
for line in open(expFile,'rU').xreadlines():
key = string.split(line,'\t')[0]
t = string.split(line,'\t')
if firstLine:
headers = line
firstLine = False
else:
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
try: values = map(lambda x: float(x), t[1:])
except Exception:
values=[]
for value in t[1:]:
try: values.append(float(value))
except Exception:pass
try:
if max(values)>inputMax: inputMax = max(values)
except Exception:
pass
if inputMax>100:
break
if inputMax>100: ### Thus, not log values
platform = 'RNASeq'
else:
platform = "3'array"
return platform
def optimizeNumberOfGenesForDiscovery(expFile,platform,expressed_uids,fold=2,samplesDiffering=2,guideGenes=[],reportOnly=False):
firstLine=True
expressed_values={}
for line in open(expFile,'rU').xreadlines():
key = string.split(line,'\t')[0]
t = string.split(line,'\t')
if firstLine:
headers = line
firstLine = False
else:
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
try: values = map(lambda x: float(x), t[1:])
except Exception:
values = t[1:]
if 'NA' in values:
values = [0 if x=='NA' else x for x in values] ### Replace NAs
values = map(lambda x: float(x), values)
else:
values=[]
for value in t[1:]:
try: values.append(float(value))
except Exception: values.append(-9999)
values = numpy.ma.masked_values(values, -9999.)
#gene = string.split(uid,':')[0]
#if uid == 'ENSMUSG00000041515': print 'IRF8'
if uid in expressed_uids:
#slope_exp_ratio = determinePattern(vs)
#if slope_exp_ratio<2 and slope_exp_ratio>0.5:
if platform == 'RNASeq':
try: values = map(lambda x: math.log(x+1,2),values)
except Exception:
if 'NA' in values:
values = [0 if x=='NA' else x for x in values] ### Replace NAs
values = map(lambda x: math.log(x+1,2),values)
elif '' in values:
values = [0 if x=='' else x for x in values] ### Replace NAs
values = map(lambda x: math.log(x+1,2),values)
vs = list(values); vs.sort()
if (vs[-1*samplesDiffering]-vs[samplesDiffering-1])>math.log(fold,2): ### Ensures that atleast 4 samples are significantly different in the set
if reportOnly==False:
expressed_values[uid] = values
else:
expressed_values[uid]=[] ### Don't store the values - datasets can contain tens of thousands of
else:
vs = list(values); vs.sort()
if (vs[-1*samplesDiffering]-vs[samplesDiffering-1])>math.log(fold,2): ### Ensures that atleast 4 samples are significantly different in the set
if reportOnly==False:
expressed_values[uid] = values
else:
expressed_values[uid]=[]
if uid in guideGenes:
expressed_values[uid] = values
#if uid == 'ENSMUSG00000062825': print (vs[-1*samplesDiffering]-vs[samplesDiffering]),math.log(fold,2);sys.exit()
if reportOnly:
print '.',
else:
print len(expressed_uids),'genes examined and', len(expressed_values),'genes expressed for a fold cutoff of', fold
if len(expressed_uids)==0 or len(expressed_values)==0:
print options_result_in_no_genes
elif len(expressed_uids) < 50 and len(expressed_values)>0:
return expressed_values, fold, samplesDiffering, headers
elif len(expressed_values)>15000:
if platform == 'exons' or platform == 'PSI':
fold+=0.1
else:
fold+=1
samplesDiffering+=1
expressed_values, fold, samplesDiffering, headers = optimizeNumberOfGenesForDiscovery(expFile,platform,expressed_uids,fold=fold,samplesDiffering=samplesDiffering,guideGenes=guideGenes,reportOnly=reportOnly)
elif fold == 1.2 and samplesDiffering == 1:
return expressed_values, fold, samplesDiffering, headers
elif len(expressed_values)<50:
fold-=0.2
samplesDiffering-=1
if samplesDiffering<1: samplesDiffering = 1
if fold < 1.1: fold = 1.2
expressed_values, fold, samplesDiffering, headers = optimizeNumberOfGenesForDiscovery(expFile,platform,expressed_uids,fold=fold,samplesDiffering=samplesDiffering,guideGenes=guideGenes,reportOnly=reportOnly)
else:
return expressed_values, fold, samplesDiffering, headers
return expressed_values, fold, samplesDiffering, headers
def intraCorrelation(expressed_values,mlp):
if mlp.cpu_count() < 3:
processors = mlp.cpu_count()
else: processors = 8
pool = mlp.Pool(processes=processors)
si = (len(expressed_values)/processors)
s = si; b=0
db_ls=[]
if len(expressed_values)<10: forceError ### will si to be zero and an infanite loop
while s<len(expressed_values):
db_ls.append(dict(expressed_values.items()[b:s]))
b+=si; s+=si
db_ls.append(dict(expressed_values.items()[b:s]))
### Create an instance of MultiZscoreWorker (store the variables to save memory)
workerMulti = MultiCorrelatePatterns(expressed_values)
results = pool.map(workerMulti,db_ls)
#for i in db_ls: workerMulti(i)
pool.close(); pool.join(); pool = None
correlated_genes={}
for a in results:
for k in a: correlated_genes[k] = a[k]
return correlated_genes
def findCommonExpressionProfiles(expFile,species,platform,expressed_uids,guide_genes,mlp,fold=2,samplesDiffering=2,parameters=None,reportOnly=False):
use_CV=False
global rho_cutoff
row_metric = 'correlation'; row_method = 'average'
column_metric = 'cosine'; column_method = 'hopach'
original_column_metric = column_metric
original_column_method = column_method
color_gradient = 'yellow_black_blue'; transpose = False; graphic_links=[]
if parameters != None:
try: excludeGuides = parameters.ExcludeGuides() ### Remove signatures
except Exception: excludeGuides = None
fold = parameters.FoldDiff()
samplesDiffering = parameters.SamplesDiffering()
amplifyGenes = parameters.amplifyGenes()
if 'Guide' in parameters.GeneSelection():
amplifyGenes = False ### This occurs when running ICGS with the BOTH option, in which Guide3 genes are retained - ignore these
parameters.setGeneSelection('')
parameters.setClusterGOElite('')
excludeCellCycle = parameters.ExcludeCellCycle()
from visualization_scripts import clustering
row_metric = 'correlation'; row_method = 'average'
column_metric = parameters.ColumnMetric(); column_method = parameters.ColumnMethod()
original_column_metric = column_metric
original_column_method = column_method
color_gradient = 'yellow_black_blue'; graphic_links=[]
if platform == 'exons' or platform =='PSI': color_gradient = 'yellow_black_blue'
guide_genes = parameters.JustShowTheseIDs()
cell_cycle_id_list = []
else:
amplifyGenes = False
excludeCellCycle = False
if platform != 'exons'and platform !='PSI':
platform = checkExpressionFileFormat(expFile,platform)
else:
if LegacyMode: pass
else:
fold = math.pow(2,0.5)
fold = 1.25
#"""
if use_CV:
expressed_values, fold, samplesDiffering, headers = CoeffVar(expFile,platform,expressed_uids,fold=2,samplesDiffering=2,guideGenes=guide_genes)
else:
if reportOnly:
print '.',
else:
print 'Finding an optimal number of genes based on differing thresholds to include for clustering...'
#fold=1; samplesDiffering=1
expressed_values, fold, samplesDiffering, headers = optimizeNumberOfGenesForDiscovery(expFile,platform,expressed_uids,fold=fold,samplesDiffering=samplesDiffering,guideGenes=guide_genes,reportOnly=reportOnly) #fold=2,samplesDiffering=2
if reportOnly:
print '.',
else:
print 'Evaluating',len(expressed_values),'genes, differentially expressed',fold,'fold for at least',samplesDiffering*2,'samples'
#sys.exit()
from import_scripts import OBO_import; import ExpressionBuilder
gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol_db)
areYouSure=False
if (excludeCellCycle == 'strict' or excludeCellCycle == True) and areYouSure:
cc_param = copy.deepcopy(parameters)
cc_param.setPathwaySelect('cell cycle')
cc_param.setGeneSet('GeneOntology')
cc_param.setGeneSelection('amplify')
transpose = cc_param
filtered_file = export.findParentDir(expFile)+'/amplify/'+export.findFilename(expFile)
writeFilteredFile(filtered_file,platform,headers,{},expressed_values,[])
if len(expressed_values)<1000:
row_method = 'hopach'; row_metric = 'correlation'
if column_method != 'hopach': row_method = 'average' ### needed due to PC errors
if len(headers)>7000: ### For very ultra-large datasets
column_method = 'average'
cc_graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
cell_cycle_id_list = genericRowIDImport(string.replace(cc_graphic_links[0][-1],'.png','.txt'))
expressed_values2 = {}
for id in expressed_values:
try: symbolID = gene_to_symbol_db[id][0]
except Exception: symbolID = id
if id not in cell_cycle_id_list and symbolID not in cell_cycle_id_list:
expressed_values2[id]=expressed_values[id]
print len(expressed_values)-len(expressed_values2),'cell-cycle associated genes removed for cluster discovery'
expressed_values = expressed_values2
if reportOnly==False:
print 'amplifyGenes:',amplifyGenes
### Write out filtered list to amplify and to filtered.YourExperiment.txt
filtered_file = export.findParentDir(expFile)+'/amplify/'+export.findFilename(expFile)
groups_file = string.replace(expFile,'exp.','groups.')
groups_filtered_file = string.replace(filtered_file,'exp.','groups.')
groups_file = string.replace(groups_file,'-steady-state','')
groups_filtered_file = string.replace(groups_filtered_file,'-steady-state','')
if reportOnly==False:
try: export.customFileCopy(groups_file,groups_filtered_file) ### if present copy over
except Exception: pass
writeFilteredFile(filtered_file,platform,headers,{},expressed_values,[])
filtered_file_new = string.replace(expFile,'exp.','filteredExp.')
try: export.customFileCopy(filtered_file,filtered_file_new) ### if present copy over
except Exception: pass
else:
filtered_file = writeFilteredFileReimport(expFile,platform,headers,expressed_values) ### expressed_values just contains the UID
print_out = '%d genes, differentially expressed %d fold for at least %d samples' % (len(expressed_values), fold, samplesDiffering*2)
return print_out, filtered_file
if len(expressed_values)<1400 and column_method == 'hopach':
row_method = 'hopach'; row_metric = 'correlation'
else:
row_method = 'weighted'; row_metric = 'cosine'
if amplifyGenes:
transpose = parameters
try:
if len(parameters.GeneSelection())>0:
parameters.setGeneSelection(parameters.GeneSelection()+' amplify')
print 'Finding correlated genes to the input geneset(s)...'
else:
print 'Finding intra-correlated genes from the input geneset(s)...'
parameters.setGeneSelection(parameters.GeneSelection()+' IntraCorrelatedOnly amplify')
except Exception:
parameters.setGeneSelection(parameters.GeneSelection()+' IntraCorrelatedOnly amplify')
print 'Finding intra-correlated genes from the input geneset(s)...'
if column_method != 'hopach': row_method = 'average' ### needed due to PC errors
graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
#return graphic_links
from visualization_scripts import clustering
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(graphic_links[-1][-1][:-4]+'.txt')
headers = ['UID']+column_header
expressed_values2={}
for i in row_header: ### Filter the expressed values for the intra-correlated queried gene set and replace
try: expressed_values2[i]=expressed_values[i]
except Exception:
try:
e = symbol_to_gene[i][0]
expressed_values2[e]=expressed_values[e]
except Exception:
pass
expressed_values = expressed_values2
print 'Looking for common gene expression profiles for class assignment...',
begin_time = time.time()
useNumpyCorr=True
negative_rho = rho_cutoff*-1
#results_file = string.replace(expFile[:-4]+'-CORRELATED-FEATURES.txt','exp.','/SamplePrediction/')
#eo = export.ExportFile(results_file[:-4]+'-genes.txt')
if useNumpyCorr:
row_ids=[]
x = []
for id in expressed_values:
row_ids.append(id)
x.append(expressed_values[id])
#if id== 'Bcl2l11': print expressed_values[id];sys.exit()
D1 = numpy.corrcoef(x)
print 'initial correlations obtained'
i=0
correlated_genes={}
if 'exons' == platform or 'PSI' == platform:
for score_ls in D1:
proceed = True
correlated = []
geneID = row_ids[i]
refgene = string.split(geneID,':')[0]
k=0
if excludeGuides!=None:
if geneID in excludeGuides: ### skip this main event
proceed=False
continue
for v in score_ls:
if v>rho_cutoff:# or v<negative_rho:
if refgene not in row_ids[k]:
correlated.append((v,row_ids[k]))
if excludeGuides!=None:
if row_ids[k] in excludeGuides: ### skip this main event
proceed=False
break
k+=1
correlated.sort()
if LegacyMode == False:
correlated.reverse()
if proceed:
correlated = map(lambda x:x[1],correlated)
correlated_genes[geneID] = correlated
i+=1
else:
for score_ls in D1:
correlated = []
geneID = row_ids[i]
k=0; temp=[]
for v in score_ls:
if v>rho_cutoff:# or v<negative_rho:
#scores.append((v,row_ids[k]))
correlated.append((v,row_ids[k]))
#temp.append((geneID,row_ids[k],str(v)))
k+=1
correlated.sort()
if LegacyMode == False:
correlated.reverse()
correlated = map(lambda x:x[1],correlated)
if len(correlated)>0:
correlated_genes[geneID] = correlated
#for (a,b,c) in temp: eo.write(a+'\t'+b+'\t'+c+'\n')
i+=1
else:
### Find common patterns now
performAllPairwiseComparisons = True
if performAllPairwiseComparisons:
correlated_genes = intraCorrelation(expressed_values,mlp)
print len(correlated_genes), 'highly correlated genes found for downstream clustering.'
else: correlated_genes={}
atleast_10={}
if len(correlated_genes)<70: connections = 0
elif len(correlated_genes)<110: connections = 4
else: connections = 5
numb_corr=[]
for i in correlated_genes:
if len(correlated_genes[i])>connections:
numb_corr.append([len(correlated_genes[i]),i])
atleast_10[i]=correlated_genes[i] ### if atleast 10 genes apart of this pattern
x=0
for k in correlated_genes[i]:
if x<30: ### cap it at 30
try: atleast_10[k]=correlated_genes[k] ### add all correlated keys and values
except Exception: pass
elif k not in atleast_10:
ADT_status = check_for_ADT(k)
if ADT_status:
try: atleast_10[k]=correlated_genes[k] ### add all correlated keys and values
except Exception: pass
x+=1
if len(atleast_10)<30:
print 'Initial correlated set too small, getting anything correlated'
for i in correlated_genes:
if len(correlated_genes[i])>0:
numb_corr.append([len(correlated_genes[i]),i])
try: atleast_10[i]=correlated_genes[i] ### if atleast 10 genes apart of this pattern
except Exception: pass
for k in correlated_genes[i]:
try: atleast_10[k]=correlated_genes[k] ### add all correlated keys and values
except Exception: pass
if len(atleast_10) == 0:
atleast_10 = expressed_values
#eo.close()
print len(atleast_10), 'genes correlated to multiple other members (initial filtering)'
### go through the list from the most linked to the least linked genes, only reported the most linked partners
if len(atleast_10)>5000:
print '\n'
try: return print_out,atleast_10
except: return [],atleast_10
removeOutlierDrivenCorrelations=True
exclude_corr=[]
numb_corr.sort(); numb_corr.reverse()
numb_corr2=[]
#print len(numb_corr)
if removeOutlierDrivenCorrelations and samplesDiffering != 1:
for key in numb_corr: ### key gene
associations,gene = key
temp_corr_matrix_db={}; rows=[]; temp_corr_matrix=[]
gene_exp_vals = list(expressed_values[gene]) ### copy the list
max_index = gene_exp_vals.index(max(gene_exp_vals))
del gene_exp_vals[max_index]
#temp_corr_matrix.append(exp_vals); rows.append(gene)
#if 'ENSG00000016082' in correlated_genes[gene] or 'ENSG00000016082' == gene: print gene_to_symbol_db[gene],associations
if gene not in exclude_corr:
#print len(correlated_genes[gene])
for k in correlated_genes[gene]:
exp_vals = list(expressed_values[k]) ### copy the list
#print exp_vals
del exp_vals[max_index]
#temp_corr_matrix.append(exp_vals); rows.append(gene)
#print exp_vals,'\n'
temp_corr_matrix_db[k]=exp_vals
temp_corr_matrix.append(exp_vals); rows.append(gene)
correlated_hits = pearsonCorrelations(gene_exp_vals,temp_corr_matrix_db)
try: avg_corr = numpyCorrelationMatrix(temp_corr_matrix,rows,gene)
except Exception: avg_corr = 0
#if gene_to_symbol_db[gene][0] == 'ISL1' or gene_to_symbol_db[gene][0] == 'CD10' or gene_to_symbol_db[gene][0] == 'POU3F2':
if len(correlated_hits)>0:
if LegacyMode:
if (float(len(correlated_hits))+1)/len(correlated_genes[gene])<0.5 or avg_corr<rho_cutoff: ### compare to the below
pass
else:
numb_corr2.append([len(correlated_hits),gene])
else:
if (float(len(correlated_hits))+1)/len(correlated_genes[gene])<0.5 or avg_corr<(rho_cutoff-0.1):
#exclude_corr.append(key)
#if gene == 'XXX': print len(correlated_hits),len(correlated_genes[gene]), avg_corr, rho_cutoff-0.1
pass
else:
numb_corr2.append([len(correlated_hits),gene])
#print (float(len(correlated_hits))+1)/len(correlated_genes[gene]), len(correlated_genes[gene]), key
numb_corr = numb_corr2
numb_corr.sort(); numb_corr.reverse()
#print len(numb_corr)
exclude_corr={}; new_filtered_set={}
limit=0
for key in numb_corr: ### key gene
associations,gene = key
#if 'ENSG00000016082' in correlated_genes[gene] or 'ENSG00000016082' == gene: print gene_to_symbol_db[gene],associations
if gene not in exclude_corr:
for k in correlated_genes[gene]:
exclude_corr[k]=[]
new_filtered_set[k]=[]
new_filtered_set[gene]=[]
limit+=1
#print key
#if limit==1: break
atleast_10 = new_filtered_set
addMultipleDrivers=True
if len(guide_genes)>0 and addMultipleDrivers: ### Artificially weight the correlated genes with known biological driverse
for gene in guide_genes:
y=1
while y<2:
if y==1:
try: atleast_10[gene]=expressed_values[gene]
except Exception: break
else:
try: atleast_10[gene+'-'+str(y)]=expressed_values[gene]
except Exception: break
expressed_values[gene+'-'+str(y)]=expressed_values[gene] ### Add this new ID to the database
#print gene+'-'+str(y)
y+=1
#atleast_10 = expressed_values
results_file = string.replace(expFile[:-4]+'-CORRELATED-FEATURES.txt','exp.','/SamplePrediction/')
writeFilteredFile(results_file,platform,headers,gene_to_symbol_db,expressed_values,atleast_10)
print len(atleast_10),'final correlated genes'
end_time = time.time()
print 'Initial clustering completed in',int(end_time-begin_time),'seconds'
results_file = string.replace(expFile[:-4]+'-CORRELATED-FEATURES.txt','exp.','/SamplePrediction/')
if len(atleast_10)<1200 and column_method == 'hopach':
row_method = 'hopach'; row_metric = 'correlation'
else:
if LegacyMode:
row_method = 'average'; row_metric = 'euclidean'
else:
row_method = 'weighted'; row_metric = 'cosine'
#print row_method, row_metric
correlateByArrayDirectly = False
if correlateByArrayDirectly:
from visualization_scripts import clustering
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(results_file)
new_column_header = map(lambda x: int(x[5:]),column_header)
matrix = [new_column_header]+matrix
matrix = zip(*matrix) ### transpose
exp_sample_db={}
for sample_data in matrix:
exp_sample_db[sample_data[0]] = sample_data[1:]
correlated_arrays = intraCorrelation(exp_sample_db,mpl)
print len(correlated_arrays), 'highly correlated arrays from gene subsets.'
mimum_corr_arrays={}
for i in correlated_arrays:
if len(correlated_arrays[i])>1:
linked_lists=correlated_arrays[i]+[i]
for k in correlated_arrays[i]:
linked_lists+=correlated_arrays[k]
linked_lists = unique.unique(linked_lists)
linked_lists.sort()
# print len(linked_lists), linked_lists
else:
try:
from visualization_scripts import clustering
if platform == 'exons': color_gradient = 'yellow_black_blue'
transpose = False
if column_method != 'hopach': row_method = 'average' ### needed due to PC errors (possibly outside of LegacyMode)
graphic_links = clustering.runHCexplicit(results_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
if len(graphic_links)==0:
graphic_links = clustering.runHCexplicit(results_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
cluster_file = string.replace(graphic_links[0][1],'.png','.txt')
except Exception: pass
#exportGroupsFromClusters(cluster_file,expFile,platform)
#"""
#filtered_file = export.findParentDir(expFile)+'/amplify/'+export.findFilename(expFile)
#graphic_links = [(1,'/Users/saljh8/Desktop/Grimes/KashishNormalization/test/ExpressionInput/SamplePrediction/DataPlots/Clustering-CombinedSingleCell_March_15_2015-CORRELATED-FEATURES-hierarchical_cosine_euclidean.txt')]
try: graphic_links,new_results_file = correlateClusteredGenes(platform,graphic_links[-1][-1][:-4]+'.txt',numSamplesClustered=samplesDiffering,excludeCellCycle=excludeCellCycle,graphics=graphic_links,ColumnMethod=column_method)
except Exception: print traceback.format_exc()
row_metric = 'correlation'; row_method = 'hopach'
#column_metric = 'cosine'
#if LegacyMode: column_method = 'hopach'
cellCycleRemove1=[]; cellCycleRemove2=[]
try:
newDriverGenes1, cellCycleRemove1 = correlateClusteredGenes(platform,graphic_links[-1][-1][:-4]+'.txt',stringency='strict',numSamplesClustered=samplesDiffering,excludeCellCycle=excludeCellCycle,ColumnMethod=column_method)
newDriverGenes1_str = 'Guide1 '+string.join(newDriverGenes1.keys(),' ')+' amplify positive'
parameters.setGeneSelection(newDriverGenes1_str) ### force correlation to these targetGenes
parameters.setGeneSet('None Selected') ### silence this
parameters.setPathwaySelect('None Selected')
if column_method != 'hopach': row_method = 'average' ### needed due to PC errors
graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, parameters, display=False, Normalize=True)
newDriverGenes2, cellCycleRemove2 = correlateClusteredGenes(platform,graphic_links[-1][-1][:-4]+'.txt',stringency='strict',numSamplesClustered=samplesDiffering,excludeCellCycle=excludeCellCycle,ColumnMethod=column_method)
newDriverGenes2_str = 'Guide2 '+string.join(newDriverGenes2.keys(),' ')+' amplify positive'
parameters.setGeneSelection(newDriverGenes2_str) ### force correlation to these targetGenes
parameters.setGeneSet('None Selected') ### silence this
parameters.setPathwaySelect('None Selected')
graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, parameters, display=False, Normalize=True)
newDriverGenes3 = unique.unique(newDriverGenes1.keys()+newDriverGenes2.keys())
cellCycleRemove=cellCycleRemove1+cellCycleRemove2 ### It is possible for a cell cycle guide-gene to be reported in both guide1 and 2, but only as cell cycle associated in one of them
newDriverGenes3_filtered=[]
for i in newDriverGenes3:
if not i in cellCycleRemove:
newDriverGenes3_filtered.append(i)
newDriverGenes3_str = 'Guide3 '+string.join(newDriverGenes3_filtered,' ')+' amplify positive'
parameters.setGeneSelection(newDriverGenes3_str)
try:
parameters.setClusterGOElite('BioMarkers')
"""
if species == 'Mm' or species == 'Hs' or species == 'Rn':
parameters.setClusterGOElite('BioMarkers')
else:
parameters.setClusterGOElite('GeneOntology')
"""
except Exception, e:
print e
graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, parameters, display=False, Normalize=True)
except Exception:
print traceback.format_exc()
try: copyICGSfiles(expFile,graphic_links)
except Exception: pass
return graphic_links,len(atleast_10)
def copyICGSfiles(expFile,graphic_links):
if 'ExpressionInput' in expFile:
root_dir = string.split(expFile,'ExpressionInput')[0]
else:
root_dir = string.split(expFile,'AltResults')[0]
destination_folder = root_dir+'/ICGS'
try: os.mkdir(destination_folder)
except Exception: pass
for (order,png) in graphic_links:
file = export.findFilename(png)
txt = string.replace(file,'.png','.txt')
pdf = string.replace(file,'.png','.pdf')
dest_png = destination_folder+'/'+file
dest_txt = destination_folder+'/'+txt
dest_pdf = destination_folder+'/'+pdf
shutil.copy(png, dest_png)
shutil.copy(png[:-4]+'.txt', dest_txt)
shutil.copy(png[:-4]+'.pdf', dest_pdf)
def pearsonCorrelations(ref_gene_exp,exp_value_db):
correlated=[]
for gene in exp_value_db:
rho,p = stats.pearsonr(ref_gene_exp,exp_value_db[gene])
if rho>rho_cutoff or rho<(rho_cutoff*-1):
if rho!= 1:
correlated.append(gene)
#print len(exp_value_db),len(correlated);sys.exit()
return correlated
def numpyCorrelationMatrix(x,rows,gene):
D1 = numpy.corrcoef(x)
gene_correlations={}
i=0
scores = []
for score_ls in D1:
for v in score_ls:
scores.append(v)
return numpy.average(scores)
def numpyCorrelationMatrixCount(x,rows,cutoff=0.4,geneTypeReport=None):
### Find which genes are most correlated
D1 = numpy.corrcoef(x)
gene_correlation_counts={}
i=0
for score_ls in D1:
correlated_genes=[]
geneID = rows[i]
k=0; genes_to_report=[]
for rho in score_ls:
if rho>cutoff:
correlated_genes.append(rows[k])
if rows[k] in geneTypeReport:
genes_to_report.append(rows[k])
k+=1
gene_correlation_counts[geneID]=len(correlated_genes),genes_to_report
i+=1
return gene_correlation_counts
def numpyCorrelationMatrixGene(x,rows,gene):
D1 = numpy.corrcoef(x)
gene_correlations={}
i=0
for score_ls in D1:
scores = []
geneID = rows[i]
k=0
for v in score_ls:
scores.append((v,rows[k]))
k+=1
scores.sort()
gene_correlations[geneID] = scores
i+=1
correlated_genes={}
rho_values = map(lambda (r,g): r,gene_correlations[gene])
genes = map(lambda (r,g): g,gene_correlations[gene])
s1 = bisect.bisect_right(rho_values,rho_cutoff)
s2 = bisect.bisect_left(rho_values,-1*rho_cutoff)
correlated = genes[:s2] ### for the right bisect, remove self correlations with -1
correlated = genes[s1:] ### for the left bisect, remove self correlations with -1
#print len(rows), len(correlated);sys.exit()
return len(correlated)/len(rows)
def numpyCorrelationMatrixGeneAlt(x,rows,genes,gene_to_symbol,rho_cutoff):
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
D1 = numpy.ma.corrcoef(x)
i=0
gene_correlations={}
for score_ls in D1:
scores = []
try: symbol = gene_to_symbol[rows[i]][0]
except Exception: symbol = '$'
if rows[i] in genes or symbol in genes:
k=0
for v in score_ls:
if str(v)!='nan':
if v > rho_cutoff:
uid = rows[k]
if uid in gene_to_symbol: uid = gene_to_symbol[uid][0]
scores.append((v,uid))
k+=1
scores.sort()
scores.reverse()
scores = map(lambda x: x[1], scores[:140]) ### grab the top 140 correlated gene symbols only
if len(symbol)==1: symbol = rows[i]
gene_correlations[symbol] = scores
i+=1
return gene_correlations
def genericRowIDImport(filename):
id_list=[]
for line in open(filename,'rU').xreadlines():
uid = string.split(line,'\t')[0]
if ' ' in uid:
for id in string.split(uid,' '):
id_list.append(id)
else:
id_list.append(uid)
return id_list
def writeFilteredFileReimport(expFile,platform,headers,expressed_values):
filtered_file=expFile[:-4]+'-VarGenes.txt'
groups_file = string.replace(expFile,'exp.','groups.')
filtered_groups = string.replace(filtered_file,'exp.','groups.')
try: shutil.copy(groups_file,filtered_groups)
except: pass
eo = export.ExportFile(filtered_file)
eo.write(headers)
for line in open(expFile,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
uid = t[0]
if uid in expressed_values:
if platform=='RNASeq': ### set to RNASeq when non-log2 data detected
values = t[1:]
try: values = map(lambda x: math.log(float(x)+1,2),values)
except Exception:
if 'NA' in values:
values = [0 if x=='NA' else x for x in values] ### Replace NAs
values = map(lambda x: math.log(x+1,2),values)
elif '' in values:
values = [0 if x=='' else x for x in values] ### Replace NAs
values = map(lambda x: math.log(x+1,2),values)
values = map(str,values)
eo.write(string.join([uid]+values,'\t')+'\n')
else:
eo.write(line)
eo.close()
return filtered_file
def writeFilteredFile(results_file,platform,headers,gene_to_symbol_db,expressed_values,atleast_10,excludeGenes=[]):
eo = export.ExportFile(results_file)
try: headers = string.replace(headers,'row_clusters-flat','UID')
except Exception:
headers = string.join(headers,'\t')+'\n'
headers = string.replace(headers,'row_clusters-flat','UID')
eo.write(headers)
keep=[]; sort_genes=False
e=0
if len(atleast_10)==0:
atleast_10 = expressed_values
sort_genes = True
for i in atleast_10:
if i in gene_to_symbol_db:
symbol = gene_to_symbol_db[i][0]
else: symbol = i
if i not in excludeGenes and symbol not in excludeGenes:
if i not in keep:
keep.append((symbol,i))
if sort_genes:
keep.sort(); keep.reverse()
for (symbol,i) in keep:
"""
if platform == 'RNASeq':
values = map(lambda x: logTransform(x), expressed_values[i])
else:
"""
values = map(str,expressed_values[i])
eo.write(string.join([symbol]+values,'\t')+'\n')
e+=1
eo.close()
def remoteGetDriverGenes(Species,platform,results_file,numSamplesClustered=3,excludeCellCycle=False,ColumnMethod='hopach'):
global species
species = Species
guideGenes, cellCycleRemove = correlateClusteredGenes(platform,results_file,stringency='strict',excludeCellCycle=excludeCellCycle,ColumnMethod=ColumnMethod)
guideGenes = string.join(guideGenes.keys(),' ')+' amplify positive'
return guideGenes
def correlateClusteredGenes(platform,results_file,stringency='medium',numSamplesClustered=3,
excludeCellCycle=False,graphics=[],ColumnMethod='hopach',rhoCutOff=0.2, transpose=False,
includeMoreCells=False):
if numSamplesClustered<1: numSamplesClustered=1
### Get all highly variably but low complexity differences, typically one or two samples that are really different
if stringency == 'medium':
new_results_file = string.replace(results_file,'.txt','-filtered.txt')
new_results_file = string.replace(new_results_file,'.cdt','-filtered.txt')
eo = export.ExportFile(new_results_file)
medVarHighComplexity=[]; medVarLowComplexity=[]; highVarHighComplexity=[]; highVarLowComplexity=[]
if transpose==False or includeMoreCells:
medVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=3,hits_to_report=6,transpose=transpose)
medVarHighComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.1,hits_cutoff=3,hits_to_report=6,transpose=transpose) #hits_cutoff=6
highVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.5,hits_cutoff=1,hits_to_report=4,transpose=transpose)
highVarHighComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.2,hits_cutoff=1,hits_to_report=6,filter=True,numSamplesClustered=numSamplesClustered,transpose=transpose)
else:
highVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.5,hits_cutoff=1,hits_to_report=4,transpose=transpose)
#combined_results = dict(medVarLowComplexity.items() + medVarLowComplexity.items() + highVarLowComplexity.items() + highVarHighComplexity.items())
combined_results={}
for i in medVarLowComplexity: combined_results[i]=[]
for i in medVarHighComplexity: combined_results[i]=[]
for i in highVarLowComplexity: combined_results[i]=[]
for i in highVarHighComplexity: combined_results[i]=[]
#combined_results = highVarHighComplexity
if stringency == 'strict':
medVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=4,hits_to_report=50,filter=True,numSamplesClustered=numSamplesClustered)
medVarHighComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.1,hits_cutoff=4,hits_to_report=50,filter=True,numSamplesClustered=numSamplesClustered) #hits_cutoff=6
highVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.5,hits_cutoff=3,hits_to_report=50,filter=True,numSamplesClustered=numSamplesClustered)
highVarHighComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=3,hits_to_report=50,filter=True,numSamplesClustered=numSamplesClustered)
#combined_results = dict(medVarLowComplexity.items() + medVarLowComplexity.items() + highVarLowComplexity.items() + highVarHighComplexity.items())
combined_results={}
for i in medVarLowComplexity: combined_results[i]=[]
for i in medVarHighComplexity: combined_results[i]=[]
for i in highVarLowComplexity: combined_results[i]=[]
for i in highVarHighComplexity: combined_results[i]=[]
guideGenes, addition_cell_cycle_associated = correlateClusteredGenesParameters(results_file,rho_cutoff=rhoCutOff,hits_cutoff=0,hits_to_report=1,geneFilter=combined_results,excludeCellCycle=excludeCellCycle)
if guideGenes == 'TooFewBlocks':
guideGenes, addition_cell_cycle_associated = correlateClusteredGenesParameters(results_file,rho_cutoff=rhoCutOff+0.1,hits_cutoff=0,hits_to_report=1,geneFilter=combined_results,excludeCellCycle=excludeCellCycle)
if guideGenes == 'TooFewBlocks':
guideGenes, addition_cell_cycle_associated = correlateClusteredGenesParameters(results_file,rho_cutoff=rhoCutOff+0.2,hits_cutoff=0,hits_to_report=1,geneFilter=combined_results,excludeCellCycle=excludeCellCycle,forceOutput=True)
if len(guideGenes)>200:
print 'Too many guides selected (>200)... performing more stringent filtering...'
guideGenes, addition_cell_cycle_associated = correlateClusteredGenesParameters(results_file,rho_cutoff=0.1,hits_cutoff=0,hits_to_report=1,geneFilter=combined_results,excludeCellCycle=excludeCellCycle,restrictTFs=True)
return guideGenes, addition_cell_cycle_associated
#B4galt6, Prom1
for tuple_ls in combined_results:
data_length = len(tuple_ls);break
if data_length == len(column_header):
eo.write(string.join(column_header,'\t')+'\n')
else:
eo.write(string.join(['UID']+column_header,'\t')+'\n')
#combined_results = highVarHighComplexity
for tuple_ls in combined_results:
eo.write(string.join(list(tuple_ls),'\t')+'\n')
eo.close()
cluster = True
if cluster == True and transpose==False:
from visualization_scripts import clustering
if ColumnMethod == 'hopach':
row_method = 'hopach'
column_method = 'hopach'
else:
column_method = ColumnMethod
row_method = 'average'
row_metric = 'correlation'
column_metric = 'cosine'
color_gradient = 'yellow_black_blue'
if platform == 'exons': color_gradient = 'yellow_black_blue'
transpose = False
try:
len(guide_genes)
except Exception:
guide_genes = []
graphics = clustering.runHCexplicit(new_results_file, graphics, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
cluster_file = string.replace(graphics[0][1],'.png','.txt')
#exportGroupsFromClusters(cluster_file,expFile,platform)
return graphics, new_results_file
def exportReDefinedClusterBlocks(results_file,block_db,rho_cutoff):
### Re-import the matrix to get the column cluster IDs
matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters = clustering.remoteImportData(results_file)
new_block_db = {}
centroid_blocks=[]
centroids = []
for block in block_db:
if len(block_db[block])>3:
new_block_db[block] = block_db[block] ### Keep track of the row_header indexes associated with each blcok
data = map(lambda x: matrix[x],block_db[block])
### Compute an expression centroid from the block (cluster)
centroid = [float(sum(col))/len(col) for col in zip(*data)]
centroids.append(centroid)
centroid_blocks.append(block)
### Compare block centroids
D1 = numpy.corrcoef(centroids)
i=0
correlated_blocks=[]
for score_ls in D1:
scores = []
block = centroid_blocks[i]
k=0
for v in score_ls:
if str(v)!='nan' and v>0.6:
if block !=centroid_blocks[k]:
blocks = [block,centroid_blocks[k]]
blocks.sort()
if blocks not in correlated_blocks:
correlated_blocks.append(blocks)
k+=1
i+=1
newBlock=0
existing=[]
updated_blocks={}
correlated_blocks.sort()
print correlated_blocks
### Build a tree of related blocks (based on the code in junctionGraph)
for (block1,block2) in correlated_blocks:
if block1 not in existing and block2 not in existing:
newBlock=newBlock+1
updated_blocks[newBlock]=[block1,]
updated_blocks[newBlock].append(block2)
existing.append(block1)
existing.append(block2)
elif block1 in existing and block2 not in existing:
for i in updated_blocks:
if block1 in updated_blocks[i]:
updated_blocks[i].append(block2)
existing.append(block2)
elif block2 in existing and block1 not in existing:
for i in updated_blocks:
if block2 in updated_blocks[i]:
updated_blocks[i].append(block1)
existing.append(block1)
elif block1 in existing and block2 in existing:
for i in updated_blocks:
if block1 in updated_blocks[i]:
b1=i
if block2 in updated_blocks[i]:
b2=i
if b1!=b2:
for b in updated_blocks[b2]:
if b not in updated_blocks[b1]:
updated_blocks[b1].append(b)
del updated_blocks[b2]
### Add blocks not correlated to other blocks (not in correlated_blocks)
#print len(existing),len(centroid_blocks)
print updated_blocks
for block in centroid_blocks:
if block not in existing:
newBlock+=1
updated_blocks[newBlock]=[block]
import collections
row_order = collections.OrderedDict()
for newBlock in updated_blocks:
events_in_block=0
for block in updated_blocks[newBlock]:
for i in new_block_db[block]:
events_in_block+=1
if events_in_block>5:
for block in updated_blocks[newBlock]:
for i in new_block_db[block]:
row_order[i] = newBlock ### i is a row_header index - row_header[i] is a UID
#if newBlock==3:
#if row_header[i]=='TAF2&ENSG00000064313&E9.1-I9.1_120807184__ENSG00000064313&E9.1-E10.1':
#print row_header[i]
print updated_blocks
### Non-clustered block results - Typically not used by good to refer back to when testing
original_block_order = collections.OrderedDict()
for block in new_block_db:
for i in new_block_db[block]:
original_block_order[i]=block
#row_order = original_block_order
### Export the results
row_header.reverse() ### Reverse order is the default
priorColumnClusters = map(str,priorColumnClusters)
new_results_file = results_file[:-4]+'-BlockIDs.txt'
eo = export.ExportFile(new_results_file)
eo.write(string.join(['UID','row_clusters-flat']+column_header,'\t')+'\n')
eo.write(string.join(['column_clusters-flat','']+priorColumnClusters,'\t')+'\n')
for i in row_order:
cluster_number = str(row_order[i])
uid = row_header[i]
values = map(str,matrix[i])
eo.write(string.join([uid,cluster_number]+values,'\t')+'\n')
eo.close()
print 'Filtered, grouped expression clusters exported to:',new_results_file
def correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=4,hits_to_report=5,
filter=False,geneFilter=None,numSamplesClustered=3,excludeCellCycle=False,restrictTFs=False,
forceOutput=False,ReDefinedClusterBlocks=False,transpose=False):
from visualization_scripts import clustering
addition_cell_cycle_associated=[]
if geneFilter != None:
geneFilter_db={}
for i in geneFilter:
geneFilter_db[i[0]]=[]
geneFilter=geneFilter_db
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(results_file,geneFilter=geneFilter)
if transpose: ### If performing reduce cluster heterogeneity on cells rather than on genes
#print 'Transposing matrix'
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
Platform = None
ADTs=[]
for i in row_header:
if 'ENS' in i and '-' in i and ':' in i: Platform = 'exons'
else:
ADT_status = check_for_ADT(i)
if ADT_status: ADTs.append(i)
#print ADTs
#print hits_to_report
if hits_to_report == 1:
### Select the best gene using correlation counts and TFs
try:
from import_scripts import OBO_import; import ExpressionBuilder
gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol_db)
try: TFs = importGeneSets('Biotypes',filterType='transcription regulator',geneAnnotations=gene_to_symbol_db)
except Exception: TFs = importGeneSets('BioTypes',filterType='transcription regulator',geneAnnotations=gene_to_symbol_db)
if excludeCellCycle == True or excludeCellCycle == 'strict':
try: cell_cycle = importGeneSets('KEGG',filterType='Cell cycle:',geneAnnotations=gene_to_symbol_db)
except Exception:
cell_cycle = {}
try: cell_cycle_go = importGeneSets('GeneOntology',filterType='GO:0022402',geneAnnotations=gene_to_symbol_db)
except Exception: cell_cycle_go={}
if len(cell_cycle_go)<10:
try: cell_cycle_go = importGeneSets('GeneOntology',filterType='GO:0007049',geneAnnotations=gene_to_symbol_db)
except: pass
for i in cell_cycle_go:
cell_cycle[i]=[]
print len(cell_cycle),'cell cycle genes being considered.'
else:
cell_cycle={}
except Exception:
print traceback.format_exc()
symbol_to_gene={}; TFs={}; cell_cycle={}
gene_corr_counts = numpyCorrelationMatrixCount(matrix,row_header,cutoff=0.4,geneTypeReport=TFs)
#try: column_header = map(lambda x: string.split(x,':')[1],column_header[1:])
#except Exception: column_header = column_header[1:]
i=0
block=0
if ReDefinedClusterBlocks:
import collections
block_db=collections.OrderedDict() ### seems benign but could alter legacy results
else:
block_db={}
for row in matrix:
if i!=0:
rho,p = stats.pearsonr(row,matrix[i-1]) ### correlate to the last ordered row
#if row_header[i] == 'Pax6': print [block],row_header[i-1],rho,rho_cutoff
"""
try:
if row_header[i] in guide_genes: print row_header[i], rho
if row_header[i-1] in guide_genes: print row_header[i-1], rho
if row_header[i+1] in guide_genes: print row_header[i+1], rho
except Exception:
pass
"""
#if hits_to_report == 1: print [block],row_header[i], row_header[i-1],rho,rho_cutoff
#print rho
if rho>0.95:
pass ### don't store this
elif rho>rho_cutoff:
try:
block_db[block].append(i) ### store the row index
except Exception:
block_db[block] = [i] ### store the row index
else:
block+=1
block_db[block] = [i] ### store the row index
else:
block_db[block] = [i] ### store the row index
i+=1
if ReDefinedClusterBlocks:
### Produces a filtered-down and centroid organized heatmap text file
exportReDefinedClusterBlocks(results_file,block_db,rho_cutoff)
if hits_to_report == 1:
if len(block_db)<4 and forceOutput==False:
return 'TooFewBlocks', None
guideGenes={}
### Select the top TFs or non-TFs with the most gene correlations
for b in block_db:
corr_counts_gene = []; cell_cycle_count=[]
#print len(block_db), b, map(lambda i: row_header[i],block_db[b])
for (gene,i) in map(lambda i: (row_header[i],i),block_db[b]):
corr_counts_gene.append((len(gene_corr_counts[gene][1]),gene_corr_counts[gene][0],gene))
if gene in cell_cycle:
cell_cycle_count.append(gene)
corr_counts_gene.sort(); tfs=[]
#print b, corr_counts_gene, '***',len(cell_cycle_count)
if (len(cell_cycle_count)>1) or (len(corr_counts_gene)<4 and (len(cell_cycle_count)>0)): pass
else:
tf_count=0
for (r,t, gene) in corr_counts_gene:
if gene in TFs:
if gene not in cell_cycle:
if restrictTFs==True and tf_count==0: pass
else:
guideGenes[gene]=[]
tf_count+=1
if len(tfs)==0:
gene = corr_counts_gene[-1][-1]
if gene in cell_cycle and LegacyMode: pass
else:
guideGenes[gene]=[]
#block_db[b]= [corr_counts_gene[-1][-1]] ### save just the selected gene indexes
### Additional filter to remove guides that will bring in cell cycle genes (the more guides the more likely)
if excludeCellCycle == 'strict':
#print 'guides',len(guideGenes)
guideCorrelated = numpyCorrelationMatrixGeneAlt(matrix,row_header,guideGenes,gene_to_symbol_db,rho_cutoff)
guideGenes={}
for gene in guideCorrelated:
cell_cycle_count=[]
for corr_gene in guideCorrelated[gene]:
if corr_gene in cell_cycle: cell_cycle_count.append(corr_gene)
#print gene, len(cell_cycle_count),len(guideCorrelated[gene])
if (float(len(cell_cycle_count))/len(guideCorrelated[gene]))>.15 or (len(guideCorrelated[gene])<4 and (len(cell_cycle_count)>0)):
print gene, cell_cycle_count
addition_cell_cycle_associated.append(gene)
pass
else:
guideGenes[gene]=[]
print 'additional Cell Cycle guide genes removed:',addition_cell_cycle_associated
for ADT in ADTs: guideGenes[ADT]=[]
print len(guideGenes), 'novel guide genes discovered:', guideGenes.keys()
return guideGenes,addition_cell_cycle_associated
def greaterThan(x,results_file,numSamplesClustered):
if 'alt_junctions' not in results_file and Platform == None:
if x>(numSamplesClustered-1): return 1
else: return 0
else:
return 1
max_block_size=0
### Sometimes the hits_cutoff is too stringent so take the largest size instead
for block in block_db:
indexes = len(block_db[block])
if indexes>max_block_size: max_block_size=indexes
max_block_size-=1
retained_ids={}; final_rows = {}
for block in block_db:
indexes = block_db[block]
#print [block], len(indexes),hits_cutoff,max_block_size
if len(indexes)>hits_cutoff or len(indexes)>max_block_size: ###Increasing this helps get rid of homogenous clusters of little significance
#if statistics.avg(matrix[indexes[0]][1:]) < -2: print statistics.avg(matrix[indexes[0]][1:]), len(indexes)
gene_names = map(lambda i: row_header[i], indexes)
#if 'Pax6' in gene_names or 'WNT8A' in gene_names: print '******',hits_to_report, gene_names
indexes = indexes[:hits_to_report]
if filter:
new_indexes = []
for index in indexes:
vs = list(matrix[index])
a = map(lambda x: greaterThan(x,results_file,numSamplesClustered),vs)
b=[1]*numSamplesClustered
c = [(i, i+len(b)) for i in range(len(a)) if a[i:i+len(b)] == b]
if len(c)>0: #http://stackoverflow.com/questions/10459493/find-indexes-of-sequence-in-list-in-python
new_indexes.append(index)
"""
vs.sort()
try:
if abs(vs[-5]-vs[5])>6: new_indexes.append(index)
except Exception:
if abs(vs[-1]-vs[1])>6: new_indexes.append(index)"""
indexes = new_indexes
#if block == 1: print map(lambda i:row_header[i],indexes)
#print indexes;sys.exit()
for ls in map(lambda i: [row_header[i]]+map(str,(matrix[i])), indexes):
final_rows[tuple(ls)]=[]
for i in indexes:
retained_ids[row_header[i]]=[]
added_genes=[]
if len(final_rows)==0:
for block in block_db:
indexes = block_db[block]
if len(indexes)>hits_cutoff or len(indexes)>max_block_size:
indexes = indexes[:hits_to_report]
for ls in map(lambda i: [row_header[i]]+map(str,(matrix[i])), indexes):
final_rows[tuple(ls)]=[]
added_genes.append(ls[0])
if len(final_rows)==0:
for block in block_db:
indexes = block_db[block]
for ls in map(lambda i: [row_header[i]]+map(str,(matrix[i])), indexes):
final_rows[tuple(ls)]=[]
added_genes.append(ls[0])
if len(ADTs)>0:
for ADT in ADTs:
if ADT not in added_genes:
ls = [ADT]+matrix[row_header.index(ADT)]
final_rows[tuple(ls)]=[]
#print 'block length:',len(block_db), 'genes retained:',len(retained_ids)
return final_rows, column_header
def exportGroupsFromClusters(cluster_file,expFile,platform,suffix=None):
lineNum=1
for line in open(cluster_file,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if lineNum==1: names = t[2:]; lineNum+=1
elif lineNum==2: clusters = t[2:]; lineNum+=1
else: break
unique_clusters=[] ### Export groups
new_groups_dir = string.replace(expFile,'exp.','groups.')
new_comps_dir = string.replace(expFile,'exp.','comps.')
if suffix != None:
new_groups_dir = new_groups_dir[:-4]+'-'+suffix+'.txt' ###Usually end in ICGS
new_comps_dir = new_comps_dir[:-4]+'-'+suffix+'.txt'
out_obj = export.ExportFile(new_groups_dir)
cluster_number=0
cluster_db={}
for name in names:
cluster = clusters[names.index(name)]
if platform == 'RNASeq':
if 'junction_quantification' not in name and '.bed' not in name:
name = name+'.bed'
elif 'junction_quantification.txt' not in name and '.txt' not in name and '.bed' not in name:
name = name+'.txt'
if ':' in name:
group,name = string.split(name,':')
if group in cluster_db:
clust_num=cluster_db[group]
else:
cluster_number+=1
cluster_db[group] = cluster_number
clust_num = cluster_number
if cluster=='NA': cluster = group
else:
clust_num = cluster
out_obj.write(name+'\t'+str(clust_num)+'\t'+cluster+'\n')
clust_num = str(clust_num)
if clust_num not in unique_clusters: unique_clusters.append(clust_num)
out_obj.close()
comps=[] #Export comps
out_obj = export.ExportFile(new_comps_dir)
""" ### All possible pairwise group comparisons
for c1 in unique_clusters:
for c2 in unique_clusters:
temp=[int(c2),int(c1)]; temp.sort(); temp.reverse()
if c1 != c2 and temp not in comps:
out_obj.write(str(temp[0])+'\t'+str(temp[1])+'\n')
comps.append(temp)
"""
### Simple method comparing each subsequent ordered cluster (HOPACH orders based on relative similarity)
last_cluster = None
for c1 in unique_clusters:
if last_cluster !=None:
out_obj.write(c1+'\t'+last_cluster+'\n')
last_cluster=c1
out_obj.close()
return new_groups_dir
def logTransform(value):
try: v = math.log(value,2)
except Exception: v = math.log(0.001,2)
return str(v)
class MultiCorrelatePatterns():
def __init__(self,expressed_values):
self.expressed_values = expressed_values
def __call__(self,features_to_correlate):
from scipy import stats
correlated_genes={}
for uid in features_to_correlate:
ref_values = self.expressed_values[uid]
for uid2 in self.expressed_values:
values = self.expressed_values[uid2]
rho,p = stats.pearsonr(values,ref_values)
if rho>rho_cutoff or rho<-1*rho_cutoff:
if uid!=uid2 and rho != 1.0:
try: correlated_genes[uid].append(uid2)
except Exception: correlated_genes[uid] = [uid]
return correlated_genes
def parseCountFile(fn,parseFeature,search_exon_db):
novel_exon_db={}; firstLine=True
unique_genes={}
for line in open(fn,'rU').xreadlines():
key = string.split(line,'\t')[0]
#t = string.split(line,'\t')
if firstLine: firstLine = False
else:
#uid, coordinates = string.split(key,'=')
#values = map(lambda x: float(x), t[1:])
#gene = string.split(uid,':')[0]
#if max(values)>5: unique_genes[gene] = []
if '_' in key: ### Only look at novel exons
#ENSG00000112695:I2.1_75953139=chr6:75953139-75953254
uid, coordinates = string.split(key,'=')
gene = string.split(uid,':')[0]
if parseFeature == 'exons':
if '-' not in uid:
chr,coordinates = string.split(coordinates,':') ### Exclude the chromosome
coord1,coord2 = string.split(coordinates,'-')
intron = string.split(uid,'_')[0]
intron = string.split(intron,':')[1]
first = intron+'_'+coord1
second = intron+'_'+coord2
proceed = True
if first in uid: search_uid = second ### if the first ID is already the one looked for, store the second with the exon ID
elif second in uid: search_uid = first
else:
proceed = False
#print uid, first, second; sys.exit()
#example: ENSG00000160785:E2.15_156170151;E2.16_156170178=chr1:156170151-156170178
if proceed:
try: novel_exon_db[gene].append((uid,search_uid))
except Exception: novel_exon_db[gene] = [(uid,search_uid)]
elif '-' in uid and 'I' in uid: ### get junctions
if gene in search_exon_db:
for (u,search_uid) in search_exon_db[gene]:
#if gene == 'ENSG00000137076': print u,search_uid,uid
if search_uid in uid:
novel_exon_db[uid] = u ### Relate the currently examined novel exon ID to the junction not current associated
#if gene == 'ENSG00000137076': print u, uid
#print uid;sys.exit()
#print len(unique_genes); sys.exit()
return novel_exon_db
def getJunctionType(species,fn):
root_dir = string.split(fn,'ExpressionInput')[0]
fn = filepath(root_dir+'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_junctions.txt')
firstLine=True
junction_type_db={}; type_db={}
for line in open(fn,'rU').xreadlines():
t = string.split(line,'\t')
if firstLine: firstLine = False
else:
id=t[0]; junction_type = t[8]
if '-' in id:
if 'trans-splicing' in line:
junction_type = 'trans-splicing'
junction_type_db[id] = junction_type
try: type_db[junction_type]+=1
except Exception: type_db[junction_type]=1
print 'Breakdown of event types'
for type in type_db:
print type, type_db[type]
return junction_type_db
def maxCount(ls):
c=0
for i in ls:
if i>0.5: c+=1
return c
def getHighExpNovelExons(species,fn):
""" Idea - if the ranking of exons based on expression changes from one condition to another, alternative splicing is occuring """
junction_type_db = getJunctionType(species,fn)
### Possible issue detected with novel exon reads: ['ENSG00000121577'] ['119364543'] cardiac
exon_max_exp_db={}; uid_key_db={}; firstLine=True
novel_intronic_junctions = {}
novel_intronic_exons = {}
cutoff = 0.2
read_threshold = 0.5
expressed_junction_types={}
features_to_export={}
exon_coord_db={}
for line in open(fn,'rU').xreadlines():
t = string.split(line,'\t')
if firstLine: firstLine = False
else:
key=t[0]
#ENSG00000112695:I2.1_75953139=chr6:75953139-75953254
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
gene = string.split(uid,':')[0]
values = map(lambda x: float(x), t[1:])
max_read_counts = max(values)
try: exon_max_exp_db[gene].append((max_read_counts,uid))
except Exception: exon_max_exp_db[gene] = [(max_read_counts,uid)]
uid_key_db[uid] = key ### retain the coordinate info
if '-' in uid and (':E' in uid or '-E' in uid):
junction_type = junction_type_db[uid]
if max_read_counts>read_threshold:
samples_expressed = maxCount(values)
if samples_expressed>2:
try: expressed_junction_types[junction_type]+=1
except Exception: expressed_junction_types[junction_type]=1
if junction_type == 'trans-splicing' and'_' not in uid:
try: expressed_junction_types['known transplicing']+=1
except Exception: expressed_junction_types['known transplicing']=1
elif junction_type == 'novel' and '_' not in uid:
try: expressed_junction_types['novel but known sites']+=1
except Exception: expressed_junction_types['novel but known sites']=1
elif junction_type == 'novel' and 'I' not in uid:
try: expressed_junction_types['novel but within 50nt of a known sites']+=1
except Exception: expressed_junction_types['novel but within 50nt of a known sites']=1
elif 'I' in uid and '_' in uid and junction_type!='trans-splicing':
#print uid;sys.exit()
try: expressed_junction_types['novel intronic junctions']+=1
except Exception: expressed_junction_types['novel intronic junctions']=1
coord = string.split(uid,'_')[-1]
if '-' in coord:
coord = string.split(coord,'-')[0]
try: novel_intronic_junctions[gene]=[coord]
except Exception: novel_intronic_junctions[gene].append(coord)
elif ('I' in uid or 'U' in uid) and '_' in uid and max_read_counts>read_threshold:
if '-' not in uid:
samples_expressed = maxCount(values)
if samples_expressed>2:
try: expressed_junction_types['novel intronic exon']+=1
except Exception: expressed_junction_types['novel intronic exon']=1
coord = string.split(uid,'_')[-1]
#print uid, coord;sys.exit()
#if 'ENSG00000269897' in uid: print [gene,coord]
try: novel_intronic_exons[gene].append(coord)
except Exception: novel_intronic_exons[gene]=[coord]
exon_coord_db[gene,coord]=uid
print 'Expressed (count>%s for at least 3 samples) junctions' % read_threshold
for junction_type in expressed_junction_types:
print junction_type, expressed_junction_types[junction_type]
expressed_junction_types={}
#print len(novel_intronic_junctions)
#print len(novel_intronic_exons)
for gene in novel_intronic_junctions:
if gene in novel_intronic_exons:
for coord in novel_intronic_junctions[gene]:
if coord in novel_intronic_exons[gene]:
try: expressed_junction_types['confirmed novel intronic exons']+=1
except Exception: expressed_junction_types['confirmed novel intronic exons']=1
uid = exon_coord_db[gene,coord]
features_to_export[uid]=[]
#else: print [gene], novel_intronic_junctions[gene]; sys.exit()
for junction_type in expressed_junction_types:
print junction_type, expressed_junction_types[junction_type]
out_file = string.replace(fn,'.txt','-highExp.txt')
print 'Exporting the highest expressed exons to:', out_file
out_obj = export.ExportFile(out_file)
### Compare the relative expression of junctions and exons separately for each gene (junctions are more comparable)
for gene in exon_max_exp_db:
junction_set=[]; exon_set=[]; junction_exp=[]; exon_exp=[]
exon_max_exp_db[gene].sort()
exon_max_exp_db[gene].reverse()
for (exp,uid) in exon_max_exp_db[gene]:
if '-' in uid: junction_set.append((exp,uid)); junction_exp.append(exp)
else: exon_set.append((exp,uid)); exon_exp.append(exp)
if len(junction_set)>0:
maxJunctionExp = junction_set[0][0]
try: lower25th,median_val,upper75th,int_qrt_range = statistics.iqr(junction_exp)
except Exception: print junction_exp;sys.exit()
if int_qrt_range>0:
maxJunctionExp = int_qrt_range
junction_percent_exp = map(lambda x: (x[1],expThreshold(x[0]/maxJunctionExp,cutoff)), junction_set)
high_exp_junctions = []
for (uid,p) in junction_percent_exp: ### ID and percentage of expression
if p!='NA':
if uid in features_to_export: ### novel exons only right now
out_obj.write(uid_key_db[uid]+'\t'+p+'\n') ### write out the original ID with coordinates
if len(exon_set)>0:
maxExonExp = exon_set[0][0]
lower25th,median_val,upper75th,int_qrt_range = statistics.iqr(exon_exp)
if int_qrt_range>0:
maxExonExp = int_qrt_range
exon_percent_exp = map(lambda x: (x[1],expThreshold(x[0]/maxExonExp,cutoff)), exon_set)
high_exp_exons = []
for (uid,p) in exon_percent_exp: ### ID and percentage of expression
if p!='NA':
if uid in features_to_export:
out_obj.write(uid_key_db[uid]+'\t'+p+'\n')
out_obj.close()
def expThreshold(ratio,cutoff):
#print [ratio,cutoff]
if ratio>cutoff: return str(ratio)
else: return 'NA'
def compareExonAndJunctionResults(species,array_type,summary_results_db,root_dir):
results_dir = root_dir +'AltResults/AlternativeOutput/'
dir_list = read_directory(results_dir)
filtered_dir_db={}
#"""
try: novel_exon_junction_db = getNovelExonCoordinates(species,root_dir)
except Exception:
#print traceback.format_exc()
print 'No counts file found.'
novel_exon_junction_db={} ### only relevant to RNA-Seq analyses
for comparison_file in summary_results_db:
for results_file in dir_list:
if (comparison_file in results_file and '-exon-inclusion-results.txt' in results_file) and ('comparison' not in results_file):
try: filtered_dir_db[comparison_file].append(results_file)
except Exception: filtered_dir_db[comparison_file] = [results_file]
try: os.remove(string.split(results_dir,'AltResults')[0]+'AltResults/Clustering/Combined-junction-exon-evidence.txt')
except Exception: pass
for comparison_file in filtered_dir_db:
alt_result_files = filtered_dir_db[comparison_file]
#print alt_result_files, comparison_file
importAltAnalyzeExonResults(alt_result_files,novel_exon_junction_db,results_dir)
#"""
### Build combined clusters of high-confidence exons
graphics2=[]; graphics=[]
import ExpressionBuilder
try:
input_dir = string.split(results_dir,'AltResults')[0]+'GO-Elite/AltExonConfirmed/'
cluster_file, rows_in_file = ExpressionBuilder.buildAltExonClusterInputs(input_dir,species,array_type,dataType='AltExonConfirmed')
if rows_in_file > 5000: useHOPACH = False
else: useHOPACH = True
if rows_in_file < 12000:
graphics = ExpressionBuilder.exportHeatmap(cluster_file,useHOPACH=useHOPACH)
except Exception: pass
try:
input_dir = string.split(results_dir,'AltResults')[0]+'GO-Elite/AltExon/'
cluster_file, rows_in_file = ExpressionBuilder.buildAltExonClusterInputs(input_dir,species,array_type,dataType='AltExon')
if rows_in_file > 5000: useHOPACH = False
else: useHOPACH = True
if rows_in_file < 12000:
graphics2 = ExpressionBuilder.exportHeatmap(cluster_file,useHOPACH=useHOPACH)
except Exception: pass
return graphics+graphics2
class SplicingData:
def __init__(self,score,symbol,description,exonid,probesets,direction,splicing_event,external_exon,genomic_loc,gene_exp,protein_annot,domain_inferred,domain_overlap,method,dataset):
self.score = score; self.dataset = dataset
self.symbol = symbol;
self.description=description;self.exonid=exonid;self.probesets=probesets;self.direction=direction
self.splicing_event=splicing_event;self.external_exon=external_exon;self.genomic_loc=genomic_loc;
self.gene_exp=gene_exp;self.protein_annot=protein_annot;self.domain_inferred=domain_inferred
self.domain_overlap=domain_overlap;self.method=method
def Score(self): return self.score
def setScore(self,score): self.score = score
def GeneExpression(self): return self.gene_exp
def Dataset(self): return self.dataset
def Symbol(self): return self.symbol
def Description(self): return self.description
def ExonID(self): return self.exonid
def appendExonID(self,exonid): self.exonid+='|'+exonid
def Probesets(self): return self.probesets
def ProbesetDisplay(self):
if len(self.Probesets()[1])>0:
return string.join(self.Probesets(),'-')
else:
return self.Probesets()[0]
def ProbesetsSorted(self):
### Don't sort the original list
a = [self.probesets[0],self.probesets[1]]
a.sort()
return a
def Direction(self): return self.direction
def setDirection(self,direction): self.direction = direction
def SplicingEvent(self): return self.splicing_event
def ProteinAnnotation(self): return self.protein_annot
def DomainInferred(self): return self.domain_inferred
def DomainOverlap(self): return self.domain_overlap
def Method(self): return self.method
def setEvidence(self,evidence): self.evidence = evidence
def Evidence(self): return self.evidence
def GenomicLocation(self): return self.genomic_loc
def setExonExpStatus(self, exon_expressed): self.exon_expressed = exon_expressed
def ExonExpStatus(self): return self.exon_expressed
def importAltAnalyzeExonResults(dir_list,novel_exon_junction_db,results_dir):
regulated_critical_exons={}; converted_db={}
includeExonJunctionComps=True ### Allow ASPIRE comparisons with the inclusion feature as an exon to count for additive reciprocal evidence
print "Reading AltAnalyze results file"
root_dir = string.split(results_dir,'AltResults')[0]
for filename in dir_list:
x=0; regulated_critical_exon_temp={}
fn=filepath(results_dir+filename)
new_filename = string.join(string.split(filename,'-')[:-5],'-')
if '_vs_' in filename and '_vs_' in new_filename: export_filename = new_filename
else: export_filename = string.join(string.split(filename,'-')[:-5],'-')
export_path = results_dir+export_filename+'-comparison-evidence.txt'
try: os.remove(filepath(export_path)) ### If we don't do this, the old results get added to the new
except Exception: null=[]
if 'AltMouse' in filename:
altmouse_ensembl_db = importAltMouseEnsembl()
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: x=1; #print t[12],t[13],t[22],t[23]
else:
converted = False ### Indicates both junction sides were regulated
geneid = t[0]; exonid = t[4]; probeset1 = t[6]; probeset2 = ''; score = t[1][:4]; symbol = t[2]; description = t[3]; regions = t[-4]; direction = t[5]
genomic_loc = t[-1]; splicing_event = t[-3]; external_exon = t[-6]; gene_exp_fold = t[-8]; protein_annot = t[14]; domain_inferred = t[15]; domain_overlap = t[17]
expressed_exon = 'NA'
if 'RNASeq' in filename: expressed_exon = 'no' ### Set by default
if ':' in geneid: geneid = string.split(geneid,':')[0] ### User reported that gene:gene was appearing and not sure exactly where or why but added this to address it
if 'FIRMA' in fn: method = 'FIRMA'
elif 'splicing-index' in fn: method = 'splicing-index'
if 'ASPIRE' in filename or 'linearregres' in filename:
f1=float(t[12]); f2=float(t[13]); probeset1 = t[8]; probeset2 = t[10]; direction = t[6]; exonid2 = t[5]; splicing_event = t[-4]
protein_annot = t[19]; domain_inferred = t[20]; domain_overlap = t[24]; method = 'linearregres'; regions = t[-5]
exon1_exp=float(t[-15]); exon2_exp=float(t[-14]); fold1=float(t[12]); fold2=float(t[13])
if fold1<0: fold1 = 1 ### don't factor in negative changes
if fold2<0: fold2 = 1 ### don't factor in negative changes
"""
if 'RNASeq' not in filename:
exon1_exp = math.pow(2,exon1_exp)
exon2_exp = math.log(2,exon2_exp)
m1 = exon1_exp*fold1
m2 = exon2_exp*fold2
max_exp = max([m1,m2])
min_exp = min([m1,m2])
percent_exon_expression = str(min_exp/max_exp)
"""
if 'ASPIRE' in filename: method = 'ASPIRE'; score = t[1][:5]
if '-' not in exonid and includeExonJunctionComps == False:
exonid=None ### Occurs when the inclusion just in an exon (possibly won't indicate confirmation so exclude)
else: exonid = exonid+' vs. '+exonid2
if 'AltMouse' in filename:
try: geneid = altmouse_ensembl_db[geneid]
except Exception: geneid = geneid
if 'RNASeq' not in filename and 'junction' not in filename: regions = string.replace(regions,'-','.')
else:
if 'RNASeq' in filename and '-' not in exonid:
fold = float(t[10]); exon_exp = float(t[18]); gene_exp = float(t[19])
if fold < 0: fold = -1.0/fold
GE_fold = float(gene_exp_fold)
if GE_fold < 0: GE_fold = -1.0/float(gene_exp_fold)
exon_psi1 = abs(exon_exp)/(abs(gene_exp))
exon_psi2 = (abs(exon_exp)*fold)/(abs(gene_exp)*GE_fold)
max_incl_exon_exp = max([exon_psi1,exon_psi2])
#if max_incl_exon_exp>0.20: expressed_exon = 'yes'
expressed_exon = max_incl_exon_exp
#if 'I2.1_75953139' in probeset1:
#print [exon_exp,gene_exp,exon_exp*fold,gene_exp*GE_fold]
#print exon_psi1, exon_psi2;sys.exit()
probesets = [probeset1,probeset2]
if (method == 'splicing-index' or method == 'FIRMA') and ('-' in exonid) or exonid == None:
pass #exclude junction IDs
else:
regions = string.replace(regions,';','|')
regions = string.replace(regions,'-','|')
regions = string.split(regions,'|')
for region in regions:
if len(region) == 0:
try: region = t[17]+t[18] ### For junction introns where no region ID exists
except Exception: null=[]
if ':' in region: region = string.split(region,':')[-1] ### User reported that gene:gene was appearing and not sure exactly where or why but added this to address it
if probeset1 in novel_exon_junction_db:
uid = novel_exon_junction_db[probeset1] ### convert the uid (alternative exon) to the annotated ID for the novel exon
converted_db[uid] = probeset1
else:
uid = geneid+':'+region
ss = SplicingData(score,symbol,description,exonid,probesets,direction,splicing_event,external_exon,genomic_loc,gene_exp_fold,protein_annot,domain_inferred,domain_overlap,method,filename)
ss.setExonExpStatus(str(expressed_exon))
try: regulated_critical_exon_temp[uid].append(ss)
except Exception: regulated_critical_exon_temp[uid] = [ss]
#print filename, len(regulated_critical_exon_temp)
for uid in regulated_critical_exon_temp:
report=None
if len(regulated_critical_exon_temp[uid])>1:
### We are only reporting one here and that's OK, since we are only reporting the top scores... won't include all inclusion junctions.
scores=[]
for ss in regulated_critical_exon_temp[uid]: scores.append((float(ss.Score()),ss))
scores.sort()
if (scores[0][0]*scores[-1][0])<0:
ss1 = scores[0][1]; ss2 = scores[-1][1]
if ss1.ProbesetsSorted() == ss2.ProbesetsSorted(): ss1.setDirection('mutual') ### same exons, hence, mutually exclusive event (or similiar)
else: ss1.setDirection('both') ### opposite directions in the same comparison-file, hence, conflicting data
report=[ss1]
else:
if abs(scores[0][0])>abs(scores[-1][0]): report=[scores[0][1]]
else: report=[scores[-1][1]]
else:
report=regulated_critical_exon_temp[uid]
### Combine data from different analysis files
try: regulated_critical_exons[uid]+=report
except Exception: regulated_critical_exons[uid]=report
"""if 'ENSG00000204120' in uid:
print uid,
for i in regulated_critical_exon_temp[uid]:
print i.Probesets(),
print ''
"""
try: report[0].setEvidence(len(regulated_critical_exon_temp[uid])) ###set the number of exons demonstrating regulation of this exons
except Exception: null=[]
clearObjectsFromMemory(regulated_critical_exon_temp)
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
header = string.join(['uid','source-IDs','symbol','description','exonids','independent confirmation','score','regulation direction','alternative exon annotations','associated isoforms','inferred regulated domains','overlapping domains','method','supporting evidence score','novel exon: high-confidence','percent exon expression of gene','differential gene-expression','genomic location'],'\t')+'\n'
export_data.write(header)
combined_export_path = string.split(results_dir,'AltResults')[0]+'AltResults/Clustering/Combined-junction-exon-evidence.txt'
combined_export_data, status= AppendOrWrite(combined_export_path)
if status == 'not found':
header = string.join(['uid','source-IDs','symbol','description','exonids','independent confirmation','score','regulation direction','alternative exon annotations','associated isoforms','inferred regulated domains','overlapping domains','method','supporting evidence score','novel exon: high-confidence','percent exon expression of gene','differential gene-expression','genomic location','comparison'],'\t')+'\n'
combined_export_data.write(header)
print len(regulated_critical_exons), 'regulated exon IDs imported.\n'
print 'writing:',export_path; n=0
# print [len(converted_db)]
### Check for alternative 3' or alternative 5' exon regions that were not matched to the right reciprocal junctions (occurs because only one of the exon regions is called alternative)
regulated_critical_exons_copy={}
for uid in regulated_critical_exons:
regulated_critical_exons_copy[uid]=regulated_critical_exons[uid]
u=0
### This is most applicable to RNA-Seq since the junction IDs correspond to the Exon Regions not the probeset Exon IDs
for uid in regulated_critical_exons_copy: ### Look through the copied version since we can't delete entries while iterating through
ls = regulated_critical_exons_copy[uid]
u+=1
#if u<20: print uid
for jd in ls:
if jd.Method() != 'splicing-index' and jd.Method() != 'FIRMA':
try: ### Applicable to RNA-Seq
gene,exonsEx = string.split(jd.Probesets()[1],':') ### Exclusion probeset will have the exon not annotated as the critical exon (although it should be as well)
gene,exonsIn = string.split(jd.Probesets()[0],':')
except Exception:
gene, ce = string.split(uid,':')
exonsIn, exonsEx = string.split(jd.ExonID(),'vs.')
if gene !=None:
critical_exon = None
five_prime,three_prime = string.split(exonsEx,'-')
try: five_primeIn,three_primeIn = string.split(exonsIn,'-')
except Exception: five_primeIn = exonsIn; three_primeIn = exonsIn ### Only should occur during testing when a exon rather than junction ID is considered
#if gene == 'ENSG00000133083': print five_prime,three_prime, five_primeIn,three_primeIn
if five_primeIn == five_prime: ### Hence, the exclusion 3' exon should be added
critical_exon = gene+':'+three_prime
exonid = three_prime
elif three_primeIn == three_prime: ### Hence, the exclusion 3' exon should be added
critical_exon = gene+':'+five_prime
exonid = five_prime
else:
if ('5' in jd.SplicingEvent()) or ('five' in jd.SplicingEvent()):
critical_exon = gene+':'+five_prime
exonid = five_prime
elif ('3' in jd.SplicingEvent()) or ('three' in jd.SplicingEvent()):
critical_exon = gene+':'+three_prime
exonid = three_prime
elif ('alt-N-term' in jd.SplicingEvent()) or ('altPromoter' in jd.SplicingEvent()):
critical_exon = gene+':'+five_prime
exonid = five_prime
elif ('alt-C-term' in jd.SplicingEvent()):
critical_exon = gene+':'+three_prime
exonid = three_prime
#print critical_exon, uid, jd.ExonID(),jd.SplicingEvent(); sys.exit()
if critical_exon != None:
if critical_exon in regulated_critical_exons:
#print uid, critical_exon; sys.exit()
if len(regulated_critical_exons[critical_exon]) == 1:
if len(ls)==1 and uid in regulated_critical_exons: ### Can be deleted by this method
if 'vs.' not in regulated_critical_exons[critical_exon][0].ExonID() and 'vs.' not in regulated_critical_exons[critical_exon][0].ExonID():
regulated_critical_exons[uid].append(regulated_critical_exons[critical_exon][0])
del regulated_critical_exons[critical_exon]
elif uid in regulated_critical_exons: ###If two entries already exit
ed = regulated_critical_exons[uid][1]
ed2 = regulated_critical_exons[critical_exon][0]
if 'vs.' not in ed.ExonID() and 'vs.' not in ed2.ExonID():
if ed.Direction() != ed2.Direction(): ### should be opposite directions
ed.appendExonID(exonid)
ed.setEvidence(ed.Evidence()+1)
ed.setScore(ed.Score()+'|'+ed2.Score())
del regulated_critical_exons[critical_exon]
firstEntry=True
for uid in regulated_critical_exons:
if uid in converted_db:
converted = True
else: converted = False
#if 'ENSG00000133083' in uid: print [uid]
exon_level_confirmation = 'no'
ls = regulated_critical_exons[uid]
jd = regulated_critical_exons[uid][0] ### We are only reporting one here and that's OK, since we are only reporting the top scores... won't include all inclusion junctions.
if len(ls)>1:
methods = []; scores = []; direction = []; exonids = []; probesets = []; evidence = 0; genomic_location = []
junctionids=[]
junction_data_found = 'no'; exon_data_found = 'no'
for jd in ls:
if jd.Method() == 'ASPIRE' or jd.Method() == 'linearregres':
junction_data_found = 'yes'
methods.append(jd.Method())
scores.append(jd.Score())
direction.append(jd.Direction())
exonids.append(jd.ExonID())
junctionids.append(jd.ExonID())
probesets.append(jd.ProbesetDisplay())
evidence+=jd.Evidence()
genomic_location.append(jd.GenomicLocation())
### Prefferentially obtain isoform annotations from the reciprocal analysis which is likely more accurate
isoform_annotations = [jd.ProteinAnnotation(), jd.DomainInferred(), jd.DomainOverlap()]
for ed in ls:
if ed.Method() == 'splicing-index' or ed.Method() == 'FIRMA':
exon_data_found = 'yes' ### pick one of them
methods.append(ed.Method())
scores.append(ed.Score())
direction.append(ed.Direction())
exonids.append(ed.ExonID())
probesets.append(ed.ProbesetDisplay())
evidence+=ed.Evidence()
genomic_location.append(ed.GenomicLocation())
#isoform_annotations = [ed.ProteinAnnotation(), ed.DomainInferred(), ed.DomainOverlap()]
if junction_data_found == 'yes' and exon_data_found == 'yes':
exon_level_confirmation = 'yes'
for junctions in junctionids:
if 'vs.' in junctions:
j1 = string.split(junctions,' vs. ')[0] ### inclusion exon or junction
if '-' not in j1: ### not a junction, hence, may not be sufficient to use for confirmation (see below)
if 'I' in j1: ### intron feature
if '_' in j1: ### novel predicted exon
exon_level_confirmation = 'no'
else:
exon_level_confirmation = 'yes'
else:
if '_' in j1:
exon_level_confirmation = 'no'
else:
exon_level_confirmation = 'partial'
method = string.join(methods,'|')
unique_direction = unique.unique(direction)
genomic_location = unique.unique(genomic_location)
if len(unique_direction) == 1: direction = unique_direction[0]
else: direction = string.join(direction,'|')
score = string.join(scores,'|')
probesets = string.join(probesets,'|')
exonids_unique = unique.unique(exonids)
if len(exonids_unique) == 1: exonids = exonids_unique[0]
else: exonids = string.join(exonids,'|')
if len(genomic_location) == 1: genomic_location = genomic_location[0]
else: genomic_location = string.join(genomic_location,'|')
evidence = str(evidence)
if 'mutual' in direction: direction = 'mutual'
if len(ls) == 1:
probesets = jd.ProbesetDisplay()
direction = jd.Direction()
score = jd.Score()
method = jd.Method()
exonids = jd.ExonID()
evidence = jd.Evidence()
genomic_location = jd.GenomicLocation()
isoform_annotations = [jd.ProteinAnnotation(), jd.DomainInferred(), jd.DomainOverlap()]
try:
#if int(evidence)>4 and 'I' in uid: novel_exon = 'yes' ### high-evidence novel exon
#else: novel_exon = 'no'
if converted == True:
novel_exon = 'yes'
splicing_event = 'cassette-exon'
else:
novel_exon = 'no'
splicing_event = jd.SplicingEvent()
values = [uid, probesets, jd.Symbol(), jd.Description(), exonids, exon_level_confirmation, score, direction, splicing_event]
values += isoform_annotations+[method, str(evidence),novel_exon,jd.ExonExpStatus(),jd.GeneExpression(),genomic_location]
values = string.join(values,'\t')+'\n'
#if 'yes' in exon_level_confirmation:
export_data.write(values); n+=1
if exon_level_confirmation != 'no' and ('|' not in direction):
geneID = string.split(uid,':')[0]
try: relative_exon_exp = float(jd.ExonExpStatus())
except Exception: relative_exon_exp = 1
if firstEntry:
### Also export high-confidence predictions for GO-Elite
elite_export_path = string.split(results_dir,'AltResults')[0]+'GO-Elite/AltExonConfirmed/'+export_filename+'-junction-exon-evidence.txt'
elite_export_data = export.ExportFile(elite_export_path)
elite_export_data.write('GeneID\tEn\tExonID\tScores\tGenomicLocation\n')
firstEntry = False
if relative_exon_exp>0.10:
elite_export_data.write(string.join([geneID,'En',uid,score,genomic_location],'\t')+'\n')
#if 'DNA' in isoform_annotations[-1]:
if '2moter' not in jd.SplicingEvent() and '2lt-N' not in jd.SplicingEvent():
values = [uid, probesets, jd.Symbol(), jd.Description(), exonids, exon_level_confirmation, score, direction, splicing_event]
values += isoform_annotations+[method, str(evidence),novel_exon,jd.ExonExpStatus(),jd.GeneExpression(),genomic_location,export_filename]
values = string.join(values,'\t')+'\n'
combined_export_data.write(values)
except Exception, e:
#print traceback.format_exc();sys.exit()
pass ### Unknown error - not evaluated in 2.0.8 - isoform_annotations not referenced
print n,'exon IDs written to file.'
export_data.close()
try: elite_export_data.close()
except Exception: pass
clearObjectsFromMemory(regulated_critical_exons)
clearObjectsFromMemory(regulated_critical_exons_copy)
#print '!!!!Within comparison evidence'
#returnLargeGlobalVars()
def FeatureCounts(bed_ref, bam_file):
output = bam_file[:-4]+'__FeatureCounts.bed'
import subprocess
#if '/bin' in kallisto_dir: kallisto_file = kallisto_dir +'/apt-probeset-summarize' ### if the user selects an APT directory
kallisto_dir= 'AltDatabase/subreads/'
if os.name == 'nt':
featurecounts_file = kallisto_dir + 'PC/featureCounts.exe'; plat = 'Windows'
elif 'darwin' in sys.platform:
featurecounts_file = kallisto_dir + 'Mac/featureCounts'; plat = 'MacOSX'
elif 'linux' in sys.platform:
featurecounts_file = kallisto_dir + '/Linux/featureCounts'; plat = 'linux'
print 'Using',featurecounts_file
featurecounts_file = filepath(featurecounts_file)
featurecounts_root = string.split(featurecounts_file,'bin/featureCounts')[0]
featurecounts_file = filepath(featurecounts_file)
print [featurecounts_file,"-a", "-F", "SAF",bed_ref, "-o", output, bam_file]
retcode = subprocess.call([featurecounts_file,"-a",bed_ref, "-F", "SAF", "-o", output, bam_file])
def filterFASTAFiles(fasta_files):
filter_fasta_files=[]
filter_dir = export.findParentDir(fasta_files[0])+'/filtered_fasta'
try: os.mkdir(filter_dir)
except Exception: pass
for file in fasta_files:
if 'filtered.fa' in file:
if file not in filter_fasta_files:
filter_fasta_files.append(file)
else:
filtered_fasta = file[:-3]+'-filtered.fa'
filter_fasta_files.append(filtered_fasta)
filename = export.findFilename(file)
eo=export.ExportFile(filtered_fasta)
for line in open(file,'rU').xreadlines():
if '>'==line[0]:
skip=False
### Exclude non-standard chromosomal transcripts
if 'PATCH' in line or '_1_' in line or '_1:' in line or ':HSCHR' in line or 'putative' in line or 'supercontig' in line or 'NOVEL_TEST' in line:
skip=True
else:
space_delim=string.split(line,' ')
space_delim=[string.split(space_delim[0],'.')[0]]+space_delim[1:]
line=string.join(space_delim,' ')
eo.write(line)
elif skip==False:
eo.write(line)
eo.close()
shutil.move(file,filter_dir+'/'+filename)
return filter_fasta_files
def getCoordinateFile(species):
geneCoordFile = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_transcript-annotations.txt'
geneCoordFile = unique.filepath(geneCoordFile)
status = verifyFile(geneCoordFile)
if status == 'not found':
try:
from build_scripts import EnsemblSQL
ensembl_version = string.replace(unique.getCurrentGeneDatabaseVersion(),'EnsMart','')
configType = 'Advanced'; analysisType = 'AltAnalyzeDBs'; externalDBName = ''; force = 'no'
EnsemblSQL.buildEnsemblRelationalTablesFromSQL(species,configType,analysisType,externalDBName,ensembl_version,force,buildCommand='exon')
except Exception:
#print traceback.format_exc()
print 'Failed to export a transcript-exon coordinate file (similar to a GTF)!!!!\n...Proceeding with standard Kallisto (no-splicing).'
geneCoordFile=None
return geneCoordFile
def runKallisto(species,dataset_name,root_dir,fastq_folder,mlp,returnSampleNames=False,customFASTA=None,log_output=True):
#print 'Running Kallisto...please be patient'
import subprocess
n_threads = mlp.cpu_count()
print 'Number of threads =',n_threads
#n_threads = 1
kallisto_dir_objects = os.listdir(unique.filepath('AltDatabase/kallisto'))
### Determine version
version = '0.43.1-splice'
alt_version = '0.46.1'
if version not in kallisto_dir_objects:
for subdir in kallisto_dir_objects: ### Use whatever version is there (can be replaced by the user)
if subdir.count('.')>1: version = subdir
if alt_version not in kallisto_dir_objects:
for subdir in kallisto_dir_objects: ### Use whatever version is there (can be replaced by the user)
if subdir.count('.')>1 and 'splice' not in subdir: alt_version = subdir
kallisto_dir= 'AltDatabase/kallisto/'+version+'/'
if os.name == 'nt':
kallisto_file = kallisto_dir + 'PC/bin/kallisto.exe'; plat = 'Windows'
elif 'darwin' in sys.platform:
kallisto_file = kallisto_dir + 'Mac/bin/kallisto'; plat = 'MacOSX'
elif 'linux' in sys.platform:
kallisto_file = kallisto_dir + '/Linux/bin/kallisto'; plat = 'linux'
print 'Using',kallisto_file
kallisto_file = filepath(kallisto_file)
kallisto_root = string.split(kallisto_file,'bin/kallisto')[0]
fn = filepath(kallisto_file)
try: os.chmod(fn,0777) ### It's rare, but this can be a write issue
except: pass
output_dir=root_dir+'/ExpressionInput/kallisto/'
try: os.mkdir(root_dir+'/ExpressionInput')
except Exception: pass
try: os.mkdir(root_dir+'/ExpressionInput/kallisto')
except Exception: pass
fastq_folder += '/'
dir_list = read_directory(fastq_folder)
fastq_paths = []
for file in dir_list:
file_lower = string.lower(file)
if 'fastq' in file_lower and '._' not in file[:4]: ### Hidden files
fastq_paths.append(fastq_folder+file)
fastq_paths,paired = findPairs(fastq_paths)
### Check to see if Kallisto files already exist and use these if so (could be problematic but allows for outside quantification)
kallisto_tsv_paths=[]
dir_list = read_directory(output_dir)
for folder in dir_list:
kallisto_outdir = output_dir+folder+'/abundance.tsv'
status = os.path.isfile(kallisto_outdir)
if status:
kallisto_tsv_paths.append(fastq_folder+file)
if returnSampleNames:
return fastq_paths
### Store/retreive the Kallisto index in the Ensembl specific SequenceData location
kallisto_index_root = 'AltDatabase/'+species+'/SequenceData/'
try: os.mkdir(filepath(kallisto_index_root))
except Exception: pass
indexFile = filepath(kallisto_index_root+species)
#indexFile = filepath(kallisto_index_root + 'Hs_intron')
indexStatus = os.path.isfile(indexFile)
if indexStatus == False or customFASTA!=None:
try: fasta_files = getFASTAFile(species)
except Exception: fasta_files = []
index_file = filepath(kallisto_index_root+species)
if len(fasta_files)==0 and customFASTA==None:
###download Ensembl fasta file to the above directory
from build_scripts import EnsemblSQL
ensembl_version = string.replace(unique.getCurrentGeneDatabaseVersion(),'EnsMart','')
try:
EnsemblSQL.getEnsemblTranscriptSequences(ensembl_version,species,restrictTo='cDNA')
fasta_files = getFASTAFile(species)
except Exception: pass
elif customFASTA!=None: ### Custom FASTA file supplied by the user
fasta_files = [customFASTA]
indexFile = filepath(kallisto_index_root+species+'-custom')
try: os.remove(indexFile) ### erase any pre-existing custom index
except Exception: pass
if len(fasta_files)>0:
print 'Building kallisto index file...'
arguments = [kallisto_file, "index","-i", indexFile]
fasta_files = filterFASTAFiles(fasta_files)
for fasta_file in fasta_files:
arguments.append(fasta_file)
try:
retcode = subprocess.call(arguments)
except Exception:
print traceback.format_exc()
if customFASTA!=None:
reimportExistingKallistoOutput = False
elif len(kallisto_tsv_paths) == len(fastq_paths):
reimportExistingKallistoOutput = True
elif len(kallisto_tsv_paths) > len(fastq_paths):
reimportExistingKallistoOutput = True ### If working with a directory of kallisto results
else:
reimportExistingKallistoOutput = False
if reimportExistingKallistoOutput:
print 'NOTE: Re-import PREVIOUSLY GENERATED kallisto output:',reimportExistingKallistoOutput
print '...To force re-analysis of FASTQ files, delete the folder "kallisto" in "ExpressionInput"'
### Just get the existing Kallisto output folders
fastq_paths = read_directory(output_dir)
kallisto_folders=[]
try:
import collections
expMatrix = collections.OrderedDict()
countMatrix = collections.OrderedDict()
countSampleMatrix = collections.OrderedDict()
sample_total_counts = collections.OrderedDict()
except Exception:
try:
import ordereddict
expMatrix = ordereddict.OrderedDict()
countMatrix = ordereddict.OrderedDict()
countSampleMatrix = ordereddict.OrderedDict()
sample_total_counts = ordereddict.OrderedDict()
except Exception:
expMatrix={}
countMatrix={}
countSampleMatrix={}
sample_total_counts={}
headers=['UID']
### Verify, import, create and/or ignore the transcript exon coordinate file for BAM file creation
if paired == 'paired':
s=[]
else:
s=["--single","-l","200","-s","20"]
geneCoordFile = getCoordinateFile(species)
for n in fastq_paths:
output_path = output_dir+n
kallisto_folders.append(output_path)
if reimportExistingKallistoOutput == False:
begin_time = time.time()
if geneCoordFile != None: ### For BAM and BED file generation
print 'Running kallisto on:',n,'...',
p=fastq_paths[n]
b=[" > "+n+'.sam']
bedFile = root_dir+ '/' + n + '__junction.bed'
kallisto_out = open(root_dir+ '/' + n + '.bam', 'ab')
if log_output:
err_out = open(output_dir + '/log.txt', 'a')
err_out.seek(0, 2) # Subprocess doesn't move the file pointer when appending!
else:
err_out = None
kallisto_out.seek(0, 2) # Subprocess doesn't move the file pointer when appending!
#geneCoordFile=None - force to run simple Kallisto
if geneCoordFile==None:
try: ### Without BAM and BED file generation
retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path]+s+p)
except Exception:
print traceback.format_exc()
else: ### Attempt to export BAM and BED files with Kallisto quantification
kallisto_command = [kallisto_file, "quant", "-i", indexFile, "-o", output_path,
"-g", geneCoordFile, "-j", bedFile, "--threads="+str(n_threads), "--sortedbam"] + s +p
kallisto_process = subprocess.Popen(kallisto_command, stdout=kallisto_out, stderr=err_out)
kallisto_process.communicate()
retcode = kallisto_process.returncode
if os.name == 'nt':
try:
sam_process = subprocess.Popen('AltDatabase\samtools\samtools.exe index ' + root_dir+ '/' + n + '.bam')
sam_process.communicate()
retcode_sam = sam_process.returncode
except: pass
#retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path,"--pseudobam"]+p+b)
#retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path]+p)
"""except Exception:
print traceback.format_exc()
kill
retcode = subprocess.call(['kallisto', "quant","-i", indexFile, "-o", output_path]+p)"""
if retcode == 0: print 'completed in', int(time.time()-begin_time), 'seconds'
else:
print 'kallisto failed due to an unknown error (report to altanalyze.org help).\nTrying without BAM file creation.'
""" The below code should be run once, when changing to the alternative kallisto version """
kallisto_file = string.replace(kallisto_file,version,alt_version)
try: os.chmod(kallisto_file,0777) ### It's rare, but this can be a write issue
except: pass
print 'Re-Building kallisto index file...'
try: os.remove(root_dir+n+'.bam') ### Remove the failed BAM file attempt - will confuse user and trigger BAM analysis later
except: pass
arguments = [kallisto_file, "index","-i", indexFile]
fasta_files = getFASTAFile(species)
fasta_files = filterFASTAFiles(fasta_files)
for fasta_file in fasta_files:
arguments.append(fasta_file)
try:
retcode = subprocess.call(arguments)
except Exception:
print traceback.format_exc()
geneCoordFile = None ### Skips BAM file generation going forward
try: ### Without BAM and BED file generation
retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path]+s+p)
except Exception:
print traceback.format_exc()
if retcode == 0: print 'completed in', int(time.time()-begin_time), 'seconds'
else: print 'kallisto failed due to an unknown error (report to altanalyze.org help).'
#"""
input_path = output_path+'/abundance.txt'
try:
try: expMatrix,countMatrix,countSampleMatrix=importTPMs(n,input_path,expMatrix,countMatrix,countSampleMatrix)
except Exception:
input_path = output_path+'/abundance.tsv'
expMatrix,countMatrix,countSampleMatrix=importTPMs(n,input_path,expMatrix,countMatrix,countSampleMatrix)
headers.append(n)
sample_total_counts = importTotalReadCounts(n,output_path+'/run_info.json',sample_total_counts)
except Exception:
print traceback.format_exc()
sys.exit()
print n, 'TPM expression import failed'
if paired == 'paired':
print '\n...Make sure the paired-end samples were correctly assigned:'
print fastq_paths
for i in fastq_paths:
print 'Common name:',i,
for x in fastq_paths[i]:
print export.findParentDir(x),
print '\n'
### Summarize alignment information
for sample in countSampleMatrix:
try: estCounts = int(float(countSampleMatrix[sample]))
except Exception: estCounts='NA'
try: totalCounts = sample_total_counts[sample]
except Exception: totalCounts = 'NA'
try: aligned = str(100*estCounts/float(totalCounts))
except Exception: aligned = 'NA'
try: aligned = string.split(aligned,'.')[0]+'.'+string.split(aligned,'.')[1][:2]
except Exception: aligned = 'NA'
countSampleMatrix[sample] = [str(estCounts),totalCounts,aligned]
dataset_name = string.replace(dataset_name,'exp.','')
dataset_name = string.replace(dataset_name,'.txt','')
to = export.ExportFile(root_dir+'/ExpressionInput/transcript.'+dataset_name+'.txt')
ico = export.ExportFile(root_dir+'/ExpressionInput/isoCounts.'+dataset_name+'.txt')
go = export.ExportFile(root_dir+'/ExpressionInput/exp.'+dataset_name+'.txt')
co = export.ExportFile(root_dir+'/ExpressionInput/counts.'+dataset_name+'.txt')
so = export.ExportFile(root_dir+'/ExpressionInput/summary.'+dataset_name+'.txt')
exportMatrix(to,headers,expMatrix) ### Export transcript expression matrix
exportMatrix(ico,headers,countMatrix,counts=True) ### Export transcript count matrix
try:
geneMatrix = calculateGeneTPMs(species,expMatrix) ### calculate combined gene level TPMs
countsGeneMatrix = calculateGeneTPMs(species,countMatrix) ### calculate combined gene level TPMs
exportMatrix(go,headers,geneMatrix) ### export gene expression matrix
exportMatrix(co,headers,countsGeneMatrix,counts=True) ### export gene expression matrix
except Exception:
print 'AltAnalyze was unable to summarize gene TPMs from transcripts, proceeding with transcripts.'
export.copyFile(root_dir+'/ExpressionInput/transcript.'+dataset_name+'.txt',root_dir+'/ExpressionInput/exp.'+dataset_name+'.txt')
exportMatrix(so,['SampleID','Estimated Counts','Total Fragments','Percent Aligned'],countSampleMatrix) ### export gene expression matrix
### Copy results to the Kallisto_Results directory
try: os.mkdir(root_dir+'/ExpressionInput/Kallisto_Results')
except: pass
try:
tf = root_dir+'/ExpressionInput/transcript.'+dataset_name+'.txt'
shutil.copyfile(tf,string.replace(tf,'ExpressionInput','ExpressionInput/Kallisto_Results'))
tf = root_dir+'/ExpressionInput/isoCounts.'+dataset_name+'.txt'
shutil.copyfile(tf,string.replace(tf,'ExpressionInput','ExpressionInput/Kallisto_Results'))
tf = root_dir+'/ExpressionInput/exp.'+dataset_name+'.txt'
shutil.copyfile(tf,string.replace(tf,'ExpressionInput','ExpressionInput/Kallisto_Results'))
tf = root_dir+'/ExpressionInput/counts.'+dataset_name+'.txt'
shutil.copyfile(tf,string.replace(tf,'ExpressionInput','ExpressionInput/Kallisto_Results'))
tf = root_dir+'/ExpressionInput/summary.'+dataset_name+'.txt'
shutil.copyfile(tf,string.replace(tf,'ExpressionInput','ExpressionInput/Kallisto_Results'))
except:
print traceback.format_exc()
pass
def calculateGeneTPMs(species,expMatrix):
import gene_associations
try:
gene_to_transcript_db = gene_associations.getGeneToUid(species,('hide','Ensembl-EnsTranscript'))
if len(gene_to_transcript_db)<10:
raise ValueError('Ensembl-EnsTranscript file missing, forcing download of this file')
except Exception:
try:
print 'Missing transcript-to-gene associations... downloading from Ensembl.'
from build_scripts import EnsemblSQL
db_version = unique.getCurrentGeneDatabaseVersion()
EnsemblSQL.getGeneTranscriptOnly(species,'Basic',db_version,'yes')
gene_to_transcript_db = gene_associations.getGeneToUid(species,('hide','Ensembl-EnsTranscript'))
except Exception:
from build_scripts import GeneSetDownloader
print 'Ensembl-EnsTranscripts required for gene conversion... downloading from the web...'
GeneSetDownloader.remoteDownloadEnsemblTranscriptAssocations(species)
gene_to_transcript_db = gene_associations.getGeneToUid(species,('hide','Ensembl-EnsTranscript'))
if len(gene_to_transcript_db)<10:
print 'NOTE: No valid Ensembl-EnsTranscripts available, proceeding with the analysis of transcripts rather than genes...'
from import_scripts import OBO_import
transcript_to_gene_db = OBO_import.swapKeyValues(gene_to_transcript_db)
gene_matrix = {}
present_gene_transcripts={}
for transcript in expMatrix:
if '.' in transcript:
transcript_alt = string.split(transcript,'.')[0]
else:
transcript_alt = transcript
if transcript_alt in transcript_to_gene_db:
gene = transcript_to_gene_db[transcript_alt][0]
try: present_gene_transcripts[gene].append(transcript)
except Exception: present_gene_transcripts[gene] = [transcript]
else: pass ### could keep track of the missing transcripts
for gene in present_gene_transcripts:
gene_values = []
for transcript in present_gene_transcripts[gene]:
gene_values.append(map(float,expMatrix[transcript]))
gene_tpms = [sum(value) for value in zip(*gene_values)] ### sum of all transcript tmp's per sample
gene_tpms = map(str,gene_tpms)
gene_matrix[gene] = gene_tpms
if len(gene_matrix)>0:
return gene_matrix
else:
print "NOTE: No valid transcript-gene associations available... proceeding with Transcript IDs rather than gene."
return expMatrix
def exportMatrix(eo,headers,matrix,counts=False):
eo.write(string.join(headers,'\t')+'\n')
for gene in matrix:
values = matrix[gene]
if counts:
values = map(str,map(int,map(float,values)))
eo.write(string.join([gene]+values,'\t')+'\n')
eo.close()
def importTPMs(sample,input_path,expMatrix,countMatrix,countSampleMatrix):
firstLine=True
for line in open(input_path,'rU').xreadlines():
data = cleanUpLine(line)
if firstLine:
firstLine=False
header = string.split(data,'\t')
else:
target_id,length,eff_length,est_counts,tpm = string.split(data,'\t')
try: float(est_counts);
except Exception: ### nan instead of float found due to lack of alignment
est_counts = '0.0'
tpm = '0.0'
if '.' in target_id:
target_id = string.split(target_id,'.')[0] ### Ensembl isoform IDs in more recent Ensembl builds
try: expMatrix[target_id].append(tpm)
except Exception: expMatrix[target_id]=[tpm]
try: countSampleMatrix[sample]+=float(est_counts)
except Exception: countSampleMatrix[sample]=float(est_counts)
try: countMatrix[target_id].append(est_counts)
except Exception: countMatrix[target_id]=[est_counts]
return expMatrix,countMatrix,countSampleMatrix
def importTotalReadCounts(sample,input_path,sample_total_counts):
### Import from Kallisto Json file
for line in open(input_path,'rU').xreadlines():
data = cleanUpLine(line)
if "n_processed: " in data:
total = string.split(data,"n_processed: ")[1]
total = string.split(total,',')[0]
sample_total_counts[sample]=total
return sample_total_counts
def findPairs(fastq_paths):
#fastq_paths = ['/Volumes/test/run0718_lane12_read1_index701=Kopan_RBP_02_14999.fastq.gz','/Volumes/run0718_lane12_read2_index701=Kopan_RBP_02_14999.fastq.gz']
import export
read_notation=0
under_suffix_notation=0
suffix_notation=0
equal_notation=0
suffix_db={}
for i in fastq_paths:
if 'read1' in i or 'read2' in i or 'pair1' in i or 'pair2' or 'R1' in i or 'R2' in i:
read_notation+=1
f = export.findFilename(i)
if 'fastq' in f:
name = string.split(f,'fastq')[0]
elif 'FASTQ' in f:
name = string.split(f,'FASTQ')[0]
elif 'fq' in f:
name = string.split(f,'fq')[0]
if '_1.' in name or '_2.' in name:
under_suffix_notation+=1
elif '1.' in name or '2.' in name:
suffix_notation+=1
suffix_db[name[-2:]]=[]
if '=' in name:
equal_notation+=1
if read_notation==0 and suffix_notation==0 and under_suffix_notation==0:
new_names={}
for i in fastq_paths:
if '/' in i or '\\' in i:
n = export.findFilename(i)
if '=' in n:
n = string.split(n,'=')[1]
new_names[n] = [i]
### likely single-end samples
return new_names, 'single'
else:
new_names={}
paired = 'paired'
if equal_notation==len(fastq_paths):
for i in fastq_paths:
name = string.split(i,'=')[-1]
name = string.replace(name,'.fastq.gz','')
name = string.replace(name,'.fastq','')
name = string.replace(name,'.FASTQ.gz','')
name = string.replace(name,'.FASTQ','')
name = string.replace(name,'.fq.gz','')
name = string.replace(name,'.fq','')
if '/' in name or '\\' in name:
name = export.findFilename(name)
if '=' in name:
name = string.split(name,'=')[1]
try: new_names[name].append(i)
except Exception: new_names[name]=[i]
else:
for i in fastq_paths:
if suffix_notation == len(fastq_paths) and len(suffix_db)==2: ### requires that files end in both .1 and .2
pairs = ['1.','2.']
else:
pairs = ['-read1','-read2','-pair1','-pair2','_read1','_read2','_pair1','_pair2','read1','read2','pair1','pair2','_1.','_2.','_R1','_R2','-R1','-R2','R1','R2']
n=str(i)
n = string.replace(n,'fastq.gz','')
n = string.replace(n,'fastq','')
n = string.replace(n,'fq.gz','')
n = string.replace(n,'fq','')
n = string.replace(n,'FASTQ.gz','')
n = string.replace(n,'FASTQ','')
for p in pairs: n = string.replace(n,p,'')
if '/' in n or '\\' in n:
n = export.findFilename(n)
if '=' in n:
n = string.split(n,'=')[1]
if n[-1]=='.':
n = n[:-1] ###remove the last decimal
try: new_names[n].append(i)
except Exception: new_names[n]=[i]
for i in new_names:
if len(new_names[i])>1:
pass
else:
paired = 'single'
new_names = checkForMultipleLanes(new_names)
return new_names, paired
def checkForMultipleLanes(new_names):
""" This function further aggregates samples run across multiple flowcells """
read_count = 0
lane_count = 0
updated_names={}
for sample in new_names:
reads = new_names[sample]
count=0
for read in reads:
read_count+=1
if '_L00' in read and '_001':
### assumes no more than 9 lanes/sample
count+=1
if len(reads) == count: ### Multiple lanes run per sample
lane_count+=count
if lane_count==read_count:
for sample in new_names:
sample_v1 = string.replace(sample,'_001','')
sample_v1 = string.split(sample_v1,'_L00')
if len(sample_v1[-1])==1: ### lane number
sample_v1 = sample_v1[0]
if sample_v1 in updated_names:
updated_names[sample_v1]+=new_names[sample]
else:
updated_names[sample_v1]=new_names[sample]
if len(updated_names)==0:
updated_names = new_names
return updated_names
def getFASTAFile(species):
fasta_folder = 'AltDatabase/'+species+'/SequenceData/'
fasta_files=[]
dir_list = read_directory(filepath(fasta_folder))
for file in dir_list:
if '.fa' in file:
fasta_files.append(filepath(fasta_folder)+file)
return fasta_files
def predictCellTypesFromClusters(icgs_groups_path, goelite_path):
### Import groups
group_db={}
clusters=[]
for line in open(icgs_groups_path,'rU').xreadlines():
line=cleanUpLine(line)
t = string.split(line,'\t')
cell_barcode=t[0]
cluster=t[-1]
try: group_db[cluster].append(cell_barcode)
except: group_db[cluster] = [cell_barcode]
if cluster not in clusters:
clusters.append(cluster)
### Import cell-type predictions
firstLine=True
celltype_db={}
top_pvalue=None
prior_cluster = None
for line in open(goelite_path,'rU').xreadlines():
line=cleanUpLine(line)
t = string.split(line,'\t')
try:
pvalue = float(t[10])
cluster = string.split(t[0][1:],'-')[0]
if cluster != prior_cluster or top_pvalue==None:
prior_cluster = cluster
top_pvalue = -1*math.log(pvalue,10)
log_pvalue = -1*math.log(pvalue,10)
if (top_pvalue-log_pvalue)<10:
#print cluster, int(top_pvalues), int(log_pvalue), t[2]
cell_type = t[2]
try: celltype_db[cluster].append([pvalue,cell_type])
except: celltype_db[cluster] = [[pvalue,cell_type]]
except:
pass ### header rows or blanks
annotatedGroupsFile = icgs_groups_path[:-4]+'-CellTypesFull.txt'
eo1=export.ExportFile(icgs_groups_path[:-4]+'-CellTypes.txt')
eo2=export.ExportFile(annotatedGroupsFile)
""" Determine the most appropriate tissue to compare to then weight higher based on that tissue """
tissue_preference = {}
for cluster in clusters:
if cluster in celltype_db:
celltype_db[cluster].sort()
x = celltype_db[cluster][0][1]
if "Adult" in x or 'Fetal'in x or 'Embryo' in x or 'Embryonic' in x or 'Term' in x:
cell_type = celltype_db[cluster][0][1]
ids = string.split(cell_type,' ')
if len(ids)>1:
tissue = ids[1]
tissue_preference[cluster] = tissue
tissue_count = {}
tissue_count_ls=[]
for cluster in tissue_preference:
tissue = tissue_preference[cluster]
if tissue in tissue_count:
tissue_count[tissue]+=1
else:
tissue_count[tissue]=1
for tissue in tissue_count:
tissue_count_ls.append([tissue_count[tissue],tissue])
try:
tissue_count_ls.sort()
tissue_count_ls.reverse()
if ((tissue_count_ls[0][0]*1.00)/len(tissue_preference))>0.33: ### At least half of the clusters
tissue_preference = tissue_count_ls[0][-1]
print 'Likely tissue based on GO-Elite =',tissue_preference
else:
tissue_preference = 'NONE'
except:
#print traceback.format_exc()
tissue_preference = 'NONE'
for cluster in clusters:
if cluster in celltype_db:
celltype_db[cluster].sort()
cell_type = None
### Preferentially select cell type names that correspond to the preferential tissue
original_cell_type = celltype_db[cluster][0][1]
for (p,term) in celltype_db[cluster]:
if tissue_preference in term and 'ICGS2' not in term:
cell_type = term
break
if cell_type == None:
cell_type = original_cell_type
cell_type = string.replace(cell_type,'/','-')
if ' (' in cell_type:
cell_type = string.split(cell_type,' (')[0]
if '(' in cell_type:
cell_type = string.split(cell_type,'(')[0]
cell_type += '_c'+cluster
### Now, shorten the name to exclude the tissue type
cell_type = string.replace(cell_type,'Adult ','')
cell_type = string.replace(cell_type,'Fetal ','')
cell_type = string.replace(cell_type,'Embryonic','')
cell_type = string.replace(cell_type,'Embryo','')
cell_type = string.replace(cell_type,'Term','')
if tissue_preference != original_cell_type:
cell_type = string.replace(cell_type,tissue_preference,'')
if cell_type[0]=='_':cell_type = original_cell_type + '_c'+cluster
if cell_type[0]==' ':cell_type = cell_type[1:]
if cell_type[0]==' ':cell_type = cell_type[1:]
if cell_type[0]=='-':cell_type = cell_type[1:]
if cell_type[0]=='_':cell_type = cell_type[1:]
eo1.write(string.join([cluster,cell_type],'\t')+'\n')
for cell in group_db[cluster]:
eo2.write(string.join([cell,cluster,cell_type],'\t')+'\n')
else:
eo1.write(string.join([cluster,'UNK-c'+cluster],'\t')+'\n')
for cell in group_db[cluster]:
eo2.write(string.join([cell,cluster,'UNK-c'+cluster],'\t')+'\n')
eo1.close()
eo2.close()
return annotatedGroupsFile
if __name__ == '__main__':
samplesDiffering = 3
column_method = 'hopach'
species = 'Hs'
excludeCellCycle = False
icgs_groups_path='/Volumes/salomonis2/CCHMC-Collaborations/Rafi-Kopan-10X-Rhesus/10X-Kopan-Monkey-Kidney-Cortex-Nuclei-20190506-3v3rhe/10X-Kopan-Monkey-Kidney-Cortex-Nuclei/outs/soupX-without_GENEL-LIST-0.5/10X-Kopan-Monkey-Kidney-Cortex-Nuclei-0.5_matrix_CPTT/ICGS-NMF_cosine_cc/FinalGroups.txt'
goelite_path='/Volumes/salomonis2/CCHMC-Collaborations/Rafi-Kopan-10X-Rhesus/10X-Kopan-Monkey-Kidney-Cortex-Nuclei-20190506-3v3rhe/10X-Kopan-Monkey-Kidney-Cortex-Nuclei/outs/soupX-without_GENEL-LIST-0.5/10X-Kopan-Monkey-Kidney-Cortex-Nuclei-0.5_matrix_CPTT/ICGS-NMF_cosine_cc/GO-Elite/clustering/exp.FinalMarkerHeatmap_all/GO-Elite_results/pruned-results_z-score_elite.txt'
predictCellTypesFromClusters(icgs_groups_path, goelite_path);sys.exit()
platform = 'RNASeq'; graphic_links=[('','/Volumes/HomeBackup/CCHMC/PBMC-10X/ExpressionInput/SamplePrediction/DataPlots/Clustering-33k_CPTT_matrix-CORRELATED-FEATURES-iterFilt-hierarchical_cosine_cosine.txt')]
"""
graphic_links,new_results_file = correlateClusteredGenes(platform,graphic_links[-1][-1][:-4]+'.txt',
numSamplesClustered=samplesDiffering,excludeCellCycle=excludeCellCycle,graphics=graphic_links,
ColumnMethod=column_method, transpose=True, includeMoreCells=True)
"""
import UI; import multiprocessing as mlp
#runKallisto('Mm','BoneMarrow','/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/altanalyze/Mm-FASTQ','/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/altanalyze/Mm-FASTQ',mlp);sys.exit()
runKallisto('Hs','BreastCancer','/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/BreastCancerDemo/FASTQs/input','/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/BreastCancerDemo/FASTQs/input',mlp);sys.exit()
results_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/l/July-2017/PSI/test/Clustering-exp.round2-Guide3-hierarchical_cosine_correlation.txt'
#correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=4,hits_to_report=50,ReDefinedClusterBlocks=True,filter=True)
#sys.exit()
#correlateClusteredGenes('exons',results_file,stringency='strict',rhoCutOff=0.6);sys.exit()
#sys.exit()
species='Hs'; platform = "3'array"; vendor = "3'array"
#FeatureCounts('/Users/saljh8/Downloads/subread-1.5.2-MaxOSX-x86_64/annotation/mm10_AltAnalyze.txt', '/Users/saljh8/Desktop/Grimes/GEC14074/Grimes_092914_Cell12.bam')
#sys.exit()
import UI; import multiprocessing as mlp
gsp = UI.GeneSelectionParameters(species,platform,vendor)
gsp.setGeneSet('None Selected')
gsp.setPathwaySelect('')
gsp.setGeneSelection('')
gsp.setJustShowTheseIDs('')
gsp.setNormalize('median')
gsp.setSampleDiscoveryParameters(1,50,4,4,
True,'gene','protein_coding',False,'cosine','hopach',0.4)
#expFile = '/Users/saljh8/Desktop/Grimes/KashishNormalization/test/Original/ExpressionInput/exp.CombinedSingleCell_March_15_2015.txt'
expFile = '/Volumes/My Passport/salomonis2/SRP042161_GBM-single-cell/bams/ExpressionInput/exp.GBM_scRNA-Seq-steady-state.txt'
#singleCellRNASeqWorkflow('Hs', "RNASeq", expFile, mlp, parameters=gsp);sys.exit()
filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/Trumpp-HSC-2017/counts.rawTrumpp.txt'
filename = '/Volumes/salomonis2/Erica-data/GSE98451/counts.GSE98451_uterus_single_cell_RNA-Seq_counts-Ensembl.txt'
#fastRPKMCalculate(filename);sys.exit()
#calculateRPKMsFromGeneCounts(filename,'Mm',AdjustExpression=False);sys.exit()
#copyICGSfiles('','');sys.exit()
import multiprocessing as mlp
import UI
species='Mm'; platform = "3'array"; vendor = 'Ensembl'
gsp = UI.GeneSelectionParameters(species,platform,vendor)
gsp.setGeneSet('None Selected')
gsp.setPathwaySelect('')
gsp.setGeneSelection('')
gsp.setJustShowTheseIDs('')
gsp.setNormalize('median')
gsp.setSampleDiscoveryParameters(0,0,1.5,3,
False,'PSI','protein_coding',False,'cosine','hopach',0.35)
#gsp.setSampleDiscoveryParameters(1,1,4,3, True,'Gene','protein_coding',False,'cosine','hopach',0.5)
filename = '/Volumes/SEQ-DATA/AML_junction/AltResults/AlternativeOutput/Hs_RNASeq_top_alt_junctions-PSI-clust.txt'
#fastRPKMCalculate(filename);sys.exit()
results_file = '/Volumes/SEQ-DATA/Grimes/14018_gmp-pro/ExpressionInput/DataPlots/400 fold for at least 4 samples/Clustering-myeloblast-steady-state-correlated-features-hierarchical_euclidean_cosine-hopach.txt'
guideGeneFile = '/Volumes/SEQ-DATA/Grimes/14018_gmp-pro/ExpressionInput/drivingTFs-symbol.txt'
expFile = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/ExpressionInput/exp.CombinedSingleCell_March_15_2015.txt'
expFile = '/Users/saljh8/Desktop/dataAnalysis/Mm_Kiddney_tubual/ExpressionInput/exp.E15.5_Adult_IRI Data-output.txt'
expFile = '/Users/saljh8/Desktop/PCBC_MetaData_Comparisons/temp/C4Meth450-filtered-SC-3_regulated.txt'
expFile = '/Volumes/SEQ-DATA/Grimeslab/TopHat/AltResults/AlternativeOutput/Mm_RNASeq_top_alt_junctions-PSI-clust-filter.txt'
expFile = '/Users/saljh8/Documents/L_TargetPSIFiles/exp.TArget_psi_noif_uncorr_03-50missing-12high.txt'
expFile = '/Volumes/BOZEMAN2015/Hs_RNASeq_top_alt_junctions-PSI-clust-filter.txt'
singleCellRNASeqWorkflow('Hs', "exons", expFile, mlp, exp_threshold=0, rpkm_threshold=0, parameters=gsp);sys.exit()
#expFile = '/Users/saljh8/Desktop/Grimes/AltSplice/Gmp-cluster-filter.txt'
#singleCellRNASeqWorkflow('Mm', "exons", expFile, mlp, exp_threshold=0, rpkm_threshold=0, parameters=gsp);sys.exit()
#expFile = '/Users/saljh8/Downloads/methylation/ExpressionInput/exp.female-steady-state.txt'
#singleCellRNASeqWorkflow('Hs', 'RNASeq', expFile, mlp, exp_threshold=50, rpkm_threshold=5) # drivers=guideGeneFile)
#sys.exit()
#correlateClusteredGenes(results_file);sys.exit()
#reformatExonFile('Hs','exon',True);sys.exit()
filename = '/Volumes/Time Machine Backups/dataAnalysis/PCBC_Sep2013/C4-reference/ExpressionInput/counts.C4.txt'
#fastRPKMCalculate(filename);sys.exit()
file1 = '/Volumes/My Passport/dataAnalysis/CardiacRNASeq/BedFiles/ExpressionInput/exp.CardiacRNASeq.txt'
file2 = '/Volumes/Time Machine Backups/dataAnalysis/PCBC_Sep2013/C4-reference/ReferenceComps/ExpressionInput/counts.C4.txt'
#getHighExpNovelExons('Hs',file1);sys.exit()
#mergeCountFiles(file1,file2); sys.exit()
import UI
test_status = 'yes'
data_type = 'ncRNA'
data_type = 'mRNA'
array_type = 'RNASeq'
array_type = 'junction'
species = 'Hs' ### edit this
summary_results_db = {}
root_dir = '/Volumes/Time Machine Backups/dataAnalysis/Human Blood/Exon/Multiple Sclerosis/Untreated_MS-analysis/'
#root_dir = '/Volumes/Time Machine Backups/dataAnalysis/Human Blood/Exon/Multiple Sclerosis/2-3rds_training-untreated/'
root_dir = '/Volumes/SEQ-DATA/Grimes/14018_gmp-pro/400-original/'
#root_dir = '/Volumes/My Passport/dataAnalysis/PCBC_Dec2013/All/bedFiles/'
root_dir = '/Users/saljh8/Desktop/dataAnalysis/HTA2.0 Files/'
#summary_results_db['Hs_Junction_d14_vs_d7.p5_average-ASPIRE-exon-inclusion-results.txt'] = [] ### edit this
#summary_results_db['Hs_Junction_d14_vs_d7.p5_average-splicing-index-exon-inclusion-results.txt'] = [] ### edit this
results_dir = root_dir +'AltResults/AlternativeOutput/'
dir_list = read_directory(results_dir)
for i in dir_list:
if '_average' in i:
comparison, end = string.split(i,'_average')
if '-exon-inclusion-results.txt' in i: summary_results_db[comparison]=[]
compareExonAndJunctionResults(species,array_type,summary_results_db,root_dir); sys.exit()
fl = UI.ExpressionFileLocationData('','','',''); fl.setCELFileDir(loc); fl.setRootDir(loc)
exp_file_location_db={}; exp_file_location_db['test']=fl
alignJunctionsToEnsembl(species,exp_file_location_db,'test'); sys.exit()
getEnsemblAssociations(species,data_type,test_status,'yes'); sys.exit()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/RNASeq.py
|
RNASeq.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import math
from stats_scripts import statistics
import sys, string
import shutil
import os.path
import unique
import update; reload(update)
import export
import ExpressionBuilder
import time
import webbrowser
import traceback
import AltAnalyze
from sys import argv
"""
import numpy
import scipy
from PIL import Image as PIL_Image
import ImageTk
import matplotlib
import matplotlib.pyplot as pylab
"""
try:
try:
from visualization_scripts import WikiPathways_webservice
except Exception:
#print traceback.format_exc()
if 'URLError' in traceback.format_exc():
print 'No internet connection found'
else:
print 'WikiPathways visualization not supported (requires installation of suds)'
try:
from PIL import Image as PIL_Image
try: import ImageTk
except Exception: from PIL import ImageTk
import PIL._imaging
import PIL._imagingft
except Exception:
print traceback.format_exc()
#print 'Python Imaging Library not installed... using default PNG viewer'
None
try:
### Only used to test if matplotlib is installed
#import matplotlib
#import matplotlib.pyplot as pylab
None
except Exception:
#print traceback.format_exc()
print 'Graphical output mode disabled (requires matplotlib, numpy and scipy)'
None
except Exception:
None
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>1 and '-' in command_args and '--GUI' not in command_args:
runningCommandLine = True
else:
runningCommandLine = False
try:
import Tkinter
#import bwidget; from bwidget import *
from Tkinter import *
from visualization_scripts import PmwFreeze
from Tkconstants import LEFT
import tkMessageBox
import tkFileDialog
except Exception: print "\nPmw or Tkinter not found... proceeding with manual input"
mac_print_mode = 'no'
if os.name == 'posix': mac_print_mode = 'yes' #os.name is 'posix', 'nt', 'os2', 'mac', 'ce' or 'riscos'
debug_mode = 'no'
def filepath(filename):
fn = unique.filepath(filename)
return fn
def osfilepath(filename):
fn = filepath(filename)
fn = string.replace(fn,'\\','/')
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir)
return dir_list
def getFolders(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Only get folder names
for entry in dir_list:
if entry[-4:] != ".txt" and entry[-4:] != ".csv" and ".zip" not in entry: dir_list2.append(entry)
return dir_list2
def returnDirectoriesNoReplace(dir):
dir_list = unique.returnDirectoriesNoReplace(dir); dir_list2 = []
for entry in dir_list:
if '.' not in entry and 'affymetrix' not in entry:
if 'EnsMart' in entry: dir_list2.append(entry)
return dir_list2
def returnFilesNoReplace(dir):
dir_list = unique.returnDirectoriesNoReplace(dir); dir_list2 = []
for entry in dir_list:
if '.' in entry: dir_list2.append(entry)
return dir_list2
def identifyCELfiles(dir,array_type,vendor):
dir_list = read_directory(dir); dir_list2=[]; full_dir_list=[]
datatype = 'arrays'
types={}
for file in dir_list:
original_file = file
file_lower = string.lower(file); proceed = 'no'
### "._" indicates a mac alias
if ('.cel' in file_lower[-4:] and '.cel.' not in file_lower) and file_lower[:2] != '._':
proceed = 'yes'
elif ('.bed' in file_lower[-4:] or '.tab' in file_lower or '.junction_quantification.txt' in file_lower or '.bam' in file_lower) and file_lower[:2] != '._' and '.bai' not in file_lower:
proceed = 'yes'
datatype = 'RNASeq'
elif array_type == "3'array" and '.cel' not in file_lower[-4:] and '.txt' in file_lower[-4:] and vendor != 'Affymetrix':
proceed = 'yes'
if proceed == 'yes':
if '__' in file and '.cel' not in string.lower(file):
#print file,string.split(file,'__'),file[-4:]
file=string.split(file,'__')[0]+file[-4:]
if '.tab' in original_file: file = string.replace(file,'.txt','.tab')
elif '.bed' in original_file: file = string.replace(file,'.txt','.bed')
if '.TAB' in original_file: file = string.replace(file,'.txt','.TAB')
elif '.BED' in original_file: file = string.replace(file,'.txt','.BED')
dir_list2.append(file)
file = dir+'/'+file
full_dir_list.append(file)
dir_list2 = unique.unique(dir_list2)
full_dir_list = unique.unique(full_dir_list)
dir_list2.sort(); full_dir_list.sort()
if datatype == 'RNASeq':
checkBEDFileFormat(dir) ### Make sure the names are wonky
dir_list3=[]
c = string.lower(string.join(dir_list2,''))
if '.bam' in c and '.bed' in c: #If bed present use bed and not bam
for i in dir_list2:
if '.bam' not in i:
dir_list3.append(i)
dir_list2 = dir_list3
elif '.bam' in c:
for i in dir_list2:
if '.bam' in i:
dir_list3.append(string.replace(i,'.bam','.bed'))
elif '.BAM' in i:
dir_list3.append(string.replace(i,'.BAM','.bed'))
dir_list2 = dir_list3
return dir_list2,full_dir_list
def checkBEDFileFormat(bed_dir):
""" This checks to see if some files have two underscores and one has none or if double underscores are missing from all."""
dir_list = read_directory(bed_dir)
condition_db={}
for filename in dir_list:
if '.tab' in string.lower(filename) or '.bed' in string.lower(filename) or '.junction_quantification.txt' in string.lower(filename):
condition_db[filename]=[]
if len(condition_db)==0: ### Occurs if BAMs present but not .bed files
for filename in dir_list:
if '.bam' in string.lower(filename):
condition_db[filename]=[]
### Check to see if exon.bed and junction.bed file names are propper or faulty (which will result in downstream errors)
double_underscores=[]
no_doubles=[]
for condition in condition_db:
if '__' in condition:
double_underscores.append(condition)
else:
no_doubles.append(condition)
exon_beds=[]
junctions_beds=[]
if len(double_underscores)>0 and len(no_doubles)>0:
### Hence, a problem is likely due to inconsistent naming
print_out = 'The input files appear to have inconsistent naming. If both exon and\njunction sample data are present, make sure they are named propperly.\n\n'
print_out += 'For example: cancer1__exon.bed, cancer1__junction.bed\n(double underscore required to match these samples up)!\n\n'
print_out += 'Exiting AltAnalyze'
IndicatorWindowSimple(print_out,'Quit')
sys.exit()
elif len(no_doubles)>0:
for condition in no_doubles:
condition = string.lower(condition)
if 'exon' in condition:
exon_beds.append(condition)
if 'junction' in condition:
junctions_beds.append(condition)
if len(exon_beds)>0 and len(junctions_beds)>0:
print_out = 'The input files appear to have inconsistent naming. If both exon and\njunction sample data are present, make sure they are named propperly.\n\n'
print_out += 'For example: cancer1__exon.bed, cancer1__junction.bed\n(double underscore required to match these samples up)!\n\n'
print_out += 'Exiting AltAnalyze'
IndicatorWindowSimple(print_out,'Quit')
sys.exit()
def identifyArrayType(full_dir_list):
#import re
arrays={}; array_type=None ### Determine the type of unique arrays in each directory
for filename in full_dir_list:
fn=filepath(filename); ln=0
for line in open(fn,'rU').xreadlines():
if '\x00' in line: ### Simple way of determining if it is a version 4 file with encoding
line = string.replace(line,'\x00\x00',' ') ### retains spaces
line = string.replace(line,'\x00','') ### returns human readable line
if ln<150:
data = cleanUpLine(line); ln+=1
if 'sq' in data:
try:
#fileencoding = "iso-8859-1"
#txt = line.decode(fileencoding); print [txt];kill ### This works but so does the above
array_info,null = string.split(data,'sq')
array_info = string.split(array_info,' ')
array_type = array_info[-1]
if '.' in array_type: array_type,null = string.split(array_type,'.')
#array_type = string.join(re.findall(r"\w",array_type),'') ### should force only alphanumeric but doesn't seem to always work
arrays[array_type]=[]
#print array_type+'\t'+filename
break
except Exception: pass
elif 'affymetrix-array-type' in data:
null, array_type = string.split(data,'affymetrix-array-type')
if '.' in array_type: array_type,null = string.split(array_type,'.')
arrays[array_type]=[]
"""else: ### some CEL file versions are encoded
fileencoding = "iso-8859-1"
txt = line.decode(fileencoding)
print txt;kill"""
else: break
array_ls = []
for array in arrays:
if len(array)<50: array_ls.append(array) ### Occurs with version 4 encoding (bad entries added)
return array_ls, array_type
def getAffyFilesRemote(array_name,arraytype,species):
global backSelect; global array_type; global debug_mode
debug_mode = 'yes'
backSelect = 'yes'
array_type = arraytype
library_dir, annotation_dir, bgp_file, clf_file = getAffyFiles(array_name,species)
return library_dir, annotation_dir, bgp_file, clf_file
def getAffyFiles(array_name,species):#('AltDatabase/affymetrix/LibraryFiles/'+library_file,species)
sa = supproted_array_db[array_name]; library_file = sa.LibraryFile(); annot_file = sa.AnnotationFile(); original_library_file = library_file
filename = 'AltDatabase/affymetrix/LibraryFiles/'+library_file
fn=filepath(filename); library_dir=filename; bgp_file = ''; clf_file = ''
local_lib_files_present = False
if backSelect == 'yes': warn = 'no'
else: warn = 'yes'
try:
for line in open(fn,'rU').xreadlines():break
### Hence, the library file was found!!!
local_lib_files_present = True
input_cdf_file = filename
if '.pgf' in input_cdf_file:
###Check to see if the clf and bgp files are present in this directory
icf_list = string.split(input_cdf_file,'/'); parent_dir = string.join(icf_list[:-1],'/'); cdf_short = icf_list[-1]
clf_short = string.replace(cdf_short,'.pgf','.clf')
if array_type == 'exon' or array_type == 'junction':
bgp_short = string.replace(cdf_short,'.pgf','.antigenomic.bgp')
else: bgp_short = string.replace(cdf_short,'.pgf','.bgp')
try: dir_list = read_directory(parent_dir)
except Exception: dir_list = read_directory('/'+parent_dir)
if clf_short in dir_list and bgp_short in dir_list:
pgf_file = input_cdf_file; clf_file = string.replace(pgf_file,'.pgf','.clf')
if array_type == 'exon' or array_type == 'junction': bgp_file = string.replace(pgf_file,'.pgf','.antigenomic.bgp')
else: bgp_file = string.replace(pgf_file,'.pgf','.bgp')
else:
try:
print_out = "The directory;\n"+parent_dir+"\ndoes not contain either a .clf or antigenomic.bgp\nfile, required for probeset summarization."
IndicatorWindow(print_out,'Continue')
except Exception: print print_out; sys.exit()
except Exception:
print_out = "AltAnalyze was not able to find a library file\nfor your arrays. Would you like AltAnalyze to\nautomatically download these files?"
try:
dw = DownloadWindow(print_out,'Download by AltAnalyze','Select Local Files')
warn = 'no' ### If already downloading the library, don't warn to download the csv too
dw_results = dw.Results(); option = dw_results['selected_option']
except Exception: option = 1 ### Occurs when Tkinter is not present - used by CommandLine mode
if option == 1:
library_file = string.replace(library_file,'.cdf','.zip')
filename = 'AltDatabase/affymetrix/LibraryFiles/'+library_file
input_cdf_file = filename
if '.pgf' in input_cdf_file:
pgf_file = input_cdf_file; clf_file = string.replace(pgf_file,'.pgf','.clf')
if array_type == 'exon' or array_type == 'junction': bgp_file = string.replace(pgf_file,'.pgf','.antigenomic.bgp')
else: bgp_file = string.replace(pgf_file,'.pgf','.bgp')
filenames = [pgf_file+'.gz',clf_file+'.gz',bgp_file+'.gz']
if 'Glue' in pgf_file:
kil_file = string.replace(pgf_file,'.pgf','.kil') ### Only applies to the Glue array
filenames.append(kil_file+'.gz')
else: filenames = [input_cdf_file]
for filename in filenames:
var_list = filename,'LibraryFiles'
if debug_mode == 'no': StatusWindow(var_list,'download')
else:
for filename in filenames:
continue_analysis = update.downloadCurrentVersion(filename,'LibraryFiles','')
try: os.remove(filepath(filename)) ### Not sure why this works now and not before
except Exception: pass
else: library_dir = ''
filename = 'AltDatabase/affymetrix/'+species+'/'+annot_file
fn=filepath(filename); annotation_dir = filename
try:
for line in open(fn,'rU').xreadlines():break
except Exception:
if warn == 'yes' and local_lib_files_present == False:
### Indicates that library file wasn't present to prior to this method
print_out = "AltAnalyze was not able to find a CSV annotation file\nfor your arrays. Would you like AltAnalyze to\nautomatically download these files?"
try:
dw = DownloadWindow(print_out,'Download by AltAnalyze','Select Local Files'); warn = 'no'
dw_results = dw.Results(); option = dw_results['selected_option']
except OSError: option = 1 ### Occurs when Tkinter is not present - used by CommandLine mode
else:
try: option = option
except Exception: option = 2
if option == 1 or debug_mode=='yes':
annot_file += '.zip'
filenames = ['AltDatabase/affymetrix/'+species+'/'+annot_file]
for filename in filenames:
var_list = filename,'AnnotationFiles'
if debug_mode == 'no': StatusWindow(var_list,'download')
else:
for filename in filenames:
try: update.downloadCurrentVersionUI(filename,'AnnotationFiles','',Tk())
except Exception:
try: update.downloadCurrentVersion(filename,'AnnotationFiles',None)
except Exception: pass ### Don't actually need Affy's annotations in most cases - GO-Elite used instead
try: os.remove(filepath(filename)) ### Not sure why this works now and not before
except Exception: pass
else: annotation_dir = ''
return library_dir, annotation_dir, bgp_file, clf_file
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
########### Status Window Functions ###########
def copyFiles(file1,file2,root):
print 'Copying files from:\n',file1
data = export.ExportFile(file2) ### Ensures the directory exists
try: shutil.copyfile(file1,file2)
except Exception: print "This file already exists in the destination directory."
root.destroy()
class StatusWindow:
def __init__(self,info_list,analysis_type,windowType='parent'):
try:
if windowType == 'child':
root = Toplevel()
else:
root = Tk()
self._parent = root
root.title('AltAnalyze version 2.1.4')
statusVar = StringVar() ### Class method for Tkinter. Description: "Value holder for strings variables."
height = 300; width = 700
if os.name != 'nt': height+=100; width+=50
self.sf = PmwFreeze.ScrolledFrame(self._parent,
labelpos = 'n', label_text = 'Download File Status Window',
usehullsize = 1, hull_width = width, hull_height = height)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
group = PmwFreeze.Group(self.sf.interior(),tag_text = 'Output')
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
Label(group.interior(),width=180,height=1000,justify=LEFT, bg='black', fg = 'white',anchor=NW,padx = 5,pady = 5, textvariable=statusVar).pack(fill=X,expand=Y)
status = StringVarFile(statusVar,root) ### Captures the stdout (or print) to the GUI instead of to the terminal
self.original_sys_out = sys.stdout ### Save the original stdout mechanism
#ProgressBar('Download',self._parent)
except Exception: None
if analysis_type == 'download':
filename,dir = info_list
try: sys.stdout = status; root.after(100,update.downloadCurrentVersionUI(filename,dir,None,self._parent))
except Exception:
update.downloadCurrentVersion(filename,dir,None)
if analysis_type == 'copy':
file1,file2 = info_list
try: sys.stdout = status; root.after(100,copyFiles(file1,file2,self._parent))
except Exception: copyFiles(file1,file2,None)
if analysis_type == 'getOnlineDBConfig':
file_location_defaults = info_list
try: sys.stdout = status; root.after(100,getOnlineDBConfig(file_location_defaults,self._parent))
except Exception,e: getOnlineDBConfig(file_location_defaults,None)
if analysis_type == 'getOnlineEliteDatabase':
file_location_defaults,db_version,new_species_codes,update_goelite_resources = info_list
try: sys.stdout = status; root.after(100,getOnlineEliteDatabase(file_location_defaults,db_version,new_species_codes,update_goelite_resources,self._parent))
except Exception,e: getOnlineEliteDatabase(file_location_defaults,db_version,new_species_codes,update_goelite_resources,None)
if analysis_type == 'getAdditionalOnlineResources':
species_code,additional_resources = info_list
try: sys.stdout = status; root.after(100,getAdditionalOnlineResources(species_code,additional_resources,self._parent))
except Exception,e: getAdditionalOnlineResources(species_code,additional_resources,None)
if analysis_type == 'createHeatMap':
filename, row_method, row_metric, column_method, column_metric, color_gradient, transpose, contrast = info_list
try: sys.stdout = status; root.after(100,createHeatMap(filename, row_method, row_metric, column_method, column_metric, color_gradient, transpose, contrast, self._parent))
except Exception,e: createHeatMap(filename, row_method, row_metric, column_method, column_metric, color_gradient, transpose,contrast,None)
if analysis_type == 'performPCA':
filename, pca_labels, dimensions, pca_algorithm, transpose, geneSetName, species, zscore, colorByGene, reimportModelScores, maskGroups = info_list
try: sys.stdout = status; root.after(100,performPCA(filename, pca_labels, pca_algorithm, transpose, self._parent, plotType = dimensions, geneSetName=geneSetName, species=species, zscore=zscore, colorByGene=colorByGene, reimportModelScores=reimportModelScores, maskGroups=maskGroups))
except Exception,e: performPCA(filename, pca_labels, pca_algorithm, transpose, None, plotType = dimensions, geneSetName=geneSetName, species=species, zscore=zscore, colorByGene=colorByGene, reimportModelScores=reimportModelScores, maskGroups=maskGroups)
if analysis_type == 'runLineageProfiler':
fl, filename, vendor, custom_markerFinder, geneModel_file, modelDiscovery = info_list
try: sys.stdout = status; root.after(100,runLineageProfiler(fl, filename, vendor, custom_markerFinder, geneModel_file, self._parent, modelSize=modelDiscovery))
except Exception,e: runLineageProfiler(fl, filename, vendor, custom_markerFinder, geneModel_file, None, modelSize=modelDiscovery)
if analysis_type == 'MergeFiles':
files_to_merge, join_option, ID_option, output_merge_dir = info_list
try: sys.stdout = status; root.after(100,MergeFiles(files_to_merge, join_option, ID_option, output_merge_dir, self._parent))
except Exception,e: MergeFiles(files_to_merge, join_option, ID_option, output_merge_dir, None)
if analysis_type == 'VennDiagram':
files_to_merge, output_venn_dir = info_list
try: sys.stdout = status; root.after(100,vennDiagram(files_to_merge, output_venn_dir, self._parent))
except Exception,e: vennDiagram(files_to_merge, output_venn_dir, None)
if analysis_type == 'AltExonViewer':
species,platform,exp_file,gene,show_introns,analysisType = info_list
try: sys.stdout = status; root.after(100,altExonViewer(species,platform,exp_file,gene,show_introns,analysisType,self._parent))
except Exception,e: altExonViewer(species,platform,exp_file,gene,show_introns,analysisType,None)
if analysis_type == 'network':
inputDir,inputType,outputdir,interactionDirs,degrees,expressionFile,gsp = info_list
try: sys.stdout = status; root.after(100,networkBuilder(inputDir,inputType,outputdir,interactionDirs,degrees,expressionFile,gsp, self._parent))
except Exception,e: networkBuilder(inputDir,inputType,outputdir,interactionDirs,degrees,expressionFile,gsp, None)
if analysis_type == 'IDConverter':
filename, species_code, input_source, output_source = info_list
try: sys.stdout = status; root.after(100,IDconverter(filename, species_code, input_source, output_source, self._parent))
except Exception,e: IDconverter(filename, species_code, input_source, output_source, None)
if analysis_type == 'predictGroups':
try: expFile, mlp_instance, gsp, reportOnly = info_list
except Exception: expFile, mlp_instance, gsp, reportOnly = info_list
try: sys.stdout = status; root.after(100,predictSampleExpGroups(expFile, mlp_instance, gsp, reportOnly, self._parent))
except Exception,e: predictSampleExpGroups(expFile, mlp_instance, gsp, reportOnly, None)
if analysis_type == 'preProcessRNASeq':
species,exp_file_location_db,dataset,mlp_instance = info_list
try: sys.stdout = status; root.after(100,preProcessRNASeq(species,exp_file_location_db,dataset,mlp_instance, self._parent))
except Exception,e: preProcessRNASeq(species,exp_file_location_db,dataset,mlp_instance, None)
try:
self._parent.protocol("WM_DELETE_WINDOW", self.deleteWindow)
self._parent.mainloop()
self._parent.destroy()
except Exception: None ### This is what typically get's called
try:
sys.stdout = self.original_sys_out ### Has to be last to work!!!
except Exception: None
def deleteWindow(self):
#tkMessageBox.showwarning("Quit Selected","Use 'Quit' button to end program!",parent=self._parent)
self._parent.destroy(); sys.exit()
def quit(self):
try: self._parent.destroy(); sys.exit() #self._parent.quit();
except Exception: sys.exit() #self._parent.quit();
def SysOut(self):
return self.original_sys_out
def preProcessRNASeq(species,exp_file_location_db,dataset,mlp_instance,root):
for dataset in exp_file_location_db:
flx = exp_file_location_db[dataset]
if root == None: display=False
else: display=True
runKallisto = False
try:
import RNASeq
from build_scripts import ExonArray
expFile = flx.ExpFile()
count = verifyFileLength(expFile)
try: fastq_folder = flx.RunKallisto()
except Exception: fastq_folder = []
try: customFASTA = flx.CustomFASTA()
except Exception: customFASTA = None
try: matrix_file = flx.ChromiumSparseMatrix()
except Exception: matrix_file = []
if len(matrix_file)>0:
print 'Exporting Chromium sparse matrix file to tab-delimited-text'
try:
#print expFile, 'expFile'
#print flx.RootDir(), 'root_dir'
output_dir = export.findParentDir(expFile)
try: os.mkdir(output_dir)
except Exception: pass
matrix_dir = export.findParentDir(matrix_file)
genome = export.findFilename(matrix_dir[:-1])
parent_dir = export.findParentDir(matrix_dir[:-1])
from import_scripts import ChromiumProcessing
ChromiumProcessing.import10XSparseMatrix(matrix_file,genome,dataset,expFile=expFile)
except Exception:
print 'Chromium export failed due to:',traceback.format_exc()
try: root.destroy()
except Exception: pass
return None
elif len(fastq_folder)>0 and count<2:
print 'Pre-processing input files'
try:
parent_dir = export.findParentDir(expFile)
flx.setRootDir(parent_dir)
RNASeq.runKallisto(species,dataset,flx.RootDir(),fastq_folder,mlp_instance,returnSampleNames=False,customFASTA=customFASTA)
except Exception:
print 'Kallisto failed due to:',traceback.format_exc()
try: root.destroy()
except Exception: pass
return None
elif len(fastq_folder)>0 and count>1:
try: root.destroy()
except Exception: pass
return None ### Already run
elif count<2:
print 'Pre-processing input BED/BAM files\n'
analyzeBAMs=False
bedFilesPresent=False
dir_list = unique.read_directory(flx.BEDFileDir())
for file in dir_list:
if '.bam' in string.lower(file):
analyzeBAMs=True
if '.bed' in string.lower(file):
bedFilesPresent=True
if analyzeBAMs and bedFilesPresent==False:
print 'No bed files present, deriving from BAM files'
from import_scripts import multiBAMtoBED
bam_dir = flx.BEDFileDir()
refExonCoordinateFile = filepath('AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt')
outputExonCoordinateRefBEDfile = bam_dir+'/BedRef/'+species+'_'+string.replace(dataset,'exp.','')
analysisType = ['exon','junction','reference']
#analysisType = ['junction']
try: multiBAMtoBED.parallelBAMProcessing(bam_dir,refExonCoordinateFile,outputExonCoordinateRefBEDfile,analysisType=analysisType,useMultiProcessing=flx.multiThreading(),MLP=mlp_instance,root=root)
except Exception:
print traceback.format_exc()
try: biotypes = RNASeq.alignExonsAndJunctionsToEnsembl(species,exp_file_location_db,dataset,Multi=mlp_instance)
except Exception:
print traceback.format_exc()
biotypes = getBiotypes(expFile)
else:
biotypes = getBiotypes(expFile)
array_linker_db,array_names = ExonArray.remoteExonProbesetData(expFile,{},'arraynames',flx.ArrayType())
steady_state_export = expFile[:-4]+'-steady-state.txt'
normalize_feature_exp = flx.FeatureNormalization()
try: excludeLowExpressionExons = flx.excludeLowExpressionExons()
except Exception: excludeLowExpressionExons = True
if flx.useJunctionsForGeneExpression():
if 'junction' in biotypes:
feature = 'junction'
else:
feature = 'exon'
else:
### Use all exons either way at this step since more specific parameters will apply to the next iteration
if 'exon' in biotypes:
feature = 'exon'
else:
feature = 'junction'
probeset_db = getAllKnownFeatures(feature,species,flx.ArrayType(),flx.Vendor(),flx)
print 'Calculating gene-level expression values from',feature+'s'
RNASeq.calculateGeneLevelStatistics(steady_state_export,species,probeset_db,normalize_feature_exp,array_names,flx,excludeLowExp=excludeLowExpressionExons,exportRPKMs=True)
#if display == False: print print_out
#try: InfoWindow(print_out, 'Continue')
#except Exception: None
try: root.destroy()
except Exception: pass
except Exception:
error = traceback.format_exc()
#print error
try:
logfile = filepath(fl.RootDir()+'Error.log')
log_report = open(logfile,'a')
log_report.write(traceback.format_exc())
except Exception:
None
print_out = 'Expression quantification failed..\n',error
if runningCommandLine==False:
try: print print_out
except Exception: pass ### Windows issue with the Tk status window stalling after pylab.show is called
try: WarningWindow(print_out,'Continue')
except Exception: pass
try: root.destroy()
except Exception: pass
def getBiotypes(filename):
biotypes={}
firstRow=True
if 'RawSpliceData' in filename: index = 2
else: index = 0
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
t = string.split(line,'\t')
if firstRow:
firstRow = False
else:
if '-' in t[index]:
biotypes['junction']=[]
else:
biotypes['exon']=[]
except Exception: pass
return biotypes
def getAllKnownFeatures(feature,species,array_type,vendor,fl):
### Simple method to extract gene features of interest
from build_scripts import ExonArrayEnsemblRules
source_biotype = 'mRNA'
if array_type == 'gene': source_biotype = 'gene'
elif array_type == 'junction': source_biotype = 'junction'
if array_type == 'AltMouse':
import ExpressionBuilder
probeset_db,constitutive_gene_db = ExpressionBuilder.importAltMerge('full')
source_biotype = 'AltMouse'
elif vendor == 'Affymetrix' or array_type == 'RNASeq':
if array_type == 'RNASeq':
source_biotype = array_type, fl.RootDir()
dbs = ExonArrayEnsemblRules.getAnnotations('no','Ensembl',source_biotype,species)
probeset_db = dbs[0]; del dbs
probeset_gene_db={}
for probeset in probeset_db:
probe_data = probeset_db[probeset]
gene = probe_data[0]; external_exonid = probe_data[-2]
if len(external_exonid)>2: ### These are known exon only (e.g., 'E' probesets)
proceed = True
if feature == 'exon': ### Restrict the analysis to exon RPKM or count data for constitutive calculation
if '-' in probeset and '_' not in probeset: proceed = False
else:
if '-' not in probeset and '_' not in probeset: proceed = False ### Use this option to override
if proceed:
try: probeset_gene_db[gene].append(probeset)
except Exception: probeset_gene_db[gene] = [probeset]
return probeset_gene_db
def RemotePredictSampleExpGroups(expFile, mlp_instance, gsp, globalVars):
global species
global array_type
species, array_type = globalVars
predictSampleExpGroups(expFile, mlp_instance, gsp, False, None, exportAdditionalResults=False)
return graphic_links
def getICGSNMFOutput(root,filename):
if 'exp.' in filename:
filename = string.replace(filename,'exp.','')
umap_scores_file=''
for file in unique.read_directory(root):
if '-UMAP_coordinates.txt' in file and filename in file:
umap_scores_file = file
return root+'/'+umap_scores_file
def exportAdditionalICGSOutputs(expFile,group_selected,outputTSNE=True):
### Remove OutlierRemoved files (they will otherwise clutter up the directory)
if 'OutliersRemoved' in group_selected and 'OutliersRemoved' not in expFile:
try: os.remove(expFile[:-4]+'-OutliersRemoved.txt')
except Exception: pass
try: os.remove(string.replace(expFile[:-4]+'-OutliersRemoved.txt','exp.','groups.'))
except Exception: pass
try: os.remove(string.replace(expFile[:-4]+'-OutliersRemoved.txt','exp.','comps.'))
except Exception: pass
### Create the new groups file but don't over-write the old
import RNASeq
new_groups_dir = RNASeq.exportGroupsFromClusters(group_selected,expFile,array_type,suffix='ICGS')
from import_scripts import sampleIndexSelection; reload(sampleIndexSelection)
### Look to see if a UMAP file exists in ICGS-NMF
if 'ICGS-NMF' in group_selected or 'NMF-SVM' in group_selected:
root = export.findParentDir(export.findParentDir(expFile)[:-1])+'/ICGS-NMF'
umap_scores_file = getICGSNMFOutput(root,export.findFilename(expFile)[:-4])
tSNE_score_file = umap_scores_file
extension = '-UMAP_scores.txt'
elif outputTSNE:
try:
### Build-tSNE plot from the selected ICGS output (maybe different than Guide-3)
tSNE_graphical_links = performPCA(group_selected, 'no', 'UMAP', False, None, plotType='2D',
display=False, geneSetName=None, species=species, zscore=True, reimportModelScores=False,
separateGenePlots=False,returnImageLoc=True)
if 'SNE' in tSNE_graphical_links[-1][-1]:
tSNE_score_file =tSNE_graphical_links[-1][-1][:-10]+'-t-SNE_scores.txt'
extension = '-t-SNE_scores.txt'
else:
tSNE_score_file =tSNE_graphical_links[-1][-1][:-9]+'-UMAP_scores.txt'
extension = '-UMAP_scores.txt'
except Exception:
print traceback.format_exc()
pass
if '-steady-state' in expFile:
newExpFile = string.replace(expFile,'-steady-state','-ICGS-steady-state')
ICGS_order = sampleIndexSelection.getFilters(new_groups_dir)
sampleIndexSelection.filterFile(expFile,newExpFile,ICGS_order)
### Copy the steady-state files for ICGS downstream-specific analyses
ssCountsFile = string.replace(expFile,'exp.','counts.')
newExpFile = string.replace(expFile,'-steady-state','-ICGS-steady-state')
newssCountsFile = string.replace(newExpFile,'exp.','counts.')
exonExpFile = string.replace(expFile,'-steady-state','')
exonCountFile = string.replace(exonExpFile,'exp.','counts.')
newExonExpFile = string.replace(newExpFile,'-steady-state','')
newExonCountsFile = string.replace(newExonExpFile,'exp.','counts.')
sampleIndexSelection.filterFile(ssCountsFile,newssCountsFile,ICGS_order)
sampleIndexSelection.filterFile(exonExpFile,newExonExpFile,ICGS_order)
sampleIndexSelection.filterFile(exonCountFile,newExonCountsFile,ICGS_order)
exonExpFile = exonExpFile[:-4]+'-ICGS.txt'
else:
newExpFile = expFile[:-4]+'-ICGS.txt'
ICGS_order = sampleIndexSelection.getFilters(new_groups_dir)
sampleIndexSelection.filterFile(expFile,newExpFile,ICGS_order)
exonExpFile = newExpFile
if outputTSNE:
try:
status = verifyFile(tSNE_score_file)
if status=='no':
tSNE_score_file = string.replace(tSNE_score_file,'Clustering-','')
### Copy the t-SNE scores to use it for gene expression analyses
if 'DataPlots' not in tSNE_score_file:
outdir = export.findParentDir(export.findParentDir(tSNE_score_file)[:-1])+'/DataPlots'
else:
outdir = export.findParentDir(tSNE_score_file)
exp_tSNE_score_file = outdir+'/'+export.findFilename(exonExpFile)[:-4]+extension
import shutil
export.customFileCopy(tSNE_score_file,exp_tSNE_score_file)
except Exception:
#print traceback.format_exc()
pass
return exonExpFile,newExpFile,new_groups_dir
def predictSampleExpGroups(expFile, mlp_instance, gsp, reportOnly, root, exportAdditionalResults=True):
global graphic_links; graphic_links=[];
if root == None: display=False
else: display=True
import RNASeq,ExpressionBuilder; reload(RNASeq) ### allows for GUI testing with restarting
try:
if gsp.FeaturestoEvaluate() != 'AltExon':
from stats_scripts import ICGS_NMF
reload(ICGS_NMF)
scaling = True ### Performs pagerank downsampling if over 2,500 cells - currently set as a hard coded default
dynamicCorrelation=True
status = verifyFile(expFile)
if status=='no':
expFile = string.replace(expFile,'-steady-state','') ### Occurs for Kallisto processed
graphic_links=ICGS_NMF.runICGS_NMF(expFile,scaling,array_type,species,gsp,enrichmentInput='',dynamicCorrelation=True)
#graphic_links = RNASeq.singleCellRNASeqWorkflow(species, array_type, expFile, mlp_instance, parameters=gsp, reportOnly=reportOnly)
if gsp.FeaturestoEvaluate() != 'Genes':
### For splice-ICGS (needs to be updated in a future version to ICGS_NMF updated code)
graphic_links2,cluster_input_file=ExpressionBuilder.unbiasedComparisonSpliceProfiles(fl.RootDir(),species,array_type,expFile=fl.CountsFile(),min_events=gsp.MinEvents(),med_events=gsp.MedEvents())
gsp.setCountsCutoff(0);gsp.setExpressionCutoff(0)
graphic_links3 = RNASeq.singleCellRNASeqWorkflow(species, 'exons', cluster_input_file, mlp_instance, parameters=gsp, reportOnly=reportOnly)
graphic_links+=graphic_links2+graphic_links3
print_out = 'Predicted sample groups saved.'
if exportAdditionalResults:
### Optionally automatically generate t-SNE and MarkerFinder Results
guide3_results = graphic_links[-1][-1][:-4]+'.txt'
exportAdditionalICGSOutputs(expFile,guide3_results)
if len(graphic_links)==0:
print_out = 'No predicted sample groups identified. Try different parameters.'
if display == False: print print_out
try: InfoWindow(print_out, 'Continue')
except Exception: None
try: root.destroy()
except Exception: pass
except Exception:
error = traceback.format_exc()
if 'score_ls' in error:
error = 'Unknown error likely due to too few genes resulting from the filtering options.'
if 'options_result_in_no_genes' in error:
error = 'ERROR: No genes differentially expressed with the input criterion'
print_out = 'Predicted sample export failed..\n'+error
try: print print_out
except Exception: pass ### Windows issue with the Tk status window stalling after pylab.show is called
try: WarningWindow(print_out,'Continue')
except Exception: pass
try: root.destroy()
except Exception: pass
try: print error
except Exception: pass
def openDirectory(output_dir):
if runningCommandLine:
pass
elif os.name == 'nt':
try: os.startfile('"'+output_dir+'"')
except Exception: os.system('open "'+output_dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+output_dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+output_dir+'"')
def networkBuilder(inputDir,inputType,outputdir,interactionDirs_short,degrees,expressionFile,gsp,root):
species = gsp.Species()
Genes = gsp.GeneSelection()
PathwaySelect = gsp.PathwaySelect()
OntologyID = gsp.OntologyID()
GeneSet = gsp.GeneSet()
IncludeExpIDs = gsp.IncludeExpIDs()
if 'Ontology' in GeneSet: directory = 'nested'
else: directory = 'gene-mapp'
interactionDirs=[]
obligatorySet=[] ### Always include interactions from these if associated with any input ID period
secondarySet=[]
print 'Species:',species, '| Algorithm:',degrees, ' | InputType:',inputType, ' | IncludeExpIDs:',IncludeExpIDs
print 'Genes:',Genes
print 'OntologyID:',gsp.OntologyID(), gsp.PathwaySelect(), GeneSet
print ''
if interactionDirs_short == None or len(interactionDirs_short)==0:
interactionDirs_short = ['WikiPathways']
for i in interactionDirs_short:
if i == None: None
else:
if 'common-' in i:
i = string.replace(i,'common-','')
secondarySet.append(i)
if 'all-' in i:
i = string.replace(i,'all-','')
obligatorySet.append(i)
fn = filepath('AltDatabase/goelite/'+species+'/gene-interactions/Ensembl-'+i+'.txt')
interactionDirs.append(fn)
print "Interaction Files:",string.join(interactionDirs_short,' ')
import InteractionBuilder
try:
output_filename = InteractionBuilder.buildInteractions(species,degrees,inputType,inputDir,outputdir,interactionDirs,Genes=Genes,
geneSetType=GeneSet,PathwayFilter=PathwaySelect,OntologyID=OntologyID,directory=directory,expressionFile=expressionFile,
obligatorySet=obligatorySet,secondarySet=secondarySet,IncludeExpIDs=IncludeExpIDs)
if output_filename==None:
print_out = 'Network creation/visualization failed..\nNo outputs produced... try different options.\n'
print_out += traceback.format_exc()
if root != None and root != '':
try: InfoWindow(print_out, 'Continue')
except Exception: None
else:
if root != None and root != '':
try: openDirectory(outputdir)
except Exception: None
else:
print 'Results saved to:',output_filename
if root != None and root != '':
GUI(root,'ViewPNG',[],output_filename) ### The last is default attributes (should be stored as defaults in the option_db var)
except Exception:
error = traceback.format_exc()
if 'queryGeneError' in error:
print_out = 'No valid gene IDs present in the input text search\n(valid IDs = FOXP1,SOX2,NANOG,TCF7L1)'
else: print_out = 'Network creation/visualization failed..\n',error
if root != None and root != '':
try: InfoWindow(print_out, 'Continue')
except Exception: None
try: root.destroy()
except Exception: None
def vennDiagram(files_to_merge, output_venn_dir, root, display=True):
from visualization_scripts import VennDiagram
if root == None and display==False: display=False
else: display=True
try:
VennDiagram.compareInputFiles(files_to_merge,output_venn_dir,display=display)
if display == False: print 'VennDiagrams saved to:',output_venn_dir
except Exception:
error = traceback.format_exc()
print_out = 'Venn Diagram export failed..\n',error
if root != None and root != '':
try: InfoWindow(print_out, 'Continue')
except Exception: None
try: root.destroy()
except Exception: None
def altExonViewer(species,platform,exp_file,gene,show_introns,analysisType,root):
from visualization_scripts import QC
transpose=True
if root == None: display = False
else: display = True
if analysisType == 'Sashimi-Plot':
showEvent = False
try:
### Create sashimi plot index
from visualization_scripts import SashimiIndex
print 'Indexing splicing-events'
SashimiIndex.remoteIndexing(species,exp_file)
from visualization_scripts import SashimiPlot
#reload(SashimiPlot)
print 'Running Sashimi-Plot...'
genes=None
if '.txt' in gene:
events_file = gene
events = None
else:
gene = string.replace(gene,',',' ')
genes = string.split(gene,' ')
events_file = None
if len(genes)==1:
showEvent = True
SashimiPlot.remoteSashimiPlot(species,exp_file,exp_file,events_file,events=genes,show=showEvent) ### assuming the bam files are in the root-dir
if root != None and root != '':
print_out = 'Sashimi-Plot results saved to:\n'+exp_file+'/SashimiPlots'
try: InfoWindow(print_out, 'Continue')
except Exception: None
except Exception:
error = traceback.format_exc()
print_out = 'AltExon Viewer failed..\n',error
if root != None and root != '':
try: WarningWindow(print_out, 'Continue')
except Exception: None
try: root.destroy()
except Exception: None
else:
#print [analysisType, species,platform,exp_file,gene,transpose,display,show_introns]
try: QC.displayExpressionGraph(species,platform,exp_file,gene,transpose,display=display,showIntrons=show_introns,analysisType=analysisType)
except Exception:
error = traceback.format_exc()
print_out = 'AltExon Viewer failed..\n',error
if root != None and root != '':
try: WarningWindow(print_out, 'Continue')
except Exception: None
try: root.destroy()
except Exception: None
def MergeFiles(files_to_merge, join_option, ID_option, output_merge_dir, root):
from import_scripts import mergeFiles
try: outputfile = mergeFiles.joinFiles(files_to_merge, join_option, ID_option, output_merge_dir)
except Exception:
outputfile = 'failed'
error = traceback.format_exc()
print traceback.format_exc()
if outputfile == 'failed':
print_out = 'File merge failed due to:\n',error
else:
print_out = 'File merge complete. See the new file:\n'+outputfile
if root != None and root!= '':
try: InfoWindow(print_out, 'Continue')
except Exception: None
try: root.destroy()
except Exception: None
if outputfile != 'failed': ### Open the folder
try: openDirectory(output_merge_dir)
except Exception: None
def IDconverter(filename, species_code, input_source, output_source, root):
import gene_associations
try: outputfile = gene_associations.IDconverter(filename, species_code, input_source, output_source)
except Exception:
outputfile = 'failed'
error = traceback.format_exc()
if outputfile == 'failed':
print_out = 'Translation failed due to:\n',error
print print_out
else:
print_out = 'ID translation complete. See the new file:\n'+outputfile
if root != None and root!= '':
try: InfoWindow(print_out, 'Continue')
except Exception: None
try: root.destroy()
except Exception: None
if outputfile != 'failed': ### Open the folder
try: openDirectory(export.findParentDir(filename))
except Exception: None
def remoteLP(fl, expr_input_dir, vendor, custom_markerFinder, geneModel, root, modelSize=None,CenterMethod='centroid'):
global species; global array_type
species = fl.Species()
array_type = fl.PlatformType()
runLineageProfiler(fl, expr_input_dir, vendor, custom_markerFinder, geneModel, root, modelSize=modelSize,CenterMethod=CenterMethod)
def runLineageProfiler(fl, expr_input_dir, vendor, custom_markerFinder, geneModel, root, modelSize=None,CenterMethod='centroid'):
try:
global logfile
root_dir = export.findParentDir(expr_input_dir)
time_stamp = AltAnalyze.timestamp()
logfile = filepath(root_dir+'/AltAnalyze_report-'+time_stamp+'.log')
except: pass
if custom_markerFinder == '': custom_markerFinder = False
if modelSize != None and modelSize != 'no':
try: modelSize = int(modelSize)
except Exception: modelSize = 'optimize'
try:
classificationAnalysis = fl.ClassificationAnalysis()
if custom_markerFinder == False and classificationAnalysis == 'cellHarmony':
classificationAnalysis = 'LineageProfiler'
except Exception:
if custom_markerFinder == False:
classificationAnalysis = 'LineageProfiler'
else:
classificationAnalysis = 'cellHarmony'
if ((geneModel == None or geneModel == False) and (modelSize == None or modelSize == 'no')) and classificationAnalysis != 'cellHarmony':
print 'LineageProfiler'
import ExpressionBuilder; reload(ExpressionBuilder)
compendium_type = fl.CompendiumType()
compendium_platform = fl.CompendiumPlatform()
if 'exp.' in expr_input_dir:
### Correct the input file to be the gene-expression version
if array_type != "3'array" and 'AltExon' not in compendium_type:
if 'steady' not in expr_input_dir:
expr_input_dir = string.replace(expr_input_dir,'.txt','-steady-state.txt')
print '****Running LineageProfiler****'
graphic_links = ExpressionBuilder.remoteLineageProfiler(fl,expr_input_dir,array_type,species,vendor,customMarkers=custom_markerFinder,specificPlatform=True,visualizeNetworks=False)
if len(graphic_links)>0:
print_out = 'Alignments and images saved to the folder "DataPlots" in the input file folder.'
try: InfoWindow(print_out, 'Continue')
except Exception: None
else:
print_out = 'Analysis error occured...\nplease see warning printouts.'
try: print print_out
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
try: WarningWindow(print_out,'Continue')
except Exception: None
try: root.destroy()
except Exception: None
else:
import LineageProfilerIterate
reload(LineageProfilerIterate)
print '****Running cellHarmony****'
codingtype = 'exon'; compendium_platform = 'exon'
platform = array_type,vendor
try: returnCentroids = fl.ReturnCentroids()
except Exception: returnCentroids = 'community'
if returnCentroids == None or returnCentroids == 'None':
""" Pre-clustered/aligned results from ICGS, Seurat or other workflows """
from stats_scripts import preAligned
try: output_dir = fl.OutputDir()
except:
output_dir = os.path.abspath(os.path.join(expr_input_dir, os.pardir))
if 'ExpressionInput' in output_dir:
output_dir = string.replace(output_dir,'ExpressionInput','')
try:
print 'Running cellHarmony differential analysis only'
preAligned.cellHarmony(species,platform,expr_input_dir,output_dir,
customMarkers=custom_markerFinder,useMulti=False,fl=fl)
except ZeroDivisionError:
print_out = traceback.format_exc()
try: InfoWindow(print_out, 'Continue') ### Causes an error when peforming heatmap visualizaiton
except Exception: None
elif returnCentroids == 'community':
""" Match cells between datasets according to their similar clusters defined by
community clustering of the two independent dataset network (same ICGS genes) """
from stats_scripts import cellHarmony
try: output_dir = fl.OutputDir()
except:
output_dir = os.path.abspath(os.path.join(expr_input_dir, os.pardir))
if 'ExpressionInput' in output_dir:
output_dir = string.replace(output_dir,'ExpressionInput','')
try:
print 'Running community cellHarmony analysis'
cellHarmony.manage_louvain_alignment(species,platform,expr_input_dir,output_dir,
customMarkers=custom_markerFinder,useMulti=False,fl=fl)
except ZeroDivisionError:
print_out = traceback.format_exc()
try: InfoWindow(print_out, 'Continue') ### Causes an error when peforming heatmap visualizaiton
except Exception: None
else:
""" Directly match cells between datasets either all cells by all cells or cells
by centroids (same ICGS genes) """
try: cellLabels = fl.Labels()
except: cellLabels = False
try: LineageProfilerIterate.runLineageProfiler(species,platform,expr_input_dir,expr_input_dir,
codingtype,compendium_platform,customMarkers=custom_markerFinder,
geneModels=geneModel,modelSize=modelSize,fl=fl,label_file=cellLabels)
except Exception:
print_out = traceback.format_exc()
try: InfoWindow(print_out, 'Continue') ### Causes an error when peforming heatmap visualizaiton
except Exception: None
print_out = 'cellHarmony classification results saved to the folder "CellClassification".'
if root!=None and root!='':
try: openDirectory(export.findParentDir(expr_input_dir)+'/cellHarmony')
except Exception: None
try: InfoWindow(print_out, 'Continue') ### Causes an error when peforming heatmap visualizaiton
except Exception: None
else:
print print_out
try: root.destroy()
except Exception: None
def performPCA(filename, pca_labels, pca_algorithm, transpose, root, plotType='3D',display=True,
geneSetName=None, species=None, zscore=True, colorByGene=None, reimportModelScores=True,
separateGenePlots=False, returnImageLoc=False, forceClusters=False, maskGroups=None):
from visualization_scripts import clustering; reload(clustering)
graphics = []
if pca_labels=='yes' or pca_labels=='true'or pca_labels=='TRUE': pca_labels=True
else: pca_labels=False
if zscore=='yes': zscore = True
elif zscore=='no': zscore = False
pca_graphical_links=[]
try:
pca_graphical_links = clustering.runPCAonly(filename, graphics, transpose, showLabels=pca_labels,
plotType=plotType,display=display, algorithm=pca_algorithm, geneSetName=geneSetName,
species=species, zscore=zscore, colorByGene=colorByGene, reimportModelScores=reimportModelScores,
separateGenePlots=separateGenePlots, forceClusters=forceClusters, maskGroups=maskGroups)
try: print'Finished building exporting plot.'
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
except Exception:
if 'importData' in traceback.format_exc():
try: print traceback.format_exc(),'\n'
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
print_out = 'Bad input file! Should be a tab-delimited text file with a single\nannotation column and row and the remaining as numeric values.'
else:
try: print traceback.format_exc(),'\n'
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
print_out = 'Analysis error occured...\nplease try again with different parameters.'
try: print print_out
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
try: WarningWindow(print_out,'Continue')
except Exception: None
try: root.destroy()
except Exception: pass
if returnImageLoc:
try: return pca_graphical_links
except Exception: pass
def createHeatMap(filename, row_method, row_metric, column_method, column_metric, color_gradient, transpose, contrast, root, display=True):
graphics = []
try:
from visualization_scripts import clustering; reload(clustering)
clustering.runHCexplicit(filename, graphics, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=display, contrast = contrast)
print_out = 'Finished building heatmap.'
try: print print_out
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
try: root.destroy()
except Exception: pass ### DO NOT PRINT HERE... CONFLICTS WITH THE STOUT
except Exception:
if 'importData' in traceback.format_exc():
try: print traceback.format_exc(),'\n'
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
print_out = 'Bad input file! Should be a tab-delimited text file with a single\nannotation column and row and the remaining as numeric values.'
else:
try: print traceback.format_exc(),'\n'
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
print_out = 'Analysis error occured...\nplease try again with different parameters.'
try: print print_out
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
try: WarningWindow(print_out,'Continue')
except Exception: None
try: root.destroy()
except Exception: pass
def getAdditionalOnlineResources(species_code,additional_resources,root):
if additional_resources[0] == 'customSet':
additional_resources = additional_resources[1]
elif additional_resources == 'All Resources':
additional_resources = importResourceList()
else: additional_resources = [additional_resources]
try:
print 'Adding supplemental GeneSet and Ontology Collections'
from build_scripts import GeneSetDownloader; force = 'yes'
GeneSetDownloader.buildAccessoryPathwayDatabases([species_code],additional_resources,force)
try: print'Finished incorporating additional resources.'
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
except Exception:
print_out = 'Download error encountered for additional ontologies and gene-sets...\nplease try again later.'
try: print print_out
except Exception: None ### Windows issue with the Tk status window stalling after pylab.show is called
try: WarningWindow(print_out,'Continue')
except Exception: None
try: root.destroy()
except Exception: pass
class StringVarFile:
def __init__(self,stringVar,window):
self.__newline = 0; self.__stringvar = stringVar; self.__window = window
def write(self,s): ### Write is called by python when any new print statement is called
new = self.__stringvar.get()
for c in s:
#if c == '\n': self.__newline = 1
if c == '\k': self.__newline = 1 ### This should not be found and thus results in a continous feed rather than replacing a single line
else:
if self.__newline: new = ""; self.__newline = 0
new = new+c
try: self.set(new)
except Exception: pass
#except Exception: None ### Not sure why this occurs
try:
log_report = open(logfile,'a')
log_report.write(s); log_report.close() ### Variable to record each print statement
except Exception: pass
def set(self,s):
try: self.__stringvar.set(s); self.__window.update()
except Exception: pass
def get(self): return self.__stringvar.get()
def flush(self): pass
################# GUI #################
class ProgressBar:
def __init__(self,method,t):
#http://tkinter.unpythonic.net/wiki/ProgressBar
self.progval = IntVar(t)
self.progmsg = StringVar(t); self.progmsg.set(method+" in Progress...")
#b = Button(t, relief=LINK, text="Quit (using bwidget)", command=t.destroy); b.pack()
self.c = ProgressDialog(t, title="Please wait...",
type="infinite",
width=30,
textvariable=self.progmsg,
variable=self.progval,
command=lambda: self.c.destroy()
)
self.update_progress()
def update_progress(self):
self.progval.set(2)
self.c.after(10, self.update_progress)
class ImageFiles:
def __init__(self,shortname,fullpath,return_gif=False):
self.shortname = shortname
self.fullpath = fullpath
self.return_gif = return_gif
def ShortName(self): return self.shortname
def FullPath(self): return self.fullpath
def returnGIF(self): return self.return_gif
def Thumbnail(self):
if self.returnGIF():
gif_path = string.replace(self.FullPath(),'.png','.gif')
return gif_path
else:
png_path = string.replace(self.FullPath(),'.png','_small.png')
return png_path
class GUI:
def PredictGroups(self):
self.button_flag = True
self.graphic_link = {}
try:
from PIL import ImageTk
from PIL import Image
except Exception:
from ImageTk import Image
self.toplevel_list=[] ### Keep track to kill later
self.filename_db={}
filenames=[]
i=1
for (name,file) in graphic_links:
self.filename_db['clusters '+str(i)]=file
filenames.append('clusters '+str(i))
i+=1
filenames_ls = list(filenames)
filenames_ls.reverse()
self.title = 'Select cluster groups for further analysis'
self.option = 'group_select' ### choose a variable name here
self.options = filenames_ls
self.default_option = 0
self.comboBox() ### This is where the cluster group gets selected and stored
# create a frame and pack it
frame1 = Tkinter.Frame(self.parent_type)
frame1.pack(side=Tkinter.TOP, fill=Tkinter.X)
### Convert PNG to GIF and re-size
assigned_index=1
#print filenames
for image_file in filenames:
file_dir = self.filename_db[image_file]
iF = ImageFiles(image_file,file_dir)
im = Image.open(file_dir)
#im.save('Gfi1.gif')
size = 128, 128
im.thumbnail(size, Image.ANTIALIAS)
im.save(iF.Thumbnail()) ### write out the small gif file
option = 'imageView'
self.option=option
#photo1 = Tkinter.PhotoImage(file=iF.Thumbnail())
try: photo1 = ImageTk.PhotoImage(file=iF.Thumbnail()) ### specifically compatible with png files
except: continue
# create the image button, image is above (top) the optional text
def view_FullImageOnClick(image_name):
tl = Toplevel() #### This is the critical location to allow multiple TopLevel instances that don't clash, that are created on demand (by click)
self.toplevel_list.append(tl)
self.graphic_link['WP'] = self.filename_db[image_name]
try: self.viewPNGFile(tl) ### ImageTK PNG viewer
except Exception:
print traceback.format_exc()
try: self.openPNGImage() ### OS default PNG viewer
except Exception: pass
#print assigned_index
if assigned_index == 1:
image_file1 = image_file; #tl1 = Toplevel() ### not good to create here if we have to destroy it, because then we can't re-invoke
button1 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file1, bg='green', command=lambda:view_FullImageOnClick(image_file1)) ### without lamda, the command is called before being clicked
button1.image = photo1; button1.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 2:
image_file2 = image_file; #tl2 = Toplevel()
button2 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file2, bg='green', command=lambda:view_FullImageOnClick(image_file2))
button2.image = photo1; button2.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 3:
image_file3 = image_file; #tl3 = Toplevel()
button3 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file3, bg='green', command=lambda:view_FullImageOnClick(image_file3))
button3.image = photo1; button3.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 4:
image_file4 = image_file; #tl4 = Toplevel()
button4 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file4, bg='green', command=lambda:view_FullImageOnClick(image_file4))
button4.image = photo1; button4.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 5:
image_file5 = image_file; #tl5 = Toplevel()
button5 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file5, bg='green', command=lambda:view_FullImageOnClick(image_file5))
button5.image = photo1; button5.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 6:
image_file6 = image_file; #tl4 = Toplevel()
button6 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file6, bg='green', command=lambda:view_FullImageOnClick(image_file6))
button6.image = photo1; button6.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 7:
image_file7 = image_file; #tl5 = Toplevel()
button7 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file7, bg='green', command=lambda:view_FullImageOnClick(image_file7))
button7.image = photo1; button7.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 8:
image_file8 = image_file; #tl5 = Toplevel()
button8 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file8, bg='green', command=lambda:view_FullImageOnClick(image_file8))
button8.image = photo1; button8.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 9:
image_file9 = image_file; #tl4 = Toplevel()
button9 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file9, bg='green', command=lambda:view_FullImageOnClick(image_file9))
button9.image = photo1; button9.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 10:
image_file10 = image_file; #tl5 = Toplevel()
button10 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file10, bg='green', command=lambda:view_FullImageOnClick(image_file10))
button10.image = photo1; button10.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 11:
image_file11 = image_file; #tl5 = Toplevel()
button11 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file11, bg='green', command=lambda:view_FullImageOnClick(image_file11))
button11.image = photo1; button11.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 12:
image_file12 = image_file; #tl5 = Toplevel()
button12 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file12, bg='green', command=lambda:view_FullImageOnClick(image_file12))
button12.image = photo1; button12.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 13:
image_file13 = image_file; #tl5 = Toplevel()
button13 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file13, bg='green', command=lambda:view_FullImageOnClick(image_file13))
button13.image = photo1; button13.pack(side=Tkinter.TOP, padx=2, pady=2)
elif assigned_index == 14:
image_file14 = image_file; #tl5 = Toplevel()
button14 = Tkinter.Button(frame1, compound=Tkinter.TOP, image=photo1,
text=image_file14, bg='green', command=lambda:view_FullImageOnClick(image_file14))
button14.image = photo1; button14.pack(side=Tkinter.TOP, padx=2, pady=2)
assigned_index+=1
# start the event loop
use_selected_button = Button(self._parent, text="Use Selected", command=self.UseSelected)
use_selected_button.pack(side = 'right', padx = 10, pady = 5)
recluster_button = Button(self._parent, text="Re-Cluster", command=self.ReCluster)
recluster_button.pack(side = 'right', padx = 10, pady = 5)
quit_button = Button(self._parent, text="Quit", command=self.quit)
quit_button.pack(side = 'right', padx = 10, pady = 5)
try: help_button = Button(self._parent, text='Help', command=self.GetHelpTopLevel); help_button.pack(side = 'left', padx = 5, pady = 5)
except Exception: help_button = Button(self._parent, text='Help', command=self.linkout); help_button.pack(side = 'left', padx = 5, pady = 5)
self._parent.protocol("WM_DELETE_WINDOW", self.deleteWindow)
self._parent.mainloop()
def UseSelected(self):
status = self.checkAllTopLevelInstances()
if status:
self.checkAllTopLevelInstances()
self._user_variables['next'] = 'UseSelected'
try: self._parent.quit(); self._parent.destroy()
except Exception: self._parent.quit()
def ReCluster(self):
status = self.checkAllTopLevelInstances()
if status:
self._user_variables['next'] = 'ReCluster'
try: self._parent.quit(); self._parent.destroy()
except Exception:
try: self._parent.destroy()
except Exception: pass
def checkAllTopLevelInstances(self):
### Ideally, we would just kill any open toplevel instances, but this was causing a "ghost" process
### to continue running even after all of the tls and roots were destroyed
if len(self.toplevel_list)>0:
removed=[]
for tl in self.toplevel_list:
try:
if 'normal' == tl.state():
InfoWindow('Please close all cluster windows before proceeding.', 'Continue')
break
except Exception:
removed.append(tl)
for tl in removed:
self.toplevel_list.remove(tl)
if len(self.toplevel_list)==0:
return True
else:
return False
def killAllTopLevelInstances(self):
### destroy's any live TopLevel instances
removed=[]
for tl in self.toplevel_list:
try: tl.quit(); tl.destroy(); removed.append(tl)
except Exception: pass
for tl in removed:
self.toplevel_list.remove(tl)
def ViewWikiPathways(self):
""" Canvas is already drawn at this point from __init__ """
global pathway_db
pathway_db={}
button_text = 'Help'
### Create a species drop-down option that can be updated
current_species_names,manufacturers_list = getSpeciesList('') ### pass the variable vendor to getSpeciesList (none in this case) --- different than the GO-Elite UI call
self.title = 'Select species to search for WikiPathways '
self.option = 'species_wp'
self.options = ['---']+current_species_names #species_list
self.default_option = 0
self.comboBox()
### Create a label that can be updated below the dropdown menu
self.label_name = StringVar()
self.label_name.set('Pathway species list may take several seconds to load')
self.invokeLabel() ### Invoke a new label indicating that the database is loading
### Create a MOD selection drop-down list
system_list,mod_list = importSystemInfo() ### --- different than the GO-Elite UI call
self.title = 'Select the ID system to translate to (MOD)'
self.option = 'mod_wp'
self.options = mod_list
try: self.default_option = mod_list.index('Ensembl') ### Get the Ensembl index number
except Exception: self.default_option = 0
self.dropDown()
### Create a file selection option
self.title = 'Select GO-Elite input ID text file'
self.notes = 'note: ID file must have a header row and at least three columns:\n'
self.notes += '(1) Identifier, (2) System Code, (3) Value to map (- OR +)\n'
self.file_option = 'goelite_input_file'
self.directory_type = 'file'
self.FileSelectionMenu()
dispaly_pathway = Button(text = 'Display Pathway', command = self.displayPathway)
dispaly_pathway.pack(side = 'right', padx = 10, pady = 10)
back_button = Button(self._parent, text="Back", command=self.goBack)
back_button.pack(side = 'right', padx =10, pady = 5)
quit_win = Button(self._parent, text="Quit", command=self.quit)
quit_win.pack(side = 'right', padx =10, pady = 5)
try: help_button = Button(self._parent, text=button_text, command=self.GetHelpTopLevel); help_button.pack(side = 'left', padx = 5, pady = 5)
except Exception: help_button = Button(self._parent, text=button_text, command=self.linkout); help_button.pack(side = 'left', padx = 5, pady = 5)
self._parent.protocol("WM_DELETE_WINDOW", self.deleteWindow)
self._parent.mainloop()
def FileSelectionMenu(self):
option = self.file_option
group = PmwFreeze.Group(self.parent_type,tag_text = self.title)
group.pack(fill = 'both', expand = 1, padx = 10, pady = 2)
def filecallback(callback=self.callback,option=option): self.getPath(option)
default_option=''
entrytxt = StringVar(); #self.entrytxt.set(self.default_dir)
entrytxt.set(default_option)
self.pathdb[option] = entrytxt
self._user_variables[option] = default_option
entry = Entry(group.interior(),textvariable=self.pathdb[option]);
entry.pack(side='left',fill = 'both', expand = 0.7, padx = 10, pady = 2)
button = Button(group.interior(), text="select "+self.directory_type, width = 10, fg="black", command=filecallback)
button.pack(side=LEFT, padx = 2,pady = 2)
if len(self.notes)>0: ln = Label(self.parent_type, text=self.notes,fg="blue"); ln.pack(padx = 10)
def dropDown(self):
def comp_callback(tag,callback=self.callbackWP,option=self.option):
callback(tag,option)
self.comp = PmwFreeze.OptionMenu(self.parent_type,
labelpos = 'w', label_text = self.title, items = self.options, command = comp_callback)
if self.option == 'wp_id_selection':
self.wp_dropdown = self.comp ### update this variable later (optional)
self.comp.pack(anchor = 'w', padx = 10, pady = 0, fill = 'x')
self.comp.invoke(self.default_option) ###Just pick the first option
def comboBox(self):
""" Alternative, more sophisticated UI than dropDown (OptionMenu).
Although it behaves similiar it requires different parameters, can not be
as easily updated with new lists (different method) and requires explict
invokation of callback when a default is set rather than selected. """
def comp_callback(tag,callback=self.callbackWP,option=self.option):
callback(tag,option)
self.comp = PmwFreeze.ComboBox(self.parent_type,
labelpos = 'w', dropdown=1, label_text = self.title,
unique = 0, history = 0,
scrolledlist_items = self.options, selectioncommand = comp_callback)
try: self.comp.component('entryfield_entry').bind('<Button-1>', lambda event, self=self: self.comp.invoke())
except Exception: None ### Above is a slick way to force the entry field to be disabled and invoke the scrolledlist
if self.option == 'wp_id_selection':
self.wp_dropdown = self.comp ### update this variable later (optional)
self.comp.pack(anchor = 'w', padx = 10, pady = 0)
try: self.comp.selectitem(self.default_option) ###Just pick the first option
except Exception: pass
try: self.callbackWP(self.options[0],self.option) ### Explicitly, invoke first option (not automatic)
except Exception: pass
def invokeLabel(self):
self.label_object = Label(self.parent_type, textvariable=self.label_name,fg="blue"); self.label_object.pack(padx = 10)
def enterMenu(self):
if len(self.notes)>0:
lb = Label(self.parent_type, text=self.notes,fg="black"); lb.pack(pady = 5)
### Create and pack a horizontal RadioSelect widget
def custom_validate(tag,custom_validate=self.custom_validate,option=self.option):
validate = custom_validate(tag,self.option)
self.entry_field = PmwFreeze.EntryField(self.parent_type,
labelpos = 'w', label_text = self.title, validate = custom_validate,
value = self.default_option, hull_borderwidth = 2)
self.entry_field.pack(fill = 'x', expand = 0.7, padx = 10, pady = 5)
def displayAnyPNG(self,png_file):
self.graphic_link={}
self.graphic_link['WP'] = png_file
self.graphic_link['quit']=None
try: tl = Toplevel()
except Exception:
import Tkinter
tl = Tkinter.Toplevel()
try: self.viewPNGFile(tl) ### ImageTK PNG viewer
except Exception:
print traceback.format_exc()
try: self.openPNGImage() ### OS default PNG viewer
except Exception:
print 'Unable to open PNG file for unknown reasons'
def displayPathway(self):
filename = self._user_variables['goelite_input_file']
mod_type = self._user_variables['mod_wp']
species = self._user_variables['species_wp']
pathway_name = self._user_variables['wp_id_selection']
wpid_selected = self._user_variables['wp_id_enter']
species_code = species_codes[species].SpeciesCode()
wpid = None
if len(wpid_selected)>0:
wpid = wpid_selected
elif len(self.pathway_db)>0:
for wpid in self.pathway_db:
if pathway_name == self.pathway_db[wpid].WPName():
break
if len(filename)==0:
print_out = 'Select an input ID file with values first'
WarningWindow(print_out,'Error Encountered!')
else:
try:
self.graphic_link = WikiPathways_webservice.visualizePathwayAssociations(filename,species_code,mod_type,wpid)
if len(self.graphic_link)==0:
force_no_matching_error
self.wp_status = 'Pathway images colored and saved to disk by webservice\n(see image title for location)'
self.label_status_name.set(self.wp_status)
tl = Toplevel()
try: self.viewPNGFile(tl) ### ImageTK PNG viewer
except Exception:
try: self.openPNGImage() ### OS default PNG viewer
except Exception:
self.wp_status = 'Unable to open PNG file using operating system'
self.label_status_name.set(self.wp_status)
except Exception,e:
try:
wp_logfile = filepath('webservice.log')
wp_report = open(wp_logfile,'a')
wp_report.write(traceback.format_exc())
except Exception:
None
try:
print traceback.format_exc()
except Exception:
null=None ### Occurs when transitioning back from the Official Database download window (not sure why) -- should be fixed in 1.2.4 (sys.stdout not re-routed)
if 'force_no_matching_error' in traceback.format_exc():
print_out = 'None of the input IDs mapped to this pathway'
elif 'force_invalid_pathway' in traceback.format_exc():
print_out = 'Invalid pathway selected'
elif 'IndexError' in traceback.format_exc():
print_out = 'Input ID file does not have at least 3 columns, with the second column being system code'
elif 'ValueError' in traceback.format_exc():
print_out = 'Input ID file error. Please check that you do not have extra rows with no data'
elif 'source_data' in traceback.format_exc():
print_out = 'Input ID file does not contain a valid system code'
else:
print_out = 'Error generating the pathway "%s"' % pathway_name
WarningWindow(print_out,'Error Encountered!')
def getSpeciesPathways(self,species_full):
pathway_list=[]
self.pathway_db = WikiPathways_webservice.getAllSpeciesPathways(species_full)
for wpid in self.pathway_db:
if self.pathway_db[wpid].WPName() != None: ### Not sure where the None comes from but will break the UI if not exlcuded
pathway_list.append(self.pathway_db[wpid].WPName())
pathway_list = unique.unique(pathway_list)
pathway_list.sort()
return pathway_list
def callbackWP(self, tag, option):
#print 'Button',[option], tag,'was pressed.'
self._user_variables[option] = tag
if option == 'group_select':
### set group_select equal to the filename
self._user_variables[option] = self.filename_db[tag]
#print option, tag
#print option, self._user_variables[option], self.filename_db[tag]
if option == 'species_wp':
### Add additional menu options based on user selection
if tag != '---':
### If this already exists from an earlier iteration
hault = False
self.label_name.set('Loading available WikiPathways')
try:
self.pathway_list=self.getSpeciesPathways(tag)
traceback_printout = ''
except Exception,e:
if 'not supported' in traceback.format_exc():
print_out = 'Species not available at WikiPathways'
WarningWindow(print_out,'Species Not Found!')
traceback_printout=''
hault = True
elif 'URLError' in traceback.format_exc():
print_out = 'Internet connection could not be established'
WarningWindow(print_out,'Internet Error')
traceback_printout=''
hault = True
else:
traceback_printout = traceback.format_exc()
try:
if len(self.pathway_list)>0: ### When true, a valid species was selected in a prior interation invoking the WP fields (need to repopulate)
hault = False
except Exception: None
self.pathway_list = ['None']; self.pathway_db={}
self.label_name.set('')
if hault == False:
try:
### If the species specific wikipathways drop down exists, just update it
self.wp_dropdown._list.setlist(self.pathway_list)
self.wp_dropdown.selectitem(self.pathway_list[0])
self.callbackWP(self.pathway_list[0],'wp_id_selection')
except Exception:
### Create a species specific wikipathways drop down
self.option = 'wp_id_selection'
self.title = 'Select WikiPathways to visualize your data'
if len(traceback_printout)>0:
self.title += traceback_printout ### Display the actual problem in the GUI (sloppy but efficient way for users to indicate the missing driver)
self.options = self.pathway_list
self.default_option = 0
self.comboBox() ### Better UI for longer lists of items (dropDown can't scroll on Linux)
### Create a species specific wikipathways ID enter option
self.notes = 'OR'
self.option = 'wp_id_enter'
self.title = 'Enter the WPID (example: WP254) '
self.default_option = ''
self.enterMenu()
try:
### Create a label that can be updated below the dropdown menu
self.wp_status = 'Pathway image may take several seconds to a minute to load...\n'
self.wp_status += '(images saved to "WikiPathways" folder in input directory)'
try: self.label_status_name.set(self.wp_status)
except Exception:
self.label_status_name = StringVar()
self.label_status_name.set(self.wp_status)
self.invokeStatusLabel() ### Invoke a new label indicating that the database is loading
except Exception:
None
if option == 'wp_id_selection':
### Reset any manually input WPID if a new pathway is selected from dropdown
try: self.entry_field.setentry('')
except Exception: pass
def ShowImageMPL(self):
png_file_dir = self.graphic_link['WP']
fig = pylab.figure()
pylab.subplots_adjust(left=0.0, right=1.0, top=1.0, bottom=0.00) ### Fill the plot area left to right
ax = fig.add_subplot(111)
ax.set_xticks([]) ### Hides ticks
ax.set_yticks([])
img= pylab.imread(png_file_dir)
imgplot = pylab.imshow(img)
pylab.show()
def viewPNGFile(self,tl):
""" View PNG file within a PMW Tkinter frame """
try: import ImageTk ### HAVE TO CALL HERE TO TRIGGER AN ERROR - DON'T WANT THE TopLevel to open otherwise
except Exception:
from PIL import ImageTk
png_file_dir = self.graphic_link['WP']
img = ImageTk.PhotoImage(file=png_file_dir)
sf = PmwFreeze.ScrolledFrame(tl, labelpos = 'n', label_text = '',
usehullsize = 1, hull_width = 800, hull_height = 550)
sf.pack(padx = 0, pady = 0, fill = 'both', expand = 1)
frame = sf.interior()
tl.title(png_file_dir)
can = Canvas(frame)
can.pack(fill=BOTH, padx = 0, pady = 0)
w = img.width()
h = height=img.height()
can.config(width=w, height=h)
can.create_image(2, 2, image=img, anchor=NW)
if 'quit' in self.graphic_link:
tl.protocol("WM_DELETE_WINDOW", lambda: self.tldeleteWindow(tl))
tl.mainloop()
else:
tl.protocol("WM_DELETE_WINDOW", lambda: self.tldeleteWindow(tl))
tl.mainloop()
def openPNGImage(self):
png_file_dir = self.graphic_link['WP']
if runningCommandLine:
pass
elif os.name == 'nt':
try: os.startfile('"'+png_file_dir+'"')
except Exception: os.system('open "'+png_file_dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+png_file_dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+png_file_dir+'"')
def centerPage(self):
# Example of how to use the yview() method of Pmw.ScrolledFrame.
top, bottom = self.sf.yview()
size = bottom - top
middle = 0.5 - size / 2
self.sf.yview('moveto', middle)
def __init__(self, parent, option_db, option_list, defaults):
if option_db == 'ViewPNG':
output_filename = defaults
self.displayAnyPNG(output_filename)
return None
self._parent = parent; self._option_list = option_list; self._option_db = option_db
self._user_variables = user_variables; self.pathdb={}; i = -1
enter_index=0; radio_index=0; dropdown_index=0; check_index=0 ### used to keep track of how many enter boxes we have
self.default_dir = PathDir; self.default_file = PathFile
self.defaults = defaults
filename = 'Config/icon.gif'; orient_type = 'left'
if 'input_cel_dir' in option_list:
filename = 'Config/aa_0.gif'
if array_type == 'RNASeq': filename = 'Config/aa_0_rs.gif'
if '10X' in vendor: filename = 'Config/aa_0_rs.gif'
if 'include_raw_data' in option_list:
filename = 'Config/aa_1.gif'; orient_type = 'top'
if array_type == 'RNASeq': filename = 'Config/aa_1_rs.gif'
if 'filter_for_AS' in option_list:
filename = 'Config/aa_2.gif'; orient_type = 'top'
if array_type == 'RNASeq': filename = 'Config/aa_2_rs.gif'
if 'pathway_permutations' in option_list: filename = 'Config/goelite.gif'
if 'GeneSelectionPredict' in option_list:
filename = 'Config/aa_3.gif'
if array_type == 'RNASeq': filename = 'Config/aa_3_rs.gif'
fn=filepath(filename); img = PhotoImage(file=fn)
self.can = Canvas(parent); self.can.pack(side='top'); self.can.config(width=img.width(), height=img.height())
try: self.can.create_image(2, 2, image=img, anchor=NW)
except Exception:
try: self.can.delete("all")
except Exception: pass
#except Exception: print filename; 'what?';kill
self.pathdb={}; use_scroll = 'no'
#if defaults == 'groups' or defaults == 'comps' or 'filter_for_AS' in option_list:
if defaults != 'null':
height = 350; width = 400
if defaults == 'groups':
notes = "For each sample, type in a name for the group it belongs to\n(e.g., 24hrs, 48hrs, 4days, etc.)."
Label(self._parent,text=notes).pack(); label_text_str = 'AltAnalyze Group Names'
if len(option_list)<15: height = 320; width = 400
elif defaults == 'batch':
notes = "For each sample, type in a name for the BATCH it belongs to\n(e.g., batch1, batch2, batch3 etc.)."
Label(self._parent,text=notes).pack(); label_text_str = 'AltAnalyze Group Names'
if len(option_list)<15: height = 320; width = 400
elif defaults == 'comps':
notes = "Experimental Group\t\t\tBaseline Group "
label_text_str = 'AltAnalyze Pairwise Group Comparisons'
if len(option_list)<5: height = 250; width = 400
elif 'filter_for_AS' in option_list:
label_text_str = 'AltAnalyze Alternative Exon Analysis Parameters'
height = 350; width = 400; use_scroll = 'yes'
if os.name != 'nt': width+=100
elif 'pathway_permutations' in option_list:
label_text_str = 'GO-Elite Parameters'
height = 350; width = 425; use_scroll = 'yes'
elif 'expression_data_format' in option_list:
label_text_str = "AltAnalyze Expression Dataset Parameters"
height = 350; width = 400; use_scroll = 'yes'
if os.name != 'nt': width+=100
elif 'input_lineage_file' in option_list:
label_text_str = "Align and Compare Distinct Single-Cell RNA-Seq Datasets"
height = 400; width = 420; use_scroll = 'yes'
#if os.name != 'nt': width+=50
elif 'Genes_network' in option_list:
label_text_str = "Network Analysis Parameters"
height = 350; width = 400; use_scroll = 'yes'
#if os.name != 'nt': width+=50
elif 'GeneSelectionPredict' in option_list:
notes = "Perform an unsupervised or supervised analysis to identify the\npredominant sample groups via expression clustering"
Label(self._parent,text=notes).pack()
label_text_str = "AltAnalyze Prediction Sample Group Parameters"
height = 310; width = 400; use_scroll = 'yes'
elif 'join_option' in option_list:
label_text_str = "AltAnalyze Merge Files Parameters"
height = 310; width = 400; use_scroll = 'yes'
else:
label_text_str = "AltAnalyze Main Dataset Parameters"
height = 310; width = 400; use_scroll = 'yes'
if os.name != 'nt':height+=75; width+=150
if os.name== 'nt':height+=25; width+=50
if 'linux' in sys.platform: offset = 25
else: offset=0
self.sf = PmwFreeze.ScrolledFrame(self._parent,
labelpos = 'n', label_text = label_text_str,
usehullsize = 1, hull_width = width-offset, hull_height = height-offset)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
if defaults == 'comps':
Label(self.frame,text=notes).pack()
create_group = 'yes'
if 'pathway_permutations' in option_list or 'expression_data_format' in option_list or 'filter_probe_types' in option_list:
if 'ge_ptype' in option_list:
self.group_tag = 'GO-Elite Gene Expression Analysis Filters'
elif 'pathway_permutations' in option_list:
self.group_tag = 'GO-Elite Over-Representation and Filtering Parameters'
if 'expression_threshold' in option_list:
self.group_tag = 'Exon/Junction Filtering Options'
od = option_db['expression_threshold']
if od.ArrayOptions() == ['NA']: create_group = 'no'
if ('rpkm_threshold' in option_list and create_group== 'no'):
create_group='yes'
self.group_tag = 'Gene Expression Filtering Options'
od = option_db['rpkm_threshold']
if od.ArrayOptions() == ['NA']: create_group = 'no'
elif 'expression_data_format' in option_list and 'rpkm_threshold' not in option_list:
self.group_tag = 'Gene Expression Analysis Options'
if 'filter_probe_types' in option_list:
self.group_tag = 'Primary Alternative Exon Parameters'
if create_group == 'yes':
custom_group = PmwFreeze.Group(self.sf.interior(),tag_text = self.group_tag)
custom_group.pack(fill = 'both', expand = 1, padx = 10, pady = 2)
insert_into_group = 'yes'
else: insert_into_group = 'no'
else: insert_into_group = 'no'
object_directions = ['top','bottom','up','down']
if option_db == 'ViewWikiPathways':
width = 520
self.parent_type = self.sf.interior()
self.ViewWikiPathways()
if option_db == 'PredictGroups':
width = 520
self.parent_type = self.sf.interior()
self.PredictGroups()
for option in option_list:
i+=1 ####Keep track of index - if options are deleted, count these to select the appropriate default from defaults
if option in option_db:
od = option_db[option]; self.title = od.Display(); notes = od.Notes()
self.display_options = od.ArrayOptions()
try: override_default = od.DefaultOption()
except Exception: override_default = ''
if 'radio' in od.DisplayObject() and self.display_options != ['NA']:
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
if 'pathway_permutations' in option_list or 'new_run' in option_list: orient_type = 'top'
if insert_into_group == 'yes': parent_type = custom_group.interior(); radio_index+=1
### Create and pack a RadioSelect widget, with radiobuttons.
self._option = option
def radiocallback(tag,callback=self.callback,option=option):
callback(tag,option)
radiobuttons = PmwFreeze.RadioSelect(parent_type,
buttontype = 'radiobutton', orient = 'vertical',
labelpos = 'w', command = radiocallback, label_text = self.title,
hull_borderwidth = 2, hull_relief = 'ridge')
if insert_into_group == 'no': radiobuttons.pack(side = orient_type, expand = 1, padx = 10, pady = 10)
elif radio_index == 1: radiobuttons1 = radiobuttons
elif radio_index == 2: radiobuttons2 = radiobuttons
### print self.display_options
### Add some buttons to the radiobutton RadioSelect.
for text in self.display_options:
if text != ['NA']: radiobuttons.add(text)
if len(override_default)>0: self.default_option = override_default
elif len(defaults) <1:
try: self.default_option = self.display_options[0]
except Exception: print option; kill
else: self.default_option = defaults[i]
radiobuttons.invoke(self.default_option)
if len(notes)>0: Label(self._parent, text=notes).pack()
if 'button' in od.DisplayObject() and self.display_options != ['NA']:
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
self._option = option
if mac_print_mode == 'yes' or 'radbutton' in od.DisplayObject(): button_type = 'radiobutton'
else: button_type = 'button'
### Create and pack a horizontal RadioSelect widget.
if len(override_default)>0: self.default_option = override_default
elif len(defaults) <1: self.default_option = self.display_options[0]
else: self.default_option = defaults[i]
def buttoncallback(tag,callback=self.callback,option=option):
callback(tag,option)
#self.sf.update_idletasks()
#self.centerPage()
orientation = 'vertical'
#if 'pathway_permutations' in option_list or 'new_run' in option_list: orientation = 'vertical'
#elif 'run_from_scratch' in option_list: orientation = 'vertical'
#else: orientation = 'vertical'
horiz = PmwFreeze.RadioSelect(parent_type, buttontype = button_type, orient = orientation,
labelpos = 'w', command = buttoncallback,
label_text = self.title, frame_borderwidth = 2,
frame_relief = 'ridge'
); horiz.pack(fill = 'x',padx = 10, pady = 10)
### Add some buttons to the horizontal RadioSelect
for text in self.display_options:
if text != ['NA']: horiz.add(text)
horiz.invoke(self.default_option)
if len(notes)>0: Label(self._parent, text=notes).pack()
if ('folder' in od.DisplayObject() or 'file' in od.DisplayObject()) and self.display_options != ['NA']:
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
if 'sparse-matrix' in self.title:
od.setDisplayObject('file') ### set the object equal to a file
proceed = 'yes'
#if option == 'raw_input': proceed = 'no'
if proceed == 'yes':
self._option = option
group = PmwFreeze.Group(parent_type,tag_text = self.title)
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
def filecallback(callback=self.callback,option=option): self.getPath(option)
entrytxt = StringVar(); #self.entrytxt.set(self.default_dir)
try: default_option = string.replace(override_default,'---','')
except Exception: default_option = ''
entrytxt.set(default_option)
self.pathdb[option] = entrytxt
self._user_variables[option] = default_option
if option == 'input_cel_dir' and '10X' in vendor:
od.setDisplayObject('file')
#l = Label(group.interior(), text=self.title); l.pack(side=LEFT)
entry = Entry(group.interior(),textvariable=self.pathdb[option]);
entry.pack(side='left',fill = 'both', expand = 1, padx = 10, pady = 0)
button = Button(group.interior(), text="select "+od.DisplayObject(), width = 10, fg="black", command=filecallback); button.pack(side=LEFT, padx = 2,pady = 0)
#print option,run_mappfinder, self.title, self.default_option
if len(notes)>0: ln = Label(parent_type, text=notes,fg="blue"); ln.pack(padx = 10, pady = 0)
if ('update-entry' in od.DisplayObject()) and self.display_options != ['NA']:
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
proceed = 'yes'
#if option == 'raw_input': proceed = 'no'
if proceed == 'yes':
self._option = option
#group = PmwFreeze.Group(parent_type,tag_text = self.title)
#group.pack(fill = 'both', expand = 1, padx = 10, pady = 2)
entrytxt = StringVar(); #self.entrytxt.set(self.default_dir)
try: default_option = defaults[i]
except Exception: default_option = ''
entrytxt.set(default_option)
self.pathdb[option] = entrytxt
self._user_variables[option] = default_option
#l = Label(parent_type, text=self.title); l.pack(side=LEFT)
#entry = Entry(parent_type,textvariable=self.pathdb[option]);
#entry.pack(side='left',fill = 'both', expand = 1, padx = 10, pady = 2)
l = Label(self.sf.interior(), text=self.title); l.pack()
entry = Entry(self.sf.interior(),textvariable=self.pathdb[option]);
entry.pack()
#print option,run_mappfinder, self.title, self.default_option
#if len(notes)>0: ln = Label(parent_type, text=notes,fg="blue"); ln.pack(padx = 10)
if 'drop-down' in od.DisplayObject() and self.display_options != ['NA']:
#print option, defaults, self.display_options
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
if insert_into_group == 'yes': parent_type = custom_group.interior(); dropdown_index+=1
self._option = option
self.default_option = self.display_options
def comp_callback1(tag,callback=self.callback,option=option):
callback(tag,option)
self.comp = PmwFreeze.OptionMenu(parent_type,
labelpos = 'w', label_text = self.title,
items = self.default_option, command = comp_callback1)
try: selected_default = od.DefaultOption()
except Exception:
if len(defaults)>0: selected_default = defaults[i]
else: selected_default = self.default_option[0] ###Just pick the first option
#print option, dropdown_index
if 'species' in option:
if 'selected_species2' in option:
self.speciescomp2 = self.comp; self.speciescomp2.pack(anchor = 'e', padx = 10, pady = 0, expand = 1, fill = 'both')
elif 'selected_species3' in option:
self.speciescomp3 = self.comp; self.speciescomp3.pack(anchor = 'e', padx = 10, pady = 0, expand = 1, fill = 'both')
else: self.speciescomp = self.comp; self.speciescomp.pack(anchor = 'e', padx = 10, pady = 0, expand = 1, fill = 'both')
self.speciescomp.invoke(selected_default)
elif 'array_type' in option:
self.arraycomp = self.comp; self.arraycomp.pack(anchor = 'e', padx = 10, pady = 0, expand = 1, fill = 'both')
self.arraycomp.invoke(selected_default)
elif 'manufacturer_selection' in option:
self.vendorcomp = self.comp; self.vendorcomp.pack(anchor = 'e', padx = 10, pady = 0, expand = 1, fill = 'both')
self.vendorcomp.invoke(selected_default)
else:
if insert_into_group == 'no':
if 'version' in option: pady_int = 0
else: pady_int = 1
self.comp.pack(anchor = 'w', padx = 10, pady = pady_int, expand = 1, fill = 'both')
elif dropdown_index == 1: comp1 = self.comp
elif dropdown_index == 2: comp2 = self.comp
elif dropdown_index == 3: comp3 = self.comp
elif dropdown_index == 4: comp4 = self.comp
elif dropdown_index == 5: comp5 = self.comp
elif dropdown_index == 6: comp6 = self.comp
elif dropdown_index == 7: comp7 = self.comp
elif dropdown_index == 8: comp8 = self.comp
elif dropdown_index == 9: comp9 = self.comp
elif dropdown_index == 10: comp10 = self.comp
elif dropdown_index == 11: comp11 = self.comp
elif dropdown_index == 12: comp12 = self.comp
elif dropdown_index == 13: comp13 = self.comp
try: self.comp.invoke(selected_default)
except Exception:
#self.comp.invoke(self.display_options[0]) # better to know the variable incase their is a conflict
print self.display_options, selected_default, option, option_list;kill
if option == 'selected_version':
notes = 'Note: Available species may vary based on database selection. Also,\n'
notes += 'different Ensembl versions will relate to different genome builds\n'
notes += '(e.g., EnsMart54-74 for hg19 and EnsMart75-current for hg38).\n'
ln = Label(parent_type, text=notes,fg="blue"); ln.pack(padx = 10)
if option == 'probability_algorithm':
notes = 'Note: Moderated tests only run for gene-expression analyses \n'
ln = Label(parent_type, text=notes,fg="blue"); ln.pack(padx = 3)
if 'comboBox' in od.DisplayObject() and self.display_options != ['NA']:
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
if insert_into_group == 'yes': parent_type = custom_group.interior(); dropdown_index+=1
self._option = option
self.default_option = self.display_options
try: selected_default = od.DefaultOption()
except Exception:
if len(defaults)>0: selected_default = defaults[i]
else: selected_default = self.default_option[0] ###Just pick the first option
listbox_selectmode = 'single'
if 'multiple' in od.DisplayObject():
listbox_selectmode = 'multiple'
def comp_callback1(tag,callback=self.callbackComboBox,option=option):
callback(tag,option)
def mult_callback(tag,callback=self.callbackComboBox,option=option):
if 'PathwaySelection' in option:
tag = self.pathwayselect.getcurselection() ### there is a conflict otherwise with another multi-comboBox multcomp object
elif 'HeatmapAdvanced' in option:
tag = self.HeatmapAdvanced.getcurselection() ### there is a conflict otherwise with another multi-comboBox multcomp object
else:
tag = self.multcomp.getcurselection() ### get the multiple item selection
callback(tag,option)
if 'selected_version' not in option_list: ### For clustering UI
label_pos = 'e' ### Orients to the text left -> east
entrywidth = 20 ### Width of entry
#entry_foreground = 'black'
hullsize = 1 #http://pmw.sourceforge.net/doc/ScrolledListBox.html -> doesn't seem to work here
else:
label_pos = 'w' ### Orients to the text right -> west
entrywidth = 20 ### Width of entry
hullsize = 1
if listbox_selectmode == 'multiple':
self.comp = PmwFreeze.ComboBox(parent_type,
labelpos = label_pos, dropdown=1, label_text = self.title,
unique = 0, history = 0, entry_background="light gray", entry_width=entrywidth,
scrolledlist_usehullsize=1,listbox_selectmode=listbox_selectmode,
scrolledlist_items = self.default_option,
selectioncommand = mult_callback)
self.multcomp = self.comp
else:
self.comp = PmwFreeze.ComboBox(parent_type,
labelpos = label_pos, dropdown=1, label_text = self.title,
unique = 0, history = 0, entry_background="light gray", entry_width=entrywidth,
scrolledlist_usehullsize=1,listbox_selectmode=listbox_selectmode,
scrolledlist_items = self.default_option,
selectioncommand = comp_callback1)
if 'HeatmapAdvanced' in option:
self.HeatmapAdvanced = self.multcomp
if 'PathwaySelection' in option or 'PathwaySelection_network' in option:
if 'network' in option:
geneset_param = 'GeneSetSelection_network' ### for network visualization
else:
geneset_param = 'GeneSetSelection' ### for heatmap visualization
self.pathwayselect = self.multcomp; self.pathwayselect.pack(anchor = 'w', padx = 10, pady = 0)
try: self.pathwayselect.component('entryfield_entry').bind('<Button-1>', lambda event, self=self: self.pathwayselect.invoke())
except Exception: None ### Above is a slick way to force the entry field to be disabled and invoke the scrolledlist
try:
### The next several lines are for a second iteration of this analysis to re-select the previously selected parameters
tag = self._user_variables[geneset_param]
if 'Ontology' in tag: directory = 'gene-go'
else: directory = 'gene-mapp'
supported_genesets = self._user_variables[tag]
#print 'loading pathways from memory A1'
#supported_genesets = listAllGeneSetCategories(species,tag,directory)
self.pathwayselect._list.setlist(supported_genesets)
self.pathwayselect.selectitem(selected_default)
self.callbackComboBox(selected_default,option)
except Exception:
try:
self.pathwayselect.selectitem(self.default_option[-1]) ###Just pick the first option
self.callbackComboBox(self.default_option[-1],option)
except Exception: pass
if 'species' in option:
if 'selected_species2' in option:
self.speciescomp2 = self.comp; self.speciescomp2.pack(anchor = 'w', padx = 10, pady = 0)
try: self.speciescomp2.component('entryfield_entry').bind('<Button-1>', lambda event, self=self: self.speciescomp2.invoke())
except Exception: None ### Above is a slick way to force the entry field to be disabled and invoke the scrolledlist
try:
self.speciescomp2.selectitem(selected_default)
self.callbackComboBox(selected_default,option)
except Exception:
self.speciescomp2.selectitem(self.default_option[0]) ###Just pick the first option
self.callbackComboBox(self.default_option[0],option)
elif 'selected_species3' in option:
self.speciescomp3 = self.comp; self.speciescomp3.pack(anchor = 'w', padx = 10, pady = 0)
try: self.speciescomp3.component('entryfield_entry').bind('<Button-1>', lambda event, self=self: self.speciescomp3.invoke())
except Exception: None ### Above is a slick way to force the entry field to be disabled and invoke the scrolledlist
try:
self.speciescomp3.selectitem(selected_default) ###Just pick the first option
self.callbackComboBox(selected_default,option)
except Exception:
self.speciescomp3.selectitem(self.default_option[0])
self.callbackComboBox(self.default_option[0],option)
else:
self.speciescomp = self.comp; self.speciescomp.pack(anchor = 'w', padx = 10, pady = 0)
try: self.speciescomp.component('entryfield_entry').bind('<Button-1>', lambda event, self=self: self.speciescomp.invoke())
except Exception: None ### Above is a slick way to force the entry field to be disabled and invoke the scrolledlist
try:
self.speciescomp.selectitem(selected_default)
self.callbackComboBox(selected_default,option)
except Exception:
self.speciescomp.selectitem(self.default_option[0])
self.callbackComboBox(self.default_option[0],option)
elif 'array_type' in option:
self.arraycomp = self.comp; self.arraycomp.pack(anchor = 'w', padx = 10, pady = 0)
try: self.arraycomp.component('entryfield_entry').bind('<Button-1>', lambda event, self=self: self.arraycomp.invoke())
except Exception: None ### Above is a slick way to force the entry field to be disabled and invoke the scrolledlist
try:
self.arraycomp.selectitem(selected_default)
self.callbackComboBox(selected_default,option)
except Exception:
try:
self.arraycomp.selectitem(self.default_option[0])
self.callbackComboBox(self.default_option[0],option)
except:
pass
elif 'manufacturer_selection' in option:
self.vendorcomp = self.comp; self.vendorcomp.pack(anchor = 'w', padx = 10, pady = 0)
try: self.vendorcomp.component('entryfield_entry').bind('<Button-1>', lambda event, self=self: self.vendorcomp.invoke())
except Exception: None ### Above is a slick way to force the entry field to be disabled and invoke the scrolledlist
try:
self.vendorcomp.selectitem(selected_default)
self.callbackComboBox(selected_default,option)
except Exception:
self.vendorcomp.selectitem(self.default_option[0])
self.callbackComboBox(self.default_option[0],option)
else:
self.combo = self.comp ### has to be a unique combo box to refer to itself in the component call below
self.combo.pack(anchor = 'w', padx = 10, pady = 1)
try: self.combo.component('entryfield_entry').bind('<Button-1>', lambda event, self=self: self.combo.invoke())
except Exception: None ### Above is a slick way to force the entry field to be disabled and invoke the scrolledlist
"""
if listbox_selectmode == 'multiple':
if len(od.DefaultOption()[0])>1: ###Hence it is a list
self.combo.ApplyTypeSelections(od.DefaultOption())
for opt in od.DefaultOption():
self.combo.invoke(opt)
self.callbackComboBox(tuple(od.DefaultOption()),option)
#self.combo.selectitem(opt)
#self.callbackComboBox(opt,option)
"""
#print selected_default
try:
if len(selected_default[0])>1: ###Hence it is a list
for opt in selected_default:
self.combo.selectitem(opt)
self.callbackComboBox(opt,option); break
else:
### This is where the default for the combobox is actually selected for GeneSets
self.combo.selectitem(selected_default)
self.callbackComboBox(selected_default,option)
except Exception:
try:
self.combo.selectitem(self.default_option[0])
self.callbackComboBox(self.default_option[0],option)
except Exception:
None
if option == 'selected_version':
notes = 'Note: Available species may vary based on database selection. Also,\n'
notes += 'different Ensembl versions will relate to different genome builds\n'
notes += '(e.g., EnsMart54-74 for hg19 and EnsMart75-current for hg38).\n'
ln = Label(parent_type, text=notes,fg="blue"); ln.pack(padx = 10)
if 'pulldown_comps' in od.DisplayObject() and self.display_options != ['NA']:
self._option = option
self.default_option = self.display_options
###From the option, create two new options, one for each group in the comparison
option1 = option+'-1'; option2 = option+'-2'
### Pack these into a groups to maintain organization
group = PmwFreeze.Group(self.sf.interior(),tag_text = self.title)
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
if check_index == -1:
check_option = 'analyze_all_conditions'
def checkbuttoncallback(tag,state,checkbuttoncallback=self.checkbuttoncallback,option=check_option):
#print tag,state,option
checkbuttoncallback(tag,state,option)
### Create and pack a vertical RadioSelect widget, with checkbuttons.
self.checkbuttons = PmwFreeze.RadioSelect(self._parent,
buttontype = 'checkbutton', command = checkbuttoncallback)
self.checkbuttons.pack(side = 'top', expand = 1, padx = 0, pady = 0)
### Add some buttons to the checkbutton RadioSelect.
self.checkbuttons.add('Analyze ALL GROUPS in addition to specifying comparisons')
self._user_variables[check_option] = 'no'
check_index+=1
def comp_callback1(tag,callback=self.callback,option1=option1):
callback(tag,option1)
def comp_callback2(tag,callback=self.callback,option2=option2):
callback(tag,option2)
#labelpos = 'w', label_text = self.title, -inside of OptionMenu
self.comp1 = PmwFreeze.OptionMenu(group.interior(),
items = self.default_option, menubutton_width = 20, command = comp_callback1)
self.comp1.pack(side = LEFT, anchor = 'w', padx = 10, pady = 0)
self.comp2 = PmwFreeze.OptionMenu (group.interior(),
items = self.default_option, menubutton_width = 20, command = comp_callback2,
); self.comp2.pack(side = LEFT, anchor = 'w', padx = 10, pady = 0)
try: self.comp1.invoke(notes[0])
except Exception: pass
try: self.comp2.invoke(notes[1])
except Exception: pass
if 'simple_entry' in od.DisplayObject() and self.display_options != ['NA']:
self._option = option
### Create and pack a horizontal RadioSelect widget.
if len(override_default)>0: self.default_option = override_default
else: self.default_option = self.display_options[0]
def enter_callback(tag,enter_callback=self.enter_callback,option=option):
enter_callback(tag,option)
#self.title = self.title + '\t ' #entry_width=entrywidth
self.entry_field = PmwFreeze.EntryField(self.sf.interior(),
labelpos = 'e', label_text = self.title,
validate = enter_callback,
value = self.default_option
); self.entry_field.pack(anchor='w',padx = 10, pady = 1)
if 'enter' in od.DisplayObject() and self.display_options != ['NA']:
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
if insert_into_group == 'yes': parent_type = custom_group.interior(); enter_index+=1
self._option = option
### Create and pack a horizontal RadioSelect widget.
if len(override_default)>0: self.default_option = override_default
elif len(defaults) <1: self.default_option = self.display_options[0]
else: self.default_option = defaults[i]
#print self.default_option, self.title; kill
### entrytxt object for alt_exon_fold_cutoff in option
def custom_validate(tag,custom_validate=self.custom_validate,option=option):
validate = custom_validate(tag,option)
def custom_validate_p(tag,custom_validate_p=self.custom_validate_p,option=option):
validate = custom_validate_p(tag,option)
#print [validate], tag, option
if 'Genes_network' in option_list:
label_pos = 'e' ### Orients to the text left -> east
entrywidth = 20 ### Width of entry entry_width=entrywidth
elif 'GeneSelection' in option_list or 'GeneSelectionPredict' in option_list: ### For clustering UI
label_pos = 'e' ### Orients to the text left -> east
entrywidth = 20 ### Width of entry entry_width=entrywidth
elif 'JustShowTheseIDs' in option_list or 'JustShowTheseIDsPredict' in option_list: ### For clustering UI
label_pos = 'e' ### Orients to the text left -> east
entrywidth = 20 ### Width of entry entry_width=entrywidth
else:
label_pos = 'e'
try:
if float(self.default_option) <= 1: use_method = 'p'
else: use_method = 'i'
except ValueError:
#self.default_option = 'CHANGE TO A NUMERIC VALUE'; use_method = 'i'
self.default_option = string.replace(self.default_option,'---','')
use_method = 'i'
if use_method == 'p':
self.entry_field = PmwFreeze.EntryField(parent_type,
labelpos = label_pos, label_text = self.title, validate = custom_validate_p,
value = self.default_option, hull_borderwidth = 1)
if use_method == 'i':
self.entry_field = PmwFreeze.EntryField(parent_type,
labelpos = label_pos, label_text = self.title, validate = custom_validate,
value = self.default_option, hull_borderwidth = 1)
#if 'GeneSelection' in option_list:
#self.entry_field.component("entry").configure(width=5)
if insert_into_group == 'no': self.entry_field.pack(anchor = 'w', padx = 10, pady = 0)
elif enter_index == 1: self.entry_field1 = self.entry_field
elif enter_index == 2: self.entry_field2 = self.entry_field
elif enter_index == 3: self.entry_field3 = self.entry_field
elif enter_index == 4: self.entry_field4 = self.entry_field
elif enter_index == 5: self.entry_field5 = self.entry_field
if len(notes)>0: Label(self._parent, text=notes).pack()
if 'multiple-checkbox' in od.DisplayObject() and self.display_options != ['NA']:
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
self._option = option
if len(override_default)>0: self.default_option = override_default
elif len(defaults) <1: self.default_option = self.display_options[0]
else: self.default_option = defaults[i]
def checkbuttoncallback(tag,state,checkbuttoncallback=self.checkbuttoncallback,option=option):
checkbuttoncallback(tag,state,option)
### Create and pack a vertical RadioSelect widget, with checkbuttons.
self.checkbuttons = PmwFreeze.RadioSelect(parent_type,
buttontype = 'checkbutton', orient = 'vertical',
labelpos = 'w', command = self.checkbuttoncallback,
label_text = self.title, hull_borderwidth = 2)
self.checkbuttons.pack(padx = 10, pady = 0)
### Add some buttons to the checkbutton RadioSelect.
for text in self.display_options:
if text != ['NA']:
self.checkbuttons.add(text)
#if 'common-' not in text and 'all-' not in text:
#self.checkbuttons.invoke(text)
if len(notes)>0: Label(self._parent, text=notes).pack()
if 'single-checkbox' in od.DisplayObject() and self.display_options != ['NA']:
if use_scroll == 'yes': parent_type = self.sf.interior()
else: parent_type = self._parent
if defaults == 'comps': parent_type = self._parent; orient_type = 'top'
if insert_into_group == 'yes': parent_type = custom_group.interior(); check_index+=1
if defaults == 'groups': parent_type = self.sf.interior(); orient_type = 'top'
self._option = option
proceed = 'yes'
"""if option == 'export_splice_index_values':
if analysis_method != 'splicing-index': proceed = 'no' ### only export corrected constitutive ratios if splicing index method chosen"""
if proceed == 'yes':
if len(override_default)>0: self.default_option = override_default
elif len(defaults) <1: self.default_option = self.display_options[0]
else: self.default_option = defaults[i]
if self.default_option != 'NA':
def checkbuttoncallback(tag,state,checkbuttoncallback=self.checkbuttoncallback,option=option):
checkbuttoncallback(tag,state,option)
### Create and pack a vertical RadioSelect widget, with checkbuttons.
self.checkbuttons = PmwFreeze.RadioSelect(parent_type,
buttontype = 'checkbutton', command = checkbuttoncallback)
#hull_borderwidth = 2, hull_relief = 'ridge')
if insert_into_group == 'no': self.checkbuttons.pack(anchor = 'w',side = orient_type, padx = 10, pady = 1)
elif check_index == 1: checkbuttons1 = self.checkbuttons
elif check_index == 2: checkbuttons2 = self.checkbuttons
elif check_index == 3: checkbuttons3 = self.checkbuttons
elif check_index == 4: checkbuttons4 = self.checkbuttons
### Add some buttons to the checkbutton RadioSelect.
self.checkbuttons.add(self.title)
if self.default_option == 'yes': self.checkbuttons.invoke(self.title)
else: self._user_variables[option] = 'no'
custom_group_endpoints = ['ge_ptype', 'get_additional', 'expression_threshold', 'run_goelite', 'gene_expression_cutoff', 'microRNA_prediction_method']
try:
eod = option_db['expression_threshold']
if eod.ArrayOptions() == ['NA']:
custom_group_endpoints.append('rpkm_threshold') ### Ensures that if analyzing pre-compiled gene expression values, only certain items are shown and in a frame
custom_group_endpoints.remove('expression_threshold')
#insert_into_group = 'yes'
except Exception: pass
if option in custom_group_endpoints and insert_into_group == 'yes':
### This is employed when we want to place several items into a group frame together.
### Since this is a generic class, we need to setup special cases to do this, however,
### this same code could be used in other instances as well
reorganize = 'no'
self.group_tag = 'GO-Elite Over-Representation and Filtering Parameters'; pady_int = 5
if 'run_goelite' in option_list: self.group_tag = 'Gene Expression Analysis Options'; pady_int = 1
if 'microRNA_prediction_method' in option_list: self.group_tag = 'Advanced Options'; pady_int = 1; reorganize = 'yes'
try: checkbuttons1.pack(anchor = 'w', side = 'top', padx = 9, pady = 0)
except Exception: pass
try: checkbuttons2.pack(anchor = 'w', side = 'top', padx = 9, pady = 0)
except Exception: pass
try: checkbuttons3.pack(anchor = 'w', side = 'top', padx = 9, pady = 0)
except Exception: pass
try: checkbuttons4.pack(anchor = 'w', side = 'top', expand = 1, padx = 9, pady = 0)
except Exception: pass
try: radiobuttons2.pack(side = orient_type, expand = 1, padx = 10, pady = 5)
except Exception: pass
try: comp1.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: radiobuttons1.pack(side = orient_type, expand = 1, padx = 10, pady = 5)
except Exception: pass
if reorganize == 'yes':
try: comp2.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: self.entry_field1.pack(anchor = 'w', padx = 10, pady = 0)
except Exception: pass
try: self.entry_field2.pack(anchor = 'w', padx = 10, pady = 0);
except Exception: pass
try: self.entry_field3.pack(anchor = 'w', padx = 10, pady = 0)
except Exception: pass
try: self.entry_field4.pack(anchor = 'w', padx = 10, pady = 0)
except Exception: pass
try: self.entry_field5.pack(anchor = 'w', padx = 10, pady = 0)
except Exception: pass
if reorganize == 'no':
try: comp2.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp3.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp4.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp5.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp6.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp7.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp8.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp9.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp10.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp11.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp12.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
try: comp13.pack(anchor = 'w', padx = 10, pady = pady_int)
except Exception: pass
enter_index=0; radio_index=0; dropdown_index=0
if 'ge_ptype' in option or 'expression_threshold' in option or 'gene_expression_cutoff' in option or 'rpkm_threshold' in option:
custom_group = PmwFreeze.Group(self.sf.interior(),tag_text = self.group_tag)
custom_group.pack(fill = 'both', expand = 1, padx = 10, pady = 10)
insert_into_group = 'yes'
#i+=1 ####Keep track of index
if len(option_list)>0: ### Used when visualizing WikiPathways (no option_list supplied - all parameters hard coded)
#def quitcommand(): parent.destroy; sys.exit()
#self.button = Button(text=" Quit ", command=quitcommand)
#self.button.pack(side = 'bottom', padx = 10, pady = 10)
if 'input_cdf_file' in option_list: ### For the CEL file selection window, provide a link to get Library files
button_text = 'Download Library Files'; d_url = 'http://www.affymetrix.com/support/technical/byproduct.affx?cat=arrays'
self.d_url = d_url; text_button = Button(self._parent, text=button_text, command=self.Dlinkout); text_button.pack(side = 'left', padx = 5, pady = 5)
if 'GeneSelectionPredict' in option_list:
run_button = Button(self._parent, text='Run Analysis', command=self.runPredictGroups)
run_button.pack(side = 'right', padx = 10, pady = 10)
else:
continue_to_next_win = Button(text = 'Continue', command = self._parent.destroy)
continue_to_next_win.pack(side = 'right', padx = 10, pady = 10)
if 'input_annotation_file' in option_list:
skip_win = Button(text = 'Skip', command = self._parent.destroy)
skip_win.pack(side = 'right', padx = 10, pady = 10)
back_button = Button(self._parent, text="Back", command=self.goBack)
back_button.pack(side = 'right', padx =10, pady = 5)
quit_win = Button(self._parent, text="Quit", command=self.quit)
quit_win.pack(side = 'right', padx =10, pady = 5)
button_text = 'Help'
url = 'http://www.altanalyze.org/help_main.htm'; self.url = url
pdf_help_file = 'Documentation/AltAnalyze-Manual.pdf'; pdf_help_file = filepath(pdf_help_file); self.pdf_help_file = pdf_help_file
try: help_button = Button(self._parent, text=button_text, command=self.GetHelpTopLevel); help_button.pack(side = 'left', padx = 5, pady = 5)
except Exception: help_button = Button(self._parent, text=button_text, command=self.linkout); help_button.pack(side = 'left', padx = 5, pady = 5)
if 'species' in option_list:
new_species_button = Button(self._parent, text='Add New Species', command=self.newSpecies)
new_species_button.pack(side = 'left', padx = 5, pady = 5)
def runPredictGroupsTest():
self.runPredictGroups(reportOnly=True)
if 'GeneSelectionPredict' in option_list:
expFilePresent = self.verifyExpressionFile()
if expFilePresent:
button_instance = Button(self._parent, text='Test Settings', command=runPredictGroupsTest)
button_instance.pack(side = 'left', padx = 5, pady = 5)
if 'build_exon_bedfile' in option_list and array_type == 'RNASeq':
self.pdf_help_file = filepath('AltDatabase/kallisto/license.txt')
button_instance = Button(self._parent, text='Kallisto License', command=self.openPDFHelp)
button_instance.pack(side = 'left', padx = 5, pady = 5)
self._parent.protocol("WM_DELETE_WINDOW", self.deleteWindow)
self._parent.mainloop()
def verifyExpressionFile(self):
continue_analysis = False ### See if the input file is already present
try:
expFile = fl.ExpFile()
count = verifyFileLength(expFile[:-4]+'-steady-state.txt')
if count>1: continue_analysis = True
else:
count = verifyFileLength(expFile)
if count>1: continue_analysis = True
except Exception: pass
return continue_analysis
def goBack(self):
self._parent.destroy()
selected_options = selected_parameters; selected_options2=[] ### If we don't do this we get variable errors
if 'Library' == selected_options[-1]: selected_options[-1] = ''
for i in selected_options:
if i!='Library': selected_options2.append(i)
selected_options = selected_options2
try:
while selected_options[-2]==selected_options[-1]:
selected_options = selected_options[:-1] ### When clicking back on the next loop of a back, makes sure you don't get looped back to the same spot
except Exception: selected_options = selected_options
if len(selected_options)<3: run_parameter = 'no'
else: run_parameter = selected_options[:-1], self._user_variables
AltAnalyze.AltAnalyzeSetup(run_parameter); sys.exit()
def newSpecies(self):
self._user_variables['species'] = 'Add Species'
self._parent.destroy()
def runPredictGroups(self,reportOnly=False):
column_metric = self.Results()['column_metric_predict']
column_method = self.Results()['column_method_predict']
GeneSetSelection = self.Results()['GeneSetSelectionPredict']
try: PathwaySelection = self.Results()['PathwaySelectionPredict']
except Exception: PathwaySelection = 'None Selected'
GeneSelection = self.Results()['GeneSelectionPredict']
JustShowTheseIDs = self.Results()['JustShowTheseIDsPredict']
ExpressionCutoff = self.Results()['ExpressionCutoff']
CountsCutoff = self.Results()['CountsCutoff']
rho_cutoff = self.Results()['rho_cutoff']
FoldDiff = self.Results()['FoldDiff']
SamplesDiffering = self.Results()['SamplesDiffering']
try: featurestoEvaluate = self.Results()['featuresToEvaluate']
except Exception: featurestoEvaluate = 'Genes'
removeOutliers = self.Results()['removeOutliers']
dynamicCorrelation = self.Results()['dynamicCorrelation']
restrictBy = self.Results()['restrictBy']
k = self.Results()['k']
excludeCellCycle = self.Results()['excludeCellCycle']
gsp = GeneSelectionParameters(species,array_type,vendor)
gsp.setGeneSet(GeneSetSelection)
gsp.setPathwaySelect(PathwaySelection)
gsp.setGeneSelection(GeneSelection)
gsp.setJustShowTheseIDs(JustShowTheseIDs)
gsp.setK(k)
gsp.setNormalize('median')
try: gsp.setCountsNormalization(fl.CountsNormalization())
except: pass
gsp.setSampleDiscoveryParameters(ExpressionCutoff,CountsCutoff,FoldDiff,SamplesDiffering,dynamicCorrelation,
removeOutliers,featurestoEvaluate,restrictBy,excludeCellCycle,column_metric,column_method,rho_cutoff)
self._user_variables['gsp'] = gsp
import RNASeq
expFile = fl.ExpFile()
mlp_instance = fl.MLP()
count = verifyFileLength(expFile[:-4]+'-steady-state.txt')
if count>1: expFile = expFile[:-4]+'-steady-state.txt'
if reportOnly:
### Only used to report back what the number of regulated genes are if the gene expression file is present
reload(RNASeq)
try: report, filteredFile = RNASeq.singleCellRNASeqWorkflow(species, array_type, expFile, mlp_instance, parameters=gsp, reportOnly=reportOnly)
except Exception: report = traceback.format_exc()
if 'options_result_in_no_genes' in report:
report = 'Options are too stringent. Try relaxing the thresholds.'
try: InfoWindow(report, 'Continue')
except Exception: print report
else:
### If the parameters look OK, or user wishes to run, collapse this GUI adn proceed (once exited it will run)
self._parent.quit()
self._parent.destroy()
"""
values = expFile, mlp_instance, gsp, reportOnly
StatusWindow(values,'predictGroups') ### display an window with download status
root = Tk()
root.title('AltAnalyze: Evaluate Sampled Groupings')
gu = GUI(root,'PredictGroups',[],'')
nextStep = gu.Results()['next']
group_selected = gu.Results()['group_select']
if nextStep == 'UseSelected':
print group_selected;sys.exit()
group_selected = group_selected
### When nothing returned here, the full analysis will run
else:
#print 're-initializing window'
AltAnalyze.AltAnalyzeSetup((selected_parameters,user_variables)); sys.exit()
"""
def GetHelpTopLevel(self):
message = ''
self.message = message; self.online_help = 'Online Documentation'; self.pdf_help = 'Local PDF File'
tl = Toplevel(); self._tl = tl; nulls = '\t\t\t\t'; tl.title('Please select one of the options')
self.sf = PmwFreeze.ScrolledFrame(self._tl,
labelpos = 'n', label_text = '',
usehullsize = 1, hull_width = 320, hull_height = 200)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
group = PmwFreeze.Group(self.sf.interior(),tag_text = 'Options')
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
filename = 'Config/icon.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(group.interior()); can.pack(side='left',padx = 10, pady = 20); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
l1 = Label(group.interior(), text=nulls); l1.pack(side = 'bottom')
text_button2 = Button(group.interior(), text=self.online_help, command=self.openOnlineHelp); text_button2.pack(side = 'top', padx = 5, pady = 5)
try: text_button = Button(group.interior(), text=self.pdf_help, command=self.openPDFHelp); text_button.pack(side = 'top', padx = 5, pady = 5)
except Exception: text_button = Button(group.interior(), text=self.pdf_help, command=self.openPDFHelp); text_button.pack(side = 'top', padx = 5, pady = 5)
tl.mainloop()
def openPDFHelp(self):
if runningCommandLine:
pass
elif os.name == 'nt':
try: os.startfile('"'+self.pdf_help_file+'"')
except Exception: os.system('open "'+self.pdf_help_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+self.pdf_help_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+self.pdf_help_file+'"')
if 'license' not in self.pdf_help_file:
try: self._tl.destroy()
except Exception: pass
def openOnlineHelp(self):
try: webbrowser.open(self.url)
except Exception: pass
self._tl.destroy()
def linkout(self):
try: webbrowser.open(self.url)
except Exception: pass
def Dlinkout(self):
try: webbrowser.open(self.d_url)
except Exception: pass
def setvscrollmode(self, tag):
self.sf.configure(vscrollmode = tag)
def info(self):
tkMessageBox.showinfo("title","message",parent=self._parent)
def deleteWindow(self):
#tkMessageBox.showwarning("Quit","Use 'Quit' button to end program!",parent=self._parent)
self._parent.destroy(); sys.exit()
def tldeleteWindow(self,tl):
try: tl.quit(); tl.destroy()#; print 1
except Exception: tl.destroy()#; print 2
def quit(self):
try: self._parent.quit(); self._parent.destroy(); sys.exit()
except Exception: self._parent.quit(); sys.exit()
def continue_win(self):
### Currently not used - can be used to check data integrity before closing a window
try: self._parent.quit(); self._parent.destroy(); sys.exit()
except Exception: self._parent.quit(); sys.exit()
def chooseDirectory(self,option):
tag = tkFileDialog.askdirectory(parent=self._parent)
self._user_variables[option] = tag
def chooseFile(self,option):
tag = tkFileDialog.askopenfile(parent=self._parent)
self._user_variables[option] = tag.name
def getPath(self,option):
try: ### Below is used to change a designated folder path to a filepath
if option == 'input_cel_dir' and '10X' in vendor:
processFile = True
else:
forceException
except Exception:
#print traceback.format_exc()
if 'dir' in option or 'folder' in option:
processFile = False
else:
processFile = True
#print option, processFile
if ('dir' in option or 'folder' in option) and processFile ==False:
try: dirPath = tkFileDialog.askdirectory(parent=self._parent,initialdir=self.default_dir)
except Exception:
self.default_dir = ''
try: dirPath = tkFileDialog.askdirectory(parent=self._parent,initialdir=self.default_dir)
except Exception:
try: dirPath = tkFileDialog.askdirectory(parent=self._parent)
except Exception:
print_out = "AltAnalyze is unable to initialize directory opening.\nThis error may be due to one of the following issues:\n"
print_out += "1) (if running directly from source code) Tkinter/PMW components are not installed or are incompatible.\n"
print_out += "2) (if running directly from source code) Python version is untested with AltAnalyze.\n"
print_out += "3) There is a conflict between the AltAnalyze packaged version of python on the OS.\n\n"
print_out += "Contact [email protected] if this error persists with your system information.\n"
print_out += "Alternatively, try AltAnalyze using command-line options (http://www.AltAnalyze.org)."
try: InfoWindow(print_out,'Continue')
except Exception: print print_out
try: self._parent.destroy(); sys.exit()
except Exception: sys.exit()
self.default_dir = dirPath
entrytxt = self.pathdb[option]; entrytxt.set(dirPath)
self._user_variables[option] = dirPath
try: file_location_defaults['PathDir'].SetLocation(dirPath) ### Possible unknown exception here... may need to correct before deployment
except Exception:
try:
### Entry was deleted from Config file - re-create it
fl = FileLocationData('local', dirPath, 'all')
file_location_defaults['PathDir'] = fl
except Exception: pass
try: exportDefaultFileLocations(file_location_defaults)
except Exception: pass
if 'file' in option or processFile:
try: tag = tkFileDialog.askopenfile(parent=self._parent,initialdir=self.default_file)
except Exception:
self.default_file = ''
try: tag = tkFileDialog.askopenfile(parent=self._parent,initialdir=self.default_file)
except Exception:
try: tag = tkFileDialog.askopenfile(parent=self._parent)
except Exception:
print_out = "AltAnalyze is unable to initialize directory opening.\nThis error may be due to one of the following issues:\n"
print_out += "1) (if running directly from source code) Tkinter/PMW components are not installed or are incompatible.\n"
print_out += "2) (if running directly from source code) Python version is untested with AltAnalyze.\n"
print_out += "3) There is a conflict between the AltAnalyze packaged version of python on the OS.\n\n"
print_out += "Contact [email protected] if this error persists with your system information.\n"
print_out += "Alternatively, try AltAnalyze using command-line options (see documentation at http://AltAnalyze.org)."
try: InfoWindow(print_out,'Continue')
except Exception: print print_out
try: self._parent.destroy(); sys.exit()
except Exception: sys.exit()
try: filePath = tag.name #initialdir=self.default_dir
except AttributeError: filePath = ''
filePath_dir = string.join(string.split(filePath,'/')[:-1],'/')
self.default_file = filePath_dir
entrytxt = self.pathdb[option]
entrytxt.set(filePath)
self._user_variables[option] = filePath
try: file_location_defaults['PathFile'].SetLocation(filePath_dir)
except Exception:
try:
### Entry was deleted from Config file - re-create it
fl = FileLocationData('local', filePath_dir, 'all')
file_location_defaults['PathFile'] = fl
except Exception: null = None
try: exportDefaultFileLocations(file_location_defaults)
except Exception: pass
def Report(self,tag,option):
output = tag
return output
def __repr__(self,tag,option): return self.Report(tag,option)
def Results(self):
for i in self._user_variables:
user_variables[i]=self._user_variables[i]
return self._user_variables
def custom_validate(self, text, option):
self._user_variables[option] = text
#try: text = float(text);return 1
#except ValueError: return -1
def enter_callback(self, tag, option):
if self.defaults == 'batch':
### Bath removal array annotation UI
self._user_variables[option,'batch'] = tag
else:
self._user_variables[option] = tag
def custom_validate_p(self, text, option):
#print [option],'text:', text
self._user_variables[option] = text
try:
text = float(text)
if text <1:return 1
else:return -1
except ValueError:return -1
def callback(self, tag, option):
#print 'Button',[option], tag,'was pressed.'
change_var = ''
self._user_variables[option] = tag
if option == 'dbase_version':
###Export new species info
exportDBversion(tag); change_var = 'all'
try: self.changeVendorSelection(); self.changeSpeciesSelection(); self.changeArraySelection()
except Exception: pass
elif option == 'species':
try: self.changeArraySelection()
except Exception: pass
elif option == 'manufacturer_selection':
try: self.changeSpeciesSelection(); self.changeArraySelection()
except Exception: pass
#elif option == 'array_type':
#self.checkSpeciesArraySelection(array_type)
elif option == 'analysis_method':
if tag == 'ASPIRE':
try: self.entry_field2.setentry('0.2')
except Exception: pass
self._user_variables['alt_exon_fold_cutoff'] = '0.2'
elif tag == 'linearregres':
try: self.entry_field2.setentry('2')
except Exception: pass
self._user_variables['alt_exon_fold_cutoff'] = '2'
elif tag == 'MultiPath-PSI':
try: self.entry_field2.setentry('0.1')
except Exception: pass
self._user_variables['alt_exon_fold_cutoff'] = '0.1'
elif option == 'selected_version':
current_species_names = db_versions[tag]
current_species_names.sort()
try: self.speciescomp.setitems(['---']+current_species_names)
except Exception: null = [] ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
try: self.speciescomp2.setitems(['---']+current_species_names)
except Exception: null = [] ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
try: self.speciescomp3.setitems(['---']+current_species_names)
except Exception: null = [] ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
"""
### Doesn't work right now because self.entry_field only has one object instance and complicated to get multiple
elif option == 'ORA_algorithm':
if tag == 'Permute p-value':
try: self.entry_field.setentry('2000')
except Exception: pass
self._user_variables['permutation'] = '2000'
elif tag == 'Fisher Exact Test':
try: self.entry_field.setentry('NA')
except Exception: pass
self._user_variables['permutation'] = '0'
"""
def callbackComboBox(self, tag, option):
""" Similiar to the above, callback, but ComboBox uses unique methods """
#print 'Button',[option], tag,'was pressed.'
if option == 'interactionDirs' or 'PathwaySelection' in option or 'HeatmapAdvanced' in option: ### Allow multiple selections
if len(tag)==0:
self._user_variables[option] = None ### no options selected
else:
if isinstance(tag, tuple) or isinstance(tag, list):
pass
else:
try: tag = self._user_variables[option] ### This indicates that this option was previously set and in the new window was not explicitly set, suggesting we should re-apply the original settings
except Exception: None
try: ### Occurs when no items selected
if len(tag[0])==1: ### Hence, just one item selected
self._user_variables[option] = [tag]
except Exception:
pass
if len(list(tag)[0]) == 1:
tag_list = [tag]
else: tag_list = list(tag)
self._user_variables[option] = tag_list
else:
self._user_variables[option] = tag
if option == 'selected_version':
current_species_names = db_versions[tag]
current_species_names.sort()
current_species_names = ['---']+current_species_names
species_option = current_species_names[0]
try:
self.speciescomp._list.setlist(current_species_names) ### This is the way we set a new list for ComboBox
### Select the best default option to display (keep existing or re-set)
if 'selected_species1' in self._user_variables: ### If this is the species downloader
species_option = 'selected_species1'
else:
for i in self._user_variables:
if 'species' in i: species_option = i
default = self.getBestDefaultSelection(species_option,current_species_names)
self.speciescomp.selectitem(default)
except Exception: None ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
try:
self.speciescomp2._list.setlist(current_species_names)
default = self.getBestDefaultSelection('selected_species2',current_species_names)
self.speciescomp2.selectitem(default)
except Exception: None ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
try:
self.speciescomp3._list.setlist(current_species_names)
default = self.getBestDefaultSelection('selected_species3',current_species_names)
self.speciescomp3.selectitem(default)
except Exception: None ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
elif option == 'dbase_version':
###Export new species info
exportDBversion(tag); change_var = 'all'
try: self.changeVendorSelection(); self.changeSpeciesSelection(); self.changeArraySelection()
except Exception: pass
elif option == 'species':
try: self.changeArraySelection()
except Exception: pass
elif option == 'manufacturer_selection':
try: self.changeSpeciesSelection(); self.changeArraySelection()
except Exception: pass
#elif option == 'array_type':
#self.checkSpeciesArraySelection(array_type)
elif option == 'analysis_method':
if tag == 'ASPIRE':
try: self.entry_field2.setentry('0.2')
except Exception: pass
self._user_variables['alt_exon_fold_cutoff'] = '0.2'
elif tag == 'linearregres':
try: self.entry_field2.setentry('2')
except Exception: pass
self._user_variables['alt_exon_fold_cutoff'] = '2'
elif tag == 'MultiPath-PSI':
try: self.entry_field2.setentry('0.1')
except Exception: pass
self._user_variables['alt_exon_fold_cutoff'] = '0.1'
elif 'GeneSetSelection' in option or 'GeneSetSelectionPredict' in option:
#print option,tag
if 'network' in option: suffix='_network'
else: suffix=''
#species = self._user_variables['species']
try:
if 'Ontology' in tag: directory = 'gene-go'
else: directory = 'gene-mapp'
if tag in self._user_variables and 'StoredGeneSets' not in tag: ### Want to reload StoredGeneSets each time
supported_genesets = self._user_variables[tag]
#print 'loading pathways from memory'
else:
#print 'importing all pathways from scratch'
supported_genesets = listAllGeneSetCategories(species,tag,directory)
self._user_variables[tag] = supported_genesets ### Store this so we don't waste time reloading it the next time
self.pathwayselect._list.setlist(supported_genesets)
##### self.pathwayselect.selectitem(supported_genesets[0]) # This sets the default for multi- or single-combo boxes... DON'T SELECT UNLESS YOU WANT TO HAVE TO DE-SELECT IT
##### self._user_variables['PathwaySelection'+suffix] = supported_genesets[0] ### store this default
##### self._user_variables['PathwaySelectionPredict'+suffix] = supported_genesets[0] ### store this default
self.pathwayselect.selectitem(0,setentry = 1) # Select the item but then re-set the list to deselect it
self.pathwayselect._list.setlist(supported_genesets)
except Exception, e:
#print e
pass ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
def getBestDefaultSelection(self,option,option_list):
default = option_list[0] ### set the default to the first option listed
if option in self._user_variables:
selected = self._user_variables[option]
if selected in option_list: ### If selected species exists in the new selected version of EnsMart
default = selected
else:
self._user_variables[option] = default ### Hence, the default has changed, so re-set it
return default
def changeSpeciesSelection(self):
vendor = self._user_variables['manufacturer_selection'] ### Get vendor (stored as global)
current_species_names = getSpeciesList(vendor) ### Need to change species, manufacturers and array_type
for i in self._option_list:
if 'species' in i: ### Necessary if the user changes dbase_version and selects continue to accept the displayed species name (since it's note directly invoked)
last_selected_species = self._user_variables[i]
if last_selected_species not in current_species_names:
try: self._user_variables[i] = current_species_names[0]
except Exception: null = []
try:
self.speciescomp._list.setlist(current_species_names)
self.speciescomp.selectitem(current_species_names[0])
except Exception:
print traceback.format_exc()
null = [] ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
def checkSpeciesArraySelection(self,array_type):
current_species_names = getSpeciesForArray(array_type)
try:
self.speciescomp._list.setlist(current_species_names)
self.speciescomp.selectitem(current_species_names[0])
except Exception: null = [] ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
for i in self._option_list:
if 'species' in i: ### Necessary if the user changes dbase_version and selects continue to accept the displayed species name (since it's note directly invoked)
try: self._user_variables[i] = current_species_names[0]
except Exception: null = []
def changeArraySelection(self):
species_name = self._user_variables['species'] ### Get species (stored as global)
vendor = self._user_variables['manufacturer_selection'] ### Get vendor (stored as global)
species = species_codes[species_name].SpeciesCode()
current_array_types, manufacturer_list = getArraysAndVendors(species,vendor)
if 'Other ID'==vendor: ### Populate the current_array_types as all Ensembl linked systems
current_array_types = getSupportedGeneSystems(species,'uid-gene')
try:
self.arraycomp._list.setlist(current_array_types)
self.arraycomp.selectitem(current_array_types[0])
except Exception:
pass ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
for i in self._option_list:
if 'array_type' in i: ### Necessary if the user changes dbase_version and selects continue to accept the displayed species name (since it's note directly invoked)
if self._user_variables[i] not in current_array_types: ### If the current array type is supported by the new species selection, keep it the same
try: self._user_variables[i] = current_array_types[0]
except Exception: null = []
def changeVendorSelection(self):
species_name = self._user_variables['species'] ### Get species (stored as global)
vendor = self._user_variables['manufacturer_selection']
current_array_types, manufacturer_list = getArraysAndVendors(species,'')
try:
self.vendorcomp._list.setlist(manufacturer_list)
self.vendorcomp.selectitem(manufacturer_list[0])
except Exception: null = [] ### Occurs before speciescomp is declared when dbase_version pulldown is first intiated
for i in self._option_list:
if 'manufacturer_selection' in i: ### Necessary if the user changes dbase_version and selects continue to accept the displayed species name (since it's note directly invoked)
if vendor in manufacturer_list: new_vendor = vendor
else: new_vendor = manufacturer_list[0]
try: self._user_variables[i] = new_vendor
except Exception: null = []
def multcallback(self, tag, state):
if state: action = 'pressed.'
else: action = 'released.'
"""print 'Button', tag, 'was', action, \
'Selection:', self.multiple.getcurselection()"""
self._user_variables[option] = tag
def checkbuttoncallback(self, tag, state, option):
if state: action = 'pressed.'
else: action = 'released.'
"""print 'Button',[option], tag, 'was', action, \
'Selection:', self.checkbuttons.getcurselection()"""
if state==0: tag2 = 'no'
else: tag2 = 'yes'
#print '---blahh', [option], [tag], [state], [action], [self.checkbuttons.getcurselection()]
self._user_variables[option] = tag2
################# Database Version Handling ##################
class PreviousResults:
def __init__(self, user_variables):
self._user_variables = user_variables
def Results(self): return self._user_variables
def getSpeciesList(vendor):
try: current_species_dirs = unique.read_directory('/AltDatabase')
except Exception: ### Occurs when the version file gets over-written with a bad directory name
try:
### Remove the version file and wipe the species file
os.remove(filepath('Config/version.txt'))
#raw = export.ExportFile('Config/species.txt'); raw.close()
os.mkdir(filepath('AltDatabase'))
AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
except Exception:
print traceback.format_exc()
print 'Cannot write Config/version.txt to the Config directory (likely Permissions Error)'
try: db_versions = returnDirectoriesNoReplace('/AltDatabase')
except Exception:
try: os.mkdir(filepath('AltDatabase'))
except Exception: pass
db_versions = returnDirectoriesNoReplace('/AltDatabase')
for db_version in db_versions:
if 'EnsMart' in db_version:
try: exportDBversion(db_version)
except Exception: exportDBversion('')
break
current_species_dirs = unique.read_directory('/AltDatabase')
current_species_names=[]; manufacturers_list=[]
for species in species_codes:
species_code = species_codes[species].SpeciesCode()
if species_code in current_species_dirs:
if len(vendor)>0:
proceed = 'no'
for array_name in array_codes:
manufacturer = array_codes[array_name].Manufacturer()
if manufacturer == vendor:
if species_code in array_codes[array_name].SpeciesCodes(): proceed = 'yes'
else:
for array_name in array_codes:
manufacturer = array_codes[array_name].Manufacturer()
if species_code in array_codes[array_name].SpeciesCodes():
manufacturers_list.append(manufacturer)
proceed = 'yes'
if proceed == 'yes': current_species_names.append(species)
current_species_names.sort(); manufacturers_list = unique.unique(manufacturers_list); manufacturers_list.sort()
if len(vendor)>0:
return current_species_names
else: return current_species_names, manufacturers_list
def exportDBversion(db_version):
import datetime
db_version = string.replace(db_version,'Plant','')
if len(db_version)==0:
db_version = unique.returnDirectoriesNoReplace('/AltDatabase',search='EnsMart')
today = str(datetime.date.today()); today = string.split(today,'-'); today = today[1]+'/'+today[2]+'/'+today[0]
try:
exportVersionData(db_version,today,'Config/')
except Exception:
try: exportVersionData(db_version,today,'Config/',force=None)
except:
print traceback.format_exc()
print 'Cannot write Config/version.txt to the Config directory (likely Permissions Error)'
return db_version
def exportVersionData(version,version_date,dir,force='application-path'):
new_file = dir+'/version.txt'
new_file_default = unique.filepath(new_file,force=force) ### can use user directory local or application local
data = export.ExportFile(new_file_default)
data.write(str(version)+'\t'+str(version_date)+'\n'); data.close()
def importResourceList():
filename = 'Config/resource_list.txt'
fn=filepath(filename); resource_list=[]
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
resource = data
resource_list.append(resource)
return resource_list
def importGeneList(filename,limit=None):
### Optionally limit the number of results imported
gene_list=[]
fn=filepath(filename); resource_list=[]; count=0
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
gene = string.split(data,'\t')[0]
if ' ' in gene:
gene = string.split(gene,' ')[0]
if ':' in gene:
gene = string.split(gene,':')[0]
if gene not in gene_list and gene != 'GeneID' and gene != 'UID' and gene != 'probesetID':
gene_list.append(gene)
count+=1
if limit != None:
if limit==count: break
gene_list = string.join(gene_list,',')
return gene_list
def exportJunctionList(filename,limit=None):
### Optionally limit the number of results imported
parent = export.findParentDir(filename)
export_file = parent+'/top'+str(limit)+'/MultiPath-PSI.txt'#+file
#export_file = filename[:-4]+'-top-'+str(limit)+'.txt'
eo = export.ExportFile(export_file)
fn=filepath(filename); count=0; firstLine=True
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine = False
elif '-' in t[0]:
junctions = string.split(data,'\t')[0]
junctions = string.replace(junctions,'|',' ')
junctions = string.join(string.split(junctions,':')[1:],':')
eo.write(junctions+'\n')
count+=1
if limit==count: break
else:
junctions = t[1] #Atg9a:ENSMUSG00000033124:E1.1-E3.1|ENSMUSG00000033124:E1.1-E3.2
junctions = string.split(junctions,'|') #ENSMUSG00000032314:I11.1_55475101-E13.1-ENSMUSG00000032314:E11.1-E13.1|ENSMUSG00000032314:I11.1_55475153;I11.1_55475101
for junction_pair in junctions:
if '-' in junction_pair:
try:
a,b = string.split(junction_pair,'-ENS')
b = 'ENS'+b
eo.write(a+' '+b+'\n')
count+=1
if limit==count: break
except Exception:
pass
if count>limit: break
if count>limit: break
eo.close()
return export_file
def importConfigFile():
#print "importing config file"
filename = 'Config/config.txt'
fn=filepath(filename); config_db={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
config_type,options = string.split(data,'\t')
config_db[config_type] = options
return config_db
def exportConfigFile(config_db):
#print "exporting config file"
new_file = 'Config/config.txt'
data = export.ExportFile(new_file)
for config in config_db:
try: data.write(config+'\t'+str(config_db[config])+'\n'); data.close()
except Exception:
print 'Cannot write Config/config.txt to the Config directory (like Permissions Error)'
def remoteOnlineDatabaseVersions():
db_versions = importOnlineDatabaseVersions()
return db_versions_vendors,db_versions
def importOnlineDatabaseVersions():
filename = 'Config/array_versions.txt'
fn=filepath(filename); global db_versions; db_versions={}; global db_versions_vendors; db_versions_vendors={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
species,version,vendors = string.split(data,'\t')
vendors = string.split(vendors,'|')
ad = ArrayData('','',vendors,'',species)
version = string.replace(version,'Plus','') ### The user won't understand the Plus which relates to the GO-Elite version (AltAnalyze does not have a plus but we want the Plus for goelite)
try: db_versions[version].append(species)
except KeyError: db_versions[version] = [species]
try: db_versions_vendors[version].append(ad)
except KeyError: db_versions_vendors[version] = [ad]
return db_versions
def getOnlineDBConfig(file_location_defaults,root):
base_url = file_location_defaults['url'].Location()
try:
fln1,status1 = update.download(base_url+'Config/species_all.txt','Config/','')
fln2,status2 = update.download(base_url+'Config/source_data.txt','Config/','')
fln3,status3 = update.download(base_url+'Config/array_versions.txt','Config/','')
except Exception:
print 'Could not download the latest online configuration files (likely Permissions Error)'
try:
if 'Internet' not in status3:
print 'Finished downloading the latest configuration files.'; root.destroy()
else:
try: WarningWindow(status3,'Error Encountered!'); root.destroy(); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
except Exception: print status3; root.destroy(); sys.exit()
except Exception: pass
def getOnlineEliteDatabase(file_location_defaults,db_version,new_species_codes,update_goelite_resources,root):
base_url = file_location_defaults['url'].Location()
goelite_url = file_location_defaults['goelite'].Location()
dbs_added = 0
AltAnalyze_folders = read_directory(''); Cytoscape_found = 'no'
for dir in AltAnalyze_folders:
if 'Cytoscape_' in dir: Cytoscape_found='yes'
if Cytoscape_found == 'no':
fln,status = update.download(goelite_url+'Cytoscape/cytoscape.tar.gz','','')
if 'Internet' not in status: print "Cytoscape program folder downloaded."
count = verifyFileLength('AltDatabase/TreeView/TreeView.jar')
if count==0:
fln,status = update.download(goelite_url+'TreeView.zip','AltDatabase/NoVersion','')
if 'Internet' not in status: print "TreeView program downloaded."
fln,status = update.download(goelite_url+'Databases/'+db_version+'Plus/OBO.zip','AltDatabase/goelite/','')
if 'Internet' not in status: print "Gene Ontology structure files downloaded."
for species_code in new_species_codes:
#print [base_url+'AltDatabase/'+db_version+'/'+species_code+'.zip']
if species_code == 'Mm' or species_code == 'Hs' or species_code == 'Rn': specific_extension=''
else: specific_extension='_RNASeq'
fln,status = update.download(base_url+'AltDatabase/updated/'+db_version+'/'+species_code+specific_extension+'.zip','AltDatabase/','') #AltDatabaseNoVersion
if 'Internet' not in status:
print 'Finished downloading the latest species database files.'
dbs_added+=1
#print goelite_url+'Databases/'+db_version+'Plus/'+species_code+'.zip'
try: fln,status = update.download(goelite_url+'Databases/'+db_version+'Plus/'+species_code+'.zip','AltDatabase/goelite/','')
except Exception: print "No species GO-Elite database found."
if update_goelite_resources == 'yes': ### Get all additional GeneSet database types (can be lengthy download times)
try: getAdditionalOnlineResources(species_code, 'All Resources',None)
except Exception: print "Unable to update additional GO-Elite resources."
if 'Internet' not in status: print "GO-Elite database installed." ; dbs_added+=1
else: print "No species GO-Elite database found."
try: os.mkdir(filepath('AltDatabase/'+species_code))
except Exception: pass
if dbs_added>0:
print_out = "New species data successfully added to database."
if root !='' and root !=None:
try: InfoWindow(print_out,'Continue')
except Exception: print print_out
else: print print_out
try: root.destroy()
except Exception: pass
else:
if root !='' and root !=None: WarningWindow(status,'Error Encountered!'); root.destroy(); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
else: print status; root.destroy(); sys.exit()
def filterExternalDBs(all_external_ids,externalDBName_list,external_ids,array_db):
filtered_external_list=[]
for name in externalDBName_list:
if name in external_ids:
id = external_ids[name]
if id in all_external_ids:
if name != 'GO': filtered_external_list.append(name)
for array in array_db:
if '\\N_' not in array: filtered_external_list.append(array)
return filtered_external_list
def updateOBOfiles(file_location_defaults,update_OBO,OBO_url,root):
run_parameter = "Create/Modify Databases"
if update_OBO == 'yes':
from import_scripts import OBO_import
c = OBO_import.GrabFiles()
c.setdirectory('/OBO'); file_dirs = c.searchdirectory('.ontology')+c.searchdirectory('.obo')
if len(OBO_url)>0: obo = OBO_url
else: ### If not present, get the gene-ontology default OBO file
obo = file_location_defaults['OBO'].Location()
fln,status = update.download(obo,'OBO/','')
run_parameter='Create/Modify Databases'
if 'Internet' not in status:
OBO_import.moveOntologyToArchiveDir()
print_out = 'Finished downloading the latest Ontology OBO files.'
print print_out
try: system_codes,source_types,mod_types = GO_Elite.getSourceData()
except Exception: null=[]
if root !='' and root !=None:
InfoWindow(print_out,'Update Complete!')
continue_to_next_win = Button(text = 'Continue', command = root.destroy)
continue_to_next_win.pack(side = 'right', padx = 10, pady = 10); root.mainloop()
GO_Elite.importGOEliteParameters(run_parameter); sys.exit()
else: null=[]
else:
if root !='' and root !=None: WarningWindow(status,'Error Encountered!'); root.destroy(); GO_Elite.importGOEliteParameters(run_parameter); sys.exit()
else: print status
else:
print_out = 'Download Aborted.'
if root !='' and root !=None: WarningWindow(print_out,print_out); root.destroy(); GO_Elite.importGOEliteParameters('Create/Modify Databases'); sys.exit()
else: print print_out
def importExternalDBs(species_full):
filename = 'Config/EnsExternalDBs.txt'
fn=filepath(filename); x = 0; external_dbs=[]; external_system={}; all_databases={}; external_ids={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
if x==0: x=1
else:
id, database, species_specific, exclude, system_code = string.split(data,'\t')
external_ids[database] = int(id)
if database != 'GO':
all_databases[database]=system_code
if (species_full == species_specific) or len(species_specific)<2:
if len(exclude)<2:
external_system[database] = system_code
filename = 'Config/external_db.txt'; external_system2={}
fn=filepath(filename)
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
try:
t = string.split(data,'\t'); id = int(t[0]); database = t[1]
external_ids[database] = id
if database in external_system:
external_system2[database] = external_system[database]
elif database not in all_databases: ### Add it if it's new
try:
try: system = database[:3]
except Exception: system = database[:2]
external_system2[database] = system
except Exception: null=[]
except Exception: null=[] ### Occurs when a bad end of line is present
filename = 'Config/array.txt'
global array_db; array_db={}
fn=filepath(filename)
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
try:
array = t[1]; vendor = t[3]
database = vendor+'_'+array; array_db[database]=[]
if database in external_system:
external_system2[database] = external_system[database]
if database in all_databases:
external_system2[database] = all_databases[database]
elif database not in all_databases: ### Add it if it's new
try:
if vendor == 'AFFY': system = 'X'
if vendor == 'ILLUMINA': system = 'Il'
if vendor == 'CODELINK': system = 'Co'
if vendor == 'AGILENT': system = 'Ag'
else: system = 'Ma' ###Miscelaneous Array type
external_system2[database] = system
except Exception: null=[]
except Exception: null=[]
external_system = external_system2
#try: del external_system['GO']
#except Exception: null=[]
for database in external_system: external_dbs.append(database)
external_dbs.append(' '); external_dbs = unique.unique(external_dbs); external_dbs.sort()
return external_dbs,external_system,array_db,external_ids
class SupprotedArrays:
def __init__(self, array_name, library_file, annotation_file, species, array_type):
self.array_name = array_name; self.library_file = library_file; self.annotation_file = annotation_file
self.species = species; self.array_type = array_type
def ArrayName(self): return self.array_name
def LibraryFile(self): return self.library_file
def AnnotationFile(self): return self.annotation_file
def Species(self): return self.species
def ArrayType(self): return self.array_type
def __repr__(self): return self.ArrayName()
def importSupportedArrayInfo():
filename = 'Config/ArrayFileInfo.txt'; x=0
fn=filepath(filename); global supproted_array_db; supproted_array_db={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
array_name,library_file,annotation_file,species,array_type = string.split(data,'\t')
if x==0: x=1
else:
sd = SupprotedArrays(array_name,library_file,annotation_file,species,array_type)
supproted_array_db[array_name] = sd
return supproted_array_db
def exportSupportedArrayInfo():
fn=filepath('Config/ArrayFileInfo.txt'); data = open(fn,'w'); x=0
header = string.join(['ArrayName','LibraryFile','AnnotationFile','Species','ArrayType'],'\t')+'\n'
data.write(header)
for array_name in supproted_array_db:
sd = supproted_array_db[array_name]
values = [array_name,sd.LibraryFile(),sd.AnnotationFile(),sd.Species(),sd.ArrayType()]
values = string.join(values,'\t')+'\n'
data.write(values)
data.close()
class SystemData:
def __init__(self, syscode, sysname, mod):
self._syscode = syscode; self._sysname = sysname; self._mod = mod
def SystemCode(self): return self._syscode
def SystemName(self): return self._sysname
def MOD(self): return self._mod
def __repr__(self): return self.SystemCode()+'|'+self.SystemName()+'|'+self.MOD()
def remoteSystemInfo():
system_codes,system_list,mod_list = importSystemInfo(returnSystemCode=True)
return system_codes,system_list,mod_list
def getSystemInfo():
importSystemInfo()
return system_codes
def importSystemInfo(returnSystemCode=False):
filename = 'Config/source_data.txt'; x=0
fn=filepath(filename); global system_list; system_list=[]; global system_codes; system_codes={}; mod_list=[]
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if '!DOCTYPE' in data:
fn2 = string.replace(fn,'.txt','_archive.txt')
import shutil; shutil.copyfile(fn2,fn) ### Bad file was downloaded (with warning)
importSystemInfo(); break
elif '<html>' in data:
print_out = "WARNING!!! Connection Error. Proxy may not be allowed from this location."
try: WarningWindow(print_out,' Continue ')
except NameError: print print_out
importSystemInfo(); break
else:
try: sysname=t[0];syscode=t[1]
except Exception: sysname=''
try: mod = t[2]
except Exception: mod = ''
if x==0: x=1
else:
system_list.append(sysname)
ad = SystemData(syscode,sysname,mod)
if len(mod)>1: mod_list.append(sysname)
system_codes[sysname] = ad
if returnSystemCode:
return system_codes,system_list,mod_list
else:
return system_list,mod_list
def exportSystemInfoRemote(system_code_db):
global system_codes; system_codes = system_code_db
exportSystemInfo()
def exportSystemInfo():
if len(system_codes)>0:
filename = 'Config/source_data.txt'
fn=filepath(filename); data = open(fn,'w')
header = string.join(['System','SystemCode','MOD_status'],'\t')+'\n'
data.write(header)
for sysname in system_codes:
ad = system_codes[sysname]
values = string.join([sysname,ad.SystemCode(),ad.MOD()],'\t')+'\n'
data.write(values)
data.close()
class SpeciesData:
def __init__(self, abrev, species, algorithms):
self._abrev = abrev; self._species = species; self._algorithms = algorithms
def SpeciesCode(self): return self._abrev
def SpeciesName(self): return self._species
def Algorithms(self): return self._algorithms
def __repr__(self): return self.Report()
def getSpeciesInfo():
### Used by AltAnalyze
global integrate_online_species; integrate_online_species = 'yes'
importSpeciesInfo(); species_names={}
for species_full in species_codes:
sc = species_codes[species_full]; abrev = sc.SpeciesCode()
species_names[abrev] = species_full
return species_names
def remoteSpeciesInfo():
global integrate_online_species; integrate_online_species = 'yes'
importSpeciesInfo()
return species_codes
def remoteSpeciesAlt():
### Replicates the output of GO-Elite's species importer
global integrate_online_species; integrate_online_species = 'yes'
importSpeciesInfo()
species_names={}
for species in species_codes:
sd = species_codes[species]
species_names[sd.SpeciesCode()] = sd
return species_names
def importSpeciesInfo():
try:
if integrate_online_species == 'yes': filename = 'Config/species_all.txt'
else: filename = 'Config/species.txt'
except Exception: filename = 'Config/species.txt'
fn=filepath(filename)
global species_list
species_list=[]
global species_codes
species_codes={}; x=0
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
try:
try: abrev,species,algorithms = string.split(data,'\t')
except Exception:
try: abrev,species = string.split(data,'\t'); algorithms = ''
except Exception:
abrev,species,taxid,compatible_mods = string.split(data,'\t')
algorithms = ''
except Exception:
if '!DOCTYPE': print_out = "A internet connection could not be established.\nPlease fix the problem before proceeding."
else: print_out = "Unknown file error encountered."
IndicatorWindow(print_out,'Continue')
raw = export.ExportFile(fn); raw.close(); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
if x==0: x=1
else:
algorithms = string.split(algorithms,'|')
species_list.append(species)
sd = SpeciesData(abrev,species,algorithms)
species_codes[species] = sd
return species_codes
def exportSpeciesInfo(species_codes):
fn=filepath('Config/species.txt'); data = open(fn,'w'); x=0
header = string.join(['species_code','species_name','compatible_algorithms'],'\t')+'\n'
data.write(header)
for species in species_codes:
sd = species_codes[species]; algorithms = string.join(sd.Algorithms(),'|')
values = [sd.SpeciesCode(),species,algorithms]
values = string.join(values,'\t')+'\n'
data.write(values)
data.close()
class ArrayGroupData:
def __init__(self, array_header, group, group_name):
self._array_header = array_header; self._group = group; self._group_name = group_name
def Array(self): return self._array_header
def Group(self): return self._group
def setGroup(self,group): self._group = group
def GroupName(self): return self._group_name
def setGroupName(self,group_name): self._group_name = group_name
def Report(self): return self.Array()
def __repr__(self): return self.Report()
def importArrayGroupsSimple(expr_group_dir,cel_files):
array_group_list = []; group_db={}
fn=filepath(expr_group_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
array_header,group,group_name = string.split(data,'\t')
if group_name == 'NA': group_name = 'None'
#print [array_header],cel_files
if (array_header in cel_files) or len(cel_files)==0: ### restrict import to array files listed in the groups file
try: group = int(group); group_db[group]=group_name
except ValueError: print group, group_name;kill
agd = ArrayGroupData(array_header,group,group_name)
array_group_list.append(agd)
if len(cel_files)>0:
if len(cel_files)!=len(array_group_list):
#print len(cel_files),len(array_group_list)
#print cel_files
array_group_list2=[]
for i in array_group_list:
if i.Array() not in cel_files:
print [i.Array()], 'not in CEL file dir (in groups file)'
array_group_list2.append(i.Array())
for i in cel_files:
if i not in array_group_list2:
print [i], 'not in groups file (in CEL file dir)'
raise NameError('Samples In Groups Not Found In Dir')
return array_group_list,group_db
class ArrayData:
def __init__(self, abrev, array, manufacturer, constitutive_source, species):
self._abrev = abrev; self._array = array; self._manufacturer = manufacturer; self._species = species
self._constitutive_source = constitutive_source
def ArrayCode(self): return self._abrev
def ArrayName(self): return self._array
def Manufacturer(self): return self._manufacturer
def ConstitutiveSource(self): return self._constitutive_source
def SpeciesCodes(self): return self._species
def setSpeciesCodes(self,species): species = self._species
def __repr__(self): return self.ArrayCode()+'|'+str(self.SpeciesCodes())+'|'+str(self.Manufacturer())
def remoteArrayInfo():
importArrayInfo()
return array_codes
def importArrayInfo():
filename = 'Config/arrays.txt'; x=0
fn=filepath(filename); global array_list; array_list=[]; global array_codes; array_codes={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
abrev,array,manufacturer,constitutive_source,species = string.split(data,'\t')
if x==0: x=1
else:
species = string.split(species,'|')
array_list.append(array)
ad = ArrayData(abrev,array,manufacturer,constitutive_source,species)
array_codes[array] = ad
return array_list
def exportArrayInfo(array_codes):
fn=filepath('Config/arrays.txt'); data = open(fn,'w'); x=0
header = string.join(['array_type','array_name','manufacturer','constitutive_source','compatible_species'],'\t')+'\n'
data.write(header)
for array in array_codes:
ad = array_codes[array]; species = string.join(ad.SpeciesCodes(),'|')
values = [ad.ArrayCode(),array,ad.Manufacturer(),ad.ConstitutiveSource(),species]
values = string.join(values,'\t')+'\n'
data.write(values)
data.close()
class FileLocationData:
def __init__(self, status, location, species):
self._status = status; self._location = location; self._species = species
def Status(self): return self._status
def Location(self): return self._location
def SetLocation(self,location): self._location = location
def Species(self): return self._species
def __repr__(self): return self.Report()
def importDefaultFileLocations():
filename = 'Config/default-files.csv'; x=0
fn=filepath(filename); file_location_defaults={}
for line in open(fn,'rU').readlines():
line = string.replace(line,',','\t') ### Make tab-delimited (had to make CSV since Excel would impoperly parse otherwise)
data = cleanUpLine(line)
###Species can be multiple species - still keep in one field
try: app,status,location,species = string.split(data,'\t')
except Exception:
try:
t = string.split(data,'\t')
app=t[0]; status=t[1]; location=t[2]; species=t[3]
except Exception:
continue
fl = FileLocationData(status, location, species)
if species == 'all': file_location_defaults[app] = fl
else:
try: file_location_defaults[app].append(fl)
except KeyError: file_location_defaults[app] = [fl]
return file_location_defaults
def exportDefaultFileLocations(file_location_defaults):
### If the user supplies new defaults, over-write the existing
fn=filepath('Config/default-files.csv'); data = open(fn,'w')
for app in file_location_defaults:
fl_list = file_location_defaults[app]
try:
for fl in fl_list:
values = [app,fl.Status(),fl.Location(),fl.Species()]
values = '"'+string.join(values,'","')+'"'+'\n'
data.write(values)
except Exception:
fl = fl_list
values = [app,fl.Status(),fl.Location(),fl.Species()]
values = '"'+string.join(values,'","')+'"'+'\n'
data.write(values)
data.close()
def exportGroups(exp_file_location_db,array_group_list,filetype='Groups'):
### If the user supplies new defaults, over-write the existing
for dataset_name in exp_file_location_db:
fl = exp_file_location_db[dataset_name]
groups_file = fl.GroupsFile()
if filetype =='Batch':
groups_file = string.replace(groups_file,'groups.','batch.')
fn=filepath(groups_file); data = open(fn,'w')
value_list = [] ### Sort grouped results based on group number
for agd in array_group_list:
values = [agd.Array(), str(agd.Group()), agd.GroupName()]
values = string.join(values,'\t')+'\n'; value_list.append(((agd.Group(),agd.Array()),values))
value_list.sort()
for values in value_list: data.write(values[-1])
data.close()
def exportComps(exp_file_location_db,comp_group_list):
### If the user supplies new defaults, over-write the existing
for dataset_name in exp_file_location_db:
fl = exp_file_location_db[dataset_name]; comps_file = fl.CompsFile()
fn=filepath(comps_file); data = open(fn,'w')
for comp_num, groups in comp_group_list:
group1, group2 = groups
values = [str(group1), str(group2)]
values = string.join(values,'\t')+'\n'; data.write(values)
data.close()
class Defaults:
def __init__(self, abrev, array, species):
self._abrev = abrev; self._array = array; self._species = species
def ArrayCode(self): return self._abrev
def ArrayName(self): return self._array
def Species(self): return self._species
def __repr__(self): return self.Report()
def verifyFile(filename):
fn=filepath(filename); file_found = 'yes'
try:
for line in open(fn,'rU').xreadlines():break
except Exception: file_found = 'no'
return file_found
def verifyFileLength(filename):
count = 0
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
count+=1
if count>9: break
except Exception: pass
return count
def getGeneSystem(filename):
firstRow=True
count=0
system = 'Symbol'
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
if firstRow: firstRow=False
else:
id = string.split(line,'\t')[0]
if 'ENS' in id: system = 'Ensembl'
count+=1
if count>9: break
except Exception: pass
return system
def determinePlatform(filename):
platform = ''
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
#print [line]
if len(line)>0: platform = line
except Exception: pass
return platform
def importDefaults(array_type,species):
filename = 'Config/defaults-expr.txt'
expr_defaults = importDefaultInfo(filename,array_type)
#perform_alt_analysis, expression_data_format, dabg_p, expression_threshold, avg_all_for_ss, include_raw_data
filename = 'Config/defaults-alt_exon.txt'
alt_exon_defaults = importDefaultInfo(filename,array_type)
#analysis_method, alt_exon_fold_variable, p_threshold, filter_probeset_types, gene_expression_cutoff, perform_permutation_analysis, permute_p_threshold,run_MiDAS, export_splice_index_values = values
filename = 'Config/defaults-funct.txt'
functional_analysis_defaults = importDefaultInfo(filename,array_type)
#analyze_functional_attributes,microRNA_prediction_method = functional_analysis_defaults
filename = 'Config/defaults-goelite.txt'
goelite_defaults = importDefaultInfo(filename,array_type)
return expr_defaults, alt_exon_defaults, functional_analysis_defaults, goelite_defaults
def importDefaultInfo(filename,array_type):
fn=filepath(filename)
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
if '-expr' in filename:
array_abrev, dabg_p, rpkm_threshold, gene_exp_threshold, exon_exp_threshold, exon_rpkm_threshold, expression_threshold, perform_alt_analysis, analyze_as_groups, expression_data_format, normalize_feature_exp, normalize_gene_data, avg_all_for_ss, include_raw_data, probability_algorithm, FDR_statistic, batch_effects, marker_finder, visualize_results, run_lineage_profiler, run_goelite = string.split(data,'\t')
if array_type == array_abrev:
return dabg_p, rpkm_threshold, gene_exp_threshold, exon_exp_threshold, exon_rpkm_threshold, expression_threshold, perform_alt_analysis, analyze_as_groups, expression_data_format, normalize_feature_exp, normalize_gene_data, avg_all_for_ss, include_raw_data, probability_algorithm, FDR_statistic, batch_effects, marker_finder, visualize_results, run_lineage_profiler, run_goelite
if '-alt' in filename:
array_abrev, analysis_method, additional_algorithms, filter_probeset_types, analyze_all_conditions, p_threshold, alt_exon_fold_variable, additional_score, permute_p_threshold, gene_expression_cutoff, remove_intronic_junctions, perform_permutation_analysis, export_splice_index_values, run_MiDAS, calculate_splicing_index_p, filter_for_AS = string.split(data,'\t')
if array_type == array_abrev:
return [analysis_method, additional_algorithms, filter_probeset_types, analyze_all_conditions, p_threshold, alt_exon_fold_variable, additional_score, permute_p_threshold, gene_expression_cutoff, remove_intronic_junctions, perform_permutation_analysis, export_splice_index_values, run_MiDAS, calculate_splicing_index_p, filter_for_AS]
if '-funct' in filename:
array_abrev, analyze_functional_attributes, microRNA_prediction_method = string.split(data,'\t')
if array_type == array_abrev:
return [analyze_functional_attributes,microRNA_prediction_method]
if '-goelite' in filename:
array_abrev, ge_fold_cutoffs, ge_pvalue_cutoffs, ge_ptype, filter_method, z_threshold, p_val_threshold, change_threshold, ORA_algorithm, resources_to_analyze, pathway_permutations, mod, returnPathways, get_additional = string.split(data,'\t')
if array_type == array_abrev:
return [ge_fold_cutoffs, ge_pvalue_cutoffs, ge_ptype, filter_method, z_threshold, p_val_threshold, change_threshold, ORA_algorithm, resources_to_analyze, pathway_permutations, mod, returnPathways, get_additional]
class OptionData:
def __init__(self,option,displayed_title,display_object,notes,array_options,global_default):
self._option = option; self._displayed_title = displayed_title; self._notes = notes
self._array_options = array_options; self._display_object = display_object
if len(global_default)>0:
if '|' in global_default:
global_default = string.split(global_default,'|') ### store as a list
self._default_option = global_default
def Option(self): return self._option
def VariableName(self): return self._option
def Display(self): return self._displayed_title
def setDisplay(self,display_title): self._displayed_title = display_title
def setDisplayObject(self,display_object): self._display_object = display_object
def DisplayObject(self): return self._display_object
def Notes(self): return self._notes
def setNotes(self,notes): self._notes = notes
def DefaultOption(self): return self._default_option
def setDefaultOption(self,default_option): self._default_option = default_option
def setNotes(self,notes): self._notes = notes
def ArrayOptions(self): return self._array_options
def setArrayOptions(self,array_options): self._array_options = array_options
def Options(self): return self._array_options
def __repr__(self): return self.Option()+'|'+self.Display()
def importUserOptions(array_type,vendor=None):
filename = 'Config/options.txt'; option_db={}; option_list_db={}
fn=filepath(filename); x=0
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
data = string.replace(data,'\k','\n') ###Used \k in the file instead of \n, since these are removed above
if array_type == 'RNASeq':
data = string.replace(data,'probeset','junction')
data = string.replace(data,'probe set','junction, exon or gene')
data = string.replace(data,'CEL file','BED, BAM, TAB or TCGA junction file')
if vendor != 'Affymetrix':
data = string.replace(data,'probe set','gene')
if vendor == 'Agilent':
if 'CEL file' in data:
data = string.replace(data,'CEL file','Feature Extraction file')
data = string.replace(data,' (required)','')
if array_type == '10XGenomics':
data = string.replace(data,'CEL file containing folder','Chromium filtered matrix.mtx or .h5 file')
try:
if '10X' in vendor:
data = string.replace(data,'CEL file containing folder','Chromium filtered matrix.mtx or .h5file')
except Exception: pass
t = string.split(data,'\t')
#option,mac_displayed_title,pc_displayed_title,pc_display2,linux_displayed_title,display_object,group,notes,description,global_default = t[:10]
option,displayed_title,display_object,group,notes,description,global_default = t[:7]
"""
if os.name == 'nt':
import platform
if '64' in platform.machine(): displayed_title = pc_display2
elif '32' in platform.machine(): displayed_title = pc_display2
elif '64bit' in platform.architecture(): displayed_title = pc_display2
else: displayed_title = pc_displayed_title
elif 'darwin' in sys.platform: displayed_title = mac_displayed_title
elif 'linux' in sys.platform: displayed_title = linux_displayed_title
else: displayed_title = linux_displayed_title
"""
"""
try:
if option == 'rho_cutoff' and '10X' in vendor:
global_default = '0.3'
if option == 'restrictBy' and '10X' in vendor:
global_default = 'yes'
if option == 'column_metric_predict' and '10X' in vendor:
global_default = 'euclidean'
except Exception:
pass
"""
if 'junction' in displayed_title: displayed_title+=' '
"""if array_type == 'RNASeq':
if option == 'dabg_p': ### substitute the text for the alternatitve text in notes
displayed_title = notes"""
if x == 0:
i = t.index(array_type) ### Index position of the name of the array_type selected by user (or arbitrary to begin with)
x = 1
else:
array_options = t[i]
if array_type == "3'array":
"""
if 'normalize_gene_data' in data and vendor != 'Agilent':
array_options = 'NA' ### only applies currently to Agilent arrays """
if 'channel_to_extract' in data and vendor != 'Agilent':
array_options = 'NA' ### only applies currently to Agilent arrays
array_options = string.split(array_options,'|')
od = OptionData(option,displayed_title,display_object,notes,array_options,global_default)
option_db[option] = od
try: option_list_db[group].append(option) ###group is the name of the GUI menu group
except KeyError: option_list_db[group] = [option]
return option_list_db,option_db
class SummaryResults:
def __init__(self):
def showLink(event):
idx= int(event.widget.tag_names(CURRENT)[1])
webbrowser.open(LINKS[idx])
LINKS=('http://www.altanalyze.org','')
self.LINKS = LINKS
try: tl = Toplevel()
except Exception: tl = Tkinter.Toplevel()
tl.title('AltAnalyze')
filename = 'Config/icon.gif'
fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(tl); can.pack(side='top'); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW); use_scroll = 'no'
label_text_str = 'AltAnalyze Result Summary'; height = 250; width = 700
self.sf = PmwFreeze.ScrolledFrame(tl,
labelpos = 'n', label_text = label_text_str,
usehullsize = 1, hull_width = width, hull_height = height)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
tl.mainloop()
class FeedbackWindow:
def __init__(self,message,button_text,button_text2):
self.message = message; self.button_text = button_text; self.button_text2 = button_text2
parent = Tk(); self._parent = parent; nulls = '\t\t\t\t\t\t\t'; parent.title('Attention!!!')
self._user_variables={}
filename = 'Config/warning_big.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
try:
can = Canvas(parent); can.pack(side='left',padx = 10); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
except Exception: pass
Label(parent, text='\n'+self.message+'\n'+nulls).pack()
text_button = Button(parent, text=self.button_text, command=self.button1); text_button.pack(side = 'bottom', padx = 5, pady = 5)
text_button2 = Button(parent, text=self.button_text2, command=self.button2); text_button2.pack(side = 'bottom', padx = 5, pady = 5)
parent.protocol("WM_DELETE_WINDOW", self.deleteWindow)
parent.mainloop()
def button1(self): self._user_variables['button']=self.button_text; self._parent.destroy()
def button2(self): self._user_variables['button']=self.button_text2; self._parent.destroy()
def ButtonSelection(self): return self._user_variables
def deleteWindow(self):
#tkMessageBox.showwarning("Quit Selected","Use 'Quit' button to end program!",parent=self._parent)
self._parent.destroy(); sys.exit()
class IndicatorWindowSimple:
def __init__(self,message,button_text):
self.message = message; self.button_text = button_text
parent = Tk(); self._parent = parent; nulls = '\t\t\t\t\t\t\t'; parent.title('Attention!!!')
filename = 'Config/warning_big.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
try:
can = Canvas(parent); can.pack(side='left',padx = 10); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
except Exception: pass
Label(parent, text='\n'+self.message+'\n'+nulls).pack()
text_button = Button(parent, text=self.button_text, command=parent.destroy); text_button.pack(side = 'bottom', padx = 5, pady = 5)
parent.mainloop()
class IndicatorWindow:
def __init__(self,message,button_text):
self.message = message; self.button_text = button_text
parent = Tk(); self._parent = parent; nulls = '\t\t\t\t\t\t\t'; parent.title('Attention!!!')
filename = 'Config/warning_big.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
try:
can = Canvas(parent); can.pack(side='left',padx = 10); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
except Exception: pass
Label(parent, text='\n'+self.message+'\n'+nulls).pack()
quit_button = Button(parent, text='Quit', command=self.quit); quit_button.pack(side = 'bottom', padx = 5, pady = 5)
text_button = Button(parent, text=self.button_text, command=parent.destroy); text_button.pack(side = 'bottom', padx = 5, pady = 5)
parent.mainloop()
def quit(self):
try: self._parent.quit(); self._parent.destroy(); sys.exit()
except Exception: self._parent.quit(); sys.exit()
class DownloadWindow:
def __init__(self,message,option1,option2):
self._user_variables = user_variables
if len(option2)==2: option2,option3 = option2; num_options = 3; self.option3 = option3
else: num_options = 2
self.message = message; self.option1 = option1; self.option2 = option2
parent = Tk(); self._parent = parent; nulls = '\t\t\t\t\t\t\t'; parent.title('Attention!!!')
filename = 'Config/warning_big.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(parent); can.pack(side='left',padx = 10); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
Label(parent, text='\n'+self.message+'\n'+nulls).pack()
text_button = Button(parent, text=self.option1, command=self.selected1); text_button.pack(side = 'bottom', padx = 5, pady = 5)
text_button2 = Button(parent, text=self.option2, command=self.selected2); text_button2.pack(side = 'bottom', padx = 5, pady = 5)
if num_options == 3:
text_button3 = Button(parent, text=self.option3, command=self.selected3); text_button3.pack(side = 'bottom', padx = 5, pady = 5)
parent.mainloop()
def selected1(self):
self._user_variables['selected_option']=1; self._parent.destroy()
def selected2(self):
self._user_variables['selected_option']=2; self._parent.destroy()
def selected3(self):
self._user_variables['selected_option']=3; self._parent.destroy()
def Results(self): return self._user_variables
class IndicatorLinkOutWindow:
def __init__(self,message,button_text,url):
self.message = message; self.button_text = button_text; nulls = '\t\t\t\t\t\t\t';
parent = Tk(); self._parent = parent; parent.title('Attention!!!'); self.url = url
filename = 'Config/warning_big.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(parent); can.pack(side='left',padx = 10); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
Label(parent, text='\n'+self.message+'\n'+nulls).pack()
continue_button = Button(parent, text='Continue', command=parent.destroy); continue_button.pack(side = 'bottom', padx = 5, pady = 5)
text_button = Button(parent, text=self.button_text, command=self.linkout); text_button.pack(side = 'bottom', padx = 5, pady = 5)
parent.mainloop()
def linkout(self):
webbrowser.open(self.url)
class IndicatorChooseWindow:
def __init__(self,message,button_text):
self.message = message; self .button_text = button_text
parent = Tk(); self._parent = parent; nulls = '\t\t\t\t\t\t\t'; parent.title('Attention!!!')
filename = 'Config/icon.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(parent); can.pack(side='left'); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
Label(parent, text='\n'+self.message+'\n'+nulls).pack()
#text_button = Button(parent, text=self.button_text, command=parent.destroy); text_button.pack(side = 'bottom', padx = 5, pady = 5)
option=''
def foldercallback(callback=self.callback,option=option): self.chooseDirectory(option)
choose_win = Button(self._parent, text=self.button_text,command=foldercallback); choose_win.pack(padx = 3, pady = 3)
quit_button = Button(parent, text='Quit', command=self.quit); quit_button.pack(padx = 3, pady = 3)
parent.mainloop()
def quit(self):
try: self._parent.quit(); self._parent.destroy(); sys.exit()
except Exception: self._parent.quit(); sys.exit()
def callback(self, tag, option): null = ''
def chooseDirectory(self,option):
tag = tkFileDialog.askdirectory(parent=self._parent)
### Below is code specific for grabbing the APT location
from import_scripts import ResultsExport_module
apt_location = ResultsExport_module.getAPTDir(tag)
if 'bin' not in apt_location:
print_out = "WARNING!!! Unable to find a valid Affymetrix Power Tools directory."
try: WarningWindow(print_out,' Continue ')
except NameError: print print_out
self._tag = ''
else: self._tag = apt_location
self.destroy_win()
def destroy_win(self):
try: self._parent.quit(); self._parent.destroy()
except Exception: self._parent.quit(); sys.exit()
def Folder(self): return self._tag
class WarningWindow:
def __init__(self,warning,window_name):
try: tkMessageBox.showerror(window_name, warning)
except Exception:
print warning
#print window_name; sys.exit()
kill
class InfoWindow:
def __init__(self,dialogue,header):
try: tkMessageBox.showinfo(header, dialogue)
except Exception:
print dialogue
#print 'Attempted to open a GUI that is not accessible...exiting program';sys.exit()
#print "Analysis finished...exiting AltAnalyze."; sys.exit()
class MacConsiderations:
def __init__(self):
parent = Tk()
self._parent = parent
parent.title('AltAnalyze: Considerations for Mac OSX')
self._user_variables={}
filename = 'Config/MacOSX.png'
try:
import ImageTk
img = ImageTk.PhotoImage(file=filepath(filename))
except Exception:
try:
from PIL import ImageTk
img = ImageTk.PhotoImage(file=filepath(filename))
except Exception:
filename = 'Config/MacOSX.gif'
fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(parent); can.pack(side='top',fill=BOTH); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
### Add some buttons to the horizontal RadioSelect
continue_to_next_win = Tkinter.Button(text = 'Continue', command = parent.destroy)
continue_to_next_win.pack(side = 'right', padx = 5, pady = 5);
info_win = Button(self._parent, text="Online Help", command=self.Linkout)
info_win.pack(side = 'left', padx = 5, pady = 5)
self.url = 'http://www.altanalyze.org/MacOSX_help.html'
parent.protocol("WM_DELETE_WINDOW", self.deleteWindow)
parent.mainloop()
def Linkout(self):
try: webbrowser.open(self.url)
except Exception,e: print e
def deleteWindow(self):
#tkMessageBox.showwarning("Quit Selected","Use 'Quit' button to end program!",parent=self._parent)
self._parent.destroy(); sys.exit()
def callback(self, tag):
#print 'Button',[option], tag,'was pressed.'
self._user_variables['continue'] = tag
class MainMenu:
def __init__(self):
parent = Tk()
self._parent = parent
parent.title('AltAnalyze: Introduction')
self._user_variables={}
filename = 'Config/logo.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(parent); can.pack(side='top',fill=BOTH); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
"""
### Create and pack a horizontal RadioSelect widget.
def buttoncallback(tag,callback=self.callback):
callback(tag)
horiz = PmwFreeze.RadioSelect(parent,
labelpos = 'w', command = buttoncallback,
label_text = 'AltAnalyze version 1.155 Main', frame_borderwidth = 2,
frame_relief = 'ridge'
); horiz.pack(fill = 'x', padx = 10, pady = 10)
for text in ['Continue']: horiz.add(text)
"""
### Add some buttons to the horizontal RadioSelect
continue_to_next_win = Tkinter.Button(text = 'Begin Analysis', command = parent.destroy)
continue_to_next_win.pack(side = 'bottom', padx = 5, pady = 5);
info_win = Button(self._parent, text="About AltAnalyze", command=self.info)
info_win.pack(side = 'bottom', padx = 5, pady = 5)
parent.protocol("WM_DELETE_WINDOW", self.deleteWindow)
parent.mainloop()
def info(self):
"""
###Display the information using a messagebox
about = 'AltAnalyze version 2.1.4.\n'
about+= 'AltAnalyze is an open-source, freely available application covered under the\n'
about+= 'Apache open-source license. Additional information can be found at:\n'
about+= 'http://www.altanalyze.org\n'
about+= '\nDeveloped by:\nNathan Salomonis Lab\nCincinnati Childrens Hospital Medical Center 2008-2016'
tkMessageBox.showinfo("About AltAnalyze",about,parent=self._parent)
"""
def showLink(event):
idx= int(event.widget.tag_names(CURRENT)[1])
webbrowser.open(LINKS[idx])
LINKS=('http://www.altanalyze.org','')
self.LINKS = LINKS
tl = Toplevel() ### Create a top-level window separate than the parent
txt=Text(tl)
#filename = 'Config/icon.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
#can = Canvas(tl); can.pack(side='left'); can.config(width=img.width(), height=img.height())
#can.create_image(2, 2, image=img, anchor=NW)
txt.pack(expand=True, fill="both")
txt.insert(END, 'AltAnalyze version 2.1.4.\n')
txt.insert(END, 'AltAnalyze is an open-source, freely available application covered under the\n')
txt.insert(END, 'Apache open-source license. Additional information can be found at:\n')
txt.insert(END, "http://www.altanalyze.org\n", ('link', str(0)))
txt.insert(END, '\nDeveloped by:\nDr. Nathan Salomonis Research Group\nCincinnati Childrens Hospital Medical Center 2008-2016')
txt.tag_config('link', foreground="blue", underline = 1)
txt.tag_bind('link', '<Button-1>', showLink)
def deleteWindow(self):
#tkMessageBox.showwarning("Quit Selected","Use 'Quit' button to end program!",parent=self._parent)
self._parent.destroy(); sys.exit()
def callback(self, tag):
#print 'Button',[option], tag,'was pressed.'
self._user_variables['continue'] = tag
class LinkOutWindow:
def __init__(self,output):
### Text window with link included
url,text_list = output
def showLink(event):
idx= int(event.widget.tag_names(CURRENT)[1])
webbrowser.open(LINKS[idx])
LINKS=(url,'')
self.LINKS = LINKS
tl = Toplevel() ### Create a top-level window separate than the parent
txt=Text(tl)
txt.pack(expand=True, fill="both")
for str_item in text_list:
txt.insert(END, str_item+'\n')
txt.insert(END, "http://www.altanalyze.org\n", ('link', str(0)))
txt.tag_config('link', foreground="blue", underline = 1)
txt.tag_bind('link', '<Button-1>', showLink)
text_button = Button(parent, text=self.button_text, command=parent.destroy); text_button.pack(side = 'bottom', padx = 5, pady = 5)
parent.mainloop()
def exportCELFileList(cel_files,cel_file_dir):
fn=cel_file_dir+'/cel_files.txt'; data = open(fn,'w')
data.write('cel_files'+'\n') ###header
for cel_file in cel_files:
data.write(cel_file+'\n')
data.close()
return fn
def predictGroupsAndComps(cel_files,output_dir,exp_name):
fn1=output_dir+'/ExpressionInput/groups.'+exp_name+'.txt'; gdata = export.ExportFile(fn1)
fn2=output_dir+'/ExpressionInput/comps.'+exp_name+'.txt'; cdata = export.ExportFile(fn2)
fn3=output_dir+'/ExpressionInput/exp.'+exp_name+'.txt'
delimited_db={}; delim_type={}; files_exported = 'no'
for cel_file in cel_files:
cel_name = cel_file
cel_file = string.replace(cel_file,'.CEL','')
cel_file = string.replace(cel_file,'.cel','')
dashed_delim = string.split(cel_file,'-')
dot_delim = string.split(cel_file,'.')
under_delim = string.split(cel_file,'_')
if len(dashed_delim) == 2:
delim_type[1]=None
try: delimited_db[dashed_delim[0]].append(cel_name)
except KeyError: delimited_db[dashed_delim[0]] = [cel_name]
elif len(under_delim) == 2:
delim_type[2]=None
try: delimited_db[under_delim[0]].append(cel_name)
except KeyError: delimited_db[under_delim[0]] = [cel_name]
elif len(dot_delim) == 2:
delim_type[3]=None
try: delimited_db[dot_delim[0]].append(cel_name)
except KeyError: delimited_db[dot_delim[0]] = [cel_name]
if len(delim_type)==1 and len(delimited_db)>1: ###only 1 type of delimiter used and at least 2 groups present
group_index=0; group_db={}; files_exported = 'yes'
for group in delimited_db:
group_index+=1; group_db[str(group_index)]=None
for array in delimited_db[group]:
gdata.write(string.join([array,str(group_index),group],'\t')+'\n')
for index1 in group_db: ### Create a comps file for all possible comps
for index2 in group_db:
if index1 != index2:
cdata.write(string.join([index1,index2],'\t')+'\n')
gdata.close(); cdata.close()
if files_exported == 'no':
os.remove(fn1); os.remove(fn2)
try: ExpressionBuilder.checkArrayHeaders(fn3,fn1) ### Create just the groups template file
except Exception: pass ### This error will more likely occur since no expression file has been created
return files_exported
def formatArrayGroupsForGUI(array_group_list, category = 'GroupArrays'):
### Format input for GUI like the imported options.txt Config file, except allow for custom fields in the GUI class
option_db={}; option_list={}
if category != 'BatchArrays':
### Add a checkbox at the top to allow for automatic assignment of groups (e.g., Single Cell Data)
option='PredictGroups';displayed_title='Run de novo cluster prediction (ICGS) to discover groups, instead';display_object='single-checkbox';notes='';array_options=['---']
od = OptionData(option,displayed_title,display_object,notes,array_options,'')
option_db[option] = od
option_list[category] = [option]
for agd in array_group_list:
option = agd.Array(); array_options = [agd.GroupName()]; displayed_title=option; display_object='simple_entry'; notes=''
od = OptionData(option,displayed_title,display_object,notes,array_options,'')
option_db[option] = od
try: option_list[category].append(option) ###group is the name of the GUI menu group
except KeyError: option_list[category] = [option]
return option_db,option_list
def importExpressionFiles():
exp_file_location_db={}; exp_files=[]; parent_dir = 'ExpressionInput'+'/'+array_type
fn =filepath(parent_dir+'/'); dir_files = read_directory('/'+parent_dir)
stats_file_dir=''
for file in dir_files:
if 'exp.' in file: exp_files.append(file)
for file in exp_files:
stats_file = string.replace(file,'exp.','stats.')
groups_file = string.replace(file,'exp.','groups.')
comps_file = string.replace(file,'exp.','comps.')
if stats_file in dir_files: stats_file_dir = fn+stats_file
if groups_file in dir_files and comps_file in dir_files:
groups_file_dir = fn+groups_file; comps_file_dir = fn+comps_file
exp_file_dir = fn+file
fl = ExpressionFileLocationData(exp_file_dir,stats_file_dir,groups_file_dir,comps_file_dir)
exp_file_location_db[file] = fl
return exp_file_location_db
class ExpressionFileLocationData:
def __init__(self, exp_file, stats_file, groups_file, comps_file):
self._exp_file = exp_file; self._stats_file = stats_file; self._groups_file = groups_file
self._comps_file = comps_file; self.biotypes='NA'
import platform; self.architecture = platform.architecture()[0]
self.normalize_feature_exp = 'NA'
self.normalize_gene_data = 'NA'
self.runKallisto =''
def setExpFile(self, exp_file):self._exp_file=exp_file
def ExpFile(self): return self._exp_file
def StatsFile(self): return self._stats_file
def CountsFile(self):
import AltAnalyze
counts_file = string.replace(self.ExpFile(),'exp.','counts.')
file_length = AltAnalyze.verifyFileLength(counts_file)
if file_length>0:
return counts_file
else:
return self.ExpFile()
def GroupsFile(self): return self._groups_file
def setGroupsFile(self, groups_file):self._groups_file=groups_file
def CompsFile(self): return self._comps_file
def setCompsFile(self, comps_file):self._comps_file=comps_file
def setArchitecture(self,architecture): self.architecture = architecture
def setAPTLocation(self,apt_location): self._apt_location = osfilepath(apt_location)
def setInputCDFFile(self,cdf_file): self._cdf_file = osfilepath(cdf_file)
def setCLFFile(self,clf_file): self._clf_file = osfilepath(clf_file)
def setBGPFile(self,bgp_file): self._bgp_file = osfilepath(bgp_file)
def setCELFileDir(self,cel_file_dir): self._cel_file_dir = osfilepath(cel_file_dir)
def setBEDFileDir(self,cel_file_dir): self._cel_file_dir = osfilepath(cel_file_dir)
def setFeatureNormalization(self,normalize_feature_exp): self.normalize_feature_exp = normalize_feature_exp
def setExcludeLowExpressionExons(self, excludeNonExpExons): self.excludeNonExpExons = excludeNonExpExons
def setNormMatrix(self,normalize_gene_data): self.normalize_gene_data = normalize_gene_data
def setProbabilityStatistic(self,probability_statistic): self.probability_statistic = probability_statistic
def setFDRStatistic(self, FDR_statistic): self.FDR_statistic = FDR_statistic
def setBatchEffectRemoval(self,batch_effects): self.batch_effects = batch_effects
def setProducePlots(self,visualize_results): self.visualize_results = visualize_results
def setPerformLineageProfiler(self, run_lineage_profiler): self.run_lineage_profiler = run_lineage_profiler
def setCompendiumType(self,compendiumType): self.compendiumType = compendiumType
def setCompendiumPlatform(self,compendiumPlatform): self.compendiumPlatform = compendiumPlatform
def set_reference_exp_file(self,exp_file): self._reference_exp_file = exp_file
def setClassificationAnalysis(self, classificationAnalysis): self.classificationAnalysis = classificationAnalysis
def setReturnCentroids(self,returnCentroids): self.returnCentroids = returnCentroids
def setMultiThreading(self, multithreading): self.multithreading = multithreading
def setVendor(self,vendor): self.vendor = vendor
def setKallistoFile(self,kallisto_exp): self.kallisto_exp = kallisto_exp
def KallistoFile(self): return self.kallisto_exp
def setPredictGroups(self, predictGroups): self.predictGroups = predictGroups
def setPredictGroupsParams(self, predictGroupsObjects): self.predictGroupsObjects = predictGroupsObjects
def setGraphicLinks(self,graphic_links): self.graphic_links = graphic_links ### file location of image files
def setSTDOUT(self, stdout): self.stdout = stdout
def setExonExpThreshold(self,exon_exp_threshold):
try: exon_exp_threshold = float(exon_exp_threshold)
except Exception: exon_exp_threshold = exon_exp_threshold
self.exon_exp_threshold = exon_exp_threshold
def setExonRPKMThreshold(self,exon_rpkm_threshold):
try: exon_rpkm_threshold = float(exon_rpkm_threshold)
except Exception: exon_rpkm_threshold = exon_rpkm_threshold
self.exon_rpkm_threshold = exon_rpkm_threshold
def setGeneExpThreshold(self,gene_exp_threshold):
try: gene_exp_threshold = float(gene_exp_threshold)
except Exception: gene_exp_threshold = gene_exp_threshold
self.gene_exp_threshold = gene_exp_threshold
def setJunctionExpThreshold(self,junction_exp_threshold):
try: junction_exp_threshold = float(junction_exp_threshold)
except Exception: junction_exp_threshold = junction_exp_threshold
self.junction_exp_threshold = junction_exp_threshold
def setRPKMThreshold(self,rpkm_threshold):
try: rpkm_threshold = float(rpkm_threshold)
except Exception: rpkm_threshold = rpkm_threshold
self.rpkm_threshold = rpkm_threshold
def setMarkerFinder(self,marker_finder): self.marker_finder = marker_finder
def reference_exp_file(self):
try:
if len(self._reference_exp_file)>1:
return self._reference_exp_file
else:
return False
except:
return False
def ReturnCentroids(self): return self.returnCentroids
def FDRStatistic(self): return self.FDR_statistic
def multiThreading(self): return self.multithreading
def STDOUT(self): return self.stdout
def ExonExpThreshold(self): return self.exon_exp_threshold
def BatchEffectRemoval(self): return self.batch_effects
def MarkerFinder(self): return self.marker_finder
def PredictGroups(self): self.predictGroups
def PredictGroupsObjects(self): self.predictGroupsObjects
def ExonRPKMThreshold(self): return self.exon_rpkm_threshold
def GeneExpThreshold(self): return self.gene_exp_threshold
def JunctionExpThreshold(self): return self.junction_exp_threshold
def RPKMThreshold(self): return self.rpkm_threshold
def ProbabilityStatistic(self): return self.probability_statistic
def ProducePlots(self): return self.visualize_results
def PerformLineageProfiler(self): return self.run_lineage_profiler
def CompendiumType(self): return self.compendiumType
def CompendiumPlatform(self): return self.compendiumPlatform
def ClassificationAnalysis(self): return self.classificationAnalysis
def GraphicLinks(self): return self.graphic_links
def setArrayType(self,array_type): self._array_type = array_type
def setOutputDir(self,output_dir): self._output_dir = output_dir
def setBiotypes(self,biotypes): self.biotypes = biotypes
def setRootDir(self,parent_dir):
### Get directory above ExpressionInput
split_dirs = string.split(parent_dir,'ExpressionInput')
root_dir = split_dirs[0]
self._root_dir = root_dir + '/'
def setXHybRemoval(self,xhyb): self._xhyb = xhyb
def XHybRemoval(self): return self._xhyb
def setExonBedBuildStatus(self,bed_build_status): self.bed_build_status = bed_build_status
def setRunKallisto(self, runKallisto): self.runKallisto = runKallisto
def RunKallisto(self): return self.runKallisto
def setCountsNormalization(self, expression_data_format): self.expression_data_format = expression_data_format
def CountsNormalization(self):
try: return self.expression_data_format
except: return 'scaled'
def setCustomFASTA(self, customFASTA): self.customFASTA = customFASTA
def CustomFASTA(self): return self.customFASTA
def setChromiumSparseMatrix(self, chromiumSparseMatrix): self.chromiumSparseMatrix = chromiumSparseMatrix
def ChromiumSparseMatrix(self): return self.chromiumSparseMatrix
def setChannelToExtract(self,channel_to_extract): self.channel_to_extract = channel_to_extract
def ExonBedBuildStatus(self): return self.bed_build_status
def ChannelToExtract(self): return self.channel_to_extract
def FeatureNormalization(self): return self.normalize_feature_exp
def setUseJunctionsForGeneExpression(self, use_junctions_for_geneexpression): self.use_junctions_for_geneexpression = use_junctions_for_geneexpression
def useJunctionsForGeneExpression(self):
try: return self.use_junctions_for_geneexpression
except Exception: return False
def excludeLowExpressionExons(self): return self.excludeNonExpExons
def NormMatrix(self): return self.normalize_gene_data
def RootDir(self): return self._root_dir
def APTLocation(self): return self._apt_location
def InputCDFFile(self): return self._cdf_file
def CLFFile(self): return self._clf_file
def BGPFile(self): return self._bgp_file
def CELFileDir(self): return self._cel_file_dir
def BEDFileDir(self): return self._cel_file_dir+'/'
def ArrayType(self):
try: return self._array_type
except Exception: return 'RNASeq'
def OutputDir(self): return self._output_dir
def Vendor(self):
try: return self.vendor
except Exception: return 'RNASeq'
def setSpecies(self, species): self.species = species
def Species(self): return self.species
def setPlatformType(self, platformType): self.platformType = platformType
def setAnalysisMode(self, analysis_mode): self.analysis_mode = analysis_mode
def setMLP(self,mlpr): self.mlp = mlpr
def setExonMapFile(self, exonMapFile): self.exonMapFile = exonMapFile
def ExonMapFile(self): return self.exonMapFile
def setCorrelationDirection(self, correlationDirection): self.correlationDirection = correlationDirection
def CorrelationDirection(self): return self.correlationDirection
def setPearsonThreshold(self, pearsonThreshold): self.pearsonThreshold = pearsonThreshold
def PearsonThreshold(self): return self.pearsonThreshold
def setUseAdjPvalue(self, useAdjPval): self.useAdjPval = useAdjPval
def UseAdjPvalue(self):
if string.lower(self.useAdjPval)=='false' or string.lower(self.useAdjPval)=='no' or self.useAdjPval==False:
return False
else:
return True
def setLabels(self,labels): self.labels = labels
def Labels(self): return self.labels
def setFoldCutoff(self, foldCutoff): self.foldCutoff = foldCutoff
def FoldCutoff(self): return self.foldCutoff
def setPvalThreshold(self, pvalThreshold): self.pvalThreshold = pvalThreshold
def PvalThreshold(self): return self.pvalThreshold
def setPeformDiffExpAnalysis(self, peformDiffExpAnalysis): self.peformDiffExpAnalysis = peformDiffExpAnalysis
def PeformDiffExpAnalysis(self):
if self.peformDiffExpAnalysis==False:
return False
if self.peformDiffExpAnalysis==True:
return True
if string.lower(self.peformDiffExpAnalysis)=='false' or string.lower(self.peformDiffExpAnalysis)=='no':
return False
else:
return True
def MLP(self): return self.mlp
def PlatformType(self): return self.platformType
def AnalysisMode(self): return self.analysis_mode
def DatasetFile(self):
if 'exp.' in self.ExpFile():
dataset_dir = string.replace(self.ExpFile(),'exp.','DATASET-')
else:
parent = export.findParentDir(self.ExpFile())
file = export.findFilename(self.ExpFile())
if 'DATASET-' not in file:
dataset_dir = parent + 'DATASET-'+file
else:
dataset_dir = self.ExpFile()
dataset_dir = string.replace(dataset_dir,'ExpressionInput','ExpressionOutput')
return dataset_dir
def Architecture(self): return self.architecture
def BioTypes(self): return self.biotypes
def Report(self): return 'fl printout'+self.ExpFile()+'|'+str(len(self.StatsFile()))+'|'+str(len(self.GroupsFile()))+'|'+str(len(self.CompsFile()))
def __repr__(self): return self.Report()
class AdditionalAlgorithms:
def __init__(self, additional_algorithm):
self._additional_algorithm = additional_algorithm
def Algorithm(self): return self._additional_algorithm
def setScore(self,score): self._score = score
def Score(self): return self._score
def __repr__(self): return self.Algorithm()
def getDirectoryFiles():
status = 'repeat'
while status == 'repeat':
if backSelect == 'no' or 'InputExpFiles' == selected_parameters[-1]:
root = Tk(); root.title('AltAnalyze: Select Expression File for Filtering')
selected_parameters.append('InputExpFiles'); backSelect = 'no'
gu = GUI(root,option_db,option_list['InputExpFiles'],'')
else: gu = PreviousResults(old_options)
try: input_exp_file = gu.Results()['input_exp_file']
except KeyError: input_exp_file = '' ### Leave this blank so that the default directory is used
try: input_stats_file = gu.Results()['input_stats_file']
except KeyError: input_stats_file = '' ### Leave this blank so that the default directory is used
#if array_type == 'exon':
if 'steady-state' in input_exp_file or 'steady-state' in input_stats_file:
print_out = "Do not select steady-state expression files.."
IndicatorWindow(print_out,'Continue'); output_dir=''
elif len(input_exp_file)>0:
try: output_dir = gu.Results()['output_dir']
except KeyError: output_dir = '' ### Leave this blank so that the default directory is used
try: cel_files, array_linker_db = ExpressionBuilder.getArrayHeaders(input_exp_file)
except Exception:
print_out = "Input Expression file does not have a valid format."
IndicatorWindow(print_out,'Continue'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
if len(cel_files)>0: status = 'continue'
else:
print_out = "The expression file:\n"+input_exp_file+"\ndoes not appear to be a valid expression file. Check to see that\nthis is the correct tab-delimited text file."
IndicatorWindow(print_out,'Continue')
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
if len(output_dir)<1:
### Set output to the same directory or parent if none selected
if 'ExpressionInput' in input_exp_file: i = -2
else: i = -1
output_dir = string.join(string.split(input_exp_file,'/')[:i],'/')
def getUpdatedParameters(array_type,species,run_from_scratch,file_dirs):
### Get default options for ExpressionBuilder and AltAnalyze
na = 'NA'; log = 'log'; no = 'no'
global user_variables; user_variables={}; global selected_parameters; selected_parameters = []
run_goelite=no; change_threshold=na;pathway_permutations=na;mod=na; ge_ptype = 'rawp';resources_to_analyze = na
ge_fold_cutoffs=2;ge_pvalue_cutoffs=0.05;filter_method=na;z_threshold=1.96;p_val_threshold=0.05
returnPathways = 'no'
option_list,option_db = importUserOptions(array_type)
global root
if run_from_scratch != 'Prefiltered': ### This is when AltAnalyze has finished an analysis
root = Tk()
root.title('AltAnalyze: Perform Additional Analyses')
selected_parameters.append('AdditionalOptions'); backSelect = 'no'
gu = GUI(root,option_db,option_list['AdditionalOptions'],'')
new_run = gu.Results()['new_run']
else: new_run = None
if new_run == 'Change Parameters and Re-Run': AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
else:
expr_defaults, alt_exon_defaults, functional_analysis_defaults, goelite_defaults = importDefaults(array_type,species)
option_db['get_additional'].setArrayOptions(['---']+importResourceList())
option_db['get_additional'].setDefaultOption('---')
default_resources = option_db['resources_to_analyze'].ArrayOptions()
import_dir1 = '/AltDatabase/goelite/'+species+'/gene-mapp'
import_dir2 = '/AltDatabase/goelite/'+species+'/gene-go'
try:
gene_mapp_list = read_directory(import_dir1)
gene_mapp_list.sort()
for file in gene_mapp_list:
resource = string.split(file,'-')[-1][:-4]
if resource != 'MAPP' and resource not in default_resources and '.txt' in file:
default_resources.append(resource)
except Exception: pass
try:
gene_go_list = read_directory(import_dir2)
gene_go_list.sort()
for file in gene_go_list:
resource = string.split(file,'-')[-1][:-4]
if resource != 'GeneOntology' and resource not in default_resources and 'version' not in resource and '.txt' in file:
default_resources.append(resource)
except Exception: pass
option_db['resources_to_analyze'].setArrayOptions(default_resources)
proceed = 'no'
while proceed == 'no':
root = Tk(); root.title('AltAnalyze: Pathway Analysis Parameters')
if 'filtered' in run_from_scratch: ### Not relevant for 'Process AltAnalyze filtered'
option_list['GOElite'] = option_list['GOElite'][3:]; goelite_defaults = goelite_defaults[3:]
selected_parameters.append('GOElite'); backSelect = 'no'
gu = GUI(root,option_db,option_list['GOElite'],goelite_defaults)
if 'filtered' not in run_from_scratch: ### Not relevant for 'Process AltAnalyze filtered'
ge_fold_cutoffs = gu.Results()['ge_fold_cutoffs']
ge_pvalue_cutoffs = gu.Results()['ge_pvalue_cutoffs']
ge_ptype = gu.Results()['ge_ptype']
filter_method = gu.Results()['filter_method']
z_threshold = gu.Results()['z_threshold']
returnPathways = gu.Results()['returnPathways']
p_val_threshold = gu.Results()['p_val_threshold']
change_threshold = gu.Results()['change_threshold']
resources_to_analyze = gu.Results()['resources_to_analyze']
pathway_permutations = gu.Results()['pathway_permutations']
ORA_algorithm = gu.Results()['ORA_algorithm']
mod = gu.Results()['mod']
get_additional = gu.Results()['get_additional']
try:
z_threshold = float(z_threshold)
change_threshold = float(change_threshold)-1 ### This reflects the > statement in the GO-Elite filtering
p_val_threshold = float(p_val_threshold)
pathway_permutations = int(pathway_permutations)
if run_from_scratch != 'Process AltAnalyze filtered':
ge_fold_cutoffs = float(ge_fold_cutoffs)
ge_pvalue_cutoffs = float(ge_pvalue_cutoffs)
proceed = 'yes'
except Exception:
print_out = "Invalid numerical entry. Try again."
IndicatorWindow(print_out,'Continue')
if get_additional != '---':
analysis = 'getAdditionalOnlineResources'
values = species,get_additional
StatusWindow(values,analysis) ### display an window with download status
try:
criterion_input_folder, criterion_denom_folder, main_output_folder = file_dirs
import GO_Elite
if run_from_scratch != 'Prefiltered': ### Only applies to AltAnalyze generated GO-Elite input
###Export dataset criterion using user-defined filters
ExpressionBuilder.buildCriterion(ge_fold_cutoffs, ge_pvalue_cutoffs, ge_ptype, main_output_folder, 'goelite')
#except Exception: null = []; # print 'No expression files to summarize'
if ORA_algorithm == 'Fisher Exact Test':
pathway_permutations = 'FisherExactTest'
goelite_var = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,''
GO_Elite.remoteAnalysis(goelite_var,'UI',Multi=mlp)
AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
except Exception:
print traceback.format_exc()
print_out = "Unexpected error encountered. Please see log file."
IndicatorWindow(print_out,'Continue'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
def addOnlineSpeciesDatabases(backSelect):
StatusWindow(file_location_defaults,'getOnlineDBConfig')
#except Exception,e: print [e]; null = []
importSystemInfo()
try: exportSystemInfo() ### By re-importing we incorporate new source data from the downloaded file
except Exception:
print 'Cannot write Config/source_data.txt to the Config directory (like Permissions Error)'
existing_species_codes = species_codes
importSpeciesInfo(); online_species = ['']
for species in species_codes: online_species.append(species)
online_species.sort()
importOnlineDatabaseVersions(); db_version_list=[]
for version in db_versions: db_version_list.append(version)
db_version_list.sort(); db_version_list.reverse(); select_version = db_version_list[0]
db_versions[select_version].sort()
option_db['selected_species1'].setArrayOptions(['---']+db_versions[select_version])
option_db['selected_species2'].setArrayOptions(['---']+db_versions[select_version])
option_db['selected_species3'].setArrayOptions(['---']+db_versions[select_version])
option_db['selected_version'].setArrayOptions(db_version_list)
proceed = 'no'
while proceed == 'no':
if backSelect == 'no' or 'OnlineDatabases' == selected_parameters[-1]:
selected_parameters.append('OnlineDatabases'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Species Databases Available for Download')
gu = GUI(root,option_db,option_list['OnlineDatabases'],'')
else: gu = PreviousResults(old_options); print 'alpha'
db_version = gu.Results()['selected_version']
exportDBversion(db_version)
try: species1 = gu.Results()['selected_species1']
except Exception: species1='---'
try: species2 = gu.Results()['selected_species2']
except Exception: species2='---'
try: species3 = gu.Results()['selected_species3']
except Exception: species3='---'
try: species_full = gu.Results()['species']
except Exception: species_full = ''
try: update_goelite_resources = gu.Results()['update_goelite_resources']
except Exception: update_goelite_resources = ''
#if species_full == 'Add Species': AltAnalyze.AltAnalyzeSetup(species_full); sys.exit()
new_species_list = [species1,species2,species3]; new_species_codes={}
for species in new_species_list:
if '---' not in species:
#try:
### Export basic species information
sc = species_codes[species].SpeciesCode()
existing_species_codes[species] = species_codes[species]
new_species_codes[sc]=[]
#except Exception: sc = None
if sc != None:
for ad in db_versions_vendors[db_version]:
if ad.SpeciesCodes() == species:
for array_system in array_codes:
ac = array_codes[array_system]
compatible_species = ac.SpeciesCodes()
if ac.Manufacturer() in ad.Manufacturer() and ('expression' in ac.ArrayName() or 'RNASeq' in ac.ArrayName() or 'RNA-seq' in ac.ArrayName()):
if sc not in compatible_species: compatible_species.append(sc)
ac.setSpeciesCodes(compatible_species)
try: exportArrayInfo(array_codes)
except Exception:
print 'Cannot write Config/arrays.txt to the Config directory (like Permissions Error)'
if len(new_species_codes) > 0:
analysis = 'getOnlineEliteDatabase'
values = file_location_defaults,db_version,new_species_codes,update_goelite_resources ### Download the online databases
StatusWindow(values,analysis)
proceed = 'yes'
else:
print_out = "Please select a species before continuing."
IndicatorWindow(print_out,'Try Again')
#db_versions_vendors
try: exportSpeciesInfo(existing_species_codes)
except Exception:
print 'Cannot write Config/species.txt to the Config directory (like Permissions Error)'
integrate_online_species = 'no'
def getArraysAndVendors(species,vendor):
array_list2=[]; manufacturer_list=[]
compatible_species,manufacturer_list_all = getSpeciesList('')
for array_name in array_list:
manufacturer = array_codes[array_name].Manufacturer()
if species in array_codes[array_name].SpeciesCodes():
manufacturer_list.append(manufacturer)
if len(vendor)>0:
if vendor == manufacturer: proceed = 'yes'
else: proceed = 'no'
else: proceed = 'yes'
if proceed == 'yes':
array_list2.append(array_name)
manufacturer_list = unique.unique(manufacturer_list)
array_list2 = unique.unique(array_list2) ### Filtered based on compatible species arrays
array_list2.sort(); manufacturer_list.sort()
if vendor == 'RNASeq':
array_list2.reverse()
return array_list2, manufacturer_list
def getSpeciesForArray(array_type):
array_list2=[]; manufacturer_list=[]; manufacturer_list_all=[]
for array_name in array_list:
current_species_codes = array_codes[array_type].SpeciesCodes()
try: current_species_dirs = unique.read_directory('/AltDatabase')
except Exception: current_species_dirs = current_species_codes
current_species_names=[]
for species in species_codes:
species_code = species_codes[species].SpeciesCode()
if species_code in current_species_codes:
if species_code in current_species_dirs: current_species_names.append(species)
current_species_names.sort()
return current_species_names
def verifyLineageProfilerDatabases(species,run_mode):
import AltAnalyze
installed = False
download_species = species
try:
gene_database = unique.getCurrentGeneDatabaseVersion()
except Exception:
gene_database = exportDBversion('')
gene_database = string.replace(gene_database,'EnsMart','')
print gene_database
try:
if int(gene_database[-2:]) < 62:
print_out = 'LineageProfiler is not supported in this database version (EnsMart62 and higher required).'
print print_out
return False
else:
if species == 'Hs':
source_file = 'AltDatabase/ensembl/'+species+'/'+species+'_exon_tissue-specific_protein_coding.txt'
download_species = 'Hs'
elif species == 'Mm':
source_file = 'AltDatabase/ensembl/'+species+'/'+species+'_gene_tissue-specific_protein_coding.txt'
download_species = 'Mm'
else: ### Use the mouse version instead - less variable data
source_file = 'AltDatabase/ensembl/'+species+'/'+species+'_gene_tissue-specific_protein_coding.txt'
download_species = 'Mm'
file_length = AltAnalyze.verifyFileLength(source_file)
if file_length>0:
installed = True
else:
print_out = 'To perform a LineageProfiler analysis AltAnalyze must\nfirst download the appropriate database.'
if run_mode == 'GUI':
IndicatorWindow(print_out,'Download')
else:
print print_out ### Occurs in command-line mode
filename = 'AltDatabase/ensembl/'+download_species+'_LineageProfiler.zip'
dir = 'AltDatabase/updated/'+gene_database ### Directory at altanalyze.org
var_list = filename,dir
if debug_mode == 'no' and run_mode == 'GUI': StatusWindow(var_list,'download')
else: update.downloadCurrentVersion(filename,dir,None)
file_length = AltAnalyze.verifyFileLength(source_file)
if file_length>0: installed = True
else:
try:
from build_scripts import GeneSetDownloader
GeneSetDownloader.translateBioMarkersBetweenSpecies('AltDatabase/ensembl/'+download_species,species)
except Exception:
None
except Exception: installed = False
return installed
def checkForLocalArraySupport(species,array_type,specific_arraytype,run_mode):
specific_arraytype = string.lower(specific_arraytype) ### Full array name
if array_type == 'junction' or array_type == 'RNASeq':
try: gene_database = unique.getCurrentGeneDatabaseVersion()
except Exception: gene_database='00'
try: int(gene_database[-2:])
except: gene_database='00'
if int(gene_database[-2:]) < 0:
print_out = 'The AltAnalyze database indicated for '+array_type+' analysis\n is not supported for alternative exon analysis.\nPlease update to EnsMart55 or greater before\nproceeding.'
if run_mode == 'GUI': IndicatorWindow(print_out,'Continue')
else: print print_out ### Occurs in command-line mode
AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
downloaded_junction_db = 'no'; file_problem='no'; wrong_junction_db = 'no'
while downloaded_junction_db == 'no': ### Used as validation in case internet connection is unavailable
try: dirs = read_directory('/AltDatabase/'+species)
except Exception: dirs=[]
if wrong_junction_db == 'yes':
print_out = 'Another junction database is installed. Select "Contine" to overwrite or manually change the name of this folder:\n'+filepath('AltDatabase/'+species+'/'+array_type)
if run_mode == 'GUI': IndicatorWindow(print_out,'Continue')
else: print print_out ### Occurs in command-line mode
if array_type not in dirs or file_problem == 'yes' or wrong_junction_db == 'yes':
if file_problem == 'yes':
print_out = 'Unknown installation error occured.\nPlease try again.'
else:
print_out = 'To perform an '+array_type+' analysis allow AltAnalyze \nto download the appropriate database now.'
if run_mode == 'GUI': IndicatorWindow(print_out,'Download')
else: print print_out ### Occurs in command-line mode
if array_type == 'RNASeq': filename = 'AltDatabase/'+species+'_'+array_type+'.zip'
elif 'glue' in specific_arraytype: filename = 'AltDatabase/'+species+'/'+species+'_'+array_type+'_Glue.zip'
elif 'hta 2.0' in specific_arraytype: filename = 'AltDatabase/'+species+'/'+species+'_'+array_type+'_HTA-2_0.zip'
elif 'mta 1.0' in specific_arraytype: filename = 'AltDatabase/'+species+'/'+species+'_'+array_type+'_MTA-1_0.zip'
else: filename = 'AltDatabase/'+species+'/'+species+'_'+array_type+'.zip'
dir = 'AltDatabase/updated/'+gene_database; var_list = filename,dir
if debug_mode == 'no' and run_mode == 'GUI':
StatusWindow(var_list,'download')
else: update.downloadCurrentVersion(filename,dir,None)
try: dirs = read_directory('/AltDatabase/'+species)
except Exception: dirs=[]
if array_type in dirs:
import AltAnalyze
file_length = AltAnalyze.verifyFileLength('AltDatabase/'+species+'/'+array_type+'/probeset-domain-annotations-exoncomp.txt')
if file_length>0: downloaded_junction_db = 'yes'
elif species == 'Mm' or species == 'Hs' or species == 'Rn': file_problem = 'yes'
else: downloaded_junction_db = 'yes' ### Occurs when no alternative exons present for species
if array_type == 'junction':
specific_platform = determinePlatform('AltDatabase/'+species+'/'+array_type+'/platform.txt')
if 'glue' in specific_arraytype and 'Glue' not in specific_platform: wrong_junction_db = 'yes'; downloaded_junction_db = 'no'
elif 'glue' not in specific_arraytype and 'Glue' in specific_platform: wrong_junction_db = 'yes'; downloaded_junction_db = 'no'
elif 'hta 2.0' in specific_arraytype and 'HTA-2_0' not in specific_platform: wrong_junction_db = 'yes'; downloaded_junction_db = 'no'
elif 'hta 2.0' not in specific_arraytype and 'HTA-2_0' in specific_platform: wrong_junction_db = 'yes'; downloaded_junction_db = 'no'
elif 'mta 1.0' in specific_arraytype and 'MTA-1_0' not in specific_platform: wrong_junction_db = 'yes'; downloaded_junction_db = 'no'
elif 'mta 1.0' not in specific_arraytype and 'MTA-1_0' in specific_platform: wrong_junction_db = 'yes'; downloaded_junction_db = 'no'
#print [specific_arraytype], [specific_platform], wrong_junction_db, downloaded_junction_db
def exportGeneList(gene_list,outputFolder):
filename = string.join(gene_list,' ')[:25]
eo = export.ExportFile(outputFolder+'/GO-Elite_input/'+filename+'.txt')
eo.write('Symbol\tSytemCode\n')
for i in gene_list:
eo.write(i+'\tSy\n')
return outputFolder+'/GO-Elite_input'
def getUserParameters(run_parameter,Multi=None):
global AltAnalyze; import AltAnalyze; global mlp; mlp=Multi ### multiprocessing support
if run_parameter == 'yes':
try: MainMenu()
except Exception:
print traceback.format_exc()
print_out = "\nCritical error encountered!!! This machine does not have either:\n"
print_out += "1) Have the required Tcl/Tk components installed.\n"
print_out += "2) Is being run from a compiled version that has critical incompatibilities your OS or hardware or\n"
print_out += "3) Is being run from source-code in the same-directory as executable code resulting in a conflict\n"
print_out += "\nIf any of these apply, we recommend downloading the Python source-code version of AltAnalyze "
print_out += "(installing necessary dependencies - see our Wiki or Documentation)."
print_out += "Otherwise, please contact AltAnalyze support (http://code.google.com/p/altanalyze/wiki/ContactUs).\n\n"
print_out += "Installation Wiki: http://code.google.com/p/altanalyze/wiki/Installation\n\n"
print print_out
try:
### Create a log report of this
try: log_file = filepath('AltAnalyze_error-report.log')
except Exception: log_file = filepath('/AltAnalyze_error-report.log')
log_report = open(log_file,'w');
log_report.write(print_out)
log_report.write(traceback.format_exc())
log_report.close()
### Open this file
if os.name == 'nt':
try: os.startfile('"'+log_file+'"')
except Exception: os.system('open "'+log_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+log_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+log_file+'/"')
except Exception: None
sys.exit()
global species; species=''; global user_variables; user_variables={}; global analysis_method; global array_type; global vendor
global PathDir; global PathFile; global file_location_defaults; global integrate_online_species; integrate_online_species = 'no'
global option_db; global option_list; global analysis_status; analysis_status = 'continue'; global selected_parameters; selected_parameters=[]
global backSelect; global fl; predictGroups = False
if os.name == 'posix' and run_parameter == 'yes':
try: MacConsiderations()
except Exception:
print traceback.format_exc()
sys.exit()
### Get default options for ExpressionBuilder and AltAnalyze
na = 'NA'; log = 'log'; no = 'no'
run_from_scratch=na; expression_threshold=na; perform_alt_analysis=na; expression_data_format=log
include_raw_data=na; avg_all_for_ss=no; dabg_p=na; normalize_feature_exp=na; normalize_gene_data = na
analysis_method=na; p_threshold=na; filter_probeset_types=na; alt_exon_fold_cutoff=na
permute_p_threshold=na; perform_permutation_analysis=na; export_splice_index_values=no
run_MiDAS=no; analyze_functional_attributes=no; microRNA_prediction_method=na
gene_expression_cutoff=na; cel_file_dir=na; input_exp_file=na; input_stats_file=na; filter_for_AS=no
remove_intronic_junctions=na; build_exon_bedfile=no; input_cdf_file = na; bgp_file = na
clf_file = na; remove_xhyb = na; multiThreading = True; input_fastq_dir = ''; sparse_matrix_file=''
compendiumType = 'protein_coding'; compendiumPlatform = 'gene'
calculate_splicing_index_p=no; run_goelite=no; ge_ptype = 'rawp'; probability_algorithm = na
ge_fold_cutoffs=2;ge_pvalue_cutoffs=0.05;filter_method=na;z_threshold=1.96;p_val_threshold=0.05
change_threshold=2;pathway_permutations=na;mod=na; analyze_all_conditions=no; resources_to_analyze=na
additional_algorithms = na; rpkm_threshold = na; exon_exp_threshold = na; run_lineage_profiler = no
gene_exp_threshold = na; exon_rpkm_threshold = na; visualize_results = no; returnPathways = 'no'
batch_effects = na; marker_finder = na
try: option_list,option_db = importUserOptions('exon') ##Initially used to just get the info for species and array_type
except IOError:
### Occurs if Config folder is absent or when the source code is run outside AltAnalyze root
print_out = '\nWarning! The Config folder in the AltAnalyze program directory cannot be found. The likely cause is:\n'
print_out +=' A): The AltAnalyze source-code is being run outside the root AltAnalyze directory or \n'
print_out +=' B): AltAnalyze was zip extracted/installed in a weird way (incommpatible zip extractor)\n'
print_out +='\nIf you beleive (B) is possible, unzip with another unzip program (e.g., default Windows unzip program).'
print_out +='\nIf neither applies, we recommend contacting our help desk (http://code.google.com/p/altanalyze/wiki/ContactUs).'
try: IndicatorWindow(print_out,'Exit')
except Exception: print printout
sys.exit()
importSpeciesInfo()
file_location_defaults = importDefaultFileLocations()
importArrayInfo()
try: elite_db_versions = returnDirectoriesNoReplace('/AltDatabase')
except Exception:
try: elite_db_versions=[]; os.mkdir(filepath('AltDatabase'))
except Exception: pass ### directory already exists
try: gene_database_dir = unique.getCurrentGeneDatabaseVersion()
except Exception: gene_database_dir=''
if len(elite_db_versions)>0 and gene_database_dir == '':
for db_version in elite_db_versions:
if 'EnsMart' in db_version:
gene_database_dir = db_version; exportDBversion(db_version)
current_species_names,manufacturer_list_all = getSpeciesList('')
option_db['species'].setArrayOptions(current_species_names)
try: PathDir = file_location_defaults['PathDir'].Location()
except Exception:
try:
### Entry was deleted from Config file - re-create it
fl = FileLocationData('local', '', 'all')
file_location_defaults['PathDir'] = fl
except Exception: null = None
PathDir = ''
try: PathFile = file_location_defaults['PathFile'].Location()
except Exception:
try:
### Entry was deleted from Config file - re-create it
fl = FileLocationData('local', '', 'all')
file_location_defaults['PathFile'] = fl
except Exception: null = None
PathFile = ''
old_options = []
try:
#### Get information from previous loop
if len(run_parameter) == 2 and run_parameter != 'no': ### Occurs when selecting "Back" from Elite parameter window
old_options = run_parameter[1]; selected_parameters = run_parameter[0]
try:
if selected_parameters[-2]==selected_parameters[-1]: selected_parameters = selected_parameters[:-1]
except Exception: selected_parameters = selected_parameters
backSelect = 'yes'
#print selected_parameters
#print old_options,'\n'
for option in old_options: ### Set options to user selected
try: option_db[option].setDefaultOption(old_options[option])
except Exception: pass
user_variables[option] = old_options[option]
if 'array_type' in old_options:
specific_array = old_options['array_type']
vendor = old_options['manufacturer_selection']
species_full = old_options['species']
species = species_codes[species_full].SpeciesCode()
if selected_parameters == []: backSelect = 'no'
else: backSelect = 'no'; old_options=[]
###Update this informatin in option_db which will be over-written after the user selects a species and array_type
option_db['species'].setArrayOptions(current_species_names)
if len(current_species_names)==0 and run_parameter != 'Add Species':
print_out = "No species databases found. Select\ncontinue to proceed with species download."
IndicatorWindow(print_out,'Continue')
integrate_online_species = 'yes'
addOnlineSpeciesDatabases(backSelect)
AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
### Set defaults based on avialable species
#default_vendor = 'RNASeq'
#default_specific_array = 'RNA-seq aligned read counts'
default_vendor = 'RNASeq'
default_specific_array='Raw sequence or processed'
"""
try: ### If the users have already analyzed Affy data, make this the default
affymetrix_library_dir = 'AltDatabase/affymetrix/LibraryFiles'
affy_dir_list = read_directory(filepath(affymetrix_library_dir))
if len(affy_dir_list)>0:
default_vendor = 'Affymetrix'
default_specific_array='Affymetrix expression array'
except Exception:
None ### Occurs if this directory is missing (possible in future versions)
"""
if run_parameter == 'Add Species':
species_full = 'Homo sapiens'; species = 'Hs'; vendor = 'Affymetrix'; specific_array = 'Exon 1.0 ST array'
if backSelect == 'yes' and 'array_type' in old_options:
pass
elif 'Homo sapiens' in current_species_names:
species_full = 'Homo sapiens'; species = 'Hs'; vendor = default_vendor; specific_array = default_specific_array
elif 'Mus musculus' in current_species_names:
species_full = 'Mus musculus'; species = 'Mm'; vendor = default_vendor; specific_array = default_specific_array
elif 'Rattus norvegicus' in current_species_names:
species_full = 'Rattus norvegicus'; species = 'Rn'; vendor = default_vendor; specific_array = default_specific_array
else:
for species_full in current_species_names:
species = species_codes[species_full].SpeciesCode()
for array_name in array_list:
vendor = array_codes[array_name].Manufacturer()
if species in array_codes[array_name].SpeciesCodes(): specific_array = array_name; break
array_list2, manufacturer_list = getArraysAndVendors(species,vendor)
#print [[array_list2]], species, vendor
option_db['species'].setDefaultOption(species_full)
option_db['array_type'].setArrayOptions(array_list2)
option_db['array_type'].setDefaultOption(specific_array)
option_db['manufacturer_selection'].setArrayOptions(manufacturer_list_all)
option_db['manufacturer_selection'].setDefaultOption(vendor)
manufacturer_list_all_possible=[]
for array_name in array_list:
manufacturer = array_codes[array_name].Manufacturer(); manufacturer_list_all_possible.append(manufacturer)
manufacturer_list_all_possible = unique.unique(manufacturer_list_all_possible); manufacturer_list_all_possible.sort()
if len(elite_db_versions)>1:
option_db['dbase_version'].setArrayOptions(elite_db_versions)
option_db['dbase_version'].setDefaultOption(gene_database_dir)
else:
### Otherwise, remove this option
del option_db['dbase_version']
### Get user array and species selections
if run_parameter != 'Add Species':
if backSelect == 'no' or 'ArrayType' == selected_parameters[-1]:
selected_parameters.append('ArrayType'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Select Species and Experimental Platform')
gu = GUI(root,option_db,option_list['ArrayType'],'')
else: gu = PreviousResults(old_options)
species_full = gu.Results()['species']
new_analysis_options=[]
try: update_dbs = gu.Results()['update_dbs']
except Exception: update_dbs = 'no'
try:
selected_parameters[-1]
except Exception:
AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
if update_dbs == 'yes' or species_full == 'Add Species' or 'NewSpecies' == selected_parameters[-1]:
integrate_online_species = 'yes'
addOnlineSpeciesDatabases(backSelect)
AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
elif species_full == 'Add Species' or 'NewSpecies' == selected_parameters[-1]: ### outdated code - bypassed by the above
species_added = 'no'
option_db['new_manufacturer'].setArrayOptions(manufacturer_list_all_possible)
while species_added == 'no':
if backSelect == 'no' or 'NewSpecies' == selected_parameters[-1]:
selected_parameters.append('NewSpecies'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Add New Species Support')
gu = GUI(root,option_db,option_list['NewSpecies'],'')
else: gu = PreviousResults(old_options)
new_species_code = gu.Results()['new_species_code']
new_species_name = gu.Results()['new_species_name']
new_manufacturer = gu.Results()['new_manufacturer']
if len(new_species_code)==2 and len(new_species_name)>0 and len(new_manufacturer)>0:
species_added = 'yes'
sd = SpeciesData(new_species_code,new_species_name,[''])
species_codes[new_species_name] = sd
try: exportSpeciesInfo(species_codes)
except Exception:
print 'Cannot write Config/species.txt to the Config directory (like Permissions Error)'
try: os.mkdir(filepath('AltDatabase/'+new_species_code))
except Exception: pass
for array_system in array_codes:
ac = array_codes[array_system]
manufacturer=ac.Manufacturer()
compatible_species = ac.SpeciesCodes()
if manufacturer == new_manufacturer and 'expression array' in ac.ArrayName():
if new_species_code not in compatible_species: compatible_species.append(new_species_code)
ac.setSpeciesCodes(compatible_species)
try: exportArrayInfo(array_codes)
except Exception:
print 'Cannot write Config/arrays.txt to the Config directory (like Permissions Error)'
fn = filepath('AltDatabase/affymetrix/'+ new_species_code)
try: os.mkdir(fn)
except OSError: null = [] ### Directory already exists
AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
else:
print_out = "Valid species data was not added. You must\nindicate a two letter species code and full species name."
IndicatorWindow(print_out,'Continue')
else: species = species_codes[species_full].SpeciesCode()
try: array_full = gu.Results()['array_type'] ### Can be 10X Genomics
except: array_full = 'RNASeq'
vendor = gu.Results()['manufacturer_selection']
if '10X' in array_full:
vendor = '10XGenomics'
try:
array_type = array_codes[array_full].ArrayCode() ### Here, 10X Genomics would be converted to RNASeq
except Exception:
if vendor == 'Other ID':
#"""
### An error occurs because this is a system name for the Other ID option
array_type = "3'array"
if array_full == "3'array" and vendor == 'RNASeq':
### Occurs when hitting the back button
### When RNASeq is selected as the platform but change to "3'array" when normalized data is imported.
vendor = 'other:Symbol'
else:
vendor = 'other:'+array_full ### Ensembl linked system name
if array_type == 'gene':
try: gene_database = unique.getCurrentGeneDatabaseVersion()
except Exception: gene_database='00'
if int(gene_database[-2:]) < 54:
print_out = 'The AltAnalyze database indicated for Gene 1.0 ST\narray analysis is not supported for alternative exon\nanalysis. Please update to EnsMart54 or greater\nbefore proceeding.'
IndicatorWindow(print_out,'Continue'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
### Examine the AltDatabase folder for directories required for specific array analyses
checkForLocalArraySupport(species,array_type,array_full,'GUI')
if array_type == 'exon' or array_type == 'AltMouse' or array_type == 'gene' or array_type == 'junction':
try: dirs = read_directory('/AltDatabase/'+species)
except Exception: dirs=[]
if len(dirs)==0:
print_out = 'Valid database directories were not found for this array.\nPlease re-install database.'
IndicatorWindow(print_out,'Continue'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
if '10X' in vendor:
### Needed when the the back button is selected for the 10X platform
array_type = '10XGenomics'
array_full == '10X Genomics sparse matrix'
option_list,option_db = importUserOptions(array_type,vendor=vendor) ##Initially used to just get the info for species and array_type
if array_type == "3'array" and '10X' not in vendor:
if species == 'Hs': compendiumPlatform = "3'array"
for i in option_db['run_from_scratch'].ArrayOptions():
if 'AltAnalyze' not in i:
if array_type == "3'array":
if 'CEL' in i and vendor != 'Affymetrix': proceed = 'no'
else: proceed = 'yes'
else: proceed = 'yes'
if proceed == 'yes': new_analysis_options.append(i)
option_db['run_from_scratch'].setArrayOptions(new_analysis_options)
proceed = 'no'
if len(new_analysis_options)!=1:
if backSelect == 'no' or 'AnalysisType' == selected_parameters[-1]:
selected_parameters.append('AnalysisType'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Select Analysis Method')
gu = GUI(root,option_db,option_list['AnalysisType'],'')
else: gu = PreviousResults(old_options)
run_from_scratch = gu.Results()['run_from_scratch']
else: run_from_scratch = 'Process Expression file'
try: vendor = array_codes[array_full].Manufacturer()
except Exception: None ### Key the existing vendor
try: constitutive_source = array_codes[array_full].ConstitutiveSource()
except Exception: constitutive_source = vendor
if '10X' in array_full or '10X' in vendor:
array_type = "3'array"
vendor = 'other:'+array_full ### Ensembl linked system name
#option_list,option_db = importUserOptions(array_type,vendor=vendor) ##Initially used to just get the info for species and array_type
if backSelect == 'yes':
for option in old_options: ### Set options to user selected
try: option_db[option].setDefaultOption(old_options[option])
except Exception: pass
if run_from_scratch == 'Interactive Result Viewer':
AltAnalyze.AltAnalyzeSetup('remoteViewer');sys.exit()
def rebootAltAnalyzeGUI(selected_parameters,user_variables):
commandline_args = ['--selected_parameters',selected_parameters[-1]]
for uv in user_variables:
if isinstance(user_variables[uv], list):
commandline_args += ['--'+uv,user_variables[uv][0]]
else:
try:
if len(user_variables[uv])>0:
commandline_args += ['--'+uv,user_variables[uv]]
except Exception: pass
commandline_args = map(lambda x: string.replace(x,' ','__'),commandline_args)
commandline_args = str(string.join(commandline_args,' '))
if os.name == 'posix' or os.name == 'nt':
try:
package_path = filepath('python')
if os.name == 'posix':
package_path = string.replace(package_path,'python','AltAnalyze.app/Contents/MacOS/AltAnalyze')
else:
package_path = string.replace(package_path,'python','AltAnalyze.exe')
package_path = 'AltAnalyze.exe'
#print [package_path+' --GUI yes '+commandline_args]
os.system(package_path+' --GUI yes '+commandline_args);sys.exit()
except Exception:
package_path = filepath('python')
package_path = string.replace(package_path,'python','AltAnalyze.py')
package_path = 'python '+package_path
if os.name == 'nt':
package_path = 'python AltAnalyze.py'
os.system(package_path+' --GUI yes '+commandline_args);sys.exit()
else:
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
if run_from_scratch == 'Additional Analyses':
if backSelect == 'no' or 'Additional Analyses' == selected_parameters[-1]:
selected_parameters.append('Additional Analyses'); backSelect = 'no'
root = Tk()
root.title('AltAnalyze: Additional Analysis Options')
gu = GUI(root,option_db,option_list['Additional Analyses'],'')
### Venn Diagram Error here with _tkinter.TclError: image "pyimage36" doesn't exist
else: gu = PreviousResults(old_options)
additional_analyses = gu.Results()['additional_analyses']
if 'nrichment' in additional_analyses:
status = 'repeat'
while status == 'repeat':
if backSelect == 'no' or 'InputGOEliteDirs' == selected_parameters[-1]:
root = Tk(); root.title('AltAnalyze: Select Expression File for Filtering')
selected_parameters.append('InputGOEliteDirs'); backSelect = 'no'
gu = GUI(root,option_db,option_list['InputGOEliteDirs'],'')
else: gu = PreviousResults(old_options)
try: criterion_input_folder = gu.Results()['criterion_input_folder']
except KeyError: criterion_input_folder = '' ### Leave this blank so that the default directory is used
try: criterion_denom_folder = gu.Results()['criterion_denom_folder']
except KeyError: criterion_denom_folder = '' ### Leave this blank so that the default directory is used
try:
try: main_output_folder = gu.Results()['main_output_folder']
except KeyError: main_output_folder = 'GO-Elite/input/' ### Leave this blank so that the default directory is
inputIDs = gu.Results()['inputIDs']
if len(inputIDs)>0:
inputIDs = string.replace(inputIDs, '\r',' ')
inputIDs = string.replace(inputIDs, '\n',' ')
inputIDs = string.split(inputIDs, ' ')
criterion_input_folder = exportGeneList(inputIDs,main_output_folder)
except Exception: inputIDs=[]
if len(criterion_input_folder)>0:# and len(criterion_denom_folder)>0:
try: main_output_folder = gu.Results()['main_output_folder']
except KeyError: main_output_folder = '' ### Leave this blank so that the default directory is
if len(main_output_folder)<1:
### Set output to the same directory or parent if none selected
i = -1 ### 1 directory up
main_output_folder = string.join(string.split(criterion_input_folder,'/')[:i],'/')
status = 'continue'
else:
print_out = "No GO-Elite input or denominator folder(s) selected."
IndicatorWindow(print_out,'Continue')
file_dirs = criterion_input_folder, criterion_denom_folder, main_output_folder
#print file_dirs
### Get GO-Elite Input Parameters
getUpdatedParameters(array_type,species,'Prefiltered',file_dirs)
if additional_analyses == 'Pathway Visualization':
root = Tk()
root.title('AltAnalyze: Visualize Data on WikiPathways')
selected_parameters.append('Pathway Visualization')
GUI(root,'ViewWikiPathways',[],'') ### The last is default attributes (should be stored as defaults in the option_db var)
if additional_analyses == 'Identifier Translation':
try:
selected_parameters.append('Identifier Translation')
supported_geneid_types = getSupportedGeneSystems(species,'uid-gene')
option_db['input_source'].setArrayOptions(['None Selected']+supported_geneid_types)
option_db['output_source'].setArrayOptions(['None Selected']+supported_geneid_types)
#option_db['PathwaySelection'].setArrayOptions(supported_genesets)
except Exception,e:
print traceback.format_exc()
status = 'repeat'
while status == 'repeat':
root = Tk()
root.title('AltAnalyze: Translate Input File Identifiers to Another System')
gu = GUI(root,option_db,option_list['IDConverter'],'')
try: input_cluster_file = gu.Results()['input_cluster_file']
except Exception: input_cluster_file = ''
input_data_file = gu.Results()['input_data_file']
input_source = gu.Results()['input_source']
output_source = gu.Results()['output_source']
if len(input_data_file)>0 and input_source != 'None Selected' and output_source != 'None Selected':
analysis = 'IDConverter'
values = input_data_file, species, input_source, output_source
StatusWindow(values,analysis) ### display an window with download status
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
if additional_analyses == 'Merge Files':
selected_parameters.append('Merge Files')
status = 'repeat'
while status == 'repeat':
root = Tk()
root.title('AltAnalyze: Merge Multiple Text Files Containing Common IDs')
gu = GUI(root,option_db,option_list['MergeFiles'],'')
input_file1 = gu.Results()['input_file1']
input_file2 = gu.Results()['input_file2']
input_file3 = gu.Results()['input_file3']
input_file4 = gu.Results()['input_file4']
join_option = gu.Results()['join_option']
ID_option = gu.Results()['ID_option']
output_merge_dir = gu.Results()['output_merge_dir']
if len(input_file1)>0 and len(input_file2)>0 and len(output_merge_dir)>0:
if ID_option == 'False': ID_option = False
if ID_option == 'True': ID_option = True
analysis = 'MergeFiles'
files_to_merge = [input_file1, input_file2]
if len(input_file3)>0: files_to_merge.append(input_file3)
if len(input_file4)>0: files_to_merge.append(input_file4)
values = files_to_merge, join_option, ID_option, output_merge_dir
StatusWindow(values,analysis) ### display an window with download status
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
if additional_analyses == 'Venn Diagram':
selected_parameters.append('Venn Diagram')
status = 'repeat'
while status == 'repeat':
root = Tk()
root.title('AltAnalyze: View Venn Diagram from AltAnalyze or Input Files')
gu = GUI(root,option_db,option_list['VennDiagram'],'')
input_file1 = gu.Results()['venn_input_file1']
input_file2 = gu.Results()['venn_input_file2']
input_file3 = gu.Results()['venn_input_file3']
input_file4 = gu.Results()['venn_input_file4']
venn_output_dir = gu.Results()['venn_output_dir']
if len(input_file1)>0 and len(input_file2)>0 and len(venn_output_dir)>0:
analysis = 'VennDiagram'
files_to_merge = [input_file1, input_file2]
if len(input_file3)>0: files_to_merge.append(input_file3)
if len(input_file4)>0: files_to_merge.append(input_file4)
values = files_to_merge, venn_output_dir
StatusWindow(values,analysis) ### display an window with download status
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
if additional_analyses == 'AltExon Viewer':
selected_parameters.append('AltExon Viewer')
status = 'repeat'
while status == 'repeat':
root = Tk()
root.title('AltAnalyze: Visualize Exon-Level Expression Results')
gu = GUI(root,option_db,option_list['AltExonViewer'],'')
altanalyze_results_folder = gu.Results()['altanalyze_results_folder']
data_type = gu.Results()['data_type']
show_introns = gu.Results()['show_introns']
gene_symbol = gu.Results()['gene_symbol']
altgenes_file = gu.Results()['altgenes_file']
analysisType = gu.Results()['analysisType']
if len(altgenes_file)>0 and analysisType != 'Sashimi-Plot':
gene_symbol = importGeneList(altgenes_file) ### list of gene IDs or symbols
if analysisType == 'Sashimi-Plot':
altanalyze_results_folder = string.split(altanalyze_results_folder,'AltResults')[0]
exp_file = altanalyze_results_folder
if len(gene_symbol)<1:
gene_symbol = altgenes_file
elif data_type == 'raw expression': ### Switch directories if expression
altanalyze_results_folder = string.replace(altanalyze_results_folder,'AltResults','ExpressionInput')
exp_file = getValidExpFile(altanalyze_results_folder)
else:
altanalyze_results_folder += '/RawSpliceData/'+species
try: exp_file = getValidSplicingScoreFile(altanalyze_results_folder)
except Exception,e:
print_out = "No files found in: "+altanalyze_results_folder
IndicatorWindow(print_out,'Continue')
if len(exp_file)>0 or ((len(exp_file)>0 or len(gene_symbol)>0) and analysisType == 'Sashimi-Plot'):
analysis = 'AltExonViewer'
values = species,array_type,exp_file,gene_symbol,show_introns,analysisType
try: StatusWindow(values,analysis) ### display an window with download status
except Exception: pass
#if len(altgenes_file)>0 or ' ' in gene_symbol or ((len(exp_file)>0 or len(gene_symbol)>0) and analysisType == 'Sashimi-Plot'):
if len(analysisType)>0:
### Typically have a Tkinter related error
rebootAltAnalyzeGUI(selected_parameters,user_variables)
else:
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "Either no gene or no AltResults folder selected."
IndicatorWindow(print_out,'Continue')
if additional_analyses == 'Network Visualization':
selected_parameters.append('Network Visualization')
supported_interaction_types = getSupportedGeneSetTypes(species,'gene-interactions')
supported_geneset_types = getSupportedGeneSetTypes(species,'gene-mapp')
supported_geneset_types += getSupportedGeneSetTypes(species,'gene-go')
option_db['GeneSetSelection_network'].setArrayOptions(['None Selected']+supported_geneset_types)
option_db['PathwaySelection_network'].setArrayOptions(['None Selected'])
#option_db['PathwaySelection'].setArrayOptions(supported_genesets)
status = 'repeat'
while status == 'repeat':
### If no databases present download and populate gene-interactions folder
if len(supported_interaction_types)==0:
print_out = 'No interaction databases available.\nPress Continue to download interaction\ndatabases for this species.'
IndicatorWindow(print_out,'Continue')
downloadInteractionDBs(species,'parent')
### Get present interaction databases (including custom added)
updated_list=[]
if 'WikiPathways' in supported_interaction_types: updated_list.append('WikiPathways')
if 'KEGG' in supported_interaction_types: updated_list.append('KEGG')
if 'TFTargets' in supported_interaction_types: updated_list.append('TFTargets')
if 'BioGRID' in supported_interaction_types: updated_list.append('BioGRID')
for db in supported_interaction_types:
if 'microRNATargets' in db:
updated_list.append('common-microRNATargets'); updated_list.append('all-microRNATargets')
elif 'DrugBank' in db:
updated_list.append('common-DrugBank'); updated_list.append('all-DrugBank')
elif db not in updated_list: updated_list.append(db)
option_db['interactionDirs'].setArrayOptions(updated_list)
root = Tk()
root.title('AltAnalyze: Create and Visualize Interaction Networks')
gu = GUI(root,option_db,option_list['network'],'')
Genes_network = gu.Results()['Genes_network']
inputDir_network = gu.Results()['input_ID_file']
GeneSetSelection_network = gu.Results()['GeneSetSelection_network']
inputType_network = gu.Results()['inputType_network']
PathwaySelection_network = gu.Results()['PathwaySelection_network']
OntologyID_network = gu.Results()['OntologyID_network']
interactionDirs = gu.Results()['interactionDirs']
degrees = gu.Results()['degrees']
update_interactions = gu.Results()['update_interactions']
expressionFile_network = gu.Results()['elite_exp_file']
outputDir_network = gu.Results()['output_net_folder']
includeExpIDs_network = gu.Results()['includeExpIDs_network']
### Set the below variables to the appropriate object types
if update_interactions == 'yes': update_interactions = True
else: update_interactions = False
if len(inputDir_network) == 0: inputDir_network = None
if len(expressionFile_network) == 0: expressionFile_network = None
if len(Genes_network) == 0: Genes_network = None
if len(outputDir_network) == 0: outputDir_network = None
if len(GeneSetSelection_network) == 'None Selected': GeneSetSelection_network = None
if includeExpIDs_network=='yes': includeExpIDs_network = True
else: includeExpIDs_network = False
### Save these as instances of GeneSelectionParameters (easier this way to add more object types in the future)
gsp = GeneSelectionParameters(species,array_type,vendor) ### only species currently neeed
gsp.setGeneSet(GeneSetSelection_network)
gsp.setPathwaySelect(PathwaySelection_network)
gsp.setGeneSelection(Genes_network)
gsp.setOntologyID(OntologyID_network)
gsp.setIncludeExpIDs(includeExpIDs_network)
if update_interactions:
downloadInteractionDBs(species,'parent')
if outputDir_network==None:
print_out = "No output directory selected."
IndicatorWindow(print_out,'Continue')
elif inputDir_network != None or GeneSetSelection_network != None or Genes_network != None:
analysis = 'network'
values = inputDir_network,inputType_network,outputDir_network,interactionDirs,degrees,expressionFile_network,gsp
StatusWindow(values,analysis,windowType='parent') ### display an window with download status
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "No input gene IDs, expression file or GeneSet selected."
IndicatorWindow(print_out,'Continue')
if additional_analyses == 'Hierarchical Clustering':
print 'Performing Hierarchical Clustering'
selected_parameters.append('Hierarchical Clustering')
supported_geneset_types = getSupportedGeneSetTypes(species,'gene-mapp')
supported_geneset_types += getSupportedGeneSetTypes(species,'gene-go')
option_db['GeneSetSelection'].setArrayOptions(['None Selected']+supported_geneset_types)
option_db['PathwaySelection'].setArrayOptions(['None Selected'])
option_db['ClusterGOElite'].setArrayOptions(['None Selected','all']+supported_geneset_types)
#option_db['PathwaySelection'].setArrayOptions(supported_genesets)
status = 'repeat'
while status == 'repeat':
root = Tk()
root.title('AltAnalyze: Create a Heatmap from an Expression Matrix')
gu = GUI(root,option_db,option_list['heatmap'],'')
try: input_cluster_file = gu.Results()['input_cluster_file']
except Exception: input_cluster_file = ''
column_metric = gu.Results()['column_metric']
column_method = gu.Results()['column_method']
row_metric = gu.Results()['row_metric']
row_method = gu.Results()['row_method']
color_selection = gu.Results()['color_selection']
cluster_rows = gu.Results()['cluster_rows']
cluster_columns = gu.Results()['cluster_columns']
GeneSetSelection = gu.Results()['GeneSetSelection']
PathwaySelection = gu.Results()['PathwaySelection']
GeneSelection = gu.Results()['GeneSelection']
ClusterGOElite = gu.Results()['ClusterGOElite']
HeatmapAdvanced = gu.Results()['HeatmapAdvanced']
JustShowTheseIDs = gu.Results()['JustShowTheseIDs']
geneSetName = gu.Results()['heatmapGeneSets']
try: CorrelationCutoff = float(gu.Results()['CorrelationCutoff'])
except Exception: CorrelationCutoff=None
OntologyID = gu.Results()['OntologyID']
transpose = gu.Results()['transpose']
normalization = gu.Results()['normalization']
contrast = gu.Results()['contrast']
if transpose == 'yes': transpose = True
else: transpose = False
translate={'None Selected':'','Exclude Cell Cycle Effects':'excludeCellCycle',
'Top Correlated Only':'top','Positive Correlations Only':'positive',
'Perform Iterative Discovery':'guide', 'Intra-Correlated Only':'IntraCorrelatedOnly',
'Correlation Only to Guides':'GuideOnlyCorrelation','Perform Monocle':'monocle'}
try:
if 'None Selected' in HeatmapAdvanced: pass
except Exception: HeatmapAdvanced = ('None Selected')
if ('None Selected' in HeatmapAdvanced and len(HeatmapAdvanced)==1) or 'None Selected' == HeatmapAdvanced: pass
else:
try:
GeneSelection += ' '+string.join(list(HeatmapAdvanced),' ')
for name in translate:
GeneSelection = string.replace(GeneSelection,name,translate[name])
GeneSelection = string.replace(GeneSelection,' ',' ')
if 'top' in GeneSelection or 'positive' in GeneSelection or 'IntraCorrelatedOnly' in GeneSelection: #or 'guide' in GeneSelection or 'excludeCellCycle' in GeneSelection - will force correlation to selected genes
GeneSelection+=' amplify'
except Exception: pass
### This variable isn't needed later, just now to indicate not to correlate in the first round
if GeneSetSelection != 'None Selected' and PathwaySelection == ['None Selected']:
PathwaySelection = [gu.Results()[GeneSetSelection][0]] ### Default this to the first selection
GeneSetSelection = string.replace(GeneSetSelection,'\n',' ')
GeneSetSelection = string.replace(GeneSetSelection,'\r',' ')
#print [GeneSetSelection, JustShowTheseIDs, GeneSelection,ClusterGOElite,normalization]
if GeneSetSelection != 'None Selected' or GeneSelection != '' or normalization != 'NA' or JustShowTheseIDs != '' or JustShowTheseIDs != 'None Selected':
gsp = GeneSelectionParameters(species,array_type,vendor)
if CorrelationCutoff!=None: #len(GeneSelection)>0 and
gsp.setRhoCutoff(CorrelationCutoff)
GeneSelection = 'amplify '+GeneSelection
if 'GuideOnlyCorrelation' in GeneSelection:
### Save the correlation cutoff for ICGS but don't get expanded correlation sets in the first round
GeneSelection = string.replace(GeneSelection,'GuideOnlyCorrelation','')
GeneSelection = string.replace(GeneSelection,'amplify','')
GeneSelection = string.replace(GeneSelection,' ','')
gsp.setGeneSet(GeneSetSelection)
gsp.setPathwaySelect(PathwaySelection)
gsp.setGeneSelection(GeneSelection)
gsp.setOntologyID(OntologyID)
gsp.setTranspose(transpose)
gsp.setNormalize(normalization)
gsp.setJustShowTheseIDs(JustShowTheseIDs)
gsp.setClusterGOElite(ClusterGOElite)
gsp.setStoreGeneSetName(geneSetName)
transpose = gsp ### this allows methods that don't transmit this object to also work
if len(input_cluster_file)>0:
analysis = 'createHeatMap'
color_selection=string.replace(color_selection, '-','_')
if cluster_rows == 'no': row_method = None
if cluster_columns == 'no': column_method = None
values = input_cluster_file, row_method, row_metric, column_method, column_metric, color_selection, transpose, contrast
StatusWindow(values,analysis) ### display an window with download status
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
if additional_analyses == 'Dimensionality Reduction':
print 'Performing Dimensionality Reduction'
selected_parameters.append('Dimensionality Reduction')
status = 'repeat'
while status == 'repeat':
root = Tk()
root.title('AltAnalyze: Perform Dimensionality Reduction from an Expression Matrix')
gu = GUI(root,option_db,option_list['PCA'],'')
try: input_cluster_file = gu.Results()['input_cluster_file']
except Exception: input_cluster_file = ''
dimensions = gu.Results()['dimensions']
pca_labels = gu.Results()['pca_labels']
pca_algorithm = gu.Results()['pca_algorithm']
zscore = gu.Results()['zscore']
transpose = gu.Results()['transpose']
geneSetName = gu.Results()['pcaGeneSets']
try:
maskGroups = gu.Results()['maskGroups']
if len(maskGroups)<1:
maskGroups=None
except:
maskGroups = None
reimportModelScores = gu.Results()['reimportModelScores']
if reimportModelScores == 'yes':
reimportModelScores = True
else:
reimportModelScores = False
try:
colorByGene = gu.Results()['colorByGene']
colorByGene_temp = string.replace(colorByGene,' ','')
if len(colorByGene_temp)==0:
colorByGene = None
else:
#Standardize the delimiter
colorByGene = string.replace(colorByGene,'|',' ')
colorByGene = string.replace(colorByGene,',',' ')
colorByGene = string.replace(colorByGene,'\r',' ')
colorByGene = string.replace(colorByGene,'\n',' ')
colorByGene = string.replace(colorByGene,' ',' ')
if colorByGene[0] == ' ': colorByGene=colorByGene[1:]
if colorByGene[-1] == ' ': colorByGene=colorByGene[:-1]
except Exception: colorByGene = None
if len(geneSetName)==0:
geneSetName = None
if len(input_cluster_file)>0:
analysis = 'performPCA'
if transpose == 'yes': transpose = True
else: transpose = False
values = input_cluster_file, pca_labels, dimensions, pca_algorithm, transpose, geneSetName, species, zscore, colorByGene, reimportModelScores, maskGroups
StatusWindow(values,analysis) ### display an window with download status
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
if additional_analyses == 'Lineage Analysis' or additional_analyses == 'Cell Classification':
selected_parameters.append('Lineage Analysis')
status = 'repeat'
while status == 'repeat':
root = Tk()
if species == 'Mm':
option_db['compendiumPlatform'].setDefaultOption('gene')
if species == 'Hs':
option_db['compendiumPlatform'].setDefaultOption('exon')
if array_type == "3'array":
option_db['compendiumType'].setArrayOptions(["protein_coding"])
root.title('AltAnalyze: Perform CellHarmony and LineageProfiler Analysis')
gu = GUI(root,option_db,option_list['LineageProfiler'],'')
input_exp_file = gu.Results()['input_lineage_file']
compendiumPlatform = gu.Results()['compendiumPlatform']
try: classificationAnalysis = gu.Results()['classificationAnalysis']
except: classificationAnalysis = 'cellHarmony'
compendiumType = gu.Results()['compendiumType']
markerFinder_file = gu.Results()['markerFinder_file']
geneModel_file = gu.Results()['geneModel_file']
modelDiscovery = gu.Results()['modelDiscovery']
pearsonThreshold = gu.Results()['PearsonThreshold']
returnCentroids = gu.Results()['returnCentroids']
performDiffExp = gu.Results()['performDiffExp']
useAdjPval = gu.Results()['UseAdjPval']
pvalThreshold = gu.Results()['pvalThreshold']
foldCutoff = gu.Results()['FoldCutoff']
labels = gu.Results()['labels']
try: referenceFull = gu.Results()['referenceFull']
except: referenceFull=None
if '.png' in markerFinder_file or '.pdf' in markerFinder_file:
markerFinder_file=markerFinder_file[:-4]+'.txt'
if len(geneModel_file) == 0: geneModel_file = None
if len(modelDiscovery) == 0: modelDiscovery = None
if len(input_exp_file)>0:
analysis = 'runLineageProfiler'
fl = ExpressionFileLocationData('','','','') ### Create this object to store additional parameters for LineageProfiler
fl.setCompendiumType(compendiumType)
fl.setCompendiumPlatform(compendiumPlatform)
fl.setClassificationAnalysis(classificationAnalysis)
fl.setPearsonThreshold(float(pearsonThreshold))
fl.setReturnCentroids(returnCentroids)
fl.setPeformDiffExpAnalysis(performDiffExp)
fl.setUseAdjPvalue(useAdjPval)
fl.setPvalThreshold(pvalThreshold)
fl.setFoldCutoff(foldCutoff)
fl.setLabels(labels)
fl.set_reference_exp_file(referenceFull)
"""
print fl.PeformDiffExpAnalysis()
print fl.CompendiumType()
print fl.CompendiumPlatform()
print fl.ClassificationAnalysis()
print fl.PearsonThreshold()
print fl.ReturnCentroids()
print fl.PeformDiffExpAnalysis()
print fl.PvalThreshold()
print fl.FoldCutoff()
print fl.UseAdjPvalue()
print fl.PearsonThreshold()"""
values = fl, input_exp_file, vendor, markerFinder_file, geneModel_file, modelDiscovery
StatusWindow(values,analysis) ### display an window with download status
### Typically have a Tkinter related error
try: rebootAltAnalyzeGUI(selected_parameters[:-1],user_variables)
except:
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
#else:
#AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
print 'here'
if additional_analyses == 'MarkerFinder Analysis':
selected_parameters.append('MarkerFinder Analysis')
status = 'repeat'
while status == 'repeat':
root = Tk()
root.title('AltAnalyze: Perform MarkerFinder Analysis from the Input in ExpressionInput')
gu = GUI(root,option_db,option_list['MarkerFinder'],'')
input_exp_file = gu.Results()['input_markerfinder_file']
genes_to_output = gu.Results()['compendiumPlatform']
if len(geneModel_file) == 0: geneModel_file = None
if len(modelDiscovery) == 0: modelDiscovery = None
if len(input_exp_file)>0:
analysis = 'runLineageProfiler'
fl = ExpressionFileLocationData('','','','') ### Create this object to store additional parameters for LineageProfiler
fl.setCompendiumType(compendiumType)
fl.setCompendiumPlatform(compendiumPlatform)
values = fl, input_exp_file, vendor, markerFinder_file, geneModel_file, modelDiscovery
StatusWindow(values,analysis) ### display an window with download status
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
if 'CEL files' in run_from_scratch or 'RNA-seq reads' in run_from_scratch or 'Feature Extraction' in run_from_scratch or 'Chromium' in run_from_scratch:
"""Designate CEL, Agilent or BED file directory, Dataset Name and Output Directory"""
assinged = 'no'
while assinged == 'no': ### Assigned indicates whether or not the CEL directory and CDF files are defined
if species == 'Rn' or array_type == 'RNASeq': del option_list['InputCELFiles'][-1] ### Don't examine xyb
#print (((backSelect,selected_parameters)))
if backSelect == 'no' or 'InputCELFiles' == selected_parameters[-1]:
selected_parameters.append('InputCELFiles'); backSelect = 'no'
root = Tk()
if array_type == 'RNASeq':
root.title('AltAnalyze: Select Exon and/or Junction files to analyze'); import_file = 'BED, BAM, TAB or TCGA'
elif '10X' in vendor:
root.title('AltAnalyze: Select Chromium Sparse Matrix Filtered Matrix'); import_file = 'Filtered Matrix'
elif vendor == 'Agilent':
root.title('AltAnalyze: Select Agilent Feature Extraction text files to analyze'); import_file = '.txt'
else:
root.title('AltAnalyze: Select CEL files for APT'); import_file = '.CEL'
gu = GUI(root,option_db,option_list['InputCELFiles'],'')
else: gu = PreviousResults(old_options)
dataset_name = gu.Results()['dataset_name']
try: remove_xhyb = gu.Results()['remove_xhyb']
except KeyError: remove_xhyb = 'no'
try:
multiThreading = gu.Results()['multithreading']
if multiThreading == 'yes': multiThreading = True
else: multiThreading = False
except KeyError: multiThreading = True
try:
build_exon_bedfile = gu.Results()['build_exon_bedfile']
try: normalize_feature_exp = 'RPKM'
except Exception: pass
except KeyError: build_exon_bedfile = 'no'
try:
input_fastq_dir = gu.Results()['input_fastq_dir']
except Exception: pass
try: channel_to_extract = gu.Results()['channel_to_extract']
except Exception: channel_to_extract = 'no'
if build_exon_bedfile == 'yes' and len(input_fastq_dir)==0:
print_out = 'Please note: AltAnalyze will exit immediately after\nimporting your junction results to allow you to build\nyour exon count files and reload this data.'
IndicatorWindowSimple(print_out,'Continue')
run_from_scratch = 'buildExonExportFiles'
if len(dataset_name)<1:
print_out = "Please provide a name for the dataset before proceeding."
IndicatorWindow(print_out,'Continue')
elif 'input_cel_dir' in gu.Results() or 'input_fastq_dir' in gu.Results():
if len(input_fastq_dir)>0:
import RNASeq
cel_files = RNASeq.runKallisto(species,'',input_fastq_dir,input_fastq_dir,mlp,returnSampleNames=True)
try: output_dir = gu.Results()['output_CEL_dir']
except KeyError: output_dir = input_fastq_dir
""" ### Change made in version 2.1.4
option_db['perform_alt_analysis'].setArrayOptions(['NA'])
option_db['exon_exp_threshold'].setArrayOptions(['NA'])
option_db['exon_rpkm_threshold'].setArrayOptions(['NA'])
option_db['expression_threshold'].setArrayOptions(['NA'])
option_db['gene_exp_threshold'].setArrayOptions(['NA'])
"""
assinged = 'yes'
else:
cel_file_dir = gu.Results()['input_cel_dir']
if '10X' in vendor:
sparse_matrix_file = gu.Results()['input_cel_dir'] # 'filtered_gene_bc_matrices'
def import10XSparseMatrixHeaders(matrix_file):
import csv
barcodes_path = string.replace(matrix_file,'matrix.mtx','barcodes.tsv' )
barcodes = [row[0] for row in csv.reader(open(barcodes_path), delimiter="\t")]
barcodes = map(lambda x: string.replace(x,'-1',''), barcodes)
return barcodes
def importH5(h5_filename):
import h5py
f = h5py.File(h5_filename, 'r')
possible_genomes = f.keys()
if len(possible_genomes) != 1:
raise Exception("{} contains multiple genomes ({}). Explicitly select one".format(h5_filename, ", ".join(possible_genomes)))
genome = possible_genomes[0]
barcodes = f[genome]['barcodes']
#barcodes = map(lambda x: string.replace(x,'-1',''), barcodes)
return barcodes
if '.mtx' in sparse_matrix_file:
barcodes = import10XSparseMatrixHeaders(sparse_matrix_file)
else:
barcodes = importH5(sparse_matrix_file)
cel_files = barcodes
else:
cel_files,cel_files_fn=identifyCELfiles(cel_file_dir,array_type,vendor)
try: output_dir = gu.Results()['output_CEL_dir']
except KeyError: output_dir = cel_file_dir
if len(output_dir)==0: output_dir = cel_file_dir
if len(cel_files)>0: assinged = 'yes' ### CEL files are present in this directory
else:
print_out = "No valid "+import_file+" files were found in the directory\n"+cel_file_dir+"\nPlease verify and try again."
IndicatorWindow(print_out,'Continue')
else:
print_out = "The directory containing "+import_file+" files has not\nbeen assigned! Select a directory before proceeding."
IndicatorWindow(print_out,'Continue')
if array_type != 'RNASeq' and vendor != 'Agilent' and len(input_fastq_dir)==0 and '10X' not in vendor:
### Specific to Affymetrix CEL files
cel_file_list_dir = exportCELFileList(cel_files_fn,cel_file_dir)
"""Determine if Library and Annotations for the array exist, if not, download or prompt for selection"""
specific_array_types,specific_array_type = identifyArrayType(cel_files_fn); num_array_types = len(specific_array_types)
#except Exception: pass; num_array_types=1; specific_array_type = None
importSupportedArrayInfo()
try:
sa = supproted_array_db[specific_array_type]; array_species = sa.Species(); cel_array_type = sa.ArrayType()
except Exception: library_dir=''; array_species=''; annotation_dir=''; cel_array_type=''
if backSelect == 'no':
### Check for issues with arrays or user input options
if num_array_types>1: ### More than one array type found in the directory
print_out = 'Warning!!!!!!!\n\nMultiple array_types found ("'+specific_array_types[0]+'" and "'+specific_array_types[1]+'").\nIt is recommended you restart, otherwise, APT will try\n to process all different array types together as "'+specific_array_types[-1]+'".'
IndicatorWindow(print_out,'Continue with Existing')
if array_species != species and len(array_species)>0:
print_out = "The CEL files indicate that the proper\nspecies is "+array_species+", however, you\nindicated "+species+ ". The species indicated by the CEL\nfiles will be used instead."
IndicatorWindow(print_out,'Continue')
species = array_species
try: spdirs = read_directory('/AltDatabase/'+species)
except Exception: spdirs = []
if len(spdirs)==0:
print_out = 'Valid database directories were not found for this species.\nPlease re-install database.'
IndicatorWindow(print_out,'Continue'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
if cel_array_type != array_type and len(cel_array_type)>0:
print_out = "The CEL files indicate that the proper\narray type is "+cel_array_type+", however, you\nindicated "+array_type+ "." #The array type indicated by the CEL\nfiles will be used instead
#IndicatorWindow(print_out,'Continue')
fw = FeedbackWindow(print_out,'Use AltAnalyze Recommended',"Use Original Selected")
choice = fw.ButtonSelection()['button']
if choice == 'Use AltAnalyze Recommended':
array_type = cel_array_type
option_list,option_db = importUserOptions(array_type) ##Initially used to just get the info for species and array_type
option_db['array_type'].setArrayOptions(array_list)
#user_variables['array_type'] = array_type
### See if the library and annotation files are on the server or are local
else: specific_array_type = ''; annotation_dir=''
if specific_array_type == None:
if array_type == 'exon':
if species == 'Hs': specific_array_type = 'HuEx-1_0-st-v2'
if species == 'Mm': specific_array_type = 'MoEx-1_0-st-v2'
if species == 'Rn': specific_array_type = 'RaEx-1_0-st-v2'
elif array_type == 'gene':
if species == 'Hs': specific_array_type = 'HuGene-1_0-st-v1'
if species == 'Mm': specific_array_type = 'MoGene-1_0-st-v1'
if species == 'Rn': specific_array_type = 'RaGene-1_0-st-v1'
elif array_type == 'AltMouse': specific_array_type = 'altMouseA'
""" ### Comment this out to allow for different junction sub-types (likely do the above in the future)
elif array_type == 'junction':
if species == 'Hs': specific_array_type = 'HJAY_v2'
if species == 'Mm': specific_array_type = 'MJAY_v2'
"""
if specific_array_type in supproted_array_db:
input_cdf_file, annotation_dir, bgp_file, clf_file = getAffyFiles(specific_array_type,species)
else: input_cdf_file=''; bgp_file = ''; clf_file = ''
### Remove the variable names for Library and Annotation file selection if these files are found
option_list_library=[]
if len(input_cdf_file)>0:
for i in option_list['InputLibraryFiles']:
if i != 'input_cdf_file': option_list_library.append(i)
if len(annotation_dir)>0:
for i in option_list['InputLibraryFiles']:
if i != 'input_annotation_file': option_list_library.append(i)
if len(option_list_library)==0:
option_list_library = option_list['InputLibraryFiles']
"""Identify and copy over any Libary or Annotation files on the computer"""
if (len(input_cdf_file)==0 and len(annotation_dir) == 0) and backSelect == 'no':
### Note: above line used to be "or" between the input_cdf_file and annotation_dir
### this was discontinued in version 2.0.9 since the annotation file is no longer needed
### unless the array type is not in the GO-elite database
assinged = 'no'
while assinged == 'no': ### Assigned indicates whether or not the CEL directory and CDF files are defined
if array_type == "3'array":
op = option_db['input_cdf_file']; input_cdf_file_label = op.Display()
op.setNotes(' note: the CDF file is apart of the standard library files for this array. ')
input_cdf_file_label = string.replace(input_cdf_file_label,'PGF','CDF')
op.setDisplay(input_cdf_file_label)
if array_type == 'exon':
op = option_db['input_annotation_file']
new_notes = string.replace(op.Notes(),'this array','the Gene 1.0 array (NOT Exon)')
new_notes = string.replace(new_notes,'annotations','transcript cluster annotations')
new_display = string.replace(op.Display(),'your array','the Gene 1.0 array')
op.setDisplay(new_display)
op.setNotes(new_notes)
#if backSelect == 'no' or 'Library' == selected_parameters[-1]:
selected_parameters.append('Library')#; backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Select Affymetrix Library and Annotation files')
gu = GUI(root,option_db,option_list_library,'')
#else: gu = PreviousResults(old_options)
if 'input_cdf_file' in option_list_library: ### Deals with Annotation Files
if 'input_cdf_file' in gu.Results():
input_cdf_file = gu.Results()['input_cdf_file']; input_cdf_file_lower = string.lower(input_cdf_file)
if array_type == "3'array":
if '.cdf' in input_cdf_file_lower:
clf_file='';bgp_file=''; assinged = 'yes'
###Thus the CDF or PDF file was confirmed, so copy it over to AltDatabase
icf_list = string.split(input_cdf_file,'/'); cdf_short = icf_list[-1]
destination_parent = 'AltDatabase/affymetrix/LibraryFiles/'
destination_parent = osfilepath(destination_parent+cdf_short)
#print destination_parent
#print input_cdf_file
if destination_parent not in input_cdf_file:
info_list = input_cdf_file,destination_parent; StatusWindow(info_list,'copy')
else:
print_out = "The file;\n"+input_cdf_file+"\ndoes not appear to be a valid Affymetix\nlibrary file. If you do not have library files, you must\ngo to the Affymetrix website to download."
IndicatorWindow(print_out,'Continue')
else:
if '.pgf' in input_cdf_file_lower:
###Check to see if the clf and bgp files are present in this directory
icf_list = string.split(input_cdf_file,'/'); parent_dir = string.join(icf_list[:-1],'/'); cdf_short = icf_list[-1]
clf_short = string.replace(cdf_short,'.pgf','.clf')
kil_short = string.replace(cdf_short,'.pgf','.kil') ### Only applies to the Glue array
if array_type == 'exon' or array_type == 'junction': bgp_short = string.replace(cdf_short,'.pgf','.antigenomic.bgp')
else: bgp_short = string.replace(cdf_short,'.pgf','.bgp')
dir_list = read_directory(parent_dir)
if clf_short in dir_list and bgp_short in dir_list:
pgf_file = input_cdf_file
clf_file = string.replace(pgf_file,'.pgf','.clf')
kil_file = string.replace(pgf_file,'.pgf','.kil') ### Only applies to the Glue array
if array_type == 'exon' or array_type == 'junction': bgp_file = string.replace(pgf_file,'.pgf','.antigenomic.bgp')
else: bgp_file = string.replace(pgf_file,'.pgf','.bgp')
assinged = 'yes'
###Thus the CDF or PDF file was confirmed, so copy it over to AltDatabase
destination_parent = 'AltDatabase/affymetrix/LibraryFiles/'
#print destination_parent
#print input_cdf_file
if destination_parent not in input_cdf_file:
info_list = input_cdf_file,osfilepath(destination_parent+cdf_short); StatusWindow(info_list,'copy')
info_list = clf_file,osfilepath(destination_parent+clf_short); StatusWindow(info_list,'copy')
info_list = bgp_file,osfilepath(destination_parent+bgp_short); StatusWindow(info_list,'copy')
if 'Glue' in pgf_file:
info_list = kil_file,osfilepath(destination_parent+kil_short); StatusWindow(info_list,'copy')
else:
print_out = "The directory;\n"+parent_dir+"\ndoes not contain either a .clf or antigenomic.bgp\nfile, required for probeset summarization."
IndicatorWindow(print_out,'Continue')
else:
print_out = "The file;\n"+input_cdf_file+"\ndoes not appear to be a valid Affymetix\nlibrary file. If you do not have library files, you must\ngo to the Affymetrix website to download."
IndicatorWindow(print_out,'Continue')
else:
print_out = "No library file has been assigned. Please\nselect a valid library file for this array."
IndicatorWindow(print_out,'Continue')
if 'input_annotation_file' in option_list_library: ### Deals with Annotation Files
assinged = 'yes'
if 'input_annotation_file' in gu.Results():
input_annotation_file = gu.Results()['input_annotation_file']; input_annotation_lower = string.lower(input_annotation_file)
if '.csv' in input_annotation_lower:
###Thus the CDF or PDF file was confirmed, so copy it over to AltDatabase
icf_list = string.split(input_annotation_file,'/'); csv_short = icf_list[-1]
destination_parent = 'AltDatabase/affymetrix/'+species+'/'
#print destination_parent
#print input_cdf_file
if destination_parent not in input_cdf_file:
info_list = input_annotation_file,filepath(destination_parent+csv_short); StatusWindow(info_list,'copy')
sd = SupprotedArrays(specific_array_type,cdf_short,csv_short,species,array_type)
supproted_array_db[specific_array_type] = sd
try: exportSupportedArrayInfo()
except Exception:
print 'Cannot write Config/ArrayFileInfo.txt to the Config directory (like Permissions Error)'
continue ### Occurs if the file is open... not critical to worry about
if run_from_scratch == 'Process Expression file':
status = 'repeat'
while status == 'repeat':
if backSelect == 'no' or 'InputExpFiles' == selected_parameters[-1]:
root = Tk(); root.title('AltAnalyze: Select Expression File for Filtering')
selected_parameters.append('InputExpFiles'); backSelect = 'no'
gu = GUI(root,option_db,option_list['InputExpFiles'],'')
else: gu = PreviousResults(old_options)
try: input_exp_file = gu.Results()['input_exp_file']
except KeyError: input_exp_file = '' ### Leave this blank so that the default directory is used
try: input_stats_file = gu.Results()['input_stats_file']
except KeyError: input_stats_file = '' ### Leave this blank so that the default directory is used
#if array_type == 'exon':
if 'steady-state' in input_exp_file or 'steady-state' in input_stats_file:
print_out = "Do not select steady-state expression files.."
IndicatorWindow(print_out,'Continue'); output_dir=''
elif len(input_exp_file)>0:
try: output_dir = gu.Results()['output_dir']
except KeyError: output_dir = '' ### Leave this blank so that the default directory is used
try: cel_files, array_linker_db = ExpressionBuilder.getArrayHeaders(input_exp_file)
except Exception:
print_out = "Input Expression file does not have a valid format."
IndicatorWindow(print_out,'Continue'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
if len(cel_files)>0: status = 'continue'
else:
if '.mtx' in input_exp_file or '.h5' in input_exp_file:
array_type = '10XGenomics'
array_full == '10X Genomics sparse matrix'
vendor = '10x'
print_out = "The expression file:\n"+input_exp_file+"\nis a 10x Genomics matrix... change the Platform to 10x Genomics Aligned in the main menu."
IndicatorWindow(print_out,'Continue')
else:
print_out = "The expression file:\n"+input_exp_file+"\ndoes not appear to be a valid expression file. Check to see that\nthis is the correct tab-delimited text file."
IndicatorWindow(print_out,'Continue')
else:
print_out = "No input expression file selected."
IndicatorWindow(print_out,'Continue')
if len(output_dir)<1:
### Set output to the same directory or parent if none selected
if 'ExpressionInput' in input_exp_file: i = -2
else: i = -1
output_dir = string.join(string.split(input_exp_file,'/')[:i],'/')
try: prior_platform = user_variables['prior_platform']
except Exception: prior_platform = None
if array_type == 'RNASeq' or prior_platform == 'RNASeq':
steady_state = string.replace(input_exp_file,'.txt','-steady-state.txt')
count = verifyFileLength(steady_state)
if count == 0 or 'exp.' not in input_exp_file: #No counts file
systm = getGeneSystem(input_exp_file)
### Wrong platform listed
array_type = "3'array"
prior_platform = 'RNASeq'
vendor = 'other:'+systm ### Ensembl linked system name
user_variables['manufacturer_selection'] = vendor
user_variables['prior_platform'] = prior_platform
if old_options==[] or 'marker_finder' not in old_options: ### If we haven't hit the back button
option_list,option_db = importUserOptions(array_type) ### will re-set the paramater values, so not good for back select
user_variables['array_type'] = array_type
if array_type == "3'array":
### This is the new option for expression filtering of non-RNASeq classified data
try:
#print option_db['rpkm_threshold'].DefaultOption(),1
if 'rpkm_threshold' in option_db:
option_db['rpkm_threshold'].setArrayOptions('1')
if "other:Symbol" in vendor or "other:Ensembl" in vendor:
option_db['rpkm_threshold'].setDefaultOption('1')
if option_db['rpkm_threshold'].DefaultOption() == ['NA']:
option_db['rpkm_threshold'].setDefaultOption('1')
option_db['rpkm_threshold'].setDisplay('Remove genes expressed below (non-log)')
else:
option_db['rpkm_threshold'].setArrayOptions('0')
option_db['rpkm_threshold'].setDefaultOption('0')
option_db['rpkm_threshold'].setDisplay('Remove genes expressed below (non-log)')
except Exception:
option_db['rpkm_threshold'].setArrayOptions('0')
option_db['rpkm_threshold'].setDefaultOption('0')
option_db['rpkm_threshold'].setDisplay('Remove genes expressed below (non-log)')
if "ExpressionInput" not in output_dir and len(input_exp_file)>1 and "ExpressionInput" not in input_exp_file:
try:
### If the user designates an output directory that doesn't contain ExpressionInput, move the exp-file there and rename
output_dir = output_dir + '/ExpressionInput' ### Store the result files here so that files don't get mixed up
try: os.mkdir(output_dir) ### Since this directory doesn't exist we have to make it
except OSError: null = [] ### Directory already exists
if 'exp.' not in input_exp_file: exp_prefix = 'exp.'
else: exp_prefix=''
moved_exp_dir = output_dir+'/'+exp_prefix+export.findFilename(input_exp_file)
alt_exp_dir = export.findParentDir(input_exp_file)+'/'+exp_prefix+export.findFilename(input_exp_file)
export.copyFile(input_exp_file, moved_exp_dir)
### Do the same thing for a groups file
try: export.copyFile(string.replace(alt_exp_dir,'exp.','groups.'), string.replace(moved_exp_dir,'exp.','groups.'))
except: pass
### Do the same thing for a comps file
try: export.copyFile(string.replace(alt_exp_dir,'exp.','comps.'), string.replace(moved_exp_dir,'exp.','comps.'))
except: pass
input_exp_file = moved_exp_dir
if len(input_stats_file)>1: ### Do the same for a stats file
if 'stats.' not in input_exp_file: stats_prefix = 'stats.'
else: stats_prefix=''
moved_stats_dir = output_dir+'/'+stats_prefix+export.findFilename(input_stats_file)
export.copyFile(input_stats_file, moved_stats_dir)
input_stats_file = moved_stats_dir
except Exception: None
if run_from_scratch != 'buildExonExportFiles': ### Update DBs is an option which has been removed from 1.1. Should be a separate menu item soon.
expr_defaults, alt_exon_defaults, functional_analysis_defaults, goelite_defaults = importDefaults(array_type,species)
#print vendor
if '10X' in vendor:
option_db['rpkm_threshold'].setDefaultOption('1')
if vendor == 'Affymetrix' or vendor == 'RNASeq':
option_db['normalize_gene_data'].setArrayOptions(['NA']) ### Only use this option when processing Feature Extraction files or non-Affy non-RNA-Seq data
if vendor == 'Agilent' and 'Feature Extraction' in run_from_scratch:
option_db['normalize_gene_data'].setDefaultOption('quantile')
option_db['normalize_gene_data'].setArrayOptions(['quantile']) ### Only set this as a default when performing Feature Extraction for Agilent data
if run_from_scratch != 'Process AltAnalyze filtered' and run_from_scratch != 'Annotate External Results':
proceed = 'no'
option_db = check_moderated_support(option_db)
while proceed == 'no':
if backSelect == 'no' or 'GeneExpression' == selected_parameters[-1]:
selected_parameters.append('GeneExpression'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Expression Analysis Parameters')
gu = GUI(root,option_db,option_list['GeneExpression'],expr_defaults)
else: gu = PreviousResults(old_options)
try: rpkm_threshold = float(gu.Results()['rpkm_threshold'])
except Exception:
if array_type == 'RNASeq': rpkm_threshold = 1
else: rpkm_threshold = 'NA'
if array_type != "3'array":
try: dabg_p = gu.Results()['dabg_p']
except Exception:
if array_type == 'RNASeq': dabg_p = 1
else: dabg_p = 'NA'
try: gene_exp_threshold = gu.Results()['gene_exp_threshold']
except Exception:
if array_type == 'RNASeq': gene_exp_threshold = 1
else: gene_exp_threshold = 'NA'
try: exon_rpkm_threshold = gu.Results()['exon_rpkm_threshold']
except Exception:
if array_type == 'RNASeq': exon_rpkm_threshold = 1
else: exon_rpkm_threshold = 'NA'
try: exon_exp_threshold = gu.Results()['exon_exp_threshold']
except Exception:
if array_type == 'RNASeq': exon_exp_threshold = 1
else: exon_exp_threshold = 'NA'
run_from_scratch = gu.Results()['run_from_scratch']
try: expression_threshold = gu.Results()['expression_threshold']
except Exception:
if array_type == 'RNASeq': expression_threshold = 0
else: expression_threshold = 'NA'
try: perform_alt_analysis = gu.Results()['perform_alt_analysis']
except Exception: perform_alt_analysis = 'just expression'
try: analyze_as_groups = gu.Results()['analyze_as_groups']
except Exception: analyze_as_groups = ''
if perform_alt_analysis == 'just expression': perform_alt_analysis = 'expression'
else: perform_alt_analysis = 'both'
try: avg_all_for_ss = gu.Results()['avg_all_for_ss']
except Exception: avg_all_for_ss = 'no'
excludeNonExpExons = True
if 'all exon aligning' in avg_all_for_ss or 'known' in avg_all_for_ss or 'expressed exons' in avg_all_for_ss:
if 'known exons' in avg_all_for_ss and array_type == 'RNASeq': excludeNonExpExons = False
if 'known junctions' in avg_all_for_ss and array_type == 'RNASeq':
fl.setUseJunctionsForGeneExpression(True)
excludeNonExpExons = False
avg_all_for_ss = 'yes'
else: avg_all_for_ss = 'no'
expression_data_format = gu.Results()['expression_data_format']
try: normalize_feature_exp = gu.Results()['normalize_feature_exp']
except Exception: normalize_feature_exp = 'NA'
try: normalize_gene_data = gu.Results()['normalize_gene_data']
except Exception: normalize_gene_data = 'NA'
include_raw_data = gu.Results()['include_raw_data']
run_goelite = gu.Results()['run_goelite']
visualize_results = gu.Results()['visualize_results']
run_lineage_profiler = gu.Results()['run_lineage_profiler']
probability_algorithm = gu.Results()['probability_algorithm']
try: FDR_statistic = gu.Results()['FDR_statistic']
except Exception: pass
try: batch_effects = gu.Results()['batch_effects']
except Exception: batch_effects = 'NA'
try: marker_finder = gu.Results()['marker_finder']
except Exception: marker_finder = 'NA'
if 'immediately' in run_goelite: run_goelite = 'yes'
else: run_goelite = 'no'
passed = 'yes'; print_out = 'Invalid threshold entered for '
if array_type != "3'array" and array_type !='RNASeq':
try:
dabg_p = float(dabg_p)
if dabg_p<=0 or dabg_p>1: passed = 'no'; print_out+= 'DABG p-value cutoff '
except Exception: passed = 'no'; print_out+= 'DABG p-value cutoff '
if array_type != "3'array":
try:
try: rpkm_threshold = float(rpkm_threshold)
except Exception:
expression_threshold = float(expression_threshold)
if expression_threshold<1: passed = 'no'; print_out+= 'expression threshold '
except Exception: passed = 'no'; print_out+= 'expression threshold '
if array_type == 'RNASeq':
try:
rpkm_threshold = float(rpkm_threshold)
if rpkm_threshold<0: passed = 'no'; print_out+= 'RPKM threshold '
except Exception: passed = 'no'; print_out+= 'RPKM threshold '
try:
exon_exp_threshold = float(exon_exp_threshold)
if exon_exp_threshold<0: passed = 'no'; print_out+= 'Exon expression threshold '
except Exception: passed = 'no'; print_out+= 'Exon expression threshold '
try:
exon_rpkm_threshold = float(exon_rpkm_threshold)
if exon_rpkm_threshold<0: passed = 'no'; print_out+= 'Exon RPKM threshold '
except Exception: passed = 'no'; print_out+= 'Exon RPKM threshold '
try:
gene_exp_threshold = float(gene_exp_threshold)
if gene_exp_threshold<0: passed = 'no'; print_out+= 'Gene expression threshold '
except Exception: passed = 'no'; print_out+= 'Gene expression threshold '
if visualize_results == 'yes':
try:
### Tests to make sure these are installed - required for visualization
import matplotlib
from numpy import array
from scipy import rand
except Exception:
passed = 'no'; print_out = 'Support for matplotlib, numpy and scipy must specifically be installed to perform data visualization.\n'
print_out += traceback.format_exc() ### useful for seeing a warning window with the actuall error
if passed == 'no': IndicatorWindow(print_out,'Continue')
else: proceed = 'yes'
if run_lineage_profiler == 'yes':
verifyLineageProfilerDatabases(species,'GUI')
if (perform_alt_analysis == 'both') or (run_from_scratch == 'Process AltAnalyze filtered') or (run_from_scratch == 'Annotate External Results'):
perform_alt_analysis = 'yes'
if run_from_scratch == 'Process AltAnalyze filtered':
input_filtered_dir = ''
while len(input_filtered_dir)<1:
if backSelect == 'no' or 'InputFilteredFiles' == selected_parameters[-1]:
selected_parameters.append('InputFilteredFiles'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Select AltAnalyze Filtered Probe set Files')
gu = GUI(root,option_db,option_list['InputFilteredFiles'],'')
else: gu = PreviousResults(old_options)
try:
input_filtered_dir = gu.Results()['input_filtered_dir']
if 'FullDataset' in input_filtered_dir: alt_exon_defaults[3] = 'all groups'
except Exception: input_filtered_dir = ''
if input_filtered_dir == '':
print_out = "The directory containing filtered probe set text files has not\nbeen assigned! Select a valid directory before proceeding."
IndicatorWindow(print_out,'Continue')
fl = ExpressionFileLocationData('','','',''); dataset_name = 'filtered-exp_dir'
dirs = string.split(input_filtered_dir,'AltExpression'); parent_dir = dirs[0]
exp_file_location_db={}; exp_file_location_db[dataset_name]=fl
if run_from_scratch == 'Annotate External Results':
input_filtered_dir = ''
while len(input_filtered_dir)<1:
if backSelect == 'no' or 'InputExternalFiles' == selected_parameters[-1]:
selected_parameters.append('InputExternalFiles'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Select AltAnalyze Filtered Probe set Files')
gu = GUI(root,option_db,option_list['InputExternalFiles'],'')
else: gu = PreviousResults(old_options)
try: input_filtered_dir = gu.Results()['input_external_dir']
except Exception: input_filtered_dir = ''
if input_filtered_dir == '':
print_out = "The directory containing external probe set text files has not\nbeen assigned! Select a valid directory before proceeding."
IndicatorWindow(print_out,'Continue')
fl = ExpressionFileLocationData('','','',''); dataset_name = 'external-results_dir'
dirs = string.split(input_filtered_dir,'AltExpression'); parent_dir = dirs[0]
exp_file_location_db={}; exp_file_location_db[dataset_name]=fl
#print option_list[i:i+len(alt_exon_defaults)+len(functional_analysis_defaults)], alt_exon_defaults+functional_analysis_defaults;kill
option_list,option_db = importUserOptions(array_type) ##Initially used to just get the info for species and array_type
if backSelect == 'yes':
for option in old_options: ### Set options to user selected
try: option_db[option].setDefaultOption(old_options[option])
except Exception: pass
if run_from_scratch == 'Process AltAnalyze filtered':
if array_type == 'RNASeq': cs_name = 'known exons'
else: cs_name = 'constitutive probesets'
functional_analysis_defaults.append(cs_name); option_list['AltAnalyze'].append('avg_all_for_ss')
if run_goelite == 'no': ### run_goelite will be set to no by default
functional_analysis_defaults.append('unpaired t-test'); option_list['AltAnalyze'].append('probability_algorithm')
functional_analysis_defaults.append('decide later'); option_list['AltAnalyze'].append('run_goelite')
if run_from_scratch == 'Annotate External Results':
### Remove options relating to expression analysis when importing filtered probeset lists
options_to_exclude = ['analysis_method','p_threshold','gene_expression_cutoff','alt_exon_fold_cutoff','run_MiDAS']
options_to_exclude+= ['export_splice_index_values','probability_algorithm','run_goelite','analyze_all_conditions','calculate_splicing_index_p']
for option in options_to_exclude: del option_db[option]
proceed = 'no'
while proceed == 'no':
if backSelect == 'no' or 'AltAnalyze' == selected_parameters[-1]:
selected_parameters.append('AltAnalyze'); backSelect = 'no'; proceed = 'no'
root = Tk(); root.title('AltAnalyze: Alternative Exon Analysis Parameters')
gu = GUI(root,option_db,option_list['AltAnalyze'],alt_exon_defaults+functional_analysis_defaults); #user_variables = {};
try: analyze_all_conditions = gu.Results()['analyze_all_conditions']
except KeyError: analyze_all_conditions = 'pairwise'
if analyze_all_conditions != 'pairwise':
print_out = 'Please note: When AltAnalyze compares all groups, the\nalternative exon fold to be filtered will be based on the\nlargest alternative exon fold for all possible comparisons.'
IndicatorWindowSimple(print_out,'Continue')
else: gu = PreviousResults(old_options)
try: analysis_method = gu.Results()['analysis_method']
except Exception: analysis_method = analysis_method
try: p_threshold = gu.Results()['p_threshold']
except Exception: p_threshold = 0.05
try: gene_expression_cutoff = gu.Results()['gene_expression_cutoff']
except Exception: gene_expression_cutoff = 3
try: remove_intronic_junctions = gu.Results()['remove_intronic_junctions']
except Exception: remove_intronic_junctions = 'NA'
try: filter_probeset_types = gu.Results()['filter_probe_types']
except Exception: filter_probeset_types = 'core'
try: alt_exon_fold_cutoff = gu.Results()['alt_exon_fold_cutoff']
except KeyError: alt_exon_fold_cutoff = 2
try: permute_p_threshold = gu.Results()['permute_p_threshold']
except KeyError: permute_p_threshold = 0.05 ### Doesn't matter, not used
try:
additional_algorithms = gu.Results()['additional_algorithms']
additional_algorithms = AdditionalAlgorithms(additional_algorithms)
except KeyError: additionalAlgorithms = AdditionalAlgorithms('')
try:
additional_score = gu.Results()['additional_score']
additional_algorithms.setScore(additional_score)
except Exception:
try: additional_algorithms.setScore(2)
except Exception: pass
try: perform_permutation_analysis = gu.Results()['perform_permutation_analysis']
except KeyError: perform_permutation_analysis = perform_permutation_analysis
try: export_splice_index_values = gu.Results()['export_splice_index_values']
except KeyError: export_splice_index_values = export_splice_index_values
try: run_MiDAS = gu.Results()['run_MiDAS']
except KeyError: run_MiDAS = run_MiDAS
try: analyze_all_conditions = gu.Results()['analyze_all_conditions']
except KeyError: analyze_all_conditions = analyze_all_conditions
try: run_goelite = gu.Results()['run_goelite']
except KeyError: run_goelite = run_goelite
try: probability_algorithm = gu.Results()['probability_algorithm']
except KeyError: probability_algorithm = probability_algorithm
try:
avg_all_for_ss = gu.Results()['avg_all_for_ss']
if 'all exon aligning' in avg_all_for_ss or 'known' in avg_all_for_ss or 'core' in avg_all_for_ss or 'expressed exons' in avg_all_for_ss:
avg_all_for_ss = 'yes'
else: avg_all_for_ss = 'no'
except Exception:
try: avg_all_for_ss = avg_all_for_ss
except Exception: avg_all_for_ss = 'no'
if 'immediately' in run_goelite: run_goelite = 'yes'
else: run_goelite = 'no'
try: calculate_splicing_index_p = gu.Results()['calculate_splicing_index_p']
except KeyError: calculate_splicing_index_p = calculate_splicing_index_p
analyze_functional_attributes = gu.Results()['analyze_functional_attributes']
filter_for_AS = gu.Results()['filter_for_AS']
microRNA_prediction_method = gu.Results()['microRNA_prediction_method']
if analysis_method == 'splicing-index': p_threshold = float(p_threshold)
else:
try: p_threshold = float(permute_p_threshold)
except ValueError: permute_p_threshold = permute_p_threshold
if analysis_method == 'linearregres-rlm':
### Test installation of rpy and/or R
x = [5.05, 6.75, 3.21, 2.66]; y = [1.65, 26.5, -5.93, 7.96]
try: s = statistics.LinearRegression(x,y,'no')
except Exception:
print_out = "The local installation of R and rpy is missing or\nis not properly configured. See the AltAnalyze ReadMe\nfor more information (may require loading AltAnalyze from source code)."
IndicatorWindow(print_out,'Continue'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
passed = 'yes'; print_out = 'Invalid threshold entered for '
try: gene_expression_cutoff = float(gene_expression_cutoff)
except Exception: passed = 'no'; print_out+= 'gene expression cutoff'
try: alt_exon_fold_cutoff = float(alt_exon_fold_cutoff)
except Exception: passed = 'no'; print_out+= 'alternative exon fold change'
try: p_threshold = float(p_threshold)
except Exception: passed = 'no'; print_out+= 'alternative exon p-value'
if gene_expression_cutoff <= 1: passed = 'no'; print_out+= 'gene expression cutoff'
elif alt_exon_fold_cutoff < 1:
if analysis_method == 'splicing-index': passed = 'no'; print_out+= 'splicing-index fold change'
elif alt_exon_fold_cutoff < 0: passed = 'no'; print_out+= 'alternative exon fold change'
elif p_threshold <= 0: passed = 'no'; print_out+= 'alternative exon p-value'
if passed == 'no': IndicatorWindow(print_out,'Continue')
else: proceed = 'yes'
if run_goelite == 'yes':
option_db['get_additional'].setArrayOptions(['---']+importResourceList())
option_db['get_additional'].setDefaultOption('---')
### Populate variables based on the existing imported data
default_resources = option_db['resources_to_analyze'].ArrayOptions() ### Include alternative ontologies and gene-lists
import_dir1 = '/AltDatabase/goelite/'+species+'/gene-mapp'
import_dir2 = '/AltDatabase/goelite/'+species+'/gene-go'
try:
gene_mapp_list = read_directory(import_dir1)
gene_mapp_list.sort()
for file in gene_mapp_list:
resource = string.split(file,'-')[-1][:-4]
if resource != 'MAPP' and resource not in default_resources and '.txt' in file:
default_resources.append(resource)
except Exception: pass
try:
gene_go_list = read_directory(import_dir2)
gene_go_list.sort()
for file in gene_go_list:
resource = string.split(file,'-')[-1][:-4]
if resource != 'GeneOntology' and resource not in default_resources and 'version' not in resource and '.txt' in file:
default_resources.append(resource)
except Exception: pass
option_db['resources_to_analyze'].setArrayOptions(default_resources)
if run_from_scratch == 'Process AltAnalyze filtered':
### Do not include gene expression analysis filters
option_list['GOElite'] = option_list['GOElite'][3:]; goelite_defaults = goelite_defaults[3:]
if backSelect == 'no' or 'GOElite' == selected_parameters[-1]:
selected_parameters.append('GOElite'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Pathway Analysis Parameters')
gu = GUI(root,option_db,option_list['GOElite'],goelite_defaults)
else: gu = PreviousResults(old_options)
if run_from_scratch != 'Process AltAnalyze filtered':
ge_fold_cutoffs = gu.Results()['ge_fold_cutoffs']
ge_pvalue_cutoffs = gu.Results()['ge_pvalue_cutoffs']
ge_ptype = gu.Results()['ge_ptype']
filter_method = gu.Results()['filter_method']
z_threshold = gu.Results()['z_threshold']
returnPathways = gu.Results()['returnPathways']
p_val_threshold = gu.Results()['p_val_threshold']
change_threshold = gu.Results()['change_threshold']
resources_to_analyze = gu.Results()['resources_to_analyze']
pathway_permutations = gu.Results()['pathway_permutations']
get_additional = gu.Results()['get_additional']
ORA_algorithm = gu.Results()['ORA_algorithm']
mod = gu.Results()['mod']
ge_fold_cutoffs = float(ge_fold_cutoffs)
change_threshold = float(change_threshold) - 1 ### This reflects the > statement in the GO-Elite filtering
if ORA_algorithm == 'Fisher Exact Test':
pathway_permutations = 'FisherExactTest'
if get_additional != '---':
analysis = 'getAdditionalOnlineResources'
values = species,get_additional
StatusWindow(values,analysis) ### display an window with download status
except OSError:
pass; sys.exit()
"""In this next section, create a set of GUI windows NOT defined by the options.txt file.
These are the groups and comps files"""
original_comp_group_list=[]; array_group_list=[]; group_name_list=[]
if run_from_scratch != 'Process AltAnalyze filtered' and run_from_scratch != 'Annotate External Results': ### Groups and Comps already defined
if run_from_scratch == 'Process CEL files' or run_from_scratch == 'Process RNA-seq reads' or 'Feature Extraction' in run_from_scratch or 'Process Chromium Matrix' in run_from_scratch:
if 'exp.' not in dataset_name: dataset_name = 'exp.'+dataset_name+'.txt'
groups_name = string.replace(dataset_name,'exp.','groups.')
comps_name = string.replace(dataset_name,'exp.','comps.')
batch_name = string.replace(groups_name,'groups.','batch.') ### may not apply
if "ExpressionInput" not in output_dir:
output_dir = output_dir + '/ExpressionInput' ### Store the result files here so that files don't get mixed up
try: os.mkdir(output_dir) ### Since this directory doesn't exist we have to make it
except OSError: null = [] ### Directory already exists
exp_file_dir = output_dir+'/'+dataset_name
### store file locations (also use these later when running APT)
stats_file_dir = string.replace(exp_file_dir,'exp.','stats.')
groups_file_dir = string.replace(exp_file_dir,'exp.','groups.')
comps_file_dir = string.replace(exp_file_dir,'exp.','comps.')
batch_file_dir = string.replace(groups_file_dir, 'groups.','batch.')
fl = ExpressionFileLocationData(exp_file_dir,stats_file_dir,groups_file_dir,comps_file_dir)
exp_file_location_db={}; exp_file_location_db[dataset_name]=fl
parent_dir = output_dir ### interchangable terms (parent_dir used with expression file import)
#print groups_file_dir
if run_from_scratch == 'Process Expression file':
if len(input_exp_file)>0:
if len(input_stats_file)>1: ###Make sure the files have the same arrays and order first
try: cel_files2, array_linker_db2 = ExpressionBuilder.getArrayHeaders(input_stats_file)
except Exception:
print_out = "Input Expression file does not have a valid format."
IndicatorWindow(print_out,'Continue'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
if cel_files2 != cel_files:
print_out = "The probe set p-value file:\n"+input_stats_file+"\ndoes not have the same array order as the\nexpression file. Correct before proceeding."
IndicatorWindow(print_out,'Continue')
### Check to see if a groups/comps file already exists and add file locations to 'exp_file_location_db'
ief_list = string.split(input_exp_file,'/'); parent_dir = string.join(ief_list[:-1],'/'); exp_name = ief_list[-1]
dataset_name = string.replace(exp_name,'exp.','')
groups_name = 'groups.'+dataset_name; comps_name = 'comps.'+dataset_name
batch_name = string.replace(groups_name,'groups.','batch.') ### may not apply
groups_file_dir = parent_dir+'/'+groups_name; comps_file_dir = parent_dir+'/'+comps_name
batch_file_dir = string.replace(groups_file_dir, 'groups.','batch.')
fl = ExpressionFileLocationData(input_exp_file,input_stats_file,groups_file_dir,comps_file_dir)
dataset_name = exp_name
exp_file_location_db={}; exp_file_location_db[exp_name]=fl
else:
### This occurs if running files in the ExpressionInput folder. However, if so, we won't allow for GUI based creation of groups and comps files (too complicated and confusing for user).
### Grab all expression file locations, where the expression, groups and comps file exist for a dataset
exp_file_location_db = importExpressionFiles() ###Don't create 'array_group_list', but pass the 'exp_file_location_db' onto ExpressionBuilder
### Import array-group and group comparisons. Only time relevant for probesetSummarization is when an error is encountered and re-running
try: dir_files = read_directory(parent_dir)
except Exception: dir_files=[]
array_group_list=[]
array_batch_list=[]
if backSelect == 'yes':
for cel_file in cel_files:
if cel_file in user_variables:
group_name = user_variables[cel_file]; group = ''
else:
group = ''; group_name = ''
agd = ArrayGroupData(cel_file,group,group_name); array_group_list.append(agd)
if batch_effects == 'yes' or normalize_gene_data == 'group': ### Used during backselect (must include a 'batch' variable in the stored var name)
if (cel_file,'batch') in user_variables:
batch_name = user_variables[cel_file,'batch']; batch = ''
else:
batch = ''; batch_name = ''
agd = ArrayGroupData(cel_file,batch,batch_name); array_batch_list.append(agd); batch_db=[]
elif run_from_scratch == 'buildExonExportFiles':
fl = ExpressionFileLocationData('','','',''); fl.setExonBedBuildStatus('yes'); fl.setFeatureNormalization('none')
fl.setCELFileDir(cel_file_dir); fl.setArrayType(array_type); fl.setOutputDir(output_dir); fl.setMultiThreading(multiThreading)
exp_file_location_db={}; exp_file_location_db[dataset_name]=fl; parent_dir = output_dir
perform_alt_analysis = 'expression'
elif groups_name in dir_files:
try:
### Try to import any current annotations and verify that the samples indicated in the input directory are in the corresponding groups file
array_group_list,group_db = importArrayGroupsSimple(groups_file_dir,cel_files) #agd = ArrayGroupData(array_header,group,group_name)
except Exception,e:
### Over-write these annotations if theres is a problem
for cel_file in cel_files:
group = ''; group_name = ''
agd = ArrayGroupData(cel_file,group,group_name); array_group_list.append(agd); group_db=[]
if batch_effects == 'yes' or normalize_gene_data == 'group':
if batch_name in dir_files: ### Almost identical format and output files (import existing if present here)
try:
array_batch_list,batch_db = importArrayGroupsSimple(batch_file_dir,cel_files) #agd = ArrayGroupData(array_header,group,group_name)
except Exception,e:
for cel_file in cel_files:
batch = ''; batch_name = ''
agd = ArrayGroupData(cel_file,batch,batch_name); array_batch_list.append(agd); batch_db=[]
else:
for cel_file in cel_files:
batch = ''; batch_name = ''
agd = ArrayGroupData(cel_file,batch,batch_name); array_batch_list.append(agd); batch_db=[]
if comps_name in dir_files and len(group_db)>0:
try:
comp_group_list, null = ExpressionBuilder.importComparisonGroups(comps_file_dir)
for group1,group2 in comp_group_list:
try:
group_name1 = group_db[int(group1)]; group_name2 = group_db[int(group2)]
original_comp_group_list.append((group_name1,group_name2)) ### If comparisons already exist, default to these
except KeyError:
print_out = 'The "comps." file for this dataset has group numbers\nnot listed in the "groups." file.'
#WarningWindow(print_out,'Exit'); AltAnalyze.AltAnalyzeSetup('no'); sys.exit()
#print print_out
original_comp_group_list=[]
except:
print_out = 'The "comps." file for this dataset has group numbers\nnot listed in the "groups." file.'
original_comp_group_list=[]
else:
for cel_file in cel_files:
group = ''; group_name = ''
agd = ArrayGroupData(cel_file,group,group_name); array_group_list.append(agd)
if len(array_group_list)>0: ### Thus we are not analyzing the default (ExpressionInput) directory of expression, group and comp data.
original_option_db,original_option_list = option_db,option_list
if len(array_group_list)>200:
### Only display the top 200 and don't record edits
option_db,option_list = formatArrayGroupsForGUI(array_group_list[:200])
else:
option_db,option_list = formatArrayGroupsForGUI(array_group_list)
###Force this GUI to repeat until the user fills in each entry, but record what they did add
user_variables_long={}
while len(user_variables_long) != len(option_db):
if backSelect == 'no' or 'GroupArrays' == selected_parameters[-1]:
selected_parameters.append('GroupArrays'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Assign files to a Group Annotation'); user_variables_long={}
#import copy; user_variables_original = copy.deepcopy(user_variables); user_variables={}
gu = GUI(root,option_db,option_list['GroupArrays'],'groups')
else: gu = PreviousResults(old_options)
try: predictGroups = gu.Results()['PredictGroups']
except Exception: predictGroups = False
for option in user_variables: ### By default, all arrays will be assigned a group of ''
try:
if len(user_variables[option])>0 and 'batch' not in option:
if option in option_db: user_variables_long[option]=[]
except Exception: pass
###Store the group names and assign group numbers
group_name_db={}; group_name_list = []; group_number = 1
if len(array_group_list)<=200:
for cel_file in option_list['GroupArrays']: ### start we these CEL files, since they are ordered according to their order in the expression dataset
group_name = gu.Results()[cel_file]
if group_name not in group_name_db:
if group_name != 'yes' and group_name !='no': ### Results for PredictGroups
group_name_db[group_name]=group_number; group_number+=1
group_name_list.append(group_name)
else:
### For very large datasets with hundreds of samples
for agd in array_group_list:
if agd.GroupName() not in group_name_list:
group_name_list.append(agd.GroupName())
group_name_db[agd.GroupName()]=agd.Group()
if len(group_name_db)==2: analyze_all_conditions = 'pairwise' ### Don't allow multiple comparison analysis if only two conditions present
###Store the group names and numbers with each array_id in memory
if len(array_group_list)<=200:
for agd in array_group_list:
cel_file = agd.Array()
group_name = gu.Results()[cel_file] ###Lookup the new group assignment entered by the user
group_number = group_name_db[group_name]
agd.setGroupName(group_name); agd.setGroup(group_number)
if predictGroups == 'yes':
predictGroups = True; break
elif predictGroups == 'no': predictGroups = False
elif (len(user_variables_long) != len(option_db)) or len(group_name_db)<2:
if len(group_name_db)<2:
print_out = "At least two array groups must be established\nbefore proceeding."
else:
print_out = "Not all arrays have been assigned a group. Please\nassign to a group before proceeding (required)."
IndicatorWindow(print_out,'Continue')
option_db,option_list = formatArrayGroupsForGUI(array_group_list) ### array_group_list at this point will be updated with any changes made in the GUI by the user
if predictGroups == False:
exported = 0 ### Export Groups file
if len(array_group_list)>200:
print 'Not storing groups due to length'
else:
while exported == 0:
try:
fl = exp_file_location_db[dataset_name]; groups_file = fl.GroupsFile()
exportGroups(exp_file_location_db,array_group_list)
exported = 1
except Exception:
print_out = "The file:\n"+groups_file+"\nis still open. This file must be closed before proceeding"
IndicatorWindow(print_out,'Continue')
exported = 0
if batch_effects == 'yes' or normalize_gene_data == 'group':
option_db,option_list = formatArrayGroupsForGUI(array_batch_list, category = 'BatchArrays')
###Force this GUI to repeat until the user fills in each entry, but record what they did add
user_variables_long={}
while len(user_variables_long) != len(option_db):
if backSelect == 'no' or 'BatchArrays' == selected_parameters[-1]:
selected_parameters.append('BatchArrays'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Indicate Which Batch a File is From'); user_variables_long={}
#import copy; user_variables_original = copy.deepcopy(user_variables); user_variables={}
gu = GUI(root,option_db,option_list['BatchArrays'],'batch')
else: gu = PreviousResults(old_options)
for option in user_variables: ### By default, all arrays will be assigned a batch of ''
try:
if len(user_variables[option])>0 and 'batch' in option:
if option[0] in option_db: user_variables_long[option]=[]
except Exception: pass
###Store the batch names and assign batch numbers
batch_name_db={}; batch_name_list = []; batch_number = 1
#print option_list['BatchArrays']
for cel_file in option_list['BatchArrays']: ### start we these CEL files, since they are ordered according to their order in the expression dataset
batch_name = gu.Results()[cel_file,'batch']
if batch_name not in batch_name_db:
batch_name_db[batch_name]=batch_number; batch_number+=1
batch_name_list.append(batch_name)
if len(batch_name_db)==2: analyze_all_conditions = 'pairwise' ### Don't allow multiple comparison analysis if only two conditions present
###Store the batch names and numbers with each array_id in memory
for agd in array_batch_list:
cel_file = agd.Array()
batch_name = gu.Results()[cel_file,'batch'] ###Lookup the new batch assignment entered by the user
batch_number = batch_name_db[batch_name]
agd.setGroupName(batch_name); agd.setGroup(batch_number)
if (len(user_variables_long) != len(option_db)) or len(batch_name_db)<2:
if len(batch_name_db)<2:
print_out = "At least two sample batchs must be established\nbefore proceeding."
else:
print_out = "Not all arrays have been assigned a batch. Please\nassign to a batch before proceeding (required)."
IndicatorWindow(print_out,'Continue')
option_db,option_list = formatArrayGroupsForGUI(array_batch_list, category = 'BatchArrays') ### array_batch_list at this point will be updated with any changes made in the GUI by the user
exported = 0 ### Export Batch file
while exported == 0:
try:
fl = exp_file_location_db[dataset_name]
exportGroups(exp_file_location_db,array_batch_list,filetype='Batch')
exported = 1
except Exception:
print_out = "The file:\n"+batch_file_dir+"\nis still open. This file must be closed before proceeding"
IndicatorWindow(print_out,'Continue')
exported = 0
i=2; px=0 ###Determine the number of possible comparisons based on the number of groups
while i<=len(group_name_list): px = px + i - 1; i+=1
group_name_list.reverse(); group_name_list.append(''); group_name_list.reverse() ### add a null entry first
if px > 150: px = 150 ### With very large datasets, AltAnalyze stalls
possible_comps = px
### Format input for GUI like the imported options.txt Config file, except allow for custom fields in the GUI class
category = 'SetupComps'; option_db={}; option_list={}; cn = 0 #; user_variables={}
while cn < px:
try: group1,group2 = original_comp_group_list[cn]
except IndexError: group1='';group2=''
cn+=1; option = 'comparison '+str(cn); array_options = group_name_list; displayed_title=option; display_object='pulldown_comps'; notes=[group1,group2]
od = OptionData(option,displayed_title,display_object,notes,array_options,'')
option_db[option] = od
try: option_list[category].append(option) ###group is the name of the GUI menu group
except KeyError: option_list[category] = [option]
proceed = 'no'
while proceed == 'no' and analyze_all_conditions != 'all groups':
identical_groups = 'no'; comp_groups_db={}; proceed = 'no'
if (backSelect == 'no' or 'SetupComps' == selected_parameters[-1]):
selected_parameters.append('SetupComps'); backSelect = 'no'
root = Tk(); root.title('AltAnalyze: Establish All Pairwise Comparisons')
gu = GUI(root,option_db,option_list['SetupComps'],'comps')
else: gu = PreviousResults(old_options)
### Sort comparisons from user for export
for comparison in gu.Results():
try:
group_name = gu.Results()[comparison]
if len(group_name)>0 and 'comparison' in comparison: ### Group_names are by default blank
cn_main,cn_minor = string.split(comparison[11:],'-') ### e.g. 1-1 and 1-2
try:
null = int(cn_main); null = int(cn_minor)
try: comp_groups_db[cn_main].append([cn_minor,group_name])
except KeyError: comp_groups_db[cn_main]=[[cn_minor,group_name]]
except Exception: pass
except Exception: pass
print_out = "You must pick at least one comparison group before proceeding."
if len(comp_groups_db)>0:
try:
comp_group_list=[]
for cn_main in comp_groups_db:
cg = comp_groups_db[cn_main]
cg.sort()
comp_group_list.append([cn_main,[group_name_db[cg[0][1]],group_name_db[cg[1][1]]]])
if cg[0][1] == cg[1][1]: identical_groups = 'yes' ### Thus the two groups in the comparisons are identical, flag
comp_group_list.sort()
proceed = 'yes'
except Exception:
print traceback.format_exc()
print_out = "You must pick at least two groups for each comparison."
if identical_groups == 'yes': proceed = 'no'; print_out = "The same group is listed as both the experimental and\ncontrol group in a comparison. Fix before proceeding."
if proceed == 'no': IndicatorWindow(print_out,'Continue')
### Export user modified comps files
while exported == 0:
try:
fl = exp_file_location_db[dataset_name]; comps_file = fl.CompsFile()
if analyze_all_conditions != 'all groups': exportComps(exp_file_location_db,comp_group_list)
exported = 1
except Exception:
print_out = "The file:\n"+comps_file+"\nis still open. This file must be closed before proceeding"
IndicatorWindow(print_out,'Continue')
### See if there are any Affymetrix annotation files for this species
import_dir = '/AltDatabase/affymetrix/'+species
try: dir_list = read_directory(import_dir); fn_dir = filepath(import_dir[1:]); species_dir_found = 'yes'
except Exception: fn_dir = filepath(import_dir); dir_list = []; species_dir_found = 'no'
### Used to check if the user has an Affymetrix CSV file around... no longer needed
"""
if (len(dir_list)<1 or species_dir_found == 'no') and array_type != 'exon':
print_out = 'No Affymetrix annnotations file found in the directory:\n'+fn_dir
print_out += '\n\nTo download, click on the below button, find your array and download the annotation CSV file'
print_out += '\nlisted under "Current NetAffx Annotation Files". Extract the compressed zip archive to the'
print_out += '\nabove listed directory and hit continue to include these annotations in your results file.'
button_text = 'Download Annotations'; url = 'http://www.affymetrix.com/support/technical/byproduct.affx?cat=arrays'
IndicatorLinkOutWindow(print_out,button_text,url)
"""
""" ### Change made in version 2.1.4
if len(input_fastq_dir)>0:
array_type = "3'array"
vendor = 'other:Ensembl' ### Ensembl linked system name
"""
if microRNA_prediction_method == 'two or more': microRNA_prediction_method = 'multiple'
else: microRNA_prediction_method = 'any'
try: permute_p_threshold = float(permute_p_threshold)
except ValueError: permute_p_threshold = permute_p_threshold
try: dabg_p = float(dabg_p)
except ValueError: dabg_p = dabg_p
try: expression_threshold = float(expression_threshold)
except ValueError: expression_threshold = expression_threshold
try: alt_exon_fold_cutoff = float(alt_exon_fold_cutoff)
except ValueError: alt_exon_fold_cutoff = alt_exon_fold_cutoff
try: gene_expression_cutoff = float(gene_expression_cutoff)
except ValueError: gene_expression_cutoff = gene_expression_cutoff
### Find the current verison of APT (if user deletes location in Config file) and set APT file locations
try: apt_location = getAPTLocations(file_location_defaults,run_from_scratch,run_MiDAS)
except Exception: pass
### Set the primary parent directory for ExpressionBuilder and AltAnalyze (one level above the ExpressionInput directory, if present)
for dataset in exp_file_location_db:
fl = exp_file_location_db[dataset_name]
try: fl.setAPTLocation(apt_location)
except Exception: pass
if run_from_scratch == 'Process CEL files' or 'Feature Extraction' in run_from_scratch:
fl.setInputCDFFile(input_cdf_file); fl.setCLFFile(clf_file); fl.setBGPFile(bgp_file); fl.setXHybRemoval(remove_xhyb)
fl.setCELFileDir(cel_file_dir); fl.setArrayType(array_type); fl.setOutputDir(output_dir)
fl.setChannelToExtract(channel_to_extract)
elif 'Chromium' in run_from_scratch:
fl.setChromiumSparseMatrix(sparse_matrix_file)
#print fl.ChromiumSparseMatrix()
elif run_from_scratch == 'Process RNA-seq reads':
fl.setCELFileDir(cel_file_dir); fl.setOutputDir(output_dir); fl.setExonBedBuildStatus(build_exon_bedfile)
fl.setRunKallisto(input_fastq_dir);
if array_type != 'gene' and array_type != "3'array":
compendiumPlatform = 'exon'
fl = exp_file_location_db[dataset]; fl.setRootDir(parent_dir)
fl.setFeatureNormalization(normalize_feature_exp)
fl.setNormMatrix(normalize_gene_data)
fl.setProbabilityStatistic(probability_algorithm)
fl.setBatchEffectRemoval(batch_effects)
fl.setMarkerFinder(marker_finder)
fl.setProducePlots(visualize_results)
fl.setPerformLineageProfiler(run_lineage_profiler)
fl.setCompendiumType(compendiumType)
fl.setCompendiumPlatform(compendiumPlatform)
fl.setVendor(vendor)
try: fl.setCountsNormalization(expression_data_format)
except: pass
try: fl.setFDRStatistic(FDR_statistic)
except Exception: pass
try: fl.setExcludeLowExpressionExons(excludeNonExpExons)
except Exception: fl.setExcludeLowExpressionExons(True)
try: fl.setPredictGroups(predictGroups)
except Exception: fl.setPredictGroups(False)
try: fl.setPredictGroupsParams(gsp)
except Exception: pass
fl.setMultiThreading(multiThreading)
if run_from_scratch == 'Process Expression file':
fl.setRootDir(output_dir) ### When the data is not primary array data files, allow for option selection of the output directory
fl.setOutputDir(output_dir)
try: fl.setRPKMThreshold(rpkm_threshold)
except Exception: pass
try: fl.setGeneExpThreshold(gene_exp_threshold)
except Exception: pass
if array_type == 'RNASeq': ### Post version 2.0, add variables in fl rather than below
fl.setRPKMThreshold(rpkm_threshold)
fl.setExonExpThreshold(exon_exp_threshold)
fl.setGeneExpThreshold(gene_exp_threshold)
fl.setExonRPKMThreshold(exon_rpkm_threshold)
fl.setJunctionExpThreshold(expression_threshold)
try: fl.setMLP(mlp)
except Exception: pass
if predictGroups:
### Single-Cell Analysis Parameters
try: option_db,option_list=original_option_db,original_option_list ### was re-set above... needed to get the propper data from the last loop
except Exception: option_list,option_db = importUserOptions(array_type,vendor=vendor)
selected_parameters.append('PredictGroups')
supported_geneset_types = getSupportedGeneSetTypes(species,'gene-mapp')
supported_geneset_types += getSupportedGeneSetTypes(species,'gene-go')
option_db['GeneSetSelectionPredict'].setArrayOptions(['None Selected']+supported_geneset_types)
option_db['PathwaySelectionPredict'].setArrayOptions(['None Selected'])
#option_db['PathwaySelection'].setArrayOptions(supported_genesets)
status = 'repeat'
while status == 'repeat':
root = Tk()
root.title('AltAnalyze: Predict Cell Populations')
### Run in GUI and wait to be executed
gu = GUI(root,option_db,option_list['PredictGroups'],'')
### Permission to run full analsyis is granted, proceed
gsp = gu.Results()['gsp']
status = 'continue'
import RNASeq
expFile = fl.ExpFile()
mlp_instance = fl.MLP()
global logfile
root_dir = export.findParentDir(expFile)
root_dir = string.replace(root_dir,'/ExpressionInput','')
time_stamp = AltAnalyze.timestamp()
logfile = filepath(root_dir+'AltAnalyze_report-'+time_stamp+'.log')
count = verifyFileLength(expFile[:-4]+'-steady-state.txt')
if count>1:
expFile = expFile[:-4]+'-steady-state.txt'
elif array_type=='RNASeq' or len(input_fastq_dir)>0 or len(sparse_matrix_file)>0:
### Indicates that the steady-state file doesn't exist. The exp. may exist, be could be junction only so need to re-build from bed files here
values = species,exp_file_location_db,dataset,mlp_instance
StatusWindow(values,'preProcessRNASeq') ### proceed to run the full discovery analysis here!!!
if array_type=='RNASeq':
expFile = expFile[:-4]+'-steady-state.txt'
"""
else:
print_out = 'WARNING... Prior to running ICGS, you must first run AltAnalyze\nusing assigned groups for this array type.'
IndicatorWindow(print_out,'Continue')
AltAnalyze.AltAnalyzeSetup((selected_parameters[:-1],user_variables)); sys.exit()"""
values = expFile, mlp_instance, gsp, False
StatusWindow(values,'predictGroups') ### proceed to run the full discovery analysis here!!!
if len(graphic_links)>0:
root = Tk()
root.title('AltAnalyze: Evaluate ICGS Clustering Results')
### Review results in custom GUI for predicting groups
gu = GUI(root,'PredictGroups',[],'')
nextStep = gu.Results()['next']
group_selected = gu.Results()['group_select']
if nextStep == 'UseSelected':
group_selected = group_selected[:-4]+'.txt'
exp_file = fl.ExpFile()
try:
exonExpFile,newExpFile,new_groups_dir = exportAdditionalICGSOutputs(expFile,group_selected,outputTSNE=False)
for exp_name in exp_file_location_db: break ### get name
fl.setExpFile(exonExpFile) ### Use the ICGS re-ordered and possibly OutlierFiltered for downstream analyses
comps_file = string.replace(newExpFile,'exp.','comps.')
fl.setGroupsFile(new_groups_dir)
fl.setCompsFile(string.replace(new_groups_dir,'groups.','comps.'))
del exp_file_location_db[exp_name]
exp_file_location_db[exp_name+'-ICGS'] = fl
except Exception:
print traceback.format_exc()
pass ### Unknown error
run_from_scratch = 'Process Expression file'
else:
#print 're-initializing window'
AltAnalyze.AltAnalyzeSetup((selected_parameters,user_variables)); sys.exit()
else:
AltAnalyze.AltAnalyzeSetup((selected_parameters,user_variables)); sys.exit()
expr_var = species,array_type,vendor,constitutive_source,dabg_p,expression_threshold,avg_all_for_ss,expression_data_format,include_raw_data, run_from_scratch, perform_alt_analysis
alt_var = analysis_method,p_threshold,filter_probeset_types,alt_exon_fold_cutoff,gene_expression_cutoff,remove_intronic_junctions,permute_p_threshold, perform_permutation_analysis, export_splice_index_values, analyze_all_conditions
additional_var = calculate_splicing_index_p, run_MiDAS, analyze_functional_attributes, microRNA_prediction_method, filter_for_AS, additional_algorithms
goelite_var = ge_fold_cutoffs,ge_pvalue_cutoffs,ge_ptype,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,pathway_permutations,mod,returnPathways
return expr_var, alt_var, additional_var, goelite_var, exp_file_location_db
def getAPTLocations(file_location_defaults,run_from_scratch,run_MiDAS):
from import_scripts import ResultsExport_module
if 'APT' in file_location_defaults:
fl = file_location_defaults['APT']
apt_location = fl.Location() ###Only one entry for all species
if len(apt_location)<1: ###If no APT version is designated, prompt the user to find the directory
if run_from_scratch == 'CEL_summarize':
print_out = 'To proceed with probeset summarization from CEL files,\nyou must select a valid Affymetrix Power Tools Directory.'
elif run_MiDAS == 'yes':
print_out = "To proceed with running MiDAS, you must select\na valid Affymetrix Power Tools Directory."
win_info = IndicatorChooseWindow(print_out,'Continue') ### Prompt the user to locate the APT directory
apt_location = win_info.Folder()
fl.SetLocation(apt_location)
try: exportDefaultFileLocations(file_location_defaults)
except Exception: pass
return apt_location
def check_moderated_support(option_db):
""" Excludes moderated t-test support when module import fails... shouldn't fail """
try:
from stats_scripts import mpmath
except Exception,e:
a = traceback.format_exc()
GUIcriticalError(a)
keep=[]
od = option_db['probability_algorithm']
for i in od.ArrayOptions():
if 'oderate' not in i: keep.append(i)
od.setArrayOptions(keep) ### remove any moderated stats
od.setDefaultOption(keep[0]) ### Change the default value to one of those in keep
return option_db
def GUIcriticalError(log_report):
log_file = filepath('GUIerror.log')
data = open(log_file,'w')
data.write(log_report); data.close()
"""
if os.name == 'nt':
try: os.startfile('"'+log_file+'"')
except Exception: os.system('open "'+log_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+log_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+log_file+'/"')
"""
class GeneSelectionParameters:
### This class specifies parameters for filtering a large dataset for downstream gene or pathway analysis/visualization
def __init__(self, species, platform, vendor):
self._species = species; self._platform = platform; self._vendor = vendor
self._PathwaySelect = False; self._gene = False; self._GeneSet = False
self._Normalize = False
def Species(self): return self._species
def Platform(self): return self._platform
def Vendor(self): return self._vendor
def setGeneSet(self,gene_set): self._GeneSet = gene_set
def GeneSet(self):
if isinstance(self._GeneSet, tuple) or isinstance(self._GeneSet, list):
return tuple(self._GeneSet)
else:
return self._GeneSet
def setPathwaySelect(self,pathway): self._PathwaySelect = pathway
def setJustShowTheseIDs(self, justShowTheseIDs): self.justShowTheseIDs = justShowTheseIDs
def setClusterGOElite(self, clusterGOElite): self.clusterGOElite = clusterGOElite
def setStoreGeneSetName(self, geneSetName): self.geneSetName = geneSetName
def StoreGeneSetName(self): return self.geneSetName
def ClusterGOElite(self):
if isinstance(self.clusterGOElite, tuple) or isinstance(self.clusterGOElite, list):
self.clusterGOElite = list(self.clusterGOElite)
if 'None Selected' in self.clusterGOElite: self.clusterGOElite.remove('None Selected')
return self.clusterGOElite
else:
self.clusterGOElite = string.replace(self.clusterGOElite,'None Selected','')
return [self.clusterGOElite]
def PathwaySelect(self):
if isinstance(self._PathwaySelect, tuple) or isinstance(self._PathwaySelect, list):
return tuple(self._PathwaySelect)
else:
return self._PathwaySelect
def setGeneSelection(self,gene): self._gene = gene
def GeneSelection(self):
try:
genes = self._gene
genes = string.replace(genes,'\r', ' ')
genes = string.replace(genes,'\n', ' ')
except Exception:
genes = self._gene
return genes
def JustShowTheseIDs(self):
if 'None Selected' in self.justShowTheseIDs:
return ''
else:
justShowTheseIDs = string.replace(self.justShowTheseIDs,'\r',' ')
justShowTheseIDs = string.replace(justShowTheseIDs,',',' ')
justShowTheseIDs = string.replace(justShowTheseIDs,' ',' ')
justShowTheseIDs = string.replace(justShowTheseIDs,'\n',' ')
justShowTheseIDs = string.split(justShowTheseIDs,' ')
try: justShowTheseIDs.remove('')
except Exception: pass
return justShowTheseIDs
def GetGeneCorrelations(self):
if len(self._gene)>0: return True
else: return False
def FilterByPathways(self):
if self._GeneSet != 'None Selected': return True
else: return False
def setTranspose(self,transpose): self._transpose = transpose
def Transpose(self):
try: return self._transpose
except Exception: return False
def setOntologyID(self,OntologyID): self._OntologyID = OntologyID
def OntologyID(self):
try:
return self._OntologyID
except Exception: return ''
def setIncludeExpIDs(self,IncludeExpIDs): self._IncludeExpIDs = IncludeExpIDs
def IncludeExpIDs(self): return self._IncludeExpIDs
def setNormalize(self,Normalize):
if Normalize == 'NA': Normalize = False
self._Normalize = Normalize
def Normalize(self): return self._Normalize
def setDownsample(self,downsample): self.downsample = downsample
def setCountsNormalization(self, expression_data_format): self.expression_data_format = expression_data_format
def CountsNormalization(self):
try: return self.expression_data_format
except: return 'scaled'
def setNumGenesExp(self,numGenesExp): self.numGenesExp = numGenesExp
def NumGenesExp(self):
return int(self.numGenesExp)
def setNumVarGenes(self,numVarGenes): self.numVarGenes = numVarGenes
def NumVarGenes(self):
return int(self.numVarGenes)
def DownSample(self):
try:
return int(self.downsample)
except:
return 2500
def setK(self,k): self.k = k
def k(self):
try: return self.k
except: return None
def K(self):
try: return self.k
except: return None
def setExcludeGuides(self,excludeGuides): self.excludeGuides = excludeGuides
def ExcludeGuides(self): return self.excludeGuides
def setSampleDiscoveryParameters(self,ExpressionCutoff,CountsCutoff,FoldDiff,SamplesDiffering,dynamicCorrelation,
removeOutliers,featurestoEvaluate,restrictBy,excludeCellCycle,column_metric,column_method,rho_cutoff):
### For single-cell RNA-Seq data
self.expressionCutoff = ExpressionCutoff
self.countsCutoff = CountsCutoff
self.rho_cutoff = rho_cutoff
self.foldDiff = FoldDiff
self.samplesDiffering = SamplesDiffering
self.featurestoEvaluate = featurestoEvaluate
self.restrictBy = restrictBy
self.excludeCellCycle = excludeCellCycle
self.column_metric = column_metric
self.column_method = column_method
self.removeOutliers = removeOutliers
self.dynamicCorrelation = dynamicCorrelation
if len(self._gene)>0:
self._gene = self._gene + ' amplify' ### always amplify the selected genes if any
def setExpressionCutoff(self,expressionCutoff):self.expressionCutoff = expressionCutoff
def setCountsCutoff(self,countsCutoff):self.countsCutoff = countsCutoff
def ExpressionCutoff(self):
try: return float(self.expressionCutoff)
except Exception: return False
def setRhoCutoff(self,rho):
self.rho_cutoff = rho
def RhoCutoff(self):
return float(self.rho_cutoff)
def CountsCutoff(self):
try: return int(float(self.countsCutoff))
except Exception: return False
def FoldDiff(self):
try: return float(self.foldDiff)
except Exception: return False
def SamplesDiffering(self):
try: return int(float(self.samplesDiffering))
except Exception: return False
def dynamicCorrelation(self):
if self.dynamicCorrelation=='yes' or self.dynamicCorrelation==True:
return True
else:
return False
def amplifyGenes(self):
if (self.FilterByPathways() != '' and self.FilterByPathways() !=False) or (self.GeneSelection() != '' and self.GeneSelection() != ' amplify'):
return True
else: return False
def FeaturestoEvaluate(self): return self.featurestoEvaluate
def RestrictBy(self):
if self.restrictBy == True or self.restrictBy == 'yes' or self.restrictBy == 'protein_coding':
return 'protein_coding'
else:
return None
def RemoveOutliers(self):
if self.removeOutliers == True or self.removeOutliers == 'yes':
return True
else:
return False
def ExcludeCellCycle(self):
if self.excludeCellCycle == 'stringent' or self.excludeCellCycle == 'strict':
return 'strict' ### Also includes removing drivers correlated to any cell cycle genes, not just in the training set
elif self.excludeCellCycle == False:
return False
elif self.excludeCellCycle == True or self.excludeCellCycle != 'no':
return True
else:
return False
def ColumnMetric(self): return self.column_metric
def ColumnMethod(self): return self.column_method
def MinEvents(self):
return self.SamplesDiffering()-1
def MedEvents(self):
return (self.SamplesDiffering()-1)*2
def getSupportedGeneSetTypes(species,directory):
try:
geneset_types=[]
current_geneset_dirs = unique.read_directory('/AltDatabase/goelite/'+species+'/'+directory)
for geneset_dir in current_geneset_dirs:
geneset_dir = string.join(string.split(geneset_dir,'-')[1:],'-')[:-4] ### remove the prefix gene system
if geneset_dir == 'MAPP': geneset_dir = 'WikiPathways'
if geneset_dir not in geneset_types:
if len(geneset_dir)>1:
geneset_types.append(geneset_dir)
except Exception:
return []
return geneset_types
def getSupportedGeneSystems(species,directory):
system_names=[]
current_system_dirs = unique.read_directory('/AltDatabase/goelite/'+species+'/'+directory)
for system_dir in current_system_dirs:
try:
system_dir = string.split(system_dir,'-')[1][:-4] ### remove the prefix gene system
if len(system_dir)>1:
if system_dir not in system_names:
system_names.append(system_dir)
except Exception: None
system_names.append('Ensembl')
system_names.append('HMDB')
system_names = unique.unique(system_names)
system_names.sort()
return system_names
def listAllGeneSetCategories(species,geneset_type,directory):
geneset_categories=[]
if directory == 'gene-go':
if geneset_type == 'GeneOntology': geneset_type = 'go'
filename = 'AltDatabase/goelite/OBO/builds/'+geneset_type+'_annotations.txt'
index = 1
else:
if geneset_type == 'WikiPathways': geneset_type = 'MAPP'
filename = 'AltDatabase/goelite/'+species+'/'+directory+'/'+'Ensembl-'+geneset_type+'.txt'
index = -1
fn=filepath(filename)
### Imports a geneset category and stores pathway-level names
i=0
for line in open(fn,'rU').xreadlines():
if i==0: i=1 ### Skip the header
else:
data = cleanUpLine(line)
geneset_category = string.split(data,'\t')[index]
if geneset_category not in geneset_categories:
geneset_categories.append(geneset_category)
geneset_categories.sort()
return geneset_categories
def getValidExpFile(altanalyze_rawexp_dir):
dir_files = read_directory(altanalyze_rawexp_dir)
valid_file = ''
for file in dir_files:
if 'exp.' in file and 'state.txt' not in file and 'highExp' not in file:
valid_file = altanalyze_rawexp_dir+'/'+file
break
return valid_file
def getValidSplicingScoreFile(altanalyze_rawsplice_dir):
valid_dirs = ['splicing-index','FIRMA','ASPIRE','linearregres']
dir_files = read_directory(altanalyze_rawsplice_dir)
valid_folder = None
for folder in valid_dirs:
if folder in dir_files:
valid_folder = folder
break
valid_file_dir = ''
primary=''
if valid_folder != None:
child_dir = altanalyze_rawsplice_dir+'/'+valid_folder
dir_files = read_directory(altanalyze_rawsplice_dir+'/'+valid_folder)
for file in dir_files:
if '.txt' in file:
valid_file_dir = child_dir+'/'+file
if '_vs_' not in file: ### You can have a folder with pairwise comps and all groups
primary = child_dir+'/'+file
if len(primary)!=0: valid_file_dir = primary
return valid_file_dir
def downloadInteractionDBs(species,windowType):
analysis = 'getAdditionalOnlineResources' ### same option as updating gene-sets
additional_resources=['Latest WikiPathways','KEGG','BioGRID','DrugBank','miRNA Targets','Transcription Factor Targets']
get_additional = 'customSet',additional_resources
values = species,get_additional
StatusWindow(values,analysis,windowType=windowType) ### open in a TopLevel TK window (don't close current option selection menu)
if __name__ == '__main__':
"""
expFile = '/Volumes/salomonis2/NICOLAS-NASSAR-Hs/Run0012-Hs/10X-Vehicle-20181005-3hg/outs/filtered_gene_bc_matrices/ExpressionInput/exp.10X-Vehicle-20181005-3hg_matrix_CPTT.txt'
group_selected = '/Volumes/salomonis2/NICOLAS-NASSAR-Hs/Run0012-Hs/10X-Vehicle-20181005-3hg/outs/filtered_gene_bc_matrices/ICGS-NMF/FinalMarkerHeatmap_all.txt'
array_type="3'array"; species='Hs'
exportAdditionalICGSOutputs(expFile,group_selected,outputTSNE=True)
sys.exit()"""
#a = exportJunctionList(dir,limit=50)
#print a;sys.exit()
try:
import multiprocessing as mlp
mlp.freeze_support()
except Exception:
print 'Note: Multiprocessing not supported for this verison python.'
mlp=None
#getUpdatedParameters(array_type,species,run_from_scratch,file_dirs)
a = getUserParameters('yes')
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/UI.py
|
UI.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys, string
import os.path
import unique
import export
import ExpressionBuilder
import copy
import traceback
try:
import warnings
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
from stats_scripts import statistics
import math
from scipy import stats
use_scipy = True
except Exception:
use_scipy = False ### scipy is not required but is used as a faster implementation of Fisher Exact Test when present
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt"or entry[-4:] == ".tab" or entry[-4:] == ".csv" or '.fa' in entry: dir_list2.append(entry)
return dir_list2
class GrabFiles:
def setdirectory(self,value):
self.data = value
def display(self):
print self.data
def searchdirectory(self,search_term):
#self is an instance while self.data is the value of the instance
file_dirs = getDirectoryFiles(self.data,str(search_term))
if len(file_dirs)<1: print search_term,'not found',self.data
return file_dirs
def getDirectoryFiles(import_dir, search_term):
exact_file = ''; exact_file_dirs=[]
dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
for data in dir_list: #loop through each file in the directory to output results
affy_data_dir = import_dir[1:]+'/'+data
if search_term in affy_data_dir: exact_file_dirs.append(affy_data_dir)
return exact_file_dirs
########## End generic file import ##########
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def createCorrelationTemplate(tissues):
i=0; tissue_template_db={}
null_template = [0]*len(tissues)
for tissue in tissues:
tissue_template = list(null_template) ### creates a modifiable copy
tissue_template[i] = correlationDirection ### make -1 reverse profile (anti-correlated)
#if 'Atrium' in tissue:
tissue_template_db[tissue] = tissue_template
i+=1
return tissue_template_db
def findHousekeepingGenes(uid,data_list1):
### Pearson won't work for no variance, so have to use an alternative approach
stdev = statistics.stdev(data_list1)
housekeeping.append([stdev,uid]) #stdev/min(data_list1)
def expressedIndexes(values):
filtered_indexes=[]
i=0
for value in values:
if value!=None:
filtered_indexes.append(i)
i+=1
return filtered_indexes
def advancedPearsonCorrelationAnalysis(uid,data_list1,tissue_template_db):
expIndexes = expressedIndexes(data_list1)
Queried[uid]=[]
if (float(len(expIndexes))/len(data_list1))>0.0: ### Atleast 50% of samples evaluated express the gene
data_list = map(lambda i: data_list1[i],expIndexes) ### Only expressed values (non-None)
max_diff = max(data_list)-statistics.avg(data_list)
if max_diff>-1000 and max(data_list)>-1000:
if correlateAllGenes:
min_rho = -1
min_p = 1
else:
min_rho = 0.3
min_p = 0.05
for tissue in tissue_template_db:
tissue_template = tissue_template_db[tissue]
c1 = tissue_template.count(1)
filtered_template = map(lambda i: tissue_template[i],expIndexes)
c2 = filtered_template.count(1)
if len(data_list)!= len(filtered_template): kill
if c1 == c2 or c1 != c2: ### If number of 1's in list1 matches list2
rho,p = rhoCalculation(data_list,filtered_template)
if tissue == 'Housekeeping':
print tissue, p, uid;sys.exit()
#if rho>min_rho:
if p<min_p and rho>min_rho:
Added[uid]=[]
try: tissue_scores[tissue].append([(rho,p),uid])
except Exception: tissue_scores[tissue] = [[(rho,p),uid]]
def PearsonCorrelationAnalysis(uid,data_list1,tissue_template_db):
if correlateAllGenes:
min_rho = -2
min_p = 1
else:
min_rho = 0.3
min_p = 0.05
for tissue in tissue_template_db:
tissue_template = tissue_template_db[tissue]
rho,p = rhoCalculation(data_list1,tissue_template)
#print rho, uid, tissue
if tissue == 'Housekeeping':
print tissue, rho, uid;sys.exit()
#if rho>min_rho:
if p<min_p and rho>min_rho:
try: tissue_scores[tissue].append([(rho,p),uid])
except Exception: tissue_scores[tissue] = [[(rho,p),uid]]
def rhoCalculation(data_list1,tissue_template):
try:
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
try:
rho,p = stats.pearsonr(data_list1,tissue_template)
return rho,p
except Exception:
#data_list_alt = [0 if x==None else x for x in data_list1]
#rho,p = stats.pearsonr(data_list1,tissue_template)
kill
except Exception:
rho = pearson(data_list1,tissue_template)
return rho, 'Null'
def simpleScipyPearson(query_lists,reference_list):
""" Get the top correlated values referenced by index of the query_lists (e.g., data matrix) """
i=0
rho_results=[]
for query_list in query_lists:
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
rho,p = stats.pearsonr(query_list,reference_list)
#if query_list == reference_list: print query_list,reference_list, rho;sys.exit()
if str(rho)!='nan':
rho_results.append([(float(rho),float(p)),i])
i+=1
rho_results.sort()
rho_results.reverse()
return rho_results
def pearson(array1,array2):
item = 0; sum_a = 0; sum_b = 0; sum_c = 0
while item < len(array1):
a = (array1[item] - avg(array1))*(array2[item] - avg(array2))
b = math.pow((array1[item] - avg(array1)),2)
c = math.pow((array2[item] - avg(array2)),2)
sum_a = sum_a + a
sum_b = sum_b + b
sum_c = sum_c + c
item = item + 1
try: r = sum_a/math.sqrt(sum_b*sum_c)
except Exception: r =0
return r
def avg(array):
return sum(array)/len(array)
def getArrayData(filename,filtered_probeset):
fn=filepath(filename); x=0; k=0; expression_data={}; annotations={}; expression_data_str={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line) #remove endline
t = string.split(data,'\t')
if x == 0:
i=0
for h in t:
if 'Definition' in h: lti = i ### last tissue index
if 'Description' in h: lti = i ### last tissue index
i+=1
x=1
try:
header = t[1:lti]
annotation_header = t[lti:]
except Exception:
lti = len(t)
header=[]
annotation_header=[]
else:
probeset = t[0]
try:
if len(filtered_probeset)>0: ### Otherwise, just get all annotations
null = filtered_probeset[probeset]
exp_values = map(float, t[1:lti])
if log_transform:
try: exp_values = map(lambda x: math.log(x,2), exp_values)
except Exception:
exp_values = map(lambda x: math.log(x+1,2), exp_values)
expression_data_str[probeset] = map(str,exp_values)
expression_data[probeset] = exp_values
try: annotations[probeset] = t[lti:]
except KeyError:
annotations[probeset] = []
except KeyError:
null=[]
sum_tissue_exp = {}
for probeset in expression_data:
i=0
for fold in expression_data[probeset]:
try: sum_tissue_exp[i].append(fold)
except Exception: sum_tissue_exp[i] = [fold]
i+=1
expression_relative={}
for probeset in expression_data:
i=0
for fold in expression_data[probeset]:
ratio = str(fold/max(sum_tissue_exp[i]))
try: expression_relative[probeset].append(ratio)
except Exception: expression_relative[probeset] = [ratio]
i+=1
return expression_data_str, annotations, header, annotation_header
def importMarkerProfiles(filename,fl):
x=0
### Import correlated marker results
fn=filepath(filename)
marker_list = []
condition_list=[]
marker_db={}
probeset_symbol_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
x=1
else:
uid,symbol,rho,p,condition=t
probeset_symbol_db[uid]=symbol
try: marker_db[condition].append(uid)
except Exception: marker_db[condition] = [uid]
if condition not in condition_list:
condition_list.append(condition)
marker_condition_db={}
try: condition_list = getOrderedGroups(fl.DatasetFile()) ### Order this set with the same way as the samples on the opposite axis
except Exception,e:
#print 'failed',e
condition_list.sort()
condition_list.reverse() ### This makes this ordered top-down for clustering
for condition in condition_list:
if condition in marker_db:
for uid in marker_db[condition]:
if uid not in marker_list:
marker_list.append(uid) ### ranked and unique marker list
marker_condition_db[uid] = condition
exportMarkersForGOElite(filename,marker_db,fl) ### Export these lists for GO-Elite
return marker_list, probeset_symbol_db, marker_condition_db
def exportMarkersForGOElite(filename,gene_db,fl):
if fl.Vendor() == 'Affymetrix': system = 'X'
elif fl.Vendor() == 'Agilent': system = 'Ag'
elif fl.Vendor() == 'Illumina': system = 'Il'
elif 'other:' in fl.Vendor():
system = string.replace(fl.Vendor(),'other:','')
if system == 'Symbol': system = 'Sy'
else: system = 'Ma'
else: system = 'Sy'
root_dir = fl.OutputDir()
if 'ReplicateBased' in filename: suffix = '-ReplicateBased'
if 'MeanBased' in filename: suffix = '-MeanBased'
for markerSet in gene_db:
header = string.join(['Gene','System','Hit'],'\t')+'\n'
filename = root_dir+'GO-Elite/MarkerFinder/'+markerSet+suffix+'.txt'
export_obj = export.ExportFile(filename)
export_obj.write(header)
for gene in gene_db[markerSet]:
if 'ENS' in gene:
system = 'En'
try: system = system
except Exception: system = 'Swiss'
values = string.join([gene,system,'1'],'\t')+'\n'
export_obj.write(values)
export_obj.close()
def reorderInputFile(custom_path,marker_list,marker_condition_db):
x=0
### Import correlated marker results
fn=filepath(custom_path)
exp_db={}
probeset_symbol_db={}
#print custom_path;sys.exit()
#print fn
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
header = line
x=1
else:
uid = string.join(string.split(t[0],' ')[0:-1],' ') ### IDs can have spaces
if '///' in uid:
uid = string.split(uid,' ')[0] ### Affy ID with multiple annotations
exp_db[uid] = line
### Over-write read in file
export_obj = export.ExportFile(custom_path)
export_obj.write(header)
marker_list.reverse() ### Reverse the order of the MarkerFinder results
for uid in marker_list:
condition = marker_condition_db[uid]
new_uid = condition+':'+uid
if uid in exp_db:
export_obj.write(condition+':'+exp_db[uid])
elif new_uid in exp_db:
export_obj.write(exp_db[new_uid])
else:
"""
print [uid], len(exp_db)
for i in exp_db:
print [i];break
print 'Error encountered with the ID:',uid, 'not in exp_db'; kill
"""
pass
export_obj.close()
def getOrderedGroups(filename):
group_list=[]
filename = string.replace(filename,'///','/')
filename = string.replace(filename,'//','/')
if 'ExpressionOutput' in filename:
filename = string.replace(filename,'ExpressionOutput','ExpressionInput')
filename = string.replace(filename,'-steady-state','')
filename = string.replace(filename,'DATASET-','exp.')
groups_dir = string.replace(filename,'exp.','groups.')
group_names_db = ExpressionBuilder.simpleGroupImport(groups_dir)[3]
for i in group_names_db: group_list.append(i)
group_list.reverse()
return group_list
def generateMarkerHeatMaps(fl,platform,convertNonLogToLog=False,graphics=[],Species=None):
from visualization_scripts import clustering
""" From the generated marker sets, output the replicate input data """
marker_root_dir = fl.OutputDir()+'/'+'ExpressionOutput/MarkerFinder'
#print 1,fl.DatasetFile()
#print 2, fl.Vendor()
for marker_dir in read_directory(marker_root_dir):
if 'MarkerGenes' in marker_dir and 'correlation' in marker_dir:
marker_dir = marker_root_dir+'/'+marker_dir
marker_list, probeset_symbol_db, marker_condition_db = importMarkerProfiles(marker_dir,fl)
custom_path = string.replace(marker_dir,'MarkerGenes','Clustering/MarkerGenes')
"""
print fl.DatasetFile()
print len(marker_list), marker_list[:3]
print len(probeset_symbol_db)
print custom_path
print convertNonLogToLog
print Species
print platform
print len(probeset_symbol_db)
print custom_path
print fl.Vendor()
print convertNonLogToLog
"""
ExpressionBuilder.exportGeometricFolds(fl.DatasetFile(),platform,marker_list,probeset_symbol_db,exportOutliers=False,exportRelative=False,customPath=custom_path,convertNonLogToLog=convertNonLogToLog)
reorderInputFile(custom_path,marker_list, marker_condition_db)
row_method = None; row_metric = 'cosine'; column_method = None; column_metric = 'euclidean'; color_gradient = 'yellow_black_blue'; transpose = False
import UI
gsp = UI.GeneSelectionParameters(Species,platform,fl.Vendor())
gsp.setPathwaySelect('None Selected')
gsp.setGeneSelection('')
gsp.setOntologyID('')
gsp.setGeneSet('None Selected')
gsp.setJustShowTheseIDs('')
gsp.setTranspose(False)
gsp.setNormalize('median')
gsp.setGeneSelection('')
gsp.setClusterGOElite('GeneOntology')
#gsp.setClusterGOElite('BioMarkers')
"""
print custom_path
print graphics
print row_method
print column_method
print column_metric
"""
reload(clustering)
try:
graphics = clustering.runHCexplicit(custom_path, graphics, row_method, row_metric,
column_method, column_metric, color_gradient, gsp, contrast=4, display=False)
except Exception:
print traceback.format_exc()
print 'Error occured in generated MarkerGene clusters... see ExpressionOutput/MarkerFinder files.'
return graphics
def reorderMultiLevelExpressionFile(input_file):
### Takes an input file and re-orders it based on the order in the groups file... needed for multi-level expression file with replicates
from import_scripts import sampleIndexSelection
output_file = input_file[:-4]+'-output.txt'
filter_file = string.replace(input_file,'-steady-state','')
filter_file = string.replace(filter_file,'exp.','groups.')
filter_file = string.replace(filter_file,'stats.','groups.')
filter_file = string.replace(filter_file,'topSplice.','groups.')
filter_file = string.replace(filter_file,'filter.','groups.')
filter_names = sampleIndexSelection.getFilters(filter_file)
sampleIndexSelection.filterFile(input_file,output_file,filter_names)
c1 = verifyFileLength(input_file)
c2 = verifyFileLength(output_file)
if c1==c2:
os.remove(input_file)
export.copyFile(output_file, input_file)
def verifyFileLength(filename):
count = 0
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
if line[0]!='#':
count+=1
except Exception: null=[]
return count
def analyzeData(filename,Species,Platform,codingType,geneToReport=60,correlateAll=True,AdditionalParameters=None,logTransform=False,binarize=False):
global genesToReport; genesToReport = geneToReport
global correlateAllGenes; correlateAllGenes = correlateAll
global all_genes_ranked; all_genes_ranked={}
global RPKM_threshold; global correlationDirection
global Added; Added={}; global Queried; Queried={}
"""
print 4,Platform, codingType, geneToReport, correlateAll, logTransform,
try:
#print AdditionalParameters.CorrelationDirection()
print AdditionalParameters.RPKMThreshold()
except Exception:
print 'nope'
"""
global AvgExpDir
if len(filename) == 2:
filename, AvgExpDir = filename #### Used when there are replicate samples: avg_exp_dir is non-replicate
if AvgExpDir==None:
AvgExpDir = string.replace(filename,'-steady-state','')
AvgExpDir = string.replace(AvgExpDir,'exp.','AVERAGE-')
AvgExpDir = string.replace(AvgExpDir,'ExpressionInput','ExpressionOutput')
if 'ExpressionOutput' in filename:
use_replicates = False
else:
use_replicates = True
import RNASeq
try: Platform = RNASeq.checkExpressionFileFormat(filename,Platform)
except Exception: Platform = "3'array"
try: RPKM_threshold = AdditionalParameters.RPKMThreshold() ### Used for exclusion of non-expressed genes
except Exception:
pass
if Platform == 'RNASeq':
try: RPKM_threshold = AdditionalParameters.RPKMThreshold() ### Used for exclusion of non-expressed genes
except Exception: RPKM_threshold = 1; logTransform = True
correlationDirection = 1.00 ### Correlate to a positive or negative idealized pattern
try:
if AdditionalParameters.CorrelationDirection() != 'up' and AdditionalParameters.CorrelationDirection() != 'positive':
correlationDirection = -1.00
except Exception: pass
#print correlationDirection
fn=filepath(filename); x=0; t2=['ID']; cluster_db={}; cluster_list = []; global coding_type; coding_type = codingType
global cluster_comps; cluster_comps = []; global compare_clusters; compare_clusters = 'no'
global housekeeping; housekeeping=[]; global analyze_housekeeping; analyze_housekeeping = 'no'
global species; global platform; species = Species; platform = Platform; global log_transform
log_transform=logTransform
#if 'topSplice.' not in fn and 'steady' not in fn and 'AVERAGE' not in fn and 'DATASET' not in fn: reorderMultiLevelExpressionFile(fn)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x == 0:
updated_names = ['ID']
correlations = 'single'
tissue_template_db,group_sample_db = getReplicateData(fn,t[1:])
if '~' in data: correlations = 'multiple'
elif t[1] in group_sample_db:
if '~' in group_sample_db[t[1]]:
correlations = 'multiple'
for i in t[1:]:
updated_names.append(group_sample_db[i])
t = updated_names
i=0
for h in t:
if correlations == 'multiple':
if '~' in h:
cluster, group_name = string.split(h,'~')
cluster = int(cluster)
try: cluster_db[cluster].append(i)
except Exception: cluster_db[cluster] = [i]
i+=1
if correlations == 'multiple':
compare_clusters = 'yes'
### If there are multiple sample group clusters
for cluster in cluster_db: cluster_list.append(cluster)
cluster_list.sort()
if cluster_list[0]==0: ### All clusters should then be compared to this first cluster if the first is named 0
cluster_comp_db={}; cluster_comp_db[0] = cluster_db[0]
cluster_comps.append(cluster_comp_db) ### Add the first by itself (cluster 0 samples only comapred among themselves initially)
for cluster in cluster_list[1:]:
cluster_comp_db={}; cluster_comp_db[0] = cluster_db[0]
cluster_comp_db[cluster] = cluster_db[cluster]
cluster_comps.append(cluster_comp_db) ### Each non-zero cluster compared to cluster 0 as pairwise group combinations
else:
for cluster in cluster_list:
cluster_comp_db={}
cluster_comp_db[cluster] = cluster_db[cluster]
cluster_comps.append(cluster_comp_db)
x = 1
break
iteration=1
if compare_clusters == 'yes':
tissue_specific_IDs_combined={}; correlations_combined={}
for cluster_comp_db in cluster_comps:
###Interate through each comparison
print 'Iteration',iteration,'of',len(cluster_comps)
tissue_specific_IDs,interim_correlations,annotation_headers,tissues = identifyMarkers(filename,cluster_comp_db,binarize=binarize)
iteration+=1
for tissue in tissue_specific_IDs:
if tissue not in tissue_specific_IDs_combined: ### Combine the tissue results from all of the cluster group analyses, not over-writing the existing
tissue_specific_IDs_combined[tissue] = tissue_specific_IDs[tissue]
correlations_combined[tissue] = interim_correlations[tissue]
tissue_specific_IDs={}; interim_correlations={}
for tissue in tissue_specific_IDs_combined:
for probeset in tissue_specific_IDs_combined[tissue]:
try: tissue_specific_IDs[probeset].append(tissue)
except Exception: tissue_specific_IDs[probeset] = [tissue]
for (probeset,symbol,(rho,p)) in correlations_combined[tissue]:
try: interim_correlations[tissue].append([probeset,symbol,(rho,p)])
except Exception: interim_correlations[tissue] = [[probeset,symbol,(rho,p)]]
analyze_housekeeping = 'yes'; compare_clusters = 'no'
original_tissue_headers2 = original_tissue_headers ### The last function will overwrite the group~ replacement
#identifyMarkers(filename,[]) ### Used to get housekeeping genes for all conditions
else:
tissue_specific_IDs,interim_correlations,annotation_headers,tissues = identifyMarkers(filename,[],binarize=binarize)
original_tissue_headers2 = original_tissue_headers
### Add a housekeeping set (genes that demonstrate expression with low variance
housekeeping.sort(); ranked_list=[]; ranked_lookup=[]; tissue = 'Housekeeping'
for (stdev,(probeset,symbol)) in housekeeping:
if probeset not in tissue_specific_IDs: ### Shouldn't be if it is a housekeeping gene
if symbol not in ranked_list:
ranked_list.append(symbol); ranked_lookup.append([probeset,symbol,(stdev,0)])
### Replicates code in identifyMarkers - but only applied to housekeeping genes to add those in addition to the existing ones in tissue_specific_IDs
for (probeset,symbol,(stdev,p)) in ranked_lookup[:genesToReport]:
try: tissue_specific_IDs[probeset].append(tissue)
except Exception: tissue_specific_IDs[probeset] = [tissue]
try: interim_correlations[tissue].append([probeset,symbol,(stdev,p)])
except Exception: interim_correlations[tissue] = [[probeset,symbol,(stdev,p)]]
### If no mean file provided
#print [use_replicates, filename, tissue]
if use_replicates:
try: filename = AvgExpDir
except Exception: pass ### For AltExon queries
try:
expression_relative,annotations,tissue_headers, annot_header = getArrayData(filename,tissue_specific_IDs)
if use_replicates:
original_tissue_headers2, annotation_headers = tissue_headers, annot_header
tissue_specific_IDs2 = copy.deepcopy(tissue_specific_IDs)
for probeset in tissue_specific_IDs2:
if probeset in annotations:
annotations[probeset]+=[string.join(list(tissue_specific_IDs[probeset]),'|')] ### Save as string
title_row = ['UID']+annotation_headers+['marker-in']+original_tissue_headers2
export_dir = exportMarkerGeneProfiles(filename,annotations,expression_relative,title_row)
except Exception,e:
#print traceback.format_exc()
pass
exportCorrelations(filename,interim_correlations)
if correlateAllGenes:
exportAllGeneCorrelations(filename,all_genes_ranked)
try: return export_dir
except Exception: pass
def getReplicateData(expr_input,t):
groups_dir = string.replace(expr_input,'exp.','groups.')
groups_dir = string.replace(groups_dir,'stats.','groups.')
groups_dir = string.replace(groups_dir,'topSplice.','groups.')
groups_dir = string.replace(groups_dir,'filter.','groups.')
groups_dir = string.replace(groups_dir,'-steady-state','') ### groups is for the non-steady-state file
#groups_dir = string.replace(groups_dir,'-average.txt','.txt') ### groups is for the non-steady-state file
if 'groups.' not in groups_dir and 'AltResults' in groups_dir:
parent_dir = string.split(expr_input,'AltResults')[0]
file = export.findFilename(expr_input)
file = string.replace(file,'AltExonConfirmed-','groups.')
file = string.replace(file,'AltExon-','groups.')
groups_dir = parent_dir+'ExpressionInput/'+file
group_index_db={}
splitHeaders=False
for i in t:
if '~' not in i:splitHeaders=True
### use comps in the future to visualize group comparison changes
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = ExpressionBuilder.simpleGroupImport(groups_dir,splitHeaders=splitHeaders, ignoreComps=True)
sample_list = t ### This is the actual order in the input expression files
for x in t:
try: group_name = group_db[x]
except Exception:
try:
y = string.split(x,':')[-1] ### for an alternative exon file with the name wt:sample1.bed
group_name = group_db[y]
except Exception: pass
try:
group_name = group_db[x]
sample_index = t.index(x)
try: group_index_db[group_name].append(sample_index)
except Exception: group_index_db[group_name] = [sample_index] ### dictionary of group to input file sample indexes
except Exception: pass
sample_template_db = createReplicateCorrelationTemplate(sample_list,group_index_db)
return sample_template_db,group_sample_db
def createReplicateCorrelationTemplate(samples,group_index_db):
### This create multiple binary indicates, but for replicates as opposed to an individual mean of multiple groups
sample_template_db={}
null_template = [0.00]*len(samples)
for group in group_index_db:
sample_template = list(null_template) ### creates a modifiable copy
group_indeces = group_index_db[group]
for index in group_indeces:
sample_template[index] = correlationDirection ### make -1 to inverse in silico pattern (anti-correlated)
sample_template_db[group] = sample_template
return sample_template_db
def selectiveFloats(values):
float_values=[]
for i in values:
try:
float_values.append(float(i))
except Exception: float_values.append(None)
return float_values
def binaryExp(value):
if value>1:
return 2
else:
return 0
def identifyMarkers(filename,cluster_comps,binarize=False):
""" This function is the real workhorse of markerFinder, which coordinates the correlation analyses and data import """
global tissue_scores; tissue_scores={}; print_interval=2000; print_limit=2000
fn=filepath(filename); x=0; k=0; probeset_db={}; tissues_with_lowest_expression={}; index_sets = []
global original_tissue_headers
global use_replicates
count=0
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
try: coding_db = ExpressionBuilder.importTranscriptBiotypeAnnotations(species)
except Exception: coding_db = {}
if 'ExpressionOutput' in filename:
use_replicates = False
else:
use_replicates = True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line) #remove endline
t = string.split(data,'\t')
if data[0] == '#':
x = 0
elif x == 0:
i=0
for h in t:
if 'Definition' in h: lti = i ### last tissue index
if 'Description' in h: lti = i ### last tissue index
if 'Select Protein Classes' in h: ct = i
i+=1
try: original_tissue_headers = t[1:lti]
except Exception:
### Occurs when analyzing a simple expression file with no annotations
original_tissue_headers = t[1:]
if len(cluster_comps) == 0: ### No group clusters to separately analyze present
tissues = list(original_tissue_headers)
else:
if len(cluster_comps)>1: ### 2 groups clusters to compare
indexes = cluster_comps[0]
tissues = t[indexes[0]:indexes[-1]+1]
index_sets = [[indexes[0],indexes[-1]+1]]
for cluster in cluster_comps:
if cluster>0:
indexes = cluster_comps[cluster]
tissues += t[indexes[0]:indexes[-1]+1]
index_sets.append([indexes[0],indexes[-1]+1])
#print tissues
print 'being analyzed now'
else: ### First reference set of tissues looked at
for cluster in cluster_comps: ### There is only one here!
indexes = cluster_comps[cluster]
tissues = t[indexes[0]:indexes[-1]+1]
index_sets = [[indexes[0],indexes[-1]+1]]
#print tissues;sys.exit()
print 'being analyzed only in round 1'
original_tissue_headers2=[]
for tissue in original_tissue_headers: ### This is the original full header for all clusters
try: cluster,tissue = string.split(tissue,'~')
except Exception: pass
original_tissue_headers2.append(tissue)
original_tissue_headers = original_tissue_headers2
try: annotation_headers = t[lti:]
except Exception: annotation_headers = []
if len(cluster_comps) > 0:
tissues2=[]
for tissue in tissues:
if '~' in tissue:
cluster, tissue = string.split(tissue,'~')
tissues2.append(tissue)
tissues = tissues2
#print tissues, cluster_comps;sys.exit()
if use_replicates:
if len(cluster_comps)>0:
tissue_template_db,group_sample_db = getReplicateData(fn,tissues)
else:
tissue_template_db,group_sample_db = getReplicateData(fn,original_tissue_headers)
try: annotation_db = getArrayData(AvgExpDir,[])[1]
except Exception: pass
else:
tissue_template_db = createCorrelationTemplate(tissues)
x = 1
else: #elif x<500:
probeset = t[0]; proceed = 'no'; symbol=''; geneID=''
try:
lti = len(tissues)+1
try: description,symbol=annotation_db[probeset][:2] ### See above annotation_db download
except Exception: symbol = probeset; description = ''
try:
probeset,geneID = string.split(probeset,':')
if 'ENS' in probeset:
geneID, probeset = probeset,geneID
probeset=geneID+':'+probeset
except Exception:
if 'ENS' in probeset:
geneID = probeset
try: symbol = gene_to_symbol[geneID][0]; description = ''
except Exception: pass
except Exception: symbol = probeset; description = ''
try: coding_class = coding_db[probeset][-1]
except Exception:
try:
geneID = symbol_to_gene[probeset][0]
symbol = probeset
coding_class = coding_db[geneID][-1]
except Exception:
coding_class = 'protein_coding'
except Exception: pass
if symbol =='' or symbol == probeset:
try: coding_class = t[ct]; symbol = t[lti+1]; description = t[lti]
except Exception: coding_class = 'protein_coding'
if coding_type == 'protein_coding':
if coding_type in coding_class:
if 'MT-' not in symbol and '.' not in symbol:
proceed = 'yes'
elif coding_type == 'AltExon':
proceed = 'yes'
else:
if 'protein_coding' not in coding_class and 'pseudogene' not in coding_class and len(description)>0:
if 'MT-' not in symbol and '.' not in symbol:
proceed = 'yes'
proceed = 'yes' ### Force it to anlayze all genes
count+=1
#if coding_class != 'protein_coding':
#print coding_class, coding_type, proceed, probeset, symbol, species, len(gene_to_symbol),coding_db[probeset];sys.exit()
#proceed = 'yes'
if len(coding_class) == 0 or proceed == 'yes':
if compare_clusters == 'yes':
exp_values=[]
for (i1,i2) in index_sets:
try: exp_values += map(float, t[i1:i2])
except Exception: exp_values+=selectiveFloats(t[i1:i2])
#print len(exp_values), len(tissues)
#print exp_values
#print tissues; kill
else:
try: exp_values = map(float, t[1:lti]) ### map allows you to apply the function to all elements in the object
except Exception: exp_values=selectiveFloats(t[1:lti])
if log_transform:
try: exp_values = map(lambda x: math.log(x,2), exp_values)
except Exception:
exp_values = map(lambda x: math.log(x+1,2), exp_values)
if binarize:
exp_values = map(lambda x: binaryExp(x), exp_values)
if analyze_housekeeping == 'yes': ### Only grab these when analyzing all tissues
findHousekeepingGenes((probeset,symbol),exp_values)
elif platform == 'RNASeq': ### Exclude low expression (RPKM) genes
if max(exp_values)>RPKM_threshold:
PearsonCorrelationAnalysis((probeset,symbol),exp_values,tissue_template_db)
else:
pass
#print max(exp_values), RPKM_threshold;sys.exit()
else:
if 'exp.' in filename:
try: PearsonCorrelationAnalysis((probeset,symbol),exp_values,tissue_template_db)
except Exception: ### For missing values
advancedPearsonCorrelationAnalysis((probeset,symbol),exp_values,tissue_template_db)
else:
advancedPearsonCorrelationAnalysis((probeset,symbol),exp_values,tissue_template_db)
x+=1
if x == print_limit:
#if print_limit == 2000: break
#print print_limit,'genes analyzed'
print '*',
print_limit+=print_interval
#print len(Added),len(Queried),len(tissue_scores),count;sys.exit()
tissue_specific_IDs={}; interim_correlations={}
gene_specific_rho_values = {}
tissue_list=[]
for tissue in tissue_scores:
tissue_scores[tissue].sort()
tissue_scores[tissue].reverse()
ranked_list=[]; ranked_lookup=[]
if tissue not in tissue_list: tissue_list.append(tissue) ### Keep track of the tissue order
for ((rho,p),(probeset,symbol)) in tissue_scores[tissue]:
if correlateAllGenes:
try: all_genes_ranked[probeset,symbol].append([(rho,p),tissue])
except Exception:all_genes_ranked[probeset,symbol] = [[(rho,p),tissue]]
### Get a matrix of all genes to correlations
try: gene_specific_rho_values[symbol].append(rho)
except Exception: gene_specific_rho_values[symbol] = [rho]
if symbol == '': symbol = probeset
#print tissue, tissue_scores[tissue];sys.exit()
if symbol not in ranked_list:
ranked_list.append(symbol); ranked_lookup.append([probeset,symbol,(rho,p)])
for (probeset,symbol,(rho,p)) in ranked_lookup[:genesToReport]: ### Here is where we would compare rho values between tissues with the same probesets
if rho>0.01 and p<0.1:
if compare_clusters == 'yes':
try: tissue_specific_IDs[tissue].append(probeset)
except Exception: tissue_specific_IDs[tissue] = [probeset]
else:
try: tissue_specific_IDs[probeset].append(tissue)
except Exception: tissue_specific_IDs[probeset] = [tissue]
try: interim_correlations[tissue].append([probeset,symbol,(rho,p)])
except Exception: interim_correlations[tissue] = [[probeset,symbol,(rho,p)]]
if correlateAllGenes: ### This was commented out - Not sure why - get an error downstream otherwise
for ID in all_genes_ranked:
ag = all_genes_ranked[ID]
ag.sort()
all_genes_ranked[ID] = ag[-1] ### topcorrelated
#"""
data = export.ExportFile(string.replace(filename[:-4]+'-all-correlations.txt','exp.','MarkerFinder.'))
data.write(string.join(tissue_list,'\t')+'\n')
for gene in gene_specific_rho_values:
data.write(string.join([gene]+map(str,gene_specific_rho_values[gene]),'\t')+'\n')
#sys.exit()
#"""
#print len(tissue_specific_IDs);sys.exit()
return tissue_specific_IDs,interim_correlations,annotation_headers,tissues
def exportMarkerGeneProfiles(original_filename,annotations,expression_relative,title_row):
destination_dir = 'AltDatabase/ensembl/'+species+'/' ### Original default
destination_dir = export.findParentDir(original_filename)
if 'AltResults' in original_filename: dataset_type = '_AltExon'
elif 'FullDatasets' in original_filename: dataset_type = '_AltExon'
else: dataset_type = ''
#filename = species+'_'+platform+'_tissue-specific'+dataset_type+'_'+coding_type+'.txt'
filename = 'MarkerFinder/MarkerGenes.txt'
try:
if use_replicates:
filename = string.replace(filename,'.txt','-ReplicateBased.txt')
else:
filename = string.replace(filename,'.txt','-MeanBased.txt')
except Exception: None
filename = destination_dir+filename
filename = string.replace(filename,'ExpressionInput','ExpressionOutput')
data = export.ExportFile(filename)
title_row = string.join(title_row,'\t')
data.write(title_row+'\n')
for probeset in expression_relative:
values = string.join([probeset]+annotations[probeset]+expression_relative[probeset],'\t')+'\n'
data.write(values)
data.close()
print '\nexported:',filepath(filename)
return filepath(filename)
def exportAllGeneCorrelations(filename,allGenesRanked):
destination_dir = export.findParentDir(filename)
filename = destination_dir+'MarkerFinder/AllGenes_correlations.txt'
filename = string.replace(filename,'ExpressionInput','ExpressionOutput')
try:
if use_replicates:
filename = string.replace(filename,'.txt','-ReplicateBased.txt')
else:
filename = string.replace(filename,'.txt','-MeanBased.txt')
except Exception: pass
data = export.ExportFile(filename)
title_row = string.join(['UID','Symbol','Pearson rho','Pearson p-value','Cell State'],'\t')
data.write(title_row+'\n')
rho_sorted=[]
for (probeset,symbol) in allGenesRanked:
try:
(rho,p),tissue = allGenesRanked[(probeset,symbol)]
except Exception:
### Applies to tiered analysis
allGenesRanked[(probeset,symbol)].sort()
(rho,p),tissue = allGenesRanked[(probeset,symbol)][-1]
values = string.join([probeset,symbol,str(rho),str(p),tissue],'\t')+'\n'
rho_sorted.append([(tissue,1.0/rho),values])
rho_sorted.sort()
for (x,values) in rho_sorted:
data.write(values)
data.close()
def exportCorrelations(original_filename,interim_correlations):
destination_dir = 'AltDatabase/ensembl/'+species+'/'
destination_dir = export.findParentDir(original_filename)
if 'AltResults' in original_filename: dataset_type = '_AltExon'
elif 'FullDatasets' in original_filename: dataset_type = '_AltExon'
else: dataset_type = ''
filename = species+'_'+platform+'_tissue-specific_correlations'+dataset_type+'_'+coding_type+'.txt'
filename = destination_dir+filename
filename = destination_dir+'MarkerFinder/MarkerGenes_correlations.txt'
filename = string.replace(filename,'ExpressionInput','ExpressionOutput')
try:
if use_replicates:
filename = string.replace(filename,'.txt','-ReplicateBased.txt')
else:
filename = string.replace(filename,'.txt','-MeanBased.txt')
except Exception: pass
data = export.ExportFile(filename)
title_row = string.join(['UID','Symbol','Pearson rho','Pearson p-value','Cell State'],'\t')
data.write(title_row+'\n')
for tissue in interim_correlations:
for key in interim_correlations[tissue]:
probeset,symbol,rho_p = key
rho,p = rho_p
values = string.join([probeset,symbol,str(rho),str(p),tissue],'\t')+'\n'
data.write(values)
data.close()
#print 'exported:',filepath(filename)
############### Second set of methods for extracting out average expression columns from initial RMA data ##########
def verifyFile(filename):
status = 'not found'
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = 'found';break
except Exception: status = 'not found'
return status
def getAverageExpressionValues(filename,platform):
""" This function imports two file sets: (A) The original raw input expression files and groups and (B) the DATASET file with annotations.
It outputs a new file with annotations and average stats for all groups (some group avgs can be missing from the DATASET file)."""
### Get the original expression input file location
if 'ExpressionInput' in filename:
exp_input_dir = filename
else:
exp_input_dir = string.replace(filename,'ExpressionOutput','ExpressionInput')
exp_input_dir = string.replace(exp_input_dir,'DATASET-','exp.')
if verifyFile(exp_input_dir) == 'not found':
exp_input_dir = string.replace(exp_input_dir,'exp.','') ### file may not have exp.
if verifyFile(exp_input_dir) == 'not found':
exp_input_dir = string.replace(exp_input_dir,'ExpressionInput/','') ### file may be in a root dir
if platform != "3'array":
exp_input_dir = string.replace(exp_input_dir,'.txt','-steady-state.txt')
### Find the DATASET file if this is not a DATASET file dir (can be complicated)
if 'ExpressionInput' in filename:
filename = string.replace(filename,'ExpressionInput','ExpressionOutput')
parent_dir = export.findParentDir(filename)
file = export.findFilename(filename)
if 'exp.' in file:
file = string.replace(file,'exp.','DATASET-')
else:
file = 'DATASET-'+file
filename = parent_dir+'/'+file
elif 'ExpressionOutput' not in filename:
parent_dir = export.findParentDir(filename)
file = export.findFilename(filename)
if 'exp.' in file:
file = string.replace(file,'exp.','DATASET-')
else:
file = 'DATASET-'+file
filename = parent_dir+'/ExpressionOutput/'+file
filename = string.replace(filename,'-steady-state.txt','.txt')
### Import the DATASET file annotations
fn=filepath(filename); x=0; k=0; expression_data={}; annotation_columns={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line) #remove endline
t = string.split(data,'\t')
if x == 0:
if 'Constitutive_exons_used' in data:
array_type = 'exon'
annotation_columns[1]=['Description']
annotation_columns[2]=['Symbol']
annotation_columns[4]=['Constitutive_exons_used']
annotation_columns[7]=['Select Cellular Compartments']
annotation_columns[8]=['Select Protein Classes']
annotation_columns[9]=['Chromosome']
annotation_columns[10]=['Strand']
annotation_columns[11]=['Genomic Gene Corrdinates']
annotation_columns[12]=['GO-Biological Process']
else:
array_type = "3'array"
annotation_columns[1]=['Description']
annotation_columns[2]=['Symbol']
annotation_columns[3]=['Ensembl_id']
annotation_columns[9]=['Pathway_info']
annotation_columns[11]=['Select Cellular Compartments']
annotation_columns[12]=['Select Protein Classes']
i=0; columns_to_save={}; title_row=[t[0]]; annotation_row=[]
for h in t:
if 'avg-' in h:
columns_to_save[i]=[]
#title_row.append(string.replace(h,'avg-',''))
if i in annotation_columns: annotation_row.append(h)
i+=1
x=1
if array_type == "3'array":
annotation_row2 = [annotation_row[1]]+[annotation_row[0]]+annotation_row[2:]### Switch Description and Symbol columns
annotation_row = annotation_row2
title_row = string.join(title_row,'\t')+'\t'
annotation_headers = annotation_row
annotation_row = string.join(annotation_row,'\t')+'\n'
title_row+=annotation_row
else:
uid = t[0]; exp_vals=[]; annotations=[]
i=0
for val in t:
try:
null=columns_to_save[i]
exp_vals.append(val)
except Exception: null=[]
try:
null = annotation_columns[i]
annotations.append(val)
except Exception: null=[]
i+=1
if array_type == "3'array":
annotations2 = [annotations[1]]+[annotations[0]]+annotations[2:]### Switch Description and Symbol columns
annotations = annotations2
expression_data[uid]=annotations # exp_vals+annotations
### This function actually takes the average of all values - not biased by groups indicated in comps.
filename = string.replace(filename,'DATASET-','AVERAGE-')
#filename = exportSimple(filename,expression_data,title_row)
importAndAverageExport(exp_input_dir,platform,annotationDB=expression_data,annotationHeader=annotation_headers,customExportPath=filename)
return filename
def getAverageExonExpression(species,platform,input_exp_file):
### Determine probesets with good evidence of expression
global max_exp_exon_db; max_exp_exon_db={}; global expressed_exon_db; expressed_exon_db={}
global alternative_exon_db; alternative_exon_db={}; global alternative_annotations; alternative_annotations={}
global exon_db
importRawExonExpData(input_exp_file)
importDABGData(input_exp_file)
importAltExonData(input_exp_file)
includeOnlyKnownAltExons = 'yes'
if includeOnlyKnownAltExons == 'yes':
### Optionally only include exons with existing alternative splicing annotations
import AltAnalyze
if platform == 'exon' or platform == 'gene': probeset_type = 'full'
else: probeset_type = 'all'
avg_all_for_ss = 'no'; root_dir = ''; filter_by_known_AE = 'no' ### Exclude exons that are not known to be alternatively expressed from cDNA databases
exon_db,constitutive_probeset_db = AltAnalyze.importSplicingAnnotations(platform,species,probeset_type,avg_all_for_ss,root_dir)
del constitutive_probeset_db
protein_coding_db = ExpressionBuilder.importTranscriptBiotypeAnnotations(species)
delete_exon_entries={}; x=0
for probeset in exon_db:
try: null = alternative_exon_db[probeset]
except Exception: delete_exon_entries[probeset]=[]
### If a splicing annotation exists
ed = exon_db[probeset]
as_call = ed.SplicingCall()
gene = ed.GeneID()
compartment, custom_class = protein_coding_db[gene]
if 'protein_coding' not in custom_class: delete_exon_entries[probeset]=[]
if filter_by_known_AE == 'yes':
if as_call == 0: delete_exon_entries[probeset]=[]
x+=1
### Delete where not expressed, alternatively expressed or known to be an alternative exon
print len(exon_db)-len(delete_exon_entries), 'out of', len(exon_db), 'with known alternative exon annotations analyzed'
for probeset in delete_exon_entries: del exon_db[probeset] ### Clear objects from memory
### Get gene-level annotations
gene_annotation_file = "AltDatabase/ensembl/"+species+"/"+species+"_Ensembl-annotations.txt"
from build_scripts import ExonAnalyze_module; global annotate_db
annotate_db = ExonAnalyze_module.import_annotations(gene_annotation_file,platform)
importRawSpliceData(input_exp_file)
AltAnalyze.clearObjectsFromMemory(exon_db)
def importAltExonData(filename):
file = export.findFilename(filename)
if 'AltResults' in filename:
root_dir = string.split(filename,'AltResults')[0]
elif 'AltExpression' in filename:
root_dir = string.split(filename,'AltExpression')[0]
alt_exon_dir = root_dir+'AltResults/AlternativeOutput/'
dir_list = read_directory(alt_exon_dir)
#print [file];kill
methods_assessed=0
for alt_exon_file in dir_list:
if file[:-4] in alt_exon_file and '-exon-inclusion-results.txt' in alt_exon_file:
alt_exon_file = alt_exon_dir+alt_exon_file
fn = filepath(alt_exon_file); x=0
print 'AltExonResults:',fn
methods_assessed+=1
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x == 0: x=1
else:
probeset = t[6]; isoform_description=t[14]; domain_description=t[15]; indirect_domain=t[17]; x+=1
#if probeset == '2681775': print '2681775 in AltResults file'
exon_annotation = t[4]; block_exon_annotation = t[26]; ens_exon = t[24]; genomic_location = t[-1]
splicing_annotation = t[27]
if len(block_exon_annotation)>0: exon_annotation = block_exon_annotation
if probeset in expressed_exon_db: ### If the probeset is expressed and in the alternative exon file
try: alternative_exon_db[probeset] += 1
except Exception: alternative_exon_db[probeset] = 1
alternative_annotations[probeset] = genomic_location,ens_exon,exon_annotation,splicing_annotation,isoform_description,domain_description,indirect_domain
print len(alternative_exon_db), 'expressed exons considered alternative out of',x
if methods_assessed > 1: ### Likely both FIRMA and splicing-index
### only include those that are present with both methods
single_algorithm = {}
for probeset in alternative_exon_db:
if alternative_exon_db[probeset]==1:
single_algorithm[probeset] = []
for probeset in single_algorithm:
del alternative_exon_db[probeset]
del alternative_annotations[probeset]
def importDABGData(filename,filterIndex=False,DABGFile=True):
try:
file = export.findFilename(filename)
if 'AltResults' in filename:
root_dir = string.split(filename,'AltResults')[0]
elif 'AltExpression' in filename:
root_dir = string.split(filename,'AltExpression')[0]
dabg_file = root_dir+'ExpressionInput/stats.'+file
except Exception:
dabg_file = filename ### when supplying this file directly
expressed_exons={}
fn = filepath(dabg_file); x=0
print 'DABGInput:',fn
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x == 0:
if '#' != data[0]:
header = t
x=1
else:
if DABGFile:
dabg_values = map(float, t[1:])
min_dabg = min(dabg_values)
if min_dabg<0.01:
proceed = True
try:
if t[0] not in max_exp_exon_db: proceed = False
except Exception: pass
#if t[0] == '2681775': print '2681775 in DABG file'
if filterIndex and proceed:
filtered_index=[]
i=0
for dabg in dabg_values:
if dabg<0.01: ### Expressed samples for that gene
filtered_index.append(i)
i+=1
if len(filtered_index)>5:
expressed_exon_db[t[0]] = filtered_index
elif proceed:
expressed_exon_db[t[0]] = []
else:
#{4: 29460, 5: 150826, 6: 249487, 7: 278714, 8: 244304, 9: 187167, 10: 135514, 11: 84828, 12: 39731, 13: 10834, 14: 500, 15: 34}
max_val = max(map(float, t[1:]))
if max_val > 10:
expressed_exons[t[0]] = []
if 'steady-state' in filename: type = 'genes'
else: type = 'exons'
print len(expressed_exon_db), type, 'meeting expression and dabg thesholds'
return expressed_exons
def filterAltExonResults(fn,filterDB=None):
### take a stats file as input
fn = string.replace(fn,'-steady-state.txt','-splicing-index-exon-inclusion-results.txt')
fn = string.replace(fn,'-steady-state-average.txt','-splicing-index-exon-inclusion-results.txt')
fn = string.replace(fn, 'ExpressionInput/stats.','AltResults/AlternativeOutput/')
firstRow=True
regulatedProbesets = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
header = t
ea = header.index('exon annotations')
fp = header.index('functional_prediction')
ps = header.index('probeset')
up = header.index('uniprot-ens_feature_predictions')
sy = header.index('symbol')
de = header.index('description')
ex = header.index('exons')
pl = header.index('probeset location')
ee = header.index('ensembl exons')
eo = header.index('ens_overlapping_domains')
en = header.index('Ensembl')
firstRow=False
else:
if filterDB!=None:
probeset = t[ps]
#UID Definition Symbol Ensembl Genomic Location ExonExternalID Exon ID Alternative Exon Annotation Isoform Associations Inferred Domain Modification Direct Domain Modification
if probeset in filterDB:
markersIn = filterDB[probeset]
markersIn = string.join(markersIn,'|')
values = string.join([probeset,t[de],t[sy],t[en],t[pl],t[ee],t[ex],t[ea],t[fp],t[up],t[eo],markersIn],'\t')
regulatedProbesets[probeset] = values
elif len(t[ea])>2: # and len(t[fp])>1 and t[up]>1:
if 'cassette' in t[ea] or 'alt-3' in t[ea] or 'alt-5' in t[ea] or 'alt-C-term' in t[ea] or 'exon' in t[ea] or 'intron' in t[ea] or 'altFive' in t[ea] or 'altThree' in t[ea]:
if 'altPromoter' not in t[ea] and 'alt-N-term' not in t[ea]:
regulatedProbesets[t[ps]]=t[up]
#regulatedProbesets[t[ps]]=t[up]
if filterDB==None: pass #regulatedProbesets[t[ps]]=t[up]
evaluatedGenes[t[en]]=[]
"""
print len(regulatedProbesets)
if filterDB==None:
import AltAnalyze
if platform == 'exon' or platform == 'gene': probeset_type = 'full'
else: probeset_type = 'all'
avg_all_for_ss = 'no'; root_dir = ''; filter_by_known_AE = 'no' ### Exclude exons that are not known to be alternatively expressed from cDNA databases
exon_db,constitutive_probeset_db = AltAnalyze.importSplicingAnnotations('exon',species,probeset_type,avg_all_for_ss,root_dir)
protein_coding_db = ExpressionBuilder.importTranscriptBiotypeAnnotations(species)
for probeset in exon_db:
if probeset not in constitutive_probeset_db:
### If a splicing annotation exists
ed = exon_db[probeset]
as_call = ed.SplicingCall()
gene = ed.GeneID()
compartment, custom_class = protein_coding_db[gene]
if 'protein_coding' in custom_class: #as_call != 0: #
regulatedProbesets[probeset]=''
evaluatedGenes[gene]=[]
print len(regulatedProbesets), 'AltExon probesets retained...'
"""
return regulatedProbesets
def averageNIValues(fn,dabg_gene_dir,regulatedProbesets):
export_data = export.ExportFile(fn)
fn = string.replace(fn, '-average','')
groups_dir = string.replace(dabg_gene_dir,'exp.','groups.')
groups_dir = string.replace(groups_dir,'stats.','groups.')
groups_dir = string.replace(groups_dir,'-steady-state','')
groups_dir = string.replace(groups_dir,'-average','')
firstRow=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
headers=[]
for h in t:
try: h = string.split(h,':')[-1]
except Exception: pass
headers.append(h)
group_index_db={}
### use comps in the future to visualize group comparison changes
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = ExpressionBuilder.simpleGroupImport(groups_dir)
for x in sample_list:
group_name = group_db[x]
sample_index = headers.index(x)
try: group_index_db[group_name].append(sample_index)
except Exception: group_index_db[group_name] = [sample_index] ### dictionary of group to input file sample indexes
groups = map(str, group_index_db) ### store group names
new_sample_list = map(lambda x: group_db[x], sample_list) ### lookup index of each sample in the ordered group sample list
groups.sort()
export_data.write(string.join(t[:3]+groups,'\t')+'\n')
firstRow=False
else:
geneID = t[0]
probeset = t[2]
if probeset in regulatedProbesets:
avg_z=[]
for group_name in groups:
group_values = map(lambda x: float(t[x]), group_index_db[group_name]) ### simple and fast way to reorganize the samples
avg = statistics.avg(group_values)
avg_z.append(str(avg))
values = string.join(t[:3]+avg_z,'\t')+'\n'
export_data.write(values)
export_data.close()
def calculateSplicingIndexForExpressedConditions(species,dabg_gene_dir,regulatedProbesets,genes_to_exclude):
protein_coding_db = ExpressionBuilder.importTranscriptBiotypeAnnotations(species)
fn = string.replace(dabg_gene_dir, 'ExpressionInput/stats.','AltResults/RawSpliceData/'+species+'/splicing-index/')
fn = string.replace(fn, '-steady-state','')
if '-average.txt' in fn:
averageNIValues(fn,dabg_gene_dir,regulatedProbesets)
firstRow=True
onlyIncludeOldExons=False
splicedExons = {}
splicedExons1={}
splicedExons2={}
splicedExons3={}
splicedExons4={}
splicedExons5={}
splicedExons6={}
splicedExons7={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
header = t[3:]
### Test to below to make sure we have the right headers - it works - only heart for a heart specific gene
#exp_indexes = expressed_exon_db['ENSG00000134571']
#filtered_headers = map(lambda i: header[i],exp_indexes)
#print filtered_headers;sys.exit()
headers=[]
for h in header:
h = string.split(h,':')[-1]
headers.append(h)
headers = string.join(headers,'\t')
firstRow = False
else:
geneID = t[0]
probeset = t[2]
compartment, custom_class = protein_coding_db[geneID]
if 'protein_coding' in custom_class and onlyIncludeOldExons == False: ### Only consider protein coding genes
if probeset in regulatedProbesets and geneID in expressed_exon_db and probeset in expressed_exon_db: ### If the probeset is associated with a protein modifying splicing event and has gene expressed conditions
exp_indexes = expressed_exon_db[geneID]
NI_values = map(float, t[3:])
exp_NI_values = map(lambda i: NI_values[i],exp_indexes)
#print len(exp_NI_values), len(exp_indexes), len(NI_values)
si = max(exp_NI_values)-statistics.avg(exp_NI_values)
if si>2 and max(NI_values)>-1: ### Ensures that the splicing event is large and highly expressed
i=0; mod_NI_values=[]
for ni in NI_values: ### Replaced non-expressed samples with empty values
if i in exp_indexes:
mod_NI_values.append(str(ni))
else:
mod_NI_values.append('')
i+=1
mod_NI_values = string.join(mod_NI_values,'\t')
if geneID not in genes_to_exclude: ### gene markers
splicedExons[probeset,geneID] = mod_NI_values
splicedExons6[probeset] = []
splicedExons7[probeset] = []
if onlyIncludeOldExons:
if probeset in prior_altExons and geneID in expressed_exon_db:
exp_indexes = expressed_exon_db[geneID]
i=0; NI_values = map(float, t[3:]); mod_NI_values=[]
for ni in NI_values: ### Replaced non-expressed samples with empty values
if i in exp_indexes:
mod_NI_values.append(str(ni))
else:
mod_NI_values.append('')
i+=1
mod_NI_values = string.join(mod_NI_values,'\t')
splicedExons[probeset,geneID] = mod_NI_values
if probeset in regulatedProbesets:
splicedExons1[probeset]=[]
if probeset in regulatedProbesets and 'protein_coding' in custom_class:
splicedExons2[probeset]=[]
if probeset in regulatedProbesets and probeset in expressed_exon_db:
splicedExons3[probeset]=[]
if probeset in regulatedProbesets and geneID in expressed_exon_db:
splicedExons4[probeset]=[]
if probeset in regulatedProbesets and geneID not in genes_to_exclude:
splicedExons5[probeset]=[]
print len(splicedExons),'top alternative exons...'
output_file = string.replace(dabg_gene_dir,'stats.','topSplice.')
eo = export.ExportFile(output_file)
eo.write('Probeset:GeneID\t'+headers+'\n')
for (probeset,geneID) in splicedExons:
mod_NI_values = splicedExons[(probeset,geneID)]
eo.write(probeset+':'+geneID+'\t'+mod_NI_values+'\n')
eo.close()
expressed_uids1 = splicedExons1.viewkeys() & prior_altExons.viewkeys()
print len(expressed_uids1)
expressed_uids1 = splicedExons1.viewkeys() & prior_altExons.viewkeys()
print len(expressed_uids1)
expressed_uids1 = splicedExons2.viewkeys() & prior_altExons.viewkeys()
print len(expressed_uids1)
expressed_uids1 = splicedExons3.viewkeys() & prior_altExons.viewkeys()
print len(expressed_uids1)
expressed_uids1 = splicedExons4.viewkeys() & prior_altExons.viewkeys()
print len(expressed_uids1)
expressed_uids1 = splicedExons5.viewkeys() & prior_altExons.viewkeys()
print len(expressed_uids1)
expressed_uids1 = splicedExons6.viewkeys() & prior_altExons.viewkeys()
print len(expressed_uids1)
expressed_uids1 = splicedExons7.viewkeys() & prior_altExons.viewkeys()
print len(expressed_uids1)
return output_file, splicedExons
def filterRNASeqSpliceEvents(Species,Platform,fl,psi_file_dir):
global species; import AltAnalyze
global platform; from import_scripts import sampleIndexSelection
global max_exp_exon_db
global evaluatedGenes; evaluatedGenes={'Gene_ID':[]}
species = Species
platform = Platform
output_file = psi_file_dir
compendiumType = 'AltExon'
#analyzeData(output_file,species,'altSplice',compendiumType,geneToReport=200,AdditionalParameters=fl,logTransform=False,correlateAll=False)
output_file = '/Volumes/SEQ-DATA/IlluminaBodyMap/exons/SplicingMarkers/exp.BodyMap.txt'
compendiumType = 'AltExon'
analyzeData(output_file,species,'RNASeq',compendiumType,geneToReport=200,AdditionalParameters=fl,logTransform=False,correlateAll=False)
def filterDetectionPvalues(Species,Platform,fl,dabg_gene_dir):
global species; import AltAnalyze
global platform; from import_scripts import sampleIndexSelection
global max_exp_exon_db; global prior_altExons
global evaluatedGenes; evaluatedGenes={'Gene_ID':[]}
global prior_altExons
species = Species
platform = Platform
averageProfiles = False
includeMarkerGeneExons = False
alsoConsiderExpression = False
filterForHighExpressionExons = True
input_exp_file = string.replace(dabg_gene_dir,'stats.','exp.')
input_exp_file = string.replace(input_exp_file,'-steady-state','')
global expressed_exon_db; expressed_exon_db={}
#"""
prior_altExons = importMarkerFinderExons(dabg_gene_dir,species=species,type='MarkerFinder-Exon')
#reorderMultiLevelExpressionFile(dabg_gene_dir) ### Re-sort stats file
regulatedProbesets = filterAltExonResults(dabg_gene_dir) ### AltResults/AlternativeOutput SI file
if filterForHighExpressionExons:
high_expression_probesets = importDABGData(input_exp_file,filterIndex=True,DABGFile=False) ### Get highly expressed exons by RMA
regulatedProbesets2={}
print len(regulatedProbesets)
for i in high_expression_probesets:
if i in regulatedProbesets:
regulatedProbesets2[i]=regulatedProbesets[i]
regulatedProbesets=regulatedProbesets2
print len(regulatedProbesets)
dabg_exon_dir = string.replace(dabg_gene_dir,'-steady-state','')
if averageProfiles:
regulatedProbesets['probeset_id']=[] ### Will force to get the header row
filtered_exon_exp = AltAnalyze.importGenericFiltered(input_exp_file,regulatedProbesets)
input_exp_file = filterAverageExonProbesets(input_exp_file,filtered_exon_exp,regulatedProbesets,exportType='expression')
filtered_gene_dabg = AltAnalyze.importGenericFiltered(dabg_gene_dir,evaluatedGenes)
dabg_gene_dir = filterAverageExonProbesets(dabg_gene_dir,filtered_gene_dabg,evaluatedGenes,exportType='expression')
filtered_exon_exp = AltAnalyze.importGenericFiltered(dabg_exon_dir,regulatedProbesets)
dabg_exon_dir = filterAverageExonProbesets(dabg_exon_dir,filtered_exon_exp,regulatedProbesets,exportType='expression')
reorderMultiLevelExpressionFile(input_exp_file)
reorderMultiLevelExpressionFile(dabg_gene_dir)
reorderMultiLevelExpressionFile(dabg_exon_dir)
input_exp_file = string.replace(input_exp_file,'.txt','-average.txt')
dabg_gene_dir = string.replace(dabg_gene_dir,'.txt','-average.txt')
dabg_exon_dir = string.replace(dabg_exon_dir,'.txt','-average.txt')
expressed_uids1 = regulatedProbesets.viewkeys() & prior_altExons.viewkeys()
importDABGData(dabg_gene_dir,filterIndex=True) ### Get expressed conditions by DABG p-value for genes
max_exp_exon_db = regulatedProbesets
importDABGData(dabg_exon_dir,filterIndex=False) ### Get expressed conditions by DABG p-value for selected exons
genes_to_exclude = importMarkerFinderExons(dabg_gene_dir,species=species,type='MarkerFinder-Gene')
output_file,splicedExons = calculateSplicingIndexForExpressedConditions(species,dabg_gene_dir,regulatedProbesets,genes_to_exclude)
output_file = string.replace(dabg_gene_dir,'stats.','topSplice.')
array_type = 'altSplice'; compendiumType = 'AltExon'
analyzeData(output_file,species,array_type,compendiumType,geneToReport=200,AdditionalParameters=fl,logTransform=False,correlateAll=False)
addSuffixToMarkerFinderFile(dabg_gene_dir,'AltExon')
#"""
splicedExons = importMarkerFinderExons(dabg_gene_dir,type='AltExon')
if alsoConsiderExpression:
exp_file = string.replace(dabg_gene_dir,'stats.','exp.')
exp_file = string.replace(exp_file,'-steady-state','')
output_file = string.replace(exp_file,'exp.','filter.')
sampleIndexSelection.filterRows(exp_file,output_file,filterDB=splicedExons)
analyzeData(output_file,species,platform,'AltExon',geneToReport=400,AdditionalParameters=fl,logTransform=False,correlateAll=False)
addSuffixToMarkerFinderFile(dabg_gene_dir,'Filtered')
filteredExons = importMarkerFinderExons(dabg_gene_dir,type='Filtered')
splicedExons = intersectMarkerFinderExons(splicedExons,filteredExons)
regulatedProbesets = filterAltExonResults(dabg_gene_dir,filterDB=splicedExons)
### Make a probeset-level average expression file
splicedExons['probeset_id']=[] ### Will force to get the header row
if includeMarkerGeneExons:
markerGenes = importMarkerFinderExons(dabg_gene_dir,species=species,type='MarkerFinder-Gene')
splicedExons, regulatedProbesets = getConstitutiveMarkerExons(species, platform, markerGenes, splicedExons, regulatedProbesets)
filtered_exon_exp = AltAnalyze.importGenericFiltered(input_exp_file,splicedExons)
filterAverageExonProbesets(input_exp_file,filtered_exon_exp,regulatedProbesets,exportType='MarkerFinder');sys.exit()
def getConstitutiveMarkerExons(species, platform, markerGenes, splicedExons, regulatedProbesets):
#Identify a single consitutive probeset for prior identified MarkerGenes
genesAdded={}; count=0
import AltAnalyze
if platform == 'exon' or platform == 'gene': probeset_type = 'full'
else: probeset_type = 'all'
avg_all_for_ss = 'no'; root_dir = ''; filter_by_known_AE = 'no' ### Exclude exons that are not known to be alternatively expressed from cDNA databases
exon_db,constitutive_probeset_db = AltAnalyze.importSplicingAnnotations(platform,species,probeset_type,avg_all_for_ss,root_dir)
filler = ['']*7
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
for probeset in constitutive_probeset_db:
geneID = constitutive_probeset_db[probeset]
if geneID in markerGenes and geneID not in genesAdded:
count+=1
splicedExons[probeset]=markerGenes[geneID]
tissues = string.join(markerGenes[geneID],'|')
try: symbol = gene_to_symbol[geneID][0]; description = ''
except Exception: symbol = ''; description = ''
regulatedProbesets[probeset]=string.join([probeset,description,symbol,geneID]+filler+[tissues],'\t')
genesAdded[geneID]=[]
print count, 'constitutive marker gene exons added'
return splicedExons, regulatedProbesets
def intersectMarkerFinderExons(splicedExons,filteredExons):
splicedExons_filtered={}
for exon in splicedExons:
tissues = splicedExons[exon]
filtered_tissues=[]
if exon in filteredExons:
tissues2 = filteredExons[exon]
for tissue in tissues:
if tissue in tissues2:
filtered_tissues.append(tissue)
if len(filtered_tissues)>0:
splicedExons_filtered[exon] = filtered_tissues
print len(splicedExons_filtered), 'exons found analyzing both NI values and raw expression values'
return splicedExons_filtered
def addSuffixToMarkerFinderFile(expr_input,suffix):
markerfinder_results = string.split(expr_input,'ExpressionInput')[0]+'ExpressionOutput/MarkerFinder/MarkerGenes_correlations-ReplicateBased.txt'
new_file = string.replace(markerfinder_results,'.txt','-'+suffix+'.txt')
export.copyFile(markerfinder_results, new_file)
def filterAverageExonProbesets(expr_input,filtered_exon_exp,regulatedProbesets,exportType='expression'):
if exportType == 'expression':
export_path = string.replace(expr_input,'.txt','-average.txt')
else:
export_path = string.split(expr_input,'ExpressionInput')[0]+'ExpressionOutput/MarkerFinder/MarkerGenes-ReplicateBased-AltExon.txt'
export_data = export.ExportFile(export_path)
groups_dir = string.replace(expr_input,'exp.','groups.')
groups_dir = string.replace(groups_dir,'stats.','groups.')
groups_dir = string.replace(groups_dir,'-steady-state','')
group_index_db={}
### use comps in the future to visualize group comparison changes
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = ExpressionBuilder.simpleGroupImport(groups_dir)
for x in sample_list:
group_name = group_db[x]
try: sample_index = filtered_exon_exp['probeset_id'].index(x)
except Exception: sample_index = filtered_exon_exp['Gene_ID'].index(x)
try: group_index_db[group_name].append(sample_index)
except Exception: group_index_db[group_name] = [sample_index] ### dictionary of group to input file sample indexes
groups = map(str, group_index_db) ### store group names
new_sample_list = map(lambda x: group_db[x], sample_list) ### lookup index of each sample in the ordered group sample list
if exportType == 'expression':
headers = 'probeset_id\t'
else:
headers = string.join(['UID','Definition','Symbol','Ensembl','Genomic Location','ExonExternalID','Exon ID','Alternative Exon Annotation','Isoform Associations','Inferred Domain Modification','Direct Domain Modification', 'marker-in'],'\t')+'\t'
headers += string.join(groups,'\t')
export_data.write(headers+'\n')
for uid in filtered_exon_exp:
if uid != 'probeset_id' and uid != 'Gene_ID':
values = filtered_exon_exp[uid]
if uid in regulatedProbesets:
annotations = regulatedProbesets[uid]
if platform == 'RNASeq':
### Convert to log2 RPKM values - or counts
values = map(lambda x: math.log(float(x),2), values)
else:
values = map(float,values)
avg_z=[]
for group_name in group_index_db:
group_values = map(lambda x: values[x], group_index_db[group_name]) ### simple and fast way to reorganize the samples
avg = statistics.avg(group_values)
avg_z.append(str(avg))
if exportType == 'expression':
values = string.join([uid]+avg_z,'\t')+'\n'
else:
values = annotations+'\t'+string.join(avg_z,'\t')+'\n'
export_data.write(values)
export_data.close()
return export_path
def importMarkerFinderExons(dabg_gene_dir,species=None,type=None):
if type=='AltExon':
fn = string.split(dabg_gene_dir,'ExpressionInput')[0]+'ExpressionOutput/MarkerFinder/MarkerGenes_correlations-ReplicateBased-AltExon.txt'
elif type=='Filtered':
fn = string.split(dabg_gene_dir,'ExpressionInput')[0]+'ExpressionOutput/MarkerFinder/MarkerGenes_correlations-ReplicateBased-Filtered.txt'
elif type == 'MarkerFinder-Gene':
coding_type = 'protein_coding'
fn = 'AltDatabase/ensembl/'+species+'/'+species+'_'+platform +'_tissue-specific_'+coding_type+'.txt'
fn = filepath(fn)
elif type == 'MarkerFinder-Exon':
coding_type = 'protein_coding'
fn = 'AltDatabase/ensembl/'+species+'/'+species+'_'+platform +'_tissue-specific_AltExon_'+coding_type+'.txt'
fn = filepath(fn)
print 'Importing',fn
firstRow=True
splicedExons = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
header = t
firstRow=False
else:
try: splicedExons[t[0]].append(t[-1])
except Exception: splicedExons[t[0]] = [t[-1]]
print 'Stored', len(splicedExons), 'markers for further evaluation.'
return splicedExons
def importRawExonExpData(filename):
file = export.findFilename(filename)
if 'AltResults' in filename:
root_dir = string.split(filename,'AltResults')[0]
elif 'AltExpression' in filename:
root_dir = string.split(filename,'AltExpression')[0]
exp_file = root_dir+'ExpressionInput/exp.'+file
fn = filepath(exp_file); x=0
log_exp_threshold = math.log(70,2)
print 'RawExpInput:',fn
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x == 0:
if '#' != data[0]: x=1
else:
max_exp = max(map(float, t[1:]))
if max_exp>log_exp_threshold:
max_exp_exon_db[t[0]] = []
#if t[0] == '2681775': print '2681775 in expression file'
print len(max_exp_exon_db), 'exons with maximal expression greater than 50'
def importRawSpliceData(filename):
"""Import the RawSplice file normalized intensity exon data (or Raw Expression - optional) and then export after filtering, averaging and annotating for each group"""
fn=filepath(filename); x=0; group_db={}
print 'RawExonInput:',fn
if 'RawSplice' in fn:
output_file = string.replace(fn,'RawSplice','AVERAGESplice')
elif 'FullDatasets' in fn:
output_file = string.replace(fn,'FullDatasets','AVERAGE-FullDatasets')
else: print 'WARNING! The text "RawSplice" must be in the input filename to perform this analysis'; sys.exit()
export_data = export.ExportFile(output_file) ### Write out the averaged data once read and processed
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x == 0:
i=0; group_list=[]; group_list2=[]; headers = ['ExonID']
for h in t:
try:
### Store the index for each group numerical ID
group_name,sample = string.split(h,':')
try: group_db[group_name].append(i)
except KeyError: group_db[group_name] = [i]
except Exception: null=[]
i+=1
for group_name in group_db:
group_list.append(group_db[group_name]) ### list of group indexes in order
group_list2.append([group_db[group_name],group_name])
group_list.sort(); group_list2.sort()
for values in group_list2: headers.append(values[-1]) ### Store the original order of group names
headers = string.join(headers+['Definition','Symbol','Ensembl','Genomic Location','ExonExternalID','Exon ID','Alternative Exon Annotation','Isoform Associations','Inferred Domain Modification','Direct Domain Modification'],'\t')+'\n'
export_data.write(headers)
x+=1
else:
avg_NI=[]
if '-' in t[0]: exonid = string.join(string.split(t[0],'-')[1:],'-') ### For the NI data
else: exonid = t[0] ### When importing the FullDataset organized expression data
try:
#if exonid == '2681775': print '2681775 is in the RawSplice file'
ed = exon_db[exonid]; gene = ed.GeneID()
#if exonid == '2681775': print '2681775 is in the filtered exon_db'
y = annotate_db[gene]; symbol = y.Symbol(); description = y.Description()
genomic_location,ens_exon,exon_annotation,splicing_annotation,isoform_description,domain_description,indirect_domain = alternative_annotations[exonid]
for indexes in group_list:
avg_NI.append(avg(map(float, t[indexes[0]:indexes[-1]+1])))
values = string.join([exonid]+map(str, avg_NI)+[description,symbol,gene,genomic_location,ens_exon,exon_annotation,splicing_annotation,isoform_description,domain_description,indirect_domain],'\t')+'\n'
export_data.write(values)
except Exception: null=[]
def getExprValsForNICorrelations(array_type,altexon_correlation_file,rawsplice_file):
"""Import the FullDatasets expression file to replace NI values from the built tissue correlation file"""
### Import the AltExon correlation file
fn = filepath(altexon_correlation_file); marker_probesets={}; x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
headers = t; x=1; index=0
for i in headers:
if 'marker-in' in i: tissue_index = index+1
index+=1
annotation_headers = headers[:tissue_index]
else:
marker_probeset = t[0]
marker_probesets[marker_probeset] = t[:tissue_index] ### Store annotations
print len(marker_probesets), 'marker probests imported'
### Import the corresponding full-dataset expression file (must average expression for replicates)
if array_type == 'exon': array_type_dir = 'ExonArray'
elif array_type == 'gene': array_type_dir = 'GeneArray'
elif array_type == 'junction': array_type_dir = 'JunctionArray'
else: array_type_dir = array_type
file = export.findFilename(rawsplice_file); x=0; group_db={}
root_dir = string.split(rawsplice_file,'AltResults')[0]
output_file = string.replace(altexon_correlation_file,'.txt','-exp.txt')
export_data = export.ExportFile(output_file) ### Write out the averaged data once read and processed
exp_file = root_dir+'AltExpression/FullDatasets/'+array_type_dir+'/'+species+'/'+file
fn = filepath(exp_file)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x == 0:
i=0; group_list=[]; group_list2=[]; headers = []
for h in t:
try:
### Store the index for each group numerical ID
group_name,sample = string.split(h,':')
if '~' in group_name:
group_name = string.split(group_name,'~')[1]
try: group_db[group_name].append(i)
except KeyError: group_db[group_name] = [i]
except Exception: null=[]
i+=1
for group_name in group_db:
group_list.append(group_db[group_name]) ### list of group indexes in order
group_list2.append([group_db[group_name],group_name])
group_list.sort(); group_list2.sort()
for values in group_list2: headers.append(values[-1]) ### Store the original order of group names
headers = string.join(annotation_headers+headers,'\t')+'\n'
export_data.write(headers)
x+=1
else:
exonid = t[0]; avg_exp=[]
try:
annotations = marker_probesets[exonid]
for indexes in group_list:
avg_exp.append(avg(map(float, t[indexes[0]:indexes[-1]+1])))
values = string.join(annotations+map(str, avg_exp),'\t')+'\n'
export_data.write(values)
except Exception: null=[]
def avg(ls):
return sum(ls)/len(ls)
def exportSimple(filename,expression_data,title_row):
filename = string.replace(filename,'DATASET-','AVERAGE-')
data = export.ExportFile(filename)
data.write(title_row)
for uid in expression_data:
values = string.join([uid]+expression_data[uid],'\t')+'\n'
data.write(values)
data.close()
#print 'exported...'
#print filename
return filename
def returnCommonProfiles(species):
###Looks at exon and gene array AltExon predictions to see which are in common
targetPlatforms = ['exon','gene']; tissue_to_gene={}; rho_threshold = 0; p_threshold = 0.2
import TissueProfiler
gene_translation_db = TissueProfiler.remoteImportExonIDTranslations('gene',species,'no','exon')
for targetPlatform in targetPlatforms:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_'+targetPlatform +'_tissue-specific_correlations_AltExon_protein_coding.txt'
#filename = 'AltDatabase/ensembl/'+species+'/'+species+'_'+targetPlatform +'_tissue-specific_correlations_'+coding_type+'.txt'
fn=filepath(filename); x=0
#print filename
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if x==0: x=1 ### Ignore header line
else:
uid,symbol,(rho,p),tissue = string.split(data,'\t')
if targetPlatform=='gene':
try: uid = gene_translation_db[uid] ### translate from gene to exon array probesets
except Exception: uid = ''
if float(rho)>rho_threshold and float(p)<p_threshold and len(uid)>0 : ### Variable used for testing different thresholds internally
try: tissue_to_gene[tissue,uid,symbol]+=1
except Exception: tissue_to_gene[tissue,uid,symbol] = 1
count=0
for (tissue,uid,symbol) in tissue_to_gene:
if tissue_to_gene[(tissue,uid,symbol)]>1:
count+=1
#print count
def importAndAverageStatsData(expr_input,compendium_filename,platform):
""" This function takes LineageProfiler z-scores and organizes the samples into groups
takes the mean results for each group and looks for changes in lineage associations """
groups_dir = string.replace(expr_input,'exp.','groups.')
groups_dir = string.replace(groups_dir,'stats.','groups.')
groups_dir = string.replace(groups_dir,'-steady-state.txt','.txt') ### groups is for the non-steady-state file
export_path = string.replace(compendium_filename,'.txt','_stats.txt')
export_data = export.ExportFile(export_path)
print 'Export LineageProfiler database dabg to:',export_path
### Import compendium file
fn=filepath(compendium_filename); row_number=0; compendium_annotation_db={}; x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
print 'Importing the tissue compedium database:',compendium_filename
headers = t; x=1; index=0
for i in headers:
if 'UID' == i: ens_index = index
if 'AltExon' in compendium_filename: ens_index = ens_index ### Assigned above when analyzing probesets
elif 'Ensembl' in i: ens_index = index
if 'marker-in' in i: tissue_index = index+1; marker_in = index
index+=1
new_headers = t[:tissue_index]
else:
uid = string.split(t[ens_index],'|')[0]
annotations = t[:tissue_index]
compendium_annotation_db[uid] = annotations
fn=filepath(expr_input); row_number=0; exp_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#': row_number = 0
elif row_number==0:
group_index_db={}
### use comps in the future to visualize group comparison changes
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = ExpressionBuilder.simpleGroupImport(groups_dir)
for x in sample_list:
group_name = group_db[x]
try: sample_index = t[1:].index(x)
except Exception, e:
print x
print t[1:]
print e; sys.exit()
try: group_index_db[group_name].append(sample_index)
except Exception: group_index_db[group_name] = [sample_index] ### dictionary of group to input file sample indexes
groups = map(str, group_index_db) ### store group names
new_sample_list = map(lambda x: group_db[x], sample_list) ### lookup index of each sample in the ordered group sample list
title = string.join(new_headers+groups,'\t')+'\n' ### output the new sample order (group file order)
export_data.write(title)
row_number=1
else:
uid = t[0]
if uid in compendium_annotation_db:
if platform == 'RNASeq':
### Convert to log2 RPKM values - or counts
values = map(lambda x: math.log(float(x),2), t[1:])
else:
try: values = map(float,t[1:])
except Exception: values = logTransformWithNAs(t[1:])
avg_z=[]
for group_name in group_index_db:
group_values = map(lambda x: values[x], group_index_db[group_name]) ### simple and fast way to reorganize the samples
avg = statistics.avg(group_values)
avg_z.append(str(avg))
export_data.write(string.join(compendium_annotation_db[uid]+avg_z,'\t')+'\n')
export_data.close()
return export_path
def floatWithNAs(values):
values2=[]
for x in values:
try: values2.append(float(x)) #values2.append(math.log(float(x),2))
except Exception:
#values2.append(0.00001)
values2.append('')
return values2
def importAndAverageExport(expr_input,platform,annotationDB=None,annotationHeader=None,customExportPath=None):
""" More simple custom used function to convert a exp. or stats. file from sample values to group means """
export_path = string.replace(expr_input,'.txt','-AVERAGE.txt')
if customExportPath != None: ### Override the above
export_path = customExportPath
groups_dir = string.replace(expr_input,'exp.','groups.')
groups_dir = string.replace(groups_dir,'stats.','groups.')
groups_dir = string.replace(groups_dir,'-steady-state.txt','.txt') ### groups is for the non-steady-state file
if 'AltExon' in expr_input:
groups_dir = string.replace(expr_input,'AltExonConfirmed-','groups.')
groups_dir = string.replace(groups_dir,'AltExon-','groups.')
groups_dir = string.replace(groups_dir,'AltResults/Clustering','ExpressionInput')
export_data = export.ExportFile(export_path)
if annotationDB == None:
annotationDB = {}
annotationHeader = []
### CRITICAL!!!! ordereddict is needed to run clustered markerFinder downstream analyses!!!
import collections
count=0
fn=filepath(expr_input); row_number=0; exp_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#' and row_number==0: row_number = 0
elif row_number==0:
if ':' in data:
tab_data = [] ### remove the group prefixes
for h in t:
h = string.split(h,':')[-1]
tab_data.append(h)
t = tab_data
try:
### OrderedDict used to return the keys in the orders added for markerFinder
group_index_db=collections.OrderedDict()
except Exception:
try:
import ordereddict
group_index_db = ordereddict.OrderedDict()
except Exception:
group_index_db={}
### use comps in the future to visualize group comparison changes
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = ExpressionBuilder.simpleGroupImport(groups_dir)
for x in sample_list:
group_name = group_db[x]
try: sample_index = t[1:].index(x)
except Exception, e:
print [x]
print t[1:]
print expr_input
print e; sys.exit()
try: group_index_db[group_name].append(sample_index)
except Exception: group_index_db[group_name] = [sample_index] ### dictionary of group to input file sample indexes
groups = map(str, group_index_db) ### store group names
new_sample_list = map(lambda x: group_db[x], sample_list) ### lookup index of each sample in the ordered group sample list
title = string.join([t[0]]+groups+annotationHeader,'\t')+'\n' ### output the new sample order (group file order)
export_data.write(title)
row_number=1
else:
uid = t[0]
try: values = map(float,t[1:])
except Exception: values = floatWithNAs(t[1:])
avg_z=[]
for group_name in group_index_db:
group_values = map(lambda x: values[x], group_index_db[group_name]) ### simple and fast way to reorganize the samples
group_values = [x for x in group_values if x != ''] ### Remove NAs from the group
five_percent_len = int(len(group_values)*0.05)
if len(group_values)>five_percent_len:
avg = statistics.avg(group_values) #stdev
else:
avg = ''
avg_z.append(str(avg))
if uid in annotationDB:
annotations = annotationDB[uid] ### If provided as an option to the function
else:
annotations=[]
export_data.write(string.join([uid]+avg_z+annotations,'\t')+'\n'); count+=1
export_data.close()
print 'Export',count,'rows to:',export_path
return export_path
if __name__ == '__main__':
Species='Mm'
filename = ('/Users/saljh8/Desktop/dataAnalysis/Damien/Revised/Guide1/PopulationComparisons/4mo/ExpressionInput/exp.restricted.txt','/Users/saljh8/Desktop/dataAnalysis/Damien/Revised/Guide1/PopulationComparisons/4mo/ExpressionOutput/AVERAGE-restricted.txt')
analyzeData(filename,Species,"RNASeq","protein_coding",geneToReport=60,correlateAll=True,AdditionalParameters=None,logTransform=True)
sys.exit()
averageNIValues('/Users/saljh8/Desktop/LineageProfiler/AltResults/RawSpliceData/Hs/splicing-index/meta-average.txt','/Users/saljh8/Desktop/LineageProfiler/ExpressionInput/stats.meta-steady-state.txt',{});sys.exit()
dabg_file_dir = '/Users/saljh8/Desktop/LineageProfiler/ExpressionInput/stats.meta-steady-state.txt'
filterDetectionPvalues(species, dabg_file_dir);sys.exit()
Platform = 'RNASeq'
codingType = 'AltExon'
expr_input = '/Users/nsalomonis/Desktop/Mm_Gene_Meta/ExpressionInput/exp.meta.txt'
#expr_input = '/home/socr/c/users2/salomoni/conklin/nsalomonis/normalization/Hs_Exon-TissueAtlas/ExpressionInput/stats.meta-steady-state.txt'
expr_input = '/Volumes/My Passport/dataAnalysis/CardiacRNASeq/BedFiles/ExpressionInput/exp.CardiacRNASeq-steady-state.txt'
expr_input = '/Volumes/My Passport/dataAnalysis/CardiacRNASeq/BedFiles/AltResults/Clustering/AltExonConfirmed-CardiacRNASeq.txt'
compendium_filename = 'Hs_exon_tissue-specific_AltExon_protein_coding.txt'
compendium_filename = '/Users/nsalomonis/Desktop/AltAnalyze/AltDatabase/EnsMart65/ensembl/Hs/Hs_exon_tissue-specific_AltExon_protein_coding.txt'
compendium_filename = '/home/socr/c/users2/salomoni/AltAnalyze_v.2.0.7-Py/AltDatabase/ensembl/Mm/Mm_gene_tissue-specific_protein_coding.txt'
#compendium_filename = '/home/socr/c/users2/salomoni/AltAnalyze_v.2.0.7-Py/AltDatabase/ensembl/Hs/Hs_exon_tissue-specific_ncRNA.txt'
compendium_filename = '/home/socr/c/users2/salomoni/AltAnalyze_v.2.0.7-Py/AltDatabase/ensembl/Mm/Mm_gene_tissue-specific_AltExon_protein_coding.txt'
importAndAverageExport(expr_input,Platform); sys.exit()
importAndAverageStatsData(expr_input,compendium_filename,Platform); sys.exit()
returnCommonProfiles(species);sys.exit()
#codingType = 'ncRNA'
"""
input_exp_file = 'C:/Users/Nathan Salomonis/Desktop/Gladstone/1-datasets/ExonArray/CP-hESC/AltResults/RawSpliceData/Hs/splicing-index/hESC_differentiation.txt'
#input_exp_file = 'C:/Users/Nathan Salomonis/Desktop/Gladstone/1-datasets/ExonArray/CP-hESC/AltExpression/FullDatasets/ExonArray/Hs/hESC_differentiation.txt'
input_exp_file = '/home/socr/c/users2/salomoni/other/boxer/normalization/Hs_Exon-TissueAtlas/AltResults/RawSpliceData/Mm/splicing-index/meta.txt'
#input_exp_file = '/home/socr/c/users2/salomoni/other/boxer/normalization/Hs_Exon-TissueAtlas/AltExpression/FullDatasets/ExonArray/Hs/meta.txt'
getAverageExonExpression(Species,Platform,input_exp_file)#;sys.exit()
dataset_file = 'DATASET-Hs_meta-Exon_101111.txt'
#getAverageExpressionValues(dataset_file); sys.exit()
group_exp_file = 'AVERAGE-Hs_meta-Exon_101111.txt'
if 'Raw' in input_exp_file:
group_exp_file = string.replace(input_exp_file,'Raw','AVERAGE')
else:
group_exp_file = string.replace(input_exp_file,'FullDatasets','AVERAGE-FullDatasets')
altexon_correlation_file = analyzeData(group_exp_file,Species,Platform,codingType)
"""
altexon_correlation_file = 'temp.txt'
input_exp_file = '/home/socr/c/users2/salomoni/conklin/nsalomonis/normalization/Hs_Exon-TissueAtlas/AltResults/RawSpliceData/Hs/meta.txt'
getExprValsForNICorrelations(Platform,altexon_correlation_file,input_exp_file)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/markerFinder.py
|
markerFinder.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"""This module contains instructions importing existing over-representation analysis (ORA)
results, pruning redundant Gene Ontology paths, reading in command-line options and coordinating
all GO-Elite analysis and update functions found in other modules."""
try:
try: import traceback
except Exception: None
import sys, string
import os.path, platform
import unique; reload(unique)
import math
import copy
import time
import shutil
import webbrowser
import gene_associations; reload(gene_associations)
try: from import_scripts import OBO_import
except Exception: pass
import export
import UI
import mappfinder; reload(mappfinder)
import datetime
from visualization_scripts import WikiPathways_webservice
except Exception:
print_out = "\nWarning!!! Critical Python incompatiblity encoutered.\nThis can occur if the users calls the GO-Elite "
print_out += "python source code WITHIN A COMPILED version directory which results in critical conflicts between "
print_out += "the compiled distributed binaries and the operating systems installed version of python. "
print_out += "If this applies, either:\n(A) double-click on the executable GO-Elite file (GO-Elite or GO-Elite.exe) "
print_out += "or\n(B) download the Source-Code version of GO-Elite and run this version (after installing any needed "
print_out += "dependencies (see our Wiki or Documentation). Otherwise, contact us at: [email protected]\n\n"
print_out += "Installation Wiki: http://code.google.com/p/go-elite/wiki/Installation\n"
print print_out
try:
### Create a log report of this
import unique
try: log_file = unique.filepath('GO-Elite_error-report.log')
except Exception: log_file = unique.filepath('/GO-Elite_error-report.log')
log_report = open(log_file,'a')
log_report.write(print_out)
try: log_report.write(traceback.format_exc())
except Exception: None
log_report.close()
print 'See log file for additional technical details:',log_file
### Open this file
if os.name == 'nt':
try: os.startfile('"'+log_file+'"')
except Exception: os.system('open "'+log_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+log_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+log_file+'/"')
except Exception: None
sys.exit()
debug_mode = 'no'
Tkinter_failure = False
start_time = time.time()
try: command_args = string.join(sys.argv,' ')
except Exception: command_args = ''
if len(sys.argv[1:])>1 and '-' in command_args:
use_Tkinter = 'no'
else:
try:
import Tkinter
from Tkinter import *
from visualization_scripts import PmwFreeze
use_Tkinter = 'yes'
except ImportError:
use_Tkinter = 'yes'
Tkinter_failure = True
###### File Import Functions ######
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv": dir_list2.append(entry)
return dir_list2
def read_directory_extended(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv" or entry[-4:] == ".tab": dir_list2.append(entry)
return dir_list2
def refDir():
reference_dir=unique.refDir()
return reference_dir
def eliminate_redundant_dict_values(database):
db1={}
for key in database: list = unique.unique(database[key]); list.sort(); db1[key] = list
return db1
###### Classes ######
class GrabFiles:
def setdirectory(self,value):
self.data = value
def display(self):
print self.data
def searchdirectory(self,search_term):
#self is an instance while self.data is the value of the instance
files,file_dir,file = gene_associations.getDirectoryFiles(self.data,str(search_term))
if len(file)<1: print search_term,'not found'
return file_dir
def searchdirectory_start(self,search_term):
#self is an instance while self.data is the value of the instance
files,file_dir,file = gene_associations.getDirectoryFiles(self.data,str(search_term))
match = ''
for file in files:
split_strs = string.split(file,'.')
split_strs = string.split(split_strs[0],'/')
if search_term == split_strs[-1]: match = file
return match
def getDirectoryFiles(import_dir, search_term):
exact_file = ''; exact_file_dir=''
dir_list = read_directory_extended(import_dir) #send a sub_directory to a function to identify all files in a directory
for data in dir_list: #loop through each file in the directory to output results
if (':' in import_dir) or ('/Users/' == import_dir[:7]) or ('Linux' in platform.system()): affy_data_dir = import_dir+'/'+data
else: affy_data_dir = import_dir[1:]+'/'+data
if search_term in affy_data_dir: exact_file = affy_data_dir
return exact_file
def getAllDirectoryFiles(import_dir, search_term):
exact_files = []
dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
for data in dir_list: #loop through each file in the directory to output results
if ':' in import_dir or ('/Users/' == import_dir[:7]) or ('Linux' in platform.system()): affy_data_dir = import_dir+'/'+data
else: affy_data_dir = import_dir[1:]+'/'+data
if search_term in affy_data_dir: exact_files.append(affy_data_dir)
return exact_files
###### GO-Elite Functions ######
def import_path_index():
global path_id_to_goid; global full_path_db; global all_nested_paths
try: built_go_paths, path_goid_db, path_dictionary = OBO_import.remoteImportOntologyTree(ontology_type)
except IOError:
### This exception was added in version 1.2 and replaces the code in OBO_import.buildNestedOntologyTree which resets the OBO version to 0/0/00 and re-runs (see unlisted_variable = kill)
print_out = "Unknown error encountered during Gene Ontology Tree import.\nPlease report to [email protected] if this error re-occurs."
try: UI.WarningWindow(print_out,'Error Encountered!'); root.destroy(); sys.exit()
except Exception: print print_out; sys.exit()
print 'Imported',ontology_type,'tree-structure for pruning'
full_path_db = built_go_paths
path_id_to_goid = path_goid_db
#all_nested_paths = path_dictionary
#print len(full_path_db), 'GO paths imported'
class MAPPFinderResults:
def __init__(self,goid,go_name,zscore,num_changed,onto_type,permute_p,result_line):
self._goid = goid; self._zscore = zscore; self._num_changed = num_changed
self._onto_type = onto_type; self._permute_p = permute_p; self._go_name= go_name
self._result_line = result_line
def GOID(self): return self._goid
def GOName(self): return self._go_name
def ZScore(self): return self._zscore
def NumChanged(self): return self._num_changed
def OntoType(self): return self._onto_type
def PermuteP(self): return self._permute_p
def ResultLine(self): return self._result_line
def Report(self):
output = self.GOID()+'|'+self.GOName()
return output
def __repr__(self): return self.Report()
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def moveMAPPFinderFiles(input_dir):
###Create new archive directory
import datetime
dir_list = read_directory_extended(input_dir)
input_dir2 = input_dir
if len(dir_list)>0: ###if there are recently run files in the directory
today = str(datetime.date.today()); today = string.split(today,'-'); today = today[0]+''+today[1]+''+today[2]
time_stamp = string.replace(time.ctime(),':','')
time_stamp = string.replace(time_stamp,' ',' ')
time_stamp = string.split(time_stamp,' ') ###Use a time-stamp as the output dir (minus the day)
time_stamp = today+'-'+time_stamp[3]
if 'archived-' not in input_dir2: ### We don't want users to select an archived directory and have the results moved to a child archive directory.
archive_dir = input_dir2 +'/archived-'+time_stamp
fn = filepath(archive_dir)
try: os.mkdir(fn) ###create new directory
except Exception:
try: archive_dir = archive_dir[1:]; fn = filepath(archive_dir); os.mkdir(fn)
except Exception: null = [] ### directory already exists
###Read all files in old directory and copy to new directory
m = GrabFiles(); m.setdirectory(input_dir)
#print 'Moving MAPPFinder results to the archived directory',archive_dir
for mappfinder_input in dir_list:
mappfinder_input_dir = m.searchdirectory(mappfinder_input)
fn = filepath(mappfinder_input_dir)
fn2 = string.split(fn,'/')
destination_dir = string.join(fn2[:-1]+['/archived-'+time_stamp+'/']+[fn2[-1]],'/')
#destination_dir = string.replace(fn,'/'+mappfinder_dir+'/','/'+mappfinder_dir+'/archived-'+time_stamp+'/');
try: shutil.copyfile(fn, destination_dir)
except Exception:
print_out = "WARNING!!! Unable to move ORA results to an archived directory."
#print traceback.format_exc()
print print_out
#try: UI.WarningWindow(print_out,' Exit ')
#except Exception: print print_out
proceed = 'no'
while proceed == 'no':
try: os.remove(fn); proceed = 'yes'
except Exception:
print 'Tried to move the file',mappfinder_input,'to an archived folder, but it is currently open.'
print 'Please close this file and hit return or quit GO-Elite'
inp = sys.stdin.readline()
def checkPathwayType(filename):
type='GeneSet'
ontology_type = string.split(filename,'-')[-1][:-4]
if ontology_type == 'local': ontology_type = 'WikiPathways'
if ontology_type == 'GO': ontology_type = 'GeneOntology'
fn=filepath(filename); x=20; y=0
for line in open(fn,'rU').readlines():
y+=1
if y<x:
if 'Ontology-ID' in line: type = 'Ontology'
return ontology_type,type
def importMAPPFinderResults(filename):
zscore_changed_path_db = {}; go_full = {}; zscore_goid = {}
global dummy_db; dummy_db = {}; global file_headers; file_headers = []
global full_go_name_db; full_go_name_db = {}; filtered_mapps=[]; filtered_mapp_list = []
run_mod = ''; run_source=''; gene_identifier_file = ''
fn=filepath(filename); x=0; y=0
#print "Importing MAPPFinder data for:",filename
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
if x == 0 and y == 0:
if 'Ontology-ID' in data:
x = 1; go_titles = data + '\n'; input_file_type = 'Ontology'
elif 'Gene-Set Name' in data:
y = 1; mapp_titles = data + '\n'; input_file_type = 'GeneSet'
elif data in species_list: ###Check species from results file
if data in species_list: species = data; species_code = species_codes[species]
elif 'source identifiers supplied' in data:
space_delimited = string.split(data,' ')
gene_input_file_data = string.split(data,'supplied in the input file:')
putative_source_type = space_delimited[1] ###Extract out uid from GO/MAPP results file
if putative_source_type in source_types: run_source = putative_source_type
elif putative_source_type in mod_types: run_source = putative_source_type
if len(gene_input_file_data)>1: gene_identifier_file = gene_input_file_data[-1]
elif 'source identifiers linked to' in data:
space_delimited = string.split(data,' ')
putative_mod_type = space_delimited[-2] ###Extract out MOD from GO/MAPP results file
if putative_mod_type in mod_types: run_mod = putative_mod_type
elif 'Probes linked to a ' in data:
space_delimited = string.split(data,' ')
putative_mod_type = space_delimited[-2] ###Extract out MOD from GO/MAPP results file (derived from GenMAPP MAPPFinder results)
if putative_mod_type in mod_types: run_mod = putative_mod_type
else:
print_out = "WARNING!!! The MOD "+putative_mod_type+"\nused by GenMAPP MAPPFinder, is not\navailable in GO-Elite. Errors may\nresult in deriving propper gene\nassociations since a different MOD has to be\nused."
try: UI.WarningWindow(print_out,' Exit ')
except Exception: print print_out
elif x == 1:
try: goid, go_name, go_type, number_changed, number_measured, number_go, percent_changed, percent_present, z_score, permutep, adjustedp = string.split(data,'\t')
except ValueError:
t = string.split(data,'\t')
print_out = "WARNING...GeneOntology results file has too few or too many columns.\nExpected 16 columns, but read"+ str(len(t))
try: UI.WarningWindow(print_out,' Exit ')
except Exception:
print print_out; print t; print 'Please correct and re-run'
sys.exit()
if goid != 'GO': ###Exclude the top level of the GO tree (not an ID number MAPPFinder output)
#go_id_full = 'GO:'+goid
###Check to make sure that the GOID is the right length (usually proceeded by zeros which can be removed)
if len(goid)<7:
extended_goid=goid
while len(extended_goid)< 7: extended_goid = '0'+extended_goid
if extended_goid in full_path_db: goid = extended_goid
if goid in full_path_db:
for path in full_path_db[goid]:
#goid = int(goid); path = path_data.PathTuple() ###Path is a tuple of integers
z_score = float(z_score); permutep = float(permutep); number_changed = int(number_changed)
#path_list = string.split(path,'.')#; path_list=[]; full_tree_index[path] = path_list ###used to created nested gene associations to GO-elite terms
full_go_name_db[goid] = go_name
#enrichment_type
if number_changed > change_threshold and permutep < p_val_threshold and int(number_go) <= max_member_count:
if (z_score > z_threshold and enrichment_type=='ORA') or (z_score < (z_threshold*-1) and enrichment_type=='URA'):
#tree_index[path] = path_list; zscore_changed_path_db[path] = z_score, number_changed; zscore_goid[goid] = z_score, go_type; go_terms[path] = goid
z = MAPPFinderResults(goid,go_name,z_score,number_changed,input_file_type,permutep,data)
zscore_changed_path_db[path] = z; zscore_goid[goid] = z
go_full[goid]= data + '\n'
### for all entries, save a dummy entry. This is needed if we want to re-examine in MAPPFinder but don't want to visualize non-GO-elite selected terms
dummy_data = str(goid) +"\t"+ go_name +"\t"+ go_type
dummy_data = dummy_data+ "\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"0"+"\t"+"1"+"\t"+"1" +"\n"
dummy_db[goid] = dummy_data
elif y == 1:
###CODE USED TO PROCESS LOCAL RESULT FILES FOR MAPPS
try:
try:
t = string.split(data,'\t')
mapp_name = t[0]; num_changed = t[1]; zscore = t[6]; permutep = t[7]; num_on_mapp = t[3]
num_changed = int(num_changed); zscore = float(zscore); permutep = float(permutep)
#print [mapp_name, num_changed, zscore,permutep]
if num_changed > change_threshold and permutep < p_val_threshold and int(num_on_mapp) <= max_member_count:
if (zscore > z_threshold and enrichment_type=='ORA') or (zscore < (z_threshold*-1) and enrichment_type=='URA'):
filtered_mapps.append((zscore, line, mapp_name))
#zscore_goid[mapp_name] = zscore, 'MAPP'
z = MAPPFinderResults(mapp_name,mapp_name,zscore,num_changed,input_file_type,permutep,data)
zscore_goid[mapp_name] = z
filtered_mapp_list.append(mapp_name) ###Use this for downstream analyses
except ValueError: continue
except IndexError: continue
if x == 1:
try: species_code = species_code ###Test to see if there is a proper species name found
except UnboundLocalError:
#print 'No proper species name found in GO results. Please change and re-run'
#inp = sys.stdin.readline(); sys.exit()
###Pick arbitrary species code
for species in species_codes: species_code = species_codes[species]
return run_mod,run_source,zscore_changed_path_db, go_full, go_titles, zscore_goid, input_file_type, gene_identifier_file, species_code
if y == 1:
filtered_mapps.sort(); filtered_mapps.reverse()
return run_mod,run_source,filtered_mapp_list, filtered_mapps, mapp_titles, zscore_goid, input_file_type, gene_identifier_file, species_code
def buildFullTreePath(go_index_list):
"""Find common parent nodes among children and parents and collapse into a single branch node"""
global path_dictionary; path_dictionary = {}; path_dictionary_filtered={}
for tuple_index in go_index_list: path_dictionary[tuple_index] = [] ###store the original top level path-indeces
for tuple_index in go_index_list:
path_len = len(tuple_index); i=-1
while path_len+i > 0:
parent_path = tuple_index[:i]
if parent_path in path_dictionary:
path_dictionary[parent_path].append(tuple_index) ###if a parent path of a
i-=1
for parent_path in path_dictionary:
if len(path_dictionary[parent_path])>0:
path_dictionary[parent_path].sort()
path_dictionary_filtered[parent_path] = path_dictionary[parent_path]
### All GOIDs NOT in path_dictionary_filtered (no significant parents) are added back later
return path_dictionary_filtered
def buildOrderedTree(go_index_list):
"""Find common parent nodes among children and parents and collapse into a single branch node"""
path_dictionary = {}; path_dictionary_filtered={}; organized_tree_db={}
original_path_dictionary={}
for tuple_index in go_index_list:
path_dictionary[tuple_index] = [] ###store the original top level path-indeces
original_path_dictionary[tuple_index] = []
for tuple_index in go_index_list:
path_len = len(tuple_index); i=-1
while path_len+i > 0:
parent_path = tuple_index[:i]
if parent_path in path_dictionary:
path_dictionary[tuple_index].append(parent_path)
original_path_dictionary[parent_path].append(tuple_index)
i-=1
for parent_path in original_path_dictionary:
if len(original_path_dictionary[parent_path])>0:
original_path_dictionary[parent_path].sort()
path_dictionary_filtered[parent_path] = original_path_dictionary[parent_path]
### All GOIDs NOT in path_dictionary_filtered (no significant parents) are added back later
keys_to_delete={}
for child in path_dictionary:
if len(path_dictionary[child])>0:
path_dictionary[child].sort()
new_path = path_dictionary[child][1:]
for path in new_path:
keys_to_delete[path] = []
for path in keys_to_delete:
try: del path_dictionary[path]
except Exception: null=[]
parent_tree_db={}
for child in path_dictionary:
if len(path_dictionary[child])>0:
path_dictionary[child].sort()
new_path = path_dictionary[child][1:]+[child]
#print new_path, path_dictionary[child][0], path_dictionary[child];kill
try: parent_tree_db[path_dictionary[child][0]].append(new_path)
except Exception: parent_tree_db[path_dictionary[child][0]] = [new_path]
else:
organized_tree_db['node',child]=['null']
for path in keys_to_delete:
try: del parent_tree_db[path]
except Exception: null=[]
for path in parent_tree_db:
del organized_tree_db['node',path]
finished = 'no'
while finished == 'no':
parent_tree_db2={}; count=0
for parent_path in parent_tree_db:
if len(parent_tree_db[parent_path])>1:
for childset in parent_tree_db[parent_path]:
top_child = childset[0]
if len(childset)>1:
if 'node' not in parent_path:
new_parent = 'node',parent_path,top_child
else: new_parent = tuple(list(parent_path)+[top_child])
try: parent_tree_db2[new_parent].append(childset[1:])
except Exception: parent_tree_db2[new_parent] = [childset[1:]]
count+=1
elif len(childset)==1:
if 'node' not in parent_path:
new_parent = 'node',parent_path,top_child
else: new_parent = tuple(list(parent_path)+[top_child])
organized_tree_db[new_parent] = ['null']
else:
childset = parent_tree_db[parent_path][0]
if 'node' not in parent_path:
new_parent = 'node',parent_path
else: new_parent = parent_path
#if len(childset)>1: print new_parent, childset;kill
organized_tree_db[new_parent] = childset
if count == 0: finished = 'yes'
parent_tree_db = parent_tree_db2
possible_sibling_paths={}; siblings_reverse_lookup={}
for path in organized_tree_db:
if len(path)>2 and organized_tree_db[path]== ['null']:
try: possible_sibling_paths[path[-1][:-1]].append(path)
except Exception: possible_sibling_paths[path[-1][:-1]] = [path]
### Since some deepest children will have siblings, along with cousins on different branches, we must exclude these
for node in path[1:-1]:
try: siblings_reverse_lookup[node].append(path)
except KeyError: siblings_reverse_lookup[node] = [path]
for node in siblings_reverse_lookup:
try:
if len(siblings_reverse_lookup[node])!=len(possible_sibling_paths[node]):
try: del possible_sibling_paths[node]
except Exception: null=[]
except Exception: null=[]
for common_path in possible_sibling_paths:
if len(possible_sibling_paths[common_path])>1:
sibling_paths=[]
for key in possible_sibling_paths[common_path]:
new_key = tuple(['sibling1']+list(key[1:]))
organized_tree_db[new_key] = ['siblings_type1']
sibling_paths.append(key[-1])
del organized_tree_db[key]
sibling_paths = unique.unique(sibling_paths)
sibling_paths.sort()
new_key = tuple(['sibling2']+list(key[1:-1]))
organized_tree_db[new_key]=sibling_paths
"""
for i in organized_tree_db:
print i, organized_tree_db[i]
kill"""
return organized_tree_db,path_dictionary_filtered
def link_score_to_all_paths(all_paths,zscore_changed_path_db):
temp_list = []
parent_highest_score = {}
for entry in all_paths:
if entry in zscore_changed_path_db:
if filter_method == 'z-score': new_entry = zscore_changed_path_db[entry].ZScore(), entry
elif filter_method == 'gene number':new_entry = zscore_changed_path_db[entry].NumChanged(), entry
elif filter_method == 'combination':
z_score_val = zscore_changed_path_db[entry].ZScore()
gene_num = math.log(zscore_changed_path_db[entry].NumChanged(),2)
new_entry = gene_num*z_score_val, entry
else:
print 'error, no filter method selected!!'
break
for item in all_paths[entry]:
if filter_method == 'z-score':
new_item = zscore_changed_path_db[item].ZScore(), item
temp_list.append(new_item)
elif filter_method == 'gene number':
new_item = zscore_changed_path_db[item].NumChanged(), item
temp_list.append(new_item)
elif filter_method == 'combination':
z_score_val = zscore_changed_path_db[item].ZScore()
gene_num = math.log(zscore_changed_path_db[item].NumChanged(),2)
new_item = gene_num*z_score_val, item
temp_list.append(new_item)
else:
print 'error, no filter method selected!!'
break
max_child_score = max(temp_list)[0]
max_child_path = max(temp_list)[1]
parent_score = new_entry[0]
parent_path = new_entry[1]
if max_child_score <= parent_score:
parent_highest_score[parent_path] = all_paths[entry] #include the elite parent and it's children
#print "parent_path",parent_path,parent_score,"max_child_path", max_child_path, max_child_score
temp_list=[]
"""for entry in parent_highest_score:
print entry,':',parent_highest_score[entry]"""
#print "Number of parents > child in tree:",len(parent_highest_score)
return parent_highest_score
def calculate_score_for_children(tree, zscore_changed_path_db):
temp_list = []
child_highest_score = {}
for entry in tree: ###The tree is a key with one or more nodes representing a branch, chewed back, with or without children
for item in tree[entry]: #get the children of the tree
if item in zscore_changed_path_db:
if filter_method == 'z-score':
new_item = zscore_changed_path_db[item].ZScore(), item
temp_list.append(new_item)
elif filter_method == 'gene number':
new_item = zscore_changed_path_db[item].NumChanged(), item
temp_list.append(new_item)
elif filter_method == 'combination':
z_score_val = zscore_changed_path_db[item].ZScore()
gene_num = math.log(zscore_changed_path_db[item].NumChanged(),2)
new_item = gene_num*z_score_val, item
temp_list.append(new_item)
#print new_item,z_score_val
else:
print 'error, no filter method selected!!'
break
"""elif item == 'siblings_type1': #this will only occur if an parent had multiple children, none of them with nested children
if filter_method == 'z-score':
parent_item = entry[-1]
new_item = zscore_changed_path_db[parent_item][0], parent_item
temp_list.append(new_item)
elif filter_method == 'gene number':
new_item = zscore_changed_path_db[parent_item][1], parent_item
temp_list.append(new_item)
elif filter_method == 'combination':
z_score_val = zscore_changed_path_db[parent_item][0]
gene_num = math.log(zscore_changed_path_db[parent_item][1],2)
new_item = gene_num*z_score_val, parent_item
temp_list.append(new_item)"""
#print new_item,z_score_val
if len(temp_list)> 0: #if there is at least one nested go_path
max_child_score = max(temp_list)[0]
max_child_path = max(temp_list)[1]
child_highest_score[entry]=max_child_path
temp_list = []
"""for entry in child_highest_score:
print entry,':',child_highest_score[entry]"""
return child_highest_score
def collapse_tree(parent_highest_score,child_highest_score,tree):
collapsed = {}
collapsed_parents = {}
collapsed_children = {}
for entry in tree:
count=0 #see next comment
for item in entry:
if item in parent_highest_score and count==0: #ensures that only the top parent is added
collapsed_parents[entry] = item, parent_highest_score[item] #include the children of the elite parent to exclude later
### aka elite_parents[parent_paths] = elite_parent_path, non_elite_children
count +=1 #see last comment
break
for entry in tree:
if entry not in collapsed_parents: ###if the highest score does not occur for one of the parent terms in the key of the tree-branch
if entry in child_highest_score: ###Entry is the parent path stored and the value is the child path with the max score (greater than parent)
max_child_path = child_highest_score[entry]
collapsed_children[entry] = max_child_path, tree[entry] #include the children to exclude from other entries later (includes the max_child_path though)
"""for entry in collapsed_children:
print entry,':',collapsed_children[entry]"""
temp_list = []
for entry in collapsed_parents:
node = collapsed_parents[entry][0]
children = collapsed_parents[entry][1]
collapsed['parent',node] = children
"""if entry in tree:
for item in entry:
temp_list.append(item)
for item in tree[entry]:
temp_list.append(item)
temp_list.sort()
temp_list2 = unique.unique(temp_list)
collapsed['parent',node] = temp_list2
temp_list=[]"""
for entry in collapsed_children:
#previously we included the blocked out code which also
#would later exclude entries that contain key entry items in the elite entries
node = collapsed_children[entry][0]
siblings = collapsed_children[entry][1]
collapsed['child',node] = siblings
"""if entry in tree:
for item in entry: temp_list.append(item)
for item in tree[entry]: temp_list.append(item)
temp_list.sort()
temp_list2 = unique.unique(temp_list)
collapsed['child',node] = temp_list2
temp_list=[]"""
for entry in tree:
nested_path = tree[entry]
if nested_path == ['null']:
collapsed['unique',entry[-1]] = 'unique',entry[-1]
#print "The length of the collapsed list is:",len(collapsed)
###This code is used to check whether or not a tree with multiple siblings listed as children was added to collapsed because the parent term(s) had a higher score
###if not, we must individually add the child sibling terms
for entry in tree:
if tree[entry] == ['siblings_type1']:
parent_root_node = 'parent',entry[-2]
if parent_root_node not in collapsed:
#use 'child' since we have to over-write the child entry created if the 'parent' was'nt more significant (thus syblings would eroneously be considered children)
collapsed['child',entry[-1]] = 'node',entry[-1]
#for entry in collapsed:
#print entry,collapsed[entry]
return collapsed
def link_goid(tree,zscore_changed_path_db,all_paths):
"""Starting with the collapsed tree, convert to goids"""
temp1=[]; temp2=[]; new_tree = {}; count = 0
for entry in tree:
for top_node in entry:
try: goid = zscore_changed_path_db[top_node].GOID(); temp1.append(goid)
except KeyError: not_valid_goid = ''
for child_node in tree[entry]:
try: goid= zscore_changed_path_db[child_node].GOID(); temp2.append(goid)
except KeyError: not_valid_goid = ''
temp2.sort()
new_top = tuple(temp1) #one elite go-term in goid form
new_child = temp2 #list of children (value items) in dictionary, in goid form
count +=1; temp_list=[]
if new_top in new_tree: #if the new dictionary, new_tree already has this key added
top_node = new_top[0]
for node in new_tree[new_top]:
if node != top_node: temp_list.append(node)
for node in new_child:
if node != top_node: temp_list.append(node)
temp_list.sort(); new_list = unique.unique(temp_list)
new_tree[new_top] = new_list; temp_list=[]
else:
#print new_child[0], new_top[0]
if new_child[0] == new_top[0]: #if the first dictionary (which should be the only) value equals the key
new_tree[new_top] = []
else: ###note: if two parents, both more significant than all children, share a child, both will still be reported. This is fine, since we don't know the parent's relationship
temp_list2 = []
for node in new_child:
if node != new_top[0]: #get rid of values that equal the key!!!
temp_list2.append(node)
temp_list2.sort()
new_child = temp_list2
new_tree[new_top] = new_child #this should be the unique term
#print new_top, new_child
temp1 = []; temp2 = []
###remove entries that are parents or children of the most 'elite' entry, in dictionary
###note: parents of the entry shouldn't be selected, since collapsing chooses
###selects the highest scoring node.
if exclude_related == 'yes':
exclude = {}
for entry in new_tree:
for item in new_tree[entry]:
exclude[item] = ''
#print item, entry, new_tree[entry]
new_tree2={}
for entry in new_tree:
if entry[0] not in exclude:
new_tree2[entry[0]] = ''
new_tree = new_tree2
#for entry in tree: print entry,':',tree[entry]
#for entry in new_tree: print entry,':',new_tree[entry]
#print "Length of input tree/output tree",len(tree),'/',len(new_tree),'count:',count
return new_tree
def exportGOResults(go_full,go_titles,collapsed_go_tree,zscore_goid,go_gene_annotation_db,go_values_db,value_headers,goids_with_redundant_genes):
reference_dir = refDir()
if len(go_elite_output_folder) == 0: output_folder = "output"; ref_dir = reference_dir +'/'+output_folder
else: output_folder = go_elite_output_folder; ref_dir = go_elite_output_folder
output_results = output_folder+'/'+mappfinder_input[0:-4]+ '_' +filter_method +'_elite.txt'
if ontology_type == 'WikiPathways':
output_results = string.replace(output_results, 'WikiPathways','local') ### Makes the output filename compatible with GenMAPP-CS plugin filenames
if ontology_type == 'GO':
output_results = string.replace(output_results, 'GeneOntology','GO') ### Makes the output filename compatible with GenMAPP-CS plugin filenames
entries_added=[] #keep track of added GOIDs - later, add back un-added for MAPPFinder examination
print_out = 'Results file: '+output_results+ '\nis open...can not re-write.\nPlease close file and select "OK".'
try: raw = export.ExportFile(output_results)
except Exception:
try: UI.WarningWindow(print_out,' OK ')
except Exception:
print print_out; print 'Please correct (hit return to continue)'; inp = sys.stdin.readline()
if export_user_summary_values == 'yes' and len(value_headers)>0:
value_headers2=[]; stdev_headers=[]
for i in value_headers: value_headers2.append('AVG-'+i); stdev_headers.append('STDEV-'+i)
value_headers = '\t'+string.join(value_headers2+stdev_headers,'\t')
else: value_headers = ''
if len(go_gene_annotation_db)>0: go_titles = go_titles[:-1]+'\t'+'redundant with terms'+'\t'+'inverse redundant'+'\t'+'gene symbols'+value_headers+'\n' ###If re-outputing results after building gene_associations
raw.write(go_titles)
combined_results[output_results] = [((ontology_type,'Ontology'),mappfinder_input,go_titles)]
#append data to a list, to sort it by go_cateogry and z-score
collapsed_go_list = []; collapsed_go_list2 = []; goids_redundant_with_others={}
for entry in collapsed_go_tree:
goid = entry
entries_added.append(goid)
if goid in zscore_goid:
z_score_val = zscore_goid[goid].ZScore()
go_type = zscore_goid[goid].OntoType()
if sort_only_by_zscore == 'yes': info = z_score_val,z_score_val,goid
else: info = go_type,z_score_val,goid
collapsed_go_list.append(info); collapsed_go_list2.append(goid)
collapsed_go_list.sort()
collapsed_go_list.reverse()
for (go_type,z_score_val,goid) in collapsed_go_list:
if goid in go_full:
data = go_full[goid]
if len(go_gene_annotation_db)>0:
symbol_ls = []; goid_vals = ''
if goid in go_gene_annotation_db:
for s in go_gene_annotation_db[goid]:
if len(s.Symbol())>0: symbol_ls.append(s.Symbol()) #s.GeneID()
else: symbol_ls.append(s.GeneID())
if goid in go_values_db:
goid_vals = string.join(go_values_db[goid][0]+go_values_db[goid][1],'\t') ###mean of values from input file, for each GO term
symbol_ls = unique.unique(symbol_ls); symbol_ls.sort()
symbols = string.join(symbol_ls,'|')
try:
rr = goids_with_redundant_genes[goid]
redundant_with_terms = rr.RedundantNames(); inverse_redundant = rr.InverseNames()
if len(redundant_with_terms)>1: goids_redundant_with_others[goid]=[]
except KeyError: redundant_with_terms = ' '; inverse_redundant =' '
if export_user_summary_values == 'yes' and len(goid_vals)>0: goid_vals = '\t'+goid_vals
else: goid_vals = ''
data = data[:-1]+'\t'+redundant_with_terms+'\t'+inverse_redundant+'\t'+symbols+goid_vals+'\n' ###If re-outputing results after building gene_associations
raw.write(data)
combined_results[output_results].append(((ontology_type,'Ontology'),mappfinder_input,data))
raw.close()
combined_results[output_results].append((('',''),'',''))
summary_data_db['redundant_go_term_count'] = len(goids_redundant_with_others)
#print "New MAPPFinder Elite file", output_results, "written...."
return collapsed_go_list2
def exportLocalResults(filtered_mapps,mapp_titles,mapp_gene_annotation_db,mapp_values_db,mapp_value_headers,mapps_with_redundant_genes):
if len(go_elite_output_folder) == 0: output_folder = "output"
else: output_folder = go_elite_output_folder
output_results = output_folder+'/'+mappfinder_input[0:-4]+ '_' +filter_method + '_elite.txt'
try: raw = export.ExportFile(output_results)
except Exception:
try: UI.WarningWindow(print_out,' OK ')
except Exception:
print print_out; print 'Please correct (hit return to continue)'; inp = sys.stdin.readline()
if export_user_summary_values == 'yes' and len(mapp_value_headers)>0:
mapp_value_headers2=[]; stdev_headers=[]
for i in mapp_value_headers: mapp_value_headers2.append('AVG-'+i); stdev_headers.append('STDEV-'+i)
mapp_value_headers = '\t'+string.join(mapp_value_headers2+stdev_headers,'\t')
else: mapp_value_headers = ''
if len(mapp_gene_annotation_db)>0: mapp_titles = mapp_titles[:-1]+'\t'+'redundant with terms'+'\t'+'inverse redundant'+'\t'+'gene symbols'+mapp_value_headers+'\n'
raw.write(mapp_titles)
combined_results[output_results] = [((ontology_type,'GeneSet'),mappfinder_input,mapp_titles)]
filtered_mapp_list = []; mapps_redundant_with_others={}
for (zscore,line,mapp_name) in filtered_mapps:
if len(mapp_gene_annotation_db)>0:
symbol_ls = []; mapp_vals=''
if mapp_name in mapp_gene_annotation_db:
for s in mapp_gene_annotation_db[mapp_name]:
if len(s.Symbol())>0: symbol_ls.append(s.Symbol()) #s.GeneID()
else: symbol_ls.append(s.GeneID())
symbol_ls = unique.unique(symbol_ls); symbol_ls.sort()
symbols = string.join(symbol_ls,'|')
if mapp_name in mapp_values_db:
mapp_vals = string.join(mapp_values_db[mapp_name][0]+mapp_values_db[mapp_name][1],'\t') ###mean of values from input file, for each MAPP
try:
rr = mapps_with_redundant_genes[mapp_name]
redundant_with_terms = rr.RedundantNames(); inverse_redundant = rr.InverseNames()
if len(redundant_with_terms)>1: mapps_redundant_with_others[mapp_name]=[]
except KeyError: redundant_with_terms = ' '; inverse_redundant = ' '
if export_user_summary_values == 'yes' and len(mapp_vals)>0: mapp_vals = '\t'+mapp_vals
else: mapp_vals = ''
line = line[:-1]+'\t'+redundant_with_terms+'\t'+inverse_redundant+'\t'+symbols+mapp_vals+'\n' ###If re-outputing results after building gene_associations
raw.write(line)
combined_results[output_results].append(((ontology_type,'GeneSet'),mappfinder_input,line))
raw.close()
combined_results[output_results].append((('',''),'',''))
#print "Local Filtered MAPPFinder file", output_results, "written...."
summary_data_db['redundant_mapp_term_count'] = len(mapps_redundant_with_others)
def importORASimple(ora_dir,elite_pathway_db,file_type):
""" This function imports pathway data for any elite pathway from the unfiltered results """
summary_results_db={}
ontology_name = file_type
if file_type == 'GeneOntology': file_type = 'GO.'
if file_type == 'WikiPathways': file_type = 'local'
dir_list = read_directory(ora_dir)
for file in dir_list:
if '.txt' in file and file_type in file:
fn=filepath(ora_dir+'/'+file)
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
try:
### Applies to Ontology ORA files
pathway_id = t[0]; pathway_name = t[1]; num_changed = t[3]; num_measured = t[4]; zscore = t[8]; permutep = t[9]; pathway_type = t[2]
except IndexError:
### Applies to Local pathway files
try:
pathway_name = t[0]; num_changed = t[1]; num_measured = t[2]; zscore = t[6]; permutep = t[7]; pathway_id = pathway_name; pathway_type = 'GeneSet'
except Exception: pathway_name='null'
### If the pathway/ontology term was considered a pruned term (elite)
if (pathway_name,ontology_name) in elite_pathway_db:
try:
int(num_changed)
### Encode this data immediately for combination with the other input files and for export
key = (pathway_id,pathway_name,ontology_name,pathway_type)
values = [num_changed,num_measured,zscore,permutep]
try: summary_results_db[pathway_name,key].append([file,values])
except Exception: summary_results_db[pathway_name,key] = [[file,values]]
except Exception: null=[]
return summary_results_db
def outputOverlappingResults(combined_results,ora_dir):
"""For any elite term/pathway from a user analysis, output all associated original ORA data"""
output_folder = go_elite_output_folder
output_results = output_folder+'/'+'overlapping-results_' +filter_method + '_elite.txt'
proceed = 'no'
while proceed == 'no':
try: raw = export.ExportFile(output_results); proceed = 'yes'
except Exception:
print_out = output_results, '\nis open. Please close and select "Continue".'
try: UI.WarningWindow(print_out,' OK ')
except Exception:
print print_out; print 'Please correct (hit return to continue)'; inp = sys.stdin.readline()
filename_list=[]; criterion={}
for filename in combined_results:
filename_list.append(filename)
criterion[string.join(string.split(filename,'-')[:-2],'-')]=None
filename_list.sort() ### rank the results alphebetically so local and GO results are sequential
ontology_to_filename={}; pathway_to_filename={}
### determine which Elite pathways are in which files and store
if len(criterion)>1:
ontology_files={}; local_files={}
for filename in filename_list:
file = export.findFilename(filename)[:-4]
for (pathway_type,mapp_name,line) in combined_results[filename]:
specific_pathway_type,pathway_type = pathway_type
t = string.split(line,'\t')
if pathway_type == 'Ontology':
go_name = t[1]
try: ontology_to_filename[go_name,specific_pathway_type].append(file)
except Exception: ontology_to_filename[go_name,specific_pathway_type]=[file]
if specific_pathway_type not in ontology_files:
ontology_files[specific_pathway_type] = [file]
elif file not in ontology_files[specific_pathway_type]:
ontology_files[specific_pathway_type].append(file)
elif pathway_type == 'GeneSet':
pathway_name = t[0]
try: pathway_to_filename[pathway_name,specific_pathway_type].append(file)
except Exception: pathway_to_filename[pathway_name,specific_pathway_type]=[file]
if specific_pathway_type not in local_files:
local_files[specific_pathway_type] = [file]
elif file not in local_files[specific_pathway_type]:
local_files[specific_pathway_type].append(file)
headers = ['number_changed','number_measured',' z_score','permuteP']
header_ontology = ['Ontology-ID','Ontology-term','Ontology-name','Ontology-type','elite_term_in']
header_local = ['GeneSet-ID','GeneSet-term','GeneSet-name','GeneSet-type','elite_term_in']
for ontology_type in ontologies_analyzed:
input_file_type = ontologies_analyzed[ontology_type]
if input_file_type == 'Ontology':
ontology_elite_db = importORASimple(ora_dir,ontology_to_filename,ontology_type)
writeOverlapLine(raw,ontology_files,headers,header_ontology,ontology_elite_db,ontology_to_filename,ontology_type)
else:
local_elite_db = importORASimple(ora_dir,pathway_to_filename,ontology_type)
writeOverlapLine(raw,local_files,headers,header_local,local_elite_db,pathway_to_filename,ontology_type)
raw.write('\n')
raw.close()
### Move to the root directory
fn = filepath(output_results)
fn2 = string.replace(fn,'CompleteResults/ORA_pruned','')
try:
export.customFileMove(fn,fn2)
from visualization_scripts import clustering
clustering.clusterPathwayZscores(fn2) ### outputs the overlapping results as a heatmap
except Exception,e:
#print e
#print fn; print fn2; print "OverlapResults failed to be copied from CompleteResults (please see CompleteResults instead)"
pass
def writeOverlapLine(raw,ontology_files,headers,header_ontology,ontology_elite_db,ontology_to_filename,ontology_type):
file_headers=[]
filenames = ontology_files[ontology_type]
filenames.sort() ### Sort by filename
for filename in filenames:
file_headers += updateHeaderName(headers,filename)
title = string.join(header_ontology+file_headers,'\t')+'\n'
raw.write(title)
for (pathway_name,key) in ontology_elite_db:
elite_files = string.join(ontology_to_filename[pathway_name,ontology_type],'|')
row = list(key)+[elite_files]
scores_data = ontology_elite_db[(pathway_name,key)]
scores_data.sort() ### Sort by filename to match above
for (file,values) in scores_data:
row += values
raw.write(string.join(row,'\t')+'\n')
def updateHeaderName(headers,filename):
headers_copy = copy.deepcopy(headers)
i=0
for header in headers_copy:
headers_copy[i]=header+'.'+filename
i+=1
return headers_copy
def output_combined_results(combined_results):
if len(go_elite_output_folder) == 0: output_folder = "output"
else: output_folder = go_elite_output_folder
output_results = output_folder+'/'+'pruned-results_' +filter_method + '_elite.txt'
proceed = 'no'
while proceed == 'no':
try: raw = export.ExportFile(output_results); proceed = 'yes'
except Exception:
print_out = output_results, '\nis open. Please close and select "Continue".'
try: UI.WarningWindow(print_out,' OK ')
except Exception:
print print_out; print 'Please correct (hit return to continue)'; inp = sys.stdin.readline()
filename_list=[]
for filename in combined_results: filename_list.append(filename)
filename_list.sort() ### rank the results alphebetically so local and GO results are seqeuntial
for filename in filename_list:
for (pathway_type,mapp_name,line) in combined_results[filename]:
specific_pathway_type,pathway_type = pathway_type
data = cleanUpLine(line)
t = string.split(data,'\t')
if pathway_type == 'Ontology':
goid = t[0]
del t[0]; #del t[2:7]
go_type = t[1]; del t[1]; specific_pathway_type = go_type
t[0] = t[0]+'('+goid+')'
t.reverse(); t.append(specific_pathway_type); t.append(mapp_name); t.reverse()
vals = string.join(t,'\t'); vals = vals + '\n'
raw.write(vals)
#print filename, len(combined_results[filename])
raw.close()
def identifyGeneFiles(import_dir, mappfinder_input):
e = GrabFiles(); e.setdirectory(import_dir)
if len(gene_identifier_file)>0: ###global variable set upon MAPPFinder result import
gene_file_dir = e.searchdirectory(gene_identifier_file)
return gene_file_dir
else:
mappfinder_input = string.replace(mappfinder_input,'-GO.txt','') ###the prefix in most cases will be the same for the MAPPFinder results and gene input filename
mappfinder_input = string.replace(mappfinder_input,'-local.txt','')
split_name = string.split(mappfinder_input,'.')
gene_file_dir = e.searchdirectory(mappfinder_input)
if len(gene_file_dir)>0: return gene_file_dir
else:
try:
index = int(split_name[0])
index_str = str(index)
gene_file_dir = e.searchdirectory_start(index_str)
except ValueError: gene_file_dir =''
return gene_file_dir
def grabAllNestedGOIDs(collapsed_go_tree,all_paths):
###Updated all_paths contains all possible paths listed in GO
nested_collapsed_go_tree={}
for goid_tuple in collapsed_go_tree:
for goid in goid_tuple:
child_goids=[]
path_id_list = full_path_db[goid]
for path_id in path_id_list: ###examine all possible paths for that goid (and different possible children)
#path_id = path_id_data.PathTuple()
if path_id in all_paths:
child_path_id_list = all_paths[path_id]
for child_path_id in child_path_id_list:
child_goid = path_id_to_goid[child_path_id]
child_goids.append(child_goid) ###append all possible children terms to a new list
nested_collapsed_go_tree[goid] = child_goids
nested_collapsed_go_tree = eliminate_redundant_dict_values(nested_collapsed_go_tree)
return nested_collapsed_go_tree
def checkGOEliteSpecies(species):
### For an external program, see if the species data is supported
speciesData()
if species in species_names: return 'yes'
else: return 'no'
def speciesData():
importSpeciesData()
if len(species_names)==0:
UI.remoteSpeciesInfo('no') ### will get the missing data from a backup file
importSpeciesData()
def importSpeciesData():
program_type,database_dir = unique.whatProgramIsThis()
if program_type == 'GO-Elite': filename = 'Config/species.txt'
else: filename = 'Config/goelite_species.txt'
x=0
fn=filepath(filename);global species_list; species_list=[]; global species_codes; species_codes={}
global species_names; species_names={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
abrev=t[0]; species=t[1]
if x==0: x=1
else:
species_list.append(species)
species_codes[species] = abrev
species_names[abrev] = species
def testFileLength(fn):
x=0
for line in open(fn,'rU').readlines(): x+=1
return x
def sourceDataCheck():
filename = 'Config/source_data.txt'; fn=filepath(filename)
file_length = testFileLength(fn)
if file_length <2:
fn2 = string.replace(fn,'.txt','_archive.txt')
import shutil; shutil.copyfile(fn2,fn) ### Bad file was downloaded (with warning)
def getSourceData():
sourceDataCheck(); sourceData()
return system_codes,source_types,mod_types
def getSourceDataNames():
sourceDataCheck(); sourceData()
system_code_names={}
for sc in system_codes:
name = system_codes[sc]
system_code_names[name]=sc
return system_code_names,source_types,mod_types
def sourceData():
filename = 'Config/source_data.txt'; x=0
fn=filepath(filename)
global source_types; source_types=[]
global system_codes; system_codes={}
global mod_types; mod_types=[]
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
t = string.split(data,'\t'); source=t[0]
try: system_code=t[1]
except IndexError: system_code = 'NuLL'
if x==0: x=1
else:
if len(t)>2: ### Therefore, this ID system is a potential MOD
if t[2] == 'MOD': mod_types.append(source)
if source not in mod_types: source_types.append(source)
system_codes[system_code] = source ###Used when users include system code data in their input file
def buildFullPathForCollapsedTree(collapsed_tree):
filtered_tree_index=[]
for (type,path) in collapsed_tree: filtered_tree_index.append(path)
all_nested_paths = buildFullTreePath(filtered_tree_index)
return all_nested_paths
def getNonEliteTerms(collapsed_goid_list,full_goid_db):
non_elite_db=[]
for goid in full_goid_db:
if goid not in collapsed_goid_list: non_elite_db.append(goid)
#non_elite_db.append(goid)
print "Out of",len(collapsed_goid_list),"GO-Elite terms for",len(full_goid_db),"terms examined,",len(non_elite_db),"terms are not-elite"
return non_elite_db
def countGOFullGenes(go_full,go_to_mod_genes):
### Count unique genes associated with each filtered pathway
unique_go_full_genes={}
for goid in go_full:
if goid in go_to_mod_genes:
for gene in go_to_mod_genes[goid]: unique_go_full_genes[gene] = []
return len(unique_go_full_genes)
def reorganizeResourceList(pathway_list):
### Cluster by resource type to minimize import of annotation and hierarchies
geneset_types = {}
for pathway in pathway_list:
geneset_type = string.split(pathway,'-')[-1][:-4]
try: geneset_types[geneset_type].append(pathway)
except Exception: geneset_types[geneset_type] = [pathway]
### Save as a list
pathway_list=[]
for geneset_type in geneset_types:
pathway_list+=geneset_types[geneset_type]
### Make sure that WikiPathways and GO are analyzed last, so that gene results are also reported last to GO_Elite.py
add_pathway=[]
pathway_list_reorganized=[]
for pathway in pathway_list:
if '-local.txt' in pathway: add_pathway.append(pathway)
elif '-GO.txt' in pathway: add_pathway.append(pathway)
else: pathway_list_reorganized.append(pathway)
pathway_list_reorganized+=add_pathway
return pathway_list_reorganized
def getAvaialbleResources(species_code):
program_type,database_dir = unique.whatProgramIsThis()
import_dir1 = '/'+database_dir+species_code+'/gene-mapp'
import_dir2 = '/'+database_dir+species_code+'/gene-go'
default_resources=[]
try:
gene_mapp_list = read_directory(import_dir1)
gene_mapp_list.sort()
for file in gene_mapp_list:
resource = string.split(file,'-')[-1][:-4]
if resource != 'MAPP' and resource not in default_resources and '.txt' in file:
default_resources.append(resource)
if resource == 'MAPP' and 'Pathways' not in default_resources and '.txt' in file:
default_resources.append('Pathways')
except Exception: pass
try:
gene_go_list = read_directory(import_dir2)
gene_go_list.sort()
for file in gene_go_list:
resource = string.split(file,'-')[-1][:-4]
if resource not in default_resources and 'version' not in resource and '.txt' in file:
default_resources.append(resource)
except Exception: pass
return default_resources
def multiMappfinder(species,species_code,source_data,mod,system_codes,permutations,resources_to_analyze,file_dirs,root):
""" Run in multiprocessing mode to run all jobs in parallel """
multiProcessing = True
multiprocessing_pipe = True
if multiprocessing_pipe:
queue = mlp.Queue()
if type(resources_to_analyze) is list: resources = resources_to_analyze
elif resources_to_analyze=='all': resources = getAvaialbleResources(species_code)
elif resources_to_analyze == 'both': resources = ['GeneOntology','Pathways']
elif isinstance(resources_to_analyze, list): resources = resources_to_analyze
else: resources = [resources_to_analyze]
variable_list=[]; incl=True
for resource in resources:
criterion_input_folder, criterion_denom_folder, output_dir, custom_sets_folder = file_dirs
input_dir_files = readDirText(criterion_input_folder)
for input_file in input_dir_files:
if incl: custom_sets_folder = custom_sets_folder; x=1 ### This file should only be processed once
else: custom_sets_folder = ''
new_file_dirs = input_file, criterion_denom_folder, output_dir, custom_sets_folder
variables = species,species_code,source_data,mod,system_codes,permutations,resource,new_file_dirs,''
variable_list.append(variables)
processes=mlp.cpu_count()
### This is approach less efficeint than pool but we print out the progress
s = processes; b=0
list_of_vl=[]
while s<len(variable_list):
list_of_vl.append(variable_list[b:s])
b+=processes; s+=processes
list_of_vl.append(variable_list[b:s])
start_time = time.time()
print 'Performing Over Representation Analyses (ORA) on input datasets... (be patient)'
go_ora_genes = {}
local_ora_genes = {}
ontology_ora_genes = {}
geneset_ora_genes = {}
if multiProcessing:
if multiprocessing_pipe:
for variable_list in list_of_vl:
procs=list()
for varset in variable_list:
species,species_code,source_data,mod,system_codes,permutations,resource,new_file_dirs,null = varset
proc = mlp.Process(target=mappfinder.generateMAPPFinderScores,args=(species,species_code,source_data,mod,system_codes,permutations,resource,new_file_dirs,None,True,queue))
procs.append(proc)
proc.start()
for _ in procs:
vals= queue.get()
if len(vals)==1:
print_out = vals[0]
if root==None:
print '\nWarning!!!',print_out,'\n'; sys.exit()
else:
try: UI.WarningWindow(print_out,'Critical Error!'); sys.exit()
except Exception: print '\nWarning!!!',print_out,'\n'; sys.exit()
else:
go_to_mod_genes, mapp_to_mod_genes, timediff, mappfinder_input, resource = vals
if 'GeneOntology' in resource:
go_ora_genes[mappfinder_input] = go_to_mod_genes
elif 'Pathways' in resource or 'MAPP' in resource:
local_ora_genes[mappfinder_input] = mapp_to_mod_genes
elif len(go_to_mod_genes)>0:
ontology_ora_genes[mappfinder_input] = go_to_mod_genes
elif len(mapp_to_mod_genes)>0:
geneset_ora_genes[mappfinder_input] = mapp_to_mod_genes
print '\tORA of',mappfinder_input[:-4],resource,'complete in %s seconds' % timediff
for proc in procs:
proc.join()
### Select a GO and local set (or other) to report for the last mappfinder_input considered
input_file = export.findFilename(input_file)
try:
if len(go_ora_genes)>0:
go_to_mod_genes = go_ora_genes[input_file]
elif len(ontology_ora_genes)>0:
go_to_mod_genes = ontology_ora_genes[input_file]
if len(go_ora_genes)>0:
mapp_to_mod_genes = local_ora_genes[input_file]
elif len(ontology_ora_genes)>0:
mapp_to_mod_genes = geneset_ora_genes[input_file]
except Exception: pass
else:
pool_size = mlp.cpu_count()
pool = mlp.Pool(processes=pool_size)
results = pool.map(runMappfinderProcesses,variable_list)
### go_to_mod_genes and mapp_to_mod_genes are only needed for the last criterion (local and GO) for screen reporting
if len(results)==1:
print results[0]; forceError
go_to_mod_genes, mapp_to_mod_genes, timediff, mappfinder_input, resource = results[-1]
else:
for variables in variable_list:
species,species_code,source_data,mod,system_codes,permutations,resource,file_dirs,r = variables
go_to_mod_genes, mapp_to_mod_genes, timediff, mappfinder_input, resource = mappfinder.generateMAPPFinderScores(species,species_code,source_data,mod,system_codes,permutations,resource,file_dirs,r,Multi=mlp)
try: pool.close(); pool.join(); pool = None
except Exception: pass
end_time = time.time()
time_diff = mappfinder.formatTime(start_time,end_time)
print 'ORA analyses finished in %s seconds' % time_diff,'\n'
return go_to_mod_genes, mapp_to_mod_genes
def runMappfinderProcesses(variables):
try:
species,species_code,source_data,mod,system_codes,permutations,resources_to_analyze,file_dirs,root = variables
go_to_mod_genes, mapp_to_mod_genes, time_diff, mappfinder_input, resource = mappfinder.generateMAPPFinderScores(species,species_code,source_data,mod,system_codes,permutations,resources_to_analyze,file_dirs,root,poolVar=True)
except Exception:
return [traceback.format_exc()]
return go_to_mod_genes, mapp_to_mod_genes, input_file, time_diff, mappfinder_input, resource
def runMappfinderPipe(variables):
try:
species,species_code,source_data,mod,system_codes,permutations,resources_to_analyze,file_dirs,queue = variables
mappfinder.generateMAPPFinderScores(species,species_code,source_data,mod,system_codes,permutations,resources_to_analyze,file_dirs,root,poolVar=True)
except Exception:
root.put([traceback.format_exc()])
def readDirText(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt": dir_list2.append(sub_dir+'/'+entry)
return dir_list2
def runGOElite(mod):
print 'Running GO-Elite version 1.2.6\n'
#UI.WarningWindow('test','test')
source_data = mod; speciesData(); sourceDataCheck(); sourceData()
try: species_code = species_codes[species]
except KeyError: species_code=''
global exclude_related; global mappfinder_input; global nested_collapsed_go_tree; global uid_to_go; global collapsed_go_tree
global path_dictionary_include; global organized_tree; global parent_highest_score; global gene_identifier_file; global main_output_folder
global ontology_type; global ontologies_analyzed; ontologies_analyzed = {}
exclude_related = 'yes'
global program_type; global log_file
program_type,database_dir = unique.whatProgramIsThis()
global combined_results; combined_results={}; global go_to_mod_genes; global mapp_to_mod_genes; global zscore_goid
global combined_associations; combined_associations={}; global combined_gene_ranking; combined_gene_ranking={}
### Add empty values for when there is no data on these values
summary_keys = ['filtered_go_term_count','elite_go_term_count','redundant_go_term_count','filtered_local_count']
summary_keys +=['redundant_mapp_term_count','go_gene_full_count','go_gene_elite_count','mapp_gene_elite_count']
for key in summary_keys: summary_data_db[key]='0'
print_items=[]
print_items.append("Primary GO-Elite Parameters")
print_items.append('\t'+'commandLineMode'+': '+commandLineMode)
print_items.append('\t'+'species'+': '+species_code)
print_items.append('\t'+'results-directory'+': '+main_output_folder)
print_items.append('\t'+'filter_method'+': '+filter_method)
print_items.append('\t'+'z_threshold'+': '+str(z_threshold))
print_items.append('\t'+'p_val_threshold'+': '+str(p_val_threshold))
print_items.append('\t'+'change_threshold'+': '+str(change_threshold))
print_items.append('\t'+'sort_only_by_zscore'+': '+sort_only_by_zscore)
print_items.append('\t'+'analysis_method'+': '+str(analysis_method))
print_items.append('\t'+'criterion_input_folder'+': '+criterion_input_folder)
print_items.append('\t'+'custom_sets_folder'+': '+custom_sets_folder)
print_items.append('\t'+'enrichment_type'+': '+enrichment_type)
universalPrintFunction(print_items)
if run_mappfinder=='yes':
global path_id_to_goid; global full_path_db; global all_nested_paths; global collapsed_tree; export_databases = 'no'
file_dirs = criterion_input_folder, criterion_denom_folder, main_output_folder, custom_sets_folder
denom_search_dir = criterion_denom_folder
multiProcessing = False ### For GO-Elite as a stand-alone app, be more conservative and allow for more printouts (less files typically run - slightly slower)
if program_type == 'AltAnalyze': multiProcessing = False ### Faster in speed tests thus far (likely memory issues for many input sets run simultaneously)
resources = resources_to_analyze ### Have to rename due to global conflict reassignment
if type(resources) is list:
if 'all' in resources: resources = 'all'
elif len(resources)==1: resources = resources[0]
else:
multiProcessing = True ### Allows for distinct resources to be run in parallel
multiProcessing = False
permute = permutations ### Have to rename due to global conflict reassignment
if denom_search_dir==None:
permute = 'FisherExactTest'
print_items = []
print_items.append("ORA Parameters")
print_items.append('\t'+'mod'+': '+mod)
print_items.append('\t'+'permutations'+': '+str(permute))
print_items.append('\t'+'resources_to_analyze'+': '+str(resources))
universalPrintFunction(print_items)
if denom_search_dir==None:
print '\nUPDATED PARAMETERS - Forcing ORA algorithm to be FisherExactTest (required with no supplied denominator)\n'
denom_search_dir='' ### Need this to be a string for downstream functions
### go_to_mod_genes and mapp_to_mod_genes are only needed for the last criterion (local and GO) for screen reporting
if multiProcessing == True:
go_to_mod_genes, mapp_to_mod_genes = multiMappfinder(species,species_code,source_data,mod,system_codes,permute,resources,file_dirs,root)
else:
try:go_to_mod_genes, mapp_to_mod_genes, timediff, mappfinder_input, resource = mappfinder.generateMAPPFinderScores(species,species_code,source_data,mod,system_codes,permute,resources,file_dirs,root,Multi=mlp)
except Exception: print traceback.format_exc()
reload(mappfinder) ### Clear memory of any retained objects
else: denom_search_dir = ''
global export_user_summary_values; global go_elite_output_folder
get_non_elite_terms_only = 'no' ### Used for internal benchmarking and QC
export_user_summary_values = 'yes'
user_defined_source = source_data
user_defined_mod = mod
if len(main_output_folder) == 0: import_dir = '/input/MAPPFinder'; go_elite_output_folder = ''
else:
if run_mappfinder == 'no':
import_dir = main_output_folder
output_dir = main_output_folder
if 'GO-Elite_results/CompleteResults/ORA_pruned' in output_dir: output_dir = string.replace(output_dir,'/GO-Elite_results/CompleteResults/ORA_pruned','')
main_output_folders = string.split(output_dir,'/')
go_elite_output_folder = string.join(main_output_folders[:-1],'/') + '/GO-Elite_results/CompleteResults/ORA_pruned' ###Get only the parent directory
else:
if 'GO-Elite_results/CompleteResults/ORA' not in main_output_folder:
import_dir = main_output_folder + '/GO-Elite_results/CompleteResults/ORA'
go_elite_output_folder = main_output_folder+ '/GO-Elite_results/CompleteResults/ORA_pruned'
else:
import_dir = main_output_folder
root_import_dir,child_dirs = string.split(main_output_folder,'/GO-Elite_results/CompleteResults/ORA')
go_elite_output_folder = root_import_dir+ '/GO-Elite_results/CompleteResults/ORA_pruned'
if len(criterion_input_folder)==0: gene_input_dir = '/input/GenesToQuery/'+species_code
else: gene_input_dir = criterion_input_folder
m = GrabFiles(); m.setdirectory(import_dir)
try: dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
except Exception:
try: dir_list = read_directory(filepath('Databases/'+species_code+'/nested'))
except Exception: dir_list = []
log_report = open(log_file,'a')
d = "File in the folder nested are: "+str(dir_list); log_report.write(d+'\n')
log_report.write('('+filepath('Databases/'+species_code+'/nested')+')\n')
mappfinder_db_input_dir = '/'+species_code+'/nested/'
#try: print os.name, platform.win32_ver()[0], platform.architecture(), platform.mac_ver(), platform.libc_ver(), platform.platform()
#except Exception: print os.name
#exportLog(log_report)
### This exception was added in version 1.2 and replaces the code in OBO_import.buildNestedOntologyTree which resets the OBO version to 0/0/00 and re-runs (see unlisted_variable = kill)
print_out = "Unknown error encountered during data processing.\nPlease see logfile in:\n\n"+log_file+"\nand report to [email protected]."
if root != None:
program,program_dir = unique.whatProgramIsThis()
if program!= 'AltAnalyze':
try: UI.WarningWindow(print_out,'Error Encountered!'); root.destroy()
except Exception: print print_out
"""
if os.name == 'nt':
try: os.startfile('"'+log_file+'"')
except Exception: os.system('open "'+log_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+log_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+log_file+'/"')
sys.exit()
"""
global dataset_name; go_ora_files=0; local_ora_files=0; oraDirTogeneDir={}
dir_list = reorganizeResourceList(dir_list)
prior_ontology_type = None
for mappfinder_input in dir_list: #loop through each file in the directory to output results
try:
dataset_name = string.join(string.split(mappfinder_input,'-')[:-1],'-')
print 'Pruning',mappfinder_input
source_data = user_defined_source ###Resets the variables back to the user defined when missing from the MAPPFinder files
mod = user_defined_mod
mappfinder_input_dir = m.searchdirectory(mappfinder_input)
ontology_type,input_file_type = checkPathwayType(mappfinder_input_dir)
ontologies_analyzed[ontology_type] = input_file_type ### Use when examining overlapping terms between criterion
if input_file_type == 'Ontology' and prior_ontology_type != ontology_type: ### Ensures we don't reimport an already imported tree-structure
import_path_index() ### need_batch_efficient
try:
run_mod,run_source,zscore_changed_path_db,go_full,go_titles,zscore_goid,input_file_type,gene_identifier_file,species_code = importMAPPFinderResults(mappfinder_input_dir)
except Exception:
print_out = 'Impropper ORA file format! Make sure to\n select the correct pre-processed input files.'
if root != None:
try: print "Analysis Failed"; UI.WarningWindow(print_out,'Critical Error!'); root.destroy()
except IOError: print "Analysis Failed\n"
else:
print "Analysis Failed\n"
sys.exit()
if len(run_mod)>1: mod = run_mod
if len(run_source)>1: source_data = run_source
if input_file_type == 'Ontology':
go_ora_files+=1
if run_mappfinder == 'yes' and 'GO.txt' in mappfinder_input_dir:
### Used for gene-level summary reporting
go_gene_full_count = countGOFullGenes(go_full,go_to_mod_genes); summary_data_db['go_gene_full_count'] = go_gene_full_count
organized_tree,all_paths = buildOrderedTree(zscore_changed_path_db) ### Improved GO-tree reconstruction method - implemented in version 1.22
parent_highest_score = link_score_to_all_paths(all_paths,zscore_changed_path_db)
child_highest_score = calculate_score_for_children(organized_tree,zscore_changed_path_db)
collapsed_tree = collapse_tree(parent_highest_score,child_highest_score,organized_tree)
collapsed_go_tree = link_goid(collapsed_tree,zscore_changed_path_db,all_paths)
collapsed_go_list = exportGOResults(go_full,go_titles,collapsed_go_tree,zscore_goid,{},{},{},{})
summary_data_db['elite_go_term_count'] = len(collapsed_go_list) ### All Elite GO-terms
summary_data_db['filtered_go_term_count'] = len(go_full)
###For testing purposes, we can export all non-Elite terms
if get_non_elite_terms_only == 'yes': collapsed_go_list = getNonEliteTerms(collapsed_go_list,go_full)
###Identify gene lists for the corresponding GO-elite results and generate nested associations
try: gene_file_dir = identifyGeneFiles(gene_input_dir, mappfinder_input)
except Exception: gene_file_dir=''
if len(gene_file_dir) > 0:
#print "Begining to re-annotate pruned results...",
nested_paths_stored = species_code
if len(denom_search_dir)==0: search_dir = export.findParentDir(gene_input_dir)
else: search_dir = denom_search_dir ### Occurs when analyzing an input list and existing pruned results
if prior_ontology_type != ontology_type:
try:
uid_to_go, uid_system, gene_annotations = gene_associations.grabNestedGeneToOntologyAssociations(species_code,
mod,source_data,system_codes,search_dir,ontology_type)
except Exception,e:
if root != None:
print_out = e
try: UI.WarningWindow(print_out,' Continue '); sys.exit()
except Exception: print print_out; sys.exit()
#print 'Annotations imported'
try:
vals = gene_associations.matchInputIDsToGOEliteTerms(gene_file_dir,
go_elite_output_folder,system_codes,mappfinder_input_dir,
collapsed_go_list,uid_to_go,gene_annotations,full_go_name_db,
uid_system,combined_associations,combined_gene_ranking)
combined_associations,combined_gene_ranking,go_gene_annotation_db,go_values_db,value_headers,goids_with_redundant_genes,go_gene_elite_count = vals
summary_data_db['go_gene_elite_count'] = go_gene_elite_count
unique_genes={}
for goid in go_gene_annotation_db:
for s in go_gene_annotation_db[goid]: unique_genes[s.GeneID()]=[]
#print len(unique_genes), "unique genes associated with GO-Elite terms"
###Re-output results, now with gene annotation data
collapsed_go_list = exportGOResults(go_full,go_titles,collapsed_go_list,zscore_goid,go_gene_annotation_db,go_values_db,value_headers,goids_with_redundant_genes)
exportFilteredSIF(mod,species_code,collapsed_go_list,mappfinder_input_dir,None)
except Exception:
continue
else:
local_ora_files+=1
if run_mappfinder == 'yes': mapp_gene_full_count = countGOFullGenes(zscore_goid,mapp_to_mod_genes); summary_data_db['mapp_gene_full_count'] = mapp_gene_full_count
filtered_mapp_list = zscore_changed_path_db
exportLocalResults(go_full,go_titles,{},{},{},{})
summary_data_db['filtered_local_count'] = len(go_full) ### All Elite GO-terms
###Identify gene lists for the corresponding GO-elite results and generate nested associations
try: gene_file_dir = identifyGeneFiles(gene_input_dir, mappfinder_input)
except Exception: gene_file_dir = ''
oraDirTogeneDir[mappfinder_input] = gene_file_dir ### Store the corresponding gene file for each ORA file
if len(gene_file_dir) > 0:
nested_paths_stored = species_code
if prior_ontology_type != ontology_type:
uid_to_mapp, uid_system, gene_annotations = gene_associations.grabNestedGeneToPathwayAssociations(species_code,
mod,source_data,system_codes,custom_sets_folder,denom_search_dir,ontology_type)
#print 'Annotations imported'
if len(uid_to_mapp)>0: ### alternative occurs if analyzing a custom_gene_set result without referencing it again (only should occur during testing)
try:
vals = gene_associations.matchInputIDsToMAPPEliteTerms(gene_file_dir,
go_elite_output_folder,system_codes,mappfinder_input_dir,
uid_to_mapp,filtered_mapp_list,gene_annotations,uid_system,
combined_associations,combined_gene_ranking)
combined_associations,combined_gene_ranking,mapp_gene_annotation_db,mapp_values_db,mapp_value_headers,mapps_with_redundant_genes,mapp_gene_elite_count = vals
summary_data_db['mapp_gene_elite_count'] = mapp_gene_elite_count
except Exception:
continue
exportLocalResults(go_full,go_titles,mapp_gene_annotation_db,mapp_values_db,mapp_value_headers,mapps_with_redundant_genes)
exportFilteredSIF(mod,species_code,mapp_gene_annotation_db,mappfinder_input_dir,oraDirTogeneDir)
if program_type != 'GO-Elite' and mappfinder_input[:3] == 'AS.':
local_filename = go_elite_output_folder+'/'+mappfinder_input[0:-4]+ '_'+filter_method+'_elite.txt'
print 'Copying GO-Elite results to DomainGraph folder...'
fn = filepath(local_filename)
fn2 = string.replace(fn,'GO-Elite_results','DomainGraph')
fn2 = string.replace(fn2,'GO-Elite','AltResults')
fn2 = string.replace(fn2,'AS.','')
fn2 = string.split(fn2,'-local'); fn2=fn2[0]+'-pathways-DomainGraph.txt'
#shutil.copyfile(fn,fn2)
prior_ontology_type = ontology_type
except Exception,e:
print traceback.format_exc()
print 'Error encountered in GO-Elite results pruning for this gene-set type'
print 'gene associations assigned'
if '/app' in filepath(import_dir): webservice = 'yes'
else: webservice = 'no'
if len(combined_results)>0:
ora_files = [local_ora_files,go_ora_files]
output_combined_results(combined_results)
if max(ora_files)>1 and 'Heatmap' not in analysis_method: ### When GO-Elite is run from the heatmap function in clustering, calling clustering below will cause extra matplotlib show occurances
outputOverlappingResults(combined_results,import_dir) ### Compare all input file results
gene_associations.exportCombinedAssociations(combined_associations,go_elite_output_folder,'gene-associations')
gene_associations.exportCombinedAssociations(combined_gene_ranking,go_elite_output_folder,'gene-ranking')
### Copy over ss from CompleteResults
combined_ora_summary = go_elite_output_folder+'/'+'pruned-results_' +filter_method + '_elite.txt'
fn = filepath(combined_ora_summary)
fn2 = string.replace(fn,'CompleteResults/ORA_pruned','')
try: export.customFileMove(fn,fn2)
except Exception: print fn; print fn2; print "SummaryResults failed to be copied from CompleteResults (please see CompleteResults instead)"
combined_ora_gene = go_elite_output_folder+'/gene_associations/pruned-gene-associations.txt'
fn = filepath(combined_ora_gene)
fn2 = string.replace(fn,'CompleteResults/ORA_pruned/gene_associations','')
try: export.customFileMove(fn,fn2)
except IOError: print fn; print fn2; print "SummaryResults failed to be copied from CompleteResults (please see CompleteResults instead)"
visualizePathways(species_code,oraDirTogeneDir,combined_results)
if webservice == 'no':
try: moveMAPPFinderFiles(import_dir)
except Exception,e: print "Could not move ORA results... this will not impact this analysis:",e
end_time = time.time(); time_diff = int(end_time-start_time)
print_out = 'Analysis completed. GO-Elite results\nexported to the specified GO-Elite\noutput and ORA output directories.'
try:
if (program_type == 'GO-Elite' or analysis_method == 'UI') and use_Tkinter == 'yes' and root != None:
print 'Analysis completed in %d seconds... Exiting GO-Elite' % time_diff
UI.InfoWindow(print_out,'Analysis Completed!')
tl = Toplevel(); SummaryResultsWindow(tl,main_output_folder)
else:
### Record results
result_list = recordGOEliteStats(); log_report.append('\n')
except Exception:
try: print 'Analysis completed in %d seconds... Exiting GO-Elite' % time_diff
except Exception: null=[]
else:
end_time = time.time(); time_diff = int(end_time-start_time)
print_out = 'No input files to summarize!'
if program_type == 'GO-Elite' and use_Tkinter == 'yes' and root != None:
print 'Analysis completed in %d seconds... Exiting GO-Elite' % time_diff
UI.WarningWindow(print_out,'Analysis Completed!')
#exportLog(log_report)
run_parameter = 'skip'
end_time = time.time(); time_diff = int(end_time-start_time)
if program_type == 'GO-Elite' and use_Tkinter == 'yes': importGOEliteParameters(run_parameter)
def exportFilteredSIF(mod,species_code,collapsed_term_list,mappfinder_input_dir,oraDirTogeneDir):
gene_association_file = string.replace(mappfinder_input_dir,'.txt','-associations.tab')
sif_output = string.replace(mappfinder_input_dir,'ORA','ORA_pruned')
sif_output = string.replace(sif_output,'.txt','.sif')
sif = export.ExportFile(sif_output)
### Import the gene associations for all GO/pathways analyzed and output Elite filtered results as a SIF
fn=filepath(gene_association_file); x = 0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if x == 0: x=1
else:
ID,symbol,term = string.split(data,'\t')
if term in collapsed_term_list:
if term in full_go_name_db: term = full_go_name_db[term]
sif.write(string.join([term,'pr',ID+':'+symbol],'\t')+'\n')
sif.close()
try:
from visualization_scripts import clustering
try:
criterion_name = export.findFilename(mappfinder_input_dir)
ora_input_dir = oraDirTogeneDir[criterion_name] ### This is ONLY needed for transcription factor graph visualization
except Exception: ora_input_dir = None
clustering.buildGraphFromSIF(mod,species_code,sif_output,ora_input_dir)
except Exception:
#print traceback.format_exc()
pass #Export from PyGraphViz not supported
class SummaryResultsWindow:
def __init__(self,tl,output_dir):
def showLink(event):
idx= int(event.widget.tag_names(CURRENT)[1])
webbrowser.open(LINKS[idx])
url = 'http://www.genmapp.org/go_elite/help_main.htm'
LINKS=(url,'')
self.LINKS = LINKS
tl.title('GO-Elite version 1.2.6'); self.tl = tl
#"""
filename = 'Config/icon.gif'
fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(tl); can.pack(side='top'); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
#"""
use_scroll = 'no'
label_text_str = 'GO-Elite Result Summary'; height = 300; width = 510
if os.name != 'nt': width = 550
elif platform.win32_ver()[0] == 'Vista': width = 550
self.sf = PmwFreeze.ScrolledFrame(tl,
labelpos = 'n', label_text = label_text_str,
usehullsize = 1, hull_width = width, hull_height = height)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
txt=Text(self.frame,bg='light gray')
txt.pack(expand=True, fill="both")
txt.insert(END, 'Primary Analysis Finished....\n')
txt.insert(END, '\nResults saved to:\n'+output_dir+'\n')
txt.insert(END, '\nFor more information see the ')
txt.insert(END, "GO-Elite Help", ('link', str(0)))
txt.insert(END, '\n\n')
try: result_list = recordGOEliteStats()
except Exception: result_list=[]
for d in result_list: txt.insert(END, d+'\n');
txt.tag_config('link', foreground="blue", underline = 1)
txt.tag_bind('link', '<Button-1>', showLink)
open_results_folder = Button(self.tl, text = 'Results Folder', command = self.openDirectory)
open_results_folder.pack(side = 'left', padx = 5, pady = 5);
self.dg_url = 'http://www.wikipathways.org'
text_button = Button(self.tl, text='WikiPathways website', command=self.WPlinkout)
text_button.pack(side = 'right', padx = 5, pady = 5)
self.output_dir = output_dir
self.whatNext_url = 'http://code.google.com/p/go-elite/wiki/Tutorial_GUI_version#Downstream_Analyses'
whatNext_pdf = 'Documentation/what_next_goelite.pdf'; whatNext_pdf = filepath(whatNext_pdf); self.whatNext_pdf = whatNext_pdf
what_next = Button(self.tl, text='What Next?', command=self.whatNextlinkout)
what_next.pack(side = 'right', padx = 5, pady = 5)
quit_buttonTL = Button(self.tl,text='Close View', command=self.close)
quit_buttonTL.pack(side = 'right', padx = 5, pady = 5)
continue_to_next_win = Button(text = 'Continue', command = root.destroy)
continue_to_next_win.pack(side = 'right', padx = 10, pady = 10)
quit_button = Button(root,text='Quit', command=self.quit)
quit_button.pack(side = 'right', padx = 5, pady = 5)
button_text = 'Help'; url = 'http://www.genmapp.org/go_elite/help_main.htm'; self.help_url = url
pdf_help_file = 'Documentation/GO-Elite_Manual.pdf'; pdf_help_file = filepath(pdf_help_file); self.pdf_help_file = pdf_help_file
help_button = Button(root, text=button_text, command=self.Helplinkout)
help_button.pack(side = 'left', padx = 5, pady = 5); root.mainloop()
tl.mainloop() ###Needed to show graphic
def openDirectory(self):
if os.name == 'nt':
try: os.startfile('"'+self.output_dir+'"')
except Exception: os.system('open "'+self.output_dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+self.output_dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+self.output_dir+'"')
def Helplinkout(self): self.GetHelpTopLevel(self.help_url,self.pdf_help_file)
def whatNextlinkout(self):
#self.GetHelpTopLevel(self.whatNext_url,self.whatNext_pdf) ### Provides the option to open a URL or PDF
webbrowser.open(self.whatNext_url) ### Just defaults to the URL
def GetHelpTopLevel(self,url,pdf_file):
self.pdf_file = pdf_file; self.url = url
message = ''; self.message = message; self.online_help = 'Online Documentation'; self.pdf_help = 'Local PDF File'
tl = Toplevel(); self._tl = tl; nulls = '\t\t\t\t'; tl.title('Please select one of the options')
self.sf = PmwFreeze.ScrolledFrame(self._tl,
labelpos = 'n', label_text = '', usehullsize = 1, hull_width = 220, hull_height = 150)
self.sf.pack(padx = 10, pady = 10, fill = 'both', expand = 1)
self.frame = self.sf.interior()
group = PmwFreeze.Group(self.sf.interior(),tag_text = 'Options')
group.pack(fill = 'both', expand = 1, padx = 20, pady = 10)
l1 = Label(group.interior(), text=nulls); l1.pack(side = 'bottom')
text_button2 = Button(group.interior(), text=self.online_help, command=self.openOnlineHelp); text_button2.pack(side = 'top', padx = 5, pady = 5)
try: text_button = Button(group.interior(), text=self.pdf_help, command=self.openPDFHelp); text_button.pack(side = 'top', padx = 5, pady = 5)
except Exception: text_button = Button(group.interior(), text=self.pdf_help, command=self.openPDFHelp); text_button.pack(side = 'top', padx = 5, pady = 5)
tl.mainloop()
def openPDFHelp(self):
if os.name == 'nt':
try: os.startfile('"'+self.pdf_file+'"')
except Exception: os.system('open "'+self.pdf_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+self.pdf_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+self.pdf_file+'"')
self._tl.destroy()
def openOnlineHelp(self):
try: webbrowser.open(self.url)
except Exception: null=[]
self._tl.destroy()
def WPlinkout(self): webbrowser.open(self.dg_url)
def quit(self):
#exportLog(log_report)
try: self.tl.quit(); self.tl.destroy()
except Exception: null=[]
root.quit(); root.destroy(); sys.exit()
def close(self):
self.tl.quit()
self.tl.destroy()
def recordGOEliteStats():
if commandLineMode == 'no': log_report = open(log_file,'a')
result_list=[]
for key in summary_data_db: summary_data_db[key] = str(summary_data_db[key])
d = 'Dataset name: '+ dataset_name; result_list.append(d+'\n')
d = summary_data_db['filtered_go_term_count']+':\tReported Filtered GO-terms'; result_list.append(d)
d = summary_data_db['elite_go_term_count']+':\tReported Elite GO-terms'; result_list.append(d)
d = summary_data_db['redundant_go_term_count']+':\tElite GO-terms redundant with other Elite GO-terms'; result_list.append(d)
d = summary_data_db['filtered_local_count']+':\tReported Filtered Pathways'; result_list.append(d)
d = summary_data_db['redundant_mapp_term_count']+':\tFiltered Pathways redundant with other Filtered Pathways'; result_list.append(d)
if run_mappfinder == 'yes':
try:
if mod == 'HMDB': name = 'metabolites'
else: name = 'genes'
except Exception: name = 'genes'
else: name = 'genes'
if run_mappfinder == 'yes':
d = summary_data_db['go_gene_full_count']+':\tNumber of '+name+' associated with Filtered GO-terms'; result_list.append(d)
d = summary_data_db['go_gene_elite_count']+':\tNumber of '+name+' associated with Elite GO-terms'; result_list.append(d)
d = summary_data_db['mapp_gene_elite_count']+':\tNumber of '+name+' associated with Filtered Pathways'; result_list.append(d)
if commandLineMode == 'no':
log_report.write('\n')
for d in result_list:
if commandLineMode == 'no': log_report.write(d+'\n')
else: print d
if commandLineMode == 'no': log_report.close()
return result_list
class StatusWindow:
def __init__(self,root,mod):
root.title('GO-Elite 1.2.6 - Status Window')
self._parent = root
statusVar = StringVar() ### Class method for Tkinter. Description: "Value holder for strings variables."
if os.name == 'nt': height = 450; width = 500
else: height = 490; width = 640
self.sf = PmwFreeze.ScrolledFrame(self._parent,
labelpos = 'n', label_text = 'Results Status Window',
usehullsize = 1, hull_width = width, hull_height = height)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
group = PmwFreeze.Group(self.sf.interior(),tag_text = 'Output')
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
Label(group.interior(),width=180,height=452,justify=LEFT, bg='black', fg = 'white',anchor=NW,padx = 5,pady = 5, textvariable=statusVar).pack(fill=X,expand=Y)
status = StringVarFile(statusVar,root) ### Likely captures the stdout
sys.stdout = status; root.after(100, runGOElite(mod))
try: self._parent.mainloop()
except Exception: null=[]
try: self._parent.destroy()
except Exception: null=[]
def deleteWindow(self):
#tkMessageBox.showwarning("Quit Selected","Use 'Quit' button to end program!",parent=self._parent)
try: self._parent.destroy(); sys.exit() ### just quit instead
except Exception: sys.exit()
def quit(self):
try: self._parent.quit(); self._parent.destroy(); sys.exit()
except Exception: sys.exit()
class StringVarFile:
def __init__(self,stringVar,window):
self.__newline = 0; self.__stringvar = stringVar; self.__window = window
def write(self,s):
log_report = open(log_file,'a')
log_report.write(s); log_report.close() ### Variable to record each print statement
new = self.__stringvar.get()
for c in s:
#if c == '\n': self.__newline = 1
if c == '\k': self.__newline = 1### This should not be found and thus results in a continous feed rather than replacing a single line
else:
if self.__newline: new = ""; self.__newline = 0
new = new+c
self.set(new)
def set(self,s):
try: self.__stringvar.set(s); self.__window.update()
except Exception: sys.exit() ### When the application is closed or exited by force
def get(self): return self.__stringvar.get()
def flush(self): pass
def timestamp():
import datetime
today = str(datetime.date.today()); today = string.split(today,'-'); today = today[0]+''+today[1]+''+today[2]
time_stamp = string.replace(time.ctime(),':','')
time_stamp = string.replace(time_stamp,' ',' ')
time_stamp = string.split(time_stamp,' ') ###Use a time-stamp as the output dir (minus the day)
time_stamp = today+'-'+time_stamp[3]
return time_stamp
def TimeStamp():
time_stamp = time.localtime()
year = str(time_stamp[0]); month = str(time_stamp[1]); day = str(time_stamp[2])
if len(month)<2: month = '0'+month
if len(day)<2: day = '0'+day
return year+month+day
def universalPrintFunction(print_items):
if commandLineMode == 'no':
log_report = open(log_file,'a')
for item in print_items:
if commandLineMode == 'no': ### Command-line has it's own log file write method (Logger)
log_report.write(item+'\n')
else: print item
if commandLineMode == 'no':
log_report.close()
class Logger(object):
def __init__(self,null):
self.terminal = sys.stdout
self.log = open(log_file, "w")
def write(self, message):
try:
self.log = open(log_file, "a")
self.terminal.write(message)
self.log.write(message)
self.log.close()
except: pass
def flush(self): pass
def importGOEliteParameters(run_parameter):
global max_member_count; global filter_method; global z_threshold; global p_val_threshold; global change_threshold
global sort_only_by_zscore; global permutations; global species; global analysis_method; global resources_to_analyze
global run_mappfinder; global criterion_input_folder; global criterion_denom_folder; global main_output_folder
global log_file; global summary_data_db; summary_data_db = {}; global custom_sets_folder; global mod; global returnPathways
global imageType; imageType = True; global commandLineMode; commandLineMode = 'no'; global enrichment_type
parameters = UI.getUserParameters(run_parameter)
species, run_mappfinder, mod, permutations, filter_method, z_threshold, p_val_threshold, change_threshold, resources_to_analyze, max_member_count, returnPathways, file_dirs, enrichment_type = parameters
change_threshold = int(change_threshold); p_val_threshold = float(p_val_threshold); z_threshold = float(z_threshold)
criterion_input_folder, criterion_denom_folder, main_output_folder, custom_sets_folder = file_dirs
time_stamp = timestamp()
log_file = filepath(main_output_folder+'/GO-Elite_report-'+time_stamp+'.log')
log_report = open(log_file,'w'); log_report.close()
sort_only_by_zscore = 'yes'; analysis_method = 'UI'
global root; root=''
if use_Tkinter == 'yes' and debug_mode == 'no':
root = Tk()
StatusWindow(root,mod)
try: root.destroy()
except Exception: null=[]
else:
sys.stdout = Logger('')
runGOElite(mod)
def remoteAnalysis(variables,run_type,Multi=None):
global max_member_count; global filter_method; global z_threshold; global p_val_threshold; global change_threshold
global sort_only_by_zscore; global permutations; global species; global root; global custom_sets_folder
global run_mappfinder; global criterion_input_folder; global criterion_denom_folder; global main_output_folder
global summary_data_db; summary_data_db = {}; global analysis_method; global log_file; global mod
global resources_to_analyze; global returnPathways; global imageType; imageType = 'both'
global commandLineMode; commandLineMode = 'no'; global enrichment_type; enrichment_type = 'ORA'
global mlp; mlp = Multi
try: species_code,mod,permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,enrichment_type,parent = variables
except Exception: species_code,mod,permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,parent = variables
#print variables
try: permutations = int(permutations)
except Exception: permutations = permutations ### For Fisher Exact
change_threshold = int(change_threshold); p_val_threshold = float(p_val_threshold); z_threshold = float(z_threshold)
if resources_to_analyze == 'WikiPathways' or resources_to_analyze == 'local':
resources_to_analyze = 'Pathways'
speciesData()
species = species_names[species_code]
max_member_count = 10000; sort_only_by_zscore = 'yes'; run_mappfinder = 'yes'
criterion_input_folder, criterion_denom_folder, main_output_folder = file_dirs; custom_sets_folder = '' ### Variable not currently used for AltAnalyze
time_stamp = timestamp()
log_file = filepath(main_output_folder+'/GO-Elite_report-'+time_stamp+'.log')
if 'non-UI' in run_type:
analysis_method = run_type
commandLineMode = 'yes'
sys.stdout = Logger('')
root = parent; runGOElite(mod)
elif run_type == 'UI':
log_report = open(log_file,'a'); log_report.close()
analysis_method = run_type
root = Tk()
StatusWindow(root,mod)
try: root.destroy()
except Exception: null=[]
def visualizePathways(species_code,oraDirTogeneDir,combined_results):
""" Sends all over-represented pathways to the WikiPathways API for visualization """
wp_start_time = time.time()
try:
failed=[]
if returnPathways != None and returnPathways != 'None' and returnPathways != 'no':
### If only the top X pathways should be returned, get this number
returnNumber = None
if 'top' in returnPathways:
returnNumber = int(string.replace(returnPathways,'top',''))
count=0
filename_list=[]
for filename in combined_results: filename_list.append(filename)
filename_list.sort() ### rank the results alphebetically so local and GO results are sequential
wp_to_visualize = []
for filename in filename_list:
if 'local' in filename:
criterion_name = string.split(export.findFilename(filename)[:-4],'-local')[0]+'-local.txt'
for (pathway_type,mapp_name,line) in combined_results[filename]:
if ':WP' in line:
gene_file_dir = oraDirTogeneDir[criterion_name]
wpid = 'WP'+string.split(line,':WP')[-1]
wpid = string.split(wpid,'\t')[0]
wp_to_visualize.append([gene_file_dir,wpid])
if len(wp_to_visualize)>0:
print 'Exporting pathway images for %s Wikipathways (expect %s minute runtime)' % (str(len(wp_to_visualize)),str(len(wp_to_visualize)/15)+'-'+str(len(wp_to_visualize)/2))
if imageType == 'png' or imageType == 'pdf':
print '...restricting to',imageType
try: wp_to_visualize = getProducedPathways(wp_to_visualize); #print len(wp_to_visualize) ### Retain pathways from older runs
except Exception: pass
try: poolWPVisualization(wp_to_visualize,species_code,mod,imageType)
except Exception,e:
#print e
try: wp_to_visualize = getProducedPathways(wp_to_visualize); #print len(wp_to_visualize)
except Exception: pass
print 'Error encountered with Multiple Processor Mode or server timeout... trying single processor mode'
for (gene_file_dir,wpid) in wp_to_visualize:
### graphic_link is a dictionary of PNG locations
try:
graphic_link = WikiPathways_webservice.visualizePathwayAssociations(gene_file_dir,species_code,mod,wpid,imageExport=imageType)
count+=1
print '.',
if returnNumber != None:
if returnNumber == count:
break
except Exception:
error_report = traceback.format_exc()
failed.append(wpid)
print 'exported to the folder "WikiPathways"'
if len(failed)>0:
print len(failed),'Wikipathways failed to be exported (e.g.,',failed[0],')'
print error_report
except Exception:
pass
wp_end_time = time.time(); time_diff = int(wp_end_time-wp_start_time)
print "Wikipathways output in %d seconds" % time_diff
def makeVariableList(wp_to_visualize,species_code,mod,imageType):
variable_list=[]
for (gene_file_dir,wpid) in wp_to_visualize:
variables = gene_file_dir,species_code,mod,wpid,imageType
variable_list.append(variables)
return variable_list
def poolWPVisualization(wp_to_visualize,species_code,mod,imageType):
wp_n = len(wp_to_visualize)
variable_list = makeVariableList(wp_to_visualize,species_code,mod,imageType)
pool_size = int(mlp.cpu_count() * 1)
pool = mlp.Pool(processes=pool_size)
try: results = pool.map(runWPProcesses,variable_list) ### try once
except Exception:
if 'forceTimeError' in traceback.format_exc():
try: pool.close(); pool.join(); pool = None
except Exception: pass
forceTimeError
try:
wp_to_visualize = getProducedPathways(wp_to_visualize)
variable_list = makeVariableList(wp_to_visualize,species_code,mod,imageType)
except Exception: pass #Occurs when the WikiPathways directory is not created yet
#if wp_n == len(wp_to_visualize): force_pool_error
#else:
try: results = pool.map(runWPProcesses,variable_list) ### try again with only failed pathways
except Exception:
try:
wp_to_visualize = getProducedPathways(wp_to_visualize)
variable_list = makeVariableList(wp_to_visualize,species_code,mod,imageType)
except Exception: pass #Occurs when the WikiPathways directory is not created yet
results = pool.map(runWPProcesses,variable_list)
try: pool.close(); pool.join(); pool = None
except Exception: pass
return results
def getProducedPathways(wp_to_visualize):
""" If a pooled run fails, try again for those pathways not output """
for (filename,wpid) in wp_to_visualize: break
root_dir = export.findParentDir(filename)
if 'GO-Elite/input' in root_dir:
root_dir = string.replace(root_dir,'GO-Elite/input','WikiPathways')
else:
root_dir+='WikiPathways/'
possible_pathways={} ### Create a list of all possible created pathways
for (gene_file_dir,wpid) in wp_to_visualize:
dataset_name = export.findFilename(gene_file_dir)[:-4]
try: possible_pathways[wpid].append((dataset_name,gene_file_dir,wpid))
except Exception: possible_pathways[wpid] = [(dataset_name,gene_file_dir,wpid)]
pathway_list = unique.read_directory(root_dir)
pathway_db={}
for pathway in pathway_list:
wpid = string.split(pathway,'_')[0]
try: pathway_db[wpid].append(pathway)
except Exception: pathway_db[wpid] = [pathway] ### pathway name contains the wpid, pathway name, dataset (pathway name from webservice)
wp_to_visualize=[]
for wpid in possible_pathways:
if wpid in pathway_db:
pathway_images = pathway_db[wpid]
for (dataset_name,gene_file_dir,wpid) in possible_pathways[wpid]:
proceed = False
for pathway in pathway_images:
if dataset_name in pathway: proceed = True#; print dataset_name, wpid
if proceed==False:
wp_to_visualize.append((gene_file_dir,wpid))
else:
for (dataset_name,gene_file_dir,wpid) in possible_pathways[wpid]:
wp_to_visualize.append((gene_file_dir,wpid))
return wp_to_visualize
def runWPProcesses(variables):
gene_file_dir,species_code,mod,wpid,imageType = variables
st = time.time()
try: graphic_link = WikiPathways_webservice.visualizePathwayAssociations(gene_file_dir,species_code,mod,wpid,imageExport=imageType)
except Exception:
try: graphic_link = WikiPathways_webservice.visualizePathwayAssociations(gene_file_dir,species_code,mod,wpid,imageExport=imageType)
except Exception: pass
if (time.time() - st)> 100: forceTimeError ### Too slow this way
return graphic_link
def displayHelp():
fn=filepath('Documentation/commandline.txt')
print '\n################################################\nGO-Elite Command-Line Help'
for line in open(fn,'rU').readlines():
print cleanUpLine(line)
print '\n################################################'
sys.exit()
class SpeciesData:
def __init__(self, abrev, species, systems, taxid):
self._abrev = abrev; self._species = species; self._systems = systems; self._taxid = taxid
def SpeciesCode(self): return self._abrev
def SpeciesName(self): return self._species
def Systems(self): return self._systems
def TaxID(self): return self._taxid
def __repr__(self): return self.SpeciesCode()+'|'+self.SpeciesName()
def importSpeciesInfo():
filename = 'Config/species_all_archive.txt'
fn=filepath(filename); global species_list; species_list=[]; global species_codes; species_codes={}; x=0
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
try:
abrev,species,taxid,compatible_mods = string.split(data,'\t')
except Exception:
if '!DOCTYPE': print_out = "A internet connection could not be established.\nPlease fix the problem before proceeding."
else: print_out = "Unknown file error encountered."
raw = export.ExportFile(fn); raw.close(); GO_Elite.importGOEliteParameters('skip'); sys.exit()
if x==0: x=1
else:
compatible_mods = string.split(compatible_mods,'|')
species_list.append(species)
sd = SpeciesData(abrev,species,compatible_mods,taxid)
species_codes[species] = sd
return species_codes
def returnDirectoriesNoReplace(dir):
dir_list = unique.returnDirectoriesNoReplace(dir); dir_list2 = []
for entry in dir_list:
if '.' not in entry: dir_list2.append(entry)
return dir_list2
###### Command Line Functions (AKA Headless Mode) ######
def commandLineRun():
import getopt
#python GO_Elite.py --species Mm --mod Ensembl --permutations 2000 --method "combination" --zscore 1.96 --pval 0.05 --num 2 --input "C:/Documents and Settings/Nathan/Desktop/GenMAPP/Mm_sample/input_list_small" --denom "C:/Documents and Settings/Nathan/Desktop/GenMAPP/Mm_sample/denominator" --output "C:/Documents and Settings/Nathan/Desktop/GenMAPP/Mm_sample"
#python GO_Elite.py --species Mm --mod Ensembl --permutations 200 --method "combination" --zscore 1.96 --pval 0.05 --num 2 --input "C:/input" --denom "C:/denominator" --output "C:/output"
#python GO_Elite.py --species Mm --input "C:/input" --denom "C:/denominator" --output "C:/output" --mod Ensembl --permutations 200
global max_member_count; global filter_method; global z_threshold; global p_val_threshold; global change_threshold
global sort_only_by_zscore; global permutations; global species; global root; global resources_to_analyze
global run_mappfinder; global criterion_input_folder; global criterion_denom_folder; global main_output_folder; global custom_sets_folder
global log_file; global summary_data_db; summary_data_db = {}; global analysis_method; global returnPathways; global mod; global imageType
global commandLineMode; commandLineMode = 'yes'; global enrichment_type
###optional
permutations = 2000; filter_method = 'z-score'; mod = 'Ensembl'; z_threshold = 1.96; enrichment_type = 'ORA'
p_val_threshold = 0.05; change_threshold = 3; main_output_folder = ''; resources_to_analyze = 'all'
###required
species_code = None
species_full = None
criterion_input_folder = None
criterion_denom_folder = None
custom_sets_folder = ''
main_output_folder = None
update_dbs = None
update_method = []
process_affygo='no'
speciesfull=None
update_ensrel = []
replaceDB = 'no'
species_taxid = ''
incorporate_previous = 'yes'
new_species = 'no'
force = 'yes'
ensembl_version = 'current'
goelite_db_version = ''
remove_download_files = 'no'
export_versions_info = 'no'
archive = 'no'
additional_resources = [None]
buildNested = 'no'
continue_build = 'no'
OBOurl = ''
buildLocalWPFiles = 'yes'
GOtype = 'GeneOntology'
input_var = sys.argv[1:]
nestTerms = 'no'
wpid = None
image_export = None
returnPathways = None
imageType = True
resources=[]
#input_var = ['--update', 'EntrezGene', '--species' ,'Mm'] ###used for testing
if '--help' in input_var or '--h' in input_var:
displayHelp()
if '--version' in input_var or '--v' in input_var:
print 'GO-Elite version 1.2.6 (http://genmapp.org/go_elite)'
try:
options, remainder = getopt.getopt(input_var,'', ['species=', 'mod=','permutations=',
'method=','zscore=','pval=','num=','input=','denom=','output=',
'update=','uaffygo=','system=', 'replaceDB=', 'speciesfull=',
'taxid=', 'addspecies=', 'force=', 'version=', 'delfiles=',
'archive=','exportinfo=','dataToAnalyze=','buildNested=',
'customSet=','OBOurl=','GOtype=','nestTerms=','additional=',
'image=','wpid=','returnPathways=','imageType=','enrichment='])
except Exception, e: print e;sys.exit()
for opt, arg in options:
if opt == '--species': species_code=arg
elif opt == '--mod': mod=arg
elif opt == '--image': image_export=arg
elif opt == '--wpid': wpid=arg
elif opt == '--imageType':
imageType=arg
if imageType=='all':
imageType = True
elif opt == '--permutations': permutations=arg
elif opt == '--method':
filter_method=arg
if filter_method == 'gene_number': filter_method = 'gene number'
if filter_method == 'gene': filter_method = 'gene number'
if 'z' in filter_method or 'Z' in filter_method: filter_method = 'z-score'
elif opt == '--zscore': z_threshold=arg
elif opt == '--pval': p_val_threshold=arg
elif opt == '--num': change_threshold=arg
elif opt == '--dataToAnalyze':
if arg == 'local' or arg == 'WikiPathways':
arg = 'Pathways'
resources.append(arg)
elif opt == '--input': criterion_input_folder=arg
elif opt == '--denom': criterion_denom_folder=arg
elif opt == '--output': main_output_folder=arg
elif opt == '--enrichment':
if string.lower(arg)=='ura' or 'under' in string.lower(arg):
enrichment_type='URA'
elif opt == '--update': update_dbs='yes'; update_method.append(arg)
elif opt == '--additional':
if additional_resources[0] == None:
additional_resources=[]
additional_resources.append(arg)
else:
additional_resources.append(arg)
elif opt == '--uaffygo': process_affygo=arg
elif opt == '--system': update_ensrel.append(arg) ### This is the related system. Multiple args to this flag are valid
elif opt == '--version': ensembl_version = arg
elif opt == '--replaceDB': replaceDB=arg
elif opt == '--speciesfull': species_full=arg
elif opt == '--taxid': species_taxid=arg
elif opt == '--addspecies': update_dbs='yes'; new_species = 'yes'
elif opt == '--force': force=arg
elif opt == '--delfiles': remove_download_files = arg
elif opt == '--archive': archive = arg
elif opt == '--exportinfo': export_versions_info = arg
elif opt == '--buildNested': buildNested = arg
elif opt == '--customSet': custom_sets_folder = arg
elif opt == '--OBOurl': OBOurl = arg
elif opt == '--GOtype': GOtype = arg
elif opt == '--nestTerms': nestTerms = arg
elif opt == '--returnPathways': returnPathways = arg
""" Build Database Outline:
python GO_Elite.py --update Ensembl --system all --version 72 --species Hs --update WikiPathways --system EntrezGene
GO_Elite.py --update EntrezGene --version 72 --species Hs
python GO_Elite.py --update metabolites --version 72 --species Hs --force no
ython GO_Elite.py --update Affymetrix --update WikiPathways --species Hs --replaceDB no --force no
"""
if len(resources)>1: resources_to_analyze = resources
elif len(resources)>0: resources_to_analyze = resources[0]
species_full_original = species_full; species_code_original = species_code
if image_export != None:
if image_export == 'WikiPathways':
#python GO_Elite.py --input /users/test/input/criterion1.txt --image WikiPathways --mod Ensembl --system arrays --species Hs --wpid WP536
if wpid==None:
print 'Please provide a valid WikiPathways ID (e.g., WP1234)';sys.exit()
if species_code==None:
print 'Please provide a valid species ID for an installed database (to install: --update Official --species Hs --version EnsMart91Plus)';sys.exit()
if criterion_input_folder==None:
print 'Please provide a valid file location for your input IDs (also needs to inlcude system code and value column)';sys.exit()
from visualization_scripts import WikiPathways_webservice
try:
print 'Attempting to output a WikiPathways colored image from user data'
print 'mod:',mod
print 'species_code:',species_code
print 'wpid:',wpid
print 'imageType',imageType
print 'input GO-Elite ID file:',criterion_input_folder
graphic_link = WikiPathways_webservice.visualizePathwayAssociations(criterion_input_folder,species_code,mod,wpid,imageExport=imageType)
except Exception,e:
print traceback.format_exc()
if 'force_no_matching_error' in traceback.format_exc():
print 'None of the input IDs mapped to this pathway'
elif 'IndexError' in traceback.format_exc():
print 'Input ID file does not have at least 3 columns, with the second column being system code'
elif 'ValueError' in traceback.format_exc():
print 'Input ID file error. Please check that you do not have extra rows with no data'
elif 'source_data' in traceback.format_exc():
print 'Input ID file does not contain a valid system code'
else:
print 'Error generating the pathway "%s"' % wpid
try: print 'Finished exporting visualized pathway to:',graphic_link['WP']
except Exception: None ### Occurs if nothing output
sys.exit()
if 'EnsMart' in ensembl_version:
import UI; UI.exportDBversion(ensembl_version)
elif 'Plant' in ensembl_version:
import UI; UI.exportDBversion(string.replace(ensembl_version,'Plant','EnsMart'))
elif 'Bacteria' in ensembl_version:
import UI; UI.exportDBversion(string.replace(ensembl_version,'Bacteria','EnsMart'))
elif 'Fung' in ensembl_version:
import UI; UI.exportDBversion(string.replace(ensembl_version,'Fungi','EnsMart'))
program_type,database_dir = unique.whatProgramIsThis()
if program_type == 'AltAnalyze': database_dir = '/AltDatabase'; goelite = '/goelite'
else: database_dir = '/Databases'; goelite = ''
if archive == 'yes':
import update; import UI
db_versions = UI.returnDirectoriesNoReplace(database_dir)
for version in db_versions:
print database_dir[1:]+'/'+version+goelite
species_dirs = returnDirectoriesNoReplace(database_dir+'/'+version+goelite)
print species_dirs
for i in species_dirs:
update.zipDirectory(database_dir[1:]+'/'+version+goelite+'/'+i); print 'Zipping',i
if export_versions_info == 'yes':
import UI; species_archive_db={}
speciesData()
db_versions = UI.returnDirectoriesNoReplace(database_dir)
### Export species names for each Official database version based on zip files in each folder
#print db_versions
for version in db_versions:
#print version
species_file_dirs = UI.returnFilesNoReplace(database_dir+'/'+version+goelite)
#print species_dirs
for file in species_file_dirs:
if '.zip' in file:
file = string.replace(file,'.zip','')
if file in species_names:
species_name = species_names[file]
try: species_archive_db[species_name].append(version)
except Exception: species_archive_db[species_name] = [version]
print 'len(species_archive_db)',len(species_archive_db)
if len(species_archive_db)>0: UI.exportSpeciesVersionInfo(species_archive_db)
### Export array systems for each species for each Official database version
species_array_db={}
for version in db_versions:
#print version
species_dirs = UI.returnDirectoriesNoReplace(database_dir+'/'+version+goelite)
for species_dir in species_dirs:
supported_arrays=[]
if species_dir in species_names:
species_name = species_names[species_dir]
species_file_dirs = UI.returnFilesNoReplace(database_dir+'/'+version+goelite+'/'+species_dir+'/uid-gene')
for file in species_file_dirs:
if 'Affymetrix' in file: supported_arrays.append('Affymetrix')
if 'MiscArray' in file: supported_arrays.append('MiscArray')
if 'Codelink' in file: supported_arrays.append('Codelink')
if 'Illumina' in file: supported_arrays.append('Illumina')
if 'Agilent' in file: supported_arrays.append('Agilent')
if len(supported_arrays)>0:
species_array_db[species_name,version] = supported_arrays
print 'len(species_array_db)',len(species_array_db)
if len(species_array_db)>0: UI.exportArrayVersionInfo(species_array_db)
if replaceDB == 'yes': incorporate_previous = 'no'
if update_dbs == 'yes' and ((species_code == None and species_full == None) and (update_method != ['Ontology'])) and update_method != ['metabolites']:
print '\nInsufficient flags entered (requires --species or --speciesfull)'; sys.exit()
elif (update_dbs == 'yes' or buildNested == 'yes') and (species_code != None or species_full != None or update_method == ['Ontology']):
from import_scripts import BuildAffymetrixAssociations; import update; from build_scripts import EnsemblSQL; import UI
file_location_defaults = UI.importDefaultFileLocations()
speciesData()
species_codes = UI.importSpeciesInfo()
species_code_list=[]
if len(species_codes) == 0:
UI.remoteSpeciesInfo('no')
species_codes = importSpeciesInfo() ### Gets the information from the backup version
if ensembl_version != 'current' and 'release-' not in ensembl_version and 'EnsMart' not in ensembl_version:
if 'Plant' not in ensembl_version and 'Fungi' not in ensembl_version:
try: version_int = int(ensembl_version); ensembl_version = 'release-'+ensembl_version
except ValueError: print 'The Ensembl version number is not formatted corrected. Please indicate the desired version number to use (e.g., "55").'; sys.exit()
if update_method == ['Ontology']: species_code_list=[]
elif species_code == 'all':
### Add all species from the current database
for species_code in species_names: species_code_list.append(species_code)
elif species_full != None and species_full != 'all':
species_full = [species_full] ###If the Ensembl species is not '' but is defined
elif species_full == 'all':
species_full = []
child_dirs, ensembl_species, ensembl_versions = EnsemblSQL.getCurrentEnsemblSpecies(ensembl_version)
for ens_species in ensembl_species:
ens_species = string.replace(ens_species,'_',' ')
species_full.append(ens_species)
else: species_code_list = [species_code]
if 'Official' in update_method:
existing_species_codes = importSpeciesInfo()
if len(existing_species_codes) == 0:
UI.remoteSpeciesInfo('no')
existing_species_codes = importSpeciesInfo() ### Gets the information from the backup version
UI.getOnlineEliteConfig(file_location_defaults,'')
if species_code != None:
### Integrate the speciescode
speciesData()
species_names_temp = UI.remoteSpeciesInfo('yes')
if species_full == 'all': species_code_ls = species_names_temp
else: species_code_ls = [species_code]
for species_code in species_names_temp:
sd = species_names_temp[species_code]
existing_species_codes[sd.SpeciesName()] = sd
UI.exportSpeciesInfo(existing_species_codes)
speciesData()
elif species_full != None:
try: ensembl_species = ensembl_species
except Exception: child_dirs, ensembl_species, ensembl_versions = EnsemblSQL.getCurrentEnsemblSpecies(ensembl_version)
for species_ens in species_full:
if species_ens in ensembl_species:
genus,species_code = string.split(species_ens,' ')
species_code = genus[0]+species_code[0]
taxid = ''; compatible_mods = ['En']
if species_code in species_names:
species = species_names[species_code]
sd = species_codes[species]
compatible_mods = sd.Systems()
taxid = sd.TaxID()
if species != species_ens: species_code = genus[:2]; species_names[species_code] = species_ens
elif 'En' not in compatible_mods: compatible_mods.append('En')
sd = UI.SpeciesData(species_code,species_ens,compatible_mods,taxid)
species_codes[species_ens] = sd
species_code_list.append(species_code) ### Add all Ensembl species (either all or single specified)
else: print "The Ensembl species",species_ens,"was not found."; sys.exit()
UI.exportSpeciesInfo(species_codes)
species_code_list = unique.unique(species_code_list)
if continue_build == 'yes' and (species_code_original == 'all' or species_full_original == 'all'):
### Only analyze species NOT in the directory
current_species_ls = unique.read_directory('/Databases'); species_code_list2=[]
for sc in species_code_list:
if sc not in current_species_ls: species_code_list2.append(sc)
species_code_list = species_code_list2
species_iteration=0
speciesData(); species_codes = importSpeciesInfo() ### Re-import the species data updated above
if len(species_codes) == 0:
UI.remoteSpeciesInfo('no'); species_codes = importSpeciesInfo() ### Gets the information from the backup version
#print species_code_list, update_ensrel
species_code_list.sort(); species_code_list.reverse()
if 'WikiPathways' in update_method and buildLocalWPFiles == 'yes':
import gene_associations; all_species = 'no'
try:
gene_associations.convertAllGPML(species_code_list,all_species) ### Downloads GPMLs and builds flat files
null=[]
except Exception:
print 'Unable to connect to http://www.wikipathways.org'; sys.exit()
for species_code in species_code_list:
species_iteration +=1
system_codes,system_list,mod_list = UI.remoteSystemInfo()
try: species = species_names[species_code]
except Exception: print 'Species code %s not found. Please add the species to the database.' % species_code; sys.exit()
print "Starting to update databases for",species, string.join(update_method,',')
### Update EntrezGene-GO Databases
if 'EntrezGene' in update_method:
ncbi_go_file = file_location_defaults['EntrezGO'].Location(); status = 'null'
if species_iteration == 1:
if force == 'yes': fln,status = update.download(ncbi_go_file,'BuildDBs/Entrez/Gene2GO/','txt')
else:
file_found = UI.verifyFile('BuildDBs/Entrez/Gene2GO/gene2go.txt')
if file_found == 'no':
fln,status = update.download(ncbi_go_file,'BuildDBs/Entrez/Gene2GO/','txt')
if 'Internet' in status: print status
else:
try: sd = species_codes[species]; species_taxid = sd.TaxID()
except KeyError: species_taxid = species_taxid
try: run_status = BuildAffymetrixAssociations.parseGene2GO(species_taxid,species_code,'over-write previous',incorporate_previous)
except Exception: run_status = 'no'
if run_status == 'run': print 'Finished building EntrezGene-GeneOntology associations files.'
else: print 'Gene2GO file not found. Select download to obtain database prior to extraction'
if remove_download_files == 'yes' and len(species_code_list)==1: export.deleteFolder('BuildDBs/Entrez/Gene2GO')
import gene_associations
try: gene_associations.swapAndExportSystems(species_code,'Ensembl','EntrezGene') ### Allows for analysis of Ensembl IDs with EntrezGene based GO annotations (which can vary from Ensembl)
except Exception: null=[] ### Occurs if EntrezGene not supported
try: gene_associations.augmentEnsemblGO(species_code)
except Exception: null=[] ### Occurs if EntrezGene not supported
if new_species == 'yes':
try:
species = species_full[0]
species_code = species_code
species_taxid = species_taxid
except Exception:
print_out = 'Additional values are needed to add a new species'
print_out +='(e.g., --speciesfull "Homo sapiens" --species Hs --taxid 9606)'
print print_out; sys.exit()
compatible_mods = ['L']
sd = UI.SpeciesData(species_code,species,compatible_mods,species_taxid)
species_codes[new_species_name] = sd
UI.exportSpeciesInfo(species_codes)
### Download Official Databases
if 'Official' in update_method:
import UI
try: os.remove(filepath('Databases/'+species_code+'/nested/version.txt'))
except Exception: null=[] ### Remove old nested file
buildNested = 'yes'
UI.getOnlineEliteConfig(file_location_defaults,'')
db_versions = UI.importOnlineDatabaseVersions(); db_version_list=[]
for version in db_versions: db_version_list.append(version)
db_version_list.sort(); db_version_list.reverse(); select_version = db_version_list[0]
db_versions[select_version].sort()
if ensembl_version != 'current':
if ensembl_version not in db_versions:
print ensembl_version, 'is not a valid version of Ensembl, while',select_version, 'is.'; sys.exit()
else: select_version = ensembl_version
if species not in db_versions[select_version]:
print species, ': This species is not available for this version %s of the Official database.' % select_version
else:
base_url = file_location_defaults['url'].Location()
#print base_url+'Databases/'+select_version+'/'+species_code+'.zip'
fln,status = update.download(base_url+'Databases/'+select_version+'/'+species_code+'.zip','Databases/','')
### Creates gene-Symbol.txt, EntrezGene-Ensembl.txt and augements gene-GO tables for between system analyses
UI.buildInferrenceTables(species_code)
### Attempt to download additional Ontologies and GeneSets
update_method.append('AdditionalResources')
### Attempt to download additional Ontologies and GeneSets
if 'AdditionalResources' in update_method:
try:
from build_scripts import GeneSetDownloader
print 'Adding supplemental GeneSet and Ontology Collections'
if 'all' in additional_resources:
additionalResources = UI.importResourceList() ### Get's all additional possible resources
else: additionalResources = additional_resources
GeneSetDownloader.buildAccessoryPathwayDatabases([species_code],additionalResources,force)
print 'Finished adding additional analysis resources.'
except Exception: print 'Download error encountered for additional Ontologies andGeneSets...\nplease try again later.'
if 'Official' in update_method:
if 'Internet' not in status:
print 'Finished downloading the latest species database files.'
### Download Ensembl Database
if 'Ensembl' in update_method:
externalDBName_list=[]
try: child_dirs, ensembl_species, ensembl_versions = EnsemblSQL.getCurrentEnsemblSpecies(ensembl_version)
except Exception: print "\nPlease try a different version. This one does not appear to be valid."; sys.exit()
try: ensembl_sql_dir,ensembl_sql_description_dir = child_dirs[species]
except Exception:
print traceback.format_exc()
print species,'species not supported in Ensembl'; continue
### Download the latest version of Ensembl
EnsemblSQL.updateFiles(ensembl_sql_dir,'Config/','external_db.txt','yes')
raw = export.ExportFile('Config/array.txt'); raw.close() ### Delete existing
try: EnsemblSQL.updateFiles(string.replace(ensembl_sql_dir,'core','funcgen'),'Config/','array.txt','yes')
except Exception: raw = export.ExportFile('Config/array.txt'); raw.close()
external_dbs, external_system, array_db, external_ids = UI.importExternalDBs(species)
if len(update_ensrel) == 0:
print "\nPlease indicate the system to update (e.g., --system all --system arrays --system Entrez)."; sys.exit()
for i in update_ensrel:
i = string.replace(i,'\x93',''); i = string.replace(i,'\x94','') ### Occurs when there is a forward slash in the system name
if i in external_system: externalDBName_list.append(i)
elif i != 'all' and i != 'arrays':
print '\nEnsembl related system',[i], 'not found!!! Check the file Config/external_db.txt for available valid system names before proceeding.'; sys.exit()
if 'all' in update_ensrel:
for i in external_system: ### Build for all possible systems
if '\\N_' not in i: externalDBName_list.append(i)
#print [i]
elif 'arrays' in update_ensrel:
externalDBName_list=[]
for array in array_db:
if '\\N_' not in array:
if 'ProbeLevel' in update_method:
if 'AFFY' in array: externalDBName_list.append(array) ### Ensures probe-level genomic coordinate assingment is restricted to Affy
else: externalDBName_list.append(array)
import datetime
today = str(datetime.date.today()); today = string.split(today,'-'); today = today[1]+'/'+today[2]+'/'+today[0]
dirversion = string.replace(ensembl_version,'release-','EnsMart')
OBO_import.exportVersionData(dirversion,today,'Config/')
overwrite_previous = 'over-write previous'
configType = 'Basic'; iteration=0
from build_scripts import EnsemblSQL; reload(EnsemblSQL)
if 'arrays' not in update_ensrel:
try: all_external_ids = EnsemblSQL.buildGOEliteDBs(species_code,ensembl_sql_dir,ensembl_sql_description_dir,'GO',configType,'GeneAndExternal',overwrite_previous,replaceDB,external_system,force); iteration+=1
except Exception, e:
print traceback.format_exc()
print 'Critical Error!!!! Exiting GO-Elite...'; sys.exit()
externalDBName_list_updated = UI.filterExternalDBs(all_external_ids,externalDBName_list,external_ids,array_db)
###Add additional systems not in our annotated Config file if the user specified parsing of all systems or all array systems
if 'arrays' in update_ensrel or 'all' in update_ensrel:
externalDBName_list = externalDBName_list_updated
for externalDBName in externalDBName_list:
if externalDBName != ' ':
if force == 'yes' and iteration == 1: force = 'no'
from build_scripts import EnsemblSQL; reload(EnsemblSQL)
output_dir = 'BuildDBs/EnsemblSQL/'+species_code+'/'
if force == 'yes': ### Delete any existing data in the destination directory that can muck up tables from a new Ensembl build
export.deleteFolder(output_dir)
if externalDBName in array_db:
#python GO_Elite.py --update Ensembl --update ProbeLevel --system arrays --version 65 --species Dr --force no
analysisType = 'FuncGen'
print [externalDBName], analysisType
if 'ProbeLevel' in update_method:
analysisType = 'ProbeLevel'
EnsemblSQL.buildGOEliteDBs(species_code,ensembl_sql_dir,ensembl_sql_description_dir,externalDBName,configType,analysisType,overwrite_previous,replaceDB,external_system,force); iteration+=1
#except Exception,e: print e;sys.exit()
else:
EnsemblSQL.buildGOEliteDBs(species_code,ensembl_sql_dir,ensembl_sql_description_dir,externalDBName,configType,analysisType,overwrite_previous,replaceDB,external_system,force); iteration+=1
#except Exception,e: print e;sys.exit()
else:
analysisType = 'GeneAndExternal'
print [externalDBName], analysisType
EnsemblSQL.buildGOEliteDBs(species_code,ensembl_sql_dir,ensembl_sql_description_dir,externalDBName,configType,analysisType,overwrite_previous,replaceDB,external_system,force); iteration+=1
#except Exception,e: print e;sys.exit()
try: swapAndExportSystems(species_code,'Ensembl','EntrezGene') ### Allows for analysis of Ensembl IDs with EntrezGene based GO annotations (which can vary from Ensembl)
except Exception: null=[] ### Occurs if EntrezGene not supported
if remove_download_files == 'yes': export.deleteFolder('BuildDBs/EnsemblSQL/'+species_code)
if 'Affymetrix' in update_method or 'WikiPathways' in update_method:
continue_analysis = 'no'
if 'WikiPathways' in update_method:
relationship_types = ['native','mapped']
for relationship_type in relationship_types:
print 'Processing',relationship_type,'relationships'
index=0
if buildLocalWPFiles == 'yes':
status = '' ### Used when building a flat file from GPML zip file
else:
### This is used when downloading a pre-built flat file from WikiPathways
UI.deleteWPFiles() ### Remove prior WP files
date = UI.TimeStamp(); file_type = ('wikipathways_'+date+'_'+species+'.tab','.txt')
if relationship_type == 'mapped':
url = 'http://wikipathways.org/wpi/cache/wikipathways_data_'+string.replace(species,' ','%20')+'.tab'
print url
else:
url = 'http://wikipathways.org/wpi/cache/wikipathways_native_data_'+string.replace(species,' ','%20')+'.tab'
print url
if force == 'yes':
fln,status = update.download(url,'BuildDBs/wikipathways/',file_type)
if 'Internet' in status: ### This file now loads for a minute or two, so this is common
print 'Connection timed out... trying again in 15 seconds.'
start_time = time.time(); end_time = time.time()
while (end_time-start_time)<15: end_time = time.time()
fln,status = update.download(url,'BuildDBs/wikipathways/',file_type)
else: status = 'null'
if 'Internet' not in status:
if 'Affymetrix' not in update_method: integrate_affy_associations = 'no'
else: integrate_affy_associations = 'yes'
counts = BuildAffymetrixAssociations.importWikipathways(system_codes,incorporate_previous,process_affygo,species,species_code,integrate_affy_associations,relationship_type,'over-write previous')
index+=1
if counts == 0: print 'No Affymetrix annotation files found and thus NO new results.'
else: print 'Finished parsing the latest Wikipathways and Affymetrix annotations.'
else: print status
else:
try:
dir = '/BuildDBs/Affymetrix'; dir_list = UI.getFolders(dir)
if species_code in dir_list: continue_analysis = 'yes'
except IOError: continue_analysis = 'yes'
if continue_analysis == 'yes':
BuildAffymetrixAssociations.buildAffymetrixCSVAnnotations(species_code,incorporate_previous,process_affygo,'no','yes','over-write previous')
if buildNested == 'yes':
#try: os.remove(filepath('OBO/version.txt')) ### Maybe not a good idea?
#except KeyError: null=[] ### Remove old nested file
species_names_temp = UI.remoteSpeciesInfo('yes')
if species_full == 'all' or species_code == 'all': species_code_ls = species_names_temp
elif species_code != None: species_code_ls = [species_code]
elif species_full != None:
species1,species2 = string.split(species_full,' ')
species_code_ls = [species1[0]+species2[0]]
for species_code in species_code_ls:
current_species_dirs = unique.returnDirectories('/Databases')
if species_code in current_species_dirs:
try: export.deleteFolder(filepath('Databases/'+species_code+'/nested')) ### Delete the existing nested folder (will force rebuilding it)
except Exception: null=[]
### Creates a nested GO (and stores objects in memory, but not needed
export_databases = 'no'; genmapp_mod = 'Ensembl'; sourceData()
print species_code,'Building Nested for mod types:',mod_types
avaialble_ontologies = OBO_import.findAvailableOntologies(species_code,mod_types)
for ontology_type in avaialble_ontologies:
try: full_path_db,path_id_to_goid,null = OBO_import.buildNestedOntologyAssociations(species_code,mod_types,ontology_type)
except Exception: None
try: UI.buildInferrenceTables(species_code)
except Exception: pass
if 'GORelationships' in update_method:
import gene_associations
import datetime
today = string.replace(str(datetime.date.today()),'-','')
go_annotations = OBO_import.importPreviousOntologyAnnotations(GOtype)
try:
#if GOtype != 'GOslim':
if nestTerms == 'yes': nested = 'nested'
else: nested = 'null'
ens_goslim = gene_associations.importGeneToOntologyData(species_code,'Ensembl',nested,ontology_type) ### The second to last last argument is gotype which can be null or nested
#else: ens_goslim = gene_associations.importUidGeneSimple(species_code,'Ensembl-goslim_goa')
print len(ens_goslim),'Ensembl-'+GOtype+ ' relationships imported...'
output_results = 'OBO/builds/'+species_code+'_'+GOtype+'_'+today+'.txt'
export_data = export.ExportFile(output_results)
title = string.join(['Ensembl','annotation.GO BIOLOGICAL_PROCESS','annotation.GO CELLULAR_COMPONENT','annotation.GO MOLECULAR_FUNCTION'],'\t')+'\n'
export_data.write(title)
except Exception: ens_goslim=[] ### GO slim relationships not supported
for gene in ens_goslim:
mf=[]; bp=[]; cc=[]
counts=0
for goid in ens_goslim[gene]:
try:
go_type = go_annotations[goid].GOType()
if go_type == 'molecular_function': mf.append(goid); counts+=1
elif go_type == 'biological_process': bp.append(goid); counts+=1
elif go_type == 'cellular_component': cc.append(goid); counts+=1
except Exception: null=[]
if counts>0:
mf = string.join(mf,','); bp = string.join(bp,','); cc = string.join(cc,',')
values = string.join([gene,bp,cc,mf],'\t')+'\n'
export_data.write(values)
try: export_data.close()
except Exception: null=[] ### If relationships not supported
if 'Ontology' in update_method:
UI.updateOBOfiles(file_location_defaults,'yes',OBOurl,'')
elif criterion_input_folder != None and main_output_folder != None and species_code != None: #and criterion_denom_folder!= None
file_dirs = criterion_input_folder, criterion_denom_folder, main_output_folder, custom_sets_folder
parent = None
time_stamp = timestamp()
log_file = filepath(main_output_folder+'/GO-Elite_report-'+time_stamp+'.log')
try: log_report = open(log_file,'a'); log_report.close(); sys.stdout = Logger('')
except Exception:
print "Warning! The output directory must exist before running GO-Elite. Since it is easy to accidently specify an invalid output directory, it is best that GO-Elite does not create this for you.\n"
sys.exit()
if ensembl_version != 'current':
### Select database version otherwise use default
goelite_db_version = ensembl_version
import UI
species_dirs = UI.returnDirectoriesNoReplace('/Databases')
if goelite_db_version in species_dirs:
import UI; UI.exportDBversion(goelite_db_version)
print 'Using database version',goelite_db_version
try: permutations = int(permutations);change_threshold = int(change_threshold)
except Exception: permutations = permutations
p_val_threshold = float(p_val_threshold); z_threshold = float(z_threshold)
try: speciesData(); species = species_names[species_code]
except Exception: print 'Species code not found. Please add the species to the database.'; sys.exit()
max_member_count = 10000; sort_only_by_zscore = 'yes'; run_mappfinder = 'yes'
#criterion_input_folder, criterion_denom_folder, main_output_folder, custom_sets_folder = file_dirs
analysis_method = 'non-UI'
change_threshold = change_threshold-1
try:
import UI
species_dirs = UI.returnDirectoriesNoReplace('/Databases')
except:
try:
species_dirs = UI.returnDirectoriesNoReplace('/AltDatabase')
except Exception:
print '\nPlease install a species database (to install: python GO_Elite.py --update Official --species Hs --version EnsMart62Plus)';sys.exit()
print ''
root = parent; runGOElite(mod)
else:
print '\nInsufficient flags entered (requires --species, --input and --output)'; sys.exit()
if 'metabolites' in update_method:
import MetabolomicsParser
try: MetabolomicsParser.buildMetabolomicsDatabase(force) ### will update any installed species
except:
print 'WARNING!!!! No metabolite database present... skipping metabolite build'
sys.exit()
if __name__ == '__main__':
try:
import multiprocessing as mlp
mlp.freeze_support()
except Exception:
print 'Note: Multiprocessing not supported for this verison python.'
mlp = None
run_parameter = 'intro'
if Tkinter_failure == True:
print "\nPmw or Tkinter not found... Tkinter print out not available"
try:
###### Determine Command Line versus GUI Control ######
program_type,database_dir = unique.whatProgramIsThis()
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>0 and '-' in command_args:
### Run in headless mode (Tkinter not required)
commandLineRun()
if use_Tkinter == 'yes':
### Run in GUI mode (Tkinter required)
importGOEliteParameters(run_parameter)
except Exception, exception:
try:
print_out = "Operating System Info: "+os.name +', '+str(platform.win32_ver()[0])+', '+str(platform.architecture())+', '+str(platform.mac_ver())+', '+str(platform.libc_ver())+', '+str(platform.platform())
except Exception:
print_out = "Operating System Info: "+os.name
trace = traceback.format_exc()
print '\nWARNING!!! Critical error encountered (see below details)\n'
print trace
print "\n...exiting GO-Elite due to unexpected error (contact [email protected] for assistance)."
time_stamp = timestamp()
if commandLineMode == 'no':
try: log_file = log_file
except Exception:
try: log_file = filepath(main_output_folder+'/GO-Elite_report-error_'+time_stamp+'.log')
except Exception:
try: log_file = filepath('GO-Elite_report-error_'+time_stamp+'.log')
except Exception: log_file = filepath('/GO-Elite_report-error_'+time_stamp+'.log')
try: log_report = open(log_file,'a') ### append
except Exception: log_report = open(log_file,'a') ### write
log_report.write(print_out+'\n')
log_report.write(trace)
print_out = "Unknown error encountered during data processing.\nPlease see logfile in:\n\n"+log_file+"\nand report to [email protected]."
if use_Tkinter == 'yes':
program,program_dir = unique.whatProgramIsThis()
if program!= 'AltAnalyze':
try: UI.WarningWindow(print_out,'Error Encountered!'); root.destroy()
except Exception: print print_out
else: print print_out
log_report.close()
if len(log_file)>0:
if os.name == 'nt':
try: os.startfile('"'+log_file+'"')
except Exception: os.system('open "'+log_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+log_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+log_file+'/"')
sys.exit()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/GO_Elite.py
|
GO_Elite.py
|
# the module "subprocess" requires Python 2.4
import os
import sys
import time
import re
import tempfile
from types import *
__version__ = '1.1.0'
if sys.version < '2.3': # actually python 2.3 is required by tempfile.mkstemp !!!
set = frozenset = tuple
basestring = str
elif sys.version < '2.4':
from sets import Set as set, ImmutableSet as frozenset
if sys.version < '3.0':
_mystr = _mybytes = lambda s:s
else:
from functools import reduce
long, basestring, unicode = int, str, str
_mybytes = lambda s:bytes(s, 'utf8') #'ascii')
_mystr = lambda s:str(s, 'utf8')
try:
import numpy
has_numpy = True
except:
has_numpy = False
_has_subp = False
if sys.platform == 'cli': # for IronPython
from System.Diagnostics import Process
PIPE, _STDOUT = None, None
def Popen(CMD, *a, **b):
'''
CMD is a list - a command and its arguments
'''
p = Process()
p.StartInfo.UseShellExecute = False
p.StartInfo.RedirectStandardInput = True
p.StartInfo.RedirectStandardOutput = True
p.StartInfo.RedirectStandardError = True
p.StartInfo.FileName = CMD[0]
p.StartInfo.Arguments = ' '.join(CMD[1:])
p.Start()
return(p)
def sendAll(p, s):
# remove ending newline since WriteLine will add newline at the end of s!
if s.endswith('\r\n'): s = s[:-2]
elif s.endswith('\n'): s = s[:-1]
p.StandardInput.WriteLine(_mybytes(s))
def readLine(p, *a, **b):
return(_mystr(p.StandardOutput.ReadLine()) + '\n') # add newline since ReadLine removed it.
else:
try:
import subprocess
import _subprocess
#try: info.dwFlags |= s
#except Exception: import _subprocess as subprocess
_has_subp = True
Popen, PIPE, _STDOUT = subprocess.Popen, subprocess.PIPE, subprocess.STDOUT
except: # Python 2.3 or older
PIPE, _STDOUT = None, None
def Popen(CMD, *a, **b):
class A: None
p = A()
p.stdin, p.stdout = os.popen4(' '.join(CMD))
return(p)
def sendAll(p, s):
p.stdin.write(_mybytes(s))
#os.write(p.stdin.fileno(), s)
try: p.stdin.flush()
except Exception: pass
def readLine(p, *a, **b):
return(_mystr(p.stdout.readline()))
def NoneStr(obj): return 'NULL'
def BoolStr(obj): return obj and 'TRUE' or 'FALSE'
def ReprStr(obj): return repr(obj)
def LongStr(obj):
rtn = repr(obj)
if rtn[-1] == 'L': rtn = rtn[:-1]
return rtn
def ComplexStr(obj):
return repr(obj).replace('j', 'i')
def SeqStr(obj, head='c(', tail=')'):
if not obj: return head + tail
# detect types
if isinstance(obj, set):
obj = list(obj)
obj0 = obj[0]
tp0 = type(obj0)
simple_types = [str, bool, int, long, float, complex]
num_types = [int, long, float, complex]
is_int = tp0 in (int, long) # token for explicit converstion to integer in R since R treat an integer from stdin as double
if tp0 not in simple_types: head = 'list('
else:
tps = isinstance(obj0, basestring) and [StringType] or num_types
for i in obj[1:]:
tp = type(i)
if tp not in tps:
head = 'list('
is_int = False
break
elif is_int and tp not in (int, long):
is_int = False
# convert
return (is_int and 'as.integer(' or '') + head + ','.join(map(Str4R, obj)) + tail + (is_int and ')' or '')
def DictStr(obj):
return 'list(' + ','.join(['%s=%s' % (Str4R(a[0]), Str4R(a[1])) for a in obj.items()]) + ')'
def OtherStr(obj):
if has_numpy:
if isinstance(obj, numpy.ndarray):
shp = obj.shape
tpdic = {'i':'as.integer(c(%s))', 'u':'as.integer(c(%s))', 'f':'as.double(c(%s))', 'c':'as.complex(c(%s))', 'b':'c(%s)', 'S':'c(%s)', 'a':'c(%s)', 'U':'c(%s)', 'V':'list(%s)'} # in order: (signed) integer, unsigned integer, float, complex, boolean, string, string, unicode, anything
def getVec(ary):
tp = ary.dtype.kind
rlt = ary.reshape(reduce(lambda a,b=1:a*b, ary.shape))
rlt = tp == 'b' and [a and 'TRUE' or 'FALSE' for a in rlt] or rlt.tolist()
if tp != 'V':
return tpdic.get(tp, 'c(%s)') % repr(rlt)[1:-1]
# record array
rlt = list(map(SeqStr, rlt)) # each record will be mapped to vector or list
return tpdic.get(tp, 'list(%s)') % (', '.join(rlt)) # use str here instead of repr since it has already been converted to str by SeqStr
if len(shp) == 1: # to vector
tp = obj.dtype
if tp.kind != 'V':
return getVec(obj)
# One-dimension record array will be converted to data.frame
def mapField(f):
ary = obj[f]
tp = ary.dtype.kind
return '"%s"=%s' % (f, tpdic.get(tp, 'list(%s)') % repr(ary.tolist())[1:-1])
return 'data.frame(%s)' % (', '.join(map(mapField, tp.names)))
elif len(shp) == 2: # two-dimenstion array will be converted to matrix
return 'matrix(%s, nrow=%d, byrow=TRUE)' % (getVec(obj), shp[0])
else: # to array
dim = list(shp[-2:]) # row, col
dim.extend(shp[-3::-1])
newaxis = list(range(len(shp)))
newaxis[-2:] = [len(shp)-1, len(shp)-2]
return 'array(%s, dim=c(%s))' % (getVec(obj.transpose(newaxis)), repr(dim)[1:-1])
# record array and char array
if hasattr(obj, '__iter__'): # for iterators
if hasattr(obj, '__len__') and len(obj) <= 10000:
return SeqStr(list(obj))
else: # waiting for better solution for huge-size containers
return SeqStr(list(obj))
return repr(obj)
base_tps = [type(None), bool, int, long, float, complex, str, unicode, list, tuple, set, frozenset, dict] # use type(None) instead of NoneType since the latter cannot be found in the types module in Python 3
base_tps.reverse()
str_func = {type(None):NoneStr, bool:BoolStr, long:LongStr, int:repr, float:repr, complex:ComplexStr, str:repr, unicode:repr, list:SeqStr, tuple:SeqStr, set:SeqStr, frozenset:SeqStr, dict:DictStr}
def Str4R(obj):
'''
convert a Python basic object into an R object in the form of string.
'''
#return str_func.get(type(obj), OtherStr)(obj)
if type(obj) in str_func:
return str_func[type(obj)](obj)
for tp in base_tps:
if isinstance(obj, tp):
return str_func[tp](obj)
return OtherStr(obj)
class RError(Exception):
def __init__(self, value):
self.value = value
def __str__(self):
return repr(self.value)
class R: # (object):
'''
A Python class to enclose an R process.
'''
__Rfun = r'''.getRvalue4Python__ <- function(x, use_dict=NULL) {
has_numpy <- %s
if (has_numpy) {
headstr <- 'numpy.array('
tailstr <- ')'}
else headstr <- tailstr <- ''
NullStr <- function(x) 'None'
VectorStr <- function(x) {
#nms <- names(x)
#if (!is.null(nms) && length(nms)>0) return(ListStr(as.list(x)))
complx <- is.complex(x)
if (is.character(x)) x <- paste('"', x, '"', sep='')
else if (is.logical(x)) x <- ifelse(x, 'True', 'False')
if (length(x)==1) x <- paste(x) # convert to character, or use "gettext", "as.character"
else x <- paste(headstr, '[', paste(x, collapse=', '), ']', tailstr, sep='')
if (complx) x <- gsub('i', 'j', x)
return(x) }
MatrixStr <- function(x) {
complx <- is.complex(x)
if (is.character(x)) x <- matrix(paste('"', x, '"', sep=''), nrow=nrow(x))
else if (is.logical(x)) x <- ifelse(x, 'True', 'False')
x <- apply(x, 1, function(r) paste('[', paste(r, collapse=', '), ']', sep=''))
x <- paste(headstr, '[', paste(x, collapse=', '), ']', tailstr, sep='')
if (complx) x <- gsub('i', 'j', x)
return(x) }
ArrayStr <- function(x) {
complx <- is.complex(x)
ndim <- length(dim(x))
if (ndim == 1) return(VectorStr(x))
if (ndim == 2) return(MatrixStr(x))
# ndim >= 3
if (is.character(x)) x <- array(paste('"', x, '"', sep=''), dim=dim(x))
else if (is.logical(x)) x <- ifelse(x, 'True', 'False')
for (i in seq(ndim-1))
x <- apply(x, seq(dim(x))[-1], function(r) paste('[', paste(r, collapse=', '), ']', sep=''))
x <- paste(headstr, '[', paste(x, collapse=', '), ']', tailstr, sep='')
if (complx) x <- gsub('i', 'j', x)
return(x) }
DataFrameStr <- function(x) {
cnms <- colnames(x) # get column names
ctp <- list()
for (i in seq(x)) {
xi <- as.vector(x[[i]])
if (is.character(xi)) {
ctp[i] <- sprintf('("%%s", "|S%%d")', cnms[i], max(nchar(xi)) )
xi <- paste('"', xi, '"', sep='') }
else if (is.logical(xi)) {
xi <- ifelse(xi, 'True', 'False')
ctp[i] <- paste('("', cnms[i], '", "<?")' ) }
else if (is.integer(xi)) {
xi <- paste(xi)
ctp[i] <- paste('("', cnms[i], '", "<q")' ) }
else if (is.double(xi)) {
xi <- paste(xi)
ctp[i] <- paste('("', cnms[i], '", "<g")' ) }
else if (is.complex(xi)) {
xi <- gsub('i', 'j', paste(xi))
ctp[i] <- paste('("', cnms[i], '", "<G")') }
x[[i]] <- xi }
x <- as.matrix(x)
x <- apply(x, 1, function(r) paste('(', paste(r, collapse=', '), ')', sep=''))
if (has_numpy) {
tailstr <- paste(', dtype=[', paste(ctp, collapse=', '), ']', tailstr, sep='')
}
x <- paste(headstr, '[', paste(x, collapse=', '), ']', tailstr, sep='')
return(x) }
ListStr <- function(x) {
nms <- names(x) # get column names
x <- sapply(x, Str4Py)
if (!is.null(nms) && length(nms)>0) {
nms <- paste('"', nms, '"', sep='')
x <- sapply(seq(nms), function(i) paste('(', nms[i], ',', x[i], ')') )
if (identical(use_dict, TRUE)) x <- paste('dict([', paste(x, collapse=', '), '])', sep='')
else if (identical(use_dict, FALSE)) x <- paste('[', paste(x, collapse=', '), ']', sep='')
else { # should be NULL or something else
if (length(nms) != length(unique(nms))) x <- paste('[', paste(x, collapse=', '), ']', sep='')
else x <- paste('dict([', paste(x, collapse=', '), '])', sep='')
}
}
else
x <- paste('[', paste(x, collapse=', '), ']', sep='')
return(x) }
Str4Py <- function(x, outmost=FALSE) {
# no considering on NA, Inf, ...
# use is.XXX, typeof, class, mode, storage.mode, sprintf
if (is.factor(x)) x <- as.vector(x)
rlt <- {
if (is.null(x)) NullStr(x)
else if (is.vector(x) && !is.list(x)) VectorStr(x)
else if (is.matrix(x) || is.array(x)) ArrayStr(x)
else if (is.data.frame(x)) DataFrameStr(x)
else if (is.list(x)) ListStr(x)
else Str4Py(as.character(x)) # other objects will be convert to character (instead of NullStr), or use "gettext"
}
if (outmost) rlt <- gsub('\\\\', '\\\\\\\\', rlt)
return(rlt)
}
Str4Py(x, outmost=TRUE)
}
# initalize library path for TCL/TK based environment on Windows, e.g. Python IDLE
.addLibs <- function() {
ruser <- Sys.getenv('R_USER')
userpath <- Sys.getenv('R_LIBS_USER')
libpaths <- .libPaths()
for (apath in userpath) {
if (length(grep(apath, libpaths)) > 0) next
if (file.exists(apath)) .libPaths(apath)
else {
d <- '/Documents'
if (substr(ruser, nchar(ruser)-nchar(d)+1, nchar(ruser)) != d) {
apath <- paste(ruser,d, substr(apath, nchar(ruser)+1, nchar(apath)), sep='')
if (file.exists(apath)) .libPaths(apath)}
}
}
}
if(identical(.Platform$OS.type, 'windows')) .addLibs()
rm(.addLibs)
'''
_DEBUG_MODE = True
def __init__(self, RCMD='R', max_len=1000, use_numpy=True, use_dict=None, host='localhost', user=None, ssh='ssh', return_err=True):
'''
RCMD: The name of a R interpreter, path information should be included
if it is not in the system search path.
use_numpy: Used as a boolean value. A False value will disable numpy
even if it has been imported.
use_dict: A R named list will be returned as a Python dictionary if
"use_dict" is True, or a list of tuples (name, value) if "use_dict"
is False. If "use_dict" is None, the return value will be a
dictionary if there is no replicated names, or a list if replicated
names found.
host: The computer name (or IP) on which the R interpreter is
installed. The value "localhost" means that R locates on the the
localhost computer. On POSIX systems (including Cygwin environment
on Windows), it is possible to use R on a remote computer if the
command "ssh" works. To do that, the user needs to set this value,
and perhaps the parameter "user".
user: The user name on the remote computer. This value needs to be set
only if the user name on the remote computer is different from the
local user. In interactive environment, the password can be input
by the user if prompted. If running in a program, the user needs to
be able to login without typing password!
ssh: The program to login to remote computer.
return_err: redict stderr to stdout
'''
# use self.__dict__.update to register variables since __setattr__ is
# used to set variables for R. tried to define __setattr in the class,
# and change it to __setattr__ for instances at the end of __init__,
# but it seems failed.
# -- maybe this only failed in Python2.5? as warned at
# http://wiki.python.org/moin/NewClassVsClassicClass:
# "Warning: In 2.5, magic names (typically those with a double
# underscore (DunderAlias) at both ends of the name) may look at the
# class rather than the instance even for old-style classes."
self.__dict__.update({
'max_len' : max_len,
'use_dict' : use_dict,
'localhost' : host=='localhost',
'newline' : sys.platform=='win32' and '\r\n' or '\n'})
RCMD = [RCMD] #shlex.split(RCMD) - shlex do not work properly on Windows! #re.split(r'\s', RCMD)
if not self.localhost:
RCMD.insert(0, host)
if user:
RCMD.insert(0, '-l%s' % user)
RCMD.insert(0, ssh)
#args = ('--vanilla',) # equal to --no-save, --no-restore, --no-site-file, --no-init-file and --no-environ
args = ('--quiet', '--no-save', '--no-restore') # "--slave" cannot be used on Windows!
for arg in args:
if arg not in RCMD: RCMD.append(arg)
if _has_subp and hasattr(subprocess, 'STARTUPINFO'):
info = subprocess.STARTUPINFO()
try: info.dwFlags |= subprocess.STARTF_USESHOWWINDOW
except Exception: info.dwFlags |= _subprocess.STARTF_USESHOWWINDOW
try: info.wShowWindow = subprocess.SW_HIDE
except Exception: info.wShowWindow = _subprocess.SW_HIDE
else: info = None
self.__dict__.update({
'prog' : Popen(RCMD, stdin=PIPE, stdout=PIPE, stderr=return_err and _STDOUT or None, startupinfo=info),
'has_numpy' : use_numpy and has_numpy,
'Rfun' : self.__class__.__Rfun % ((use_numpy and has_numpy) and 'TRUE' or 'FALSE')})
self.__call__(self.Rfun)
#to_discard = recv_some(self.prog, e=0, t=wait0)
def __runOnce(self, CMD, use_try=None):
'''
CMD: a R command string
'''
use_try = use_try or self._DEBUG_MODE
newline = self.newline
tail_token = 'R command at time: %s' % repr(time.time())
#tail_token_r = re.sub(r'[\(\)\.]', r'\\\1', tail_token)
tail_cmd = 'print("%s")%s' % (tail_token, newline)
re_tail = re.compile(r'>\sprint\("%s"\)\r?\n\[1\]\s"%s"\r?\n$' % (tail_token.replace(' ', '\\s'), tail_token.replace(' ', '\\s')) )
if len(CMD) <= self.max_len or not self.localhost:
fn = None
else:
fh, fn = tempfile.mkstemp()
os.fdopen(fh, 'wb').write(_mybytes(CMD))
if sys.platform == 'cli': os.close(fh) # this is necessary on IronPython
CMD = 'source("%s")' % fn.replace('\\', '/')
CMD = (use_try and 'try({%s})%s%s' or '%s%s%s') % (CMD, newline, tail_cmd)
sendAll(self.prog, CMD)
rlt = ''
while not re_tail.search(rlt):
try:
rltonce = readLine(self.prog)
if rltonce: rlt = rlt + rltonce
except: break
else:
rlt = re_tail.sub('', rlt)
if rlt.startswith('> '): rlt = rlt[2:]
if fn is not None:
os.unlink(fn)
return rlt
def __call__(self, CMDS=[], use_try=None):
'''
Run a (list of) R command(s), and return the output message from the STDOUT of R.
CMDS: an R command string or a list of R commands
'''
rlt = []
if isinstance(CMDS, basestring): # a single command
rlt.append(self.__runOnce(CMDS, use_try=use_try))
else: # should be a list of commands
for CMD in CMDS:
rlt.append(self.__runOnce(CMD, use_try=use_try))
if len(rlt) == 1: rlt = rlt[0]
return rlt
def __getitem__(self, obj, use_try=None, use_dict=None): # to model r['XXX']
'''
Get the value of an R variable or expression. The return value is
converted to the corresponding Python object.
obj: a string - the name of an R variable, or an R expression
use_try: use "try" function to wrap the R expression. This can avoid R
crashing if the obj does not exist in R.
use_dict: named list will be returned a dict if use_dict is True,
otherwise it will be a list of tuples (name, value)
'''
if obj.startswith('_'):
raise RError('Leading underscore ("_") is not permitted in R variable names!')
use_try = use_try or self._DEBUG_MODE
if use_dict is None: use_dict = self.use_dict
cmd = '.getRvalue4Python__(%s, use_dict=%s)' % (obj, use_dict is None and 'NULL' or use_dict and 'TRUE' or 'FALSE')
rlt = self.__call__(cmd, use_try=use_try)
head = (use_try and 'try({%s})%s[1] ' or '%s%s[1] ') % (cmd, self.newline)
# sometimes (e.g. after "library(fastICA)") the R on Windows uses '\n' instead of '\r\n'
head = rlt.startswith(head) and len(head) or len(head) - 1
tail = rlt.endswith(self.newline) and len(rlt) - len(self.newline) or len(rlt) - len(self.newline) + 1 # - len('"')
try:
rlt = eval(eval(rlt[head:tail])) # The inner eval remove quotes and recover escaped characters.
except:
raise RError(rlt)
return rlt
def __setitem__(self, obj, val): # to model r['XXX']
'''
Assign a value (val) to an R variable (obj).
obj: a string - the name of an R variable
val: a python object - the value to be passed to an R object
'''
if obj.startswith('_'):
raise RError('Leading underscore ("_") is not permitted in R variable names!')
self.__call__('%s <- %s' % (obj, Str4R(val)))
def __delitem__(self, obj):
if obj.startswith('_'):
raise RError('Leading underscore ("_") is not permitted in R variable names!')
self.__call__('rm(%s)' % obj)
def __del__(self):
sendAll(self.prog, 'q("no")'+self.newline)
self.prog = None
def __getattr__(self, obj, use_dict=None): # to model r.XXX
'''
obj: a string - the name of an R variable
use_dict: named list will be returned a dict if use_dict is True,
otherwise it will be a list of tuples (name, value)
'''
# Overriding __getattr__ is safer than __getattribute__ since it is
# only called as a last resort i.e. if there are no attributes in the
# instance that match the name
try:
if use_dict is None: use_dict = self.use_dict
rlt = self.__getitem__(obj, use_dict=use_dict)
except:
raise RError('No this object!')
return rlt
def __setattr__(self, obj, val): # to model r.XXX
if obj in self.__dict__ or obj in self.__class__.__dict__: # or obj.startswith('_'):
self.__dict__[obj] = val # for old-style class
#object.__setattr__(self, obj, val) # for new-style class
else:
self.__setitem__(obj, val)
def __delattr__(self, obj):
if obj in self.__dict__:
del self.__dict__[obj]
else:
self.__delitem__(obj)
def get(self, obj, default=None, use_dict=None):
'''
obj: a string - the name of an R variable, or an R expression
default: a python object - the value to be returned if failed to get data from R
use_dict: named list will be returned a dict if use_dict is True,
otherwise it will be a list of tuples (name, value). If use_dict is
None, the value of self.use_dict will be used instead.
'''
try:
rlt = self.__getitem__(obj, use_try=True, use_dict=use_dict)
except:
if True: #val is not None:
rlt = default
else:
raise RError('No this object!')
return rlt
run, assign, remove = __call__, __setitem__, __delitem__
# for a single-round duty:
def runR(CMDS, Robj='R', max_len=1000, use_numpy=True, use_dict=None, host='localhost', user=None, ssh='ssh'):
'''
Run a (list of) R command(s), and return the output from the STDOUT.
CMDS: a R command string or a list of R commands.
Robj: can be a shell command (like /usr/bin/R), or the R class.
max_len: define the upper limitation for the length of command string. A
command string will be passed to R by a temporary file if it is longer
than this value.
use_numpy: Used as a boolean value. A False value will disable numpy even
if it has been imported.
use_dict: named list will be returned a dict if use_dict is True, otherwise
it will be a list of tuples (name, value).
host: The computer name (or IP) on which the R interpreter is
installed. The value "localhost" means that the R locates on the
the localhost computer. On POSIX systems (including Cygwin
environment on Windows), it is possible to use R on a remote
computer if the command "ssh" works. To do that, the user need set
this value, and perhaps the parameter "user".
user: The user name on the remote computer. This value need to be set
only if the user name is different on the remote computer. In
interactive environment, the password can be input by the user if
prompted. If running in a program, the user need to be able to
login without typing password!
ssh: The program to login to remote computer.
'''
if isinstance(Robj, basestring):
Robj = R(RCMD=Robj, max_len=max_len, use_numpy=use_numpy, use_dict=use_dict, host=host, user=user, ssh=ssh)
rlt = Robj.run(CMDS=CMDS)
if len(rlt) == 1: rlt = rlt[0]
return rlt
if __name__ == '__main__':
import unique
path = unique.filepath("AltDatabase/R/Contents/MacOS/R")
r = R(RCMD='R',use_numpy=True)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/pyper.py
|
pyper.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import math
#import pkg_resources
#import distutils
from stats_scripts import statistics
import sys, string
import os.path
import unique
import update
import UI
import copy
import export; reload(export)
import ExpressionBuilder; reload(ExpressionBuilder)
from build_scripts import ExonAnalyze_module; reload(ExonAnalyze_module)
from import_scripts import ExonAnnotate_module; reload(ExonAnnotate_module)
from import_scripts import ResultsExport_module
from build_scripts import FeatureAlignment
import GO_Elite
import time
import webbrowser
import random
import traceback
import shutil
try:
import multiprocessing as mlp
except Exception:
mlp=None
print 'Note: Multiprocessing not supported for this verison python.'
try:
from scipy import stats
except Exception:
pass ### scipy is not required but is used as a faster implementation of Fisher Exact Test when present
try:
from PIL import Image as PIL_Image
try: import ImageTk
except Exception: from PIL import ImageTk
import PIL._imaging
import PIL._imagingft
except Exception:
print traceback.format_exc()
pass #print 'Python Imaging Library not installed... using default PNG viewer'
use_Tkinter = 'no'
debug_mode = 'no'
analysis_start_time = time.time()
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>1 and '-' in command_args and '--GUI' not in command_args:
runningCommandLine = True
else:
runningCommandLine = False
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir)
dir_list2 = [] #add in code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv" or entry[-4:] == ".TXT":
dir_list2.append(entry)
return dir_list2
def eliminate_redundant_dict_values(database):
db1={}
for key in database:
list = unique.unique(database[key])
list.sort()
db1[key] = list
return db1
def makeUnique(item):
db1={}; list1=[]; k=0
for i in item:
try: db1[i]=[]
except TypeError: db1[tuple(i)]=[]; k=1
for i in db1:
if k==0: list1.append(i)
else: list1.append(list(i))
list1.sort()
return list1
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def returnLargeGlobalVars():
### Prints all large global variables retained in memory (taking up space)
all = [var for var in globals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(globals()[var])>500:
print var, len(globals()[var])
except Exception: null=[]
def clearObjectsFromMemory(db_to_clear):
db_keys={}
try:
for key in db_to_clear: db_keys[key]=[]
except Exception:
for key in db_to_clear: del key ### if key is a list
for key in db_keys:
try: del db_to_clear[key]
except Exception:
try:
for i in key: del i ### For lists of tuples
except Exception: del key ### For plain lists
def importGeneric(filename):
fn=filepath(filename); key_db = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
key_db[t[0]] = t[1:]
return key_db
def importGenericFiltered(filename,filter_db):
fn=filepath(filename); key_db = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
key = t[0]
if key in filter_db: key_db[key] = t[1:]
return key_db
def importGenericFilteredDBList(filename,filter_db):
fn=filepath(filename); key_db = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
try:
null=filter_db[t[0]]
try: key_db[t[0]].append(t[1])
except KeyError: key_db[t[0]] = [t[1]]
except Exception: null=[]
return key_db
def importGenericDBList(filename):
fn=filepath(filename); key_db = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
try: key_db[t[0]].append(t[1])
except KeyError: key_db[t[0]] = [t[1]]
return key_db
def importExternalDBList(filename):
fn=filepath(filename); key_db = {}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
try: key_db[t[0]].append(t[1:])
except Exception: key_db[t[0]] = [t[1:]]
return key_db
def FindDir(dir,term):
dir_list = unique.read_directory(dir)
dir_list2=[]
dir_list.sort()
for i in dir_list:
if term == i: dir_list2.append(i)
if len(dir_list2)==0:
for i in dir_list:
if term in i: dir_list2.append(i)
dir_list2.sort(); dir_list2.reverse()
if len(dir_list2)>0: return dir_list2[0]
else: return ''
def openFile(file_dir):
if runningCommandLine:
pass
elif os.name == 'nt':
try: os.startfile('"'+file_dir+'"')
except Exception: os.system('open "'+file_dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+file_dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+file_dir+'"')
def openCytoscape(parent_dir,application_dir,application_name):
cytoscape_dir = FindDir(parent_dir,application_dir); cytoscape_dir = filepath(parent_dir+'/'+cytoscape_dir)
app_dir = FindDir(cytoscape_dir,application_name)
app_dir = cytoscape_dir+'/'+app_dir
if 'linux' in sys.platform:
app_dir = app_dir
app_dir2 = cytoscape_dir+'/Cytoscape'
try: createCytoscapeDesktop(cytoscape_dir)
except Exception: null=[]
dir_list = unique.read_directory('/usr/bin/') ### Check to see that JAVA is installed
if 'java' not in dir_list: print 'Java not referenced in "usr/bin/. If not installed,\nplease install and re-try opening Cytoscape'
try:
jar_path = cytoscape_dir+'/cytoscape.jar'
main_path = cytoscape_dir+'/cytoscape.CyMain'
plugins_path = cytoscape_dir+'/plugins'
os.system('java -Dswing.aatext=true -Xss5M -Xmx512M -jar '+jar_path+' '+main_path+' -p '+plugins_path+' &')
print 'Cytoscape jar opened:',jar_path
except Exception:
print 'OS command to open Java failed.'
try:
try: openFile(app_dir2); print 'Cytoscape opened:',app_dir2
except Exception:
os.chmod(app_dir,0777)
openFile(app_dir2)
except Exception:
try: openFile(app_dir)
except Exception:
os.chmod(app_dir,0777)
openFile(app_dir)
else:
try: openFile(app_dir)
except Exception:
os.chmod(app_dir,0777)
openFile(app_dir)
def createCytoscapeDesktop(cytoscape_dir):
cyto_ds_output = cytoscape_dir+'/Cytoscape.desktop'
data = export.ExportFile(cyto_ds_output)
cytoscape_desktop = cytoscape_dir+'/Cytoscape'; #cytoscape_desktop = '/hd3/home/nsalomonis/Cytoscape_v2.6.1/Cytoscape'
cytoscape_png = cytoscape_dir+ '/.install4j/Cytoscape.png'; #cytoscape_png = '/hd3/home/nsalomonis/Cytoscape_v2.6.1/.install4j/Cytoscape.png'
data.write('[Desktop Entry]'+'\n')
data.write('Type=Application'+'\n')
data.write('Name=Cytoscape'+'\n')
data.write('Exec=/bin/sh "'+cytoscape_desktop+'"'+'\n')
data.write('Icon='+cytoscape_png+'\n')
data.write('Categories=Application;'+'\n')
data.close()
########### Parse Input Annotations ###########
def ProbesetCalls(array_type,probeset_class,splice_event,constitutive_call,external_exonid):
include_probeset = 'yes'
if array_type == 'AltMouse':
exonid = splice_event
if filter_probesets_by == 'exon':
if '-' in exonid or '|' in exonid: ###Therfore the probeset represents an exon-exon junction or multi-exon probeset
include_probeset = 'no'
if filter_probesets_by != 'exon':
if '|' in exonid: include_probeset = 'no'
if constitutive_call == 'yes': include_probeset = 'yes'
else:
if avg_all_for_ss == 'yes' and (probeset_class == 'core' or len(external_exonid)>2): constitutive_call = 'yes'
#if len(splice_event)>2 and constitutive_call == 'yes' and avg_all_for_ss == 'no': constitutive_call = 'no'
if constitutive_call == 'no' and len(splice_event)<2 and len(external_exonid)<2: ###otherwise these are interesting probesets to keep
if filter_probesets_by != 'full':
if filter_probesets_by == 'extended':
if probeset_class == 'full': include_probeset = 'no'
elif filter_probesets_by == 'core':
if probeset_class != 'core': include_probeset = 'no'
return include_probeset,constitutive_call
def EvidenceOfAltSplicing(slicing_annot):
splice_annotations = ["ntron","xon","strangeSplice","Prime","3","5","C-term"]; as_call = 0
splice_annotations2 = ["ntron","assette","strangeSplice","Prime","3","5"]
for annot in splice_annotations:
if annot in slicing_annot: as_call = 1
if as_call == 1:
if "C-term" in slicing_annot and ("N-" in slicing_annot or "Promoter" in slicing_annot):
as_call = 0
for annot in splice_annotations2:
if annot in slicing_annot: as_call = 1
elif "bleed" in slicing_annot and ("N-" in slicing_annot or "Promoter" in slicing_annot):
as_call = 0
for annot in splice_annotations2:
if annot in slicing_annot: as_call = 1
return as_call
########### Begin Analyses ###########
class SplicingAnnotationData:
def ArrayType(self):
self._array_type = array_type
return self._array_type
def Probeset(self): return self._probeset
def setProbeset(self,probeset): self._probeset = probeset
def ExonID(self): return self._exonid
def setDisplayExonID(self,exonid): self._exonid = exonid
def GeneID(self): return self._geneid
def Symbol(self):
symbol = ''
if self.GeneID() in annotate_db:
y = annotate_db[self.GeneID()]
symbol = y.Symbol()
return symbol
def ExternalGeneID(self): return self._external_gene
def ProbesetType(self):
###e.g. Exon, junction, constitutive(gene)
return self._probeset_type
def GeneStructure(self): return self._block_structure
def SecondaryExonID(self): return self._block_exon_ids
def setSecondaryExonID(self,ids): self._block_exon_ids = ids
def setLocationData(self, chromosome, strand, probeset_start, probeset_stop):
self._chromosome = chromosome; self._strand = strand
self._start = probeset_start; self._stop = probeset_stop
def LocationSummary(self):
location = self.Chromosome()+':'+self.ProbeStart()+'-'+self.ProbeStop()+'('+self.Strand()+')'
return location
def Chromosome(self): return self._chromosome
def Strand(self): return self._strand
def ProbeStart(self): return self._start
def ProbeStop(self): return self._stop
def ProbesetClass(self):
###e.g. core, extendended, full
return self._probest_class
def ExternalExonIDs(self): return self._external_exonids
def ExternalExonIDList(self):
external_exonid_list = string.split(self.ExternalExonIDs(),'|')
return external_exonid_list
def Constitutive(self): return self._constitutive_status
def setTranscriptCluster(self,secondary_geneid): self._secondary_geneid = secondary_geneid
def setNovelExon(self,novel_exon): self._novel_exon = novel_exon
def NovelExon(self): return self._novel_exon
def SecondaryGeneID(self): return self._secondary_geneid
def setExonRegionID(self,exon_region): self._exon_region = exon_region
def ExonRegionID(self): return self._exon_region
def SplicingEvent(self):
splice_event = self._splicing_event
if len(splice_event)!=0:
if splice_event[0] == '|': splice_event = splice_event[1:]
return splice_event
def SplicingCall(self): return self._splicing_call
def SpliceJunctions(self): return self._splice_junctions
def Delete(self): del self
def Report(self):
output = self.ArrayType() +'|'+ self.ExonID() +'|'+ self.ExternalGeneID()
return output
def __repr__(self): return self.Report()
class AltMouseData(SplicingAnnotationData):
def __init__(self,affygene,exons,ensembl,block_exon_ids,block_structure,probe_type_call):
self._geneid = affygene; self._external_gene = ensembl; self._exonid = exons; self._secondary_geneid = ensembl
self._probeset_type = probe_type_call; self._block_structure = block_structure; self._block_exon_ids = block_exon_ids
self._external_exonids = 'NA';
self._constitutive_status = 'no'
self._splicing_event = ''
self._secondary_geneid = 'NA'
self._exon_region = ''
if self._probeset_type == 'gene': self._constitutive_status = 'yes'
else: self._constitutive_status = 'no'
class AffyExonSTData(SplicingAnnotationData):
def __init__(self,ensembl_gene_id,exon_id,ens_exon_ids, constitutive_call_probeset, exon_region, splicing_event, splice_junctions, splicing_call):
self._geneid = ensembl_gene_id; self._external_gene = ensembl_gene_id; self._exonid = exon_id
self._constitutive_status = constitutive_call_probeset#; self._start = probeset_start; self._stop = probeset_stop
self._external_exonids = ens_exon_ids; #self._secondary_geneid = transcript_cluster_id#; self._chromosome = chromosome; self._strand = strand
self._exon_region=exon_region; self._splicing_event=splicing_event; self._splice_junctions=splice_junctions; self._splicing_call = splicing_call
if self._exonid[0] == 'U': self._probeset_type = 'UTR'
elif self._exonid[0] == 'E': self._probeset_type = 'exonic'
elif self._exonid[0] == 'I': self._probeset_type = 'intronic'
class AffyExonSTDataAbbreviated(SplicingAnnotationData):
def __init__(self,ensembl_gene_id,exon_id,splicing_call):
self._geneid = ensembl_gene_id; self._exonid = exon_id; self._splicing_call = splicing_call
def importSplicingAnnotations(array_type,Species,probeset_type,avg_ss_for_all,root_dir):
global filter_probesets_by; filter_probesets_by = probeset_type
global species; species = Species; global avg_all_for_ss; avg_all_for_ss = avg_ss_for_all; global exon_db; exon_db={}
global summary_data_db; summary_data_db={}; global remove_intronic_junctions; remove_intronic_junctions = 'no'
if array_type == 'RNASeq':
probeset_annotations_file = root_dir+'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_junctions.txt'
else: probeset_annotations_file = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_probesets.txt'
filtered_arrayids={};filter_status='no'
constitutive_probeset_db,exon_db,genes_being_analyzed = importSplicingAnnotationDatabase(probeset_annotations_file,array_type,filtered_arrayids,filter_status)
return exon_db, constitutive_probeset_db
def importSplicingAnnotationDatabase(filename,array_type,filtered_arrayids,filter_status):
begin_time = time.time()
probesets_included_by_new_evidence = 0; export_exon_regions = 'yes'
if 'fake' in array_type: array_type = string.replace(array_type,'-fake',''); original_arraytype = 'RNASeq'
else: original_arraytype = array_type
if filter_status == 'no': global gene_transcript_cluster_db; gene_transcript_cluster_db={}; gene_transcript_cluster_db2={}; global last_exon_region_db; last_exon_region_db = {}
else: new_exon_db={}
fn=filepath(filename)
last_gene = ' '; last_exon_region = ''
constitutive_probeset_db = {}; constitutive_gene = {}
count = 0; x = 0; constitutive_original = {}
#if filter_status == 'yes': exon_db = {}
if array_type == 'AltMouse':
for line in open(fn,'rU').xreadlines():
probeset_data = cleanUpLine(line) #remove endline
probeset,affygene,exons,transcript_num,transcripts,probe_type_call,ensembl,block_exon_ids,block_structure,comparison_info = string.split(probeset_data,'\t')
###note: currently exclude comparison_info since not applicable for existing analyses
if x == 0: x = 1
else:
if exons[-1] == '|': exons = exons[0:-1]
if affygene[-1] == '|': affygene = affygene[0:-1]; constitutive_gene[affygene]=[]
if probe_type_call == 'gene': constitutive_call = 'yes' #looked through the probe annotations and the gene seems to be the most consistent constitutive feature
else: constitutive_call = 'no'
include_call,constitutive_call = ProbesetCalls(array_type,'',exons,constitutive_call,'')
if include_call == 'yes':
probe_data = AltMouseData(affygene,exons,ensembl,block_exon_ids,block_structure,probe_type_call) #this used to just have affygene,exon in the values (1/17/05)
exon_db[probeset] = probe_data
if filter_status == 'yes': new_exon_db[probeset] = probe_data
if constitutive_call == 'yes': constitutive_probeset_db[probeset] = affygene
genes_being_analyzed = constitutive_gene
else:
for line in open(fn,'rU').xreadlines():
probeset_data = cleanUpLine(line) #remove endline
if x == 0: x = 1
else:
try: probeset_id, exon_id, ensembl_gene_id, transcript_cluster_id, chromosome, strand, probeset_start, probeset_stop, affy_class, constitutive_call_probeset, external_exonid, ens_const_exons, exon_region, exon_region_start, exon_region_stop, splicing_event, splice_junctions = string.split(probeset_data,'\t')
except Exception: print probeset_data;force_error
if affy_class == 'free': affy_class = 'full' ### Don't know what the difference is
include_call,constitutive_call = ProbesetCalls(array_type,affy_class,splicing_event,constitutive_call_probeset,external_exonid)
#if 'ENSG00000163904:E11.5' in probeset_id: print probeset_data
#print array_type,affy_class,splicing_event,constitutive_call_probeset,external_exonid,constitutive_call,include_call;kill
if array_type == 'junction' and '.' not in exon_id: exon_id = string.replace(exon_id,'-','.'); exon_region = string.replace(exon_region,'-','.')
if ensembl_gene_id != last_gene: new_gene = 'yes'
else: new_gene = 'no'
if filter_status == 'no' and new_gene == 'yes':
if '.' in exon_id: ### Exclude junctions
if '-' not in last_exon_region and 'E' in last_exon_region: last_exon_region_db[last_gene] = last_exon_region
else: last_exon_region_db[last_gene] = last_exon_region
last_gene = ensembl_gene_id
if len(exon_region)>1: last_exon_region = exon_region ### some probeset not linked to an exon region
###Record the transcript clusters assoicated with each gene to annotate the results later on
if constitutive_call_probeset!=constitutive_call: probesets_included_by_new_evidence +=1#; print probeset_id,[splicing_event],[constitutive_call_probeset];kill
proceed = 'no'; as_call = 0
if array_type == 'RNASeq' or array_type == 'junction': include_call = 'yes' ### Constitutive expression is not needed
if remove_intronic_junctions == 'yes':
if 'E' not in exon_id: include_call = 'no' ### Remove junctions that only have splice-sites within an intron or UTR
if include_call == 'yes' or constitutive_call == 'yes':
#if proceed == 'yes':
as_call = EvidenceOfAltSplicing(splicing_event)
if filter_status == 'no':
probe_data = AffyExonSTDataAbbreviated(ensembl_gene_id, exon_id, as_call)
if array_type != 'RNASeq':
probe_data.setTranscriptCluster(transcript_cluster_id)
try:
if export_exon_regions == 'yes':
probe_data.setExonRegionID(exon_region)
except Exception: null=[]
else:
probe_data = AffyExonSTData(ensembl_gene_id, exon_id, external_exonid, constitutive_call, exon_region, splicing_event, splice_junctions, as_call)
probe_data.setLocationData(chromosome, strand, probeset_start, probeset_stop)
if array_type != 'RNASeq':
probe_data.setTranscriptCluster(transcript_cluster_id)
else:
probe_data.setNovelExon(affy_class)
if filter_status == 'yes':
try: ### saves memory
null = filtered_arrayids[probeset_id]
new_exon_db[probeset_id] = probe_data
except KeyError: null = []
else: exon_db[probeset_id] = probe_data
if constitutive_call == 'yes' and filter_status == 'no': ###only perform function when initially running
constitutive_probeset_db[probeset_id] = ensembl_gene_id
try: constitutive_gene[ensembl_gene_id].append(probeset_id)
except Exception: constitutive_gene[ensembl_gene_id] = [probeset_id]
###Only consider transcript clusters that make up the constitutive portion of the gene or that are alternatively regulated
if array_type != 'RNASeq':
try: gene_transcript_cluster_db[ensembl_gene_id].append(transcript_cluster_id)
except KeyError: gene_transcript_cluster_db[ensembl_gene_id] = [transcript_cluster_id]
if constitutive_call_probeset == 'yes' and filter_status == 'no': ###only perform function when initially running
try: constitutive_original[ensembl_gene_id].append(probeset_id)
except KeyError: constitutive_original[ensembl_gene_id] = [probeset_id]
if array_type != 'RNASeq':
try: gene_transcript_cluster_db2[ensembl_gene_id].append(transcript_cluster_id)
except KeyError: gene_transcript_cluster_db2[ensembl_gene_id] = [transcript_cluster_id]
###If no constitutive probesets for a gene as a result of additional filtering (removing all probesets associated with a splice event), add these back
original_probesets_add = 0; genes_being_analyzed = {}
for gene in constitutive_gene: genes_being_analyzed[gene]=[]
for gene in constitutive_original:
if gene not in constitutive_gene:
genes_being_analyzed[gene] = [gene]
constitutive_gene[gene]=[]
original_probesets_add +=1
gene_transcript_cluster_db[gene] = gene_transcript_cluster_db2[gene]
for probeset in constitutive_original[gene]: constitutive_probeset_db[probeset] = gene
#if array_type == 'junction' or array_type == 'RNASeq':
### Added the below in 1.16!!!
### If no constitutive probesets for a gene assigned, assign all gene probesets
for probeset in exon_db:
gene = exon_db[probeset].GeneID()
proceed = 'no'
exonid = exon_db[probeset].ExonID()
### Rather than add all probesets, still filter based on whether the probeset is in an annotated exon
if 'E' in exonid and 'I' not in exonid and '_' not in exonid: proceed = 'yes'
if proceed == 'yes':
if gene not in constitutive_gene:
constitutive_probeset_db[probeset] = gene
genes_being_analyzed[gene] = [gene]
### DO NOT ADD TO constitutive_gene SINCE WE WANT ALL mRNA ALIGNING EXONS/JUNCTIONS TO BE ADDED!!!!
#constitutive_gene[gene]=[]
gene_transcript_cluster_db = eliminate_redundant_dict_values(gene_transcript_cluster_db)
#if affygene == 'ENSMUSG00000023089': print [abs(fold_change_log)],[log_fold_cutoff];kill
if array_type == 'RNASeq':
import RNASeq
try: last_exon_region_db = RNASeq.importExonAnnotations(species,'distal-exon','')
except Exception: null=[]
constitutive_original=[]; constitutive_gene=[]
#clearObjectsFromMemory(exon_db); constitutive_probeset_db=[];genes_being_analyzed=[] ### used to evaluate how much memory objects are taking up
#print 'remove_intronic_junctions:',remove_intronic_junctions
#print constitutive_gene['ENSMUSG00000031170'];kill ### Determine if avg_ss_for_all is working
if original_arraytype == 'RNASeq': id_name = 'exon/junction IDs'
else: id_name = 'array IDs'
print len(exon_db),id_name,'stored as instances of SplicingAnnotationData in memory'
#print len(constitutive_probeset_db),'array IDs stored as constititive'
#print probesets_included_by_new_evidence, 'array IDs were re-annotated as NOT constitutive based on mRNA evidence'
if array_type != 'AltMouse': print original_probesets_add, 'genes not viewed as constitutive as a result of filtering',id_name,'based on splicing evidence, added back'
end_time = time.time(); time_diff = int(end_time-begin_time)
#print filename,"import finished in %d seconds" % time_diff
if filter_status == 'yes': return new_exon_db
else:
summary_data_db['gene_assayed'] = len(genes_being_analyzed)
try: exportDenominatorGenes(genes_being_analyzed)
except Exception: null=[]
return constitutive_probeset_db,exon_db,genes_being_analyzed
def exportDenominatorGenes(genes_being_analyzed):
goelite_output = root_dir+'GO-Elite/denominator/AS.denominator.txt'
goelite_data = export.ExportFile(goelite_output)
systemcode = 'En'
goelite_data.write("GeneID\tSystemCode\n")
for gene in genes_being_analyzed:
if array_type == 'AltMouse':
try: gene = annotate_db[gene].ExternalGeneID()
except KeyError: null = []
goelite_data.write(gene+'\t'+systemcode+'\n')
try: goelite_data.close()
except Exception: null=[]
def performExpressionAnalysis(filename,constitutive_probeset_db,exon_db,annotate_db,dataset_name):
#if analysis_method == 'splicing-index': returnLargeGlobalVars();kill ### used to ensure all large global vars from the reciprocal junction analysis have been cleared from memory
#returnLargeGlobalVars()
"""import list of expression values for arrayids and calculates statistics"""
global fold_dbase; global original_conditions; global normalization_method
stats_dbase = {}; fold_dbase={}; ex_db={}; si_db=[]; bad_row_import = {}; count=0
global array_group_name_db; array_group_name_db = {}
global array_group_db; array_group_db = {};
global array_raw_group_values; array_raw_group_values = {}; global original_array_names; original_array_names=[]
global max_replicates; global equal_replicates; global array_group_list
array_index_list = [] ###Use this list for permutation analysis
fn=filepath(filename); line_num = 1
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line); t = string.split(data,'\t'); probeset = t[0]
if t[0]== '#': null=[] ### Don't import line
elif line_num == 1:
line_num += 1 #makes this value null for the next loop of actual array data
###Below ocucrs if the data is raw opposed to precomputed
if ':' in t[1]:
array_group_list = []; x=0 ###gives us an original index value for each entry in the group
for entry in t[1:]:
original_array_names.append(entry)
aa = string.split(entry,':')
try: array_group,array_name = aa
except Exception: array_name = string.join(aa[1:],':'); array_group = aa[0]
try:
array_group_db[array_group].append(x)
array_group_name_db[array_group].append(array_name)
except KeyError:
array_group_db[array_group] = [x]
array_group_name_db[array_group] = [array_name]
### below only occurs with a new group addition
array_group_list.append(array_group) #use this to generate comparisons in the below linked function
x += 1
else:
#try: print data_type
#except Exception,exception:
#print exception
#print traceback.format_exc()
print_out = 'The AltAnalyze filtered expression file "'+filename+'" is not propperly formatted.\n Review formatting requirements if this file was created by another application.\n'
print_out += "\nFirst line\n"+line
try: UI.WarningWindow(print_out,'Exit'); print print_out
except Exception: print print_out
badExit()
else:
#if probeset in exon_db:
#if exon_db[probeset].GeneID() == 'ENSG00000139970':
###Use the index values from above to assign each expression value to a new database
temp_group_array = {}
line_num+=1
for group in array_group_db:
if count == 0: array_index_list.append(array_group_db[group])
for array_index in array_group_db[group]:
try: exp_val = float(t[array_index+1])
except Exception:
if 'Gene_ID' not in line: bad_row_import[probeset]=line; exp_val = 1
###appended is the numerical expression value for each array in the group (temporary array)
try: temp_group_array[group].append(exp_val) #add 1 since probeset is the first column
except KeyError: temp_group_array[group] = [exp_val]
if count == 0: array_index_list.sort(); count = 1
####store the group database within the probeset database entry
try:
null = exon_db[probeset] ###To conserve memory, don't store any probesets not used for downstream analyses (e.g. not linked to mRNAs)
#if 'ENSG00000139970' in probeset:
#print [max_exp]
#print t[1:];kill
#max_exp = max(map(float, t[1:]))
#if len(array_raw_group_values)>10000: break
#if max_exp>math.log(70,2):
array_raw_group_values[probeset] = temp_group_array
except KeyError:
#print probeset
pass
print len(array_raw_group_values), 'sequence identifiers imported out of', line_num-1
if len(bad_row_import)>0:
print len(bad_row_import), "Rows with an unexplained import error processed and deleted."
print "Example row:"; x=0
for i in bad_row_import:
if x==0: print bad_row_import[i]
try: del array_raw_group_values[i]
except Exception: null=[]
x+=1
### If no gene expression reporting probesets were imported, update constitutive_probeset_db to include all mRNA aligning probesets
cs_genedb={}; missing_genedb={}; addback_genedb={}; rnaseq_cs_gene_db={}
for probeset in constitutive_probeset_db:
gene = constitutive_probeset_db[probeset]
#if gene == 'ENSG00000185008': print [probeset]
try:
null=array_raw_group_values[probeset]; cs_genedb[gene]=[]
if gene == probeset: rnaseq_cs_gene_db[gene]=[] ### If RPKM normalization used, use the gene expression values already calculated
except Exception: missing_genedb[gene]=[] ### Collect possible that are missing from constitutive database (verify next)
for gene in missing_genedb:
try: null=cs_genedb[gene]
except Exception: addback_genedb[gene]=[]
for probeset in array_raw_group_values:
try:
gene = exon_db[probeset].GeneID()
try:
null=addback_genedb[gene]
if 'I' not in probeset and 'U' not in probeset: ### No intron or UTR containing should be used for constitutive expression
null=string.split(probeset,':')
if len(null)<3: ### No trans-gene junctions should be used for constitutive expression
constitutive_probeset_db[probeset]=gene
except Exception: null=[]
except Exception: null=[]
for probeset in constitutive_probeset_db:
gene = constitutive_probeset_db[probeset]
#if gene == 'ENSG00000185008': print [[probeset]]
### Only examine values for associated exons when determining RNASeq constitutive expression (when exon data is present)
normalization_method = 'raw'
if array_type == 'RNASeq':
junction_count=0; constitutive_probeset_db2={}
for uid in constitutive_probeset_db:
if '-' in uid: junction_count+=1
if len(rnaseq_cs_gene_db)>0: ### If filtered RPKM gene-level expression data present, use this instead (and only this)
normalization_method = 'RPKM'
constitutive_probeset_db={} ### Re-set this database
for gene in rnaseq_cs_gene_db:
constitutive_probeset_db[gene]=gene
elif junction_count !=0 and len(constitutive_probeset_db) != junction_count:
### occurs when there is a mix of junction and exon IDs
for uid in constitutive_probeset_db:
if '-' not in uid: constitutive_probeset_db2[uid] = constitutive_probeset_db[uid]
constitutive_probeset_db = constitutive_probeset_db2; constitutive_probeset_db2=[]
"""
for probeset in constitutive_probeset_db:
gene = constitutive_probeset_db[probeset]
if gene == 'ENSG00000185008': print [probeset]
"""
###Build all putative splicing events
global alt_junction_db; global exon_dbase; global critical_exon_db; critical_exon_db={}
if array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
### Applies to reciprocal junction analyses only
if array_type == 'AltMouse':
alt_junction_db,critical_exon_db,exon_dbase,exon_inclusion_db,exon_db = ExonAnnotate_module.identifyPutativeSpliceEvents(exon_db,constitutive_probeset_db,array_raw_group_values,agglomerate_inclusion_probesets,onlyAnalyzeJunctions)
print 'Number of Genes with Examined Splice Events:',len(alt_junction_db)
elif (array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null':
from build_scripts import JunctionArray
alt_junction_db,critical_exon_db,exon_dbase,exon_inclusion_db,exon_db = JunctionArray.getPutativeSpliceEvents(species,array_type,exon_db,agglomerate_inclusion_probesets,root_dir)
print 'Number of Genes with Examined Splice Events:',len(alt_junction_db)
#alt_junction_db=[]; critical_exon_db=[]; exon_dbase=[]; exon_inclusion_db=[]
if agglomerate_inclusion_probesets == 'yes':
array_raw_group_values = agglomerateInclusionProbesets(array_raw_group_values,exon_inclusion_db)
exon_inclusion_db=[]
### For datasets with high memory requirements (RNASeq), filter the current and new databases
### Begin this function after agglomeration to ensure agglomerated probesets are considered
reciprocal_probesets = {}
if array_type == 'junction' or array_type == 'RNASeq':
for affygene in alt_junction_db:
for event in alt_junction_db[affygene]:
reciprocal_probesets[event.InclusionProbeset()]=[]
reciprocal_probesets[event.ExclusionProbeset()]=[]
not_evalutated={}
for probeset in array_raw_group_values:
try: null=reciprocal_probesets[probeset]
except Exception:
### Don't remove constitutive probesets
try: null=constitutive_probeset_db[probeset]
except Exception: not_evalutated[probeset]=[]
#print 'Removing',len(not_evalutated),'exon/junction IDs not evaulated for splicing'
for probeset in not_evalutated:
del array_raw_group_values[probeset]
###Check to see if we have precomputed expression data or raw to be analyzed
x=0; y=0; array_raw_group_values2={}; probesets_to_delete=[] ### Record deleted probesets
if len(array_raw_group_values)==0:
print_out = "No genes were considered 'Expressed' based on your input options. Check to make sure that the right species database is indicated and that the right data format has been selected (e.g., non-log versus log expression)."
try: UI.WarningWindow(print_out,'Exit')
except Exception: print print_out; print "Exiting program"
badExit()
elif len(array_raw_group_values)>0:
###array_group_list should already be unique and correctly sorted (see above)
for probeset in array_raw_group_values:
data_lists=[]
for group_name in array_group_list:
data_list = array_raw_group_values[probeset][group_name] ###nested database entry access - baseline expression
if global_addition_factor > 0: data_list = addGlobalFudgeFactor(data_list,'log')
data_lists.append(data_list)
if len(array_group_list)==2:
data_list1 = data_lists[0]; data_list2 = data_lists[-1]; avg1 = statistics.avg(data_list1); avg2 = statistics.avg(data_list2)
log_fold = avg2 - avg1
try:
#t,df,tails = statistics.ttest(data_list1,data_list2,2,3) #unpaired student ttest, calls p_value function
#t = abs(t); df = round(df) #Excel doesn't recognize fractions in a DF
#p = statistics.t_probability(t,df)
p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic)
if p == -1:
if len(data_list1)>1 and len(data_list2)>1:
print_out = "The probability statistic selected ("+probability_statistic+") is not compatible with the\nexperimental design. Please consider an alternative statistic or correct the problem.\nExiting AltAnalyze."
try: UI.WarningWindow(print_out,'Exit')
except Exception: print print_out; print "Exiting program"
badExit()
else: p = 1
except Exception: p = 1
fold_dbase[probeset] = [0]; fold_dbase[probeset].append(log_fold)
stats_dbase[probeset]=[avg1]; stats_dbase[probeset].append(p)
###replace entries with the two lists for later permutation analysis
if p == -1: ### should by p == 1: Not sure why this filter was here, but mistakenly removes probesets where there is just one array for each group
del fold_dbase[probeset];del stats_dbase[probeset]; probesets_to_delete.append(probeset); x += 1
if x == 1: print 'Bad data detected...', data_list1, data_list2
elif (avg1 < expression_threshold and avg2 < expression_threshold and p > p_threshold) and array_type != 'RNASeq': ### Inserted a filtering option to exclude small variance, low expreession probesets
del fold_dbase[probeset];del stats_dbase[probeset]; probesets_to_delete.append(probeset); x += 1
else: array_raw_group_values2[probeset] = [data_list1,data_list2]
else: ###Non-junction analysis can handle more than 2 groups
index=0
for data_list in data_lists:
try: array_raw_group_values2[probeset].append(data_list)
except KeyError: array_raw_group_values2[probeset] = [data_list]
if len(array_group_list)>2: ### Thus, there is some variance for this probeset
### Create a complete stats_dbase containing all fold changes
if index==0:
avg_baseline = statistics.avg(data_list); stats_dbase[probeset] = [avg_baseline]
else:
avg_exp = statistics.avg(data_list)
log_fold = avg_exp - avg_baseline
try: fold_dbase[probeset].append(log_fold)
except KeyError: fold_dbase[probeset] = [0,log_fold]
index+=1
if array_type == 'RNASeq': id_name = 'exon/junction IDs'
else: id_name = 'array IDs'
array_raw_group_values = array_raw_group_values2; array_raw_group_values2=[]
print x, id_name,"excluded prior to analysis... predicted not detected"
global original_avg_const_exp_db; global original_fold_dbase
global avg_const_exp_db; global permute_lists; global midas_db
if len(array_raw_group_values)>0:
adj_fold_dbase, nonlog_NI_db, conditions, gene_db, constitutive_gene_db, constitutive_fold_change, original_avg_const_exp_db = constitutive_exp_normalization(fold_dbase,stats_dbase,exon_db,constitutive_probeset_db)
stats_dbase=[] ### No longer needed after this point
original_fold_dbase = fold_dbase; avg_const_exp_db = {}; permute_lists = []; y = 0; original_conditions = conditions; max_replicates,equal_replicates = maxReplicates()
gene_expression_diff_db = constitutive_expression_changes(constitutive_fold_change,annotate_db) ###Add in constitutive fold change filter to assess gene expression for ASPIRE
while conditions > y:
avg_const_exp_db = constitutive_exp_normalization_raw(gene_db,constitutive_gene_db,array_raw_group_values,exon_db,y,avg_const_exp_db); y+=1
#print len(avg_const_exp_db),constitutive_gene_db['ENSMUSG00000054850']
###Export Analysis Results for external splicing analysis (e.g. MiDAS format)
if run_MiDAS == 'yes' and normalization_method != 'RPKM': ### RPKM has negative values which will crash MiDAS
status = ResultsExport_module.exportTransitResults(array_group_list,array_raw_group_values,array_group_name_db,avg_const_exp_db,adj_fold_dbase,exon_db,dataset_name,apt_location)
print "Finished exporting input data for MiDAS analysis"
try: midas_db = ResultsExport_module.importMidasOutput(dataset_name)
except Exception: midas_db = {} ### Occurs if there are not enough samples to calculate a MiDAS p-value
else: midas_db = {}
###Provides all pairwise permuted group comparisons
if array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
permute_lists = statistics.permute_arrays(array_index_list)
### Now remove probesets from the analysis that were used to evaluate gene expression
for probeset in constitutive_probeset_db:
try: null = reciprocal_probesets[probeset]
except Exception:
try: del array_raw_group_values[probeset]
except Exception: null=[]
not_evalutated=[]; reciprocal_probesets=[]
constitutive_probeset_db=[]
### Above, all conditions were examined when more than 2 are present... change this so that only the most extreeem are analyzed further
if len(array_group_list)>2 and analysis_method == 'splicing-index' and (array_type == 'exon' or array_type == 'gene' or explicit_data_type != 'null'): ### USED FOR MULTIPLE COMPARISONS
print 'Calculating splicing-index values for multiple group comparisons (please be patient)...',
"""
if len(midas_db)==0:
print_out = 'Warning!!! MiDAS failed to run for multiple groups. Please make\nsure there are biological replicates present for your groups.\nAltAnalyze requires replicates for multi-group (more than two) analyses.'
try: UI.WarningWindow(print_out,'Exit')
except Exception: print print_out; print "Exiting program"
badExit()"""
if filter_for_AS == 'yes':
for probeset in exon_db:
as_call = exon_db[probeset].SplicingCall()
if as_call == 0:
try: del nonlog_NI_db[probeset]
except KeyError: null=[]
if export_NI_values == 'yes':
export_exon_regions = 'yes'
### Currently, we don't deal with raw adjusted expression values, just group, so just export the values for each group
summary_output = root_dir+'AltResults/RawSpliceData/'+species+'/'+analysis_method+'/'+dataset_name[:-1]+'.txt'
print "Exporting all normalized intensities to:\n"+summary_output
adjoutput = export.ExportFile(summary_output)
title = string.join(['Gene\tExonID\tprobesetID']+original_array_names,'\t')+'\n'; adjoutput.write(title)
### Pick which data lists have the most extreem values using the NI_dbase (adjusted folds for each condition)
original_increment = int(len(nonlog_NI_db)/20); increment = original_increment; interaction = 0
for probeset in nonlog_NI_db:
if interaction == increment: increment+=original_increment; print '*',
interaction +=1
geneid = exon_db[probeset].GeneID(); ed = exon_db[probeset]
index=0; NI_list=[] ### Add the group_name to each adj fold value
for NI in nonlog_NI_db[probeset]:
NI_list.append((NI,index)); index+=1 ### setup to sort for the extreeme adj folds and get associated group_name using the index
raw_exp_vals = array_raw_group_values[probeset]
adj_exp_lists={} ### Store the adjusted expression values for each group
if geneid in avg_const_exp_db:
k=0; gi=0; adj_exp_vals = []
for exp_list in raw_exp_vals:
for exp in exp_list:
adj_exp_val = exp-avg_const_exp_db[geneid][k]
try: adj_exp_lists[gi].append(adj_exp_val)
except Exception: adj_exp_lists[gi] = [adj_exp_val]
if export_NI_values == 'yes': adj_exp_vals.append(str(adj_exp_val))
k+=1
gi+=1
if export_NI_values == 'yes':
#print geneid+'-'+probeset, adj_exp_val, [ed.ExonID()];kill
if export_exon_regions == 'yes':
try: ### Thid will only work if ExonRegionID is stored in the abreviated AffyExonSTData object - useful in comparing results between arrays (exon-region centric)
if (array_type == 'exon' or array_type == 'gene') or '-' not in ed.ExonID(): ### only include exon entries not junctions
exon_regions = string.split(ed.ExonRegionID(),'|')
for er in exon_regions:
if len(er)>0: er = er
else:
try: er = ed.ExonID()
except Exception: er = 'NA'
ev = string.join([geneid+'\t'+er+'\t'+probeset]+adj_exp_vals,'\t')+'\n'
if len(filtered_probeset_db)>0:
if probeset in filtered_probeset_db: adjoutput.write(ev) ### This is used when we want to restrict to only probesets known to already by changed
else: adjoutput.write(ev)
except Exception:
ev = string.join([geneid+'\t'+'NA'+'\t'+probeset]+adj_exp_vals,'\t')+'\n'; adjoutput.write(ev)
NI_list.sort()
examine_pairwise_comparisons = 'yes'
if examine_pairwise_comparisons == 'yes':
k1=0; k2=0; filtered_NI_comps = []
NI_list_rev = list(NI_list); NI_list_rev.reverse()
NI1,index1 = NI_list[k1]; NI2,index2 = NI_list_rev[k2]; abs_SI = abs(math.log(NI1/NI2,2))
if abs_SI<alt_exon_logfold_cutoff:
### Indicates that no valid matches were identified - hence, exit loop and return an NI_list with no variance
NI_list = [NI_list[0],NI_list[0]]
else:
### Indicates that no valid matches were identified - hence, exit loop and return an NI_list with no variance
constit_exp1 = original_avg_const_exp_db[geneid][index1]
constit_exp2 = original_avg_const_exp_db[geneid][index2]
ge_fold = constit_exp2-constit_exp1
#print 'original',abs_SI,k1,k2, ge_fold, constit_exp1, constit_exp2
if abs(ge_fold) < log_fold_cutoff: filtered_NI_comps.append([abs_SI,k1,k2])
else:
for i1 in NI_list:
k2=0
for i2 in NI_list_rev:
NI1,index1 = i1; NI2,index2 = i2; abs_SI = abs(math.log(NI1/NI2,2))
#constit_exp1 = original_avg_const_exp_db[geneid][index1]
#constit_exp2 = original_avg_const_exp_db[geneid][index2]
#ge_fold = constit_exp2-constit_exp1
#if abs(ge_fold) < log_fold_cutoff: filtered_NI_comps.append([abs_SI,k1,k2])
#print k1,k2, i1, i2, abs_SI, abs(ge_fold), log_fold_cutoff, alt_exon_logfold_cutoff
if abs_SI<alt_exon_logfold_cutoff: break
else:
constit_exp1 = original_avg_const_exp_db[geneid][index1]
constit_exp2 = original_avg_const_exp_db[geneid][index2]
ge_fold = constit_exp2-constit_exp1
if abs(ge_fold) < log_fold_cutoff:
filtered_NI_comps.append([abs_SI,k1,k2])
#if k1 == 49 or k1 == 50 or k1 == 51: print probeset, abs_SI, k1, k2, abs(ge_fold),log_fold_cutoff, index1, index2, NI1, NI2, constit_exp1,constit_exp2
k2+=1
k1+=1
if len(filtered_NI_comps)>0:
#print filtered_NI_comps
#print NI_list_rev
#print probeset,geneid
#print len(filtered_NI_comps)
#print original_avg_const_exp_db[geneid]
filtered_NI_comps.sort()
si,k1,k2 = filtered_NI_comps[-1]
NI_list = [NI_list[k1],NI_list_rev[k2]]
"""
NI1,index1 = NI_list[0]; NI2,index2 = NI_list[-1]
constit_exp1 = original_avg_const_exp_db[geneid][index1]
constit_exp2 = original_avg_const_exp_db[geneid][index2]
ge_fold = constit_exp2-constit_exp1
print probeset, si, ge_fold, NI_list"""
#print k1,k2;sys.exit()
index1 = NI_list[0][1]; index2 = NI_list[-1][1]
nonlog_NI_db[probeset] = [NI_list[0][0],NI_list[-1][0]] ### Update the values of this dictionary
data_list1 = array_raw_group_values[probeset][index1]; data_list2 = array_raw_group_values[probeset][index2]
avg1 = statistics.avg(data_list1); avg2 = statistics.avg(data_list2); log_fold = avg2 - avg1
group_name1 = array_group_list[index1]; group_name2 = array_group_list[index2]
try:
#t,df,tails = statistics.ttest(data_list1,data_list2,2,3) #unpaired student ttest, calls p_value function
#t = abs(t); df = round(df); ttest_exp_p = statistics.t_probability(t,df)
ttest_exp_p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic)
except Exception: ttest_exp_p = 1
fold_dbase[probeset] = [0]; fold_dbase[probeset].append(log_fold)
if ttest_exp_p == -1: del fold_dbase[probeset]; probesets_to_delete.append(probeset); x += 1
elif avg1 < expression_threshold and avg2 < expression_threshold and (ttest_exp_p > p_threshold and ttest_exp_p != 1): ### Inserted a filtering option to exclude small variance, low expreession probesets
del fold_dbase[probeset]; probesets_to_delete.append(probeset); x += 1
else:
constit_exp1 = original_avg_const_exp_db[geneid][index1]
constit_exp2 = original_avg_const_exp_db[geneid][index2]
ge_fold = constit_exp2-constit_exp1
normInt1 = (avg1-constit_exp1); normInt2 = (avg2-constit_exp2)
adj_fold = normInt2 - normInt1
splicing_index = -1*adj_fold; abs_splicing_index = abs(splicing_index)
#print probeset, splicing_index, ge_fold, index1, index2
#normIntList1 = adj_exp_lists[index1]; normIntList2 = adj_exp_lists[index2]
all_nI=[]
for g_index in adj_exp_lists: all_nI.append(adj_exp_lists[g_index])
try: normIntensityP = statistics.OneWayANOVA(all_nI) #[normIntList1,normIntList2] ### This stays an ANOVA independent of the algorithm choosen since groups number > 2
except Exception: normIntensityP = 'NA'
if (normInt1*normInt2)<0: opposite_SI_log_mean = 'yes'
else: opposite_SI_log_mean = 'no'
abs_log_ratio = abs(ge_fold)
if probeset in midas_db:
try: midas_p = float(midas_db[probeset])
except ValueError: midas_p = 'NA'
else: midas_p = 'NA'
#if 'ENSG00000059588' in geneid: print probeset, splicing_index, constit_exp1, constit_exp2, ge_fold,group_name2+'_vs_'+group_name1, index1, index2
if abs_splicing_index>alt_exon_logfold_cutoff and (midas_p < p_threshold or midas_p == 'NA'): #and abs_log_ratio>1 and ttest_exp_p<0.05: ###and ge_threshold_count==2
exonid = ed.ExonID(); critical_exon_list = [1,[exonid]]
ped = ProbesetExpressionData(avg1, avg2, log_fold, adj_fold, ttest_exp_p, group_name2+'_vs_'+group_name1)
sid = ExonData(splicing_index,probeset,critical_exon_list,geneid,normInt1,normInt2,normIntensityP,opposite_SI_log_mean)
sid.setConstitutiveExpression(constit_exp1); sid.setConstitutiveFold(ge_fold); sid.setProbesetExpressionData(ped)
si_db.append((splicing_index,sid))
else:
### Also record the data for probesets that are excluded... Used by DomainGraph
eed = ExcludedExonData(splicing_index,geneid,normIntensityP)
ex_db[probeset] = eed
if array_type == 'RNASeq': id_name = 'exon/junction IDs'
else: id_name = 'array IDs'
print len(si_db),id_name,"with evidence of Alternative expression"
original_fold_dbase = fold_dbase; si_db.sort()
summary_data_db['denominator_exp_events']=len(nonlog_NI_db)
del avg_const_exp_db; del gene_db; del constitutive_gene_db; gene_expression_diff_db={}
if export_NI_values == 'yes': adjoutput.close()
### Above, all conditions were examined when more than 2 are present... change this so that only the most extreeem are analyzed further
elif len(array_group_list)>2 and (array_type == 'junction' or array_type == 'RNASeq' or array_type == 'AltMouse'): ### USED FOR MULTIPLE COMPARISONS
excluded_probeset_db={}
group_sizes = []; original_array_indices = permute_lists[0] ###p[0] is the original organization of the group samples prior to permutation
for group in original_array_indices: group_sizes.append(len(group))
if analysis_method == 'linearregres': ### For linear regression, these scores are non-long
original_array_raw_group_values = copy.deepcopy(array_raw_group_values)
for probeset in array_raw_group_values:
ls_concatenated=[]
for group in array_raw_group_values[probeset]: ls_concatenated+=group
ls_concatenated = statistics.log_fold_conversion_fraction(ls_concatenated)
array_raw_group_values[probeset] = ls_concatenated
pos1=0; pos2=0; positions=[]
for group in group_sizes:
if pos1 == 0: pos2 = group; positions.append((pos1,pos2))
else: pos2 = pos1+group; positions.append((pos1,pos2))
pos1 = pos2
if export_NI_values == 'yes':
export_exon_regions = 'yes'
### Currently, we don't deal with raw adjusted expression values, just group, so just export the values for each group
summary_output = root_dir+'AltResults/RawSpliceData/'+species+'/'+analysis_method+'/'+dataset_name[:-1]+'.txt'
print "Exporting all normalized intensities to:\n"+summary_output
adjoutput = export.ExportFile(summary_output)
title = string.join(['gene\tprobesets\tExonRegion']+original_array_names,'\t')+'\n'; adjoutput.write(title)
events_examined= 0; denominator_events=0; fold_dbase=[]; adj_fold_dbase=[]; scores_examined=0
splice_event_list=[]; splice_event_list_mx=[]; splice_event_list_non_mx=[]; event_mx_temp = []; permute_p_values={}; probeset_comp_db={}#use this to exclude duplicate mx events
for geneid in alt_junction_db:
affygene = geneid
for event in alt_junction_db[geneid]:
if array_type == 'AltMouse':
#event = [('ei', 'E16-E17'), ('ex', 'E16-E18')]
#critical_exon_db[affygene,tuple(critical_exons)] = [1,'E'+str(e1a),'E'+str(e2b)] --- affygene,tuple(event) == key, 1 indicates both are either up or down together
event_call = event[0][0] + '-' + event[1][0]
exon_set1 = event[0][1]; exon_set2 = event[1][1]
probeset1 = exon_dbase[affygene,exon_set1]
probeset2 = exon_dbase[affygene,exon_set2]
critical_exon_list = critical_exon_db[affygene,tuple(event)]
if array_type == 'junction' or array_type == 'RNASeq':
event_call = 'ei-ex' ### Below objects from JunctionArrayEnsemblRules - class JunctionInformation
probeset1 = event.InclusionProbeset(); probeset2 = event.ExclusionProbeset()
exon_set1 = event.InclusionJunction(); exon_set2 = event.ExclusionJunction()
try: novel_event = event.NovelEvent()
except Exception: novel_event = 'known'
critical_exon_list = [1,event.CriticalExonSets()]
key,jd = formatJunctionData([probeset1,probeset2],geneid,critical_exon_list[1])
if array_type == 'junction' or array_type == 'RNASeq':
try: jd.setSymbol(annotate_db[geneid].Symbol())
except Exception: null=[]
#if '|' in probeset1: print probeset1, key,jd.InclusionDisplay();kill
probeset_comp_db[key] = jd ### This is used for the permutation analysis and domain/mirBS import
dI_scores=[]
if probeset1 in nonlog_NI_db and probeset2 in nonlog_NI_db and probeset1 in array_raw_group_values and probeset2 in array_raw_group_values:
events_examined+=1
if analysis_method == 'ASPIRE':
index1=0; NI_list1=[]; NI_list2=[] ### Add the group_name to each adj fold value
for NI in nonlog_NI_db[probeset1]: NI_list1.append(NI)
for NI in nonlog_NI_db[probeset2]: NI_list2.append(NI)
for NI1_g1 in NI_list1:
NI2_g1 = NI_list2[index1]; index2=0
for NI1_g2 in NI_list1:
try: NI2_g2 = NI_list2[index2]
except Exception: print index1, index2, NI_list1, NI_list2;kill
if index1 != index2:
b1 = NI1_g1; e1 = NI1_g2
b2 = NI2_g1; e2 = NI2_g2
try:
dI = statistics.aspire_stringent(b1,e1,b2,e2); Rin = b1/e1; Rex = b2/e2
if (Rin>1 and Rex<1) or (Rin<1 and Rex>1):
if dI<0: i1,i2 = index2,index1 ### all scores should indicate upregulation
else: i1,i2=index1,index2
dI_scores.append((abs(dI),i1,i2))
except Exception:
#if array_type != 'RNASeq': ### RNASeq has counts of zero and one that can cause the same result between groups and probesets
#print probeset1, probeset2, b1, e1, b2, e2, index1, index2, events_examined;kill
### Exception - Occurs for RNA-Seq but can occur for array data under extreemly rare circumstances (Rex=Rin even when different b1,e1 and b2,ed values)
null=[]
index2+=1
index1+=1
dI_scores.sort()
if analysis_method == 'linearregres':
log_fold,i1,i2 = getAllPossibleLinearRegressionScores(probeset1,probeset2,positions,group_sizes)
dI_scores.append((log_fold,i1,i2))
raw_exp_vals1 = original_array_raw_group_values[probeset1]; raw_exp_vals2 = original_array_raw_group_values[probeset2]
else: raw_exp_vals1 = array_raw_group_values[probeset1]; raw_exp_vals2 = array_raw_group_values[probeset2]
adj_exp_lists1={}; adj_exp_lists2={} ### Store the adjusted expression values for each group
if geneid in avg_const_exp_db:
gi=0; l=0; adj_exp_vals = []; anova_test=[]
for exp_list in raw_exp_vals1:
k=0; anova_group=[]
for exp in exp_list:
adj_exp_val1 = exp-avg_const_exp_db[geneid][l]
try: adj_exp_lists1[gi].append(adj_exp_val1)
except Exception: adj_exp_lists1[gi] = [adj_exp_val1]
adj_exp_val2 = raw_exp_vals2[gi][k]-avg_const_exp_db[geneid][l]
try: adj_exp_lists2[gi].append(adj_exp_val2)
except Exception: adj_exp_lists2[gi] = [adj_exp_val2]
anova_group.append(adj_exp_val2-adj_exp_val1)
if export_NI_values == 'yes':
#if analysis_method == 'ASPIRE':
adj_exp_vals.append(str(adj_exp_val2-adj_exp_val1))
### BELOW CODE PRODUCES THE SAME RESULT!!!!
"""folds1 = statistics.log_fold_conversion_fraction([exp])
folds2 = statistics.log_fold_conversion_fraction([raw_exp_vals2[gi][k]])
lr_score = statistics.convert_to_log_fold(statistics.simpleLinRegress(folds1,folds2))
adj_exp_vals.append(str(lr_score))"""
k+=1; l+=0
gi+=1; anova_test.append(anova_group)
if export_NI_values == 'yes':
if export_exon_regions == 'yes':
exon_regions = string.join(critical_exon_list[1],'|')
exon_regions = string.split(exon_regions,'|')
for er in exon_regions:
ev = string.join([geneid+'\t'+probeset1+'-'+probeset2+'\t'+er]+adj_exp_vals,'\t')+'\n'
if len(filtered_probeset_db)>0:
if probeset1 in filtered_probeset_db and probeset2 in filtered_probeset_db:
adjoutput.write(ev) ### This is used when we want to restrict to only probesets known to already by changed
else: adjoutput.write(ev)
try: anovaNIp = statistics.OneWayANOVA(anova_test) ### This stays an ANOVA independent of the algorithm choosen since groups number > 2
except Exception: anovaNIp='NA'
if len(dI_scores)>0 and geneid in avg_const_exp_db:
dI,index1,index2 = dI_scores[-1]; count=0
probesets = [probeset1, probeset2]; index=0
key,jd = formatJunctionData([probeset1,probeset2],affygene,critical_exon_list[1])
if array_type == 'junction' or array_type == 'RNASeq':
try: jd.setSymbol(annotate_db[affygene].Symbol())
except Exception:null=[]
probeset_comp_db[key] = jd ### This is used for the permutation analysis and domain/mirBS import
if max_replicates >2 or equal_replicates==2: permute_p_values[(probeset1,probeset2)] = [anovaNIp, 'NA', 'NA', 'NA']
index=0
for probeset in probesets:
if analysis_method == 'linearregres':
data_list1 = original_array_raw_group_values[probeset][index1]; data_list2 = original_array_raw_group_values[probeset][index2]
else: data_list1 = array_raw_group_values[probeset][index1]; data_list2 = array_raw_group_values[probeset][index2]
baseline_exp = statistics.avg(data_list1); experimental_exp = statistics.avg(data_list2); fold_change = experimental_exp - baseline_exp
group_name1 = array_group_list[index1]; group_name2 = array_group_list[index2]
try:
ttest_exp_p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic)
except Exception: ttest_exp_p = 'NA'
if ttest_exp_p==1: ttest_exp_p = 'NA'
if index == 0:
try: adj_fold = statistics.avg(adj_exp_lists1[index2]) - statistics.avg(adj_exp_lists1[index1])
except Exception:
print raw_exp_vals1,raw_exp_vals2, avg_const_exp_db[geneid]
print probeset,probesets,adj_exp_lists1,adj_exp_lists2,index1,index2;kill
ped1 = ProbesetExpressionData(baseline_exp, experimental_exp, fold_change, adj_fold, ttest_exp_p, group_name2+'_vs_'+group_name1)
else:
adj_fold = statistics.avg(adj_exp_lists2[index2]) - statistics.avg(adj_exp_lists2[index1])
ped2 = ProbesetExpressionData(baseline_exp, experimental_exp, fold_change, adj_fold, ttest_exp_p, group_name2+'_vs_'+group_name1)
constit_exp1 = original_avg_const_exp_db[geneid][index1]
constit_exp2 = original_avg_const_exp_db[geneid][index2]
ge_fold = constit_exp2-constit_exp1
index+=1
try:
pp1 = statistics.runComparisonStatistic(adj_exp_lists1[index1], adj_exp_lists1[index2],probability_statistic)
pp2 = statistics.runComparisonStatistic(adj_exp_lists2[index1], adj_exp_lists2[index2],probability_statistic)
except Exception: pp1 = 'NA'; pp2 = 'NA'
if analysis_method == 'ASPIRE' and len(dI_scores)>0:
p1 = JunctionExpressionData(adj_exp_lists1[index1], adj_exp_lists1[index2], pp1, ped1)
p2 = JunctionExpressionData(adj_exp_lists2[index1], adj_exp_lists2[index2], pp2, ped2)
### ANOVA p-replaces the below p-value
"""try: baseline_scores, exp_scores, pairwiseNIp = calculateAllASPIREScores(p1,p2)
except Exception: baseline_scores = [0]; exp_scores=[dI]; pairwiseNIp = 0 """
#if pairwiseNIp == 'NA': pairwiseNIp = 0 ### probably comment out
if len(dI_scores)>0:
scores_examined+=1
if probeset in midas_db:
try: midas_p = float(midas_db[probeset])
except ValueError: midas_p = 'NA'
else: midas_p = 'NA'
if dI>alt_exon_logfold_cutoff and (anovaNIp < p_threshold or perform_permutation_analysis == 'yes' or anovaNIp == 'NA' or anovaNIp == 1): #and abs_log_ratio>1 and ttest_exp_p<0.05: ###and ge_threshold_count==2
#print [dI, probeset1,probeset2, anovaNIp, alt_exon_logfold_cutoff];kill
ejd = ExonJunctionData(dI,probeset1,probeset2,pp1,pp2,'upregulated',event_call,critical_exon_list,affygene,ped1,ped2)
ejd.setConstitutiveFold(ge_fold); ejd.setConstitutiveExpression(constit_exp1)
if array_type == 'RNASeq':
ejd.setNovelEvent(novel_event)
splice_event_list.append((dI,ejd))
else: excluded_probeset_db[affygene+':'+critical_exon_list[1][0]] = probeset1, affygene, dI, 'NA', anovaNIp
statistics.adjustPermuteStats(permute_p_values)
ex_db = splice_event_list, probeset_comp_db, permute_p_values, excluded_probeset_db
original_fold_dbase = fold_dbase; original_avg_const_exp_db=[]; nonlog_NI_db = []; fold_dbase=[]
summary_data_db['denominator_exp_events']=events_examined
del avg_const_exp_db; del gene_db; del constitutive_gene_db; gene_expression_diff_db={}
if export_NI_values == 'yes': adjoutput.close()
print len(splice_event_list), 'alternative exons out of %s exon events examined' % events_examined
fold_dbase=[]; original_fold_dbase=[]; exon_db=[]; constitutive_gene_db=[]; addback_genedb=[]
gene_db=[]; missing_genedb=[]
"""
print 'local vars'
all = [var for var in locals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(locals()[var])>500: print var, len(locals()[var])
except Exception: null=[]
"""
return conditions,adj_fold_dbase,nonlog_NI_db,dataset_name,gene_expression_diff_db,midas_db,ex_db,si_db
class ProbesetExpressionData:
def __init__(self, baseline_exp, experimental_exp, fold_change, adj_fold, ttest_raw_exp, annotation):
self.baseline_exp = baseline_exp; self.experimental_exp = experimental_exp
self.fold_change = fold_change; self.adj_fold = adj_fold
self.ttest_raw_exp = ttest_raw_exp; self.annotation = annotation
def BaselineExp(self): return str(self.baseline_exp)
def ExperimentalExp(self): return str(self.experimental_exp)
def FoldChange(self): return str(self.fold_change)
def AdjFold(self): return str(self.adj_fold)
def ExpPval(self): return str(self.ttest_raw_exp)
def Annotation(self): return self.annotation
def __repr__(self): return self.BaselineExp()+'|'+FoldChange()
def agglomerateInclusionProbesets(array_raw_group_values,exon_inclusion_db):
###Combine expression profiles for inclusion probesets that correspond to the same splice event
for excl_probeset in exon_inclusion_db:
inclusion_event_profiles = []
if len(exon_inclusion_db[excl_probeset])>1:
for incl_probeset in exon_inclusion_db[excl_probeset]:
if incl_probeset in array_raw_group_values and excl_probeset in array_raw_group_values:
array_group_values = array_raw_group_values[incl_probeset]
inclusion_event_profiles.append(array_group_values)
#del array_raw_group_values[incl_probeset] ###Remove un-agglomerated original entry
if len(inclusion_event_profiles) > 0: ###Thus, some probesets for this splice event in input file
combined_event_profile = combine_profiles(inclusion_event_profiles)
###Combine inclusion probesets into a single ID (identical manner to that in ExonAnnotate_module.identifyPutativeSpliceEvents
incl_probesets = exon_inclusion_db[excl_probeset]
incl_probesets_str = string.join(incl_probesets,'|')
array_raw_group_values[incl_probesets_str] = combined_event_profile
return array_raw_group_values
def combine_profiles(profile_list):
profile_group_sizes={}
for db in profile_list:
for key in db: profile_group_sizes[key] = len(db[key])
break
new_profile_db={}
for key in profile_group_sizes:
x = profile_group_sizes[key] ###number of elements in list for key
new_val_list=[]; i = 0
while i<x:
temp_val_list=[]
for db in profile_list:
if key in db: val = db[key][i]; temp_val_list.append(val)
i+=1; val_avg = statistics.avg(temp_val_list); new_val_list.append(val_avg)
new_profile_db[key] = new_val_list
return new_profile_db
def constitutive_exp_normalization(fold_db,stats_dbase,exon_db,constitutive_probeset_db):
"""For every expression value, normalize to the expression of the constitutive gene features for that condition,
then store those ratios (probeset_exp/avg_constitutive_exp) and regenerate expression values relative only to the
baseline avg_constitutive_exp, for all conditions, to normalize out gene expression changes"""
#print "\nParameters:"
#print "Factor_out_expression_changes:",factor_out_expression_changes
#print "Only_include_constitutive_containing_genes:",only_include_constitutive_containing_genes
#print "\nAdjusting probeset average intensity values to factor out condition specific expression changes for optimal splicing descrimination"
gene_db = {}; constitutive_gene_db = {}
### organize everything by gene
for probeset in fold_db: conditions = len(fold_db[probeset]); break
remove_diff_exp_genes = remove_transcriptional_regulated_genes
if conditions > 2: remove_diff_exp_genes = 'no'
for probeset in exon_db:
affygene = exon_db[probeset].GeneID() #exon_db[probeset] = affygene,exons,ensembl,block_exon_ids,block_structure,comparison_info
if probeset in fold_db:
try: gene_db[affygene].append(probeset)
except KeyError: gene_db[affygene] = [probeset]
if probeset in constitutive_probeset_db and (only_include_constitutive_containing_genes == 'yes' or factor_out_expression_changes == 'no'):
#the second conditional is used to exlcude constitutive data if we wish to use all probesets for
#background normalization rather than just the designated 'gene' probesets.
if probeset in stats_dbase:
try: constitutive_gene_db[affygene].append(probeset)
except KeyError: constitutive_gene_db[affygene] = [probeset]
if len(constitutive_gene_db)>0:
###This is blank when there are no constitutive and the above condition is implemented
gene_db2 = constitutive_gene_db
else: gene_db2 = gene_db
avg_const_exp_db = {}
for affygene in gene_db2:
probeset_list = gene_db2[affygene]
x = 0
while x < conditions:
### average all exp values for constitutive probesets for each condition
exp_list=[]
for probeset in probeset_list:
probe_fold_val = fold_db[probeset][x]
baseline_exp = stats_dbase[probeset][0]
exp_val = probe_fold_val + baseline_exp
exp_list.append(exp_val)
avg_const_exp = statistics.avg(exp_list)
try: avg_const_exp_db[affygene].append(avg_const_exp)
except KeyError: avg_const_exp_db[affygene] = [avg_const_exp]
x += 1
adj_fold_dbase={}; nonlog_NI_db={}; constitutive_fold_change={}
for affygene in avg_const_exp_db: ###If we only wish to include propper constitutive probes, this will ensure we only examine those genes and probesets that are constitutive
probeset_list = gene_db[affygene]
x = 0
while x < conditions:
exp_list=[]
for probeset in probeset_list:
expr_to_subtract = avg_const_exp_db[affygene][x]
baseline_const_exp = avg_const_exp_db[affygene][0]
probe_fold_val = fold_db[probeset][x]
baseline_exp = stats_dbase[probeset][0]
exp_val = probe_fold_val + baseline_exp
exp_val_non_log = statistics.log_fold_conversion_fraction(exp_val)
expr_to_subtract_non_log = statistics.log_fold_conversion_fraction(expr_to_subtract)
baseline_const_exp_non_log = statistics.log_fold_conversion_fraction(baseline_const_exp)
if factor_out_expression_changes == 'yes':
exp_splice_valff = exp_val_non_log/expr_to_subtract_non_log
else: #if no, then we just normalize to the baseline constitutive expression in order to keep gene expression effects (useful if you don't trust constitutive feature expression levels)
exp_splice_valff = exp_val_non_log/baseline_const_exp_non_log
constitutive_fold_diff = expr_to_subtract_non_log/baseline_const_exp_non_log
###To calculate adjusted expression, we need to get the fold change in the constitutive avg (expr_to_subtract/baseline_const_exp) and divide the experimental expression
###By this fold change.
ge_adj_exp_non_log = exp_val_non_log/constitutive_fold_diff #gives a GE adjusted expression
try: ge_adj_exp = math.log(ge_adj_exp_non_log,2)
except ValueError: print probeset,ge_adj_exp_non_log,constitutive_fold_diff,exp_val_non_log,exp_val,baseline_exp, probe_fold_val, dog
adj_probe_fold_val = ge_adj_exp - baseline_exp
### Here we normalize probeset expression to avg-constitutive expression by dividing probe signal by avg const.prove sig (should be < 1)
### refered to as steady-state normalization
if array_type != 'AltMouse' or (probeset not in constitutive_probeset_db):
"""Can't use constitutive gene features since these have no variance for pearson analysis
Python will approximate numbers to a small decimal point range. If the first fold value is
zero, often, zero will be close to but not exactly zero. Correct below """
try:
adj_fold_dbase[probeset].append(adj_probe_fold_val)
except KeyError:
if abs(adj_probe_fold_val - 0) < 0.0000001: #make zero == exactly to zero
adj_probe_fold_val = 0
adj_fold_dbase[probeset] = [adj_probe_fold_val]
try: nonlog_NI_db[probeset].append(exp_splice_valff) ###ratio of junction exp relative to gene expression at that time-point
except KeyError: nonlog_NI_db[probeset] = [exp_splice_valff]
n = 0
#if expr_to_subtract_non_log != baseline_const_exp_non_log: ###otherwise this is the first value in the expression array
if x!=0: ###previous expression can produce errors when multiple group averages have identical values
fold_change = expr_to_subtract_non_log/baseline_const_exp_non_log
fold_change_log = math.log(fold_change,2)
constitutive_fold_change[affygene] = fold_change_log
### If we want to remove any genes from the analysis with large transcriptional changes
### that may lead to false positive splicing calls (different probeset kinetics)
if remove_diff_exp_genes == 'yes':
if abs(fold_change_log) > log_fold_cutoff:
del constitutive_fold_change[affygene]
try: del adj_fold_dbase[probeset]
except KeyError: n = 1
try: del nonlog_NI_db[probeset]
except KeyError: n = 1
"""elif expr_to_subtract_non_log == baseline_const_exp_non_log: ###This doesn't make sense, since n can't equal 1 if the conditional is false (check this code again later 11/23/07)
if n == 1:
del adj_fold_dbase[probeset]
del nonlog_NI_db[probeset]"""
x += 1
print "Intensity normalization complete..."
if factor_out_expression_changes == 'no':
adj_fold_dbase = fold_db #don't change expression values
print len(constitutive_fold_change), "genes undergoing analysis for alternative splicing/transcription"
summary_data_db['denominator_exp_genes']=len(constitutive_fold_change)
"""
mir_gene_count = 0
for gene in constitutive_fold_change:
if gene in gene_microRNA_denom: mir_gene_count+=1
print mir_gene_count, "Genes with predicted microRNA binding sites undergoing analysis for alternative splicing/transcription"
"""
global gene_analyzed; gene_analyzed = len(constitutive_gene_db)
return adj_fold_dbase, nonlog_NI_db, conditions, gene_db, constitutive_gene_db,constitutive_fold_change, avg_const_exp_db
class TranscriptionData:
def __init__(self, constitutive_fold, rna_processing_annotation):
self._constitutive_fold = constitutive_fold; self._rna_processing_annotation = rna_processing_annotation
def ConstitutiveFold(self): return self._constitutive_fold
def ConstitutiveFoldStr(self): return str(self._constitutive_fold)
def RNAProcessing(self): return self._rna_processing_annotation
def __repr__(self): return self.ConstitutiveFoldStr()+'|'+RNAProcessing()
def constitutive_expression_changes(constitutive_fold_change,annotate_db):
###Add in constitutive fold change filter to assess gene expression for ASPIRE
gene_expression_diff_db = {}
for affygene in constitutive_fold_change:
constitutive_fold = constitutive_fold_change[affygene]; rna_processing_annotation=''
if affygene in annotate_db:
if len(annotate_db[affygene].RNAProcessing()) > 4: rna_processing_annotation = annotate_db[affygene].RNAProcessing()
###Add in evaluation of RNA-processing/binding factor
td = TranscriptionData(constitutive_fold,rna_processing_annotation)
gene_expression_diff_db[affygene] = td
return gene_expression_diff_db
def constitutive_exp_normalization_raw(gene_db,constitutive_gene_db,array_raw_group_values,exon_db,y,avg_const_exp_db):
"""normalize expression for raw expression data (only for non-baseline data)"""
#avg_true_const_exp_db[affygene] = [avg_const_exp]
temp_avg_const_exp_db={}
for probeset in array_raw_group_values:
conditions = len(array_raw_group_values[probeset][y]); break #number of raw expresson values to normalize
for affygene in gene_db:
###This is blank when there are no constitutive or the above condition is implemented
if affygene in constitutive_gene_db:
probeset_list = constitutive_gene_db[affygene]
z = 1
else: ###so we can analyze splicing independent of gene expression even if no 'gene' feature is present
probeset_list = gene_db[affygene]
z = 0
x = 0
while x < conditions:
### average all exp values for constitutive probesets for each conditionF
exp_list=[]
for probeset in probeset_list:
try: exp_val = array_raw_group_values[probeset][y][x] ### try statement is used for constitutive probes that were deleted due to filtering in performExpressionAnalysis
except KeyError: continue
exp_list.append(exp_val)
try: avg_const_exp = statistics.avg(exp_list)
except Exception: avg_const_exp = 'null'
if only_include_constitutive_containing_genes == 'yes' and avg_const_exp != 'null':
if z == 1:
try: avg_const_exp_db[affygene].append(avg_const_exp)
except KeyError: avg_const_exp_db[affygene] = [avg_const_exp]
try: temp_avg_const_exp_db[affygene].append(avg_const_exp)
except KeyError: temp_avg_const_exp_db[affygene] = [avg_const_exp]
elif avg_const_exp != 'null': ###***
try: avg_const_exp_db[affygene].append(avg_const_exp)
except KeyError: avg_const_exp_db[affygene] = [avg_const_exp]
try: temp_avg_const_exp_db[affygene].append(avg_const_exp)
except KeyError: temp_avg_const_exp_db[affygene] = [avg_const_exp]
x += 1
if analysis_method == 'ANOVA':
global normalized_raw_exp_ratios; normalized_raw_exp_ratios = {}
for affygene in gene_db:
probeset_list = gene_db[affygene]
for probeset in probeset_list:
while x < group_size:
new_ratios = [] ### Calculate expression ratios relative to constitutive expression
exp_val = array_raw_group_values[probeset][y][x]
const_exp_val = temp_avg_const_exp_db[affygene][x]
###Since the above dictionary is agglomerating all constitutive expression values for permutation,
###we need an unbiased way to grab just those relevant const. exp. vals. (hence the temp dictionary)
#non_log_exp_val = statistics.log_fold_conversion_fraction(exp_val)
#non_log_const_exp_val = statistics.log_fold_conversion_fraction(const_exp_val)
#non_log_exp_ratio = non_log_exp_val/non_log_const_exp_val
log_exp_ratio = exp_val - const_exp_val
try: normalized_raw_exp_ratios[probeset].append(log_exp_ratio)
except KeyError: normalized_raw_exp_ratios[probeset] = [log_exp_ratio]
return avg_const_exp_db
######### Z Score Analyses #######
class ZScoreData:
def __init__(self,element,changed,measured,zscore,null_z,gene_symbols):
self._element = element; self._changed = changed; self._measured = measured
self._zscore = zscore; self._null_z = null_z; self._gene_symbols = gene_symbols
def ElementID(self): return self._element
def Changed(self): return str(self._changed)
def Measured(self): return str(self._measured)
def AssociatedWithElement(self): return str(self._gene_symbols)
def ZScore(self): return str(self._zscore)
def SetP(self,p): self._permute_p = p
def PermuteP(self): return str(self._permute_p)
def SetAdjP(self,adjp): self._adj_p = adjp
def AdjP(self): return str(self._adj_p)
def PercentChanged(self):
try: pc = float(self.Changed())/float(self.Measured())*100
except Exception: pc = 0
return str(pc)
def NullZ(self): return self._null_z
def Report(self):
output = self.ElementID()
return output
def __repr__(self): return self.Report()
class FDRStats(ZScoreData):
def __init__(self,p): self._permute_p = p
def AdjP(self): return str(self._adj_p)
def countGenesForElement(permute_input_list,probeset_to_gene,probeset_element_db):
element_gene_db={}
for probeset in permute_input_list:
try:
element_list = probeset_element_db[probeset]
gene = probeset_to_gene[probeset]
for element in element_list:
try: element_gene_db[element].append(gene)
except KeyError: element_gene_db[element] = [gene]
except KeyError: null=[]
### Count the number of unique genes per element
for element in element_gene_db:
t = {}
for i in element_gene_db[element]: t[i]=[]
element_gene_db[element] = len(t)
return element_gene_db
def formatGeneSymbolHits(geneid_list):
symbol_list=[]
for geneid in geneid_list:
symbol = ''
if geneid in annotate_db: symbol = annotate_db[geneid].Symbol()
if len(symbol)<1: symbol = geneid
symbol_list.append(symbol)
symbol_str = string.join(symbol_list,', ')
return symbol_str
def zscore(r,n,N,R):
z = (r - n*(R/N))/math.sqrt(n*(R/N)*(1-(R/N))*(1-((n-1)/(N-1)))) #z = statistics.zscore(r,n,N,R)
return z
def calculateZScores(hit_count_db,denom_count_db,total_gene_denom_count,total_gene_hit_count,element_type):
N = float(total_gene_denom_count) ###Genes examined
R = float(total_gene_hit_count) ###AS genes
for element in denom_count_db:
element_denom_gene_count = denom_count_db[element]
n = float(element_denom_gene_count) ###all genes associated with element
if element in hit_count_db:
element_hit_gene_count = len(hit_count_db[element])
gene_symbols = formatGeneSymbolHits(hit_count_db[element])
r = float(element_hit_gene_count) ###regulated genes associated with element
else: r = 0; gene_symbols = ''
try: z = zscore(r,n,N,R)
except Exception: z = 0; #print 'error:',element,r,n,N,R; kill
try: null_z = zscore(0,n,N,R)
except Exception: null_z = 0; #print 'error:',element,r,n,N,R; kill
zsd = ZScoreData(element,r,n,z,null_z,gene_symbols)
if element_type == 'domain': original_domain_z_score_data[element] = zsd
elif element_type == 'microRNA': original_microRNA_z_score_data[element] = zsd
permuted_z_scores[element] = [z]
if perform_element_permutation_analysis == 'no':
### The below is an alternative to the permute t-statistic that is more effecient
p = FishersExactTest(r,n,R,N)
zsd.SetP(p)
return N,R
######### Begin Permutation Analysis #######
def calculatePermuteZScores(permute_element_inputs,element_denominator_gene_count,N,R):
###Make this code as efficient as possible
for element_input_gene_count in permute_element_inputs:
for element in element_input_gene_count:
r = element_input_gene_count[element]
n = element_denominator_gene_count[element]
try: z = statistics.zscore(r,n,N,R)
except Exception: z = 0
permuted_z_scores[element].append(abs(z))
#if element == '0005488':
#a.append(r)
def calculatePermuteStats(original_element_z_score_data):
for element in original_element_z_score_data:
zsd = original_element_z_score_data[element]
z = abs(permuted_z_scores[element][0])
permute_scores = permuted_z_scores[element][1:] ###Exclude the true value
nullz = zsd.NullZ()
if abs(nullz) == z: ###Only add the nullz values if they can count towards the p-value (if equal to the original z)
null_z_to_add = permutations - len(permute_scores)
permute_scores+=[abs(nullz)]*null_z_to_add ###Add null_z's in proportion to the amount of times there were not genes found for that element
if len(permute_scores)>0:
p = permute_p(permute_scores,z)
else: p = 1
#if p>1: p=1
zsd.SetP(p)
def FishersExactTest(r,n,R,N):
a = r; b = n-r; c=R-r; d=N-R-b
table = [[int(a),int(b)], [int(c),int(d)]]
try: ### Scipy version - cuts down rutime by ~1/3rd the time
oddsratio, pvalue = stats.fisher_exact(table)
return pvalue
except Exception:
ft = fishers_exact_test.FishersExactTest(table)
return ft.two_tail_p()
def adjustPermuteStats(original_element_z_score_data):
#1. Sort ascending the original input p value vector. Call this spval. Keep the original indecies so you can sort back.
#2. Define a new vector called tmp. tmp= spval. tmp will contain the BH p values.
#3. m is the length of tmp (also spval)
#4. i=m-1
#5 tmp[ i ]=min(tmp[i+1], min((m/i)*spval[ i ],1)) - second to last, last, last/second to last
#6. i=m-2
#7 tmp[ i ]=min(tmp[i+1], min((m/i)*spval[ i ],1))
#8 repeat step 7 for m-3, m-4,... until i=1
#9. sort tmp back to the original order of the input p values.
spval=[]
for element in original_element_z_score_data:
zsd = original_element_z_score_data[element]
p = float(zsd.PermuteP())
spval.append([p,element])
spval.sort(); tmp = spval; m = len(spval); i=m-2; x=0 ###Step 1-4
while i > -1:
tmp[i]=min(tmp[i+1][0], min((float(m)/(i+1))*spval[i][0],1)),tmp[i][1]; i -= 1
for (adjp,element) in tmp:
zsd = original_element_z_score_data[element]
zsd.SetAdjP(adjp)
spval=[]
def permute_p(null_list,true_value):
y = 0; z = 0; x = permutations
for value in null_list:
if value >= true_value: y += 1
#if true_value > 8: global a; a = null_list; print true_value,y,x;kill
return (float(y)/float(x)) ###Multiply probabilty x2?
######### End Permutation Analysis #######
def exportZScoreData(original_element_z_score_data,element_type):
element_output = root_dir+'AltResults/AlternativeOutput/' + dataset_name + analysis_method+'-'+element_type+'-zscores.txt'
data = export.ExportFile(element_output)
headers = [element_type+'-Name','Number Changed','Number Measured','Percent Changed', 'Zscore','PermuteP','AdjP','Changed GeneSymbols']
headers = string.join(headers,'\t')+'\n'
data.write(headers); sort_results=[]
#print "Results for",len(original_element_z_score_data),"elements exported to",element_output
for element in original_element_z_score_data:
zsd=original_element_z_score_data[element]
try: results = [zsd.Changed(), zsd.Measured(), zsd.PercentChanged(), zsd.ZScore(), zsd.PermuteP(), zsd.AdjP(), zsd.AssociatedWithElement()]
except AttributeError: print element,len(permuted_z_scores[element]);kill
results = [element] + results
results = string.join(results,'\t') + '\n'
sort_results.append([float(zsd.PermuteP()),-1/float(zsd.Measured()),results])
sort_results.sort()
for values in sort_results:
results = values[2]
data.write(results)
data.close()
def getInputsForPermutationAnalysis(exon_db):
### Filter fold_dbase, which is the proper denominator
probeset_to_gene = {}; denominator_list = []
for probeset in exon_db:
proceed = 'no'
if filter_for_AS == 'yes':
as_call = exon_db[probeset].SplicingCall()
if as_call == 1: proceed = 'yes'
else: proceed = 'yes'
if proceed == 'yes':
gene = exon_db[probeset].GeneID()
probeset_to_gene[probeset] = gene
denominator_list.append(probeset)
return probeset_to_gene,denominator_list
def getJunctionSplicingAnnotations(regulated_exon_junction_db):
filter_status = 'yes'
########### Import critical exon annotation for junctions, build through the exon array analysis pipeline - link back to probesets
filtered_arrayids={}; critical_probeset_annotation_db={}
if array_type == 'RNASeq' and explicit_data_type == 'null':
critical_exon_annotation_file = root_dir+'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_exons.txt'
elif array_type == 'RNASeq' and explicit_data_type != 'null':
critical_exon_annotation_file = root_dir+'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_junctions.txt'
else:
critical_exon_annotation_file = "AltDatabase/"+species+"/"+array_type+"/"+species+"_Ensembl_"+array_type+"_probesets.txt"
critical_exon_annotation_file = filename=getFilteredFilename(critical_exon_annotation_file)
for uid in regulated_exon_junction_db:
gene = regulated_exon_junction_db[uid].GeneID()
critical_exons = regulated_exon_junction_db[uid].CriticalExons()
"""### It appears that each critical exon for junction arrays can be a concatenation of multiple exons, making this unnecessary
if len(critical_exons)>1 and array_type == 'junction':
critical_exons_joined = string.join(critical_exons,'|')
filtered_arrayids[gene+':'+critical_exon].append(uid)"""
for critical_exon in critical_exons:
try:
try: filtered_arrayids[gene+':'+critical_exon].append(uid)
except TypeError: print gene, critical_exon, uid;kill
except KeyError: filtered_arrayids[gene+':'+critical_exon]=[uid]
critical_exon_annotation_db = importSplicingAnnotationDatabase(critical_exon_annotation_file,'exon-fake',filtered_arrayids,filter_status);null=[] ###The file is in exon centric format, so designate array_type as exon
for key in critical_exon_annotation_db:
ced = critical_exon_annotation_db[key]
for junction_probesets in filtered_arrayids[key]:
try: critical_probeset_annotation_db[junction_probesets].append(ced) ###use for splicing and Exon annotations
except KeyError: critical_probeset_annotation_db[junction_probesets] = [ced]
for junction_probesets in critical_probeset_annotation_db:
if len(critical_probeset_annotation_db[junction_probesets])>1: ###Thus multiple exons associated, must combine annotations
exon_ids=[]; external_exonids=[]; exon_regions=[]; splicing_events=[]
for ed in critical_probeset_annotation_db[junction_probesets]:
ensembl_gene_id = ed.GeneID(); transcript_cluster_id = ed.ExternalGeneID()
exon_ids.append(ed.ExonID()); external_exonids.append(ed.ExternalExonIDs()); exon_regions.append(ed.ExonRegionID()); se = string.split(ed.SplicingEvent(),'|')
for i in se: splicing_events.append(i)
splicing_events = unique.unique(splicing_events) ###remove duplicate entries
exon_id = string.join(exon_ids,'|'); external_exonid = string.join(external_exonids,'|'); exon_region = string.join(exon_regions,'|'); splicing_event = string.join(splicing_events,'|')
probe_data = AffyExonSTData(ensembl_gene_id, exon_id, external_exonid, '', exon_region, splicing_event, '','')
if array_type != 'RNASeq': probe_data.setTranscriptCluster(transcript_cluster_id)
critical_probeset_annotation_db[junction_probesets] = probe_data
else:
critical_probeset_annotation_db[junction_probesets] = critical_probeset_annotation_db[junction_probesets][0]
return critical_probeset_annotation_db
def determineExternalType(external_probeset_db):
external_probeset_db2={}
if 'TC' in external_probeset_db:
temp_index={}; i=0; type = 'JETTA'
for name in external_probeset_db['TC'][0]: temp_index[i]=i; i+=1
if 'PS:norm_expr_fold_change' in temp_index: NI_fold_index = temp_index['PS:norm_expr_fold_change']
if 'MADS:pv_1over2' in temp_index: MADS_p1_index = temp_index['MADS:pv_1over2']
if 'MADS:pv_2over1' in temp_index: MADS_p2_index = temp_index['MADS:pv_2over1']
if 'TC:expr_fold_change' in temp_index: MADS_p2_index = temp_index['MADS:pv_2over1']
if 'PsId' in temp_index: ps_index = temp_index['PsId']
for tc in external_probeset_db:
for list in external_probeset_db[tc]:
try: NI_fold = float(list[NI_fold_index])
except Exception: NI_fold = 1
try: MADSp1 = float(list[MADS_p1_index])
except Exception: MADSp1 = 1
try: MADSp2 = float(list[MADS_p2_index])
except Exception: MADSp1 = 1
if MADSp1<MADSp2: pval = MADSp1
else: pval = MADSp2
probeset = list[ps_index]
external_probeset_db2[probeset] = NI_fold,pval
else:
type = 'generic'
a = []; b = []
for id in external_probeset_db:
#print external_probeset_db[id]
try: a.append(abs(float(external_probeset_db[id][0][0])))
except Exception: null=[]
try: b.append(abs(float(external_probeset_db[id][0][1])))
except Exception: null=[]
a.sort(); b.sort(); pval_index = None; score_index = None
if len(a)>0:
if max(a) > 1: score_index = 0
else: pval_index = 0
if len(b)>0:
if max(b) > 1: score_index = 1
else: pval_index = 1
for id in external_probeset_db:
if score_index != None: score = external_probeset_db[id][0][score_index]
else: score = 1
if pval_index != None: pval = external_probeset_db[id][0][pval_index]
else: pval = 1
external_probeset_db2[id] = score,pval
return external_probeset_db2, type
def importExternalProbesetData(dataset_dir):
excluded_probeset_db={}; splice_event_list=[]; p_value_call={}; permute_p_values={}; gene_expression_diff_db={}
analyzed_probeset_db = {}
external_probeset_db = importExternalDBList(dataset_dir)
external_probeset_db, ext_type = determineExternalType(external_probeset_db)
for probeset in exon_db: analyzed_probeset_db[probeset] = []
### Used to restrict the analysis to a pre-selected set of probesets (e.g. those that have a specifc splicing pattern)
if len(filtered_probeset_db)>0:
temp_db={}
for probeset in analyzed_probeset_db: temp_db[probeset]=[]
for probeset in temp_db:
try: filtered_probeset_db[probeset]
except KeyError: del analyzed_probeset_db[probeset]
### Used to restrict the analysis to a pre-selected set of probesets (e.g. those that have a specifc splicing annotation)
if filter_for_AS == 'yes':
for probeset in exon_db:
as_call = exon_db[probeset].SplicingCall()
if as_call == 0:
try: del analyzed_probeset_db[probeset]
except KeyError: null=[]
for probeset in analyzed_probeset_db:
ed = exon_db[probeset]; geneid = ed.GeneID()
td = TranscriptionData('',''); gene_expression_diff_db[geneid] = td
if probeset in external_probeset_db:
exonid = ed.ExonID(); critical_exon_list = [1,[exonid]]
splicing_index,normIntensityP = external_probeset_db[probeset]
group1_ratios=[]; group2_ratios=[];exp_log_ratio=''; ttest_exp_p='';normIntensityP='';opposite_SI_log_mean=''
sid = ExonData(splicing_index,probeset,critical_exon_list,geneid,group1_ratios,group2_ratios,normIntensityP,opposite_SI_log_mean)
splice_event_list.append((splicing_index,sid))
else:
### Also record the data for probesets that are excluded... Used by DomainGraph
eed = ExcludedExonData(0,geneid,'NA')
excluded_probeset_db[probeset] = eed
print len(splice_event_list), 'pre-filtered external results imported...\n'
return splice_event_list, p_value_call, permute_p_values, excluded_probeset_db, gene_expression_diff_db
def splicingAnalysisAlgorithms(nonlog_NI_db,fold_dbase,dataset_name,gene_expression_diff_db,exon_db,ex_db,si_db,dataset_dir):
protein_exon_feature_db={}; global regulated_exon_junction_db; global critical_exon_annotation_db; global probeset_comp_db; probeset_comp_db={}
if original_conditions == 2: print "Beginning to run", analysis_method, "algorithm on",dataset_name[0:-1],"data"
if run_from_scratch == 'Annotate External Results':
splice_event_list, p_value_call, permute_p_values, excluded_probeset_db, gene_expression_diff_db = importExternalProbesetData(dataset_dir)
elif analysis_method == 'ASPIRE' or analysis_method == 'linearregres':
original_exon_db = exon_db
if original_conditions > 2:
splice_event_list, probeset_comp_db, permute_p_values, excluded_probeset_db = ex_db
splice_event_list, p_value_call, permute_p_values, exon_db, regulated_exon_junction_db = furtherProcessJunctionScores(splice_event_list, probeset_comp_db, permute_p_values)
else:
splice_event_list, probeset_comp_db, permute_p_values, excluded_probeset_db = analyzeJunctionSplicing(nonlog_NI_db)
splice_event_list, p_value_call, permute_p_values, exon_db, regulated_exon_junction_db = furtherProcessJunctionScores(splice_event_list, probeset_comp_db, permute_p_values)
elif analysis_method == 'splicing-index':
regulated_exon_junction_db = {}
if original_conditions > 2:
excluded_probeset_db = ex_db; splice_event_list = si_db;
clearObjectsFromMemory(ex_db); clearObjectsFromMemory(si_db)
ex_db=[]; si_db=[]; permute_p_values={}; p_value_call=''
else: splice_event_list, p_value_call, permute_p_values, excluded_probeset_db = analyzeSplicingIndex(fold_dbase)
elif analysis_method == 'FIRMA':
regulated_exon_junction_db = {}
splice_event_list, p_value_call, permute_p_values, excluded_probeset_db = FIRMAanalysis(fold_dbase)
global permuted_z_scores; permuted_z_scores={}; global original_domain_z_score_data; original_domain_z_score_data={}
global original_microRNA_z_score_data; original_microRNA_z_score_data={}
nonlog_NI_db=[] ### Clear memory of this large dictionary
try: clearObjectsFromMemory(original_avg_const_exp_db); clearObjectsFromMemory(array_raw_group_values)
except Exception: null=[]
try: clearObjectsFromMemory(avg_const_exp_db)
except Exception: null=[]
try: clearObjectsFromMemory(alt_junction_db)
except Exception: null=[]
try: clearObjectsFromMemory(fold_dbase); fold_dbase=[]
except Exception: null=[]
microRNA_full_exon_db,microRNA_count_db,gene_microRNA_denom = ExonAnalyze_module.importmicroRNADataExon(species,array_type,exon_db,microRNA_prediction_method,explicit_data_type,root_dir)
#print "MicroRNA data imported"
if use_direct_domain_alignments_only == 'yes':
protein_ft_db_len,domain_associated_genes = importProbesetAligningDomains(exon_db,'gene')
else: protein_ft_db_len,domain_associated_genes = importProbesetProteinCompDomains(exon_db,'gene','exoncomp')
if perform_element_permutation_analysis == 'yes':
probeset_to_gene,denominator_list = getInputsForPermutationAnalysis(exon_db)
if array_type == 'gene' or array_type == 'junction' or array_type == 'RNASeq':
exon_gene_array_translation_file = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_'+array_type+'-exon_probesets.txt'
try: exon_array_translation_db = importGeneric(exon_gene_array_translation_file)
except Exception: exon_array_translation_db={} ### Not present for all species
exon_hits={}; clearObjectsFromMemory(probeset_comp_db); probeset_comp_db=[]
###Run analyses in the ExonAnalyze_module module to assess functional changes
for (score,ed) in splice_event_list:
geneid = ed.GeneID()
if analysis_method == 'ASPIRE' or 'linearregres' in analysis_method:
pl = string.split(ed.Probeset1(),'|'); probeset1 = pl[0] ### When agglomerated, this is important
uid = (probeset1,ed.Probeset2())
else: uid = ed.Probeset1()
gene_exon = geneid,uid; exon_hits[gene_exon] = ed
#print probeset1,ed.Probeset1(),ed.Probeset2(),gene_exon,ed.CriticalExons()
dataset_name_original = analysis_method+'-'+dataset_name[8:-1]
global functional_attribute_db; global protein_features
### Possibly Block-out code for DomainGraph export
########### Re-import the exon_db for significant entries with full annotaitons
exon_db={}; filtered_arrayids={}; filter_status='yes' ###Use this as a means to save memory (import multiple times - only storing different types relevant information)
for (score,entry) in splice_event_list:
try: probeset = original_exon_db[entry.Probeset1()].Probeset()
except Exception: probeset = entry.Probeset1()
pl = string.split(probeset,'|'); probeset = pl[0]; filtered_arrayids[probeset] = [] ### When agglomerated, this is important
if array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
try: probeset = entry.Probeset2(); filtered_arrayids[probeset] = []
except AttributeError: null =[] ###occurs when running Splicing
exon_db = importSplicingAnnotationDatabase(probeset_annotations_file,array_type,filtered_arrayids,filter_status);null=[] ###replace existing exon_db (probeset_annotations_file should be a global)
###domain_gene_changed_count_db is the number of genes for each domain that are found for regulated probesets
if array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
if use_direct_domain_alignments_only == 'yes':
protein_features,domain_gene_changed_count_db,functional_attribute_db = importProbesetAligningDomains(regulated_exon_junction_db,'probeset')
else: protein_features,domain_gene_changed_count_db,functional_attribute_db = importProbesetProteinCompDomains(regulated_exon_junction_db,'probeset','exoncomp')
else:
if use_direct_domain_alignments_only == 'yes':
protein_features,domain_gene_changed_count_db,functional_attribute_db = importProbesetAligningDomains(exon_db,'probeset')
else: protein_features,domain_gene_changed_count_db,functional_attribute_db = importProbesetProteinCompDomains(exon_db,'probeset','exoncomp')
filtered_microRNA_exon_db = ExonAnalyze_module.filterMicroRNAProbesetAssociations(microRNA_full_exon_db,exon_hits)
microRNA_full_exon_db=[]
###add microRNA data to functional_attribute_db
microRNA_hit_gene_count_db = {}; all_microRNA_gene_hits={}; microRNA_attribute_db={}; probeset_mirBS_db={}
for (affygene,uid) in filtered_microRNA_exon_db: ###example ('G7091354', 'E20|') [('hsa-miR-130a', 'Pbxip1'), ('hsa-miR-130a', 'Pbxip1'
###3-1-08
miR_list = []
microRNA_symbol_list = filtered_microRNA_exon_db[(affygene,uid)]
for mir_key in microRNA_symbol_list:
microRNA,gene_symbol,miR_seq, miR_sources = mir_key
#if 'ENS' in microRNA: print microRNA; kill ### bug in some miRNA annotations introduced in the build process
specific_microRNA_tuple = (microRNA,'~')
try: microRNA_hit_gene_count_db[microRNA].append(affygene)
except KeyError: microRNA_hit_gene_count_db[microRNA] = [affygene]
###Create a database with the same structure as "protein_exon_feature_db"(below) for over-representation analysis (direction specific), after linking up splice direction data
try: microRNA_attribute_db[(affygene,uid)].append(specific_microRNA_tuple)
except KeyError: microRNA_attribute_db[(affygene,uid)] = [specific_microRNA_tuple]
miR_data = microRNA+':'+miR_sources
miR_list.append(miR_data) ###Add miR information to the record
function_type = ('miR-sequence: ' +'('+miR_data+')'+miR_seq,'~') ###Add miR sequence information to the sequence field of the report
try: functional_attribute_db[(affygene,uid)].append(function_type)
except KeyError: functional_attribute_db[(affygene,uid)]=[function_type]
#print (affygene,uid), [function_type];kill
if perform_element_permutation_analysis == 'yes':
try: probeset_mirBS_db[uid].append(microRNA)
except KeyError: probeset_mirBS_db[uid] = [microRNA]
miR_str = string.join(miR_list,','); miR_str = '('+miR_str+')'
function_type = ('microRNA-target'+miR_str,'~')
try: functional_attribute_db[(affygene,uid)].append(function_type)
except KeyError: functional_attribute_db[(affygene,uid)]=[function_type]
all_microRNA_gene_hits[affygene] = []
###Replace the gene list for each microRNA hit with count data
microRNA_hit_gene_count_db = eliminate_redundant_dict_values(microRNA_hit_gene_count_db)
###Combines any additional feature alignment info identified from 'ExonAnalyze_module.characterizeProteinLevelExonChanges' (e.g. from Ensembl or junction-based queries rather than exon specific) and combines
###this with this database of (Gene,Exon)=[(functional element 1,'~'),(functional element 2,'~')] for downstream result file annotatations
domain_hit_gene_count_db = {}; all_domain_gene_hits = {}; probeset_domain_db={}
for entry in protein_features:
gene,uid = entry
for data_tuple in protein_features[entry]:
domain,call = data_tuple
try: protein_exon_feature_db[entry].append(data_tuple)
except KeyError: protein_exon_feature_db[entry] = [data_tuple]
try: domain_hit_gene_count_db[domain].append(gene)
except KeyError: domain_hit_gene_count_db[domain] = [gene]
all_domain_gene_hits[gene]=[]
if perform_element_permutation_analysis == 'yes':
try: probeset_domain_db[uid].append(domain)
except KeyError: probeset_domain_db[uid] = [domain]
protein_features=[]; domain_gene_changed_count_db=[]
###Replace the gene list for each microRNA hit with count data
domain_hit_gene_count_db = eliminate_redundant_dict_values(domain_hit_gene_count_db)
############ Perform Element Over-Representation Analysis ############
"""Domain/FT Fishers-Exact test: with "protein_exon_feature_db" (transformed to "domain_hit_gene_count_db") we can analyze over-representation of domain/features WITHOUT taking into account exon-inclusion or exclusion
Do this using: "domain_associated_genes", which contains domain tuple ('Tyr_pkinase', 'IPR001245') as a key and count in unique genes as the value in addition to
Number of genes linked to splice events "regulated" (SI and Midas p<0.05), number of genes with constitutive probesets
MicroRNA Fishers-Exact test: "filtered_microRNA_exon_db" contains gene/exon to microRNA data. For each microRNA, count the representation in spliced genes microRNA (unique gene count - make this from the mentioned file)
Do this using: "microRNA_count_db"""
domain_gene_counts = {} ### Get unique gene counts for each domain
for domain in domain_associated_genes:
domain_gene_counts[domain] = len(domain_associated_genes[domain])
total_microRNA_gene_hit_count = len(all_microRNA_gene_hits)
total_microRNA_gene_denom_count = len(gene_microRNA_denom)
Nm,Rm = calculateZScores(microRNA_hit_gene_count_db,microRNA_count_db,total_microRNA_gene_denom_count,total_microRNA_gene_hit_count,'microRNA')
gene_microRNA_denom =[]
summary_data_db['miRNA_gene_denom'] = total_microRNA_gene_denom_count
summary_data_db['miRNA_gene_hits'] = total_microRNA_gene_hit_count
summary_data_db['alt_events']=len(splice_event_list)
total_domain_gene_hit_count = len(all_domain_gene_hits)
total_domain_gene_denom_count = protein_ft_db_len ###genes connected to domain annotations
Nd,Rd = calculateZScores(domain_hit_gene_count_db,domain_gene_counts,total_domain_gene_denom_count,total_domain_gene_hit_count,'domain')
microRNA_hit_gene_counts={}; gene_to_miR_db={} ### Get unique gene counts for each miR and the converse
for microRNA in microRNA_hit_gene_count_db:
microRNA_hit_gene_counts[microRNA] = len(microRNA_hit_gene_count_db[microRNA])
for gene in microRNA_hit_gene_count_db[microRNA]:
try: gene_to_miR_db[gene].append(microRNA)
except KeyError: gene_to_miR_db[gene] = [microRNA]
gene_to_miR_db = eliminate_redundant_dict_values(gene_to_miR_db)
if perform_element_permutation_analysis == 'yes':
###Begin Domain/microRNA Permute Analysis
input_count = len(splice_event_list) ### Number of probesets or probeset pairs (junction array) alternatively regulated
original_increment = int(permutations/20); increment = original_increment
start_time = time.time(); print 'Permuting the Domain/miRBS analysis %d times' % permutations
x=0; permute_domain_inputs=[]; permute_miR_inputs=[]
while x<permutations:
if x == increment: increment+=original_increment; print '*',
permute_input_list = random.sample(denominator_list,input_count); x+=1
permute_domain_input_gene_counts = countGenesForElement(permute_input_list,probeset_to_gene,probeset_domain_db)
permute_domain_inputs.append(permute_domain_input_gene_counts)
permute_miR_input_gene_counts = countGenesForElement(permute_input_list,probeset_to_gene,probeset_mirBS_db)
permute_miR_inputs.append(permute_miR_input_gene_counts)
calculatePermuteZScores(permute_domain_inputs,domain_gene_counts,Nd,Rd)
calculatePermuteZScores(permute_miR_inputs,microRNA_hit_gene_counts,Nm,Rm)
calculatePermuteStats(original_domain_z_score_data)
calculatePermuteStats(original_microRNA_z_score_data)
adjustPermuteStats(original_domain_z_score_data)
adjustPermuteStats(original_microRNA_z_score_data)
exportZScoreData(original_domain_z_score_data,'ft-domain')
exportZScoreData(original_microRNA_z_score_data,'microRNA')
end_time = time.time(); time_diff = int(end_time-start_time)
print "Enrichment p-values for Domains/miRBS calculated in %d seconds" % time_diff
denominator_list=[]
try: clearObjectsFromMemory(original_microRNA_z_score_data)
except Exception: null=[]
microRNA_hit_gene_count_db={}; microRNA_hit_gene_counts={};
clearObjectsFromMemory(permuted_z_scores); permuted_z_scores=[]; original_domain_z_score_data=[]
if (array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null')) and analysis_method != 'splicing-index':
critical_probeset_annotation_db = getJunctionSplicingAnnotations(regulated_exon_junction_db)
probeset_aligning_db = importProbesetAligningDomains(regulated_exon_junction_db,'perfect_match')
else: probeset_aligning_db = importProbesetAligningDomains(exon_db,'perfect_match')
############ Export exon/junction level results ############
splice_event_db={}; protein_length_list=[]; aspire_gene_results={}
critical_gene_exons={}; unique_exon_event_db={}; comparison_count={}; direct_domain_gene_alignments={}
functional_attribute_db2={}; protein_exon_feature_db2={}; microRNA_exon_feature_db2={}
external_exon_annot={}; gene_exon_region={}; gene_smallest_p={}; gene_splice_event_score={}; alternatively_reg_tc={}
aspire_output = root_dir+'AltResults/AlternativeOutput/' + dataset_name + analysis_method+'-exon-inclusion-results.txt'
data = export.ExportFile(aspire_output)
goelite_output = root_dir+'GO-Elite/AltExon/AS.'+ dataset_name + analysis_method+'.txt'
goelite_data = export.ExportFile(goelite_output); gcn=0
#print 'LENGTH OF THE GENE ANNOTATION DATABASE',len(annotate_db)
if array_type != 'AltMouse':
DG_output = root_dir+'AltResults/DomainGraph/' + dataset_name + analysis_method+'-DomainGraph.txt'
DG_data = export.ExportFile(DG_output)
### Write out only the inclusion hits to a subdir
SRFinder_inclusion = root_dir+'GO-Elite/exon/' + dataset_name + analysis_method+'-inclusion.txt'
SRFinder_in_data = export.ExportFile(SRFinder_inclusion)
SRFinder_in_data.write('probeset\tSystemCode\tdeltaI\tp-value\n')
### Write out only the exclusion hits to a subdir
SRFinder_exclusion = root_dir+'GO-Elite/exon/' + dataset_name + analysis_method+'-exclusion.txt'
SRFinder_ex_data = export.ExportFile(SRFinder_exclusion)
SRFinder_ex_data.write('probeset\tSystemCode\tdeltaI\tp-value\n')
### Write out only the denominator set to a subdir
SRFinder_denom = root_dir+'GO-Elite/exon_denominator/' + species+'-'+array_type+'.txt'
SRFinder_denom_data = export.ExportFile(SRFinder_denom)
SRFinder_denom_data.write('probeset\tSystemCode\n')
ens_version = unique.getCurrentGeneDatabaseVersion()
ProcessedSpliceData_output = string.replace(DG_output,'DomainGraph','ProcessedSpliceData') ### This is the same as the DG export but without converting the probeset IDs for non-exon arrays
ProcessedSpliceData_data = export.ExportFile(ProcessedSpliceData_output)
if ens_version == '':
try:
elite_db_versions = UI.returnDirectoriesNoReplace('/AltDatabase')
if len(elite_db_versions)>0: ens_version = elite_db_versions[0]
except Exception: null=[]
ens_version = string.replace(ens_version,'EnsMart','ENS_')
DG_data.write(ens_version+"\n")
DG_data.write("Probeset\tGeneID\tRegulation call\tSI\tSI p-value\tMiDAS p-value\n")
ProcessedSpliceData_data.write("ExonID(s)\tGeneID\tRegulation call\t"+analysis_method+"\t"+analysis_method+" p-value\tMiDAS p-value\n")
if analysis_method == 'ASPIRE' or 'linearregres' in analysis_method:
if perform_permutation_analysis == 'yes': p_value_type = 'permutation-values'
else: p_value_type = 'FDR-'+p_value_call
if array_type == 'AltMouse': gene_name = 'AffyGene'; extra_transcript_annotation = 'block_structure'; extra_exon_annotation = 'splice_event_description'
if array_type == 'junction' or array_type == 'RNASeq':
gene_name = 'Ensembl'; extra_transcript_annotation = 'transcript cluster ID'; extra_exon_annotation = 'distal exon-region-ID'
goelite_data.write("GeneID\tSystemCode\tscore\tp-value\tSymbol\tExonIDs\n")
if array_type == 'RNASeq':
id1='junctionID-1'; id2='junctionID-2'; loc_column='exon/junction locations'
extra_transcript_annotation = 'Known/Novel Feature'
else: id1='probeset1'; id2='probeset2'; loc_column='probeset locations'
title = [gene_name,analysis_method,'symbol','description','exons1','exons2','regulation_call','event_call',id1,'norm-p1',id2,'norm-p2','fold1','fold2']
title +=['adj-fold1' ,'adj-fold2' ,extra_transcript_annotation,'critical_up_exons','critical_down_exons','functional_prediction','uniprot-ens_feature_predictions']
title +=['peptide_predictions','exp1','exp2','ens_overlapping_domains','constitutive_baseline_exp',p_value_call,p_value_type,'permutation-false-positives']
title +=['gene-expression-change', extra_exon_annotation ,'ExternalExonIDs','ExonRegionID','SplicingEvent','ExonAnnotationScore','large_splicing_diff',loc_column]
else:
goelite_data.write("GeneID\tSystemCode\tSI\tSI p-value\tMiDAS p-value\tSymbol\tExonID\n")
if analysis_method == 'splicing-index':
NIpval = 'SI_rawp'; splicing_score = 'Splicing-Index'; lowestp = 'lowest_p (MIDAS or SI)'; AdjPcolumn = 'Deviation-Value'; #AdjPcolumn = 'SI_adjp'
else:
NIpval = 'FIRMA_rawp'; splicing_score = 'FIRMA_fold'; lowestp = 'lowest_p (MIDAS or FIRMA)'; AdjPcolumn = 'Deviation-Value'; #AdjPcolumn = 'FIRMA_adjp'
if array_type == 'RNASeq':
id1='junctionID'; pval_column='junction p-value'; loc_column='junction location'
else: id1='probeset'; pval_column='probeset p-value'; loc_column='probeset location'
if array_type == 'RNASeq': secondary_ID_title = 'Known/Novel Feature'
else: secondary_ID_title = 'alternative gene ID'
title= ['Ensembl',splicing_score,'symbol','description','exons','regulation_call',id1,pval_column,lowestp,'midas p-value','fold','adjfold']
title+=['up_exons','down_exons','functional_prediction','uniprot-ens_feature_predictions','peptide_predictions','ens_overlapping_domains','baseline_probeset_exp']
title+=['constitutive_baseline_exp',NIpval,AdjPcolumn,'gene-expression-change']
title+=[secondary_ID_title, 'ensembl exons', 'consitutive exon', 'exon-region-ID', 'exon annotations','distal exon-region-ID',loc_column]
title = string.join(title,'\t') + '\n'
try:
if original_conditions>2: title = string.replace(title,'regulation_call','conditions_compared')
except Exception: null=[]
data.write(title)
### Calculate adjusted normalized intensity p-values
fdr_exon_stats={}
if analysis_method != 'ASPIRE' and 'linearregres' not in analysis_method:
for (score,entry) in splice_event_list: ### These are all "significant entries"
fds = FDRStats(entry.TTestNormalizedRatios())
fdr_exon_stats[entry.Probeset1()] = fds
for probeset in excluded_probeset_db: ### These are all "non-significant entries"
fds = FDRStats(excluded_probeset_db[probeset].TTestNormalizedRatios())
fdr_exon_stats[probeset] = fds
try: adjustPermuteStats(fdr_exon_stats)
except Exception: null=[]
### Calculate score average and stdev for each gene to alter get a Deviation Value
gene_deviation_db={}
for (score,entry) in splice_event_list:
dI = entry.Score(); geneID = entry.GeneID()
try: gene_deviation_db[geneID].append(dI)
except Exception: gene_deviation_db[geneID] = [dI]
for i in excluded_probeset_db:
entry = excluded_probeset_db[i]
try: dI = entry.Score(); geneID = entry.GeneID()
except Exception: geneID = entry[1]; dI = entry[-1]
try: gene_deviation_db[geneID].append(dI)
except Exception: None ### Don't include genes with no hits
for geneID in gene_deviation_db:
try:
avg_dI=statistics.avg(gene_deviation_db[geneID])
stdev_dI=statistics.stdev(gene_deviation_db[geneID])
gene_deviation_db[geneID] = avg_dI,stdev_dI
except Exception:
gene_deviation_db[geneID] = 'NA','NA'
event_count = 0
for (score,entry) in splice_event_list:
event_count += 1
dI = entry.Score(); probeset1 = entry.Probeset1(); regulation_call = entry.RegulationCall(); event_call = entry.EventCall();critical_exon_list = entry.CriticalExonTuple()
probeset1_display = probeset1; selected_probeset = probeset1
if agglomerate_inclusion_probesets == 'yes':
if array_type == 'AltMouse':
exons1 = original_exon_db[probeset1].ExonID()
try: probeset1 = original_exon_db[probeset1].Probeset()
except Exception: null=[]
else:
probeset1 = probeset1; exons1 = original_exon_db[probeset1].ExonID()
try: selected_probeset = original_exon_db[probeset1].Probeset()
except Exception: selected_probeset = probeset1
else:
try: exons1 = exon_db[probeset1].ExonID()
except Exception:
print probeset1, len(exon_db)
for i in exon_db: print i; break
kill
critical_probeset_list = [selected_probeset]
affygene = entry.GeneID()
### Calculate deviation value for each exon
avg_dI,stdev_dI = gene_deviation_db[affygene]
try: DV = deviation(dI,avg_dI,stdev_dI) ### Note: the dI values are always in log2 space, independent of platform
except Exception: DV = 'NA'
if affygene in annotate_db: description = annotate_db[affygene].Description(); symbol = annotate_db[affygene].Symbol()
else: description = ''; symbol = ''
ped1 = entry.ProbesetExprData1(); adjfold1 = ped1.AdjFold(); exp1 = ped1.BaselineExp(); fold1 = ped1.FoldChange(); rawp1 = ped1.ExpPval()
### Get Constitutive expression values
baseline_const_exp = entry.ConstitutiveExpression() ### For multiple group comparisosn
#if affygene in gene_expression_diff_db: mean_fold_change = gene_expression_diff_db[affygene].ConstitutiveFoldStr()
try: mean_fold_change = str(entry.ConstitutiveFold()) ### For multi-condition analyses, the gene expression is dependent on the conditions compared
except Exception: mean_fold_change = gene_expression_diff_db[affygene].ConstitutiveFoldStr()
if analysis_method == 'ASPIRE' or 'linearregres' in analysis_method:
probeset2 = entry.Probeset2(); exons2 = exon_db[probeset2].ExonID(); rawp1 = str(entry.TTestNormalizedRatios()); rawp2 = str(entry.TTestNormalizedRatios2()); critical_probeset_list.append(probeset2)
ped2 = entry.ProbesetExprData2(); adjfold2 = ped2.AdjFold(); exp2 = ped2.BaselineExp(); fold2 = ped2.FoldChange()
try: location_summary=original_exon_db[selected_probeset].LocationSummary()+'|'+original_exon_db[probeset2].LocationSummary()
except Exception:
try: location_summary=exon_db[selected_probeset].LocationSummary()+'|'+exon_db[probeset2].LocationSummary()
except Exception: location_summary=''
if array_type == 'AltMouse':
extra_transcript_annotation = exon_db[probeset1].GeneStructure()
else:
try: extra_exon_annotation = last_exon_region_db[affygene]
except KeyError: extra_exon_annotation = ''
try:
tc1 = original_exon_db[probeset1].SecondaryGeneID()
tc2 = original_exon_db[probeset2].SecondaryGeneID() ### Transcript Cluster
probeset_tc = makeUnique([tc1,tc2])
extra_transcript_annotation = string.join(probeset_tc,'|')
try: alternatively_reg_tc[affygene] += probeset_tc
except KeyError: alternatively_reg_tc[affygene] = probeset_tc
except Exception: extra_transcript_annotation=''
if array_type == 'RNASeq':
try: extra_transcript_annotation = entry.NovelEvent() ### Instead of secondary gene ID, list known vs. novel reciprocal junction annotation
except Exception: None
exp_list = [float(exp1),float(exp2),float(exp1)+float(fold1),float(exp2)+float(fold2)]; exp_list.sort(); exp_list.reverse()
probeset_tuple = (probeset1,probeset2)
else:
try: exp_list = [float(exp1),float(exp1)+float(fold1)]; exp_list.sort(); exp_list.reverse()
except Exception: exp_list = ['']
probeset_tuple = (probeset1)
highest_exp = exp_list[0]
###Use permuted p-value or lowest expression junction p-value based on the situtation
###This p-value is used to filter out aspire events for further analyses
if len(p_value_call)>0:
if probeset_tuple in permute_p_values:
lowest_raw_p, pos_permute, total_permute, false_pos = permute_p_values[probeset_tuple]
else: lowest_raw_p = "NA"; pos_permute = "NA"; total_permute = "NA"; false_pos = "NA"
else:
if analysis_method == 'ASPIRE' or 'linearregres' in analysis_method: raw_p_list = [entry.TTestNormalizedRatios(),entry.TTestNormalizedRatios2()] #raw_p_list = [float(rawp1),float(rawp2)]; raw_p_list.sort()
else:
try: raw_p_list = [float(entry.TTestNormalizedRatios())] ###Could also be rawp1, but this is more appropriate
except Exception: raw_p_list = [1] ### Occurs when p='NA'
raw_p_list.sort()
lowest_raw_p = raw_p_list[0]; pos_permute = "NA"; total_permute = "NA"; false_pos = "NA"
if perform_permutation_analysis == 'yes':
p_value_extra = str(pos_permute)+' out of '+str(total_permute)
else: p_value_extra = str(pos_permute)
up_exons = ''; down_exons = ''; up_exon_list = []; down_exon_list = []; gene_exon_list=[]
exon_data = critical_exon_list
variable = exon_data[0]
if variable == 1 and regulation_call == 'upregulated':
for exon in exon_data[1]:
up_exons = up_exons + exon + ',';up_exon_list.append(exon)
key = affygene,exon+'|'; gene_exon_list.append(key)
elif variable == 1 and regulation_call == 'downregulated':
for exon in exon_data[1]:
down_exons = down_exons + exon + ',';down_exon_list.append(exon)
key = affygene,exon+'|';gene_exon_list.append(key)
else:
try: exon1 = exon_data[1][0]; exon2 = exon_data[1][1]
except Exception: print exon_data;kill
if adjfold1 > 0:
up_exons = up_exons + exon1 + ',';down_exons = down_exons + exon2 + ','
up_exon_list.append(exon1); down_exon_list.append(exon2)
key = affygene,exon1+'|'; gene_exon_list.append(key);key = affygene,exon2+'|'; gene_exon_list.append(key)
else:
up_exons = up_exons + exon2 + ',';down_exons = down_exons + exon1 + ','
up_exon_list.append(exon2); down_exon_list.append(exon1)
key = affygene,exon1+'|'; gene_exon_list.append(key); key = affygene,exon2+'|'; gene_exon_list.append(key)
up_exons = up_exons[0:-1];down_exons = down_exons[0:-1]
try: ### Get comparisons group annotation data for multigroup comparison analyses
if original_conditions>2:
try: regulation_call = ped1.Annotation()
except Exception: null=[]
except Exception: null=[]
###Format functional results based on exon level fold change
null = []
#global a; a = exon_hits; global b; b=microRNA_attribute_db; kill
"""if 'G7100684@J934332_RC@j_at' in critical_probeset_list:
print probeset1, probeset2, gene, critical_probeset_list, 'blah'
if ('G7100684', ('G7100684@J934333_RC@j_at', 'G7100684@J934332_RC@j_at')) in functional_attribute_db:
print functional_attribute_db[('G7100684', ('G7100684@J934333_RC@j_at', 'G7100684@J934332_RC@j_at'))];blah
blah"""
new_functional_attribute_str, functional_attribute_list2, seq_attribute_str,protein_length_list = format_exon_functional_attributes(affygene,critical_probeset_list,functional_attribute_db,up_exon_list,down_exon_list,protein_length_list)
new_uniprot_exon_feature_str, uniprot_exon_feature_list, null, null = format_exon_functional_attributes(affygene,critical_probeset_list,protein_exon_feature_db,up_exon_list,down_exon_list,null)
null, microRNA_exon_feature_list, null, null = format_exon_functional_attributes(affygene,critical_probeset_list,microRNA_attribute_db,up_exon_list,down_exon_list,null)
if len(new_functional_attribute_str) == 0: new_functional_attribute_str = ' '
if len(new_uniprot_exon_feature_str) == 0: new_uniprot_exon_feature_str = ' '
if len(seq_attribute_str) > 12000: seq_attribute_str = 'The sequence is too long to report for spreadsheet analysis'
### Add entries to a database to quantify the number of reciprocal isoforms regulated
reciprocal_isoform_data = [len(critical_exon_list[1]),critical_exon_list[1],event_call,regulation_call]
try: float((lowest_raw_p))
except ValueError: lowest_raw_p=0
if (float((lowest_raw_p))<=p_threshold or false_pos < 2) or lowest_raw_p == 1 or lowest_raw_p == 'NA':
try: unique_exon_event_db[affygene].append(reciprocal_isoform_data)
except KeyError: unique_exon_event_db[affygene] = [reciprocal_isoform_data]
### Add functional attribute information to a new database
for item in uniprot_exon_feature_list:
attribute = item[0]
exon = item[1]
if (float((lowest_raw_p))<=p_threshold or false_pos < 2) or lowest_raw_p == 1 or lowest_raw_p == 'NA':
try: protein_exon_feature_db2[affygene,attribute].append(exon)
except KeyError: protein_exon_feature_db2[affygene,attribute]=[exon]
### Add functional attribute information to a new database
"""Database not used for exon/junction data export but for over-representation analysis (direction specific)"""
for item in microRNA_exon_feature_list:
attribute = item[0]
exon = item[1]
if (float((lowest_raw_p))<=p_threshold or false_pos < 2) or lowest_raw_p == 1 or lowest_raw_p == 'NA':
try: microRNA_exon_feature_db2[affygene,attribute].append(exon)
except KeyError: microRNA_exon_feature_db2[affygene,attribute]=[exon]
### Add functional attribute information to a new database
for item in functional_attribute_list2:
attribute = item[0]
exon = item[1]
if (float((lowest_raw_p))<=p_threshold or false_pos < 2) or lowest_raw_p == 1 or lowest_raw_p == 'NA':
try: functional_attribute_db2[affygene,attribute].append(exon)
except KeyError: functional_attribute_db2[affygene,attribute]=[exon]
try:
abs_fold = abs(float(mean_fold_change)); fold_direction = 'down'; fold1_direction = 'down'; fold2_direction = 'down'
large_splicing_diff1 = 0; large_splicing_diff2 = 0; large_splicing_diff = 'null'; opposite_splicing_pattern = 'no'
if float(mean_fold_change)>0: fold_direction = 'up'
if float(fold1)>0: fold1_direction = 'up'
if fold1_direction != fold_direction:
if float(fold1)>float(mean_fold_change): large_splicing_diff1 = float(fold1)-float(mean_fold_change)
except Exception:
fold_direction = ''; large_splicing_diff = ''; opposite_splicing_pattern = ''
if analysis_method != 'ASPIRE' and 'linearregres' not in analysis_method: ed = exon_db[probeset1]
else:
try: ed = critical_probeset_annotation_db[selected_probeset,probeset2]
except KeyError:
try: ed = exon_db[selected_probeset] ###not useful data here, but the objects need to exist
except IOError: ed = original_exon_db[probeset1]
ucsc_splice_annotations = ["retainedIntron","cassetteExon","strangeSplice","altFivePrime","altThreePrime","altPromoter","bleedingExon"]
custom_annotations = ["alt-3'","alt-5'","alt-C-term","alt-N-term","cassette-exon","cassette-exon","exon-region-exclusion","intron-retention","mutually-exclusive-exon","trans-splicing"]
custom_exon_annotations_found='no'; ucsc_annotations_found = 'no'; exon_annot_score=0
if len(ed.SplicingEvent())>0:
for annotation in ucsc_splice_annotations:
if annotation in ed.SplicingEvent(): ucsc_annotations_found = 'yes'
for annotation in custom_annotations:
if annotation in ed.SplicingEvent(): custom_exon_annotations_found = 'yes'
if custom_exon_annotations_found == 'yes' and ucsc_annotations_found == 'no': exon_annot_score = 3
elif ucsc_annotations_found == 'yes' and custom_exon_annotations_found == 'no': exon_annot_score = 4
elif ucsc_annotations_found == 'yes' and custom_exon_annotations_found == 'yes': exon_annot_score = 5
else: exon_annot_score = 2
try: gene_splice_event_score[affygene].append(exon_annot_score) ###store for gene level results
except KeyError: gene_splice_event_score[affygene] = [exon_annot_score]
try: gene_exon_region[affygene].append(ed.ExonRegionID()) ###store for gene level results
except KeyError: gene_exon_region[affygene] = [ed.ExonRegionID()]
if analysis_method == 'ASPIRE' or 'linearregres' in analysis_method:
if float(fold2)>0: fold2_direction = 'up'
if fold2_direction != fold_direction:
if float(fold2)>float(mean_fold_change):
large_splicing_diff2 = float(fold2)-float(mean_fold_change)
if abs(large_splicing_diff2) > large_splicing_diff1: large_splicing_diff = str(large_splicing_diff2)
else: large_splicing_diff = str(large_splicing_diff1)
if fold1_direction != fold2_direction and abs(float(fold1))>0.4 and abs(float(fold2))>0.4 and abs(float(mean_fold_change))< max([float(fold2),float(fold1)]):
opposite_splicing_pattern = 'yes'
### Annotate splicing events based on exon_strucuture data
if array_type == 'AltMouse':
extra_exon_annotation = ExonAnnotate_module.annotate_splice_event(exons1,exons2,extra_transcript_annotation)
try: splice_event_db[extra_exon_annotation] += 1
except KeyError: splice_event_db[extra_exon_annotation] = 1
try:
direct_domain_alignments = probeset_aligning_db[selected_probeset,probeset2]
try: direct_domain_gene_alignments[affygene]+=', '+direct_domain_alignments
except KeyError: direct_domain_gene_alignments[affygene]=direct_domain_alignments
except KeyError: direct_domain_alignments = ' '
splicing_event = ed.SplicingEvent()
if array_type == 'RNASeq':
splicing_event = checkForTransSplicing(probeset1_display,splicing_event)
splicing_event = checkForTransSplicing(probeset2,splicing_event)
exp1 = covertLogExpressionToNonLog(exp1)
exp2 = covertLogExpressionToNonLog(exp2)
baseline_const_exp = covertLogExpressionToNonLog(baseline_const_exp)
fold1 = covertLogFoldToNonLog(fold1)
fold2 = covertLogFoldToNonLog(fold2)
adjfold1 = covertLogFoldToNonLog(adjfold1)
adjfold2 = covertLogFoldToNonLog(adjfold2)
mean_fold_change = covertLogFoldToNonLog(mean_fold_change)
### Annotate splicing events based on pre-computed and existing annotations
values= [affygene,dI,symbol,fs(description),exons1,exons2,regulation_call,event_call,probeset1_display,rawp1,probeset2,rawp2,fold1,fold2,adjfold1,adjfold2]
values+=[extra_transcript_annotation,up_exons,down_exons,fs(new_functional_attribute_str),fs(new_uniprot_exon_feature_str),fs(seq_attribute_str),exp1,exp2,fs(direct_domain_alignments)]
values+=[str(baseline_const_exp),str(lowest_raw_p),p_value_extra,str(false_pos),mean_fold_change,extra_exon_annotation]
values+=[ed.ExternalExonIDs(),ed.ExonRegionID(),splicing_event,str(exon_annot_score),large_splicing_diff,location_summary]
exon_sets = abs(float(dI)),regulation_call,event_call,exons1,exons2,''
### Export significant reciprocol junction pairs and scores
values_ps = [probeset1+'|'+probeset2,affygene,'changed',dI,'NA',str(lowest_raw_p)]; values_ps = string.join(values_ps,'\t')+'\n'
try: ProcessedSpliceData_data.write(values_ps)
except Exception: None
values_ge = [affygene,'En',dI,str(lowest_raw_p),symbol,probeset1_display+' | '+probeset2]; values_ge = string.join(values_ge,'\t')+'\n'
if array_type == 'junction' or array_type == 'RNASeq': ### Only applies to reciprocal junction sensitive platforms (but not currently AltMouse)
goelite_data.write(values_ge)
if array_type == 'junction' or array_type == 'RNASeq': ### Only applies to reciprocal junction sensitive platforms (but not currently AltMouse)
try: exon_probeset = exon_array_translation_db[affygene+':'+exon_data[1][0]][0]; probeset1 = exon_probeset; gcn+=1
except Exception: probeset1 = None #probeset1 = affygene+':'+exon_data[1][0]
try:
null = int(probeset1) ### Must be an int to work in DomainGraph
values_dg = [probeset1,affygene,'changed',dI,'NA',str(lowest_raw_p)]; values_dg = string.join(values_dg,'\t')+'\n'
if array_type == 'junction' or array_type == 'RNASeq':
DG_data.write(values_dg)
values_srf = string.join([probeset1,'Ae',dI,str(lowest_raw_p)],'\t')+'\n'
if float(dI)>0:
SRFinder_ex_data.write(values_srf)
elif float(dI)<0:
SRFinder_in_data.write(values_srf)
except Exception: null=[]
else:
si_pvalue = lowest_raw_p
if si_pvalue == 1: si_pvalue = 'NA'
if probeset1 in midas_db:
midas_p = str(midas_db[probeset1])
if float(midas_p)<lowest_raw_p: lowest_raw_p = float(midas_p) ###This is the lowest and SI-pvalue
else: midas_p = ''
###Determine what type of exon-annotations are present to assign a confidence score
if affygene in annotate_db: ###Determine the transcript clusters used to comprise a splice event (genes and exon specific)
try:
gene_tc = annotate_db[affygene].TranscriptClusterIDs()
try: probeset_tc = [ed.SecondaryGeneID()]
except Exception: probeset_tc = [affygene]
for transcript_cluster in gene_tc: probeset_tc.append(transcript_cluster)
probeset_tc = makeUnique(probeset_tc)
except Exception: probeset_tc = ''; gene_tc=''
else:
try:
try: probeset_tc = [ed.SecondaryGeneID()]
except Exception: probeset_tc = [affygene]
probeset_tc = makeUnique(probeset_tc)
except Exception: probeset_tc = ''; gene_tc=''
cluster_number = len(probeset_tc)
try: alternatively_reg_tc[affygene] += probeset_tc
except KeyError: alternatively_reg_tc[affygene] = probeset_tc
try: last_exon_region = last_exon_region_db[affygene]
except KeyError: last_exon_region = ''
if cluster_number>1: exon_annot_score = 1
direct_domain_alignments = ' '
if array_type == 'exon' or array_type == 'gene' or explicit_data_type != 'null':
try:
direct_domain_alignments = probeset_aligning_db[probeset1]
try: direct_domain_gene_alignments[affygene]+=', '+direct_domain_alignments
except KeyError: direct_domain_gene_alignments[affygene]=direct_domain_alignments
except KeyError: direct_domain_alignments = ' '
else:
try: direct_domain_alignments = probeset_aligning_db[affygene+':'+exons1]
except KeyError: direct_domain_alignments = ''
if array_type == 'RNASeq':
exp1 = covertLogExpressionToNonLog(exp1)
baseline_const_exp = covertLogExpressionToNonLog(baseline_const_exp)
fold1 = covertLogFoldToNonLog(fold1)
adjfold1 = covertLogFoldToNonLog(adjfold1)
mean_fold_change = covertLogFoldToNonLog(mean_fold_change)
try: adj_SIp=fdr_exon_stats[probeset1].AdjP()
except Exception: adj_SIp = 'NA'
try: secondary_geneid = ed.SecondaryGeneID()
except Exception: secondary_geneid = affygene
if array_type == 'RNASeq':
secondary_geneid = ed.NovelExon()
### Write Splicing Index results
values= [affygene,dI,symbol,fs(description),exons1,regulation_call,probeset1,rawp1,str(lowest_raw_p),midas_p,fold1,adjfold1]
values+=[up_exons,down_exons,fs(new_functional_attribute_str),fs(new_uniprot_exon_feature_str),fs(seq_attribute_str),fs(direct_domain_alignments),exp1]
values+=[str(baseline_const_exp),str(si_pvalue),DV,mean_fold_change,secondary_geneid, ed.ExternalExonIDs()]
values+=[ed.Constitutive(),ed.ExonRegionID(),ed.SplicingEvent(),last_exon_region,ed.LocationSummary()] #str(exon_annot_score)
if probeset1 in filtered_probeset_db: values += filtered_probeset_db[probeset1]
exon_sets = abs(float(dI)),regulation_call,event_call,exons1,exons1,midas_p
probeset = probeset1 ### store original ID (gets converted below)
### Write DomainGraph results
try: midas_p = str(midas_db[probeset1])
except KeyError: midas_p = 'NA'
### Export significant exon/junction IDs and scores
values_ps = [probeset1,affygene,'changed',dI,'NA',str(lowest_raw_p)]; values_ps = string.join(values_ps,'\t')+'\n'
try: ProcessedSpliceData_data.write(values_ps)
except Exception: None
if array_type == 'gene' or array_type == 'junction' or array_type == 'RNASeq':
if (array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null':
try: exon_probeset = exon_array_translation_db[affygene+':'+exon_data[1][0]][0]; probeset1 = exon_probeset; gcn+=1
except Exception: probeset1 = None ### don't write out a line
else:
try: exon_probeset = exon_array_translation_db[probeset1][0]; probeset1 = exon_probeset; gcn+=1
except Exception: probeset1=None; #null=[]; #print gcn, probeset1;kill - force an error - new in version 2.0.8
try:
null = int(probeset1)
values_dg = [probeset1,affygene,'changed',dI,str(si_pvalue),midas_p]; values_dg = string.join(values_dg,'\t')+'\n'
DG_data.write(values_dg)
values_srf = string.join([probeset1,'Ae',dI,str(lowest_raw_p)],'\t')+'\n'
if float(dI)>0:
SRFinder_ex_data.write(values_srf)
elif float(dI)<0:
SRFinder_in_data.write(values_srf)
except Exception: null=[]
values_ge = [affygene,'En',dI,str(si_pvalue),midas_p,symbol,probeset]; values_ge = string.join(values_ge,'\t')+'\n'
goelite_data.write(values_ge)
if len(ed.SplicingEvent())>2:
try: external_exon_annot[affygene].append(ed.SplicingEvent())
except KeyError: external_exon_annot[affygene] = [ed.SplicingEvent()]
try: values = string.join(values,'\t')+'\n'
except Exception: print values;kill
data.write(values)
###Process data for gene level reports
if float((lowest_raw_p))<=p_threshold or false_pos < 2 or lowest_raw_p == 1:
try: comparison_count[affygene] += 1
except KeyError: comparison_count[affygene] = 1
try: aspire_gene_results[affygene].append(exon_sets)
except KeyError: aspire_gene_results[affygene] = [exon_sets]
for exon in up_exon_list:
exon_info = exon,'upregulated'
try: critical_gene_exons[affygene].append(exon_info)
except KeyError: critical_gene_exons[affygene] = [exon_info]
for exon in down_exon_list:
exon_info = exon,'downregulated'
try: critical_gene_exons[affygene].append(exon_info)
except KeyError: critical_gene_exons[affygene] = [exon_info]
data.close()
print event_count, analysis_method, "results written to:", aspire_output,'\n'
try: clearObjectsFromMemory(original_exon_db)
except Exception: null=[]
exon_array_translation_db=[]; original_exon_db=[]; probeset_to_gene=[]
### Finish writing the DomainGraph export file with non-significant probesets
if array_type != 'AltMouse':
for probeset in excluded_probeset_db:
eed = excluded_probeset_db[probeset]
try: midas_p = str(midas_db[probeset])
except KeyError: midas_p = 'NA'
### Export significant exon/junction IDs and scores
try: values_ps = [probeset,eed.GeneID(),'UC',eed.Score(),str(eed.TTestNormalizedRatios()),midas_p]
except Exception: excl_probeset, geneid, score, rawp, pvalue = eed; values_ps = [probeset,geneid,'UC', str(score), str(rawp), str(pvalue)]
values_ps = string.join(values_ps,'\t')+'\n'; ProcessedSpliceData_data.write(values_ps)
### Write DomainGraph results
if array_type == 'gene' or array_type == 'junction' or array_type == 'RNASeq':
try: exon_probeset = exon_array_translation_db[probeset][0]; probeset = exon_probeset; gcn+=1
except Exception: probeset=None; # null=[] - force an error - new in version 2.0.8
try: values_dg = [probeset,eed.GeneID(),'UC',eed.Score(),str(eed.TTestNormalizedRatios()),midas_p]
except Exception:
try:
excl_probeset, geneid, score, rawp, pvalue = eed
if ':' in probeset: probeset = excl_probeset ### Example: ENSMUSG00000029213:E2.1, make this just the numeric exclusion probeset - Not sure if DG handles non-numeric
values_dg = [probeset,geneid,'UC', str(score), str(rawp), str(pvalue)]
except Exception: None
try:
null=int(probeset)
values_dg = string.join(values_dg,'\t')+'\n'; DG_data.write(values_dg)
except Exception: null=[]
if array_type == 'gene' or array_type == 'junction' or array_type == 'RNASeq':
for id in exon_array_translation_db:
SRFinder_denom_data.write(exon_array_translation_db[id]+'\tAe\n')
else:
for probeset in original_exon_db:
SRFinder_denom_data.write(probeset+'\tAe\n')
DG_data.close()
SRFinder_in_data.close()
SRFinder_ex_data.close()
SRFinder_denom_data.close()
for affygene in direct_domain_gene_alignments:
domains = string.split(direct_domain_gene_alignments[affygene],', ')
domains = unique.unique(domains); domains = string.join(domains,', ')
direct_domain_gene_alignments[affygene] = domains
### functional_attribute_db2 will be reorganized so save the database with another. Use this
functional_attribute_db = functional_attribute_db2
functional_attribute_db2 = reorganize_attribute_entries(functional_attribute_db2,'no')
external_exon_annot = eliminate_redundant_dict_values(external_exon_annot)
protein_exon_feature_db = protein_exon_feature_db2
protein_exon_feature_db2 = reorganize_attribute_entries(protein_exon_feature_db2,'no')
############ Export Gene Data ############
up_splice_val_genes = 0; down_dI_genes = 0; diff_exp_spliced_genes = 0; diff_spliced_rna_factor = 0
ddI = 0; udI = 0
summary_data_db['direct_domain_genes']=len(direct_domain_gene_alignments)
summary_data_db['alt_genes']=len(aspire_gene_results)
critical_gene_exons = eliminate_redundant_dict_values(critical_gene_exons)
aspire_output_gene = root_dir+'AltResults/AlternativeOutput/' + dataset_name + analysis_method + '-exon-inclusion-GENE-results.txt'
data = export.ExportFile(aspire_output_gene)
if array_type == 'AltMouse': goelite_data.write("GeneID\tSystemCode\n")
title = ['AffyGene','max_dI','midas-p (corresponding)','symbol','external gene ID','description','regulation_call','event_call']
title +=['number_of_comparisons','num_effected_exons','up_exons','down_exons','functional_attribute','uniprot-ens_exon_features','direct_domain_alignments']
title +=['pathways','mean_fold_change','exon-annotations','exon-region IDs','alternative gene ID','splice-annotation score']
title = string.join(title,'\t')+'\n'
data.write(title)
for affygene in aspire_gene_results:
if affygene in annotate_db:
description = annotate_db[affygene].Description()
symbol = annotate_db[affygene].Symbol()
ensembl = annotate_db[affygene].ExternalGeneID()
if array_type != 'AltMouse' and array_type != 'RNASeq': transcript_clusters = alternatively_reg_tc[affygene]; transcript_clusters = makeUnique(transcript_clusters); transcript_clusters = string.join(transcript_clusters,'|')
else: transcript_clusters = affygene
rna_processing_factor = annotate_db[affygene].RNAProcessing()
else: description='';symbol='';ensembl=affygene;rna_processing_factor=''; transcript_clusters=''
if ensembl in go_annotations: wpgo = go_annotations[ensembl]; goa = wpgo.Combined()
else: goa = ''
if array_type == 'AltMouse':
if len(ensembl) >0: goelite_data.write(ensembl+'\tL\n')
try: gene_splice_event_score[affygene].sort(); top_se_score = str(gene_splice_event_score[affygene][-1])
except KeyError: top_se_score = 'NA'
try: gene_regions = gene_exon_region[affygene]; gene_regions = makeUnique(gene_regions); gene_regions = string.join(gene_regions,'|')
except KeyError: gene_regions = 'NA'
if analysis_method == 'ASPIRE' or analysis_method == 'linearregres': number_of_comparisons = str(comparison_count[affygene])
else: number_of_comparisons = 'NA'
results_list = aspire_gene_results[affygene]
results_list.sort(); results_list.reverse()
max_dI = str(results_list[0][0])
regulation_call = results_list[0][1]
event_call = results_list[0][2]
midas_p = results_list[0][-1]
num_critical_exons = str(len(critical_gene_exons[affygene]))
try: direct_domain_annots = direct_domain_gene_alignments[affygene]
except KeyError: direct_domain_annots = ' '
down_exons = ''; up_exons = ''; down_list=[]; up_list=[]
for exon_info in critical_gene_exons[affygene]:
exon = exon_info[0]; call = exon_info[1]
if call == 'downregulated':
down_exons = down_exons + exon + ','
down_list.append(exon)
ddI += 1
if call == 'upregulated':
up_exons = up_exons + exon + ','
up_list.append(exon)
udI += 1
down_exons = down_exons[0:-1]
up_exons = up_exons[0:-1]
up_exons = add_a_space(up_exons); down_exons = add_a_space(down_exons)
functional_annotation =''
if affygene in functional_attribute_db2:
number_of_functional_attributes = str(len(functional_attribute_db2[affygene]))
attribute_list = functional_attribute_db2[affygene]
attribute_list.sort()
for attribute_exon_info in attribute_list:
exon_attribute = attribute_exon_info[0]
exon_list = attribute_exon_info[1]
functional_annotation = functional_annotation + exon_attribute
exons = '('
for exon in exon_list: exons = exons + exon + ','
exons = exons[0:-1] + '),'
if add_exons_to_annotations == 'yes': functional_annotation = functional_annotation + exons
else: functional_annotation = functional_annotation + ','
functional_annotation = functional_annotation[0:-1]
uniprot_exon_annotation = ''
if affygene in protein_exon_feature_db2:
number_of_functional_attributes = str(len(protein_exon_feature_db2[affygene]))
attribute_list = protein_exon_feature_db2[affygene]; attribute_list.sort()
for attribute_exon_info in attribute_list:
exon_attribute = attribute_exon_info[0]
exon_list = attribute_exon_info[1]
uniprot_exon_annotation = uniprot_exon_annotation + exon_attribute
exons = '('
for exon in exon_list: exons = exons + exon + ','
exons = exons[0:-1] + '),'
if add_exons_to_annotations == 'yes': uniprot_exon_annotation = uniprot_exon_annotation + exons
else: uniprot_exon_annotation = uniprot_exon_annotation + ','
uniprot_exon_annotation = uniprot_exon_annotation[0:-1]
if len(uniprot_exon_annotation) == 0: uniprot_exon_annotation = ' '
if len(functional_annotation) == 0: functional_annotation = ' '
if affygene in gene_expression_diff_db:
mean_fold_change = gene_expression_diff_db[affygene].ConstitutiveFoldStr()
try:
if abs(float(mean_fold_change)) > log_fold_cutoff: diff_exp_spliced_genes += 1
except Exception: diff_exp_spliced_genes = diff_exp_spliced_genes
else: mean_fold_change = 'NC'
if len(rna_processing_factor) > 2: diff_spliced_rna_factor +=1
###Add annotations for where in the gene structure these exons are (according to Ensembl)
if affygene in external_exon_annot: external_gene_annot = string.join(external_exon_annot[affygene],', ')
else: external_gene_annot = ''
if array_type == 'RNASeq':
mean_fold_change = covertLogFoldToNonLog(mean_fold_change)
values =[affygene,max_dI,midas_p,symbol,ensembl,fs(description),regulation_call,event_call,number_of_comparisons]
values+=[num_critical_exons,up_exons,down_exons,functional_annotation]
values+=[fs(uniprot_exon_annotation),fs(direct_domain_annots),fs(goa),mean_fold_change,external_gene_annot,gene_regions,transcript_clusters,top_se_score]
values = string.join(values,'\t')+'\n'
data.write(values)
### Use results for summary statistics
if len(up_list)>len(down_list): up_splice_val_genes +=1
else: down_dI_genes +=1
data.close()
print "Gene-level results written"
###yes here indicates that although the truncation events will initially be filtered out, later they will be added
###back in without the non-truncation annotations....if there is no second database (in this case functional_attribute_db again)
###IF WE WANT TO FILTER OUT NON-NMD ENTRIES WHEN NMD IS PRESENT (FOR A GENE) MUST INCLUDE functional_attribute_db AS THE SECOND VARIABLE!!!!
###Currently, yes does nothing
functional_annotation_db, null = grab_summary_dataset_annotations(functional_attribute_db,'','yes')
upregulated_genes = 0; downregulated_genes = 0
###Calculate the number of upregulated and downregulated genes
for affygene in gene_expression_diff_db:
fold_val = gene_expression_diff_db[affygene].ConstitutiveFold()
try:
if float(fold_val) > log_fold_cutoff: upregulated_genes += 1
elif abs(float(fold_val)) > log_fold_cutoff: downregulated_genes += 1
except Exception: null=[]
upregulated_rna_factor = 0; downregulated_rna_factor = 0
###Calculate the total number of putative RNA-processing/binding factors differentially regulated
for affygene in gene_expression_diff_db:
gene_fold = gene_expression_diff_db[affygene].ConstitutiveFold()
rna_processing_factor = gene_expression_diff_db[affygene].RNAProcessing()
if len(rna_processing_factor) > 1:
if gene_fold>log_fold_cutoff: upregulated_rna_factor += 1
elif abs(gene_fold)>log_fold_cutoff: downregulated_rna_factor += 1
###Generate three files for downstream functional summary
### functional_annotation_db2 is output to the same function as functional_annotation_db, ranked_uniprot_list_all to get all ranked uniprot annotations,
### and ranked_uniprot_list_coding_only to get only coding ranked uniprot annotations
functional_annotation_db2, ranked_uniprot_list_all = grab_summary_dataset_annotations(protein_exon_feature_db,'','') #functional_attribute_db
null, ranked_uniprot_list_coding_only = grab_summary_dataset_annotations(protein_exon_feature_db,functional_attribute_db,'') #functional_attribute_db
functional_attribute_db=[]; protein_exon_feature_db=[]
###Sumarize changes in avg protein length for each splice event
up_protein_list=[];down_protein_list=[]; protein_length_fold_diff=[]
for [down_protein,up_protein] in protein_length_list:
up_protein = float(up_protein); down_protein = float(down_protein)
down_protein_list.append(down_protein); up_protein_list.append(up_protein)
if up_protein > 10 and down_protein > 10:
fold_change = up_protein/down_protein; protein_length_fold_diff.append(fold_change)
median_fold_diff = statistics.median(protein_length_fold_diff)
try: down_avg=int(statistics.avg(down_protein_list)); up_avg=int(statistics.avg(up_protein_list))
except Exception: down_avg=0; up_avg=0
try:
try:
down_std=int(statistics.stdev(down_protein_list)); up_std=int(statistics.stdev(up_protein_list))
except ValueError: ###If 'null' is returned fro stdev
down_std = 0;up_std = 0
except Exception:
down_std = 0;up_std = 0
if len(down_protein_list)>1 and len(up_protein_list)>1:
try:
#t,df,tails = statistics.ttest(down_protein_list,up_protein_list,2,3)
#t = abs(t);df = round(df)
#print 'ttest t:',t,'df:',df
#p = str(statistics.t_probability(t,df))
p = str(statistics.runComparisonStatistic(down_protein_list,up_protein_list,probability_statistic))
#print dataset_name,p
except Exception: p = 'NA'
if p == 1: p = 'NA'
else: p = 'NA'
###Calculate unique reciprocal isoforms for exon-inclusion, exclusion and mutual-exclusive events
unique_exon_inclusion_count=0;unique_exon_exclusion_count=0;unique_mutual_exclusive_count=0;
unique_exon_event_db = eliminate_redundant_dict_values(unique_exon_event_db)
for affygene in unique_exon_event_db:
isoform_entries = unique_exon_event_db[affygene]
possibly_redundant=[]; non_redundant=[]; check_for_redundant=[]
for entry in isoform_entries:
if entry[0] == 1: ### If there is only one regulated exon
possibly_redundant.append(entry)
else:
non_redundant.append(entry)
critical_exon_list = entry[1]
for exon in critical_exon_list:
check_for_redundant.append(exon)
for entry in possibly_redundant:
exon = entry[1][0]
if exon not in check_for_redundant:
non_redundant.append(entry)
for entry in non_redundant:
if entry[2] == 'ei-ex':
if entry[3] == 'upregulated': unique_exon_inclusion_count += 1
else: unique_exon_exclusion_count += 1
else: unique_mutual_exclusive_count += 1
udI = unique_exon_inclusion_count; ddI = unique_exon_exclusion_count; mx = unique_mutual_exclusive_count
###Add splice event information to the functional_annotation_db
for splice_event in splice_event_db:count = splice_event_db[splice_event]; functional_annotation_db.append((splice_event,count))
if analysis_method == 'splicing-index' or analysis_method == 'FIRMA': udI='NA'; ddI='NA'
summary_results_db[dataset_name[0:-1]] = udI,ddI,mx,up_splice_val_genes,down_dI_genes,(up_splice_val_genes + down_dI_genes),upregulated_genes, downregulated_genes, diff_exp_spliced_genes, upregulated_rna_factor,downregulated_rna_factor,diff_spliced_rna_factor,down_avg,down_std,up_avg,up_std,p,median_fold_diff,functional_annotation_db
result_list = exportComparisonSummary(dataset_name,summary_data_db,'log')
###Re-set this variable (useful for testing purposes)
clearObjectsFromMemory(gene_expression_diff_db)
clearObjectsFromMemory(splice_event_list); clearObjectsFromMemory(si_db); si_db=[]
clearObjectsFromMemory(fdr_exon_stats)
try: clearObjectsFromMemory(excluded_probeset_db); clearObjectsFromMemory(ex_db); ex_db=[]
except Exception: ex_db=[]
clearObjectsFromMemory(exon_db)
#clearObjectsFromMemory(annotate_db)
critical_probeset_annotation_db=[]; gene_expression_diff_db=[]; domain_associated_genes=[]; permute_p_values=[]
permute_miR_inputs=[]; seq_attribute_str=[]; microRNA_count_db=[]; excluded_probeset_db=[]; fdr_exon_stats=[]
splice_event_list=[]; critical_exon_db_len=len(critical_exon_db)#; critical_exon_db=[] deleting here will cause a global instance problem
all_domain_gene_hits=[]; gene_splice_event_score=[]; unique_exon_event_db=[]; probeset_aligning_db=[]; ranked_uniprot_list_all=[];
filtered_microRNA_exon_db=[]; permute_domain_inputs=[]; functional_annotation_db2=[]; functional_attribute_db2=[]; protein_length_list=[];
ranked_uniprot_list_coding_only=[]; miR_str=[]; permute_input_list=[]; microRNA_exon_feature_db2=[]; alternatively_reg_tc=[];
direct_domain_gene_alignments=[]; aspire_gene_results=[]; domain_gene_counts=[]; functional_annotation=[]; protein_exon_feature_db2=[];
microRNA_attribute_db=[]; probeset_mirBS_db=[]; exon_hits=[]; critical_gene_exons=[]; gene_exon_region=[]; exon_db=[]; external_exon_annot=[];
values=[]; down_protein_list=[]; functional_annotation_db=[]; protein_length_fold_diff=[]; comparison_count=[]; filtered_arrayids=[];
domain_hit_gene_count_db=[]; up_protein_list=[]; probeset_domain_db=[]
try: goelite_data.close()
except Exception: null=[]
"""
print 'local vars'
all = [var for var in locals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(locals()[var])>500: print var, len(locals()[var])
except Exception: null=[]
"""
return summary_results_db, summary_results_db2, aspire_output, aspire_output_gene, critical_exon_db_len
def deviation(dI,avg_dI,stdev_dI):
dI = covertLogFoldToNonLogFloat(dI)
avg_dI = covertLogFoldToNonLogFloat(avg_dI)
stdev_dI = covertLogFoldToNonLogFloat(stdev_dI)
return str(abs((dI-avg_dI)/stdev_dI))
def covertLogExpressionToNonLog(log_val):
if normalization_method == 'RPKM':
nonlog_val = (math.pow(2,float(log_val)))
else:
nonlog_val = (math.pow(2,float(log_val)))-1
return str(nonlog_val)
def covertLogFoldToNonLog(log_val):
try:
if float(log_val)<0: nonlog_val = (-1/math.pow(2,(float(log_val))))
else: nonlog_val = (math.pow(2,float(log_val)))
except Exception: nonlog_val = log_val
return str(nonlog_val)
def covertLogFoldToNonLogFloat(log_val):
if float(log_val)<0: nonlog_val = (-1/math.pow(2,(float(log_val))))
else: nonlog_val = (math.pow(2,float(log_val)))
return nonlog_val
def checkForTransSplicing(uid,splicing_event):
pl = string.split(uid,':')
if len(pl)>2:
if pl[0] not in pl[1]: ### Two different genes
if len(splicing_event)>0: splicing_event+= '|trans-splicing'
else: splicing_event = '|trans-splicing'
return splicing_event
def fs(text):
### Formats a text entry to prevent delimiting a comma
return '"'+text+'"'
def analyzeSplicingIndex(fold_dbase):
"""The Splicing Index (SI) represents the log ratio of the exon intensities between the two tissues after normalization
to the gene intensities in each sample: SIi = log2((e1i/g1j)/(e2i/g2j)), for the i-th exon of the j-th gene in tissue
type 1 or 2. The splicing indices are then subjected to a t-test to probe for differential inclusion of the exon into the gene.
In order to determine if the change in isoform expression was statistically significant, a simple two-tailed t-test was carried
out on the isoform ratios by grouping the 10 samples from either "tumor" or "normal" tissue.
The method ultimately producing the highest proportion of true positives was to retain only: a) exons with a DABG p-value < 0.05,
b) genes with a signal > 70, c) exons with a log ratio between tissues (i.e., the gene-level normalized fold change) > 0.5,
d) Splicing Index p-values < 0.005 and e) Core exons.
Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, Veitch J, Schweitzer A, Awad T, Sugnet C, Dee S, Davies C, Williams A, Turpaz Y.
Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array.
BMC Genomics. 2006 Dec 27;7:325. PMID: 17192196
"""
### Used to restrict the analysis to a pre-selected set of probesets (e.g. those that have a specifc splicing pattern)
if len(filtered_probeset_db)>0:
temp_db={}
for probeset in fold_dbase: temp_db[probeset]=[]
for probeset in temp_db:
try: filtered_probeset_db[probeset]
except KeyError: del fold_dbase[probeset]
### Used to restrict the analysis to a pre-selected set of probesets (e.g. those that have a specifc splicing annotation)
if filter_for_AS == 'yes':
proceed = 0
for probeset in exon_db:
as_call = exon_db[probeset].SplicingCall()
if as_call == 0:
try: del fold_dbase[probeset]
except KeyError: null=[]
### Used to the export relative individual adjusted probesets fold changes used for splicing index values
if export_NI_values == 'yes':
summary_output = root_dir+'AltResults/RawSpliceData/'+species+'/'+analysis_method+'/'+dataset_name[:-1]+'.txt'
data = export.ExportFile(summary_output)
title = string.join(['gene\tExonID\tprobesets']+original_array_names,'\t')+'\n'; data.write(title)
print 'Calculating splicing-index values (please be patient)...',
if array_type == 'RNASeq': id_name = 'exon/junction IDs'
else: id_name = 'array IDs'
print len(fold_dbase),id_name,'beging examined'
###original_avg_const_exp_db contains constitutive mean expression values per group: G6953871 [7.71, 7.66]
###array_raw_group_values: Raw expression values in list of groups: G7072464@J935416_RC@j_at ([1.79, 2.16, 2.22], [1.68, 2.24, 1.97, 1.92, 2.12])
###avg_const_exp_db contains the raw constitutive expression values in a single list
splicing_index_hash=[]; excluded_probeset_db={}; denominator_probesets=0; interaction = 0
original_increment = int(len(exon_db)/20); increment = original_increment
for probeset in exon_db:
ed = exon_db[probeset]
#include_probeset = ed.IncludeProbeset()
if interaction == increment: increment+=original_increment; print '*',
interaction +=1
include_probeset = 'yes' ###Moved this filter to import of the probeset relationship file
###Examines user input parameters for inclusion of probeset types in the analysis
if include_probeset == 'yes':
geneid = ed.GeneID()
if probeset in fold_dbase and geneid in original_avg_const_exp_db: ###used to search for array_raw_group_values, but when filtered by expression changes, need to filter by adj_fold_dbase
denominator_probesets+=1
###Includes probesets with a calculated constitutive expression value for each gene and expression data for that probeset
group_index = 0; si_interim_group_db={}; si_interim_group_str_db={}; ge_threshold_count=0; value_count = 0
for group_values in array_raw_group_values[probeset]:
"""gene_expression_value = math.pow(2,original_avg_const_exp_db[geneid][group_index])
###Check to see if gene expression is > threshod for both conditions
if gene_expression_value>gene_expression_threshold:ge_threshold_count+=1"""
value_index = 0; ratio_hash=[]; ratio_str_hash=[]
for value in group_values: ###Calculate normalized ratio's for each condition and save raw values for later permutation
#exp_val = math.pow(2,value);ge_val = math.pow(2,avg_const_exp_db[geneid][value_count]) ###To calculate a ttest we need the raw constitutive expression values, these are not in group list form but are all in a single list so keep count.
exp_val = value;ge_val = avg_const_exp_db[geneid][value_count]
exp_ratio = exp_val-ge_val; ratio_hash.append(exp_ratio); ratio_str_hash.append(str(exp_ratio))
value_index +=1; value_count +=1
si_interim_group_db[group_index] = ratio_hash
si_interim_group_str_db[group_index] = ratio_str_hash
group_index+=1
group1_ratios = si_interim_group_db[0]; group2_ratios = si_interim_group_db[1]
group1_mean_ratio = statistics.avg(group1_ratios); group2_mean_ratio = statistics.avg(group2_ratios)
if export_NI_values == 'yes':
try: er = ed.ExonID()
except Exception: er = 'NA'
ev = string.join([geneid+'\t'+er+'\t'+probeset]+si_interim_group_str_db[0]+si_interim_group_str_db[1],'\t')+'\n'; data.write(ev)
#if ((math.log(group1_mean_ratio,2))*(math.log(group2_mean_ratio,2)))<0: opposite_SI_log_mean = 'yes'
if (group1_mean_ratio*group2_mean_ratio)<0: opposite_SI_log_mean = 'yes'
else: opposite_SI_log_mean = 'no'
try:
if calculate_normIntensity_p == 'yes':
try:
normIntensityP = statistics.runComparisonStatistic(group1_ratios,group2_ratios,probability_statistic)
except Exception: normIntensityP = 'NA' ### Occurs when analyzing two groups with no variance
else: normIntensityP = 'NA' ### Set to an always signficant value
if normIntensityP == 1: normIntensityP = 'NA'
splicing_index = group1_mean_ratio-group2_mean_ratio; abs_splicing_index = abs(splicing_index)
#if probeset == '3061323': print abs_splicing_index,normIntensityP,ed.ExonID(),group1_mean_ratio,group2_mean_ratio,math.log(group1_mean_ratio,2),math.log(group2_mean_ratio,2),((math.log(group1_mean_ratio,2))*(math.log(group2_mean_ratio,2))),opposite_SI_log_mean; kill
if probeset in midas_db:
try: midas_p = float(midas_db[probeset])
except ValueError:
midas_p = 0
#if abs_splicing_index>1 and normIntensityP < 0.05: print probeset,normIntensityP, abs_splicing_index;kill
else: midas_p = 0
#print ed.GeneID(),ed.ExonID(),probeset,splicing_index,normIntensityP,midas_p,group1_ratios,group2_ratios
if abs_splicing_index>alt_exon_logfold_cutoff and (normIntensityP < p_threshold or normIntensityP == 'NA' or normIntensityP == 1) and midas_p < p_threshold:
exonid = ed.ExonID(); critical_exon_list = [1,[exonid]]
constit_exp1 = original_avg_const_exp_db[geneid][0]
constit_exp2 = original_avg_const_exp_db[geneid][1]
ge_fold=constit_exp2-constit_exp1
### Re-define all of the pairwise values now that the two Splicing-Index groups to report have been determined
data_list1 = array_raw_group_values[probeset][0]; data_list2 = array_raw_group_values[probeset][1]
baseline_exp = statistics.avg(data_list1); experimental_exp = statistics.avg(data_list2); fold_change = experimental_exp - baseline_exp
try:
ttest_exp_p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic)
except Exception: ttest_exp_p = 1
normInt1 = (baseline_exp-constit_exp1); normInt2 = (experimental_exp-constit_exp2); adj_fold = normInt2 - normInt1
ped = ProbesetExpressionData(baseline_exp, experimental_exp, fold_change, adj_fold, ttest_exp_p, '')
sid = ExonData(splicing_index,probeset,critical_exon_list,geneid,group1_ratios,group2_ratios,normIntensityP,opposite_SI_log_mean)
sid.setConstitutiveExpression(constit_exp1); sid.setConstitutiveFold(ge_fold); sid.setProbesetExpressionData(ped)
splicing_index_hash.append((splicing_index,sid))
else:
### Also record the data for probesets that are excluded... Used by DomainGraph
eed = ExcludedExonData(splicing_index,geneid,normIntensityP)
excluded_probeset_db[probeset] = eed
except Exception:
null = [] ###If this occurs, then most likely, the exon and constitutive probeset are the same
print 'Splicing Index analysis complete'
if export_NI_values == 'yes': data.close()
splicing_index_hash.sort(); splicing_index_hash.reverse()
print len(splicing_index_hash),id_name,"with evidence of Alternative expression"
p_value_call=''; permute_p_values = {}; summary_data_db['denominator_exp_events']=denominator_probesets
return splicing_index_hash,p_value_call,permute_p_values, excluded_probeset_db
def importResiduals(filename,probe_probeset_db):
fn=filepath(filename); key_db = {}; x=0; prior_uid = ''; uid_gene_db={}
for line in open(fn,'rU').xreadlines():
if x == 0 and line[0] == '#': null=[]
elif x == 0: x+=1
else:
data = cleanUpLine(line)
t = string.split(data,'\t')
uid = t[0]; uid,probe = string.split(uid,'-')
try:
probeset = probe_probeset_db[probe]; residuals = t[1:]
if uid == prior_uid:
try: uid_gene_db[probeset].append(residuals) ### Don't need to keep track of the probe ID
except KeyError: uid_gene_db[probeset] = [residuals]
else: ### Hence, we have finished storing all residual data for that gene
if len(uid_gene_db)>0: calculateFIRMAScores(uid_gene_db); uid_gene_db={}
try: uid_gene_db[probeset].append(residuals) ### Don't need to keep track of the probe ID
except KeyError: uid_gene_db[probeset] = [residuals]
prior_uid = uid
except Exception: null=[]
### For the last gene imported
if len(uid_gene_db)>0: calculateFIRMAScores(uid_gene_db)
def calculateFIRMAScores(uid_gene_db):
probeset_residuals={}; all_gene_residuals=[]; total_probes=0
for probeset in uid_gene_db:
residuals_list = uid_gene_db[probeset]; sample_db={}; total_probes+=len(residuals_list)
### For all probes in a probeset, calculate the median residual for each sample
for residuals in residuals_list:
index=0
for residual in residuals:
try: sample_db[index].append(float(residual))
except KeyError: sample_db[index] = [float(residual)]
all_gene_residuals.append(float(residual))
index+=1
for index in sample_db:
median_residual = statistics.median(sample_db[index])
sample_db[index] = median_residual
probeset_residuals[probeset] = sample_db
### Calculate the Median absolute deviation
"""http://en.wikipedia.org/wiki/Absolute_deviation
The median absolute deviation (also MAD) is the median absolute deviation from the median. It is a robust estimator of dispersion.
For the example {2, 2, 3, 4, 14}: 3 is the median, so the absolute deviations from the median are {1, 1, 0, 1, 11} (or reordered as
{0, 1, 1, 1, 11}) with a median absolute deviation of 1, in this case unaffected by the value of the outlier 14.
Here, the global gene median will be expressed as res_gene_median.
"""
res_gene_median = statistics.median(all_gene_residuals); subtracted_residuals=[]
for residual in all_gene_residuals: subtracted_residuals.append(abs(res_gene_median-residual))
gene_MAD = statistics.median(subtracted_residuals)
#if '3263614' in probeset_residuals: print len(all_gene_residuals),all_gene_residuals
for probeset in probeset_residuals:
sample_db = probeset_residuals[probeset]
for index in sample_db:
median_residual = sample_db[index]
try:
firma_score = median_residual/gene_MAD
sample_db[index] = firma_score
except Exception: null=[]
#if probeset == '3263614': print index, median_residual, firma_score, gene_MAD
firma_scores[probeset] = sample_db
def importProbeToProbesets(fold_dbase):
#print "Importing probe-to-probeset annotations (please be patient)..."
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_probeset-probes.txt'
probeset_to_include={}
gene2examine={}
### Although we want to restrict the analysis to probesets in fold_dbase, we don't want to effect the FIRMA model - filter later
for probeset in fold_dbase:
try: ed = exon_db[probeset]; gene2examine[ed.GeneID()]=[]
except Exception: null=[]
for gene in original_avg_const_exp_db: gene2examine[gene]=[]
for probeset in exon_db:
ed = exon_db[probeset]; geneid = ed.GeneID()
if geneid in gene2examine:
gene2examine[geneid].append(probeset) ### Store these so we can break things up
probeset_to_include[probeset]=[]
probeset_probe_db = importGenericFilteredDBList(filename,probeset_to_include)
### Get Residuals filename and verify it's presence
#print "Importing comparison residuals..."
filename_objects = string.split(dataset_name[:-1],'.p'); filename = filename_objects[0]+'.txt'
if len(array_group_list)==2:
filename = import_dir = root_dir+'AltExpression/FIRMA/residuals/'+array_type+'/'+species+'/'+filename
else: filename = import_dir = root_dir+'AltExpression/FIRMA/FullDatasets/'+array_type+'/'+species+'/'+filename
status = verifyFile(filename)
if status != 'found':
print_out = 'The residual file:'; print_out+= filename
print_out+= 'was not found in the default location.\nPlease make re-run the analysis from the Beginning.'
try: UI.WarningWindow(print_out,'Exit')
except Exception: print print_out
print traceback.format_exc(); badExit()
print "Calculating FIRMA scores..."
input_count = len(gene2examine) ### Number of probesets or probeset pairs (junction array) alternatively regulated
original_increment = int(input_count/20); increment = original_increment
start_time = time.time(); x=0
probe_probeset_db={}; gene_count=0; total_gene_count = 0; max_gene_count=3000; round = 1
for gene in gene2examine:
gene_count+=1; total_gene_count+=1; x+=1
#if x == increment: increment+=original_increment; print '*',
for probeset in gene2examine[gene]:
for probe in probeset_probe_db[probeset]: probe_probeset_db[probe] = probeset
if gene_count == max_gene_count:
### Import residuals and calculate primary sample/probeset FIRMA scores
importResiduals(filename,probe_probeset_db)
#print max_gene_count*round,"genes"
print '*',
gene_count=0; probe_probeset_db={}; round+=1 ### Reset these variables and re-run
probeset_probe_db={}
### Analyze residuals for the remaining probesets (< max_gene_count)
importResiduals(filename,probe_probeset_db)
end_time = time.time(); time_diff = int(end_time-start_time)
print "FIRMA scores calculted for",total_gene_count, "genes in %d seconds" % time_diff
def FIRMAanalysis(fold_dbase):
"""The FIRMA method calculates a score for each probeset and for each samples within a group of arrays, independent
of group membership. However, in AltAnalyze, these analyses are performed dependent on group. The FIRMA score is calculated
by obtaining residual values (residuals is a variable for each probe that can't be explained by the GC content or intensity
of that probe) from APT, for all probes corresponding to a metaprobeset (Ensembl gene in AltAnalyze). These probe residuals
are imported and the ratio of the median residual per probeset per sample divided by the absolute standard deviation of the
median of all probes for all samples for that gene."""
### Used to restrict the analysis to a pre-selected set of probesets (e.g. those that have a specifc splicing pattern)
if len(filtered_probeset_db)>0:
temp_db={}
for probeset in fold_dbase: temp_db[probeset]=[]
for probeset in temp_db:
try: filtered_probeset_db[probeset]
except KeyError: del fold_dbase[probeset]
### Used to restrict the analysis to a pre-selected set of probesets (e.g. those that have a specifc splicing annotation)
if filter_for_AS == 'yes':
proceed = 0
for probeset in exon_db:
as_call = exon_db[probeset].SplicingCall()
if as_call == 0:
try: del fold_dbase[probeset]
except KeyError: null=[]
#print 'Beginning FIRMA analysis (please be patient)...'
### Used to the export relative individual adjusted probesets fold changes used for splicing index values
if export_NI_values == 'yes':
sample_names_ordered = [] ### note: Can't use original_array_names since the order is potentially different (FIRMA stores sample data as indeces within dictionary keys)
for group_name in array_group_list: ### THIS LIST IS USED TO MAINTAIN CONSISTENT GROUP ORDERING DURING ANALYSIS
for sample_name in array_group_name_db[group_name]: sample_names_ordered.append(sample_name)
summary_output = root_dir+'AltResults/RawSpliceData/'+species+'/'+analysis_method+'/'+dataset_name[:-1]+'.txt'
data = export.ExportFile(summary_output)
title = string.join(['gene-probesets']+sample_names_ordered,'\t')+'\n'; data.write(title)
### Import probes for probesets to be analyzed
global firma_scores; firma_scores = {}
importProbeToProbesets(fold_dbase)
print 'FIRMA scores obtained for',len(firma_scores),'probests.'
### Group sample scores for each probeset and calculate statistics
firma_hash=[]; excluded_probeset_db={}; denominator_probesets=0; interaction = 0
original_increment = int(len(firma_scores)/20); increment = original_increment
for probeset in firma_scores:
if probeset in fold_dbase: ### Filter based on expression
ed = exon_db[probeset]; geneid = ed.GeneID()
if interaction == increment: increment+=original_increment; print '*',
interaction +=1; denominator_probesets+=1
sample_db = firma_scores[probeset]
###Use the index values from performExpressionAnalysis to assign each expression value to a new database
firma_group_array = {}
for group_name in array_group_db:
for array_index in array_group_db[group_name]:
firma_score = sample_db[array_index]
try: firma_group_array[group_name].append(firma_score)
except KeyError: firma_group_array[group_name] = [firma_score]
###array_group_list should already be unique and correctly sorted (see above)
firma_lists=[]; index=0
for group_name in array_group_list:
firma_list = firma_group_array[group_name]
if len(array_group_list)>2: firma_list = statistics.avg(firma_list), firma_list, index
firma_lists.append(firma_list); index+=1
if export_NI_values == 'yes': ### DO THIS HERE SINCE firma_lists IS SORTED BELOW!!!!
try: er = ed.ExonID()
except Exception: er = 'NA'
export_list = [geneid+'\t'+er+'\t'+probeset]; export_list2=[]
for firma_ls in firma_lists:
if len(array_group_list)>2: firma_ls =firma_ls[1] ### See above modification of firma_list object for multiple group anlaysis
export_list+=firma_ls
for i in export_list: export_list2.append(str(i))
ev = string.join(export_list2,'\t')+'\n'; data.write(ev)
if len(array_group_list)==2:
firma_list1 = firma_lists[0]; firma_list2 = firma_lists[-1]; firma_avg1 = statistics.avg(firma_list1); firma_avg2 = statistics.avg(firma_list2)
index1=0; index2=1 ### Only two groups, thus only two indeces
else: ### The below code deals with identifying the comparisons which yeild the greatest FIRMA difference
firma_lists.sort(); index1=firma_lists[0][-1]; index2 = firma_lists[-1][-1]
firma_list1 = firma_lists[0][1]; firma_list2 = firma_lists[-1][1]; firma_avg1 = firma_lists[0][0]; firma_avg2 = firma_lists[-1][0]
if calculate_normIntensity_p == 'yes':
try:
normIntensityP = statistics.runComparisonStatistic(firma_list1,firma_list2,probability_statistic)
except Exception: normIntensityP = 'NA' ### Occurs when analyzing two groups with no variance
else: normIntensityP = 'NA'
if normIntensityP == 1: normIntensityP = 'NA'
firma_fold_change = firma_avg2 - firma_avg1
firma_fold_change = -1*firma_fold_change ### Make this equivalent to Splicing Index fold which is also relative to experimental not control
if (firma_avg2*firma_avg1)<0: opposite_FIRMA_scores = 'yes'
else: opposite_FIRMA_scores = 'no'
if probeset in midas_db:
try: midas_p = float(midas_db[probeset])
except ValueError: midas_p = 0
else: midas_p = 0
#if probeset == '3263614': print firma_fold_change, normIntensityP, midas_p,'\n',firma_list1, firma_list2, [p_threshold];kill
if abs(firma_fold_change)>alt_exon_logfold_cutoff and (normIntensityP < p_threshold or normIntensityP == 'NA') and midas_p < p_threshold:
exonid = ed.ExonID(); critical_exon_list = [1,[exonid]]
#gene_expression_values = original_avg_const_exp_db[geneid]
constit_exp1 = original_avg_const_exp_db[geneid][index1]
constit_exp2 = original_avg_const_exp_db[geneid][index2]
ge_fold = constit_exp2-constit_exp1
### Re-define all of the pairwise values now that the two FIRMA groups to report have been determined
data_list1 = array_raw_group_values[probeset][index1]; data_list2 = array_raw_group_values[probeset][index2]
baseline_exp = statistics.avg(data_list1); experimental_exp = statistics.avg(data_list2); fold_change = experimental_exp - baseline_exp
group_name1 = array_group_list[index1]; group_name2 = array_group_list[index2]
try:
ttest_exp_p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic)
except Exception: ttest_exp_p = 1
normInt1 = (baseline_exp-constit_exp1); normInt2 = (experimental_exp-constit_exp2); adj_fold = normInt2 - normInt1
ped = ProbesetExpressionData(baseline_exp, experimental_exp, fold_change, adj_fold, ttest_exp_p, group_name2+'_vs_'+group_name1)
fid = ExonData(firma_fold_change,probeset,critical_exon_list,geneid,data_list1,data_list2,normIntensityP,opposite_FIRMA_scores)
fid.setConstitutiveExpression(constit_exp1); fid.setConstitutiveFold(ge_fold); fid.setProbesetExpressionData(ped)
firma_hash.append((firma_fold_change,fid))
#print [[[probeset,firma_fold_change,normIntensityP,p_threshold]]]
else:
### Also record the data for probesets that are excluded... Used by DomainGraph
eed = ExcludedExonData(firma_fold_change,geneid,normIntensityP)
excluded_probeset_db[probeset] = eed
print 'FIRMA analysis complete'
if export_NI_values == 'yes': data.close()
firma_hash.sort(); firma_hash.reverse()
print len(firma_hash),"Probesets with evidence of Alternative expression out of",len(excluded_probeset_db)+len(firma_hash)
p_value_call=''; permute_p_values = {}; summary_data_db['denominator_exp_events']=denominator_probesets
return firma_hash,p_value_call,permute_p_values, excluded_probeset_db
def getFilteredFilename(filename):
if array_type == 'junction':
filename = string.replace(filename,'.txt','-filtered.txt')
return filename
def getExonVersionFilename(filename):
original_filename = filename
if array_type == 'junction' or array_type == 'RNASeq':
if explicit_data_type != 'null':
filename = string.replace(filename,array_type,array_type+'/'+explicit_data_type)
### Make sure the file exists, otherwise, use the original
file_status = verifyFile(filename)
#print [[filename,file_status]]
if file_status != 'found': filename = original_filename
return filename
def importProbesetAligningDomains(exon_db,report_type):
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_domain_aligning_probesets.txt'
filename=getFilteredFilename(filename)
probeset_aligning_db = importGenericDBList(filename)
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_indirect_domain_aligning_probesets.txt'
filename=getFilteredFilename(filename)
probeset_indirect_aligning_db = importGenericDBList(filename)
if array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
new_exon_db={}; splicing_call_db={}
for probeset_pair in exon_db:
### For junction analyses exon_db is really regulated_exon_junction_db, containing the inclusion,exclusion probeset tuple and an object as values
ed = exon_db[probeset_pair]; geneid = ed.GeneID(); critical_exons = ed.CriticalExons()
for exon in critical_exons:
new_key = geneid+':'+exon
try: new_exon_db[new_key].append(probeset_pair)
except KeyError: new_exon_db[new_key] = [probeset_pair]
try: splicing_call_db[new_key].append(ed.SplicingCall())
except KeyError: splicing_call_db[new_key] = [ed.SplicingCall()]
for key in new_exon_db:
probeset_pairs = new_exon_db[key]; probeset_pair = probeset_pairs[0] ### grab one of the probeset pairs
ed = exon_db[probeset_pair]; geneid = ed.GeneID()
jd = SimpleJunctionData(geneid,'','','',probeset_pairs) ### use only those necessary fields for this function (probeset pairs will be called as CriticalExons)
splicing_call_db[key].sort(); splicing_call = splicing_call_db[key][-1]; jd.setSplicingCall(splicing_call) ### Bug from 1.15 to have key be new_key?
new_exon_db[key] = jd
exon_db = new_exon_db
gene_protein_ft_db={};domain_gene_count_db={};protein_functional_attribute_db={}; probeset_aligning_db2={}
splicing_call_db=[]; new_exon_db=[] ### Clear memory
for probeset in exon_db:
#if probeset == '107650':
#if probeset in probeset_aligning_db: print probeset_aligning_db[probeset];kill
if probeset in probeset_aligning_db:
proceed = 'no'
if filter_for_AS == 'yes':
as_call = exon_db[probeset].SplicingCall()
if as_call == 1: proceed = 'yes'
else: proceed = 'yes'
gene = exon_db[probeset].GeneID()
new_domain_list=[]; new_domain_list2=[]
if report_type == 'gene' and proceed == 'yes':
for domain in probeset_aligning_db[probeset]:
try: domain_gene_count_db[domain].append(gene)
except KeyError: domain_gene_count_db[domain] = [gene]
try: gene_protein_ft_db[gene].append(domain)
except KeyError: gene_protein_ft_db[gene]=[domain]
elif proceed == 'yes':
if array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
probeset_list = exon_db[probeset].CriticalExons()
else: probeset_list = [probeset]
for id in probeset_list:
for domain in probeset_aligning_db[probeset]:
new_domain_list.append('(direct)'+domain)
new_domain_list2.append((domain,'+'))
new_domain_list = unique.unique(new_domain_list)
new_domain_list_str = string.join(new_domain_list,', ')
gene_protein_ft_db[gene,id] = new_domain_list2
probeset_aligning_db2[id] = new_domain_list_str
#print exon_db['107650']
for probeset in exon_db:
if probeset in probeset_indirect_aligning_db:
proceed = 'no'
if filter_for_AS == 'yes':
as_call = exon_db[probeset].SplicingCall()
if as_call == 1: proceed = 'yes'
else: proceed = 'yes'
gene = exon_db[probeset].GeneID()
new_domain_list=[]; new_domain_list2=[]
if report_type == 'gene' and proceed == 'yes':
for domain in probeset_indirect_aligning_db[probeset]:
try: domain_gene_count_db[domain].append(gene)
except KeyError: domain_gene_count_db[domain] = [gene]
try: gene_protein_ft_db[gene].append(domain)
except KeyError: gene_protein_ft_db[gene]=[domain]
elif proceed == 'yes':
if array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
probeset_list = exon_db[probeset].CriticalExons()
else: probeset_list = [probeset]
for id in probeset_list:
for domain in probeset_indirect_aligning_db[probeset]:
new_domain_list.append('(indirect)'+domain)
new_domain_list2.append((domain,'-'))
new_domain_list = unique.unique(new_domain_list)
new_domain_list_str = string.join(new_domain_list,', ')
gene_protein_ft_db[gene,id] = new_domain_list2
probeset_aligning_db2[id] = new_domain_list_str
domain_gene_count_db = eliminate_redundant_dict_values(domain_gene_count_db)
gene_protein_ft_db = eliminate_redundant_dict_values(gene_protein_ft_db)
if analysis_method == 'ASPIRE' or analysis_method == 'linearregres':
clearObjectsFromMemory(exon_db);exon_db=[]
try: clearObjectsFromMemory(new_exon_db)
except Exception: null=[]
probeset_indirect_aligning_db=[]; probeset_aligning_db=[]
if report_type == 'perfect_match':
gene_protein_ft_db=[];domain_gene_count_db=[];protein_functional_attribute_db=[]
return probeset_aligning_db2
elif report_type == 'probeset':
probeset_aligning_db2=[]
return gene_protein_ft_db,domain_gene_count_db,protein_functional_attribute_db
else:
probeset_aligning_db2=[]; protein_functional_attribute_db=[]; probeset_aligning_db2=[]
len_gene_protein_ft_db = len(gene_protein_ft_db); gene_protein_ft_db=[]
return len_gene_protein_ft_db,domain_gene_count_db
def importProbesetProteinCompDomains(exon_db,report_type,comp_type):
filename = 'AltDatabase/'+species+'/'+array_type+'/probeset-domain-annotations-'+comp_type+'.txt'
if (array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type != 'null': filename=getFilteredFilename(filename)
filename=getExonVersionFilename(filename)
probeset_aligning_db = importGeneric(filename)
filename = 'AltDatabase/'+species+'/'+array_type+'/probeset-protein-annotations-'+comp_type+'.txt'
if (array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type != 'null': filename=getFilteredFilename(filename)
filename=getExonVersionFilename(filename)
gene_protein_ft_db={};domain_gene_count_db={}
for probeset in exon_db:
initial_proceed = 'no'; original_probeset = probeset
if probeset in probeset_aligning_db: initial_proceed = 'yes'
elif array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
### For junction analyses exon_db is really regulated_exon_junction_db, containing the inclusion,exclusion probeset tuple and an object as values
if '|' in probeset[0]: probeset1 = string.split(probeset[0],'|')[0]; probeset = probeset1,probeset[1]
try: alternate_probeset_id = exon_db[probeset].InclusionLookup(); probeset = alternate_probeset_id,probeset[1]
except Exception: null=[]
probeset_joined = string.join(probeset,'|')
#print [probeset_joined],[probeset]
if probeset_joined in probeset_aligning_db: initial_proceed = 'yes'; probeset = probeset_joined
elif probeset[0] in probeset_aligning_db: initial_proceed = 'yes'; probeset = probeset[0]
elif probeset[1] in probeset_aligning_db: initial_proceed = 'yes'; probeset = probeset[1]
#else: for i in probeset_aligning_db: print [i];kill
if initial_proceed == 'yes':
proceed = 'no'
if filter_for_AS == 'yes':
as_call = exon_db[original_probeset].SplicingCall()
if as_call == 1: proceed = 'yes'
else: proceed = 'yes'
new_domain_list = []
gene = exon_db[original_probeset].GeneID()
if report_type == 'gene' and proceed == 'yes':
for domain_data in probeset_aligning_db[probeset]:
try:
domain,call = string.split(domain_data,'|')
except Exception:
values = string.split(domain_data,'|')
domain = values[0]; call = values[-1] ### occurs when a | exists in the annotations from UniProt
try: domain_gene_count_db[domain].append(gene)
except KeyError: domain_gene_count_db[domain] = [gene]
try: gene_protein_ft_db[gene].append(domain)
except KeyError: gene_protein_ft_db[gene]=[domain]
elif proceed == 'yes':
for domain_data in probeset_aligning_db[probeset]:
try: domain,call = string.split(domain_data,'|')
except Exception:
values = string.split(domain_data,'|')
domain = values[0]; call = values[-1]
new_domain_list.append((domain,call))
#new_domain_list = string.join(new_domain_list,', ')
gene_protein_ft_db[gene,original_probeset] = new_domain_list
domain_gene_count_db = eliminate_redundant_dict_values(domain_gene_count_db)
probeset_aligning_db=[] ### Clear memory
probeset_aligning_protein_db = importGeneric(filename)
probeset_pairs={} ### Store all possible probeset pairs as single probesets for protein-protein associations
for probeset in exon_db:
if len(probeset)==2:
for p in probeset: probeset_pairs[p] = probeset
if report_type == 'probeset':
### Below code was re-written to be more memory efficient by not storing all data in probeset-domain-annotations-*comp*.txt via generic import
protein_functional_attribute_db={}; probeset_protein_associations={}; protein_db={}
for probeset in exon_db:
initial_proceed = 'no'; original_probeset = probeset
if probeset in probeset_aligning_protein_db: initial_proceed = 'yes'
elif array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
if '|' in probeset[0]: probeset1 = string.split(probeset[0],'|')[0]; probeset = probeset1,probeset[1]
try: alternate_probeset_id = exon_db[probeset].InclusionLookup(); probeset = alternate_probeset_id,probeset[1]
except Exception: null=[]
probeset_joined = string.join(probeset,'|')
#print [probeset_joined],[probeset]
if probeset_joined in probeset_aligning_protein_db: initial_proceed = 'yes'; probeset = probeset_joined
elif probeset[0] in probeset_aligning_protein_db: initial_proceed = 'yes'; probeset = probeset[0]
elif probeset[1] in probeset_aligning_protein_db: initial_proceed = 'yes'; probeset = probeset[1]
#else: for i in probeset_aligning_db: print [i];kill
if initial_proceed == 'yes':
protein_data_list=probeset_aligning_protein_db[probeset]
new_protein_list = []
gene = exon_db[original_probeset].GeneID()
for protein_data in protein_data_list:
protein_info,call = string.split(protein_data,'|')
if 'AA:' in protein_info:
protein_info_r = string.replace(protein_info,')','*')
protein_info_r = string.replace(protein_info_r,'(','*')
protein_info_r = string.split(protein_info_r,'*')
null_protein = protein_info_r[1]; hit_protein = protein_info_r[3]
probeset_protein_associations[original_probeset] = null_protein,hit_protein,call
protein_db[null_protein] = []; protein_db[hit_protein] = []
new_protein_list.append((protein_info,call))
#new_protein_list = string.join(new_domain_list,', ')
protein_functional_attribute_db[gene,original_probeset] = new_protein_list
filename = 'AltDatabase/'+species+'/'+array_type+'/SEQUENCE-protein-dbase_'+comp_type+'.txt'
filename=getExonVersionFilename(filename)
protein_seq_db = importGenericFiltered(filename,protein_db)
for key in protein_functional_attribute_db:
gene,probeset = key
try:
null_protein,hit_protein,call = probeset_protein_associations[probeset]
null_seq = protein_seq_db[null_protein][0]; hit_seq = protein_seq_db[hit_protein][0]
seq_attr = 'sequence: ' +'('+null_protein+')'+null_seq +' -> '+'('+hit_protein+')'+hit_seq
protein_functional_attribute_db[key].append((seq_attr,call))
except KeyError: null=[]
protein_seq_db=[]; probeset_aligning_protein_db=[]
return gene_protein_ft_db,domain_gene_count_db,protein_functional_attribute_db
else:
probeset_aligning_protein_db=[]; len_gene_protein_ft_db = len(gene_protein_ft_db); gene_protein_ft_db=[]
return len_gene_protein_ft_db,domain_gene_count_db
class SimpleJunctionData:
def __init__(self, geneid, probeset1, probeset2, probeset1_display, critical_exon_list):
self._geneid = geneid; self._probeset1 = probeset1; self._probeset2 = probeset2
self._probeset1_display = probeset1_display; self._critical_exon_list = critical_exon_list
def GeneID(self): return self._geneid
def Probeset1(self): return self._probeset1
def Probeset2(self): return self._probeset2
def InclusionDisplay(self): return self._probeset1_display
def CriticalExons(self): return self._critical_exon_list
def setSplicingCall(self,splicing_call):
#self._splicing_call = EvidenceOfAltSplicing(slicing_annot)
self._splicing_call = splicing_call
def setSymbol(self,symbol): self.symbol = symbol
def Symbol(self): return self.symbol
def SplicingCall(self): return self._splicing_call
def setInclusionLookup(self,incl_junction_probeset): self.incl_junction_probeset = incl_junction_probeset
def InclusionLookup(self): return self.incl_junction_probeset
def formatJunctionData(probesets,affygene,critical_exon_list):
if '|' in probesets[0]: ### Only return the first inclusion probeset (agglomerated probesets)
incl_list = string.split(probesets[0],'|')
incl_probeset = incl_list[0]; excl_probeset = probesets[1]
else: incl_probeset = probesets[0]; excl_probeset = probesets[1]
jd = SimpleJunctionData(affygene,incl_probeset,excl_probeset,probesets[0],critical_exon_list)
key = incl_probeset,excl_probeset
return key,jd
class JunctionExpressionData:
def __init__(self, baseline_norm_exp, exper_norm_exp, pval, ped):
self.baseline_norm_exp = baseline_norm_exp; self.exper_norm_exp = exper_norm_exp; self.pval = pval; self.ped = ped
def ConNI(self):
ls=[]
for i in self.logConNI():
ls.append(math.pow(2,i))
return ls
def ExpNI(self):
ls=[]
for i in self.logExpNI():
ls.append(math.pow(2,i))
return ls
def ConNIAvg(self): return math.pow(2,statistics.avg(self.logConNI()))
def ExpNIAvg(self): return math.pow(2,statistics.avg(self.logExpNI()))
def logConNI(self): return self.baseline_norm_exp
def logExpNI(self): return self.exper_norm_exp
def Pval(self): return self.pval
def ProbesetExprData(self): return self.ped
def __repr__(self): return self.ConNI()+'|'+self.ExpNI()
def calculateAllASPIREScores(p1,p2):
b1o = p1.ConNIAvg(); b2o = p2.ConNIAvg()
e1o = p1.ExpNIAvg(); e2o = p2.ExpNIAvg(); original_score = statistics.aspire_stringent(b1o,e1o,b2o,e2o)
index=0; baseline_scores=[] ### Loop through each control ratio and compare to control ratio mean
for b1 in p1.ConNI():
b2 = p2.ConNI()[index]
score = statistics.aspire_stringent(b2,e2o,b1,e1o); index+=1
baseline_scores.append(score)
index=0; exp_scores=[] ### Loop through each experimental ratio and compare to control ratio mean
for e1 in p1.ExpNI():
e2 = p2.ExpNI()[index]
score = statistics.aspire_stringent(b1o,e1,b2o,e2); index+=1
exp_scores.append(score)
try:
aspireP = statistics.runComparisonStatistic(baseline_scores,exp_scores,probability_statistic)
except Exception: aspireP = 'NA' ### Occurs when analyzing two groups with no variance
if aspireP == 1: aspireP = 'NA'
"""
if aspireP<0.05 and oscore>0.2 and statistics.avg(exp_scores)<0:
index=0
for e1 in p1.ExpNI():
e2 = p2.ExpNI()[index]
score = statistics.aspire_stringent(b1,e1,b2,e2)
print p1.ExpNI(), p2.ExpNI(); print e1, e2
print e1o,e2o; print b1, b2; print score, original_score
print exp_scores, statistics.avg(exp_scores); kill"""
return baseline_scores, exp_scores, aspireP
def stringListConvert(ls):
ls2=[]
for i in ls: ls2.append(str(i))
return ls2
def analyzeJunctionSplicing(nonlog_NI_db):
group_sizes = []; original_array_indices = permute_lists[0] ###p[0] is the original organization of the group samples prior to permutation
for group in original_array_indices: group_sizes.append(len(group))
### Used to restrict the analysis to a pre-selected set of probesets (e.g. those that have a specifc splicing pattern)
if len(filtered_probeset_db)>0:
temp_db={}
for probeset in nonlog_NI_db: temp_db[probeset]=[]
for probeset in temp_db:
try: filtered_probeset_db[probeset]
except KeyError: del nonlog_NI_db[probeset]
### Used to the export relative individual adjusted probesets fold changes used for splicing index values
if export_NI_values == 'yes':
global NIdata_export
summary_output = root_dir+'AltResults/RawSpliceData/'+species+'/'+analysis_method+'/'+dataset_name[:-1]+'.txt'
NIdata_export = export.ExportFile(summary_output)
title = string.join(['inclusion-probeset','exclusion-probeset']+original_array_names,'\t')+'\n'; NIdata_export.write(title)
### Calculate a probeset p-value adjusted for constitutive expression levels (taken from splicing index method)
xl=0
probeset_normIntensity_db={}
for probeset in array_raw_group_values:
ed = exon_db[probeset]; geneid = ed.GeneID(); xl+=1
#if geneid in alt_junction_db and geneid in original_avg_const_exp_db: ### Don't want this filter since it causes problems for Trans-splicing
group_index = 0; si_interim_group_db={}; ge_threshold_count=0; value_count = 0
### Prepare normalized expression lists for recipricol-junction algorithms
if geneid in avg_const_exp_db:
for group_values in array_raw_group_values[probeset]:
value_index = 0; ratio_hash=[]
for value in group_values: ###Calculate normalized ratio's for each condition and save raw values for later permutation
exp_val = value;ge_val = avg_const_exp_db[geneid][value_count]; exp_ratio = exp_val-ge_val
ratio_hash.append(exp_ratio); value_index +=1; value_count +=1
si_interim_group_db[group_index] = ratio_hash
group_index+=1
group1_ratios = si_interim_group_db[0]; group2_ratios = si_interim_group_db[1]
### Calculate and store simple expression summary stats
data_list1 = array_raw_group_values[probeset][0]; data_list2 = array_raw_group_values[probeset][1]
baseline_exp = statistics.avg(data_list1); experimental_exp = statistics.avg(data_list2); fold_change = experimental_exp - baseline_exp
#group_name1 = array_group_list[0]; group_name2 = array_group_list[1]
try:
ttest_exp_p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic)
except Exception: ttest_exp_p = 'NA'
if ttest_exp_p == 1: ttest_exp_p = 'NA'
adj_fold = statistics.avg(group2_ratios) - statistics.avg(group1_ratios)
ped = ProbesetExpressionData(baseline_exp, experimental_exp, fold_change, adj_fold, ttest_exp_p, '')
try:
try:
normIntensityP = statistics.runComparisonStatistic(group1_ratios,group2_ratios,probability_statistic)
except Exception:
#print group1_ratios,group2_ratios,array_raw_group_values[probeset],avg_const_exp_db[geneid];kill
normIntensityP = 'NA' ###occurs for constitutive probesets
except Exception: normIntensityP = 0
if normIntensityP == 1: normIntensityP = 'NA'
ji = JunctionExpressionData(group1_ratios, group2_ratios, normIntensityP, ped)
probeset_normIntensity_db[probeset]=ji ### store and access this below
#if probeset == 'G6899622@J916374@j_at': print normIntensityP,group1_ratios,group2_ratios;kill
###Concatenate the two raw expression groups into a single list for permutation analysis
ls_concatenated = []
for group in array_raw_group_values[probeset]:
for entry in group: ls_concatenated.append(entry)
if analysis_method == 'linearregres': ###Convert out of log space
ls_concatenated = statistics.log_fold_conversion_fraction(ls_concatenated)
array_raw_group_values[probeset] = ls_concatenated
s = 0; t = 0; y = ''; denominator_events=0; excluded_probeset_db = {}
splice_event_list=[]; splice_event_list_mx=[]; splice_event_list_non_mx=[]; event_mx_temp = []; permute_p_values={} #use this to exclude duplicate mx events
for affygene in alt_junction_db:
if affygene in original_avg_const_exp_db:
constit_exp1 = original_avg_const_exp_db[affygene][0]
constit_exp2 = original_avg_const_exp_db[affygene][1]
ge_fold=constit_exp2-constit_exp1
for event in alt_junction_db[affygene]:
if array_type == 'AltMouse':
#event = [('ei', 'E16-E17'), ('ex', 'E16-E18')]
#critical_exon_db[affygene,tuple(critical_exons)] = [1,'E'+str(e1a),'E'+str(e2b)] --- affygene,tuple(event) == key, 1 indicates both are either up or down together
event_call = event[0][0] + '-' + event[1][0]
exon_set1 = event[0][1]; exon_set2 = event[1][1]
probeset1 = exon_dbase[affygene,exon_set1]
probeset2 = exon_dbase[affygene,exon_set2]
critical_exon_list = critical_exon_db[affygene,tuple(event)]
if array_type == 'junction' or array_type == 'RNASeq':
event_call = 'ei-ex' ### Below objects from JunctionArrayEnsemblRules - class JunctionInformation
probeset1 = event.InclusionProbeset(); probeset2 = event.ExclusionProbeset()
exon_set1 = event.InclusionJunction(); exon_set2 = event.ExclusionJunction()
try: novel_event = event.NovelEvent()
except Exception: novel_event = 'known'
critical_exon_list = [1,event.CriticalExonSets()]
key,jd = formatJunctionData([probeset1,probeset2],affygene,critical_exon_list[1])
if array_type == 'junction' or array_type == 'RNASeq':
try: jd.setSymbol(annotate_db[affygene].Symbol())
except Exception:null=[]
#if '|' in probeset1: print probeset1, key,jd.InclusionDisplay();kill
probeset_comp_db[key] = jd ### This is used for the permutation analysis and domain/mirBS import
#print probeset1,probeset2, critical_exon_list,event_call,exon_set1,exon_set2;kill
if probeset1 in nonlog_NI_db and probeset2 in nonlog_NI_db:
denominator_events+=1
try: p1 = probeset_normIntensity_db[probeset1]; p2 = probeset_normIntensity_db[probeset2]
except Exception:
print probeset1, probeset2
p1 = probeset_normIntensity_db[probeset1]
p2 = probeset_normIntensity_db[probeset2]
#if '|' in probeset1: print
pp1 = p1.Pval(); pp2 = p2.Pval()
baseline_ratio1 = p1.ConNIAvg()
experimental_ratio1 = p1.ExpNIAvg()
baseline_ratio2 = p2.ConNIAvg()
experimental_ratio2 = p2.ExpNIAvg()
ped1 = p1.ProbesetExprData()
ped2 = p2.ProbesetExprData()
Rin = ''; Rex = ''
r = 0 ###Variable used to determine if we should take the absolute value of dI for mutually exlcusive events
if event_call == 'ei-ex': #means probeset1 is an exon inclusion and probeset2 is an exon exclusion
Rin = baseline_ratio1/experimental_ratio1 # Rin=A/C
Rex = baseline_ratio2/experimental_ratio2 # Rin=B/D
I1=baseline_ratio1/(baseline_ratio1+baseline_ratio2)
I2=experimental_ratio1/(experimental_ratio1+experimental_ratio2)
###When Rex is larger, the exp_ratio for exclusion is decreased in comparison to baseline.
###Thus, increased inclusion (when Rin is small, inclusion is big)
if (Rin>1 and Rex<1): y = 'downregulated'
elif (Rin<1 and Rex>1): y = 'upregulated'
elif (Rex<Rin): y = 'downregulated'
else: y = 'upregulated'
temp_list = []
if event_call == 'mx-mx':
temp_list.append(exon_set1); temp_list.append(exon_set2);temp_list.sort()
if (affygene,temp_list) not in event_mx_temp: #use this logic to prevent mx entries being added more than once
event_mx_temp.append((affygene,temp_list))
###Arbitrarily choose which exon-set will be Rin or Rex, does matter for mutually exclusive events
Rin = baseline_ratio1/experimental_ratio1 # Rin=A/C
Rex = baseline_ratio2/experimental_ratio2 # Rin=B/D
I1=baseline_ratio1/(baseline_ratio1+baseline_ratio2)
I2=experimental_ratio1/(experimental_ratio1+experimental_ratio2)
y = 'mutually-exclusive'; r = 1
if analysis_method == 'ASPIRE' and Rex != '':
#if affygene == 'ENSMUSG00000000126': print Rin, Rex, probeset1, probeset2
if (Rin>1 and Rex<1) or (Rin<1 and Rex>1):
s +=1
in1=((Rex-1.0)*Rin)/(Rex-Rin); in2=(Rex-1.0)/(Rex-Rin)
dI = ((in2-in1)+(I2-I1))/2.0 #modified to give propper exon inclusion
dI = dI*(-1) ### Reverse the fold to make equivalent to splicing-index and FIRMA scores
try: baseline_scores, exp_scores, aspireP = calculateAllASPIREScores(p1,p2)
except Exception: baseline_scores = [0]; exp_scores=[dI]; aspireP = 0
if export_NI_values == 'yes':
baseline_scores = stringListConvert(baseline_scores); exp_scores = stringListConvert(exp_scores)
ev = string.join([probeset1,probeset2]+baseline_scores+exp_scores,'\t')+'\n'; NIdata_export.write(ev)
if max_replicates >2 or equal_replicates==2:
permute_p_values[(probeset1,probeset2)] = [aspireP, 'NA', 'NA', 'NA']
if r == 1: dI = abs(dI) ###Occurs when event is mutually exclusive
#if abs(dI)>alt_exon_logfold_cutoff: print [dI],pp1,pp2,aspireP;kill
#print [affygene,dI,pp1,pp2,aspireP,event.CriticalExonSets(),probeset1,probeset2,alt_exon_logfold_cutoff,p_threshold]
if ((pp1<p_threshold or pp2<p_threshold) or pp1==1 or pp1=='NA') and abs(dI) > alt_exon_logfold_cutoff: ###Require that the splice event have a constitutive corrected p less than the user defined threshold
ejd = ExonJunctionData(dI,probeset1,probeset2,pp1,pp2,y,event_call,critical_exon_list,affygene,ped1,ped2)
"""if probeset1 == 'ENSMUSG00000033335:E16.1-E17.1' and probeset2 == 'ENSMUSG00000033335:E16.1-E19.1':
print [dI,pp1,pp2,p_threshold,alt_exon_logfold_cutoff]
print baseline_scores, exp_scores, [aspireP]#;sys.exit()"""
ejd.setConstitutiveExpression(constit_exp1); ejd.setConstitutiveFold(ge_fold)
if perform_permutation_analysis == 'yes': splice_event_list.append((dI,ejd))
elif aspireP < permute_p_threshold or aspireP=='NA': splice_event_list.append((dI,ejd))
#if abs(dI)>.2: print probeset1, probeset2, critical_exon_list, [exon_set1], [exon_set2]
#if dI>.2 and aspireP<0.05: print baseline_scores,exp_scores,aspireP, statistics.avg(exp_scores), dI
elif array_type == 'junction' or array_type == 'RNASeq':
excluded_probeset_db[affygene+':'+event.CriticalExonSets()[0]] = probeset1, affygene, dI, 'NA', aspireP
if array_type == 'RNASeq':
try: ejd.setNovelEvent(novel_event)
except Exception: None
if analysis_method == 'linearregres' and Rex != '':
s+=1
log_fold,linregressP,rsqrd_status = getLinearRegressionScores(probeset1,probeset2,group_sizes)
log_fold = log_fold ### Reverse the fold to make equivalent to splicing-index and FIRMA scores
if max_replicates >2 or equal_replicates==2: permute_p_values[(probeset1,probeset2)] = [linregressP, 'NA', 'NA', 'NA']
if rsqrd_status == 'proceed':
if ((pp1<p_threshold or pp2<p_threshold) or pp1==1 or pp1=='NA') and abs(log_fold) > alt_exon_logfold_cutoff: ###Require that the splice event have a constitutive corrected p less than the user defined threshold
ejd = ExonJunctionData(log_fold,probeset1,probeset2,pp1,pp2,y,event_call,critical_exon_list,affygene,ped1,ped2)
ejd.setConstitutiveExpression(constit_exp1); ejd.setConstitutiveFold(ge_fold)
if perform_permutation_analysis == 'yes': splice_event_list.append((log_fold,ejd))
elif linregressP < permute_p_threshold: splice_event_list.append((log_fold,ejd))
#if probeset1 == 'G6990053@762121_762232_at' and probeset2 == 'G6990053@J926254@j_at':
#print event_call, critical_exon_list,affygene, Rin, Rex, y, temp_list;kill
elif array_type == 'junction' or array_type == 'RNASeq':
excluded_probeset_db[affygene+':'+event.CriticalExonSets()[0]] = probeset1, affygene, log_fold, 'NA', linregressP
if array_type == 'RNASeq':
try: ejd.setNovelEvent(novel_event)
except Exception: None
else: t +=1
clearObjectsFromMemory(probeset_normIntensity_db)
probeset_normIntensity_db={}; ### Potentially large memory object containing summary stats for all probesets
statistics.adjustPermuteStats(permute_p_values)
summary_data_db['denominator_exp_events']=denominator_events
print "Number of exon-events analyzed:", s
print "Number of exon-events excluded:", t
return splice_event_list, probeset_comp_db, permute_p_values, excluded_probeset_db
def maxReplicates():
replicates=0; greater_than_two=0; greater_than_one=0; group_sizes=[]
for probeset in array_raw_group_values:
for group_values in array_raw_group_values[probeset]:
try:
replicates+=len(group_values); group_sizes.append(len(group_values))
if len(group_values)>2: greater_than_two+=1
elif len(group_values)>1: greater_than_one+=1
except Exception: replicates+=len(array_raw_group_values[probeset]); break
break
group_sizes = unique.unique(group_sizes)
if len(group_sizes) == 1: equal_replicates = group_sizes[0]
else: equal_replicates = 0
max_replicates = replicates/float(original_conditions)
if max_replicates<2.01:
if greater_than_two>0 and greater_than_one>0: max_replicates=3
return max_replicates, equal_replicates
def furtherProcessJunctionScores(splice_event_list, probeset_comp_db, permute_p_values):
splice_event_list.sort(); splice_event_list.reverse()
print "filtered %s scores:" % analysis_method, len(splice_event_list)
if perform_permutation_analysis == 'yes':
###*********BEGIN PERMUTATION ANALYSIS*********
if max_replicates >2 or equal_replicates==2:
splice_event_list, p_value_call, permute_p_values = permuteSplicingScores(splice_event_list)
else:
print "WARNING...Not enough replicates to perform permutation analysis."
p_value_call=''; permute_p_values = {}
else:
if max_replicates >2 or equal_replicates==2:
if probability_statistic == 'unpaired t-test':
p_value_call=analysis_method+'-OneWayAnova'
else:
p_value_call=analysis_method+'-'+probability_statistic
else:
if probability_statistic == 'unpaired t-test':
p_value_call='OneWayAnova'; permute_p_values = {}
else:
p_value_call=probability_statistic; permute_p_values = {}
print len(splice_event_list), 'alternative events after subsequent filtering (optional)'
### Get ExonJunction annotaitons
junction_splicing_annot_db = getJunctionSplicingAnnotations(probeset_comp_db)
regulated_exon_junction_db={}; new_splice_event_list=[]
if filter_for_AS == 'yes': print "Filtering for evidence of Alternative Splicing"
for (fold,ejd) in splice_event_list:
proceed = 'no'
if filter_for_AS == 'yes':
try:
ja = junction_splicing_annot_db[ejd.Probeset1(),ejd.Probeset2()]; splicing_call = ja.SplicingCall()
if splicing_call == 1: proceed = 'yes'
except KeyError: proceed = 'no'
else: proceed = 'yes'
if proceed == 'yes':
key,jd = formatJunctionData([ejd.Probeset1(),ejd.Probeset2()],ejd.GeneID(),ejd.CriticalExons())
regulated_exon_junction_db[key] = jd ### This is used for the permutation analysis and domain/mirBS import
new_splice_event_list.append((fold,ejd))
### Add junction probeset lookup for reciprocal junctions composed of an exonid (not in protein database currently)
if array_type == 'RNASeq' and '-' not in key[0]: ### Thus, it is an exon compared to a junction
events = alt_junction_db[ejd.GeneID()]
for ji in events:
if (ji.InclusionProbeset(),ji.ExclusionProbeset()) == key:
jd.setInclusionLookup(ji.InclusionLookup()) ### This is the source junction from which the exon ID comes from
probeset_comp_db[ji.InclusionLookup(),ji.ExclusionProbeset()]=jd
#print ji.InclusionProbeset(),ji.ExclusionProbeset(),' ',ji.InclusionLookup()
if filter_for_AS == 'yes': print len(new_splice_event_list), "remaining after filtering for evidence of Alternative splicing"
filtered_exon_db = {}
for junctions in probeset_comp_db:
rj = probeset_comp_db[junctions] ### Add splicing annotations to the AltMouse junction DBs (needed for permutation analysis statistics and filtering)
try: ja = junction_splicing_annot_db[junctions]; splicing_call = ja.SplicingCall(); rj.setSplicingCall(ja.SplicingCall())
except KeyError: rj.setSplicingCall(0)
if filter_for_AS == 'yes': filtered_exon_db[junctions] = rj
for junctions in regulated_exon_junction_db:
rj = regulated_exon_junction_db[junctions]
try: ja = junction_splicing_annot_db[junctions]; rj.setSplicingCall(ja.SplicingCall())
except KeyError: rj.setSplicingCall(0)
if filter_for_AS == 'yes': probeset_comp_db = filtered_exon_db
try: clearObjectsFromMemory(alt_junction_db)
except Exception: null=[]
return new_splice_event_list, p_value_call, permute_p_values, probeset_comp_db, regulated_exon_junction_db
class SplicingScoreData:
def Method(self):
###e.g. ASPIRE
return self._method
def Score(self): return str(self._score)
def Probeset1(self): return self._probeset1
def Probeset2(self): return self._probeset2
def RegulationCall(self): return self._regulation_call
def GeneID(self): return self._geneid
def CriticalExons(self): return self._critical_exon_list[1]
def CriticalExonTuple(self): return self._critical_exon_list
def TTestNormalizedRatios(self): return self._normIntensityP
def TTestNormalizedRatios2(self): return self._normIntensityP2
def setConstitutiveFold(self,exp_log_ratio): self._exp_log_ratio = exp_log_ratio
def ConstitutiveFold(self): return str(self._exp_log_ratio)
def setConstitutiveExpression(self,const_baseline): self.const_baseline = const_baseline
def ConstitutiveExpression(self): return str(self.const_baseline)
def setProbesetExpressionData(self,ped): self.ped1 = ped
def ProbesetExprData1(self): return self.ped1
def ProbesetExprData2(self): return self.ped2
def setNovelEvent(self,novel_event): self._novel_event = novel_event
def NovelEvent(self): return self._novel_event
def EventCall(self):
###e.g. Exon inclusion (ei) Exon exclusion (ex), ei-ex, reported in that direction
return self._event_call
def Report(self):
output = self.Method() +'|'+ self.GeneID() +'|'+ string.join(self.CriticalExons(),'|')
return output
def __repr__(self): return self.Report()
class ExonJunctionData(SplicingScoreData):
def __init__(self,score,probeset1,probeset2,probeset1_p,probeset2_p,regulation_call,event_call,critical_exon_list,affygene,ped1,ped2):
self._score = score; self._probeset1 = probeset1; self._probeset2 = probeset2; self._regulation_call = regulation_call
self._event_call = event_call; self._critical_exon_list = critical_exon_list; self._geneid = affygene
self._method = analysis_method; self._normIntensityP = probeset1_p; self._normIntensityP2 = probeset2_p
self.ped1 = ped1; self.ped2=ped2
class ExonData(SplicingScoreData):
def __init__(self,splicing_index,probeset,critical_exon_list,geneid,group1_ratios,group2_ratios,normIntensityP,opposite_SI_log_mean):
self._score = splicing_index; self._probeset1 = probeset; self._opposite_SI_log_mean = opposite_SI_log_mean
self._critical_exon_list = critical_exon_list; self._geneid = geneid
self._baseline_ratio1 = group1_ratios; self._experimental_ratio1 = group2_ratios
self._normIntensityP = normIntensityP
self._method = analysis_method; self._event_call = 'exon-inclusion'
if splicing_index > 0: regulation_call = 'downregulated' ###Since baseline is the numerator ratio
else: regulation_call = 'upregulated'
self._regulation_call = regulation_call
def OppositeSIRatios(self): return self._opposite_SI_log_mean
class ExcludedExonData(ExonData):
def __init__(self,splicing_index,geneid,normIntensityP):
self._score = splicing_index; self._geneid = geneid; self._normIntensityP = normIntensityP
def getAllPossibleLinearRegressionScores(probeset1,probeset2,positions,group_sizes):
### Get Raw expression values for the two probests
p1_exp = array_raw_group_values[probeset1]
p2_exp = array_raw_group_values[probeset2]
all_possible_scores=[]; index1=0 ### Perform all possible pairwise comparisons between groups (not sure how this will work for 10+ groups)
for (pos1a,pos2a) in positions:
index2=0
for (pos1b,pos2b) in positions:
if pos1a != pos1b:
p1_g1 = p1_exp[pos1a:pos2a]; p1_g2 = p1_exp[pos1b:pos2b]
p2_g1 = p2_exp[pos1a:pos2a]; p2_g2 = p2_exp[pos1b:pos2b]
#log_fold, linregressP, rsqrd = getAllLinearRegressionScores(probeset1,probeset2,p1_g1,p2_g1,p1_g2,p2_g2,len(group_sizes)) ### Used to calculate a pairwise group pvalue
log_fold, rsqrd = performLinearRegression(p1_g1,p2_g1,p1_g2,p2_g2)
if log_fold<0: i1,i2 = index2,index1 ### all scores should indicate upregulation
else: i1,i2=index1,index2
all_possible_scores.append((abs(log_fold),i1,i2))
index2+=1
index1+=1
all_possible_scores.sort()
try: log_fold,index1,index2 = all_possible_scores[-1]
except Exception: log_fold=0; index1=0; index2=0
return log_fold, index1, index2
def getLinearRegressionScores(probeset1,probeset2,group_sizes):
### Get Raw expression values for the two probests
p1_exp = array_raw_group_values[probeset1]
p2_exp = array_raw_group_values[probeset2]
try:
p1_g1 = p1_exp[:group_sizes[0]]; p1_g2 = p1_exp[group_sizes[0]:]
p2_g1 = p2_exp[:group_sizes[0]]; p2_g2 = p2_exp[group_sizes[0]:]
except Exception:
print probeset1,probeset2
print p1_exp
print p2_exp
print group_sizes
force_kill
log_fold, linregressP, rsqrd = getAllLinearRegressionScores(probeset1,probeset2,p1_g1,p2_g1,p1_g2,p2_g2,2)
return log_fold, linregressP, rsqrd
def getAllLinearRegressionScores(probeset1,probeset2,p1_g1,p2_g1,p1_g2,p2_g2,groups):
log_fold, rsqrd = performLinearRegression(p1_g1,p2_g1,p1_g2,p2_g2)
try:
### Repeat for each sample versus baselines to calculate a p-value
index=0; group1_scores=[]
for p1_g1_sample in p1_g1:
p2_g1_sample = p2_g1[index]
log_f, rs = performLinearRegression(p1_g1,p2_g1,[p1_g1_sample],[p2_g1_sample])
group1_scores.append(log_f); index+=1
index=0; group2_scores=[]
for p1_g2_sample in p1_g2:
p2_g2_sample = p2_g2[index]
log_f, rs = performLinearRegression(p1_g1,p2_g1,[p1_g2_sample],[p2_g2_sample])
group2_scores.append(log_f); index+=1
try:
linregressP = statistics.runComparisonStatistic(group1_scores,group2_scores,probability_statistic)
except Exception:
linregressP = 0; group1_scores = [0]; group2_scores = [log_fold]
if linregressP == 1: linregressP = 0
except Exception:
linregressP = 0; group1_scores = [0]; group2_scores = [log_fold]
if export_NI_values == 'yes' and groups==2:
group1_scores = stringListConvert(group1_scores)
group2_scores = stringListConvert(group2_scores)
ev = string.join([probeset1,probeset2]+group1_scores+group2_scores,'\t')+'\n'; NIdata_export.write(ev)
return log_fold, linregressP, rsqrd
def performLinearRegression(p1_g1,p2_g1,p1_g2,p2_g2):
return_rsqrd = 'no'
if use_R == 'yes': ###Uses the RLM algorithm
#print "Performing Linear Regression analysis using rlm."
g1_slope = statistics.LinearRegression(p1_g1,p2_g1,return_rsqrd)
g2_slope = statistics.LinearRegression(p1_g2,p2_g2,return_rsqrd)
else: ###Uses a basic least squared method
#print "Performing Linear Regression analysis using python specific methods."
g1_slope = statistics.simpleLinRegress(p1_g1,p2_g1)
g2_slope = statistics.simpleLinRegress(p1_g2,p2_g2)
log_fold = statistics.convert_to_log_fold(g2_slope/g1_slope)
rsqrd = 'proceed'
#if g1_rsqrd > 0 and g2_rsqrd > 0: rsqrd = 'proceed'
#else: rsqrd = 'hault'
return log_fold, rsqrd
########### Permutation Analysis Functions ###########
def permuteLinearRegression(probeset1,probeset2,p):
p1_exp = array_raw_group_values[probeset1]
p2_exp = array_raw_group_values[probeset2]
p1_g1, p1_g2 = permute_samples(p1_exp,p)
p2_g1, p2_g2 = permute_samples(p2_exp,p)
return_rsqrd = 'no'
if use_R == 'yes': ###Uses the RLM algorithm
g1_slope = statistics.LinearRegression(p1_g1,p2_g1,return_rsqrd)
g2_slope = statistics.LinearRegression(p1_g2,p2_g2,return_rsqrd)
else: ###Uses a basic least squared method
g1_slope = statistics.simpleLinRegress(p1_g1,p2_g1)
g2_slope = statistics.simpleLinRegress(p1_g2,p2_g2)
log_fold = statistics.convert_to_log_fold(g2_slope/g1_slope)
return log_fold
def permuteSplicingScores(splice_event_list):
p_value_call = 'lowest_raw_p'
permute_p_values = {}; splice_event_list2=[]
if len(permute_lists) > 0:
#tuple_data in splice_event_list = dI,probeset1,probeset2,y,event_call,critical_exon_list
all_samples = []; a = 0
for (score,x) in splice_event_list:
###NOTE: This reference dI differs slightly from the below calculated, since the values are calculated from raw relative ratios rather than the avg
###Solution: Use the first calculated dI as the reference
score = score*(-1) ### Reverse the score to make equivalent to splicing-index and FIRMA scores
ref_splice_val = score; probeset1 = x.Probeset1(); probeset2 = x.Probeset2(); affygene = x.GeneID()
y = 0; p_splice_val_dist = []; count = 0; return_rsqrd = 'no'
for p in permute_lists: ###There are two lists in each entry
count += 1
permute = 'yes'
if analysis_method == 'ASPIRE':
p_splice_val = permute_ASPIRE_filtered(affygene, probeset1,probeset2,p,y,ref_splice_val,x)
elif analysis_method == 'linearregres':
slope_ratio = permuteLinearRegression(probeset1,probeset2,p)
p_splice_val = slope_ratio
if p_splice_val != 'null': p_splice_val_dist.append(p_splice_val)
y+=1
p_splice_val_dist.sort()
new_ref_splice_val = str(abs(ref_splice_val)); new_ref_splice_val = float(new_ref_splice_val[0:8]) #otherwise won't match up the scores correctly
if analysis_method == 'linearregres':
if ref_splice_val<0:
p_splice_val_dist2=[]
for val in p_splice_val_dist: p_splice_val_dist2.append(-1*val)
p_splice_val_dist=p_splice_val_dist2; p_splice_val_dist.reverse()
p_val, pos_permute, total_permute, greater_than_true_permute = statistics.permute_p(p_splice_val_dist,new_ref_splice_val,len(permute_lists))
#print p_val,ref_splice_val, pos_permute, total_permute, greater_than_true_permute,p_splice_val_dist[-3:];kill
###When two groups are of equal size, there will be 2 pos_permutes rather than 1
if len(permute_lists[0][0]) == len(permute_lists[0][1]): greater_than_true_permute = (pos_permute/2) - 1 #size of the two groups are equal
else:greater_than_true_permute = (pos_permute) - 1
if analysis_method == 'linearregres': greater_than_true_permute = (pos_permute) - 1 ###since this is a one sided test, unlike ASPIRE
###Below equation is fine if the population is large
permute_p_values[(probeset1,probeset2)] = [p_val, pos_permute, total_permute, greater_than_true_permute]
###Remove non-significant linear regression results
if analysis_method == 'linearregres':
if p_val <= permute_p_threshold or greater_than_true_permute < 2: splice_event_list2.append((score,x)) ###<= since many p=0.05
print "Number of permutation p filtered splice event:",len(splice_event_list2)
if len(permute_p_values)>0: p_value_call = 'permuted_aspire_p-value'
if analysis_method == 'linearregres': splice_event_list = splice_event_list2
return splice_event_list, p_value_call, permute_p_values
def permute_ASPIRE_filtered(affygene,probeset1,probeset2,p,y,ref_splice_val,x):
### Get raw expression values for each permuted group for the two probesets
b1,e1 = permute_dI(array_raw_group_values[probeset1],p)
try: b2,e2 = permute_dI(array_raw_group_values[probeset2],p)
except IndexError: print probeset2, array_raw_group_values[probeset2],p; kill
### Get the average constitutive expression values (averaged per-sample across probesets) for each permuted group
try: bc,ec = permute_dI(avg_const_exp_db[affygene],p)
except IndexError: print affygene, avg_const_exp_db[affygene],p; kill
if factor_out_expression_changes == 'no':
ec = bc
### Analyze the averaged ratio's of junction expression relative to permuted constitutive expression
try: p_splice_val = abs(statistics.aspire_stringent(b1/bc,e1/ec,b2/bc,e2/ec)) ### This the permuted ASPIRE score
except Exception: p_splice_val = 0
#print p_splice_val, ref_splice_val, probeset1, probeset2, affygene; dog
if y == 0: ###The first permutation is always the real one
### Grab the absolute number with small number of decimal places
try:
new_ref_splice_val = str(p_splice_val); new_ref_splice_val = float(new_ref_splice_val[0:8])
ref_splice_val = str(abs(ref_splice_val)); ref_splice_val = float(ref_splice_val[0:8]); y += 1
except ValueError:
###Only get this error if your ref_splice_val is a null
print y, probeset1, probeset2; print ref_splice_val, new_ref_splice_val, p
print b1/bc,e1/ec,b2/bc,e2/ec; print (b1/bc)/(e1/ec), (b2/bc)/(e2/ec)
print x[7],x[8],x[9],x[10]; kill
return p_splice_val
def permute_samples(a,p):
baseline = []; experimental = []
for p_index in p[0]:
baseline.append(a[p_index]) ###Append expression values for each permuted list
for p_index in p[1]:
experimental.append(a[p_index])
return baseline, experimental
def permute_dI(all_samples,p):
baseline, experimental = permute_samples(all_samples,p)
#if get_non_log_avg == 'no':
gb = statistics.avg(baseline); ge = statistics.avg(experimental) ###Group avg baseline, group avg experimental value
gb = statistics.log_fold_conversion_fraction(gb); ge = statistics.log_fold_conversion_fraction(ge)
#else:
#baseline = statistics.log_fold_conversion_fraction(baseline); experimental = statistics.log_fold_conversion_fraction(experimental)
#gb = statistics.avg(baseline); ge = statistics.avg(experimental) ###Group avg baseline, group avg experimental value
return gb,ge
def format_exon_functional_attributes(affygene,critical_probeset_list,functional_attribute_db,up_exon_list,down_exon_list,protein_length_list):
### Add functional attributes
functional_attribute_list2=[]
new_functional_attribute_str=''
new_seq_attribute_str=''
new_functional_attribute_list=[]
if array_type == 'exon' or array_type == 'gene' or explicit_data_type != 'null': critical_probesets = critical_probeset_list[0]
else: critical_probesets = tuple(critical_probeset_list)
key = affygene,critical_probesets
if key in functional_attribute_db:
###Grab exon IDs corresponding to the critical probesets
if analysis_method == 'ASPIRE' or 'linearregres' in analysis_method:
try: critical_exons = regulated_exon_junction_db[critical_probesets].CriticalExons() ###For junction arrays
except Exception: print key, functional_attribute_db[key];kill
else: critical_exons = [exon_db[critical_probesets].ExonID()] ###For exon arrays
for exon in critical_exons:
for entry in functional_attribute_db[key]:
x = 0
functional_attribute = entry[0]
call = entry[1] # +, -, or ~
if ('AA:' in functional_attribute) or ('ref' in functional_attribute):
x = 1
if exon in up_exon_list:
### design logic to determine whether up or down regulation promotes the functional change (e.g. NMD)
if 'ref' in functional_attribute:
new_functional_attribute = '(~)'+functional_attribute
data_tuple = new_functional_attribute,exon
elif call == '+' or call == '~':
new_functional_attribute = '(+)'+functional_attribute
data_tuple = new_functional_attribute,exon
elif call == '-':
new_functional_attribute = '(-)'+functional_attribute
data_tuple = new_functional_attribute,exon
if 'AA:' in functional_attribute and '?' not in functional_attribute:
functional_attribute_temp = functional_attribute[3:]
if call == '+' or call == '~':
val1,val2 = string.split(functional_attribute_temp,'->')
else:
val2,val1 = string.split(functional_attribute_temp,'->')
val1,null = string.split(val1,'(')
val2,null = string.split(val2,'(')
protein_length_list.append([val1,val2])
elif exon in down_exon_list:
if 'ref' in functional_attribute:
new_functional_attribute = '(~)'+functional_attribute
data_tuple = new_functional_attribute,exon
elif call == '+' or call == '~':
new_functional_attribute = '(-)'+functional_attribute
data_tuple = new_functional_attribute,exon
elif call == '-':
new_functional_attribute = '(+)'+functional_attribute
data_tuple = new_functional_attribute,exon
if 'AA:' in functional_attribute and '?' not in functional_attribute:
functional_attribute_temp = functional_attribute[3:]
if call == '+' or call == '~':
val2,val1 = string.split(functional_attribute_temp,'->')
else:
val1,val2 = string.split(functional_attribute_temp,'->')
val1,null = string.split(val1,'(')
val2,null = string.split(val2,'(')
protein_length_list.append([val1,val2])
if x == 0 or (exclude_protein_details != 'yes'):
try: new_functional_attribute_list.append(new_functional_attribute)
except UnboundLocalError:
print entry
print up_exon_list,down_exon_list
print exon, critical_exons
print critical_probesets, (key, affygene,critical_probesets)
for i in functional_attribute_db:
print i, functional_attribute_db[i]; kill
###remove protein sequence prediction_data
if 'sequence' not in data_tuple[0]:
if x == 0 or exclude_protein_details == 'no':
functional_attribute_list2.append(data_tuple)
###Get rid of duplicates, but maintain non-alphabetical order
new_functional_attribute_list2=[]
for entry in new_functional_attribute_list:
if entry not in new_functional_attribute_list2:
new_functional_attribute_list2.append(entry)
new_functional_attribute_list = new_functional_attribute_list2
#new_functional_attribute_list = unique.unique(new_functional_attribute_list)
#new_functional_attribute_list.sort()
for entry in new_functional_attribute_list:
if 'sequence' in entry: new_seq_attribute_str = new_seq_attribute_str + entry + ','
else: new_functional_attribute_str = new_functional_attribute_str + entry + ','
new_seq_attribute_str = new_seq_attribute_str[0:-1]
new_functional_attribute_str = new_functional_attribute_str[0:-1]
return new_functional_attribute_str, functional_attribute_list2, new_seq_attribute_str,protein_length_list
def grab_summary_dataset_annotations(functional_attribute_db,comparison_db,include_truncation_results_specifically):
###If a second filtering database present, filter the 1st database based on protein length changes
fa_db={}; cp_db={} ###index the geneids for efficient recall in the next segment of code
for (affygene,annotation) in functional_attribute_db:
try: fa_db[affygene].append(annotation)
except KeyError: fa_db[affygene]= [annotation]
for (affygene,annotation) in comparison_db:
try: cp_db[affygene].append(annotation)
except KeyError: cp_db[affygene]= [annotation]
functional_attribute_db_exclude = {}
for affygene in fa_db:
if affygene in cp_db:
for annotation2 in cp_db[affygene]:
if ('trunc' in annotation2) or ('frag' in annotation2) or ('NMDs' in annotation2):
try: functional_attribute_db_exclude[affygene].append(annotation2)
except KeyError: functional_attribute_db_exclude[affygene] = [annotation2]
functional_annotation_db = {}
for (affygene,annotation) in functional_attribute_db:
### if we wish to filter the 1st database based on protein length changes
if affygene not in functional_attribute_db_exclude:
try: functional_annotation_db[annotation] += 1
except KeyError: functional_annotation_db[annotation] = 1
elif include_truncation_results_specifically == 'yes':
for annotation_val in functional_attribute_db_exclude[affygene]:
try: functional_annotation_db[annotation_val] += 1
except KeyError: functional_annotation_db[annotation_val] = 1
annotation_list = []
annotation_list_ranked = []
for annotation in functional_annotation_db:
if 'micro' not in annotation:
count = functional_annotation_db[annotation]
annotation_list.append((annotation,count))
annotation_list_ranked.append((count,annotation))
annotation_list_ranked.sort(); annotation_list_ranked.reverse()
return annotation_list, annotation_list_ranked
def reorganize_attribute_entries(attribute_db1,build_attribute_direction_databases):
attribute_db2 = {}; inclusion_attributes_hit_count={}; exclusion_attributes_hit_count={}
genes_with_inclusion_attributes={}; genes_with_exclusion_attributes={};
###This database has unique gene, attribute information. No attribute will now be represented more than once per gene
for key in attribute_db1:
###Make gene the key and attribute (functional elements or protein information), along with the associated exons the values
affygene = key[0];exon_attribute = key[1];exon_list = attribute_db1[key]
exon_list = unique.unique(exon_list);exon_list.sort()
attribute_exon_info = exon_attribute,exon_list #e.g. 5'UTR, [E1,E2,E3]
try: attribute_db2[affygene].append(attribute_exon_info)
except KeyError: attribute_db2[affygene] = [attribute_exon_info]
###Separate out attribute data by direction for over-representation analysis
if build_attribute_direction_databases == 'yes':
direction=exon_attribute[1:2];unique_gene_attribute=exon_attribute[3:]
if direction == '+':
try: inclusion_attributes_hit_count[unique_gene_attribute].append(affygene)
except KeyError: inclusion_attributes_hit_count[unique_gene_attribute] = [affygene]
genes_with_inclusion_attributes[affygene]=[]
if direction == '-':
try: exclusion_attributes_hit_count[unique_gene_attribute].append(affygene)
except KeyError: exclusion_attributes_hit_count[unique_gene_attribute] = [affygene]
genes_with_exclusion_attributes[affygene]=[]
inclusion_attributes_hit_count = eliminate_redundant_dict_values(inclusion_attributes_hit_count)
exclusion_attributes_hit_count = eliminate_redundant_dict_values(exclusion_attributes_hit_count)
"""for key in inclusion_attributes_hit_count:
inclusion_attributes_hit_count[key] = len(inclusion_attributes_hit_count[key])
for key in exclusion_attributes_hit_count:
exclusion_attributes_hit_count[key] = len(exclusion_attributes_hit_count[key])"""
if build_attribute_direction_databases == 'yes': return attribute_db2,inclusion_attributes_hit_count,genes_with_inclusion_attributes,exclusion_attributes_hit_count,genes_with_exclusion_attributes
else: return attribute_db2
########### Misc. Functions ###########
def eliminate_redundant_dict_values(database):
db1={}
for key in database:
list = unique.unique(database[key])
list.sort()
db1[key] = list
return db1
def add_a_space(string):
if len(string)<1:
string = ' '
return string
def convertToLog2(data_list):
return map(lambda x: math.log(float(x), 2), data_list)
def addGlobalFudgeFactor(data_list,data_type):
new_list = []
if data_type == 'log':
for item in data_list:
new_item = statistics.log_fold_conversion_fraction(item)
new_list.append(float(new_item) + global_addition_factor)
new_list = convertToLog2(new_list)
else:
for item in data_list: new_list.append(float(item) + global_addition_factor)
return new_list
def copyDirectoryPDFs(root_dir,AS='AS'):
directories = ['AltResults/AlternativeOutputDirectoryDescription.pdf',
'AltResultsDirectoryDescription.pdf',
'ClusteringDirectoryDescription.pdf',
'ExpressionInputDirectoryDescription.pdf',
'ExpressionOutputDirectoryDescription.pdf',
'GO-Elite/GO-Elite_resultsDirectoryDescription.pdf',
'GO-EliteDirectoryDescription.pdf',
'RootDirectoryDescription.pdf']
import shutil
for dir in directories:
file = string.split(dir,'/')[-1]
proceed=True
if 'AltResult' in dir and AS!='AS': proceed=False
if proceed:
try: shutil.copyfile(filepath('Documentation/DirectoryDescription/'+file), filepath(root_dir+dir))
except Exception: pass
def restrictProbesets(dataset_name):
### Take a file with probesets and only perform the splicing-analysis on these (e.g. those already identified from a previous run with a specific pattern)
### Allows for propper denominator when calculating z-scores for microRNA and protein-domain ORA
probeset_list_filename = import_dir = '/AltDatabaseNoVersion/filtering'; filtered_probeset_db={}
if array_type == 'RNASeq': id_name = 'exon/junction IDs'
else: id_name = 'array IDs'
try:
dir_list = read_directory(import_dir)
fn_dir = filepath(import_dir[1:])
except Exception: dir_list=[]; fn_dir=''
if len(dir_list)>0:
for file in dir_list:
if file[:-4] in dataset_name:
fn = fn_dir+'/'+file; fn = string.replace(fn,'AltDatabase','AltDatabaseNoVersion')
filtered_probeset_db = importGeneric(fn)
print len(filtered_probeset_db), id_name,"will be used to restrict analysis..."
return filtered_probeset_db
def RunAltAnalyze():
#print altanalyze_files
#print '!!!!!starting to run alt-exon analysis'
#returnLargeGlobalVars()
global annotate_db; annotate_db={}; global splice_event_list; splice_event_list=[]; residuals_dirlist=[]
global dataset_name; global constitutive_probeset_db; global exon_db; dir_list2=[]; import_dir2=''
if array_type == 'AltMouse': import_dir = root_dir+'AltExpression/'+array_type
elif array_type == 'exon':
import_dir = root_dir+'AltExpression/ExonArray/'+species+'/'
elif array_type == 'gene':
import_dir = root_dir+'AltExpression/GeneArray/'+species+'/'
elif array_type == 'junction':
import_dir = root_dir+'AltExpression/JunctionArray/'+species+'/'
else:
import_dir = root_dir+'AltExpression/'+array_type+'/'+species+'/'
#if analysis_method == 'ASPIRE' or analysis_method == 'linearregres' or analysis_method == 'splicing-index':
if array_type != 'AltMouse': gene_annotation_file = "AltDatabase/ensembl/"+species+"/"+species+"_Ensembl-annotations.txt"
else: gene_annotation_file = "AltDatabase/"+species+"/"+array_type+"/"+array_type+"_gene_annotations.txt"
annotate_db = ExonAnalyze_module.import_annotations(gene_annotation_file,array_type)
###Import probe-level associations
exon_db={}; filtered_arrayids={};filter_status='no'
try: constitutive_probeset_db,exon_db,genes_being_analyzed = importSplicingAnnotationDatabase(probeset_annotations_file,array_type,filtered_arrayids,filter_status)
except IOError:
print_out = 'The annotation database: \n'+probeset_annotations_file+'\nwas not found. Ensure this file was not deleted and that the correct species has been selected.'
try: UI.WarningWindow(print_out,'Exit'); print print_out
except Exception: print print_out
print traceback.format_exc()
badExit()
run=0
### Occurs when analyzing multiple conditions rather than performing a simple pair-wise comparison
if run_from_scratch == 'Annotate External Results': import_dir = root_dir
elif analyze_all_conditions == 'all groups':
import_dir = string.replace(import_dir,'AltExpression','AltExpression/FullDatasets')
if array_type == 'AltMouse':
import_dir = string.replace(import_dir,'FullDatasets/AltMouse','FullDatasets/AltMouse/Mm')
elif analyze_all_conditions == 'both':
import_dir2 = string.replace(import_dir,'AltExpression','AltExpression/FullDatasets')
if array_type == 'AltMouse':
import_dir2 = string.replace(import_dir2,'FullDatasets/AltMouse','FullDatasets/AltMouse/Mm')
try: dir_list2 = read_directory(import_dir2) #send a sub_directory to a function to identify all files in a directory
except Exception:
try:
if array_type == 'exon': array_type_dir = 'ExonArray'
elif array_type == 'gene': array_type_dir = 'GeneArray'
elif array_type == 'junction': array_type_dir = 'GeneArray'
else: array_type_dir = array_type
import_dir2 = string.replace(import_dir2,'AltExpression/'+array_type_dir+'/'+species+'/','')
import_dir2 = string.replace(import_dir2,'AltExpression/'+array_type_dir+'/','');
dir_list2 = read_directory(import_dir2)
except Exception:
print_out = 'The expression files were not found. Please make\nsure you selected the correct species and array type.\n\nselected species: '+species+'\nselected array type: '+array_type+'\nselected directory:'+import_dir2
try: UI.WarningWindow(print_out,'Exit'); print print_out
except Exception: print print_out
print traceback.format_exc()
badExit()
try: dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
except Exception:
try:
if array_type == 'exon': array_type_dir = 'ExonArray'
elif array_type == 'gene': array_type_dir = 'GeneArray'
elif array_type == 'junction': array_type_dir = 'JunctionArray'
else: array_type_dir = array_type
import_dir = string.replace(import_dir,'AltExpression/'+array_type_dir+'/'+species+'/','')
import_dir = string.replace(import_dir,'AltExpression/'+array_type_dir+'/','');
try: dir_list = read_directory(import_dir)
except Exception:
import_dir = root_dir
dir_list = read_directory(root_dir) ### Occurs when reading in an AltAnalyze filtered file under certain conditions
except Exception:
print_out = 'The expression files were not found. Please make\nsure you selected the correct species and array type.\n\nselected species: '+species+'\nselected array type: '+array_type+'\nselected directory:'+import_dir
try: UI.WarningWindow(print_out,'Exit')
except Exception: print print_out
print traceback.format_exc()
badExit()
dir_list+=dir_list2
### Capture the corresponding files in the residual dir to make sure these files exist for all comparisons - won't if FIRMA was run on some files
if analysis_method == 'FIRMA':
try:
residual_dir = root_dir+'AltExpression/FIRMA/residuals/'+array_type+'/'+species+'/'
residuals_dirlist = read_directory(residual_dir)
except Exception: null=[]
try:
residual_dir = root_dir+'AltExpression/FIRMA/FullDatasets/'+array_type+'/'+species+'/'
residuals_dirlist += read_directory(residual_dir)
except Exception: null=[]
dir_list_verified=[]
for file in residuals_dirlist:
for filename in dir_list:
if file[:-4] in filename: dir_list_verified.append(filename)
dir_list = unique.unique(dir_list_verified)
junction_biotype = 'no'
if array_type == 'RNASeq':
### Check to see if user data includes junctions or just exons
for probeset in exon_db:
if '-' in probeset: junction_biotype = 'yes'; break
if junction_biotype == 'no' and analysis_method != 'splicing-index' and array_type == 'RNASeq':
dir_list=[] ### DON'T RUN ALTANALYZE WHEN JUST ANALYZING EXON DATA
print 'No junction data to summarize... proceeding with exon analysis\n'
elif len(dir_list)==0:
print_out = 'No expression files available in the input directory:\n'+root_dir
try: UI.WarningWindow(print_out,'Exit'); print print_out
except Exception: print print_out
badExit()
dir_list = filterAltExpressionFiles(dir_list,altanalyze_files) ### Looks to see if the AltExpression files are for this run or from an older run
for altanalyze_input in dir_list: #loop through each file in the directory to output results
###Import probe-level associations
if 'cel_files' in altanalyze_input:
print_out = 'The AltExpression directory containing the necessary import file(s) is missing. Please verify the correct parameters and input directory were selected. If this error persists, contact us.'
try: UI.WarningWindow(print_out,'Exit'); print print_out
except Exception: print print_out
badExit()
if run>0: ### Only re-set these databases after the run when batch analysing multiple files
exon_db={}; filtered_arrayids={};filter_status='no' ###Use this as a means to save memory (import multiple times - only storing different types relevant information)
constitutive_probeset_db,exon_db,genes_being_analyzed = importSplicingAnnotationDatabase(probeset_annotations_file,array_type,filtered_arrayids,filter_status)
if altanalyze_input in dir_list2: dataset_dir = import_dir2 +'/'+ altanalyze_input ### Then not a pairwise comparison
else: dataset_dir = import_dir +'/'+ altanalyze_input
dataset_name = altanalyze_input[:-4] + '-'
print "Beginning to process",dataset_name[0:-1]
### If the user want's to restrict the analysis to preselected probesets (e.g., limma or FIRMA analysis selected)
global filtered_probeset_db; filtered_probeset_db={}
try: filtered_probeset_db = restrictProbesets(dataset_name)
except Exception: null=[]
if run_from_scratch != 'Annotate External Results':
###Import expression data and stats and filter the expression data based on fold and p-value OR expression threshold
try: conditions,adj_fold_dbase,nonlog_NI_db,dataset_name,gene_expression_diff_db,midas_db,ex_db,si_db = performExpressionAnalysis(dataset_dir,constitutive_probeset_db,exon_db,annotate_db,dataset_name)
except IOError:
#except Exception,exception:
#print exception
print traceback.format_exc()
print_out = 'The AltAnalyze filtered expression file "'+dataset_name+'" is not propperly formatted. Review formatting requirements if this file was created by another application.'
try: UI.WarningWindow(print_out,'Exit'); print print_out
except Exception: print print_out
badExit()
else:
conditions = 0; adj_fold_dbase={}; nonlog_NI_db={}; gene_expression_diff_db={}; ex_db={}; si_db={}
defineEmptyExpressionVars(exon_db); adj_fold_dbase = original_fold_dbase
###Run Analysis
summary_results_db, summary_results_db2, aspire_output, aspire_output_gene, number_events_analyzed = splicingAnalysisAlgorithms(nonlog_NI_db,adj_fold_dbase,dataset_name,gene_expression_diff_db,exon_db,ex_db,si_db,dataset_dir)
aspire_output_list.append(aspire_output); aspire_output_gene_list.append(aspire_output_gene)
try: clearObjectsFromMemory(exon_db); clearObjectsFromMemory(constitutive_probeset_db); constitutive_probeset_db=[]
except Exception: null=[]
try: clearObjectsFromMemory(last_exon_region_db);last_exon_region_db=[]
except Exception: null=[]
try: clearObjectsFromMemory(adj_fold_dbase);adj_fold_dbase=[]; clearObjectsFromMemory(nonlog_NI_db);nonlog_NI_db=[]
except Exception: null=[]
try: clearObjectsFromMemory(gene_expression_diff_db);gene_expression_diff_db=[]; clearObjectsFromMemory(midas_db);midas_db=[]
except Exception: null=[]
try: clearObjectsFromMemory(ex_db);ex_db=[]; clearObjectsFromMemory(si_db);si_db=[]
except Exception: null=[]
try: run+=1
except Exception: run = 1
if run>0: ###run = 0 if no filtered expression data present
try: return summary_results_db, aspire_output_gene_list, number_events_analyzed
except Exception:
print_out = 'AltAnalyze was unable to find an expression dataset to analyze in:\n',import_dir,'\nor\n',import_dir2,'\nPlease re-run and select a valid input directory.'
try: UI.WarningWindow(print_out,'Exit'); print print_out
except Exception: print print_out
badExit()
else:
try: clearObjectsFromMemory(exon_db); clearObjectsFromMemory(constitutive_probeset_db); constitutive_probeset_db=[]
except Exception: null=[]
try: clearObjectsFromMemory(last_exon_region_db);last_exon_region_db=[]
except Exception: null=[]
return None
def filterAltExpressionFiles(dir_list,current_files):
dir_list2=[]
try:
if len(current_files) == 0: current_files = dir_list ###if no filenames input
for altanalzye_input in dir_list: #loop through each file in the directory to output results
if altanalzye_input in current_files:
dir_list2.append(altanalzye_input)
dir_list = dir_list2
except Exception: dir_list = dir_list
return dir_list
def defineEmptyExpressionVars(exon_db):
global fold_dbase; fold_dbase={}; global original_fold_dbase; global critical_exon_db; critical_exon_db={}
global midas_db; midas_db = {}; global max_replicates; global equal_replicates; max_replicates=0; equal_replicates=0
for probeset in exon_db: fold_dbase[probeset]='',''
original_fold_dbase = fold_dbase
def universalPrintFunction(print_items):
log_report = open(log_file,'a')
for item in print_items:
if commandLineMode == 'no': ### Command-line has it's own log file write method (Logger)
log_report.write(item+'\n')
else: print item
log_report.close()
class StatusWindow:
def __init__(self,root,expr_var,alt_var,goelite_var,additional_var,exp_file_location_db):
root.title('AltAnalyze version 2.1.4')
statusVar = StringVar() ### Class method for Tkinter. Description: "Value holder for strings variables."
self.root = root
height = 450; width = 500
if os.name != 'nt': height = 500; width = 600
self.sf = PmwFreeze.ScrolledFrame(root,
labelpos = 'n', label_text = 'Results Status Window',
usehullsize = 1, hull_width = width, hull_height = height)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
group = PmwFreeze.Group(self.sf.interior(),tag_text = 'Output')
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
Label(group.interior(),width=190,height=1000,justify=LEFT, bg='black', fg = 'white',anchor=NW,padx = 5,pady = 5, textvariable=statusVar).pack(fill=X,expand=Y)
status = StringVarFile(statusVar,root) ### Likely captures the stdout
sys.stdout = status
for dataset in exp_file_location_db:
fl = exp_file_location_db[dataset]; fl.setSTDOUT(sys.stdout)
root.after(100, AltAnalyzeMain(expr_var, alt_var, goelite_var, additional_var, exp_file_location_db, root))
try:
root.protocol("WM_DELETE_WINDOW", self.deleteWindow)
root.mainloop()
except Exception: pass
def deleteWindow(self):
try: self.root.destroy()
except Exception: pass
def quit(self):
try:
self.root.quit()
self.root.destroy()
except Exception: pass
sys.exit()
def exportComparisonSummary(dataset_name,summary_data_dbase,return_type):
log_report = open(log_file,'a')
result_list=[]
for key in summary_data_dbase:
if key != 'QC': ### The value is a list of strings
summary_data_dbase[key] = str(summary_data_dbase[key])
d = 'Dataset name: '+ dataset_name[:-1]; result_list.append(d+'\n')
try:
d = summary_data_dbase['gene_assayed']+':\tAll genes examined'; result_list.append(d)
d = summary_data_dbase['denominator_exp_genes']+':\tExpressed genes examined for AS'; result_list.append(d)
if explicit_data_type == 'exon-only':
d = summary_data_dbase['alt_events']+':\tAlternatively regulated probesets'; result_list.append(d)
d = summary_data_dbase['denominator_exp_events']+':\tExpressed probesets examined'; result_list.append(d)
elif (array_type == 'AltMouse' or array_type == 'junction' or array_type == 'RNASeq') and (explicit_data_type == 'null' or return_type == 'print'):
d = summary_data_dbase['alt_events']+':\tAlternatively regulated junction-pairs'; result_list.append(d)
d = summary_data_dbase['denominator_exp_events']+':\tExpressed junction-pairs examined'; result_list.append(d)
else:
d = summary_data_dbase['alt_events']+':\tAlternatively regulated probesets'; result_list.append(d)
d = summary_data_dbase['denominator_exp_events']+':\tExpressed probesets examined'; result_list.append(d)
d = summary_data_dbase['alt_genes']+':\tAlternatively regulated genes (ARGs)'; result_list.append(d)
d = summary_data_dbase['direct_domain_genes']+':\tARGs - overlaping with domain/motifs'; result_list.append(d)
d = summary_data_dbase['miRNA_gene_hits']+':\tARGs - overlaping with microRNA binding sites'; result_list.append(d)
except Exception:
pass
result_list2=[]
for d in result_list:
if explicit_data_type == 'exon-only': d = string.replace(d,'probeset','exon')
elif array_type == 'RNASeq': d = string.replace(d,'probeset','junction')
result_list2.append(d)
result_list = result_list2
if return_type == 'log':
for d in result_list: log_report.write(d+'\n')
log_report.write('\n')
log_report.close()
return result_list
class SummaryResultsWindow:
def __init__(self,tl,analysis_type,output_dir,dataset_name,output_type,summary_data_dbase):
def showLink(event):
try:
idx = int(event.widget.tag_names(CURRENT)[1]) ### This is just the index provided below (e.g., str(0))
#print [self.LINKS[idx]]
if 'http://' in self.LINKS[idx]:
webbrowser.open(self.LINKS[idx])
elif self.LINKS[idx][-1] == '/':
self.openSuppliedDirectory(self.LINKS[idx])
else:
### Instead of using this option to open a hyperlink (which is what it should do), we can open another Tk window
try: self.viewPNGFile(self.LINKS[idx]) ### ImageTK PNG viewer
except Exception:
try: self.ShowImageMPL(self.LINKS[idx]) ### MatPlotLib based dispaly
except Exception:
self.openPNGImage(self.LINKS[idx]) ### Native OS PNG viewer
#self.DisplayPlots(self.LINKS[idx]) ### GIF based display
except Exception:
null=[] ### anomalous error
self.emergency_exit = False
self.LINKS = []
self.tl = tl
self.tl.title('AltAnalyze version 2.1.4')
self.analysis_type = analysis_type
filename = 'Config/icon.gif'
fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(tl); can.pack(side='top'); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
use_scroll = 'yes'
try: runGOElite = run_GOElite
except Exception: runGOElite='decide_later'
if 'QC' in summary_data_dbase:
graphic_links = summary_data_dbase['QC'] ### contains hyperlinks to QC and Clustering plots
if len(graphic_links)==0: del summary_data_dbase['QC'] ### This can be added if an analysis fails
else:
graphic_links = []
label_text_str = 'AltAnalyze Result Summary'; height = 150; width = 500
if analysis_type == 'AS' or 'QC' in summary_data_dbase: height = 330
if analysis_type == 'AS' and 'QC' in summary_data_dbase: height = 330
self.sf = PmwFreeze.ScrolledFrame(tl,
labelpos = 'n', label_text = label_text_str,
usehullsize = 1, hull_width = width, hull_height = height)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
txt=Text(self.frame,bg='light gray',width=150, height=80)
txt.pack(expand=True, fill="both")
#txt.insert(END, 'Primary Analysis Finished....\n')
txt.insert(END, 'Results saved to:\n'+output_dir+'\n')
f = Font(family="System", size=12, weight="bold")
txt.tag_config("font", font=f)
i=0
copyDirectoryPDFs(output_dir,AS=analysis_type)
if analysis_type == 'AS':
txt.insert(END, '\n')
result_list = exportComparisonSummary(dataset_name,summary_data_dbase,'print')
for d in result_list: txt.insert(END, d+'\n')
if 'QC' in summary_data_dbase and len(graphic_links)>0:
txt.insert(END, '\nQC and Expression Clustering Plots',"font")
txt.insert(END, '\n\n 1) ')
for (name,file_dir) in graphic_links:
txt.insert(END, name, ('link', str(i)))
if len(graphic_links) > (i+1):
txt.insert(END, '\n %s) ' % str(i+2))
self.LINKS.append(file_dir)
i+=1
txt.insert(END, '\n\nView all primary plots in the folder ')
txt.insert(END, 'DataPlots',('link', str(i))); i+=1
self.LINKS.append(output_dir+'DataPlots/')
else:
url = 'http://altanalyze.readthedocs.io/en/latest/'
self.LINKS=(url,'')
txt.insert(END, '\nFor more information see the ')
txt.insert(END, "AltAnalyze Online Help", ('link', str(0)))
txt.insert(END, '\n\n')
if runGOElite == 'run-immediately':
txt.insert(END, '\n\nView all pathway enrichment results in the folder ')
txt.insert(END, 'GO-Elite',('link', str(i))); i+=1
self.LINKS.append(output_dir+'GO-Elite/')
if analysis_type == 'AS':
txt.insert(END, '\n\nView all splicing plots in the folder ')
txt.insert(END, 'ExonPlots',('link', str(i))); i+=1
try: self.LINKS.append(output_dir+'ExonPlots/')
except Exception: pass
txt.tag_config('link', foreground="blue", underline = 1)
txt.tag_bind('link', '<Button-1>', showLink)
txt.insert(END, '\n\n')
open_results_folder = Button(tl, text = 'Results Folder', command = self.openDirectory)
open_results_folder.pack(side = 'left', padx = 5, pady = 5);
if analysis_type == 'AS':
#self.dg_url = 'http://www.altanalyze.org/domaingraph.htm'
self.dg_url = 'http://www.altanalyze.org/domaingraph.htm'
dg_pdf_file = 'Documentation/domain_graph.pdf'; dg_pdf_file = filepath(dg_pdf_file); self.dg_pdf_file = dg_pdf_file
text_button = Button(tl, text='Start DomainGraph in Cytoscape', command=self.SelectCytoscapeTopLevel)
text_button.pack(side = 'right', padx = 5, pady = 5)
self.output_dir = output_dir + "AltResults"
self.whatNext_url = 'http://altanalyze.readthedocs.io/en/latest/' #http://www.altanalyze.org/what_next_altexon.htm'
whatNext_pdf = 'Documentation/what_next_alt_exon.pdf'; whatNext_pdf = filepath(whatNext_pdf); self.whatNext_pdf = whatNext_pdf
if output_type == 'parent': self.output_dir = output_dir ###Used for fake datasets
else:
if pathway_permutations == 'NA':
self.output_dir = output_dir + "ExpressionOutput"
else: self.output_dir = output_dir
self.whatNext_url = 'http://altanalyze.readthedocs.io/en/latest/' #'http://www.altanalyze.org/what_next_expression.htm'
whatNext_pdf = 'Documentation/what_next_GE.pdf'; whatNext_pdf = filepath(whatNext_pdf); self.whatNext_pdf = whatNext_pdf
what_next = Button(tl, text='What Next?', command=self.whatNextlinkout)
what_next.pack(side = 'right', padx = 5, pady = 5)
quit_buttonTL = Button(tl,text='Close View', command=self.close)
quit_buttonTL.pack(side = 'right', padx = 5, pady = 5)
continue_to_next_win = Button(text = 'Continue', command = self.continue_win)
continue_to_next_win.pack(side = 'right', padx = 10, pady = 10)
quit_button = Button(root,text='Quit', command=self.quit)
quit_button.pack(side = 'right', padx = 5, pady = 5)
button_text = 'Help'; help_url = 'http://www.altanalyze.org/help_main.htm'; self.help_url = filepath(help_url)
pdf_help_file = 'Documentation/AltAnalyze-Manual.pdf'; pdf_help_file = filepath(pdf_help_file); self.pdf_help_file = pdf_help_file
help_button = Button(root, text=button_text, command=self.Helplinkout)
help_button.pack(side = 'left', padx = 5, pady = 5)
if self.emergency_exit == False:
self.tl.protocol("WM_DELETE_WINDOW", self.tldeleteWindow)
self.tl.mainloop() ###Needed to show graphic
else:
""" This shouldn't have to be called, but is when the topLevel window isn't closed first
specifically if a PNG file is opened. the sys.exitfunc() should work but doesn't.
work on this more later """
#AltAnalyzeSetup('no')
try: self._tls.quit(); self._tls.destroy()
except Exception: None
try: self._tlx.quit(); self._tlx.destroy()
except Exception: None
try: self._tlx.quit(); self._tlx.destroy()
except Exception: None
try: self.tl.quit(); self.tl.destroy()
except Exception: None
try: root.quit(); root.destroy()
except Exception: None
UI.getUpdatedParameters(array_type,species,'Process Expression file',output_dir)
sys.exit() ### required when opening PNG files on Windows to continue (not sure why)
#sys.exitfunc()
def tldeleteWindow(self):
try: self.tl.quit(); self.tl.destroy()
except Exception: self.tl.destroy()
def deleteTLWindow(self):
self.emergency_exit = True
try: self._tls.quit(); self._tls.destroy()
except Exception: None
try: self._tlx.quit(); self._tlx.destroy()
except Exception: None
self.tl.quit()
self.tl.destroy()
sys.exitfunc()
def deleteWindow(self):
self.emergency_exit = True
try: self._tls.quit(); self._tls.destroy()
except Exception: None
try: self._tlx.quit(); self._tlx.destroy()
except Exception: None
try:
self.tl.quit()
self.tl.destroy()
except Exception: None
sys.exitfunc()
def continue_win(self):
self.emergency_exit = True
try: self._tls.quit(); self._tls.destroy()
except Exception: None
try: self._tlx.quit(); self._tlx.destroy()
except Exception: None
try: self.tl.quit(); self.tl.destroy()
except Exception: pass
root.quit()
root.destroy()
try: self.tl.grid_forget()
except Exception: None
try: root.grid_forget()
except Exception: None
sys.exitfunc()
def openDirectory(self):
if runningCommandLine:
pass
elif os.name == 'nt':
try: os.startfile('"'+self.output_dir+'"')
except Exception: os.system('open "'+self.output_dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+self.output_dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+self.output_dir+'/"')
def openSuppliedDirectory(self,dir):
if runningCommandLine:
pass
elif os.name == 'nt':
try: os.startfile('"'+self.output_dir+'"')
except Exception: os.system('open "'+dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+dir+'/"')
def DGlinkout(self):
try:
altanalyze_path = filepath('') ### Find AltAnalye's path
altanalyze_path = altanalyze_path[:-1]
except Exception: null=[]
if os.name == 'nt':
parent_dir = 'C:/Program Files'; application_dir = 'Cytoscape_v'; application_name = 'Cytoscape.exe'
elif 'darwin' in sys.platform:
parent_dir = '/Applications'; application_dir = 'Cytoscape_v'; application_name = 'Cytoscape.app'
elif 'linux' in sys.platform:
parent_dir = '/opt'; application_dir = 'Cytoscape_v'; application_name = 'Cytoscape'
try: openCytoscape(altanalyze_path,application_dir,application_name)
except Exception: null=[]
try: self._tls.destroy()
except Exception: None
try: ###Remove this cytoscape as the default
file_location_defaults = UI.importDefaultFileLocations()
del file_location_defaults['CytoscapeDir']
UI.exportDefaultFileLocations(file_location_defaults)
except Exception: null=[]
self.GetHelpTopLevel(self.dg_url,self.dg_pdf_file)
def Helplinkout(self): self.GetHelpTopLevel(self.help_url,self.pdf_help_file)
def whatNextlinkout(self): self.GetHelpTopLevel(self.whatNext_url,self.whatNext_pdf)
def ShowImageMPL(self,file_location):
""" Visualization method using MatPlotLib """
try:
import matplotlib
import matplotlib.pyplot as pylab
except Exception:
#print 'Graphical output mode disabled (requires matplotlib, numpy and scipy)'
None
fig = pylab.figure()
pylab.subplots_adjust(left=0.0, right=1.0, top=1.0, bottom=0.00) ### Fill the plot area left to right
ax = fig.add_subplot(111)
ax.set_xticks([]) ### Hides ticks
ax.set_yticks([])
img= pylab.imread(file_location)
imgplot = pylab.imshow(img)
pylab.show()
def viewPNGFile(self,png_file_dir):
""" View PNG file within a PMW Tkinter frame """
try: import ImageTk
except Exception:
from PIL import ImageTk
from PIL import Image
tlx = Toplevel(); self._tlx = tlx
sf = PmwFreeze.ScrolledFrame(tlx, labelpos = 'n', label_text = '',
usehullsize = 1, hull_width = 800, hull_height = 550)
sf.pack(padx = 0, pady = 0, fill = 'both', expand = 1)
frame = sf.interior()
tlx.title(png_file_dir)
img = ImageTk.PhotoImage(file=png_file_dir)
can = Canvas(frame)
can.pack(fill=BOTH, padx = 0, pady = 0)
w = img.width()
h = height=img.height()
can.config(width=w, height=h)
can.create_image(2, 2, image=img, anchor=NW)
tlx.mainloop()
def openPNGImage(self,png_file_dir):
if runningCommandLine:
pass
elif os.name == 'nt':
try: os.startfile('"'+png_file_dir+'"')
except Exception: os.system('open "'+png_file_dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+png_file_dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+png_file_dir+'"')
def DisplayPlots(self,file_location):
""" Native Tkinter method - Displays a gif file in a standard TopLevel window (nothing fancy) """
tls = Toplevel(); self._tls = tls; nulls = '\t\t\t\t'; tls.title('AltAnalyze Plot Visualization')
self.sf = PmwFreeze.ScrolledFrame(self._tls,
labelpos = 'n', label_text = '', usehullsize = 1, hull_width = 520, hull_height = 500)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
group = PmwFreeze.Group(self.sf.interior(),tag_text = file_location)
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
img = PhotoImage(file=filepath(file_location))
can = Canvas(group.interior()); can.pack(side='left',padx = 10, pady = 20); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
tls.mainloop()
def GetHelpTopLevel(self,url,pdf_file):
try:
config_db = UI.importConfigFile()
ask_for_help = config_db['help'] ### hide_selection_option
except Exception: ask_for_help = 'null'; config_db={}
self.pdf_file = pdf_file; self.url = url
if ask_for_help == 'null':
message = ''; self.message = message; self.online_help = 'Online Documentation'; self.pdf_help = 'Local PDF File'
tls = Toplevel(); self._tls = tls; nulls = '\t\t\t\t'; tls.title('Please select one of the options')
self.sf = PmwFreeze.ScrolledFrame(self._tls,
labelpos = 'n', label_text = '', usehullsize = 1, hull_width = 320, hull_height = 200)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
group = PmwFreeze.Group(self.sf.interior(),tag_text = 'Options')
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
filename = 'Config/icon.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(group.interior()); can.pack(side='left',padx = 10, pady = 20); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
l1 = Label(group.interior(), text=nulls); l1.pack(side = 'bottom')
text_button2 = Button(group.interior(), text=self.online_help, command=self.openOnlineHelp); text_button2.pack(side = 'top', padx = 5, pady = 5)
try: text_button = Button(group.interior(), text=self.pdf_help, command=self.openPDFHelp); text_button.pack(side = 'top', padx = 5, pady = 5)
except Exception: text_button = Button(group.interior(), text=self.pdf_help, command=self.openPDFHelp); text_button.pack(side = 'top', padx = 5, pady = 5)
text_button3 = Button(group.interior(), text='No Thanks', command=self.skipHelp); text_button3.pack(side = 'top', padx = 5, pady = 5)
c = Checkbutton(group.interior(), text = "Apply these settings each time", command=self.setHelpConfig); c.pack(side = 'bottom', padx = 5, pady = 0)
tls.mainloop()
try: tls.destroy()
except Exception: None
else:
file_location_defaults = UI.importDefaultFileLocations()
try:
help_choice = file_location_defaults['HelpChoice'].Location()
if help_choice == 'PDF': self.openPDFHelp()
elif help_choice == 'http': self.openOnlineHelp()
else: self.skip()
except Exception: self.openPDFHelp() ### Open PDF if there's a problem
def SelectCytoscapeTopLevel(self):
try:
config_db = UI.importConfigFile()
cytoscape_type = config_db['cytoscape'] ### hide_selection_option
except Exception: cytoscape_type = 'null'; config_db={}
if cytoscape_type == 'null':
message = ''; self.message = message
tls = Toplevel(); self._tls = tls; nulls = '\t\t\t\t'; tls.title('Cytoscape Automatic Start Options')
self.sf = PmwFreeze.ScrolledFrame(self._tls,
labelpos = 'n', label_text = '', usehullsize = 1, hull_width = 420, hull_height = 200)
self.sf.pack(padx = 5, pady = 1, fill = 'both', expand = 1)
self.frame = self.sf.interior()
group = PmwFreeze.Group(self.sf.interior(),tag_text = 'Options')
group.pack(fill = 'both', expand = 1, padx = 10, pady = 0)
filename = 'Config/cyto-logo-smaller.gif'; fn=filepath(filename); img = PhotoImage(file=fn)
can = Canvas(group.interior()); can.pack(side='left',padx = 10, pady = 5); can.config(width=img.width(), height=img.height())
can.create_image(2, 2, image=img, anchor=NW)
#"""
self.local_cytoscape = 'AltAnalyze Bundled Version'; self.custom_cytoscape = 'Previously Installed Version'
l1 = Label(group.interior(), text=nulls); l1.pack(side = 'bottom')
l3 = Label(group.interior(), text='Select version of Cytoscape to open:'); l3.pack(side = 'top', pady = 5)
"""
self.local_cytoscape = ' No '; self.custom_cytoscape = ' Yes '
l1 = Label(group.interior(), text=nulls); l1.pack(side = 'bottom')
l2 = Label(group.interior(), text='Note: Cytoscape can take up-to a minute to initalize', fg="red"); l2.pack(side = 'top', padx = 5, pady = 0)
"""
text_button2 = Button(group.interior(), text=self.local_cytoscape, command=self.DGlinkout); text_button2.pack(padx = 5, pady = 5)
try: text_button = Button(group.interior(), text=self.custom_cytoscape, command=self.getPath); text_button.pack(padx = 5, pady = 5)
except Exception: text_button = Button(group.interior(), text=self.custom_cytoscape, command=self.getPath); text_button.pack(padx = 5, pady = 5)
l2 = Label(group.interior(), text='Note: Cytoscape can take up-to a minute to initalize', fg="blue"); l2.pack(side = 'bottom', padx = 5, pady = 0)
c = Checkbutton(group.interior(), text = "Apply these settings each time and don't show again", command=self.setCytoscapeConfig); c.pack(side = 'bottom', padx = 5, pady = 0)
#c2 = Checkbutton(group.interior(), text = "Open PDF of DomainGraph help rather than online help", command=self.setCytoscapeConfig); c2.pack(side = 'bottom', padx = 5, pady = 0)
tls.mainloop()
try: tls.destroy()
except Exception: None
else:
file_location_defaults = UI.importDefaultFileLocations()
try: cytoscape_app_dir = file_location_defaults['CytoscapeDir'].Location(); openFile(cytoscape_app_dir)
except Exception:
try: altanalyze_path = filepath(''); altanalyze_path = altanalyze_path[:-1]
except Exception: altanalyze_path=''
application_dir = 'Cytoscape_v'
if os.name == 'nt': application_name = 'Cytoscape.exe'
elif 'darwin' in sys.platform: application_name = 'Cytoscape.app'
elif 'linux' in sys.platform: application_name = 'Cytoscape'
try: openCytoscape(altanalyze_path,application_dir,application_name)
except Exception: null=[]
def setCytoscapeConfig(self):
config_db={}; config_db['cytoscape'] = 'hide_selection_option'
UI.exportConfigFile(config_db)
def setHelpConfig(self):
config_db={}; config_db['help'] = 'hide_selection_option'
UI.exportConfigFile(config_db)
def getPath(self):
file_location_defaults = UI.importDefaultFileLocations()
if os.name == 'nt': parent_dir = 'C:/Program Files'; application_dir = 'Cytoscape_v'; application_name = 'Cytoscape.exe'
elif 'darwin' in sys.platform: parent_dir = '/Applications'; application_dir = 'Cytoscape_v'; application_name = 'Cytoscape.app'
elif 'linux' in sys.platform: parent_dir = '/opt'; application_dir = 'Cytoscape_v'; application_name = 'Cytoscape'
try:
self.default_dir = file_location_defaults['CytoscapeDir'].Location()
self.default_dir = string.replace(self.default_dir,'//','/')
self.default_dir = string.replace(self.default_dir,'\\','/')
self.default_dir = string.join(string.split(self.default_dir,'/')[:-1],'/')
except Exception:
dir = FindDir(parent_dir,application_dir); dir = filepath(parent_dir+'/'+dir)
self.default_dir = filepath(parent_dir)
try: dirPath = tkFileDialog.askdirectory(parent=self._tls,initialdir=self.default_dir)
except Exception:
self.default_dir = ''
try: dirPath = tkFileDialog.askdirectory(parent=self._tls,initialdir=self.default_dir)
except Exception:
try: dirPath = tkFileDialog.askdirectory(parent=self._tls)
except Exception: dirPath=''
try:
#print [dirPath],application_name
app_dir = dirPath+'/'+application_name
if 'linux' in sys.platform:
try: createCytoscapeDesktop(cytoscape_dir)
except Exception: null=[]
dir_list = unique.read_directory('/usr/bin/') ### Check to see that JAVA is installed
if 'java' not in dir_list: print 'Java not referenced in "usr/bin/. If not installed,\nplease install and re-try opening Cytoscape'
try:
jar_path = dirPath+'/cytoscape.jar'
main_path = dirPath+'/cytoscape.CyMain'
plugins_path = dirPath+'/plugins'
os.system('java -Dswing.aatext=true -Xss5M -Xmx512M -jar '+jar_path+' '+main_path+' -p '+plugins_path+' &')
print 'Cytoscape jar opened:',jar_path
except Exception:
print 'OS command to open Java failed.'
try: openFile(app_dir2); print 'Cytoscape opened:',app_dir2
except Exception: openFile(app_dir)
else: openFile(app_dir)
try: file_location_defaults['CytoscapeDir'].SetLocation(app_dir)
except Exception:
fl = UI.FileLocationData('', app_dir, 'all')
file_location_defaults['CytoscapeDir'] = fl
UI.exportDefaultFileLocations(file_location_defaults)
except Exception: null=[]
try: self._tls.destroy()
except Exception: None
self.GetHelpTopLevel(self.dg_url,self.dg_pdf_file)
def openOnlineHelp(self):
file_location_defaults = UI.importDefaultFileLocations()
try:file_location_defaults['HelpChoice'].SetLocation('http')
except Exception:
fl = UI.FileLocationData('', 'http', 'all')
file_location_defaults['HelpChoice'] = fl
UI.exportDefaultFileLocations(file_location_defaults)
webbrowser.open(self.url)
#except Exception: null=[]
try: self._tls.destroy()
except Exception: None
def skipHelp(self):
file_location_defaults = UI.importDefaultFileLocations()
try: file_location_defaults['HelpChoice'].SetLocation('skip')
except Exception:
fl = UI.FileLocationData('', 'skip', 'all')
file_location_defaults['HelpChoice'] = fl
UI.exportDefaultFileLocations(file_location_defaults)
try: self._tls.destroy()
except Exception: None
def openPDFHelp(self):
file_location_defaults = UI.importDefaultFileLocations()
try:file_location_defaults['HelpChoice'].SetLocation('PDF')
except Exception:
fl = UI.FileLocationData('', 'PDF', 'all')
file_location_defaults['HelpChoice'] = fl
UI.exportDefaultFileLocations(file_location_defaults)
if runningCommandLine:
pass
elif os.name == 'nt':
try: os.startfile('"'+self.pdf_file+'"')
except Exception: os.system('open "'+self.pdf_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+self.pdf_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+self.pdf_file+'"')
try: self._tls.destroy()
except Exception: None
def quit(self):
root.quit()
root.destroy()
sys.exit()
def close(self):
#self.tl.quit() #### This was causing multiple errors in 2.0.7 - evaluate more!
self.tl.destroy()
class StringVarFile:
def __init__(self,stringVar,window):
self.__newline = 0; self.__stringvar = stringVar; self.__window = window
def write(self,s):
try:
log_report = open(log_file,'a')
log_report.write(s); log_report.close() ### Variable to record each print statement
new = self.__stringvar.get()
for c in s:
#if c == '\n': self.__newline = 1
if c == '\k': self.__newline = 1### This should not be found and thus results in a continous feed rather than replacing a single line
else:
if self.__newline: new = ""; self.__newline = 0
new = new+c
self.set(new)
except Exception: pass
def set(self,s):
try: self.__stringvar.set(s); self.__window.update()
except Exception: pass
def get(self):
try:
return self.__stringvar.get()
except Exception: pass
def flush(self):
pass
def timestamp():
import datetime
today = str(datetime.date.today()); today = string.split(today,'-'); today = today[0]+''+today[1]+''+today[2]
time_stamp = string.replace(time.ctime(),':','')
time_stamp = string.replace(time_stamp,' ',' ')
time_stamp = string.split(time_stamp,' ') ###Use a time-stamp as the output dir (minus the day)
time_stamp = today+'-'+time_stamp[3]
return time_stamp
def callWXPython():
import wx
import AltAnalyzeViewer
app = wx.App(False)
AltAnalyzeViewer.remoteViewer(app)
def AltAnalyzeSetup(skip_intro):
global apt_location; global root_dir;global log_file; global summary_data_db; summary_data_db={}; reload(UI)
global probability_statistic; global commandLineMode; commandLineMode = 'no'
if 'remoteViewer' == skip_intro:
if os.name == 'nt':
callWXPython()
elif os.name == 'ntX':
package_path = filepath('python')
win_package_path = string.replace(package_path,'python','AltAnalyzeViewer.exe')
import subprocess
subprocess.call([win_package_path]);sys.exit()
elif os.name == 'posix':
package_path = filepath('python')
#mac_package_path = string.replace(package_path,'python','AltAnalyze.app/Contents/MacOS/python')
#os.system(mac_package_path+' RemoteViewer.py');sys.exit()
mac_package_path = string.replace(package_path,'python','AltAnalyzeViewer.app/Contents/MacOS/AltAnalyzeViewer')
import subprocess
subprocess.call([mac_package_path]);sys.exit()
"""
import threading
import wx
app = wx.PySimpleApp()
t = threading.Thread(target=callWXPython)
t.setDaemon(1)
t.start()
s = 1
queue = mlp.Queue()
proc = mlp.Process(target=callWXPython) ### passing sys.stdout unfortunately doesn't work to pass the Tk string
proc.start()
sys.exit()
"""
reload(UI)
expr_var, alt_var, additional_var, goelite_var, exp_file_location_db = UI.getUserParameters(skip_intro,Multi=mlp)
"""except Exception:
if 'SystemExit' not in str(traceback.format_exc()):
expr_var, alt_var, additional_var, goelite_var, exp_file_location_db = UI.getUserParameters('yes')
else: sys.exit()"""
for dataset in exp_file_location_db:
fl = exp_file_location_db[dataset]
apt_location = fl.APTLocation()
root_dir = fl.RootDir()
try: probability_statistic = fl.ProbabilityStatistic()
except Exception: probability_statistic = 'unpaired t-test'
time_stamp = timestamp()
log_file = filepath(root_dir+'AltAnalyze_report-'+time_stamp+'.log')
log_report = open(log_file,'w'); log_report.close()
if use_Tkinter == 'yes' and debug_mode == 'no':
try:
global root; root = Tk()
StatusWindow(root,expr_var, alt_var, goelite_var, additional_var, exp_file_location_db)
root.destroy()
except Exception, exception:
try:
print traceback.format_exc()
badExit()
except Exception: sys.exit()
else: AltAnalyzeMain(expr_var, alt_var, goelite_var, additional_var, exp_file_location_db,'')
def badExit():
print "\n...exiting AltAnalyze due to unexpected error"
try:
time_stamp = timestamp()
print_out = "Unknown error encountered during data processing.\nPlease see logfile in:\n\n"+log_file+"\nand report to [email protected]."
try:
if len(log_file)>0:
if commandLineMode == 'no':
if os.name == 'nt':
try: os.startfile('"'+log_file+'"')
except Exception: os.system('open "'+log_file+'"')
elif 'darwin' in sys.platform: os.system('open "'+log_file+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+log_file+'"')
if commandLineMode == 'no':
try: UI.WarningWindow(print_out,'Error Encountered!'); root.destroy()
except Exception: print print_out
except Exception: sys.exit()
except Exception: sys.exit()
sys.exit()
kill
def AltAnalyzeMain(expr_var,alt_var,goelite_var,additional_var,exp_file_location_db,root):
### Hard-coded defaults
w = 'Agilent'; x = 'Affymetrix'; y = 'Ensembl'; z = 'any'; data_source = y; constitutive_source = z; manufacturer = x ### Constitutive source, is only really paid attention to if Ensembl, otherwise Affymetrix is used (even if default)
### Get default options for ExpressionBuilder and AltAnalyze
start_time = time.time()
test_goelite = 'no'; test_results_pannel = 'no'
global species; global array_type; global expression_data_format; global use_R; use_R = 'no'
global analysis_method; global p_threshold; global filter_probeset_types
global permute_p_threshold; global perform_permutation_analysis; global export_NI_values
global run_MiDAS; global analyze_functional_attributes; global microRNA_prediction_method
global calculate_normIntensity_p; global pathway_permutations; global avg_all_for_ss; global analyze_all_conditions
global remove_intronic_junctions
global agglomerate_inclusion_probesets; global expression_threshold; global factor_out_expression_changes
global only_include_constitutive_containing_genes; global remove_transcriptional_regulated_genes; global add_exons_to_annotations
global exclude_protein_details; global filter_for_AS; global use_direct_domain_alignments_only; global run_from_scratch
global explicit_data_type; explicit_data_type = 'null'
global altanalyze_files; altanalyze_files = []
species,array_type,manufacturer,constitutive_source,dabg_p,raw_expression_threshold,avg_all_for_ss,expression_data_format,include_raw_data, run_from_scratch, perform_alt_analysis = expr_var
analysis_method,p_threshold,filter_probeset_types,alt_exon_fold_variable,gene_expression_cutoff,remove_intronic_junctions,permute_p_threshold,perform_permutation_analysis, export_NI_values, analyze_all_conditions = alt_var
calculate_normIntensity_p, run_MiDAS, use_direct_domain_alignments_only, microRNA_prediction_method, filter_for_AS, additional_algorithms = additional_var
ge_fold_cutoffs,ge_pvalue_cutoffs,ge_ptype,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,pathway_permutations,mod,returnPathways = goelite_var
original_remove_intronic_junctions = remove_intronic_junctions
if run_from_scratch == 'Annotate External Results': analysis_method = 'external'
if returnPathways == 'no' or returnPathways == 'None':
returnPathways = None
for dataset in exp_file_location_db:
fl = exp_file_location_db[dataset]
try: exon_exp_threshold = fl.ExonExpThreshold()
except Exception: exon_exp_threshold = 'NA'
try: gene_exp_threshold = fl.GeneExpThreshold()
except Exception: gene_exp_threshold = 'NA'
try: exon_rpkm_threshold = fl.ExonRPKMThreshold()
except Exception: exon_rpkm_threshold = 'NA'
try: rpkm_threshold = fl.RPKMThreshold() ### Gene-Level
except Exception: rpkm_threshold = 'NA'
fl.setJunctionExpThreshold(raw_expression_threshold) ### For RNA-Seq, this specifically applies to exon-junctions
try: predictGroups = fl.predictGroups()
except Exception: predictGroups = False
try:
if fl.excludeLowExpressionExons(): excludeLowExpExons = 'yes'
else: excludeLowExpExons = 'no'
except Exception: excludeLowExpExons = 'no'
if test_goelite == 'yes': ### It can be difficult to get error warnings from GO-Elite, unless run here
results_dir = filepath(fl.RootDir())
elite_input_dirs = ['AltExonConfirmed','AltExon','regulated','upregulated','downregulated'] ### Run GO-Elite multiple times to ensure heatmaps are useful and to better organize results
for elite_dir in elite_input_dirs:
file_dirs = results_dir+'GO-Elite/'+elite_dir,results_dir+'GO-Elite/denominator',results_dir+'GO-Elite/'+elite_dir
variables = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,root
GO_Elite.remoteAnalysis(variables,'non-UI',Multi=mlp)
global perform_element_permutation_analysis; global permutations
perform_element_permutation_analysis = 'yes'; permutations = 2000
analyze_functional_attributes = 'yes' ### Do this by default (shouldn't substantially increase runtime)
if run_from_scratch != 'Annotate External Results' and (array_type != "3'array" and array_type!='RNASeq'):
if run_from_scratch !='Process AltAnalyze filtered':
try: raw_expression_threshold = float(raw_expression_threshold)
except Exception: raw_expression_threshold = 1
if raw_expression_threshold<1:
raw_expression_threshold = 1
print "Expression threshold < 1, forcing to be a minimum of 1."
try: dabg_p = float(dabg_p)
except Exception: dabg_p = 0
if dabg_p == 0 or dabg_p > 1:
print "Invalid dabg-p value threshold entered,(",dabg_p,") setting to default of 0.05"
dabg_p = 0.05
if use_direct_domain_alignments_only == 'direct-alignment': use_direct_domain_alignments_only = 'yes'
if run_from_scratch == 'Process CEL files': expression_data_format = 'log'
print "Beginning AltAnalyze Analysis... Format:", expression_data_format
if array_type == 'RNASeq': id_name = 'exon/junction IDs'
else: id_name = 'array IDs'
print_items=[]; #print [permute_p_threshold]; sys.exit()
if 'array' in array_type:
dataType='Gene Expression'
else:
dataType=array_type
print_items.append("AltAnalyze version 2.1.4 - Expression Analysis Parameters Being Used...")
print_items.append('\t'+'database'+': '+unique.getCurrentGeneDatabaseVersion())
print_items.append('\t'+'species'+': '+species)
print_items.append('\t'+'method'+': '+dataType)
print_items.append('\t'+'manufacturer'+': '+manufacturer)
print_items.append('\t'+'probability_statistic'+': '+probability_statistic)
print_items.append('\t'+'constitutive_source'+': '+constitutive_source)
print_items.append('\t'+'dabg_p'+': '+str(dabg_p))
if array_type == 'RNASeq':
print_items.append('\t'+'junction expression threshold'+': '+str(raw_expression_threshold))
print_items.append('\t'+'exon_exp_threshold'+': '+str(exon_exp_threshold))
print_items.append('\t'+'gene_exp_threshold'+': '+str(gene_exp_threshold))
print_items.append('\t'+'exon_rpkm_threshold'+': '+str(exon_rpkm_threshold))
print_items.append('\t'+'gene_rpkm_threshold'+': '+str(rpkm_threshold))
print_items.append('\t'+'exclude low expressing exons for RPKM'+': '+excludeLowExpExons)
else:
print_items.append('\t'+'raw_expression_threshold'+': '+str(raw_expression_threshold))
print_items.append('\t'+'avg_all_for_ss'+': '+avg_all_for_ss)
print_items.append('\t'+'expression_data_format'+': '+expression_data_format)
print_items.append('\t'+'include_raw_data'+': '+include_raw_data)
print_items.append('\t'+'run_from_scratch'+': '+run_from_scratch)
print_items.append('\t'+'perform_alt_analysis'+': '+perform_alt_analysis)
if avg_all_for_ss == 'yes': cs_type = 'core'
else: cs_type = 'constitutive'
print_items.append('\t'+'calculate_gene_expression_using'+': '+cs_type)
print_items.append("Alternative Exon Analysis Parameters Being Used..." )
print_items.append('\t'+'analysis_method'+': '+analysis_method)
print_items.append('\t'+'p_threshold'+': '+str(p_threshold))
print_items.append('\t'+'filter_data_types'+': '+filter_probeset_types)
print_items.append('\t'+'alt_exon_fold_variable'+': '+str(alt_exon_fold_variable))
print_items.append('\t'+'gene_expression_cutoff'+': '+str(gene_expression_cutoff))
print_items.append('\t'+'remove_intronic_junctions'+': '+remove_intronic_junctions)
print_items.append('\t'+'avg_all_for_ss'+': '+avg_all_for_ss)
print_items.append('\t'+'permute_p_threshold'+': '+str(permute_p_threshold))
print_items.append('\t'+'perform_permutation_analysis'+': '+perform_permutation_analysis)
print_items.append('\t'+'export_NI_values'+': '+export_NI_values)
print_items.append('\t'+'run_MiDAS'+': '+run_MiDAS)
print_items.append('\t'+'use_direct_domain_alignments_only'+': '+use_direct_domain_alignments_only)
print_items.append('\t'+'microRNA_prediction_method'+': '+microRNA_prediction_method)
print_items.append('\t'+'analyze_all_conditions'+': '+analyze_all_conditions)
print_items.append('\t'+'filter_for_AS'+': '+filter_for_AS)
if pathway_permutations == 'NA': run_GOElite = 'decide_later'
else: run_GOElite = 'run-immediately'
print_items.append('\t'+'run_GOElite'+': '+ run_GOElite)
universalPrintFunction(print_items)
if commandLineMode == 'yes': print 'Running command line mode:',commandLineMode
summary_data_db['gene_assayed'] = 0
summary_data_db['denominator_exp_genes']=0
summary_data_db['alt_events'] = 0
summary_data_db['denominator_exp_events'] = 0
summary_data_db['alt_genes'] = 0
summary_data_db['direct_domain_genes'] = 0
summary_data_db['miRNA_gene_denom'] = 0
summary_data_db['miRNA_gene_hits'] = 0
if test_results_pannel == 'yes': ### It can be difficult to get error warnings from GO-Elite, unless run here
graphic_links = []
graphic_links.append(['test','Config/AltAnalyze_structure-RNASeq.jpg'])
summary_data_db['QC']=graphic_links
print_out = 'Analysis complete. AltAnalyze results\nexported to "AltResults/AlternativeOutput".'
dataset = 'test'; results_dir=''
print "Analysis Complete\n";
if root !='' and root !=None:
UI.InfoWindow(print_out,'Analysis Completed!')
tl = Toplevel(); SummaryResultsWindow(tl,'GE',results_dir,dataset,'parent',summary_data_db)
root.destroy(); sys.exit()
global export_go_annotations; global aspire_output_list; global aspire_output_gene_list
global filter_probesets_by; global global_addition_factor; global onlyAnalyzeJunctions
global log_fold_cutoff; global aspire_cutoff; global annotation_system; global alt_exon_logfold_cutoff
"""dabg_p = 0.75; data_type = 'expression' ###used for expression analysis when dealing with AltMouse arrays
a = "3'array"; b = "exon"; c = "AltMouse"; e = "custom"; array_type = c
l = 'log'; n = 'non-log'; expression_data_format = l
hs = 'Hs'; mm = 'Mm'; dr = 'Dr'; rn = 'Rn'; species = mm
include_raw_data = 'yes'; expression_threshold = 70 ### Based on suggestion from BMC Genomics. 2006 Dec 27;7:325. PMID: 17192196, for hu-exon 1.0 st array
avg_all_for_ss = 'no' ###Default is 'no' since we don't want all probes averaged for the exon arrays"""
###### Run ExpressionBuilder ######
"""ExpressionBuilder is used to:
(1) extract out gene expression values, provide gene annotations, and calculate summary gene statistics
(2) filter probesets based DABG p-values and export to pair-wise comparison files
(3) build array annotations files matched to gene structure features (e.g. exons, introns) using chromosomal coordinates
options 1-2 are executed in remoteExpressionBuilder and option 3 is by running ExonArrayEnsembl rules"""
try:
additional_algorithm = additional_algorithms.Algorithm()
additional_score = additional_algorithms.Score()
except Exception: additional_algorithm = 'null'; additional_score = 'null'
if analysis_method == 'FIRMA': analyze_metaprobesets = 'yes'
elif additional_algorithm == 'FIRMA': analyze_metaprobesets = 'yes'
else: analyze_metaprobesets = 'no'
### Check to see if this is a real or FAKE (used for demonstration purposes) dataset
if run_from_scratch == 'Process CEL files' or 'Feature Extraction' in run_from_scratch:
for dataset in exp_file_location_db:
if run_from_scratch == 'Process CEL files':
fl = exp_file_location_db[dataset]
pgf_file=fl.InputCDFFile()
results_dir = filepath(fl.RootDir())
if '_demo' in pgf_file: ### Thus we are running demo CEL files and want to quit immediately
print_out = 'Analysis complete. AltAnalyze results\nexported to "AltResults/AlternativeOutput".'
try:
print "Analysis Complete\n";
if root !='' and root !=None:
UI.InfoWindow(print_out,'Analysis Completed!')
tl = Toplevel(); SummaryResultsWindow(tl,'AS',results_dir,dataset,'parent',summary_data_db)
except Exception: null=[]
skip_intro = 'yes'
if pathway_permutations == 'NA' and run_from_scratch != 'Annotate External Results':
reload(UI)
UI.getUpdatedParameters(array_type,species,run_from_scratch,results_dir)
try: AltAnalyzeSetup('no')
except Exception: sys.exit()
if 'CEL files' in run_from_scratch:
from import_scripts import APT
try:
try:
APT.probesetSummarize(exp_file_location_db,analyze_metaprobesets,filter_probeset_types,species,root)
if analyze_metaprobesets == 'yes':
analyze_metaprobesets = 'no' ### Re-run the APT analysis to obtain probeset rather than gene-level results (only the residuals are needed from a metaprobeset run)
APT.probesetSummarize(exp_file_location_db,analyze_metaprobesets,filter_probeset_types,species,root)
except Exception:
import platform
print "Trying to change APT binary access privileges"
for dataset in exp_file_location_db: ### Instance of the Class ExpressionFileLocationData
fl = exp_file_location_db[dataset]; apt_dir =fl.APTLocation()
if '/bin' in apt_dir: apt_file = apt_dir +'/apt-probeset-summarize' ### if the user selects an APT directory
elif os.name == 'nt': apt_file = apt_dir + '/PC/'+platform.architecture()[0]+'/apt-probeset-summarize.exe'
elif 'darwin' in sys.platform: apt_file = apt_dir + '/Mac/apt-probeset-summarize'
elif 'linux' in sys.platform:
if '32bit' in platform.architecture(): apt_file = apt_dir + '/Linux/32bit/apt-probeset-summarize'
elif '64bit' in platform.architecture(): apt_file = apt_dir + '/Linux/64bit/apt-probeset-summarize'
apt_file = filepath(apt_file)
os.chmod(apt_file,0777)
midas_dir = string.replace(apt_file,'apt-probeset-summarize','apt-midas')
os.chmod(midas_dir,0777)
APT.probesetSummarize(exp_file_location_db,analysis_method,filter_probeset_types,species,root)
except Exception:
print_out = 'AltAnalyze encountered an un-expected error while running Affymetrix\n'
print_out += 'Power Tools (APT). Additional information may be found in the directory\n'
print_out += '"ExpressionInput/APT" in the output directory. You may also encounter issues\n'
print_out += 'if you are logged into an account with restricted priveledges.\n\n'
print_out += 'If this issue can not be resolved, contact AltAnalyze help or run RMA outside\n'
print_out += 'of AltAnalyze and import the results using the analysis option "expression file".\n'
print traceback.format_exc()
try:
UI.WarningWindow(print_out,'Exit')
root.destroy(); sys.exit()
except Exception:
print print_out; sys.exit()
elif 'Feature Extraction' in run_from_scratch:
from import_scripts import ProcessAgilentArrays
try: ProcessAgilentArrays.agilentSummarize(exp_file_location_db)
except Exception:
print_out = 'Agilent array import and processing failed... see error log for details...'
print traceback.format_exc()
try:
UI.WarningWindow(print_out,'Exit')
root.destroy(); sys.exit()
except Exception:
print print_out; sys.exit()
reload(ProcessAgilentArrays)
if run_from_scratch == 'Process RNA-seq reads' or run_from_scratch == 'buildExonExportFiles':
import RNASeq; reload(RNASeq); import RNASeq
for dataset in exp_file_location_db: fl = exp_file_location_db[dataset]
### The below function aligns splice-junction coordinates to Ensembl exons from BED Files and
### exports AltAnalyze specific databases that are unique to this dataset to the output directory
try: fastq_folder = fl.RunKallisto()
except Exception: print traceback.format_exc()
try: customFASTA = fl.CustomFASTA()
except Exception: customFASTA = None
processBEDfiles = True
if len(fastq_folder)>0:
### Perform pseudoalignment with Kallisto on FASTQ files
processBEDfiles=False
try:
RNASeq.runKallisto(species,dataset,root_dir,fastq_folder,mlp,returnSampleNames=False,customFASTA=customFASTA)
biotypes = 'ran'
dir_list = unique.read_directory(root_dir)
### If we are performing a splicing analysis
if perform_alt_analysis != 'no' and perform_alt_analysis != 'expression':
print '...Performing analyses on junction-RPKM versus Kallisto-TPM.'
for file in dir_list:
if '.bam' in string.lower(file):
processBEDfiles=True
if '.bed' in string.lower(file):
processBEDfiles=True
if processBEDfiles:
try: rpkm_threshold = fl.RPKMThreshold()
except Exception: rpkm_threshold = []
if isinstance(rpkm_threshold, int) ==False:
array_type = 'RNASeq'
fl.setArrayType(array_type)
fl.setBEDFileDir(root_dir)
fl.setRPKMThreshold(1.0)
fl.setExonExpThreshold(5.0)
fl.setGeneExpThreshold(200.0)
fl.setExonRPKMThreshold(0.5)
fl.setJunctionExpThreshold(5.0)
fl.setVendor('RNASeq')
### Export BAM file indexes
try:
from import_scripts import BAMtoJunctionBED
try: BAMtoJunctionBED.exportIndexes(root_dir)
except:
print 'BAM file indexing failed...'
print traceback.format_exc()
except: print 'BAM file support missing due to lack of pysam...'
else:
print '...Performing analyses on Kallisto-TPM values directly.'
array_type = "3'array"
fl.setArrayType(array_type)
vendor = 'other:Ensembl' ### Ensembl linked system name
fl.setVendor(vendor)
else:
print '...Performing analyses on Kallisto-TPM values directly.'
array_type = "3'array"
fl.setArrayType(array_type)
vendor = 'other:Ensembl' ### Ensembl linked system name
fl.setVendor(vendor)
except Exception:
print traceback.format_exc()
biotypes='failed'
if processBEDfiles:
analyzeBAMs = False; bedFilesPresent = False
dir_list = unique.read_directory(fl.BEDFileDir())
for file in dir_list:
if '.bam' in string.lower(file):
analyzeBAMs=True
if '.bed' in string.lower(file):
bedFilesPresent=True
if analyzeBAMs and bedFilesPresent==False:
from import_scripts import multiBAMtoBED
bam_dir = fl.BEDFileDir()
refExonCoordinateFile = filepath('AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt')
outputExonCoordinateRefBEDfile = bam_dir+'/BedRef/'+species+'_'+string.replace(dataset,'exp.','')
analysisType = ['exon','junction','reference']
#analysisType = ['junction']
#print [fl.multiThreading()]
multiBAMtoBED.parallelBAMProcessing(bam_dir,refExonCoordinateFile,outputExonCoordinateRefBEDfile,analysisType=analysisType,useMultiProcessing=fl.multiThreading(),MLP=mlp,root=root)
biotypes = RNASeq.alignExonsAndJunctionsToEnsembl(species,exp_file_location_db,dataset,Multi=mlp)
if biotypes == 'failed':
print_out = 'No valid chromosomal positions in the input BED or BioScope files. Exiting AltAnalyze.'
if len(fastq_folder)>0:
if 'FTP' in traceback.format_exc():
print_out = 'AltAnlayze was unable to retreive a transcript fasta sequence file from the Ensembl website. '
print_out += 'Ensure you are connected to the internet and that the website http://ensembl.org is live.'
else:
print_out = 'An unexplained error was encountered with Kallisto analysis:\n'
print_out += traceback.format_exc()
try:
UI.WarningWindow(print_out,'Exit')
root.destroy(); sys.exit()
except Exception:
print print_out; sys.exit()
reload(RNASeq)
if root_dir in biotypes:
print_out = 'Exon-level BED coordinate predictions exported to:\n'+biotypes
print_out+= '\n\nAfter obtaining exon expression estimates, rename exon BED files to\n'
print_out+= 'match the junction name (e.g., Sample1__exon.bed and Sample1__junction.bed)\n'
print_out+= 'and re-run AltAnalyze (see tutorials at http://altanalyze.org for help).'
UI.InfoWindow(print_out,'Export Complete')
try: root.destroy(); sys.exit()
except Exception: sys.exit()
if predictGroups == True:
expFile = fl.ExpFile()
if array_type == 'RNASeq':
exp_threshold=100; rpkm_threshold=10
else:
exp_threshold=200; rpkm_threshold=8
RNASeq.singleCellRNASeqWorkflow(species, array_type, expFile, mlp, exp_threshold=exp_threshold, rpkm_threshold=rpkm_threshold)
goelite_run = False
if run_from_scratch == 'Process Expression file' or run_from_scratch == 'Process CEL files' or run_from_scratch == 'Process RNA-seq reads' or 'Feature Extraction' in run_from_scratch:
if (fl.NormMatrix()=='quantile' or fl.NormMatrix()=='group') and 'Feature Extraction' not in run_from_scratch:
from stats_scripts import NormalizeDataset
try: NormalizeDataset.normalizeDataset(fl.ExpFile(),normalization=fl.NormMatrix(),platform=array_type)
except Exception: print "Normalization failed for unknown reasons..."
#"""
status = ExpressionBuilder.remoteExpressionBuilder(species,array_type,
dabg_p,raw_expression_threshold,avg_all_for_ss,expression_data_format,
manufacturer,constitutive_source,data_source,include_raw_data,
perform_alt_analysis,ge_fold_cutoffs,ge_pvalue_cutoffs,ge_ptype,
exp_file_location_db,root)
reload(ExpressionBuilder) ### Clears Memory
#"""
graphics=[]
if fl.MarkerFinder() == 'yes':
### Identify putative condition-specific marker genees
import markerFinder
fl.setOutputDir(root_dir) ### This needs to be set here
exp_file = fl.ExpFile()
if array_type != "3'array": exp_file = string.replace(exp_file,'.txt','-steady-state.txt')
try:
exp_file = fl.KallistoFile() ### Override with the Kallisto expression file if present
print 'Using the Kallisto expressio file for MarkerFinder...'
except: pass
markerFinder_inputs = [exp_file,fl.DatasetFile()] ### Output a replicate and non-replicate version
markerFinder_inputs = [exp_file] ### Only considers the replicate and not mean analysis (recommended)
for input_exp_file in markerFinder_inputs:
### This applies to an ExpressionOutput DATASET file compoosed of gene expression values (averages already present)
try:
output_dir = markerFinder.getAverageExpressionValues(input_exp_file,array_type) ### Either way, make an average annotated file from the DATASET file
except Exception:
print "Unknown MarkerFinder failure (possible filename issue or data incompatibility)..."
print traceback.format_exc()
continue
if 'DATASET' in input_exp_file:
group_exp_file = string.replace(input_exp_file,'DATASET','AVERAGE')
else:
group_exp_file = (input_exp_file,output_dir) ### still analyze the primary sample
compendiumType = 'protein_coding'
if expression_data_format == 'non-log': logTransform = True
else: logTransform = False
try: markerFinder.analyzeData(group_exp_file,species,array_type,compendiumType,AdditionalParameters=fl,logTransform=logTransform)
except Exception: None
### Generate heatmaps (unclustered - order by markerFinder)
try: graphics = markerFinder.generateMarkerHeatMaps(fl,array_type,graphics=graphics,Species=species)
except Exception: print traceback.format_exc()
remove_intronic_junctions = original_remove_intronic_junctions ### This var gets reset when running FilterDABG
try:
summary_data_db['QC'] = fl.GraphicLinks()+graphics ### provides links for displaying QC and clustering plots
except Exception:
null=[] ### Visualization support through matplotlib either not present or visualization options excluded
#print '!!!!!finished expression builder'
#returnLargeGlobalVars()
expression_data_format = 'log' ### This variable is set from non-log in FilterDABG when present (version 1.16)
try:
parent_dir = fl.RootDir()+'/GO-Elite/regulated/'
dir_list = read_directory(parent_dir)
for file in dir_list:
input_file_dir = parent_dir+'/'+file
inputType = 'IDs'
interactionDirs = ['WikiPathways','KEGG','BioGRID','TFTargets']
output_dir = parent_dir
degrees = 'direct'
input_exp_file = input_file_dir
gsp = UI.GeneSelectionParameters(species,array_type,manufacturer)
gsp.setGeneSet('None Selected')
gsp.setPathwaySelect('')
gsp.setGeneSelection('')
gsp.setOntologyID('')
gsp.setIncludeExpIDs(True)
UI.networkBuilder(input_file_dir,inputType,output_dir,interactionDirs,degrees,input_exp_file,gsp,'')
except Exception:
#print traceback.format_exc()
pass
if status == 'stop':
### See if the array and species are compatible with GO-Elite analysis
system_codes = UI.getSystemInfo()
go_elite_analysis_supported = 'yes'
species_names = UI.getSpeciesInfo()
for dataset in exp_file_location_db:
fl = exp_file_location_db[dataset]; results_dir = filepath(fl.RootDir())
### Perform GO-Elite Analysis
if pathway_permutations != 'NA':
try:
print '\nBeginning to run GO-Elite analysis on alternative exon results'
elite_input_dirs = ['AltExonConfirmed','AltExon','regulated','upregulated','downregulated'] ### Run GO-Elite multiple times to ensure heatmaps are useful and to better organize results
for elite_dir in elite_input_dirs:
file_dirs = results_dir+'GO-Elite/'+elite_dir,results_dir+'GO-Elite/denominator',results_dir+'GO-Elite/'+elite_dir
input_dir = results_dir+'GO-Elite/'+elite_dir
variables = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,root
try: input_files = read_directory(input_dir) ### Are there any files to analyze?
except Exception: input_files=[]
if len(input_files)>0:
try: GO_Elite.remoteAnalysis(variables,'non-UI',Multi=mlp); goelite_run = True
except Exception,e:
print e
print "GO-Elite analysis failed"
try: GO_Elite.moveMAPPFinderFiles(file_dirs[0])
except Exception: print 'Input GO-Elite files could NOT be moved.'
try: GO_Elite.moveMAPPFinderFiles(file_dirs[1])
except Exception: print 'Input GO-Elite files could NOT be moved.'
except Exception: pass
if goelite_run == False:
print 'No GO-Elite input files to analyze (check your criterion).'
print_out = 'Analysis complete. Gene expression\nsummary exported to "ExpressionOutput".'
try:
if use_Tkinter == 'yes':
print "Analysis Complete\n"; UI.InfoWindow(print_out,'Analysis Completed!')
tl = Toplevel(); SummaryResultsWindow(tl,'GE',results_dir,dataset,'parent',summary_data_db)
if pathway_permutations == 'NA' and run_from_scratch != 'Annotate External Results':
if go_elite_analysis_supported == 'yes':
UI.getUpdatedParameters(array_type,species,run_from_scratch,file_dirs)
try: AltAnalyzeSetup('no')
except Exception:
print traceback.format_exc()
sys.exit()
else: print '\n'+print_out; sys.exit()
except Exception:
#print 'Failed to report status through GUI.'
sys.exit()
else: altanalyze_files = status[1] ### These files are the comparison files to analyze
elif run_from_scratch == 'update DBs':
null=[] ###Add link to new module here (possibly)
#updateDBs(species,array_type)
sys.exit()
if perform_alt_analysis != 'expression': ###Thus perform_alt_analysis = 'both' or 'alt' (default when skipping expression summary step)
###### Run AltAnalyze ######
global dataset_name; global summary_results_db; global summary_results_db2
summary_results_db={}; summary_results_db2={}; aspire_output_list=[]; aspire_output_gene_list=[]
onlyAnalyzeJunctions = 'no'; agglomerate_inclusion_probesets = 'no'; filter_probesets_by = 'NA'
if array_type == 'AltMouse' or ((array_type == 'junction' or array_type == 'RNASeq') and explicit_data_type == 'null'):
if filter_probeset_types == 'junctions-only': onlyAnalyzeJunctions = 'yes'
elif filter_probeset_types == 'combined-junctions': agglomerate_inclusion_probesets = 'yes'; onlyAnalyzeJunctions = 'yes'
elif filter_probeset_types == 'exons-only': analysis_method = 'splicing-index'; filter_probesets_by = 'exon'
if filter_probeset_types == 'combined-junctions' and array_type == 'junction' or array_type == 'RNASeq': filter_probesets_by = 'all'
else: filter_probesets_by = filter_probeset_types
c = 'Ensembl'; d = 'Entrez Gene'
annotation_system = c
expression_threshold = 0 ###This is different than the raw_expression_threshold (probably shouldn't filter so set to 0)
if analysis_method == 'linearregres-rlm': analysis_method = 'linearregres';use_R = 'yes'
if gene_expression_cutoff<1:
gene_expression_cutoff = 2 ### A number less than one is invalid
print "WARNING!!!! Invalid gene expression fold cutoff entered,\nusing the default value of 2, must be greater than 1."
log_fold_cutoff = math.log(float(gene_expression_cutoff),2)
if analysis_method != 'ASPIRE' and analysis_method != 'none' and analysis_method != 'MultiPath-PSI':
if p_threshold <= 0 or p_threshold >1:
p_threshold = 0.05 ### A number less than one is invalid
print "WARNING!!!! Invalid alternative exon p-value threshold entered,\nusing the default value of 0.05."
if alt_exon_fold_variable<1:
alt_exon_fold_variable = 1 ### A number less than one is invalid
print "WARNING!!!! Invalid alternative exon fold cutoff entered,\nusing the default value of 2, must be greater than 1."
try: alt_exon_logfold_cutoff = math.log(float(alt_exon_fold_variable),2)
except Exception: alt_exon_logfold_cutoff = 1
else: alt_exon_logfold_cutoff = float(alt_exon_fold_variable)
global_addition_factor = 0
export_junction_comparisons = 'no' ### No longer accessed in this module - only in update mode through a different module
factor_out_expression_changes = 'yes' ### Use 'no' if data is normalized already or no expression normalization for ASPIRE desired
only_include_constitutive_containing_genes = 'yes'
remove_transcriptional_regulated_genes = 'yes'
add_exons_to_annotations = 'no'
exclude_protein_details = 'no'
if analysis_method == 'ASPIRE' or 'linearregres' in analysis_method: annotation_system = d
if 'linear' in analysis_method: analysis_method = 'linearregres'
if 'aspire' in analysis_method: analysis_method = 'ASPIRE'
if array_type == 'AltMouse': species = 'Mm'
#if export_NI_values == 'yes': remove_transcriptional_regulated_genes = 'no'
###Saves run-time while testing the software (global variable stored)
#import_dir = '/AltDatabase/affymetrix/'+species
#dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
### Get Ensembl-GO and pathway annotations from GO-Elite files
universalPrintFunction(["Importing GO-Elite pathway/GO annotations"])
global go_annotations; go_annotations={}
from import_scripts import BuildAffymetrixAssociations
go_annotations = BuildAffymetrixAssociations.getEnsemblAnnotationsFromGOElite(species)
global probeset_annotations_file
if array_type == 'RNASeq': probeset_annotations_file = root_dir+'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_junctions.txt'
elif array_type == 'AltMouse': probeset_annotations_file = 'AltDatabase/'+species+'/'+array_type+'/'+'MASTER-probeset-transcript.txt'
else: probeset_annotations_file = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_probesets.txt'
#"""
if analysis_method != 'none' and analysis_method != 'MultiPath-PSI':
analysis_summary = RunAltAnalyze() ### Only run if analysis methods is specified (only available for RNA-Seq and junction analyses)
else: analysis_summary = None
if analysis_summary != None:
summary_results_db, aspire_output_gene_list, number_events_analyzed = analysis_summary
summary_data_db2 = copy.deepcopy(summary_data_db)
for i in summary_data_db2: del summary_data_db[i] ### If we reset the variable it violates it's global declaration... do this instead
#universalPrintFunction(['Alternative Exon Results for Junction Comparisons:'])
#for i in summary_data_db: universalPrintFunction([i+' '+ str(summary_data_db[i])])
exportSummaryResults(summary_results_db,analysis_method,aspire_output_list,aspire_output_gene_list,annotate_db,array_type,number_events_analyzed,root_dir)
else:
### Occurs for RNASeq when no junctions are present
summary_data_db2={}
if array_type == 'junction' or array_type == 'RNASeq':
#Reanalyze junction array data separately for individual probests rather than recipricol junctions
if array_type == 'junction': explicit_data_type = 'exon'
elif array_type == 'RNASeq': explicit_data_type = 'junction'
else: report_single_probeset_results = 'no'
### Obtain exon analysis defaults
expr_defaults, alt_exon_defaults, functional_analysis_defaults, goelite_defaults = UI.importDefaults('exon',species)
analysis_method, null, filter_probeset_types, null, null, alt_exon_fold_variable, null, null, null, null, null, null, null, calculate_normIntensity_p, null = alt_exon_defaults
filter_probesets_by = filter_probeset_types
if additional_algorithm == 'splicing-index' or additional_algorithm == 'FIRMA':
analysis_method = additional_algorithm
#print [analysis_method], [filter_probeset_types], [p_threshold], [alt_exon_fold_variable]
try: alt_exon_logfold_cutoff = math.log(float(additional_score),2)
except Exception: alt_exon_logfold_cutoff = 1
agglomerate_inclusion_probesets = 'no'
try:
summary_results_db, aspire_output_gene_list, number_events_analyzed = RunAltAnalyze()
exportSummaryResults(summary_results_db,analysis_method,aspire_output_list,aspire_output_gene_list,annotate_db,'exon',number_events_analyzed,root_dir)
if len(summary_data_db2)==0: summary_data_db2 = summary_data_db; explicit_data_type = 'exon-only'
#universalPrintFunction(['Alternative Exon Results for Individual Probeset Analyses:'])
#for i in summary_data_db: universalPrintFunction([i+' '+ str(summary_data_db[i])])
except Exception:
print traceback.format_exc()
None
#"""
### Perform dPSI Analysis
try:
if 'counts.' in fl.CountsFile(): pass
else:
dir_list = read_directory(fl.RootDir()+'ExpressionInput')
for file in dir_list:
if 'exp.' in file and 'steady-state' not in file:
fl.setExpFile(fl.RootDir()+'ExpressionInput/'+file)
#print [fl.RootDir()+'ExpressionInput/'+file]
if 'groups.' in file:
fl.setGroupsFile(search_dir+'/'+file)
except Exception:
search_dir = fl.RootDir()+'/ExpressionInput'
files = unique.read_directory(fl.RootDir()+'/ExpressionInput')
for file in files:
if 'exp.' in file and 'steady-state.txt' not in file:
fl.setExpFile(search_dir+'/'+file)
if 'groups.' in file:
fl.setGroupsFile(search_dir+'/'+file)
try:
#"""
try:
#"""
graphic_links2,cluster_input_file=ExpressionBuilder.unbiasedComparisonSpliceProfiles(fl.RootDir(),
species,array_type,expFile=fl.CountsFile(),min_events=1,med_events=1)
#"""
from import_scripts import AugmentEventAnnotations
psi_annotated = AugmentEventAnnotations.parse_junctionfiles(fl.RootDir()+'/AltResults/AlternativeOutput/',species,array_type) ### Added in 2.1.1 - adds cassette and domains annotations
except Exception:
print traceback.format_exc()
pass
#"""
inputpsi = fl.RootDir()+'AltResults/AlternativeOutput/'+species+'_'+array_type+'_top_alt_junctions-PSI-clust.txt'
useAdvancedMetaDataAnalysis = True
### Calculate ANOVA p-value stats based on groups
if array_type !='gene' and array_type != 'exon':
if useAdvancedMetaDataAnalysis:
from stats_scripts import metaDataAnalysis
if ge_ptype == 'adjp':
use_adjusted_pval = True
else:
use_adjusted_pval = False
try:
log_fold_cutoff = float(alt_exon_fold_variable)
if log_fold_cutoff == 0.1:
log_fold_cutoff = 0.1 ### For significant digits
except: log_fold_cutoff = 0.1
try:
if p_threshold <= 0 or p_threshold >1:
pvalThreshold = 0.05 ### A number less than one is invalid
else: pvalThreshold = p_threshold
except: pvalThreshold = ge_pvalue_cutoffs ### Use the gene expression p-value cutoff if NA
try:
graphics_alt = metaDataAnalysis.remoteAnalysis(species,psi_annotated,fl.GroupsFile(),
platform='PSI',log_fold_cutoff=0.1,use_adjusted_pval=use_adjusted_pval,
pvalThreshold=ge_pvalue_cutoffs)
try: summary_data_db['QC'] += graphics_alt
except Exception: summary_data_db['QC'] = graphics_alt
try: summary_data_db['QC'] += graphic_links2
except Exception: pass
except Exception:
print traceback.format_exc()
else:
matrix,compared_groups,original_data = statistics.matrixImport(inputpsi)
matrix_pvalues=statistics.runANOVA(inputpsi,matrix,compared_groups)
significantFilteredDir = statistics.returnANOVAFiltered(inputpsi,original_data,matrix_pvalues)
graphic_link1 = ExpressionBuilder.exportHeatmap(significantFilteredDir)
try: summary_data_db['QC']+=graphic_link1
except Exception: summary_data_db['QC']=graphic_link1
except Exception:
print traceback.format_exc()
import RNASeq
try:
graphic_link = RNASeq.compareExonAndJunctionResults(species,array_type,summary_results_db,root_dir)
try: summary_data_db['QC']+=graphic_link
except Exception: summary_data_db['QC']=graphic_link
except Exception:
print traceback.format_exc()
#"""
### Export the top 15 spliced genes
try:
altresult_dir = fl.RootDir()+'/AltResults/'
splicing_results_root = altresult_dir+'/Clustering/'
dir_list = read_directory(splicing_results_root)
gene_string=''
altanalyze_results_folder = altresult_dir+'/RawSpliceData/'+species
### Lookup the raw expression dir
expression_results_folder = string.replace(altresult_dir,'AltResults','ExpressionInput')
expression_dir = UI.getValidExpFile(expression_results_folder)
show_introns=False
try: altresult_dir = UI.getValidSplicingScoreFile(altanalyze_results_folder)
except Exception,e:
print traceback.format_exc()
analysisType='plot'
for file in dir_list:
if 'AltExonConfirmed' in file:
gene_dir = splicing_results_root+'/'+file
genes = UI.importGeneList(gene_dir,limit=50) ### list of gene IDs or symbols
gene_string = gene_string+','+genes
print 'Imported genes from',file,'\n'
analysisType='plot'
for file in dir_list:
if 'Combined-junction-exon-evidence' in file and 'top' not in file:
gene_dir = splicing_results_root+'/'+file
try: isoform_dir = UI.exportJunctionList(gene_dir,limit=50) ### list of gene IDs or symbols
except Exception: print traceback.format_exc()
UI.altExonViewer(species,array_type,expression_dir, gene_string, show_introns, analysisType, None); print 'completed'
UI.altExonViewer(species,array_type,altresult_dir, gene_string, show_introns, analysisType, None); print 'completed'
except Exception:
#print traceback.format_exc()
pass
if array_type != 'exon' and array_type != 'gene':
### SashimiPlot Visualization
try:
expression_results_folder = fl.RootDir()+'/ExpressionInput/'
expression_dir = UI.getValidExpFile(expression_results_folder)
show_introns=False
analysisType='plot'
top_PSI_junction = inputpsi
#isoform_dir2 = UI.exportJunctionList(top_PSI_junction,limit=50) ### list of gene IDs or symbols
altoutput_dir = export.findParentDir(top_PSI_junction)
isoform_dir2 = altoutput_dir+'/top'+str(50)+'/MultiPath-PSI.txt'
gene_string = UI.importGeneList(isoform_dir2,limit=50)
UI.altExonViewer(species,array_type,expression_dir, gene_string, show_introns, analysisType, None); print 'completed'
UI.altExonViewer(species,array_type,altresult_dir, gene_string, show_introns, analysisType, None); print 'completed'
except Exception:
print traceback.format_exc()
try:
analyzeBAMs = False
dir_list = unique.read_directory(fl.RootDir())
for file in dir_list:
if '.bam' in string.lower(file):
analyzeBAMs=True
if analyzeBAMs:
### Create sashimi plot index
from visualization_scripts import SashimiIndex
SashimiIndex.remoteIndexing(species,fl)
from visualization_scripts import SashimiPlot
print 'Exporting Sashimi Plots for the top-predicted splicing events... be patient'
try: SashimiPlot.remoteSashimiPlot(species,fl,fl.RootDir(),isoform_dir) ### assuming the bam files are in the root-dir
except Exception: pass # print traceback.format_exc()
print 'completed'
try: SashimiPlot.remoteSashimiPlot(species,fl,fl.RootDir(),isoform_dir2) ### assuming the bam files are in the root-dir
except Exception: pass #print traceback.format_exc()
print 'completed'
### Try again, in case the symbol conversion failed
SashimiPlot.justConvertFilenames(species,fl.RootDir()+'/SashimiPlots')
else:
print 'No BAM files present in the root directory... skipping SashimiPlot analysis...'
except Exception:
print traceback.format_exc()
try:
clearObjectsFromMemory(exon_db); clearObjectsFromMemory(constitutive_probeset_db)
clearObjectsFromMemory(go_annotations); clearObjectsFromMemory(original_microRNA_z_score_data)
clearObjectsFromMemory(last_exon_region_db)
"""
print 'local vars'
all = [var for var in locals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(locals()[var])>500: print var, len(locals()[var])
except Exception: null=[]
"""
except Exception: null=[]
#print '!!!!!finished'
#returnLargeGlobalVars()
end_time = time.time(); time_diff = int(end_time-start_time)
universalPrintFunction(["Analyses finished in %d seconds" % time_diff])
#universalPrintFunction(["Hit Enter/Return to exit AltAnalyze"])
for dataset in exp_file_location_db:
fl = exp_file_location_db[dataset]; results_dir = filepath(fl.RootDir())
### Perform GO-Elite Analysis
if pathway_permutations != 'NA':
goelite_run = False
print '\nBeginning to run GO-Elite analysis on alternative exon results'
elite_input_dirs = ['AltExonConfirmed','AltExon','regulated','upregulated','downregulated'] ### Run GO-Elite multiple times to ensure heatmaps are useful and to better organize results
for elite_dir in elite_input_dirs:
file_dirs = results_dir+'GO-Elite/'+elite_dir,results_dir+'GO-Elite/denominator',results_dir+'GO-Elite/'+elite_dir
input_dir = results_dir+'GO-Elite/'+elite_dir
try: input_files = read_directory(input_dir) ### Are there any files to analyze?
except Exception: input_files = []
if len(input_files)>0:
variables = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,root
try: GO_Elite.remoteAnalysis(variables,'non-UI',Multi=mlp); goelite_run = True
except Exception,e:
print e
print "GO-Elite analysis failed"
try: GO_Elite.moveMAPPFinderFiles(file_dirs[0])
except Exception: print 'Input GO-Elite files could NOT be moved.'
try: GO_Elite.moveMAPPFinderFiles(file_dirs[1])
except Exception: print 'Input GO-Elite files could NOT be moved.'
if goelite_run == False:
print 'No GO-Elite input files to analyze (check your criterion).'
print_out = 'Analysis complete. AltAnalyze results\nexported to "AltResults/AlternativeOutput".'
try:
if root !='' and root !=None:
print "Analysis Complete\n";
UI.InfoWindow(print_out,'Analysis Completed!')
try: dataset_name = dataset_name
except: dataset_name = dataset
#tl = Toplevel(); SummaryResultsWindow(tl,'AS',results_dir,dataset_name,'specific',summary_data_db2)
tl = Toplevel(); SummaryResultsWindow(tl,'GE',results_dir,dataset_name,'specific',summary_data_db)
except Exception:
print traceback.format_exc()
pass #print 'Failed to open GUI.'
skip_intro = 'yes'
if root !='' and root !=None:
if pathway_permutations == 'NA' and run_from_scratch != 'Annotate External Results':
try: UI.getUpdatedParameters(array_type,species,run_from_scratch,file_dirs)
except Exception: pass
try: AltAnalyzeSetup('no')
except Exception: sys.exit()
def exportSummaryResults(summary_results_db,analysis_method,aspire_output_list,aspire_output_gene_list,annotate_db,array_type,number_events_analyzed,root_dir):
try:
ResultsExport_module.outputSummaryResults(summary_results_db,'',analysis_method,root_dir)
#ResultsExport_module.outputSummaryResults(summary_results_db2,'-uniprot_attributes',analysis_method)
ResultsExport_module.compareAltAnalyzeResults(aspire_output_list,annotate_db,number_events_analyzed,'no',analysis_method,array_type,root_dir)
ResultsExport_module.compareAltAnalyzeResults(aspire_output_gene_list,annotate_db,'','yes',analysis_method,array_type,root_dir)
except UnboundLocalError: print "...No results to summarize" ###Occurs if there is a problem parsing these files
def checkGOEliteProbesets(fn,species):
### Get all probesets in GO-Elite files
mod_source = 'Ensembl'+'-'+'Affymetrix'
import gene_associations
try: ensembl_to_probeset_id = gene_associations.getGeneToUid(species,mod_source)
except Exception: ensembl_to_probeset_id={}
mod_source = 'EntrezGene'+'-'+'Affymetrix'
try: entrez_to_probeset_id = gene_associations.getGeneToUid(species,mod_source)
except Exception: entrez_to_probeset_id={}
probeset_db={}
for gene in ensembl_to_probeset_id:
for probeset in ensembl_to_probeset_id[gene]: probeset_db[probeset]=[]
for gene in entrez_to_probeset_id:
for probeset in entrez_to_probeset_id[gene]: probeset_db[probeset]=[]
###Import an Affymetrix array annotation file (from http://www.affymetrix.com) and parse out annotations
csv_probesets = {}; x=0; y=0
fn=filepath(fn); status = 'no'
for line in open(fn,'r').readlines():
probeset_data = string.replace(line,'\n','') #remove endline
probeset_data = string.replace(probeset_data,'---','')
affy_data = string.split(probeset_data[1:-1],'","')
if x==0 and line[0]!='#':
x=1; affy_headers = affy_data
for header in affy_headers:
y = 0
while y < len(affy_headers):
if 'Probe Set ID' in affy_headers[y] or 'probeset_id' in affy_headers[y]: ps = y
y+=1
elif x == 1:
try: probeset = affy_data[ps]; csv_probesets[probeset]=[]
except Exception: null=[]
for probeset in csv_probesets:
if probeset in probeset_db: status = 'yes';break
return status
class SpeciesData:
def __init__(self, abrev, species, systems, taxid):
self._abrev = abrev; self._species = species; self._systems = systems; self._taxid = taxid
def SpeciesCode(self): return self._abrev
def SpeciesName(self): return self._species
def Systems(self): return self._systems
def TaxID(self): return self._taxid
def __repr__(self): return self.SpeciesCode()+'|'+SpeciesName
def getSpeciesInfo():
### Used by AltAnalyze
UI.importSpeciesInfo(); species_names={}
for species_full in species_codes:
sc = species_codes[species_full]; abrev = sc.SpeciesCode()
species_names[abrev] = species_full
return species_codes,species_names
def importGOEliteSpeciesInfo():
filename = 'Config/goelite_species.txt'; x=0
fn=filepath(filename); species_codes={}
for line in open(fn,'rU').readlines():
data = cleanUpLine(line)
abrev,species,taxid,compatible_mods = string.split(data,'\t')
if x==0: x=1
else:
compatible_mods = string.split(compatible_mods,'|')
sd = SpeciesData(abrev,species,compatible_mods,taxid)
species_codes[species] = sd
return species_codes
def exportGOEliteSpeciesInfo(species_codes):
fn=filepath('Config/goelite_species.txt'); data = open(fn,'w'); x=0
header = string.join(['species_code','species_name','tax_id','compatible_algorithms'],'\t')+'\n'
data.write(header)
for species in species_codes:
if 'other' not in species and 'all-' not in species:
sd = species_codes[species]
mods = string.join(sd.Systems(),'|')
values = [sd.SpeciesCode(),sd.SpeciesName(),sd.TaxID(),mods]
values = string.join(values,'\t')+'\n'
data.write(values)
data.close()
def TimeStamp():
time_stamp = time.localtime()
year = str(time_stamp[0]); month = str(time_stamp[1]); day = str(time_stamp[2])
if len(month)<2: month = '0'+month
if len(day)<2: day = '0'+day
return year+month+day
def verifyFile(filename):
status = 'not found'
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = 'found';break
except Exception: status = 'not found'
return status
def verifyFileLength(filename):
count = 0
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
count+=1
if count>9: break
except Exception: null=[]
return count
def verifyGroupFileFormat(filename):
correct_format = False
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if len(string.split(data,'\t'))==3:
correct_format = True
break
except Exception: correct_format = False
return correct_format
def parseExcludeGuides(excludeGuides):
guides=[]
for line in open(excludeGuides,'rU').readlines():
data = cleanUpLine(line)
if 'Guide' not in data:
guides.append(data)
return guides
def displayHelp():
fn=filepath('Documentation/commandline.txt')
print '\n################################################\nAltAnalyze Command-Line Help'
for line in open(fn,'rU').readlines():
print cleanUpLine(line)
print '\n################################################ - END HELP'
sys.exit()
def searchDirectory(directory,var):
directory = unique.filepath(directory)
files = unique.read_directory(directory)
version = unique.getCurrentGeneDatabaseVersion()
for file in files:
if var in file:
location = string.split(directory+'/'+file,version)[1][1:]
return [location]
break
###### Command Line Functions (AKA Headless Mode) ######
def commandLineRun():
print 'Running commandline options'
import getopt
#/hd3/home/nsalomonis/normalization/mir1 - boxer
#python AltAnalyze.py --species Mm --arraytype "3'array" --celdir "C:/CEL" --output "C:/CEL" --expname miR1_column --runGOElite yes --GEelitepval 0.01
#python AltAnalyze.py --species Hs --arraytype "3'array" --FEdir "C:/FEfiles" --output "C:/FEfiles" --channel_to_extract "green/red ratio" --expname cancer --runGOElite yes --GEelitepval 0.01
#python AltAnalyze.py --celdir "C:/CEL" --output "C:/CEL" --expname miR1_column
#open ./AltAnalyze.app --celdir "/Users/nsalomonis/Desktop" --output "/Users/nsalomonis/Desktop" --expname test
#python AltAnalyze.py --species Mm --arraytype "3'array" --expdir "C:/CEL/ExpressionInput/exp.miR1_column.txt" --output "C:/CEL" --runGOElite yes --GEelitepval 0.01
#python AltAnalyze.py --species Mm --platform RNASeq --bedDir "/Users/nsalomonis/Desktop/code/AltAnalyze/datasets/BedFiles" --groupdir "/Users/nsalomonis/Desktop/code/AltAnalyze/datasets/BedFiles/ExpressionInput/groups.test.txt" --compdir "/Users/nsalomonis/Desktop/code/AltAnalyze/datasets/BedFiles/ExpressionInput/comps.test.txt" --output "/Users/nsalomonis/Desktop/code/AltAnalyze/datasets/BedFiles" --expname "test"
#python AltAnalyze.py --species Mm --platform RNASeq --filterdir "/Users/nsalomonis/Desktop/code/AltAnalyze/datasets/BedFiles/" --output "/Users/nsalomonis/Desktop/code/AltAnalyze/datasets/BedFiles"
#python AltAnalyze.py --expdir "/Users/nsalomonis/Desktop/Nathan/ExpressionInput/exp.test.txt" --exonMapFile "/Users/nsalomonis/Desktop/Nathan/hgu133_probe.txt" --species Hs --platform "3'array" --output "/Users/nsalomonis/Desktop/Nathan"
#python AltAnalyze.py --species Hs --platform "3'array" --expname test --channelToExtract green --FEdir /Users/saljh8/Downloads/AgllentTest/ --output /Users/saljh8/Downloads/AgllentTest/
global apt_location; global root_dir; global probability_statistic; global log_file; global summary_data_db; summary_data_db={}
###required
marker_finder='no'
manufacturer='Affymetrix'
constitutive_source='Ensembl'
ensembl_version = 'current'
species_code = None
species = None
main_input_folder = None
output_dir = None
array_type = None
input_annotation_file = None
groups_file = None
comps_file = None
input_cdf_file = None
exp_name = None
run_GOElite = 'yes'
visualize_qc_results = 'yes'
run_lineage_profiler = 'yes'
input_exp_file = ''
cel_file_dir = ''
input_stats_file = ''
input_filtered_dir = ''
external_annotation_dir = ''
xhyb_remove = 'no'
update_method = []
update_dbs = 'no'
analyze_all_conditions = 'no'
return_all = 'no'
additional_array_types = []
remove_intronic_junctions = 'no'
ignore_built_species = 'no'
build_exon_bedfile = 'no'
compendiumType = 'protein_coding'
probability_statistic = 'unpaired t-test'
specific_array_type = None
additional_resources = [None]
wpid = None
mod = 'Ensembl'
transpose = False
input_file_dir = None
denom_file_dir = None
image_export = []
selected_species = ['Hs','Mm','Rn'] ### These are the species that additional array types are currently supported
selected_platforms = ['AltMouse','exon','gene','junction']
returnPathways = 'no'
compendiumPlatform = 'gene'
exonMapFile = None
platformType = None ### This option is used to store the orignal platform type
perform_alt_analysis = 'no'
mappedExonAnalysis = False ### Map the original IDs to the RNA-Seq exon database (when True)
microRNA_prediction_method = None
pipelineAnalysis = True
OntologyID=''
PathwaySelection=''
GeneSetSelection=''
interactionDirs=[]
inputType='ID list'
Genes=''; genes=''
degrees='direct'
includeExpIDs=True
update_interactions=False
data_type = 'raw expression'
batch_effects = 'no'
channel_to_extract = None
normalization = False
justShowTheseIDs = ''
display=False
accessoryAnalysis=''
modelSize=None
geneModel=False
run_from_scratch = None
systemToUse = None ### For other IDs
custom_reference = False
multiThreading = True
genesToReport = 60
correlateAll = True
expression_data_format='log'
runICGS=False
IDtype=None
runKallisto = False
input_fastq_dir = ''
ChromiumSparseMatrix=''
perform_tests=False
testType = "fast"
inputTestData = "text"
customFASTA = None
filterFile = None
PearsonThreshold = 0.1
returnCentroids = 'community'
runCompleteWorkflow=True
referenceFull=None
k=None
labels=None
original_arguments = sys.argv
arguments=[]
for arg in original_arguments:
arg = string.replace(arg,'\xe2\x80\x9c','') ### These are non-standard forward quotes
arg = string.replace(arg,'\xe2\x80\x9d','') ### These are non-standard reverse quotes
arg = string.replace(arg,'\xe2\x80\x93','-') ### These are non-standard dashes
arg = string.replace(arg,'\x96','-') ### These are non-standard dashes
arg = string.replace(arg,'\x93','') ### These are non-standard forward quotes
arg = string.replace(arg,'\x94','') ### These are non-standard reverse quotes
arguments.append(arg)
print '\nArguments input:',arguments,'\n'
if '--help' in arguments[1:] or '--h' in arguments[1:]:
try: displayHelp() ### Print out a help file and quit
except Exception: print 'See: http://www.altanalyze.org for documentation and command-line help';sys.exit()
if 'AltAnalyze' in arguments[1]:
arguments = arguments[1:] ### Occurs on Ubuntu with the location of AltAnalyze being added to sys.argv (exclude this since no argument provided for this var)
try:
options, remainder = getopt.getopt(arguments[1:],'', ['species=', 'mod=','elitepval=', 'elitepermut=',
'method=','zscore=','pval=','num=',
'runGOElite=','denom=','output=','arraytype=',
'celdir=','expdir=','output=','statdir=',
'filterdir=','cdfdir=','csvdir=','expname=',
'dabgp=','rawexp=','avgallss=','logexp=',
'inclraw=','runalt=','altmethod=','altp=',
'probetype=','altscore=','GEcutoff=',
'exportnormexp=','calcNIp=','runMiDAS=',
'GEcutoff=','GEelitepval=','mirmethod=','ASfilter=',
'vendor=','GEelitefold=','update=','version=',
'analyzeAllGroups=','GEeliteptype=','force=',
'resources_to_analyze=', 'dataToAnalyze=','returnAll=',
'groupdir=','compdir=','annotatedir=','additionalScore=',
'additionalAlgorithm=','noxhyb=','platform=','bedDir=',
'altpermutep=','altpermute=','removeIntronOnlyJunctions=',
'normCounts=','buildExonExportFile=','groupStat=',
'compendiumPlatform=','rpkm=','exonExp=','specificArray=',
'ignoreBuiltSpecies=','ORAstat=','outputQCPlots=',
'runLineageProfiler=','input=','image=', 'wpid=',
'additional=','row_method=','column_method=',
'row_metric=','column_metric=','color_gradient=',
'transpose=','returnPathways=','compendiumType=',
'exonMapFile=','geneExp=','labels=','contrast=',
'plotType=','geneRPKM=','exonRPKM=','runMarkerFinder=',
'update_interactions=','includeExpIDs=','degrees=',
'genes=','inputType=','interactionDirs=','GeneSetSelection=',
'PathwaySelection=','OntologyID=','dataType=','combat=',
'channelToExtract=','showIntrons=','display=','join=',
'uniqueOnly=','accessoryAnalysis=','inputIDType=','outputIDType=',
'FEdir=','channelToExtract=','AltResultsDir=','geneFileDir=',
'AltResultsDir=','modelSize=','geneModel=','reference=',
'multiThreading=','multiProcessing=','genesToReport=',
'correlateAll=','normalization=','justShowTheseIDs=',
'direction=','analysisType=','algorithm=','rho=',
'clusterGOElite=','geneSetName=','runICGS=','IDtype=',
'CountsCutoff=','FoldDiff=','SamplesDiffering=','removeOutliers=',
'featurestoEvaluate=','restrictBy=','ExpressionCutoff=',
'excludeCellCycle=','runKallisto=','fastq_dir=','FDR=',
'reimportModelScores=','separateGenePlots=','ChromiumSparseMatrix=',
'test=','testType=','inputTestData=','customFASTA=','i=',
'excludeGuides=','cellHarmony=','BAM_dir=','filterFile=',
'correlationCutoff=','referenceType=','DE=','cellHarmonyMerge=',
'o=','dynamicCorrelation=','runCompleteWorkflow=','adjp=',
'fold=','performDiffExp=','centerMethod=', 'k=','bamdir=',
'downsample=','query=','referenceFull=', 'maskGroups=',
'elite_dir=','numGenesExp=','numVarGenes=','accessoryAnalyses=',
'dataFormat='])
except Exception:
print traceback.format_exc()
print "There is an error in the supplied command-line arguments (each flag requires an argument)"; sys.exit()
for opt, arg in options:
#print [opt, arg]
if opt == '--species': species=arg
elif opt == '--arraytype':
if array_type != None: additional_array_types.append(arg)
else: array_type=arg; platform = array_type
if specific_array_type == None: specific_array_type = platform
elif opt == '--test':
try: perform_tests.append(arg)
except Exception: perform_tests = [arg]
elif opt == '--testType':
testType = arg
elif opt == '--inputTestData':
inputTestData = arg
elif opt == '--exonMapFile':
perform_alt_analysis = 'yes' ### Perform alternative exon analysis
exonMapFile = arg
elif opt == '--specificArray': specific_array_type = arg ### e.g., hGlue
elif opt == '--celdir':
arg = verifyPath(arg)
cel_file_dir=arg
elif opt == '--bedDir' or opt == '--BAM_dir' or opt == 'bamdir=' or opt == 'bamDir':
arg = verifyPath(arg)
cel_file_dir=arg
elif opt == '--ChromiumSparseMatrix':
arg = verifyPath(arg)
ChromiumSparseMatrix=arg
elif opt == '--FEdir':
arg = verifyPath(arg)
cel_file_dir = arg
elif opt == '--expdir':
arg = verifyPath(arg)
input_exp_file=arg
elif opt == '--statdir':
arg = verifyPath(arg)
input_stats_file=arg
elif opt == '--filterdir':
arg = verifyPath(arg)
input_filtered_dir=arg
elif opt == '--groupdir':
arg = verifyPath(arg)
groups_file=arg
elif opt == '--compdir':
arg = verifyPath(arg)
comps_file=arg
elif opt == '--cdfdir':
arg = verifyPath(arg)
input_cdf_file=arg
elif opt == '--csvdir':
arg = verifyPath(arg)
input_annotation_file=arg
elif opt == '--expname': exp_name=arg
elif opt == '--output' or opt == '--o':
arg = verifyPath(arg)
output_dir=arg
elif opt == '--vendor': manufacturer=arg
elif opt == '--runICGS': runICGS=True
elif opt == '--IDtype': IDtype=arg
elif opt == '--ignoreBuiltSpecies': ignore_built_species=arg
elif opt == '--platform':
if array_type != None: additional_array_types.append(arg)
else: array_type=arg; platform = array_type
if specific_array_type == None: specific_array_type = platform
elif opt == '--update': update_dbs='yes'; update_method.append(arg)
elif opt == '--version': ensembl_version = arg
elif opt == '--compendiumPlatform': compendiumPlatform=arg ### platform for which the LineageProfiler compendium is built on
elif opt == '--force': force=arg
elif opt == '--input' or opt == '--i' or opt == '--query':
arg = verifyPath(arg)
input_file_dir=arg
#input_exp_file=arg
pipelineAnalysis = False ### If this option is entered, only perform the indicated analysis
elif opt == '--image': image_export.append(arg)
elif opt == '--wpid': wpid=arg
elif opt == '--mod': mod=arg
elif opt == '--runKallisto':
if arg == 'yes' or string.lower(arg) == 'true':
runKallisto = True
elif opt == '--fastq_dir':
input_fastq_dir = arg
elif opt == '--customFASTA':
customFASTA = arg
elif opt == '--additional':
if additional_resources[0] == None:
additional_resources=[]
additional_resources.append(arg)
else:
additional_resources.append(arg)
elif opt == '--transpose':
if arg == 'True': transpose = True
elif opt == '--runLineageProfiler' or opt == '--cellHarmony': ###Variable declared here and later (independent analysis here or pipelined with other analyses later)
run_lineage_profiler=arg
elif opt == '--compendiumType': ### protein-coding, ncRNA, or exon
compendiumType=arg
elif opt == '--denom':
denom_file_dir=arg ### Indicates that GO-Elite is run independent from AltAnalyze itself
elif opt == '--accessoryAnalysis' or opt == '--accessoryAnalyses':
accessoryAnalysis = arg
elif opt == '--channelToExtract': channel_to_extract=arg
elif opt == '--genesToReport': genesToReport = int(arg)
elif opt == '--correlateAll': correlateAll = True
elif opt == '--direction': direction = arg
elif opt == '--logexp' or opt == '--dataFormat': expression_data_format=arg
elif opt == '--geneRPKM': rpkm_threshold=arg
elif opt == '--correlationCutoff': PearsonThreshold=float(arg)
elif opt == '--DE':
if string.lower(arg) == 'true':
DE = True
else:
DE = False
elif opt == '--referenceType':
if string.lower(arg) == 'centroid' or string.lower(arg) == 'mean':
returnCentroids = True; CenterMethod='centroid'
elif string.lower(arg) == 'medoid' or string.lower(arg) == 'median':
returnCentroids = True; CenterMethod='median'
elif string.lower(arg) == 'community' or string.lower(arg) == 'louvain':
returnCentroids = 'community'; CenterMethod='community'
elif string.lower(arg) == 'cells' or string.lower(arg) == 'cell':
returnCentroids = False; CenterMethod='centroid'
elif string.lower(arg) == 'none' or string.lower(arg) == ' ':
returnCentroids = 'None'; CenterMethod='None'
else:
returnCentroids = 'community'; CenterMethod='community'
elif opt == '--multiThreading' or opt == '--multiProcessing':
multiThreading=arg
if multiThreading == 'yes': multiThreading = True
elif 'rue' in multiThreading: multiThreading = True
else: multiThreading = False
if perform_tests != False:
### Requires the mouse RNASeq database
### python AltAnalyze.py --test --testType ICGS --inputTestData text
### python AltAnalyze.py --test --testType ICGS --inputTestData BAM
### python AltAnalyze.py --test --testType ICGS --inputTestData FASTQ
### python AltAnalyze.py --test --testType ICGS --inputTestData 10X
count = verifyFileLength('AltDatabase/demo_data/ReadMe.txt')
if count==0:
file_location_defaults = UI.importDefaultFileLocations()
goelite_url = file_location_defaults['goelite'].Location()
fln,status = update.download(goelite_url+'TestData/demo_data.zip','AltDatabase/NoVersion','')
if 'Internet' not in status: print "Demo data downloaded."
if 'ICGS' in perform_tests:
from tests.scripts import ICGS_test
if runKallisto:
inputTestData = "FASTQ"
ICGS_test.runICGStest(testType=testType,inputData=inputTestData)
sys.exit()
if 'other' in manufacturer or 'Other' in manufacturer:
### For other IDs
systemToUse = array_type
if array_type == None:
print 'Please indicate a ID type as --platform when setting vendor equal to "Other IDs"'; sys.exit()
array_type = "3'array"
if array_type == 'RNASeq': manufacturer = array_type
if platformType == None: platformType = array_type
if perform_alt_analysis == 'yes':
if platform == "3'array":
mappedExonAnalysis = True
cel_file_dir = input_exp_file
exp_name = export.findFilename(input_exp_file)
exp_name = string.replace(exp_name,'.txt','')
exp_name = string.replace(exp_name,'exp.','')
input_exp_file = ''
### To perform alternative exon analyses for platforms without a dedicated database, must happing appropriate mapping info or array type data
### (will need to perform downstream testing for unsupported Affymetrix exon, gene and junction arrays)
if exonMapFile == None and specific_array_type == None and cel_file_dir == '':
print_out = "\nUnable to run!!! Please designate either a specific platfrom (e.g., --specificArray hgU133_2), select CEL files, or an "
print_out += "exon-level mapping file location (--exonMapFile C:/mapping.txt) to perform alternative exon analyses for this platform."
### Will need to check here to see if the platform is supported (local or online files) OR wait until an error is encountered later
""" Check to see if a database is already installed """
try: current_species_dirs = unique.read_directory('/AltDatabase')
except Exception: current_species_dirs=[]
if len(current_species_dirs)==0 and update_dbs != 'yes':
print "Please install a database before running AltAnalyze. Please note, AltAnalyze may need to install additional files later for RNASeq and LineageProfiler for some species, automatically. Make sure to list your platform as RNASeq if analyzing RNA-Seq data (--platform RNASeq)."
print "Example:\n"
print 'python AltAnalyze.py --species Hs --update Official --version EnsMart72';sys.exit()
######## Perform analyses independent from AltAnalyze database centric analyses that require additional parameters
if len(image_export) > 0 or len(accessoryAnalysis)>0 or runICGS:
""" Annotate existing ICGS groups with selected GO-Elite results """
if 'annotateICGS' in accessoryAnalysis:
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--elite_dir':
goelite_path = arg
import RNASeq
RNASeq.predictCellTypesFromClusters(groups_file, goelite_path)
sys.exit()
if runICGS:
#python AltAnalyze.py --runICGS yes --platform "RNASeq" --species Hs --column_method hopach --column_metric euclidean --rho 0.3 --ExpressionCutoff 1 --FoldDiff 4 --SamplesDiffering 3 --restrictBy protein_coding --excludeCellCycle conservative --removeOutliers yes --expdir /RNA-Seq/run1891_normalized.txt
#python AltAnalyze.py --runICGS yes --expdir "/Users/saljh8/Desktop/demo/Myoblast/ExpressionInput/exp.myoblast.txt" --platform "3'array" --species Hs --GeneSetSelection BioMarkers --PathwaySelection Heart --column_method hopach --rho 0.4 --ExpressionCutoff 200 --justShowTheseIDs "NKX2-5 T TBX5" --FoldDiff 10 --SamplesDiffering 3 --excludeCellCycle conservative
try: species = species
except Exception: 'Please designate a species before continuing (e.g., --species Hs)'
try: array_type = array_type
except Exception: 'Please designate a species before continuing (e.g., --species Hs)'
if len(cel_file_dir)>0: ### For BED files or BAM files
if len(cel_file_dir) > 0: pass
else: 'Please indicate a source folder (e.g., --bedDir /data/BAMFiles)'
else:
if len(input_exp_file) > 0: pass
else: 'Please indicate a source folder or expression file (e.g., --expdir /dataset/singleCells.txt)'
if array_type == 'Other' or 'Other' in array_type:
if ':' in array_type:
array_type, IDtype = string.split(array_type)
array_type == "3'array"
if IDtype == None: IDtype = manufacturer
row_method = 'hopach'
column_method = 'hopach'
row_metric = 'cosine'
column_metric = 'cosine'
color_gradient = 'yellow_black_blue'
contrast=3
vendor = manufacturer
GeneSelection = ''
PathwaySelection = ''
GeneSetSelection = 'None Selected'
excludeCellCycle = True
rho_cutoff = 0.2
restrictBy = None
featurestoEvaluate = 'Genes'
ExpressionCutoff = 1
CountsCutoff = 0.9
FoldDiff = 4
SamplesDiffering = 4
JustShowTheseIDs=''
removeOutliers = False
excludeGuides = None
PathwaySelection=[]
dynamicCorrelation=True
runCompleteWorkflow=False
downsample=2500
numGenesExp=500
numVarGenes=500
if ChromiumSparseMatrix != '':
rho_cutoff = 0.2
column_metric = 'euclidean'
restrictBy = 'protein_coding'
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--row_method':
row_method=arg
if row_method == 'None': row_method = None
elif opt == '--column_method':
column_method=arg
if column_method == 'None': column_method = None
elif opt == '--row_metric': row_metric=arg
elif opt == '--column_metric': column_metric=arg
elif opt == '--color_gradient': color_gradient=arg
elif opt == '--GeneSetSelection': GeneSetSelection=arg
elif opt == '--PathwaySelection': PathwaySelection.append(arg)
elif opt == '--genes': GeneSelection=arg
elif opt == '--ExpressionCutoff': ExpressionCutoff=arg
elif opt == '--normalization': normalization=arg
elif opt == '--justShowTheseIDs': justShowTheseIDs=arg
elif opt == '--rho': rho_cutoff=float(arg)
elif opt == '--clusterGOElite':clusterGOElite=float(arg)
elif opt == '--CountsCutoff':CountsCutoff=int(float(arg))
elif opt == '--FoldDiff':FoldDiff=float(arg)
elif opt == '--SamplesDiffering':SamplesDiffering=int(float(arg))
elif opt == '--excludeGuides': excludeGuides=arg
elif opt == '--dynamicCorrelation': dynamicCorrelation=arg
elif opt == '--k':
try: k=int(arg)
except:
print 'Invalid k... setting to None'
k=None
elif opt == '--downsample': downsample=int(arg) ### Number of cells to downsample to
elif opt == '--numVarGenes': numVarGenes=int(arg) ### Number of cells to downsample to
elif opt == '--numGenesExp': numGenesExp=int(arg) ### For barcode filtering
elif opt == '--runCompleteWorkflow':
runCompleteWorkflow=arg
if string.lower(arg)=='false' or string.lower(arg)=='no':
runCompleteWorkflow = False
else:
runCompleteWorkflow = True
elif opt == '--removeOutliers':
removeOutliers=arg
if removeOutliers=='yes' or removeOutliers=='True':
removeOutliers = True
elif opt == '--featurestoEvaluate':featurestoEvaluate=arg
elif opt == '--restrictBy':
if arg == 'None': restrictBy = None
else: restrictBy=arg
elif opt == '--excludeCellCycle':
excludeCellCycle=arg
if excludeCellCycle == 'False' or excludeCellCycle == 'no': excludeCellCycle = False
elif excludeCellCycle == 'True' or excludeCellCycle == 'yes' or excludeCellCycle == 'conservative': excludeCellCycle = True
elif opt == '--contrast':
try: contrast=float(arg)
except Exception: print '--contrast not a valid float';sys.exit()
elif opt == '--vendor': vendor=arg
elif opt == '--display':
if arg=='yes':
display=True
elif arg=='True':
display=True
else:
display=False
if excludeGuides!=None:
if '.txt' in excludeGuides:
try: excludeGuides = parseExcludeGuides(excludeGuides)
except Exception:
print 'Failure to parse input excludeGuides text file. Check to see if the correct file location is provided.'
sys.exit()
if len(PathwaySelection)==0: PathwaySelection=''
if len(GeneSetSelection)>0 or GeneSelection != '':
gsp = UI.GeneSelectionParameters(species,array_type,vendor)
gsp.setGeneSet(GeneSetSelection)
gsp.setPathwaySelect(PathwaySelection)
gsp.setGeneSelection(GeneSelection)
gsp.setJustShowTheseIDs(JustShowTheseIDs)
gsp.setNormalize('median')
gsp.setExcludeGuides(excludeGuides)
gsp.setK(k)
gsp.setDownsample(downsample)
gsp.setNumGenesExp(numGenesExp)
gsp.setNumVarGenes(numVarGenes)
try: gsp.setCountsNormalization(expression_data_format)
except: pass
gsp.setSampleDiscoveryParameters(ExpressionCutoff,CountsCutoff,FoldDiff,SamplesDiffering, dynamicCorrelation,
removeOutliers,featurestoEvaluate,restrictBy,excludeCellCycle,column_metric,column_method,rho_cutoff)
import RNASeq
mlp_instance = mlp
if exp_name == None:
exp_name = export.findFilename(input_exp_file)
exp_name = string.replace(exp_name,'.txt','')
exp_name = string.replace(exp_name,'exp.','')
if cel_file_dir != '':
expFile = output_dir + '/ExpressionInput/'+ 'exp.'+exp_name+'.txt' ### cel_file_dir will point to the input directory
elif ChromiumSparseMatrix != '':
expFile = output_dir + '/ExpressionInput/'+ 'exp.'+exp_name+'.txt'
elif input_exp_file !='':
if 'ExpressionInput' in input_exp_file: expFile = input_exp_file
else:
### Copy over expression file to ExpressionInput
expdir2 = string.replace(input_exp_file,'exp.','')
if output_dir == None:
### Not suppplied, define relative to the input expression file
root_dir = export.findParentDir(input_exp_file)
else:
root_dir = output_dir
expFile = root_dir+'/ExpressionInput/exp.'+export.findFilename(expdir2)
export.copyFile(input_exp_file, expFile)
### Groups file if present to the output directory ExpressionInput folder
if 'exp.' in input_exp_file:
initial_groups = string.replace(input_exp_file,'exp.','groups.')
else:
initial_groups = export.findParentDir(input_exp_file)+'/groups.'+export.findFilename(input_exp_file)
try:
groups_file = string.replace(expFile,'exp.','groups.') ### destination file
export.copyFile(initial_groups, groups_file)
print 'Copied the groups file to ExpressionInput folder in the output directory'
except Exception:
print 'No groups file present in the input file folder.'
global log_file
try:
if len(output_dir)>0:
root_dir = output_dir
else:
forceError
except Exception:
try: root_dir = export.findParentDir(expFile)
except Exception:
print 'Please include an output directory for the AltAnalyze results (e.g., --output /Data/Results)';sys.exit()
root_dir = string.replace(root_dir,'/ExpressionInput','')
fl = UI.ExpressionFileLocationData('','','',''); fl.setFeatureNormalization('none')
try: fl.setExpFile(expFile)
except Exception:
expFile = root_dir+'/ExpressionInput/exp.'+exp_name+'.txt'
fl.setExpFile(expFile)
fl.setArrayType(array_type)
fl.setOutputDir(root_dir)
fl.setMultiThreading(multiThreading)
exp_file_location_db={}; exp_file_location_db[exp_name]=fl
### Assign variables needed to run Kallisto from FASTQ files
if runKallisto and len(input_fastq_dir)==0:
#python AltAnalyze.py --runICGS yes --platform "RNASeq" --species Mm --column_method hopach --rho 0.4 --ExpressionCutoff 1 --FoldDiff 4 --SamplesDiffering 1 --excludeCellCycle strict --output /Users/saljh8/Desktop/Grimes/GEC14074 --expname test --fastq_dir /Users/saljh8/Desktop/Grimes/GEC14074
print 'Please include the flag "--fastq_dir" in the command-line arguments with an appropriate path';sys.exit()
elif len(input_fastq_dir)>0:
fl.setRunKallisto(input_fastq_dir)
fl.setArrayType("3'array")
fl.setMultiThreading(multiThreading)
array_type = "3'array"
if customFASTA!=None:
fl.setCustomFASTA(customFASTA)
### Assign variables needed to run BAMtoBED and/or BED file count analysis
if len(cel_file_dir)>0 and array_type=='RNASeq':
#python AltAnalyze.py --runICGS yes --platform "RNASeq" --species Mm --column_method hopach --rho 0.4 --ExpressionCutoff 1 --FoldDiff 4 --SamplesDiffering 1 --excludeCellCycle strict --output /Users/saljh8/Desktop/Grimes/GEC14074 --expname test --bedDir /Users/saljh8/Desktop/Grimes/GEC14074 --multiProcessing no
fl.setCELFileDir(cel_file_dir)
fl.setMultiThreading(multiThreading)
fl.setExonBedBuildStatus('no')
fl.setFeatureNormalization('RPKM')
fl.setArrayType(array_type)
fl.setRootDir(root_dir)
### Import expression defaults
expr_defaults, alt_exon_defaults, functional_analysis_defaults, goelite_defaults = UI.importDefaults(array_type,species)
dabg_p, rpkm_threshold, gene_exp_threshold, exon_exp_threshold, exon_rpkm_threshold, expression_threshold, perform_alt_analysis, analyze_as_groups, expression_data_format, normalize_feature_exp, normalize_gene_data, avg_all_for_ss, include_raw_data, probability_statistic, FDR_statistic, batch_effects, marker_finder, visualize_qc_results, run_lineage_profiler, null = expr_defaults
fl.setRPKMThreshold(rpkm_threshold)
fl.setExonExpThreshold(exon_exp_threshold)
fl.setGeneExpThreshold(gene_exp_threshold)
fl.setExonRPKMThreshold(exon_rpkm_threshold)
fl.setJunctionExpThreshold(expression_threshold)
elif len(ChromiumSparseMatrix)>0:
#python AltAnalyze.py --runICGS yes --platform "RNASeq" --species Mm --column_method hopach --rho 0.4 --ExpressionCutoff 1 --FoldDiff 4 --SamplesDiffering 1 --excludeCellCycle strict --output /Users/saljh8/Desktop/Grimes/GEC14074 --expname test --ChromiumSparseMatrix /Users/saljh8/Desktop/Grimes/GEC14074 --multiProcessing no
fl.setChromiumSparseMatrix(ChromiumSparseMatrix)
fl.setMultiThreading(multiThreading)
fl.setArrayType("3'array")
array_type = "3'array"
fl.setRootDir(root_dir)
elif len(input_exp_file)>0:
### Dealing with an expression file which should not be treated as RNASeq workflow
count = verifyFileLength(expFile[:-4]+'-steady-state.txt')
if count<2:
if array_type != 'PSI' and array_type != 'exons':
array_type = "3'array" ### No steady-state file, must be an standard gene-level analysis
time_stamp = timestamp()
log_file = filepath(root_dir+'/AltAnalyze_report-'+time_stamp+'.log')
log_report = open(log_file,'w'); log_report.close()
sys.stdout = Logger('')
print "\nFull commandline:"
try: print string.join(arguments,' ')
except Exception: pass
print ''
count = verifyFileLength(expFile[:-4]+'-steady-state.txt')
exonExpFile = str(expFile)
if count>1:
expFile = expFile[:-4]+'-steady-state.txt'
elif array_type=='RNASeq' or len(ChromiumSparseMatrix)>0 or len(input_fastq_dir)>0:
try:
### Indicates that the steady-state file doesn't exist. The exp. may exist, be could be junction only so need to re-build from bed files here
values = species,exp_file_location_db,exp_name,mlp_instance
### proceed to run the full discovery analysis here (Kallisto, BAM, BED, Chromium matrix)
UI.StatusWindow(values,'preProcessRNASeq') ### proceed to run the full discovery analysis here!!!
SteadyStateFile = expFile[:-4]+'-steady-state.txt'
status = verifyFile(SteadyStateFile)
if status == "found":
#fl.setExpFile(SteadyStateFile) ### This should not be over-written by a steady-state file
expFile = SteadyStateFile
except Exception:
### RNASeq is an official datatype that requires a steady-state file. However, for scRNA-Seq, usually the input is a text file or FASTQ which gets
### changed to "3'array". We correct for this by excepting this error without doing anything else
#print traceback.format_exc();sys.exit()
pass
if excludeCellCycle != False:
print "Excluding Cell Cycle effects status:",excludeCellCycle
### Run ICGS through the GUI
graphic_links = UI.RemotePredictSampleExpGroups(expFile, mlp_instance, gsp,(species,array_type)) ### proceed to run the full discovery analysis here!!!
### Export Guide3 Groups automatically
Guide3_results = graphic_links[-1][-1][:-4]+'.txt'
new_groups_dir = RNASeq.exportGroupsFromClusters(Guide3_results,fl.ExpFile(),array_type,suffix='ICGS')
exonExpFile,newExpFile,new_groups_dir = UI.exportAdditionalICGSOutputs(expFile,Guide3_results,outputTSNE=True)
fl.setExpFile(newExpFile) ### set this to the outlier removed version
comps_file = string.replace(new_groups_dir,'groups.','comps.')
fl.setGroupsFile(new_groups_dir)
fl.setCompsFile(comps_file)
exp_file_location_db[exp_name+'-ICGS'] = fl
### force MarkerFinder to be run
input_exp_file = newExpFile ### Point MarkerFinder to the new ICGS ordered copied expression file
runMarkerFinder=True ### Not necessary for ICGS2 as MarkerFinder will already have been run - but good for other ICGS outputs
if runMarkerFinder:
update_method = ['markers']
if runCompleteWorkflow == False:
print 'ICGS run complete... halted prior to full differential comparison analysis'
sys.exit()
if 'WikiPathways' in image_export:
#python AltAnalyze.py --input /Users/test/input/criterion1.txt --image WikiPathways --mod Ensembl --species Hs --wpid WP536
if wpid==None:
print 'Please provide a valid WikiPathways ID (e.g., WP1234)';sys.exit()
if species==None:
print 'Please provide a valid species ID for an installed database (to install: --update Official --species Hs --version EnsMart72Plus)';sys.exit()
if input_file_dir==None:
print 'Please provide a valid file location for your input IDs (also needs to inlcude system code and value column)';sys.exit()
from visualization_scripts import WikiPathways_webservice
try:
print 'Attempting to output a WikiPathways colored image from user data'
print 'mod:',mod
print 'species_code:',species
print 'wpid:',wpid
print 'input GO-Elite ID file:',input_file_dir
graphic_link = WikiPathways_webservice.visualizePathwayAssociations(input_file_dir,species,mod,wpid)
except Exception,e:
if 'force_no_matching_error' in traceback.format_exc():
print '\nUnable to run!!! None of the input IDs mapped to this pathway\n'
elif 'IndexError' in traceback.format_exc():
print '\nUnable to run!!! Input ID file does not have at least 3 columns, with the second column being system code\n'
elif 'ValueError' in traceback.format_exc():
print '\nUnable to run!!! Input ID file error. Please check that you do not have extra rows with no data\n'
elif 'source_data' in traceback.format_exc():
print '\nUnable to run!!! Input ID file does not contain a valid system code\n'
elif 'goelite' in traceback.format_exc():
print '\nUnable to run!!! A valid species database needs to first be installed. For example, run:'
print 'python AltAnalyze.py --update Official --species Hs --version EnsMart72\n'
else:
print traceback.format_exc()
print '\nError generating the pathway "%s"' % wpid,'\n'
try:
printout = 'Finished exporting visualized pathway to:',graphic_link['WP']
print printout,'\n'
except Exception: None
sys.exit()
if 'FilterFile' in accessoryAnalysis:
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--filterFile':
filterFile = arg
if opt == '--input':
input_file = verifyPath(arg)
output_file = input_file[:-4]+'-filtered.txt'
from import_scripts import sampleIndexSelection
filter_order = sampleIndexSelection.getFilters(filterFile)
sampleIndexSelection.filterFile(input_file,output_file,filter_order)
sys.exit()
if 'MergeFiles' in accessoryAnalysis:
#python AltAnalyze.py --accessoryAnalysis MergeFiles --input "C:\file1.txt" --input "C:\file2.txt" --output "C:\tables"
files_to_merge=[]
join_option='Intersection'
uniqueOnly=False
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--input':
arg = verifyPath(arg)
files_to_merge.append(arg)
if opt == '--join': join_option = arg
if opt == '--uniqueOnly': unique_only = arg
if len(files_to_merge)<2:
print 'Please designate two or more files to merge (--input)';sys.exit()
UI.MergeFiles(files_to_merge, join_option, uniqueOnly, output_dir, None)
sys.exit()
if 'IDTranslation' in accessoryAnalysis:
#python AltAnalyze.py --accessoryAnalysis IDTranslation --inputIDType Symbol --outputIDType RefSeq --input "C:\file1.txt" --species Hs
inputIDType=None
outputIDType=None
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--inputIDType': inputIDType = arg
if opt == '--outputIDType': outputIDType = arg
if inputIDType==None or outputIDType==None:
print 'Please designate an input ID type and and output ID type (--inputIDType Ensembl --outputIDType Symbol)'; sys.exit()
if species==None:
print "Please enter a valide species (--species)"; sys.exit()
UI.IDconverter(input_file_dir, species, inputIDType, outputIDType, None)
sys.exit()
if 'hierarchical' in image_export:
#python AltAnalyze.py --input "/Users/test/pluri.txt" --image hierarchical --row_method average --column_method single --row_metric cosine --column_metric euclidean --color_gradient red_white_blue --transpose False --PathwaySelection Apoptosis:WP254 --GeneSetSelection WikiPathways --species Hs --platform exon --display false
if input_file_dir==None:
print 'Please provide a valid file location for your input data matrix (must have an annotation row and an annotation column)';sys.exit()
row_method = 'weighted'
column_method = 'average'
row_metric = 'cosine'
column_metric = 'cosine'
color_gradient = 'red_black_sky'
contrast=2.5
vendor = 'Affymetrix'
GeneSelection = ''
PathwaySelection = ''
GeneSetSelection = 'None Selected'
rho = None
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--row_method':
row_method=arg
if row_method == 'None': row_method = None
elif opt == '--column_method':
column_method=arg
if column_method == 'None': column_method = None
elif opt == '--row_metric': row_metric=arg
elif opt == '--column_metric': column_metric=arg
elif opt == '--color_gradient': color_gradient=arg
elif opt == '--GeneSetSelection': GeneSetSelection=arg
elif opt == '--PathwaySelection': PathwaySelection=arg
elif opt == '--genes': GeneSelection=arg
elif opt == '--OntologyID': OntologyID=arg
elif opt == '--normalization': normalization=arg
elif opt == '--justShowTheseIDs': justShowTheseIDs=arg
elif opt == '--rho': rho=arg
elif opt == '--clusterGOElite':clusterGOElite=arg
elif opt == '--contrast':
try: contrast=float(arg)
except Exception: print '--contrast not a valid float';sys.exit()
elif opt == '--vendor': vendor=arg
elif opt == '--display':
if arg=='yes':
display=True
elif arg=='True':
display=True
else:
display=False
if len(GeneSetSelection)>0 or GeneSelection != '':
gsp = UI.GeneSelectionParameters(species,array_type,vendor)
gsp.setGeneSet(GeneSetSelection)
gsp.setPathwaySelect(PathwaySelection)
gsp.setGeneSelection(GeneSelection)
gsp.setOntologyID(OntologyID)
gsp.setTranspose(transpose)
gsp.setNormalize(normalization)
gsp.setJustShowTheseIDs(justShowTheseIDs)
try: gsp.setClusterGOElite(clusterGOElite)
except Exception: pass
if rho!=None:
try:
float(rho)
gsp.setRhoCutoff(rho)
except Exception: print 'Must enter a valid Pearson correlation cutoff (float)'
transpose = gsp ### this allows methods that don't transmit this object to also work
if row_method == 'no': row_method = None
if column_method == 'no': column_method = None
if len(GeneSetSelection)>0:
if species == None:
print "Please enter a valide species (--species)"; sys.exit()
try:
files = unique.read_directory(input_file_dir+'/')
dir = input_file_dir
for file in files:
filename = dir+'/'+file
UI.createHeatMap(filename, row_method, row_metric, column_method, column_metric, color_gradient, transpose, contrast, None, display=display)
except Exception:
UI.createHeatMap(input_file_dir, row_method, row_metric, column_method, column_metric, color_gradient, transpose, contrast, None, display=display)
#from visualization_scripts import clustering; clustering.outputClusters([input_file_dir],[])
sys.exit()
if 'PCA' in image_export or 't-SNE' in image_export or 'UMAP' in image_export or 'umap' in image_export:
#AltAnalyze.py --input "/Users/nsalomonis/Desktop/folds.txt" --image PCA --plotType 3D --display True --labels yes
#python AltAnalyze.py --input "/Users/nsalomonis/Desktop/log2_expression.txt" --image "t-SNE" --plotType 2D --display True --labels no --genes "ACTG2 ARHDIA KRT18 KRT8 ATP2B1 ARHGDIB" --species Hs --platform RNASeq --separateGenePlots True --zscore no
#--algorithm "t-SNE"
include_labels = 'yes'
plotType = '2D'
pca_algorithm = 'SVD'
geneSetName = None
zscore = True
colorByGene=None
separateGenePlots = False
reimportModelScores = True
maskGroups = None
if 't-SNE' in image_export:
pca_algorithm = 't-SNE'
if 'UMAP' in image_export or 'umap' in image_export:
pca_algorithm = 'UMAP'
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
#print opt,arg
if opt == '--labels':
include_labels=arg
if include_labels == 'True' or include_labels == 'yes':
include_labels = 'yes'
else:
include_labels = 'no'
if opt == '--plotType': plotType=arg
if opt == '--algorithm': pca_algorithm=arg
if opt == '--geneSetName': geneSetName=arg
if opt == '--genes': colorByGene=arg
if opt == '--maskGroups': maskGroups=arg
if opt == '--reimportModelScores':
if arg == 'yes' or arg == 'True' or arg == 'true':
reimportModelScores = True
else:
reimportModelScores = False
if opt == '--separateGenePlots':
if arg=='yes' or arg=='True' or arg == 'true':
separateGenePlots = True
else:
separateGenePlots = False
if opt == '--zscore':
if arg=='yes' or arg=='True' or arg == 'true':
zscore=True
else:
zscore=False
if opt == '--display':
if arg=='yes' or arg=='True' or arg == 'true':
display=True
if input_file_dir==None:
print 'Please provide a valid file location for your input data matrix (must have an annotation row and an annotation column)';sys.exit()
UI.performPCA(input_file_dir, include_labels, pca_algorithm, transpose, None,
plotType=plotType, display=display, geneSetName=geneSetName, species=species, zscore=zscore,
colorByGene=colorByGene, reimportModelScores=reimportModelScores, separateGenePlots=separateGenePlots,
maskGroups=maskGroups)
sys.exit()
if 'VennDiagram' in image_export:
# AltAnalyze.py --image "VennDiagram" --input "C:\file1.txt" --input "C:\file2.txt" --output "C:\graphs"
files_to_merge=[]
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--input':
arg = verifyPath(arg)
files_to_merge.append(arg)
if opt == '--display':
if arg=='yes' or arg=='True' or arg == 'true':
display=True
if len(files_to_merge)<2:
print 'Please designate two or more files to compare (--input)';sys.exit()
UI.vennDiagram(files_to_merge, output_dir, None, display=display)
sys.exit()
if 'AltExonViewer' in image_export:
#python AltAnalyze.py --image AltExonViewer --AltResultsDir "C:\CP-hESC" --genes "ANXA7 FYN TCF3 NAV2 ETS2 MYLK ATP2A2" --species Hs --platform exon --dataType "splicing-index"
genes=[]
show_introns='no'
geneFileDir=''
analysisType='plot'
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--genes':genes=arg
elif opt == '--dataType': data_type = arg
elif opt == '--showIntrons': show_introns = arg
elif opt == '--AltResultsDir': altresult_dir = arg
elif opt == '--geneFileDir': geneFileDir = arg
elif opt == '--analysisType': analysisType=arg
if altresult_dir == None:
print 'Please include the location of the AltResults directory (--AltResultsDir)'; sys.exit()
if len(genes)==0 and len(geneFileDir)==0:
print "Please indicate the genes (--genes) or gene file location (--geneFileDir) for AltExonViewer";sys.exit()
if species == None:
print "Please enter a valide species (--species)"; sys.exit()
if array_type == None:
print "Please enter a valide platform (--platform)"; sys.exit()
if 'AltResults' not in altresult_dir:
altresult_dir+='/AltResults/'
if 'Sashimi' in analysisType:
#python AltAnalyze.py --image AltExonViewer --AltResultsDir "/Users/saljh8/Desktop/Grimes/GEC14074/AltResults/" --genes "Dgat1 Dgat2 Tcf7l1" --species Mm --platform RNASeq --analysisType SashimiPlot
analysisType = 'Sashimi-Plot'
altresult_dir = string.split(altresult_dir,'AltResults')[0]
if len(geneFileDir)>0: genes = geneFileDir
geneFileDir=''
elif 'raw' in data_type: ### Switch directories if expression
altanalyze_results_folder = string.replace(altresult_dir,'AltResults','ExpressionInput')
altresult_dir = UI.getValidExpFile(altanalyze_results_folder)
if len(altresult_dir)==0:
print 'No valid expression input file (e.g., exp.MyExperiment.txt) found in',altanalyze_results_folder;sys.exit()
else:
altanalyze_results_folder = altresult_dir+'/RawSpliceData/'+species
try: altresult_dir = UI.getValidSplicingScoreFile(altanalyze_results_folder)
except Exception,e:
print "No files found in: "+altanalyze_results_folder; sys.exit()
if len(geneFileDir)>0:
try:
genes = UI.importGeneList(geneFileDir) ### list of gene IDs or symbols
except Exception:
### Can occur if a directory of files is selected
try:
files = unique.read_directory(geneFileDir+'/')
gene_string=''
for file in files:
if '.txt' in file:
filename = geneFileDir+'/'+file
genes = UI.importGeneList(filename) ### list of gene IDs or symbols
gene_string = gene_string+','+genes
print 'Imported genes from',file,'\n'
#print [altresult_dir];sys.exit()
UI.altExonViewer(species,platform,altresult_dir, gene_string, show_introns, analysisType, False)
except Exception: pass
sys.exit()
if len(genes)==0:
print 'Please list one or more genes (--genes "ANXA7 FYN TCF3 NAV2 ETS2 MYLK ATP2A2")'; sys.exit()
try: UI.altExonViewer(species,platform,altresult_dir, genes, show_introns, analysisType, False)
except Exception:
print traceback.format_exc()
sys.exit()
if 'network' in image_export:
#AltAnalyze.py --image network --species Hs --output "C:\GSE9440_RAW" --PathwaySelection Apoptosis:WP254 --GeneSetSelection WikiPathways
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--update_interactions': update_interactions=arg
elif opt == '--includeExpIDs': includeExpIDs=arg
elif opt == '--degrees': degrees=arg
elif opt == '--genes':
Genes=arg
inputType = 'IDs'
elif opt == '--inputType': inputType=arg
elif opt == '--interactionDirs': interactionDirs.append(arg)
elif opt == '--GeneSetSelection': GeneSetSelection=arg
elif opt == '--PathwaySelection': PathwaySelection=arg
elif opt == '--OntologyID': OntologyID=arg
elif opt == '--display': display=arg
if update_interactions == 'yes': update_interactions = True
else: update_interactions = False
if input_file_dir == None: pass
elif len(input_file_dir) == 0: input_file_dir = None
if len(input_exp_file) == 0: input_exp_file = None
if len(interactionDirs) == 0: interactionDirs=['WikiPathways']
if interactionDirs == ['all']:
interactionDirs = ['WikiPathways','KEGG','BioGRID','TFTargets','common-microRNATargets','all-microRNATargets','common-DrugBank','all-DrugBank']
if interactionDirs == ['main']:
interactionDirs = ['WikiPathways','KEGG','BioGRID','TFTargets']
if interactionDirs == ['confident']:
interactionDirs = ['WikiPathways','KEGG','TFTargets']
if len(Genes) == 0: Genes = None
if output_dir == None: pass
elif len(output_dir) == 0: output_dir = None
if len(GeneSetSelection) == 'None Selected': GeneSetSelection = None
if includeExpIDs=='yes': includeExpIDs = True
else: includeExpIDs = False
gsp = UI.GeneSelectionParameters(species,array_type,manufacturer)
gsp.setGeneSet(GeneSetSelection)
gsp.setPathwaySelect(PathwaySelection)
gsp.setGeneSelection(Genes)
gsp.setOntologyID(OntologyID)
gsp.setIncludeExpIDs(includeExpIDs)
root = ''
if species == None:
print 'Please designate a species (--species).'; sys.exit()
if output_dir == None:
print 'Please designate an ouput directory (--output)'; sys.exit()
if input_file_dir !=None:
if '.txt' in input_file_dir or '.sif' in input_file_dir:
UI.networkBuilder(input_file_dir,inputType,output_dir,interactionDirs,degrees,input_exp_file,gsp,root)
else:
parent_dir = input_file_dir
dir_list = read_directory(parent_dir)
for file in dir_list:
input_file_dir = parent_dir+'/'+file
try:
UI.networkBuilder(input_file_dir,inputType,output_dir,interactionDirs,degrees,input_exp_file,gsp,root)
except Exception:
print file, 'failed to produce network'
else:
UI.networkBuilder(None,inputType,output_dir,interactionDirs,degrees,input_exp_file,gsp,root)
sys.exit()
########## Begin database dependent AltAnalyze workflows
if ensembl_version != 'current' and 'markers' not in update_method:
dbversion = string.replace(ensembl_version,'EnsMart','')
UI.exportDBversion('EnsMart'+dbversion)
gene_database = unique.getCurrentGeneDatabaseVersion()
print 'Current database version:',gene_database
if array_type == None and update_dbs != 'yes' and denom_file_dir == None:
print "Please specify an array or data type (e.g., RNASeq, exon, gene, junction, AltMouse, 3'array)."; sys.exit()
if 'archive' in update_method:
###
print 'Archiving databases', ensembl_version
try: archive_dir = 'ArchiveDBs/EnsMart'+ensembl_version+'/archive'; export.createDirPath(filepath(archive_dir))
except Exception: null = [] ### directory already exists
dirs = unique.read_directory('/ArchiveDBs/EnsMart'+ensembl_version)
print len(dirs), dirs
import shutil
for species_dir in dirs:
try:
#print '/ArchiveDBs/EnsMart'+ensembl_version+'/'+species_dir+'/'+species_dir+'_RNASeq.zip'
src = filepath('ArchiveDBs/EnsMart'+ensembl_version+'/'+species_dir+'/'+species_dir+'_RNASeq.zip')
dstn = filepath('ArchiveDBs/EnsMart'+ensembl_version+'/archive/'+species_dir+'_RNASeq.zip')
#export.copyFile(src, dstn)
shutil.move(src, dstn)
try:
srcj = string.replace(src,'RNASeq.','junction.'); dstnj = string.replace(dstn,'RNASeq.','junction.')
shutil.move(srcj, dstnj)
except Exception: null=[]
try:
src = string.replace(src,'_RNASeq.','.'); dstn = string.replace(dstn,'_RNASeq.','.')
shutil.move(src, dstn)
except Exception:
print traceback.format_exc()
pass
except Exception:
print traceback.format_exc()
pass
sys.exit()
if update_dbs == 'yes' and 'Official' not in update_method:
if 'cleanup' in update_method:
existing_species_dirs = unique.read_directory('/AltDatabase/ensembl')
print 'Deleting EnsemblSQL directory for all species, ensembl version',ensembl_version
for species in existing_species_dirs:
export.deleteFolder('AltDatabase/ensembl/'+species+'/EnsemblSQL')
existing_species_dirs = unique.read_directory('/AltDatabase')
print 'Deleting SequenceData directory for all species, ensembl version',ensembl_version
for species in existing_species_dirs:
export.deleteFolder('AltDatabase/'+species+'/SequenceData')
print 'Finished...exiting'
sys.exit()
if 'package' not in update_method and 'markers' not in update_method:
### Example:
### python AltAnalyze.py --species all --arraytype all --update all --version 60
### tr -d \\r < AltAnalyze.py > AltAnalyze_new.py
### chmod +x AltAnalyze_new.py
### nohup ./AltAnalyze.py --update all --species Mm --arraytype gene --arraytype exon --version 60 2>&1 > nohup_v60_Mm.txt
if array_type == 'all' and (species == 'Mm' or species == 'all'): array_type = ['AltMouse','exon','gene','junction','RNASeq']
elif array_type == 'all' and (species == 'Hs' or species == 'Rn'): array_type = ['exon','gene','junction','RNASeq']
else: array_type = [array_type]+additional_array_types
if species == 'all' and 'RNASeq' not in array_type: species = selected_species ### just analyze the species for which multiple platforms are supported
if species == 'selected': species = selected_species ### just analyze the species for which multiple platforms are supported
elif species == 'all':
all_supported_names = {}; all_species_names={}
species_names = UI.getSpeciesInfo()
for species in species_names: all_supported_names[species_names[species]]=species
from build_scripts import EnsemblSQL
child_dirs, ensembl_species, ensembl_versions = EnsemblSQL.getCurrentEnsemblSpecies('release-'+ensembl_version)
for ens_species in ensembl_species:
ens_species = string.replace(ens_species,'_',' ')
if ens_species in all_supported_names:
all_species_names[all_supported_names[ens_species]]=[]
del all_species_names['Hs']
del all_species_names['Mm']
del all_species_names['Rn']
"""
del all_species_names['Go']
del all_species_names['Bt']
del all_species_names['Sc']
del all_species_names['Ss']
del all_species_names['Pv']
del all_species_names['Pt']
del all_species_names['La']
del all_species_names['Tt']
del all_species_names['Tr']
del all_species_names['Ts']
del all_species_names['Pb']
del all_species_names['Pc']
del all_species_names['Ec']
del all_species_names['Tb']
del all_species_names['Tg']
del all_species_names['Dn']
del all_species_names['Do']
del all_species_names['Tn']
del all_species_names['Dm']
del all_species_names['Oc']
del all_species_names['Og']
del all_species_names['Fc']
del all_species_names['Dr']
del all_species_names['Me']
del all_species_names['Cp']
del all_species_names['Tt']
del all_species_names['La']
del all_species_names['Tr']
del all_species_names['Ts']
del all_species_names['Et'] ### No alternative isoforms?
del all_species_names['Pc']
del all_species_names['Tb']
del all_species_names['Fc']
del all_species_names['Sc']
del all_species_names['Do']
del all_species_names['Dn']
del all_species_names['Og']
del all_species_names['Ga']
del all_species_names['Me']
del all_species_names['Ml']
del all_species_names['Mi']
del all_species_names['St']
del all_species_names['Sa']
del all_species_names['Cs']
del all_species_names['Vp']
del all_species_names['Ch']
del all_species_names['Ee']
del all_species_names['Ac']"""
sx=[]; all_species_names2=[] ### Ensure that the core selected species are run first
for species in selected_species:
if species in all_species_names: sx.append(species)
for species in all_species_names:
if species not in selected_species: all_species_names2.append(species)
all_species_names = sx+all_species_names2
species = all_species_names
else: species = [species]
update_uniprot='no'; update_ensembl='no'; update_probeset_to_ensembl='no'; update_domain='no'; update_miRs = 'no'; genomic_build = 'new'; update_miR_seq = 'yes'
if 'all' in update_method:
update_uniprot='yes'; update_ensembl='yes'; update_probeset_to_ensembl='yes'; update_domain='yes'; update_miRs = 'yes'
if 'UniProt' in update_method: update_uniprot = 'yes'
if 'Ensembl' in update_method: update_ensembl = 'yes'
if 'Probeset' in update_method or 'ExonAnnotations' in update_method: update_probeset_to_ensembl = 'yes'
if 'Domain' in update_method:
update_domain = 'yes'
try: from Bio import Entrez #test this
except Exception: print 'The dependent module Bio is not installed or not accessible through the default python interpretter. Existing AltAnalyze.'; sys.exit()
if 'miRBs' in update_method or 'miRBS' in update_method: update_miRs = 'yes'
if 'NewGenomeBuild' in update_method: genomic_build = 'new'
if 'current' in ensembl_version: print "Please specify an Ensembl version number (e.g., 60) before proceeding with the update.";sys.exit()
try: force = force ### Variable is not declared otherwise
except Exception: force = 'yes'; print 'force:',force
existing_species_dirs={}
update_all = 'no' ### We don't pass this as yes, in order to skip certain steps when multiple array types are analyzed (others are specified above)
try: print "Updating AltDatabase the following array_types",string.join(array_type),"for the species",string.join(species)
except Exception: print 'Please designate a valid platform/array_type (e.g., exon) and species code (e.g., Mm).'
for specific_species in species:
for platform_name in array_type:
if platform_name == 'AltMouse' and specific_species == 'Mm': proceed = 'yes'
elif platform_name == 'exon' or platform_name == 'gene':
from build_scripts import ExonArrayEnsemblRules
#### Check to see if the probeset.csv file is present
#try: probeset_transcript_file = ExonArrayEnsemblRules.getDirectoryFiles('/AltDatabase/'+specific_species+'/'+platform_name)
#except Exception: print "Affymetrix probeset.csv anotation file is not found. You must save this to",'/AltDatabase/'+specific_species+'/'+platform_name,'before updating (unzipped).'; sys.exit()
proceed = 'yes'
elif platform_name == 'junction' and (specific_species == 'Hs' or specific_species == 'Mm'): proceed = 'yes'
elif platform_name == 'RNASeq': proceed = 'yes'
else: proceed = 'no'
if proceed == 'yes':
print "Analyzing", specific_species, platform_name
if (platform_name != array_type[0]) and len(species)==1:
update_uniprot = 'no'; update_ensembl = 'no'; update_miR_seq = 'no' ### Don't need to do this twice in a row
print 'Skipping ensembl, uniprot and mir-sequence file import updates since already completed for this species',array_type,platform_name
if ignore_built_species == 'yes': ### Useful for when building all species for a new database build
try:
### call this here to update with every species - if running multiple instances
existing_species_dirs = unique.read_directory('/AltDatabase/ensembl')
except Exception: #ZeroDivisionError
try: os.mkdir(unique.filepath('AltDatabase/'))
except Exception: pass #already exists
existing_species_dirs = []
if specific_array_type != None and specific_array_type != platform_name: platform_name+='|'+specific_array_type ### For the hGlue vs. JAY arrays
if specific_species not in existing_species_dirs: ### Useful when running multiple instances of AltAnalyze to build all species
print 'update_ensembl',update_ensembl
print 'update_uniprot',update_uniprot
print 'update_probeset_to_ensembl',update_probeset_to_ensembl
print 'update_domain',update_domain
print 'update_miRs',update_miRs
update.executeParameters(specific_species,platform_name,force,genomic_build,update_uniprot,update_ensembl,update_probeset_to_ensembl,update_domain,update_miRs,update_all,update_miR_seq,ensembl_version)
else: print 'ignoring',specific_species
sys.exit()
if 'package' in update_method:
### Example: python AltAnalyze.py --update package --species all --platform all --version 65
if ensembl_version == 'current': print '\nPlease specify version of the database to package (e.g., --version 60).'; sys.exit()
ensembl_version = 'EnsMart'+ensembl_version
### Get all possible species
species_names = UI.getSpeciesInfo(); possible_species={}
possible_species = species_names
possible_arrays = ['exon','gene','junction','AltMouse','RNASeq']
try:
if species == 'all': possible_species = possible_species
elif species == 'selected': possible_species = selected_species
else: possible_species = [species]
except Exception: species = possible_species
if array_type == None or array_type == 'all': possible_arrays = possible_arrays
else: possible_arrays = [array_type]+additional_array_types
species_to_package={}
dirs = unique.read_directory('/AltDatabase/'+ensembl_version)
#print possible_arrays, possible_species; sys.exit()
for species_code in dirs:
if species_code in possible_species:
array_types = unique.read_directory('/AltDatabase/'+ensembl_version+'/'+species_code)
for arraytype in array_types:
if arraytype in possible_arrays:
if species_code in possible_species:
array_types = unique.read_directory('/AltDatabase/'+ensembl_version+'/'+species_code)
try: species_to_package[species_code].append(arraytype)
except Exception: species_to_package[species_code] = [arraytype]
species_to_package = eliminate_redundant_dict_values(species_to_package)
for species in species_to_package:
files_to_copy =[species+'_Ensembl_domain_aligning_probesets.txt']
files_to_copy+=[species+'_Ensembl_indirect_domain_aligning_probesets.txt']
files_to_copy+=[species+'_Ensembl_probesets.txt']
files_to_copy+=[species+'_Ensembl_exons.txt']
#files_to_copy+=[species+'_Ensembl_junctions.txt']
files_to_copy+=[species+'_exon_core.mps']
files_to_copy+=[species+'_exon_extended.mps']
files_to_copy+=[species+'_exon_full.mps']
files_to_copy+=[species+'_gene_core.mps']
files_to_copy+=[species+'_gene_extended.mps']
files_to_copy+=[species+'_gene_full.mps']
files_to_copy+=[species+'_gene-exon_probesets.txt']
files_to_copy+=[species+'_probes_to_remove.txt']
files_to_copy+=[species+'_probeset-probes.txt']
files_to_copy+=[species+'_probeset_microRNAs_any.txt']
files_to_copy+=[species+'_probeset_microRNAs_multiple.txt']
files_to_copy+=['probeset-domain-annotations-exoncomp.txt']
files_to_copy+=['probeset-protein-annotations-exoncomp.txt']
#files_to_copy+=['probeset-protein-dbase_exoncomp.txt']
files_to_copy+=['SEQUENCE-protein-dbase_exoncomp.txt']
files_to_copy+=[species+'_Ensembl_junction_probesets.txt']
files_to_copy+=[species+'_Ensembl_AltMouse_probesets.txt']
files_to_copy+=[species+'_RNASeq-exon_probesets.txt']
files_to_copy+=[species+'_junction-exon_probesets.txt']
files_to_copy+=[species+'_junction_all.mps']
files_to_copy+=['platform.txt'] ### Indicates the specific platform for an array type (e.g., HJAY for junction or hGlue for junction)
files_to_copy+=[species+'_junction_comps_updated.txt']
files_to_copy+=['MASTER-probeset-transcript.txt']
files_to_copy+=['AltMouse-Ensembl.txt']
files_to_copy+=['AltMouse_junction-comparisons.txt']
files_to_copy+=['AltMouse_gene_annotations.txt']
files_to_copy+=['AltMouse_annotations.txt']
common_to_copy =['uniprot/'+species+'/custom_annotations.txt']
common_to_copy+=['ensembl/'+species+'/'+species+'_Ensembl-annotations_simple.txt']
common_to_copy+=['ensembl/'+species+'/'+species+'_Ensembl-annotations.txt']
common_to_copy+=['ensembl/'+species+'/'+species+'_microRNA-Ensembl.txt']
common_to_copy+=['ensembl/'+species+'/'+species+'_Ensembl_transcript-biotypes.txt']
common_to_copy+=['ensembl/'+species+'/'+species+'_Ensembl_transcript-annotations.txt']
common_to_copy+= searchDirectory("AltDatabase/ensembl/"+species+"/",'Ensembl_Protein')
common_to_copy+= searchDirectory("AltDatabase/ensembl/"+species+"/",'ProteinFeatures')
common_to_copy+= searchDirectory("AltDatabase/ensembl/"+species+"/",'ProteinCoordinates')
common_to_copy+= searchDirectory("AltDatabase/uniprot/"+species+"/",'FeatureCoordinate')
supported_arrays_present = 'no'
for arraytype in selected_platforms:
if arraytype in species_to_package[species]: supported_arrays_present = 'yes' #Hence a non-RNASeq platform is present
if supported_arrays_present == 'yes':
for file in common_to_copy:
ir = 'AltDatabase/'+ensembl_version+'/'
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+ensembl_version+'/'
export.copyFile(ir+file, er+file)
if 'RNASeq' in species_to_package[species]:
common_to_copy+=['ensembl/'+species+'/'+species+'_Ensembl_junction.txt']
common_to_copy+=['ensembl/'+species+'/'+species+'_Ensembl_exon.txt']
for file in common_to_copy:
ir = 'AltDatabase/'+ensembl_version+'/'
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+ensembl_version+'/'
if species in selected_species:
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/RNASeq/'+ensembl_version+'/' ### This allows us to build the package archive in a separate directory for selected species, so separate but overlapping content can be packaged
export.copyFile(ir+file, er+file)
for array_type in species_to_package[species]:
ir = 'AltDatabase/'+ensembl_version+'/'+species+'/'+array_type+'/'
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+ensembl_version+'/'+species+'/'+array_type+'/'
if array_type == 'junction':
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+array_type+'/'
if array_type == 'RNASeq' and species in selected_species:
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/RNASeq/'+ensembl_version+'/'+species+'/'+array_type+'/'
for file in files_to_copy:
if array_type == 'RNASeq': file=string.replace(file,'_updated.txt','.txt')
filt_file = string.replace(file ,'.txt','-filtered.txt')
try: export.copyFile(ir+filt_file, er+filt_file); export_path = er+filt_file
except Exception:
try: export.copyFile(ir+file, er+file); export_path = er+file
except Exception: null = [] ### File not found in directory
if len(export_path)>0:
if 'AltMouse' in export_path or 'probes_' in export_path:
export.cleanFile(export_path)
if array_type == 'junction':
subdir = '/exon/'
ir = 'AltDatabase/'+ensembl_version+'/'+species+'/'+array_type+subdir
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+array_type+subdir
for file in files_to_copy:
export_path=[]
filt_file = string.replace(file ,'.txt','-filtered.txt')
try: export.copyFile(ir+filt_file, er+filt_file); export_path = er+filt_file
except Exception:
try: export.copyFile(ir+file, er+file); export_path = er+file
except Exception: null = [] ### File not found in directory
if array_type == 'RNASeq':
subdir = '/junction/'
ir = 'AltDatabase/'+ensembl_version+'/'+species+'/'+array_type+subdir
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+ensembl_version+'/'+species+'/'+array_type+subdir
if species in selected_species:
er = 'ArchiveDBs/'+ensembl_version+'/'+species+'/RNASeq/'+ensembl_version+'/'+species+'/'+array_type+subdir
for file in files_to_copy:
if 'SEQUENCE-protein-dbase' not in file and 'domain_aligning' not in file: ### This data is now combined into the main file
export_path=[]
filt_file = string.replace(file ,'.txt','-filtered.txt')
try: export.copyFile(ir+filt_file, er+filt_file); export_path = er+filt_file
except Exception:
try: export.copyFile(ir+file, er+file); export_path = er+file
except Exception: null = [] ### File not found in directory
if 'RNASeq' in species_to_package[species]:
src = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+ensembl_version
dst = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+species+'_RNASeq.zip'
if species in selected_species:
src = 'ArchiveDBs/'+ensembl_version+'/'+species+'/RNASeq/'+ensembl_version
update.zipDirectory(src); print 'Zipping',species, array_type, dst
os.rename(src+'.zip', dst)
if supported_arrays_present == 'yes':
src = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+ensembl_version
dst = 'ArchiveDBs/'+ensembl_version+'/'+species+'/'+species+'.zip'
update.zipDirectory(src); print 'Zipping',species, array_type, dst
os.rename(src+'.zip', dst)
if 'junction' in species_to_package[species]:
src = 'ArchiveDBs/'+ensembl_version+'/'+species+'/junction'
dst = string.replace(src,'junction',species+'_junction.zip')
update.zipDirectory(src); print 'Zipping',species+'_junction'
os.rename(src+'.zip', dst)
sys.exit()
if 'markers' in update_method:
if species == None or platform == None:
print "WARNING! A species and platform (e.g., exon, junction, 3'array or RNASeq) must be defined to identify markers.";sys.exit()
elif input_exp_file == '':
print "WARNING! A input expression file must be supplied (e.g., ExpressionOutput/DATASET.YourExperimentName.txt) for this analysis.";sys.exit()
else:
#python AltAnalyze.py --update markers --platform gene --expdir "/home/socr/c/users2/salomoni/other/boxer/normalization/Mm_Gene-TissueAtlas/ExpressionInput/exp.meta.txt"
#python AltAnalyze.py --update markers --platform gene --expdir "/home/socr/c/users2/salomoni/other/boxer/normalization/Mm_Gene-TissueAtlas/AltResults/RawSpliceData/Mm/splicing-index/meta.txt"
#python AltAnalyze.py --update markers --platform "3'array" --expdir "/home/socr/c/users2/salomoni/other/boxer/normalization/U133/ExpressionOutput/DATASET-meta.txt"
#python AltAnalyze.py --update markers --compendiumType ncRNA --platform "exon" --expdir "/home/socr/c/users2/salomoni/conklin/nsalomonis/normalization/Hs_Exon-TissueAtlas/ExpressionOutput/DATASET-meta.txt"
#python AltAnalyze.py --update markers --platform RNASeq --species Mm --geneRPKM 1 --expdir /Users/saljh8/Desktop/Grimes/MergedRSEM/DN-Analysis/ExpressionInput/exp.DN.txt --genesToReport 200
"""The markerFinder module:
1) takes an input ExpressionOutput file (DATASET.YourExperimentName.txt)
2) extracts group average expression and saves to AVERAGE.YourExperimentName.txt to the ExpressionOutput directory
3) re-imports AVERAGE.YourExperimentName.txt
4) correlates the average expression of each gene to an idealized profile to derive a Pearson correlation coefficient
5) identifies optimal markers based on these correlations for each tissue
6) exports an expression file with just these marker genes and tissues
This module can peform these analyses on protein coding or ncRNAs and can segregate the cell/tissue groups into clusters
when a group notation is present in the sample name (e.g., 0~Heart, 0~Brain, 1~Stem Cell)"""
import markerFinder
if 'AltResults' in input_exp_file and 'Clustering' not in input_exp_file:
### This applies to a file compoosed of exon-level normalized intensities (calculae average group expression)
markerFinder.getAverageExonExpression(species,platform,input_exp_file)
if 'Raw' in input_exp_file:
group_exp_file = string.replace(input_exp_file,'Raw','AVERAGE')
else:
group_exp_file = string.replace(input_exp_file,'FullDatasets','AVERAGE-FullDatasets')
altexon_correlation_file = markerFinder.analyzeData(group_exp_file,species,platform,compendiumType,geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl)
markerFinder.getExprValsForNICorrelations(platform,altexon_correlation_file,group_exp_file)
else:
### This applies to an ExpressionOutput DATASET file compoosed of gene expression values (averages already present)
import collections
try: test_ordereddict=collections.OrderedDict()
except Exception:
try: import ordereddict
except Exception:
### This is needed to re-order the average file so that the groups are sequentially ordered when analyzing clustered groups (0~)
print 'Warning!!!! To run markerFinder correctly call python version 2.7x or greater (python 3.x not supported)'
print 'Requires ordereddict (also can install the library ordereddict). To call 2.7: /usr/bin/python2.7'
sys.exit()
try:
output_dir = markerFinder.getAverageExpressionValues(input_exp_file,platform) ### Either way, make an average annotated file from the DATASET file
if 'DATASET' in input_exp_file:
group_exp_file = string.replace(input_exp_file,'DATASET','AVERAGE')
else:
group_exp_file = (input_exp_file,output_dir) ### still analyze the primary sample
except Exception:
#print traceback.format_exc()
print 'No DATASET file present (used to obtain gene annotations)...'
### Work around when performing this analysis on an alternative exon input cluster file
group_exp_file = input_exp_file
fl = UI.ExpressionFileLocationData(input_exp_file,'','',''); fl.setOutputDir(export.findParentDir(export.findParentDir(input_exp_file)[:-1]))
try: fl.setSpecies(species); fl.setVendor(vendor)
except Exception: pass
try:
rpkm_threshold = float(rpkm_threshold) ### If supplied, for any platform, use it
fl.setRPKMThreshold(rpkm_threshold)
except Exception: pass
if platform=='RNASeq':
try: rpkm_threshold = float(rpkm_threshold)
except Exception: rpkm_threshold = 1.0
fl.setRPKMThreshold(rpkm_threshold)
try: correlationDirection = direction ### correlate to a positive or inverse negative in silico artificial pattern
except Exception: correlationDirection = 'up'
fl.setCorrelationDirection(correlationDirection)
if expression_data_format == 'non-log': logTransform = True
else: logTransform = False
if 'topSplice' in input_exp_file:
markerFinder.filterRNASeqSpliceEvents(species,platform,fl,input_exp_file)
sys.exit()
if 'stats.' in input_exp_file:
markerFinder.filterDetectionPvalues(species,platform,fl,input_exp_file)
sys.exit()
else:
markerFinder.analyzeData(group_exp_file,species,platform,compendiumType,geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl,logTransform=logTransform)
try: fl.setVendor(manufacturer)
except Exception:
print '--vendor not indicated by user... assuming Affymetrix'
fl.setVendor('Affymetrix')
try: markerFinder.generateMarkerHeatMaps(fl,array_type,convertNonLogToLog=logTransform,Species=species)
except Exception: print traceback.format_exc()
print 'Cell/Tissue marker classification analysis finished';sys.exit()
if 'EnsMart' in ensembl_version:
UI.exportDBversion(ensembl_version)
annotation_found = verifyFile(input_annotation_file)
proceed = 'no'
if 'Official' not in update_method and denom_file_dir == None: ### If running GO-Elite independent of AltAnalyze (see below GO_Elite call)
try:
time_stamp = timestamp()
if len(cel_file_dir)>0:
if output_dir == None:
output_dir = cel_file_dir
print "Setting output directory to the input path:", output_dir
if output_dir == None and input_filtered_dir>0:
output_dir = input_filtered_dir
try:
if '/' == output_dir[-1] or '\\' in output_dir[-2]: null=[]
else: output_dir +='/'
except:
try: output_dir = export.findParentDir(input_file_dir)
except:
output_dir = input_fastq_dir
log_file = filepath(output_dir+'/AltAnalyze_report-'+time_stamp+'.log')
log_report = open(log_file,'w'); log_report.close()
sys.stdout = Logger('')
except Exception,e:
print traceback.format_exc()
print 'Please designate an output directory before proceeding (e.g., --output "C:\RNASeq)';sys.exit()
try: print string.join(arguments,' ')
except Exception: pass
if mappedExonAnalysis:
array_type = 'RNASeq' ### Although this is not the actual platform, the resulting data will be treated as RNA-Seq with parameters most suitable for arrays
if len(external_annotation_dir)>0:
run_from_scratch = 'Annotate External Results'
if channel_to_extract != None:
run_from_scratch = 'Process Feature Extraction files' ### Agilent Feature Extraction files as input for normalization
manufacturer = 'Agilent'
constitutive_source = 'Agilent'
expression_threshold = 'NA'
perform_alt_analysis = 'NA'
if len(input_filtered_dir)>0:
run_from_scratch ='Process AltAnalyze filtered'; proceed='yes'
if len(input_exp_file)>0:
run_from_scratch = 'Process Expression file'; proceed='yes'
input_exp_file = string.replace(input_exp_file,'\\','/') ### Windows convention is \ rather than /, but works with /
ief_list = string.split(input_exp_file,'/')
if len(output_dir)>0: parent_dir = output_dir
else: parent_dir = string.join(ief_list[:-1],'/')
exp_name = ief_list[-1]
if len(cel_file_dir)>0 or runKallisto == True:
# python AltAnalyze.py --species Mm --platform RNASeq --runKallisto yes --expname test
if exp_name == None:
print "No experiment name defined. Please sumbit a name (e.g., --expname CancerComp) before proceeding."; sys.exit()
else:
dataset_name = 'exp.'+exp_name+'.txt'; exp_file_dir = filepath(output_dir+'/ExpressionInput/'+dataset_name)
if runKallisto:
run_from_scratch == 'Process RNA-seq reads'
elif run_from_scratch!= 'Process Feature Extraction files':
run_from_scratch = 'Process CEL files'; proceed='yes'
if array_type == 'RNASeq': file_ext = '.BED'
else: file_ext = '.CEL'
try: cel_files,cel_files_fn = UI.identifyCELfiles(cel_file_dir,array_type,manufacturer)
except Exception,e:
print e
if mappedExonAnalysis: pass
else: print "No",file_ext,"files found in the directory:",cel_file_dir;sys.exit()
if array_type != 'RNASeq': cel_file_list_dir = UI.exportCELFileList(cel_files_fn,cel_file_dir)
if groups_file != None and comps_file != None:
try: export.copyFile(groups_file, string.replace(exp_file_dir,'exp.','groups.'))
except Exception: print 'Groups file already present in target location OR bad input path.'
try: export.copyFile(comps_file, string.replace(exp_file_dir,'exp.','comps.'))
except Exception: print 'Comparison file already present in target location OR bad input path.'
groups_file = string.replace(exp_file_dir,'exp.','groups.')
comps_file = string.replace(exp_file_dir,'exp.','comps.')
if verifyGroupFileFormat(groups_file) == False:
print "\nWarning! The format of your groups file is not correct. For details, see:\nhttp://altanalyze.readthedocs.io/en/latest/ManualGroupsCompsCreation\n"
sys.exit()
if array_type != 'RNASeq' and manufacturer!= 'Agilent':
"""Determine if Library and Annotations for the array exist, if not, download or prompt for selection"""
try:
### For the HGLUE and HJAY arrays, this step is critical in order to have the commond-line AltAnalyze downloadthe appropriate junction database (determined from specific_array_type)
specific_array_types,specific_array_type = UI.identifyArrayType(cel_files_fn)
num_array_types = len(specific_array_types)
except Exception:
null=[]; num_array_types=1; specific_array_type=None
if array_type == 'exon':
if species == 'Hs': specific_array_type = 'HuEx-1_0-st-v2'
if species == 'Mm': specific_array_type = 'MoEx-1_0-st-v2'
if species == 'Rn': specific_array_type = 'RaEx-1_0-st-v2'
elif array_type == 'gene':
if species == 'Hs': specific_array_type = 'HuGene-1_0-st-v1'
if species == 'Mm': specific_array_type = 'MoGene-1_0-st-v1'
if species == 'Rn': specific_array_type = 'RaGene-1_0-st-v1'
elif array_type == 'AltMouse': specific_array_type = 'altMouseA'
"""
elif array_type == 'junction':
if species == 'Mm': specific_array_type = 'MJAY'
if species == 'Hs': specific_array_type = 'HJAY'
"""
supproted_array_db = UI.importSupportedArrayInfo()
if specific_array_type in supproted_array_db and input_cdf_file == None and input_annotation_file == None:
sa = supproted_array_db[specific_array_type]; species = sa.Species(); array_type = sa.ArrayType()
input_cdf_file, input_annotation_file, bgp_file, clf_file = UI.getAffyFilesRemote(specific_array_type,array_type,species)
else: array_type = "3'array"
cdf_found = verifyFile(input_cdf_file)
annotation_found = verifyFile(input_annotation_file)
if input_cdf_file == None:
print [specific_array_type], 'not currently supported... Please provide CDF to AltAnalyze (commandline or GUI) or manually add to AltDatabase/affymetrix/LibraryFiles'; sys.exit()
if cdf_found != "found":
### Copy valid Library files to a local AltAnalyze database directory
input_cdf_file_lower = string.lower(input_cdf_file)
if array_type == "3'array":
if '.cdf' in input_cdf_file_lower:
clf_file='';bgp_file=''; assinged = 'yes'
###Thus the CDF or PDF file was confirmed, so copy it over to AltDatabase
icf_list = string.split(input_cdf_file,'/'); cdf_short = icf_list[-1]
destination_parent = 'AltDatabase/affymetrix/LibraryFiles/'
destination_parent = osfilepath(destination_parent+cdf_short)
info_list = input_cdf_file,destination_parent; UI.StatusWindow(info_list,'copy')
else: print "Valid CDF file not found. Exiting program.";sys.exit()
else:
if '.pgf' in input_cdf_file_lower:
###Check to see if the clf and bgp files are present in this directory
icf_list = string.split(input_cdf_file,'/'); parent_dir = string.join(icf_list[:-1],'/'); cdf_short = icf_list[-1]
clf_short = string.replace(cdf_short,'.pgf','.clf')
kil_short = string.replace(cdf_short,'.pgf','.kil') ### Only applies to the Glue array
if array_type == 'exon' or array_type == 'junction': bgp_short = string.replace(cdf_short,'.pgf','.antigenomic.bgp')
else: bgp_short = string.replace(cdf_short,'.pgf','.bgp')
dir_list = read_directory(parent_dir)
if clf_short in dir_list and bgp_short in dir_list:
pgf_file = input_cdf_file
clf_file = string.replace(pgf_file,'.pgf','.clf')
kil_file = string.replace(pgf_file,'.pgf','.kil') ### Only applies to the Glue array
if array_type == 'exon' or array_type == 'junction': bgp_file = string.replace(pgf_file,'.pgf','.antigenomic.bgp')
else: bgp_file = string.replace(pgf_file,'.pgf','.bgp')
assinged = 'yes'
###Thus the CDF or PDF file was confirmed, so copy it over to AltDatabase
destination_parent = 'AltDatabase/affymetrix/LibraryFiles/'
info_list = input_cdf_file,osfilepath(destination_parent+cdf_short); UI.StatusWindow(info_list,'copy')
info_list = clf_file,osfilepath(destination_parent+clf_short); UI.StatusWindow(info_list,'copy')
info_list = bgp_file,osfilepath(destination_parent+bgp_short); UI.StatusWindow(info_list,'copy')
if 'Glue' in pgf_file:
info_list = kil_file,osfilepath(destination_parent+kil_short); UI.StatusWindow(info_list,'copy')
if annotation_found != "found" and update_dbs == 'no' and array_type != 'RNASeq' and denom_file_dir == None and manufacturer != 'Agilent':
### Copy valid Annotation files to a local AltAnalyze database directory
try:
input_annotation_lower = string.lower(input_annotation_file)
if '.csv' in input_annotation_lower:
assinged = 'yes'
###Thus the CDF or PDF file was confirmed, so copy it over to AltDatabase
icf_list = string.split(input_annotation_file,'/'); csv_short = icf_list[-1]
destination_parent = 'AltDatabase/affymetrix/'+species+'/'
info_list = input_annotation_file,filepath(destination_parent+csv_short); UI.StatusWindow(info_list,'copy')
except Exception: print "No Affymetrix annotation file provided. AltAnalyze will use any .csv annotations files in AltDatabase/Affymetrix/"+species
if array_type == 'PSI':
array_type = "3'array"
vendor = 'PSI'
if 'Official' in update_method and species != None:
proceed = 'yes'
elif array_type != None and species != None:
expr_defaults, alt_exon_defaults, functional_analysis_defaults, goelite_defaults = UI.importDefaults(array_type,species)
ge_fold_cutoffs, ge_pvalue_cutoffs, ge_ptype, filter_method, z_threshold, p_val_threshold, change_threshold, ORA_algorithm, resources_to_analyze, goelite_permutations, mod, returnPathways, NA = goelite_defaults
use_direct_domain_alignments_only,microRNA_prediction_method = functional_analysis_defaults
analysis_method, additional_algorithms, filter_probeset_types, analyze_all_conditions, p_threshold, alt_exon_fold_variable, additional_score, permute_p_threshold, gene_expression_cutoff, remove_intronic_junctions, perform_permutation_analysis, export_NI_values, run_MiDAS, calculate_normIntensity_p, filter_for_AS = alt_exon_defaults
dabg_p, rpkm_threshold, gene_exp_threshold, exon_exp_threshold, exon_rpkm_threshold, expression_threshold, perform_alt_analysis, analyze_as_groups, expression_data_format, normalize_feature_exp, normalize_gene_data, avg_all_for_ss, include_raw_data, probability_statistic, FDR_statistic, batch_effects, marker_finder, visualize_qc_results, run_lineage_profiler, null = expr_defaults
elif denom_file_dir != None and species != None:
proceed = 'yes' ### Only run GO-Elite
expr_defaults, alt_exon_defaults, functional_analysis_defaults, goelite_defaults = UI.importDefaults('RNASeq',species) ### platform not relevant
ge_fold_cutoffs, ge_pvalue_cutoffs, ge_ptype, filter_method, z_threshold, p_val_threshold, change_threshold, ORA_algorithm, resources_to_analyze, goelite_permutations, mod, returnPathways, NA = goelite_defaults
else:
print 'No species defined. Please include the species code (e.g., "--species Hs") and array type (e.g., "--arraytype exon") before proceeding.'
print '\nAlso check the printed arguments above to see if there are formatting errors, such as bad quotes.'; sys.exit()
array_type_original = array_type
#if array_type == 'gene': array_type = "3'array"
for opt, arg in options:
if opt == '--runGOElite': run_GOElite=arg
elif opt == '--outputQCPlots': visualize_qc_results=arg
elif opt == '--runLineageProfiler' or opt == '--cellHarmony' or opt == '--cellHarmonyMerge':
if string.lower(arg) == 'yes' or string.lower(arg) == 'true':
run_lineage_profiler = 'yes'
elif opt == '--elitepermut': goelite_permutations=arg
elif opt == '--method': filter_method=arg
elif opt == '--zscore': z_threshold=arg
elif opt == '--elitepval': p_val_threshold=arg
elif opt == '--num': change_threshold=arg
elif opt == '--dataToAnalyze': resources_to_analyze=arg
elif opt == '--GEelitepval': ge_pvalue_cutoffs=arg
elif opt == '--GEelitefold': ge_fold_cutoffs=arg
elif opt == '--GEeliteptype': ge_ptype=arg
elif opt == '--ORAstat': ORA_algorithm=arg
elif opt == '--returnPathways': returnPathways=arg
elif opt == '--FDR': FDR_statistic=arg
elif opt == '--dabgp': dabg_p=arg
elif opt == '--rawexp': expression_threshold=arg
elif opt == '--geneRPKM': rpkm_threshold=arg
elif opt == '--exonRPKM': exon_rpkm_threshold=arg
elif opt == '--geneExp': gene_exp_threshold=arg
elif opt == '--exonExp': exon_exp_threshold=arg
elif opt == '--groupStat': probability_statistic=arg
elif opt == '--avgallss': avg_all_for_ss=arg
elif opt == '--logexp': expression_data_format=arg
elif opt == '--inclraw': include_raw_data=arg
elif opt == '--combat': batch_effects=arg
elif opt == '--runalt': perform_alt_analysis=arg
elif opt == '--altmethod': analysis_method=arg
elif opt == '--altp': p_threshold=arg
elif opt == '--probetype': filter_probeset_types=arg
elif opt == '--altscore': alt_exon_fold_variable=arg
elif opt == '--GEcutoff': gene_expression_cutoff=arg
elif opt == '--removeIntronOnlyJunctions': remove_intronic_junctions=arg
elif opt == '--normCounts': normalize_feature_exp=arg
elif opt == '--normMatrix': normalize_gene_data=arg
elif opt == '--altpermutep': permute_p_threshold=arg
elif opt == '--altpermute': perform_permutation_analysis=arg
elif opt == '--exportnormexp': export_NI_values=arg
elif opt == '--buildExonExportFile': build_exon_bedfile = 'yes'
elif opt == '--runMarkerFinder': marker_finder = arg
elif opt == '--calcNIp': calculate_normIntensity_p=arg
elif opt == '--runMiDAS': run_MiDAS=arg
elif opt == '--analyzeAllGroups':
analyze_all_conditions=arg
if analyze_all_conditions == 'yes': analyze_all_conditions = 'all groups'
elif opt == '--GEcutoff': use_direct_domain_alignments_only=arg
elif opt == '--mirmethod': microRNA_prediction_method=arg
elif opt == '--ASfilter': filter_for_AS=arg
elif opt == '--noxhyb': xhyb_remove=arg
elif opt == '--returnAll': return_all=arg
elif opt == '--annotatedir': external_annotation_dir=arg
elif opt == '--additionalScore': additional_score=arg
elif opt == '--additionalAlgorithm': additional_algorithms=arg
elif opt == '--modelSize':
modelSize=arg
try: modelSize = int(modelSize)
except Exception: modelSize = None
elif opt == '--geneModel':
geneModel=arg # file location
if geneModel == 'no' or 'alse' in geneModel:
geneModel = False
elif opt == '--reference':
custom_reference = arg
if run_from_scratch == 'Process Feature Extraction files': ### Agilent Feature Extraction files as input for normalization
normalize_gene_data = 'quantile' ### required for Agilent
proceed = 'yes'
if returnPathways == 'no' or returnPathways == 'None':
returnPathways = None
if pipelineAnalysis == False:
proceed = 'yes'
if len(input_fastq_dir)>0:
proceed = 'yes'
run_from_scratch = 'Process RNA-seq reads'
fl = UI.ExpressionFileLocationData('','','',''); fl.setFeatureNormalization('none')
try: root_dir = root_dir
except: root_dir = output_dir
try: fl.setExpFile(expFile)
except Exception:
expFile = root_dir+'/ExpressionInput/exp.'+exp_name+'.txt'
fl.setExpFile(expFile)
fl.setArrayType(array_type)
fl.setOutputDir(root_dir)
fl.setMultiThreading(multiThreading)
exp_file_location_db={}; exp_file_location_db[exp_name]=fl
### Assign variables needed to run Kallisto from FASTQ files
if runKallisto and len(input_fastq_dir)==0:
#python AltAnalyze.py --runICGS yes --platform "RNASeq" --species Mm --column_method hopach --rho 0.4 --ExpressionCutoff 1 --FoldDiff 4 --SamplesDiffering 1 --excludeCellCycle strict --output /Users/saljh8/Desktop/Grimes/GEC14074 --expname test --fastq_dir /Users/saljh8/Desktop/Grimes/GEC14074
print 'Please include the flag "--fastq_dir" in the command-line arguments with an appropriate path';sys.exit()
elif len(input_fastq_dir)>0:
fl.setRunKallisto(input_fastq_dir)
fl.setArrayType("3'array")
array_type = "3'array"
if customFASTA!=None:
fl.setCustomFASTA(customFASTA)
if proceed == 'yes':
species_codes = UI.remoteSpeciesInfo()
### Update Ensembl Databases
if 'Official' in update_method:
file_location_defaults = UI.importDefaultFileLocations()
db_versions_vendors,db_versions = UI.remoteOnlineDatabaseVersions()
array_codes = UI.remoteArrayInfo()
UI.getOnlineDBConfig(file_location_defaults,'')
if len(species)==2:
species_names = UI.getSpeciesInfo()
species_full = species_names[species]
else: species_full = species
print 'Species name to update:',species_full
db_version_list=[]
for version in db_versions: db_version_list.append(version)
db_version_list.sort(); db_version_list.reverse(); select_version = db_version_list[0]
db_versions[select_version].sort()
print 'Ensembl version',ensembl_version
if ensembl_version != 'current':
if len(ensembl_version) < 4: ensembl_version = 'EnsMart'+ensembl_version
if ensembl_version not in db_versions:
try: UI.getOnlineEliteDatabase(file_location_defaults,ensembl_version,[species],'no',''); sys.exit()
except Exception:
### This is only for database that aren't officially released yet for prototyping
print ensembl_version, 'is not a valid version of Ensembl, while',select_version, 'is.'; sys.exit()
else: select_version = ensembl_version
### Export basic species information
sc = species; db_version = ensembl_version
if sc != None:
for ad in db_versions_vendors[db_version]:
if ad.SpeciesCodes() == species_full:
for array_system in array_codes:
ac = array_codes[array_system]
compatible_species = ac.SpeciesCodes()
if ac.Manufacturer() in ad.Manufacturer() and ('expression' in ac.ArrayName() or 'RNASeq' in ac.ArrayName() or 'RNA-seq' in ac.ArrayName()):
if sc not in compatible_species: compatible_species.append(sc)
ac.setSpeciesCodes(compatible_species)
UI.exportArrayInfo(array_codes)
if species_full not in db_versions[select_version]:
print db_versions[select_version]
print species_full, ': This species is not available for this version %s of the Official database.' % select_version
else:
update_goelite_resources = 'no' ### This is handled separately below
UI.getOnlineEliteDatabase(file_location_defaults,ensembl_version,[species],update_goelite_resources,'');
### Attempt to download additional Ontologies and GeneSets
if additional_resources[0] != None: ### Indicates that the user requested the download of addition GO-Elite resources
try:
from build_scripts import GeneSetDownloader
print 'Adding supplemental GeneSet and Ontology Collections'
if 'all' in additional_resources:
additionalResources = UI.importResourceList() ### Get's all additional possible resources
else: additionalResources = additional_resources
GeneSetDownloader.buildAccessoryPathwayDatabases([species],additionalResources,'yes')
print 'Finished adding additional analysis resources.'
except Exception:
print 'Download error encountered for additional Ontologies and GeneSets...\nplease try again later.'
status = UI.verifyLineageProfilerDatabases(species,'command-line')
if status == False:
print 'Please note: LineageProfiler not currently supported for this species...'
if array_type == 'junction' or array_type == 'RNASeq': ### Download junction databases
try: UI.checkForLocalArraySupport(species,array_type,specific_array_type,'command-line')
except Exception:
print 'Please install a valid gene database before proceeding.\n'
print 'For example: python AltAnalyze.py --species Hs --update Official --version EnsMart72';sys.exit()
status = UI.verifyLineageProfilerDatabases(species,'command-line')
print "Finished adding database"
sys.exit()
try:
#print ge_fold_cutoffs,ge_pvalue_cutoffs, change_threshold, resources_to_analyze, goelite_permutations, p_val_threshold, z_threshold
change_threshold = int(change_threshold)-1
goelite_permutations = int(goelite_permutations);change_threshold = change_threshold
p_val_threshold = float(p_val_threshold); z_threshold = float(z_threshold)
if ORA_algorithm == 'Fisher Exact Test':
goelite_permutations = 'FisherExactTest'
except Exception,e:
print e
print 'One of the GO-Elite input values is inapporpriate. Please review and correct.';sys.exit()
if run_GOElite == None or run_GOElite == 'no': goelite_permutations = 'NA' ### This haults GO-Elite from running
else:
if output_dir == None:
print "\nPlease specify an output directory using the flag --output"; sys.exit()
try: expression_threshold = float(expression_threshold)
except Exception: expression_threshold = 1
try: dabg_p = float(dabg_p)
except Exception: dabg_p = 1 ### Occurs for RNASeq
if microRNA_prediction_method == 'two or more': microRNA_prediction_method = 'multiple'
else: microRNA_prediction_method = 'any'
### Run GO-Elite directly from user supplied input and denominator ID folders (outside of the normal workflows)
if run_GOElite == 'yes' and pipelineAnalysis == False and '--runGOElite' in arguments:# and denom_file_dir != None:
#python AltAnalyze.py --input "/Users/nsalomonis/Desktop/Mm_sample/input_list_small" --runGOElite yes --denom "/Users/nsalomonis/Desktop/Mm_sample/denominator" --mod Ensembl --species Mm
"""if denom_file_dir == None:
print 'Please include a folder containing a valid denominator ID list for the input ID sets.'; sys.exit()"""
try:
if output_dir==None:
### Set output to the same directory or parent if none selected
i = -1 ### 1 directory up
output_dir = string.join(string.split(input_file_dir,'/')[:i],'/')
file_dirs = input_file_dir, denom_file_dir, output_dir
import GO_Elite
if ORA_algorithm == 'Fisher Exact Test':
goelite_permutations = 'FisherExactTest'
goelite_var = species,mod,goelite_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,''
GO_Elite.remoteAnalysis(goelite_var,'non-UI',Multi=mlp)
sys.exit()
except Exception:
print traceback.format_exc()
print "Unexpected error encountered. Please see log file."; sys.exit()
if run_lineage_profiler == 'yes':
status = UI.verifyLineageProfilerDatabases(species,'command-line')
if status == False:
print 'Please note: LineageProfiler not currently supported for this species...'
if run_lineage_profiler == 'yes' and input_file_dir != None and pipelineAnalysis == False and ('--runLineageProfiler' in arguments or '--cellHarmony' in arguments or '--cellHarmonyMerge' in arguments):
#python AltAnalyze.py --input "/Users/arrays/test.txt" --runLineageProfiler yes --vendor Affymetrix --platform "3'array" --species Mm --output "/Users/nsalomonis/Merrill"
#python AltAnalyze.py --input "/Users/qPCR/samples.txt" --runLineageProfiler yes --geneModel "/Users/qPCR/models.txt" --reference "Users/qPCR/reference_profiles.txt"
if array_type==None:
print "Please include a platform name (e.g., --platform RNASeq)";sys.exit()
if species==None:
print "Please include a species name (e.g., --species Hs)";sys.exit()
try:
status = UI.verifyLineageProfilerDatabases(species,'command-line')
except ValueError:
### Occurs due to if int(gene_database[-2:]) < 65: - ValueError: invalid literal for int() with base 10: ''
print '\nPlease install a valid gene database before proceeding.\n'
print 'For example: python AltAnalyze.py --species Hs --update Official --version EnsMart72\n';sys.exit()
if status == False:
print 'Please note: LineageProfiler not currently supported for this species...'
try:
FoldDiff=1.5
performDiffExp=True
pval = 0.05
adjp = True
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--fold': FoldDiff=float(arg)
elif opt == '--pval': pval = float(arg)
elif opt == '--adjp': adjp = arg
elif opt == '--performDiffExp': performDiffExp = arg
elif opt == '--centerMethod': CenterMethod = arg
elif opt == '--labels': labels = arg
elif opt == '--genes': genes = arg
elif opt == '--referenceFull': referenceFull = arg
fl = UI.ExpressionFileLocationData('','','','')
fl.setSpecies(species)
fl.setVendor(manufacturer)
fl.setPlatformType(array_type)
fl.setCompendiumType('protein_coding')
if '--cellHarmony' in arguments:
fl.setClassificationAnalysis('cellHarmony')
fl.setPearsonThreshold(PearsonThreshold)
fl.setReturnCentroids(returnCentroids)
fl.setPeformDiffExpAnalysis(performDiffExp)
fl.setUseAdjPvalue(adjp)
fl.setPvalThreshold(pval)
fl.setFoldCutoff(FoldDiff)
fl.setLabels(labels)
else:
fl.setClassificationAnalysis('LineageProfiler')
#fl.setCompendiumType('AltExon')
fl.setCompendiumPlatform(array_type)
try: expr_input_dir
except Exception: expr_input_dir = input_file_dir
if '--cellHarmonyMerge' in arguments:
ICGS_files=[]
for opt, arg in options: ### Accept user input for these hierarchical clustering variables
if opt == '--input' or opt == '--i':
input_file = verifyPath(arg)
ICGS_files.append(input_file)
import LineageProfilerIterate
print 'center method =',CenterMethod
LineageProfilerIterate.createMetaICGSResults(ICGS_files,output_dir,CenterMethod=CenterMethod,species=species,PearsonThreshold=PearsonThreshold)
#except: LineageProfilerIterate.createMetaICGSResults(ICGS_files,output_dir,CenterMethod=CenterMethod)
sys.exit()
print 'center method =',CenterMethod
try: CenterMethod=CenterMethod
except: CenterMethod='community'
""" Only align sparse matrix files and skip other analyses """
if len(genes)>0 and ('h5' in custom_reference or 'mtx' in custom_reference):
fl.set_reference_exp_file(custom_reference)
custom_reference = genes
if referenceFull != None:
fl.set_reference_exp_file(referenceFull)
UI.remoteLP(fl, expr_input_dir, manufacturer, custom_reference, geneModel, None, modelSize=modelSize, CenterMethod=CenterMethod) #,display=display
#graphic_links = ExpressionBuilder.remoteLineageProfiler(fl,input_file_dir,array_type,species,manufacturer)
print_out = 'Alignments and images saved to the folder "DataPlots" in the input file folder.'
print print_out
except Exception:
print traceback.format_exc()
print_out = 'Analysis error occured...\nplease see warning printouts.'
print print_out
sys.exit()
if array_type == 'junction' or array_type == 'RNASeq': ### Download junction databases
try: UI.checkForLocalArraySupport(species,array_type,specific_array_type,'command-line')
except Exception:
print 'Please install a valid gene database before proceeding.\n'
print 'For example: python AltAnalyze.py --species Hs --update Official --version EnsMart72';sys.exit()
probeset_types = ['full','core','extended']
if return_all == 'yes': ### Perform no alternative exon filtering when annotating existing FIRMA or MADS results
dabg_p = 1; expression_threshold = 1; p_threshold = 1; alt_exon_fold_variable = 1
gene_expression_cutoff = 10000; filter_probeset_types = 'full'; exon_exp_threshold = 1; rpkm_threshold = 0
gene_exp_threshold = 1; exon_rpkm_threshold = 0
if array_type == 'RNASeq':
gene_exp_threshold = 0
else:
if array_type != "3'array":
try:
p_threshold = float(p_threshold); alt_exon_fold_variable = float(alt_exon_fold_variable)
expression_threshold = float(expression_threshold); gene_expression_cutoff = float(gene_expression_cutoff)
dabg_p = float(dabg_p); additional_score = float(additional_score)
gene_expression_cutoff = float(gene_expression_cutoff)
except Exception:
try: gene_expression_cutoff = float(gene_expression_cutoff)
except Exception: gene_expression_cutoff = 0
try: rpkm_threshold = float(rpkm_threshold)
except Exception: rpkm_threshold = -1
try: exon_exp_threshold = float(exon_exp_threshold)
except Exception: exon_exp_threshold = 0
try: gene_exp_threshold = float(gene_exp_threshold)
except Exception: gene_exp_threshold = 0
try: exon_rpkm_threshold = float(exon_rpkm_threshold)
except Exception: exon_rpkm_threshold = 0
if filter_probeset_types not in probeset_types and array_type == 'exon':
print "Invalid probeset-type entered:",filter_probeset_types,'. Must be "full", "extended" or "core"'; sys.exit()
elif array_type == 'gene' and filter_probeset_types == 'NA': filter_probeset_types = 'core'
if dabg_p > 1 or dabg_p <= 0:
print "Invalid DABG p-value entered:",dabg_p,'. Must be > 0 and <= 1'; sys.exit()
if expression_threshold <1:
print "Invalid expression threshold entered:",expression_threshold,'. Must be > 1'; sys.exit()
if p_threshold > 1 or p_threshold <= 0:
print "Invalid alternative exon p-value entered:",p_threshold,'. Must be > 0 and <= 1'; sys.exit()
if alt_exon_fold_variable < 1 and analysis_method != 'ASPIRE' and analysis_method != 'MultiPath-PSI':
print "Invalid alternative exon threshold entered:",alt_exon_fold_variable,'. Must be > 1'; sys.exit()
if gene_expression_cutoff < 1:
print "Invalid gene expression threshold entered:",gene_expression_cutoff,'. Must be > 1'; sys.exit()
if additional_score < 1:
print "Invalid additional score threshold entered:",additional_score,'. Must be > 1'; sys.exit()
if array_type == 'RNASeq':
if rpkm_threshold < 0:
print "Invalid gene RPKM threshold entered:",rpkm_threshold,'. Must be >= 0'; sys.exit()
if exon_exp_threshold < 1:
print "Invalid exon expression threshold entered:",exon_exp_threshold,'. Must be > 1'; sys.exit()
if exon_rpkm_threshold < 0:
print "Invalid exon RPKM threshold entered:",exon_rpkm_threshold,'. Must be >= 0'; sys.exit()
if gene_exp_threshold < 1:
print "Invalid gene expression threshold entered:",gene_exp_threshold,'. Must be > 1'; sys.exit()
if 'FIRMA' in additional_algorithms and array_type == 'RNASeq':
print 'FIRMA is not an available option for RNASeq... Changing this to splicing-index.'
additional_algorithms = 'splicing-index'
additional_algorithms = UI.AdditionalAlgorithms(additional_algorithms); additional_algorithms.setScore(additional_score)
if array_type == 'RNASeq':
try:
if 'CEL' in run_from_scratch: run_from_scratch = 'Process RNA-seq reads'
if build_exon_bedfile == 'yes': run_from_scratch = 'buildExonExportFiles'
manufacturer = 'RNASeq'
except Exception:
### When technically 3'array format
array_type = "3'array"
if run_from_scratch == 'Process AltAnalyze filtered': expression_data_format = 'log' ### This is switched to log no matter what, after initial import and analysis of CEL or BED files
### These variables are modified from the defaults in the module UI as below
excludeNonExpExons = True
if avg_all_for_ss == 'yes': avg_all_for_ss = 'yes'
elif 'all exon aligning' in avg_all_for_ss or 'known exons' in avg_all_for_ss or 'expressed exons' in avg_all_for_ss:
if 'known exons' in avg_all_for_ss and array_type == 'RNASeq': excludeNonExpExons = False
avg_all_for_ss = 'yes'
else: avg_all_for_ss = 'no'
if run_MiDAS == 'NA': run_MiDAS = 'no'
if perform_alt_analysis == 'yes': perform_alt_analysis = 'yes'
elif perform_alt_analysis == 'expression': perform_alt_analysis = 'expression'
elif perform_alt_analysis == 'just expression': perform_alt_analysis = 'expression'
elif perform_alt_analysis == 'no': perform_alt_analysis = 'expression'
elif platform != "3'array": perform_alt_analysis = 'both'
if systemToUse != None: array_type = systemToUse
try: permute_p_threshold = float(permute_p_threshold)
except Exception: permute_p_threshold = permute_p_threshold
### Store variables for AltAnalyzeMain
expr_var = species,array_type,manufacturer,constitutive_source,dabg_p,expression_threshold,avg_all_for_ss,expression_data_format,include_raw_data,run_from_scratch,perform_alt_analysis
alt_var = analysis_method,p_threshold,filter_probeset_types,alt_exon_fold_variable,gene_expression_cutoff,remove_intronic_junctions,permute_p_threshold,perform_permutation_analysis, export_NI_values, analyze_all_conditions
additional_var = calculate_normIntensity_p, run_MiDAS, use_direct_domain_alignments_only, microRNA_prediction_method, filter_for_AS, additional_algorithms
goelite_var = ge_fold_cutoffs,ge_pvalue_cutoffs,ge_ptype,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,goelite_permutations,mod,returnPathways
if run_from_scratch == 'buildExonExportFiles':
fl = UI.ExpressionFileLocationData('','','',''); fl.setExonBedBuildStatus('yes'); fl.setFeatureNormalization('none')
fl.setCELFileDir(cel_file_dir); fl.setArrayType(array_type); fl.setOutputDir(output_dir)
fl.setMultiThreading(multiThreading)
exp_file_location_db={}; exp_file_location_db[dataset_name]=fl; parent_dir = output_dir
perform_alt_analysis = 'expression'
if run_from_scratch == 'Process Expression file':
if len(input_exp_file)>0:
if groups_file != None and comps_file != None:
if 'exp.' in input_exp_file: new_exp_file = input_exp_file
else:
new_exp_file = export.findParentDir(input_exp_file)+'exp.'+export.findFilename(input_exp_file)
if 'ExpressionInput' not in new_exp_file:
### This expression file is not currently used (could make it the default after copying to this location)
if output_dir[-1] != '/' and output_dir[-1] != '\\':
output_dir += '/'
new_exp_file = output_dir+'ExpressionInput/'+export.findFilename(new_exp_file)
try: export.copyFile(input_exp_file, new_exp_file)
except Exception: print 'Expression file already present in target location.'
try: export.copyFile(groups_file, string.replace(new_exp_file,'exp.','groups.'))
except Exception: print 'Groups file already present in target location OR bad input path.'
try: export.copyFile(comps_file, string.replace(new_exp_file,'exp.','comps.'))
except Exception: print 'Comparison file already present in target location OR bad input path.'
groups_file = string.replace(new_exp_file,'exp.','groups.')
comps_file = string.replace(new_exp_file,'exp.','comps.')
input_exp_file = new_exp_file
if verifyGroupFileFormat(groups_file) == False:
print "\nWarning! The format of your groups file is not correct. For details, see:\nhttp://altanalyze.readthedocs.io/en/latest/ManualGroupsCompsCreation\n"
sys.exit()
try:
cel_files, array_linker_db = ExpressionBuilder.getArrayHeaders(input_exp_file)
if len(input_stats_file)>1: ###Make sure the files have the same arrays and order first
cel_files2, array_linker_db2 = ExpressionBuilder.getArrayHeaders(input_stats_file)
if cel_files2 != cel_files:
print "The probe set p-value file:\n"+input_stats_file+"\ndoes not have the same array order as the\nexpression file. Correct before proceeding."; sys.exit()
except Exception: print '\nWARNING...Expression file not found: "'+input_exp_file+'"\n\n'; sys.exit()
exp_name = string.replace(exp_name,'exp.',''); dataset_name = exp_name; exp_name = string.replace(exp_name,'.txt','')
groups_name = 'ExpressionInput/groups.'+dataset_name; comps_name = 'ExpressionInput/comps.'+dataset_name
groups_file_dir = output_dir+'/'+groups_name; comps_file_dir = output_dir+'/'+comps_name
groups_found = verifyFile(groups_file_dir)
comps_found = verifyFile(comps_file_dir)
if ((groups_found != 'found' or comps_found != 'found') and analyze_all_conditions != 'all groups') or (analyze_all_conditions == 'all groups' and groups_found != 'found'):
files_exported = UI.predictGroupsAndComps(cel_files,output_dir,exp_name)
if files_exported == 'yes': print "AltAnalyze inferred a groups and comps file from the CEL file names."
elif run_lineage_profiler == 'yes' and input_file_dir != None and pipelineAnalysis == False and ('--runLineageProfiler' in arguments or '--cellHarmony' in arguments): pass
else: print '...groups and comps files not found. Create before running AltAnalyze in command line mode.';sys.exit()
fl = UI.ExpressionFileLocationData(input_exp_file,input_stats_file,groups_file_dir,comps_file_dir)
dataset_name = exp_name
if analyze_all_conditions == "all groups":
try: array_group_list,group_db = UI.importArrayGroupsSimple(groups_file_dir,cel_files)
except Exception:
print '...groups and comps files not found. Create before running AltAnalyze in command line mode.';sys.exit()
print len(group_db), 'groups found'
if len(group_db) == 2: analyze_all_conditions = 'pairwise'
exp_file_location_db={}; exp_file_location_db[exp_name]=fl
elif run_from_scratch == 'Process CEL files' or run_from_scratch == 'Process RNA-seq reads' or run_from_scratch == 'Process Feature Extraction files':
if groups_file != None and comps_file != None:
try: shutil.copyfile(groups_file, string.replace(exp_file_dir,'exp.','groups.'))
except Exception: print 'Groups file already present in target location OR bad input path.'
try: shutil.copyfile(comps_file, string.replace(exp_file_dir,'exp.','comps.'))
except Exception: print 'Comparison file already present in target location OR bad input path.'
stats_file_dir = string.replace(exp_file_dir,'exp.','stats.')
groups_file_dir = string.replace(exp_file_dir,'exp.','groups.')
comps_file_dir = string.replace(exp_file_dir,'exp.','comps.')
groups_found = verifyFile(groups_file_dir)
comps_found = verifyFile(comps_file_dir)
if ((groups_found != 'found' or comps_found != 'found') and analyze_all_conditions != 'all groups') or (analyze_all_conditions == 'all groups' and groups_found != 'found'):
if mappedExonAnalysis: pass
elif len(input_fastq_dir)<0: ### Don't check for FASTQ to allow for fast expression quantification even if groups not present
files_exported = UI.predictGroupsAndComps(cel_files,output_dir,exp_name)
if files_exported == 'yes': print "AltAnalyze inferred a groups and comps file from the CEL file names."
#else: print '...groups and comps files not found. Create before running AltAnalyze in command line mode.';sys.exit()
fl = UI.ExpressionFileLocationData(exp_file_dir,stats_file_dir,groups_file_dir,comps_file_dir)
exp_file_location_db={}; exp_file_location_db[dataset_name]=fl
parent_dir = output_dir ### interchangable terms (parent_dir used with expression file import)
if analyze_all_conditions == "all groups":
array_group_list,group_db = UI.importArrayGroupsSimple(groups_file_dir,cel_files)
UI.exportGroups(exp_file_location_db,array_group_list)
print len(group_db), 'groups found'
if len(group_db) == 2: analyze_all_conditions = 'pairwise'
try: fl.setRunKallisto(input_fastq_dir)
except Exception: pass
elif run_from_scratch == 'Process AltAnalyze filtered':
if '.txt' in input_filtered_dir: ### Occurs if the user tries to load a specific file
dirs = string.split(input_filtered_dir,'/')
input_filtered_dir = string.join(dirs[:-1],'/')
fl = UI.ExpressionFileLocationData('','','',''); dataset_name = 'filtered-exp_dir'
dirs = string.split(input_filtered_dir,'AltExpression'); parent_dir = dirs[0]
exp_file_location_db={}; exp_file_location_db[dataset_name]=fl
for dataset in exp_file_location_db:
fl = exp_file_location_db[dataset_name]
file_location_defaults = UI.importDefaultFileLocations()
apt_location = UI.getAPTLocations(file_location_defaults,run_from_scratch,run_MiDAS)
fl.setAPTLocation(apt_location)
if run_from_scratch == 'Process CEL files':
if xhyb_remove == 'yes' and (array_type == 'gene' or array_type == 'junction'): xhyb_remove = 'no' ### This is set when the user mistakenly selects exon array, initially
fl.setInputCDFFile(input_cdf_file); fl.setCLFFile(clf_file); fl.setBGPFile(bgp_file); fl.setXHybRemoval(xhyb_remove)
fl.setCELFileDir(cel_file_dir); fl.setArrayType(array_type_original); fl.setOutputDir(output_dir)
elif run_from_scratch == 'Process RNA-seq reads':
fl.setCELFileDir(cel_file_dir); fl.setOutputDir(output_dir)
elif run_from_scratch == 'Process Feature Extraction files':
fl.setCELFileDir(cel_file_dir); fl.setOutputDir(output_dir)
fl = exp_file_location_db[dataset]; fl.setRootDir(parent_dir)
try: apt_location = fl.APTLocation()
except Exception: apt_location = ''
root_dir = fl.RootDir(); fl.setExonBedBuildStatus(build_exon_bedfile)
fl.setMarkerFinder(marker_finder)
fl.setFeatureNormalization(normalize_feature_exp)
fl.setNormMatrix(normalize_gene_data)
fl.setProbabilityStatistic(probability_statistic)
fl.setProducePlots(visualize_qc_results)
fl.setPerformLineageProfiler(run_lineage_profiler)
fl.setCompendiumType(compendiumType)
fl.setCompendiumPlatform(compendiumPlatform)
fl.setVendor(manufacturer)
try: fl.setFDRStatistic(FDR_statistic)
except Exception: pass
fl.setAnalysisMode('commandline')
fl.setBatchEffectRemoval(batch_effects)
fl.setChannelToExtract(channel_to_extract)
fl.setMultiThreading(multiThreading)
try: fl.setExcludeLowExpressionExons(excludeNonExpExons)
except Exception: fl.setExcludeLowExpressionExons(True)
if 'other' in manufacturer or 'Other' in manufacturer:
### For data without a primary array ID key
manufacturer = "other:3'array"
fl.setVendor(manufacturer)
if array_type == 'RNASeq': ### Post version 2.0, add variables in fl rather than below
fl.setRPKMThreshold(rpkm_threshold)
fl.setExonExpThreshold(exon_exp_threshold)
fl.setGeneExpThreshold(gene_exp_threshold)
fl.setExonRPKMThreshold(exon_rpkm_threshold)
fl.setJunctionExpThreshold(expression_threshold)
fl.setExonMapFile(exonMapFile)
fl.setPlatformType(platformType)
### Verify database presence
try: dirs = unique.read_directory('/AltDatabase')
except Exception: dirs=[]
if species not in dirs:
print '\n'+species,'species not yet installed. Please install before proceeding (e.g., "python AltAnalyze.py --update Official --species',species,'--version EnsMart75").'
global commandLineMode; commandLineMode = 'yes'
AltAnalyzeMain(expr_var, alt_var, goelite_var, additional_var, exp_file_location_db,None)
else:
print traceback.format_exc()
print 'Insufficient Flags entered (requires --species and --output)'
def cleanUpCommandArguments():
### Needed on PC
command_args = string.join(sys.argv,' ')
arguments = string.split(command_args,' --')
for argument in arguments:
"""
argument_list = string.split(argument,' ')
if len(argument_list)>2:
filename = string.join(argument_list[1:],' ')
argument = argument_list[0]+' '+string.replace(filename,' ','$$$')
"""
argument_list = string.split(argument,' ')
#argument = string.join(re.findall(r"\w",argument),'')
if ':' in argument: ### Windows OS
z = string.find(argument_list[1],':')
if z!= -1 and z!=1: ### Hence, it is in the argument but not at the second position
print 'Illegal parentheses found. Please re-type these and re-run.'; sys.exit()
def runCommandLineVersion():
### This code had to be moved to a separate function to prevent iterative runs upon AltAnalyze.py re-import
command_args = string.join(sys.argv,' ')
#try: cleanUpCommandArguments()
#except Exception: null=[]
#print 3,[sys.argv],
if len(sys.argv[1:])>0 and '--' in command_args:
if '--GUI' in command_args:
### Hard-restart of AltAnalyze while preserving the prior parameters
command_arguments = string.split(command_args,' --')
if len(command_arguments)>2:
command_arguments = map(lambda x: string.split(x,' '),command_arguments)
command_arguments = map(lambda (x,y): (x,string.replace(y,'__',' ')),command_arguments[2:])
selected_parameters = [command_arguments[0][1]]
user_variables={}
for (o,v) in command_arguments: user_variables[o]=v
AltAnalyzeSetup((selected_parameters,user_variables))
else:
AltAnalyzeSetup('no') ### a trick to get back to the main page of the GUI (if AltAnalyze has Tkinter conflict)
try:
commandLineRun()
except Exception:
print traceback.format_exc()
###### Determine Command Line versus GUI Control ######
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>1 and '-' in command_args: null=[]
else:
try:
import Tkinter
from Tkinter import *
from visualization_scripts import PmwFreeze
import tkFileDialog
from tkFont import Font
use_Tkinter = 'yes'
except ImportError: use_Tkinter = 'yes'; print "\nPmw or Tkinter not found... Tkinter print out not available";
def testResultsPanel():
from visualization_scripts import QC
file = "/Users/nsalomonis/Desktop/code/AltAnalyze/datasets/3'Array/Merrill/ExpressionInput/exp.test.txt"
#QC.outputArrayQC(file)
global root; root = Tk()
global pathway_permutations; pathway_permutations = 'NA'
global log_file; log_file = 'null.txt'
global array_type; global explicit_data_type
global run_GOElite; run_GOElite = 'run-immediately'
explicit_data_type = 'exon-only'
array_type = 'RNASeq'
fl = UI.ExpressionFileLocationData('','','','')
graphic_links = []
graphic_links.append(['PCA','PCA.png'])
graphic_links.append(['HC','HC.png'])
graphic_links.append(['PCA1','PCA.png'])
graphic_links.append(['HC1','HC.png'])
graphic_links.append(['PCA2','PCA.png'])
graphic_links.append(['HC2','HC.png'])
graphic_links.append(['PCA3','PCA.png'])
graphic_links.append(['HC3','HC.png'])
graphic_links.append(['PCA4','PCA.png'])
graphic_links.append(['HC4','HC.png'])
summary_db={}
summary_db['QC'] = graphic_links
#summary_db={}
fl.setGraphicLinks(graphic_links)
summary_db['gene_assayed'] = 1
summary_db['denominator_exp_genes'] = 1
summary_db['alt_events'] = 1
summary_db['denominator_exp_events'] = 1
summary_db['alt_events'] = 1
summary_db['denominator_exp_events'] = 1
summary_db['alt_events'] = 1
summary_db['denominator_exp_events'] = 1
summary_db['alt_genes'] = 1
summary_db['direct_domain_genes'] = 1
summary_db['miRNA_gene_hits'] = 1
#summary_db={}
print_out = 'Analysis complete. AltAnalyze results\nexported to "AltResults/AlternativeOutput".'
dataset = 'test'; results_dir=''
print "Analysis Complete\n";
if root !='' and root !=None:
UI.InfoWindow(print_out,'Analysis Completed!')
tl = Toplevel(); SummaryResultsWindow(tl,'GE',results_dir,dataset,'parent',summary_db)
#sys.exit()
class Logger(object):
def __init__(self,null):
self.terminal = sys.stdout
self.log = open(log_file, "w")
def write(self, message):
self.terminal.write(message)
self.log = open(log_file, "a")
self.log.write(message)
self.log.close()
def flush(self): pass
class SysLogger(object):
def __init__(self,null):
self.terminal = sys.stdout
self.log = open(sys_log_file, "w")
def write(self, message):
self.terminal.write(message)
self.log = open(sys_log_file, "a")
self.log.write(message)
self.log.close()
def flush(self): pass
def verifyPath(filename):
### See if the file is in the current working directory
new_filename = filename
cwd = os.getcwd()
try:
files = unique.read_directory(cwd)
if filename in files:
new_filename = cwd+'/'+new_filename
except Exception:
pass
try:
### For local AltAnalyze directories
if os.path.isfile(cwd+'/'+filename):
new_filename = cwd+'/'+filename
else:
files = unique.read_directory(cwd+'/'+filename)
new_filename = cwd+'/'+filename
except Exception:
#print traceback.format_exc()
pass
return new_filename
def dependencyCheck():
### Make sure core dependencies for AltAnalyze are met and if not report back
from pkgutil import iter_modules
modules = set(x[1] for x in iter_modules()) ### all installed modules
dependent_modules = ['string','csv','base64','getpass','requests']
dependent_modules += ['warnings','sklearn','os','webbrowser']
dependent_modules += ['scipy','numpy','matplotlib','igraph','pandas','patsy']
dependent_modules += ['ImageTk','PIL','cairo','wx','fastcluster','pysam', 'Tkinter']
dependent_modules += ['networkx','numba','umap','nimfa','lxml','annoy','llvmlite']
print ''
count=0
for module in dependent_modules:
if module not in modules:
if 'ImageTk' != module and 'PIL' != module:
print 'AltAnalyze depedency not met for:',module
if 'fastcluster' == module:
print '...Faster hierarchical cluster not supported without fastcluster'
if 'pysam' == module:
print '...BAM file access not supported without pysam'
if 'scipy' == module:
print '...Many required statistical routines not supported without scipy'
if 'numpy' == module:
print '...Many required statistical routines not supported without numpy'
if 'matplotlib' == module:
print '...Core graphical outputs not supported without matplotlib'
if 'requests' == module:
print '...Wikipathways visualization not supported without requests'
if 'lxml' == module:
print '...Wikipathways visualization not supported without lxml'
if 'wx' == module:
print '...The AltAnalyze Results Viewer requires wx'
if 'ImageTk' == module or 'PIL' == module:
if 'PIL' not in dependent_modules:
print 'AltAnalyze depedency not met for:',module
print '...Some graphical results displays require ImageTk and PIL'
if 'Tkinter' == module:
print '...AltAnalyze graphical user interface mode requires Tkinter'
if 'igraph' == module or 'cairo' == module:
print '...Network visualization requires igraph and cairo'
if 'sklearn' == module:
print '...t-SNE analysis requires sklearn'
if 'pandas' == module or 'patsy' == module:
print '...Combat batch effects correction requires pandas and patsy'
count+=1
if count>1:
print '\nWARNING!!!! Some dependencies are not currently met.'
print "This may impact AltAnalyze's performance\n"
def unpackConfigFiles():
""" When pypi installs AltAnalyze in site-packages, a zip file for the Config
and AltDatabase in the pypi installed AltAnalyze library directory. To allow
for different flexible database versions to be written, AltDatabase and Config
are written to the user home directory in the folder 'altanalyze'."""
fn = filepath('Config/options.txt') ### See if a Config folder is already available
fileExists = os.path.isfile(fn)
if fileExists == False:
import subprocess
import shutil
import site
import zipfile
from os.path import expanduser
userdir = expanduser("~")
try:
os.mkdir(userdir+"/altanalyze")
except:
pass
config_filepath = filepath("Config.zip")
altdatabase_filepath = string.replace(config_filepath,'Config.zip','AltDatabase.zip')
print '...Creating Config directory in:',userdir+"/altanalyze",
with zipfile.ZipFile(config_filepath,"r") as zip_ref:
zip_ref.extractall(userdir+"/altanalyze")
with zipfile.ZipFile(altdatabase_filepath,"r") as zip_ref:
zip_ref.extractall(userdir+"/altanalyze")
print '...written'
def systemLog():
global sys_log_file
sys_log_file = filepath('Config/report.log')
sys_report = open(sys_log_file,'w'); sys_report.close()
sys.stdout = SysLogger('')
def versionCheck():
import platform
if platform.system()=='Darwin':
if platform.mac_ver()[0] == '10.14.6':
print 'CRITICAL ERROR. AltAanlyze has a critical incompatibility with this specific OS version.'
url = 'http://www.altanalyze.org/MacOS-critical-error.html'
try: webbrowser.open(url)
except Exception: pass
sys.exit()
if __name__ == '__main__':
try: mlp.freeze_support()
except Exception: pass
try:
systemLog()
sys_log_file = filepath('Config/report.log')
except:
pass
print 'Using the Config location:',sys_log_file
versionCheck()
try: unpackConfigFiles()
except: pass
#testResultsPanel()
skip_intro = 'yes'; #sys.exit()
#skip_intro = 'remoteViewer'
dependencyCheck()
try:
runCommandLineVersion()
if use_Tkinter == 'yes':
AltAnalyzeSetup(skip_intro)
except:
if 'SystemExit' not in traceback.format_exc():
print traceback.format_exc()
""" To do list:
3) SQLite for gene-set databases prior to clustering and network visualization
7) (partially) Integrate splicing factor enrichment analysis (separate module?)
17) Support R check (and response that they need it) along with GUI gcrma, agilent array, hopach, combat
19) Update the software from the software
Advantages of this tool kit:
0) Easiest to use, hands down
1) Established and novel functionality for transcriptome/proteomics analysis built in
2) Independent and cooperative options for RNA-Seq and array analysis (splicing and gene expression)
3) Superior functional analyses (TF-target, splicing-factor target, lineage markers, WikiPathway visualization)
4) Options for different levels of users with different integration options (multiple statistical method options, option R support)
5) Built in secondary analysis options for already processed data (graphing, clustering, biomarker discovery, pathway analysis, network visualization)
6) Incorporates highly validated alternative exon identification methods, independent and jointly
Primary Engineer Work:
0) C-library calls and/or multithreading where applicable to improve peformance.
1) MySQL or equivalent transition for all large database queries (e.g., HuEx 2.1 on-the-fly coordinate mapping).
3) Isoform-domain network visualization and WP overlays.
4) Webservice calls to in silico protein translation, domain prediction, splicing factor regulation.
"""
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/AltAnalyze.py
|
AltAnalyze.py
|
import sys, string
import export
import math
import random
import copy
import os
import os.path
import unique
import traceback
R_present=True
try:
### If file is present use this location
loc = unique.filepath('Config/R_location.txt')
s = open(loc,'r')
useStaticLocation=s.read()
#print useStaticLocation
#print 'Using the Config designated location
for p in os.environ['PATH'].split(':'): ### For Unix cluster environments
if os.path.exists(p + '/R'):
#path = p + '/R'
useStaticLocation = False
except Exception:
#print 'NOT using the Config designated location'
useStaticLocation = False
try:
forceError ### This doesn't currently work with the compiled version of AltAnalyze
import rpy2.robjects as robjects
r = robjects.r
print "\n---------Using RPY2---------\n"
except Exception:
from pyper import *
#print "\n---------Using PypeR---------\n"
### Running the wrong one once is fine, but multiple times causes it to stall in a single session
try:
try:
if 'Xdarwin' in sys.platform: ### Xdarwin is indicated since this if statement is invalid without a stand-alone Mac R package (ideal)
#print 'Using AltAnalyze local version of R'
#print 'A'
path = unique.filepath("AltDatabase/tools/R/Mac/R")
r = R(RCMD=path,use_numpy=True)
elif os.name == 'nt':
path = unique.filepath("AltDatabase/tools/R/PC/bin/x64/R.exe")
r = R(RCMD=path,use_numpy=True)
else:
#print 'B'
if useStaticLocation == False or useStaticLocation=='no':
print 'NOT using static location'
r = R(use_numpy=True)
else:
print 'Using static location'
path = '/usr/local/bin/R'
if os.path.exists(path): pass
else:
path = '/usr/bin/R'
if os.path.exists(path):
print 'Using the R path:',path
r = R(RCMD=path,use_numpy=True)
else:
r = None
R_present=False
print 'R does not appear to be installed... Please install first.'
except Exception:
#print 'C'
r = R(use_numpy=True)
except Exception:
print traceback.format_exc()
r = None
R_present=False
pass
LegacyMode = True
### Create a Directory for R packages in the AltAnalyze program directory (in non-existant)
r_package_path = string.replace(os.getcwd()+'/Config/R','\\','/') ### R doesn't link \\
r_package_path = unique.filepath(r_package_path) ### Remove the AltAnalyze.app location
try: os.mkdir(r_package_path)
except Exception: None
if R_present:
### Set an R-package installation path
command = '.libPaths("'+r_package_path+'")'; r(command) ### doesn't work with %s for some reason
#print_out = r('.libPaths()');print print_out; sys.exit()
def remoteMonocle(input_file,expPercent,pval,numGroups):
#input_file="Altanalyze"
setWorkingDirectory(findParentDir(input_file)[:-1])
try: os.mkdir(findParentDir(input_file)[:-1])
except Exception: None
z = RScripts(input_file)
setWorkingDirectory(input_file)
z.Monocle(input_file,expPercent,pval,numGroups)
def remoteHopach(input_file,cluster_method,metric_gene,metric_array,force_array='',force_gene=''):
""" Run Hopach via a call from an external clustering and visualizaiton module """
#input_file = input_file[1:] #not sure why, but the '\' needs to be there while reading initally but not while accessing the file late
row_order = []
column_order = []
if 'ICGS-SubCluster' in input_file:
force_array=2
input_file = checkForDuplicateIDs(input_file) ### Duplicate IDs will cause R to exit when creating the data matrix
z = RScripts(input_file)
setWorkingDirectory(input_file)
z.Hopach(cluster_method,metric_gene,force_gene,metric_array,force_array)
if cluster_method == 'both' or cluster_method == 'gene':
filename = findParentDir(input_file)+'/hopach/rows.'+findFileName(input_file)
row_order = importHopachOutput(filename)
if cluster_method == 'both' or cluster_method == 'array':
filename = findParentDir(input_file)+'/hopach/columns.'+findFileName(input_file)
column_order = importHopachOutput(filename)
#print row_order; sys.exit()
return input_file, row_order, column_order
def remoteAffyNormalization(input_file,normalization_method,probe_level,batch_effects):
### Input file is the path of the expression output from normalization
setWorkingDirectory(findParentDir(input_file)[:-1])
try: os.mkdir(findParentDir(input_file)[:-1])
except Exception: None #Already exists
z = RScripts(input_file)
z.AffyNormalization(normalization_method,probe_level,batch_effects)
def checkForDuplicateIDs(input_file, useOrderedDict=True):
if 'SamplePrediction' in input_file or '-Guide' in input_file:
### OrderedDict is prefered but will alter prior ICGS results
useOrderedDict = False
first_row = True
import collections
if useOrderedDict:
try: key_db = collections.OrderedDict()
except Exception:
try:
import ordereddict
key_db = ordereddict.OrderedDict()
except Exception:
key_db={}
else:
key_db={}
key_list=[]
fn=filepath(input_file)
offset=0
nonNumericsPresent=False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if first_row == True:
if ('row_clusters-flat' in t and 'row_clusters-flat' not in t[0]):
headers = string.join(['uid']+t[2:],'\t')+'\n'
offset = 1
elif '-filtered.txt' in fn and ".R2." in t[1] and LegacyMode:
headers = string.join(['uid']+t[2:],'\t')+'\n'
offset = 1
else:
headers = line
first_row = False
else:
key = t[0]
try:
k1,k2string.split(key,' ')
print [k1, k2],
if k1==k2: key = k1
print key
except Exception: pass
if key!='column_clusters-flat':
key_list.append(key)
try: s = map(float,t[offset+1:])
except Exception:
nonNumericsPresent=True
key_db[key]=t
if nonNumericsPresent:
import numpy
for key in key_db:
t = key_db[key]
s=[key]
if offset ==1: s.append('')
temp=[]
for value in t[offset+1:]:
try: temp.append(float(value))
except Exception: pass
avg=numpy.mean(temp)
for value in t[offset+1:]:
try: s.append(str(float(value)-avg))
except Exception: s.append('0.000101')
key_db[key]=s
if len(key_db) != len(key_list) or offset>0 or nonNumericsPresent:
print 'Writing a cleaned-up version of the input file:'
### Duplicate IDs present
input_file = input_file[:-4]+'-clean.txt'
export_text = export.ExportFile(input_file) ### create a new input file
export_text.write(headers) ### Header is the same for each file
for key in key_db:
t = key_db[key]
if offset > 0:
t = [t[0]]+t[1+offset:]
export_text.write(string.join(t,'\t')+'\n') ### Write z-score values and row names
export_text.close()
print 'File written...'
return input_file
def importHopachOutput(filename):
#print filename
""" Import the ID order information """
db={} ### Used to store the cluster data
hopach_clusters=[]
cluster_level=[]
cluster_level2=[]
cluster_level3=[]
hopach_db={}
cluster_db={}
level2_level1={}
firstLine = True
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if firstLine: firstLine = False
else:
t = string.split(data,'\t')
final_level_order = int(t[-1])
index, uid, cluster_number, cluster_label, cluster_level_order, final_label, final_level_order = string.split(data,'\t')
try: l2 = str(int(round(float(cluster_label),0)))[:2]
except Exception: l2 = int(cluster_label[0])
try: l3 = str(int(round(float(cluster_label),0)))[:3]
except Exception: l3 = int(cluster_label[0])
hopach_clusters.append((int(final_level_order),int(index)-1)) ### Need to order according to the original index, sorted by the clustered order
cluster_level.append(int(cluster_label[0])) ### This is the root cluster number
cluster_level2.append(l2) ### Additional cluster levels
cluster_level3.append(l3)
hopach_db[uid] = cluster_label
level2_level1[l2] = int(cluster_label[0])
level2_level1[l3] = int(cluster_label[0])
try: cluster_db[int(float(cluster_label[0]))].append(uid)
except Exception: cluster_db[int(cluster_label[0])] = [uid]
try: cluster_db[l2].append(uid)
except Exception: cluster_db[l2] = [uid]
try: cluster_db[l3].append(uid)
except Exception: cluster_db[l3] = [uid]
split_cluster=[]
if 'column' in fn:
cluster_limit = 50 ### typically less columns than rows
else:
cluster_limit = 75
for cluster in cluster_db:
#print cluster,len(cluster_db[cluster]),(float(len(cluster_db[cluster]))/len(hopach_db))
if len(cluster_db[cluster])>cluster_limit and (float(len(cluster_db[cluster]))/len(hopach_db))>0.2:
#print cluster
if cluster<10:
split_cluster.append(cluster)
import unique
levels1 = unique.unique(cluster_level)
already_split={}
updated_indexes={}
if len(split_cluster)>0:
print 'Splitting large hopach clusters:',split_cluster
i=0
for l2 in cluster_level2:
l1 = level2_level1[l2]
if l1 in split_cluster:
cluster_level[i] = l2
try:
l2_db = already_split[l1]
l2_db[l2]=[]
except Exception: already_split[l1] = {l2:[]}
i+=1
### Check and see if the l1 was split or not (might need 3 levels)
i=0
for l3 in cluster_level3:
l1 = level2_level1[l3]
if l1 in already_split:
#l1_members = len(cluster_db[l1])
l2_members = len(already_split[l1])
#print l1, l3, l1_members, l2_members
if l2_members == 1: ### Thus, not split
cluster_level[i] = l3
#print l1, l3, 'split'
i+=1
else:
if len(cluster_level) > 50: ### Decide to use different hopach levels
if len(levels1)<3:
cluster_level = cluster_level2
if len(cluster_level) > 200:
if len(levels1)<4:
cluster_level = cluster_level2
hopach_clusters.sort()
hopach_clusters = map(lambda x: x[1], hopach_clusters) ### Store the original file indexes in order based the cluster final order
### Change the cluster_levels from non-integers to integers for ICGS comparison group simplicity and better coloring of the color bar
cluster_level2 = []
### Rename the sorted cluster IDs as integers
cluster_level_sort = []
for i in cluster_level:
if str(i) not in cluster_level_sort:
cluster_level_sort.append(str(i))
cluster_level2.append(str(i))
cluster_level_sort.sort()
cluster_level = cluster_level2
cluster_level2=[]
i=1; cluster_conversion={}
for c in cluster_level_sort:
cluster_conversion[str(c)] = str(i)
i+=1
for c in cluster_level:
cluster_level2.append(cluster_conversion[c])
#print string.join(map(str,cluster_level2),'\t');sys.exit()
db['leaves'] = hopach_clusters ### This mimics Scipy's cluster output data structure
db['level'] = cluster_level2
return db
class RScripts:
def __init__(self,file):
self._file = file
def format_value_for_R(self,value):
value = '"'+value+'"'
return value
def File(self):
filename = self._file
filename_list = string.split(filename,'/')
filename = filename_list[-1]
filename = self.format_value_for_R(filename)
#root_dir = string.join(filename_list[:-1],'/')
return filename
def Monocle(self,samplelogfile,expPercent,p_val,numGroups):
#samplelogfile='C:/Users/venz6v/Documents/Altanalyze R/data.txt'
#grp_list="C:/Users/venz6v/Documents/Altanalyze R/grous.txt"
#gene_list="C:/Users/venz6v/Documents/Altanalyze R/gene.txt"
filename=self.File()
samplelogfile=findParentDir(filename)+'Monocle/expressionFile.txt"'
grp_list=findParentDir(filename)+'Monocle/sampleGroups.txt"'
gene_list=findParentDir(filename)+'Monocle/geneAnnotations.txt"'
pseudo_tree=findParentDir(filename)+'Monocle/monoclePseudotime.pdf"'
pseudo_txt=findParentDir(filename)+'Monocle/monoclePseudotime.txt"'
#try: os.mkdir(findParentDir(samplelogfile)) ### create "hopach" dir if not present
#except Exception: None
#try: os.mkdir(findParentDir(grp_list)) ### create "hopach" dir if not present
#except Exception: None
#try: os.mkdir(findParentDir(gene_list)) ### create "hopach" dir if not present
#except Exception: None
#self._file = samplelogfile
#samplelogfile = self.File()
#self._file = grp_list
#grp_list = self.File()
#self._file = gene_list
#gene_list = self.File()
print 'Loading monocle package in R'
print_out = r('library("monocle")')
if "Error" in print_out:
print 'Installing the R package "monocle" in Config/R'
print_out = r('source("http://bioconductor.org/biocLite.R"); biocLite("monocle")')
print print_out
print_out = r('library("monocle")')
if "Error" in print_out: print 'unable to download the package "monocle"';
print_out = r('library("monocle")')
print "Reading Monocle data..."
data_import = 'fpkm_matrix<-read.delim(%s,row.names=1,check.names=FALSE)' % samplelogfile
#print [data_import]
print_out = r(data_import);
print print_out
data_import = 'sample_sheet<-read.delim(%s,row.names=1,check.names=FALSE)' % grp_list
#print [data_import]
print_out = r(data_import);
print print_out
data_import = 'gene_ann<-read.delim(%s,row.names=1,check.names=FALSE)' % gene_list
#print [data_import]
print_out = r(data_import);
print print_out
print_out= r('pd <- new("AnnotatedDataFrame",data=sample_sheet)');
print_out=r('fd <- new("AnnotatedDataFrame",data=gene_ann)');
print_out=r('URMM <- newCellDataSet(as.matrix(fpkm_matrix),phenoData = pd,featureData =fd)');
print print_out
#colname(a) == colname(b)
print_out=r('URMM<- detectGenes(URMM, min_expr = 0)')
gene_exp='expressed_genes <- row.names(subset(fData(URMM), num_cells_expressed >=%s ))'% expPercent
#print [gene_exp]
try:print_out = r(gene_exp)
except Exception:
print "expression genes"
print_out=r('length(expressed_genes)')
print print_out
# specify the grouping column for finding differential genes
import multiprocessing
cores = multiprocessing.cpu_count()
print 'using', cores, 'cores'
k = 'diff_test_res <- differentialGeneTest(URMM[expressed_genes, ], fullModelFormulaStr = "expression~Group",cores=%s)' % cores
print [k]
print_out=r(k)
print print_out
gene_ord='ordering_genes <- row.names(subset(diff_test_res, pval < %s))' %p_val
print_out=r(gene_ord); print print_out
print_out=r('write.table(ordering_genes,file="ordering_genes.txt")') ### Writing out the informative genes used
print print_out
print_out=r('length(ordering_genes)'); print 'number or ordering genes',print_out
print_out=r('ordering_genes <- intersect(ordering_genes, expressed_genes)'); print print_out
print_out=r('URMM <- setOrderingFilter(URMM, ordering_genes)'); print print_out
print_out=r('URMM <- reduceDimension(URMM, use_irlba = F)'); print print_out
for i in range(numGroups,1,-1):
span='URMM <- orderCells(URMM, num_paths = %s, reverse = F)'% i;
print_out=r(span);
print print_out
if "Error" in print_out:
continue
else:
print_out=r(span);print i
print print_out
break
print_out=r('png("Monocle/monoclePseudotime.png")');
print print_out
print_out=r('plot_spanning_tree(URMM)'); print print_out
print_out=r('dev.off()')
print_out=r('pdf("Monocle/monoclePseudotime.pdf")');
print print_out
print_out=r('plot_spanning_tree(URMM)'); print print_out
print_out=r('dev.off()')
"""
print_out=r('pdf("Monocle/monoclePseudotimeOriginalGroups.pdf")');
print print_out
print_out=r('plot_spanning_tree(URMM), color_by = "originalGroups"'); print print_out
print_out=r('dev.off()')
"""
print_out=r('write.table(pData(URMM),file="Monocle/monoclePseudotime.txt")')
print " completed"
def AffyNormalization(self,normalization_method,probe_level,batch_effects):
print 'Loading affy package in R'
print_out = r('library("affy")')
if "Error" in print_out:
#print_out = r('install.packages("ggplot2", repos="http://cran.us.r-project.org")')
print 'Installing the R package "affy" in Config/R'
print_out = r('source("http://bioconductor.org/biocLite.R"); biocLite("affy")')
if "Error" in print_out: print 'unable to download the package "affy"'; forceError
print_out = r('library("affy")')
if 'gcrma' in normalization_method:
print 'Loading gcrma package in R'
print_out = r('library("gcrma")')
if "Error" in print_out:
print 'Installing the R package "gcrma" in Config/R'
print_out = r('source("http://bioconductor.org/biocLite.R"); biocLite("gcrma")')
if "Error" in print_out: print 'unable to download the package "gcrma"'; forceError
print_out = r('library("gcrma")')
if batch_effects == 'remove':
### Import or download support for SVA/Combat
print 'Loading sva package in R'
print_out = r('library("sva")')
if "Error" in print_out:
print 'Installing the R package "sva" in Config/R'
print_out = r('source("http://bioconductor.org/biocLite.R"); biocLite("sva")')
if "Error" in print_out: print 'unable to download the package "sva"'; forceError
print_out = r('library("sva")')
print "Reading Affy files..."
print_out = r('rawdata<-ReadAffy()')
print print_out
r('setwd("ExpressionInput")')
if probe_level: ### normalize at the level of probes rahter than probeset (e.g., alt.exon analysis of 3' array)
print_out = r('PM<-probes(rawdata,which="pm")'); print print_out
print_out = r('AffyInfo<-dimnames(PM)[[1]]'); print print_out
print_out = r('cutpos<-regexpr("\\d+$",AffyInfo,perl=T)'); print print_out
print_out = r('AffyID<-substr(AffyInfo,1,cutpos-1)'); print print_out
print_out = r('probe<-as.numeric(substr(AffyInfo,cutpos,nchar(AffyInfo)))'); print print_out
print_out = r('data.bgc<-bg.correct(rawdata,method="rma")'); print print_out
print_out = r('data.bgc.q<-normalize.AffyBatch.quantiles(data.bgc,type="pmonly")'); print print_out
print_out = r('pm.bgc.q<-probes(data.bgc.q,which="pm")'); print print_out
print_out = r('normalized<-cbind(AffyID,probe,pm.bgc.q)'); print print_out
command = 'write.table(normalized,file='+self.File()+',sep="\t",row.names=FALSE, quote=FALSE)'
print_out = r(command)
print print_out
print 'probe-level normalization complete'
else:
print "Begining %s normalization (will install array annotations if needed)... be patient" % normalization_method
print_out = r('normalized<-%s(rawdata)') % normalization_method
print print_out
command = 'write.exprs(normalized,'+self.File()+')'; print_out = r(command)
print print_out
print self.File(), 'written...'
if batch_effects == 'remove':
### Import data
command = 'mod = model.matrix(~as.factor(cancer) + age, data=pheno)'
print_out = r(command)
command = 'cdata = ComBat(dat=normalized, batch=as.factor(pheno$batch), mod=mod, numCov=match("age", colnames(mod)))'
print_out = r(command)
command = 'write.table(cdata,file='+self.File()+',sep="\t",row.names=FALSE, quote=FALSE)'
print_out = r(command)
output_file = string.replace(self.File(),'exp.','stats.')
print_out = r('calls<-mas5calls(rawdata)')
#print_out = r('pvals<-se.exprs(calls)') ### outdated?
print_out = r('pvals<-assayData(calls)[["se.exprs"]]')
command = 'write.table(pvals,'+output_file+',sep = "\t", col.names = NA)'; print_out = r(command)
print output_file, 'written...'
def Limma(self,test_type):
r('library("limma")')
filename = self.File()
try: output_file = string.replace(filename,'input','output-'+test_type)
except ValueError: output_file = filename[0:-4]+'-output.txt'
print "Begining to process",filename
data_import = 'data<-read.table(%s,sep="\t",header=T,row.names=1,as.is=T)' % filename
print_out = r(data_import)
design_matrix_file = string.replace(filename,'input','design')
design_import = 'design<-read.table(%s,sep="\t",header=T,row.names=1,as.is=T)' % design_matrix_file
design_matrix = r(design_import)
print_out = r('fit<-lmFit(data,design)')
fit_data = r['fit']
print_out = r('fit<-eBayes(fit)')
fit_data = r['fit']
contrast_matrix_file = string.replace(filename,'input','contrast')
contrast_import = 'contrast<-read.table(%s,sep="\t",header=T,row.names=1,as.is=T)' % contrast_matrix_file
print_out = r(contrast_import)
contrast_matrix = r['contrast']
r('contrast<-as.matrix(contrast)')
r('fit.contrast<-contrasts.fit(fit,contrast)')
r('fit.contrast<-eBayes(fit.contrast)')
r('nonadj<-fit.contrast$F.p.value')
if test_type == 'fdr':
print_out = r('results<-p.adjust(fit.contrast$F.p.value,method="fdr")')
else:
print_out = r('results<-nonadj')
result = r['results']
print 'test_type=',test_type
print_out = r('sum(results<0.05)')
summary = r['sum']
print "Number of probeset with a p<0.05",summary,"using",test_type
r('output<-cbind(data,results)')
output = 'write.table(output,%s,sep="\t")' % output_file
print_out = r(output)
print output_file, 'written...'
def Multtest(self,test_type):
r('library("multtest")')
filename = self.File()
try: output_file = string.replace(filename,'input','output')
except ValueError: output_file = filename[0:-4]+'-output.txt'
print "Begining to process",filename
parse_line = 'job<-read.table(%s,sep="\t", row.names=1, as.is=T)' % filename
print_out = r(parse_line)
print_out = r('matrix_size<-dim(job)')
print_out = r('label<-job[1,2:matrix_size[2]]')
print_out = r('jobdata<-job[2:matrix_size[1],2:matrix_size[2]]')
if test_type == "f":
print_out = r('ttest<-mt.maxT(jobdata,label, test="f", B=50000)')
if test_type == "t":
print_out = r('ttest<-mt.maxT(jobdata,label)')
print_out = r('ttest2<-ttest[order(ttest[,1]),]')
write_file = 'write.table(ttest2,%s,sep="\t")' % output_file
print_out = r(write_file)
print "Results written to:",output_file
def check_hopach_file_type(self):
if 'hopach.input' in self.File():
return 'continue'
else: return 'break'
def check_multtest_file_type(self):
if 'output' not in self.File():
return 'continue'
else: return 'break'
def check_limma_file_type(self):
if 'input' in self.File():
return 'continue'
else: return 'break'
def Hopach(self,cluster_method,metric_gene,force_gene,metric_array,force_array):
if R_present==False:
rNotPresent
print_out = r('library("Biobase")')
if "Error" in print_out:
print 'Installing the R package "Biobase" in Config/R'
print_out = r('source("http://bioconductor.org/biocLite.R"); biocLite("Biobase")')
if "Error" in print_out: print 'unable to download the package "Biobase"'; forceError
print_out = r('library("Biobase")')
print_out = r('library("hopach")')
if "Error" in print_out:
print 'Installing the R package "hopach" in Config/R'
print_out = r('source("http://bioconductor.org/biocLite.R"); biocLite("hopach")')
if "Error" in print_out: print 'unable to download the package "hopach"'; forceError
print_out = r('library("hopach")')
filename = self.File()
#r('memory.limit(2000)')
print "Begining to process",filename,"using HOPACH"
metric_g = self.format_value_for_R(metric_gene)
metric_a = self.format_value_for_R(metric_array)
parse_line = 'data<-read.table(%s,sep="\t",as.is=T,row.names=1,header=T)' % filename
checklinelengths(self._file)
print_out = r(parse_line)
dat = r['data']
print_out = r(parse_line)
#print "Number of columns in input file:",len(dat)
print_out = r('data<-as.matrix(data)')
dat = r['data']
#print "Number of columns in matrix:",len(dat)
force1=''; force2=''; hopg='NULL'; hopa='NULL'; distmatg='NULL'; distmata = 'NULL' ### defaults for tree export
if force_gene != '' and force_gene != 0: force1=',kmax='+str(force_gene)+', khigh='+str(force_gene)+', K='+str(force_array)
if force_array != '' and force_array != 0: force2=',kmax='+str(force_array)+', khigh='+str(force_array)+', K='+str(force_array)
if cluster_method == 'both' or cluster_method == 'gene':
distance_matrix_line = 'distmatg<-distancematrix(data,d=%s)' % metric_g
#print distance_matrix_line
if len(dat) > 1:
print_out1 = r(distance_matrix_line)
print_out2 = r('hopg<-hopach(data,dmat=distmatg,ord="own"'+force1+')')
#print 'hopg<-hopach(data,dmat=distmatg,ord="own"'+force1+')'
try: hopach_run = r['hopg']
except Exception:
print print_out1
print print_out2
hopg = 'hopg'
distmatg = 'distmatg'
gene_output = self.HopachGeneOutputFilename(metric_gene,str(force_gene))
output = 'out<-makeoutput(data,hopg,file=%s)' % gene_output
#print output
print_out = r(output)
#print print_out
output_file = r['out']
status = 'stop'
if 'clustering' in hopach_run:
if 'order' in hopach_run['clustering']:
try:
if len(hopach_run['clustering']['order']) > 10: status = 'continue'
except TypeError:
error = 'file: '+filename+": Hopach returned the array of cluster orders as blank while clustering GENES... can not process cluster... continuing with other files"
print error; errors.append(error)
if status == 'continue':
r(output_file); print 'hopach output written'
else:
error = 'file: '+filename+" Hopach returned data-matrix length zero...ARRAY clusters can not be generated"
print error; errors.append(error)
if cluster_method == 'both' or cluster_method == 'array':
distance_matrix_line = 'distmata<-distancematrix(t(data),d=%s)' % metric_a
if len(dat) > 1:
dist = r(distance_matrix_line)
#print distance_matrix_line
print_out = r('hopa<-hopach(t(data),dmat=distmata,ord="own"'+force1+')') #,coll="all"
#print ['hopa<-hopach(t(data),dmat=distmata,ord="own",'+force2+')']
#print 'hopa<-hopach(t(data),dmat=distmata,ord="own"'+force2+')'
hopach_run = r['hopa']
hopa = 'hopa'
distmata = 'distmata'
array_output = self.HopachArrayOutputFilename(metric_array,str(force_array))
output = 'out<-makeoutput(t(data),hopa,file=%s)' % array_output
print_out = r(output)
output_file = r['out']
status = 'stop'
if 'clustering' in hopach_run:
if 'order' in hopach_run['clustering']:
try:
if len(hopach_run['clustering']['order']) > 10: status = 'continue'
except TypeError:
error = 'file: '+filename+": Hopach returned the array of cluster orders as blank while clustering ARRAYS... can not process cluster"
print error; errors.append(error)
if status == 'continue':
r(output_file); print 'hopach output written'
else:
error = 'file: '+filename+"data-matrix length zero...ARRAY clusters can not be generated...continuing analysis"
print error; errors.append(error)
if len(metric_g)==0: metric_g = 'NULL'
if len(metric_a)==0: metric_a = 'NULL'
try:
output_filename = string.replace(gene_output,'rows.','')
cdt_output_line = 'hopach2tree(data, file = %s, hopach.genes = %s, hopach.arrays = %s, dist.genes = %s, dist.arrays = %s, d.genes = %s, d.arrays = %s, gene.wts = NULL, array.wts = NULL, gene.names = NULL)' % (output_filename,hopg,hopa,distmatg,distmata,metric_g,metric_a) ###7 values
except Exception: None
make_tree_line = 'makeTree(labels, ord, medoids, dist, side = "GENE")' ### Used internally by HOPACH
#print cdt_output_line
try: print_out = r(cdt_output_line)
except Exception: None
#print print_out
def HopachGeneOutputFilename(self,value,force):
filename = self.File() ### Relative to the set working directory
if 'hopach.input' in filename: ### When running this module on it's own (requires nown filetypes)
new_filename = string.replace(filename,'hopach.input','hopach.output')
if len(value)>1: new_filename = string.replace(new_filename,'.txt','-'+value+'.txt')
if len(force)>0: new_filename = string.replace(new_filename,'.txt','-'+'force_'+str(force)+'c.txt')
else: ### When called from an external heatmap visualization module
filename = self._file ### full path
new_filename = findParentDir(filename)+'/hopach/rows.'+findFileName(filename)
try: os.mkdir(findParentDir(new_filename)) ### create "hopach" dir if not present
except Exception: None
new_filename = '"'+new_filename+'"'
return new_filename
def HopachArrayOutputFilename(self,value,force):
filename = self.File()
if 'hopach.input' in filename: ### When running this module on it's own (requires nown filetypes)
new_filename = string.replace(filename,'hopach.input','arrays.output')
if len(value)>1: new_filename = string.replace(new_filename,'.txt','-'+value+'.txt')
if len(force)>0: new_filename = string.replace(new_filename,'.txt','-'+'force_'+str(force)+'c.txt')
else:
filename = self._file ### full path
filename = self._file ### full path
new_filename = findParentDir(filename)+'/hopach/columns.'+findFileName(filename)
try: os.mkdir(findParentDir(new_filename)) ### create "hopach" dir if not present
except Exception: None
new_filename = '"'+new_filename+'"'
return new_filename
def display(self):
print self.data
class FormatData:
def setdata(self,value):
self.data = value
def transform(self):
self.data = checktype(self.data)
def display(self):
print self.data
def returndata(self):
return self.data
def checktype(object):
###Checks to see if item is a list or dictionary. If dictionary, convert to list
import types
if type(object) is types.DictType:
object = converttolist(object)
elif type(object) is types.ListType:
object = object
elif type(object) is types.TupleType:
object = list(object)
elif type(object) is types.StringType:
object = importtable(object)
return object
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def checklinelengths(filename):
fn=filepath(filename); first_row='yes'; line_number=0
for line in open(fn,'rU').xreadlines():
try: data = cleanUpLine(line)
except Exception: print 'error parsing the line:',[line], line_number
t = string.split(data,'\t')
if first_row == 'yes':
elements = len(t)
first_row = 'no'
else:
if len(t) != elements:
print "Line number", line_number, "contains",len(t),"elements, when",elements,"expected...kill program"
print filename; kill
line_number+=1
def converttolist(dictionary):
###Converts dictionary to list by appending the dictionary key as the first item in the list
converted_lists=[]
for key in dictionary:
dictionary_list = dictionary[key]
dictionary_list.reverse(); dictionary_list.append(key); dictionary_list.reverse()
converted_lists.append(dictionary_list)
return converted_lists
############ IMPORT FILES BEGIN ############
def importtable(filename):
fn=filepath(filename); tab_db = []
for line in open(fn,'rU').readlines():
data,null = string.split(line,'\n')
t = string.split(data,'\t')
tab_db.append(t)
return tab_db
def filepath(filename):
dir=os.path.dirname(__file__) #directory file is input as a variable
status = verifyFile(filename)
if status:
fn = filename
else:
fn=os.path.join(dir,filename)
return fn
def verifyFile(filename):
status = False
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = True;break
except Exception: status = False
return status
def findFileName(filename):
filename = string.replace(filename,'\\','/')
dataset_name = string.split(filename,'/')[-1]
return dataset_name
def findParentDir(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1
return filename[:x]
def setWorkingDirectory(filename):
### Set R's working directory when calling this module remotely
working_dir = findParentDir(filename)
setwd = 'setwd("%s")' % working_dir
try: r(setwd)
except Exception:
print [filename]
print [working_dir]
print traceback.format_exc()
kill
def read_directory(sub_dir):
dir=os.path.dirname(__file__)
#print "Working Directory:", r('getwd()')
working_dir = dir+'/'+sub_dir[1:]
setwd = 'setwd("%s")' % working_dir
r(setwd)
#print "Working Directory:", r('getwd()')
dir_list = os.listdir(dir +'/'+ sub_dir[1:]); dir_list2 = []
for entry in dir_list: #add in code to prevent folder names from being included
if entry[-4:] == ".txt" or entry[-4:] == ".csv": dir_list2.append(entry)
return dir_list2
def CreateFilesMonocle(filename,rawExpressionFile,species='Hs'):
first_row = True
key_db={}
key_list=[]
fn=filepath(filename)
offset=0
nonNumericsPresent=False
try:
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
except Exception:
print "gene_symbols present"
gene_to_symbol={}
setWorkingDirectory(findParentDir(filename)[:-1])
try: os.mkdir(findParentDir(filename)+'/Monocle')
except Exception: None
#filename=self.File()
x = 0
data_name=findParentDir(filename)+'/Monocle/expressionFile.txt'
gene_name=findParentDir(filename)+'/Monocle/geneAnnotations.txt'
sample_name=findParentDir(filename)+'/Monocle/sampleGroups.txt'
gene_names = [];
gene_list=[];
dat=[];
export_cdt = open(sample_name,'w')
export_gene=open(gene_name,'w')
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if first_row == True:
if 'row_clusters-flat' in t and 'row_clusters-flat' not in t[0]:
headers = string.join(t[2:],'\t')+'\n'
offset = 1
else:
headers = string.join(t[1:],'\t')+'\n'
first_row = False
else:
key = t[0]
if key!='column_clusters-flat':
key_list.append(key)
try: s = map(float,t[offset+1:])
except Exception:
nonNumericsPresent=True
key_db[key]=t
else:
clusters = map(str,t[offset+1:])
for key in key_list:
t = key_db[key]
s=[key]
if offset ==1: s.append('')
temp=[]
for value in t[offset+1:]:
try: temp.append(float(value))
except Exception: pass
min1=min(temp)
for value in t[offset+1:]:
try: s.append(str(float(value)-min1))
except Exception: s.append('0.000101')
key_db[key]=s
export_object = open(data_name,'w')
export_object.write(''+'\t'+headers) ### Header is the same for each file
for key in key_list:
t = key_db[key]
if offset > 0:
t = [t[0]]+t[1+offset:]
export_object.write(string.join(t,'\t')+'\n') ### Write z-score values and row names
export_object.close()
print 'File written...'
#return input_file
array_names = []; array_linker_db = {}; d = 0; i = 0
for entry in headers.split('\t'):
entry=cleanUpLine(entry)
if '::' in entry:
a = (entry.split("::"))
elif ':' in entry:
a = (entry.split(":"))
else:
a = (clusters[i],entry)
#entry=string.join(a,'.')
ent=entry+'\t'+a[0];
#if(ent[0].isdigit()):
# ent='X'+ent[0:]
#if '-' in ent:
# ent=string.replace(ent,'-','.')
#if '+' in ent:
# ent=string.replace(ent,'+','.')
#print j
array_names.append(ent);
i+=1
i=0
eheader = string.join(['']+['Group'],'\t')+'\n' ### format column-flat-clusters for export
export_cdt.write(eheader)
for row in array_names:
export_cdt.write(row+'\n')
i+=1
export_cdt.close()
gheader = string.join(['']+ ['gene_short_name'],'\t')+'\n' ### format column-flat-clusters for export
export_gene.write(gheader)
for key in key_list:
proceed=False
### The commented out code just introduces errors and is not needed - re-evaluate in the future if needed
"""
if key in gene_to_symbol:
symbol = gene_to_symbol[key][0]
if symbol in gene_list:
nid = symbol
proceed = True
if proceed:
k=gene_list.index(nid)
export_object.write(line)
export_gene.write(key+'\n')
else:
export_gene.write(key+'\t'+key+'\n')"""
export_gene.write(key+'\t'+key+'\n')
export_object.close()
export_gene.close()
def reformatHeatmapFile(input_file):
import unique
export_file=string.replace(input_file,'Clustering-','Input-')
eo = export.ExportFile(export_file)
first_row = True
fn=filepath(input_file)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if first_row == True:
if 'column_clusters-flat' not in t:
array_names = []
for i in t[2:]:
array_names.append(string.replace(i,':','-'))
#print array_names;sys.exit()
#array_names.append(i)
elif 'column_clusters-flat' in t:
array_clusters = t[2:]
unique_clusters = unique.unique(array_clusters)
ind=0; headers=[]
for c in array_clusters:
headers.append(c+'::'+array_names[ind])
ind+=1
headers = string.join(['uid']+headers,'\t')+'\n'
eo.write(headers)
first_row = False
else:
values = string.join([t[0]]+t[2:],'\t')+'\n'
eo.write(values)
return export_file, len(unique_clusters)
def run_JTKcycle(expFile,annotFile,Time_range1, Time_range2,No_of_Timepoints,No_of_replicates,timepoint_difference):
print 'Loading JTK-Cycle package in R'
path='"'+r_package_path+'/JTK_CYCLE.R"'
#print [path]
line = 'source(%s)' % path
print_out = r(line)
"""
if "Error" in print_out:
print 'Installing the R package "JTK_CYCLE.R" in Config/R'
print_out = r('install.packages("devtools")')
print print_out
print_out = r('library(devtools)')
print print_out
print_out = r('install_github("mfcovington/jtk-cycle")')
#print_out = r('source("http://bioconductor.org/biocLite.R"); biocLite("jtk-cycle")')
print print_out
print_out = r('library("JTK_CYCLE.R")')
sys,exit()
print_out = r('source("/Users/ram5ge/Desktop/Krithika/JTK_Cycle/JTK_CYCLE.R")');print print_out
if "Error" in print_out: print "JTK_CYCLE.R is missing"
else: print 'Loading JTK Cycle'
"""
print_out = r('project <- "JTK_output"')
print_out = r('options(stringsAsFactors=FALSE)');print print_out
a = '"'+annotFile+'"'
read_annot = 'annot <- read.delim(%s)' % a
print [read_annot]
print_out = r(read_annot);#print print_out
v = '"'+expFile+'"'
read_data = 'input_data <- read.delim(%s)' % v
print [read_data]
print_out = r(read_data);#print print_out
print_out = r('rownames(input_data) <- input_data[,1]');#print print_out
print_out = r('input_data <- input_data[,-1]');#print print_out
#dist_calc = r('jtkdist(24,1)')
dist_calc = 'jtkdist(%s,%s)' % (str(No_of_Timepoints), str(No_of_replicates))
print [dist_calc]
print_out = r(dist_calc);#print print_out
period_calc = 'periods <- %s:%s' %(str(Time_range1), str(Time_range2))
print [period_calc]
print_out = r(period_calc);#print print_out
j = str(timepoint_difference)
jtk_calc = 'jtk.init(periods,%s)' % j
print [jtk_calc]
print_out = r(jtk_calc);#print print_out
v = 'cat("JTK analysis started on",date(),"\n")'
print [v]
print_out = r(v);#print print_out
print_out = r('flush.console()');#print print_out
v = 'st <- system.time({res <- apply(data,1,function(z)'
v+= ' {jtkx(z); c(JTK.ADJP,JTK.PERIOD,JTK.LAG,JTK.AMP)});'
v+= ' res <- as.data.frame(t(res)); bhq <- p.adjust(unlist(res[,1]),"BH");'
v+= ' res <- cbind(bhq,res); colnames(res) <- c("BH.Q","ADJ.P","PER","LAG","AMP");'
v+= ' results <- cbind(annot,res,data); results <- results[order(res$ADJ.P,-res$AMP),]})'
print [v]
print_out = r(v); print print_out
#print_out = r('dim(X)');print print_out
print_out = r('print(st)');print #print_out
print_out = r('save(results,file=paste("JTK",project,"rda",sep="."))');#print print_out
print_out = r('write.table(results,file=paste("JTK",project,"txt",sep="."),row.names=F,col.names=T,quote=F,sep="\t")');#print print_out
def performMonocleAnalysisFromHeatmap(species,heatmap_output_dir,rawExpressionFile):
numGroups=10
if 'Clustering-' in heatmap_output_dir:
export_file,numGroups = reformatHeatmapFile(heatmap_output_dir)
#else:
export_file = heatmap_output_dir;
CreateFilesMonocle(export_file,rawExpressionFile,species=species)
print 'Looking for',numGroups, 'Monocle groups in the input expression file.'
remoteMonocle(export_file,expPercent=5,pval=0.05,numGroups=numGroups)
if __name__ == '__main__':
expFile = '/Users/saljh8/Downloads/Liver_Smoothed_exp_steady_state.txt'
annotFile = '/Users/saljh8/Downloads/Liver_annot.txt'
Time_range1 = '10'
Time_range2 = '12'
No_of_Timepoints = '24'
No_of_replicates = '1'
timepoint_difference = '2'
run_JTKcycle(expFile,annotFile,Time_range1, Time_range2,No_of_Timepoints,No_of_replicates,timepoint_difference);sys.exit()
errors = []
cluster_method='array';metric_gene="";force_gene='';metric_array="euclid";force_array=''
analysis_method='hopach'; multtest_type = 'f'
#Sample log File
#Input-exp.MixedEffectsThanneer-DPF3%20DMRT3%20FOXA1%20SMAD6%20TBX3%20amplify%20monocle-hierarchical_cosine_correlated.txt
filename='/Users/saljh8/Desktop/cardiacRNASeq/DataPlots/Clustering-additionalExpressionSingleCell-annotated-hierarchical_cosine_cosine2.txt'
rawExpressionFile = filename
#filename = "/Volumes/SEQ-DATA/Eric/embryonic_singlecell_kidney/ExpressionOutput/Clustering/SampleLogFolds-Kidney.txt"
#filename = "/Volumes/SEQ-DATA/SingleCell-Churko/Filtered/Unsupervised-AllExons/NewCardiacMarkers1/FullDataset/ExpressionOutput/Clustering/SampleLogFolds-CM.txt"
#rawExpressionFile = '/Volumes/SEQ-DATA/SingleCell-Churko/Filtered/Unsupervised-AllExons/NewCardiacMarkers1/FullDataset/ExpressionInput/exp.CM-steady-state.txt'
#filename = '/Users/saljh8/Desktop/Stanford/ExpressionInput/amplify/DataPlots/Clustering-exp.EB-SingleCell-GPCR-hierarchical_cosine_correlation.txt'
#rawExpressionFile = '/Users/saljh8/Desktop/Stanford/ExpressionInput/exp.EB-SingleCell.txt'
performMonocleAnalysisFromHeatmap('Hs',filename,rawExpressionFile);sys.exit()
CreateFilesMonocle(filename,rawExpressionFile)
remoteMonocle(filename,expPercent=0,pval=0.01,numGroups=5);sys.exit()
filename = '/Users/nsalomonis/Downloads/GSE9440_RAW/ExpressionInput/exp.differentiation.txt'
remoteAffyNormalization(filename,'rma',True,'remove'); sys.exit()
print "******Analysis Method*******"
print "Options:"
print "1) Multtest (permutation ftest/ttest)"
print "2) HOPACH clustering"
print "3) limma 2-way ANOVA"
inp = sys.stdin.readline(); inp = inp.strip()
analysis_method_val = int(inp)
if analysis_method_val == 1: analysis_method = "multtest"
if analysis_method_val == 2: analysis_method = "hopach"
if analysis_method_val == 3: analysis_method = "limma"
if analysis_method == "hopach":
print "******Analysis Options*******"
print "Cluster type:"
print "1) genes only (cluster rows)"
print "2) arrays only (cluster columns)"
print "3) both"
inp = sys.stdin.readline(); inp = inp.strip()
cluster_type_call = int(inp)
if cluster_type_call == 1: cluster_method = "gene"
if cluster_type_call == 2: cluster_method = "array"
if cluster_type_call == 3: cluster_method = "both"
if cluster_method == "array" or cluster_method == "both":
print "******Analysis Options For Array Clustering*******"
print "Cluster metrics:"
print "1) euclidian distance (sensitive to magnitude)"
print "2) cosine angle distance (not sensitive to magnitude)"
print "3) correlation distance"
inp = sys.stdin.readline(); inp = inp.strip()
if cluster_method == "array" or cluster_method == "both":
metric_array_call = int(inp)
if metric_array_call == 1: metric_array = "euclid"
if metric_array_call == 2: metric_array = "cosangle"
if metric_array_call == 3: metric_array = "cor"
if cluster_method == "gene" or cluster_method == "both":
print "******Analysis Options For Gene Clustering*******"
print "Cluster metrics:"
print "1) euclidian distance (sensitive to magnitude)"
print "2) cosine angle distance (not sensitive to magnitude)"
print "3) correlation distance"
inp = sys.stdin.readline(); inp = inp.strip()
if cluster_method == "gene" or cluster_method == "both":
try: metric_gene_call = int(inp)
except ValueError: print [inp], 'not a valid option'; sys.exit()
if metric_gene_call == 1: metric_gene = "euclid"
if metric_gene_call == 2: metric_gene = "cosangle"
if metric_gene_call == 3: metric_gene = "cor"
if metric_gene == "cosangle":
print "******Analysis Options*******"
print "Absolute Clustering:"
print "1) yes"
print "2) no"
inp = sys.stdin.readline(); inp = inp.strip()
if inp == "1": metric_gene = "abscosangle"
print "Force Cluster Number for Arrays:"
print "Enter 'n' if you don't want to "
print "Enter number of clusters of arrays if you do"
inp = sys.stdin.readline(); inp = inp.strip()
if inp == 'n' or inp == 'N': force_array = ''
else:force_array = int(inp)
working_dir = '/hopach_input'
if analysis_method == "multtest":
print "******Analysis Options*******"
print "Statistical test:"
print "1) ftest (for multiple groups)"
print "2) ttest (for two groups)"
inp = sys.stdin.readline(); inp = inp.strip()
multtest_type_call = int(inp)
if multtest_type_call == 1: multtest_type = "f"
if multtest_type_call == 2: multtest_type = "t"
working_dir = '/multtest_input'
if analysis_method == "limma":
working_dir = '/limma_input'
print "******Analysis Options*******"
print "Statistical test:"
print "1) Non-adjusted"
print "2) FDR"
inp = sys.stdin.readline(); inp = inp.strip()
limma_type_call = int(inp)
if limma_type_call == 1: limma_type = "nonadj"
if limma_type_call == 2: limma_type = "fdr"
dir_list = read_directory(working_dir)
for input in dir_list: #loop through each file in the directory to output results
input_file = working_dir + "/"+ input
input_file = input_file[1:] #not sure why, but the '\' needs to be there while reading initally but not while accessing the file late
z = RScripts(input_file)
if analysis_method == "hopach":
status = z.check_hopach_file_type()
if status == 'continue':
z.Hopach(cluster_method,metric_gene,force_gene,metric_array,force_array)
if analysis_method == "multtest":
status = z.check_multtest_file_type()
if status == 'continue':
z.Multtest(multtest_type)
if analysis_method == "limma":
status = z.check_limma_file_type()
if status == 'continue':
design_matrix_file = string.replace(input,'input','design')
contrast_matrix_file = string.replace(input,'input','contrast')
if design_matrix_file in dir_list and contrast_matrix_file in dir_list:
z.Limma(limma_type)
if analysis_method == "hopach":
if len(errors)>0:
print "**************ALL ERRORS**************"
for entry in errors:
print entry
else: print 'Execution complete... check outputs for verification'
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/R_interface.py
|
R_interface.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys, string
import os.path
import unique
from stats_scripts import statistics
import math
def filepath(filename):
fn = unique.filepath(filename)
return fn
def reorderArrayHeaders(data_headers,array_order,comp_group_list,array_linker_db):
###array_order gives the final level order sorted, followed by the original index order as a tuple
data_headers2 = {}; array_linker_db2 = {}; ranked_array_headers = []; raw_data_comps={}; group_name_db = {}
for x in array_order:
y = x[1] ### this is the new first index
group = x[2]; group_name = x[3]
group_name_db[group] = group_name
### for example y = 5, therefore the data[row_id][5] entry is now the first
try: data_headers2[group].append(data_headers[y])
except KeyError: data_headers2[group]= [data_headers[y]]
raw_data_comp_headers = {}
for comp in comp_group_list:
temp_raw = []
group1 = int(comp[0]);group2 = int(comp[1])
comp = str(comp[0]),str(comp[1])
g1_headers = data_headers2[group1]
g2_headers = data_headers2[group2]
g1_name = group_name_db[group1]
g2_name = group_name_db[group2]
for header in g2_headers: temp_raw.append(g2_name+':'+header)
for header in g1_headers: temp_raw.append(g1_name+':'+header)
raw_data_comp_headers[comp] = temp_raw
for array_name in array_linker_db: array_linker_db2[array_linker_db[array_name]]=array_name
###Determine the number of arrays in each group for f-test analysis
group_count={}
for x in array_order:
original_index = x[1]; group = x[2]; group_name = x[3]
array_name = array_linker_db2[original_index]; ranked_array_headers.append(group_name+':'+array_name)
try: group_count[group] += 1
except KeyError: group_count[group] = 1
group_count_list=[]; group_count_list2=[]
for group_number in group_count:
count = group_count[group_number]
group_count_list.append((group_number,count))
group_count_list.sort()
#print group_count_list
for (group_number,count) in group_count_list: group_count_list2.append(count)
#return expbuilder_value_db,group_count_list2,ranked_array_headers,raw_data_comps,raw_data_comp_headers
return group_count_list2,raw_data_comp_headers
def filterBlanks(data_list):
data_list_new=[]
for i in data_list:
if i=='':pass
else: data_list_new.append(i)
return data_list_new
def reorder(data,data_headers,array_order,comp_group_list,probeset_db,include_raw_data,array_type,norm,fl,logvalues=True,blanksPresent=False):
###array_order gives the final level order sorted, followed by the original index order as a tuple
expbuilder_value_db = {}; group_name_db = {}; summary_filtering_stats = {}; pval_summary_db= {}
replicates = 'yes'
stat_result_names = ['avg-','log_fold-','fold-','rawp-','adjp-']
group_summary_result_names = ['avg-']
### Define expression variables
try: probability_statistic = fl.ProbabilityStatistic()
except Exception: probability_statistic = 'unpaired t-test'
try: gene_exp_threshold = math.log(fl.GeneExpThreshold(),2)
except Exception: gene_exp_threshold = 0
try: gene_rpkm_threshold = float(fl.RPKMThreshold())
except Exception: gene_rpkm_threshold = 0
try: FDR_statistic = fl.FDRStatistic()
except Exception: FDR_statistic = 'Benjamini-Hochberg'
calculateAsNonLog=True
if blanksPresent:
calculateAsNonLog=False
### Begin processing sample expression values according to the organized groups
for row_id in data:
try: gene = probeset_db[row_id][0]
except: gene = '' #not needed if not altsplice data
data_headers2 = {} #reset each time
grouped_ordered_array_list = {}
for x in array_order:
y = x[1] #this is the new first index
group = x[2]
group_name = x[3]
group_name_db[group] = group_name
#for example y = 5, therefore the data[row_id][5] entry is now the first
try:
try: new_item = data[row_id][y]
except IndexError: print row_id,data[row_id],len(data[row_id]),y,len(array_order),array_order;kill
if logvalues==False and calculateAsNonLog and array_type == 'RNASeq':
new_item = math.pow(2,new_item)
except TypeError: new_item = '' #this is for a spacer added in the above function
try: grouped_ordered_array_list[group].append(new_item)
except KeyError: grouped_ordered_array_list[group] = [new_item]
try: data_headers2[group].append(data_headers[y])
except KeyError: data_headers2[group]= [data_headers[y]]
#perform statistics on each group comparison - comp_group_list: [(1,2),(3,4)]
stat_results = {}
group_summary_results = {}
for comp in comp_group_list:
group1 = int(comp[0])
group2 = int(comp[1])
group1_name = group_name_db[group1]
group2_name = group_name_db[group2]
groups_name = group1_name + "_vs_" + group2_name
data_list1 = grouped_ordered_array_list[group1]
data_list2 = grouped_ordered_array_list[group2] #baseline expression
if blanksPresent: ### Allows for empty cells
data_list1 = filterBlanks(data_list1)
data_list2 = filterBlanks(data_list2)
try: avg1 = statistics.avg(data_list1)
except Exception: avg1 = ''
try: avg2 = statistics.avg(data_list2)
except Exception: avg2=''
try:
if (logvalues == False and array_type != 'RNASeq') or (logvalues==False and calculateAsNonLog):
fold = avg1/avg2
log_fold = math.log(fold,2)
if fold<1: fold = -1.0/fold
else:
log_fold = avg1 - avg2
fold = statistics.log_fold_conversion(log_fold)
except Exception:
log_fold=''; fold=''
try:
#t,df,tails = statistics.ttest(data_list1,data_list2,2,3) #unpaired student ttest, calls p_value function
#t = abs(t); df = round(df); p = str(statistics.t_probability(t,df))
p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic)
except Exception: p = 1; sg = 1; N1=0; N2=0
comp = group1,group2
if array_type == 'RNASeq': ### Also non-log but treated differently
if 'RPKM' == norm: adj = 0
else: adj = 1
if calculateAsNonLog == False:
try: avg1 = math.pow(2,avg1)-adj; avg2 = math.pow(2,avg2)-adj
except Exception: avg1=''; avg2=''
if 'RPKM' == norm:
if avg1 < gene_rpkm_threshold and avg2 < gene_rpkm_threshold:
log_fold = 'Insufficient Expression'
fold = 'Insufficient Expression'
else:
if avg1 < gene_exp_threshold and avg2 < gene_exp_threshold:
log_fold = 'Insufficient Expression'
fold = 'Insufficient Expression'
#if row_id=='ENSG00000085514':
#if fold=='Insufficient Expression':
#print [norm, avg1, avg2, fold, comp, gene_exp_threshold, gene_rpkm_threshold, row_id]
#5.96999111075 7.72930768675 Insufficient Expression (3, 1) 1.0 ENSG00000085514
if gene_rpkm_threshold!=0 and calculateAsNonLog: ### Any other data
a1 = nonLogAvg(data_list1)
a2 = nonLogAvg(data_list2)
#print [a1,a2,gene_rpkm_threshold]
if a1<gene_rpkm_threshold and a2<gene_rpkm_threshold:
log_fold = 'Insufficient Expression'
fold = 'Insufficient Expression'
#print log_fold;kill
try:
gs = statistics.GroupStats(log_fold,fold,p)
stat_results[comp] = groups_name,gs,group2_name
if probability_statistic == 'moderated t-test':
gs.setAdditionalStats(data_list1,data_list2) ### Assuming equal variance
if probability_statistic == 'moderated Welch-test':
gs.setAdditionalWelchStats(data_list1,data_list2) ### Assuming unequal variance
except Exception:
null=[]; replicates = 'no' ### Occurs when not enough replicates
#print comp, len(stat_results); kill_program
group_summary_results[group1] = group1_name,[avg1]
group_summary_results[group2] = group2_name,[avg2]
### Replaces the below method to get the largest possible comparison fold and ftest p-value
grouped_exp_data = []; avg_exp_data = []
for group in grouped_ordered_array_list:
data_list = grouped_ordered_array_list[group]
if blanksPresent: ### Allows for empty cells
data_list = filterBlanks(data_list)
if len(data_list)>0: grouped_exp_data.append(data_list)
try: avg = statistics.avg(data_list); avg_exp_data.append(avg)
except Exception:
avg = ''
#print row_id, group, data_list;kill
try: avg_exp_data.sort(); max_fold = avg_exp_data[-1]-avg_exp_data[0]
except Exception: max_fold = 'NA'
try: ftestp = statistics.OneWayANOVA(grouped_exp_data)
except Exception: ftestp = 1
gs = statistics.GroupStats(max_fold,0,ftestp)
summary_filtering_stats[row_id] = gs
stat_result_list = []
for entry in stat_results:
data_tuple = entry,stat_results[entry]
stat_result_list.append(data_tuple)
stat_result_list.sort()
grouped_ordered_array_list2 = []
for group in grouped_ordered_array_list:
data_tuple = group,grouped_ordered_array_list[group]
grouped_ordered_array_list2.append(data_tuple)
grouped_ordered_array_list2.sort() #now the list is sorted by group number
###for each rowid, add in the reordered data, and new statistics for each group and for each comparison
for entry in grouped_ordered_array_list2:
group_number = entry[0]
original_data_values = entry[1]
if include_raw_data == 'yes': ###optionally exclude the raw values
for value in original_data_values:
if array_type == 'RNASeq':
if norm == 'RPKM': adj = 0
else: adj = 1
if calculateAsNonLog == False:
value = math.pow(2,value)-adj
try: expbuilder_value_db[row_id].append(value)
except KeyError: expbuilder_value_db[row_id] = [value]
if group_number in group_summary_results:
group_summary_data = group_summary_results[group_number][1] #the group name is listed as the first entry
for value in group_summary_data:
try: expbuilder_value_db[row_id].append(value)
except KeyError: expbuilder_value_db[row_id] = [value]
for info in stat_result_list:
if info[0][0] == group_number: #comp,(groups_name,[avg1,log_fold,fold,ttest])
comp = info[0]; gs = info[1][1]
expbuilder_value_db[row_id].append(gs.LogFold())
expbuilder_value_db[row_id].append(gs.Fold())
expbuilder_value_db[row_id].append(gs.Pval())
### Create a placeholder and store the position of the adjusted p-value to be calculated
expbuilder_value_db[row_id].append('')
gs.SetAdjPIndex(len(expbuilder_value_db[row_id])-1)
gs.SetPvalIndex(len(expbuilder_value_db[row_id])-2)
pval_summary_db[(row_id,comp)] = gs
###do the same for the headers, but at the dataset level (redundant processes)
array_fold_headers = []; data_headers3 = []
try:
for group in data_headers2:
data_tuple = group,data_headers2[group] #e.g. 1, ['X030910_25_hl.CEL', 'X030910_29R_hl.CEL', 'X030910_45_hl.CEL'])
data_headers3.append(data_tuple)
data_headers3.sort()
except UnboundLocalError:
print data_headers,'\n',array_order,'\n',comp_group_list,'\n'; kill_program
for entry in data_headers3:
x = 0 #indicates the times through a loop
y = 0 #indicates the times through a loop
group_number = entry[0]
original_data_values = entry[1]
if include_raw_data == 'yes': ###optionally exclude the raw values
for value in original_data_values:
array_fold_headers.append(value)
if group_number in group_summary_results:
group_name = group_summary_results[group_number][0]
group_summary_data = group_summary_results[group_number][1]
for value in group_summary_data:
combined_name = group_summary_result_names[x] + group_name #group_summary_result_names = ['avg-']
array_fold_headers.append(combined_name)
x += 1 #increment the loop index
for info in stat_result_list:
if info[0][0] == group_number: #comp,(groups_name,[avg1,log_fold,fold,ttest],group2_name)
groups_name = info[1][0]
only_add_these = stat_result_names[1:]
for value in only_add_these:
new_name = value + groups_name
array_fold_headers.append(new_name)
###For the raw_data only export we need the headers for the different groups (data_headers2) and group names (group_name_db)
raw_data_comp_headers = {}
for comp in comp_group_list:
temp_raw = []
group1 = int(comp[0]);group2 = int(comp[1])
comp = str(comp[0]),str(comp[1])
g1_headers = data_headers2[group1]
g2_headers = data_headers2[group2]
g1_name = group_name_db[group1]
g2_name = group_name_db[group2]
for header in g2_headers: temp_raw.append(g2_name+':'+header)
for header in g1_headers: temp_raw.append(g1_name+':'+header)
raw_data_comp_headers[comp] = temp_raw
###Calculate adjusted ftest p-values using BH95 sorted method
statistics.adjustPermuteStats(summary_filtering_stats)
### Calculate adjusted p-values for all p-values using BH95 sorted method
round=0
for info in comp_group_list:
compid = int(info[0]),int(info[1]); pval_db={}
for (rowid,comp) in pval_summary_db:
if comp == compid:
gs = pval_summary_db[(rowid,comp)]
pval_db[rowid] = gs
if 'moderated' in probability_statistic and replicates == 'yes':
### Moderates the original reported test p-value prior to adjusting
try: statistics.moderateTestStats(pval_db,probability_statistic)
except Exception:
if round == 0:
if replicates == 'yes':
print 'Moderated test failed due to issue with mpmpath or out-of-range values\n ... using unmoderated unpaired test instead!'
null=[] ### Occurs when not enough replicates
round+=1
if FDR_statistic == 'Benjamini-Hochberg':
statistics.adjustPermuteStats(pval_db)
else:
### Calculate a qvalue (https://github.com/nfusi/qvalue)
import numpy; from stats_scripts import qvalue; pvals = []; keys = []
for key in pval_db: pvals.append(pval_db[key].Pval()); keys.append(key)
pvals = numpy.array(pvals)
pvals = qvalue.estimate(pvals)
for i in range(len(pvals)): pval_db[keys[i]].SetAdjP(pvals[i])
for rowid in pval_db:
gs = pval_db[rowid]
expbuilder_value_db[rowid][gs.AdjIndex()] = gs.AdjP() ### set the place holder to the calculated value
if 'moderated' in probability_statistic:
expbuilder_value_db[rowid][gs.RawIndex()] = gs.Pval() ### Replace the non-moderated with a moderated p-value
pval_summary_db=[]
###Finished re-ordering lists and adding statistics to expbuilder_value_db
return expbuilder_value_db, array_fold_headers, summary_filtering_stats, raw_data_comp_headers
def nonLogAvg(data_list):
return statistics.avg(map(lambda x: math.pow(2,x)-1,data_list))
if __name__ == '__main__':
print array_cluster_final
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/reorder_arrays.py
|
reorder_arrays.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"""
This script iterates the LineageProfiler algorithm (correlation based classification method) to identify sample types relative to one
of two references given one or more gene models. The main function is runLineageProfiler.
The program performs the following actions:
1) Import a tab-delimited reference expression file with three columns (ID, biological group 1, group 2) and a header row (biological group names)
2) Import a tab-delimited expression file with gene IDs (column 1), sample names (row 1) and normalized expression values (e.g., delta CT values)
3) (optional - import existing models) Import a tab-delimited file with comma delimited gene-models for analysis
4) (optional - find new models) Identify all possible combinations of gene models for a supplied model size variable (e.g., --s 7)
5) Iterate through any supplied or identified gene models to obtain predictions for novel or known sample types
6) Export prediction results for all analyzed models to the folder CellClassification.
7) (optional) Print the top 20 scores and models for all possible model combinations of size --s
"""
import sys, string, shutil
import math
import os.path
import copy
import time
import getopt
try: import scipy
except Exception: pass
import traceback
import warnings
import random
import collections
try: import unique ### Not required (used in AltAnalyze)
except Exception: None
try: import export ### Not required (used in AltAnalyze)
except Exception: None
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>1 and '-' in command_args and '--GUI' not in command_args:
runningCommandLine = True
else:
runningCommandLine = False
#from stats_scripts import salstat_stats; reload(salstat_stats)
try:
from scipy import stats
use_scipy = True
except Exception:
use_scipy = False ### scipy is not required but is used as a faster implementation of Fisher Exact Test when present
def filepath(filename):
try: fn = unique.filepath(filename)
except Exception: fn = filename
return fn
def exportFile(filename):
try: export_data = export.ExportFile(filename)
except Exception: export_data = open(filename,'w')
return export_data
def makeUnique(item):
db1={}; list1=[]; k=0
for i in item:
try: db1[i]=[]
except TypeError: db1[tuple(i)]=[]; k=1
for i in db1:
if k==0: list1.append(i)
else: list1.append(list(i))
list1.sort()
return list1
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def verifyFile(filename):
status = True
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = True;break
except Exception: status = False
return status
def getHeader(filename):
header=[]
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
header = string.split(data,'\t')[1:];break
if header[0] == 'row_clusters-flat': ### remove this cluster annotation column
header = header[1:]
return header
def getGroupsFromExpFile(filename):
""" Import cells and clusters from an Expression Heatmap """
cluster_db = collections.OrderedDict()
fn=filepath(filename)
count=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')[1:]
if count==0: header = t
else: clusters = t
count+=1
if count == 2: ### Exit on the 3rd row
break
if header[0] == 'row_clusters-flat': ### remove this cluster annotation column
header = header[1:]
clusters = clusters[1:]
index = 0
for cell in header:
cluster_db[cell] = clusters[index]
index+=1
return cluster_db
def getDataMatrix(filename):
header=[]
matrix={}
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if len(header)==0:
header = string.split(data,'\t')[1:]
else:
t = string.split(data,'\t')
values = map(float,t[1:])
matrix[t[0]]=values
return header,matrix
def returnLargeGlobalVars():
### Prints all large global variables retained in memory (taking up space)
all = [var for var in globals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(globals()[var])>1:
print var, len(globals()[var])
except Exception: null=[]
def clearObjectsFromMemory(db_to_clear):
db_keys={}
for key in db_to_clear: db_keys[key]=[]
for key in db_keys:
try: del db_to_clear[key]
except Exception:
try:
for i in key: del i ### For lists of tuples
except Exception: del key ### For plain lists
def int_check(value):
val_float = float(value)
val_int = int(value)
if val_float == val_int:
integer_check = 'yes'
if val_float != val_int:
integer_check = 'no'
return integer_check
def IQR(array):
k1 = 75
k2 = 25
array.sort()
n = len(array)
value1 = float((n*k1)/100)
value2 = float((n*k2)/100)
if int_check(value1) == 'no':
k1_val = int(value1) + 1
if int_check(value1) == 'yes':
k1_val = int(value1)
if int_check(value2) == 'no':
k2_val = int(value2) + 1
if int_check(value2) == 'yes':
k2_val = int(value2)
try: median_val = scipy.median(array)
except Exception: median_val = Median(array)
upper75th = array[k1_val]
lower25th = array[k2_val]
int_qrt_range = upper75th - lower25th
T1 = lower25th-(1.5*int_qrt_range)
T2 = upper75th+(1.5*int_qrt_range)
return lower25th,median_val,upper75th,int_qrt_range,T1,T2
class IQRData:
def __init__(self,maxz,minz,medz,iq1,iq3):
self.maxz = maxz; self.minz = minz
self.medz = medz; self.iq1 = iq1
self.iq3 = iq3
def Max(self): return self.maxz
def Min(self): return self.minz
def Medium(self): return self.medz
def IQ1(self): return self.iq1
def IQ3(self): return self.iq3
def SummaryValues(self):
vals = string.join([str(self.IQ1()),str(self.Min()),str(self.Medium()),str(self.Max()),str(self.IQ3())],'\t')
return vals
def importGeneModels(filename):
x=0
geneModels={}; thresholds=None
#filename = None ### Override file import with default reference data (hard-coded)
if filename != None:
fn=filepath(filename)
fileRead = open(fn,'rU').xreadlines()
else:
fileRead = defaultGeneModels()
for line in fileRead:
try:
data = cleanUpLine(line)
t = string.split(data,'\t')
except Exception:
t = line
genes = t[0]
genes = string.replace(genes,"'",'')
genes = string.replace(genes,' ',',')
genes = string.split(genes,',')
if t>1:
try: thresholds = map(float,t[1:])
except Exception: thresholds = None
try:
if len(thresholds)==0: thresholds = None
except Exception: pass
models=[]
for gene in genes:
if len(gene)>0:
models.append(gene)
if len(models)>0:
geneModels[tuple(models)] = thresholds
return geneModels
def convertClusterOrderToGroupLabels(filename,refCells=None):
""" Lookup for cluster number to assigned cell-type name (used for cellHarmony centroid alignment)"""
clusterNumber = 0
fn=filepath(filename)
prior_label=None
groupNumber_label_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
label = t[-1]
cell = t[0]
if refCells != None:
if cell not in refCells: ### skip this cell/header/extra notation
alt_cell = cell+'.Reference'
if alt_cell not in refCells:
continue
else:
ICGS_cluster_number = refCells[alt_cell] ### Use the original cluster number if avaialble
clusterNumber = ICGS_cluster_number
else:
ICGS_cluster_number = refCells[cell] ### Use the original cluster number if avaialble
clusterNumber = ICGS_cluster_number
else:
try: ICGS_cluster_number = t[-2]
except: ICGS_cluster_number = t[-1]
if label != prior_label:
clusterNumber+=1
if str(ICGS_cluster_number) not in groupNumber_label_db:
if label == ICGS_cluster_number:
ICGS_cluster_number = clusterNumber ### If the two columns are the same in the labels file
groupNumber_label_db[str(ICGS_cluster_number)]=label
prior_label = label
#for i in groupNumber_label_db: print [i,groupNumber_label_db[i]]
return groupNumber_label_db
######### Below code deals is specific to this module #########
def runLineageProfiler(species,array_type,exp_input,exp_output,
codingtype,compendium_platform,modelSize=None,customMarkers=False,
geneModels=False,permute=False,useMulti=False,fl=None,label_file=None):
""" This code differs from LineageProfiler.py in that it is able to iterate through the LineageProfiler functions with distinct geneModels
that are either supplied by the user or discovered from all possible combinations. """
#global inputType
global exp_output_file; exp_output_file = exp_output; global targetPlatform
global tissues; global sample_headers; global collapse_override
global analysis_type; global coding_type; coding_type = codingtype
global tissue_to_gene; tissue_to_gene = {}; global platform; global cutoff
global customMarkerFile; global delim; global keyed_by; global pearson_list
global Permute; Permute=permute; global useMultiRef; useMultiRef = useMulti
pearson_list={}
cellHarmony=True
#global tissue_specific_db
try: returnCentroids = fl.ReturnCentroids()
except Exception: returnCentroids = False
print 'platform:',array_type
collapse_override = True
if 'ICGS' in customMarkers or 'MarkerGene' in customMarkers or returnCentroids:
""" When performing cellHarmony, build an ICGS expression reference with log2 TPM values rather than fold """
print 'Converting ICGS folds to ICGS expression values as a reference first...'
try: customMarkers = convertICGSClustersToExpression(customMarkers,exp_input,returnCentroids=returnCentroids,species=species,fl=fl)
except:
print traceback.format_exc()
print "Using the supplied reference file only (not importing raw expression)...Proceeding without differential expression analsyes..."
pass
if label_file != None:
refCells = None
reference_exp_file = string.replace(customMarkers,'-centroid.txt','.txt')
status = verifyFile(reference_exp_file) ### Organized cells in the final cluster order
if status==True:
refCells = getGroupsFromExpFile(reference_exp_file)
label_file = convertClusterOrderToGroupLabels(label_file,refCells=refCells)
#print label_file
customMarkerFile = customMarkers
if geneModels == False or geneModels == None: geneModels = []
else:
geneModels = importGeneModels(geneModels)
exp_input = string.replace(exp_input,'\\','/')
exp_input = string.replace(exp_input,'//','/')
exp_output = string.replace(exp_output,'\\','/')
exp_output = string.replace(exp_output,'//','/')
delim = "/"
print '\nRunning cellHarmony analysis on',string.split(exp_input,delim)[-1][:-4]
global correlate_by_order; correlate_by_order = 'no'
global rho_threshold; rho_threshold = -1
global correlate_to_tissue_specific; correlate_to_tissue_specific = 'no'
cutoff = 0.01
global value_type
platform = array_type
if 'stats.' in exp_input:
value_type = 'calls'
else:
value_type = 'expression'
tissue_specific_db={}; sample_headers=[]; tissues=[]
if len(array_type)==2:
### When a user-supplied expression is provided (no ExpressionOutput files provided - importGeneIDTranslations)
array_type, vendor = array_type
if ':' in vendor:
vendor = string.split(vendor,':')[1]
if array_type == 'RNASeq':
vendor = 'Symbol'
platform = array_type
else: vendor = 'Symbol'
if 'RawSplice' in exp_input or 'FullDatasets' in exp_input or coding_type == 'AltExon':
analysis_type = 'AltExon'
if platform != compendium_platform: ### If the input IDs are not Affymetrix Exon 1.0 ST probesets, then translate to the appropriate system
translate_to_genearray = 'no'
targetPlatform = compendium_platform
translation_db = importExonIDTranslations(array_type,species,translate_to_genearray)
keyed_by = 'translation'
else: translation_db=[]; keyed_by = 'primaryID'; targetPlatform = compendium_platform
else:
try:
### Get arrayID to Ensembl associations
if vendor != 'Not needed':
### When no ExpressionOutput files provided (user supplied matrix)
translation_db = importVendorToEnsemblTranslations(species,vendor,exp_input)
else:
translation_db = importGeneIDTranslations(exp_output)
keyed_by = 'translation'
targetPlatform = compendium_platform
analysis_type = 'geneLevel'
except Exception:
translation_db=[]; keyed_by = 'primaryID'; targetPlatform = compendium_platform; analysis_type = 'geneLevel'
targetPlatform = compendium_platform ### Overides above
try: importTissueSpecificProfiles(species,tissue_specific_db)
except Exception:
try:
try:
targetPlatform = 'exon'
importTissueSpecificProfiles(species,tissue_specific_db)
except Exception:
try:
targetPlatform = 'gene'
importTissueSpecificProfiles(species,tissue_specific_db)
except Exception:
targetPlatform = "3'array"
importTissueSpecificProfiles(species,tissue_specific_db)
except Exception:
print 'No compatible compendiums present...'
print traceback.format_exc()
forceError
gene_expression_db, sample_headers = importGeneExpressionValuesSimple(exp_input,translation_db)
### Make sure each sample ID is unique
samples_added=[]; i=1
### Organize sample expression, with the same gene order as the tissue expression set
for s in sample_headers:
if s in samples_added:
samples_added.append(s+"."+str(i)) ### Ensure only unique sample IDs exist
i+=1
else:
samples_added.append(s)
sample_headers = samples_added
pruneTissueSpecific=False
for gene in tissue_specific_db:
if gene not in gene_expression_db:
pruneTissueSpecific = True
break
if pruneTissueSpecific:
tissue_specific_db2={}
for gene in gene_expression_db:
if gene in tissue_specific_db:
tissue_specific_db2[gene] = tissue_specific_db[gene]
elif gene in translation_db:
altGeneID = translation_db[gene]
if altGeneID in tissue_specific_db:
tissue_specific_db2[gene] = tissue_specific_db[altGeneID]
tissue_specific_db = tissue_specific_db2
all_marker_genes=[]
for gene in tissue_specific_db:
all_marker_genes.append(gene)
#print [modelSize]
if len(geneModels)>0:
allPossibleClassifiers = geneModels
elif modelSize == None or modelSize == 'optimize' or modelSize == 'no':
allPossibleClassifiers={}
allPossibleClassifiers[tuple(all_marker_genes)]=None
else:
### A specific model size has been specified (e.g., find all 10-gene models)
allPossibleClassifiers = getRandomSets(all_marker_genes,modelSize)
num=1
all_models=[]
if len(allPossibleClassifiers)<16:
print 'Using:'
for model in allPossibleClassifiers:
print 'model',num, 'with',len(model),'genes' #model
num+=1
all_models+=model
#all_models = unique.unique(all_models)
#print len(all_models);sys.exit()
### This is the main analysis function
print 'Number of reference samples to compare to:',len(tissues)
if len(tissues)<20:
print tissues
if modelSize != 'optimize':
hit_list, hits, fails, prognostic_class_db,sample_diff_z, evaluate_size, prognostic_class1_db, prognostic_class2_db = iterateLineageProfiler(exp_input,
tissue_specific_db, allPossibleClassifiers,translation_db,compendium_platform,modelSize,species,gene_expression_db, sample_headers)
else:
summary_hit_list=[]
try: evaluate_size = len(allPossibleClassifiers[0])
except Exception:
### Occurs when custom models loaded
for i in allPossibleClassifiers:
evaluate_size = len(i); break
hit_list, hits, fails, prognostic_class_db,sample_diff_z, evaluate_size, prognostic_class1_db, prognostic_class2_db = iterateLineageProfiler(exp_input,
tissue_specific_db, allPossibleClassifiers,translation_db,compendium_platform,None,species,gene_expression_db, sample_headers)
while evaluate_size > 4:
hit_list.sort()
top_model = hit_list[-1][-1]
top_model_score = hit_list[-1][0]
"""
try: ### Used for evaluation only - gives the same top models
second_model = hit_list[-2][-1]
second_model_score = hit_list[-2][0]
if second_model_score == top_model_score:
top_model = second_model_score ### Try this
print 'selecting secondary'
except Exception: None
"""
allPossibleClassifiers = [hit_list[-1][-1]]
hit_list, hits, fails, prognostic_class_db,sample_diff_z, evaluate_size, prognostic_class1_db, prognostic_class2_db = iterateLineageProfiler(exp_input,
tissue_specific_db, allPossibleClassifiers,translation_db,compendium_platform,modelSize,species,gene_expression_db, sample_headers)
summary_hit_list+=hit_list
hit_list = summary_hit_list
root_dir = string.join(string.split(exp_output_file,'/')[:-1],'/')+'/'
dataset_name = string.replace(string.split(exp_input,'/')[-1][:-4],'exp.','')
output_classification_file = root_dir+'CellClassification/'+dataset_name+'-CellClassification.txt'
try: os.mkdir(root_dir+'CellClassification')
except Exception: None
export_summary = exportFile(output_classification_file)
models = []
for i in allPossibleClassifiers:
i = string.replace(str(i),"'",'')[1:-1]
models.append(i)
### If multiple class-headers with a common phenotype (different source), combine into a single report
class_list=[]
processed=0
for h in tissues:
if ':' in h and collapse_override==False:
try:
phenotype, source = string.split(h,':')
processed+=1
if phenotype not in class_list:
class_list.append(phenotype)
except Exception: pass
if len(class_list)==2 and len(tissues) == processed and collapse_override==False and cellHarmony==False: ### Ensures all reference headers have : in them
tissue_list = class_list
collapse = True
else:
tissue_list = tissues
collapse = False
print ''
class_headers = map(lambda x: x+' Predicted Hits',tissue_list)
export_header = ['Samples']+class_headers+['Composite Classification Score','Combined Correlation DiffScore','Predicted Class','Max-Rho']
if label_file != None:
export_header.append('CentroidLabel')
headers = string.join(export_header,'\t')+'\n'
export_summary.write(headers)
sorted_results=[] ### sort the results
try: numberOfModels = len(allPossibleClassifiers)
except Exception: numberOfModels = 1
accuracy=[]
ar=[]
non=[]
no_intermediate_accuracy=[]
verboseReport = False
for sample in prognostic_class_db:
if len(tissues)==2:
class1_score = prognostic_class1_db[sample]
class2_score = prognostic_class2_db[sample]
zscore_distribution = map(lambda x: str(x[0]), sample_diff_z[sample])
pearson_max_values = map(lambda x: str(x[1]), sample_diff_z[sample])
dist_list=[]
for i in zscore_distribution:
try: dist_list.append(float(i))
except Exception: None ### Occurs for 'NA's
#try: median_score = scipy.median(dist_list)
#except Exception: median_score = Median(dist_list)
try: sum_score = sum(dist_list) #scipy.median
except Exception: sum_score = sum(dist_list)
correlations=[]
for i in pearson_max_values:
try: correlations.append(float(i))
except Exception: None ### Occurs for 'NA's
median_correlation = scipy.median(correlations)
if median_correlation<0.8 and verboseReport:
print 'Sample: %s has a low median model Pearson correlation coefficient (%s)' % (sample,str(median_correlation))
if verboseReport==False:
print '.',
class_db = prognostic_class_db[sample]
class_scores=[]; class_scores_str=[]; class_scores_refs=[]; collapsed_pheno_scores={}
for tissue in tissues:
if collapse and collapse_override==False:
phenotype,source = string.split(tissue,':')
try: collapsed_pheno_scores[phenotype]+=class_db[tissue]
except Exception: collapsed_pheno_scores[phenotype]=class_db[tissue]
else:
class_scores_str.append(str(class_db[tissue]))
class_scores.append(class_db[tissue])
class_scores_refs.append((class_db[tissue],tissue))
if collapse and collapse_override == False:
for phenotype in tissue_list:
class_scores.append(collapsed_pheno_scores[phenotype]) ### Collapse the scores and report in the original phenotype order
class_scores_str.append(str(collapsed_pheno_scores[phenotype]))
class_scores_refs.append((collapsed_pheno_scores[phenotype],phenotype))
"""
for tissue in tissues:
class_scores_str.append(str(class_db[tissue]))
class_scores.append(class_db[tissue])
class_scores_refs.append((class_db[tissue],tissue))
"""
overall_prog_score = str(max(class_scores)-min(class_scores))
if len(tissues)==2 and cellHarmony==False:
class_scores_str = [str(class1_score),str(class2_score)] ### range of positive and negative scores for a two-class test
if class1_score == 0 and class2_score == 0:
call = 'Intermediate Risk '+ tissues[0]
elif class1_score == numberOfModels:
call = 'High Risk '+ tissues[0]
elif class2_score == numberOfModels:
call = 'Low Risk '+ tissues[0]
elif class1_score == 0:
call = 'Intermediate Risk '+ tissues[0]
elif class2_score == 0:
call = 'Intermediate Risk '+ tissues[0]
else:
call = 'Itermediate Risk '+ tissues[0]
overall_prog_score = str(class1_score-class2_score)
else:
class_scores_refs.sort()
call=class_scores_refs[-1][1] ### This is the reference with the max reported score
if call == tissue_list[-1]: ### Usually the classifier of interest should be listed first in the reference file, not second
overall_prog_score = str(float(overall_prog_score)*-1)
sum_score = sum_score*-1
values = [sample]+class_scores_str+[overall_prog_score,str(sum_score),call]
if label_file != None:
sampleLabel=''
if call in label_file:
groupLabel = label_file[call] ### This is the unique label for the cluster number
else:
print [call]
for c in label_file:
print [c]
kill
values = string.join(values+zscore_distribution[:-1]+[str(max(correlations)),groupLabel],'\t')+'\n' ### Export line for cellHarmony classification results
else:
values = string.join(values+zscore_distribution[:-1]+[str(max(correlations))],'\t')+'\n' ### Export line for cellHarmony classification results
if ':' in sample:
sample = string.split(sample,':')[0]
if ':' in call:
call = string.split(call,':')[0]
if call==sample:
accuracy.append(float(1))
if float(overall_prog_score) > 10 or float(overall_prog_score) < -10:
no_intermediate_accuracy.append(float(1))
if 'non' in call: non.append(float(1))
else: ar.append(float(1))
else:
accuracy.append(float(0))
if float(overall_prog_score) > 10 or float(overall_prog_score) < -10:
no_intermediate_accuracy.append(float(0))
if 'non' in call: non.append(float(0))
else: ar.append(float(0))
sorted_results.append([float(overall_prog_score),sum_score,values])
sample_diff_z[sample] = dist_list
if verboseReport:
print len(no_intermediate_accuracy)
print no_intermediate_accuracy
print 'Overall Acuracy:',Average(accuracy)*100
print 'Sensititivity:', sum(ar), len(ar)
print 'Specificity:', sum(non), len(non)
print str(Average(accuracy)*100)+'\t'+str(Average(ar)*100)+'\t'+str(Average(non)*100)+'\t'+str(Average(no_intermediate_accuracy)*100)+'\t'+str(sum(ar))+'\t'+str(len(ar))+'\t'+str(sum(non))+'\t'+str(len(non))
else:
print '\nClassification analysis completed...'
sorted_results.sort()
sorted_results.reverse()
for i in sorted_results:
export_summary.write(i[-1])
export_summary.close()
print '\nResults file written to:',root_dir+'CellClassification/'+dataset_name+'-CellClassification.txt','\n'
hit_list.sort(); hit_list.reverse()
top_hit_list=[]
top_hit_db={}
hits_db={}; fails_db={}
### Only look at the max correlation for each sample
max_pearson_list=[]
for sample in pearson_list:
pearson_list[sample].sort()
for rho in pearson_list[sample][-2:]: ### get the top two correlations
max_pearson_list.append(rho)
avg_pearson_rho = Average(max_pearson_list)
maxPearson = max(max_pearson_list)
try:
for i in sample_diff_z:
zscore_distribution = sample_diff_z[i]
maxz = max(zscore_distribution); minz = min(zscore_distribution)
sample_diff_z[i] = string.join(map(str,zscore_distribution),'\t')
try:
lower25th,medz,upper75th,int_qrt_range,T1,T2 = IQR(zscore_distribution)
if float(maxz)>float(T2): maxz = T2
if float(minz) < float(T1): minz = T1
#iqr = IQRData(maxz,minz,medz,lower25th,upper75th)
#sample_diff_z[i] = iqr
except Exception:
pass
for i in hits:
try: hits_db[i]+=1
except Exception: hits_db[i]=1
for i in fails:
try: fails_db[i]+=1
except Exception: fails_db[i]=1
for i in fails_db:
if i not in hits:
try:
#print i+'\t'+'0\t'+str(fails_db[i])+'\t'+ sample_diff_z[i]
None
except Exception:
#print i
None
except Exception:
pass
exportModelScores = True
if modelSize != False:
#print 'Returning all model overall scores'
hits=[]
for i in hits_db:
hits.append([hits_db[i],i])
hits.sort()
hits.reverse()
for i in hits:
if i[1] in fails_db: fail = fails_db[i[1]]
else: fail = 0
try:
#print i[1]+'\t'+str(i[0])+'\t'+str(fail)+'\t'+sample_diff_z[i[1]]
None
except Exception:
#print i[1]
None
if modelSize == 'optimize': threshold = 80
else: threshold = 0
#print 'threshold:',threshold
for i in hit_list:
if i[0]>threshold:
top_hit_list.append(i[-1])
top_hit_db[tuple(i[-1])]=i[0]
if len(geneModels) > 0 and exportModelScores==False:
for i in hit_list:
#print i[:5],i[-1],i[-2] ### print all
pass
else:
"""
print 'Returning all over 90'
for i in hit_list:
if i[0]>85:
print i[:5],i[-1],i[-2] ### print all
sys.exit()"""
#print 'Top hits'
output_model_file = root_dir+'CellClassification/'+dataset_name+'-ModelScores.txt'
export_summary = exportFile(output_model_file)
print 'Exporting top-scoring models to:',output_model_file
title = 'Classification-Rate\tClass1-Hits\tClass1-Total\tClass2-Hits\tClass2-Total\tModel\tModel-Gene-Number\n'
export_summary.write(title)
for i in hit_list: #hit_list[:500]
overall_scores=[]
for x in i[:5]: overall_scores.append(str(x))
model = string.replace(str(i[-1])[1:-1],"'",'')
values = string.join(overall_scores+[model]+[str(i[-2])],'\t')+'\n'
export_summary.write(values)
export_summary.close()
"""
try:
if hit_list[0][0] == hit_list[20][0]:
for i in hit_list[20:]:
if hit_list[0][0] == i[0]:
print i[:5],i[-1],i[-2]
else: sys.exit()
except Exception: None ### Occurs if less than 20 entries here
"""
print 'Average Pearson correlation coefficient:', avg_pearson_rho
if avg_pearson_rho<0.9 and verboseReport:
print '\n\nWARNING!!!!!!!!!'
print '\tThe average Pearson correlation coefficient for all example models is less than 0.9.'
print '\tYour data may not be comparable to the provided reference (quality control may be needed).\n\n'
elif verboseReport:
print 'No unusual warning.\n'
reference_exp_file = customMarkers
query_exp_file = exp_input
classification_file = output_classification_file
harmonizeClassifiedSamples(species,reference_exp_file, query_exp_file, classification_file,fl=fl)
return top_hit_db
def iterateLineageProfiler(exp_input,tissue_specific_db,allPossibleClassifiers,translation_db,compendium_platform,
modelSize,species,gene_expression_db,sampleHeaders):
classifyBasedOnRho=True
hit_list=[]
### Iterate through LineageProfiler for all gene models (allPossibleClassifiers)
times = 1; k=1000; l=1000; hits=[]; fails=[]; f=0; s=0; sample_diff_z={}; prognostic_class1_db={}; prognostic_class2_db={}
prognostic_class_db={}
begin_time = time.time()
try: evaluate_size=len(allPossibleClassifiers[0]) ### Number of reference markers to evaluate
except Exception:
for i in allPossibleClassifiers: evaluate_size = len(i); break
if modelSize=='optimize':
evaluate_size -= 1
allPossibleClassifiers = getRandomSets(allPossibleClassifiers[0],evaluate_size)
### Determine if we should collapse the entries or not based on common phenotype references
class_list=[]; processed=0; alternate_class={}
for h in tissues:
if ':' in h and 'ENS' not in h:
try:
phenotype, source = string.split(h,':'); processed+=1
if phenotype not in class_list: class_list.append(phenotype)
except Exception: pass
try:
alternate_class[class_list[0]] = class_list[1]
alternate_class[class_list[1]] = class_list[0]
except Exception: pass
if len(class_list)==2 and len(tissues) == processed and collapse_override==False: ### Ensures all reference headers have : in them
tissue_list = class_list; collapse = True
else: tissue_list = tissues; collapse = False
cellHarmony=True
mean_percent_positive=[]
for classifiers in allPossibleClassifiers:
try: thresholds = allPossibleClassifiers[classifiers]
except Exception: thresholds = None
tissue_to_gene={}; expession_subset=[]; sample_headers=[]; classifier_specific_db={}
for gene in classifiers:
try: classifier_specific_db[gene] = tissue_specific_db[gene]
except Exception: None
#print len(gene_expression_db), len(classifier_specific_db), len(expession_subset), len(translation_db)
expession_subset = filterGeneExpressionValues(gene_expression_db,classifier_specific_db,translation_db,expession_subset)
### If the incorrect gene system was indicated re-run with generic parameters
if len(expession_subset)==0:
translation_db=[]; keyed_by = 'primaryID'; targetPlatform = compendium_platform; analysis_type = 'geneLevel'
tissue_specific_db={}
importTissueSpecificProfiles(species,tissue_specific_db)
expession_subset = filterGeneExpressionValues(gene_expression_db,tissue_specific_db,translation_db,expession_subset)
if len(sample_diff_z)==0: ### Do this for the first model examine only
for h in sampleHeaders:
sample_diff_z[h]=[] ### Create this before any data is added, since some models will exclude data for some samples (missing dCT values)
if len(expession_subset)!=len(classifiers): f+=1
#if modelSize=='optimize': print len(expession_subset), len(classifiers);sys.exit()
if (len(expession_subset) != len(classifiers)) and modelSize=='optimize':
print "Please provide a reference set of equal length or smaller to the input analysis set"; kill
#print len(expession_subset), len(classifiers);sys.exit()
if len(expession_subset)==len(classifiers): ### Sometimes a gene or two are missing from one set
s+=1
#print classifiers,'\t',
zscore_output_dir,tissue_scores,sampleHeaders = analyzeTissueSpecificExpressionPatterns(tissue_specific_db,expession_subset,sampleHeaders)
#except Exception: print len(classifier_specific_db), classifiers; error
headers = list(tissue_scores['headers']); del tissue_scores['headers']
if times == k:
end_time = time.time()
print int(end_time-begin_time),'seconds'
k+=l
times+=1; index=0; positive=0; positive_score_diff=0
sample_number = (len(headers)-1)
population1_denom=0; population1_pos=0; population2_pos=0; population2_denom=0
diff_positive=[]; diff_negative=[]
while index < sample_number:
### The scores are now a tuple of (Z-score,original_pearson_rho)
scores = map(lambda x: tissue_scores[x][index], tissue_scores)
zscores_only = map(lambda x: tissue_scores[x][index][0], tissue_scores)
scores_copy = list(scores); scores_copy.sort()
max_pearson_model = scores_copy[-1][1] ### e.g., tumor rho (this is the one we want to QC on later)
min_pearson_model = scores_copy[-2][1] ### e.g., non-tumor rho
diff_rho = max_pearson_model - min_pearson_model
diff_z = (scores_copy[-1][0]-scores_copy[-2][0])*100 ### Diff between the top two scores (z-scores are the first item)
if classifyBasedOnRho == True:
diff_z = diff_rho*10
positive_class=None
j=0
for tissue in tissue_scores:
if ':' in tissue and 'ENS' not in tissue:
group_name = string.split(tissue,':')[0]
else:
group_name = tissue
if scores[j][0] == max(zscores_only):
hit_score = 1; positive_class = tissue
else: hit_score = 0
if len(tissues)>2 or cellHarmony==True:
if group_name+':' in headers[index+1] and hit_score==1:
g = string.split(headers[index+1],':')[0]+':'
if g in group_name+':': ### reciprocol of above
positive+=1
try:
class_db = prognostic_class_db[headers[index+1]]
try: class_db[tissue]+=hit_score
except Exception: class_db[tissue]=hit_score
except Exception:
class_db={}
class_db[tissue]=hit_score
prognostic_class_db[headers[index+1]] = class_db
j+=1
if collapse and collapse_override==False:
phenotype, source = string.split(positive_class,':')
baseline_positive_class = alternate_class[phenotype]+':'+source
denom_score,denom_rho = tissue_scores[baseline_positive_class][index]
old_diff = diff_z
diff_z = scores_copy[-1][0]-denom_score ### Diff between the top two scores of the SAME SOURCE
diff_rho = (max_pearson_model - denom_rho)
if classifyBasedOnRho == True:
diff_z = diff_rho*10
#print headers[index+1], scores_copy[-1]
if len(tissues)==2:
if ':' in headers[index+1]:
pheno = string.split(headers[index+1],':')[0]
else:
pheno = None
diff_z = tissue_scores[tissues[0]][index][0]-tissue_scores[tissues[-1]][index][0] ### z-scores are the first item and pearson-rho is the second
diff_rho = (tissue_scores[tissues[0]][index][1]-tissue_scores[tissues[-1]][index][1])
if classifyBasedOnRho == True:
diff_z = diff_rho*10
if thresholds == None:
threshold1 = 0; threshold2 = 0
else:
threshold1, threshold2 = thresholds ### emperically derived cutoffs provided by the user for each model (e.g., mean+2SD of diff_rho)
if headers[index+1] not in prognostic_class1_db:
prognostic_class1_db[headers[index+1]]=0 ### Create a default value for each sample
if headers[index+1] not in prognostic_class2_db:
prognostic_class2_db[headers[index+1]]=0 ### Create a default value for each sample
if diff_z>threshold1:
prognostic_class1_db[headers[index+1]]+=1
elif diff_z<threshold2:
prognostic_class2_db[headers[index+1]]+=1
if diff_z>0 and (tissues[0] == pheno):
positive+=1; positive_score_diff+=abs(diff_z)
population1_pos+=1; diff_positive.append(abs(diff_z))
hits.append(headers[index+1]) ### see which are correctly classified
elif diff_z<0 and (tissues[-1] == pheno):
positive+=1; positive_score_diff+=abs(diff_z)
population2_pos+=1; diff_positive.append(abs(diff_z))
hits.append(headers[index+1]) ### see which are correctly classified
elif diff_z>0 and (tissues[-1] == pheno): ### Incorrectly classified
diff_negative.append(abs(diff_z))
fails.append(headers[index+1])
elif diff_z<0 and (tissues[0] == pheno): ### Incorrectly classified
#print headers[index+1]
diff_negative.append(abs(diff_z))
fails.append(headers[index+1])
if (tissues[0] == pheno):
population1_denom+=1
else:
population2_denom+=1
sample_diff_z[headers[index+1]].append((diff_z,max([max_pearson_model,min_pearson_model]))) ### Added pearson max here
index+=1
try: percent_positive = (float(positive)/float(index))*100
except ZeroDivisionError:
print 'WARNING!!!! No matching genes. Make sure your gene IDs are the same ID type as your reference.'
forceNoMatchingGeneError
mean_percent_positive.append(percent_positive)
if len(tissues)==2 and cellHarmony==False:
try:
pos = float(population1_pos)/float(population1_denom)
neg = float(population2_pos)/float(population2_denom)
#percent_positive = (pos+neg)/2
except Exception: pos = 0; neg = 0
hit_list.append([percent_positive,population1_pos, population1_denom,population2_pos,population2_denom,[Average(diff_positive),Average(diff_negative)],positive_score_diff,len(classifiers),classifiers])
else:
hit_list.append([percent_positive,len(classifiers),classifiers])
for sample in sample_diff_z:
if len(sample_diff_z[sample]) != (times-1): ### Occurs when there is missing data for a sample from the analyzed model
sample_diff_z[sample].append(('NA','NA')) ### add a null result
#print Average(mean_percent_positive), '\tAverage'
return hit_list, hits, fails, prognostic_class_db, sample_diff_z, evaluate_size, prognostic_class1_db, prognostic_class2_db
def factorial(n):
### Code from http://docs.python.org/lib/module-doctest.html
if not n >= 0:
raise ValueError("n must be >= 0")
if math.floor(n) != n:
raise ValueError("n must be exact integer")
if n+1 == n: # catch a value like 1e300
raise OverflowError("n too large")
result = 1
factor = 2
while factor <= n:
result *= factor
factor += 1
return result
def choose(n,x):
"""Equation represents the number of ways in which x objects can be selected from a total of n objects without regard to order."""
#(n x) = n!/(x!(n-x)!)
f = factorial
result = f(n)/(f(x)*f(n-x))
return result
def getRandomSets(a,size):
#a = ['a','b','c','d','e','f','g','h','i','j','k','l','m','n','o','p','q','r','s','t','u','v','w','x','y','z']
#size = 4
select_set={'ENSG00000140678':'ITGAX','ENSG00000105835':'NAMPT','ENSG00000027697':'IFNGR1','ENSG00000120129':'DUSP1','ENSG00000003402':'CFLAR','ENSG00000113269':'RNF130'}
select_set={}
select_set2={'ENSG00000163602': 'RYBP'}
negative_select = {'ENSG00000105352':'CEACAM4'}
negative_select={}
possible_sets = choose(len(a),size)
print 'Possible',size,'gene combinations to test',possible_sets
permute_ls = []; done = 0; permute_db={}
while done == 0:
b = list(tuple(a)); random.shuffle(b)
bx_set={}
i = 0
while i < len(b):
try:
bx = b[i:i+size]; bx.sort()
if len(bx)==size: permute_db[tuple(bx)]=None
else: break
except Exception: break
i+=1
if len(permute_db) == possible_sets:
done=1; break
for i in permute_db:
add=0; required=0; exclude=0
for l in i:
if len(select_set)>0:
if l in select_set: add+=1
#if l in select_set2: required+=1
#if l in negative_select: exclude+=1
else: add = 1000
if add>2 and exclude==0:# and required==1:
permute_ls.append(i)
#print len(permute_ls)
return permute_ls
def importVendorToEnsemblTranslations(species,vendor,exp_input):
translation_db={}
"""
### Faster method but possibly not as good
uid_db = simpleUIDImport(exp_input)
import gene_associations
### Use the same annotation method that is used to create the ExpressionOutput annotations
array_to_ens = gene_associations.filterGeneToUID(species,'Ensembl',vendor,associated_IDs)
for arrayid in array_to_ens:
ensembl_list = array_to_ens[arrayid]
try: translation_db[arrayid] = ensembl_list[0] ### This first Ensembl is ranked as the most likely valid based on various metrics in getArrayAnnotationsFromGOElite
except Exception: None
"""
translation_db={}
### Use the same annotation method that is used to create the ExpressionOutput annotations
use_go = 'yes'
conventional_array_db={}
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-'+vendor))
for EnsGeneID in gene_to_symbol:
vendorID = gene_to_symbol[EnsGeneID][0]
try:
translation_db[vendorID] = EnsGeneID
translation_db[EnsGeneID] = vendorID
except Exception: None
return translation_db
def importTissueSpecificProfiles(species,tissue_specific_db):
if analysis_type == 'AltExon':
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_'+targetPlatform +'_tissue-specific_AltExon_protein_coding.txt'
else:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_'+targetPlatform +'_tissue-specific_'+coding_type+'.txt'
if customMarkerFile != False:
filename = customMarkerFile
if value_type == 'calls':
filename = string.replace(filename,'.txt','_stats.txt')
fn=filepath(filename); x=0
tissues_added={}
dataType = 'matrix'
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
#print 'Importing the tissue compedium database:',string.split(filename,delim)[-1][:-4]
headers = t; x=1; index=0
for i in headers:
if 'UID' == i or 'uid' == i: ens_index = index; uid_index = index
if analysis_type == 'AltExon': ens_index = ens_index ### Assigned above when analyzing probesets
elif 'Ensembl' in i: ens_index = index
if 'marker-in' in i:
tissue_index = index+1; marker_in = index
dataType = 'MarkerFinder'
if 'row_clusters-flat' in i:
tissue_index = 2 ### start reading fold data at column 3
dataType = 'clustering'
index+=1
try:
for i in t[tissue_index:]: tissues.append(i)
except Exception:
for i in t[1:]: tissues.append(i)
if keyed_by == 'primaryID':
try: ens_index = uid_index
except Exception: None
else:
if 'column_clusters-flat' == t[0]: ### clustered heatmap - skip this row
continue
if dataType=='clustering':
gene = t[0]
tissue_exp = map(float, t[2:])
tissue_specific_db[gene]=x,tissue_exp
else:
try:
gene = t[0]
tissue_exp = map(float, t[1:])
tissue_specific_db[gene]=x,tissue_exp ### Use this to only grab relevant gene expression profiles from the input dataset
except Exception:
gene = string.split(t[ens_index],'|')[0] ### Only consider the first listed gene - this gene is the best option based on ExpressionBuilder rankings
#if 'Pluripotent Stem Cells' in t[marker_in] or 'Heart' in t[marker_in]:
#if t[marker_in] not in tissues_added: ### Only add the first instance of a gene for that tissue - used more for testing to quickly run the analysis
tissue_exp = map(float, t[tissue_index:])
if value_type == 'calls':
tissue_exp = produceDetectionCalls(tissue_exp,platform) ### 0 or 1 calls
tissue_specific_db[gene]=x,tissue_exp ### Use this to only grab relevant gene expression profiles from the input dataset
tissues_added[t[marker_in]]=[]
x+=1
#print len(tissue_specific_db), 'genes in the reference database'
if correlate_to_tissue_specific == 'yes':
try: importTissueCorrelations(filename)
except Exception:
null=[]
#print '\nNo tissue-specific correlations file present. Skipping analysis.'; kill
return tissue_specific_db
def importTissueCorrelations(filename):
filename = string.replace(filename,'specific','specific_correlations')
fn=filepath(filename); x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if x==0: x=1 ### Ignore header line
else:
uid,symbol,rho,tissue = string.split(data,'\t')
if float(rho)>rho_threshold: ### Variable used for testing different thresholds internally
try: tissue_to_gene[tissue].append(uid)
except Exception: tissue_to_gene[tissue] = [uid]
def simpleUIDImport(filename):
"""Import the UIDs in the gene expression file"""
uid_db={}
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
uid_db[string.split(data,'\t')[0]]=[]
return uid_db
def importGeneExpressionValuesSimple(filename,translation_db,translate=False):
### Import gene-level expression raw values
fn=filepath(filename); x=0; genes_added={}; gene_expression_db={}
dataset_name = string.split(filename,delim)[-1][:-4]
#print 'importing:',dataset_name
sampleIndex=1
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
if 'row_clusters-flat' in t:
sampleIndex = 2 ### start reading fold data at column 3
dataType = 'clustering'
if '#' not in data[0]:
for i in t[sampleIndex:]: sample_headers.append(i)
x=1
else:
gene = t[0]
if 'column_clusters-flat' == t[0]: ### clustered heatmap - skip this row
continue
#if '-' not in gene and ':E' in gene: print gene;sys.exit()
if analysis_type == 'AltExon':
try: ens_gene,exon = string.split(gene,'-')[:2]
except Exception: exon = gene
gene = exon
if keyed_by == 'translation': ### alternative value is 'primaryID'
"""if gene == 'ENSMUSG00000025915-E19.3':
for i in translation_db: print [i], len(translation_db); break
print gene, [translation_db[gene]];sys.exit()"""
if translate:
### This caused two many problems so we removed it with the translate default above
try: gene = translation_db[gene] ### Ensembl annotations
except Exception: pass
try: genes_added[gene]+=1
except Exception: genes_added[gene]=1
try: exp_vals = map(float, t[sampleIndex:])
except Exception:
### If a non-numeric value in the list
exp_vals=[]
for i in t[sampleIndex:]:
try: exp_vals.append(float(i))
except Exception: exp_vals.append(i)
gene_expression_db[gene] = exp_vals
#print len(gene_expression_db), 'matching genes in the dataset and tissue compendium database'
return gene_expression_db, sample_headers
def filterGeneExpressionValues(all_gene_expression_db,tissue_specific_db,translation_db,expession_subset):
### Filter all imported gene expression values
gene_expression_db={}; genes_added={}
for gene in all_gene_expression_db:
exp_vals = all_gene_expression_db[gene]
if gene in tissue_specific_db:
index,tissue_exp=tissue_specific_db[gene]
try: genes_added[gene]+=1
except Exception: genes_added[gene]=1
if value_type == 'calls': ### Hence, this is a DABG or RNA-Seq expression
exp_vals = produceDetectionCalls(exp_vals,targetPlatform) ### 0 or 1 calls
gene_expression_db[gene] = [index,exp_vals]
#print len(gene_expression_db), 'matching genes in the dataset and tissue compendium database'
for gene in genes_added:
if genes_added[gene]>1: del gene_expression_db[gene] ### delete entries that are present in the input set multiple times (not trustworthy)
else: expession_subset.append(gene_expression_db[gene]) ### These contain the rank order and expression
#print len(expession_subset);sys.exit()
expession_subset.sort() ### This order now matches that of
gene_expression_db=[]
return expession_subset
def importGeneExpressionValues(filename,tissue_specific_db,translation_db,expession_subset):
### Import gene-level expression raw values
fn=filepath(filename); x=0; genes_added={}; gene_expression_db={}
dataset_name = string.split(filename,delim)[-1][:-4]
#print 'importing:',dataset_name
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
if '#' not in data[0]:
for i in t[1:]: sample_headers.append(i)
x=1
else:
gene = t[0]
#if '-' not in gene and ':E' in gene: print gene;sys.exit()
if analysis_type == 'AltExon':
try: ens_gene,exon = string.split(gene,'-')[:2]
except Exception: exon = gene
gene = exon
if keyed_by == 'translation': ### alternative value is 'primaryID'
"""if gene == 'ENSMUSG00000025915-E19.3':
for i in translation_db: print [i], len(translation_db); break
print gene, [translation_db[gene]];sys.exit()"""
try: gene = translation_db[gene] ### Ensembl annotations
except Exception: gene = 'null'
if gene in tissue_specific_db:
index,tissue_exp=tissue_specific_db[gene]
try: genes_added[gene]+=1
except Exception: genes_added[gene]=1
try: exp_vals = map(float, t[1:])
except Exception:
### If a non-numeric value in the list
exp_vals=[]
for i in t[1:]:
try: exp_vals.append(float(i))
except Exception: exp_vals.append(i)
if value_type == 'calls': ### Hence, this is a DABG or RNA-Seq expression
exp_vals = produceDetectionCalls(exp_vals,targetPlatform) ### 0 or 1 calls
gene_expression_db[gene] = [index,exp_vals]
#print len(gene_expression_db), 'matching genes in the dataset and tissue compendium database'
for gene in genes_added:
if genes_added[gene]>1: del gene_expression_db[gene] ### delete entries that are present in the input set multiple times (not trustworthy)
else: expession_subset.append(gene_expression_db[gene]) ### These contain the rank order and expression
#print len(expession_subset);sys.exit()
expession_subset.sort() ### This order now matches that of
gene_expression_db=[]
return expession_subset, sample_headers
def produceDetectionCalls(values,Platform):
# Platform can be the compendium platform (targetPlatform) or analyzed data platform (platform or array_type)
new=[]
for value in values:
if Platform == 'RNASeq':
if value>1:
new.append(1) ### expressed
else:
new.append(0)
else:
if value<cutoff: new.append(1)
else: new.append(0)
return new
def importGeneIDTranslations(filename):
### Import ExpressionOutput/DATASET file to obtain Ensembl associations (typically for Affymetrix 3' arrays)
fn=filepath(filename); x=0; translation_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
headers = t; x=1; index=0
for i in headers:
if 'Ensembl' in i: ens_index = index; break
index+=1
else:
uid = t[0]
ens_geneids = t[ens_index]
ens_geneid = string.split(ens_geneids,'|')[0] ### In v.2.0.5, the first ID is the best protein coding candidate
if len(ens_geneid)>0:
translation_db[uid] = ens_geneid
return translation_db
def remoteImportExonIDTranslations(array_type,species,translate_to_genearray,targetplatform):
global targetPlatform; targetPlatform = targetplatform
translation_db = importExonIDTranslations(array_type,species,translate_to_genearray)
return translation_db
def importExonIDTranslations(array_type,species,translate_to_genearray):
gene_translation_db={}; gene_translation_db2={}
if targetPlatform == 'gene' and translate_to_genearray == 'no':
### Get gene array to exon array probeset associations
gene_translation_db = importExonIDTranslations('gene',species,'yes')
for geneid in gene_translation_db:
exonid = gene_translation_db[geneid]
gene_translation_db2[exonid] = geneid
#print exonid, geneid
translation_db = gene_translation_db2
else:
filename = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_'+array_type+'-exon_probesets.txt'
### Import exon array to target platform translations (built for DomainGraph visualization)
fn=filepath(filename); x=0; translation_db={}
print 'Importing the translation file',string.split(fn,delim)[-1][:-4]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: x=1
else:
platform_id,exon_id = t
if targetPlatform == 'gene' and translate_to_genearray == 'no':
try:
translation_db[platform_id] = gene_translation_db[exon_id] ### return RNA-Seq to gene array probeset ID
#print platform_id, exon_id, gene_translation_db[exon_id];sys.exit()
except Exception: null=[]
else:
translation_db[platform_id] = exon_id
del gene_translation_db; del gene_translation_db2
return translation_db
def analyzeTissueSpecificExpressionPatterns(tissue_specific_db,expession_subset,sampleHeaders):
tissue_specific_sorted = []; genes_present={}; tissue_exp_db={}; gene_order_db={}; gene_order=[]
gene_list=[]
for (index,vals) in expession_subset: genes_present[index]=[]
for gene in tissue_specific_db:
gene_list.append(gene)
tissue_specific_sorted.append(tissue_specific_db[gene])
gene_order_db[tissue_specific_db[gene][0]] = gene ### index order (this index was created before filtering)
tissue_specific_sorted.sort()
new_index=0
for (index,tissue_exp) in tissue_specific_sorted:
try:
null=genes_present[index]
i=0
gene_order.append([new_index,gene_order_db[index]]); new_index+=1
for f in tissue_exp:
### The order of the tissue specific expression profiles is based on the import gene order
try: tissue_exp_db[tissues[i]].append(f)
except Exception: tissue_exp_db[tissues[i]] = [f]
i+=1
except Exception: null=[] ### Gene is not present in the input dataset
sample_exp_db={};
for (index,exp_vals) in expession_subset:
i=0
for f in exp_vals:
try: s = sampleHeaders[i]
except Exception: print i, len(sampleHeaders); print index, len(exp_vals), f;sys.exit()
### The order of the tissue specific expression profiles is based on the import gene order
try: sample_exp_db[s].append(f)
except Exception: sample_exp_db[s] = [f]
i+=1
if correlate_by_order == 'yes':
### Rather than correlate to the absolute expression order, correlate to the order of expression (lowest to highest)
sample_exp_db = replaceExpressionWithOrder(sample_exp_db)
tissue_exp_db = replaceExpressionWithOrder(tissue_exp_db)
global tissue_comparison_scores; tissue_comparison_scores={}
if correlate_to_tissue_specific == 'yes':
### Create a gene_index that reflects the current position of each gene
gene_index={}
for (i,gene) in gene_order: gene_index[gene] = i
### Create a tissue to gene-index from the gene_index
tissue_to_index={}
for tissue in tissue_to_gene:
for gene in tissue_to_gene[tissue]:
if gene in gene_index: ### Some are not in both tissue and sample datasets
index = gene_index[gene] ### Store by index, since the tissue and expression lists are sorted by index
try: tissue_to_index[tissue].append(index)
except Exception: tissue_to_index[tissue] = [index]
tissue_to_index[tissue].sort()
sample_exp_db,tissue_exp_db = returnTissueSpecificExpressionProfiles(sample_exp_db,tissue_exp_db,tissue_to_index)
distributionNull = True
if Permute:
import copy
sample_exp_db_original = copy.deepcopy(sample_exp_db)
tissue_exp_db_original = copy.deepcopy(tissue_exp_db)
group_list=[]; group_db={}
for sample in sample_exp_db:
group = string.split(sample,':')[0]
try: group_db[group].append(sample)
except Exception: group_db[group] = [sample]
if distributionNull:
group_lengths=[]
for group in group_db:
group_lengths.append(len(group_db[group]))
group_db={}
for sample in sample_exp_db:
group = 'null1'
try: group_db[group].append(sample)
except Exception: group_db[group] = [sample]
group_db['null2'] = group_db['null1']
choice = random.sample
tissue_groups = ['null1','null2']
else:
choice = random.choice
tissue_groups = tuple(tissues)
permute_groups=[]
groups=[]
gn=0
for group in group_db:
samples = group_db[group]
permute_db={}; x=0
while x<200:
if distributionNull:
size = group_lengths[gn]
psamples = choice(samples,size)
else: psamples = [choice(samples) for _ in xrange(len(samples))] ### works for random.sample or choice (with replacement)
permute_db[tuple(psamples)]=None
x+=1
permute_groups.append(permute_db)
groups.append(group); gn+=1 ### for group sizes
groups.sort()
permute_group1 = permute_groups[0]
permute_group2 = permute_groups[1]
permute_group1_list=[]
permute_group2_list=[]
for psamples in permute_group1:
permute_group1_list.append(psamples)
for psamples in permute_group2:
permute_group2_list.append(psamples)
i=0; diff_list=[]
group_zdiff_means={}
sample_diff_zscores=[]
for psamples1 in permute_group1_list:
psamples2 = permute_group2_list[i] #this is the second group to compare to
x=0; permute_sample_exp_db={}
for sample in psamples1:
if distributionNull:
nsample = 'null1:'+string.split(sample,':')[1] ### reassign group ID
new_sampleID=nsample+str(x)
else: new_sampleID=sample+str(x)
try: permute_sample_exp_db[new_sampleID]=sample_exp_db[sample]
except Exception: print 'Error:', sample, new_sampleID, sample_exp_db[sample];sys.exit()
x+=1
for sample in psamples2:
if distributionNull:
nsample = 'null2:'+string.split(sample,':')[1] ### reassign group ID
new_sampleID=nsample+str(x)
else: new_sampleID=sample+str(x)
permute_sample_exp_db[new_sampleID]=sample_exp_db[sample]
x+=1
i+=1
new_tissue_exp_db={}
### Create a new reference from the permuted data
for sample in permute_sample_exp_db:
group = string.split(sample,':')[0]
try: new_tissue_exp_db[group].append(permute_sample_exp_db[sample])
except Exception: new_tissue_exp_db[group] = [permute_sample_exp_db[sample]]
for group in new_tissue_exp_db:
k = new_tissue_exp_db[group]
new_tissue_exp_db[group] = [Average(value) for value in zip(*k)] ### create new reference from all same group sample values
PearsonCorrelationAnalysis(permute_sample_exp_db,new_tissue_exp_db)
zscore_output_dir,tissue_scores = exportCorrelationResults()
tissue_comparison_scores={}
headers = list(tissue_scores['headers']); del tissue_scores['headers']
index=0; positive=0; positive_score_diff=0
sample_number = (len(headers)-1)
diff_z_list=[]
population1_denom=0; population1_pos=0; population2_pos=0; population2_denom=0
group_diff_z_scores={} ### Keep track of the differences between the z-scores between the two groups
while index < sample_number:
j=0
#ref1 = tissue_groups[0]+':'; ref2 = tissue_groups[-1]+':'
sample = headers[index+1]
diff_z = tissue_scores[tissue_groups[0]][index]-tissue_scores[tissue_groups[-1]][index]
diff_list.append([diff_z,sample])
group = string.split(sample,':')[0]
try: group_diff_z_scores[group].append(diff_z)
except Exception: group_diff_z_scores[group] = [diff_z]
sample_diff_zscores.append(diff_z)
index+=1
for group in group_diff_z_scores:
avg_group_zdiff = Average(group_diff_z_scores[group])
try: group_zdiff_means[group].append(avg_group_zdiff)
except Exception: group_zdiff_means[group] = [avg_group_zdiff]
diff_list.sort()
all_group_zdiffs=[]
for group in group_zdiff_means:
all_group_zdiffs += group_zdiff_means[group]
all_group_zdiffs.sort()
#print sample_diff_zscores;sys.exit()
#for i in diff_list: print i
#sys.exit()
i=1
groups.reverse()
group1,group2 = groups[:2]
group1+=':'; group2+=':'
scores=[]
#print max(diff_list), min(diff_list);sys.exit()
while i < len(diff_list):
g1_hits=0; g2_hits=0
list1 = diff_list[:i]
list2 = diff_list[i:]
for (z,s) in list1:
if group1 in s: g1_hits+=1
for (z,s) in list2:
if group2 in s: g2_hits+=1
sensitivity = float(g1_hits)/len(list1)
specificity = float(g2_hits)/len(list2)
accuracy = sensitivity+specificity
#accuracy = g1_hits+g2_hits
#print g1_hits, len(list1)
#print g2_hits, len(list2)
#print sensitivity, specificity;sys.exit()
z_cutoff = Average([list1[-1][0],list2[0][0]])
scores.append([accuracy,z_cutoff])
i+=1
scores.sort(); scores.reverse()
print scores[0][0],'\t',scores[0][1]
sample_exp_db = sample_exp_db_original
tissue_exp_db = tissue_exp_db_original
PearsonCorrelationAnalysis(sample_exp_db,tissue_exp_db)
sample_exp_db=[]; tissue_exp_db=[]
zscore_output_dir,tissue_scores = exportCorrelationResults()
return zscore_output_dir, tissue_scores, sampleHeaders
def returnTissueSpecificExpressionProfiles(sample_exp_db,tissue_exp_db,tissue_to_index):
tissue_exp_db_abreviated={}
sample_exp_db_abreviated={} ### This db is designed differently than the non-tissue specific (keyed by known tissues)
### Build the tissue specific expression profiles
for tissue in tissue_exp_db:
tissue_exp_db_abreviated[tissue] = []
for index in tissue_to_index[tissue]:
tissue_exp_db_abreviated[tissue].append(tissue_exp_db[tissue][index]) ### populate with just marker expression profiles
### Build the sample specific expression profiles
for sample in sample_exp_db:
sample_tissue_exp_db={}
sample_exp_db[sample]
for tissue in tissue_to_index:
sample_tissue_exp_db[tissue] = []
for index in tissue_to_index[tissue]:
sample_tissue_exp_db[tissue].append(sample_exp_db[sample][index])
sample_exp_db_abreviated[sample] = sample_tissue_exp_db
return sample_exp_db_abreviated, tissue_exp_db_abreviated
def replaceExpressionWithOrder(sample_exp_db):
for sample in sample_exp_db:
sample_exp_sorted=[]; i=0
for exp_val in sample_exp_db[sample]: sample_exp_sorted.append([exp_val,i]); i+=1
sample_exp_sorted.sort(); sample_exp_resort = []; order = 0
for (exp_val,i) in sample_exp_sorted: sample_exp_resort.append([i,order]); order+=1
sample_exp_resort.sort(); sample_exp_sorted=[] ### Order lowest expression to highest
for (i,o) in sample_exp_resort: sample_exp_sorted.append(o) ### The expression order replaces the expression, in the original order
sample_exp_db[sample] = sample_exp_sorted ### Replace exp with order
return sample_exp_db
def PearsonCorrelationAnalysis(sample_exp_db,tissue_exp_db):
#print "Beginning LineageProfiler analysis"
k=0
original_increment = int(len(tissue_exp_db)/15.00); increment = original_increment
p = 1 ### Default value if not calculated
for tissue in tissue_exp_db:
#print k,"of",len(tissue_exp_db),"classifier tissue/cell-types"
if k == increment: increment+=original_increment; #print '*',
k+=1
tissue_expression_list = tissue_exp_db[tissue]
for sample in sample_exp_db:
if correlate_to_tissue_specific == 'yes':
### Keyed by tissue specific sample profiles
sample_expression_list = sample_exp_db[sample][tissue] ### dictionary as the value for sample_exp_db[sample]
#print tissue, sample_expression_list
#print tissue_expression_list; sys.exit()
else: sample_expression_list = sample_exp_db[sample]
try:
### p-value is likely useful to report (not supreemly accurate but likely sufficient)
if len(tissue_expression_list) != len(sample_expression_list):
print len(tissue_expression_list), len(sample_expression_list)
print "Program Error!!! The length of the input expression list does not match the reference expression list (could indicate duplicate sample names)"
pass
rho,p = stats.pearsonr(tissue_expression_list,sample_expression_list)
if p == 1: ### When rho == nan... will break the program and result in miss-asignment of cells to clusters
rho = 0.0
#print rho,p
#rho,p = stats.spearmanr(tissue_expression_list,sample_expression_list)
try: pearson_list[sample].append(rho)
except Exception: pearson_list[sample] = [rho]
try: tissue_comparison_scores[tissue].append([rho,p,sample])
except Exception: tissue_comparison_scores[tissue] = [[rho,p,sample]]
except Exception:
### simple pure python implementation - no scipy required (not as fast though and no p-value)
try:
rho = pearson(tissue_expression_list,sample_expression_list); p=0
try: pearson_list[sample].append(rho)
except Exception: pearson_list[sample] = [rho]
try: tissue_comparison_scores[tissue].append([rho,p,sample])
except Exception: tissue_comparison_scores[tissue] = [[rho,p,sample]]
pearson_list.append(rho)
except Exception: None ### Occurs when an invalid string is present - ignore and move onto the next model
"""
from stats_scripts import salstat_stats
tst = salstat_stats.TwoSampleTests(tissue_expression_list,sample_expression_list)
pp,pr = tst.PearsonsCorrelation()
sp,sr = tst.SpearmansCorrelation()
print tissue, sample
if rho>.5: print [rho, pr, sr],[pp,sp];sys.exit()
if rho<.5: print [rho, pr, sr],[pp,sp];sys.exit()
"""
sample_exp_db=[]; tissue_exp_db=[]
#print 'Correlation analysis finished'
def pearson(array1,array2):
item = 0; sum_a = 0; sum_b = 0; sum_c = 0
while item < len(array1):
a = (array1[item] - Average(array1))*(array2[item] - Average(array2))
b = math.pow((array1[item] - Average(array1)),2)
c = math.pow((array2[item] - Average(array2)),2)
sum_a = sum_a + a
sum_b = sum_b + b
sum_c = sum_c + c
item = item + 1
r = sum_a/math.sqrt(sum_b*sum_c)
return r
def Median(array):
array.sort()
len_float = float(len(array))
len_int = int(len(array))
if (len_float/2) == (len_int/2):
try: median_val = avg([array[(len_int/2)-1],array[(len_int/2)]])
except IndexError: median_val = ''
else:
try: median_val = array[len_int/2]
except IndexError: median_val = ''
return median_val
def Average(array):
try: return sum(array)/len(array)
except Exception: return 0
def adjustPValues():
""" Can be applied to calculate an FDR p-value on the p-value reported by scipy.
Currently this method is not employed since the p-values are not sufficiently
stringent or appropriate for this type of analysis """
from stats_scripts import statistics
all_sample_data={}
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]:
all_sample_data[sample] = db = {} ### populate this dictionary and create sub-dictionaries
break
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]:
gs = statistics.GroupStats('','',p)
all_sample_data[sample][tissue] = gs
for sample in all_sample_data:
statistics.adjustPermuteStats(all_sample_data[sample])
for tissue in tissue_comparison_scores:
scores = []
for (r,p,sample) in tissue_comparison_scores[tissue]:
p = all_sample_data[sample][tissue].AdjP()
scores.append([r,p,sample])
tissue_comparison_scores[tissue] = scores
def stdev(array):
sum_dev = 0
try: x_bar = scipy.average(array)
except Exception: x_bar=Average(array)
n = float(len(array))
for x in array:
x = float(x)
sq_deviation = math.pow((x-x_bar),2)
sum_dev += sq_deviation
try:
s_sqr = (1.0/(n-1.0))*sum_dev #s squared is the variance
s = math.sqrt(s_sqr)
except Exception:
s = 'null'
return s
def replacePearsonPvalueWithZscore():
adjust_rho=False
all_sample_data={}
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]:
all_sample_data[sample] = [] ### populate this dictionary and create sub-dictionaries
break
for tissue in tissue_comparison_scores:
for (r,p,sample) in tissue_comparison_scores[tissue]:
if adjust_rho:
try: r = 0.5*math.log(((1+r)/(1-r)))
except Exception: print 'Error1:',tissue, sample, r, p; sys.exit()
all_sample_data[sample].append(r)
#print tissue, sample, r
#sample_stats={}
all_dataset_rho_values=[]
### Get average and standard deviation for all sample rho's
for sample in all_sample_data:
try:
all_sample_data[sample].sort() ### Sort, since when collapsing references, only the top two matter
all_dataset_rho_values+=all_sample_data[sample][-2:]
#try: avg=scipy.average(all_sample_data[sample])
#except Exception: avg=Average(all_sample_data[sample])
except Exception:
all_dataset_rho_values+=all_sample_data[sample]
#try: avg=scipy.average(all_sample_data[sample])
#except Exception: avg=Average(all_sample_data[sample])
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
st_dev=stdev(all_sample_data[sample])
#sample_stats[sample]=avg,st_dev
try: global_rho_avg = scipy.average(all_dataset_rho_values)
except Exception: global_rho_avg=Average(all_sample_data[sample])
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
global_rho_stdev = stdev(all_dataset_rho_values)
### Replace the p-value for each rho
for tissue in tissue_comparison_scores:
scores = []
for (r,p,sample) in tissue_comparison_scores[tissue]:
pearson_rho = r
if adjust_rho:
try: r = 0.5*math.log(((1+r)/(1-r)))
except Exception: print tissue, sample, r, p; sys.exit()
#u,s=sample_stats[sample]
#z = (r-u)/s
z = (r-global_rho_avg)/global_rho_stdev ### Instead of doing this for the sample background, do it relative to all analyzed samples
#z_alt = (r-global_rho_avg)/global_rho_stdev
scores.append([pearson_rho, r,z,sample])
#print sample, r, global_rho_avg, global_rho_stdev, z
tissue_comparison_scores[tissue] = scores
def exportCorrelationResults():
corr_output_file = string.replace(exp_output_file,'DATASET','LineageCorrelations')
corr_output_file = string.replace(corr_output_file,'.txt','-'+coding_type+'.txt')
if analysis_type == 'AltExon':
corr_output_file = string.replace(corr_output_file,coding_type,'AltExon')
filename = string.split(corr_output_file,delim)[-1][:-4]
#score_data = exportFile(corr_output_file)
zscore_output_dir = string.replace(corr_output_file,'.txt','-zscores.txt')
#probability_data = exportFile(zscore_output_dir)
#adjustPValues()
#sample_pearson_db = copy.deepcopy(tissue_comparison_scores) ### prior to pearson normalization
replacePearsonPvalueWithZscore()
### Make title row
headers=['Sample_name']
for tissue in tissue_comparison_scores:
for (pearson_rho, r,z,sample) in tissue_comparison_scores[tissue]: headers.append(sample)
break
title_row = string.join(headers,'\t')+'\n'
#score_data.write(title_row)
#if use_scipy: probability_data.write(title_row)
### Export correlation data
tissue_scores = {}; tissue_probabilities={}; tissue_score_list = [] ### store and rank tissues according to max(score)
tissue_scores2={}
for tissue in tissue_comparison_scores:
correlations=[]
probabilities=[]
for (pearson_rho, r,z,sample) in tissue_comparison_scores[tissue]:
correlations.append(pearson_rho) ### un-adjusted correlation
probabilities.append((z,pearson_rho))
tissue_score_list.append((max(correlations),tissue))
tissue_scores[tissue] = probabilities ### These are actually z-scores
tissue_scores2[tissue] = string.join(map(str,[tissue]+correlations),'\t')+'\n' ### export line
if use_scipy:
tissue_probabilities[tissue] = string.join(map(str,[tissue]+probabilities),'\t')+'\n'
tissue_score_list.sort()
tissue_score_list.reverse()
#for (score,tissue) in tissue_score_list: score_data.write(tissue_scores2[tissue])
#if use_scipy: probability_data.write(tissue_probabilities[tissue])
#score_data.close()
#if use_scipy: probability_data.close()
#print filename,'exported...'
tissue_scores['headers'] = headers
return zscore_output_dir, tissue_scores
def visualizeLineageZscores(zscore_output_dir,grouped_lineage_zscore_dir,graphic_links):
from visualization_scripts import clustering
### Perform hierarchical clustering on the LineageProfiler Zscores
graphic_links = clustering.runHCOnly(zscore_output_dir,graphic_links)
return graphic_links
def crossValidation(filename,setsToOutput=10,outputName=None):
""" Function for performing 2-level cross-validation. This entails randomly dividing the input set into 2/3rds
of the samples from each indicated biological group represented (equal prortion of disease and control each time)
calculating a mean reference for each gene in the 2/3rds set and then exporting the remaining 1/3rd of samples """
print 'Importing data to permute for 2-level cross-validation'
fn = filepath(filename)
dct_db={}; target_db={}; sample_list=[]; genes=[]
### Get the known group types (only works on two groups right now)
firstLine = True
group_list=[]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
headers = t[1:]
firstLine = False
for h in headers:
if ':' in h:
group = string.split(h,':')[0]
if group not in group_list:
group_list.append(group)
group_samples=collections.OrderedDict()
firstLine = True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine = False
headers = t[1:]
group1=[]; group2=[]
for h in headers:
group = string.split(h,':')[0]
try: group_samples[group].append(headers.index(h))
except Exception: group_samples[group] = [headers.index(h)]
else:
genes.append(t[0])
for sample in headers:
i = headers.index(sample)
try:
dct_db[sample].append(t[i+1])
except Exception:
dct_db[sample] = [t[i+1]]
permute_set = 1
#for i in dct_db: print i, dct_db[i]
#sys.exit()
inputTwoThirdsFiles=[]; inputOneThirdFiles=[]; referenceFiles=[]
while permute_set < (setsToOutput+1):
if setsToOutput == 1:
output_file = string.replace(filename,'.txt','_training.txt')
else:
output_file = string.replace(filename,'.txt','-'+str(permute_set)+'_2-3rds'+'.txt')
inputTwoThirdsFiles.append(output_file)
export_obj = export.ExportFile(output_file)
group_samples_two_thirds={}
if setsToOutput == 1:
ref_file = string.replace(filename,'.txt','-reference.txt')
else:
ref_file = string.replace(filename,'.txt','-'+str(permute_set)+'-reference'+'.txt')
referenceFiles.append(ref_file)
ref_obj = export.ExportFile(ref_file)
for group in group_list:
group_names = group_samples[group]
two_thirds = int((2.0/3.0)*len(group_names)) ### e.g., AR
group_random = random.sample(group_names,two_thirds)
group_samples_two_thirds[group] = group_random
permute_db={}; group_permute_db={}
for group in group_list:
for i in group_samples_two_thirds[group]:
s = headers[i]
values = dct_db[s]
permute_db[s] = values
try:
db = group_permute_db[group]
db[s] = values
except Exception:
db = {}
db[s] = values
group_permute_db[group] = db
### Get the 1/3rd remain samples for export to a separate file
if setsToOutput == 1:
outputR_file = string.replace(filename,'.txt','_test.txt')
else:
outputR_file = string.replace(filename,'.txt','-'+str(permute_set)+'_1-3rds'+'.txt')
inputOneThirdFiles.append(outputR_file)
exportR_obj = export.ExportFile(outputR_file)
remaining_headers=[]; remaining_sample_db={}
for s in dct_db:
if s not in permute_db:
remaining_sample_db[s] = dct_db[s]
remaining_headers.append(s)
exportR_obj.write(string.join(['UID']+remaining_headers,'\t')+'\n')
for gene in genes:
i = genes.index(gene)
values = [gene]
for s in remaining_headers:
values.append(remaining_sample_db[s][i])
values = string.join(values,'\t')+'\n'
exportR_obj.write(values)
exportR_obj.close()
### Export samples and references fro the 2/3rds set
updated_headers = []
for s in permute_db:
updated_headers.append(s)
export_obj.write(string.join(['UID']+updated_headers,'\t')+'\n')
group_headers={}
for group in group_list:
for s in group_permute_db[group]:
try: group_headers[group].append(s)
except Exception: group_headers[group] = [s]
for gene in genes:
i = genes.index(gene)
values = [gene]
for s in updated_headers:
values.append(permute_db[s][i])
values = string.join(values,'\t')+'\n'
export_obj.write(values)
export_obj.close()
ref_obj.write(string.join(['UID']+group_list,'\t')+'\n')
for gene in genes:
i = genes.index(gene)
group_avgs=[]
for group in group_list:
group_values = []
gdb = group_permute_db[group]
for s in gdb:
try: group_values.append(float(gdb[s][i]))
except Exception: pass ### Exclude columns with NA from mean calculation
group_avg = str(Average(group_values))
group_avgs.append(group_avg)
values = string.join([gene]+group_avgs,'\t')+'\n'
ref_obj.write(values)
ref_obj.close()
permute_set+=1
return inputTwoThirdsFiles, inputOneThirdFiles, referenceFiles
def crossValidationAnalysis(species,platform,exp_input,exp_output,codingtype,compendium_platform,
modelSize,geneModels,permute, useMulti, finalNumberSetsToOutput):
inputTwoThirdsFiles, inputOneThirdFiles, referenceFiles = crossValidation(exp_input,setsToOutput=1,outputName=None) ### This is the set to validate
#inputTwoThirdsFiles = '/Users/saljh8/Desktop/Sarwal-New/UCRM_bx-transposed-train8.txt'
#inputOneThirdFiles = '/Users/saljh8/Desktop/Sarwal-New/UCRM_bx-transposed-test_most.txt'
#referenceFiles = ['/Users/saljh8/Desktop/Sarwal-New/manual-ref.txt']
inputTwoThirdsFile = inputTwoThirdsFiles[0]; inputOneThirdFile = inputOneThirdFiles[0]; reference = referenceFiles[0]
exp_input = string.replace(exp_input,'.txt','_training.txt')
inputTwoThirdsFiles, inputOneThirdFiles, referenceFiles = crossValidation(exp_input,setsToOutput=finalNumberSetsToOutput) ### This is the set to validate
#referenceFiles = ['/Users/saljh8/Desktop/Sarwal-New/manual-ref.txt']*len(referenceFiles)
index = 0
comparison_db={} ### store all of the 1/3rd validation results
all_models={}
### Iterate through each 2/3rds set -> discover the best models over 80% -> evalute in 1/3rd set -> store those above 80%
for exp_in in inputTwoThirdsFiles:
exp_out = exp_in
customMarkers = referenceFiles[index]
top_hit_db = runLineageProfiler(species,platform,exp_in,exp_out,codingtype,compendium_platform,modelSize='optimize',
customMarkers=customMarkers,geneModels=geneModels,permute=permute,useMulti=useMulti)
model_file = exportModels(exp_in,top_hit_db)
exp_in = string.replace(exp_in,'_2-3rds','_1-3rds')
try:
top_hit_db = runLineageProfiler(species,platform,exp_in,exp_out,codingtype,compendium_platform,
customMarkers=customMarkers,geneModels=model_file,permute=permute)
except Exception: top_hit_db={}
#comparison_db[exp_in] = top_hit_db
all_models.update(top_hit_db)
index+=1
### Create a combined models file
exp_in = string.replace(exp_input,'.txt','-combinedModels.txt')
model_file = exportModels(exp_in,all_models)
index = 0
### Re-analyze all of the 2/3rd and 1/3rd files with these
for exp_in in inputTwoThirdsFiles:
customMarkers = referenceFiles[index]
top_hit_db = runLineageProfiler(species,platform,exp_in,exp_out,codingtype,compendium_platform,modelSize=modelSize,
customMarkers=customMarkers,geneModels=model_file,permute=permute)
comparison_db[exp_in] = top_hit_db
exp_in = string.replace(exp_in,'_2-3rds','_1-3rds')
top_hit_db = runLineageProfiler(species,platform,exp_in,exp_out,codingtype,compendium_platform,modelSize=modelSize,
customMarkers=customMarkers,geneModels=model_file,permute=permute)
comparison_db[exp_in] = top_hit_db
index+=1
score_db={}
for one_third_input_name in comparison_db:
top_hit_db = comparison_db[one_third_input_name]
for model in top_hit_db:
score = top_hit_db[model]
try: score_db[model].append(float(score))
except Exception: score_db[model] = [float(score)]
score_ranked_list=[]
for model in score_db:
avg_score = Average(score_db[model])
min_score = min(score_db[model])
score = avg_score*min_score
score_ranked_list.append([score,avg_score,min_score,model])
score_ranked_list.sort()
score_ranked_list.reverse()
for s in score_ranked_list:
print s
def exportModels(exp_in,top_hit_db):
model_file = string.replace(exp_in,'.txt','_topModels.txt')
export_obj = export.ExportFile(model_file)
for m in top_hit_db:
if float(top_hit_db[m])>70:
m = list(m)
m = string.join(m,',')+'\n'
export_obj.write(m)
export_obj.close()
return model_file
def modelScores(results_dir):
score_db={}
files = unique.read_directory(results_dir+'/')
for file in files:
firstLine = True
if '-ModelScore' in file:
filename = results_dir+'/'+file
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if firstLine == True: firstLine = False
else:
t = string.split(data,'\t')
model=t[-2]
score = t[0]
sensitivity = float(t[1])/float(t[2])*100
specificity = float(t[3])/float(t[4])*100
score = sensitivity+specificity
try: score_db[model].append(float(score))
except Exception: score_db[model] = [float(score)]
score_ranked_list=[]
for model in score_db:
avg_score = Average(score_db[model])
min_score = min(score_db[model])
stdev_min = stdev(score_db[model])
l = (avg_score*min_score)
score_ranked_list.append([l,avg_score,min_score,stdev_min,model,score_db[model]])
score_ranked_list.sort()
score_ranked_list.reverse()
x = 100; y=1
output_dir = results_dir+'/overview.txt'
oe = export.ExportFile(output_dir)
for s in score_ranked_list:
y+=1
s = map(lambda x: str(x), s)
#if y==x: sys.exit()
#print s
oe.write(string.join(s,'\t')+'\n')
oe.close()
#sys.exit()
def allPairwiseSampleCorrelation(fn):
import numpy
firstLine = True
group_list=[]
control_set={}
exp_set={}
all_samples_names=[]
all_sample_values=[]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine=False
else:
values = map(float,t[1:])
group = string.split(t[0],':')[0]
if 'no' in group:
#control_set[t[0]] = []
s = 0 ### control
else:
#exp_set[t[0]] = []
s = 1 ### experiment
all_samples_names.append((s,t[0]))
all_sample_values.append(values)
all_sample_values = numpy.array(all_sample_values)
### calculate all pairwise comparisons and store exp and control correlations in two separate dictionaries
D1 = numpy.ma.corrcoef(all_sample_values)
i=0
for score_ls in D1:
scores = []
s,sample = all_samples_names[i]
if s==0: control_set[sample] = score_ls
if s==1: exp_set[sample] = score_ls
i+=1
k=0
#for rho in score_ls: sample2 = all_samples_names[k]; k+=1
def subtract(values):
return values[0]-values[1]
from stats_scripts import statistics
results=[]
n = len(all_samples_names)
e = len(exp_set)
c = len(control_set)
for cs in control_set:
ccor = control_set[cs] ### all sample correlations for the control
for es in exp_set:
ecor = exp_set[es] ### all sample correlations for the exp
diff_corr = [subtract(value) for value in zip(*[ecor,ccor])]
k=0
score=0; sensitivity=0; specificity=0
for diff in diff_corr:
s,sample = all_samples_names[k]; k+=1
if s==1 and diff>0: score+=1; sensitivity+=1
elif s==0 and diff<0: score+=1; specificity+=1
sens = float(sensitivity)/e
spec = float(specificity)/c
accuracy = float(score)/n
avg_accuracy = statistics.avg([sens,spec])
results.append([avg_accuracy,accuracy,sens,spec,es,cs])
results.sort()
results.reverse()
for i in results[:20]:
#if i[1]>.70: print i
print i
def harmonizeClassifiedSamples(species,reference_exp_file, query_exp_file, classification_file,fl=None):
"""
The goal of this function is to take LineageProfilerIterative classified samples to a reference matrix,
combine the reference matrix and the query matrix at the gene symbol level, retain the original reference
column and row orders, then re-order the query samples within the context of the reference samples the
correlations were derived from. In doing so, the harmonization occurs by attempting to map the expression
values within a common numerical format to minimize dataset specific effects (folds, non-log expression).
The function culminates in a heatmap showing the genes and re-ordered samples in thier listed orders.
Outlier samples with low correlations will ultimately need to be represented outside of the reference
sample continuum. The function attempts to propagate sample group labels from the reference and query sets
(if avaialble, in the sample headers or in a groups file), and indicate original gene and column clusters
if available. We call this approach cellHarmony.
"""
"""
Alternative description: To represent new cells/samples within the continuum of established scRNA-Seq profiles
we have developed a robust and fast data harmonization function. This data harmonization approach applies a
k-nearest neighbor classification approach to order submitted cells along the continuum of reference gene expression
profiles, without alternating the order of the reference cells and cluster groups in original reference matrix.
The established group labels are in effect propagated to new queried samples. The final data is represented in an
intuitive queriable heatmap and dimensionality reduction scatter plot (t-SNE, PCA). To harmonize the input exp. data,
the input file formats are emperically determined (log, non-log, log-fold) and standardized to log2 expression.
Submitted FASTQ files to AltAnalyze will be optionally pseudoaligned and normalized to maximize compatibility with the
existing reference datasets. Alternative classification algoriths and options for submission of new reference sets will
be supported. Issues may arrise in the expression harmonization step, however, profiles are already biased towards genes
with cell population restricted expression, largely abrogating this problem.
"""
### Set the user or default options for cellHarmony Differential Expression Analyses
try: pearsonThreshold=fl.PearsonThreshold()
except: pearsonThreshold = 0.1
try: peformDiffExpAnalysis=fl.PeformDiffExpAnalysis()
except: peformDiffExpAnalysis = True
try: use_adjusted_pval=fl.UseAdjPvalue()
except: use_adjusted_pval = False
try: pvalThreshold=float(fl.PvalThreshold())
except: pvalThreshold = 0.05
try: FoldCutoff = fl.FoldCutoff()
except: FoldCutoff = 1.5
customLabels = None
try:
if len(fl.Labels())>0:
customLabels = fl.Labels()
except: pass
### Output the alignment results and perform the differential expression analysis
output_file,query_output_file,folds_file,DEGs_combined = importAndCombineExpressionFiles(species,reference_exp_file,
query_exp_file,classification_file,pearsonThreshold=pearsonThreshold,peformDiffExpAnalysis=peformDiffExpAnalysis,
pvalThreshold=pvalThreshold,fold_cutoff=FoldCutoff,use_adjusted_pval=use_adjusted_pval,customLabels=customLabels)
output_dir = export.findParentDir(output_file)
if len(folds_file)<1:
""" If performDiffExp==False, see if a prior folds file exists """
folds_file = string.replace(output_file,'-ReOrdered','-AllCells-folds')
### Output the cellHarmony heatmaps
from visualization_scripts import clustering
row_method = None; row_metric = 'cosine'; column_method = None; column_metric = 'euclidean'; color_gradient = 'yellow_black_blue'
transpose = False; Normalize='median'
if runningCommandLine:
display = False
else:
display = True
display = False
print 'Exporting cellHarmony heatmaps...'
heatmaps_dir = output_dir+'/heatmaps/'
try: os.mkdir(heatmaps_dir)
except: pass
try:
graphics = clustering.runHCexplicit(query_output_file, [], row_method, row_metric, column_method,
column_metric, color_gradient, transpose, Normalize=Normalize, contrast=5, display=display)
plot = graphics[-1][-1][:-4]+'.pdf'
file = graphics[-1][-1][:-4]+'.txt'
shutil.copy(plot,output_dir+'/heatmaps/heatmap-query-aligned.pdf')
shutil.copy(file,output_dir+'/heatmaps/heatmap-query-aligned.txt')
graphics = clustering.runHCexplicit(output_file, [], row_method, row_metric, column_method,
column_metric, color_gradient, transpose, Normalize=Normalize, contrast=5, display=display)
plot = graphics[-1][-1][:-4]+'.pdf'
file = graphics[-1][-1][:-4]+'.txt'
shutil.copy(plot,output_dir+'/heatmaps/heatmap-all-cells-combined.pdf')
shutil.copy(file,output_dir+'/heatmaps/heatmap-all-cells-combined.txt')
except:
print traceback.format_exc()
zscore = True
graphics=[]
transpose='no'
try: fl.setSpecies(species); fl.setVendor("3'array")
except:
import UI
fl = UI.ExpressionFileLocationData(folds_file,'','','')
fl.setSpecies(species); fl.setVendor("3'array")
fl.setOutputDir(output_dir)
try: platform=platform
except:
platform = 'RNASeq'
### Build-UMAP plot
import UI
import warnings
warnings.filterwarnings('ignore')
try:
try: os.mkdir(fl.OutputDir()+'/UMAP-plots')
except: pass
""" Output UMAP combined plot colored by reference and query cell identity """
plot = UI.performPCA(output_file, 'no', 'UMAP', False, None, plotType='2D',
display=False, geneSetName=None, species=species, zscore=False, reimportModelScores=False,
separateGenePlots=False, returnImageLoc=True)
plot = plot[-1][-1][:-4]+'.pdf'
shutil.copy(plot,fl.OutputDir()+'/UMAP-plots/UMAP-query-vs-ref.pdf')
""" Output UMAP combined plot colored by cell tates """
plot = UI.performPCA(output_file, 'no', 'UMAP', False, None, plotType='2D',
display=False, geneSetName=None, species='Mm', zscore=False, reimportModelScores=True,
separateGenePlots=False, returnImageLoc=True, forceClusters=True)
plot = plot[-1][-1][:-4]+'.pdf'
shutil.copy(plot,fl.OutputDir()+'/UMAP-plots/UMAP-query-vs-ref-clusters.pdf')
""" Output individual UMAP plots colored by cell tates """
groups_file = string.replace(output_file,'exp.','groups.')
plots = UI.performPCA(output_file, 'no', 'UMAP', False, None, plotType='2D',
display=False, geneSetName=None, species='Mm', zscore=False, reimportModelScores=True,
separateGenePlots=False, returnImageLoc=True, forceClusters=True, maskGroups=groups_file)
for plot in plots:
plot = plot[-1][:-4]+'.pdf'
if '-cellHarmony-Reference-' in plot:
shutil.copy(plot,fl.OutputDir()+'/UMAP-plots/UMAP-ref-clusters.pdf')
else:
shutil.copy(plot,fl.OutputDir()+'/UMAP-plots/UMAP-query-clusters.pdf')
except:
try:
print traceback.format_exc()
print 'UMAP error encountered (dependency not met), trying t-SNE'
UI.performPCA(output_file, 'no', 't-SNE', False, None, plotType='2D',
display=False, geneSetName=None, species=species, zscore=True, reimportModelScores=False,
separateGenePlots=False, returnImageLoc=True)
except: pass
useMarkerFinder=False
### Run MarkerFinder
if len(DEGs_combined) and useMarkerFinder:
exportCombinedMarkerFinderResults(species,platform,fl,folds_file,DEGs_combined)
elif len(DEGs_combined):
exportPvalueRankedGenes(species,platform,fl,folds_file,DEGs_combined)
### Cleanup directory
cleanupOutputFolder(output_dir)
def cleanupOutputFolder(output_dir):
""" Reorganizes cellHarmony output folder to be easier to navigate """
other_files_dir = output_dir+'/OtherFiles/'
def createFolder(folder):
try: os.mkdir(folder)
except: pass
createFolder(other_files_dir)
createFolder(other_files_dir+'DataPlots/')
createFolder(other_files_dir+'GO-Elite/')
createFolder(other_files_dir+'PValues/')
dir_list = unique.read_directory(output_dir)
for file in dir_list:
if 'exp.' in file or 'groups.' in file or 'comps.' in file:
shutil.move(output_dir+file,other_files_dir+file)
def moveFolder(folder):
try:
dir_list = unique.read_directory(output_dir+folder)
for file in dir_list:
shutil.move(output_dir+folder+file,other_files_dir+folder+file)
export.deleteFolder(output_dir+folder)
except:
pass
moveFolder('DataPlots/')
moveFolder('GO-Elite/')
moveFolder('PValues/')
def exportPvalueRankedGenes(species,platform,fl,folds_file,DEGs_combined):
""" Produce a hierarchically ordered heatmap of differential expression differences
across cell-states to provide a hollistic representation of impacted genes """
""" Write out headers for OrganizedDifferential results """
export_file = fl.OutputDir()+'/OrganizedDifferentials.txt'
export_object = export.ExportFile(export_file)
""" Import the folds and headers for all genes/comparisons """
headers,matrix = getDataMatrix(folds_file)
display_headers=[]
cell_states=[]
for h in headers:
h = string.replace(h,'-fold','')
display_headers.append(h+':'+h)
cell_states.append(h)
export_object.write(string.join(['UID']+display_headers,'\t')+'\n')
top_comparison_genes={}
comparisons={}
for gene in DEGs_combined:
pval,direction,comparison,uid = DEGs_combined[gene]
### Group by comparison and remove ".txt" suffix
comparison = string.split(comparison,'_vs_')[0]
comparison = string.replace(comparison,'GE.','')
comparison = string.replace(comparison,'PSI.','')
try: top_comparison_genes[comparison+"--"+direction].append((pval,gene))
except: top_comparison_genes[comparison+"--"+direction] = [(pval,gene)]
comparisons[comparison]=[]
""" Augment the ordered cell-state header list with the other broader comparisons """
cluster_comparisons=[]
for comparison in comparisons:
if comparison not in cell_states:
if '-Query' not in comparison:
bulk_comparison = comparison
else:
first_group = string.split(comparison,'__')[0]
index = cell_states.index(first_group)
cluster_comparisons.append([index,comparison])
cluster_comparisons.sort()
cell_states.reverse()
for (rank,comparison) in cluster_comparisons:
cell_states.append(comparison)
try: cell_states.append(bulk_comparison)
except: pass ### If no bulk differences are ranked better than cell-state differences
cell_states.reverse()
for h in cell_states:
for direction in ('positive','negative'):
signature = h+'--'+direction
if signature in top_comparison_genes: ### Not all comparisons will yield significant genes
top_comparison_genes[signature].sort()
for (pval,gene) in top_comparison_genes[signature]:
if gene in matrix:
export_object.write(string.join([signature+':'+gene]+map(str,matrix[gene]),'\t')+'\n')
export_object.close()
from visualization_scripts import clustering
row_method = None; row_metric = 'cosine'; column_method = None; column_metric = 'euclidean'; color_gradient = 'yellow_black_blue'
transpose = False; Normalize=False; display = False
import UI
gsp = UI.GeneSelectionParameters(species,"RNASeq","RNASeq")
gsp.setPathwaySelect('None Selected')
gsp.setGeneSelection('')
gsp.setOntologyID('')
gsp.setGeneSet('None Selected')
gsp.setJustShowTheseIDs('')
gsp.setTranspose(False)
gsp.setNormalize(False)
gsp.setGeneSelection('')
if species == 'Mm' or species == 'Hs':
gsp.setClusterGOElite('PathwayCommons') #GeneOntology
else:
gsp.setClusterGOElite('GeneOntology')
graphics = clustering.runHCexplicit(export_file, [], row_method, row_metric,
column_method, column_metric, color_gradient, gsp, Normalize=Normalize,
contrast=3, display=display)
shutil.copy(graphics[-1][-1][:-4]+'.pdf',export_file[:-4]+'.pdf')
from import_scripts import OBO_import; import ExpressionBuilder; import RNASeq
gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol_db)
try:
TFs = RNASeq.importGeneSets('Biotypes',filterType='transcription regulator',geneAnnotations=gene_to_symbol_db,speciesName = species)
gsp.setJustShowTheseIDs(string.join(TFs.keys(),' '))
gsp.setClusterGOElite('MergedTFTargets') #GeneOntology
graphics = clustering.runHCexplicit(export_file, [], row_method, row_metric,
column_method, column_metric, color_gradient, gsp, Normalize=Normalize,
contrast=3, display=display)
shutil.copy(graphics[-1][-1][:-4]+'.pdf',export_file[:-4]+'-TFs.pdf')
except ZeroDivisionError:
pass
def editFoldsGroups(folds_file):
folds_copy = string.replace(folds_file,'-AllCells-folds','-AllCells-folds2')
original_groups_file = string.replace(folds_file,'exp.','groups.')
output_groups_file = string.replace(folds_copy,'exp.','groups.')
eo = export.ExportFile(output_groups_file)
eo.write('Null\t1\tNull\n')
for line in open(original_groups_file,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
eo.write(t[0]+'\t2\tall\n')
eo.close()
eo = export.ExportFile(string.replace(output_groups_file,'groups.','comps.'))
eo.close()
eo = export.ExportFile(folds_copy)
header=True
for line in open(folds_file,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
header=False
eo.write(string.join([t[0],'Null']+t[1:],'\t')+'\n')
else:
eo.write(string.join([t[0],'0']+t[1:],'\t')+'\n')
eo.close()
return folds_copy
def exportCombinedMarkerFinderResults(species,platform,fl,folds_file,DEGs_combined):
""" Use MarkerFinder to define the major gene expression associated patterns """
fl.setRPKMThreshold(0.00)
fl.setCorrelationDirection('up')
compendiumType = 'protein_coding'
genesToReport = 100000
correlateAll = True
import markerFinder
directories=[]
markerFinder.analyzeData(folds_file,species,platform,compendiumType,geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl,logTransform=False)
export.deleteFolder(fl.OutputDir()+'/MarkerFinder-positive')
os.rename(fl.OutputDir()+'/MarkerFinder',fl.OutputDir()+'/MarkerFinder-positive')
directories.append(fl.OutputDir()+'/MarkerFinder-positive')
fl.setCorrelationDirection('down')
markerFinder.analyzeData(folds_file,species,platform,compendiumType,geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl,logTransform=False)
export.deleteFolder(fl.OutputDir()+'/MarkerFinder-negative')
os.rename(fl.OutputDir()+'/MarkerFinder',fl.OutputDir()+'/MarkerFinder-negative')
directories.append(fl.OutputDir()+'/MarkerFinder-negative')
#try: shutil.copyfile(src,dst)
#except: pass
fl.setCorrelationDirection('up')
folds_copy = editFoldsGroups(folds_file)
markerFinder.analyzeData(folds_copy,species,platform,compendiumType,geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl,logTransform=False)
export.deleteFolder(fl.OutputDir()+'/MarkerFinder-all-up')
os.rename(fl.OutputDir()+'/MarkerFinder',fl.OutputDir()+'/MarkerFinder-all-up')
directories.append(fl.OutputDir()+'/MarkerFinder-all-up')
fl.setCorrelationDirection('down')
markerFinder.analyzeData(folds_copy,species,platform,compendiumType,geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl,logTransform=False)
export.deleteFolder(fl.OutputDir()+'/MarkerFinder-all-down')
os.rename(fl.OutputDir()+'/MarkerFinder',fl.OutputDir()+'/MarkerFinder-all-down')
directories.append(fl.OutputDir()+'/MarkerFinder-all-down')
marker_db=collections.OrderedDict()
def importMarkerFinderResults(input_file,direction):
header_row=True
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
data = string.replace(data,'"','')
geneID, symbol, rho, p, cell_state = string.split(data,'\t')
if header_row:
header_row=False
else:
rho = float(rho)
if geneID in marker_db:
### Positive marker genes
alt_cell_state,alt_direction,alt_rho = marker_db[geneID]
if rho>alt_rho:
marker_db[geneID]=cell_state, direction, rho
elif geneID in DEGs_combined:
marker_db[geneID]=cell_state, direction, rho
for file in directories:
file+='/AllGenes_correlations-ReplicateBased.txt'
if 'up' in file:
direction = 'postive'
else:
direction = 'negative'
importMarkerFinderResults(file,direction)
ordered_markers={}
for gene in marker_db:
cell_state, direction, rho = marker_db[gene]
try: ordered_markers[cell_state+'_'+direction].append([direction, rho, gene])
except Exception: ordered_markers[cell_state+'_'+direction]= [[direction, rho, gene]]
for cell_state in ordered_markers:
ordered_markers[cell_state].sort()
ordered_markers[cell_state].reverse()
headers,matrix = getDataMatrix(folds_file)
export_file = folds_file[:-4]+'-MarkerFinder.txt'
export_object = export.ExportFile(export_file)
headers2=[]
for h in headers:
headers2.append(h+':'+h)
export_object.write(string.join(['UID']+headers2,'\t')+'\n')
for h in headers:
h2 = h+'_positive'
if h2 in ordered_markers:
for (direction, rho, gene) in ordered_markers[h2]:
export_object.write(string.join([h2+':'+gene]+map(str,matrix[gene]),'\t')+'\n')
h2 = h+'_negative'
if h2 in ordered_markers:
for (direction, rho, gene) in ordered_markers[h2]:
export_object.write(string.join([h2+':'+gene]+map(str,matrix[gene]),'\t')+'\n')
export_object.close()
from visualization_scripts import clustering
row_method = None; row_metric = 'cosine'; column_method = None; column_metric = 'euclidean'; color_gradient = 'yellow_black_blue'
transpose = False; Normalize=False; display = False
import UI
gsp = UI.GeneSelectionParameters(species,"RNASeq","RNASeq")
gsp.setPathwaySelect('None Selected')
gsp.setGeneSelection('')
gsp.setOntologyID('')
gsp.setGeneSet('None Selected')
gsp.setJustShowTheseIDs('')
gsp.setTranspose(False)
gsp.setNormalize(False)
gsp.setGeneSelection('')
gsp.setClusterGOElite('GeneOntology')
graphics = clustering.runHCexplicit(export_file, [], row_method, row_metric,
column_method, column_metric, color_gradient, gsp, Normalize=Normalize,
contrast=3, display=display)
class ClassificationData:
def __init__(self,sample,score,assigned_class):
self.sample = sample; self.score = score; self.assigned_class = assigned_class
def Sample(self): return self.sample
def Score(self): return self.score
def AssignedClass(self): return self.assigned_class
def importAndCombineExpressionFiles(species,reference_exp_file,query_exp_file,classification_file,platform='RNASeq',pearsonThreshold=0.1,
peformDiffExpAnalysis=True, pvalThreshold=0.05,fold_cutoff=1.5, use_adjusted_pval=False, customLabels = None):
"""Harmonize the numerical types and feature IDs of the input files, then combine """
from visualization_scripts import clustering
original_reference_exp_file = reference_exp_file
reference_exp_file_alt = string.replace(reference_exp_file,'-centroid.txt','.txt')
status = verifyFile(reference_exp_file_alt)
if status==True:
reference_exp_file = reference_exp_file_alt
includeAllReferences=True
else:
includeAllReferences=False
ref_filename = export.findFilename(reference_exp_file)
ordered_ref_cells = getGroupsFromExpFile(reference_exp_file) ### get the original reference heatmap cell order with clusters
query_filename = export.findFilename(query_exp_file)
root_dir = string.replace(export.findParentDir(query_exp_file),'ExpressionInput','')+'/cellHarmony/'
output_dir = ref_filename[:-4]+'__'+query_filename ### combine the filenames and query path
output_dir = root_dir+'/exp.'+string.replace(output_dir,'exp.','')
output_dir =string.replace(output_dir,'-OutliersRemoved','')
groups_dir = string.replace(output_dir,'exp.','groups.')
ref_exp_db,ref_headers,ref_col_clusters,cluster_format_reference = importExpressionFile(reference_exp_file,customLabels=customLabels)
cluster_results = clustering.remoteImportData(query_exp_file,geneFilter=ref_exp_db)
if len(cluster_results[0])>0: filterIDs = ref_exp_db
else: filterIDs = False
query_exp_db,query_headers,query_col_clusters,cluster_format_query = importExpressionFile(query_exp_file,ignoreClusters=True,filterIDs=False)
ref_filename_clean = string.replace(ref_filename,'exp.','')
ref_filename_clean = string.replace(ref_filename_clean,'.txt','')
ref_filename_clean = 'cellHarmony-Reference'
query_filename_clean = string.replace(query_filename,'exp.','')
query_filename_clean = string.replace(query_filename_clean,'.txt','')
### Define Reference groups and import prior reference file
groups_file = groups_dir[:-4]+'-AllCells.txt'
groups_file = string.replace(groups_file,'OutliersRemoved-','')
if includeAllReferences:
groups_file = string.replace(groups_file,'-centroid__','__')
comps_file = string.replace(groups_file,'groups.','comps.')
groups_reference_file = string.replace(groups_file,'-AllCells.txt','-Reference.txt')
""" If reference labels provided, use these instead of cluster labels """
if customLabels!=None:
groups_reference_file = customLabels
try:
import ExpressionBuilder ### If using centroids for classification, merge with original reference groups
sample_group_ref_db = ExpressionBuilder.simplerGroupImport(groups_reference_file)
groups_to_reference_cells = {}
sample_group_ref_db2=collections.OrderedDict()
if '-centroid' in original_reference_exp_file:
for cell in sample_group_ref_db:
group = sample_group_ref_db[cell]
if cell in ordered_ref_cells:
try: groups_to_reference_cells[group].append(cell)
except: groups_to_reference_cells[group]=[cell]
else: ### Have to add .Reference to the name to make valid
try: groups_to_reference_cells[group].append(cell+'.Reference')
except: groups_to_reference_cells[group]=[cell+'.Reference']
sample_group_ref_db2[cell+'.Reference'] = group
else:
for cell in sample_group_ref_db:
group = sample_group_ref_db[cell]
if cell not in ordered_ref_cells:
try: groups_to_reference_cells[group].append(cell+'.Reference')
except: groups_to_reference_cells[group]=[cell+'.Reference']
sample_group_ref_db2[cell+'.Reference'] = group
if len(sample_group_ref_db2)>0:
sample_group_ref_db = sample_group_ref_db2
except:
print traceback.format_exc()
sample_group_ref_db={}
groups_to_reference_cells={}
column_clusters=[]
final_clusters=[]
numeric_cluster=True
comps=[]
### If using all cells as a reference, rather than centroid, then len(ref_col_clusters)==len(sample_group_ref_db)
if len(sample_group_ref_db)>len(ref_col_clusters):
for sample in ordered_ref_cells:
column_clusters.append(sample_group_ref_db[sample]) ### this is an ordered dictionary
try: final_clusters.append([ref_filename_clean,int(float(sample_group_ref_db[sample])),sample]) ### used later for creating the apples-to-apples group file
except Exception:
final_clusters.append([ref_filename_clean,sample_group_ref_db[sample],sample])
numeric_cluster=False
elif len(ref_col_clusters)>0:
for sample in ref_col_clusters:
column_clusters.append(ref_col_clusters[sample]) ### this is an ordered dictionary
try: final_clusters.append([ref_filename_clean,int(float(ref_col_clusters[sample])),sample]) ### used later for creating the apples-to-apples group file
except Exception:
final_clusters.append([ref_filename_clean,ref_col_clusters[sample],sample])
numeric_cluster=False
""" Store an alternative reference for each header """
### In case a ":" is in one header but not in another, create an alternative reference set
alt_ref_headers = []
for sample in ref_headers:
if ':' in sample:
alt_sample_id = string.split(sample,':')[1]
alt_ref_headers.append(alt_sample_id)
if sample in ref_col_clusters: ### If clusters assigned in original file
cluster = ref_col_clusters[sample]
ref_col_clusters[alt_sample_id]=cluster ### append to the original
""" Store alternative query sample names with groups added """
original_sampleID_translation={}
for sample in query_headers:
if ':' in sample:
original_sampleID = string.split(sample,':')[1]
original_sampleID_translation[original_sampleID] = sample
""" Import the Classification data """
input_dir = output_dir
sample_classes={}
new_headers=[]
query_header_proppegated_clusters={}
firstLine = True
exclude=[]
for line in open(classification_file,'rU').xreadlines():
data = line.rstrip()
data = string.replace(data,'"','')
values = string.split(data,'\t')
if firstLine:
firstLine=False
header_row = values
try: score_index = header_row.index('Max-Rho')
except: score_index = header_row.index('Correlation')
try: class_index = header_row.index('CentroidLabel')
except:
try: class_index = header_row.index('Predicted Class')
except: class_index = header_row.index('Ref Barcode')
else:
sample = values[0]
score = float(values[score_index])
assigned_class = values[class_index]
if sample in original_sampleID_translation:
sample = original_sampleID_translation[sample]
### groups_to_reference_cells is from the labels file - will have the labels as the assigned_class
if len(groups_to_reference_cells)>0: ### Hence, centroids - replace centroids with individual reference cells
try:
assigned_class = groups_to_reference_cells[assigned_class][-1]
except:
print assigned_class, 'not found in the groups_to_reference_cells database'
"""
print [assigned_class]
for ac in groups_to_reference_cells: print [ac]
"""
cd = ClassificationData(sample,score,assigned_class)
try: sample_classes[assigned_class].append([score,cd])
except Exception: sample_classes[assigned_class] = [[score,cd]]
query_header_proppegated_clusters[sample]=assigned_class
if score<pearsonThreshold: ### Minimum allowed correlation threshold
exclude.append(sample)
print len(exclude), 'cells excluded due to correlation below the indicated threshold'
""" Assign a cluster label to the query sample if applicable """
query_clusters=[]
classified_samples={}
if numeric_cluster: prefix = 'c'
else: prefix = ''
selected_query_headers=[]
for sample in query_headers:
original_sample_id=sample
if ':' in sample:
sample_alt = string.split(sample,':')[1]
else:
sample_alt = sample
if sample in query_header_proppegated_clusters or sample_alt in query_header_proppegated_clusters:
try: ref_sample = query_header_proppegated_clusters[sample]
except: ref_sample = query_header_proppegated_clusters[sample_alt]
if ref_sample in ref_col_clusters:
if ':' in sample:
sample = string.split(sample,':')[1]
cluster = ref_col_clusters[ref_sample]
#column_clusters.append(cluster) ### Assign a predicted cluster label to the query sample
try: final_clusters.append([query_filename_clean,int(float(cluster)),sample]) ### used later for creating the apples-to-apples group file
except Exception: final_clusters.append([query_filename_clean,cluster,sample])
try: ranked_cluster = int(cluster)
except Exception: ranked_cluster = cluster
comps.append([ranked_cluster,prefix+cluster+'_'+query_filename_clean,prefix+cluster+'_'+ref_filename_clean])
classified_samples[sample]=cluster
selected_query_headers.append(original_sample_id)
for assigned_class in sample_classes:
### Ordered by score
sample_classes[assigned_class].sort()
sample_classes[assigned_class].reverse()
""" Integrate the queried samples in with the reference continuum """
### Loop through the reference headers and add those in the query where they arise
new_query_headers = []
for sample in ref_headers:
if sample not in new_headers:
new_headers.append(sample)
if sample in sample_classes:
sample_data = sample_classes[sample]
for (score,cd) in sample_data:
if cd.Sample() not in new_headers and cd.Sample() not in alt_ref_headers: ### In case some of the same reference samples were added to the query
if cd.Sample() not in new_headers:
new_headers.append(cd.Sample())
new_query_headers.append(cd.Sample())
if len(classified_samples)>0:
for sample in query_headers:
if ':' in sample:
sample_alt = string.split(sample,':')[1]
try: cluster = classified_samples[sample]
except: cluster = classified_samples[sample_alt]
column_clusters.append(cluster)
""" Combine the two datasets, before re-ordering """
### In case the UIDs in the two datasets are in different formats (we assume either Ensembl or Symbol)
### we allow conversion between these systems
for refUID in ref_exp_db: break
for queryUID in query_exp_db: break
if ('ENS' in refUID or 'ENS' in queryUID) and '.' not in refUID:
### Covert one or both to gene symbol
try:
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
except Exception: gene_to_symbol={}
if 'ENS' in refUID:
ref_exp_db2 = convertFromEnsemblToSymbol(ref_exp_db,gene_to_symbol)
ratio = len(ref_exp_db2)/(len(ref_exp_db)*1.00)
if ratio>0.7: ### Sometimes the UID are a mix of Ensembl and symbols, if so check to make sure they actually convert
ref_exp_db=ref_exp_db2
if 'ENS' in queryUID:
query_exp_db2 = convertFromEnsemblToSymbol(query_exp_db,gene_to_symbol)
ratio = len(query_exp_db2)/(len(query_exp_db)*1.00)
if ratio>0.7: ### Sometimes the UID are a mix of Ensembl and symbols, if so check to make sure they actually convert
query_exp_db=query_exp_db2
### Write out the combined data
export_object = export.ExportFile(output_dir)
if len(column_clusters)>0:
### Output with gene and column clusters
export_object.write(string.join(['UID','row_clusters-flat']+ref_headers+query_headers,'\t')+'\n')
export_object.write(string.join(['column_clusters-flat','']+column_clusters,'\t')+'\n')
new_headers = ['row_clusters-flat']+new_headers
new_query_headers = ['row_clusters-flat']+new_query_headers
else:
export_object.write(string.join(['UID']+ref_headers+query_headers,'\t')+'\n')
for uid in ref_exp_db:
if uid in query_exp_db:
if cluster_format_reference: ### If the reference is an AltAnalyze heatmap format text file
export_object.write(string.join([uid]+ref_exp_db[uid]+query_exp_db[uid],'\t')+'\n')
else:
export_object.write(string.join([uid,'1']+ref_exp_db[uid]+query_exp_db[uid],'\t')+'\n')
export_object.close()
""" Write out a groups file that lists samples per file for optional visualization """
reference_query_groups = groups_dir[:-4]+'-ReOrdered.txt'
group_object = export.ExportFile(reference_query_groups)
comps_object = export.ExportFile(string.replace(reference_query_groups,'groups.','comps.'))
comps_object.write('2\t1\n')
comps_object.close()
for sample in ref_headers:
if ':' in sample:
sample = string.split(sample,':')[1]
group_object.write(sample+'\t1\t'+ref_filename_clean+'\n')
for sample in query_headers:
if ':' in sample:
group_object = string.split(sample,':')[1]
group_object.write(sample+'\t2\t'+query_filename_clean+'\n')
group_object.close()
""" Re-order the samples based on the classification analysis """
### The ref_headers has the original reference sample order used to guide the query samples
from import_scripts import sampleIndexSelection
input_file=output_dir
output_file = input_file[:-4]+'-ReOrdered.txt'
query_output_file = input_file[:-4]+'-ReOrdered-Query.txt'
filter_names = new_headers
""" Remove the correlation outliers """
group_export_object = export.ExportFile(root_dir+'/QueryGroups.cellHarmony.txt')
filter_names2=[]
new_query_headers2=[]
added=[]
order=0
ordering=[]
ordered_clusters=[]
for sample_name in filter_names:
if sample_name not in added:
added.append(sample_name)
if sample_name not in exclude:
filter_names2.append(sample_name)
if sample_name in new_query_headers:
new_query_headers2.append(sample_name)
if ':' in sample_name:
sample_name = string.split(sample_name,':')[1]
try:
cluster = classified_samples[sample_name]
group_export_object.write(sample_name+'\t'+cluster+'\t'+prefix+cluster+'\n')
if cluster not in ordered_clusters:
ordered_clusters.append(cluster) ### Needed to organize the clusters in the reference order
except Exception: pass
group_export_object.close()
sampleIndexSelection.filterFile(input_file,output_file,filter_names2,force=True)
sampleIndexSelection.filterFile(input_file,query_output_file,new_query_headers2,force=True)
"""Export a combined groups and comps file to perform apples-to-apples comparisons"""
folds_file=''
all_DEGs=[]
comps = unique.unique(comps)
comps.sort()
if len(final_clusters)>0:
try:
""" Re-order the groups the original ICGS order """
cells_lookup_temp={}
comps_lookup_temp={}
#final_clusters.sort() ### Here is where the groups are ordered for export
for (source,cluster,sample) in final_clusters:
try: cells_lookup_temp[cluster].append((source,cluster,sample))
except: cells_lookup_temp[cluster] = [(source,cluster,sample)]
for (group,g1,g2) in comps:
comps_lookup_temp[group] = (group,g1,g2)
final_clusters2=[]
comps2=[]
for group in ordered_clusters:
try: cells_lookup_temp[group].sort()
except:
group = int(group) ### Occurs if the group number is an integer
cells_lookup_temp[group].sort()
final_clusters2+=cells_lookup_temp[group]
try: comps2.append(comps_lookup_temp[group])
except:
comps2.append(comps_lookup_temp[str(group)])
final_clusters = final_clusters2
comps = comps2
new_query_headers=[]
expression_file = string.replace(groups_file,'groups.','exp.')
export_object = export.ExportFile(groups_file)
group_counter=0
added_groups={}
group_numbers={}
try: ### If using centroids for classification, merge with original reference groups
import ExpressionBuilder
sample_group_ref_db = ExpressionBuilder.simplerGroupImport(groups_reference_file)
except:
pass
### The below tuple order controls which clusters and groups are listed in which orders
groups_to_samples={}
cluster_lookup={}
for (source,cluster,sample) in final_clusters:
cluster_name = prefix+str(cluster)+'_'+source
cluster_lookup[cluster_name]=prefix+str(cluster)
if cluster_name in added_groups:
added_groups[cluster_name]+=1
group_number = group_counter
else:
added_groups[cluster_name]=1
group_counter += 1
group_number = group_counter
group_numbers[cluster_name]=group_number
if ':' in sample:
sample = string.split(sample,':')[1]
export_object.write(sample+'\t'+str(group_number)+'\t'+cluster_name+'\n')
new_query_headers.append(sample)
### For summary plot export of cell cluster frequencies
try: groups_to_samples[cluster_name].append(sample)
except: groups_to_samples[cluster_name] = [sample]
export_object.close()
expression_file_reordered = expression_file[:-4]+'-reordered.txt'
status = verifyFile(expression_file)
if status==False: ### If not AllCells expression file, use the merged file (assuming all genes are in both the query and reference)
export_object = export.ExportFile(expression_file)
export_object.write(string.join(['UID']+ref_headers+query_headers,'\t')+'\n') #
for uid in ref_exp_db:
if uid in query_exp_db:
export_object.write(string.join([uid]+ref_exp_db[uid]+query_exp_db[uid],'\t')+'\n')
export_object.close()
sampleIndexSelection.filterFile(expression_file,expression_file_reordered,new_query_headers)
shutil.move(expression_file_reordered,expression_file) ### replace our temporary file
export_object = export.ExportFile(comps_file)
freq_object = export.ExportFile(root_dir+'/cell-frequency-stats.txt')
### Export the comps and frequency results
freq_object.write('Cluster-name\t'+ref_filename_clean+'\t'+query_filename_clean+'\tFisher exact test p-value\t'+'% '+ref_filename_clean+'\t'+'% '+query_filename_clean+'\n')
added=[]
g2_all = len(ref_headers)
g1_all = len(query_headers)
if len(query_header_proppegated_clusters) > 600:
min_cells = 19
else:
min_cells = 3
for (null,group1,group2) in comps:
g = cluster_lookup[group1]
g1_len = len(groups_to_samples[group1])
g2_len = len(groups_to_samples[group2])
g1_frq = (g1_len*100.00)/g1_all
g2_frq = (g2_len*100.00)/g2_all
oddsratio, pvalue = stats.fisher_exact([[g2_len, g1_len], [g2_all-g2_len, g1_all-g1_len]])
#print g2_len, g1_len, g2_all-g2_len, g1_all-g1_len, g2_all, g1_all, pvalue
added.append(group1); added.append(group2)
freq_object.write(g+'\t'+str(g2_len)+'\t'+str(g1_len)+'\t'+str(pvalue)+'\t'+str(g2_frq)[:4]+'\t'+str(g1_frq)[:4]+'\n')
if added_groups[group1]>min_cells and added_groups[group2]>min_cells:
### Atleast 10 cells present in the two compared groups
g1 = str(group_numbers[group1])
g2 = str(group_numbers[group2])
export_object.write(g1+'\t'+g2+'\n')
for group in groups_to_samples:
g = cluster_lookup[group]
if group not in added:
g2_len = len(groups_to_samples[group])
g2_frq = (g2_len*100.00)/g2_all
freq_object.write(g+'\t'+str(g2_len)+'\t'+str(0)+'\t'+str(pvalue)+'\t'+str(g2_frq)[:4]+'\t'+str(0)+'\n')
export_object.close()
freq_object.close()
try:
index1=-2;index2=-1; x_axis='Cell-State Percentage'; y_axis = 'Reference clusters'; title='Assigned Cell Frequencies'
clustering.barchart(root_dir+'/cell-frequency-stats.txt',index1,index2,x_axis,y_axis,title)
except Exception:
#print traceback.format_exc()
pass
from stats_scripts import metaDataAnalysis
"""
strictCutoff = True
pvalThreshold=0.05
use_adjusted_pval = False
if platform == 'RNASeq':
log_fold_cutoff=0.585
output_dir = root_dir+'/DEGs-LogFold_0.585_rawp'
if strictCutoff:
use_adjusted_pval = True
log_fold_cutoff = 1
pvalThreshold = 0.05
output_dir = root_dir+'/DEGs-LogFold_1_adjp'
else:
log_fold_cutoff=0.1
output_dir = root_dir+'Events-LogFold_0.1_rawp'
"""
if peformDiffExpAnalysis:
gene_summaries=[]
if use_adjusted_pval:
pval_type = '_adjp_'+str(pvalThreshold)
else:
pval_type = '_rawp_'+str(pvalThreshold)
use_custom_output_dir = 'DifferentialExpression_Fold_'+str(fold_cutoff)[:4]+pval_type
output_dir = root_dir+use_custom_output_dir
log_fold_cutoff = math.log(float(fold_cutoff),2)
""" Re-run the differential analysis comparing all cell states in the query vs. reference """
metaDataAnalysis.remoteAnalysis(species,expression_file,groups_file,platform=platform,use_custom_output_dir=use_custom_output_dir,
log_fold_cutoff=log_fold_cutoff,use_adjusted_pval=use_adjusted_pval,pvalThreshold=pvalThreshold,suppressPrintOuts=True)
gene_summary_file = output_dir+'/gene_summary.txt'
moved_summary_file = gene_summary_file[:-4]+'-cell-states.txt'
try: os.remove(moved_summary_file)
except: pass
shutil.move(gene_summary_file,moved_summary_file)
gene_summaries.append(moved_summary_file)
cellstate_DEGs = aggregateRegulatedGenes(output_dir) ### Collect these genes here rather than below to avoid cell-type frequency bias
""" Re-run the differential analysis comparing all cells in the query vs. reference (not cell states) """
try:
metaDataAnalysis.remoteAnalysis(species,expression_file,reference_query_groups,platform=platform,use_custom_output_dir=use_custom_output_dir,
log_fold_cutoff=log_fold_cutoff,use_adjusted_pval=use_adjusted_pval,pvalThreshold=pvalThreshold,suppressPrintOuts=True)
gene_summary_file = output_dir+'/gene_summary.txt'
moved_summary_file = gene_summary_file[:-4]+'-query-reference.txt'
try: os.remove(moved_summary_file)
except: pass
shutil.move(gene_summary_file,moved_summary_file)
gene_summaries.append(moved_summary_file)
except:
print traceback.format_exc()
pass ### Unknown errors - skip this analysis
global_DEGs = aggregateRegulatedGenes(output_dir) ### Collect these genes here rather than below to avoid cell-type frequency bias
""" Export a folds-file with the average fold differences per cell state (query vs. reference) """
try:
folds_file = expression_file[:-4]+'-folds.txt'
comparisons = sampleIndexSelection.getComparisons(groups_file)
filter_names,group_index_db = sampleIndexSelection.getFilters(groups_file,calculateCentroids=True)
sampleIndexSelection.filterFile(expression_file,folds_file,(filter_names,group_index_db),force=False,calculateCentroids=True,comparisons=comparisons)
export_object = export.ExportFile(string.replace(folds_file,'exp.','groups.'))
header = getHeader(folds_file)
i=0
for h in header:
i+=1
export_object.write(h+'\t'+str(i)+'\t'+h+'\n')
export_object.close()
export_object = export.ExportFile(string.replace(folds_file,'exp.','comps.'))
export_object.close()
except:
pass
""" Find similar clusters """
try:
clustered_groups_file = findSimilarImpactedCellStates(folds_file,cellstate_DEGs)
except:
### Unknown error - likely extra files in the PValues folder from a prior error
clustered_groups_file=None
if clustered_groups_file!=None:
try:
""" Re-run the differential analysis comparing all cells in the query vs. reference (not cell states) """
metaDataAnalysis.remoteAnalysis(species,expression_file,clustered_groups_file,platform=platform,use_custom_output_dir=use_custom_output_dir,
log_fold_cutoff=log_fold_cutoff,use_adjusted_pval=use_adjusted_pval,pvalThreshold=pvalThreshold,suppressPrintOuts=True)
gene_summary_file = output_dir+'/gene_summary.txt'
moved_summary_file = gene_summary_file[:-4]+'-clustered-states.txt'
try: os.remove(moved_summary_file)
except: pass
shutil.move(gene_summary_file,moved_summary_file)
gene_summaries.append(moved_summary_file)
except:
print traceback.format_exc()
pass ### Unknown errors - skip this analysis
#sys.exit()
""" Clean up cellHarmony output directory """
export.deleteFolder(root_dir+'ExpressionProfiles')
#export.deleteFolder(root_dir+'PValues')
export.deleteFolder(root_dir+'top50')
gene_summary_combined = string.replace(gene_summary_file,use_custom_output_dir,'')
combineSummaryFiles(gene_summaries,gene_summary_combined)
index1=2;index2=3; x_axis='Number of DEGs'; y_axis = 'Reference clusters'; title='cellHarmony Differentially Expressed Genes'
clustering.barchart(gene_summary_combined,index1,index2,x_axis,y_axis,title,color1='IndianRed',color2='SkyBlue')
import InteractionBuilder
print 'Generating gene regulatory networks...'
pdfs = InteractionBuilder.remoteBuildNetworks(species, output_dir)
networks_dir = root_dir+'/networks/'
try: os.mkdir(networks_dir)
except: pass
for pdf in pdfs:
file = export.findFilename(pdf)
file = string.replace(file,'AltAnalyze-network-WKT_GE.','')
file = string.replace(file,'_cellHarmony-Reference-interactions','')
shutil.copy(pdf,root_dir+'/networks/'+file)
#""" Union of all differentially expressed genes """
all_DEGs = aggregateRegulatedGenes(output_dir,filterGenes=cellstate_DEGs) ### do not include additional genes as these may represent cell frequency bias
display_genes = string.join(list(all_DEGs),' ')
ICGS_DEGs_combined = ref_exp_db.keys()
for gene in all_DEGs:
if gene not in ICGS_DEGs_combined:
ICGS_DEGs_combined.append(gene) ### Add these genes at the end
ICGS_DEGs_combined.reverse()
all_DEGs2 = string.join(ICGS_DEGs_combined,' ')
import UI
vendor = 'Ensembl'
gsp = UI.GeneSelectionParameters(species,platform,vendor)
gsp.setGeneSet('None Selected')
gsp.setPathwaySelect('')
gsp.setGeneSelection(all_DEGs2)
gsp.setJustShowTheseIDs(display_genes)
gsp.setNormalize('median')
transpose = gsp
column_method = None #'hopach'
column_metric = 'cosine'
row_method = None #'hopach'
row_metric = 'correlation'
graphic_links=[]
color_gradient = 'yellow_black_blue'
from visualization_scripts import clustering
if runningCommandLine:
display = False
else:
display = True
display = False
outputDEGheatmap=False
if outputDEGheatmap:
graphic_links = clustering.runHCexplicit(expression_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=display, Normalize=True)
except IndexError:
print traceback.format_exc()
print '!!!!! NO merged expression file availble for differential expression analyis (apples-to-apples).'
print 'Completed cellHarmony file creation...'
return output_file, query_output_file, folds_file, all_DEGs
def combineSummaryFiles(gene_summaries,gene_summary_combined):
""" Combine the Summary output files from the differential expression analyses """
eo = export.ExportFile(gene_summary_combined)
header = None
for file in gene_summaries:
firstLine = True
for line in open(file,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
firstLine = False
if header == None:
header= line
eo.write(line)
else:
eo.write(line)
eo.close()
def findSimilarImpactedCellStates(folds_file,cellstate_DEGs):
import numpy, scipy
similar_groups=collections.OrderedDict()
folds_header_clean=[]
expression_db, folds_header = simpleExpressionFileImport(folds_file,filterUID=cellstate_DEGs)
matrix=[]
for gene in expression_db:
matrix.append(map(float,expression_db[gene]))
matrix = numpy.array(matrix)
matrix = numpy.transpose(matrix)
for h in folds_header:
folds_header_clean.append(string.replace(h,'-fold',''))
output_dir = os.path.abspath(os.path.join(folds_file, os.pardir))
pvalue_dir = output_dir+'/PValues/'
dir_list = unique.read_directory(pvalue_dir)
import UI
pval_all_db={}
genes={}
valid_comparisons=[]
for file in dir_list:
if '.txt' in file and '|' not in file:
pval_file_dir = pvalue_dir+'/'+file
pval_db, header = simpleExpressionFileImport(pval_file_dir,filterUID=cellstate_DEGs)
for gene in pval_db: genes[gene]=[]
for h in folds_header_clean:
if h in file:
valid_comparisons.append(h) ### Use the simple name of this cluster
pval_all_db[h] = pval_db
break
"""Create a binarized matrix of p-values"""
combined_pvalues={}
for file in valid_comparisons:
for gene in genes:
if gene in pval_all_db[file]:
if float(pval_all_db[file][gene][0])<0.1:
val=1
else:
val=0
else:
val=0 ### If no variance detected in either cell-state for that gene
try: combined_pvalues[gene].append(val)
except: combined_pvalues[gene]=[val]
pval_patterns={}
for gene in combined_pvalues:
i=0
pattern = list(combined_pvalues[gene])
uid = cellstate_DEGs[gene][-1]
for value in combined_pvalues[gene]:
if value == 1:
try:
if '-' in expression_db[uid][i]:
pattern[i] = -1
except:
#print pattern
#print i,expression_db[uid];kill
pass
i+=1
try: pval_patterns[tuple(pattern)]+=1
except: pval_patterns[tuple(pattern)]=1
patterns_ranked=[]
for pattern in pval_patterns:
s = pattern.count(0)
n = pattern.count(-1)
p = pattern.count(1)
counts = pval_patterns[pattern]
if s == (len(valid_comparisons)-1) or s == (len(valid_comparisons)) or s==0:
if s==0:
if n==len(valid_comparisons) or p == len(valid_comparisons):
pass
else:
### complex pattern of 1 and -1 only
patterns_ranked.append([counts,pattern])
else:
patterns_ranked.append([counts,pattern])
patterns_ranked.sort()
patterns_ranked.reverse()
new_aggregate_clusters=[]
for (count,pattern) in patterns_ranked[:8]: ### restrict to the top 4 patterns
pattern_groups=[]
if count > 9: ### require that atleast 10 genes have that pattern
i=0
for cluster in pattern:
if cluster == 1 or cluster == -1:
pattern_groups.append(valid_comparisons[i])
i+=1
if pattern_groups not in new_aggregate_clusters:
new_aggregate_clusters.append(pattern_groups)
if len(new_aggregate_clusters)==4:
break
"""
index=0
prior_cell_state_array=[]
cluster=1
for cell_state_array in matrix:
if len(prior_cell_state_array)>0:
rho,p = scipy.stats.pearsonr(cell_state_array,prior_cell_state_array)
#print cluster, header[index], rho
if rho>0.3:
if cluster in similar_groups:
similar_groups[cluster].append(header[index])
else:
similar_groups[cluster]=[header[index-1],header[index]]
else:
if len(similar_groups)==0:
### occurs for the first group if not similar to the next
similar_groups[cluster] = [header[index-1]]
cluster+=1
similar_groups[cluster] = [header[index]]
prior_cell_state_array = cell_state_array
index+=1
num_new_clusters=0
if len(similar_groups)!=len(folds_header): ### If not clusters combined
exp_file = string.replace(folds_file,'-folds.txt','.txt')
groups_file = string.replace(exp_file,'exp.','groups.')
output_groups_file = string.replace(groups_file,'.txt','-clusters.txt')
output_comps_file = string.replace(output_groups_file,'groups.','comps.')
og = export.ExportFile(output_groups_file)
oc = export.ExportFile(output_comps_file)
import ExpressionBuilder
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = ExpressionBuilder.simpleGroupImport(groups_file,reverseOrder=True)
for cluster in similar_groups:
if len(similar_groups[cluster])>1:
### Combine the cells from different groups
exp_cells=[]
control_cells=[]
for group in similar_groups[cluster]:
exp_group = string.replace(group,'-fold','')
exp_cells += group_name_sample_db[exp_group]
control_group = comps_name_db[exp_group][1]
control_cells += group_name_sample_db[control_group]
cluster_number = cluster*2-1
if len(similar_groups[cluster]) != len(folds_header): ### If ALL clusters combined
num_new_clusters+=1
cluster_name = string.replace(string.join(similar_groups[cluster],'|'),'-fold','')
for cell in exp_cells:
og.write(cell+'\t'+str(cluster_number)+'\t'+cluster_name+'-Query'+'\n')
for cell in control_cells:
og.write(cell+'\t'+str(cluster_number+1)+'\t'+cluster_name+'-Ref'+'\n')
oc.write(str(cluster_number)+'\t'+str(cluster_number+1)+'\n')
og.close()
oc.close()
"""
num_new_clusters=0
if len(new_aggregate_clusters)>0: ### If not clusters combined
exp_file = string.replace(folds_file,'-folds.txt','.txt')
groups_file = string.replace(exp_file,'exp.','groups.')
output_groups_file = string.replace(groups_file,'.txt','-clusters.txt')
output_comps_file = string.replace(output_groups_file,'groups.','comps.')
output_dir = os.path.abspath(os.path.join(output_comps_file, os.pardir))
summary_file = output_dir+'/corregulated-clusters.txt'
og = export.ExportFile(output_groups_file)
oc = export.ExportFile(output_comps_file)
ox = export.ExportFile(summary_file)
print summary_file
ox.write('Combined Clusters\tFilename\n')
import ExpressionBuilder
sample_list,group_sample_db,group_db,group_name_sample_db,comp_groups,comps_name_db = ExpressionBuilder.simpleGroupImport(groups_file,reverseOrder=True)
c=0
for clustered_groups in new_aggregate_clusters:
c+=1
### Combine the cells from different groups
exp_cells=[]
control_cells=[]
for exp_group in clustered_groups:
exp_cells += group_name_sample_db[exp_group]
control_group = comps_name_db[exp_group][1]
control_cells += group_name_sample_db[control_group]
num_new_clusters+=1
#cluster_name = string.replace(string.join(clustered_groups,'|'),'-fold','')
cluster_name = string.replace(string.join(clustered_groups[:1]+['co-'+str(c)],'__'),'-fold','')
ox.write(string.join(clustered_groups,'__')+'\t'+string.join(clustered_groups[:1]+['co-'+str(c)],'|')+'\n')
for cell in exp_cells:
og.write(cell+'\t'+str(num_new_clusters)+'\t'+cluster_name+'-Query'+'\n')
for cell in control_cells:
og.write(cell+'\t'+str(num_new_clusters+1)+'\t'+cluster_name+'-Ref'+'\n')
oc.write(str(num_new_clusters)+'\t'+str(num_new_clusters+1)+'\n')
num_new_clusters = num_new_clusters*2
og.close()
oc.close()
ox.close()
if num_new_clusters>0:
return output_groups_file
else:
return None
def aggregateRegulatedGenes(folder,filterGenes=None):
""" Find the best representative comparison for each gene
dependent on the observed rawp """
import UI
geneIDs={}
files = UI.read_directory(folder)
for file in files:
if '.txt' in file and ('PSI.' in file or 'GE.' in file):
ls=[]
fn = folder+'/'+file
firstLine = True
if os.path.isfile(fn): ### When file paths are too long - will error
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
if 'Event-Direction' in t:
uid_index = t.index('Event-Direction')
fold_index = t.index('dPSI')
else:
uid_index = t.index('GeneID')
symbol_index = t.index('Symbol')
fold_index = t.index('LogFold')
p_index = t.index('rawp')
firstLine= False
continue
uid = t[uid_index]
symbol = t[symbol_index]
pval = float(t[p_index])
fold = t[fold_index]
if '-' in fold:
direction = 'negative'
else:
direction = 'positive'
if filterGenes!=None:
if uid not in filterGenes and symbol not in filterGenes:
continue
try:
geneIDs[uid].append([pval,direction,file,symbol])
except:
geneIDs[uid] = [[pval,direction,file,symbol]]
if symbol != uid:
""" Support both ID systems for different databases """
try:
geneIDs[symbol].append([pval,direction,file,uid])
except:
geneIDs[symbol] = [[pval,direction,file,uid]]
def check_if_globally_regulated(hits):
""" Determine if the global regulation pattern is supported by multiple
comparisons """
if len(hits)>2:
direction_db={}
for (pval,direction,file,uid) in hits:
try: direction_db[direction]+=1
except: direction_db[direction]=1
if len(direction_db)==1:
pass
else:
del hits[0]
else:
del hits[0]
return hits
geneIDs2={}
for uid in geneIDs:
hits = geneIDs[uid]
hits.sort()
if 'vs_cellHarmony-Reference' in hits[0][-2]:
hits = check_if_globally_regulated(hits)
if len(hits)>0:
geneIDs2[uid] = hits[0] ### all hits removed
return geneIDs2
def convertFromEnsemblToSymbol(exp_db,gene_to_symbol):
### covert primary ID to symbol
exp_db_symbol=collections.OrderedDict()
for UID in exp_db:
if UID in gene_to_symbol:
symbol = gene_to_symbol[UID][0]
exp_db_symbol[symbol] = exp_db[UID]
return exp_db_symbol
def checkForGroupsFile(filename,headers):
new_headers = headers
groups_db=collections.OrderedDict()
if ('exp.' in filename or 'filteredExp.' in filename):
filename = string.replace(filename,'-steady-state.txt','.txt')
try:
import ExpressionBuilder
sample_group_db = ExpressionBuilder.simplerGroupImport(filename)
new_headers = []
for v in headers:
if v in sample_group_db:
groups_db[v]=sample_group_db[v]
v = sample_group_db[v]+':'+v
new_headers.append(v)
except Exception:
#print traceback.format_exc()
pass
return headers, new_headers, groups_db
def importExpressionFile(input_file,ignoreClusters=False,filterIDs=False,customLabels=None):
""" Import the expression value and harmonize (cellHarmony) to log, non-fold values """
if customLabels!=None:
try:
import ExpressionBuilder
customLabels = ExpressionBuilder.simplerGroupImport(customLabels)
except:
print 'WARNING!!! Custom labels failed to import due to formatting error.'
customLabels={}
else:
customLabels={}
expression_db=collections.OrderedDict()
column_cluster_index=collections.OrderedDict()
row_cluster_index=collections.OrderedDict()
### Check the file format (log, non-log, fold) - assume log is log2
import ExpressionBuilder
expressionDataFormat,increment,convertNonLogToLog = ExpressionBuilder.checkExpressionFileFormat(input_file,reportNegatives=True,filterIDs=filterIDs)
inputFormat = 'NumericMatrix'
firstLine = True
cluster_format_file = True
if filterIDs!=False:
try:
### Conserve memory by only importing reference matching IDs
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception:
gene_to_symbol={}
symbol_to_gene={}
row_count=0
for line in open(input_file,'rU').xreadlines():
data = line.rstrip()
data = string.replace(data,'"','')
row_count+=1
if '.csv' in input_file:
values = string.split(data,',')
else:
values = string.split(data,'\t')
if firstLine:
firstLine=False
if 'row_clusters-flat' in values[1]:
numStart = 2 ### Start numeric import at column 3
inputFormat = 'Clustering'
header_row = values[numStart:]
if ':' in line:
### For a MarkerFinder format file with folds (no separate cluster columns/rows)
column_cluster_index_alt=collections.OrderedDict()
new_headers=[]
for header in header_row:
cluster,header = header.split(':')
column_cluster_index_alt[header] = cluster
new_headers.append(header)
original_header = header_row
header_row = new_headers
else:
numStart = 1 ### Start numeric import at column 2
header_row = values[numStart:]
original_header, header_row, column_cluster_index_alt=checkForGroupsFile(input_file,header_row) ### in case there is a groups file
else:
### store the column to cluster relationships (reference only - propegated to query)
### We can simply retain this column for the output
if 'column_clusters-flat' in values:
clusters = values[numStart:]
if 'NA' not in clusters:
i=0
for header in header_row:
cluster = clusters[i]
""" If labels provided by the user """
try:
alt_header = string.replace(header,'.Reference','')
cluster = customLabels[alt_header]
except:
try:
h=string.split(alt_header,':')[1]
cluster = customLabels[h]
except: pass
column_cluster_index[header]=cluster
i+=1
### Replace the cluster with the sample name if there is only one sample per cluster (centroid name)
if len(unique.unique(clusters)) == len(column_cluster_index):
for header in column_cluster_index:
column_cluster_index[header]=header
continue
else:
#header_row=original_header
column_cluster_index = column_cluster_index_alt
cluster_format_file = True
continue
elif row_count==2:
header_row=original_header
column_cluster_index = column_cluster_index_alt
cluster_format_file = False
uid = values[0]
cluster = values[1]
if ':' in uid:
cluster = string.split(uid,':')[0]
uid = string.split(uid,':')[1]
if ' ' in uid:
uid = string.split(uid,' ')[0]
if filterIDs !=False:
if uid not in filterIDs:
if uid in gene_to_symbol:
alt_id = gene_to_symbol[uid][0]
if alt_id not in filterIDs:
continue ### Skip additional processing of this line
elif uid in symbol_to_gene:
alt_id = symbol_to_gene[uid][0]
if alt_id not in filterIDs:
continue ### Skip additional processing of this line
else:
continue ### Skip additional processing of this line
if inputFormat == 'Clustering':
### store the row cluster ID
row_cluster_index[uid]=cluster
numericVals = map(float, values[numStart:])
if increment<-1 and convertNonLogToLog == False:
### Indicates the data is really fold changes (if the increment is a small negative,
### it could be due to quantile normalization and the values are really raw expression values)
row_min = min(numericVals)
numericVals = map(lambda x: x-row_min, numericVals) ### move into positive log expression space
elif increment<-1 and convertNonLogToLog: ### shouldn't be encountered since fold values should all be logged
row_min = min(numericVals)
numericVals = map(lambda x: math.log(x-row_min+1,2), numericVals) ### move into positive log expression space
elif convertNonLogToLog:
try: numericVals = map(lambda x: math.log(x+increment,2), numericVals) ### log2 and increment
except Exception:
print 'increment',increment
print numericVals[0:10],numericVals[-10:]
print traceback.format_exc()
kill
numericVals = map(str,numericVals) ### we are saving to a file
if inputFormat == 'Clustering' and ignoreClusters==False:
expression_db[uid] = [cluster]+numericVals
else:
expression_db[uid] = numericVals
print len(expression_db),'IDs imported'
return expression_db, header_row, column_cluster_index, cluster_format_file
def createMetaICGSAllCells(ICGS_files,outputDir,CenterMethod='median',
species='Hs',platform='RNASeq',PearsonThreshold=0.9):
"""This function combines ICGS or MarkerFinder results from multiple outputs
after createMetaICGSResults has been run"""
""" Rename the Expanded CellHarmonyReference files based on the dataset """
files_to_merge, all_cells, groups_db = renameICGSfiles(ICGS_files,CenterMethod,ReturnAllCells=True)
final_output_dir = outputDir+'/CellHarmonyReference/ICGS-merged-reference.txt'
genes, ordered_barcodes, ordered_clusters = importMergedICGS(final_output_dir,outputDir,groups_db,CenterMethod)
join_option = 'Intersection'
ID_option = False ### Require unique-matches only when True
output_merge_dir = outputDir
# output a combined centroid file, combining all input ICGS or MarkerFinder results with raw expression values
from import_scripts import mergeFiles
""" Merge the full cell matrix results for the union of ICGS-NMF genes """
outputfile=output_merge_dir+'/MergedFiles.txt'
outputfile = mergeFiles.joinFiles(files_to_merge, join_option, ID_option, output_merge_dir) # Comment out if already run
revised_outputfile = output_merge_dir+'/ICGS-merged-all-temp.txt'
from import_scripts import sampleIndexSelection
print 'Reorganizing the full-cell merged-ICGS results'
sampleIndexSelection.filterFile(outputfile,revised_outputfile,ordered_barcodes) # Comment out if already run
""" Re-output the ordered cell expression matrix with ordered genes in heatmap format """
exportFullMergedMarkerFile(revised_outputfile,genes,ordered_clusters)
def exportFullMergedMarkerFile(filename,genes,ordered_clusters):
""" Re-order the genes in the genes dictionary order"""
reordered_expression_file = filename[:-4]+'-sorted'
reordered_expression_file = exportSorted(filename, genes) # Comment out if already run
output = filename[:-4]+'-temp.txt'
eo = export.ExportFile(output)
rowNumber=1
for line in open(reordered_expression_file,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if rowNumber==1:
header = t[1:]
eo.write(string.join(['UID','row_clusters-flat']+t[1:],'\t')+'\n')
elif rowNumber==2:
eo.write(string.join(['column_clusters-flat','']+ordered_clusters,'\t')+'\n')
else:
cluster = str(genes[t[0]])
eo.write(string.join([t[0],cluster]+t[1:],'\t')+'\n')
rowNumber+=1
eo.close()
print 'Merged-ICGS all-cells file exported to:',output
def importMergedICGS(final_output_dir,outputDir,groups_db,CenterMethod):
""" Reimport the final ICGS centroids and genes for merging the all cell results """
expression_db = {}
rowNumber = 1
genes=collections.OrderedDict()
for line in open(final_output_dir,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if rowNumber==1:
original_cluster_names = t[2:]
elif rowNumber==2:
clusters = t[2:]
else:
genes[t[0]]=t[1] ### Store the gene and cluster number
rowNumber+=1
index = 0
cluster_db = collections.OrderedDict()
ordered_barcodes=[]
ordered_clusters=[]
names_added=[]
cluster_names=[]
for original_name in original_cluster_names:
names = string.split(original_name,'|')
cluster = clusters[index]
barcodes=[]
for name in names:
if name in names_added:
continue ### Looks like a bug or issue with how the data is loaded
names_added.append(name)
name = string.replace(name,'-'+CenterMethod,'')
ordered_barcodes+=groups_db[name]
ordered_clusters+=[cluster]*len(groups_db[name])
cluster_names+=[original_name]*len(groups_db[name])
cluster_db[cluster]=barcodes
index+=1
eo = export.ExportFile(outputDir+'/groups.all-cell-ICGS.txt')
i=0
for b in ordered_barcodes:
eo.write(b+'\t'+ordered_clusters[i]+'\t'+cluster_names[i]+'\n')
i+=1
eo.close()
return genes, ordered_barcodes, ordered_clusters
def exportSorted(filename, uids, sort_col=0, excludeHeader=True):
### efficient method to sort a big file without storing everything in memory
### http://stackoverflow.com/questions/7079473/sorting-large-text-data
ouput_file = filename[:-4]+'-sorted' ### temporary
index = []
f = open(filename)
index_db={}
firstLine = True
while True:
offset = f.tell()
line = f.readline()
if not line: break
length = len(line)
col = line.split('\t')[sort_col].strip()
if firstLine:
header = line
firstLine = False
if excludeHeader == False:
index.append((col, offset, length))
else:
if col in uids:
#index.append((col, offset, length))
index_db[col] = (col, offset, length)
f.close()
for uid in uids:
if uid in index_db:
index.append(index_db[uid]) ### Order to seek to
o = open(ouput_file,'w')
f = open(filename)
if excludeHeader:
o.write(header)
for col, offset, length in index:
#print col, offset, length
f.seek(offset)
o.write(f.read(length))
o.close()
"""
try:
### Error occurs when the file can't be deleted due to system permissions
os.remove(filename)
os.rename(ouput_file,filename)
return filename
except Exception:
return ouput_file
"""
print filename,'...sorted'
return ouput_file
def retreive_groups_from_file(filename,groups_db):
root_dir = os.path.abspath(os.path.join(filename, os.pardir))
root_dir = os.path.abspath(os.path.join(root_dir[:-1], os.pardir))
dataset= os.path.basename(root_dir)
fn=filepath(filename)
expression_db = {}
rowNumber = 1
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if rowNumber==1:
header = t[2:]
clusters = collections.OrderedDict()
cluster_number=1
for h in header:
cluster,barcode = string.split(h,':')
if cluster in clusters:
cn = clusters[cluster]
groups_db[str(cn)+'.'+dataset].append(barcode+'.'+dataset)
else:
clusters[cluster]=cluster_number
groups_db[str(cluster_number)+'.'+dataset]=[barcode+'.'+dataset]
cluster_number+=1
firstRow = False
else:
break
rowNumber+=1
### Verify that there is not redundancy
temp={}
for cluster in groups_db:
for barcode in groups_db[cluster]:
if barcode in temp:
print barcode, temp[barcode], cluster
print 'Warning!!!!! Redundant barcodes by merged';sys.exit()
temp[barcode]=cluster
return groups_db
def renameICGSfiles(ICGS_files,CenterMethod,ReturnAllCells=False):
""" Rename the input files """
files_to_merge = []
all_cells={}
groups_db = collections.OrderedDict()
for heatmap_file in ICGS_files:
root_dir = os.path.abspath(os.path.join(heatmap_file, os.pardir))
if 'ICGS' in root_dir:
print 'Importing prior produced ICGS-results from:',root_dir
root_dir = os.path.abspath(os.path.join(root_dir[:-1], os.pardir))
cellHarmonyReferenceFile = root_dir+'/CellHarmonyReference/MarkerFinder-cellHarmony-reference-centroid.txt'
cellHarmonyReferenceFileFull = root_dir+'/CellHarmonyReference/MarkerFinder-cellHarmony-reference.txt'
dataset= os.path.basename(root_dir)
src=cellHarmonyReferenceFile
dst=root_dir+'/CellHarmonyReference/'+dataset+'-'+CenterMethod+'.txt'
try: shutil.copyfile(src,dst) # Coment out if already run
except: pass
if ReturnAllCells == False:
files_to_merge.append(dst)
src=cellHarmonyReferenceFileFull
dst=root_dir+'/CellHarmonyReference/'+dataset+'.txt'
try: shutil.copyfile(src,dst) # Coment out if already run
except: pass
all_cells[dataset+'-'+CenterMethod]=dst
groups_db = retreive_groups_from_file(heatmap_file,groups_db)
if ReturnAllCells:
files_to_merge.append(dst)
return files_to_merge, all_cells, groups_db
def createMetaICGSResults(ICGS_files,outputDir,CenterMethod='median',
species='Hs',platform='RNASeq',PearsonThreshold=0.9,force=True):
"""This function combines ICGS or MarkerFinder results from multiple outputs"""
#### If you only want to merge existing results, run this
#createMetaICGSAllCells(ICGS_files,outputDir,CenterMethod=CenterMethod,
# species=species,platform=platform,PearsonThreshold=PearsonThreshold)
#sys.exit()
# import raw expression values for each ICGS or MarkerFinder
files_to_merge = []
all_cells={}
# Check to see if the output already exist
# If not, create the MarkerFinder-cellHarmony-reference with the union of all genes
#if len(files_to_merge) != len(ICGS_files) or force==True:
if force==True:
# import all ICGS or MarkerFinder variable genes
gene_db = simpleICGSGeneImport(ICGS_files)
print len(gene_db), 'unique genes from ICGS results imported...'
for heatmap_file in ICGS_files:
### returnCentroids = False to return all cells
cellHarmonyReferenceFile = convertICGSClustersToExpression(heatmap_file,heatmap_file,returnCentroids=True,
CenterMethod=CenterMethod,geneOverride=gene_db,combineFullDatasets=False,species=species)
files_to_merge.append(cellHarmonyReferenceFile)
""" Rename the Expanded CellHarmonyReference files based on the dataset """
files_to_merge, all_cells, groups_db = renameICGSfiles(ICGS_files,CenterMethod)
join_option = 'Intersection'
ID_option = False ### Require unique-matches only when True
output_merge_dir = outputDir
# output a combined centroid file, combining all input ICGS or MarkerFinder results with raw expression values
from import_scripts import mergeFiles
outputfile = mergeFiles.joinFiles(files_to_merge, join_option, ID_option, output_merge_dir)
# collapse similar medoids into centroids to remove redundant references
query_output_file, unclustered_collapsed = collapseSimilarMedoids(outputfile,cutoff=PearsonThreshold)
# re-cluster this merged file with HOPACH to produce the final combined medoid reference
from visualization_scripts import clustering
row_method = None; row_metric = 'correlation'; column_method = None; column_metric = 'cosine'; color_gradient = 'yellow_black_blue'
transpose = False; Normalize=False
graphics = clustering.runHCexplicit(query_output_file, [], row_method, row_metric,
column_method, column_metric, color_gradient, transpose, Normalize=Normalize,
contrast=3, display=False)
print 'Completed clustering'
revised_cellHarmony_reference = graphics[-1][-1][:-4]+'.txt'
final_output_dir = outputDir+'/CellHarmonyReference/ICGS-merged-reference.txt'
exportMergedReference(unclustered_collapsed,revised_cellHarmony_reference,final_output_dir,outputDir,species,platform)
createMetaICGSAllCells(ICGS_files,outputDir,CenterMethod=CenterMethod,
species=species,platform=platform,PearsonThreshold=PearsonThreshold)
return final_output_dir
def exportMergedReference(unclustered_centroids,input,output,outputDir,species,platform):
import UI
fl = UI.ExpressionFileLocationData('','','','')
fl.setOutputDir(outputDir)
fl.setSpecies(species)
fl.setPlatformType(platform)
fl.setRPKMThreshold(0.00)
fl.setCorrelationDirection('up')
compendiumType = 'protein_coding'
genesToReport = 50
correlateAll = True
import markerFinder
print [unclustered_centroids]
print [input]
print 'Running MarkerFinder'
markerFinder.analyzeData(unclustered_centroids,species,platform,compendiumType,
geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl,
logTransform=False)
markerfinder_dir= outputDir+'CellHarmonyReference/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
marker_db = collections.OrderedDict()
def importMarkerFinderResults(input_file):
header_row=True
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
data = string.replace(data,'"','')
geneID, symbol, rho, p, cell_state = string.split(data,'\t')
if header_row:
header_row=False
else:
rho = float(rho)
if cell_state in marker_db:
if len(marker_db[cell_state])>49:
continue
elif rho>0.2:
marker_db[cell_state].append(geneID)
else:
marker_db[cell_state] = [geneID]
importMarkerFinderResults(markerfinder_dir)
row_count=0
eo = export.ExportFile(output)
clustered_genes={}
for line in open(input,'rU').xreadlines():
row_count+=1
data = cleanUpLine(line)
t = string.split(data,'\t')
if row_count==1:
clusters = len(t)-1
#eo.write(string.join(['UID','row_clusters-flat']+map(str,range(1,clusters)),'\t')+'\n')
headers = t[2:]
eo.write(line)
elif row_count==2:
clusters = len(t)-1
eo.write(string.join(['column_clusters-flat','']+map(str,range(1,clusters)),'\t')+'\n')
else:
#eo.write(line)
clustered_genes[t[0]]=t[2:]
clusters=1
for cell_state in headers:
if cell_state in marker_db:
for gene in marker_db[cell_state]:
eo.write(string.join([gene,str(clusters)]+clustered_genes[gene],'\t')+'\n')
clusters+=1
eo.close()
def collapseSimilarMedoids(outputfile,cutoff=0.9):
print 'correlation cutoff =',[cutoff]
from visualization_scripts import clustering
import numpy
from stats_scripts import statistics
matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters = clustering.remoteImportData(outputfile)
# Filter for rows with median values
filteredMatrix = []
filtered_row_header = []
i=0
for values in matrix:
if (max(values)-min(values))==0:
pass
else:
filteredMatrix.append(values)
filtered_row_header.append(row_header[i])
i+=1
matrix = numpy.array(filteredMatrix)
row_header=filtered_row_header
Tmatrix = zip(*matrix) ### transpose this back to normal
D1 = numpy.corrcoef(Tmatrix)
i=0
combine={}
combined_list=[]
for score_ls in D1:
k=0
for v in score_ls:
if v!=1:
if v>cutoff: ### Pearson cutoff
c1=column_header[i]
c2=column_header[k]
clusters = [c1,c2]
clusters.sort()
a,b = clusters
if a in combine:
if b not in combine[a]:
if b not in combine:
combine[a].append(b)
combined_list.append(b)
else:
if b in combine:
if a not in combine[b]:
combine[b].append(a)
combined_list.append(a)
else:
present=False
for x in combine:
if b in combine[x]:
present = True
if present==False:
try: combine[a].append(b)
except Exception: combine[a] = [a,b]
combined_list.append(a)
combined_list.append(b)
k+=1
i+=1
# Add each cluster not in correlated set to obtain all final clusters
for cluster in column_header:
if cluster not in combined_list:
combine[cluster]=[cluster]
group_index_db={} ### Order is not important yet
for cluster in combine:
ci_list=[]
for c in combine[cluster]:
ci = column_header.index(c) ### get the index of the cluster name
ci_list.append(ci)
combined_cluster_name = string.join(combine[cluster],'|')
group_index_db[combined_cluster_name] = ci_list
root_dir = export.findParentDir(outputfile)
collapsed_dir = root_dir+'/CellHarmonyReference/collapsedMedoids.txt'
eo = export.ExportFile(collapsed_dir)
eo.write(string.join(['UID']+map(str,group_index_db),'\t')+'\n')
i=0
for values in matrix:
avg_matrix=[]
for cluster in group_index_db:
try: avg_matrix.append(str(statistics.avg(map(lambda x: matrix[i][x], group_index_db[cluster]))))
except Exception: ### Only one value
try: avg_matrix.append(str(map(lambda x: matrix[i][x], group_index_db[cluster])[0]))
except Exception:
print matrix[i]
print group_index_db[cluster];sys.exit()
eo.write(string.join([row_header[i]]+avg_matrix,'\t')+'\n')
i+=1
eo.close()
# Copy to a file for MarkerFinder analysis
unclustered_collapsed = string.replace(collapsed_dir,'collapsedMedoids.txt','exp.unclustered.txt')
try: shutil.copyfile(collapsed_dir,unclustered_collapsed)
except: pass
eo = export.ExportFile(string.replace(unclustered_collapsed,'exp.','groups.'))
number=1
for cluster in group_index_db:
eo.write(cluster+'\t'+str(number)+'\t'+cluster+'\n')
number+=1
eo.close()
eo = export.ExportFile(string.replace(unclustered_collapsed,'exp.','comps.'))
eo.close()
return collapsed_dir, unclustered_collapsed
def convertICGSClustersToExpression(heatmap_file,query_exp_file,returnCentroids=False,
CenterMethod='median',geneOverride=None,combineFullDatasets=True,species='Hs',fl=None):
"""This function will import an ICGS row normalized heatmap and return raw
expression values substituted for the values. """
from visualization_scripts import clustering
graphic_links=[]
filename = export.findFilename(heatmap_file)
ICGS_dir = export.findParentDir(heatmap_file)
if 'DataPlots' in ICGS_dir: ### For MarkerFinder input
### Go one more level up
ICGS_dir = export.findParentDir(ICGS_dir[:-1])
root_dir = export.findParentDir(ICGS_dir[:-1])
try: files = unique.read_directory(root_dir+'/ExpressionInput')
except: files=[]
exp_dir_prefix = string.split(string.replace(filename,'Clustering-',''),'-')[0]
### Look for the best expression file match
specific_matches = []
steady_state_files = []
exp_files = []
filteredExp_files = []
for file in files:
if 'exp.' in file and '~exp.' not in file:
exp_files.append([os.path.getsize(root_dir+'/ExpressionInput/'+file),file])
if 'steady-state' in file:
steady_state_files.append([os.path.getsize(root_dir+'/ExpressionInput/'+file),file])
expression_files_ss
if exp_dir_prefix in file:
specific_matches.append([os.path.getsize(root_dir+'/ExpressionInput/'+file),file])
if 'filteredExp.' in file:
filteredExp_files.append([os.path.getsize(root_dir+'/ExpressionInput/'+file),file])
specific_matches.sort()
steady_state_files.sort()
exp_files.sort()
filteredExp_files.sort()
if len(specific_matches)>0:
expdir = root_dir+'/ExpressionInput/'+specific_matches[-1][1]
elif len(steady_state_files)>0:
expdir = root_dir+'/ExpressionInput/'+steady_state_files[-1][1]
else:
try: expdir = root_dir+'/ExpressionInput/'+exp_files[-1][1]
except: expdir = ''
try: filtered_expdir = root_dir+'/ExpressionInput/'+filteredExp_files[-1][1]
except: filtered_expdir = ''
try:
### Allow for custom expression file paths
full_ref_exp_path = fl.reference_exp_file()
if full_ref_exp_path != False and full_ref_exp_path != '':
expdir = full_ref_exp_path
filtered_expdir = ''
root_dir = ICGS_dir
except:
pass
print 'Selected the full expression file:',expdir
if '-Guide' in filename:
guide = string.split(filename,'-Guide')[1][:1]
cellHarmonyReferenceFile = root_dir+'/CellHarmonyReference/Guide'+guide+'-cellHarmony-reference.txt'
elif 'Marker' in filename:
cellHarmonyReferenceFile = root_dir+'/CellHarmonyReference/MarkerFinder-cellHarmony-reference.txt'
else:
cellHarmonyReferenceFile = root_dir+'/CellHarmonyReference/ICGS-cellHarmony-reference.txt'
eo = export.ExportFile(cellHarmonyReferenceFile)
### Import the heatmap and expression files
matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters = clustering.remoteImportData(heatmap_file,reverseOrder=False)
gene_cluster_db={}
if len(priorRowClusters)==0:
clusters=[]
cluster_number=0
new_row_header = []
for uid in row_header:
if ':' in uid:
try: cluster,uid = string.split(uid,':')
except:
### Occurs with zebrafish IDs
vals = string.split(uid,':')
cluster = vals[0]
uid = string.join(vals[1:],':')
if cluster not in clusters:
clusters.append(cluster)
cluster_number+=1
if ' ' in uid:
uid = string.split(uid,' ')[0] ### Typically source ID and then symbol
gene_cluster_db[uid] = str(cluster_number)
new_row_header.append(uid)
clusters=[]
cluster_number=0
new_column_header = []
for uid in column_header:
if ':' in uid:
try: cluster,uid = string.split(uid,':')
except:
### Occurs with zebrafish IDs
vals = string.split(uid,':')
cluster = vals[0]
uid = string.join(vals[1:],':')
if cluster not in clusters:
clusters.append(cluster)
cluster_number+=1
priorColumnClusters.append(str(cluster_number))
new_column_header.append(uid)
row_header = new_row_header
column_header = new_column_header
else:
### Store the row clusters in a dictionary rather than a list since matching IDs in the expression file may change (e.g., different ID systems in the ICGS results)
index=0
for gene in row_header:
gene_cluster_db[gene] = priorRowClusters[index]
index+=1
### Correct fileheader if group prefix present
try: column_header = map(lambda x: string.split(x,':')[-1],column_header)
except Exception: pass ### If no ":" in cell library names
updated_column_header=[]
for i in column_header:
if ':' in i:
i = string.split(i,':')[1]
updated_column_header.append(i)
column_header = updated_column_header
### Record the index for each sample name in the ICGS result order in the original expression file (exp.*)
priorColumnClusters = map(str,priorColumnClusters)
if geneOverride != None:
row_header = geneOverride.keys() ### Replace with a custom or expanded list of gene IDs to use
### Expand the possible set of usable IDs
matrix_exp, column_header_exp, row_header_exp, dataset_name, group_db_exp = clustering.importData(expdir,geneFilter=row_header)
percent_found = (len(row_header_exp)*1.00)/len(row_header)
if percent_found<0.5:
print "...Incompatible primary ID (Symbol), converting to Ensembl"
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
### Assume the original IDs were Ensembl IDs and get IDs from the filtered Expression File
filteredIDs = simpleICGSGeneImport([filtered_expdir])
row_header2=[]
index=0
for symbol in row_header:
if symbol in symbol_to_gene:
for gene in symbol_to_gene[symbol]:
if 'LRG' not in gene and gene in filteredIDs:
row_header2.append(gene)
gene_cluster_db[gene]=priorRowClusters[index]
index+=1
row_header = row_header2
matrix_exp, column_header_exp, row_header_exp, dataset_name, group_db_exp = clustering.importData(expdir,geneFilter=row_header)
### Correct fileheader if group prefix present
try: column_header_exp = map(lambda x: string.split(x,':')[1],column_header_exp)
except Exception: pass ### If no ":" in cell library names
try: sample_index_list = map(lambda x: column_header_exp.index(x), column_header)
except Exception: ### Hence, there are missing cells in the expression file
sample_index_list=[]
for cell in column_header:
if cell in column_header_exp:
sample_index_list.append(column_header_exp.index(cell))
print "WARNING... only",len(sample_index_list), "out of",len(column_header), "cells found in the expression file."
if '.Reference' not in column_header[-1]:
### Separately denote reference IDs from query sample IDs
column_header = map(lambda x: x+'.Reference',column_header)
if geneOverride != None:
eo.write(string.join(['UID']+column_header,'\t')+'\n')
else:
eo.write(string.join(['UID','row_clusters-flat']+column_header,'\t')+'\n')
eo.write(string.join(['column_clusters-flat','']+priorColumnClusters,'\t')+'\n')
index=0
reference_matrix = collections.OrderedDict() ### store the reordered data for later medioid calculation
for uid in row_header:
if uid in row_header_exp:
### Get the heatmap ordered gene entries
exp_row_index = row_header_exp.index(uid)
### Re-order the samples according to the heatmap
try: reordered_values = map(lambda x: str(matrix_exp[exp_row_index][x]), sample_index_list) ### simple and fast way to reorganize the samples
except Exception:
### For PSI files with missing values at the end of each line, often
if len(column_header_exp) != len(matrix_exp[exp_row_index]):
diff = len(column_header_exp)-len(matrix_exp[exp_row_index])
matrix_exp[exp_row_index]+=diff*['']
reordered_values = map(lambda x: str(matrix_exp[exp_row_index][x]), sample_index_list)
if geneOverride != None:
eo.write(string.join([uid]+reordered_values,'\t')+'\n')
reference_matrix[uid,0]=map(float,reordered_values)
else:
eo.write(string.join([uid,str(gene_cluster_db[uid])]+reordered_values,'\t')+'\n')
reference_matrix[uid,str(gene_cluster_db[uid])]=map(float,reordered_values)
index+=1
eo.close()
print 'New ICGS CellHarmony reference saved to:',cellHarmonyReferenceFile
### Create the groups for median calculation
group_index_db = collections.OrderedDict()
index=0
for cluster in priorColumnClusters:
try: group_index_db[cluster].append(index) #group_index_db['cluster-'+cluster].append(index)
except Exception: group_index_db[cluster]=[index] #group_index_db['cluster-'+cluster]=[index]
index+=1
cellHarmonyReferenceFileMediod = cellHarmonyReferenceFile[:-4]+'-centroid.txt'
eo = export.ExportFile(cellHarmonyReferenceFileMediod)
if geneOverride != None:
eo.write(string.join(['UID']+map(str,group_index_db),'\t')+'\n')
else:
eo.write(string.join(['UID','row_clusters-flat']+map(str,group_index_db),'\t')+'\n')
eo.write(string.join(['column_clusters-flat','']+map(lambda x: string.replace(x,'cluster-',''),group_index_db),'\t')+'\n')
from stats_scripts import statistics
try:
for uid in reference_matrix:
median_matrix=[]
for cluster in group_index_db:
if CenterMethod == 'mean' or CenterMethod == 'centroid':
try: median_matrix.append(str(statistics.avg(map(lambda x: reference_matrix[uid][x], group_index_db[cluster]))))
except Exception: ### Only one value
median_matrix.append(str(map(lambda x: reference_matrix[uid][x], group_index_db[cluster])))
else: ### Median
try: median_matrix.append(str(statistics.median(map(lambda x: reference_matrix[uid][x], group_index_db[cluster]))))
except Exception: ### Only one value
median_matrix.append(str(map(lambda x: reference_matrix[uid][x], group_index_db[cluster])))
if geneOverride != None:
eo.write(string.join([uid[0]]+median_matrix,'\t')+'\n')
else:
eo.write(string.join([uid[0],uid[1]]+median_matrix,'\t')+'\n')
eo.close()
except:
returnCentroids = False ### When an error occurs, use community alignment
try:
if combineFullDatasets:
""" Merge the query and the reference expression files """
print 'Producing a merged input and query expression file (be patient)...'
query_exp_matrix, query_header, query_row_header, null, group_db_exp = clustering.importData(query_exp_file)
reference_exp_matrix, reference_header, ref_row_header, null, group_db_exp = clustering.importData(expdir)
if ':' in reference_header[-1]:
reference_header = map(lambda x: string.split(x,':')[1],reference_header)
if '.Reference' not in reference_header[-1]:
### Separately denote reference IDs from query sample IDs
reference_header = map(lambda x: x+'.Reference',reference_header)
""" Simple combine of the two expression files without normalization """
try:
matchingUIDs=0
#ref_filename = export.findFilename(expdir)
ref_filename = export.findFilename(cellHarmonyReferenceFile)
query_filename = export.findFilename(query_exp_file)
root_dir = export.findParentDir(query_exp_file)
output_dir = ref_filename[:-4]+'__'+query_filename[:-4]+'-AllCells.txt' ### combine the filenames and query path
output_dir = string.replace(output_dir,'OutliersRemoved-','')
output_dir = string.replace(output_dir,'-centroid__','__')
### Matches the outputdir name from the function importAndCombineExpressionFiles
output_dir = string.replace(root_dir,'/ExpressionInput','')+'/cellHarmony/exp.'+string.replace(output_dir,'exp.','')
group_ref_export_dir = string.replace(output_dir,'exp.','groups.')
group_ref_export_dir = string.replace(group_ref_export_dir,'-AllCells.','-Reference.')
eo = export.ExportFile(output_dir)
eo.write(string.join(['UID']+reference_header+query_header,'\t')+'\n')
for uid in ref_row_header:
ri=ref_row_header.index(uid)
if uid in query_row_header:
qi=query_row_header.index(uid)
eo.write(string.join([uid]+map(str,reference_exp_matrix[ri])+map(str,query_exp_matrix[qi]),'\t')+'\n')
matchingUIDs+=1
eo.close()
print matchingUIDs,'Combined query and reference expression file exported to:',output_dir
eo = export.ExportFile(group_ref_export_dir)
index=0
for cell in column_header:
eo.write(cell+'\t'+str(priorColumnClusters[index])+'\t'+str(priorColumnClusters[index])+'\n')
index+=1
eo.close()
except:
print 'Failed to join the query and reference expression files due to:'
print traceback.format_exc()
except:
print 'Failed to join the query and reference expression files due to:'
print traceback.format_exc()
if returnCentroids == True or returnCentroids == 'yes' or returnCentroids == 'centroid':
print 'Using centroids rather than individual cells for alignment.'
return cellHarmonyReferenceFileMediod
else:
print 'Using invidual cells rather than cell centroids for alignment.'
return cellHarmonyReferenceFile
def simpleExpressionFileImport(filename,filterUID={}):
fn=filepath(filename)
expression_db = {}
firstRow = True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
header = t[1:]
firstRow = False
else:
if len(filterUID)>0:
if t[0] not in filterUID:
continue
expression_db[t[0]] = t[1:]
return expression_db, header
def simpleICGSGeneImport(files):
""" Import the gene IDs from different ICGS or MarkerFinder results prior
to combining to derive combined ICGS results and making combined medoid file"""
gene_db=collections.OrderedDict()
for file in files:
fn=filepath(file)
firstRow = True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
header = t[1:]
firstRow = False
else:
gene = t[0]
if '-flat' not in gene:
if gene not in gene_db:
if ':' in gene: ### If MarkerFinder as the input
geneID = string.split(gene,':')[1]
gene = string.split(geneID,' ')[0]
gene_db[gene]={}
return gene_db
def compareICGSpopulationFrequency(folder):
ea = export.ExportFile(folder+'/overlap.tsv')
files = unique.read_directory(folder)
cluster_counts={}
datasets = []
clusters=[]
from collections import Counter
for file in files:
if '.txt' in file:
datasets.append(file[:-4])
fn = folder+'/'+file
LineCount = 0
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
LineCount += 1
if LineCount<3:
if LineCount==2:
""" Examine the second row only for cluster counts """
dataset_cluster_counts = Counter(t[2:])
for cluster in dataset_cluster_counts:
try: cluster_counts[cluster].append(str(dataset_cluster_counts[cluster]))
except Exception: cluster_counts[cluster] = [str(dataset_cluster_counts[cluster])]
if cluster not in clusters:
clusters.append(cluster)
else:
break
clusters.sort()
ea.write(string.join(['Cluster']+datasets,'\t')+'\n')
for cluster in clusters:
ea.write(string.join([cluster]+cluster_counts[cluster],'\t')+'\n')
ea.close()
if __name__ == '__main__':
import UI
#### Begin post-alignment test
folds_file = '/Users/saljh8/Desktop/CCHMC-Board/exp.MarkerFinder-cellHarmony-reference__MergedFiles-ReOrdered.txt'
output = '/Users/saljh8/Dropbox/Collaborations/Jayati/Thpok/10X_Grimes_RR44_-l-_20191119_3v3mm-LGGEX8/'
labels = '/Users/saljh8/Dropbox/Collaborations/Jayati/Thpok/10X_Grimes_WT_20191119_3v3mm-LGGEX7/cellHarmony/QueryGroups.cellHarmony.txt'
platform = 'RNASeq'
fl = UI.ExpressionFileLocationData(folds_file,'','',''); species='Mm'; platform = 'RNASeq'
fl.setSpecies(species); fl.setVendor(platform)
fl.setOutputDir(output)
fl.setPeformDiffExpAnalysis(False)
fl.setLabels(labels)
species = 'Mm'
reference_exp_file = '/Users/saljh8/Dropbox/Collaborations/Jayati/Thpok/10X_Grimes_WT_20191119_3v3mm-LGGEX7/cellHarmony/OtherFiles/CellHarmonyReference/MarkerFinder-cellHarmony-reference-centroid.txt'
query_exp_file = '/Users/saljh8/Dropbox/Collaborations/Jayati/Thpok/10X_Grimes_RR44_-l-_20191119_3v3mm-LGGEX8/exp.10X_Grimes_RR44-OutliersRemoved-filtered.txt'
classification_file = '/Users/saljh8/Dropbox/Collaborations/Jayati/Thpok/10X_Grimes_RR44_-l-_20191119_3v3mm-LGGEX8/CellClassification/10X_Grimes_RR44-OutliersRemoved-CellClassification.txt'
harmonizeClassifiedSamples(species,reference_exp_file,query_exp_file,classification_file,fl=fl);sys.exit()
#### End post-alignment test
folds_file = '/Users/saljh8/Desktop/CCHMC-Board/exp.MarkerFinder-cellHarmony-reference__MergedFiles-ReOrdered.txt'
output = '/Users/saljh8/Desktop/CCHMC-Board/'
#DEGs_combined = aggregateRegulatedGenes('/Users/saljh8/Desktop/DemoData/cellHarmony/Mouse_BoneMarrow/inputFile/cellHarmony/DifferentialExpression_Fold_2.0_adjp_0.05')
#folds_file = '/Volumes/salomonis2/LabFiles/Dan-Schnell/To_cellHarmony/MIToSham/Input/cellHarmony/exp.ICGS-cellHarmony-reference__MI-AllCells-folds.txt'
#output = '/Volumes/salomonis2/LabFiles/Dan-Schnell/To_cellHarmony/MIToSham/Input/cellHarmony/'
#DEGs_combined = aggregateRegulatedGenes('/Volumes/salomonis2/LabFiles/Dan-Schnell/To_cellHarmony/MIToSham/Input/cellHarmony/DifferentialExpression_Fold_1.5_adjp_0.05_less')
#DEGs_combined = aggregateRegulatedGenes('/Volumes/salomonis2/CCHMC-Collaborations/Harinder-singh/scRNASeq/Day_35_Plasma_Cell_Precursors/Day_35/outs/filtered_feature_bc_matrix/cellHarmony/DifferentialExpression_Fold_1.2_adjp_0.05_less')
#DEGs_combined = aggregateRegulatedGenes('/Volumes/salomonis2/CCHMC-Collaborations/Harinder-singh/scRNASeq/Day_35_Plasma_Cell_Precursors/Day_35/outs/filtered_feature_bc_matrix/cellHarmony/DifferentialExpression_Fold_1.2_adjp_0.05',filterGenes=DEGs_combined)
platform = 'RNASeq'
fl = UI.ExpressionFileLocationData(folds_file,'','',''); species='Mm'; platform = 'RNASeq'
fl.setSpecies(species); fl.setVendor(platform)
fl.setOutputDir(output)
species = 'Mm'
#clustered_groups_file = findSimilarImpactedCellStates(folds_file,DEGs_combined)
#sys.exit()
#exportPvalueRankedGenes(species,platform,fl,folds_file,DEGs_combined)
#sys.exit()
"""
root_dir = output+'/DifferentialExpression_Fold_1.2_adjp_0.05/'
print 'Generating gene regulatory networks...'
import InteractionBuilder
pdfs=[]
pdfs = InteractionBuilder.remoteBuildNetworks(species, root_dir)
networks_dir = output+'/networks/'
try: os.mkdir(networks_dir)
except: pass
for pdf in pdfs:
file = export.findFilename(pdf)
file = string.replace(file,'AltAnalyze-network-WKT_GE.','')
file = string.replace(file,'_cellHarmony-Reference-interactions','')
shutil.copy(pdf,output+'/networks/'+file)
"""
#sys.exit()
"""
species = 'Hs'
reference_exp_file = '/Users/saljh8/Desktop/DemoData/sample_data/tempRef/FinalMarkerHeatmap_all.txt'
query_exp_file = '/Users/saljh8/Desktop/DemoData/sample_data/tempRef/cellHarmony-query_matrix_CPTT.txt'
classification_file = '/Users/saljh8/Desktop/DemoData/sample_data/tempRef/CellClassification/cellHarmony-query_matrix_CPTT-CellClassification.txt'
pearsonThreshold=0.3
peformDiffExpAnalysis=True
pvalThreshold=0.05
FoldCutoff=2
use_adjusted_pval=True
customLabels=None
output_file,query_output_file,folds_file,DEGs_combined = importAndCombineExpressionFiles(species,reference_exp_file,
query_exp_file,classification_file,pearsonThreshold=pearsonThreshold,peformDiffExpAnalysis=peformDiffExpAnalysis,
pvalThreshold=pvalThreshold,fold_cutoff=FoldCutoff,use_adjusted_pval=use_adjusted_pval,customLabels=customLabels)
sys.exit()
"""
output_file = '/Users/saljh8/Desktop/CCHMC-Board/exp.MarkerFinder-cellHarmony-reference__MergedFiles-ReOrdered.txt'
"""
if len(folds_file)<1:
folds_file = string.replace(output_file,'-ReOrdered','-AllCells-folds')
### Output the cellHarmony heatmaps
from visualization_scripts import clustering
row_method = None; row_metric = 'cosine'; column_method = None; column_metric = 'euclidean'; color_gradient = 'yellow_black_blue'
transpose = False; Normalize='median'
if runningCommandLine:
display = False
else:
display = True
display = False
query_output_file = '/Volumes/salomonis2/CCHMC-Collaborations/Harinder-singh/scRNASeq/Day_35_Plasma_Cell_Precursors/Day_35/outs/filtered_feature_bc_matrix/cellHarmony/exp.MarkerFinder-cellHarmony-reference__Day_35-ReOrdered-Query.txt'
print 'Exporting cellHarmony heatmaps...'
heatmaps_dir = output+'/heatmaps/'
try: os.mkdir(heatmaps_dir)
except: pass
try:
graphics = clustering.runHCexplicit(query_output_file, [], row_method, row_metric, column_method,
column_metric, color_gradient, transpose, Normalize=Normalize, contrast=5, display=display)
plot = graphics[-1][-1][:-4]+'.pdf'
file = graphics[-1][-1][:-4]+'.txt'
shutil.copy(plot,output+'/heatmaps/heatmap-query-aligned.pdf')
shutil.copy(file,output+'/heatmaps/heatmap-query-aligned.txt')
graphics = clustering.runHCexplicit(output_file, [], row_method, row_metric, column_method,
column_metric, color_gradient, transpose, Normalize=Normalize, contrast=5, display=display)
plot = graphics[-1][-1][:-4]+'.pdf'
file = graphics[-1][-1][:-4]+'.txt'
shutil.copy(plot,output+'/heatmaps/heatmap-all-cells-combined.pdf')
shutil.copy(file,output+'/heatmaps/heatmap-all-cells-combined.txt')
except:
print traceback.format_exc()
sys.exit()
"""
### Build-UMAP plot
import UI
import warnings
warnings.filterwarnings('ignore')
try:
try: os.mkdir(output+'/UMAP-plots')
except: pass
""" Output UMAP combined plot colored by reference and query cell identity """
plot = UI.performPCA(output_file, 'no', 'UMAP', False, None, plotType='2D',
display=False, geneSetName=None, species=species, zscore=False, reimportModelScores=True,
separateGenePlots=False, returnImageLoc=True)
plot = plot[-1][-1][:-4]+'.pdf'
shutil.copy(plot,output+'/UMAP-plots/UMAP-query-vs-ref.pdf')
""" Output UMAP combined plot colored by cell states """
plot = UI.performPCA(output_file, 'no', 'UMAP', False, None, plotType='2D',
display=False, geneSetName=None, species='Dr', zscore=False, reimportModelScores=True,
separateGenePlots=False, returnImageLoc=True, forceClusters=True)
plot = plot[-1][-1][:-4]+'.pdf'
shutil.copy(plot,output+'/UMAP-plots/UMAP-query-vs-ref-clusters.pdf')
""" Output individual UMAP plots colored by cell tates """
groups_file = string.replace(output_file,'exp.','groups.')
plots = UI.performPCA(output_file, 'no', 'UMAP', False, None, plotType='2D',
display=False, geneSetName=None, species='Mm', zscore=False, reimportModelScores=True,
separateGenePlots=False, returnImageLoc=True, forceClusters=True, maskGroups=groups_file)
for plot in plots:
plot = plot[-1][:-4]+'.pdf'
if '-cellHarmony-Reference-' in plot:
shutil.copy(plot,output+'/UMAP-plots/UMAP-ref-clusters.pdf')
else:
shutil.copy(plot,output+'/UMAP-plots/UMAP-query-clusters.pdf')
except ZeroDivisionError:
print traceback.format_exc()
pass
sys.exit()
exportPvalueRankedGenes(species,platform,fl,folds_file,DEGs_combined)
sys.exit()
icgs_dir = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/ICGS/Clustering-exp.NaturePanorma-Ly6G-Guide3-Augment-F2r-hierarchical_cosine_correlation.txt'
exp_dir = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/ExpressionInput/exp.Guide3-cellHarmony-revised.txt'
#convertICGSClustersToExpression(icgs_dir,exp_dir);sys.exit()
"""compareICGSpopulationFrequency('/Users/saljh8/Desktop/dataAnalysis/Collaborative/Jose/NewTranscriptome/cellHarmonyResults/');sys.exit()
"""
reference_exp_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/cellHarmony-evaluation/Grimes/exp.WT.txt'
query_exp_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/cellHarmony-evaluation/Grimes/exp.AML.txt'
classification_file= '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/cellHarmony-evaluation/Grimes/CellClassification/AML-CellClassification.txt'
folds_file = '/Users/saljh8/Desktop/DemoData/cellHarmony/Mouse_BoneMarrow/inputFile/cellHarmony/exp.ICGS-cellHarmony-reference__AML-AllCells-folds.txt'
output = '/Users/saljh8/Desktop/DemoData/cellHarmony/Mouse_BoneMarrow/inputFile/cellHarmony/'
species = 'Mm'
array_type = 'RNASeq'
parent_dir = output+'/DifferentialExpression_Fold_1.5_rawp_0.05/'
dir_list = unique.read_directory(parent_dir)
import UI
for file in dir_list:
input_file_dir = parent_dir+'/'+file
inputType = 'IDs'
interactionDirs = ['WikiPathways','KEGG','BioGRID','TFTargets']
output_dir = parent_dir
degrees = 'direct'
input_exp_file = input_file_dir
gsp = UI.GeneSelectionParameters(species,array_type,array_type)
gsp.setGeneSet('None Selected')
gsp.setPathwaySelect('')
gsp.setGeneSelection('')
gsp.setOntologyID('')
gsp.setIncludeExpIDs(True)
UI.networkBuilder(input_file_dir,inputType,output_dir,interactionDirs,degrees,input_exp_file,gsp,'')
sys.exit()
DEGs_combined = aggregateRegulatedGenes(output+'/DifferentialExpression_Fold_1.5_rawp_0.05')
exportCombinedMarkerFinderResults(folds_file,output+'/MarkerFinder-positive',output+'/MarkerFinder-negative',DEGs_combined,species)
sys.exit()
harmonizeClassifiedSamples('Hs',reference_exp_file,query_exp_file,classification_file);sys.exit()
#modelScores('/Users/saljh8/Desktop/dataAnalysis/LineageProfiler/Training/CellClassification');sys.exit()
#allPairwiseSampleCorrelation('/Users/saljh8/Desktop/Sarwal-New/UCRM_bx.txt');sys.exit()
try:
import multiprocessing as mlp
mlp.freeze_support()
except Exception:
mpl = None
################ Default Variables ################
species = 'Hs'
platform = "exon"
vendor = 'Affymetrix'
compendium_platform = "exon"
codingtype = 'protein_coding'
platform = vendor, platform
exp_output = None
geneModels = False
modelSize = None
permute = False
useMulti = False
finalNumberSetsToOutput = 10
cross_validation = False
""" This script iterates the LineageProfiler algorithm (correlation based classification method) to identify sample types relative
two one of two references given one or more gene models. The program '
"""
#python LineageProfilerIterate.py --i "/Users/nsalomonis/Desktop/dataAnalysis/qPCR/ExpressionInput/exp.ABI_Pediatric.txt" --r "/Users/nsalomonis/Desktop/dataAnalysis/qPCR/ExpressionOutput/MarkerFinder/MarkerFinder-ABI_Pediatric.txt" --m "/Users/nsalomonis/Desktop/dataAnalysis/qPCR/ExpressionInput/7GeneModels.txt"
#python LineageProfilerIterate.py --i "/Users/nsalomonis/Desktop/dataAnalysis/qPCR/deltaCT/LabMeeting/ExpressionInput/exp.ABI_PediatricSNS.txt" --r "/Users/nsalomonis/Desktop/dataAnalysis/qPCR/ExpressionOutput/MarkerFinder/MarkerFinder-ABI_PediatricSNS.txt" --s 4
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Please designate a tab-delimited input expression file in the command-line"
print 'Example: python LineageProfilerIterate.py --i "/Users/me/qPCR.txt" --r "/Users/me/reference.txt" --m "/Users/me/models.txt"'
else:
try:
options, remainder = getopt.getopt(sys.argv[1:],'', ['i=','species=','o=','platform=','codingtype=',
'compendium_platform=','r=','m=','v=','s=','permute=','useMulti=',
'cross_validation=','setsToOutput='])
except Exception,e:
print ""
for opt, arg in options:
if opt == '--i': exp_input=arg
elif opt == '--o': exp_output=arg
elif opt == '--platform': platform=arg
elif opt == '--codingtype': codingtype=arg
elif opt == '--compendium_platform': compendium_platform=arg
elif opt == '--r': customMarkers=arg
elif opt == '--m': geneModels=arg
elif opt == '--v': vendor=arg
elif opt == '--permute': permute=True
elif opt == '--useMulti': useMulti=True
elif opt == '--cross_validation': cross_validation = True
elif opt == '--setsToOutput': finalNumberSetsToOutput = int(arg)
elif opt == '--s':
try: modelSize = int(arg)
except Exception:
modelSize = arg
if modelSize != 'optimize':
print 'Please specify a modelSize (e.g., 7-gene model search) as a single integer (e.g., 7)'
sys.exit()
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
if exp_output == None: exp_output = exp_input
if cross_validation == True:
### Generate 2/3rd and 1/3rd sets for testing and validation
crossValidationAnalysis(species,platform,exp_input,exp_output,codingtype,compendium_platform,modelSize,
geneModels,permute,useMulti,finalNumberSetsToOutput)
sys.exit()
runLineageProfiler(species,platform,exp_input,exp_output,codingtype,compendium_platform,modelSize=modelSize,
customMarkers=customMarkers,geneModels=geneModels,permute=permute,useMulti=useMulti)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/LineageProfilerIterate.py
|
LineageProfilerIterate.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from Tkinter import *
import webbrowser
MOVE_LINES = 0
MOVE_PAGES = 1
MOVE_TOEND = 2
class TableViewer(Frame):
def __init__(self, master, lists, command=None, **options):
defaults = {
'background': None,
'borderwidth': 2,
'font': None,
'foreground': None,
'height': 10,
'highlightcolor': None,
'highlightthickness': 1,
'relief': SUNKEN,
'takefocus': 1,
}
aliases = {'bg':'background', 'fg':'foreground', 'bd':'borderwidth'}
for k in aliases.keys ():
if options.has_key (k):
options [aliases[k]] = options [k]
for key in defaults.keys():
if not options.has_key (key):
options [key] = defaults [key]
apply (Frame.__init__, (self, master), options)
self.lists = []
# MH (05/20049
# These are needed for sorting
self.colmapping={}
self.origData = None
# Keyboard navigation.
self.bind ('<Up>', lambda e, s=self: s._move (-1, MOVE_LINES))
self.bind ('<Down>', lambda e, s=self: s._move (+1, MOVE_LINES))
self.bind ('<Prior>', lambda e, s=self: s._move (-1, MOVE_PAGES))
self.bind ('<Next>', lambda e, s=self: s._move (+1, MOVE_PAGES))
self.bind ('<Home>', lambda e, s=self: s._move (-1, MOVE_TOEND))
self.bind ('<End>', lambda e, s=self: s._move (+1, MOVE_TOEND))
if command:
self.bind ('<Return>', command)
# Columns are a frame with listbox and label in it.
# MH (05/2004):
# Introduced a PanedWindow to make the columns resizable
m = PanedWindow(self, orient=HORIZONTAL, bd=0,
background=options['background'], showhandle=0, sashpad=1)
m.pack(side=LEFT, fill=BOTH, expand=1)
for label, width in lists:
lbframe = Frame(m)
m.add(lbframe, width=width)
# MH (05/2004)
# modified this, click to sort
b = Label(lbframe, text=label, borderwidth=1, relief=RAISED)
b.pack(fill=X)
b.bind('<Button-1>', self._sort)
self.colmapping[b]=(len(self.lists),1)
lb = Listbox (lbframe,
width=width,
height=options ['height'],
borderwidth=0,
font=options ['font'],
background=options ['background'],
selectborderwidth=0,
relief=SUNKEN,
takefocus=FALSE,
exportselection=FALSE)
lb.pack (expand=YES, fill=BOTH)
self.lists.append (lb)
# Mouse features
lb.bind ('<B1-Motion>', lambda e, s=self: s._select (e.y))
lb.bind ('<Button-1>', lambda e, s=self: s._select (e.y))
lb.bind ('<Leave>', lambda e: 'break')
lb.bind ('<B2-Motion>', lambda e, s=self: s._b2motion (e.x, e.y))
lb.bind ('<Button-2>', lambda e, s=self: s._button2 (e.x, e.y))
if command:
lb.bind ('<Double-Button-1>', command)
sbframe = Frame (self)
sbframe.pack (side=LEFT, fill=Y)
l = Label (sbframe, borderwidth=1, relief=RAISED)
l.bind ('<Button-1>', lambda e, s=self: s.focus_set ())
l.pack(fill=X)
sb = Scrollbar (sbframe,
takefocus=FALSE,
orient=VERTICAL,
command=self._scroll)
sb.pack (expand=YES, fill=Y)
self.lists[0]['yscrollcommand']=sb.set
return
# MH (05/2004)
# Sort function, adopted from Rick Lawson
# http://tkinter.unpythonic.net/wiki/SortableTable
def _sort(self, e):
# get the listbox to sort by (mapped by the header button)
b=e.widget
col, direction = self.colmapping[b]
# get the entire table data into mem
tableData = self.get(0,END)
if self.origData == None:
import copy
self.origData = copy.deepcopy(tableData)
rowcount = len(tableData)
#remove old sort indicators if it exists
for btn in self.colmapping:
lab = btn.cget('text')
if lab[0]=='[': btn.config(text=lab[4:])
btnLabel = b.cget('text')
#sort data based on direction
if direction==0:
tableData = self.origData
else:
if direction==1: b.config(text='[+] ' + btnLabel)
else: b.config(text='[-] ' + btnLabel)
# sort by col
tableData.sort(key=lambda x: x[col], reverse=direction<0)
#clear widget
self.delete(0,END)
# refill widget
for row in range(rowcount):
self.insert(END, tableData[row])
# toggle direction flag
if direction==1: direction=-1
else: direction += 1
self.colmapping[b] = (col, direction)
def _move (self, lines, relative=0):
"""
Move the selection a specified number of lines or pages up or
down the list. Used by keyboard navigation.
"""
selected = self.lists [0].curselection ()
try:
selected = map (int, selected)
except ValueError:
pass
try:
sel = selected [0]
except IndexError:
sel = 0
old = sel
size = self.lists [0].size ()
if relative == MOVE_LINES:
sel = sel + lines
elif relative == MOVE_PAGES:
sel = sel + (lines * int (self.lists [0]['height']))
elif relative == MOVE_TOEND:
if lines < 0:
sel = 0
elif lines > 0:
sel = size - 1
else:
print "TableViewer._move: Unknown move type!"
if sel < 0:
sel = 0
elif sel >= size:
sel = size - 1
self.selection_clear (old, old)
self.see (sel)
self.selection_set (sel)
return 'break'
def _select (self, y):
"""
User clicked an item to select it.
"""
row = self.lists[0].nearest (y)
label = labels[row]
try: webbrowser.open('http://www.genecards.org/cgi-bin/carddisp.pl?gene='+label)
except Exception: null=[]
self.selection_clear (0, END)
self.selection_set (row)
self.focus_set ()
return 'break'
def _button2 (self, x, y):
"""
User selected with button 2 to start a drag.
"""
for l in self.lists:
l.scan_mark (x, y)
return 'break'
def _b2motion (self, x, y):
"""
User is dragging with button 2.
"""
for l in self.lists:
l.scan_dragto (x, y)
return 'break'
def _scroll (self, *args):
"""
Scrolling with the scrollbar.
"""
for l in self.lists:
apply(l.yview, args)
def curselection (self):
"""
Return index of current selection.
"""
return self.lists[0].curselection()
def delete (self, first, last=None):
"""
Delete one or more items from the list.
"""
for l in self.lists:
l.delete(first, last)
def get (self, first, last=None):
"""
Get items between two indexes, or one item if second index
is not specified.
"""
result = []
for l in self.lists:
result.append (l.get (first,last))
if last:
return apply (map, [None] + result)
return result
def index (self, index):
"""
Adjust the view so that the given index is at the top.
"""
for l in self.lists:
l.index (index)
def insert (self, index, *elements):
"""
Insert list or tuple of items.
"""
for e in elements:
i = 0
for l in self.lists:
l.insert (index, e[i])
i = i + 1
if self.size () == 1:
self.selection_set (0)
def size (self):
"""
Return the total number of items.
"""
return self.lists[0].size ()
def see (self, index):
"""
Make sure given index is visible.
"""
for l in self.lists:
l.see (index)
def selection_anchor (self, index):
"""
Set selection anchor to index.
"""
for l in self.lists:
l.selection_anchor (index)
def selection_clear (self, first, last=None):
"""
Clear selections between two indexes.
"""
for l in self.lists:
l.selection_clear (first, last)
def selection_includes (self, index):
"""
Determine if given index is selected.
"""
return self.lists[0].selection_includes (index)
def selection_set (self, first, last=None):
"""
Select a range of indexes.
"""
for l in self.lists:
l.selection_set (first, last)
def viewTable(title,header,list_values):
global labels
tk = Toplevel()
Label(tk, text=title).pack()
column_values = []
labels=[]
import string
for i in header:
width = 120
column_values.append((i,width))
mlb = TableViewer (tk,
tuple(column_values),
height=20,
bg='white')
for i in list_values:
mlb.insert (END, i)
labels.append(i[0])
mlb.pack (expand=YES,fill=BOTH)
def deleteWindow():
tk.quit()
tk.destroy() ### just quit instead
try: tk.clipboard_clear()
except Exception: pass
try: tk.clipboard_append(string.join(labels,'\n'))
except Exception: pass
Button (tk, text="Close", command = deleteWindow).pack ()
tk.protocol("WM_DELETE_WINDOW", deleteWindow)
tk.mainloop()
if __name__ == '__main__':
title = 'test'
list_values = [(1,2,3)]
header = ['1','2','3']
header = ['Associated Genes']
list_values = [[('CAMK2D')],[('TTN')]]
viewTable(title,header,list_values)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/TableViewer.py
|
TableViewer.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import platform
useDefaultBackend=False
if platform.system()=='Darwin':
if platform.mac_ver()[0] == '10.14.6':
useDefaultBackend=True
import sys,string,os,copy
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>0 and '--' in command_args: commandLine=True
else: commandLine=False
display_label_names = True
benchmark = False
cluster_colors = 'Paired' #Paired #gist_ncar
import traceback
try:
import math
import warnings
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
import matplotlib
if commandLine and 'linux' in sys.platform:
### TkAgg doesn't work when AltAnalyze is run remotely (ssh or sh script)
try: matplotlib.use('Agg');
except Exception: pass
try:
matplotlib.rcParams['backend'] = 'Agg'
except Exception: pass
else:
try:
if useDefaultBackend == False:
import matplotlib.backends.backend_tkagg
if platform.system()=='Darwin':
matplotlib.use('macosx')
else:
matplotlib.use('TkAgg')
except Exception: pass
if useDefaultBackend == False:
if platform.system()=='Darwin':
try: matplotlib.rcParams['backend'] = 'macosx'
except Exception: pass
else:
try: matplotlib.rcParams['backend'] = 'TkAgg'
except Exception: pass
try:
import matplotlib.pyplot as pylab
import matplotlib.colors as mc
import matplotlib.mlab as mlab
import matplotlib.ticker as tic
from matplotlib.patches import Circle
import mpl_toolkits
from mpl_toolkits import mplot3d
from mpl_toolkits.mplot3d import Axes3D
try: from matplotlib.cbook import _string_to_bool
except: pass
matplotlib.rcParams['axes.linewidth'] = 0.5
matplotlib.rcParams['pdf.fonttype'] = 42
matplotlib.rcParams['font.family'] = 'sans-serif'
matplotlib.rcParams['font.sans-serif'] = 'Arial'
matplotlib.rcParams['figure.facecolor'] = 'white' ### Added in 2.1.2
#matplotlib.rcParams['figure.dpi'] = 200 ### Control the image resolution for pylab.show()
except Exception:
print traceback.format_exc()
print 'Matplotlib support not enabled'
import scipy
try: from scipy.sparse.csgraph import _validation
except Exception: pass
try: from scipy import stats
except: pass
try:
from scipy.linalg import svd
import scipy.special._ufuncs_cxx
from scipy.spatial import _voronoi
from scipy.spatial import _spherical_voronoi
from scipy.spatial import qhull
import scipy._lib.messagestream
except Exception:
pass
import scipy.cluster.hierarchy as sch
import scipy.spatial.distance as dist
#import scipy.interpolate.interpnd
#from scipy import optimize
try: import numpy; np = numpy
except:
print 'Numpy import error...'
print traceback.format_exc()
### The below is used for binary freeze dependency identification
if 'darwin' in sys.platform:
### The below is used for binary freeze dependency identification
try: import umap
except: pass
try:
from cairo import ImageSurface
except: pass
try:
import igraph.vendor.texttable
except ImportError: pass
try:
from sklearn.decomposition import PCA, FastICA
except Exception: pass
try: from sklearn.neighbors import quad_tree ### supported in sklearn>18.2
except: pass
try: import sklearn.utils.sparsetools._graph_validation
except: pass
try: import sklearn.utils.weight_vector
except: pass
from sklearn.neighbors import *
from sklearn.manifold.t_sne import *
from sklearn.tree import *; from sklearn.tree import _utils
from sklearn.manifold.t_sne import _utils
from sklearn.manifold import TSNE
from sklearn.neighbors import NearestNeighbors
import sklearn.linear_model.sgd_fast
import sklearn.utils.lgamma
try: import scipy.special.cython_special
except: pass
import sklearn.neighbors.typedefs
import sklearn.neighbors.ball_tree
try:
import numba
import numba.config
import llvmlite; from llvmlite import binding; from llvmlite.binding import *
from llvmlite.binding import ffi; from llvmlite.binding import dylib
except:
pass
#pylab.ion() # closes Tk window after show - could be nice to include
except Exception:
print traceback.format_exc()
pass
try: import numpy
except: pass
import time
import unique
from stats_scripts import statistics
import os
import export
import webbrowser
import warnings
import UI
use_default_colors = False
try:
warnings.simplefilter("ignore", numpy.ComplexWarning)
warnings.simplefilter("ignore", DeprecationWarning) ### Annoying depreciation warnings (occurs in sch somewhere)
#This shouldn't be needed in python 2.7 which suppresses DeprecationWarning - Larsson
except Exception: None
from visualization_scripts import WikiPathways_webservice
try:
import fastcluster as fc
#print 'Using fastcluster instead of scipy hierarchical cluster'
#fc = sch
except Exception:
#print 'Using scipy insteady of fastcluster (not installed)'
try: fc = sch ### fastcluster uses the same convention names for linkage as sch
except Exception: print 'Scipy support not present...'
def getColorRange(x):
""" Determines the range of colors, centered at zero, for normalizing cmap """
vmax=x.max()
vmin=x.min()
if vmax<0 and vmin<0: direction = 'negative'
elif vmax>0 and vmin>0: direction = 'positive'
else: direction = 'both'
if direction == 'both':
vmax = max([vmax,abs(vmin)])
vmin = -1*vmax
return vmax,vmin
else:
return vmax,vmin
def heatmap(x, row_header, column_header, row_method, column_method, row_metric, column_metric, color_gradient,
dataset_name, display=False, contrast=None, allowAxisCompression=True,Normalize=True,
PriorColumnClusters=None, PriorRowClusters=None):
print "Performing hieararchical clustering using %s for columns and %s for rows" % (column_metric,row_metric)
show_color_bars = True ### Currently, the color bars don't exactly reflect the dendrogram colors
try: ExportCorreleationMatrix = exportCorreleationMatrix
except Exception: ExportCorreleationMatrix = False
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
if display == False:
pylab.figure() ### Add this to avoid a Tkinter bug after running MarkerFinder (not sure why it is needed) - creates a second empty window when display == True
if row_method == 'hopach' or column_method == 'hopach':
### Test R and hopach
"""
try:
import R_test
except Exception,e:
#print traceback.format_exc()
print 'Failed to install hopach or R not installed (install R before using hopach)'
row_method = 'average'; column_method = 'average'
if len(column_header)==2: column_method = 'average'
if len(row_header)==2: row_method = 'average'
"""
pass
"""
Prototype methods:
http://old.nabble.com/How-to-plot-heatmap-with-matplotlib--td32534593.html
http://stackoverflow.com/questions/7664826/how-to-get-flat-clustering-corresponding-to-color-clusters-in-the-dendrogram-cre
Scaling the color gradient so that zero is white:
http://stackoverflow.com/questions/2369492/generate-a-heatmap-in-matplotlib-using-a-scatter-data-set
Other cluster methods:
http://stackoverflow.com/questions/9362304/how-to-get-centroids-from-scipys-hierarchical-agglomerative-clustering
x is a m by n ndarray, m observations, n genes
"""
### Perform the associated clustering by HOPACH via PYPE or Rpy to R
if row_method == 'hopach' or column_method == 'hopach':
try:
""" HOPACH is a clustering method implemented in R that builds a hierarchical tree of clusters by recursively
partitioning a data set, while ordering and possibly collapsing clusters at each level:
http://www.bioconductor.org/packages/release/bioc/html/hopach.html
"""
import R_interface
#reload(R_interface)
if row_method == 'hopach' and column_method == 'hopach': cluster_method = 'both'
elif row_method == 'hopach': cluster_method = 'gene'
else: cluster_method = 'array'
if row_metric == 'cosine': metric_gene = "euclid"
elif row_metric == 'euclidean': metric_gene = "cosangle"
elif row_metric == 'correlation': metric_gene = "cor"
else: metric_gene = "cosangle"
if column_metric == 'cosine': metric_array = "euclid"
elif column_metric == 'euclidean': metric_array = "cosangle"
elif column_metric == 'correlation': metric_array = "cor"
else: metric_array = "euclid"
### Returned are the row_order and column_order in the Scipy clustering output format
newFilename, Z1, Z2 = R_interface.remoteHopach(inputFilename,cluster_method,metric_gene,metric_array)
if newFilename != inputFilename:
### If there were duplicates, re-import the matrix data for the cleaned up filename
try:
matrix, column_header, row_header, dataset_name, group_db = importData(newFilename,Normalize=normalize,reverseOrder=False)
except Exception:
matrix, column_header, row_header, dataset_name, group_db = importData(newFilename)
x = numpy.array(matrix)
except Exception:
row_method = 'average'; column_method = 'average'
print traceback.format_exc()
print 'hopach failed... continue with an alternative method'
skipClustering = False
try:
if len(PriorColumnClusters)>0 and len(PriorRowClusters)>0 and row_method==None and column_method == None:
print 'Prior generated clusters being used rather re-clustering'
"""
try:
if len(targetGeneIDs)>0:
PriorColumnClusters=[] ### If orderded genes input, we want to retain this order rather than change
except Exception: pass
"""
if len(PriorColumnClusters)>0: ### this corresponds to the above line
Z1={}; Z2={}
Z1['level'] = PriorRowClusters; Z1['level'].reverse()
Z2['level'] = PriorColumnClusters; #Z2['level'].reverse()
Z1['leaves'] = range(0,len(row_header)); #Z1['leaves'].reverse()
Z2['leaves'] = range(0,len(column_header)); #Z2['leaves'].reverse()
skipClustering = True
### When clusters are imported, you need something other than None, otherwise, you need None (need to fix here)
row_method = None
column_method = None
row_method = 'hopach'
column_method = 'hopach'
except Exception,e:
#print traceback.format_exc()
pass
n = len(x[0]); m = len(x)
if color_gradient == 'red_white_blue':
cmap=pylab.cm.bwr
if color_gradient == 'red_black_sky':
cmap=RedBlackSkyBlue()
if color_gradient == 'red_black_blue':
cmap=RedBlackBlue()
if color_gradient == 'red_black_green':
cmap=RedBlackGreen()
if color_gradient == 'yellow_black_blue':
cmap=YellowBlackBlue()
if color_gradient == 'black_yellow_blue':
cmap=BlackYellowBlue()
if color_gradient == 'seismic':
cmap=pylab.cm.seismic
if color_gradient == 'green_white_purple':
cmap=pylab.cm.PiYG_r
if color_gradient == 'coolwarm':
cmap=pylab.cm.coolwarm
if color_gradient == 'Greys':
cmap=pylab.cm.Greys
if color_gradient == 'yellow_orange_red':
cmap=pylab.cm.YlOrRd
if color_gradient == 'Spectral':
cmap = pylab.cm.Spectral_r
vmin=x.min()
vmax=x.max()
vmax = max([vmax,abs(vmin)])
if Normalize != False:
vmin = vmax*-1
elif 'Clustering-Zscores-' in dataset_name:
vmin = vmax*-1
elif vmin<0 and vmax>0 and Normalize==False:
vmin = vmax*-1
#vmin = vmax*-1
#print vmax, vmin
default_window_hight = 8.5
default_window_width = 12
if len(column_header)>80:
default_window_width = 14
if len(column_header)>100:
default_window_width = 16
if contrast == None:
scaling_factor = 2.5 #2.5
else:
try: scaling_factor = float(contrast)
except Exception: scaling_factor = 2.5
#print vmin/scaling_factor
norm = matplotlib.colors.Normalize(vmin/scaling_factor, vmax/scaling_factor) ### adjust the max and min to scale these colors by 2.5 (1 scales to the highest change)
#fig = pylab.figure(figsize=(default_window_width,default_window_hight)) ### could use m,n to scale here
fig = pylab.figure() ### could use m,n to scale here - figsize=(12,10)
fig.set_figwidth(12)
fig.set_figheight(7)
fig.patch.set_facecolor('white')
pylab.rcParams['font.size'] = 7.5
#pylab.rcParams['axes.facecolor'] = 'white' ### Added in 2.1.2
if show_color_bars == False:
color_bar_w = 0.000001 ### Invisible but not gone (otherwise an error persists)
else:
color_bar_w = 0.0125 ### Sufficient size to show
bigSampleDendrogram = True
if bigSampleDendrogram == True and row_method==None and column_method != None and allowAxisCompression == True:
dg2 = 0.30
dg1 = 0.43
else: dg2 = 0.1; dg1 = 0.63
try:
if EliteGeneSets != [''] and EliteGeneSets !=[]:
matrix_horiz_pos = 0.27
elif skipClustering:
if len(row_header)<100:
matrix_horiz_pos = 0.20
else:
matrix_horiz_pos = 0.27
else:
matrix_horiz_pos = 0.14
except Exception:
matrix_horiz_pos = 0.14
""" Adjust the position of the heatmap based on the number of columns """
if len(column_header)<50:
matrix_horiz_pos+=0.1
## calculate positions for all elements
# ax1, placement of dendrogram 1, on the left of the heatmap
[ax1_x, ax1_y, ax1_w, ax1_h] = [0.05,0.235,matrix_horiz_pos,dg1] ### The last controls matrix hight, second value controls the position of the matrix relative to the bottom of the view [0.05,0.22,0.2,0.6]
width_between_ax1_axr = 0.004
height_between_ax1_axc = 0.004 ### distance between the top color bar axis and the matrix
# axr, placement of row side colorbar
[axr_x, axr_y, axr_w, axr_h] = [0.31,0.1,color_bar_w-0.002,0.6] ### second to last controls the width of the side color bar - 0.015 when showing [0.31,0.1,color_bar_w,0.6]
axr_x = ax1_x + ax1_w + width_between_ax1_axr
axr_y = ax1_y; axr_h = ax1_h
width_between_axr_axm = 0.004
# axc, placement of column side colorbar (3rd value controls the width of the matrix!)
[axc_x, axc_y, axc_w, axc_h] = [0.5,0.63,0.5,color_bar_w] ### last one controls the hight of the top color bar - 0.015 when showing [0.4,0.63,0.5,color_bar_w]
""" Adjust the width of the heatmap based on the number of columns """
if len(column_header)<50:
axc_w = 0.3
if len(column_header)<20:
axc_w = 0.2
axc_x = axr_x + axr_w + width_between_axr_axm
axc_y = ax1_y + ax1_h + height_between_ax1_axc
height_between_axc_ax2 = 0.004
# axm, placement of heatmap for the data matrix
[axm_x, axm_y, axm_w, axm_h] = [0.4,0.9,2.5,0.5] #[0.4,0.9,2.5,0.5]
axm_x = axr_x + axr_w + width_between_axr_axm
axm_y = ax1_y; axm_h = ax1_h
axm_w = axc_w
# ax2, placement of dendrogram 2, on the top of the heatmap
[ax2_x, ax2_y, ax2_w, ax2_h] = [0.3,0.72,0.6,dg2] ### last one controls hight of the dendrogram [0.3,0.72,0.6,0.135]
ax2_x = axr_x + axr_w + width_between_axr_axm
ax2_y = ax1_y + ax1_h + height_between_ax1_axc + axc_h + height_between_axc_ax2
ax2_w = axc_w
# axcb - placement of the color legend
[axcb_x, axcb_y, axcb_w, axcb_h] = [0.02,0.938,0.17,0.025] ### Last one controls the hight [0.07,0.88,0.18,0.076]
# axcc - placement of the colum colormap legend colormap (distinct map)
[axcc_x, axcc_y, axcc_w, axcc_h] = [0.02,0.12,0.17,0.025] ### Last one controls the hight [0.07,0.88,0.18,0.076]
# Compute and plot top dendrogram
if column_method == 'hopach':
ind2 = numpy.array(Z2['level']) ### from R_interface - hopach root cluster level
elif column_method != None:
start_time = time.time()
#print x;sys.exit()
d2 = dist.pdist(x.T)
#print d2
#import mdistance2
#d2 = mdistance2.mpdist(x.T)
#print d2;sys.exit()
D2 = dist.squareform(d2)
ax2 = fig.add_axes([ax2_x, ax2_y, ax2_w, ax2_h], frame_on=False)
if ExportCorreleationMatrix:
new_matrix=[]
for i in D2:
#string.join(map(inverseDist,i),'\t')
log2_data = map(inverseDist,i)
avg = statistics.avg(log2_data)
log2_norm = map(lambda x: x-avg,log2_data)
new_matrix.append(log2_norm)
x = numpy.array(new_matrix)
row_header = column_header
#sys.exit()
Y2 = fc.linkage(D2, method=column_method, metric=column_metric) ### array-clustering metric - 'average', 'single', 'centroid', 'complete'
#Y2 = sch.fcluster(Y2, 10, criterion = "maxclust")
try: Z2 = sch.dendrogram(Y2)
except Exception:
if column_method == 'average':
column_metric = 'euclidean'
else: column_method = 'average'
Y2 = fc.linkage(D2, method=column_method, metric=column_metric)
Z2 = sch.dendrogram(Y2)
#ind2 = sch.fcluster(Y2,0.6*D2.max(), 'distance') ### get the correlations
#ind2 = sch.fcluster(Y2,0.2*D2.max(), 'maxclust') ### alternative method biased based on number of clusters to obtain (like K-means)
ind2 = sch.fcluster(Y2,0.7*max(Y2[:,2]),'distance') ### This is the default behavior of dendrogram
ax2.set_xticks([]) ### Hides ticks
ax2.set_yticks([])
time_diff = str(round(time.time()-start_time,1))
print 'Column clustering completed in %s seconds' % time_diff
else:
ind2 = ['NA']*len(column_header) ### Used for exporting the flat cluster data
# Compute and plot left dendrogram
if row_method == 'hopach':
ind1 = numpy.array(Z1['level']) ### from R_interface - hopach root cluster level
elif row_method != None:
start_time = time.time()
d1 = dist.pdist(x)
D1 = dist.squareform(d1) # full matrix
# postion = [left(x), bottom(y), width, height]
#print D1;sys.exit()
Y1 = fc.linkage(D1, method=row_method, metric=row_metric) ### gene-clustering metric - 'average', 'single', 'centroid', 'complete'
no_plot=False ### Indicates that we want to show the dendrogram
try:
if runGOElite: no_plot = True
elif len(PriorColumnClusters)>0 and len(PriorRowClusters)>0 and row_method==None and column_method == None:
no_plot = True ### If trying to instantly view prior results, no dendrogram will be display, but prior GO-Elite can
else:
ax1 = fig.add_axes([ax1_x, ax1_y, ax1_w, ax1_h], frame_on=False) # frame_on may be False - this window conflicts with GO-Elite labels
except Exception:
ax1 = fig.add_axes([ax1_x, ax1_y, ax1_w, ax1_h], frame_on=False) # frame_on may be False
try: Z1 = sch.dendrogram(Y1, orientation='left',no_plot=no_plot) ### This is where plotting occurs - orientation 'right' in old matplotlib
except Exception:
row_method = 'average'
try:
Y1 = fc.linkage(D1, method=row_method, metric=row_metric)
Z1 = sch.dendrogram(Y1, orientation='right',no_plot=no_plot)
except Exception:
row_method = 'ward'
Y1 = fc.linkage(D1, method=row_method, metric=row_metric)
Z1 = sch.dendrogram(Y1, orientation='right',no_plot=no_plot)
#ind1 = sch.fcluster(Y1,0.6*D1.max(),'distance') ### get the correlations
#ind1 = sch.fcluster(Y1,0.2*D1.max(),'maxclust')
ind1 = sch.fcluster(Y1,0.7*max(Y1[:,2]),'distance') ### This is the default behavior of dendrogram
if ExportCorreleationMatrix:
Z1 = sch.dendrogram(Y2, orientation='right')
Y1 = Y2
d1 = d2
D1 = D2
ind1 = ind2
try: ax1.set_xticks([]); ax1.set_yticks([]) ### Hides ticks
except Exception: pass
time_diff = str(round(time.time()-start_time,1))
print 'Row clustering completed in %s seconds' % time_diff
else:
ind1 = ['NA']*len(row_header) ### Used for exporting the flat cluster data
# Plot distance matrix.
axm = fig.add_axes([axm_x, axm_y, axm_w, axm_h]) # axes for the data matrix
xt = x
if column_method != None:
idx2 = Z2['leaves'] ### apply the clustering for the array-dendrograms to the actual matrix data
xt = xt[:,idx2]
#ind2 = ind2[:,idx2] ### reorder the flat cluster to match the order of the leaves the dendrogram
""" Error can occur here if hopach was selected in a prior run but now running NONE """
try: ind2 = [ind2[i] for i in idx2] ### replaces the above due to numpy specific windows version issue
except Exception:
column_method=None
xt = x
ind2 = ['NA']*len(column_header) ### Used for exporting the flat cluster data
ind1 = ['NA']*len(row_header) ### Used for exporting the flat cluster data
if row_method != None:
idx1 = Z1['leaves'] ### apply the clustering for the gene-dendrograms to the actual matrix data
prior_xt = xt
xt = xt[idx1,:] # xt is transformed x
#ind1 = ind1[idx1,:] ### reorder the flat cluster to match the order of the leaves the dendrogram
try: ind1 = [ind1[i] for i in idx1] ### replaces the above due to numpy specific windows version issue
except Exception:
if 'MarkerGenes' in dataset_name:
ind1 = ['NA']*len(row_header) ### Used for exporting the flat cluster data
row_method = None
### taken from http://stackoverflow.com/questions/2982929/plotting-results-of-hierarchical-clustering-ontop-of-a-matrix-of-data-in-python/3011894#3011894
im = axm.matshow(xt, aspect='auto', origin='lower', cmap=cmap, norm=norm) ### norm=norm added to scale coloring of expression with zero = white or black
axm.set_xticks([]) ### Hides x-ticks
axm.set_yticks([])
#axm.set_axis_off() ### Hide border
#fix_verts(ax1,1)
#fix_verts(ax2,0)
### Adjust the size of the fonts for genes and arrays based on size and character length
row_fontsize = 5
column_fontsize = 5
column_text_max_len = max(map(lambda x: len(x), column_header)) ### Get the maximum length of a column annotation
if len(row_header)<75:
row_fontsize = 6.5
if len(row_header)<50:
row_fontsize = 8
if len(row_header)<25:
row_fontsize = 11
if len(column_header)<75:
column_fontsize = 6.5
if len(column_header)<50:
column_fontsize = 8
if len(column_header)<25:
column_fontsize = 11
if column_text_max_len < 15:
column_fontsize = 15
elif column_text_max_len > 30:
column_fontsize = 6.5
else:
column_fontsize = 10
try:
if len(justShowTheseIDs)>50:
column_fontsize = 7
elif len(justShowTheseIDs)>0:
column_fontsize = 10
if len(justShowTheseIDs)>0:
additional_symbols=[]
import gene_associations
try:
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
except Exception: gene_to_symbol={}; symbol_to_gene={}
JustShowTheseIDs = copy.deepcopy(justShowTheseIDs)
except Exception:
JustShowTheseIDs=[]
# Add text
new_row_header=[]
new_column_header=[]
for i in range(x.shape[0]):
if row_method != None:
new_row_header.append(row_header[idx1[i]])
else:
new_row_header.append(row_header[i])
for i in range(x.shape[1]):
if column_method != None:
new_column_header.append(column_header[idx2[i]])
else: ### When not clustering columns
new_column_header.append(column_header[i])
dataset_name = string.replace(dataset_name,'Clustering-','')### clean up the name if already a clustered file
if '-hierarchical' in dataset_name:
dataset_name = string.split(dataset_name,'-hierarchical')[0]
filename = 'Clustering-%s-hierarchical_%s_%s.pdf' % (dataset_name,column_metric,row_metric)
if 'MarkerGenes' in dataset_name:
time_stamp = timestamp() ### Don't overwrite the previous version
filename = string.replace(filename,'hierarchical',time_stamp)
elite_dir, cdt_file, markers, SystemCode = exportFlatClusterData(root_dir + filename, root_dir, dataset_name, new_row_header,new_column_header,xt,ind1,ind2,vmax,display)
def ViewPNG(png_file_dir):
if os.name == 'nt':
try: os.startfile('"'+png_file_dir+'"')
except Exception: os.system('open "'+png_file_dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+png_file_dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+png_file_dir+'"')
try:
try:
temp1=len(justShowTheseIDs)
if 'monocle' in justShowTheseIDs and ('guide' not in justShowTheseIDs):
import R_interface
print 'Running Monocle through R (be patient, this can take 20 minutes+)'
R_interface.performMonocleAnalysisFromHeatmap(species,cdt_file[:-3]+'txt',cdt_file[:-3]+'txt')
png_file_dir = root_dir+'/Monocle/monoclePseudotime.png'
#print png_file_dir
ViewPNG(png_file_dir)
except Exception: pass # no justShowTheseIDs
except Exception:
print '...Monocle error:'
print traceback.format_exc()
pass
cluster_elite_terms={}; ge_fontsize=11.5; top_genes=[]; proceed=True
try:
try:
if 'guide' in justShowTheseIDs: proceed = False
except Exception: pass
if proceed:
try:
cluster_elite_terms,top_genes = remoteGOElite(elite_dir,SystemCode=SystemCode)
if cluster_elite_terms['label-size']>40: ge_fontsize = 9.5
except Exception:
pass
except Exception: pass #print traceback.format_exc()
if len(cluster_elite_terms)<1:
try:
elite_dirs = string.split(elite_dir,'GO-Elite')
old_elite_dir = elite_dirs[0]+'GO-Elite'+elite_dirs[-1] ### There are actually GO-Elite/GO-Elite directories for the already clustered
old_elite_dir = string.replace(old_elite_dir,'ICGS/','')
if len(PriorColumnClusters)>0 and len(PriorRowClusters)>0 and skipClustering:
cluster_elite_terms,top_genes = importGOEliteResults(old_elite_dir)
except Exception,e:
#print traceback.format_exc()
pass
try:
if len(justShowTheseIDs)<1 and len(markers) > 0 and column_fontsize < 9: ### substituted top_genes with markers
column_fontsize = 10
if len(justShowTheseIDs)<1:
additional_symbols=[]
import gene_associations; from import_scripts import OBO_import
try:
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
#symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception: gene_to_symbol={}; symbol_to_gene={}
except Exception: pass
def formatpval(p):
if '-' in p: p1=p[:1]+p[-4:]
else:
p1 = '{number:.{digits}f}'.format(number=float(p), digits=3)
p1=str(p1)
#print traceback.format_exc();sys.exit()
return p1
# Add text
new_row_header=[]
new_column_header=[]
ci=0 ### index of entries in the cluster
last_cluster=1
""" The below interval variable determines the spacing of GO-Elite labels """
interval = int(float(string.split(str(len(row_header)/35.0),'.')[0]))+1 ### for enrichment term labels with over 100 genes
increment=interval-2
if len(row_header)<100: increment = interval-1
cluster_num={}
for i in cluster_elite_terms: cluster_num[i[0]]=[]
cluster_num = len(cluster_num)
if cluster_num>15:
interval = int(float(string.split(str(len(row_header)/40.0),'.')[0]))+1 ### for enrichment term labels with over 100 genes
increment=interval-2
ge_fontsize = 7
column_fontsize = 7
if cluster_num>25:
interval = int(float(string.split(str(len(row_header)/50.0),'.')[0]))+1 ### for enrichment term labels with over 100 genes
increment=interval-2
ge_fontsize = 6
column_fontsize = 6
if cluster_num>40:
ge_fontsize = 4
column_fontsize = 4
label_pos=-0.03*len(column_header)-.8
alternate=1
#print ge_fontsize, cluster_num
#print label_pos
try:
if 'top' in justShowTheseIDs: justShowTheseIDs.remove('top')
if 'positive' in justShowTheseIDs: justShowTheseIDs.remove('positive')
if 'amplify' in justShowTheseIDs: justShowTheseIDs.remove('amplify')
if 'IntraCorrelatedOnly' in justShowTheseIDs: justShowTheseIDs.remove('IntraCorrelatedOnly')
if 'GuideOnlyCorrelation' in justShowTheseIDs: justShowTheseIDs.remove('GuideOnlyCorrelation')
except Exception:
pass
for i in range(x.shape[0]):
if len(row_header)<40:
radj = len(row_header)*0.009 ### row offset value to center the vertical position of the row label
elif len(row_header)<70:
radj = len(row_header)*0.007 ### row offset value to center the vertical position of the row label
else:
radj = len(row_header)*0.005
try: cluster = str(ind1[i])
except Exception: cluster = 'NA'
if cluster == 'NA':
new_index = i
try: cluster = 'cluster-'+string.split(row_header[new_index],':')[0]
except Exception: pass
if cluster != last_cluster:
ci=0
increment=0
#print cluster,i,row_header[idx1[i]]
color = 'black'
if row_method != None:
try:
if row_header[idx1[i]] in JustShowTheseIDs:
if len(row_header)>len(justShowTheseIDs):
color = 'red'
else: color = 'black'
except Exception: pass
if len(row_header)<106: ### Don't visualize gene associations when more than 100 rows
if display_label_names == False or 'ticks' in JustShowTheseIDs:
if color=='red':
axm.text(x.shape[1]-0.5, i-radj, ' '+'-',fontsize=row_fontsize, color=color, picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+'',fontsize=row_fontsize, color=color, picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+row_header[idx1[i]],fontsize=row_fontsize, color=color, picker=True)
new_row_header.append(row_header[idx1[i]])
new_index = idx1[i]
else:
try:
feature_id = row_header[i]
if ':' in feature_id:
feature_id = string.split(feature_id,':')[1]
if feature_id[-1]==' ': feature_id = feature_id[:-1]
if feature_id in JustShowTheseIDs:
color = 'red'
else: color = 'black'
except Exception: pass
if len(row_header)<106: ### Don't visualize gene associations when more than 100 rows
if display_label_names == False or 'ticks' in JustShowTheseIDs:
if color=='red':
axm.text(x.shape[1]-0.5, i-radj, ' '+'-',fontsize=row_fontsize, color=color, picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+'',fontsize=row_fontsize, color=color, picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+row_header[i],fontsize=row_fontsize, color=color, picker=True) ### When not clustering rows
new_row_header.append(row_header[i])
new_index = i ### This is different when clustering rows versus not
if len(row_header)<106:
"""
if cluster in cluster_elite_terms:
try:
term = cluster_elite_terms[cluster][ci][1]
axm.text(-1.5, i-radj, term,horizontalalignment='right',fontsize=row_fontsize)
except Exception: pass
ci+=1
"""
pass
else:
feature_id = row_header[new_index]
original_feature_id = feature_id
if ':' in feature_id:
if 'ENS' != feature_id[:3] or 'G0000' in feature_id:
feature_id = string.split(feature_id,':')[1]
if feature_id[-1]==' ': feature_id = feature_id[:-1]
else:
feature_id = string.split(feature_id,':')[0]
try: feature_id = gene_to_symbol[feature_id][0]
except Exception: pass
if (' ' in feature_id and ('ENS' in feature_id or 'G0000' in feature_id)):
feature_id = string.split(feature_id,' ')[1]
try:
if feature_id in JustShowTheseIDs or original_feature_id in JustShowTheseIDs: color = 'red'
else: color = 'black'
except Exception: pass
try:
if feature_id in justShowTheseIDs or (len(justShowTheseIDs)<1 and feature_id in markers) or original_feature_id in justShowTheseIDs: ### substitutes top_genes with markers
if 'ENS' in feature_id or 'G0000' in feature_id:
if feature_id in gene_to_symbol:
feature_id = gene_to_symbol[feature_id][0]
if original_feature_id in justShowTheseIDs:
feature_id = original_feature_id
if display_label_names and 'ticks' not in justShowTheseIDs:
if alternate==1: buffer=1.2; alternate=2
elif alternate==2: buffer=2.4; alternate=3
elif alternate==3: buffer=3.6; alternate=4
elif alternate==4: buffer=0; alternate=1
axm.text(x.shape[1]-0.4+buffer, i-radj, feature_id,fontsize=column_fontsize, color=color,picker=True) ### When not clustering rows
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+"-",fontsize=column_fontsize, color=color,picker=True) ### When not clustering rows
elif ' ' in row_header[new_index]:
symbol = string.split(row_header[new_index], ' ')[-1]
if len(symbol)>0:
if symbol in justShowTheseIDs:
if display_label_names and 'ticks' not in justShowTheseIDs:
axm.text(x.shape[1]-0.5, i-radj, ' '+row_header[new_index],fontsize=column_fontsize, color=color,picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+"-",fontsize=column_fontsize, color=color,picker=True)
except Exception: pass
if cluster in cluster_elite_terms or 'cluster-'+cluster in cluster_elite_terms:
if 'cluster-'+cluster in cluster_elite_terms:
new_cluster_id = 'cluster-'+cluster
else:
new_cluster_id = cluster
if cluster != last_cluster:
cluster_intialized = False
try:
increment+=1
#print [increment,interval,cluster],cluster_elite_terms[cluster][ci][1];sys.exit()
#if increment == interval or (
#print increment,interval,len(row_header),cluster_intialized
if (increment == interval) or (len(row_header)>200 and increment == (interval-9) and cluster_intialized==False): ### second argument brings the label down
cluster_intialized=True
atypical_cluster = False
if ind1[i+9] == 'NA': ### This occurs for custom cluster, such MarkerFinder (not cluster numbers)
atypical_cluster = True
cluster9 = 'cluster-'+string.split(row_header[new_index+9],':')[0]
if (len(row_header)>200 and str(cluster9)!=cluster): continue
elif (len(row_header)>200 and str(ind1[i+9])!=cluster): continue ### prevents the last label in a cluster from overlapping with the first in the next cluster
pvalue,original_term = cluster_elite_terms[new_cluster_id][ci]
term = original_term
if 'GO:' in term:
term = string.split(term, '(')[0]
if ':WP' in term:
term = string.split(term, ':WP')[0]
pvalue = formatpval(str(pvalue))
term += ' p='+pvalue
if atypical_cluster == False:
term += ' (c'+str(cluster)+')'
try: cluster_elite_terms[term] = cluster_elite_terms[cluster,original_term] ### store the new term name with the associated genes
except Exception: pass
axm.text(label_pos, i-radj, term,horizontalalignment='right',fontsize=ge_fontsize, picker=True, color = 'blue')
increment=0
ci+=1
except Exception,e:
#print traceback.format_exc();sys.exit()
increment=0
last_cluster = cluster
def onpick1(event):
text = event.artist
print('onpick1 text:', text.get_text())
if 'TreeView' in text.get_text():
try: openTreeView(cdt_file)
except Exception: print 'Failed to open TreeView'
elif 'p=' not in text.get_text():
webbrowser.open('http://www.genecards.org/cgi-bin/carddisp.pl?gene='+string.replace(text.get_text(),' ',''))
else:
#"""
from visualization_scripts import TableViewer
header = ['Associated Genes']
tuple_list = []
for gene in cluster_elite_terms[text.get_text()]:
tuple_list.append([(gene)])
if matplotlib.rcParams['backend'] != 'MacOSX': ### Throws an error when macosx is the backend for matplotlib
try: TableViewer.viewTable(text.get_text(),header,tuple_list)
except: pass ### Due to an an issue using a nonTkAgg backend
#"""
cluster_prefix = 'c'+string.split(text.get_text(),'(c')[-1][:-1]+'-'
for geneSet in EliteGeneSets:
if geneSet == 'GeneOntology':
png_file_dir = elite_dir+'/GO-Elite_results/networks/'+cluster_prefix+'GO'+'.png'
elif geneSet == 'WikiPathways':
png_file_dir = elite_dir+'/GO-Elite_results/networks/'+cluster_prefix+'local'+'.png'
elif len(geneSet)>1:
png_file_dir = elite_dir+'/GO-Elite_results/networks/'+cluster_prefix+geneSet+'.png'
#try: UI.GUI(root_dir,'ViewPNG',[],png_file_dir)
#except Exception: print traceback.format_exc()
try:
alt_png_file_dir = elite_dir+'/GO-Elite_results/networks/'+cluster_prefix+eliteGeneSet+'.png'
png_file_dirs = string.split(alt_png_file_dir,'GO-Elite/')
alt_png_file_dir = png_file_dirs[0]+'GO-Elite/'+png_file_dirs[-1]
except Exception: pass
if os.name == 'nt':
try: os.startfile('"'+png_file_dir+'"')
except Exception:
try: os.system('open "'+png_file_dir+'"')
except Exception: os.startfile('"'+alt_png_file_dir+'"')
elif 'darwin' in sys.platform:
try: os.system('open "'+png_file_dir+'"')
except Exception: os.system('open "'+alt_png_file_dir+'"')
elif 'linux' in sys.platform:
try: os.system('xdg-open "'+png_file_dir+'"')
except Exception: os.system('xdg-open "'+alt_png_file_dir+'"')
#print cluster_elite_terms[text.get_text()]
fig.canvas.mpl_connect('pick_event', onpick1)
""" Write x-axis labels """
for i in range(x.shape[1]):
adji = i
### Controls the vertical position of the column (array) labels
if len(row_header)<3:
cadj = len(row_header)*-0.26 ### column offset value
elif len(row_header)<4:
cadj = len(row_header)*-0.23 ### column offset value
elif len(row_header)<6:
cadj = len(row_header)*-0.18 ### column offset value
elif len(row_header)<10:
cadj = len(row_header)*-0.08 ### column offset value
elif len(row_header)<15:
cadj = len(row_header)*-0.04 ### column offset value
elif len(row_header)<20:
cadj = len(row_header)*-0.05 ### column offset value
elif len(row_header)<22:
cadj = len(row_header)*-0.06 ### column offset value
elif len(row_header)<23:
cadj = len(row_header)*-0.08 ### column offset value
elif len(row_header)>200:
cadj = -2
else:
cadj = -0.9
#cadj = -1
if len(column_header)>15:
adji = i-0.1 ### adjust the relative position of the column label horizontally
if len(column_header)>20:
adji = i-0.2 ### adjust the relative position of the column label horizontally
if len(column_header)>25:
adji = i-0.2 ### adjust the relative position of the column label horizontally
if len(column_header)>30:
adji = i-0.25 ### adjust the relative position of the column label horizontally
if len(column_header)>35:
adji = i-0.3 ### adjust the relative position of the column label horizontally
if len(column_header)>200:
column_fontsize = 2
if column_method != None:
if len(column_header)<150: ### Don't show the headers when too many values exist
axm.text(adji, cadj, ''+column_header[idx2[i]], rotation=270, verticalalignment="top",fontsize=column_fontsize) # rotation could also be degrees
new_column_header.append(column_header[idx2[i]])
else: ### When not clustering columns
if len(column_header)<300: ### Don't show the headers when too many values exist
axm.text(adji, cadj, ''+column_header[i], rotation=270, verticalalignment="top",fontsize=column_fontsize)
new_column_header.append(column_header[i])
# Plot colside colors
# axc --> axes for column side colorbar
group_name_list=[]
ind1_clust,ind2_clust = ind1,ind2
ind1,ind2,group_name_list,cb_status = updateColorBarData(ind1,ind2,new_column_header,new_row_header,row_method)
if (column_method != None or 'column' in cb_status) and show_color_bars == True:
axc = fig.add_axes([axc_x, axc_y, axc_w, axc_h]) # axes for column side colorbar
cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF', '#CCCCE0','#000066','#FFFF00', '#FF1493'])
if use_default_colors:
cmap_c = PairedColorMap()
#cmap_c = pylab.cm.gist_ncar
else:
#cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF','#FFFF00', '#FF1493'])
if len(unique.unique(ind2))==2: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
#cmap_c = matplotlib.colors.ListedColormap(['#7CFC00','k'])
cmap_c = matplotlib.colors.ListedColormap(['w', 'k'])
elif len(unique.unique(ind2))>0: ### cmap_c is too few colors
#cmap_c = pylab.cm.Paired
cmap_c = PairedColorMap()
"""
elif len(unique.unique(ind2))==3: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
#cmap_c = matplotlib.colors.ListedColormap(['r', 'y', 'b'])
elif len(unique.unique(ind2))==4: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C','#FEBC18']) #['#FEBC18','#EE2C3C','#3D3181','#88BF47']
#cmap_c = matplotlib.colors.ListedColormap(['k', 'w', 'w', 'w'])
elif len(unique.unique(ind2))==5: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2))==6: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
#cmap_c = matplotlib.colors.ListedColormap(['black', '#1DA532', '#88BF47','b', 'grey','r'])
#cmap_c = matplotlib.colors.ListedColormap(['w', '#0B9B48', 'w', '#5D82C1','#4CB1E4','#71C065'])
#cmap_c = matplotlib.colors.ListedColormap(['w', 'w', 'k', 'w','w','w'])
elif len(unique.unique(ind2))==7: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
#cmap_c = matplotlib.colors.ListedColormap(['w', 'w', 'w', 'k', 'w','w','w'])
#cmap_c = matplotlib.colors.ListedColormap(['w','w', '#0B9B48', 'w', '#5D82C1','#4CB1E4','#71C065'])
#elif len(unique.unique(ind2))==9: cmap_c = matplotlib.colors.ListedColormap(['k', 'w', 'w', 'w', 'w', 'w', 'w', 'w', 'w'])
#elif len(unique.unique(ind2))==11:
#cmap_c = matplotlib.colors.ListedColormap(['w', '#DC2342', '#0B9B48', '#FDDF5E', '#E0B724', 'k', '#5D82C1', '#F79020', '#4CB1E4', '#983894', '#71C065'])
"""
try: dc = numpy.array(ind2, dtype=int)
except:
### occurs with the cluster numbers are cluster annotation names (cell types)
ind2 = convertClusterNameToInt(ind2)
dc = numpy.array(ind2, dtype=int)
dc.shape = (1,len(ind2))
im_c = axc.matshow(dc, aspect='auto', origin='lower', cmap=cmap_c)
axc.set_xticks([]) ### Hides ticks
if 'hopach' == column_method and len(group_name_list)>0:
axc.set_yticklabels(['','Groups'],fontsize=10)
else:
axc.set_yticks([])
#axc.set_frame_on(False) ### Hide border
if len(group_name_list)>0: ### Add a group color legend key
if 'hopach' == column_method: ### allows us to add the second color bar
axcd = fig.add_axes([ax2_x, ax2_y, ax2_w, color_bar_w]) # dendrogram coordinates with color_bar_w substituted - can use because dendrogram is not used
cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF', '#CCCCE0','#000066','#FFFF00', '#FF1493'])
#cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF','#FFFF00', '#FF1493'])
if use_default_colors:
#cmap_c = pylab.cm.Paired
cmap_c = PairedColorMap()
else:
if len(unique.unique(ind2_clust))==2: ### cmap_c is too few colors
#cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
cmap_c = matplotlib.colors.ListedColormap(['w', 'k'])
elif len(unique.unique(ind2_clust))>0: ### cmap_c is too few colors
#cmap_c = pylab.cm.Paired
cmap_c = PairedColorMap()
"""
elif len(unique.unique(ind2_clust))==3: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
#cmap_c = matplotlib.colors.ListedColormap(['r', 'y', 'b'])
elif len(unique.unique(ind2_clust))==4: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
#cmap_c = matplotlib.colors.ListedColormap(['black', '#1DA532', 'b','r'])
elif len(unique.unique(ind2_clust))==5: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2_clust))==6: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2_clust))==7: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
"""
try: dc = numpy.array(ind2_clust, dtype=int)
except:
### occurs with the cluster numbers are cluster annotation names (cell types)
ind2_clust = convertClusterNameToInt(ind2_clust)
dc = numpy.array(ind2_clust, dtype=int)
dc.shape = (1,len(ind2_clust))
im_cd = axcd.matshow(dc, aspect='auto', origin='lower', cmap=cmap_c)
#axcd.text(-1,-1,'clusters')
axcd.set_yticklabels(['','Clusters'],fontsize=10)
#pylab.yticks(range(1),['HOPACH clusters'])
axcd.set_xticks([]) ### Hides ticks
#axcd.set_yticks([])
axd = fig.add_axes([axcc_x, axcc_y, axcc_w, axcc_h])
group_name_list.sort()
group_colors = map(lambda x: x[0],group_name_list)
group_names = map(lambda x: x[1],group_name_list)
cmap_d = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF', '#CCCCE0','#000066','#FFFF00', '#FF1493'])
#cmap_d = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF','#FFFF00', '#FF1493'])
if len(unique.unique(ind2))==2: ### cmap_c is too few colors
#cmap_d = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
cmap_d = matplotlib.colors.ListedColormap(['w', 'k'])
elif len(unique.unique(ind2))>0: ### cmap_c is too few colors
#cmap_d = pylab.cm.Paired
cmap_d = PairedColorMap()
"""
elif len(unique.unique(ind2))==3: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
#cmap_d = matplotlib.colors.ListedColormap(['r', 'y', 'b'])
elif len(unique.unique(ind2))==4: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
elif len(unique.unique(ind2))==5: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2))==6: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
#cmap_d = matplotlib.colors.ListedColormap(['black', '#1DA532', '#88BF47','b', 'grey','r'])
#cmap_d = matplotlib.colors.ListedColormap(['w', '#0B9B48', 'w', '#5D82C1','#4CB1E4','#71C065'])
#cmap_d = matplotlib.colors.ListedColormap(['w', 'w', 'k', 'w', 'w','w','w'])
elif len(unique.unique(ind2))==7: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
#cmap_d = matplotlib.colors.ListedColormap(['w', 'w', 'w', 'k', 'w','w','w'])
#cmap_d = matplotlib.colors.ListedColormap(['w','w', '#0B9B48', 'w', '#5D82C1','#4CB1E4','#71C065'])
#elif len(unique.unique(ind2))==10: cmap_d = matplotlib.colors.ListedColormap(['w', 'w', 'w', 'w', 'w', 'w', 'w', 'w', 'w', 'k'])
"""
dc = numpy.array(group_colors, dtype=int)
dc.shape = (1,len(group_colors))
im_c = axd.matshow(dc, aspect='auto', origin='lower', cmap=cmap_d)
axd.set_yticks([])
#axd.set_xticklabels(group_names, rotation=45, ha='left')
#if len(group_names)<200:
pylab.xticks(range(len(group_names)),group_names,rotation=90,ha='left') #rotation = 45
#cmap_c = matplotlib.colors.ListedColormap(map(lambda x: GroupDB[x][-1], new_column_header))
if show_color_bars == False:
axc = fig.add_axes([axc_x, axc_y, axc_w, axc_h]) # axes for column side colorbar
axc.set_frame_on(False)
# Plot rowside colors
# axr --> axes for row side colorbar
if (row_method != None or 'row' in cb_status) and show_color_bars == True:
axr = fig.add_axes([axr_x, axr_y, axr_w, axr_h]) # axes for column side colorbar
try:
dr = numpy.array(ind1, dtype=int)
dr.shape = (len(ind1),1)
cmap_r = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF', '#FFFF00', '#FF1493'])
if len(unique.unique(ind1))>4: ### cmap_r is too few colors
#cmap_r = pylab.cm.Paired
cmap_r = PairedColorMap()
if 'MarkerGenes' in dataset_name: ### reverse the order of the colors to match the top color bar
#cmap_r = pylab.cm.Paired_r
cmap_r = PairedColorMap().reversed()
if len(unique.unique(ind1))==2:
cmap_r = matplotlib.colors.ListedColormap(['w', 'k'])
im_r = axr.matshow(dr, aspect='auto', origin='lower', cmap=cmap_r)
axr.set_xticks([]) ### Hides ticks
axr.set_yticks([])
#axr.set_frame_on(False) ### Hide border
except Exception:
row_method = None
pass ### likely occurs for some reason when row_method should be None
if show_color_bars == False:
axr = fig.add_axes([axr_x, axr_y, axr_w, axr_h]) # axes for column side colorbar
axr.set_frame_on(False)
""" write x-axis group labels """
groupNames_to_cell={}
cluster_to_cell={}
try: ### Group names (from groups file or embeded groups annotations)
for i in range(x.shape[1]):
cluster = str(ind2[i])
try: groupNames_to_cell[cluster].append(i)
except: groupNames_to_cell[cluster]=[i]
except: pass
try: ### Cluster names from clustering
for i in range(x.shape[1]):
cluster = str(ind2_clust[i])
try: cluster_to_cell[cluster].append(i)
except: cluster_to_cell[cluster]=[i]
except: pass
### Use the groups rather than clusters if not clustered
cluster_group_matching = False
group_length=[]
cluster_length=[]
try:
index=0
for c in cluster_to_cell:
cluster_length.append(len(cluster_to_cell[c]))
for c in groupNames_to_cell:
group_length.append(len(groupNames_to_cell[c]))
### if the clusters and groups are the same size
if max(cluster_length) == max(group_length) and (len(cluster_to_cell) == len(groupNames_to_cell)):
cluster_group_matching = True
except: pass
clusterType = 'Numbers'
if (len(cluster_to_cell) < 2) or cluster_group_matching:
cluster_to_cell = groupNames_to_cell
ind2_clust = ind2
clusterType = 'Groups'
try:
last_cluster = None
group_index=0
cluster_count = 0
cluster_borders=[]
if len(column_header)>1:
for i in range(x.shape[1]):
adji = i
cadj = 0.6
try: cluster = str(ind2_clust[i])
except Exception: cluster = 'NA'
fontsize = 5
middle_cluster_index = len(cluster_to_cell[cluster])/3
if cluster != last_cluster:
cluster_count=0
#ax.plot([70, 70], [100, 250], 'w-', lw=0.5)
if i>0: ### Don't need to draw a white line at 0
cluster_borders.append(i-0.5)
if cluster_count == middle_cluster_index:
if clusterType == 'Numbers':
try:
axcd.text(adji, cadj, ''+cluster, rotation=45, verticalalignment="bottom",fontsize=5) # rotation could also be degrees
except:
axc.text(adji, cadj, ''+cluster, rotation=45, verticalalignment="bottom",fontsize=5) # rotation could also be degrees
else:
try:
axcd.text(adji, cadj, ''+group_name_list[group_index][1], rotation=45, verticalalignment="bottom",fontsize=fontsize) # rotation could also be degrees
except:
try:
axc.text(adji, cadj, ''+group_name_list[group_index][1], rotation=45, verticalalignment="bottom",fontsize=fontsize) # rotation could also be degrees
except:
try:
axcd.text(adji, cadj, ''+cluster, rotation=45, verticalalignment="bottom",fontsize=5) # rotation could also be degrees
except:
axc.text(adji, cadj, ''+cluster, rotation=45, verticalalignment="bottom",fontsize=5) # rotation could also be degrees
group_index+=1
last_cluster = cluster
cluster_count+=1
except:
#print group_name_list
#print len(group_name_list), group_index
#print traceback.format_exc()
pass
try:
#print cluster_borders
axm.vlines(cluster_borders, color='w',lw=0.3, *axm.get_ylim())
except:
pass
# Plot color legend
axcb = fig.add_axes([axcb_x, axcb_y, axcb_w, axcb_h], frame_on=False) # axes for colorbar
cb = matplotlib.colorbar.ColorbarBase(axcb, cmap=cmap, norm=norm, orientation='horizontal')
#axcb.set_title("colorkey",fontsize=14)
if 'LineageCorrelations' in dataset_name:
cb.set_label("Lineage Correlation Z Scores",fontsize=11)
elif 'Heatmap' in root_dir:
cb.set_label("GO-Elite Z Scores",fontsize=11)
else:
cb.set_label("Differential Expression (log2)",fontsize=10)
### Add filename label to the heatmap
if len(dataset_name)>30:fontsize = 10
else: fontsize = 12.5
fig.text(0.015, 0.970, dataset_name, fontsize = fontsize)
### Render and save the graphic
pylab.savefig(root_dir + filename,dpi=1000)
#print 'Exporting:',filename
filename = filename[:-3]+'png'
pylab.savefig(root_dir + filename, dpi=100) #,dpi=200
includeBackground=False
try:
if 'TkAgg' != matplotlib.rcParams['backend']:
includeBackground = False
except Exception: pass
if includeBackground:
fig.text(0.020, 0.070, 'Open heatmap in TreeView (click here)', fontsize = 11.5, picker=True,color = 'red', backgroundcolor='white')
else:
fig.text(0.020, 0.070, 'Open heatmap in TreeView (click here)', fontsize = 11.5, picker=True,color = 'red')
if 'Outlier' in dataset_name and 'Removed' not in dataset_name:
graphic_link.append(['Hierarchical Clustering - Outlier Genes Genes',root_dir+filename])
elif 'Relative' in dataset_name:
graphic_link.append(['Hierarchical Clustering - Significant Genes (Relative comparisons)',root_dir+filename])
elif 'LineageCorrelations' in filename:
graphic_link.append(['Hierarchical Clustering - Lineage Correlations',root_dir+filename])
elif 'MarkerGenes' in filename:
graphic_link.append(['Hierarchical Clustering - MarkerFinder',root_dir+filename])
elif 'AltExonConfirmed' in filename:
graphic_link.append(['Hierarchical Clustering - AltExonConfirmed',root_dir+filename])
elif 'AltExon' in filename:
graphic_link.append(['Hierarchical Clustering - AltExon',root_dir+filename])
elif 'alt_junction' in filename:
graphic_link.append(['Hierarchical Clustering - Variable Splice-Events',root_dir+filename])
else:
graphic_link.append(['Hierarchical Clustering - Significant Genes',root_dir+filename])
if display:
proceed=True
try:
if 'guide' in justShowTheseIDs:
proceed = False
except Exception: pass
if proceed:
print 'Exporting:',filename
try: pylab.show()
except Exception: None ### when run in headless mode
fig.clf()
#fig.close() causes segfault
#pylab.close() causes segfault
def openTreeView(filename):
import subprocess
fn = filepath("AltDatabase/TreeView/TreeView.jar")
print 'java', "-Xmx4000m", '-jar', fn, "-r", filename
retcode = subprocess.Popen(['java', "-Xmx4000m", '-jar', fn, "-r", filename])
def remoteGOElite(elite_dir,SystemCode = None):
mod = 'Ensembl'
if SystemCode == 'Ae':
mod = 'AltExon'
pathway_permutations = 'FisherExactTest'
filter_method = 'z-score'
z_threshold = 1.96
p_val_threshold = 0.005
change_threshold = 2
if runGOElite:
resources_to_analyze = EliteGeneSets
if 'all' in resources_to_analyze:
resources_to_analyze = 'all'
returnPathways = 'no'
root = None
import GO_Elite
reload(GO_Elite)
input_files = dir_list = unique.read_directory(elite_dir) ### Are there any files to analyze?
if len(input_files)>0 and resources_to_analyze !=['']:
print '\nBeginning to run GO-Elite analysis on all results'
file_dirs = elite_dir,None,elite_dir
enrichmentAnalysisType = 'ORA'
#enrichmentAnalysisType = 'URA'
variables = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,enrichmentAnalysisType,root
try:
GO_Elite.remoteAnalysis(variables, 'non-UI Heatmap')
except Exception:
print 'GO-Elite failed for:', elite_dir
print traceback.format_exc()
if commandLine==False:
try: UI.openDirectory(elite_dir+'/GO-Elite_results')
except Exception: None
cluster_elite_terms,top_genes = importGOEliteResults(elite_dir)
return cluster_elite_terms,top_genes
else:
return {},[]
else:
return {},[]
def importGOEliteResults(elite_dir):
global eliteGeneSet
pruned_results = elite_dir+'/GO-Elite_results/CompleteResults/ORA_pruned/pruned-results_z-score_elite.txt' ### This is the exception (not moved)
if os.path.isfile(pruned_results) == False:
pruned_results = elite_dir+'/GO-Elite_results/pruned-results_z-score_elite.txt'
firstLine=True
cluster_elite_terms={}
all_term_length=[0]
for line in open(pruned_results,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
try: symbol_index = values.index('gene symbols')
except Exception: symbol_index = None
else:
try: symbol_index = values.index('gene symbols')
except Exception: pass
try:
eliteGeneSet = string.split(values[0][1:],'-')[1][:-4]
try: cluster = str(int(float(string.split(values[0][1:],'-')[0])))
except Exception:
cluster = string.join(string.split(values[0],'-')[:-1],'-')
term = values[2]
num_genes_changed = int(values[3])
all_term_length.append(len(term))
pval = float(values[9]) ### adjusted is values[10]
#pval = float(values[10]) ### adjusted is values[10]
if num_genes_changed>2:
try: cluster_elite_terms[cluster].append([pval,term])
except Exception: cluster_elite_terms[cluster] = [[pval,term]]
if symbol_index!=None:
symbols = string.split(values[symbol_index],'|')
cluster_elite_terms[cluster,term] = symbols
except Exception,e: pass
for cluster in cluster_elite_terms:
cluster_elite_terms[cluster].sort()
cluster_elite_terms['label-size'] = max(all_term_length)
top_genes = []; count=0
ranked_genes = elite_dir+'/GO-Elite_results/CompleteResults/ORA_pruned/gene_associations/pruned-gene-ranking.txt'
for line in open(ranked_genes,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
count+=1
if len(values)>2:
if values[2]!='Symbol':
try: top_genes.append((int(values[4]),values[2]))
except Exception: pass
top_genes.sort(); top_genes.reverse()
top_genes = map(lambda x: x[1],top_genes[:21])
return cluster_elite_terms,top_genes
def mergeRotateAroundPointPage(page, page2, rotation, tx, ty):
from pyPdf import PdfFileWriter, PdfFileReader
translation = [[1, 0, 0],
[0, 1, 0],
[-tx,-ty,1]]
rotation = math.radians(rotation)
rotating = [[math.cos(rotation), math.sin(rotation),0],
[-math.sin(rotation),math.cos(rotation), 0],
[0, 0, 1]]
rtranslation = [[1, 0, 0],
[0, 1, 0],
[tx,ty,1]]
ctm = numpy.dot(translation, rotating)
ctm = numpy.dot(ctm, rtranslation)
return page.mergeTransformedPage(page2, [ctm[0][0], ctm[0][1],
ctm[1][0], ctm[1][1],
ctm[2][0], ctm[2][1]])
def mergePDFs2(pdf1,pdf2,outPdf):
from pyPdf import PdfFileWriter, PdfFileReader
input1 = PdfFileReader(file(pdf1, "rb"))
page1 = input1.getPage(0)
input2 = PdfFileReader(file(pdf2, "rb"))
page2 = input2.getPage(0)
page3 = mergeRotateAroundPointPage(page1, page2,
page1.get('/Rotate') or 0,
page2.mediaBox.getWidth()/2, page2.mediaBox.getWidth()/2)
output = PdfFileWriter()
output.addPage(page3)
outputStream = file(outPdf, "wb")
output.write(outputStream)
outputStream.close()
def mergePDFs(pdf1,pdf2,outPdf):
# http://stackoverflow.com/questions/6041244/how-to-merge-two-landscape-pdf-pages-using-pypdf
from pyPdf import PdfFileWriter, PdfFileReader
input1 = PdfFileReader(file(pdf1, "rb"))
page1 = input1.getPage(0)
page1.mediaBox.upperRight = (page1.mediaBox.getUpperRight_x(), page1.mediaBox.getUpperRight_y())
input2 = PdfFileReader(file(pdf2, "rb"))
page2 = input2.getPage(0)
page2.mediaBox.getLowerLeft_x = (page2.mediaBox.getLowerLeft_x(), page2.mediaBox.getLowerLeft_y())
# Merge
page2.mergePage(page1)
# Output
output = PdfFileWriter()
output.addPage(page1)
outputStream = file(outPdf, "wb")
output.write(outputStream)
outputStream.close()
"""
def merge_horizontal(out_filename, left_filename, right_filename):
#Merge the first page of two PDFs side-to-side
import pyPdf
# open the PDF files to be merged
with open(left_filename) as left_file, open(right_filename) as right_file, open(out_filename, 'w') as output_file:
left_pdf = pyPdf.PdfFileReader(left_file)
right_pdf = pyPdf.PdfFileReader(right_file)
output = pyPdf.PdfFileWriter()
# get the first page from each pdf
left_page = left_pdf.pages[0]
right_page = right_pdf.pages[0]
# start a new blank page with a size that can fit the merged pages side by side
page = output.addBlankPage(
width=left_page.mediaBox.getWidth() + right_page.mediaBox.getWidth(),
height=max(left_page.mediaBox.getHeight(), right_page.mediaBox.getHeight()),
)
# draw the pages on that new page
page.mergeTranslatedPage(left_page, 0, 0)
page.mergeTranslatedPage(right_page, left_page.mediaBox.getWidth(), 0)
# write to file
output.write(output_file)
"""
def inverseDist(value):
if value == 0: value = 1
return math.log(value,2)
def getGOEliteExportDir(root_dir,dataset_name):
if 'AltResults' in root_dir:
root_dir = string.split(root_dir,'AltResults')[0]
if 'ExpressionInput' in root_dir:
root_dir = string.split(root_dir,'ExpressionInput')[0]
if 'ExpressionOutput' in root_dir:
root_dir = string.split(root_dir,'ExpressionOutput')[0]
if 'DataPlots' in root_dir:
root_dir = string.replace(root_dir,'DataPlots','GO-Elite')
elite_dir = root_dir
else:
elite_dir = root_dir+'/GO-Elite'
try: os.mkdir(elite_dir)
except Exception: pass
return elite_dir+'/clustering/'+dataset_name
def systemCodeCheck(IDs):
import gene_associations
id_type_db={}
for id in IDs:
id_type = gene_associations.predictIDSourceSimple(id)
try: id_type_db[id_type]+=1
except Exception: id_type_db[id_type]=1
id_type_count=[]
for i in id_type_db:
id_type_count.append((id_type_db[i],i))
id_type_count.sort()
id_type = id_type_count[-1][-1]
return id_type
def exportFlatClusterData(filename, root_dir, dataset_name, new_row_header,new_column_header,xt,ind1,ind2,vmax,display):
""" Export the clustered results as a text file, only indicating the flat-clusters rather than the tree """
filename = string.replace(filename,'.pdf','.txt')
export_text = export.ExportFile(filename)
column_header = string.join(['UID','row_clusters-flat']+new_column_header,'\t')+'\n' ### format column-names for export
export_text.write(column_header)
column_clusters = string.join(['column_clusters-flat','']+ map(str, ind2),'\t')+'\n' ### format column-flat-clusters for export
export_text.write(column_clusters)
### The clusters, dendrogram and flat clusters are drawn bottom-up, so we need to reverse the order to match
#new_row_header = new_row_header[::-1]
#xt = xt[::-1]
try: elite_dir = getGOEliteExportDir(root_dir,dataset_name)
except Exception: elite_dir = None
elite_columns = string.join(['InputID','SystemCode'])
try: sy = systemCodeCheck(new_row_header)
except Exception: sy = None
### Export each row in the clustered data matrix xt
i=0
cluster_db={}
export_lines = []
last_cluster=None
cluster_top_marker={}
for row in xt:
try:
id = new_row_header[i]
new_id = id
original_id = str(id)
if sy == 'Ae' and '--' in id:
cluster = 'cluster-' + string.split(id, ':')[0]
elif sy == '$En:Sy':
cluster = 'cluster-'+string.split(id,':')[0]
elif sy == 'S' and ':' in id:
cluster = 'cluster-'+string.split(id,':')[0]
elif sy == 'Sy' and ':' in id:
cluster = 'cluster-'+string.split(id,':')[0]
else:
cluster = 'c'+str(ind1[i])
if ':' in id:
new_id = string.split(id,':')[1]
if ' ' in new_id:
new_id = string.split(new_id,' ')[0]
#if cluster not in cluster_top_marker: ### Display the top marker gene
cluster_top_marker[cluster] = new_id
last_cluster = cluster
except Exception:
pass
try:
if 'MarkerGenes' in originalFilename:
cluster = 'cluster-' + string.split(id, ':')[0]
id = string.split(id, ':')[1]
if ' ' in id:
id = string.split(id, ' ')[0]
if 'G000' in id:
sy = 'En'
else:
sy = 'Sy'
except Exception: pass
try: cluster_db[cluster].append(id)
except Exception: cluster_db[cluster] = [id]
try: export_lines.append(string.join([original_id,str(ind1[i])]+map(str, row),'\t')+'\n')
except Exception:
export_lines.append(string.join([original_id,'NA']+map(str, row),'\t')+'\n')
i+=1
### Reverse the order of the file
export_lines.reverse()
for line in export_lines:
export_text.write(line)
export_text.close()
### Export GO-Elite input files
allGenes={}
sc=sy
for cluster in cluster_db:
export_elite = export.ExportFile(elite_dir + '/' + cluster + '.txt')
if sy == None:
export_elite.write('ID\n')
else:
export_elite.write('ID\tSystemCode\n')
for id in cluster_db[cluster]:
if ' ' in id and ':' not in id:
ids = string.split(id, ' ')
if ids[0] == ids[1]:
id = ids[0]
elif ' ' in id and ':' in id:
id = string.split(id, ':')[-1]
id = string.split(id, ' ')[0]
if sy == '$En:Sy':
try: id = string.split(id, ':')[1]
except:
if 'ENS' in id:
sy = 'En'
continue
ids = string.split(id, ' ')
if 'ENS' in ids[0] or 'G0000' in ids[0]:
id = ids[0]
else:
id = ids[(-1)]
sc = 'En'
elif sy == 'Sy' and ':' in id:
id = string.split(id, ':')[1]
ids = string.split(id, ' ')
sc = 'Sy'
elif sy == 'En:Sy':
id = string.split(id, ' ')[0]
sc = 'En'
elif sy == 'En' and ':' in id:
ids = string.split(id,':')
if len(ids) == 2:
id = ids[1]
else:
id = ids[1]
elif sy == 'Ae':
if '--' in id:
sc = 'En'
id = string.split(id, ':')[(-1)]
else:
l = string.split(id, ':')
if len(l) == 2:
id = string.split(id, ':')[0]
if len(l) == 3:
id = string.split(id, ':')[1]
sc = 'En'
if ' ' in id:
ids = string.split(id, ' ')
if 'ENS' in ids[(-1)] or 'G0000' in ids[(-1)]:
id = ids[(-1)]
else:
id = ids[0]
elif sy == 'En' and '&' in id:
for i in string.split(id, '&'):
if 'G0000' in i:
id = i
sc = 'En'
break
elif sy == 'Sy' and 'EFN' in id:
sc = 'En'
else:
sc = sy
if sy == 'S':
if ':' in id:
id = string.split(id, ':')[(-1)]
sc = 'Ae'
if ' ' in id:
id = string.split(id, ' ')[(0)]
if '&' in id:
sc = 'Ae'
if len(id) == 9 and 'SRS' in id or len(id) == 15 and 'TCGA-' in id:
sc = 'En'
try:
export_elite.write(id + '\t' + sc + '\n')
except Exception:
export_elite.write(id + '\n')
else:
allGenes[id] = []
export_elite.close()
try:
if storeGeneSetName != None:
if len(storeGeneSetName)>0 and ('guide' not in justShowTheseIDs):
exportCustomGeneSet(storeGeneSetName,species,allGenes)
print 'Exported geneset to "StoredGeneSets"'
except Exception: pass
### Export as CDT file
filename = string.replace(filename,'.txt','.cdt')
if display:
try: exportJTV(filename, new_column_header, new_row_header,vmax=vmax)
except Exception: pass
export_cdt = export.ExportFile(filename)
column_header = string.join(['UNIQID','NAME','GWEIGHT']+new_column_header,'\t')+'\n' ### format column-names for export
export_cdt.write(column_header)
eweight = string.join(['EWEIGHT','','']+ ['1']*len(new_column_header),'\t')+'\n' ### format column-flat-clusters for export
export_cdt.write(eweight)
### Export each row in the clustered data matrix xt
i=0; cdt_lines=[]
for row in xt:
cdt_lines.append(string.join([new_row_header[i]]*2+['1']+map(str, row),'\t')+'\n')
i+=1
### Reverse the order of the file
cdt_lines.reverse()
for line in cdt_lines:
export_cdt.write(line)
### Save the top marker gene IDs
markers=[]
for cluster in cluster_top_marker:
markers.append(cluster_top_marker[cluster])
export_cdt.close()
return elite_dir, filename, markers, sc
def exportJTV(cdt_dir, column_header, row_header,vmax=None):
### This is a config file for TreeView
filename = string.replace(cdt_dir,'.cdt','.jtv')
export_jtv = export.ExportFile(filename)
cscale = '3'
if len(column_header)>100:
cscale = '1.5'
if len(column_header)>200:
cscale = '1.1'
if len(column_header)>300:
cscale = '0.6'
if len(column_header)>400:
cscale = '0.3'
hscale = '5'
if len(row_header)< 50:
hscale = '10'
if len(row_header)>100:
hscale = '3'
if len(row_header)>500:
hscale = '1'
if len(row_header)>1000:
hscale = '0.5'
contrast = str(float(vmax)/4)[:4] ### base the contrast on the heatmap vmax variable
"""
config = '<DocumentConfig><UrlExtractor/><ArrayUrlExtractor/><MainView><ColorExtractor>'
config+= '<ColorSet down="#00FFFF"/></ColorExtractor><ArrayDrawer/><GlobalXMap>'
config+= '<FixedMap type="Fixed" scale="'+cscale+'"/><FillMap type="Fill"/><NullMap type="Null"/>'
config+= '</GlobalXMap><GlobalYMap><FixedMap type="Fixed" scale="'+hscale+'"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></GlobalYMap><ZoomXMap><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></ZoomXMap><ZoomYMap><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></ZoomYMap><TextView><TextView><GeneSummary/></TextView><TextView>'
config+= '<GeneSummary/></TextView><TextView><GeneSummary/></TextView></TextView><ArrayNameView>'
config+= '<ArraySummary included="0"/></ArrayNameView><AtrSummary/><GtrSummary/></MainView></DocumentConfig>'
export_jtv.write(config)
"""
config = '<DocumentConfig><UrlExtractor/><ArrayUrlExtractor/><MainView><ColorExtractor>'
config+= '<ColorSet down="#00FFFF"/></ColorExtractor><ArrayDrawer/><GlobalXMap>'
config+= '<FixedMap type="Fixed" scale="'+cscale+'"/><FillMap type="Fill"/><NullMap type="Null"/>'
config+= '</GlobalXMap><GlobalYMap><FixedMap type="Fixed" scale="'+hscale+'"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></GlobalYMap><ZoomXMap><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></ZoomXMap><ZoomYMap><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></ZoomYMap><TextView><TextView><GeneSummary/></TextView><TextView>'
config+= '<GeneSummary/></TextView><TextView><GeneSummary/></TextView></TextView><ArrayNameView>'
config+= '<ArraySummary included="0"/></ArrayNameView><AtrSummary/><GtrSummary/></MainView><Views>'
config+= '<View type="Dendrogram" dock="1"><ColorExtractor contrast="'+contrast+'"><ColorSet up="#FFFF00" down="#00CCFF"/>'
config+= '</ColorExtractor><ArrayDrawer/><GlobalXMap current="Fill"><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></GlobalXMap><GlobalYMap current="Fill"><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></GlobalYMap><ZoomXMap><FixedMap type="Fixed"/><FillMap type="Fill"/><NullMap type="Null"/>'
config+= '</ZoomXMap><ZoomYMap current="Fixed"><FixedMap type="Fixed"/><FillMap type="Fill"/><NullMap type="Null"/></ZoomYMap>'
config+= '<TextView><TextView><GeneSummary/></TextView><TextView><GeneSummary/></TextView><TextView><GeneSummary/></TextView>'
config+= '</TextView><ArrayNameView><ArraySummary included="0"/></ArrayNameView><AtrSummary/><GtrSummary/></View></Views></DocumentConfig>'
export_jtv.write(config)
### How to create custom colors - http://matplotlib.sourceforge.net/examples/pylab_examples/custom_cmap.html
def updateColorBarData(ind1,ind2,column_header,row_header,row_method):
""" Replace the top-level cluster information with group assignments for color bar coloring (if group data present)"""
cb_status = 'original'
group_number_list=[]
group_name_list=[]
try: ### Error if GroupDB not recognized as global
if column_header[0] in GroupDB: ### Thus group assignments exist for column headers
cb_status = 'column'
for header in column_header:
group,color,color_num = GroupDB[header]
group_number_list.append(color_num) ### will replace ind2
if (color_num,group) not in group_name_list:
group_name_list.append((color_num,group))
ind2 = group_number_list
if row_header[0] in GroupDB and row_method == None: ### Thus group assignments exist for row headers
group_number_list=[]
if cb_status == 'column': cb_status = 'column-row'
else: cb_status = 'row'
for header in row_header:
group,color,color_num = GroupDB[header]
group_number_list.append(color_num) ### will replace ind2
#group_number_list.reverse()
ind1 = group_number_list
except Exception: None
return ind1,ind2,group_name_list,cb_status
def ConvertFromHex(color1,color2,color3):
c1tuple = tuple(ord(c) for c in color1.lsstrip('0x').decode('hex'))
c2tuple = tuple(ord(c) for c in color2.lsstrip('0x').decode('hex'))
c3tuple = tuple(ord(c) for c in color3.lsstrip('0x').decode('hex'))
def RedBlackSkyBlue():
cdict = {'red': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0)),
'green': ((0.0, 0.0, 0.9),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0)),
'blue': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0))
}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def RedBlackBlue():
cdict = {'red': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0)),
'green': ((0.0, 0.0, 0.0),
(1.0, 0.0, 0.0)),
'blue': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0))
}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def RedBlackGreen():
cdict = {'red': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0)),
'blue': ((0.0, 0.0, 0.0),
(1.0, 0.0, 0.0)),
'green': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0))
}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def YellowBlackBlue():
cdict = {'red': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0)),
'green': ((0.0, 0.0, 0.8),
(0.5, 0.1, 0.0),
(1.0, 1.0, 1.0)),
'blue': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0))
}
### yellow is created by adding y = 1 to RedBlackSkyBlue green last tuple
### modulate between blue and cyan using the last y var in the first green tuple
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def BlackYellowBlue():
cdict = {'red': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0)),
'green': ((0.0, 0.0, 0.8),
(0.5, 0.1, 0.0),
(1.0, 1.0, 1.0)),
'blue': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0))
}
### yellow is created by adding y = 1 to RedBlackSkyBlue green last tuple
### modulate between blue and cyan using the last y var in the first green tuple
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def PairedColorMap():
### Taken from Matplotlib versions 1.3 as a smooth rather than segmented map
cdict = {'blue': [(0.0, 0.89019608497619629,
0.89019608497619629), (0.090909090909090912, 0.70588237047195435,
0.70588237047195435), (0.18181818181818182, 0.54117649793624878,
0.54117649793624878), (0.27272727272727271, 0.17254902422428131,
0.17254902422428131), (0.36363636363636365, 0.60000002384185791,
0.60000002384185791), (0.45454545454545453, 0.10980392247438431,
0.10980392247438431), (0.54545454545454541, 0.43529412150382996,
0.43529412150382996), (0.63636363636363635, 0.0, 0.0),
(0.72727272727272729, 0.83921569585800171, 0.83921569585800171),
(0.81818181818181823, 0.60392159223556519, 0.60392159223556519),
(0.90909090909090906, 0.60000002384185791, 0.60000002384185791), (1.0,
0.15686275064945221, 0.15686275064945221)],
'green': [(0.0, 0.80784314870834351, 0.80784314870834351),
(0.090909090909090912, 0.47058823704719543, 0.47058823704719543),
(0.18181818181818182, 0.87450981140136719, 0.87450981140136719),
(0.27272727272727271, 0.62745100259780884, 0.62745100259780884),
(0.36363636363636365, 0.60392159223556519, 0.60392159223556519),
(0.45454545454545453, 0.10196078568696976, 0.10196078568696976),
(0.54545454545454541, 0.74901962280273438, 0.74901962280273438),
(0.63636363636363635, 0.49803921580314636, 0.49803921580314636),
(0.72727272727272729, 0.69803923368453979, 0.69803923368453979),
(0.81818181818181823, 0.23921568691730499, 0.23921568691730499),
(0.90909090909090906, 1.0, 1.0), (1.0, 0.3490196168422699,
0.3490196168422699)],
'red': [(0.0, 0.65098041296005249, 0.65098041296005249),
(0.090909090909090912, 0.12156862765550613, 0.12156862765550613),
(0.18181818181818182, 0.69803923368453979, 0.69803923368453979),
(0.27272727272727271, 0.20000000298023224, 0.20000000298023224),
(0.36363636363636365, 0.9843137264251709, 0.9843137264251709),
(0.45454545454545453, 0.89019608497619629, 0.89019608497619629),
(0.54545454545454541, 0.99215686321258545, 0.99215686321258545),
(0.63636363636363635, 1.0, 1.0), (0.72727272727272729,
0.7921568751335144, 0.7921568751335144), (0.81818181818181823,
0.41568627953529358, 0.41568627953529358), (0.90909090909090906,
1.0, 1.0), (1.0, 0.69411766529083252, 0.69411766529083252)]}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def Pastel1ColorMap():
### Taken from Matplotlib versions 1.3 as a smooth rather than segmented map
cdict = {'blue': [(0.0, 0.68235296010971069,
0.68235296010971069), (0.125, 0.89019608497619629,
0.89019608497619629), (0.25, 0.77254903316497803,
0.77254903316497803), (0.375, 0.89411765336990356,
0.89411765336990356), (0.5, 0.65098041296005249, 0.65098041296005249),
(0.625, 0.80000001192092896, 0.80000001192092896), (0.75,
0.74117648601531982, 0.74117648601531982), (0.875,
0.92549020051956177, 0.92549020051956177), (1.0, 0.94901961088180542,
0.94901961088180542)],
'green': [(0.0, 0.70588237047195435, 0.70588237047195435), (0.125,
0.80392158031463623, 0.80392158031463623), (0.25,
0.92156863212585449, 0.92156863212585449), (0.375,
0.79607844352722168, 0.79607844352722168), (0.5,
0.85098040103912354, 0.85098040103912354), (0.625, 1.0, 1.0),
(0.75, 0.84705883264541626, 0.84705883264541626), (0.875,
0.85490196943283081, 0.85490196943283081), (1.0,
0.94901961088180542, 0.94901961088180542)],
'red': [(0.0, 0.9843137264251709, 0.9843137264251709), (0.125,
0.70196080207824707, 0.70196080207824707), (0.25,
0.80000001192092896, 0.80000001192092896), (0.375,
0.87058824300765991, 0.87058824300765991), (0.5,
0.99607843160629272, 0.99607843160629272), (0.625, 1.0, 1.0),
(0.75, 0.89803922176361084, 0.89803922176361084), (0.875,
0.99215686321258545, 0.99215686321258545), (1.0,
0.94901961088180542, 0.94901961088180542)]}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def Pastel2ColorMap():
### Taken from Matplotlib versions 1.3 as a smooth rather than segmented map
cdict = {'blue': [(0.0, 0.80392158031463623,
0.80392158031463623), (0.14285714285714285, 0.67450982332229614,
0.67450982332229614), (0.2857142857142857, 0.90980392694473267,
0.90980392694473267), (0.42857142857142855, 0.89411765336990356,
0.89411765336990356), (0.5714285714285714, 0.78823530673980713,
0.78823530673980713), (0.7142857142857143, 0.68235296010971069,
0.68235296010971069), (0.8571428571428571, 0.80000001192092896,
0.80000001192092896), (1.0, 0.80000001192092896,
0.80000001192092896)],
'green': [(0.0, 0.88627451658248901, 0.88627451658248901),
(0.14285714285714285, 0.80392158031463623, 0.80392158031463623),
(0.2857142857142857, 0.83529412746429443, 0.83529412746429443),
(0.42857142857142855, 0.7921568751335144, 0.7921568751335144),
(0.5714285714285714, 0.96078431606292725, 0.96078431606292725),
(0.7142857142857143, 0.94901961088180542, 0.94901961088180542),
(0.8571428571428571, 0.88627451658248901, 0.88627451658248901),
(1.0, 0.80000001192092896, 0.80000001192092896)],
'red': [(0.0, 0.70196080207824707, 0.70196080207824707),
(0.14285714285714285, 0.99215686321258545, 0.99215686321258545),
(0.2857142857142857, 0.79607844352722168, 0.79607844352722168),
(0.42857142857142855, 0.95686274766921997, 0.95686274766921997),
(0.5714285714285714, 0.90196079015731812, 0.90196079015731812),
(0.7142857142857143, 1.0, 1.0), (0.8571428571428571,
0.94509804248809814, 0.94509804248809814), (1.0,
0.80000001192092896, 0.80000001192092896)]}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def filepath(filename):
fn = unique.filepath(filename)
return fn
def remoteImportData(filename,geneFilter=None,reverseOrder=True):
matrix, column_header, row_header, dataset_name, group_db = importData(filename,geneFilter=geneFilter,reverseOrder=reverseOrder)
try:
return matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters
except:
return matrix, column_header, row_header, dataset_name, group_db, [], []
def convertClusterNameToInt(cluster_ids):
index=0
c=1; prior=[]; clusters={}
for i in cluster_ids:
if i in clusters:
c1 = clusters[i]
else:
c1 = c; clusters[i]=c1
c+=1
prior.append(c1)
index+=1
return prior
def importData(filename,Normalize=False,reverseOrder=True,geneFilter=None,
zscore=False,forceClusters=False):
global priorColumnClusters
global priorRowClusters
try:
if len(priorColumnClusters)>0:
priorColumnClusters = None
priorRowClusters = None
except Exception: pass
getRowClusters=False
start_time = time.time()
fn = filepath(filename)
matrix=[]
original_matrix=[]
row_header=[]
overwriteGroupNotations=True
x=0; inputMax=0; inputMin=100
filename = string.replace(filename,'\\','/')
dataset_name = string.split(filename,'/')[-1][:-4]
if '.cdt' in filename: start = 3
else: start = 1
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
if '.cdt' in filename: t = [t[0]]+t[3:]
if t[1] == 'row_clusters-flat':
t = t = [t[0]]+t[2:]
### color samples by annotated groups if an expression file
new_headers=[]
temp_groups={}
original_headers=t[1:]
if ('exp.' in filename or 'filteredExp.' in filename or 'MarkerGene' in filename) and forceClusters==False:# and ':' not in data:
if overwriteGroupNotations:
### Use groups file annotations over any header sample separation with a ":"
for i in t:
if ':' in i: ### Don't annotate groups according to the clusters
group,i = string.split(i,':')
new_headers.append(i)
temp_groups[i] = group
else: new_headers.append(i)
filename = string.replace(filename,'-steady-state.txt','.txt')
try:
import ExpressionBuilder
try: sample_group_db = ExpressionBuilder.simplerGroupImport(filename)
except Exception: sample_group_db={}
if len(temp_groups)>0 and len(sample_group_db)==0:
sample_group_db = temp_groups
if len(new_headers)>0:
t = new_headers
new_headers = []
for v in t:
if v in sample_group_db:
v = sample_group_db[v]+':'+v
new_headers.append(v)
t = new_headers
except Exception:
#print traceback.format_exc()
pass
group_db, column_header = assignGroupColors(t[1:])
x=1
elif 'column_clusters-flat' in t:
try:
if 'NA' in t:
kill
try:
if forceClusters==False:
try:
prior = map(lambda x: int(float(x)),t[2:])
except:
if 'Query.txt' in filename:
forceClusterIntError
else:
prior = map(lambda x: x,t[2:])
else:
prior = map(lambda x: x,t[2:])
except Exception:
### Replace the cluster string with number
index=0
c=1; prior=[]; clusters={}
for i in t[2:]:
original_headers[index] = i+':'+original_headers[index]
if i in clusters:
c1 = clusters[i]
else:
c1 = c; clusters[i]=c1
c+=1
prior.append(c1)
index+=1
#prior=[]
if len(temp_groups)==0: ### Hence, no secondary group label combined with the sample name
if '-ReOrdered.txt' not in filename: ### Applies to cellHarmony UMAP and heatmap visualization
group_db, column_header = assignGroupColors(original_headers)
#priorColumnClusters = dict(zip(column_header, prior))
priorColumnClusters = prior
except Exception:
#print traceback.format_exc()
pass
start = 2
getRowClusters = True
priorRowClusters=[]
elif 'EWEIGHT' in t: pass
else:
gene = t[0]
if geneFilter==None:
proceed = True
elif gene in geneFilter:
proceed = True
else:
proceed = False
if proceed:
nullsPresent = False
#if ' ' not in t and '' not in t: ### Occurs for rows with missing data
try: s = map(float,t[start:])
except Exception:
nullsPresent=True
s=[]
for value in t[start:]:
try: s.append(float(value))
except Exception: s.append(0.000101)
#s = numpy.ma.masked_values(s, 0.000101)
original_matrix.append(s)
try:
if max(s)>inputMax: inputMax = max(s)
except:
continue ### empty row
if min(s)<inputMin: inputMin = min(s)
#if (abs(max(s)-min(s)))>2:
if Normalize!=False:
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
if Normalize=='row mean':
#avg = min(s)
avg = numpy.mean(s)
else:
avg = numpy.median(s)
if nullsPresent:
s=[] ### Needs to be done to zero out the values
for value in t[start:]:
try: s.append(float(value)-avg)
except Exception: s.append(0.000101)
#s = numpy.ma.masked_values(s, 0.000101)
else:
s = map(lambda x: x-avg,s) ### normalize to the mean
if ' ' in gene:
try:
g1,g2 = string.split(gene,' ')
if g1 == g2: gene = g1
except Exception: pass
if getRowClusters:
try:
#priorRowClusters[gene]=int(float(t[1]))
priorRowClusters.append(int(float(t[1])))
except Exception: pass
if zscore:
### convert to z-scores for normalization prior to PCA
avg = numpy.mean(s)
std = numpy.std(s)
if std ==0:
std = 0.1
try: s = map(lambda x: (x-avg)/std,s)
except Exception: pass
if geneFilter==None:
matrix.append(s)
row_header.append(gene)
else:
if gene in geneFilter:
matrix.append(s)
row_header.append(gene)
x+=1
if inputMax>100: ### Thus, not log values
print 'Converting values to log2...'
matrix=[]
k=0
if inputMin==0: increment = 1#0.01
else: increment = 1
for s in original_matrix:
if 'counts.' in filename:
s = map(lambda x: math.log(x+1,2),s)
else:
try: s = map(lambda x: math.log(x+increment,2),s)
except Exception:
print filename
print Normalize
print row_header[k], min(s),max(s); kill
if Normalize!=False:
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
if Normalize=='row mean':
avg = numpy.average(s)
else: avg = avg = numpy.median(s)
s = map(lambda x: x-avg,s) ### normalize to the mean
if zscore: ### The above z-score does not impact the original_matrix which is analyzed
### convert to z-scores for normalization prior to PCA
avg = numpy.mean(s)
std = numpy.std(s)
if std ==0:
std = 0.1
try: s = map(lambda x: (x-avg)/std,s)
except Exception: pass
matrix.append(s)
k+=1
del original_matrix
if zscore: print 'Converting values to normalized z-scores...'
#reverseOrder = True ### Cluster order is background (this is a temporary workaround)
if reverseOrder == True:
matrix.reverse(); row_header.reverse()
time_diff = str(round(time.time()-start_time,1))
try:
print '%d rows and %d columns imported for %s in %s seconds...' % (len(matrix),len(column_header),dataset_name,time_diff)
except Exception:
print 'No data in input file.'; force_error
### Add groups for column pre-clustered samples if there
group_db2, row_header2 = assignGroupColors(list(row_header)) ### row_header gets sorted in this function and will get permenantly screwed up if not mutated
#if '.cdt' in filename: matrix.reverse(); row_header.reverse()
for i in group_db2:
if i not in group_db: group_db[i] = group_db2[i]
##print group_db;sys.exit()
return matrix, column_header, row_header, dataset_name, group_db
def importSIF(filename):
fn = filepath(filename)
edges=[]
x=0
if '/' in filename:
dataset_name = string.split(filename,'/')[-1][:-4]
else:
dataset_name = string.split(filename,'\\')[-1][:-4]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
parent,type,child = string.split(data,'\t')
if 'AltAnalyze' in dataset_name:
### This is the order for proper directed interactions in the AltAnalyze-interaction viewer
edges.append([parent,child,type])
else:
if '(' in parent: ### for TF-target annotations
parent = string.split(parent,'(')[0]
if ':' in child:
child = string.split(child,':')[1]
if 'TF' in dataset_name or 'UserSuppliedAssociations' in dataset_name or 'WGRV' in dataset_name:
edges.append([parent,child,type]) ### Do this to indicate that the TF is regulating the target
else:
edges.append([child,parent,type])
edges = unique.unique(edges)
return edges
def customShuffle(ls):
index=0
shuffled=[]
for i in ls:
if i not in shuffled:
shuffled.append(i)
try: alt_i = ls[(1+index)*-1]
except:
alt_i = ls[-1]
if alt_i not in shuffled:
shuffled.append(alt_i)
try: alt_i = ls[int((index+len(ls))/2)]
except:
alt_i = ls[-1]
if alt_i not in shuffled:
shuffled.append(alt_i)
index+=1
return shuffled
def cmap_map(function, cmap):
#https://scipy-cookbook.readthedocs.io/items/Matplotlib_ColormapTransformations.html
""" Applies function (which should operate on vectors of shape 3: [r, g, b]), on colormap cmap.
This routine will break any discontinuous points in a colormap.
"""
cdict = cmap._segmentdata
step_dict = {}
# Firt get the list of points where the segments start or end
for key in ('red', 'green', 'blue'):
step_dict[key] = list(map(lambda x: x[0], cdict[key]))
step_list = sum(step_dict.values(), [])
step_list = np.array(list(set(step_list)))
# Then compute the LUT, and apply the function to the LUT
reduced_cmap = lambda step : np.array(cmap(step)[0:3])
old_LUT = np.array(list(map(reduced_cmap, step_list)))
new_LUT = np.array(list(map(function, old_LUT)))
# Now try to make a minimal segment definition of the new LUT
cdict = {}
for i, key in enumerate(['red','green','blue']):
this_cdict = {}
for j, step in enumerate(step_list):
if step in step_dict[key]:
this_cdict[step] = new_LUT[j, i]
elif new_LUT[j,i] != old_LUT[j, i]:
this_cdict[step] = new_LUT[j, i]
colorvector = list(map(lambda x: x + (x[1], ), this_cdict.items()))
colorvector.sort()
cdict[key] = colorvector
return matplotlib.colors.LinearSegmentedColormap('colormap',cdict,1024)
def remoteAssignGroupColors(groups_file):
import ExpressionBuilder
### Import an ordered groups dictionary
sample_group_db = ExpressionBuilder.simplerGroupImport(groups_file)
group_header = []
for sample in sample_group_db:
group = sample_group_db[sample]
group_header.append(group+':'+sample)
group_db, column_header = assignGroupColors(group_header)
return group_db
def assignGroupColors(t):
""" Assign a unique color to each group. Optionally used for cluster display. """
column_header=[]; group_number_db={}
groupNamesPresent=False # Some samples may have missing group names which will result in a clustering error
for i in t:
if ':' in i: groupNamesPresent = True
for i in t:
repls = {'.2txt' : '', '.2bed' : '', '.2tab' : ''}
i=reduce(lambda a, kv: a.replace(*kv), repls.iteritems(), i)
if ':' in i:
group,j = string.split(i,':')[:2]
group_number_db[group]=[]
elif groupNamesPresent:
group_number_db['UNK']=[]
i = 'UNK:'+i
column_header.append(i)
import random
k = 0
group_db={}; color_db={}
color_list = ['r', 'b', 'y', 'g', 'w', 'k', 'm']
n = len(group_number_db)
from_list = matplotlib.colors.LinearSegmentedColormap.from_list
if len(group_number_db)>3:
color_list = []
cm=PairedColorMap()
#cm = pylab.cm.get_cmap('Paired') #Paired # binary #Paired
#cm = cmap_map(lambda x: x/2 + 0.5,cm)
sorted_range = range(len(group_number_db))
sorted_range = customShuffle(sorted_range)
random.seed(0)
random.shuffle(sorted_range)
for i in sorted_range:
rgb = cm(1.0*i/(len(group_number_db)-1)) # color will now be an RGBA tuple
color_list.append(rgb)
#color_list=[]
#color_template = [1,1,1,0,0,0,0.5,0.5,0.5,0.25,0.25,0.25,0.75,0.75,0.75]
t.sort() ### Ensure that all clusters have the same order of groups
for i in column_header:
repls = {'.2txt' : '', '.2bed' : '', '.2tab' : ''}
i=reduce(lambda a, kv: a.replace(*kv), repls.iteritems(), i)
if ':' in i:
group,j = string.split(i,':')[:2]
try: color,ko = color_db[group]
except Exception:
try: color_db[group] = color_list[k],k
except Exception:
### If not listed in the standard color set add a new random color
rgb = tuple(scipy.rand(3)) ### random color
#rgb = tuple(random.sample(color_template,3)) ### custom alternative method
color_list.append(rgb)
color_db[group] = color_list[k], k
color,ko = color_db[group]
k+=1
group_db[i] = group, color, ko
#column_header.append(i)
return group_db, column_header
def verifyFile(filename):
status = 'not found'
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = 'found';break
except Exception: status = 'not found'
return status
def AppendOrWrite(export_path):
export_path = filepath(export_path)
status = verifyFile(export_path)
if status == 'not found':
export_data = export.ExportFile(export_path) ### Write this new file
else:
export_data = open(export_path,'a') ### Appends to existing file
return export_path, export_data, status
def exportCustomGeneSet(geneSetName,species,allGenes):
for gene in allGenes:break
if 'ENS' not in gene:
try:
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception: symbol_to_gene={}
if species != None:
export_path, export_data, status = AppendOrWrite('AltDatabase/goelite/'+species+'/gene-mapp/Ensembl-StoredGeneSets.txt')
stored_lines=[]
for line in open(export_path,'rU').xreadlines(): stored_lines.append(line)
if status == 'not found':
export_data.write('GeneID\tEmpty\tGeneSetName\n')
for gene in allGenes:
if ' ' in gene:
a,b=string.split(gene,' ')
if 'ENS' in a: gene = a
else: gene = b
if 'ENS' not in gene and gene in symbol_to_gene:
gene = symbol_to_gene[gene][0]
line = gene+'\t\t'+geneSetName+'\n'
if line not in stored_lines:
export_data.write(line)
export_data.close()
else:
print 'Could not store since no species name provided.'
def writetSNEScores(scores,outputdir):
export_obj = export.ExportFile(outputdir)
for matrix_row in scores:
matrix_row = map(str,matrix_row)
export_obj.write(string.join(matrix_row,'\t')+'\n')
export_obj.close()
def importtSNEScores(inputdir):
#print inputdir
scores=[]
### Imports tSNE scores to allow for different visualizations of the same scatter plot
for line in open(inputdir,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
t=map(float,t)
scores.append(t)
return scores
def runUMAP(matrix, column_header,dataset_name,group_db,display=False,showLabels=False,
row_header=None,colorByGene=None,species=None,reimportModelScores=True,method="UMAP",
rootDir='',finalOutputDir='',group_alt=None):
global root_dir
global graphic_link
graphic_link=[]
root_dir = rootDir
tSNE(matrix, column_header,dataset_name,group_db,display=False,showLabels=False,
row_header=None,colorByGene=None,species=None,reimportModelScores=reimportModelScores,method="UMAP")
import shutil
filename = 'Clustering-'+dataset_name+'-'+method+'.pdf'
filename = string.replace(filename,'Clustering-Clustering','Clustering')
new_file=finalOutputDir + filename
new_file=string.replace(new_file,'Clustering-','')
new_file=string.replace(new_file,'exp.','')
old_file=root_dir+filename
shutil.move(old_file,new_file)
filename = filename[:-3]+'png'
new_file=finalOutputDir + filename
new_file=string.replace(new_file,'Clustering-','')
new_file=string.replace(new_file,'exp.','')
old_file=root_dir+filename
shutil.move(old_file,new_file)
if group_alt != None:
tSNE(matrix, column_header,dataset_name,group_alt,display=False,showLabels=False,
row_header=None,colorByGene=None,species=None,reimportModelScores=True,method="UMAP")
filename = filename[:-3]+'pdf'
new_file=finalOutputDir + filename[:-4]+'-CellType.pdf'
new_file=string.replace(new_file,'Clustering-','')
new_file=string.replace(new_file,'exp.','')
old_file=root_dir+filename
shutil.move(old_file,new_file)
filename = filename[:-3]+'png'
new_file=finalOutputDir + filename[:-4]+'-CellType.png'
new_file=string.replace(new_file,'Clustering-','')
new_file=string.replace(new_file,'exp.','')
old_file=root_dir+filename
shutil.move(old_file,new_file)
old_file=root_dir+dataset_name+'-'+method+'_scores.txt'
new_file=finalOutputDir+dataset_name+'-'+method+'_coordinates.txt'
new_file=string.replace(new_file,'exp.','')
shutil.copy(old_file,new_file)
def tSNE(matrix, column_header,dataset_name,group_db,display=True,showLabels=False,
row_header=None,colorByGene=None,species=None,reimportModelScores=True,
method="tSNE",maskGroups=None):
try: prior_clusters = priorColumnClusters
except Exception: prior_clusters=[]
try:
if priorColumnClusters==None: prior_clusters=[]
except:
pass
try:
if len(prior_clusters)>0 and len(group_db)==0:
newColumnHeader=[]
i=0
for sample_name in column_header:
newColumnHeader.append(str(prior_clusters[i])+':'+sample_name)
i+=1
group_db, column_header = assignGroupColors(newColumnHeader)
elif len(group_db)>0: ### When a non-group prefix groups file is supplied
try:
newColumnHeader=[]
i=0
alt_name_db={}
for orig_sample_name in group_db:
sample_name = orig_sample_name
if ':' in orig_sample_name:
sample_name = string.split(orig_sample_name,':')[1]
alt_name_db[sample_name] = orig_sample_name
for sample_name in column_header:
if ':' in sample_name:
revised_sample_name = string.split(sample_name,':')[1]
if revised_sample_name in alt_name_db:
sample_name = alt_name_db[revised_sample_name]
newColumnHeader.append(sample_name)
i+=1
group_db, column_header = assignGroupColors(newColumnHeader)
except:
pass
except Exception,e:
print traceback.format_exc()
#print e
group_db={}
if reimportModelScores:
start = time.time()
print 'Re-importing',method,'model scores rather than calculating from scratch',
print root_dir+dataset_name+'-'+method+'_scores.txt'
try: scores = importtSNEScores(root_dir+dataset_name+'-'+method+'_scores.txt'); print '...import finished'
except Exception:
reimportModelScores=False; print '...no existing score file found'
if benchmark:
print 0,time.time() - start, 'seconds'
if reimportModelScores==False:
start = time.time()
X=matrix.T
"""
from tsne import bh_sne
X = np.asarray(X).astype('float64')
X = X.reshape((X.shape[0], -1))
x_data = x_data.reshape((x_data.shape[0], -1))
scores = bh_sne(X)"""
#model = TSNE(n_components=2, random_state=0,init='pca',early_exaggeration=4.0,perplexity=20)
print "Performing",method,
if method=="tSNE" or method=="t-SNE":
from sklearn.manifold import TSNE
model = TSNE(n_components=2)
if method=="UMAP":
try:
import umap
model=umap.UMAP(n_neighbors=50,min_dist=0.75,metric='correlation')
except:
try:
from visualization_scripts.umap_learn import umap ### Bypasses issues with Py2app importing umap (and secondarily numba/llvmlite)
model=umap.UMAP(n_neighbors=50,min_dist=0.75,metric='correlation')
except: ### requires single-threading for Windows platforms (possibly others)
from visualization_scripts.umap_learn_single import umap ### Bypasses issues with Py2app importing umap (and secondarily numba/llvmlite)
model=umap.UMAP(n_neighbors=50,min_dist=0.75,metric='correlation')
print '... UMAP run'
#model = TSNE(n_components=2,init='pca', random_state=0, verbose=1, perplexity=40, n_iter=300)
#model = TSNE(n_components=2,verbose=1, perplexity=40, n_iter=300)
#model = TSNE(n_components=2, random_state=0, n_iter=10000, early_exaggeration=10)
scores=model.fit_transform(X)
### Export the results for optional re-import later
writetSNEScores(scores,root_dir+dataset_name+'-'+method+'_scores.txt')
#pylab.scatter(scores[:,0], scores[:,1], 20, labels);
if benchmark:
print 0,time.time() - start, 'seconds'
if maskGroups != None:
group_name,restricted_samples = maskGroups
dataset_name += '-'+group_name ### indicate the restricted group
start = time.time()
### Exclude samples with high TSNE deviations
scoresT = zip(*scores)
exclude={}
if benchmark:
print 1,time.time() - start, 'seconds'
start = time.time()
try:
for vector in scoresT:
lower1th,median_val,upper99th,int_qrt_range = statistics.iqr(list(vector),k1=99.9,k2=0.1)
index=0
for i in vector:
if (i > upper99th+1) or (i<lower1th-1):
exclude[index]=None
index+=1
except Exception:
pass
print 'Not showing',len(exclude),'outlier samples.'
if benchmark:
print 2,time.time() - start, 'seconds'
start = time.time()
fig = pylab.figure()
ax = fig.add_subplot(111)
pylab.xlabel(method.upper()+'-X')
pylab.ylabel(method.upper()+'-Y')
axes = getAxesTransposed(scores,exclude=exclude) ### adds buffer space to the end of each axis and creates room for a legend
pylab.axis(axes)
if benchmark:
print 3,time.time() - start, 'seconds'
start = time.time()
marker_size = 15
if len(column_header)>20:
marker_size = 12
if len(column_header)>40:
marker_size = 10
if len(column_header)>150:
marker_size = 7
if len(column_header)>500:
marker_size = 5
if len(column_header)>1000:
marker_size = 4
if len(column_header)>2000:
marker_size = 3
if len(column_header)>4000:
marker_size = 2
if len(column_header)>6000:
marker_size = 1
### Color By Gene
if colorByGene != None and len(matrix)==0:
print 'Gene %s not found in the imported dataset... Coloring by groups.' % colorByGene
if colorByGene != None and len(matrix)>0:
gene_translation_db={}
matrix = numpy.array(matrix)
min_val = matrix.min() ### min val
if ' ' in colorByGene:
genes = string.split(colorByGene,' ')
else:
genes = [colorByGene]
genePresent=False
numberGenesPresent=[]
for gene in genes:
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
### Translate symbol to Ensembl
if benchmark:
print 4,time.time() - start, 'seconds'
start = time.time()
if len(numberGenesPresent)==0:
try:
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
for symbol in genes:
if symbol in symbol_to_gene:
gene = symbol_to_gene[symbol][0]
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
gene_translation_db[symbol]=gene
except Exception: pass
numberGenesPresent = len(numberGenesPresent)
if numberGenesPresent==1:
cm = pylab.cm.get_cmap('Reds')
else:
if numberGenesPresent==2:
cm = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
#cm = matplotlib.colors.ListedColormap(['w', 'k']) ### If you want to hide one of the groups
else:
cm = pylab.cm.get_cmap('gist_rainbow')
cm = pylab.cm.get_cmap('Paired')
"""
elif numberGenesPresent==3:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
elif numberGenesPresent==4:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
elif numberGenesPresent==5:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif numberGenesPresent==6:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif numberGenesPresent==7:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
"""
def get_cmap(n, name='hsv'):
'''Returns a function that maps each index in 0, 1, ..., n-1 to a distinct
RGB color; the keyword argument name must be a standard mpl colormap name.'''
return pylab.cm.get_cmap(name, n)
if benchmark:
print 5,time.time() - start, 'seconds'
start = time.time()
if genePresent:
dataset_name+='-'+colorByGene
group_db={}
bestGeneAssociated={}
k=0
for gene in genes:
try:
try: i = row_header.index(gene)
except Exception: i = row_header.index(gene_translation_db[gene])
values = map(float,matrix[i])
min_val = min(values)
bin_size = (max(values)-min_val)/8
max_val = max(values)
ranges = []
iz=min_val
while iz < (max(values)-bin_size/100):
r = iz,iz+bin_size
if len(ranges)==7:
r = iz,max_val
ranges.append(r)
iz+=bin_size
color_db = {}
colors = get_cmap(len(genes))
for i in range(len(ranges)):
if i==0:
color = '#C0C0C0'
else:
if numberGenesPresent==1:
### use a single color gradient
color = cm(1.*i/len(ranges))
#color = cm(1.*(i+1)/len(ranges))
else:
if i>2:
if len(genes)<8:
color = cm(k)
else:
color = colors(k)
else:
color = '#C0C0C0'
color_db[ranges[i]] = color
i=0
for val in values:
sample = column_header[i]
for (l,u) in color_db:
range_index = ranges.index((l,u)) ### what is the ranking of this range
if val>=l and val<=u:
color = color_db[(l,u)]
color_label = [gene+'-range: '+str(l)[:4]+'-'+str(u)[:4],color,'']
group_db[sample] = color_label
try: bestGeneAssociated[sample].append([range_index,val,color_label])
except Exception: bestGeneAssociated[sample] = [[range_index,val,color_label]]
i+=1
#print min(values),min_val,bin_size,max_val
if len(genes)>1:
### Collapse and rank multiple gene results
for sample in bestGeneAssociated:
bestGeneAssociated[sample].sort()
color_label = bestGeneAssociated[sample][-1][-1]
if numberGenesPresent>1:
index = bestGeneAssociated[sample][-1][0]
if index > 2:
gene = string.split(color_label[0],'-')[0]
else:
gene = 'Null'
color_label[0] = gene
group_db[sample] = color_label
except Exception:
print [gene], 'not found in rows...'
#print traceback.format_exc()
k+=1
else:
print [colorByGene], 'not found in rows...'
if benchmark:
print 6,time.time() - start, 'seconds'
start = time.time()
pylab.title(method+' - '+dataset_name)
import collections
group_names = collections.OrderedDict()
group_scores={}
i=0
if showLabels:
### plot each dot separately
for sample_name in column_header: #scores[0]
if maskGroups != None:
base_name = sample_name
if ':' in sample_name:
base_name = string.split(base_name,':')[1]
if base_name not in restricted_samples:
exclude[i]=None ### Don't visualize this sample
if i not in exclude:
### Add the text labels for each
try:
### Get group name and color information
group_name,color,k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color
### Get the mean coordinates for each group label
try: group_scores[group_name].append([scores[i][0],scores[i][1]])
except: group_scores[group_name] = [[scores[i][0],scores[i][1]]]
except Exception:
color = 'r'; label=None
ax.plot(scores[i][0],scores[i][1],color=color,marker='o',markersize=marker_size,label=label,markeredgewidth=0,picker=False)
#except Exception: print i, len(scores[pcB]);kill
if showLabels:
try: sample_name = ' '+string.split(sample_name,':')[1]
except Exception: pass
ax.text(scores[i][0],scores[i][1],sample_name,fontsize=11)
i+=1
else:
### Plot the dots for each group
for sample_name in column_header: #scores[0]
if maskGroups != None:
base_name = sample_name
if ':' in sample_name:
base_name = string.split(base_name,':')[1]
if base_name not in restricted_samples:
exclude[i]=None ### Don't visualize this sample
if i not in exclude:
try:
### Get group name and color information
group_name,color,k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color
### Get the mean coordinates for each group label
try: group_scores[group_name].append([scores[i][0],scores[i][1]])
except: group_scores[group_name] = [[scores[i][0],scores[i][1]]]
except Exception:
color = 'r'; label=None
i+=1
""" Plot separately for efficency """
for group_name in group_names:
color = group_names[group_name]
label = group_name
scores = group_scores[group_name]
x_coords = map(lambda s: s[0],scores)
y_coords = map(lambda s: s[1],scores)
ax.scatter(x_coords,y_coords,color=color,s=marker_size,label=label,linewidths=0,alpha=None)
### Set the legend label size
markerscale = 2; ncol = 1
try:
if len(group_names)>15:
markerscale = 4
if len(group_names)>30:
markerscale = 4
#ncol = 2
except: pass
if benchmark:
print 7,time.time() - start, 'seconds'
start = time.time()
### Compute the mode coordinate pair to assign the group label to a fragmented population
if colorByGene == None:
try:
median_or_mode = 'median'
font_size = 10
if len(group_scores)>10:
font_size = 8
if len(group_scores)>20:
font_size = 4
if len(group_scores)>40:
font_size = 3
for group_name in group_scores:
coords = group_scores[group_name]
coords.sort()
new_font_size = font_size
#avg = [float(sum(col))/len(col) for col in zip(*coords)] ### separate average values
#avg = [float(numpy.median(col)) for col in zip(*coords)] ### separate median values
if median_or_mode == 'median':
coord1,coord2 = coords[len(coords)/2] ### median list
else:
coord1 = stats.mode(map(lambda x: int(x[0]), coords))[0][0]
coord2 = stats.mode(map(lambda x: int(x[1]), coords))[0][0]
for coord in coords:
if int(coord[0]) == coord1:
coord1 = coord[0]
coord2 = coord[1]
break
#if len(group_name)>15: new_font_size = font_size-2
#ax.text(coord1+0.02,coord2+0.02,group_name,fontsize=new_font_size,color='white',ha='center')
#ax.text(coord1-0.02,coord2-0.02,group_name,fontsize=new_font_size,color='white',ha='center')
ax.text(coord1,coord2,group_name,fontsize=new_font_size,color='black',ha='center')
except:
#print traceback.format_exc()
pass
group_count = []
for i in group_db:
if group_db[i][0] not in group_count:
group_count.append(group_db[i][0])
if benchmark:
print 8,time.time() - start, 'seconds'
start = time.time()
#print len(group_count)
Lfontsize = 6
if len(group_count)>20:
Lfontsize = 5
if len(group_count)>30:
Lfontsize = 4
if len(group_count)>40:
Lfontsize = 4
if len(group_count)>50:
Lfontsize = 3
i=0
box = ax.get_position()
if len(group_count) > 0: ### Make number larger to get the legend in the plot -- BUT, the axis buffer above has been disabled
# Shink current axis by 20%
ax.set_position([box.x0, box.y0, box.width * 0.8, box.height])
""" Plot the legend to the right of the plot """
ax.legend(ncol=ncol,loc='center left', bbox_to_anchor=(1, 0.5),fontsize = Lfontsize, markerscale = markerscale) ### move the legend over to the right of the plot
#except Exception: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
else:
ax.set_position([box.x0, box.y0, box.width, box.height])
pylab.legend(loc="upper left", prop={'size': 10})
if benchmark:
print 9,time.time() - start, 'seconds'
start = time.time()
filename = 'Clustering-'+dataset_name+'-'+method+'.pdf'
filename = string.replace(filename,'Clustering-Clustering','Clustering')
try: pylab.savefig(root_dir + filename)
except Exception: None ### Rare error
#print 'Exporting:',filename
filename = filename[:-3]+'png'
if benchmark:
print 10,time.time() - start, 'seconds'
start = time.time()
try: pylab.savefig(root_dir + filename) #dpi=200, transparent=True
except Exception: None ### Rare error
graphic_link.append(['Principal Component Analysis',root_dir+filename])
if benchmark:
print 11,time.time() - start, 'seconds'
if display:
print 'Exporting:',filename
try:
pylab.show()
except Exception:
#print traceback.format_exc()
pass### when run in headless mode
def excludeHighlyCorrelatedHits(x,row_header):
### For methylation data or other data with redundant signatures, remove these and only report the first one
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
D1 = numpy.corrcoef(x)
i=0
exclude={}
gene_correlations={}
include = []
for score_ls in D1:
k=0
for v in score_ls:
if str(v)!='nan':
if v>1.00 and k!=i:
#print row_header[i], row_header[k], v
if row_header[i] not in exclude:
exclude[row_header[k]]=[]
#if k not in exclude: include.append(row_header[k])
k+=1
#if i not in exclude: include.append(row_header[i])
i+=1
#print len(exclude),len(row_header);sys.exit()
return exclude
def PrincipalComponentAnalysis(matrix, column_header, row_header, dataset_name,
group_db, display=False, showLabels=True, algorithm='SVD', geneSetName=None,
species=None, pcA=1,pcB=2, colorByGene=None, reimportModelScores=True):
print "Performing Principal Component Analysis..."
from numpy import mean,cov,double,cumsum,dot,linalg,array,rank
try: prior_clusters = priorColumnClusters
except Exception: prior_clusters=[]
if prior_clusters == None: prior_clusters=[]
try:
if len(prior_clusters)>0 and len(group_db)==0:
newColumnHeader=[]
i=0
for sample_name in column_header:
newColumnHeader.append(str(prior_clusters[i])+':'+sample_name)
i+=1
group_db, column_header = assignGroupColors(newColumnHeader)
except Exception,e:
print traceback.format_exc()
group_db={}
pcA-=1
pcB-=1
label1=''
label2=''
""" Based in part on code from:
http://glowingpython.blogspot.com/2011/07/principal-component-analysis-with-numpy.html
Performs performs principal components analysis
(PCA) on the n-by-p data matrix A
Rows of A correspond to observations, columns to variables.
Returns :
coeff :
is a p-by-p matrix, each column containing coefficients
for one principal component.
score :
the principal component scores; that is, the representation
of A in the principal component space. Rows of SCORE
correspond to observations, columns to components.
latent :
a vector containing the eigenvalues
of the covariance matrix of A.
"""
# computing eigenvalues and eigenvectors of covariance matrix
if algorithm == 'SVD': use_svd = True
else: use_svd = False
if reimportModelScores:
print 'Re-importing PCA model scores rather than calculating from scratch',
print root_dir+dataset_name+'-PCA_scores.txt'
try:
scores = importtSNEScores(root_dir+dataset_name+'-PCA_scores.txt'); print '...import finished'
matrix = zip(*matrix)
except Exception:
reimportModelScores=False; print '...no existing score file found'
if reimportModelScores==False:
#Mdif = matrix-matrix.mean(axis=0)# subtract the mean (along columns)
#M = (matrix-mean(matrix.T,axis=1)).T # subtract the mean (along columns)
Mdif = matrix/matrix.std()
Mdif = Mdif.T
u, s, vt = svd(Mdif, 0)
fracs = s**2/np.sum(s**2)
entropy = -sum(fracs*np.log(fracs))/np.log(np.min(vt.shape))
label1 = 'PC%i (%2.1f%%)' %(pcA+1, fracs[0]*100)
label2 = 'PC%i (%2.1f%%)' %(pcB+1, fracs[1]*100)
#http://docs.scipy.org/doc/scipy/reference/sparse.html
#scipy.sparse.linalg.svds - sparse svd
#idx = numpy.argsort(vt[0,:])
#print idx;sys.exit() # Use this as your cell order or use a density analysis to get groups
#### FROM LARSSON ########
#100 most correlated Genes with PC1
#print vt
PCsToInclude = 4
correlated_db={}
allGenes={}
new_matrix = []
new_headers = []
added_indexes=[]
x = 0
#100 most correlated Genes with PC1
print 'exporting PCA loading genes to:',root_dir+'/PCA/correlated.txt'
exportData = export.ExportFile(root_dir+'/PCA/correlated.txt')
matrix = zip(*matrix) ### transpose this back to normal
try:
while x<PCsToInclude:
idx = numpy.argsort(u[:,x])
correlated = map(lambda i: row_header[i],idx[:300])
anticorrelated = map(lambda i: row_header[i],idx[-300:])
correlated_db[x] = correlated,anticorrelated
### Create a new filtered matrix of loading gene indexes
fidx = list(idx[:300])+list(idx[-300:])
for i in fidx:
if i not in added_indexes:
added_indexes.append(i)
new_headers.append(row_header[i])
new_matrix.append(matrix[i])
x+=1
#redundant_genes = excludeHighlyCorrelatedHits(numpy.array(new_matrix),new_headers)
redundant_genes = []
for x in correlated_db:
correlated,anticorrelated = correlated_db[x]
count=0
for gene in correlated:
if gene not in redundant_genes and count<100:
exportData.write(gene+'\tcorrelated-PC'+str(x+1)+'\n'); allGenes[gene]=[]
count+=1
count=0
for gene in anticorrelated:
if gene not in redundant_genes and count<100:
exportData.write(gene+'\tanticorrelated-PC'+str(x+1)+'\n'); allGenes[gene]=[]
count+=1
exportData.close()
if geneSetName != None:
if len(geneSetName)>0:
exportCustomGeneSet(geneSetName,species,allGenes)
print 'Exported geneset to "StoredGeneSets"'
except Exception:
pass
###########################
#if len(row_header)>20000:
#print '....Using eigenvectors of the real symmetric square matrix for efficiency...'
#[latent,coeff] = scipy.sparse.linalg.eigsh(cov(M))
#scores=mlab.PCA(scores)
if use_svd == False:
[latent,coeff] = linalg.eig(cov(M))
scores = dot(coeff.T,M) # projection of the data in the new space
else:
### transform u into the same structure as the original scores from linalg.eig coeff
scores = vt
writetSNEScores(scores,root_dir+dataset_name+'-PCA_scores.txt')
fig = pylab.figure()
ax = fig.add_subplot(111)
pylab.xlabel(label1)
pylab.ylabel(label2)
axes = getAxes(scores) ### adds buffer space to the end of each axis and creates room for a legend
pylab.axis(axes)
marker_size = 15
if len(column_header)>20:
marker_size = 12
if len(column_header)>40:
marker_size = 10
if len(column_header)>150:
marker_size = 7
if len(column_header)>500:
marker_size = 5
if len(column_header)>1000:
marker_size = 4
if len(column_header)>2000:
marker_size = 3
#marker_size = 9
#samples = list(column_header)
### Color By Gene
if colorByGene != None:
print 'Coloring based on feature expression.'
gene_translation_db={}
matrix = numpy.array(matrix)
min_val = matrix.min() ### min val
if ' ' in colorByGene:
genes = string.split(colorByGene,' ')
else:
genes = [colorByGene]
genePresent=False
numberGenesPresent=[]
for gene in genes:
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
### Translate symbol to Ensembl
if len(numberGenesPresent)==0:
try:
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
for symbol in genes:
if symbol in symbol_to_gene:
gene = symbol_to_gene[symbol][0]
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
gene_translation_db[symbol]=gene
except Exception: pass
numberGenesPresent = len(numberGenesPresent)
if numberGenesPresent==1:
cm = pylab.cm.get_cmap('Reds')
else:
if numberGenesPresent==2:
cm = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
#cm = matplotlib.colors.ListedColormap(['w', 'k']) ### If you want to hide one of the groups
else:
cm = pylab.cm.get_cmap('gist_rainbow')
cm = pylab.cm.get_cmap('Paired')
"""
elif numberGenesPresent==3:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
elif numberGenesPresent==4:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
elif numberGenesPresent==5:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif numberGenesPresent==6:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif numberGenesPresent==7:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
"""
def get_cmap(n, name='hsv'):
'''Returns a function that maps each index in 0, 1, ..., n-1 to a distinct
RGB color; the keyword argument name must be a standard mpl colormap name.'''
return pylab.cm.get_cmap(name, n)
if genePresent:
dataset_name+='-'+colorByGene
group_db={}
bestGeneAssociated={}
k=0
for gene in genes:
try:
try: i = row_header.index(gene)
except Exception: i = row_header.index(gene_translation_db[gene])
values = map(float,matrix[i])
min_val = min(values)
bin_size = (max(values)-min_val)/8
max_val = max(values)
ranges = []
iz=min_val
while iz < (max(values)-bin_size/100):
r = iz,iz+bin_size
if len(ranges)==7:
r = iz,max_val
ranges.append(r)
iz+=bin_size
color_db = {}
colors = get_cmap(len(genes))
for i in range(len(ranges)):
if i==0:
color = '#C0C0C0'
else:
if numberGenesPresent==1:
### use a single color gradient
color = cm(1.*i/len(ranges))
#color = cm(1.*(i+1)/len(ranges))
else:
if i>2:
if len(genes)<8:
color = cm(k)
else:
color = colors(k)
else:
color = '#C0C0C0'
color_db[ranges[i]] = color
i=0
for val in values:
sample = column_header[i]
for (l,u) in color_db:
range_index = ranges.index((l,u)) ### what is the ranking of this range
if val>=l and val<=u:
color = color_db[(l,u)]
color_label = [gene+'-range: '+str(l)[:4]+'-'+str(u)[:4],color,'']
group_db[sample] = color_label
try: bestGeneAssociated[sample].append([range_index,val,color_label])
except Exception: bestGeneAssociated[sample] = [[range_index,val,color_label]]
i+=1
#print min(values),min_val,bin_size,max_val
if len(genes)>1:
### Collapse and rank multiple gene results
for sample in bestGeneAssociated:
bestGeneAssociated[sample].sort()
color_label = bestGeneAssociated[sample][-1][-1]
if numberGenesPresent>1:
index = bestGeneAssociated[sample][-1][0]
if index > 2:
gene = string.split(color_label[0],'-')[0]
else:
gene = 'Null'
color_label[0] = gene
group_db[sample] = color_label
except Exception:
print [gene], 'not found in rows...'
#print traceback.format_exc()
k+=1
else:
print [colorByGene], 'not found in rows...'
pylab.title('Principal Component Analysis - '+dataset_name)
group_names={}
i=0
for sample_name in column_header: #scores[0]
### Add the text labels for each
try:
### Get group name and color information
group_name,color,k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color
except Exception:
color = 'r'; label=None
try: ax.plot(scores[pcA][i],scores[1][i],color=color,marker='o',markersize=marker_size,label=label,markeredgewidth=0,picker=True)
except Exception, e: print e; print i, len(scores[pcB]);kill
if showLabels:
try: sample_name = ' '+string.split(sample_name,':')[1]
except Exception: pass
ax.text(scores[pcA][i],scores[pcB][i],sample_name,fontsize=11)
i+=1
group_count = []
for i in group_db:
if group_db[i][0] not in group_count:
group_count.append(group_db[i][0])
#print len(group_count)
Lfontsize = 8
if len(group_count)>20:
Lfontsize = 10
if len(group_count)>30:
Lfontsize = 8
if len(group_count)>40:
Lfontsize = 6
if len(group_count)>50:
Lfontsize = 5
i=0
#group_count = group_count*10 ### force the legend box out of the PCA core plot
box = ax.get_position()
if len(group_count) > 0:
# Shink current axis by 20%
ax.set_position([box.x0, box.y0, box.width * 0.8, box.height])
try: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5),fontsize = Lfontsize) ### move the legend over to the right of the plot
except Exception: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
else:
ax.set_position([box.x0, box.y0, box.width, box.height])
pylab.legend(loc="upper left", prop={'size': 10})
filename = 'Clustering-%s-PCA.pdf' % dataset_name
try: pylab.savefig(root_dir + filename)
except Exception: None ### Rare error
#print 'Exporting:',filename
filename = filename[:-3]+'png'
try: pylab.savefig(root_dir + filename) #dpi=200
except Exception: None ### Rare error
graphic_link.append(['Principal Component Analysis',root_dir+filename])
if display:
print 'Exporting:',filename
try:
pylab.show()
except Exception:
pass### when run in headless mode
fig.clf()
def ViolinPlot():
def readData(filename):
all_data = {}
headers={}
groups=[]
firstRow=True
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow=False
i=0
for x in t[1:]:
try: g,h = string.split(x,':')
except Exception: g=x; h=x
headers[i] = g
if g not in groups: groups.append(g)
i+=1
else:
#all_data.append(map(lambda x: math.log(math.pow(2,float(x))-1+0.001,2), t[1:]))
t = map(lambda x: float(x), t[1:])
i = 0
for x in t:
try: g = headers[i]
except Exception: print i;sys.exit()
try: all_data[g].append(x)
except Exception: all_data[g] = [x]
i+=1
all_data2=[]
print groups
for group in groups:
all_data2.append(all_data[group])
return all_data2
def violin_plot(ax, data, pos, bp=False):
'''
create violin plots on an axis
'''
from scipy.stats import gaussian_kde
from numpy import arange
dist = max(pos)-min(pos)
w = min(0.15*max(dist,1.0),0.5)
for d,p in zip(data,pos):
k = gaussian_kde(d) #calculates the kernel density
m = k.dataset.min() #lower bound of violin
M = k.dataset.max() #upper bound of violin
x = arange(m,M,(M-m)/100.) # support for violin
v = k.evaluate(x) #violin profile (density curve)
v = v/v.max()*w #scaling the violin to the available space
ax.fill_betweenx(x,p,v+p,facecolor='y',alpha=0.3)
ax.fill_betweenx(x,p,-v+p,facecolor='y',alpha=0.3)
if bp:
ax.boxplot(data,notch=1,positions=pos,vert=1)
def draw_all(data, output):
pos = [1,2,3]
fig = pylab.figure()
ax = fig.add_subplot(111)
violin_plot(ax, data, pos)
pylab.show()
pylab.savefig(output+'.pdf')
all_data = []
all_data = readData('/Users/saljh8/Downloads/TPM_cobound.txt')
import numpy
#all_data = map(numpy.array, zip(*all_data))
#failed_data = map(numpy.array, zip(*failed_data))
draw_all(all_data, 'alldata')
def simpleScatter(fn):
import matplotlib.patches as mpatches
values=[]
legends={}
colors={}
skip=True
scale = 100.0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if skip:
x_header, y_header, color_header,label_header, shape_header = string.split(data,'\t')
skip=False
else:
x, y, color,label,shape = string.split(data,'\t')
if color in colors:
xval,yval,label,shape = colors[color]
xval.append(float(x)); yval.append(float(y))
else:
xval = [float(x)]; yval = [float(y)]
colors[color] = xval,yval,label,shape
for color in colors:
xval,yval,label,shape = colors[color]
pylab.scatter(xval, yval, s=scale, c=color, alpha=0.75, label=label, marker=shape,edgecolor="none")
pylab.legend(loc='upper left')
pylab.title(fn)
pylab.xlabel(x_header, fontsize=15)
pylab.ylabel(y_header, fontsize=15)
marker_size = 7
#pylab.grid(True)
pylab.show()
def ica(filename):
showLabels=True
X, column_header, row_header, dataset_name, group_db = importData(filename)
X = map(numpy.array, zip(*X)) ### coverts these to tuples
column_header, row_header = row_header, column_header
ica = FastICA()
scores = ica.fit(X).transform(X) # Estimate the sources
scores /= scores.std(axis=0)
fig = pylab.figure()
ax = fig.add_subplot(111)
pylab.xlabel('ICA-X')
pylab.ylabel('ICA-Y')
pylab.title('ICA - '+dataset_name)
axes = getAxes(scores) ### adds buffer space to the end of each axis and creates room for a legend
pylab.axis(axes)
marker_size = 15
if len(column_header)>20:
marker_size = 12
if len(column_header)>40:
marker_size = 10
if len(column_header)>150:
marker_size = 7
if len(column_header)>500:
marker_size = 5
if len(column_header)>1000:
marker_size = 4
if len(column_header)>2000:
marker_size = 3
group_names={}
i=0
for sample_name in row_header: #scores[0]
### Add the text labels for each
try:
### Get group name and color information
group_name,color,k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color
except Exception:
color = 'r'; label=None
ax.plot(scores[0][i],scores[1][i],color=color,marker='o',markersize=marker_size,label=label)
if showLabels:
ax.text(scores[0][i],scores[1][i],sample_name,fontsize=8)
i+=1
pylab.title('ICA recovered signals')
pylab.show()
def plot_samples(S, axis_list=None):
pylab.scatter(S[:, 0], S[:, 1], s=20, marker='o', linewidths=0, zorder=10,
color='red', alpha=0.5)
if axis_list is not None:
colors = ['orange', 'red']
for color, axis in zip(colors, axis_list):
axis /= axis.std()
x_axis, y_axis = axis
# Trick to get legend to work
pylab.plot(0.1 * x_axis, 0.1 * y_axis, linewidth=2, color=color)
pylab.quiver(0, 0, x_axis, y_axis, zorder=11, width=2, scale=6,
color=color)
pylab.xlabel('x')
pylab.ylabel('y')
def PCA3D(matrix, column_header, row_header, dataset_name, group_db,
display=False, showLabels=True, algorithm='SVD',geneSetName=None,
species=None,colorByGene=None):
from numpy import mean,cov,double,cumsum,dot,linalg,array,rank
fig = pylab.figure()
ax = fig.add_subplot(111, projection='3d')
start = time.time()
#M = (matrix-mean(matrix.T,axis=1)).T # subtract the mean (along columns)
try: prior_clusters = priorColumnClusters
except Exception: prior_clusters=[]
if prior_clusters == None: prior_clusters=[]
try:
if len(prior_clusters)>0 and len(group_db)==0:
newColumnHeader=[]
i=0
for sample_name in column_header:
newColumnHeader.append(str(prior_clusters[i])+':'+sample_name)
i+=1
group_db, column_header = assignGroupColors(newColumnHeader)
except Exception,e:
#print e
group_db={}
if algorithm == 'SVD': use_svd = True
else: use_svd = False
Mdif = matrix/matrix.std()
Mdif = Mdif.T
u, s, vt = svd(Mdif, 0)
fracs = s**2/np.sum(s**2)
entropy = -sum(fracs*np.log(fracs))/np.log(np.min(vt.shape))
label1 = 'PC%i (%2.1f%%)' %(0+1, fracs[0]*100)
label2 = 'PC%i (%2.1f%%)' %(1+1, fracs[1]*100)
label3 = 'PC%i (%2.1f%%)' %(2+1, fracs[2]*100)
PCsToInclude = 4
correlated_db={}
allGenes={}
new_matrix = []
new_headers = []
added_indexes=[]
x = 0
#100 most correlated Genes with PC1
print 'exporting PCA loading genes to:',root_dir+'/PCA/correlated.txt'
exportData = export.ExportFile(root_dir+'/PCA/correlated.txt')
matrix = zip(*matrix) ### transpose this back to normal
try:
while x<PCsToInclude:
idx = numpy.argsort(u[:,x])
correlated = map(lambda i: row_header[i],idx[:300])
anticorrelated = map(lambda i: row_header[i],idx[-300:])
correlated_db[x] = correlated,anticorrelated
### Create a new filtered matrix of loading gene indexes
fidx = list(idx[:300])+list(idx[-300:])
for i in fidx:
if i not in added_indexes:
added_indexes.append(i)
new_headers.append(row_header[i])
new_matrix.append(matrix[i])
x+=1
#redundant_genes = excludeHighlyCorrelatedHits(numpy.array(new_matrix),new_headers)
redundant_genes = []
for x in correlated_db:
correlated,anticorrelated = correlated_db[x]
count=0
for gene in correlated:
if gene not in redundant_genes and count<100:
exportData.write(gene+'\tcorrelated-PC'+str(x+1)+'\n'); allGenes[gene]=[]
count+=1
count=0
for gene in anticorrelated:
if gene not in redundant_genes and count<100:
exportData.write(gene+'\tanticorrelated-PC'+str(x+1)+'\n'); allGenes[gene]=[]
count+=1
exportData.close()
if geneSetName != None:
if len(geneSetName)>0:
exportCustomGeneSet(geneSetName,species,allGenes)
print 'Exported geneset to "StoredGeneSets"'
except ZeroDivisionError:
pass
#numpy.Mdiff.toFile(root_dir+'/PCA/correlated.txt','\t')
if use_svd == False:
[latent,coeff] = linalg.eig(cov(M))
scores = dot(coeff.T,M) # projection of the data in the new space
else:
### transform u into the same structure as the original scores from linalg.eig coeff
scores = vt
end = time.time()
print 'PCA completed in', end-start, 'seconds.'
### Hide the axis number labels
#ax.w_xaxis.set_ticklabels([])
#ax.w_yaxis.set_ticklabels([])
#ax.w_zaxis.set_ticklabels([])
#"""
#ax.set_xticks([]) ### Hides ticks
#ax.set_yticks([])
#ax.set_zticks([])
ax.set_xlabel(label1)
ax.set_ylabel(label2)
ax.set_zlabel(label3)
#"""
#pylab.title('Principal Component Analysis\n'+dataset_name)
"""
pylab.figure()
pylab.xlabel('Principal Component 1')
pylab.ylabel('Principal Component 2')
"""
axes = getAxes(scores,PlotType='3D') ### adds buffer space to the end of each axis and creates room for a legend
pylab.axis(axes)
Lfontsize = 8
group_count = []
for i in group_db:
if group_db[i][0] not in group_count:
group_count.append(group_db[i][0])
### Color By Gene
if colorByGene != None:
gene_translation_db={}
matrix = numpy.array(matrix)
min_val = matrix.min() ### min val
if ' ' in colorByGene:
genes = string.split(colorByGene,' ')
else:
genes = [colorByGene]
genePresent=False
numberGenesPresent=[]
for gene in genes:
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
### Translate symbol to Ensembl
if len(numberGenesPresent)==0:
try:
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
for symbol in genes:
if symbol in symbol_to_gene:
gene = symbol_to_gene[symbol][0]
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
gene_translation_db[symbol]=gene
except Exception: pass
numberGenesPresent = len(numberGenesPresent)
if numberGenesPresent==1:
cm = pylab.cm.get_cmap('Reds')
else:
if numberGenesPresent==2:
cm = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
cm = matplotlib.colors.ListedColormap(['w', 'k'])
elif numberGenesPresent==3:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
elif numberGenesPresent==4:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
elif numberGenesPresent==5:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif numberGenesPresent==6:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif numberGenesPresent==7:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
else:
cm = pylab.cm.get_cmap('gist_rainbow')
if genePresent:
dataset_name+='-'+colorByGene
group_db={}
bestGeneAssociated={}
k=0
for gene in genes:
try:
try: i = row_header.index(gene)
except Exception: i = row_header.index(gene_translation_db[gene])
values = map(float,matrix[i])
min_val = min(values)
bin_size = (max(values)-min_val)/8
max_val = max(values)
ranges = []
iz=min_val
while iz < (max(values)-bin_size/100):
r = iz,iz+bin_size
if len(ranges)==7:
r = iz,max_val
ranges.append(r)
iz+=bin_size
color_db = {}
for i in range(len(ranges)):
if i==0:
color = '#C0C0C0'
else:
if numberGenesPresent==1:
### use a single color gradient
color = cm(1.*i/len(ranges))
#color = cm(1.*(i+1)/len(ranges))
else:
if i>2:
color = cm(k)
else:
color = '#C0C0C0'
color_db[ranges[i]] = color
i=0
for val in values:
sample = column_header[i]
for (l,u) in color_db:
range_index = ranges.index((l,u)) ### what is the ranking of this range
if val>=l and val<=u:
color = color_db[(l,u)]
color_label = [gene+'-range: '+str(l)[:4]+'-'+str(u)[:4],color,'']
group_db[sample] = color_label
try: bestGeneAssociated[sample].append([range_index,val,color_label])
except Exception: bestGeneAssociated[sample] = [[range_index,val,color_label]]
i+=1
#print min(values),min_val,bin_size,max_val
if len(genes)>1:
### Collapse and rank multiple gene results
for sample in bestGeneAssociated:
bestGeneAssociated[sample].sort()
color_label = bestGeneAssociated[sample][-1][-1]
if numberGenesPresent>1:
index = bestGeneAssociated[sample][-1][0]
if index > 2:
gene = string.split(color_label[0],'-')[0]
else:
gene = 'Null'
color_label[0] = gene
group_db[sample] = color_label
except Exception:
print [gene], 'not found in rows...'
#print traceback.format_exc()
k+=1
else:
print [colorByGene], 'not found in rows...'
#print len(group_count)
if len(group_count)>20:
Lfontsize = 10
if len(group_count)>30:
Lfontsize = 8
if len(group_count)>40:
Lfontsize = 6
if len(group_count)>50:
Lfontsize = 5
if len(scores[0])>150:
markersize = 7
else:
markersize = 10
i=0
group_names={}
for x in scores[0]:
### Add the text labels for each
sample_name = column_header[i]
try:
### Get group name and color information
group_name,color, k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color, k
except Exception:
color = 'r'; label=None
ax.plot([scores[0][i]],[scores[1][i]],[scores[2][i]],color=color,marker='o',markersize=markersize,label=label,markeredgewidth=0,picker=True) #markeredgecolor=color
if showLabels:
#try: sample_name = ' '+string.split(sample_name,':')[1]
#except Exception: pass
ax.text(scores[0][i],scores[1][i],scores[2][i], ' '+sample_name,fontsize=9)
i+=1
# Shink current axis by 20%
box = ax.get_position()
ax.set_position([box.x0, box.y0, box.width * 0.8, box.height])
#pylab.legend(loc="upper left", prop={'size': 10})
try: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5),fontsize = Lfontsize) ### move the legend over to the right of the plot
except Exception: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
filename = 'Clustering-%s-3D-PCA.pdf' % dataset_name
pylab.savefig(root_dir + filename)
#print 'Exporting:',filename
filename = filename[:-3]+'png'
pylab.savefig(root_dir + filename) #dpi=200
graphic_link.append(['Principal Component Analysis',root_dir+filename])
if display:
print 'Exporting:',filename
try: pylab.show()
except Exception: None ### when run in headless mode
fig.clf()
def getAxes1(scores,PlotType=None):
""" Adjust these axes to account for (A) legend size (left hand upper corner)
and (B) long sample name extending to the right
"""
try:
x_range = max(scores[0])-min(scores[0])
y_range = max(scores[1])-min(scores[1])
if PlotType == '3D':
x_axis_min = min(scores[0])-(x_range/10)
x_axis_max = max(scores[0])+(x_range/10)
y_axis_min = min(scores[1])-(y_range/10)
y_axis_max = max(scores[1])+(y_range/10)
else:
x_axis_min = min(scores[0])-(x_range/10)
x_axis_max = max(scores[0])+(x_range/10)
y_axis_min = min(scores[1])-(y_range/10)
y_axis_max = max(scores[1])+(y_range/10)
except KeyError:
None
return [x_axis_min, x_axis_max, y_axis_min, y_axis_max]
def getAxes(scores,PlotType=None):
""" Adjust these axes to account for (A) legend size (left hand upper corner)
and (B) long sample name extending to the right
"""
try:
x_range = max(scores[0])-min(scores[0])
y_range = max(scores[1])-min(scores[1])
if PlotType == '3D':
x_axis_min = min(scores[0])-(x_range/1.5)
x_axis_max = max(scores[0])+(x_range/1.5)
y_axis_min = min(scores[1])-(y_range/5)
y_axis_max = max(scores[1])+(y_range/5)
else:
x_axis_min = min(scores[0])-(x_range/10)
x_axis_max = max(scores[0])+(x_range/10)
y_axis_min = min(scores[1])-(y_range/10)
y_axis_max = max(scores[1])+(y_range/10)
except KeyError:
None
return [x_axis_min, x_axis_max, y_axis_min, y_axis_max]
def getAxesTransposed(scores,exclude={}):
""" Adjust these axes to account for (A) legend size (left hand upper corner)
and (B) long sample name extending to the right
"""
scores_filtered=[]
for i in range(len(scores)):
if i not in exclude:
scores_filtered.append(scores[i])
scores = scores_filtered
scores = map(numpy.array, zip(*scores))
try:
x_range = max(scores[0])-min(scores[0])
y_range = max(scores[1])-min(scores[1])
x_axis_min = min(scores[0])-int((float(x_range)/7))
x_axis_max = max(scores[0])+int((float(x_range)/7))
y_axis_min = min(scores[1])-int(float(y_range/7))
y_axis_max = max(scores[1])+int(float(y_range/7))
except KeyError:
None
return [x_axis_min, x_axis_max, y_axis_min, y_axis_max]
def Kmeans(features, column_header, row_header):
#http://www.janeriksolem.net/2009/04/clustering-using-scipys-k-means.html
#class1 = numpy.array(numpy.random.standard_normal((100,2))) + numpy.array([5,5])
#class2 = 1.5 * numpy.array(numpy.random.standard_normal((100,2)))
features = numpy.vstack((class1,class2))
centroids,variance = scipy.cluster.vq.kmeans(features,2)
code,distance = scipy.cluster.vq.vq(features,centroids)
"""
This generates two normally distributed classes in two dimensions. To try and cluster the points, run k-means with k=2 like this.
The variance is returned but we don't really need it since the SciPy implementation computes several runs (default is 20) and selects the one with smallest variance for us. Now you can check where each data point is assigned using the vector quantization function in the SciPy package.
By checking the value of code we can see if there are any incorrect assignments. To visualize, we can plot the points and the final centroids.
"""
pylab.plot([p[0] for p in class1],[p[1] for p in class1],'*')
pylab.plot([p[0] for p in class2],[p[1] for p in class2],'r*')
pylab.plot([p[0] for p in centroids],[p[1] for p in centroids],'go')
pylab.show()
"""
def displaySimpleNetworkX():
import networkx as nx
print 'Graphing output with NetworkX'
gr = nx.Graph(rotate=90,bgcolor='white') ### commands for neworkx and pygraphviz are the same or similiar
edges = importSIF('Config/TissueFateMap.sif')
### Add nodes and edges
for (node1,node2,type) in edges:
gr.add_edge(node1,node2)
draw_networkx_edges
#gr['Myometrium']['color']='red'
# Draw as PNG
nx.draw_shell(gr) #wopi, gvcolor, wc, ccomps, tred, sccmap, fdp, circo, neato, acyclic, nop, gvpr, dot, sfdp. - fdp
pylab.savefig('LineageNetwork.png')
def displaySimpleNetwork(sif_filename,fold_db,pathway_name):
import pygraphviz as pgv
#print 'Graphing output with PygraphViz'
gr = pgv.AGraph(bgcolor='white',directed=True) ### Graph creation and setting of attributes - directed indicates arrows should be added
#gr = pgv.AGraph(rotate='90',bgcolor='lightgray')
### Set graph attributes
gr.node_attr['style']='filled'
gr.graph_attr['label']='%s Network' % pathway_name
edges = importSIF(sif_filename)
if len(edges) > 700:
print sif_filename, 'too large to visualize...'
else:
### Add nodes and edges
for (node1,node2,type) in edges:
nodes = (node1,node2)
gr.add_edge(nodes)
child, parent = nodes
edge = gr.get_edge(nodes[0],nodes[1])
if 'TF' in pathway_name or 'WGRV' in pathway_name:
node = child ### This is the regulating TF
else:
node = parent ### This is the pathway
n=gr.get_node(node)
### http://www.graphviz.org/doc/info/attrs.html
n.attr['penwidth'] = 4
n.attr['fillcolor']= '#FFFF00' ### yellow
n.attr['shape']='rectangle'
#n.attr['weight']='yellow'
#edge.attr['arrowhead'] = 'diamond' ### set the arrow type
id_color_db = WikiPathways_webservice.getHexadecimalColorRanges(fold_db,'Genes')
for gene_symbol in id_color_db:
color_code = id_color_db[gene_symbol]
try:
n=gr.get_node(gene_symbol)
n.attr['fillcolor']= '#'+string.upper(color_code) #'#FF0000'
#n.attr['rotate']=90
except Exception: None
# Draw as PNG
#gr.layout(prog='dot') #fdp (spring embedded), sfdp (OK layout), neato (compressed), circo (lots of empty space), dot (hierarchical - linear)
gr.layout(prog='neato')
output_filename = '%s.png' % sif_filename[:-4]
#print output_filename
gr.draw(output_filename)
"""
def findParentDir(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1 ### get just the parent directory
return filename[:x]
def findFilename(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1 ### get just the parent directory
return filename[x:]
def runHierarchicalClustering(matrix, row_header, column_header, dataset_name,
row_method, row_metric, column_method, column_metric,
color_gradient, display=False, contrast=None,
allowAxisCompression=True,Normalize=True):
""" Running with cosine or other distance metrics can often produce negative Z scores
during clustering, so adjustments to the clustering may be required.
=== Options Include ===
row_method = 'average'
column_method = 'single'
row_metric = 'cosine'
column_metric = 'euclidean'
color_gradient = 'red_white_blue'
color_gradient = 'red_black_sky'
color_gradient = 'red_black_blue'
color_gradient = 'red_black_green'
color_gradient = 'yellow_black_blue'
color_gradient == 'coolwarm'
color_gradient = 'seismic'
color_gradient = 'green_white_purple'
"""
try:
if allowLargeClusters: maxSize = 50000
else: maxSize = 7000
except Exception: maxSize = 7000
try:
PriorColumnClusters=priorColumnClusters
PriorRowClusters=priorRowClusters
except Exception:
PriorColumnClusters=None
PriorRowClusters=None
run = False
print 'max allowed cluster size:',maxSize
if len(matrix)>0 and (len(matrix)<maxSize or row_method == None):
#if len(matrix)>5000: row_metric = 'euclidean'
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
try:
### Default for display is False, when set to True, Pylab will render the image
heatmap(numpy.array(matrix), row_header, column_header, row_method, column_method,
row_metric, column_metric, color_gradient, dataset_name, display=display,
contrast=contrast,allowAxisCompression=allowAxisCompression,Normalize=Normalize,
PriorColumnClusters=PriorColumnClusters,PriorRowClusters=PriorRowClusters)
run = True
except Exception:
print traceback.format_exc()
try:
pylab.clf()
pylab.close() ### May result in TK associated errors later on
import gc
gc.collect()
except Exception: None
if len(matrix)<10000:
print 'Error using %s ... trying euclidean instead' % row_metric
row_metric = 'cosine'; row_method = 'average' ### cityblock
else:
print 'Error with hierarchical clustering... only clustering arrays'
row_method = None ### Skip gene clustering
try:
heatmap(numpy.array(matrix), row_header, column_header, row_method, column_method,
row_metric, column_metric, color_gradient, dataset_name, display=display,
contrast=contrast,allowAxisCompression=allowAxisCompression,Normalize=Normalize,
PriorColumnClusters=PriorColumnClusters,PriorRowClusters=PriorRowClusters)
run = True
except Exception:
print traceback.format_exc()
print 'Unable to generate cluster due to dataset incompatibilty.'
elif len(matrix)==0:
print_out = 'SKIPPING HIERARCHICAL CLUSTERING!!! - Your dataset file has no associated rows.'
print print_out
else:
print_out = 'SKIPPING HIERARCHICAL CLUSTERING!!! - Your dataset file is over the recommended size limit for clustering ('+str(maxSize)+' rows). Please cluster later using "Additional Analyses"'
print print_out
try:
pylab.clf()
pylab.close() ### May result in TK associated errors later on
import gc
gc.collect()
except Exception: None
return run
def debugTKBug():
return None
def runHCexplicit(filename, graphics, row_method, row_metric, column_method, column_metric, color_gradient,
extra_params, display=True, contrast=None, Normalize=False, JustShowTheseIDs=[],compressAxis=True):
""" Explicit method for hieararchical clustering with defaults defined by the user (see below function) """
#print [filename, graphics, row_method, row_metric, column_method, column_metric, color_gradient, contrast, Normalize]
global root_dir
global inputFilename
global originalFilename
global graphic_link
global allowLargeClusters
global GroupDB
global justShowTheseIDs
global targetGeneIDs
global normalize
global rho_cutoff
global species
global runGOElite
global EliteGeneSets
global storeGeneSetName
EliteGeneSets=[]
targetGene=[]
filterByPathways=False
runGOElite = False
justShowTheseIDs = JustShowTheseIDs
allowLargeClusters = True
if compressAxis:
allowAxisCompression = True
else:
allowAxisCompression = False
graphic_link=graphics ### Store all locations of pngs
inputFilename = filename ### Used when calling R
filterIDs = False
normalize = Normalize
try:
### Specific additional optional parameters for filtering
transpose = extra_params.Transpose()
try:
rho_cutoff = extra_params.RhoCutoff()
print 'Setting correlation cutoff to a rho of',rho_cutoff
except Exception:
rho_cutoff = 0.5 ### Always done if no rho, but only used if getGeneCorrelations == True
#print 'Setting correlation cutoff to a rho of',rho_cutoff
PathwayFilter = extra_params.PathwaySelect()
GeneSet = extra_params.GeneSet()
OntologyID = extra_params.OntologyID()
Normalize = extra_params.Normalize()
normalize = Normalize
filterIDs = True
species = extra_params.Species()
platform = extra_params.Platform()
vendor = extra_params.Vendor()
newInput = findParentDir(inputFilename)+'/GeneSetClustering/'+findFilename(inputFilename)
targetGene = extra_params.GeneSelection() ### Select a gene or ID to get the top correlating genes
getGeneCorrelations = extra_params.GetGeneCorrelations() ### Select a gene or ID to get the top correlating genes
filterByPathways = extra_params.FilterByPathways()
PathwayFilter, filterByPathways = verifyPathwayName(PathwayFilter,GeneSet,OntologyID,filterByPathways)
justShowTheseIDs_var = extra_params.JustShowTheseIDs()
if len(justShowTheseIDs_var)>0:
justShowTheseIDs = justShowTheseIDs_var
elif len(targetGene)>0:
targetGene = string.replace(targetGene,'\n',' ')
targetGene = string.replace(targetGene,'\r',' ')
justShowTheseIDs = string.split(targetGene,' ')
try:
EliteGeneSets = extra_params.ClusterGOElite()
if EliteGeneSets != ['']: runGOElite = True
except Exception:
#print traceback.format_exc()
pass
try:
storeGeneSetName = extra_params.StoreGeneSetName()
except Exception:
storeGeneSetName = ''
except Exception,e:
#print traceback.format_exc();sys.exit()
transpose = extra_params
root_dir = findParentDir(filename)
if 'ExpressionOutput/Clustering' in root_dir:
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
elif 'ExpressionOutput' in root_dir:
root_dir = string.replace(root_dir,'ExpressionOutput','DataPlots') ### Applies to clustering of LineageProfiler results
root_dir = string.replace(root_dir,'/Clustering','') ### Applies to clustering of MarkerFinder results
else:
root_dir += '/DataPlots/'
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
if row_method == 'hopach': reverseOrder = False
else: reverseOrder = True
#"""
matrix, column_header, row_header, dataset_name, group_db = importData(filename,Normalize=Normalize,reverseOrder=reverseOrder)
GroupDB = group_db
inputFilename = string.replace(inputFilename,'.cdt','.txt')
originalFilename = inputFilename
if len(justShowTheseIDs)==0:
try:
if len(priorColumnClusters)>0 and priorRowClusters>0 and row_method==None and column_method == None:
try: justShowTheseIDs = importPriorDrivers(inputFilename)
except Exception: pass #justShowTheseIDs=[]
except Exception:
#print traceback.format_exc()
pass
#print len(matrix),;print len(column_header),;print len(row_header)
if filterIDs:
transpose_update = True ### Since you can filterByPathways and getGeneCorrelations, only transpose once
if filterByPathways: ### Restrict analyses to only a single pathway/gene-set/ontology term
if isinstance(PathwayFilter, tuple) or isinstance(PathwayFilter, list):
FileName = string.join(list(PathwayFilter),' ')
FileName = string.replace(FileName,':','-')
else: FileName = PathwayFilter
if len(FileName)>40:
FileName = FileName[:40]
try: inputFilename = string.replace(newInput,'.txt','_'+FileName+'.txt') ### update the pathway reference for HOPACH
except Exception: inputFilename = string.replace(newInput,'.txt','_GeneSets.txt')
vars = filterByPathway(matrix,row_header,column_header,species,platform,vendor,GeneSet,PathwayFilter,OntologyID,transpose)
try: dataset_name += '-'+FileName
except Exception: dataset_name += '-GeneSets'
transpose_update = False
if 'amplify' in targetGene:
targetGene = string.join(vars[1],' ')+' amplify '+targetGene ### amplify the gene sets, but need the original matrix and headers (not the filtered)
else: matrix,row_header,column_header = vars
try:
alt_targetGene = string.replace(targetGene,'amplify','')
alt_targetGene = string.replace(alt_targetGene,'amplify','')
alt_targetGene = string.replace(alt_targetGene,'driver','')
alt_targetGene = string.replace(alt_targetGene,'guide','')
alt_targetGene = string.replace(alt_targetGene,'top','')
alt_targetGene = string.replace(alt_targetGene,'positive','')
alt_targetGene = string.replace(alt_targetGene,'excludeCellCycle','')
alt_targetGene = string.replace(alt_targetGene,'monocle','')
alt_targetGene = string.replace(alt_targetGene,'GuideOnlyCorrelation','')
alt_targetGene = string.replace(alt_targetGene,' ','')
except Exception:
alt_targetGene = ''
if getGeneCorrelations and targetGene != 'driver' and targetGene != 'GuideOnlyCorrelation' and \
targetGene != 'guide' and targetGene !='excludeCellCycle' and \
targetGene !='top' and targetGene != ' monocle' and \
targetGene !='positive' and len(alt_targetGene)>0: ###Restrict analyses to only genes that correlate with the target gene of interest
allowAxisCompression = False
if transpose and transpose_update == False: transpose_update = False ### If filterByPathways selected
elif transpose and transpose_update: transpose_update = True ### If filterByPathways not selected
else: transpose_update = False ### If transpose == False
if '\r' in targetGene or '\n' in targetGene:
targetGene = string.replace(targetGene, '\r',' ')
targetGene = string.replace(targetGene, '\n',' ')
if len(targetGene)>15:
inputFilename = string.replace(newInput,'.txt','-'+targetGene[:50]+'.txt') ### update the pathway reference for HOPACH
dataset_name += '-'+targetGene[:50]
else:
inputFilename = string.replace(newInput,'.txt','-'+targetGene+'.txt') ### update the pathway reference for HOPACH
dataset_name += '-'+targetGene
inputFilename = root_dir+'/'+string.replace(findFilename(inputFilename),'|',' ')
inputFilename = root_dir+'/'+string.replace(findFilename(inputFilename),':',' ') ### need to be careful of C://
dataset_name = string.replace(dataset_name,'|',' ')
dataset_name = string.replace(dataset_name,':',' ')
try:
matrix,row_header,column_header,row_method = getAllCorrelatedGenes(matrix,row_header,column_header,species,platform,vendor,targetGene,row_method,transpose_update)
except Exception:
print traceback.format_exc()
print targetGene, 'not found in input expression file. Exiting. \n\n'
badExit
targetGeneIDs = targetGene
exportTargetGeneList(targetGene,inputFilename)
else:
if transpose: ### Transpose the data matrix
print 'Transposing the data matrix'
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
#print len(matrix),;print len(column_header),;print len(row_header)
if len(column_header)>1000 or len(row_header)>1000:
print 'Performing hierarchical clustering (please be patient)...'
runHierarchicalClustering(matrix, row_header, column_header, dataset_name, row_method, row_metric,
column_method, column_metric, color_gradient, display=display,contrast=contrast,
allowAxisCompression=allowAxisCompression, Normalize=Normalize)
#"""
#graphic_link = [root_dir+'Clustering-exp.myeloid-steady-state-amplify positive Mki67 Clec4a2 Gria3 Ifitm6 Gfi1b -hierarchical_cosine_cosine.txt']
if 'guide' in targetGene:
import RNASeq
input_file = graphic_link[-1][-1][:-4]+'.txt'
if 'excludeCellCycle' in targetGene: excludeCellCycle = True
else: excludeCellCycle = False
print 'excludeCellCycle',excludeCellCycle
targetGene = RNASeq.remoteGetDriverGenes(species,platform,input_file,excludeCellCycle=excludeCellCycle,ColumnMethod=column_method)
extra_params.setGeneSelection(targetGene) ### force correlation to these
extra_params.setGeneSet('None Selected') ### silence this
graphic_link= runHCexplicit(filename, graphic_link, row_method, row_metric, column_method, column_metric, color_gradient,
extra_params, display=display, contrast=contrast, Normalize=Normalize, JustShowTheseIDs=JustShowTheseIDs,compressAxis=compressAxis)
return graphic_link
def importPriorDrivers(inputFilename):
filename = string.replace(inputFilename,'Clustering-','')
filename = string.split(filename,'-hierarchical')[0]+'-targetGenes.txt'
genes = open(filename, "rU")
genes = map(lambda x: cleanUpLine(x),genes)
return genes
def exportTargetGeneList(targetGene,inputFilename):
exclude=['positive','top','driver', 'guide', 'amplify','GuideOnlyCorrelation']
exportFile = inputFilename[:-4]+'-targetGenes.txt'
eo = export.ExportFile(root_dir+findFilename(exportFile))
targetGenes = string.split(targetGene,' ')
for gene in targetGenes:
if gene not in exclude:
try: eo.write(gene+'\n')
except Exception: print 'Error export out gene (bad ascii):', [gene]
eo.close()
def debugPylab():
pylab.figure()
pylab.close()
pylab.figure()
def verifyPathwayName(PathwayFilter,GeneSet,OntologyID,filterByPathways):
import gene_associations
### If the user supplied an Ontology ID rather than a Ontology term name, lookup the term name and return this as the PathwayFilter
if len(OntologyID)>0:
PathwayFilter = gene_associations.lookupOntologyID(GeneSet,OntologyID,type='ID')
filterByPathways = True
return PathwayFilter, filterByPathways
def filterByPathway(matrix,row_header,column_header,species,platform,vendor,GeneSet,PathwayFilter,OntologyID,transpose):
### Filter all the matrix and header entries for IDs in the selected pathway
import gene_associations
from import_scripts import OBO_import
exportData = export.ExportFile(inputFilename)
matrix2=[]; row_header2=[]
if 'Ontology' in GeneSet: directory = 'nested'
else: directory = 'gene-mapp'
print "GeneSet(s) to analyze:",PathwayFilter
if isinstance(PathwayFilter, tuple) or isinstance(PathwayFilter, list): ### see if it is one or more pathways
associated_IDs={}
for p in PathwayFilter:
associated = gene_associations.simpleGenePathwayImport(species,GeneSet,p,OntologyID,directory)
for i in associated:associated_IDs[i]=[]
else:
associated_IDs = gene_associations.simpleGenePathwayImport(species,GeneSet,PathwayFilter,OntologyID,directory)
gene_annotations = gene_associations.importGeneData(species,'Ensembl')
vendor = string.replace(vendor,'other:','') ### For other IDs
try: array_to_ens = gene_associations.filterGeneToUID(species,'Ensembl',vendor,associated_IDs)
except Exception: array_to_ens={}
if platform == "3'array":
### IDs thus won't be Ensembl - need to translate
try:
#ens_to_array = gene_associations.getGeneToUidNoExon(species,'Ensembl-'+vendor); print vendor, 'IDs imported...'
array_to_ens = gene_associations.filterGeneToUID(species,'Ensembl',vendor,associated_IDs)
except Exception:
pass
#print platform, vendor, 'not found!!! Exiting method'; badExit
#array_to_ens = gene_associations.swapKeyValues(ens_to_array)
try:
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception:
pass
i=0
original_rows={} ### Don't add the same original ID twice if it associates with different Ensembl IDs
for row_id in row_header:
original_id = row_id; symbol = row_id
if 'SampleLogFolds' in inputFilename or 'RelativeLogFolds' in inputFilename or 'AltConfirmed' in inputFilename or 'MarkerGenes' in inputFilename or 'blah' not in inputFilename:
try: row_id,symbol = string.split(row_id,' ')[:2] ### standard ID convention is ID space symbol
except Exception:
try: symbol = gene_to_symbol[row_id][0]
except Exception: None
if len(symbol)==0: symbol = row_id
if ':' in row_id:
try:
cluster,row_id = string.split(row_id,':')
updated_row_id = cluster+':'+symbol
except Exception:
pass
else:
updated_row_id = symbol
try: original_id = updated_row_id
except Exception: pass
if platform == "3'array":
try:
try: row_ids = array_to_ens[row_id]
except Exception: row_ids = symbol_to_gene[symbol]
except Exception:
row_ids = [row_id]
else:
try:
try: row_ids = array_to_ens[row_id]
except Exception: row_ids = symbol_to_gene[symbol]
except Exception:
row_ids = [row_id]
for row_id in row_ids:
if row_id in associated_IDs:
if 'SampleLogFolds' in inputFilename or 'RelativeLogFolds' in inputFilename:
if original_id != symbol:
row_id = original_id+' '+symbol
else: row_id = symbol
else:
try: row_id = gene_annotations[row_id].Symbol()
except Exception: None ### If non-Ensembl data
if original_id not in original_rows: ### Don't add the same ID twice if associated with mult. Ensembls
matrix2.append(matrix[i])
#row_header2.append(row_id)
row_header2.append(original_id)
original_rows[original_id]=None
i+=1
if transpose:
matrix2 = map(numpy.array, zip(*matrix2)) ### coverts these to tuples
column_header, row_header2 = row_header2, column_header
exportData.write(string.join(['UID']+column_header,'\t')+'\n') ### title row export
i=0
for row_id in row_header2:
exportData.write(string.join([row_id]+map(str,matrix2[i]),'\t')+'\n') ### export values
i+=1
print len(row_header2), 'filtered IDs'
exportData.close()
return matrix2,row_header2,column_header
def getAllCorrelatedGenes(matrix,row_header,column_header,species,platform,vendor,targetGene,row_method,transpose):
### Filter all the matrix and header entries for IDs in the selected targetGene
resort_by_ID_name=False
if resort_by_ID_name:
index=0; new_row_header=[]; new_matrix=[]; temp_row_header = []
for name in row_header: temp_row_header.append((name,index)); index+=1
temp_row_header.sort()
for (name,index) in temp_row_header:
new_row_header.append(name)
new_matrix.append(matrix[index])
matrix = new_matrix
row_header = new_row_header
exportData = export.ExportFile(inputFilename)
try:
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
#from import_scripts import OBO_import; symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception:
print 'No Ensembl-Symbol database available for',species
if platform == "3'array":
### IDs thus won't be Ensembl - need to translate
try:
if ':' in vendor:
vendor = string.split(vendor,':')[1]
#ens_to_array = gene_associations.getGeneToUidNoExon(species,'Ensembl-'+vendor); print vendor, 'IDs imported...'
array_to_ens = gene_associations.filterGeneToUID(species,'Ensembl',vendor,{})
except Exception,e:
array_to_ens={}
for uid in array_to_ens:
for gid in array_to_ens[uid]:
if gid in gene_to_symbol:
symbol = gene_to_symbol[gid][0]
try: gene_to_symbol[uid].append(symbol)
except Exception: gene_to_symbol[uid] = [symbol]
matrix2=[]
row_header2=[]
matrix_db={} ### Used to optionally sort according to the original order
multipleGenes = False
intersecting_ids=[]
i=0
### If multiple genes entered, just display these
targetGenes=[targetGene]
if ' ' in targetGene or ',' in targetGene or '|' in targetGene or '\n' in targetGene or '\r' in targetGene:
multipleGenes = True
if ' ' in targetGene: targetGene = string.replace(targetGene,' ', ' ')
if ',' in targetGene: targetGene = string.replace(targetGene,',', ' ')
#if '|' in targetGene and 'alt_junction' not in originalFilename: targetGene = string.replace(targetGene,'|', ' ')
if '\n' in targetGene: targetGene = string.replace(targetGene,'\n', ' ')
if '\r' in targetGene: targetGene = string.replace(targetGene,'\r', ' ')
targetGenes = string.split(targetGene,' ')
if row_method != None: targetGenes.sort()
intersecting_ids = [val for val in targetGenes if val in row_header]
for row_id in row_header:
original_rowid = row_id
symbol=row_id
new_symbol = symbol
rigorous_search = True
if ':' in row_id and '|' in row_id:
rigorous_search = False
elif ':' in row_id and '|' not in row_id:
a,b = string.split(row_id,':')[:2]
if 'ENS' in a or len(a)==17:
try:
row_id = a
symbol = gene_to_symbol[row_id][0]
except Exception:
symbol =''
elif 'ENS' not in b and len(a)!=17:
row_id = b
elif 'ENS' in b:
symbol = original_rowid
row_id = a
if rigorous_search:
try: row_id,symbol = string.split(row_id,' ')[:2] ### standard ID convention is ID space symbol
except Exception:
try: symbol = gene_to_symbol[row_id][0]
except Exception:
if 'ENS' not in original_rowid:
row_id, symbol = row_id, row_id
new_symbol = symbol
if 'ENS' not in original_rowid and len(original_rowid)!=17:
if original_rowid != symbol:
symbol = original_rowid+' '+symbol
for gene in targetGenes:
if string.lower(gene) == string.lower(row_id) or string.lower(gene) == string.lower(symbol) or string.lower(original_rowid)==string.lower(gene) or string.lower(gene) == string.lower(new_symbol):
matrix2.append(matrix[i]) ### Values for the row
row_header2.append(symbol)
matrix_db[symbol]=matrix[i]
else:
if row_id in targetGenes:
matrix2.append(matrix[i])
row_header2.append(row_id)
matrix_db[row_id]=matrix[i]
i+=1
i=0
#for gene in targetGenes:
# if gene not in matrix_db: print gene
else:
i=0
original_rows={} ### Don't add the same original ID twice if it associates with different Ensembl IDs
for row_id in row_header:
original_id = row_id
symbol = 'NA'
if 'SampleLogFolds' in inputFilename or 'RelativeLogFolds' in inputFilename or 'blah' not in inputFilename:
try: row_id,symbol = string.split(row_id,' ')[:2] ### standard ID convention is ID space symbol
except Exception:
try: symbol = gene_to_symbol[row_id][0]
except Exception:
row_id, symbol = row_id, row_id
original_id = row_id
if row_id == targetGene or symbol == targetGene:
targetGeneValues = matrix[i] ### Values for the row
break
i+=1
i=0
if multipleGenes==False: limit = 50
else: limit = 140 # lower limit is 132
print 'limit:',limit
#print len(intersecting_ids),len(targetGenes), multipleGenes
if multipleGenes==False or 'amplify' in targetGene or 'correlated' in targetGene:
row_header3=[] ### Convert to symbol if possible
if multipleGenes==False:
targetGeneValue_array = [targetGeneValues]
else:
targetGeneValue_array = matrix2
if (len(row_header2)>4) or len(row_header)>15000: # and len(row_header)<20000
print 'Performing all iterative pairwise corelations...',
corr_matrix = numpyCorrelationMatrixGene(matrix,row_header,row_header2,gene_to_symbol)
print 'complete'
matrix2=[]; original_headers=row_header2; row_header2 = []
matrix2_alt=[]; row_header2_alt=[]
### If one gene entered, display the most positive and negative correlated
import markerFinder; k=0
for targetGeneValues in targetGeneValue_array:
correlated=[]
anticorrelated=[]
try: targetGeneID = original_headers[k]
except Exception: targetGeneID=''
try:
rho_results = list(corr_matrix[targetGeneID])
except Exception:
#print traceback.format_exc()
rho_results = markerFinder.simpleScipyPearson(matrix,targetGeneValues)
correlated_symbols={}
#print targetGeneID, rho_results[:100]
#print targetGeneID, rho_results[-100:];sys.exit()
for (rho,ind) in rho_results[:limit]: ### Get the top-50 correlated plus the gene of interest
proceed = True
try:
if len(rho)==2: rho = rho[0]
except: pass
if 'top' in targetGene:
if rho_results[4][0]<rho_cutoff: proceed = False
if rho>rho_cutoff and proceed: #and rho_results[3][0]>rho_cutoff:# ensures only clustered genes considered
rh = row_header[ind]
#if gene_to_symbol[rh][0] in targetGenes:correlated.append(gene_to_symbol[rh][0])
#correlated.append(gene_to_symbol[rh][0])
if len(row_header2)<100 or multipleGenes:
rh = row_header[ind]
#print rh, rho # Ly6c1, S100a8
if matrix[ind] not in matrix2:
if 'correlated' in targetGene:
if rho!=1:
matrix2.append(matrix[ind])
row_header2.append(rh)
if targetGeneValues not in matrix2: ### gene ID systems can be different between source and query
matrix2.append(targetGeneValues)
row_header2.append(targetGeneID)
try:correlated_symbols[gene_to_symbol[rh][0]]=ind
except Exception: correlated_symbols[rh]=ind
#print targetGeneValues, targetGene;sys.exit()
else:
matrix2.append(matrix[ind])
row_header2.append(rh)
try: correlated_symbols[gene_to_symbol[rh][0]]=ind
except Exception: correlated_symbols[rh]=ind
#if rho!=1: print gene_to_symbol[rh][0],'pos',targetGeneID
#sys.exit()
rho_results.reverse()
for (rho,ind) in rho_results[:limit]: ### Get the top-50 anti-correlated plus the gene of interest
try:
if len(rho)==2: rho = rho[0]
except: pass
if rho<-1*rho_cutoff and 'positive' not in targetGene:
rh = row_header[ind]
#if gene_to_symbol[rh][0] in targetGenes:anticorrelated.append(gene_to_symbol[rh][0])
#anticorrelated.append(gene_to_symbol[rh][0])
if len(row_header2)<100 or multipleGenes:
rh = row_header[ind]
if matrix[ind] not in matrix2:
if 'correlated' in targetGene:
if rho!=1:
matrix2.append(matrix[ind])
row_header2.append(rh)
if targetGeneValues not in matrix2:
matrix2.append(targetGeneValues)
row_header2.append(targetGeneID)
try: correlated_symbols[gene_to_symbol[rh][0]]=ind
except Exception: correlated_symbols[rh]=ind
#print targetGeneValues, targetGene;sys.exit()
else:
matrix2.append(matrix[ind])
row_header2.append(rh)
try: correlated_symbols[gene_to_symbol[rh][0]]=ind
except Exception: correlated_symbols[rh]=ind
#if rho!=1: print gene_to_symbol[rh][0],'neg',targetGeneID
try:
### print overlapping input genes that are correlated
if len(correlated_symbols)>0:
potentially_redundant=[]
for i in targetGenes:
if i in correlated_symbols:
if i != targetGeneID: potentially_redundant.append((i,correlated_symbols[i]))
if len(potentially_redundant)>0:
### These are intra-correlated genes based on the original filtered query
#print targetGeneID, potentially_redundant
for (rh,ind) in potentially_redundant:
matrix2_alt.append(matrix[ind])
row_header2_alt.append(rh)
rho_results.reverse()
#print targetGeneID, correlated_symbols, rho_results[:5]
except Exception:
pass
k+=1
#print targetGeneID+'\t'+str(len(correlated))+'\t'+str(len(anticorrelated))
#sys.exit()
if 'IntraCorrelatedOnly' in targetGene:
matrix2 = matrix2_alt
row_header2 = row_header2_alt
for r in row_header2:
try:
row_header3.append(gene_to_symbol[r][0])
except Exception: row_header3.append(r)
row_header2 = row_header3
#print len(row_header2),len(row_header3),len(matrix2);sys.exit()
matrix2.reverse() ### Display from top-to-bottom rather than bottom-to-top (this is how the clusters are currently ordered in the heatmap)
row_header2.reverse()
if 'amplify' not in targetGene:
row_method = None ### don't cluster the rows (row_method)
if 'amplify' not in targetGene and 'correlated' not in targetGene:
### reorder according to orignal
matrix_temp=[]
header_temp=[]
#print targetGenes
for symbol in targetGenes:
if symbol in matrix_db:
matrix_temp.append(matrix_db[symbol]); header_temp.append(symbol)
#print len(header_temp), len(matrix_db)
if len(header_temp) >= len(matrix_db): ### Hence it worked and all IDs are the same type
matrix2 = matrix_temp
row_header2 = header_temp
if transpose:
matrix2 = map(numpy.array, zip(*matrix2)) ### coverts these to tuples
column_header, row_header2 = row_header2, column_header
exclude=[]
#exclude = excludeHighlyCorrelatedHits(numpy.array(matrix2),row_header2)
exportData.write(string.join(['UID']+column_header,'\t')+'\n') ### title row export
i=0
for row_id in row_header2:
if ':' in row_id and '|' not in row_id:
a,b = string.split(row_id,':')[:2]
if 'ENS' in a:
try: row_id=string.replace(row_id,a,gene_to_symbol[a][0])
except Exception,e: pass
row_header2[i] = row_id
elif 'ENS' in row_id and ' ' in row_id and '|' not in row_id:
row_id = string.split(row_id, ' ')[1]
row_header2[i] = row_id
elif ' ' in row_id:
try: a,b = string.split(row_id, ' ')
except Exception: a = 1; b=2
if a==b:
row_id = a
if row_id not in exclude:
exportData.write(string.join([row_id]+map(str,matrix2[i]),'\t')+'\n') ### export values
i+=1
if 'amplify' not in targetGene and 'correlated' not in targetGene:
print len(row_header2), 'input gene IDs found'
else:
print len(row_header2), 'top-correlated IDs'
exportData.close()
return matrix2,row_header2,column_header,row_method
def numpyCorrelationMatrixGeneStore(x,rows,genes,gene_to_symbol):
### Decided not to use since it would require writing out the whole correlation matrix which is huge (1+GB) and time-intensive to import
start = time.time()
output_file = string.replace(originalFilename,'.txt','.corrmatrix')
status = verifyFile(output_file)
gene_correlations={}
if status == 'found':
try: symbol = gene_to_symbol[rows[i]][0]
except Exception: symbol = '$'
def splitInt(x):
rho,ind = string.split(x,'|')
return (float(rho),int(float(ind)))
for line in open(output_file,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
scores = map(lambda x: splitInt(x), t[1:])
gene_correlations[t[0]] = scores
else:
eo=export.ExportFile(output_file)
#D1 = numpy.ma.corrcoef(x)
D1 = numpy.corrcoef(x)
i=0
for score_ls in D1:
scores = []
try: symbol = gene_to_symbol[rows[i]][0]
except Exception: symbol = '$'
if rows[i] in genes or symbol in genes:
k=0
for v in score_ls:
if str(v)!='nan':
scores.append((v,k))
k+=1
scores.sort()
scores.reverse()
if len(symbol)==1: symbol = rows[i]
gene_correlations[symbol] = scores
export_values = [symbol]
for (v,k) in scores: ### re-import next time to save time
export_values.append(str(v)[:5]+'|'+str(k))
eo.write(string.join(export_values,'\t')+'\n')
i+=1
eo.close()
print len(gene_correlations)
print time.time() - start, 'seconds';sys.exit()
return gene_correlations
def numpyCorrelationMatrixGene(x,rows,genes,gene_to_symbol):
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
#D1 = numpy.ma.corrcoef(x)
D1 = numpy.corrcoef(x)
i=0
gene_correlations={}
for score_ls in D1:
scores = []
try: symbol = gene_to_symbol[rows[i]][0]
except Exception: symbol = '$'
if rows[i] in genes or symbol in genes:
k=0
for v in score_ls:
if str(v)!='nan':
scores.append((v,k))
k+=1
scores.sort()
scores.reverse()
if len(symbol)==1: symbol = rows[i]
gene_correlations[symbol] = scores
i+=1
return gene_correlations
def runHCOnly(filename,graphics,Normalize=False):
""" Simple method for hieararchical clustering with defaults defined by the function rather than the user (see above function) """
global root_dir
global graphic_link
global inputFilename
global GroupDB
global allowLargeClusters
global runGOElite
global EliteGeneSets
runGOElite = False
EliteGeneSets=[]
allowLargeClusters = False
###############
global inputFilename
global originalFilename
global GroupDB
global justShowTheseIDs
global targetGeneIDs
global normalize
global species
global storeGeneSetName
targetGene=[]
filterByPathways=False
justShowTheseIDs=[]
###############
graphic_link=graphics ### Store all locations of pngs
inputFilename = filename ### Used when calling R
root_dir = findParentDir(filename)
if 'ExpressionOutput/Clustering' in root_dir:
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
elif 'ExpressionOutput' in root_dir:
root_dir = string.replace(root_dir,'ExpressionOutput','DataPlots') ### Applies to clustering of LineageProfiler results
else:
root_dir += '/DataPlots/'
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
row_method = 'average'
column_method = 'weighted'
row_metric = 'cosine'
column_metric = 'cosine'
if 'Lineage' in filename or 'Elite' in filename:
color_gradient = 'red_white_blue'
else:
color_gradient = 'yellow_black_blue'
color_gradient = 'red_black_sky'
matrix, column_header, row_header, dataset_name, group_db = importData(filename,Normalize=Normalize)
GroupDB = group_db
runHierarchicalClustering(matrix, row_header, column_header, dataset_name,
row_method, row_metric, column_method, column_metric, color_gradient, display=False, Normalize=Normalize)
return graphic_link
def timestamp():
import datetime
today = str(datetime.date.today()); today = string.split(today,'-'); today = today[0]+''+today[1]+''+today[2]
time_stamp = string.replace(time.ctime(),':','')
time_stamp = string.replace(time_stamp,' ',' ')
time_stamp = string.split(time_stamp,' ') ###Use a time-stamp as the output dir (minus the day)
time_stamp = today+'-'+time_stamp[3]
return time_stamp
def runPCAonly(filename,graphics,transpose,showLabels=True,plotType='3D',display=True,
algorithm='SVD',geneSetName=None, species=None, zscore=True, colorByGene=None,
reimportModelScores=True, separateGenePlots=False, forceClusters=False, maskGroups=None):
global root_dir
global graphic_link
graphic_link=graphics ### Store all locations of pngs
root_dir = findParentDir(filename)
root_dir = string.replace(root_dir,'/DataPlots','')
root_dir = string.replace(root_dir,'/amplify','')
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
root_dir = string.replace(root_dir,'ExpressionInput','DataPlots')
root_dir = string.replace(root_dir,'ICGS-NMF','DataPlots')
if 'DataPlots' not in root_dir:
root_dir += '/DataPlots/'
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
### Transpose matrix and build PCA
geneFilter=None
if (algorithm == 't-SNE' or algorithm == 'UMAP') and reimportModelScores:
dataset_name = string.split(filename,'/')[-1][:-4]
try:
### if the scores are present, we only need to import the genes of interest (save time importing large matrices)
if algorithm == 't-SNE':
importtSNEScores(root_dir+dataset_name+'-t-SNE_scores.txt')
if algorithm == 'UMAP':
importtSNEScores(root_dir+dataset_name+'-UMAP_scores.txt')
if len(colorByGene)==None:
geneFilter = [''] ### It won't import the matrix, basically
elif ' ' in colorByGene or ',' in colorByGene:
colorByGene = string.replace(colorByGene,',',' ')
geneFilter = string.split(colorByGene,' ')
else:
geneFilter = [colorByGene]
except Exception:
#print traceback.format_exc();sys.exit()
geneFilter = None ### It won't import the matrix, basically
matrix, column_header, row_header, dataset_name, group_db = importData(filename,zscore=zscore,geneFilter=geneFilter,forceClusters=forceClusters)
if transpose == False: ### We normally transpose the data, so if True, we don't transpose (I know, it's confusing)
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
if (len(column_header)>1000 or len(row_header)>1000) and algorithm != 't-SNE' and algorithm != 'UMAP':
print 'Performing Principal Component Analysis (please be patient)...'
#PrincipalComponentAnalysis(numpy.array(matrix), row_header, column_header, dataset_name, group_db, display=True)
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
if algorithm == 't-SNE' or algorithm == 'UMAP':
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
if separateGenePlots and (len(colorByGene)>0 or colorByGene==None):
for gene in geneFilter:
tSNE(numpy.array(matrix),column_header,dataset_name,group_db,display=False,
showLabels=showLabels,row_header=row_header,colorByGene=gene,species=species,
reimportModelScores=reimportModelScores,method=algorithm)
if display:
### Show the last one
tSNE(numpy.array(matrix),column_header,dataset_name,group_db,display=True,
showLabels=showLabels,row_header=row_header,colorByGene=gene,species=species,
reimportModelScores=reimportModelScores,method=algorithm)
elif maskGroups!=None:
""" Mask the samples not present in each examined group below """
import ExpressionBuilder
sample_group_db = ExpressionBuilder.simplerGroupImport(maskGroups)
##print maskGroups
#print sample_group_db;sys.exit()
group_sample_db = {}
for sample in sample_group_db:
try: group_sample_db[sample_group_db[sample]].append(sample)
except: group_sample_db[sample_group_db[sample]] = [sample]
for group in group_sample_db:
restricted_samples = group_sample_db[group]
tSNE(numpy.array(matrix),column_header,dataset_name,group_db,display=display,
showLabels=showLabels,row_header=row_header,colorByGene=colorByGene,species=species,
reimportModelScores=reimportModelScores,method=algorithm,maskGroups=(group,restricted_samples))
else:
tSNE(numpy.array(matrix),column_header,dataset_name,group_db,display=display,
showLabels=showLabels,row_header=row_header,colorByGene=colorByGene,species=species,
reimportModelScores=reimportModelScores,method=algorithm)
elif plotType == '3D':
try: PCA3D(numpy.array(matrix), row_header, column_header, dataset_name, group_db,
display=display, showLabels=showLabels, algorithm=algorithm, geneSetName=geneSetName,
species=species, colorByGene=colorByGene)
except Exception:
print traceback.format_exc()
PrincipalComponentAnalysis(numpy.array(matrix), row_header, column_header,
dataset_name, group_db, display=display, showLabels=showLabels, algorithm=algorithm,
geneSetName=geneSetName, species=species, colorByGene=colorByGene,
reimportModelScores=reimportModelScores)
else:
PrincipalComponentAnalysis(numpy.array(matrix), row_header, column_header, dataset_name,
group_db, display=display, showLabels=showLabels, algorithm=algorithm,
geneSetName=geneSetName, species=species, colorByGene=colorByGene,
reimportModelScores=reimportModelScores)
return graphic_link
def outputClusters(filenames,graphics,Normalize=False,Species=None,platform=None,vendor=None):
""" Peforms PCA and Hiearchical clustering on exported log-folds from AltAnalyze """
global root_dir
global graphic_link
global inputFilename
global GroupDB
global allowLargeClusters
global EliteGeneSets
EliteGeneSets=[]
global runGOElite
runGOElite = False
allowLargeClusters=False
graphic_link=graphics ### Store all locations of pngs
filename = filenames[0] ### This is the file to cluster with "significant" gene changes
inputFilename = filename ### Used when calling R
root_dir = findParentDir(filename)
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
### Transpose matrix and build PCA
original = importData(filename,Normalize=Normalize)
matrix, column_header, row_header, dataset_name, group_db = original
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
if len(row_header)<700000 and len(column_header)<700000 and len(column_header)>2:
PrincipalComponentAnalysis(numpy.array(matrix), row_header, column_header, dataset_name, group_db)
else:
print 'SKIPPING PCA!!! - Your dataset file is over or under the recommended size limit for clustering (>7000 rows). Please cluster later using "Additional Analyses".'
row_method = 'average'
column_method = 'average'
row_metric = 'cosine'
column_metric = 'cosine'
color_gradient = 'red_white_blue'
color_gradient = 'red_black_sky'
global species
species = Species
if 'LineageCorrelations' not in filename and 'Zscores' not in filename:
EliteGeneSets=['GeneOntology']
runGOElite = True
### Generate Significant Gene HeatMap
matrix, column_header, row_header, dataset_name, group_db = original
GroupDB = group_db
runHierarchicalClustering(matrix, row_header, column_header, dataset_name, row_method, row_metric, column_method, column_metric, color_gradient, Normalize=Normalize)
### Generate Outlier and other Significant Gene HeatMap
for filename in filenames[1:]:
inputFilename = filename
matrix, column_header, row_header, dataset_name, group_db = importData(filename,Normalize=Normalize)
GroupDB = group_db
try:
runHierarchicalClustering(matrix, row_header, column_header, dataset_name, row_method, row_metric, column_method, column_metric, color_gradient, Normalize=Normalize)
except Exception: print 'Could not cluster',inputFilename,', file not found'
return graphic_link
def importEliteGeneAssociations(gene_filename):
fn = filepath(gene_filename)
x=0; fold_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#': x=0
elif x==0: x=1
else:
geneid=t[0];symbol=t[1]
fold = 0
try:
if '|' in t[6]:
fold = float(string.split(t[6])[0]) ### Sometimes there are multiple folds for a gene (multiple probesets)
except Exception:
None
try: fold=float(t[6])
except Exception: None
fold_db[symbol] = fold
return fold_db
def importPathwayLevelFolds(filename):
fn = filepath(filename)
x=0
folds_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if len(data)==0: x=0
elif x==0:
z_score_indexes = []; i=0
z_headers = []
for header in t:
if 'z_score.' in header:
z_score_indexes.append(i)
header = string.split(header,'z_score.')[1] ### Get rid of z_score.
if 'AS.' in header:
header = string.split(header,'.p')[0] ### Remove statistics details
header = 'AS.'+string.join(string.split(header,'_')[2:],'_') ### species and array type notation
else:
header = string.join(string.split(header,'-')[:-2],'-')
if '-fold' in header:
header = string.join(string.split(header,'-')[:-1],'-')
z_headers.append(header)
i+=1
headers = string.join(['Gene-Set Name']+z_headers,'\t')+'\n'
x=1
else:
term_name=t[1];geneset_type=t[2]
zscores = map(lambda x: t[x], z_score_indexes)
max_z = max(map(float, zscores)) ### If there are a lot of terms, only show the top 70
line = string.join([term_name]+zscores,'\t')+'\n'
try: zscore_db[geneset_type].append((max_z,line))
except Exception: zscore_db[geneset_type] = [(max_z,line)]
exported_files = []
for geneset_type in zscore_db:
### Create an input file for hierarchical clustering in a child directory (Heatmaps)
clusterinput_filename = findParentDir(filename)+'/Heatmaps/Clustering-Zscores-'+geneset_type+'.txt'
exported_files.append(clusterinput_filename)
export_text = export.ExportFile(clusterinput_filename)
export_text.write(headers) ### Header is the same for each file
zscore_db[geneset_type].sort()
zscore_db[geneset_type].reverse()
i=0 ### count the entries written
for (max_z,line) in zscore_db[geneset_type]:
if i<60:
export_text.write(line) ### Write z-score values and row names
i+=1
export_text.close()
return exported_files
def importOverlappingEliteScores(filename):
fn = filepath(filename)
x=0
zscore_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if len(data)==0: x=0
elif x==0:
z_score_indexes = []; i=0
z_headers = []
for header in t:
if 'z_score.' in header:
z_score_indexes.append(i)
header = string.split(header,'z_score.')[1] ### Get rid of z_score.
if 'AS.' in header:
header = string.split(header,'.p')[0] ### Remove statistics details
header = 'AS.'+string.join(string.split(header,'_')[2:],'_') ### species and array type notation
else:
header = string.join(string.split(header,'-')[:-2],'-')
if '-fold' in header:
header = string.join(string.split(header,'-')[:-1],'-')
z_headers.append(header)
i+=1
headers = string.join(['Gene-Set Name']+z_headers,'\t')+'\n'
x=1
else:
term_name=t[1];geneset_type=t[2]
zscores = map(lambda x: t[x], z_score_indexes)
max_z = max(map(float, zscores)) ### If there are a lot of terms, only show the top 70
line = string.join([term_name]+zscores,'\t')+'\n'
try: zscore_db[geneset_type].append((max_z,line))
except Exception: zscore_db[geneset_type] = [(max_z,line)]
exported_files = []
for geneset_type in zscore_db:
### Create an input file for hierarchical clustering in a child directory (Heatmaps)
clusterinput_filename = findParentDir(filename)+'/Heatmaps/Clustering-Zscores-'+geneset_type+'.txt'
exported_files.append(clusterinput_filename)
export_text = export.ExportFile(clusterinput_filename)
export_text.write(headers) ### Header is the same for each file
zscore_db[geneset_type].sort()
zscore_db[geneset_type].reverse()
i=0 ### count the entries written
for (max_z,line) in zscore_db[geneset_type]:
if i<60:
export_text.write(line) ### Write z-score values and row names
i+=1
export_text.close()
return exported_files
def buildGraphFromSIF(mod,species,sif_filename,ora_input_dir):
""" Imports a SIF and corresponding gene-association file to get fold changes for standardized gene-symbols """
global SpeciesCode; SpeciesCode = species
mod = 'Ensembl'
if sif_filename == None:
### Used for testing only
sif_filename = '/Users/nsalomonis/Desktop/dataAnalysis/collaborations/WholeGenomeRVista/Alex-Figure/GO-Elite_results/CompleteResults/ORA_pruned/up-2f_p05-WGRV.sif'
ora_input_dir = '/Users/nsalomonis/Desktop/dataAnalysis/collaborations/WholeGenomeRVista/Alex-Figure/up-stringent/up-2f_p05.txt'
#sif_filename = 'C:/Users/Nathan Salomonis/Desktop/Endothelial_Kidney/GO-Elite/GO-Elite_results/CompleteResults/ORA_pruned/GE.b_vs_a-fold2.0_rawp0.05-local.sif'
#ora_input_dir = 'C:/Users/Nathan Salomonis/Desktop/Endothelial_Kidney/GO-Elite/input/GE.b_vs_a-fold2.0_rawp0.05.txt'
gene_filename = string.replace(sif_filename,'.sif','_%s-gene-associations.txt') % mod
gene_filename = string.replace(gene_filename,'ORA_pruned','ORA_pruned/gene_associations')
pathway_name = string.split(sif_filename,'/')[-1][:-4]
output_filename = None
try: fold_db = importEliteGeneAssociations(gene_filename)
except Exception: fold_db={}
if ora_input_dir != None:
### This is an optional accessory function that adds fold changes from genes that are NOT in the GO-Elite pruned results (TFs regulating these genes)
try: fold_db = importDataSimple(ora_input_dir,species,fold_db,mod)
except Exception: None
try:
### Alternative Approaches dependening on the availability of GraphViz
#displaySimpleNetXGraph(sif_filename,fold_db,pathway_name)
output_filename = iGraphSimple(sif_filename,fold_db,pathway_name)
except Exception:
print 'igraph export failed due to an unknown error (not installed)'
print traceback.format_exc()
try: displaySimpleNetwork(sif_filename,fold_db,pathway_name)
except Exception: pass ### GraphViz problem
return output_filename
def iGraphSimple(sif_filename,fold_db,pathway_name):
""" Build a network export using iGraph and Cairo """
edges = importSIF(sif_filename)
id_color_db = WikiPathways_webservice.getHexadecimalColorRanges(fold_db,'Genes')
output_filename = iGraphDraw(edges,pathway_name,filePath=sif_filename,display=True,graph_layout='spring',colorDB=id_color_db)
return output_filename
def iGraphDraw(edges, pathway_name, labels=None, graph_layout='shell', display=False,
node_size=700, node_color='yellow', node_alpha=0.5, node_text_size=7,
edge_color='black', edge_alpha=0.5, edge_thickness=2, edges_pos=.3,
text_font='sans-serif',filePath='test',colorDB=None):
### Here node = vertex
output_filename=None
if len(edges) > 700 and 'AltAnalyze' not in pathway_name:
print findFilename(filePath), 'too large to visualize...'
elif len(edges) > 3000:
print findFilename(filePath), 'too large to visualize...'
else:
arrow_scaler = 1 ### To scale the arrow
if edges>40: arrow_scaler = .9
vars = formatiGraphEdges(edges,pathway_name,colorDB,arrow_scaler)
vertices,iGraph_edges,vertice_db,label_list,shape_list,vertex_size, color_list, vertex_label_colors, arrow_width, edge_colors = vars
if vertices>0:
import igraph
gr = igraph.Graph(vertices, directed=True)
canvas_scaler = 0.8 ### To scale the canvas size (bounding box)
if vertices<15: canvas_scaler = 0.5
elif vertices<25: canvas_scaler = .70
elif vertices>35:
canvas_scaler += len(iGraph_edges)/400.00
filePath,canvas_scaler = correctedFilePath(filePath,canvas_scaler) ### adjust for GO-Elite
#print vertices, len(iGraph_edges), pathway_name, canvas_scaler
canvas_size = (600*canvas_scaler,600*canvas_scaler)
gr.add_edges(iGraph_edges)
gr.vs["label"] = label_list
gr.vs["shape"] = shape_list
gr.vs["size"] = vertex_size
gr.vs["label_dist"] = [1.3]*vertices
gr.vs["label_size"] = [12]*vertices
gr.vs["color"]=color_list
gr.vs["label_color"]=vertex_label_colors
gr.es["color"] = edge_colors
gr.es["arrow_size"]=arrow_width
output_filename = '%s.pdf' % filePath[:-4]
output_filename = output_filename.encode('ascii','ignore') ### removes the damned unicode u proceeding the filename
layout = "kk"
visual_style = {}
#visual_style["layout"] = layout #The default is auto, which selects a layout algorithm automatically based on the size and connectedness of the graph
visual_style["margin"] = 50 ### white-space around the network (see vertex size)
visual_style["bbox"] = canvas_size
igraph.plot(gr,output_filename, **visual_style)
output_filename = '%s.png' % filePath[:-4]
output_filename = output_filename.encode('ascii','ignore') ### removes the damned unicode u proceeding the filename
if vertices <15: gr,visual_style = increasePlotSize(gr,visual_style)
igraph.plot(gr,output_filename, **visual_style)
#surface = cairo.ImageSurface(cairo.FORMAT_ARGB32, width, height)
return output_filename
def correctedFilePath(filePath,canvas_scaler):
""" Move this file to it's own network directory for GO-Elite """
if 'ORA_pruned' in filePath:
filePath = string.replace(filePath,'CompleteResults/ORA_pruned','networks')
try: os.mkdir(findParentDir(filePath))
except Exception: pass
canvas_scaler = canvas_scaler*1.3 ### These graphs tend to be more dense and difficult to read
return filePath,canvas_scaler
def increasePlotSize(gr,visual_style):
### To display the plot better, need to manually increase the size of everything
factor = 2
object_list = ["size","label_size"]
for i in object_list:
new=[]
for k in gr.vs[i]:
new.append(k*factor)
gr.vs[i] = new
new=[]
for i in gr.es["arrow_size"]:
new.append(i*factor)
new=[]
for i in visual_style["bbox"]:
new.append(i*factor)
visual_style["bbox"] = new
visual_style["margin"]=visual_style["margin"]*factor
return gr,visual_style
def getHMDBDataSimple():
### Determine which IDs are metabolites
program_type,database_dir = unique.whatProgramIsThis()
filename = database_dir+'/'+SpeciesCode+'/gene/HMDB.txt'
symbol_hmdb_db={}
x=0
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if x==0: x=1
else:
t = string.split(data,'\t')
hmdb_id = t[0]; symbol = t[1]; ProteinNames = t[-1]
symbol_hmdb_db[symbol]=hmdb_id
return symbol_hmdb_db
def formatiGraphEdges(edges,pathway_name,colorDB,arrow_scaler):
### iGraph appears to require defined vertice number and edges as numbers corresponding to these vertices
edge_db={}
edges2=[]
vertice_db={}
shape_list=[] ### node shape in order
label_list=[] ### Names of each vertix aka node
vertex_size=[]
color_list=[]
vertex_label_colors=[]
arrow_width=[] ### Indicates the presence or absence of an arrow
edge_colors=[]
k=0
try: symbol_hmdb_db = getHMDBDataSimple()
except Exception: symbol_hmdb_db={}
for (node1,node2,type) in edges:
edge_color = 'grey'
### Assign nodes to a numeric vertix ID
if 'TF' in pathway_name or 'WGRV' in pathway_name:
pathway = node1 ### This is the regulating TF
else:
pathway = node2 ### This is the pathway
if 'drugInteraction' == type: edge_color = "purple"
elif 'TBar' == type: edge_color = 'blue'
elif 'microRNAInteraction' == type: edge_color = '#53A26D'
elif 'transcription' in type: edge_color = '#FF7D7D'
if 'AltAnalyze' in pathway_name: default_node_color = 'grey'
else: default_node_color = "yellow"
if node1 in vertice_db: v1=vertice_db[node1]
else: #### Left hand node
### Only time the vertex is added to the below attribute lists
v1=k; label_list.append(node1)
rs = 1 ### relative size
if 'TF' in pathway_name or 'WGRV' in pathway_name and 'AltAnalyze' not in pathway_name:
shape_list.append('rectangle')
vertex_size.append(15)
vertex_label_colors.append('blue')
else:
if 'drugInteraction' == type:
rs = 0.75
shape_list.append('rectangle')
vertex_label_colors.append('purple')
default_node_color = "purple"
elif 'Metabolic' == type and node1 in symbol_hmdb_db:
shape_list.append('triangle-up')
vertex_label_colors.append('blue') #dark green
default_node_color = 'grey' #'#008000'
elif 'microRNAInteraction' == type:
rs = 0.75
shape_list.append('triangle-up')
vertex_label_colors.append('#008000') #dark green
default_node_color = 'grey' #'#008000'
else:
shape_list.append('circle')
vertex_label_colors.append('black')
vertex_size.append(10*rs)
vertice_db[node1]=v1; k+=1
try:
color = '#'+string.upper(colorDB[node1])
color_list.append(color) ### Hex color
except Exception:
color_list.append(default_node_color)
if node2 in vertice_db: v2=vertice_db[node2]
else: #### Right hand node
### Only time the vertex is added to the below attribute lists
v2=k; label_list.append(node2)
if 'TF' in pathway_name or 'WGRV' in pathway_name:
shape_list.append('circle')
vertex_size.append(10)
vertex_label_colors.append('black')
default_node_color = "grey"
elif 'AltAnalyze' not in pathway_name:
shape_list.append('rectangle')
vertex_size.append(15)
vertex_label_colors.append('blue')
default_node_color = "grey"
elif 'Metabolic' == type and node2 in symbol_hmdb_db:
shape_list.append('triangle-up')
vertex_label_colors.append('blue') #dark green
default_node_color = 'grey' #'#008000'
else:
shape_list.append('circle')
vertex_size.append(10)
vertex_label_colors.append('black')
default_node_color = "grey"
vertice_db[node2]=v2; k+=1
try:
color = '#'+string.upper(colorDB[node2])
color_list.append(color) ### Hex color
except Exception: color_list.append(default_node_color)
edges2.append((v1,v2))
if type == 'physical': arrow_width.append(0)
else: arrow_width.append(arrow_scaler)
try: edge_db[v1].append(v2)
except Exception: edge_db[v1]=[v2]
try: edge_db[v2].append(v1)
except Exception: edge_db[v2]=[v1]
edge_colors.append(edge_color)
vertices = len(edge_db) ### This is the number of nodes
edge_db = eliminate_redundant_dict_values(edge_db)
vertice_db2={} ### Invert
for node in vertice_db:
vertice_db2[vertice_db[node]] = node
#print len(edges2), len(edge_colors)
print vertices, 'and', len(edges2),'edges in the iGraph network.'
return vertices,edges2,vertice_db2, label_list, shape_list, vertex_size, color_list, vertex_label_colors, arrow_width, edge_colors
def eliminate_redundant_dict_values(database):
db1={}
for key in database: list = unique.unique(database[key]); list.sort(); db1[key] = list
return db1
def importDataSimple(filename,species,fold_db,mod):
""" Imports an input ID file and converts those IDs to gene symbols for analysis with folds """
import GO_Elite
from import_scripts import OBO_import
import gene_associations
fn = filepath(filename)
x=0
metabolite_codes = ['Ck','Ca','Ce','Ch','Cp']
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#': x=0
elif x==0:
si=None; symbol_present = False
try:
si= t.index('Symbol')
symbol_present = True
except: pass
x=1
else:
if x == 1:
system_code = t[1]
if system_code in metabolite_codes:
mod = 'HMDB'
system_codes,source_types,mod_types = GO_Elite.getSourceData()
try: source_data = system_codes[system_code]
except Exception:
source_data = None
if 'ENS' in t[0]: source_data = system_codes['En']
else: ### Assume the file is composed of gene symbols
source_data = system_codes['Sy']
if source_data == mod:
source_is_mod = True
elif source_data==None:
None ### Skip this
else:
source_is_mod = False
mod_source = mod+'-'+source_data+'.txt'
gene_to_source_id = gene_associations.getGeneToUid(species,('hide',mod_source))
source_to_gene = OBO_import.swapKeyValues(gene_to_source_id)
try: gene_to_symbol = gene_associations.getGeneToUid(species,('hide',mod+'-Symbol'))
except Exception: gene_to_symbol={}
try: met_to_symbol = gene_associations.importGeneData(species,'HMDB',simpleImport=True)
except Exception: met_to_symbol={}
for i in met_to_symbol: gene_to_symbol[i] = met_to_symbol[i] ### Add metabolite names
x+=1
if source_is_mod == True:
if t[0] in gene_to_symbol:
symbol = gene_to_symbol[t[0]][0]
try: fold_db[symbol] = float(t[2])
except Exception: fold_db[symbol] = 0
else:
fold_db[t[0]] = 0 ### If not found (wrong ID with the wrong system) still try to color the ID in the network as yellow
elif symbol_present:
fold_db[t[si]] = 0
try: fold_db[t[si]] = float(t[2])
except Exception:
try: fold_db[t[si]] = 0
except: fold_db[t[0]] = 0
elif t[0] in source_to_gene:
mod_ids = source_to_gene[t[0]]
try: mod_ids+=source_to_gene[t[2]] ###If the file is a SIF
except Exception:
try: mod_ids+=source_to_gene[t[1]] ###If the file is a SIF
except Exception: None
for mod_id in mod_ids:
if mod_id in gene_to_symbol:
symbol = gene_to_symbol[mod_id][0]
try: fold_db[symbol] = float(t[2]) ### If multiple Ensembl IDs in dataset, only record the last associated fold change
except Exception: fold_db[symbol] = 0
else: fold_db[t[0]] = 0
return fold_db
def clusterPathwayZscores(filename):
""" Imports a overlapping-results file and exports an input file for hierarchical clustering and clusters """
### This method is not fully written or in use yet - not sure if needed
if filename == None:
### Only used for testing
filename = '/Users/nsalomonis/Desktop/dataAnalysis/r4_Bruneau_TopHat/GO-Elite/TF-enrichment2/GO-Elite_results/overlapping-results_z-score_elite.txt'
exported_files = importOverlappingEliteScores(filename)
graphic_links=[]
for file in exported_files:
try: graphic_links = runHCOnly(file,graphic_links)
except Exception,e:
#print e
print 'Unable to generate cluster due to dataset incompatibilty.'
print 'Clustering of overlapping-results_z-score complete (see "GO-Elite_results/Heatmaps" directory)'
def clusterPathwayMeanFolds():
""" Imports the pruned-results file and exports an input file for hierarchical clustering and clusters """
filename = '/Users/nsalomonis/Desktop/User Diagnostics/Mm_spinal_cord_injury/GO-Elite/GO-Elite_results/pruned-results_z-score_elite.txt'
exported_files = importPathwayLevelFolds(filename)
def VennDiagram():
f = pylab.figure()
ax = f.gca()
rad = 1.4
c1 = Circle((-1,0),rad, alpha=.2, fc ='red',label='red')
c2 = Circle((1,0),rad, alpha=.2, fc ='blue',label='blue')
c3 = Circle((0,1),rad, alpha=.2, fc ='green',label='g')
#pylab.plot(c1,color='green',marker='o',markersize=7,label='blue')
#ax.add_patch(c1)
ax.add_patch(c2)
ax.add_patch(c3)
ax.set_xlim(-3,3)
ax.set_ylim(-3,3)
pylab.show()
def plotHistogram(filename):
matrix, column_header, row_header, dataset_name, group_db = importData(filename)
transpose=True
if transpose: ### Transpose the data matrix
print 'Transposing the data matrix'
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
pylab.figure()
for i in matrix:
pylab.hist(i, 200, normed=0, histtype='step', cumulative=-1)
#pylab.hist(matrix, 50, cumulative=-1)
pylab.show()
def stackedbarchart(filename,display=False,output=False):
header=[]
conditions = []
data_matrix=[]
for line in open(filename,'rU').xreadlines():
cd = cleanUpLine(line)
t = string.split(cd,'\t')
if len(header)==0:
header = t[4:]
exc_indexes = [0,2,4,6,8,10,12]
inc_indexes = [1,3,5,7,9,11,13]
inlc_header = map(lambda i: string.split(header[i],'_')[0],inc_indexes)
header = inlc_header
else:
condition = t[0]
data = t[4:]
conditions.append(condition+'-inclusion ')
data_matrix.append(map(lambda i: float(data[i]),inc_indexes))
conditions.append(condition+'-exclusion ')
data_matrix.append(map(lambda i: float(data[i]),exc_indexes))
data_matrix = map(numpy.array, zip(*data_matrix))
#https://www.w3resource.com/graphics/matplotlib/barchart/matplotlib-barchart-exercise-16.php
# multi-dimensional data_matrix
y_pos = np.arange(len(conditions))
fig, ax = pylab.subplots()
#fig = pylab.figure(figsize=(10,8))
#ax = fig.add_subplot(111)
#pos1 = ax.get_position() # get the original position
#pos2 = [pos1.x0 + 0.2, pos1.y0 - 0.2, pos1.width / 1.2, pos1.height / 1.2 ]
#ax.set_position(pos2) # set a new position
colors =['royalblue','salmon','grey','gold','cornflowerblue','mediumseagreen','navy']
patch_handles = []
# left alignment of data_matrix starts at zero
left = np.zeros(len(conditions))
index=0
for i, d in enumerate(data_matrix):
patch_handles.append(ax.barh(y_pos, d, 0.3,
color=colors[index], align='center',
left=left,label = header[index]))
left += d
index+=1
# search all of the bar segments and annotate
"""
for j in range(len(patch_handles)):
for i, patch in enumerate(patch_handles[j].get_children()):
bl = patch.get_xy()
x = 0.5*patch.get_width() + bl[0]
y = 0.5*patch.get_height() + bl[1]
#ax.text(x,y, "%d%%" % (percentages[i,j]), ha='center')
"""
ax.set_yticks(y_pos)
ax.set_yticklabels(conditions)
ax.set_xlabel('Events')
ax.legend(loc="best", bbox_to_anchor=(1.0, 1.0))
box = ax.get_position()
# Shink current axis by 20%
ax.set_position([box.x0+0.2, box.y0, box.width * 0.6, box.height])
try: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5),fontsize = 10) ### move the legend over to the right of the plot
except Exception: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
ax.set_title('MultiPath-PSI Splicing Event Types')
#pylab.tight_layout(pad=0.4, w_pad=0.5, h_pad=1.0)
if output==False:
pylab.savefig(filename[:-4]+'.pdf')
pylab.savefig(filename[:-4]+'.png')
else:
pylab.savefig(output[:-4]+'.pdf')
pylab.savefig(output[:-4]+'.png')
if display:
print 'Exporting:',filename
try: pylab.show()
except Exception: None ### when run in headless mode
def barchart(filename,index1,index2,x_axis,y_axis,title,display=False,color1='gold',color2='darkviolet',output=False):
header=[]
reference_data=[]
query_data=[]
groups=[]
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if len(header)==0:
header = t
header1=header[index1]
header2=header[index2]
else:
reference_data.append(float(t[index1]))
q_value = float(t[index2])
if 'frequen' not in filename:
q_value = q_value*-1
query_data.append(q_value)
name = t[0]
if '_vs_' in name and 'event_summary' not in filename:
name = string.split(name,'_vs_')[0]
suffix=None
if '__' in name:
suffix = string.split(name,'__')[-1]
if '_' in name:
name = string.split(name,'_')[:-1]
name = string.join(name,'_')
if len(name)>20:
name = string.split(name,'_')[0]
if suffix !=None:
name+='_'+suffix
groups.append(name)
fig, ax = pylab.subplots()
pos1 = ax.get_position() # get the original position
pos2 = [pos1.x0 + 0.2, pos1.y0 + 0.1, pos1.width / 1.2, pos1.height / 1.2 ]
ax.set_position(pos2) # set a new position
ind = np.arange(len(groups)) # the x locations for the groups
width = 0.35 # the width of the bars
query_data.reverse()
reference_data.reverse()
groups.reverse()
ax.barh(ind - width/2, query_data, width, color=color2, label=header2)
ax.barh(ind + width/2, reference_data, width,color=color1, label=header1)
ax.set_xlabel(x_axis)
ax.set_ylabel(y_axis)
ax.set_yticks(ind+0.175)
ax.set_yticklabels(groups)
ax.set_title(title)
ax.legend()
if output==False:
pylab.savefig(filename[:-4]+'.pdf')
#pylab.savefig(filename[:-4]+'.png')
else:
pylab.savefig(output[:-4]+'.pdf')
#pylab.savefig(output[:-4]+'.png')
if display:
print 'Exporting:',filename
try: pylab.show()
except Exception: None ### when run in headless mode
def multipleSubPlots(filename,uids,SubPlotType='column',n=20):
#uids = [uids[-1]]+uids[:-1]
str_uids = string.join(uids,'_')
matrix, column_header, row_header, dataset_name, group_db = importData(filename,geneFilter=uids)
for uid in uids:
if uid not in row_header:
print uid,"is missing from the expression file."
fig = pylab.figure()
def ReplaceZeros(val,min_val):
if val == 0:
return min_val
else: return val
### Order the graphs based on the original gene order
new_row_header=[]
matrix2 = []
for uid in uids:
if uid in row_header:
ind = row_header.index(uid)
new_row_header.append(uid)
try: update_exp_vals = map(lambda x: ReplaceZeros(x,0.0001),matrix[ind])
except Exception: print uid, len(matrix[ind]);sys.exit()
#update_exp_vals = map(lambda x: math.pow(2,x+1),update_exp_vals) #- nonlog transform
matrix2.append(update_exp_vals)
matrix = numpy.array(matrix2)
row_header = new_row_header
#print row_header
color_list = ['r', 'b', 'y', 'g', 'w', 'k', 'm']
groups=[]
for sample in column_header:
try: group = group_db[sample][0]
except: group = '1'
if group not in groups:
groups.append(group)
fontsize=10
if len(groups)>0:
color_list = []
if len(groups)==9:
cm = matplotlib.colors.ListedColormap(['#80C241', '#118943', '#6FC8BB', '#ED1D30', '#F26E21','#8051A0', '#4684C5', '#FBD019','#3A52A4'])
elif len(groups)==3:
cm = matplotlib.colors.ListedColormap(['#4684C4','#FAD01C','#7D7D7F'])
#elif len(groups)==5: cm = matplotlib.colors.ListedColormap(['#41449B','#6182C1','#9DDAEA','#42AED0','#7F7F7F'])
else:
cm = pylab.cm.get_cmap('gist_rainbow') #Paired
for i in range(len(groups)):
color_list.append(cm(1.*i/(len(groups)-1))) # color will now be an RGBA tuple
for i in range(len(matrix)):
ax = pylab.subplot(n,1,1+i)
OY = matrix[i]
pylab.xlim(0,len(OY))
pylab.subplots_adjust(right=0.85)
ind = np.arange(len(OY))
index_list = []
v_list = []
colors_list = []
if SubPlotType=='column':
index=-1
for v in OY:
index+=1
try: group = group_db[column_header[index]][0]
except: group = '1'
index_list.append(index)
v_list.append(v)
colors_list.append(color_list[groups.index(group)])
#pylab.bar(index, v,edgecolor='black',linewidth=0,color=color_list[groups.index(group)])
width = .35
#print i ,row_header[i]
print 1
barlist = pylab.bar(index_list, v_list,edgecolor='black',linewidth=0)
ci = 0
for cs in barlist:
barlist[ci].set_color(colors_list[ci])
ci+=1
if SubPlotType=='plot':
pylab.plot(x,y)
ax.text(matrix.shape[1]-0.5, i, ' '+row_header[i],fontsize=8)
fig.autofmt_xdate()
pylab.subplots_adjust(hspace = .001)
temp = tic.MaxNLocator(3)
ax.yaxis.set_major_locator(temp)
ax.set_xticks([])
#ax.title.set_visible(False)
#pylab.xticks(ind + width / 2, column_header)
#ax.set_xticklabels(column_header)
#ax.xaxis.set_ticks([-1]+range(len(OY)+1))
#xtickNames = pylab.setp(pylab.gca(), xticklabels=['']+column_header)
#pylab.setp(xtickNames, rotation=90, fontsize=10)
#pylab.show()
if len(str_uids)>50:
str_uids = str_uids[:50]
pylab.savefig(filename[:-4]+'-1'+str_uids+'.pdf')
def simpleTranspose(filename):
fn = filepath(filename)
matrix = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,' ')
matrix.append(t)
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
filename = filename[:-4]+'-transposed.txt'
ea = export.ExportFile(filename)
for i in matrix:
ea.write(string.join(i,'\t')+'\n')
ea.close()
def CorrdinateToBed(filename):
fn = filepath(filename)
matrix = []
translation={}
multiExon={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
data = string.replace(data,' ','')
t = string.split(data,'\t')
if '.gtf' in filename:
if 'chr' not in t[0]: chr = 'chr'+t[0]
else: chr = t[0]
start = t[3]; end = t[4]; strand = t[6]; annotation = t[8]
annotation = string.replace(annotation,'gene_id','')
annotation = string.replace(annotation,'transcript_id','')
annotation = string.replace(annotation,'gene_name','')
geneIDs = string.split(annotation,';')
geneID = geneIDs[0]; symbol = geneIDs[3]
else:
chr = t[4]; strand = t[5]; start = t[6]; end = t[7]
#if 'ENS' not in annotation:
t = [chr,start,end,geneID,'0',strand]
#matrix.append(t)
translation[geneID] = symbol
try: multiExon[geneID]+=1
except Exception: multiExon[geneID]=1
filename = filename[:-4]+'-new.bed'
ea = export.ExportFile(filename)
for i in translation:
#ea.write(string.join(i,'\t')+'\n')
ea.write(i+'\t'+translation[i]+'\t'+str(multiExon[i])+'\n')
ea.close()
def SimpleCorrdinateToBed(filename):
fn = filepath(filename)
matrix = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
data = string.replace(data,' ','')
t = string.split(data,'\t')
if '.bed' in filename:
print t;sys.exit()
chr = t[4]; strand = t[5]; start = t[6]; end = t[7]
if 'ENS' in t[0]:
t = [chr,start,end,t[0],'0',strand]
matrix.append(t)
filename = filename[:-4]+'-new.bed'
ea = export.ExportFile(filename)
for i in matrix:
ea.write(string.join(i,'\t')+'\n')
ea.close()
def simpleIntegrityCheck(filename):
fn = filepath(filename)
matrix = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
data = string.replace(data,' ','')
t = string.split(data,'\t')
matrix.append(t)
filename = filename[:-4]+'-new.bed'
ea = export.ExportFile(filename)
for i in matrix:
ea.write(string.join(i,'\t')+'\n')
ea.close()
def BedFileCheck(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.bed'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow = False
else:
#if len(t) != 12: print len(t);sys.exit()
ea.write(string.join(t,'\t')+'\n')
ea.close()
def simpleFilter(filename):
fn = filepath(filename)
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
matrix = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,',')
uid = t[0]
#if '=chr' in t[0]:
if 1==2:
a,b = string.split(t[0],'=')
b = string.replace(b,'_',':')
uid = a+ '='+b
matrix.append(t)
ea.write(string.join([uid]+t[1:],'\t')+'\n')
ea.close()
def test(filename):
symbols2={}
firstLine=True
fn = filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine=False
header = t
i=0; start=None; alt_start=None
value_indexes=[]
groups = {}
group = 0
for h in header:
if h == 'WikiPathways': start=i
if h == 'Select Protein Classes': alt_start=i
i+=1
if start == None: start = alt_start
for h in header:
if h>i:
group[i]
i+=1
if start == None: start = alt_start
else:
uniprot = t[0]
symbols = string.replace(t[-1],';;',';')
symbols = string.split(symbols,';')
for s in symbols:
if len(s)>0:
symbols2[string.upper(s),uniprot]=[]
for (s,u) in symbols2:
ea.write(string.join([s,u],'\t')+'\n')
ea.close()
def coincentIncedenceTest(exp_file,TFs):
fn = filepath(TFs)
tfs={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
tfs[data]=[]
comparisons={}
for tf1 in tfs:
for tf2 in tfs:
if tf1!=tf2:
temp = [tf1,tf2]
temp.sort()
comparisons[tuple(temp)]=[]
gene_data={}
firstLine=True
fn = filepath(exp_file)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if firstLine:
firstLine=False
header = string.split(data,'\t')[1:]
else:
t = string.split(data,'\t')
gene = t[0]
values = map(float,t[1:])
gene_data[gene] = values
filename = TFs[:-4]+'-all-coincident-5z.txt'
ea = export.ExportFile(filename)
comparison_db={}
for comparison in comparisons:
vals1 = gene_data[comparison[0]]
vals2 = gene_data[comparison[1]]
i=0
coincident=[]
for v1 in vals1:
v2 = vals2[i]
#print v1,v2
if v1>1 and v2>1:
coincident.append(i)
i+=1
i=0
population_db={}; coincident_db={}
for h in header:
population=string.split(h,':')[0]
if i in coincident:
try: coincident_db[population]+=1
except Exception: coincident_db[population]=1
try: population_db[population]+=1
except Exception: population_db[population]=1
i+=1
import mappfinder
final_population_percent=[]
for population in population_db:
d = population_db[population]
try: c = coincident_db[population]
except Exception: c = 0
N = float(len(header)) ### num all samples examined
R = float(len(coincident)) ### num all coincedent samples for the TFs
n = float(d) ### num all samples in cluster
r = float(c) ### num all coincident samples in cluster
try: z = mappfinder.Zscore(r,n,N,R)
except Exception: z=0
#if 'Gfi1b' in comparison and 'Gata1' in comparison: print N, R, n, r, z
final_population_percent.append([population,str(c),str(d),str(float(c)/float(d)),str(z)])
comparison_db[comparison]=final_population_percent
filtered_comparison_db={}
top_scoring_population={}
for comparison in comparison_db:
max_group=[]
for population_stat in comparison_db[comparison]:
z = float(population_stat[-1])
c = float(population_stat[1])
population = population_stat[0]
max_group.append([z,population])
max_group.sort()
z = max_group[-1][0]
pop = max_group[-1][1]
if z>(1.96)*2 and c>3:
filtered_comparison_db[comparison]=comparison_db[comparison]
top_scoring_population[comparison] = pop,z
firstLine = True
for comparison in filtered_comparison_db:
comparison_alt = string.join(list(comparison),'|')
all_percents=[]
for line in filtered_comparison_db[comparison]:
all_percents.append(line[3])
if firstLine:
all_headers=[]
for line in filtered_comparison_db[comparison]:
all_headers.append(line[0])
ea.write(string.join(['gene-pair']+all_headers+['Top Population','Top Z'],'\t')+'\n')
firstLine=False
pop,z = top_scoring_population[comparison]
ea.write(string.join([comparison_alt]+all_percents+[pop,str(z)],'\t')+'\n')
ea.close()
def getlastexon(filename):
filename2 = filename[:-4]+'-last-exon.txt'
ea = export.ExportFile(filename2)
firstLine=True
fn = filepath(filename)
last_gene = 'null'; last_exon=''
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine=False
else:
gene = t[2]
if gene != last_gene:
if ':E' in last_exon:
gene,exon = last_exon = string.split(':E')
block,region = string.split(exon,'.')
try: ea.write(last_exon+'\n')
except: pass
last_gene = gene
last_exon = t[0]
ea.close()
def replaceWithBinary(filename):
filename2 = filename[:-4]+'-binary.txt'
ea = export.ExportFile(filename2)
firstLine=True
fn = filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
ea.write(line)
firstLine=False
else:
try: values = map(float,t[1:])
except Exception: print t[1:];sys.exit()
values2=[]
for v in values:
if v == 0: values2.append('0')
else: values2.append('1')
ea.write(string.join([t[0]]+values2,'\t')+'\n')
ea.close()
def geneMethylationOutput(filename):
filename2 = filename[:-4]+'-binary.txt'
ea = export.ExportFile(filename2)
firstLine=True
fn = filepath(filename)
db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
values = (t[20],t[3]+'-methylation')
db[values]=[]
for value in db:
ea.write(string.join(list(value),'\t')+'\n')
ea.close()
def coincidentIncedence(filename,genes):
exportPairs=True
gene_data=[]
firstLine=True
fn = filepath(filename)
if exportPairs:
filename = filename[:-4]+'_'+genes[0]+'-'+genes[1]+'2.txt'
ea = export.ExportFile(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if firstLine:
firstLine=False
header = string.split(data,'\t')[1:]
else:
t = string.split(data,'\t')
gene = t[0]
if gene in genes:
values = map(float,t[1:])
gene_data.append(values)
vals1 = gene_data[0]
vals2 = gene_data[1]
i=0
coincident=[]
for v1 in vals1:
v2 = vals2[i]
#print v1,v2
if v1>1 and v2>1:
coincident.append(i)
i+=1
i=0
population_db={}; coincident_db={}
for h in header:
population=string.split(h,':')[0]
if i in coincident:
try: coincident_db[population]+=1
except Exception: coincident_db[population]=1
try: population_db[population]+=1
except Exception: population_db[population]=1
i+=1
import mappfinder
final_population_percent=[]
for population in population_db:
d = population_db[population]
try: c = coincident_db[population]
except Exception: c = 0
N = float(len(header)) ### num all samples examined
R = float(len(coincident)) ### num all coincedent samples for the TFs
n = d ### num all samples in cluster
r = c ### num all coincident samples in cluster
try: z = mappfinder.zscore(r,n,N,R)
except Exception: z = 0
final_population_percent.append([population,str(c),str(d),str(float(c)/float(d)),str(z)])
if exportPairs:
for line in final_population_percent:
ea.write(string.join(line,'\t')+'\n')
ea.close()
else:
return final_population_percent
def extractFeatures(countinp,IGH_gene_file):
import export
ExonsPresent=False
igh_genes=[]
firstLine = True
for line in open(IGH_gene_file,'rU').xreadlines():
if firstLine: firstLine=False
else:
data = cleanUpLine(line)
gene = string.split(data,'\t')[0]
igh_genes.append(gene)
if 'counts.' in countinp:
feature_file = string.replace(countinp,'counts.','IGH.')
fe = export.ExportFile(feature_file)
firstLine = True
for line in open(countinp,'rU').xreadlines():
if firstLine:
fe.write(line)
firstLine=False
else:
feature_info = string.split(line,'\t')[0]
gene = string.split(feature_info,':')[0]
if gene in igh_genes:
fe.write(line)
fe.close()
def filterForJunctions(countinp):
import export
ExonsPresent=False
igh_genes=[]
firstLine = True
count = 0
if 'counts.' in countinp:
feature_file = countinp[:-4]+'-output.txt'
fe = export.ExportFile(feature_file)
firstLine = True
for line in open(countinp,'rU').xreadlines():
if firstLine:
fe.write(line)
firstLine=False
else:
feature_info = string.split(line,'\t')[0]
junction = string.split(feature_info,'=')[0]
if '-' in junction:
fe.write(line)
count+=1
fe.close()
print count
def countIntronsExons(filename):
import export
exon_db={}
intron_db={}
firstLine = True
last_transcript=None
for line in open(filename,'rU').xreadlines():
if firstLine:
firstLine=False
else:
line = line.rstrip()
t = string.split(line,'\t')
transcript = t[-1]
chr = t[1]
strand = t[2]
start = t[3]
end = t[4]
exon_db[chr,start,end]=[]
if transcript==last_transcript:
if strand == '1':
intron_db[chr,last_end,start]=[]
else:
intron_db[chr,last_start,end]=[]
last_end = end
last_start = start
last_transcript = transcript
print len(exon_db)+1, len(intron_db)+1
def importGeneList(gene_list_file,n=20):
genesets=[]
genes=[]
for line in open(gene_list_file,'rU').xreadlines():
gene = line.rstrip()
gene = string.split(gene,'\t')[0]
genes.append(gene)
if len(genes)==n:
genesets.append(genes)
genes=[]
if len(genes)>0 and len(genes)<(n+1):
genes+=(n-len(genes))*[gene]
genesets.append(genes)
return genesets
def simpleListImport(filename):
genesets=[]
genes=[]
for line in open(filename,'rU').xreadlines():
gene = line.rstrip()
gene = string.split(gene,'\t')[0]
genes.append(gene)
return genes
def customClean(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow = False
#print len(t)
ea.write(string.join(['UID']+t,'\t')+'\n')
else:
if ';' in t[0]:
uid = string.split(t[0],';')[0]
else:
uid = t[0]
values = map(lambda x: float(x),t[1:])
values.sort()
if values[3]>=1:
ea.write(string.join([uid]+t[1:],'\t')+'\n')
ea.close()
def MakeJunctionFasta(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'.fasta'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
probeset, seq = string.split(data,'\t')[:2]
ea.write(">"+probeset+'\n')
ea.write(string.upper(seq)+'\n')
ea.close()
def ToppGeneFilter(filename):
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid('Hs',('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow = False
#print len(t)
ea.write(string.join(['Ensembl\t\tCategory'],'\t')+'\n')
else:
symbol = t[1]; category = t[3]
symbol = symbol[0]+string.lower(symbol[1:]) ### Mouse
category = category[:100]
if symbol in symbol_to_gene:
ensembl = symbol_to_gene[symbol][0]
ea.write(string.join([ensembl,symbol,category],'\t')+'\n')
ea.close()
def CountKallistoAlignedJunctions(filename):
fn = filepath(filename)
firstRow=True
#filename = filename[:-4]+'.fasta'
ea = export.ExportFile(filename)
found = False
counts=0
unique={}
ea = export.ExportFile(filename[:-4]+'-Mpo.txt')
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if 'ENS' in line and 'JUNC1201' in line:
ea.write(line)
unique[t[0]]=[]
counts+=1
print counts, len(unique)
ea.close()
def filterRandomFile(filename,col1,col2):
fn = filepath(filename)
firstRow=True
counts=0
ea = export.ExportFile(filename[:-4]+'-columns.txt')
for line in open(fn,'rU').xreadlines():
if line[0]!='#':
data = line.rstrip()
t = string.split(data,',')
#print t[col1-1]+'\t'+t[col2-1];sys.exit()
if ' ' in t[col2-1]:
t[col2-1] = string.split(t[col2-1],' ')[2]
ea.write(t[col1-1]+'\t'+t[col2-1]+'\n')
counts+=1
#print counts, len(unique)
ea.close()
def getBlockExonPositions():
fn = '/Users/saljh8/Desktop/Code/AltAnalyze/AltDatabase/EnsMart65/ensembl/Mm/Mm_Ensembl_exon.txt'
firstRow=True
filename = fn[:-4]+'.block.txt'
ea = export.ExportFile(filename)
found = False
lines=0
exon_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
gene,exonid,chromosome,strand,start,stop, a, b, c, d = string.split(data,'\t')
exonid = string.split(exonid,'.')[0]
uid = gene+':'+exonid
if lines>0:
try:
exon_db[uid,strand].append(int(start))
exon_db[uid,strand].append(int(stop))
except Exception:
exon_db[uid,strand] = [int(start)]
exon_db[uid,strand].append(int(stop))
lines+=1
print len(exon_db)
for (uid,strand) in exon_db:
exon_db[uid,strand].sort()
if strand == '-':
exon_db[uid,strand].reverse()
start = str(exon_db[uid,strand][0])
stop = str(exon_db[uid,strand][1])
coord = [start,stop]; coord.sort()
ea.write(uid+'\t'+strand+'\t'+coord[0]+'\t'+coord[1]+'\n')
ea.close()
def combineVariants(fn):
firstRow=True
filename = fn[:-4]+'.gene-level.txt'
ea = export.ExportFile(filename)
found = False
lines=0
gene_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
gene = t[9]
if lines == 0:
header = ['UID']+t[16:]
header = string.join(header,'\t')+'\n'
ea.write(header)
lines+=1
else:
var_calls = map(float,t[16:])
if gene in gene_db:
count_sum_array = gene_db[gene]
count_sum_array = [sum(value) for value in zip(*[count_sum_array,var_calls])]
gene_db[gene] = count_sum_array
else:
gene_db[gene] = var_calls
for gene in gene_db:
var_calls = gene_db[gene]
var_calls2=[]
for i in var_calls:
if i==0: var_calls2.append('0')
else: var_calls2.append('1')
ea.write(gene+'\t'+string.join(var_calls2,'\t')+'\n')
ea.close()
def compareFusions(fn):
firstRow=True
filename = fn[:-4]+'.matrix.txt'
ea = export.ExportFile(filename)
found = False
lines=0
fusion_db={}
sample_list=[]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if 'Gene_Fusion_Pair' in line:
headers = string.split(data,'\t')[1:]
try:
sample, fusion = string.split(data,'\t')
try: fusion_db[fusion].append(sample)
except Exception: fusion_db[fusion] = [sample]
if sample not in sample_list: sample_list.append(sample)
except Exception:
t = string.split(data,'\t')
fusion = t[0]
index=0
for i in t[1:]:
if i=='1':
sample = headers[index]
try: fusion_db[fusion].append(sample)
except Exception: fusion_db[fusion] = [sample]
if sample not in sample_list: sample_list.append(sample)
index+=1
fusion_db2=[]
for fusion in fusion_db:
samples = fusion_db[fusion]
samples2=[]
for s in sample_list:
if s in samples: samples2.append('1')
else: samples2.append('0')
fusion_db[fusion] = samples2
ea.write(string.join(['Fusion']+sample_list,'\t')+'\n')
for fusion in fusion_db:
print [fusion]
ea.write(fusion+'\t'+string.join(fusion_db[fusion],'\t')+'\n')
ea.close()
def customCleanSupplemental(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
line = string.split(data,', ')
gene_data=[]
for gene in line:
gene = string.replace(gene,' ','')
if '/' in gene:
genes = string.split(gene,'/')
gene_data.append(genes[0])
for i in genes[1:]:
gene_data.append(genes[0][:len(genes[1])*-1]+i)
elif '(' in gene:
genes = string.split(gene[:-1],'(')
gene_data+=genes
else:
gene_data.append(gene)
ea.write(string.join(gene_data,' ')+'\n')
ea.close()
def customCleanBinomial(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
from stats_scripts import statistics
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
headers = t
firstRow = False
ea.write(string.join(['uid']+headers,'\t')+'\n')
else:
gene = t[0]
values = map(float,t[1:])
min_val = abs(min(values))
values = map(lambda x: x+min_val,values)
values = map(str,values)
ea.write(string.join([gene]+values,'\t')+'\n')
ea.close()
class MarkerFinderInfo:
def __init__(self,gene,rho,tissue):
self.gene = gene
self.rho = rho
self.tissue = tissue
def Gene(self): return self.gene
def Rho(self): return self.rho
def Tissue(self): return self.tissue
def ReceptorLigandCellInteractions(species,lig_receptor_dir,cell_type_gene_dir):
ligand_db={}
receptor_db={}
fn = filepath(lig_receptor_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
ligand,receptor = string.split(data,'\t')
if species=='Mm':
ligand = ligand[0]+string.lower(ligand[1:])
receptor = receptor[0]+string.lower(receptor[1:])
try: ligand_db[ligand].apepnd(receptor)
except Exception: ligand_db[ligand] = [receptor]
try: receptor_db[receptor].append(ligand)
except Exception: receptor_db[receptor] = [ligand]
firstRow=True
filename = cell_type_gene_dir[:-4]+'-new.txt'
ea = export.ExportFile(filename)
found = False
cell_specific_ligands={}
cell_specific_receptor={}
fn = filepath(cell_type_gene_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
gene, rho, tissue, notes, order = string.split(data,'\t')
mf = MarkerFinderInfo(gene, rho, tissue)
if gene in ligand_db:
cell_specific_ligands[gene]=mf
if gene in receptor_db:
cell_specific_receptor[gene]=mf
ligand_receptor_pairs=[]
for gene in cell_specific_ligands:
receptors = ligand_db[gene]
for receptor in receptors:
if receptor in cell_specific_receptor:
rmf = cell_specific_receptor[receptor]
lmf = cell_specific_ligands[gene]
gene_data = [gene,lmf.Tissue(),lmf.Rho(),receptor,rmf.Tissue(),rmf.Rho()]
pair = gene,receptor
if pair not in ligand_receptor_pairs:
ea.write(string.join(gene_data,'\t')+'\n')
ligand_receptor_pairs.append(pair)
for receptor in cell_specific_receptor:
ligands = receptor_db[receptor]
for gene in ligands:
if gene in cell_specific_ligands:
rmf = cell_specific_receptor[receptor]
lmf = cell_specific_ligands[gene]
gene_data = [gene,lmf.Tissue(),lmf.Rho(),receptor,rmf.Tissue(),rmf.Rho()]
pair = gene,receptor
if pair not in ligand_receptor_pairs:
ea.write(string.join(gene_data,'\t')+'\n')
ligand_receptor_pairs.append(pair)
ea.close()
def findReciprocal(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-filtered.txt'
ea = export.ExportFile(filename)
found = False
gene_ko={}; gene_oe={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow = False
headers={}
TFs={}
i=0
for v in t[1:]:
TF,direction = string.split(v,'-')
headers[i]=TF,direction,v
i+=1
if v not in TFs:
f = filename[:-4]+'-'+v+'-up.txt'
tea = export.ExportFile(f)
TFs[v+'-up']=tea
tea.write('GeneID\tEn\n')
f = filename[:-4]+'-'+v+'-down.txt'
tea = export.ExportFile(f)
TFs[v+'-down']=tea
tea.write('GeneID\tEn\n')
else:
values = map(float,t[1:])
gene = t[0]
i=0
for v in values:
TF,direction,name = headers[i]
if 'KO' in direction:
if v > 1:
gene_ko[gene,TF,1]=[]
tea = TFs[name+'-up']
tea.write(gene+'\tEn\n')
else:
gene_ko[gene,TF,-1]=[]
tea = TFs[name+'-down']
tea.write(gene+'\tEn\n')
if 'OE' in direction:
if v > 1:
gene_oe[gene,TF,1]=[]
tea = TFs[name+'-up']
tea.write(gene+'\tEn\n')
else:
gene_oe[gene,TF,-1]=[]
tea = TFs[name+'-down']
tea.write(gene+'\tEn\n')
i+=1
print len(gene_oe)
for (gene,TF,direction) in gene_oe:
alt_dir=direction*-1
if (gene,TF,alt_dir) in gene_ko:
ea.write(string.join([TF,gene,str(direction)],'\t')+'\n')
ea.close()
for TF in TFs:
TFs[TF].close()
def effectsPrioritization(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
from stats_scripts import statistics
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
headers = t[1:]
firstRow = False
else:
gene = t[0]
values = map(float,t[1:])
max_val = abs(max(values))
max_header = headers[values.index(max_val)]
ea.write(gene+'\t'+max_header+'\t'+str(max_val)+'\n')
ea.close()
def simpleCombine(folder):
filename = folder+'/combined/combined.txt'
ea = export.ExportFile(filename)
headers=['UID']
data_db={}
files = UI.read_directory(folder)
for file in files: #:70895507-70895600
if '.txt' in file:
fn = filepath(folder+'/'+file)
print fn
firstRow=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
for i in t[1:]:
headers.append(i+'.'+file[:-4])
firstRow = False
else:
gene = t[0]
try: data_db[gene]+=t[1:]
except Exception: data_db[gene] = t[1:]
len_db={}
ea.write(string.join(headers,'\t')+'\n')
for gene in data_db:
if len(data_db[gene])==(len(headers)-1):
values = map(float,data_db[gene])
count=0
for i in values:
if i>0.9: count+=1
if count>7:
ea.write(string.join([gene]+data_db[gene],'\t')+'\n')
len_db[len(data_db[gene])]=[]
print len(len_db)
for i in len_db:
print i
ea.close()
def simpleCombineFiles(folder, elite_output=True, uniqueOnly=False):
filename = folder + '/combined/combined.txt'
ea = export.ExportFile(filename)
files = UI.read_directory(folder)
unique_entries = []
firstRow = True
for file in files:
if '.txt' in file:
fn = filepath(folder + '/' + file)
firstRow = True
for line in open(fn, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if elite_output:
if firstRow:
t = 'UID\tSystemCode\tCategory'
ea.write(t + '\n')
firstRow = False
elif uniqueOnly:
if t[0] not in unique_entries:
ea.write(t[0] + '\t' + t[0] + '\t\n')
unique_entries.append(t[0])
else:
ea.write(string.join([string.split(t[0], '|')[0], 'Ae', string.replace(file[:-4], 'PSI.', '')], '\t') + '\n')
elif firstRow:
t.append('Comparison')
ea.write(string.join(t, '\t') + '\n')
firstRow = False
else:
t.append(file[:-4])
ea.write(string.join(t, '\t') + '\n')
ea.close()
def simpleCombineBedFiles(folder):
filename = folder + '/combined/annotations.bed'
ea = export.ExportFile(filename)
files = UI.read_directory(folder)
for file in files:
if '.bed' in file:
fn = filepath(folder + '/' + file)
for line in open(fn, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
t[3]+=';'+file[5:-4]
ea.write(string.join(t, '\t') + '\n')
ea.close()
def advancedCombineBedFiles(folder):
filename = folder + '/combined/annotations.bed'
ea = export.ExportFile(filename)
files = UI.read_directory(folder)
annotations=[]
for file in files:
if '.bed' in file:
fn = filepath(folder + '/' + file)
for line in open(fn, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
t[3]+=';'+file[5:-4]
uid = (t[0],int(t[1]),int(t[2]))
#ea.write(string.join(t, '\t') + '\n')
ea.close()
def evaluateMultiLinRegulatoryStructure(all_genes_TPM,MarkerFinder,SignatureGenes,state,query=None):
"""Predict multi-lineage cells and their associated coincident lineage-defining TFs"""
ICGS_State_as_Row = True
### Import all genes with TPM values for all cells
matrix, column_header, row_header, dataset_name, group_db = importData(all_genes_TPM)
group_index={}
all_indexes=[]
for sampleName in group_db:
ICGS_state = group_db[sampleName][0]
try: group_index[ICGS_state].append(column_header.index(sampleName))
except Exception: group_index[ICGS_state] = [column_header.index(sampleName)]
all_indexes.append(column_header.index(sampleName))
for ICGS_state in group_index:
group_index[ICGS_state].sort()
all_indexes.sort()
def importGeneLists(fn):
genes={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
gene,cluster = string.split(data,'\t')[0:2]
genes[gene]=cluster
return genes
def importMarkerFinderHits(fn):
genes={}
skip=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if skip: skip=False
else:
gene,symbol,rho,ICGS_State = string.split(data,'\t')
#if ICGS_State!=state and float(rho)>0.0:
if float(rho)>0.0:
genes[gene]=float(rho),ICGS_State ### Retain all population specific genes (lax)
genes[symbol]=float(rho),ICGS_State
return genes
def importQueryDataset(fn):
matrix, column_header, row_header, dataset_name, group_db = importData(fn)
return matrix, column_header, row_header, dataset_name, group_db
signatureGenes = importGeneLists(SignatureGenes)
markerFinderGenes = importMarkerFinderHits(MarkerFinder)
#print len(signatureGenes),len(markerFinderGenes)
### Determine for each gene, its population frequency per cell state
index=0
expressedGenesPerState={}
def freqCutoff(x,cutoff):
if x>cutoff: return 1 ### minimum expression cutoff
else: return 0
for row in matrix:
ICGS_state_gene_frq={}
gene = row_header[index]
for ICGS_state in group_index:
state_values = map(lambda i: row[i],group_index[ICGS_state])
def freqCheck(x):
if x>1: return 1 ### minimum expression cutoff
else: return 0
expStateCells = sum(map(lambda x: freqCheck(x),state_values))
statePercentage = (float(expStateCells)/len(group_index[ICGS_state]))
ICGS_state_gene_frq[ICGS_state] = statePercentage
multilin_frq = ICGS_state_gene_frq[state]
datasets_values = map(lambda i: row[i],all_indexes)
all_cells_frq = sum(map(lambda x: freqCheck(x),datasets_values))/(len(datasets_values)*1.0)
all_states_frq = map(lambda x: ICGS_state_gene_frq[x],ICGS_state_gene_frq)
all_states_frq.sort() ### frequencies of all non-multilin states
rank = all_states_frq.index(multilin_frq)
states_expressed = sum(map(lambda x: freqCutoff(x,0.5),all_states_frq))/(len(all_states_frq)*1.0)
if multilin_frq > 0.25 and rank>0: #and states_expressed<0.75 #and all_cells_frq>0.75
if 'Rik' not in gene and 'Gm' not in gene:
if gene in signatureGenes:# and gene in markerFinderGenes:
if ICGS_State_as_Row:
ICGS_State = signatureGenes[gene]
if gene in markerFinderGenes:
if ICGS_State_as_Row == False:
rho, ICGS_State = markerFinderGenes[gene]
else:
rho, ICGS_Cell_State = markerFinderGenes[gene]
score = int(rho*100*multilin_frq)*(float(rank)/len(all_states_frq))
try: expressedGenesPerState[ICGS_State].append((score,gene))
except Exception: expressedGenesPerState[ICGS_State]=[(score,gene)] #(rank*multilin_frq)
index+=1
if query!=None:
matrix, column_header, row_header, dataset_name, group_db = importQueryDataset(query)
createPseudoCell=True
### The expressedGenesPerState defines genes and modules co-expressed in the multi-Lin
### Next, find the cells that are most frequent in mulitple states
representativeMarkers={}
for ICGS_State in expressedGenesPerState:
expressedGenesPerState[ICGS_State].sort()
expressedGenesPerState[ICGS_State].reverse()
if '1Multi' not in ICGS_State:
markers = expressedGenesPerState[ICGS_State][:5]
print ICGS_State,":",string.join(map(lambda x: x[1],list(markers)),', ')
if createPseudoCell:
for gene in markers:
def getBinary(x):
if x>1: return 1
else: return 0
if gene[1] in row_header: ### Only for query datasets
row_index = row_header.index(gene[1])
binaryValues = map(lambda x: getBinary(x), matrix[row_index])
#if gene[1]=='S100a8': print binaryValues;sys.exit()
try: representativeMarkers[ICGS_State].append(binaryValues)
except Exception: representativeMarkers[ICGS_State] = [binaryValues]
else:
representativeMarkers[ICGS_State]=markers[0][-1]
#int(len(markers)*.25)>5:
#print ICGS_State, markers
#sys.exit()
for ICGS_State in representativeMarkers:
if createPseudoCell:
signature_values = representativeMarkers[ICGS_State]
signature_values = [int(numpy.median(value)) for value in zip(*signature_values)]
representativeMarkers[ICGS_State] = signature_values
else:
gene = representativeMarkers[ICGS_State]
row_index = row_header.index(gene)
gene_values = matrix[row_index]
representativeMarkers[ICGS_State] = gene_values
### Determine for each gene, its population frequency per cell state
expressedStatesPerCell={}
for ICGS_State in representativeMarkers:
gene_values = representativeMarkers[ICGS_State]
index=0
for cell in column_header:
log2_tpm = gene_values[index]
if log2_tpm>=1:
try: expressedStatesPerCell[cell].append(ICGS_State)
except Exception: expressedStatesPerCell[cell] = [ICGS_State]
index+=1
cell_mutlilin_ranking=[]
for cell in expressedStatesPerCell:
lineageCount = expressedStatesPerCell[cell]
cell_mutlilin_ranking.append((len(lineageCount),cell))
cell_mutlilin_ranking.sort()
cell_mutlilin_ranking.reverse()
for cell in cell_mutlilin_ranking:
print cell[0], cell[1], string.join(expressedStatesPerCell[cell[1]],'|')
def compareGenomicLocationAndICGSClusters():
species = 'Mm'
array_type = 'RNASeq'
from build_scripts import EnsemblImport
gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,array_type,'key_by_array')
markerfinder = '/Users/saljh8/Desktop/Old Mac/Desktop/Grimes/Kallisto/ExpressionOutput/MarkerFinder/AllCorrelationsAnnotated-ProteinCodingOnly.txt'
eo = export.ExportFile(markerfinder[:-4]+'-bidirectional_promoters.txt')
firstRow=True
chr_cellTypeSpecific={}
for line in open(markerfinder,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
symbol = t[1]
ensembl = t[0]
try: rho = float(t[6])
except Exception: pass
cellType = t[7]
if firstRow:
firstRow = False
else:
if ensembl in gene_location_db and rho>0.2:
chr,strand,start,end = gene_location_db[ensembl]
start = int(start)
end = int(end)
#region = start[:-5]
try:
db = chr_cellTypeSpecific[chr,cellType]
try: db[strand].append([start,end,symbol,ensembl])
except Exception: db[strand] = [[start,end,symbol,ensembl]]
except Exception:
db={}
db[strand] = [[start,end,symbol,ensembl]]
chr_cellTypeSpecific[chr,cellType] = db
bidirectional={}
eo.write(string.join(['CellType','Chr','Ensembl1','Symbol1','Start1','End1','Strand1','Ensembl2','Symbol2','Start2','End2','Strand2'],'\t')+'\n')
for (chr,cellType) in chr_cellTypeSpecific:
db = chr_cellTypeSpecific[chr,cellType]
if len(db)>1: ### hence two strands
for (start,end,symbol,ens) in db['+']:
for (start2,end2,symbol2,ens2) in db['-']:
if abs(start-end2)<100000 and start>end2:
eo.write(string.join([cellType,chr,ens,symbol,str(start),str(end),'+',ens2,symbol2,str(end2),str(start2),'-'],'\t')+'\n')
try: bidirectional[chr,cellType].append([start,end,symbol,ens,start2,end2,symbol2,ens2])
except Exception: bidirectional[chr,cellType] = [[start,end,symbol,ens,start2,end2,symbol2,ens2]]
eo.close()
def filterCountsFile(filename):
fn = filepath(filename)
firstRow=True
def countif(value,cutoff=9):
if float(value)>cutoff: return 1
else: return 0
header = True
unique_genes = {}
ea = export.ExportFile(filename[:-4]+'-filtered.txt')
for line in open(fn,'rU').xreadlines():
if header:
header = False
ea.write(line)
else:
data = line.rstrip()
t = string.split(data,'\t')
gene = string.split(t[0],':')[0]
unique_genes[gene]=[]
expressedSamples = map(countif,t[1:])
if sum(expressedSamples)>2:
ea.write(line)
ea.close()
print len(unique_genes),'unique genes.'
def filterPSIValues(filename):
fn = filepath(filename)
firstRow=True
header = True
rows=0
filtered=0
new_file = filename[:-4]+'-75p.txt'
new_file_clust = new_file[:-4]+'-clustID.txt'
ea = export.ExportFile(new_file)
eac = export.ExportFile(new_file_clust)
added=[]
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
header = False
t = [t[1]]+t[8:]
header_length = len(t)-1
minimum_values_present = int(0.75*int(header_length))
not_detected = header_length-minimum_values_present
new_line = string.join(t,'\t')+'\n'
ea.write(new_line)
else:
cID = t[5]
t = [t[1]]+t[8:]
missing_values_at_the_end = (header_length+1)-len(t)
missing = missing_values_at_the_end+t.count('')
if missing<not_detected:
#if cID not in added:
added.append(cID)
new_line = string.join(t,'\t')+'\n'
ea.write(new_line)
eac.write(t[0]+'\t'+cID+'\n')
filtered+=1
rows+=1
print rows, filtered
ea.close()
eac.close()
#removeRedundantCluster(new_file,new_file_clust)
def removeRedundantCluster(filename,clusterID_file):
from scipy import stats
import ExpressionBuilder
sort_col=0
export_count=0
### Sort the filtered PSI model by gene name
ExpressionBuilder.exportSorted(filename, sort_col, excludeHeader=True)
new_file = filename[:-4]+'-unique.txt'
ea = export.ExportFile(new_file)
event_clusterID_db={}
for line in open(clusterID_file,'rU').xreadlines():
data = line.rstrip()
eventID,clusterID = string.split(data,'\t')
event_clusterID_db[eventID]=clusterID
def compareEvents(events_to_compare,export_count):
### This is where we compare the events and write out the unique entries
if len(events_to_compare)==1:
ea.write(events_to_compare[0][-1])
export_count+=1
else:
exclude={}
compared={}
for event1 in events_to_compare:
if event1[0] not in exclude:
ea.write(event1[-1])
exclude[event1[0]]=[]
export_count+=1
for event2 in events_to_compare:
if event2[0] not in exclude:
if event1[0] != event2[0] and (event1[0],event2[0]) not in compared:
uid1,values1,line1 = event1
uid2,values2,line2 = event2
coefr=numpy.ma.corrcoef(values1,values2)
#rho,p = stats.pearsonr(values1,values2)
rho = coefr[0][1]
if rho>0.6 or rho<-0.6:
exclude[event2[0]]=[]
compared[event1[0],event2[0]]=[]
compared[event2[0],event1[0]]=[]
for event in events_to_compare:
if event[0] not in exclude:
ea.write(event[-1]) ### write out the line
exclude.append(event[0])
export_count+=1
return export_count
header = True
rows=0
filtered=0
prior_cID = 0
events_to_compare=[]
for line in open(filename,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
ea.write(line)
header_row = t
header=False
else:
uid = t[0]
cID = event_clusterID_db[uid]
empty_offset = len(header_row)-len(t)
t+=['']*empty_offset
values = ['0.000101' if x=='' else x for x in t[1:]]
values = map(float,values)
values = numpy.ma.masked_values(values,0.000101)
if prior_cID==0: prior_cID = cID ### Occurs for the first entry
if cID == prior_cID:
### Replace empty values with 0
events_to_compare.append((uid,values,line))
else:
export_count = compareEvents(events_to_compare,export_count)
events_to_compare=[(uid,values,line)]
prior_cID = cID
if len(events_to_compare)>0: ### If the laster cluster set not written out yet
export_count = compareEvents(events_to_compare,export_count)
ea.close()
print export_count,'Non-redundant splice-events exported'
def correlateIsoformPSIvalues(isoform_data,psi_data,psi_annotations):
""" Determine if isoform predictions are valid, based on corresponding correlated PSI event """
from scipy import stats
import ExpressionBuilder
sort_col=0
export_count=0
new_file = isoform_data[:-4]+'-VerifiedEvents.txt'
print new_file
ea = export.ExportFile(new_file)
header = True
event_annotations = {}
for line in open(psi_annotations,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
proteinPrediction = t[5]
clusterID = t[7]
event = t[8]
eventType = t[10]
event_annotations[event] = proteinPrediction, clusterID, eventType
def importIsoData(filedir):
gene_to_event = {}
event_to_values = {}
header = True
for line in open(filedir,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
uid = t[0]
if header:
l = len(t)
header = False
else:
if ':' in uid:
gene = string.split(uid,':')[0]
else:
uids = string.split(uid,'-')
if len(uids)==2:
gene = uids[0]
else:
gene = string.join(uids[:-1],'-')
try: gene_to_event[gene].append(uid)
except: gene_to_event[gene] = [uid]
values = t[1:]
empty_offset = l-len(t)
t+=['']*empty_offset
values = ['0.000101' if x=='' else x for x in t[1:]]
values = map(float,values)
values = numpy.ma.masked_values(values,0.000101)
event_to_values[uid] = values
return gene_to_event, event_to_values
gene_to_isoform,isoform_to_values = importIsoData(isoform_data)
gene_to_event,event_to_values = importIsoData(psi_data)
print len(gene_to_isoform)
print len(gene_to_event)
common_genes = 0
for gene in gene_to_isoform:
if gene in gene_to_event:
common_genes+=1
print common_genes
export_count=0
gene_isoforms_confirmed = {}
event_type_confirmed = {}
event_protein_confirmed = {}
for gene in gene_to_event:
if gene in gene_to_isoform:
events = gene_to_event[gene]
isoforms = gene_to_isoform[gene]
for event in events:
psi_values = event_to_values[event]
for isoform in isoforms:
iso_values = isoform_to_values[isoform]
coefr=numpy.ma.corrcoef(iso_values,psi_values)
rho = coefr[0][1]
#print rho
#print event, isoform;sys.exit()
if rho>0.6 or rho<-0.6:
if rho>0.6: cor = '+'
else: cor = '-'
proteinPrediction, clusterID, eventType = event_annotations[event]
try:
if eventType not in event_type_confirmed[gene,isoform]:
event_type_confirmed[gene,isoform].append(eventType)
except: event_type_confirmed[gene,isoform] = [eventType]
if len(proteinPrediction)>1:
try:
if proteinPrediction not in event_protein_confirmed[gene,isoform]:
event_protein_confirmed[gene,isoform].append(proteinPrediction)
except: event_protein_confirmed[gene,isoform] = [proteinPrediction]
try: gene_isoforms_confirmed[gene,isoform].append(event+'('+cor+')')
except: gene_isoforms_confirmed[gene,isoform] = [event+'('+cor+')']
for (gene,isoform) in gene_isoforms_confirmed:
events = string.join(gene_isoforms_confirmed[(gene,isoform)],',')
eventTypes = string.join(event_type_confirmed[gene,isoform],'|')
try: proteinPredictions = string.join(event_protein_confirmed[gene,isoform],'|')
except: proteinPredictions = ''
try:
if eventTypes[0] == '|': eventTypes = eventTypes[1:]
if eventTypes[-1] == '|': eventTypes = eventTypes[:-1]
except: pass
try:
if proteinPredictions[0] == '|': proteinPredictions = proteinPredictions[1:]
if proteinPredictions[-1] == '|': proteinPredictions = proteinPredictions[:-1]
except: pass
ea.write(string.join([gene,isoform,events,eventTypes,proteinPredictions],'\t')+'\n')
export_count+=1
ea.close()
print export_count,'Correlated splicing events-isoforms reported'
def convertToGOElite(folder):
files = UI.read_directory(folder)
for file in files:
if '.txt' in file:
gene_count=0; up_count=0; down_count=0
new_filename = string.split(file[3:],"_")[0]+'.txt'
ea = export.ExportFile(folder+'/GO-Elite/'+new_filename)
fn = folder+'/'+file
ea.write('GeneID\tSystemCode\n')
firstLine = True
for line in open(fn,'rU').xreadlines():
if firstLine:
firstLine= False
continue
data = line.rstrip()
t = string.split(data,'\t')
if ':' in t[0]:
ea.write(string.split(t[0],':')[0]+'\tSy\n')
else:
gene_count+=1
if '-' in t[2]: down_count+=1
else: up_count+=1
ea.close()
print file,'\t',gene_count,'\t',up_count,'\t',down_count
def geneExpressionSummary(folder):
import collections
event_db = collections.OrderedDict()
groups_list=['']
files = UI.read_directory(folder)
for file in files:
if '.txt' in file and 'GE.' in file:
ls=[]
event_db[file[:-4]]=ls
groups_list.append(file[:-4])
fn = folder+'/'+file
firstLine = True
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
fold_index = t.index('LogFold')
firstLine= False
continue
uid = t[0]
if float(t[fold_index])>0:
fold_dir = 1
else:
fold_dir = -1
ls.append((uid,fold_dir))
for file in event_db:
print file,'\t',len(event_db[file])
def compareEventLists(folder):
import collections
event_db = collections.OrderedDict()
groups_list=['']
files = UI.read_directory(folder)
file_headers = {}
for file in files:
if '.txt' in file and 'PSI.' in file:
ls={}
event_db[file[:-4]]=ls
groups_list.append(file[:-4])
fn = folder+'/'+file
firstLine = True
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
file_headers[file[:-4]] = t ### Store the headers
cid = t.index('ClusterID')
try: event_index = t.index('Event-Direction')
except:
try: event_index = t.index('Inclusion-Junction') ### legacy
except: print file, 'Event-Direction error';sys.exit()
firstLine= False
continue
uid = t[0]
uid = string.split(uid,'|')[0]
#uid = t[cid]
if 'U2AF1-l' in file or 'U2AF1-E' in file:
if t[2] == "inclusion":
ls[(uid,t[event_index])]=t ### Keep the event data for output
else:
ls[(uid,t[event_index])]=t ### Keep the event data for output
def convertEvents(events):
opposite_events=[]
for (event,direction) in events:
if direction == 'exclusion':
direction = 'inclusion'
else:
direction = 'exclusion'
opposite_events.append((event,direction))
return opposite_events
ea1 = export.ExportFile(folder+'/overlaps-same-direction.txt')
ea2 = export.ExportFile(folder+'/overlaps-opposite-direction.txt')
ea3 = export.ExportFile(folder+'/concordance.txt')
#ea4 = export.ExportFile(folder+'/overlap-same-direction-events.txt')
ea1.write(string.join(groups_list,'\t')+'\n')
ea2.write(string.join(groups_list,'\t')+'\n')
ea3.write(string.join(groups_list,'\t')+'\n')
comparison_db={}
best_hits={}
for comparison1 in event_db:
events1 = event_db[comparison1]
hits1=[comparison1]
hits2=[comparison1]
hits3=[comparison1]
best_hits[comparison1]=[]
for comparison2 in event_db:
events2 = event_db[comparison2]
events3 = convertEvents(events2)
overlapping_events = list(set(events1).intersection(events2))
overlap = len(overlapping_events)
inverse_overlap = len(set(events1).intersection(events3)) ### Get opposite events
### Calculate ratios based on the size of the smaller set
min_events1 = min([len(events1),len(events2)])
min_events2 = min([len(events1),len(events3)])
denom = overlap+inverse_overlap
if denom == 0: denom = 0.00001
#comparison_db[comparison1,comparison2]=overlap
if min_events1 == 0: min_events1 = 1
if (overlap+inverse_overlap)<20:
hits1.append('0.5')
hits2.append('0.5')
hits3.append('0.5|0.5')
else:
hits1.append(str((1.00*overlap)/min_events1))
hits2.append(str((1.00*inverse_overlap)/min_events1))
hits3.append(str(1.00*overlap/denom)+'|'+str(1.00*inverse_overlap/denom)+':'+str(overlap+inverse_overlap))
if 'Leu' not in comparison2:
comp_name = string.split(comparison2,'_vs')[0]
best_hits[comparison1].append([abs(1.00*overlap/denom),'cor',comp_name])
best_hits[comparison1].append([abs(1.00*inverse_overlap/denom),'anti',comp_name])
if comparison1 != comparison2:
if len(overlapping_events)>0:
#ea4.write(string.join(['UID',comparison1]+file_headers[comparison1]+[comparison2]+file_headers[comparison2],'\t')+'\n')
pass
overlapping_events.sort()
for event in overlapping_events:
vals = string.join([event[0],comparison1]+event_db[comparison1][event]+[comparison2]+event_db[comparison2][event],'\t')
#ea4.write(vals+'\n')
pass
ea1.write(string.join(hits1,'\t')+'\n')
ea2.write(string.join(hits2,'\t')+'\n')
ea3.write(string.join(hits3,'\t')+'\n')
ea1.close()
ea2.close()
ea3.close()
#ea4.close()
for comparison in best_hits:
best_hits[comparison].sort()
best_hits[comparison].reverse()
hits = best_hits[comparison][:10]
hits2=[]
for (score,dir,comp) in hits:
h = str(score)[:4]+'|'+dir+'|'+comp
hits2.append(h)
print comparison,'\t',string.join(hits2,', ')
def convertGroupsToBinaryMatrix(groups_file,sample_order,cellHarmony=False):
eo = export.ExportFile(groups_file[:-4]+'-matrix.txt')
print groups_file[:-4]+'-matrix.txt'
firstRow=True
samples = []
### Import a file with the sample names in the groups file in the correct order
for line in open(sample_order,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if 'row_clusters-flat' in t:
samples=[]
samples1 = t[2:]
for name in samples1:
if ':' in name:
group,name = string.split(name,':')
samples.append(name)
if cellHarmony==False:
break
elif 'column_clusters-flat' in t and cellHarmony:
clusters = t[2:]
elif groups_file == sample_order:
samples.append(t[0])
elif firstRow:
samples = t[1:]
firstRow=False
### Import a groups file
import collections
sample_groups = collections.OrderedDict()
for line in open(groups_file,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
sample, groupNum, groupName = t[:3]
if cellHarmony == False: ### JUST USE THE PROVIDED GROUPS FOR SAMPLES FOUND IN BOTH FILES
if sample in samples:
si=samples.index(sample) ### Index of the sample
try: sample_groups[groupName][si] = '1' ### set that sample to 1
except Exception:
sample_groups[groupName] = ['0']*len(samples)
sample_groups[groupName][si] = '1' ### set that sample to 1
else: ### JUST GRAB THE GROUP NAMES FOR THE SAMPLE GROUPS NOT THE SAMPES
sample_groups[groupNum]=groupName
if cellHarmony:
i=0
for sample in samples1:
cluster = clusters[i]
group_name = sample_groups[cluster]
eo.write(sample+'\t'+cluster+'\t'+group_name+'\n')
i+=1
eo.close()
else:
eo.write(string.join(['GroupName']+samples,'\t')+'\n')
for group in sample_groups:
eo.write(string.join([group]+sample_groups[group],'\t')+'\n')
eo.close()
def returnIntronJunctionRatio(counts_file,species = 'Mm'):
eo = export.ExportFile(counts_file[:-4]+'-intron-ratios.txt')
### Import a groups file
header=True
prior_gene=[]
exon_junction_values=[]
intron_junction_values=[]
eoi = export.ExportFile(counts_file[:-4]+'-intron-ratios-gene.txt')
rows=0
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
def logratio(list):
try: return list[0]/list[1]
except Exception: return 0
for line in open(counts_file,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
junctionID = t[0]
if header:
eoi.write(line)
samples = t[1:]
#zero_ref =[0]*len(samples)
global_intron_ratios={}
i=0
for val in samples:
global_intron_ratios[i]=[]
i+=1
header = False
continue
else:
uid,coords = string.split(junctionID,'=')
genes = string.split(uid,':') ### can indicate trans-splicing
if len(genes)>2: trans_splicing = True
else: trans_splicing = False
coords = string.split(coords,':')[1]
coords = string.split(coords,'-')
coords = map(int,coords)
coord_diff = abs(coords[1]-coords[0])
#ENSMUSG00000027770:I23.1-E24.1=chr3:62470748-62470747
gene = string.split(junctionID,':')[0]
rows+=1
if rows == 1:
prior_gene = gene
if gene != prior_gene:
#print gene
### merge all of the gene level counts for all samples
if len(intron_junction_values)==0:
#global_intron_ratios = [sum(value) for value in zip(*[global_intron_ratios,zero_ref])]
pass
else:
intron_junction_values_original = list(intron_junction_values)
exon_junction_values_original = list(exon_junction_values)
intron_junction_values = [sum(i) for i in zip(*intron_junction_values)]
exon_junction_values = [sum(i) for i in zip(*exon_junction_values)]
intron_ratios = [logratio(value) for value in zip(*[intron_junction_values,exon_junction_values])]
#if sum(intron_ratios)>3:
intron_ratios2=[]
if prior_gene in gene_to_symbol:
symbol = gene_to_symbol[prior_gene][0]
else:
symbol = prior_gene
i=0
#"""
if symbol == 'Pi4ka':
print samples[482:487]
for x in exon_junction_values_original:
print x[482:487]
print exon_junction_values[482:487]
print intron_ratios[482:487]
#"""
for val in intron_ratios:
if exon_junction_values[i]>9:
if val>0:
### stringent requirement - make sure it's not just a few reads
if intron_junction_values[i]>9:
intron_ratios2.append(val)
else:
intron_ratios2.append(0)
else:
intron_ratios2.append(0)
else:
"""
if val>0:
print val
print intron_junction_values
print exon_junction_values;sys.exit()"""
intron_ratios2.append('')
i+=1
eoi.write(string.join([symbol]+map(str,intron_ratios2),'\t')+'\n')
i = 0
for val in intron_ratios:
if exon_junction_values[i]!=0: ### Only consider values with a non-zero denominator
global_intron_ratios[i].append(intron_ratios[i])
i+=1
exon_junction_values = []
intron_junction_values = []
prior_gene = gene
values = map(float,t[1:])
if 'I' in junctionID and '_' not in junctionID and coord_diff==1 and trans_splicing == False:
intron_junction_values.append(values)
exon_junction_values.append(values)
elif trans_splicing == False:
exon_junction_values.append(values)
print rows, 'processed'
import numpy
i=0; global_intron_ratios_values=[]
for val in samples:
global_intron_ratios_values.append(100*numpy.mean(global_intron_ratios[i])) ### list of lists
i+=1
eo.write(string.join(['UID']+samples,'\t')+'\n')
eo.write(string.join(['Global-Intron-Retention-Ratio']+map(str,global_intron_ratios_values),'\t')+'\n')
eo.close()
eoi.close()
def convertSymbolLog(input_file,ensembl_symbol,species=None,logNormalize=True):
gene_symbol_db={}
try:
for line in open(ensembl_symbol,'rU').xreadlines():
data = cleanUpLine(line)
ensembl,symbol = string.split(data,'\t')
gene_symbol_db[ensembl]=symbol
except:
pass
if species != None and len(gene_symbol_db)==0:
import gene_associations
gene_symbol_db = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
for i in gene_symbol_db:
gene_symbol_db[i] = gene_symbol_db[i][0]
#print [i], gene_to_symbol_db[i];sys.exit()
convert = True
if logNormalize:
eo = export.ExportFile(input_file[:-4]+'-log2.txt')
else:
eo = export.ExportFile(input_file[:-4]+'-symbol.txt')
header=0
added_symbols=[]
not_found=[]
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
gene = values[0]
if header == 0:
#eo.write(line)
data = cleanUpLine(line)
headers = []
values = string.split(data,'\t')
for v in values:
if "exp." in v:
headers.append(string.split(v,'.exp.')[0])
else:
headers.append(v)
eo.write(string.join(headers,'\t')+'\n')
header+=1
elif 'column' in values[0]:
eo.write(line)
else:
header +=1
if gene in gene_symbol_db:
symbol = gene_symbol_db[gene]
if symbol not in added_symbols:
added_symbols.append(symbol)
if logNormalize:
values = map(lambda x: math.log(float(x)+1,2),values[1:])
if max(values)> 0.5:
values = map(lambda x: str(x)[:5],values)
eo.write(string.join([symbol]+values,'\t')+'\n')
else:
max_val = max(map(float,values[1:]))
if max_val>0.5:
eo.write(string.join([symbol]+values[1:],'\t')+'\n')
elif convert==False and header>1:
values = map(lambda x: math.log(float(x)+1,2),values[1:])
if max(values)> 0.5:
values = map(lambda x: str(x)[:5],values)
eo.write(string.join([gene]+values,'\t')+'\n')
else:
not_found.append(gene)
print len(not_found),not_found[:10]
eo.close()
def convertXenaBrowserIsoformDataToStandardRatios(input_file):
eo = open(input_file[:-4]+'-log2.txt','w')
header=0
count=0
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
uid = string.split(values[0],'.')[0]
isoform = values[0]
if header == 0:
eo.write(line)
header +=1
else:
values = map(lambda x: math.pow(2,float(x)),values[1:]) # convert list out of log space
values = map(lambda x: math.log(float(x)+1,2),values) # convert to value+1 log2
def percentExp(x):
if x>1: return 1
else: return 0
counts = map(lambda x: percentExp(x),values) # find how many values > 1
if sum(counts)/(len(values)*1.000)>0.1: # only write out genes with >10% of values > 1
values = map(str,values)
values = string.join([uid]+values,'\t')
eo.write(values+'\n')
count+=1
eo.close()
print count,'genes written'
def outputForGOElite(folds_dir):
matrix, column_header, row_header, dataset_name, group_db = importData(folds_dir,Normalize=False)
matrix = zip(*matrix) ### transpose
ci=0
root_dir = findParentDir(folds_dir)
for group_data in matrix:
group_name = column_header[ci]
eo = export.ExportFile(root_dir+'/folds/'+group_name+'.txt')
gi=0
eo.write('geneID'+'\tSy\t'+'log2-fold'+'\n')
for fold in group_data:
gene = row_header[gi]
if fold>0:
eo.write(gene+'\tSy\t'+str(fold)+'\n')
gi+=1
eo.close()
ci+=1
def transposeMatrix(input_file):
arrays=[]
eo = export.ExportFile(input_file[:-4]+'-transposed.txt')
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
arrays.append(values)
t_arrays = zip(*arrays)
for t in t_arrays:
eo.write(string.join(t,'\t')+'\n')
eo.close()
def simpleStatsSummary(input_file):
cluster_counts={}
header=True
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
if header:
header = False
else:
sample,cluster,counts = string.split(data,'\t')
try: cluster_counts[cluster].append(float(counts))
except Exception: cluster_counts[cluster]=[float(counts)]
for cluster in cluster_counts:
avg = statistics.avg(cluster_counts[cluster])
stdev = statistics.stdev(cluster_counts[cluster])
print cluster+'\t'+str(avg)+'\t'+str(stdev)
def latteralMerge(file1, file2):
import collections
cluster_db = collections.OrderedDict()
eo = export.ExportFile(file2[:-4]+'combined.txt')
for line in open(file1,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
cluster_db[t[0]]=t
for line in open(file2,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if t[0] in cluster_db:
t1=cluster_db[t[0]]
eo.write(string.join(t1+t[2:],'\t')+'\n')
eo.close()
def removeMarkerFinderDoublets(heatmap_file,diff=1):
matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters = remoteImportData(heatmap_file)
priorRowClusters.reverse()
if len(priorColumnClusters)==0:
for c in column_header:
cluster = string.split(c,':')[0]
priorColumnClusters.append(cluster)
for r in row_header:
cluster = string.split(r,':')[0]
priorRowClusters.append(cluster)
import collections
cluster_db = collections.OrderedDict()
i=0
for cluster in priorRowClusters:
try: cluster_db[cluster].append(matrix[i])
except: cluster_db[cluster] = [matrix[i]]
i+=1
transposed_data_matrix=[]
clusters=[]
for cluster in cluster_db:
cluster_cell_means = numpy.mean(cluster_db[cluster],axis=0)
cluster_db[cluster] = cluster_cell_means
transposed_data_matrix.append(cluster_cell_means)
if cluster not in clusters:
clusters.append(cluster)
transposed_data_matrix = zip(*transposed_data_matrix)
i=0
cell_max_scores=[]
cell_max_score_db = collections.OrderedDict()
for cell_scores in transposed_data_matrix:
cluster = priorColumnClusters[i]
cell = column_header[i]
ci = clusters.index(cluster)
#print ci, cell, cluster, cell_scores;sys.exit()
cell_state_score = cell_scores[ci] ### This is the score for that cell for it's assigned MarkerFinder cluster
alternate_state_scores=[]
for score in cell_scores:
if score != cell_state_score:
alternate_state_scores.append(score)
alt_max_score = max(alternate_state_scores)
alt_sum_score = sum(alternate_state_scores)
cell_max_scores.append([cell_state_score,alt_max_score,alt_sum_score]) ### max and secondary max score - max for the cell-state should be greater than secondary max
try: cell_max_score_db[cluster].append(([cell_state_score,alt_max_score,alt_sum_score]))
except: cell_max_score_db[cluster] = [[cell_state_score,alt_max_score,alt_sum_score]]
i+=1
for cluster in cell_max_score_db:
cluster_cell_means = numpy.median(cell_max_score_db[cluster],axis=0)
cell_max_score_db[cluster] = cluster_cell_means ### This is the cell-state mean score for all cells in that cluster and the alternative max mean score (difference gives you the threshold for detecting double)
i=0
print len(cell_max_scores)
keep=['row_clusters-flat']
keep_alt=['row_clusters-flat']
remove = ['row_clusters-flat']
remove_alt = ['row_clusters-flat']
min_val = 1000
for (cell_score,alt_score,alt_sum) in cell_max_scores:
cluster = priorColumnClusters[i]
cell = column_header[i]
ref_max, ref_alt, ref_sum = cell_max_score_db[cluster]
ci = clusters.index(cluster)
ref_diff= math.pow(2,(ref_max-ref_alt))*diff #1.1
ref_alt = math.pow(2,(ref_alt))
cell_diff = math.pow(2,(cell_score-alt_score))
cell_score = math.pow(2,cell_score)
if cell_diff<min_val: min_val = cell_diff
if cell_diff>ref_diff and cell_diff>diff: #cell_score cutoff removes some, but cell_diff is more crucial
#if alt_sum<cell_score:
assignment=0 #1.2
keep.append(cell)
try: keep_alt.append(string.split(cell,':')[1]) ### if prefix added
except Exception:
keep_alt.append(cell)
else:
remove.append(cell)
try: remove_alt.append(string.split(cell,':')[1])
except Exception: remove_alt.append(cell)
assignment=1
#print assignment
i+=1
print min_val
print len(keep), len(remove)
from import_scripts import sampleIndexSelection
input_file=heatmap_file
output_file = heatmap_file[:-4]+'-Singlets.txt'
try: sampleIndexSelection.filterFile(input_file,output_file,keep)
except: sampleIndexSelection.filterFile(input_file,output_file,keep_alt)
output_file = heatmap_file[:-4]+'-Multiplets.txt'
try: sampleIndexSelection.filterFile(input_file,output_file,remove)
except: sampleIndexSelection.filterFile(input_file,output_file,remove_alt)
def exportTFcorrelations(filename,TF_file,threshold,anticorrelation=False):
eo = export.ExportFile(filename[:-4]+'-TF-correlations.txt')
TFs = simpleListImport(TF_file)
x, column_header, row_header, dataset_name, group_db = importData(filename)
### For methylation data or other data with redundant signatures, remove these and only report the first one
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
D1 = numpy.corrcoef(x)
i=0
correlation_pairs=[]
for score_ls in D1:
k=0
for v in score_ls:
if str(v)!='nan':
if k!=i:
#print row_header[i], row_header[k], v
if row_header[i] in TFs or row_header[k] in TFs:
#correlation_pairs.append([row_header[i],row_header[k],v])
if anticorrelation:
if v<(-1*threshold):
eo.write(row_header[i]+'\t'+row_header[k]+'\t'+str(v)+'\n')
elif v<(-1*threshold) or v>threshold:
eo.write(row_header[i]+'\t'+row_header[k]+'\t'+str(v)+'\n')
k+=1
i+=1
eo.close()
def TFisoformImport(filename):
isoform_db = {}
for line in open(filename, 'rU').xreadlines():
data = line.rstrip()
trans, prot, gene, symbol, uid, uid2, uid3 = string.split(data, '\t')
isoform_db[trans] = (symbol, prot)
return isoform_db
def exportIntraTFIsoformCorrelations(filename, TF_file, threshold, anticorrelation=False):
eo = export.ExportFile(filename[:-4] + '-TF-correlations.txt')
isoform_db = TFisoformImport(TF_file)
x, column_header, row_header, dataset_name, group_db = importData(filename)
### For methylation data or other data with redundant signatures, remove these and only report the first one
with warnings.catch_warnings():
warnings.filterwarnings('ignore', category=RuntimeWarning)
D1 = numpy.corrcoef(x)
i = 0
correlation_pairs = []
for score_ls in D1:
k = 0
for v in score_ls:
if str(v) != 'nan':
if k != i:
if row_header[i] in isoform_db or row_header[k] in isoform_db:
try:
gene1, prot1 = isoform_db[row_header[i]]
gene2, prot2 = isoform_db[row_header[k]]
if gene1 == gene2:
if anticorrelation:
if v < -1 * threshold:
eo.write(row_header[i] + '\t' + row_header[k] + '\t' + str(v) + '\n')
elif v < -1 * threshold or v > threshold:
eo.write(row_header[i] + '\t' + row_header[k] + '\t' + str(v) + '\n')
except:
pass
k += 1
i += 1
eo.close()
def PSIfilterAndImpute(folder):
### Filter a PSI file and impute missing values based on neighbors
files = UI.read_directory(folder)
for file in files:
filename = folder + '/' + file
if '.txt' in file:
eo = export.ExportFile(filename[:-4] + '-impute.txt')
header = True
count = 0
for line in open(filename, 'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data, '\t')
t0 = values[1]
tl = values[(-1)]
vs = values[1:]
if header:
header = False
eo.write(line)
elif len(vs) == len(vs) - vs.count(''):
sum_val = sum(map(float, vs)) / len(vs)
if sum_val != 1 and sum_val != 0:
eo.write(line)
count += 1
elif len(vs) - vs.count('') > len(vs) - 3:
new_values = []
i = 0
for v in vs:
if v=='':
if i==0: ### if the first element is null
try: new_values.append((float(vs[i+1])+float(tl))/2)
except: new_values.append(None) ### If two nulls occur in a row
elif i==len(vs)-1: ### if the last element is null
try: new_values.append((float(vs[i-1])+float(t0))/2)
except: new_values.append(None) ### If two nulls occur in a row
else: ### if the another element is null
try: new_values.append((float(vs[i-1])+float(vs[i+1]))/2)
except: new_values.append(None) ### If two nulls occur in a row
else:
new_values.append(v)
i += 1
if None not in new_values:
sum_val = sum(map(float, new_values)) / len(new_values)
if sum_val != 1 and sum_val != 0:
eo.write(string.join([values[0]] + map(str, new_values), '\t') + '\n')
count += 1
eo.close()
print count, '\t', fileg
def summarizePSIresults(folder, TF_file):
#TFs = simpleListImport(TF_file)
### Import PSI results and report number of impacted TFs
files = UI.read_directory(folder)
#eo = export.ExportFile(folder + '/TF_events.txt')
all_TFs = []
for file in files:
TFs_in_file = []
filename = folder + '/' + file
if '.txt' in file and 'PSI.' in file:
header = True
count = 0
header = True
for line in open(filename, 'rU').xreadlines():
if header:
header = False
else:
data = cleanUpLine(line)
t = string.split(data, '\t')
symbol = string.split(t[0], ':')[0]
dPSI = abs(float(t[(-5)]))
if symbol=='HOXA1':
"""
if symbol in TFs and symbol not in TFs_in_file and dPSI > 0.2:
eo.write(string.join(t + [file], '\t') + '\n')
TFs_in_file.append(symbol)
if symbol not in all_TFs:
all_TFs.append(symbol)
count += 1
"""
print file+"\t"+t[-5]+"\t"+t[-4]+"\t"+t[0]
##print file, count, len(all_TFs), string.join(TFs_in_file, ',')
#eo.close()
def convertPSICoordinatesToBED(folder):
files = UI.read_directory(folder)
eo = export.ExportFile(folder + '/combined.bed')
all_TFs = []
for file in files:
TFs_in_file = []
filename = folder + '/' + file
if '.txt' in file:
header = True
count = 0
header = True
for line in open(filename, 'rU').xreadlines():
if header:
header = False
else:
data = cleanUpLine(line)
t = string.split(data, '\t')
symbol = string.split(t[0], ':')[0]
try:
coordinates = t[7]
except:
print t
sys.exit()
else:
j1, j2 = string.split(coordinates, '|')
c1a, c1b = map(int, string.split(j1.split(':')[1], '-'))
strand = '+'
if c1a > c1b:
c1a, c1b = c1b, c1a
strand = '-'
c2a, c2b = map(int, string.split(j2.split(':')[1], '-'))
if c2a > c2b:
c2a, c2b = c2b, c2a
chr = string.split(coordinates, ':')[0]
uid = string.replace(t[0], ':', '__')
eo.write(string.join([chr, str(c1a), str(c1b), uid + '--' + file, strand, str(c1a), str(c1b), '0'], '\t') + '\n')
eo.write(string.join([chr, str(c2a), str(c2b), uid + '--' + file, strand, str(c2a), str(c2b), '0'], '\t') + '\n')
eo.close()
def convertPSIConservedCoordinatesToBED(Mm_Ba_coordinates, Ba_events):
if 'Baboon' in Mm_Ba_coordinates:
equivalencies={'Heart':['Heart'],
'Kidney':['Kidney-cortex','Kidney-medulla'],
'WFAT':['White-adipose-pericardial','White-adipose-mesenteric','White-adipose-subcutaneous','Omental-fat'],
'BFAT':['White-adipose-pericardial','White-adipose-mesenteric','White-adipose-subcutaneous','Omental-fat'],
'Lung':['Lungs'],
'Cere':['Cerebellum','Ventromedial-hypothalamus','Habenula','Pons','Pineal-gland','Visual-cortex','Lateral-globus-pallidus',
'Paraventricular-nuclei','Arcuate-nucleus','Suprachiasmatic-nuclei','Putamen','Optic-nerve-head', 'Medial-globus-pallidus',
'Amygdala','Prefontal-cortex','Dorsomedial-hypothalamus'],
'BS':['Cerebellum','Ventromedial-hypothalamus','Habenula','Pons','Pineal-gland','Visual-cortex','Lateral-globus-pallidus',
'Paraventricular-nuclei','Arcuate-nucleus','Suprachiasmatic-nuclei','Putamen','Optic-nerve-head', 'Medial-globus-pallidus',
'Amygdala','Prefontal-cortex','Dorsomedial-hypothalamus'],
'Hypo':['Cerebellum','Ventromedial-hypothalamus','Habenula','Pons','Pineal-gland','Visual-cortex','Lateral-globus-pallidus',
'Paraventricular-nuclei','Arcuate-nucleus','Suprachiasmatic-nuclei','Putamen','Optic-nerve-head', 'Medial-globus-pallidus',
'Amygdala','Prefontal-cortex','Dorsomedial-hypothalamus'],
'Adrenal':['Adrenal-cortex','Adrenal-medulla'],
'SM':['Muscle-gastrocnemian','Muscle-abdominal'],
'Liver':['Liver'],
}
else:
equivalencies={'Heart':['Heart'],
'Kidney':['Kidney','Kidney'],
'WFAT':['WFAT'],
'BFAT':['BFAT'],
'Lung':['Lungs'],
'Adrenal':['Adrenal'],
'Liver':['Liver'],
}
eo = export.ExportFile(Mm_Ba_coordinates[:-4] + '-matched.txt')
eo2 = export.ExportFile(Mm_Ba_coordinates[:-4] + '-matrix.txt')
mouse_events = {}
baboon_events = {}
baboon_corridinates = {}
### This mouse circadian events file has been lifted over to baboon coordinates
countX = 0
for line in open(Mm_Ba_coordinates, 'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data, '\t')
chr, c1, c2, event, strand, null, null, null = values
event = string.replace(event, '__', ':')
event, tissue = event.split('--')
junctions = string.split(event, ':')[1:]
junctions = string.join(junctions, ':')
junctions = string.split(junctions, '|')
junctions.sort() ### make a unique event
junctions = string.join(junctions, '|')
symbol = string.split(event, ':')[0]
event = symbol + ':' + junctions
countX += 1
tissue = string.replace(tissue, '_event_annot_file.txt', '')
tissue = string.replace(tissue, 'PSI.', '')
tissue = string.replace(tissue, '_Mm', '')
junction = chr + ':' + c2 + '-' + c1
alt_junction1 = chr + ':' + str(int(c2) + 1) + '-' + str(int(c1) + 1)
alt_junction2 = chr + ':' + str(int(c2) - 1) + '-' + str(int(c1) - 1)
try:
mouse_events[junction].append([event, tissue])
except:
mouse_events[junction] = [[event, tissue]]
else:
try:
mouse_events[alt_junction1].append([event, tissue])
except:
mouse_events[alt_junction1] = [[event, tissue]]
else:
try:
mouse_events[alt_junction2].append([event, tissue])
except:
mouse_events[alt_junction2] = [[event, tissue]]
else:
junction = chr + ':' + c1 + '-' + c2
alt_junction1 = chr + ':' + str(int(c1) + 1) + '-' + str(int(c2) + 1)
alt_junction2 = chr + ':' + str(int(c1) - 1) + '-' + str(int(c2) - 1)
try:
mouse_events[junction].append([event, tissue])
except:
mouse_events[junction] = [[event, tissue]]
try:
mouse_events[alt_junction1].append([event, tissue])
except:
mouse_events[alt_junction1] = [[event, tissue]]
try:
mouse_events[alt_junction2].append([event, tissue])
except:
mouse_events[alt_junction2] = [[event, tissue]]
for line in open(Ba_events, 'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data, '\t')
event, tissue_num, tissues, coordinates = values
junctions = string.split(event, ':')[1:]
junctions = string.join(junctions, ':')
junctions = string.split(junctions, '|')
junctions.sort()
junctions = string.join(junctions, '|')
symbol = string.split(event, ':')[0]
event = symbol + ':' + junctions
baboon_corridinates[event] = coordinates
try:
j1, j2 = string.split(coordinates, '|')
except:
continue
else:
tissues = tissues.split('|')
try:
baboon_events[j1].append([event, tissues])
except:
baboon_events[j1] = [[event, tissues]]
try:
baboon_events[j2].append([event, tissues])
except:
baboon_events[j2] = [[event, tissues]]
print len(mouse_events), len(baboon_events)
common = 0
matched_events = {}
matched_mm_events = {}
tissue_matrix = {}
mm_single_tissue_counts = {}
ba_single_tissue_counts = {}
for junction in mouse_events:
if junction in baboon_events:
common += 1
mm_events = {}
for mm_event, mm_tissue in mouse_events[junction]:
try:
mm_events[mm_event].append(mm_tissue)
except:
mm_events[mm_event] = [mm_tissue]
for mm_event in mm_events:
mm_tissues = mm_events[mm_event]
mm_tissues = unique.unique(mm_tissues)
for ba_event, ba_tissues in baboon_events[junction]:
ba_tissues = unique.unique(ba_tissues)
matched_events[(mm_event, ba_event)] = (mm_tissues, ba_tissues)
matched_mm_events[mm_event] = []
def matchingTissues(mouse, baboon):
m_matches = []
b_matches = []
for m in mouse:
for b in baboon:
if m in equivalencies:
if b in equivalencies[m]:
m_matches.append(m)
b_matches.append(b)
if len(m_matches) == 0:
return ''
m_matches = string.join(unique.unique(m_matches), ', ')
b_matches = string.join(unique.unique(b_matches), ', ')
return m_matches + ':' + b_matches
for mm_event, ba_event in matched_events:
mm_tissues, ba_tissues = matched_events[(mm_event, ba_event)]
matching_tissues = matchingTissues(mm_tissues, ba_tissues)
eo.write(string.join([mm_event, ba_event, string.join(mm_tissues, '|'), string.join(ba_tissues, '|'), str(len(mm_tissues)), str(len(ba_tissues)), matching_tissues], '\t') + '\n')
for mt in mm_tissues:
for bt in ba_tissues:
try:
tissue_matrix[(mt, bt)] += 1
except:
tissue_matrix[(mt, bt)] = 1
else:
try:
mm_single_tissue_counts[mt] += 1
except:
mm_single_tissue_counts[mt] = 1
try:
ba_single_tissue_counts[bt] += 1
except:
ba_single_tissue_counts[bt] = 1
print mm_single_tissue_counts['Heart']
print tissue_matrix[('Heart', 'Heart')]
tissue_matrix_table = []
ba_tissues = ['Tissues']
for bt in ba_single_tissue_counts:
ba_tissues.append(bt)
eo2.write(string.join(ba_tissues, '\t') + '\n')
for mt in mm_single_tissue_counts:
table = []
for bt in ba_single_tissue_counts:
if bt == 'Thyroid' and mt == 'Heart':
print tissue_matrix[(mt, bt)]
print tissue_matrix[(mt, bt)] / (1.0 * ba_single_tissue_counts[bt])
try:
table.append(str(tissue_matrix[(mt, bt)] / (1.0 * ba_single_tissue_counts[bt])))
except:
table.append('0')
eo2.write(string.join([mt] + table, '\t') + '\n')
print common, len(matched_events), len(matched_mm_events)
eo.close()
eo2.close()
def rankExpressionRescueFromCellHarmony(organized_diff_ref, repair1_folds, repair2_folds, reference_fold_dir, repair_dir1, repair_dir2):
def importCellHarmonyDEGs(folder, repair=False):
print folder
files = os.listdir(folder)
DEG_db = {}
for file in files:
filename = folder + '/' + file
if '.txt' in file and 'GE.' in file:
header = True
count = 0
header = True
file = file[:-4]
file = string.split(file[3:], '_')[0]
if file[:2] == 'DM':
file = 'global'
for line in open(filename, 'rU').xreadlines():
if header:
header = False
else:
data = cleanUpLine(line)
t = string.split(data, '\t')
GeneID, SystemCode, LogFold, rawp, adjp, Symbol, avg_g2, avg_g1 = t
rawp = float(rawp)
adjp = float(adjp)
if float(LogFold) > 0:
direction = 'positive'
else:
direction = 'negative'
if repair:
if float(LogFold) > 0:
fold = math.pow(2, float(LogFold))
else:
fold = -1 / math.pow(2, float(LogFold))
if Symbol == 'BC049762':
print 'BC049762', file, LogFold, fold
if abs(fold) > 1.5 and adjp < 0.05:
try:
DEG_db[Symbol].append([file, direction])
except:
DEG_db[Symbol] = [[file, direction]]
else:
try:
DEG_db[Symbol].append([file, direction])
except:
DEG_db[Symbol] = [[file, direction]]
return DEG_db
ref_DEGs = importCellHarmonyDEGs(reference_fold_dir)
repaired_DEGs = importCellHarmonyDEGs(repair_dir1, repair=True)
repaired2_DEGs = importCellHarmonyDEGs(repair_dir2, repair=True)
def importCellHarmonyPseudoBulkFolds(filename):
fold_db = {}
header = True
for line in open(filename, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if header:
fold_db['header'] = t[1:]
header = False
else:
uid = t[0]
folds = t[1:]
fold_db[uid] = folds
return fold_db
repaired_fold_db = importCellHarmonyPseudoBulkFolds(repair1_folds)
repaired2_fold_db = importCellHarmonyPseudoBulkFolds(repair2_folds)
import collections
ordered_ref_degs = collections.OrderedDict()
ordered_cluster_genes = collections.OrderedDict()
repair_verified = collections.OrderedDict()
repair2_verified = collections.OrderedDict()
cluster_ordered_ref_db = collections.OrderedDict()
header = True
for line in open(organized_diff_ref, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if header:
ref_header = t
header = False
else:
cluster, geneID = string.split(t[0], ':')
cluster = string.split(cluster, '_')[0]
if cluster[:2] == 'DM':
cluster = 'global'
ordered_ref_degs[(geneID, cluster)] = t
try:
cluster_ordered_ref_db[cluster].append(geneID)
except:
cluster_ordered_ref_db[cluster] = [geneID]
repaired_verified = {}
verified = {}
for geneID, ref_cluster in ordered_ref_degs:
for cluster, ref_direction in ref_DEGs[geneID]:
if geneID in repaired_DEGs:
for repair_cluster, repair_direction in repaired_DEGs[geneID]:
if repair_cluster == cluster and ref_direction != repair_direction and ('Neu' in repair_cluster or 'global' in repair_cluster):
try:
repair_verified[repair_cluster].append(geneID)
except:
repair_verified[repair_cluster] = [geneID]
else:
print geneID + '\t' + repair_direction + '\t' + repair_cluster + '\tR412X-HMZ'
try:
verified[geneID].append('R412X-HMZ')
except:
verified[geneID] = ['R412X-HMZ']
if geneID in repaired2_DEGs:
for repair_cluster, repair_direction in repaired2_DEGs[geneID]:
if repair_cluster == cluster and ref_direction != repair_direction and ('Neu' in cluster or 'global' in cluster):
try:
repair2_verified[repair_cluster].append(geneID)
except:
repair2_verified[repair_cluster] = [geneID]
else:
print geneID + '\t' + repair_direction + '\t' + repair_cluster + '\t' + 'R412X-Irf8'
try:
verified[geneID].append('R412X-Irf8')
except:
verified[geneID] = ['R412X-Irf8']
for gene in verified:
verified[gene] = unique.unique(verified[gene])
eo1 = export.ExportFile(organized_diff_ref[:-4] + '-Repair-Sorted.txt')
eo2 = export.ExportFile(organized_diff_ref[:-4] + '-Repaired-Only.txt')
header = ref_header + repaired_fold_db['header'] + repaired2_fold_db['header']
eo1.write(string.join(header, '\t') + '\n')
eo2.write(string.join(header, '\t') + '\n')
print len(ordered_ref_degs)
print len(repaired_fold_db)
print len(repaired2_fold_db)
print len(repair_verified)
print len(repair2_verified)
print len(verified)
print len(ordered_ref_degs)
prior_cluster = None
added_genes = []
for geneID, cluster in ordered_ref_degs:
try:
folds = ordered_ref_degs[(geneID, cluster)] + repaired_fold_db[geneID] + repaired2_fold_db[geneID]
except:
print '...Error in identifying match UID for:', geneID
added_genes.append(geneID)
continue
else:
if geneID not in verified:
eo1.write(string.join(folds, '\t') + '\n')
elif len(verified[geneID]) > 1:
added_genes.append(geneID)
elif 'R412X-HMZ' in verified[geneID]:
added_genes.append(geneID)
else:
eo2.write(string.join(folds, '\t') + '\n')
added_genes.append(geneID)
eo1.close()
eo2.close()
def exportSeuratMarkersToClusters(filename):
prior_cluster = None
for line in open(filename, 'rU').xreadlines():
data = cleanUpLine(line)
cluster,gene = string.split(data, '\t')
if cluster!= prior_cluster:
try: eo.close()
except: pass
path = filename[:-4]+'_'+cluster+'.txt'
eo = export.ExportFile(path)
eo.write('UID\tSy\n')
eo.write(gene+'\tSy\n')
prior_cluster = cluster
eo.close()
def reorganizeMetaData(filename):
path = filename[:-4]+'_reorganized'+'.txt'
eo = export.ExportFile(path)
firstRow=True
for line in open(filename, 'rU').xreadlines():
if firstRow:
firstRow=False
else:
data = cleanUpLine(line)
t = string.split(data, '\t')
uid = t[0]
for val in t[1:]:
if len(val)>1:
eo.write(uid+'\t'+val+'\n')
eo.close()
def formatMetaData(filename):
""" Export metadata annotations from a matrix that consist of mutations, numerical values
categorical values and true/false values to a simple two column format for OncoSplice"""
export_path = filename[:-4]+'_reorganized'+'.txt'
eo = export.ExportFile(export_path)
metadata=[]
quantitative_values={}
quantitative_sample_values={}
row_count=1
for line in open(filename, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if row_count==1:
headers = t[1:]
elif row_count==2:
if t[0] == 'FORMAT':
dataFormat = t[1:]
else:
sampleID = t[0]
index=0
for value in t[1:]:
if dataFormat[index] == 'TRUE-FALSE':
if value == 'TRUE' or value == 'y' or value == 'yes' or value == 'true' or value == 'YES' or value == 'True':
### The sample is labeled as the column header (e.g., Alive)
metadata.append([sampleID,headers[index]])
if dataFormat[index] == 'QUANTITATIVE':
if len(headers[index])>0 and value != '':
### Prior to export, define low, median and high clusters
try:
value = float(value)
try:
quantitative_values[headers[index]].append(value)
except:
quantitative_values[headers[index]] = [value]
try:
quantitative_sample_values[headers[index]].append([sampleID,value])
except:
quantitative_sample_values[headers[index]] = [[sampleID,value]]
except:
pass ### Invalid non-numeric
if dataFormat[index] == 'VERBOSE':
if len(headers[index])>0 and value != '':
metadata.append([sampleID,headers[index]+'('+value+')'])
if dataFormat[index] == 'MUTATION':
if len(headers[index])>0 and value != '':
metadata.append([sampleID,headers[index]])
if 'p.' in value:
value = string.replace(value,'p.','')
if '; ' in value:
value = string.split(value,';')[0]
if len(value)>1:
metadata.append([sampleID,headers[index]+'-'+value[:-1]])
index+=1
row_count+=1
for annotation in quantitative_values:
values = quantitative_values[annotation]
one_third = len(values)/3
bottom = values[:one_third]
middle = values[one_third:-1*one_third]
top = values[-1*one_third:]
for (sampleID, value) in quantitative_sample_values[annotation]:
if value in bottom:
metadata.append([sampleID,annotation+'-low'])
elif value in middle:
metadata.append([sampleID,annotation+'-mid'])
elif value in top:
metadata.append([sampleID,annotation+'-high'])
else:
print value,'value is out-of-range!!!'; sys.exit()
### Write these metadata annotations out to a two column file
for (sampleID,annotation) in metadata:
if len(sampleID)>1:
eo.write(sampleID+'\t'+annotation+'\n')
eo.close()
def reformatCellDistanceNetworks(filename):
path = filename[:-4]+'_reformated'+'.txt'
eo = export.ExportFile(path)
firstRow=True
for line in open(filename, 'rU').xreadlines():
if firstRow:
data = cleanUpLine(line)
t = string.split(data, '\t')
headers = t[1:]
firstRow=False
else:
data = cleanUpLine(line)
t = string.split(data, '\t')
cell_type = t[0]
index=0
for val in t[1:]:
try:
cell_type2 = headers[index]
float(val)
eo.write(cell_type+'\t'+cell_type2+'\t'+val+'\n')
except:
pass
index+=1
eo.close()
def parseCellMarkerDB(filename):
path = filename[:-4]+'_reformated'+'.txt'
eo = export.ExportFile(path)
firstRow=True
gene_to_cell_db={}
for line in open(filename, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if firstRow:
headers = t
firstRow=False
else:
tissueType = t[1]
cellName = t[5]
geneSymbols = t[8]
species = t[0]
pmid = t[13]
if pmid == 'Company':
pmid = t[14]
else:
pmid = 'PMID'+pmid
geneSymbols = string.replace(geneSymbols,'[','')
geneSymbols = string.replace(geneSymbols,']','')
genes = string.split(geneSymbols,', ')
cellName = string.replace(cellName,'(',' ')
cellName = string.replace(cellName,')','')
cell_type_name = tissueType+ ' ' + cellName + ' ('+pmid+' markers - CellMarkerDB)'
if cell_type_name in gene_to_cell_db:
gene_to_cell_db[cell_type_name,species]+=genes
else:
gene_to_cell_db[cell_type_name,species]=genes
for (cell_type_name,species) in gene_to_cell_db:
genes = unique.unique(gene_to_cell_db[cell_type_name,species])
for gene in genes:
eo.write(gene+'\t'+cell_type_name+'\t'+species+'\n')
eo.close()
def findGTEXsubsets(all_samples,selected_samples):
path = all_samples[:-4]+'_additional_samples.txt'
eo = export.ExportFile(path)
firstRow=True
sample_to_tissue={}
downloaded=[]
old_sample_to_tissue={}
for line in open(selected_samples, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if firstRow:
headers = t
firstRow=False
else:
downloaded.append(t[0])
for line in open(all_samples, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if firstRow:
headers = t
firstRow=False
else:
sraID = t[0]
sampleID = t[1]
tissue = t[3]
if sraID not in downloaded:
try: sample_to_tissue[tissue].append(sraID)
except: sample_to_tissue[tissue] = [sraID]
else:
try: old_sample_to_tissue[tissue].append(sraID)
except: old_sample_to_tissue[tissue] = [sraID]
for tissue in sample_to_tissue:
if tissue in old_sample_to_tissue:
existing = len(old_sample_to_tissue[tissue])
possible = len(sample_to_tissue[tissue])
if existing < 24:
new = 24-existing
for sample in sample_to_tissue[tissue][:new]:
eo.write(sample+'\n')
else:
for sample in sample_to_tissue[tissue][:25]:
eo.write(sample+'\n')
eo.close()
def massMarkerFinder(groups_file,exp_dir,class_type=1):
import collections
firstRow=True
tissues = collections.OrderedDict()
for line in open(exp_dir, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
valid_cellIDs = t
break
print len(valid_cellIDs),'valid cellIDs'
for line in open(groups_file, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if firstRow:
headers = t
firstRow=False
else:
cellID = t[1]
tissue = t[2]
class1 = t[-2]
class2 = t[-1]
if cellID in valid_cellIDs:
if class_type == 2:
class1 = class2
if tissue in tissues:
db = tissues[tissue]
try: db[class1].append(cellID)
except: db[class1] = [cellID]
else:
db = collections.OrderedDict()
try: db[class1].append(cellID)
except: db[class1] = [cellID]
tissues[tissue] = db
### Write out tissue group files
from import_scripts import sampleIndexSelection
for tissue in tissues:
filter_IDs=[]
path = export.findParentDir(exp_dir)+'/groups.'+tissue+'.txt'
eo = export.ExportFile(path)
path2 = export.findParentDir(exp_dir)+'/comps.'+tissue+'.txt'
eo2 = export.ExportFile(path2)
eo2.write('\n')
db = tissues[tissue]
for category in db:
for cellID in db[category]:
eo.write(cellID+'\t'+category+'\t'+category+'\n')
filter_IDs.append(cellID)
eo.close()
eo2.close()
path = export.findParentDir(exp_dir)+'/exp.'+tissue+'.txt'
sampleIndexSelection.filterFile(exp_dir,path,filter_IDs)
def aggregateMarkerFinderResults(folder):
eo = export.ExportFile(folder+'/ConsolidatedMarkers.txt')
files = UI.read_directory(folder)
for tissue in files:
fn = folder+tissue+'/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
prior_cell_type = None
cell_type_count=0
firstRow = True
if '.' not in tissue:
for line in open(fn, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if firstRow:
headers = t
firstRow=False
else:
gene = t[0]
cell_type = t[-1]
rho = float(t[2])
if prior_cell_type == cell_type:
cell_type_count+=1
else:
cell_type_count = 0
if cell_type_count<100 and rho>0.1:
eo.write(gene+'\t'+cell_type+'\n')
prior_cell_type = cell_type
eo.close()
def summarizeCovariates(fn):
eo = export.ExportFile(fn[:-4]+'-summary.txt')
eo2 = export.ExportFile(fn[:-4]+'-cells.txt')
cluster_patient={}
clusters=[]
individuals = []
firstRow=True
patient_cells={}
for line in open(fn, 'rU').xreadlines():
data = cleanUpLine(line)
Barcode,Cluster,ClusterName,PublishedClusterName,individual,region,age,sex,diagnosis = string.split(data, '\t')
individual = diagnosis+' '+sex+' '+region+' '+age+' '+individual+''
if firstRow:
#headers = t
firstRow=False
else:
if individual not in individuals:
individuals.append(individual)
if ClusterName not in clusters:
clusters.append(ClusterName)
if ClusterName in cluster_patient:
patients = cluster_patient[ClusterName]
try: patients[individual]+=1
except: patients[individual]=1
else:
patients={}
patients[individual]=1
cluster_patient[ClusterName]=patients
try: patient_cells[individual,ClusterName].append(Barcode)
except: patient_cells[individual,ClusterName]= [Barcode]
eo.write(string.join(['ClusterName']+individuals,'\t')+'\n')
for ClusterName in clusters:
values = []
patients = cluster_patient[ClusterName]
for i in individuals:
if i in patients:
if patients[i]>29:
if (i,ClusterName) in patient_cells:
for barcode in patient_cells[(i,ClusterName)]:
eo2.write(barcode+'\t'+ClusterName+'--'+i+'\n')
values.append(str(patients[i]))
else:
values.append('0')
eo.write(string.join([ClusterName]+values,'\t')+'\n')
eo.close()
def computeIsoformRatio(gene_exp_file, isoform_exp_file):
path = isoform_exp_file[:-4]+'_ratios.txt'
eo = export.ExportFile(path)
firstRow=True
gene_exp_db={}
for line in open(gene_exp_file, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if firstRow:
samples = t[1:]
firstRow=False
else:
uid = t[0]
values = map(float,t[1:])
if '.' in uid:
uid = string.split(uid,'.')[0]
gene_exp_db[uid] = values
firstRow=True
isoform_exp_db={}
gene_to_isoform={}
for line in open(isoform_exp_file, 'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data, '\t')
if firstRow:
iso_samples = t[1:]
eo.write(line)
firstRow=False
else:
uid = t[0]
genes=None
original_uid = uid
uids = string.split(uid,'-')
if len(uids)>2:
gene = string.join(uids[:2],'-')
else:
gene = uids[0]
values = map(float,t[1:])
isoform_exp_db[original_uid]=values
if '.' in gene:
gene = string.split(gene,'.')[0]
gene = string.split(gene,': ')[1]
if '|' in gene:
gene = string.split(gene,'|')[0]
if '-' in gene:
genes = string.split(gene,'-')
if 'NKX' in genes[0]:
gene = string.join(genes,'-')
elif len(genes)>2:
gene = string.join(genes[:2],'-')
else:
gene = genes[0]
try: gene_exp = gene_exp_db[gene]
except:
gene = string.join(genes,'-')
try:
gene_exp = gene_exp_db[gene]
except:
gene = string.split(gene,'-')[0]
gene_exp = gene_exp_db[gene]
try: gene_to_isoform[gene].append(original_uid)
except: gene_to_isoform[gene] = [original_uid]
for gene in gene_to_isoform:
if len(gene_to_isoform[gene])>1:
for isoform in gene_to_isoform[gene]:
values = isoform_exp_db[isoform]
gene_exp = gene_exp_db[gene]
index=0
ratios=[]
for i in values:
#v = math.log(i+1,2)-math.log(gene_exp[index]+1,2)
k = gene_exp[index]
if k>1:
try: v = i/k
except: v = 1
else:
v=''
index+=1
try: ratios.append(str(round(v,2)))
except: ratios.append('')
"""
if 'MYRFL' in isoform:
print isoform
print gene_exp[:10]
print values[:10]
print ratios[:10]"""
eo.write(string.join([isoform]+ratios,'\t')+'\n')
#max_ratios = max(map(float,ratios))
eo.close()
if __name__ == '__main__':
input_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Autism/PRJNA434002/ICGS-NMF/CellFrequencies/FinalGroups-CellTypesFull-Author.txt'
summarizeCovariates(input_file);sys.exit()
psi_data = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Isoform-U01/AS/ExpressionInput/exp.PSI-filtered.txt'
isoform_data = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Isoform-U01/Alt-Analyze/ExpressionInput/exp.GC30-basic-MainTissues_ratios-sparse-filtered.txt'
psi_annotations = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/GTEx/Hs_RNASeq_top_alt_junctions-PSI_EventAnnotation.txt'
#correlateIsoformPSIvalues(isoform_data,psi_data,psi_annotations);sys.exit()
isoform_exp = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Isoform-U01/protein.GC30-basic-MainTissues.txt'
gene_exp = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Isoform-U01/gene.TF.GC30-basic-MainTissues.txt'
#computeIsoformRatio(gene_exp,isoform_exp);sys.exit()
#aggregateMarkerFinderResults('/Volumes/salomonis2/LabFiles/TabulaMuris/Smart-Seq2_Nextera/CPTT-Files/all-comprehensive/');sys.exit()
groups_file = '/data/salomonis2/LabFiles/TabulaMuris/Smart-Seq2_Nextera/CPTT-Files/all-comprehensive/FACS_annotation-edit.txt'
exp_dir = '/data/salomonis2/LabFiles/TabulaMuris/Smart-Seq2_Nextera/CPTT-Files/all-comprehensive/MergedFiles.txt'
#massMarkerFinder(groups_file,exp_dir);sys.exit()
all_samples = '/Users/saljh8/Dropbox/Collaborations/Isoform-U01/GTEX-30-sample/SraRunTable-All-SamplesRnaSeq.txt'
selected_samples = '/Users/saljh8/Dropbox/Collaborations/Isoform-U01/GTEX-30-sample/summary.GC30.txt'
#findGTEXsubsets(all_samples,selected_samples);sys.exit()
#remoteAssignGroupColors('/Users/saljh8/Documents/GitHub/altanalyze/DemoData/cellHarmony/Mouse_BoneMarrow/inputFile/ICGS-NMF/FinalGroups-CellTypesFull.txt');sys.exit()
#parseCellMarkerDB('/Users/saljh8/Dropbox/scRNA-Seq Markers/Human/Markers/SourceFiles/Cross-Tissue/Single_cell_markers.txt');sys.exit()
#reformatCellDistanceNetworks('/Users/saljh8/Desktop/dataAnalysis/Collaborative/Lucas/pvalue.txt');sys.exit()
#formatMetaData('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/BEAT-AML/metadata/BEAT-AML_MetaData-STRUCTURED.txt');sys.exit()
#reorganizeMetaData('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/BEAT-AML/metadata/metadata_same-format.txt');sys.exit()
folder = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/RBM20/eCLIP/ENCODE/annotations'
#simpleCombineBedFiles(folder);sys.exit()
PSI_dir = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/PSI/SpliceICGS.R1.Depleted.12.27.17/all-depleted-and-KD/temp/'
##summarizePSIresults(PSI_dir,PSI_dir);sys.exit()
#tempFunction('/Users/saljh8/Downloads/LungCarcinoma/HCC.S5063.TPM.txt');sys.exit()
a = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/PSI/SpliceICGS.R1.Depleted.12.27.17/all-depleted-and-KD/temp/'
#compareEventLists(a);sys.exit()
filename = '/Users/saljh8/Downloads/Kerscher_lists_mouse_versus_mouse_and_human_gene_lists/Top50MouseandHuman1-clusters.txt'
#exportSeuratMarkersToClusters(filename); sys.exit()
organized_diff_ref = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/WuXi-David-Nature-Revision/PROJ-00584/fastqs/DM-4-Gfi1-R412X-ModGMP-1694-ADT/outs/filtered_gene_bc_matrices/Merged-Cells/centroid-revised/custom/cellHarmony/OrganizedDifferentials.txt'
repair1_folds = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/WuXi-David-Nature-Revision/PROJ-00584/fastqs/DM-5-Gfi1-R412X-R412X-ModGMP-1362-ADT/outs/filtered_gene_bc_matrices/Merged-Cells/hybrid/cellHarmony-vs-DM2-1.2-fold-adjp/OtherFiles/exp.ICGS-cellHarmony-reference__DM-5-Gfi1-R412X-R412X-ModGMP-1362-D7Cells-ADT-Merged_matrix_CPTT-AllCells-folds.txt'
repair2_folds = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/WuXi-David-Nature-Revision/PROJ-00584/fastqs/DM-6-Gfi1-R412X-Irf8-ModGMP-1499-ADT/outs/filtered_gene_bc_matrices/Merged-Cells-iseq/cellHarmony-centroid-revsied/hybrid/cellHarmony/OtherFiles/exp.ICGS-cellHarmony-reference__DM-6-Gfi1-R412X-Irf8-ModGMP-1499-ADT_matrix-3_matrix_CPTT-hybrid-AllCells-folds.txt'
reference_fold_dir = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/WuXi-David-Nature-Revision/PROJ-00584/fastqs/DM-4-Gfi1-R412X-ModGMP-1694-ADT/outs/filtered_gene_bc_matrices/Merged-Cells/centroid-revised/custom/cellHarmony/DifferentialExpression_Fold_1.2_adjp_0.05'
repair_dir1 = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/WuXi-David-Nature-Revision/PROJ-00584/fastqs/DM-5-Gfi1-R412X-R412X-ModGMP-1362-ADT/outs/filtered_gene_bc_matrices/Merged-Cells/hybrid/vs-R412X-het/cellHarmony/DifferentialExpression_Fold_1.2_adjp_0.05'
repair_dir1 = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/WuXi-David-Nature-Revision/PROJ-00584/fastqs/DM-5-Gfi1-R412X-R412X-ModGMP-1362-ADT/outs/filtered_gene_bc_matrices/Merged-Cells/hybrid/vs-R412X-het/cellHarmony/OtherFiles/DEGs-LogFold_0.0_rawp'
repair_dir2 = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/WuXi-David-Nature-Revision/PROJ-00584/fastqs/DM-6-Gfi1-R412X-Irf8-ModGMP-1499-ADT/outs/filtered_gene_bc_matrices/Merged-Cells-iseq/cellHarmony-centroid-revsied/hybrid/vs-R412X-Het/cellHarmony/DifferentialExpression_Fold_1.2_adjp_0.05'
repair_dir2 = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/WuXi-David-Nature-Revision/PROJ-00584/fastqs/DM-6-Gfi1-R412X-Irf8-ModGMP-1499-ADT/outs/filtered_gene_bc_matrices/Merged-Cells-iseq/cellHarmony-centroid-revsied/hybrid/vs-R412X-Het/cellHarmony/OtherFiles/DEGs-LogFold_0.0_rawp'
TF_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/NCI-R01/CCSB_TFIso_Clones.txt'
PSI_dir = '/Volumes/salomonis2/NCI-R01/TCGA-BREAST-CANCER/TCGA-files-Ens91/bams/AltResults/AlternativeOutput/OncoSPlice-All-Samples-filtered-names/SubtypeAnalyses-Results/round1/Events-dPSI_0.1_adjp/'
#simpleCombineFiles('/Volumes/salomonis2/NCI-R01/Harvard/BRC_PacBio_Seq/metadataanalysis/PSICluster/TCGA/FilteredTF')
#sys.exit()
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Anukana/Breast-Cancer/TF-isoform/TF_ratio_correlation-analysis/tcga_rsem_isopct_filtered-filtered.2-filtered.txt'
TF_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Anukana/Breast-Cancer/TF-isoform/Ensembl-isoform-key-CCSB.txt'
input_file = '/Volumes/salomonis2/NCI-R01/TCGA-BREAST-CANCER/Anukana/UO1analysis/xenabrowserFiles/tcga_rsem_isoform_tpm_filtered.txt'
Mm_Ba_coordinates = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Krithika/Baboon-Mouse/mm10-circadian_liftOverTo_baboon.txt'
Ba_events = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Krithika/Baboon-Mouse/Baboon_metacycle-significant-AS-coordinates.txt'
Mm_Ba_coordinates = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Krithika/Human-Mouse/hg19-mm10-12-tissue-circadian.txt'
Ba_events = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Krithika/Human-Mouse/Human_CYCLOPS-significant-AS-coordinates.txt'
filename = '/Users/saljh8/Desktop/DemoData/Venetoclax/D4/cellHarmony-rawp-stringent/gene_summary.txt'
filename = '/Volumes/salomonis2/LabFiles/Nathan/10x-PBMC-CD34+/AML-p27-pre-post/pre/cellHarmony-latest/gene_summary-p27.txt'
filename = '/Volumes/salomonis2/LabFiles/Dan-Schnell/To_cellHarmony/MIToSham/Input/cellHarmony/cell-frequency-stats.txt'
TF_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/NCI-R01/CCSB_TFIso_Clones.txt'
PSI_dir = '/Volumes/salomonis2/NCI-R01/TCGA-BREAST-CANCER/TCGA-files-Ens91/bams/AltResults/AlternativeOutput/OncoSPlice-All-Samples-filtered-names/SubtypeAnalyses-Results/round1/Events-dPSI_0.1_adjp/'
#convertPSICoordinatesToBED(PSI_dir);sys.exit()
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Anukana/Breast-Cancer/TF-isoform/TF_ratio_correlation-analysis/tcga_rsem_isopct_filtered-filtered.2-filtered.txt'
TF_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Anukana/Breast-Cancer/TF-isoform/Ensembl-isoform-key-CCSB.txt'
#exportIntraTFIsoformCorrelations(filename,TF_file,0.3,anticorrelation=True);sys.exit()
input_file= '/Volumes/salomonis2/NCI-R01/TCGA-BREAST-CANCER/Anukana/UO1analysis/xenabrowserFiles/tcga_rsem_isoform_tpm_filtered.txt'
#convertXenaBrowserIsoformDataToStandardRatios(input_file);sys.exit()
#Mm_Ba_coordinates = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Krithika/Baboon-Mouse/mm10-circadian_liftOverTo_baboon.txt'
Ba_events = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Krithika/Baboon-Mouse/Baboon_metacycle-significant-AS-coordinates.txt'
#convertPSICoordinatesToBED(Mm_Ba_coordinates,Ba_events);sys.exit()
#PSIfilterAndImpute('/Volumes/salomonis2/LabFiles/krithika_circadian/GSE98965-Papio_Anubis/files/grp-files/Filtered-Psi-groups-files'); sys.exit()
filename='/Users/saljh8/Desktop/DemoData/Venetoclax/D4/cellHarmony-rawp-stringent/gene_summary.txt'
filename = '/Volumes/salomonis2/LabFiles/Nathan/10x-PBMC-CD34+/AML-p27-pre-post/pre/cellHarmony-latest/gene_summary-p27.txt'
filename = '/Volumes/salomonis2/LabFiles/Dan-Schnell/To_cellHarmony/MIToSham/Input/cellHarmony/cell-frequency-stats.txt'
index1=2;index2=3; x_axis='Number of Differentially Expressed Genes'; y_axis = 'Comparisons'; title='Hippocampus - Number of Differentially Expressed Genes'
#OutputFile = export.findParentDir(filename)
#OutputFile = export.findParentDir(OutputFile[:-1])+'/test.pdf'
#exportTFcorrelations('/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/SuperPan/ExpressionInput/exp.Cdt1-2139-genes.txt','/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Marie.Dominique/TF-to-gene/228-tfs.txt',0.1);sys.exit()
#stackedbarchart(filename,display=True,output=OutputFile);sys.exit()
index1=2;index2=3; x_axis='Number of DEGs'; y_axis = 'Reference clusters'; title='cellHarmony Differentially Expressed Genes'
index1=-2;index2=-1; x_axis='Cell-State Percentage'; y_axis = 'Reference clusters'; title='Assigned Cell Frequencies'
#barchart(filename,index1,index2,x_axis,y_axis,title,display=True)
#barchart(filename,index1,index2,x_axis,y_axis,title,display=True,color1='IndianRed',color2='SkyBlue');sys.exit()
diff=0.7
print 'diff:',diff
#latteralMerge(file1, file2);sys.exit()
#removeMarkerFinderDoublets('/Volumes/salomonis2/Nancy_ratner/2mo-NF/exp.Figure_SX-ICGS-MarkerFinder.filt.txt',diff=diff);sys.exit()
#outputForGOElite('/Users/saljh8/Desktop/R412X/completed/centroids.WT.R412X.median.txt');sys.exit()
#simpleStatsSummary('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/HCA/Mean-Comparisons/ExpressionInput/MergedFiles.Counts.UMI.txt');sys.exit()
a = '/Users/saljh8/Downloads/groups.CellTypes-Predicted-Label-Transfer-For-Nuclei-matrix.txt'
b = '/Volumes/salomonis2/Immune-10x-data-Human-Atlas/Bone-Marrow/Stuart/Browser/ExpressionInput/HS-compatible_symbols.txt'
b = '/data/salomonis2/GSE107727_RAW-10X-Mm/filtered-counts/ExpressionInput/Mm_compatible_symbols.txt'
input_file = '/Users/saljh8/Downloads/exprMatrix.txt'
##transposeMatrix(a);sys.exit()
convertSymbolLog(input_file,b,species='Hs',logNormalize=False); sys.exit()
#returnIntronJunctionRatio('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Fluidigm_scRNA-Seq/12.09.2107/counts.WT-R412X.txt');sys.exit()
#geneExpressionSummary('/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/ExpressionInput/DEGs-LogFold_1.0_rawp');sys.exit()
b = '/Users/saljh8/Dropbox/scRNA-Seq Markers/Human/Expression/Lung/Adult/PRJEB31843/1k-T0-cellHarmony-groups2.txt'
a = '/Users/saljh8/Dropbox/scRNA-Seq Markers/Human/Expression/Lung/Adult/Perl-CCHMC/FinalMarkerHeatmap_all.txt'
convertGroupsToBinaryMatrix(b,b,cellHarmony=False);sys.exit()
a = '/Users/saljh8/Desktop/temp/groups.TNBC.txt'
b = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/tests/clusters.txt'
#simpleCombineFiles('/Users/saljh8/Desktop/dataAnalysis/Collaborative/Jose/NewTranscriptome/CombinedDataset/ExpressionInput/Events-LogFold_0.58_rawp')
#removeRedundantCluster(a,b);sys.exit()
a = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/PSI/SpliceICGS.R1.Depleted.12.27.17/all-depleted-and-KD'
#a = '/Users/saljh8/Desktop/Ashish/all/Events-dPSI_0.1_rawp-0.01/'
#filterPSIValues('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/PSI/CORNEL-AML/PSI/exp.Cornell-Bulk.txt');sys.exit()
#compareGenomicLocationAndICGSClusters();sys.exit()
#ViolinPlot();sys.exit()
#simpleScatter('/Users/saljh8/Downloads/CMdiff_paper/calcium_data-KO4.txt');sys.exit()
query_dataset = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/exp.GSE81682_HTSeq-cellHarmony-filtered.txt'
all_tpm = '/Users/saljh8/Desktop/demo/BoneMarrow/ExpressionInput/exp.BoneMarrow-scRNASeq.txt'
markerfinder = '/Users/saljh8/Desktop/demo/BoneMarrow/ExpressionOutput1/MarkerFinder/AllGenes_correlations-ReplicateBasedOriginal.txt'
signature_genes = '/Users/saljh8/Desktop/Grimes/KashishNormalization/test/Panorama.txt'
state = 'Multi-Lin'
#evaluateMultiLinRegulatoryStructure(all_tpm,markerfinder,signature_genes,state);sys.exit()
query_dataset = None
all_tpm = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/MultiLin/Gottgens_HarmonizeReference.txt'
signature_genes = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/MultiLin/Gottgens_HarmonizeReference.txt'
markerfinder = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
state = 'Eryth_Multi-Lin'
#evaluateMultiLinRegulatoryStructure(all_tpm,markerfinder,signature_genes,state,query = query_dataset);sys.exit()
#simpleCombine("/Volumes/My Passport/Ari-10X/input");sys.exit()
#effectsPrioritization('/Users/saljh8/Documents/1-dataAnalysis/RBM20-collaboration/RBM20-BAG3_splicing/Missing Values-Splicing/Effects.txt');sys.exit()
#customCleanBinomial('/Volumes/salomonis2-1/Lab backup/Theresa-Microbiome-DropSeq/NegBinomial/ExpressionInput/exp.Instesinal_microbiome2.txt');sys.exit()
#findReciprocal('/Volumes/HomeBackup/CCHMC/Jared-KO/BatchCorrectedFiltered/exp.CM-KO-steady-state.txt');sys.exit()
#ReceptorLigandCellInteractions('Mm','/Users/saljh8/Downloads/ncomms8866-s3.txt','/Users/saljh8/Downloads/Round3-MarkerFinder_All-Genes.txt');sys.exit()
#compareFusions('/Volumes/salomonis2-2/CPMC_Melanoma-GBM/Third-batch-files/Complete_analysis/temp/Combined_Fusion_GBM.txt');sys.exit()
#combineVariants('/Volumes/salomonis2/CPMC_Melanoma-GBM/Third-batch-files/Complete_analysis/Variant_results/GBM/Variants_HighModerate-GBM_selected.txt');sys.exit()
#customCleanSupplemental('/Users/saljh8/Desktop/dataAnalysis/CPMC/TCGA_MM/MM_genes_published.txt');sys.exit()
#customClean('/Users/saljh8/Desktop/dataAnalysis/Driscoll/R3/2000_run1708A_normalized.txt');sys.exit()
#simpleFilter('/Volumes/SEQ-DATA 1/all_10.5_mapped_norm_GC.csv');sys.exit()
#filterRandomFile('/Users/saljh8/Downloads/HuGene-1_1-st-v1.na36.hg19.transcript2.csv',1,8);sys.exit()
filename = '/Users/saljh8/Desktop/Grimes/GEC14078/MergedFiles.txt'
#CountKallistoAlignedJunctions(filename);sys.exit()
filename = '/Users/saljh8/Desktop/Code/AltAnalyze/AltDatabase/EnsMart72/Mm/junction1/junction_critical-junction-seq.txt'
#MakeJunctionFasta(filename);sys.exit()
filename = '/Users/saljh8/Downloads/CoexpressionAtlas.txt'
#ToppGeneFilter(filename); sys.exit()
#countIntronsExons(filename);sys.exit()
#filterForJunctions(filename);sys.exit()
#filename = '/Users/saljh8/Desktop/Grimes/GEC14074/ExpressionOutput/LineageCorrelations-test-protein_coding-zscores.txt'
#runHCOnly(filename,[]); sys.exit()
folder = '/Users/saljh8/Desktop/Code/AltAnalyze/AltDatabase/EnsMart72/ensembl/Hs'
try:
files = UI.read_directory(folder)
for file in files: #:70895507-70895600
if '.bed' in file:
#BedFileCheck(folder+'/'+file)
pass
except Exception: pass
#sys.exit()
#runPCAonly(filename,[],False,showLabels=False,plotType='2D');sys.exit()
countinp = '/Volumes/salomonis2/SinghLab/20150715_single_GCBCell/bams/ExpressionInput/counts.Bcells.txt'
IGH_gene_file = '/Volumes/salomonis2/SinghLab/20150715_single_GCBCell/bams/ExpressionInput/IGH_genes.txt'
#extractFeatures(countinp,IGH_gene_file);sys.exit()
import UI
#geneMethylationOutput(filename);sys.exit()
#ica(filename);sys.exit()
#replaceWithBinary('/Users/saljh8/Downloads/Neg_Bi_wholegenome.txt');sys.exit()
#simpleFilter('/Volumes/SEQ-DATA/AML-TCGA/ExpressionInput/counts.LAML1.txt');sys.exit()
filename = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/genes.tpm_tracking-ordered.txt'
#filename = '/Users/saljh8/Desktop/Grimes/KashishNormalization/6-5-2015/ExpressionInput/amplify/exp.All-wt-output.txt'
#getlastexon(filename);sys.exit()
TFs = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/TF-by-gene_matrix/all-TFs2.txt'
folder = '/Users/saljh8/Downloads/BLASTX2_Gecko.tab'
genes = ['Gfi1', 'Irf8'] #'Cebpe', 'Mecom', 'Vwf', 'Itga2b', 'Meis1', 'Gata2','Ctsg','Elane', 'Klf4','Gata1']
#genes = ['Gata1','Gfi1b']
#coincentIncedenceTest(filename,TFs);sys.exit()
#coincidentIncedence(filename,genes);sys.exit()
#test(folder);sys.exit()
#files = UI.read_directory(folder)
#for file in files: SimpleCorrdinateToBed(folder+'/'+file)
#filename = '/Users/saljh8/Desktop/bed/RREs0.5_exons_unique.txt'
#simpleIntegrityCheck(filename);sys.exit()
gene_list = ['S100a8','Chd7','Ets1','Chd7','S100a8']
gene_list_file = '/Users/saljh8/Desktop/demo/Amit/ExpressionInput/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/Grimes/Comb-plots/AML_genes-interest.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Grimes/Mm_Sara-single-cell-AML/alt/AdditionalHOPACH/ExpressionInput/AML_combplots.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Grimes/MDS-array/Comb-plot genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Grimes/All-Fluidigm/ExpressionInput/comb_plot3.txt'
gene_list_file = '/Users/saljh8/Desktop/Grimes/MultiLin-Code/MultiLin-TFs.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/ExpressionInput/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/10X-DropSeq-comparison/Final-Classifications/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/TFs/Myelo_TFs2.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/R412X/customGenes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/ExpressionInput/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/R412X/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/HCA/BM1-8_CD34+/ExpressionInput/MixedLinPrimingGenes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Churko/ExpressionInput/genes.txt'
genesets = importGeneList(gene_list_file,n=22)
filename = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/comb-plots/exp.IG2_GG1-extended-output.txt'
filename = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/comb-plots/genes.tpm_tracking-ordered.txt'
filename = '/Users/saljh8/Desktop/demo/Amit/ExpressedCells/GO-Elite_results/3k_selected_LineageGenes-CombPlotInput2.txt'
filename = '/Users/saljh8/Desktop/Grimes/Comb-plots/exp.AML_single-cell-output.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Grimes/Mm_Sara-single-cell-AML/alt/AdditionalHOPACH/ExpressionInput/exp.AML.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Grimes/MDS-array/comb-plot/input.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Grimes/All-Fluidigm/ExpressionInput/exp.Lsk_panorama.txt'
filename = '/Users/saljh8/Desktop/demo/BoneMarrow/ExpressionInput/exp.BoneMarrow-scRNASeq.txt'
filename = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/exp.GSE81682_HTSeq-cellHarmony-filtered.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Harinder/scRNASeq_Mm-Plasma/PCA-loading/ExpressionInput/exp.PCA-Symbol.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/10X-DropSeq-comparison/Final-Classifications/cellHarmony/MF-analysis/ExpressionInput/exp.Fluidigm-log2-NearestNeighbor-800.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/10X-DropSeq-comparison/Final-Classifications/cellHarmony/MF-analysis/ExpressionInput/exp.10X-log2-NearestNeighbor.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/10X-DropSeq-comparison/DropSeq/MultiLinDetect/ExpressionInput/DataPlots/exp.DropSeq-2k-log2.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/R412X/exp.allcells-v2.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/HCA/BM1-8_CD34+/ExpressionInput/exp.CD34+.v5-log2.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Churko/ExpressionInput/exp.10x-Multi-CCA-iPS-CM-CPTT-non-log.txt'
#filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/CITE-Seq-MF-indexed/ExpressionInput/exp.cellHarmony.v3.txt'
#filename = '/Volumes/salomonis2/Theodosia-Kalfa/Combined-10X-CPTT/ExpressionInput/exp.MergedFiles-ICGS.txt'
#filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/R412X/exp.cellHarmony-WT-R412X-relative.txt'
#filename = '/Users/saljh8/Desktop/Old Mac/Desktop/Grimes/Kallisto/Ly6g/CodingOnly/Guide3-Kallisto-Coding-NatureAugmented/SubClustering/Nov-27-Final-version/ExpressionInput/exp.wt-panorama.txt'
#filename = '/Volumes/salomonis2/Harinder-singh/Run2421-10X/10X_IRF4_Lo/outs/filtered_gene_bc_matrices/ExpressionInput/exp.10X_IRF4_Lo_matrix_CPTT-ICGS.txt'
#filename = '/Users/saljh8/Desktop/Old Mac/Desktop/Grimes/Kallisto/Ly6g/CodingOnly/Guide3-Kallisto-Coding-NatureAugmented/SubClustering/Nov-27-Final-version/R412X/exp.R412X-RSEM-order.txt'
print genesets
for gene_list in genesets:
multipleSubPlots(filename,gene_list,SubPlotType='column',n=22)
sys.exit()
plotHistogram(filename);sys.exit()
filename = '/Users/saljh8/Desktop/Grimes/Expression_final_files/ExpressionInput/amplify-wt/DataPlots/Clustering-exp.myeloid-steady-state-PCA-all_wt_myeloid_SingleCell-Klhl7 Dusp7 Slc25a33 H6pd Bcorl1 Sdpr Ypel3 251000-hierarchical_cosine_cosine.cdt'
openTreeView(filename);sys.exit()
pdf1 = "/Users/saljh8/Desktop/Grimes/1.pdf"
pdf2 = "/Users/saljh8/Desktop/Grimes/2.pdf"
outPdf = "/Users/saljh8/Desktop/Grimes/3.pdf"
merge_horizontal(outPdf, pdf1, pdf2);sys.exit()
mergePDFs(pdf1,pdf2,outPdf);sys.exit()
filename = '/Volumes/SEQ-DATA/CardiacRNASeq/BedFiles/ExpressionOutput/Clustering/SampleLogFolds-CardiacRNASeq.txt'
ica(filename);sys.exit()
features = 5
matrix, column_header, row_header, dataset_name, group_db = importData(filename)
Kmeans(features, column_header, row_header); sys.exit()
#graphViz();sys.exit()
filename = '/Users/saljh8/Desktop/delete.txt'
filenames = [filename]
outputClusters(filenames,[]); sys.exit()
#runPCAonly(filename,[],False);sys.exit()
#VennDiagram(); sys.exit()
#buildGraphFromSIF('Ensembl','Mm',None,None); sys.exit()
#clusterPathwayZscores(None); sys.exit()
pruned_folder = '/Users/nsalomonis/Desktop/CBD/LogTransformed/GO-Elite/GO-Elite_results/CompleteResults/ORA_pruned/'
input_ora_folder = '/Users/nsalomonis/Desktop/CBD/LogTransformed/GO-Elite/input/'
files = UI.read_directory(pruned_folder)
for file in files:
if '.sif' in file:
input_file = string.join(string.split(file,'-')[:-1],'-')+'.txt'
sif_file = pruned_folder+file
input_file = input_ora_folder+input_file
buildGraphFromSIF('Ensembl','Hs',sif_file,input_file)
sys.exit()
filenames = [filename]
outputClusters(filenames,[])
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/clustering.py
|
clustering.py
|
import numpy as np
import pylab as pl
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import misopy
from misopy.sashimi_plot import sashimi_plot as ssp
import subprocess
import multiprocessing
import time
import subprocess
import random
import argparse
import math
import unique
import traceback
import anydbm
import dbhash
count_sum_array_db={}
sampleReadDepth={}
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def update_plot_settings(bamdir,group_psi_values,sample_headers):
### This functions writes out the sample orders, colors and sequence coverage for each BAM files for SashimiPlot
bams=[]
sample_colors=[]
sample_coverage=[]
colors = ['red','blue','green','grey','orange','purple','yellow','peach','pink','violet','magenta','navy']
colors = colors*300
color_index=0
for group in group_psi_values:
for index in group_psi_values[group]:
g=sample_headers[index].replace('.bed','.bam')
bams.append('"'+g+'"')
sample_colors.append('"'+colors[color_index]+'"')
sample_coverage.append(str(int(sampleReadDepth[index])))
color_index+=1 ### reset for the new group
bams = string.join(bams,',')
sample_colors = string.join(sample_colors,',')
sample_coverage = string.join(sample_coverage,',')
export_pl=open(unique.filepath('Config/sashimi_plot_settings.txt'),'w')
export_pl.write('[data]\n')
export_pl.write('bam_prefix = '+bamdir+'\n')
export_pl.write('bam_files =['+bams+']\n')
export_pl.write('\n')
export_pl.write('[plotting]')
export_pl.write('\n')
export_pl.write('fig_width = 7 \nfig_height = 7 \nintron_scale = 30 \nexon_scale = 4 \nlogged = False\n')
export_pl.write('font_size = 6 \nbar_posteriors = False \nnyticks = 4 \nnxticks = 4 \n')
export_pl.write('show_ylabel = False \nshow_xlabel = True \nshow_posteriors = False \nnumber_junctions = True \n')
export_pl.write('resolution = .5 \nposterior_bins = 40 \ngene_posterior_ratio = 5 \n')
export_pl.write('colors =['+sample_colors+']\n')
export_pl.write('coverages =['+sample_coverage+']\n')
export_pl.write('bar_color = "b" \nbf_thresholds = [0, 1, 2, 5, 10, 20]')
export_pl.close()
def importSplicingEventsToVisualize(eventsToVisualizeFilename):
splicing_events=[]
### Import the splicing events to visualize from an external text file (multiple formats supported)
type = None
expandedSearch = False
firstLine = True
for line in open(eventsToVisualizeFilename,'rU').xreadlines():
line = cleanUpLine(line)
t = string.split(line,'\t')
if firstLine:
if 'junctionID-1' in t:
j1i = t.index('junctionID-1')
j2i = t.index('junctionID-2')
type='ASPIRE'
expandedSearch = True
if 'ANOVA' in t:
type='PSI'
elif 'independent confirmation' in t:
type='confirmed'
expandedSearch = True
elif 'ANOVA' in eventsToVisualizeFilename:
type = 'ANOVA'
firstLine=False
if '|' in t[0]:
type = 'ANOVA'
if ' ' in t[0] and ':' in t[0]:
splicing_events.append(t[0])
elif type=='ASPIRE':
splicing_events.append(t[j1i] +' '+ t[j2i])
splicing_events.append(t[j2i] +' '+ t[j1i])
elif type=='ANOVA':
try:
a,b = string.split(t[0],'|')
a = string.split(a,':')
a = string.join(a[1:],':')
splicing_events.append(a +' '+ b)
splicing_events.append(b +' '+ a)
except Exception: pass
elif type=='PSI':
try:
j1,j2 = string.split(t[0],'|')
a,b,c = string.split(j1,':')
j1 = b+':'+c
splicing_events.append(j1 +' '+ j2)
splicing_events.append(j2 +' '+ j1)
except Exception:
#print traceback.format_exc();sys.exit()
pass
elif type=='confirmed':
try:
event_pair1 = string.split(t[1],'|')[0]
a,b,c,d = string.split(event_pair1,'-')
splicing_events.append(a+'-'+b +' '+ c+'-'+d)
splicing_events.append(c+'-'+d +' '+ a+'-'+b)
except Exception: pass
else:
splicing_events.append(t[0])
splicing_events = unique.unique(splicing_events)
return splicing_events,expandedSearch
def sashmi_plot_list(bamdir,eventsToVisualizeFilename,PSIFilename,events=None):
try:
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
from import_scripts import OBO_import; symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception:
symbol_to_gene={}
if events==None:
splicing_events,expandedSearch = importSplicingEventsToVisualize(eventsToVisualizeFilename)
else:
### Replace any ":" from the input events
#for i in range(len(events)): events[i] = string.replace(events[i],':','__')
expandedSearch = True
for i in range(len(events)):
gene = string.split(events[i],'__')[0]
if gene in gene_to_symbol:
symbol = gene_to_symbol[gene][0]
elif 'ENS' not in gene or 'G0000' in gene:
if gene in symbol_to_gene:
ensID = symbol_to_gene[gene][0]
symbol = gene
events[i] = ensID ### translate this ID to an Ensembl gene ID for propper SashimiPlot lookup
splicing_events = events ### optionally get from supplied variable
if len(splicing_events)==0:
print eventsToVisualizeFilename
forceNoCompatibleEventsInFile
print 'Exporting plots',
### Determine Groups for Coloring
groups_file = 'None'
dir_list = unique.read_directory(root_dir+'/ExpressionInput')
for file in dir_list:
if 'groups.' in file:
groups_file = root_dir+'/ExpressionInput/'+file
if groups_file != None:
try:
import ExpressionBuilder
sample_group_db = ExpressionBuilder.simplerGroupImport(groups_file)
groups=[]
for sample in sample_group_db:
if sample_group_db[sample] not in groups:
groups.append(sample_group_db[sample]) ### create an ordered list of unique group
except Exception:
groups = ['None']
#print traceback.format_exc()
pass
processed_events = formatAndSubmitSplicingEventsToSashimiPlot(PSIFilename, bamdir, splicing_events, sample_group_db, groups, False)
mopup_events = getMopUpEvents(splicing_events, processed_events)
### Do the same for supplied gene queries or junctions that didn't map above using the gene expression values as a guide
#print len(splicing_events),len(processed_events),len(mopup_events)
processed_events = formatAndSubmitSplicingEventsToSashimiPlot(steady_state_exp_file,bamdir,mopup_events,sample_group_db,groups,expandedSearch)
if len(processed_events)>0:
mopup_events = getMopUpEvents(mopup_events, processed_events)
processed_events = formatAndSubmitSplicingEventsToSashimiPlot(PSIFilename, bamdir, mopup_events, sample_group_db, groups, True)
return gene_to_symbol
def getMopUpEvents(splicing_events, processed_events):
mopup_events = []
for event in splicing_events:
add = True
if event in processed_events:
add = False
if ' ' in event:
try:
j1, j2 = string.split(event, ' ')
if j1 in processed_events:
add = False
if j2 in processed_events:
add = False
except Exception:
pass
if add:
mopup_events.append(event)
return mopup_events
def reorderEvents(events):
splicing_events = events
index = 0
for e in events:
j1o, j2o = string.split(e, ' ')
gene, j1 = string.split(j1o, ':')
gene, j2 = string.split(j2o, ':')
if '-' in j1 and '-' in j2:
j1a, j1b = string.split(j1, '-')
j2a, j2b = string.split(j2, '-')
j1a_block, j1a_region = string.split(j1a[1:], '.')
j2a_block, j2a_region = string.split(j2a[1:], '.')
j1b_block, j1b_region = string.split(j1b[1:], '.')
j2b_block, j2b_region = string.split(j2b[1:], '.')
if int(j1b_block) < int(j2b_block) and int(j1a_block) < int(j2a_block):
pass ### Occurs for complex cassette exon splicing events but matches SashimiIndex's selection for exclusion
elif int(j1b_block) > int(j2b_block):
new_e = j2o + ' ' + j1o
splicing_events[index] = new_e
elif int(j1a_block) < int(j2a_block):
new_e = j2o + ' ' + j1o
splicing_events[index] = new_e
elif int(j1b_region) > int(j2b_region):
new_e = j2o + ' ' + j1o
splicing_events[index] = new_e
elif int(j1a_region) < int(j2a_region):
new_e = j2o + ' ' + j1o
splicing_events[index] = new_e
index += 1
return splicing_events
def formatAndSubmitSplicingEventsToSashimiPlot(filename,bamdir,splicing_events,sample_group_db,groups,expandedSearch):
### Begin exporting parameters and events for SashimiPlot visualization
firstLine = True
setting = unique.filepath("Config/sashimi_plot_settings.txt")
psi_parent_dir=findParentDir(filename)
if 'PSI' not in filename:
index_dir=string.split(psi_parent_dir,'ExpressionInput')[0]+"AltResults/AlternativeOutput/sashimi_index/"
else:
index_dir=psi_parent_dir+"sashimi_index/"
spliced_junctions=[] ### Alternatively, compare to just one of the junctions
for splicing_event in splicing_events:
try:
j1,j2 = string.split(splicing_event,' ')
spliced_junctions.append(j1)
spliced_junctions.append(j2)
except Exception:
spliced_junctions.append(splicing_event) ### single gene ID or junction
if 'PSI' not in filename:
splicing_events_db = {}
for event in splicing_events:
event = string.replace(event,':','__')
if ' ' in event:
event = string.split(event,' ')[-1]
gene = string.split(event,"__")[0]
try: splicing_events_db[gene].append(event)
except Exception: splicing_events_db[gene] = [event]
splicing_events = splicing_events_db
import collections
analyzed_junctions=[]
processed_events=[]
#restrictToTheseGroups=['WT','R636S_Het','R636S_Homo'] #Meg HSCP-1 , Myelocyte Mono
restrictToTheseGroups = None
for line in open(filename,'rU').xreadlines():
line = cleanUpLine(line)
t = string.split(line,'\t')
if firstLine:
if 'PSI' in filename:
sampleIndexBegin = 11
sample_headers = t[sampleIndexBegin:]
else:
sampleIndexBegin = 1
sample_headers = t[sampleIndexBegin:]
if '.bed' not in sample_headers[0]: ### Add .bed if removed manually
sample_headers = map(lambda s: s+'.bed',sample_headers)
index=0
sample_group_index={}
for s in sample_headers:
group = sample_group_db[s]
sample_group_index[index]=group
try: sampleReadDepth[index]=count_sum_array_db[s]
except Exception: sampleReadDepth[index]=count_sum_array_db[s]
index+=1
firstLine = False
else:
if 'PSI' in filename:
splicing_event = val=t[2]+' '+t[3]
j1=t[2]
j2=t[3]
if t[2] in analyzed_junctions and t[3] in analyzed_junctions:
continue
else:
splicing_event = t[0] ### The gene ID
j1 = t[0]
j2 = t[0]
if ":U" in splicing_event or "-U" in splicing_event:
continue
else:
### First check to see if the full splicing event matches the entry
### If not (and not a PSI regulation hits list), look for an individual junction match
if splicing_event in splicing_events or (expandedSearch and (j1 in spliced_junctions or j2 in spliced_junctions)):
if splicing_event in processed_events: continue
if j2 in processed_events: continue
if j1 in processed_events: continue
processed_events.append(splicing_event)
processed_events.append(j1)
processed_events.append(j2)
#print processed_events, splicing_event
if 'PSI' in filename:
geneID = string.split(t[2],':')[0]
symbol = t[0]
analyzed_junctions.append(t[2])
analyzed_junctions.append(t[3])
else: ### For exp.dataset-steady-state.txt files
geneID = splicing_event
events = splicing_events[geneID]
index=0
import collections
initial_group_psi_values={}
try: group_psi_values = collections.OrderedDict()
except Exception:
try:
import ordereddict
group_psi_values = ordereddict.OrderedDict()
except Exception:
group_psi_values={}
for i in t[sampleIndexBegin:]: ### Value PSI range in the input file
try: group = sample_group_index[index]
except Exception: group=None
try:
try: initial_group_psi_values[group].append([float(i),index])
except Exception: initial_group_psi_values[group] = [[float(i),index]]
except Exception:
#print traceback.format_exc();sys.exit()
pass ### Ignore the NULL values
index+=1
if restrictToTheseGroups !=None: ### Exclude unwanted groups
initial_group_psi_values2={}
groups2 = collections.OrderedDict()
for group in groups:
if group in initial_group_psi_values:
if group in restrictToTheseGroups:
initial_group_psi_values2[group]=initial_group_psi_values[group]
groups2[group]=[]
initial_group_psi_values = initial_group_psi_values2
groups = groups2
### limit the number of events reported and sort based on the PSI values in each group
if 'None' in groups and len(groups)==1:
initial_group_psi_values['None'].sort()
group_size = len(initial_group_psi_values['None'])/2
filtered_group_index1 = map(lambda x: x[1], initial_group_psi_values['None'][:group_size])
filtered_group_index2 = map(lambda x: x[1], initial_group_psi_values['None'][group_size:])
group_psi_values['low']=filtered_group_index1
group_psi_values['high']=filtered_group_index2
else:
gn=0
for group in groups:
#print group
gn+=1
#if gn>4: break
if group in initial_group_psi_values:
initial_group_psi_values[group].sort()
if len(groups)>7:
filtered_group_indexes = map(lambda x: x[1], initial_group_psi_values[group][:1])
elif len(groups)>5:
filtered_group_indexes = map(lambda x: x[1], initial_group_psi_values[group][:2])
elif len(groups)>3:
filtered_group_indexes = map(lambda x: x[1], initial_group_psi_values[group][:4])
else:
filtered_group_indexes = map(lambda x: x[1], initial_group_psi_values[group][:5])
group_psi_values[group]=filtered_group_indexes
try: update_plot_settings(bamdir,group_psi_values,sample_headers)
except Exception:
print 'Cannot update the settings file. Likely permissions issue.'
try:
reordered = reorderEvents([t[2] + ' ' + t[3]])
reordered = string.split(reordered[0], ' ')
except Exception:
reordered = [t[2] + ' ' + t[3]]
reordered = string.split(reordered[0], ' ')
#print reordered
if 'PSI' in filename:
try: formatted_splice_event = string.replace(reordered[1], ':', '__')
except Exception: pass
### Submit the query
try: ssp.plot_event(formatted_splice_event,index_dir,setting,outputdir); success = True
except Exception:
success = False
#print traceback.format_exc()
else:
for event in events:
try:
ssp.plot_event(event,index_dir,setting,outputdir)
#print 'success' #formatted_splice_event='ENSMUSG00000000355__E4.1-E5.1'
except Exception: ### If it fails, output the gene-level plot
try: ssp.plot_event(geneID,index_dir,setting,outputdir); success = True
except Exception:
success = False
#print traceback.format_exc()
"""
### Second attempt
if 'PSI' in filename and success==False: ### Only relevant when parsing the junction pairs but not genes
try: formatted_splice_event=string.replace(reordered[0],':','__')
except Exception: pass
try: ssp.plot_event(formatted_splice_event,index_dir,setting,outputdir); # print 'success'
except Exception: pass
"""
return processed_events
def findParentDir(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1
return filename[:x]
def Sashimiplottting(bamdir,countsin,PSIFilename,eventsToVisualizeFilename,events=None):
PSIFilename = unique.filepath(PSIFilename)
header=True
junction_max=[]
countsin = unique.filepath(countsin)
count_sum_array=[]
count=0
for line in open(countsin,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
samples = []
for s in t[1:]:
if '.bed' not in s: s+='.bed'
samples.append(s)
header=False
count_sum_array=[0]*len(samples)
else:
values = map(float,t[1:])
count_sum_array = [sum(value) for value in zip(*[count_sum_array,values])]
count+=1
if count >30000 and 'salomonis' in bamdir: break
index=0
for sample in samples:
count_sum_array_db[sample] = count_sum_array[index]
index+=1
if events==None:
#print 'Preparing Sashimi-Input:',eventsToVisualizeFilename
eventsToVisualizeFilename = unique.filepath(eventsToVisualizeFilename)
gene_to_symbol=sashmi_plot_list(bamdir,eventsToVisualizeFilename,PSIFilename,events=events)
return gene_to_symbol
def remoteSashimiPlot(Species,fl,bamdir,eventsToVisualizeFilename,events=None,show=False):
global PSIFilename
global outputdir
global root_dir
global steady_state_exp_file
global species
species = Species
try:
countinp = fl.CountsFile()
root_dir = fl.RootDir()
except Exception:
root_dir = fl
search_dir = root_dir+'/ExpressionInput'
files = unique.read_directory(search_dir)
for file in files:
if 'counts.' in file and 'steady-state.txt' not in file:
countinp = search_dir+'/'+file
### Export BAM file indexes
from import_scripts import BAMtoJunctionBED
try: BAMtoJunctionBED.exportIndexes(root_dir)
except:
print 'BAM file indexing failed...'
print traceback.format_exc()
PSIFilename = root_dir+'/AltResults/AlternativeOutput/'+species+'_RNASeq_top_alt_junctions-PSI.txt'
import ExpressionBuilder
dir_list = unique.read_directory(root_dir+'/ExpressionInput')
for file in dir_list:
if 'exp.' in file and 'steady-state' not in file:
exp_file = root_dir+'/ExpressionInput/'+file
elif 'exp.' in file and 'steady-state' in file:
steady_state_exp_file = root_dir+'/ExpressionInput/'+file
global sample_group_db
sample_group_db = ExpressionBuilder.simplerGroupImport(exp_file)
#outputdir=findParentDir(PSIFilename)+"sashimiplots"
outputdir = root_dir+'/ExonPlots'
outputdir = root_dir+'/SashimiPlots'
try: os.mkdir(unique.filepath(outputdir))
except Exception: pass
if show:
s = open(outputdir+'/show.txt','w')
s.write('TRUE'); s.close()
else:
s = open(outputdir+'/show.txt','w')
s.write('FALSE'); s.close()
geneSymbol_db=Sashimiplottting(bamdir,countinp,PSIFilename,eventsToVisualizeFilename,events=events)
for filename in os.listdir(outputdir):
if '.pdf' in filename or '.png' in filename:
fn = string.replace(filename,'.pdf','')
fn = string.replace(fn,'.png','')
newname=string.split(fn,'__')
if newname[0] in geneSymbol_db:
new_filename = str(filename)
if '__' in filename:
new_filename = string.split(filename,'__')[1]
elif '\\' in filename:
new_filename = string.split(filename,'\\')[1]
elif '/' in filename:
new_filename = string.split(filename,'/')[1]
nnname=geneSymbol_db[newname[0]][0]+'-SashimiPlot_'+new_filename
try: os.rename(os.path.join(outputdir, filename), os.path.join(outputdir,nnname))
except Exception:
if 'already exists' in traceback.format_exc():
### File already exists, delete the new one
try: os.remove(os.path.join(outputdir,nnname))
except Exception: pass
### Now right the new one
try: os.rename(os.path.join(outputdir, filename), os.path.join(outputdir,nnname))
except Exception: pass
pass
else:
continue
print ''
def justConvertFilenames(species,outputdir):
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
from import_scripts import OBO_import; symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
for filename in os.listdir(outputdir):
if '.pdf' in filename or '.png' in filename:
fn = string.replace(filename,'.pdf','')
fn = string.replace(fn,'.png','')
newname=string.split(fn,'__')
if newname[0] in gene_to_symbol:
new_filename = str(filename)
if '__' in filename:
new_filename = string.split(filename,'__')[1]
elif '\\' in filename:
new_filename = string.split(filename,'\\')[1]
elif '/' in filename:
new_filename = string.split(filename,'/')[1]
nnname=gene_to_symbol[newname[0]][0]+'-SashimiPlot_'+new_filename
try: os.rename(os.path.join(outputdir, filename), os.path.join(outputdir,nnname))
except Exception: pass
else:
continue
if __name__ == '__main__':
root_dir = '/Volumes/salomonis2/Bruce_conklin_data/Pig-RNASeq/bams/'
events = ['ENSSSCG00000024564']
events = None
eventsToVisualizeFilename = None
eventsToVisualizeFilename = '/Volumes/salomonis2/Bruce_conklin_data/Pig-RNASeq/events.txt'
bamdir = root_dir
remoteSashimiPlot('Ss', root_dir, bamdir, eventsToVisualizeFilename, events=events, show=False)
sys.exit()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/SashimiPlot.py
|
SashimiPlot.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import base64
import export
import time
import unique
import traceback
try:
import gene_associations
"""
import suds
from suds.client import Client
wsdl = 'http://www.wikipathways.org/wpi/webservice/webservice.php?wsdl'
client = Client(wsdl) """
from wikipathways_api_client import WikipathwaysApiClient
wikipathways_api_client_instance = WikipathwaysApiClient()
except Exception:
#print traceback.format_exc()
None ### Occurs when functions in this module are resued in different modules
def filepath(filename):
fn = unique.filepath(filename)
return fn
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
class PathwayData:
def __init__(self,wpname):
self._wpname = wpname
def WPName(self): return self._wpname
def setSourceIDs(self,id_db): self.source_ids = id_db
def SourceIDs(self): return self.source_ids
def Report(self):
output = self.WPName()
return output
def __repr__(self): return self.Report()
def getPathwayAs(pathway_db,species_code,mod):
begin_time = time.time()
for wpid in pathway_db:
#print [wpid],'pathway_db',len(pathway_db)
file_type = 'gpml'
#file_type = 'application/gpml+xml'
processor_time = str(time.clock())
#try: export.deleteFolder('BuildDBs/WPs') ### clear any remaining pathway files
#except Exception: pass
#wp_id_data = client.service.getPathwayAs(fileType = file_type,pwId = wpid, revision = 0)
kwargs = {
'identifier': 'WP2062',
'version': 0,
'file_format': 'application/gpml+xml'}
#wp_id_data = wikipathways_api_client_instance.get_pathway_as(**kwargs)
wp_id_data = wikipathways_api_client_instance.get_pathway_as(file_format = file_type,identifier = wpid, version = 0)
#wp_id_data = base64.b64decode(wp_id_data)
gpml_path = filepath('BuildDBs/WPs/'+processor_time+'/'+wpid+'.gpml')
#print gpml_path
outfile = export.ExportFile(gpml_path)
outfile.write(wp_id_data); outfile.close()
gene_system_list = string.split(wp_id_data,'\n')
parent_path = export.findParentDir(gpml_path)
pathway_db = gene_associations.getGPMLGraphData(parent_path,species_code,mod) ### get GPML data back
#os.remove(gpml_path) ### Only store the file temporarily
try: export.deleteFolder('BuildDBs/WPs/'+processor_time) ### clear any remaining pathway files
except Exception: pass
end_time = time.time(); time_diff = float(end_time-begin_time)
"""
try: print "WikiPathways data imported in %d seconds" % time_diff
except Exception: null=None ### Occurs when transitioning back from the Official Database download window (not sure why) -- TclError: can't invoke "update" command
"""
return pathway_db
def getHexadecimalColorRanges(fold_db,analysis_type):
all_folds=[]
folds_to_gene={}
for gene in fold_db:
fold = fold_db[gene]
all_folds.append(fold)
try: folds_to_gene[fold].append(gene)
except Exception: folds_to_gene[fold] = [gene]
all_folds.sort() ### Sorted range of folds
if analysis_type == 'Lineage':
vmax = max(all_folds) ### replaces the old method of getting centered colors
else:
try: vmax,vmin=getColorRange(all_folds) ### We want centered colors for this (unlike with Lineage analysis)
except Exception: vmax,vmin = 0,0
### Normalize these values from 0 to 1
norm_folds_db={}; norm_folds=[]; color_all_yellow = False
for f in all_folds:
if analysis_type != 'Lineage':
try: n=(f-vmin)/(vmax-vmin) ### normalized
except Exception: n = 1; color_all_yellow = True
else:
n=(f-1.0)/(vmax-1.0) ### customized -> 1 is the lowest
norm_folds_db[n]=f
norm_folds.append(n)
### Calculate the color tuple for R, G and then B (blue to red)
### 00,00,255 (blue) -> 255,00,00 (red)
### 207, 207, 207 (grey) -> 255, 64, 64 (indian red 2) -> http://web.njit.edu/~kevin/rgb.txt.html
r0 = 207; g0 = 207; b0 = 207
rn = 255; gn = 64; bn = 64
if color_all_yellow:
for gene in fold_db:
fold_db[gene] = 'FFFF00'
else:
if analysis_type != 'Lineage':
### blue -> grey
r0 = 0; g0 = 191; b0 = 255
rn = 207; gn = 207; bn = 207
### grey -> red
r20 = 207; g20 = 207; b20 = 207
r2n = 255; g2n = 0; b2n = 0
gene_colors_hex = {}
for n in norm_folds:
ri=int(r0+(n*(rn-r0)))
gi=int(g0+(n*(gn-g0)))
bi=int(b0+(n*(bn-b0)))
rgb=ri,gi,bi ###blue to grey for non-lineage analyses
#rgb = (255,0,0)
if analysis_type != 'Lineage':
r2i=int(r20+(n*(r2n-r20)))
g2i=int(g20+(n*(g2n-g20)))
b2i=int(b20+(n*(b2n-b20)))
rgb2=r2i,g2i,b2i ### grey->red
f = norm_folds_db[n] ### get the original fold
genes = folds_to_gene[f] ## look up the gene(s) for that fold
if f<=1 and analysis_type == 'Lineage': ### only show positive z-scores with color
rgb = (207, 207, 207)
if f>0 and analysis_type == 'Genes':
rgb = rgb2
hex = '#%02x%02x%02x' % rgb
#print f,n,rgb,hex
for gene in genes:
fold_db[gene] = hex[1:]
return fold_db
def getColorRange(x):
""" Determines the range of colors, centered at zero, for normalizing cmap """
vmax=max(x)
vmin=min(x)
vmax = max([vmax,abs(vmin)])
vmin = -1*vmax
return vmax,vmin
def getGraphIDAssociations(id_color_db,pathway_db,key_by):
graphID_pathway_db={}
for pathway in pathway_db:
wpi = pathway_db[pathway] ### all data for the pathway is stored in this single object - wpi.Pathway() is the pathway name
graphID_pathway_db[pathway,wpi.Pathway()]=db={} ### add a new dictionary key (pathway) and initialize a new dictionary inside
for gi in wpi.PathwayGeneData():
if key_by == 'Label':
if gi.Label() in id_color_db:
hex_color = id_color_db[gi.Label()]
graphID_pathway_db[pathway,wpi.Pathway()][gi.GraphID()] = hex_color ### set the key,value of the child dictionary
elif len(wpi.PathwayGeneData())<277: ### more than this and parser.pxi thows an error - memory associated?
### add it as white node
graphID_pathway_db[pathway,wpi.Pathway()][gi.GraphID()] = 'FFFFFF'
else:
try:
for mod_id in gi.ModID():
if mod_id in id_color_db:
hex_color = id_color_db[mod_id]
graphID_pathway_db[pathway,wpi.Pathway()][gi.GraphID()] = hex_color ### set the key,value of the child dictionary
except Exception: None ### No MOD translation for this ID
return graphID_pathway_db
def viewLineageProfilerResults(filename,graphic_links):
global graphic_link
graphic_link=graphic_links ### This is a list of tuples containing name and file location
### Log any potential problems
log_file = filepath('webservice.log')
log_report = open(log_file,'w')
root_dir = export.findParentDir(filename)
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
if 'DataPlots' not in root_dir: ### Occurs when directly supplying an input matrix by the user
root_dir += '/DataPlots/'
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
id_db,column_headers = importDataSimple(filename,'LineageProfiler')
log_report.write('LineageProfiler input ID file imported successfully\n')
pathway_db={}
pathway_db['WP2062'] = PathwayData('TissueFateMap')
### MOD and species are not particularly important for Lineage analysis
pathway_db = getPathwayAs(pathway_db,'Hs','Ensembl')
log_report.write('Pathway data imported from GPML files obtained from webservice\n')
i=0
group_id_db={} ### store the results separately for each sample
### When analyzing z-scores, you can have multiple samples you wish to visualize results for (not so for regulated gene lists)
for biological_group in column_headers:
group_id_db[biological_group]=db={}
for gene in id_db:
group_id_db[biological_group][gene] = id_db[gene][i] ### get the index value of that biological group (z-score change)
i+=1
for biological_group in group_id_db:
group_specific = group_id_db[biological_group]
analysis_type = 'Lineage'
id_color_db = getHexadecimalColorRanges(group_specific,analysis_type) ### example "id_db" is key:tissue, value:z-score
graphID_db = getGraphIDAssociations(id_color_db,pathway_db,'Label')
file_type = 'png' ### svg, pdf, png
getColoredPathway(root_dir,graphID_db,file_type,'-'+biological_group)
file_type = 'pdf' ### svg, pdf, png
getColoredPathway(root_dir,graphID_db,file_type,'-'+biological_group)
log_report.write('Pathways colored and images saved to disk. Exiting webservice.\n')
log_report.close()
return graphic_link
def visualizePathwayAssociations(filename,species,mod_type,wpid,imageExport=True):
### Log any potential problems
log_file = filepath('webservice.log')
log_report = open(log_file,'w')
if wpid == None:
force_invalid_pathway
global mod
global species_code
global graphic_link
graphic_link={}
mod = mod_type
species_code = species
root_dir = export.findParentDir(filename)
criterion_name = export.findFilename(filename)[:-4]
log_report.write('Filename: %s and WPID %s\n' % (filename,wpid))
if 'GO-Elite/input' in root_dir:
root_dir = string.replace(root_dir,'GO-Elite/input','WikiPathways')
else:
root_dir+='WikiPathways/'
analysis_type = 'Genes'
id_db,column_headers = importDataSimple(filename,'GO-Elite')
log_report.write('GO-Elite input ID file imported successfully\n')
log_report.write('%d IDs imported\n' % len(id_db))
pathway_db={}
pathway_db[wpid] = PathwayData(None) ### only need to analyze object (method allows for analysis of any number)
pathway_db = getPathwayAs(pathway_db,species_code,mod)
log_report.write('Pathway data imported from GPML files obtained from webservice\n')
id_color_db = getHexadecimalColorRanges(id_db,analysis_type) ### example id_db" is key:gene, value:fold
graphID_db = getGraphIDAssociations(id_color_db,pathway_db,'MOD')
if imageExport != 'png':
file_type = 'pdf' ### svg, pdf, png
getColoredPathway(root_dir,graphID_db,file_type,'-'+criterion_name,WPID=wpid)
if imageExport != 'pdf':
file_type = 'png' ### svg, pdf, png
getColoredPathway(root_dir,graphID_db,file_type,'-'+criterion_name,WPID=wpid)
log_report.write('Pathways colored and image data returned. Exiting webservice.\n')
log_report.close()
return graphic_link
def getColoredPathway(root_dir,graphID_db,file_type,dataset_name,WPID=None):
for (wpid,name) in graphID_db:
### Example: graphId="ffffff90"; wpid = "WP2062"; color = "0000ff"
if WPID==wpid or WPID==None:
graphID_list = []
hex_color_list = []
for graphID in graphID_db[(wpid,name)]:
graphID_list.append(graphID)
hex_color_list.append(graphID_db[(wpid,name)][graphID]) ### order is thus the same for both
#hex_color_list = ["0000ff"]*11
#print len(graphID_list),graphID_list
#print len(hex_color_list),hex_color_list
#print file_type
if len(graphID_list)==0:
continue
### revision = 0 is the most current version
#file = client.service.getColoredPathway(pwId=wpid,revision=0,graphId=graphID_list,color=hex_color_list,fileType=file_type)
file = wikipathways_api_client_instance.get_colored_pathway(identifier=wpid,version=0,
element_identifiers=graphID_list,colors=hex_color_list,file_format=file_type)
#file = base64.b64decode(file) ### decode this file
name = string.replace(name,':','-')
name = string.replace(name,'/','-')
name = string.replace(name,'\\','-') ### will otherwise create a sub-directory
output_filename = root_dir+wpid+'_'+name+dataset_name+'.'+file_type
outfile = export.ExportFile(output_filename)
if file_type == 'png':
if wpid == 'WP2062': ### This is the LineageMapp
graphic_link.append(('LineageProfiler'+dataset_name,output_filename))
else:
graphic_link['WP'] = output_filename
outfile.write(file); outfile.close()
#http://au.answers.yahoo.com/question/index?qid=20111029100100AAqxS8l
#http://stackoverflow.com/questions/2374427/python-2-x-write-binary-output-to-stdout
def importDataComplex(filename,input_type,MOD=None,Species=None):
### If multiple system codes and mods exist
None
def importDataSimple(filename,input_type,MOD=None,Species=None):
id_db={}
fn = filepath(filename)
x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#' and x==0: x=0
elif x==0:
column_headers = t[1:]
if input_type != 'LineageProfiler':
try: column_headers = t[2] ### exclude the ID, system code and p-value column headers
except Exception: column_headers = 'NA'
x=1
else:
if x==1 and input_type != 'LineageProfiler':
### get system conversions
system_code = t[1]
import GO_Elite
from import_scripts import OBO_import
system_codes,source_types,mod_types = GO_Elite.getSourceData()
source_data = system_codes[system_code]
try:
Mod=mod ### global established in upstream functions
speciescode = species_code
except Exception:
Mod=MOD
speciescode = Species
if source_data == Mod:
source_is_mod = True
else:
source_is_mod = False
mod_source = Mod+'-'+source_data+'.txt'
gene_to_source_id = gene_associations.getGeneToUid(speciescode,('hide',mod_source))
source_to_gene = OBO_import.swapKeyValues(gene_to_source_id)
if input_type != 'LineageProfiler':
if source_is_mod == True:
try: id_db[t[0]] = float(t[2])
except Exception: id_db[t[0]] = 'NA'
elif t[0] in source_to_gene:
mod_ids = source_to_gene[t[0]]
for mod_id in mod_ids:
try: value = t[2]
except Exception: value = 'NA'
if value == '+': value = 1
elif value == '-': value = -1
try: id_db[mod_id] = float(value) ### If multiple Ensembl IDs in dataset, only record the last associated fold change
except Exception: id_db[mod_id] = 'NA'
break
else:
id_db[t[0]]= map(float,t[1:]) ### Applies to LineageProfiler
x+=1
#print len(id_db),column_headers
return id_db,column_headers
def getColoredPathwayTest():
fileType = 'png' ### svg, pdf
graphId="ffffff90"; wpid = "WP2062"; color = "0000ff"
graphId=["ffffff90","ffffffe5"]
color = ["0000ff","0000ff"]
### revision = 0 is the most current version
#file = client.service.getColoredPathway(pwId=wpid,revision=0,graphId=graphId,color=color,fileType=fileType)
kwargs = {
'identifier': 'WP2062',
'version': 0,
'element_identifiers': ["ffffff90","ffffffe5"],
'colors': ["#0000FF","#0000FF"],
'file_format': 'image/svg+xml'}
file = wikipathways_api_client_instance.get_colored_pathway(identifier=wpid,version=0,element_identifiers=graphId,colors=color,file_format=fileType)
#file = base64.b64decode(file) ### decode this file
outfile = export.ExportFile(wpid+'.png')
outfile.write(file); outfile.close()
def getAllSpeciesPathways(species_full):
#import GO_Elite
#species_names = GO_Elite.remoteSpeciesData()
#species_full = string.replace(species_names[species_code],'_',' ')
#species_full = 'Mycobacterium tuberculosis'; pathway_db = {}
#pathways_all = client.service.listPathways(organism = species_full)
pathways_all = wikipathways_api_client_instance.list_pathways(organism = species_full)
pathway_db={}
for pathway in pathways_all:
#wpid = pathway[0]; wpname = pathway[2]
wpid = pathway['identifier']
wpname = pathway['name']
pathway_db[wpid] = PathwayData(wpname)
return pathway_db
if __name__ == '__main__':
pathway_db = getAllSpeciesPathways('Homo sapiens');
for i in pathway_db:
print i
getPathwayAs(pathway_db,'','');sys.exit()
getColoredPathwayTest();sys.exit()
filename = "/Users/saljh8/Desktop/PCBC_MetaData_Comparisons/AltAnalyzeExon/Methylation_Variance/GO-Elite_adjp-2fold/regulated/GE.poor_vs_good-fold2.0_adjp0.05.txt"
visualizePathwayAssociations(filename,'Hs','Ensembl','WP2857')
#viewLineageProfilerResults(filename,[]); sys.exit()
filename = "/Users/nsalomonis/Desktop/code/AltAnalyze/datasets/3'Array/Merrill/GO-Elite/input/GE.ko_vs_wt.txt"
pathway_db = getAllSpeciesPathways('Homo sapiens')
for i in pathway_db:
print i, pathway_db[i].WPName(), len(pathway_db)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/WikiPathways_webservice.py
|
WikiPathways_webservice.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
#import matplotlib
#matplotlib.use('GTKAgg')
import sys,string,os,copy
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>0 and '--' in command_args: commandLine=True
else: commandLine=False
display_label_names = True
import platform
useDefaultBackend=False
if platform.system()=='Darwin':
if platform.mac_ver()[0] == '10.14.6':
useDefaultBackend=True
import traceback
try:
import math
import warnings
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
import matplotlib
if commandLine and 'linux' in sys.platform:
### TkAgg doesn't work when AltAnalyze is run remotely (ssh or sh script)
try: matplotlib.use('Agg');
except Exception: pass
try:
matplotlib.rcParams['backend'] = 'Agg'
except Exception: pass
else:
try:
if useDefaultBackend == False:
import matplotlib.backends.backend_tkagg
matplotlib.use('TkAgg')
except Exception: pass
if useDefaultBackend == False:
try: matplotlib.rcParams['backend'] = 'TkAgg'
except Exception: pass
try:
import matplotlib.pyplot as pylab
import matplotlib.colors as mc
import matplotlib.mlab as mlab
import matplotlib.ticker as tic
from matplotlib.patches import Circle
import mpl_toolkits
from mpl_toolkits import mplot3d
from mpl_toolkits.mplot3d import Axes3D
try: from matplotlib.cbook import _string_to_bool
except: pass
matplotlib.rcParams['axes.linewidth'] = 0.5
matplotlib.rcParams['pdf.fonttype'] = 42
matplotlib.rcParams['font.family'] = 'sans-serif'
matplotlib.rcParams['font.sans-serif'] = 'Arial'
matplotlib.rcParams['figure.facecolor'] = 'white' ### Added in 2.1.2
except Exception:
print traceback.format_exc()
print 'Matplotlib support not enabled'
import scipy
try: from scipy.sparse.csgraph import _validation
except Exception: pass
try:
from scipy.linalg import svd
import scipy.special._ufuncs_cxx
from scipy.spatial import _voronoi
from scipy.spatial import _spherical_voronoi
from scipy.spatial import qhull
import scipy._lib.messagestream
except Exception:
pass
import scipy.cluster.hierarchy as sch
import scipy.spatial.distance as dist
#import scipy.interpolate.interpnd
#from scipy import optimize
try: import numpy; np = numpy
except:
print 'Numpy import error...'
print traceback.format_exc()
### The below is used for binary freeze dependency identification
if 'darwin' in sys.platform:
### The below is used for binary freeze dependency identification
try: import umap
except: pass
try:
from cairo import ImageSurface
except: pass
try:
import igraph.vendor.texttable
except ImportError: pass
try:
from sklearn.decomposition import PCA, FastICA
except Exception: pass
try: from sklearn.neighbors import quad_tree ### supported in sklearn>18.2
except: pass
try: import sklearn.utils.sparsetools._graph_validation
except: pass
try: import sklearn.utils.weight_vector
except: pass
from sklearn.neighbors import *
from sklearn.manifold.t_sne import *
from sklearn.tree import *; from sklearn.tree import _utils
from sklearn.manifold.t_sne import _utils
from sklearn.manifold import TSNE
from sklearn.neighbors import NearestNeighbors
import sklearn.linear_model.sgd_fast
import sklearn.utils.lgamma
try: import scipy.special.cython_special
except: pass
import sklearn.neighbors.typedefs
import sklearn.neighbors.ball_tree
try:
import numba
import numba.config
import llvmlite; from llvmlite import binding; from llvmlite.binding import *
from llvmlite.binding import ffi; from llvmlite.binding import dylib
except:
pass
#pylab.ion() # closes Tk window after show - could be nice to include
except Exception:
print traceback.format_exc()
pass
try: import numpy
except: pass
import time
import unique
from stats_scripts import statistics
import os
import export
import webbrowser
import warnings
import UI
use_default_colors = False
try:
warnings.simplefilter("ignore", numpy.ComplexWarning)
warnings.simplefilter("ignore", DeprecationWarning) ### Annoying depreciation warnings (occurs in sch somewhere)
#This shouldn't be needed in python 2.7 which suppresses DeprecationWarning - Larsson
except Exception: None
from visualization_scripts import WikiPathways_webservice
try:
import fastcluster as fc
#print 'Using fastcluster instead of scipy hierarchical cluster'
#fc = sch
except Exception:
#print 'Using scipy insteady of fastcluster (not installed)'
try: fc = sch ### fastcluster uses the same convention names for linkage as sch
except Exception: print 'Scipy support not present...'
def getColorRange(x):
""" Determines the range of colors, centered at zero, for normalizing cmap """
vmax=x.max()
vmin=x.min()
if vmax<0 and vmin<0: direction = 'negative'
elif vmax>0 and vmin>0: direction = 'positive'
else: direction = 'both'
if direction == 'both':
vmax = max([vmax,abs(vmin)])
vmin = -1*vmax
return vmax,vmin
else:
return vmax,vmin
def heatmap(x, row_header, column_header, row_method, column_method, row_metric, column_metric, color_gradient,
dataset_name, display=False, contrast=None, allowAxisCompression=True,Normalize=True,
PriorColumnClusters=None, PriorRowClusters=None):
print "Performing hieararchical clustering using %s for columns and %s for rows" % (column_metric,row_metric)
show_color_bars = True ### Currently, the color bars don't exactly reflect the dendrogram colors
try: ExportCorreleationMatrix = exportCorreleationMatrix
except Exception: ExportCorreleationMatrix = False
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
if display == False:
pylab.figure() ### Add this to avoid a Tkinter bug after running MarkerFinder (not sure why it is needed) - creates a second empty window when display == True
if row_method == 'hopach' or column_method == 'hopach':
### Test R and hopach
"""
try:
import R_test
except Exception,e:
#print traceback.format_exc()
print 'Failed to install hopach or R not installed (install R before using hopach)'
row_method = 'average'; column_method = 'average'
if len(column_header)==2: column_method = 'average'
if len(row_header)==2: row_method = 'average'
"""
pass
"""
Prototype methods:
http://old.nabble.com/How-to-plot-heatmap-with-matplotlib--td32534593.html
http://stackoverflow.com/questions/7664826/how-to-get-flat-clustering-corresponding-to-color-clusters-in-the-dendrogram-cre
Scaling the color gradient so that zero is white:
http://stackoverflow.com/questions/2369492/generate-a-heatmap-in-matplotlib-using-a-scatter-data-set
Other cluster methods:
http://stackoverflow.com/questions/9362304/how-to-get-centroids-from-scipys-hierarchical-agglomerative-clustering
x is a m by n ndarray, m observations, n genes
"""
### Perform the associated clustering by HOPACH via PYPE or Rpy to R
if row_method == 'hopach' or column_method == 'hopach':
try:
""" HOPACH is a clustering method implemented in R that builds a hierarchical tree of clusters by recursively
partitioning a data set, while ordering and possibly collapsing clusters at each level:
http://www.bioconductor.org/packages/release/bioc/html/hopach.html
"""
import R_interface
#reload(R_interface)
if row_method == 'hopach' and column_method == 'hopach': cluster_method = 'both'
elif row_method == 'hopach': cluster_method = 'gene'
else: cluster_method = 'array'
if row_metric == 'cosine': metric_gene = "euclid"
elif row_metric == 'euclidean': metric_gene = "cosangle"
elif row_metric == 'correlation': metric_gene = "cor"
else: metric_gene = "cosangle"
if column_metric == 'cosine': metric_array = "euclid"
elif column_metric == 'euclidean': metric_array = "cosangle"
elif column_metric == 'correlation': metric_array = "cor"
else: metric_array = "euclid"
### Returned are the row_order and column_order in the Scipy clustering output format
newFilename, Z1, Z2 = R_interface.remoteHopach(inputFilename,cluster_method,metric_gene,metric_array)
if newFilename != inputFilename:
### If there were duplicates, re-import the matrix data for the cleaned up filename
try:
matrix, column_header, row_header, dataset_name, group_db = importData(newFilename,Normalize=normalize,reverseOrder=False)
except Exception:
matrix, column_header, row_header, dataset_name, group_db = importData(newFilename)
x = numpy.array(matrix)
except Exception:
row_method = 'average'; column_method = 'average'
print traceback.format_exc()
print 'hopach failed... continue with an alternative method'
skipClustering = False
try:
if len(PriorColumnClusters)>0 and len(PriorRowClusters)>0 and row_method==None and column_method == None:
print 'Prior generated clusters being used rather re-clustering'
"""
try:
if len(targetGeneIDs)>0:
PriorColumnClusters=[] ### If orderded genes input, we want to retain this order rather than change
except Exception: pass
"""
if len(PriorColumnClusters)>0: ### this corresponds to the above line
Z1={}; Z2={}
Z1['level'] = PriorRowClusters; Z1['level'].reverse()
Z2['level'] = PriorColumnClusters; #Z2['level'].reverse()
Z1['leaves'] = range(0,len(row_header)); #Z1['leaves'].reverse()
Z2['leaves'] = range(0,len(column_header)); #Z2['leaves'].reverse()
skipClustering = True
### When clusters are imported, you need something other than None, otherwise, you need None (need to fix here)
row_method = None
column_method = None
row_method = 'hopach'
column_method = 'hopach'
except Exception,e:
#print traceback.format_exc()
pass
n = len(x[0]); m = len(x)
if color_gradient == 'red_white_blue':
cmap=pylab.cm.bwr
if color_gradient == 'red_black_sky':
cmap=RedBlackSkyBlue()
if color_gradient == 'red_black_blue':
cmap=RedBlackBlue()
if color_gradient == 'red_black_green':
cmap=RedBlackGreen()
if color_gradient == 'yellow_black_blue':
cmap=YellowBlackBlue()
if color_gradient == 'black_yellow_blue':
cmap=BlackYellowBlue()
if color_gradient == 'seismic':
cmap=pylab.cm.seismic
if color_gradient == 'green_white_purple':
cmap=pylab.cm.PiYG_r
if color_gradient == 'coolwarm':
cmap=pylab.cm.coolwarm
if color_gradient == 'Greys':
cmap=pylab.cm.Greys
if color_gradient == 'yellow_orange_red':
cmap=pylab.cm.YlOrRd
vmin=x.min()
vmax=x.max()
vmax = max([vmax,abs(vmin)])
if Normalize != False:
vmin = vmax*-1
elif 'Clustering-Zscores-' in dataset_name:
vmin = vmax*-1
elif vmin<0 and vmax>0 and Normalize==False:
vmin = vmax*-1
#vmin = vmax*-1
#print vmax, vmin
default_window_hight = 8.5
default_window_width = 12
if len(column_header)>80:
default_window_width = 14
if len(column_header)>100:
default_window_width = 16
if contrast == None:
scaling_factor = 2.5 #2.5
else:
try: scaling_factor = float(contrast)
except Exception: scaling_factor = 2.5
#print vmin/scaling_factor
norm = matplotlib.colors.Normalize(vmin/scaling_factor, vmax/scaling_factor) ### adjust the max and min to scale these colors by 2.5 (1 scales to the highest change)
#fig = pylab.figure(figsize=(default_window_width,default_window_hight)) ### could use m,n to scale here
fig = pylab.figure() ### could use m,n to scale here - figsize=(12,10)
fig.set_figwidth(12)
fig.set_figheight(7)
fig.patch.set_facecolor('white')
pylab.rcParams['font.size'] = 7.5
#pylab.rcParams['axes.facecolor'] = 'white' ### Added in 2.1.2
if show_color_bars == False:
color_bar_w = 0.000001 ### Invisible but not gone (otherwise an error persists)
else:
color_bar_w = 0.0125 ### Sufficient size to show
bigSampleDendrogram = True
if bigSampleDendrogram == True and row_method==None and column_method != None and allowAxisCompression == True:
dg2 = 0.30
dg1 = 0.43
else: dg2 = 0.1; dg1 = 0.63
try:
if EliteGeneSets != [''] and EliteGeneSets !=[]:
matrix_horiz_pos = 0.27
elif skipClustering:
if len(row_header)<100:
matrix_horiz_pos = 0.20
else:
matrix_horiz_pos = 0.27
else:
matrix_horiz_pos = 0.14
except Exception:
matrix_horiz_pos = 0.14
""" Adjust the position of the heatmap based on the number of columns """
if len(column_header)<50:
matrix_horiz_pos+=0.1
## calculate positions for all elements
# ax1, placement of dendrogram 1, on the left of the heatmap
[ax1_x, ax1_y, ax1_w, ax1_h] = [0.05,0.235,matrix_horiz_pos,dg1] ### The last controls matrix hight, second value controls the position of the matrix relative to the bottom of the view [0.05,0.22,0.2,0.6]
width_between_ax1_axr = 0.004
height_between_ax1_axc = 0.004 ### distance between the top color bar axis and the matrix
# axr, placement of row side colorbar
[axr_x, axr_y, axr_w, axr_h] = [0.31,0.1,color_bar_w-0.002,0.6] ### second to last controls the width of the side color bar - 0.015 when showing [0.31,0.1,color_bar_w,0.6]
axr_x = ax1_x + ax1_w + width_between_ax1_axr
axr_y = ax1_y; axr_h = ax1_h
width_between_axr_axm = 0.004
# axc, placement of column side colorbar (3rd value controls the width of the matrix!)
[axc_x, axc_y, axc_w, axc_h] = [0.5,0.63,0.5,color_bar_w] ### last one controls the hight of the top color bar - 0.015 when showing [0.4,0.63,0.5,color_bar_w]
""" Adjust the width of the heatmap based on the number of columns """
if len(column_header)<50:
axc_w = 0.3
if len(column_header)<20:
axc_w = 0.2
axc_x = axr_x + axr_w + width_between_axr_axm
axc_y = ax1_y + ax1_h + height_between_ax1_axc
height_between_axc_ax2 = 0.004
# axm, placement of heatmap for the data matrix
[axm_x, axm_y, axm_w, axm_h] = [0.4,0.9,2.5,0.5] #[0.4,0.9,2.5,0.5]
axm_x = axr_x + axr_w + width_between_axr_axm
axm_y = ax1_y; axm_h = ax1_h
axm_w = axc_w
# ax2, placement of dendrogram 2, on the top of the heatmap
[ax2_x, ax2_y, ax2_w, ax2_h] = [0.3,0.72,0.6,dg2] ### last one controls hight of the dendrogram [0.3,0.72,0.6,0.135]
ax2_x = axr_x + axr_w + width_between_axr_axm
ax2_y = ax1_y + ax1_h + height_between_ax1_axc + axc_h + height_between_axc_ax2
ax2_w = axc_w
# axcb - placement of the color legend
[axcb_x, axcb_y, axcb_w, axcb_h] = [0.02,0.938,0.17,0.025] ### Last one controls the hight [0.07,0.88,0.18,0.076]
# axcc - placement of the colum colormap legend colormap (distinct map)
[axcc_x, axcc_y, axcc_w, axcc_h] = [0.02,0.12,0.17,0.025] ### Last one controls the hight [0.07,0.88,0.18,0.076]
# Compute and plot top dendrogram
if column_method == 'hopach':
ind2 = numpy.array(Z2['level']) ### from R_interface - hopach root cluster level
elif column_method != None:
start_time = time.time()
#print x;sys.exit()
d2 = dist.pdist(x.T)
#print d2
#import mdistance2
#d2 = mdistance2.mpdist(x.T)
#print d2;sys.exit()
D2 = dist.squareform(d2)
ax2 = fig.add_axes([ax2_x, ax2_y, ax2_w, ax2_h], frame_on=False)
if ExportCorreleationMatrix:
new_matrix=[]
for i in D2:
#string.join(map(inverseDist,i),'\t')
log2_data = map(inverseDist,i)
avg = statistics.avg(log2_data)
log2_norm = map(lambda x: x-avg,log2_data)
new_matrix.append(log2_norm)
x = numpy.array(new_matrix)
row_header = column_header
#sys.exit()
Y2 = fc.linkage(D2, method=column_method, metric=column_metric) ### array-clustering metric - 'average', 'single', 'centroid', 'complete'
#Y2 = sch.fcluster(Y2, 10, criterion = "maxclust")
try: Z2 = sch.dendrogram(Y2)
except Exception:
if column_method == 'average':
column_metric = 'euclidean'
else: column_method = 'average'
Y2 = fc.linkage(D2, method=column_method, metric=column_metric)
Z2 = sch.dendrogram(Y2)
#ind2 = sch.fcluster(Y2,0.6*D2.max(), 'distance') ### get the correlations
#ind2 = sch.fcluster(Y2,0.2*D2.max(), 'maxclust') ### alternative method biased based on number of clusters to obtain (like K-means)
ind2 = sch.fcluster(Y2,0.7*max(Y2[:,2]),'distance') ### This is the default behavior of dendrogram
ax2.set_xticks([]) ### Hides ticks
ax2.set_yticks([])
time_diff = str(round(time.time()-start_time,1))
print 'Column clustering completed in %s seconds' % time_diff
else:
ind2 = ['NA']*len(column_header) ### Used for exporting the flat cluster data
# Compute and plot left dendrogram
if row_method == 'hopach':
ind1 = numpy.array(Z1['level']) ### from R_interface - hopach root cluster level
elif row_method != None:
start_time = time.time()
d1 = dist.pdist(x)
D1 = dist.squareform(d1) # full matrix
# postion = [left(x), bottom(y), width, height]
#print D1;sys.exit()
Y1 = fc.linkage(D1, method=row_method, metric=row_metric) ### gene-clustering metric - 'average', 'single', 'centroid', 'complete'
no_plot=False ### Indicates that we want to show the dendrogram
try:
if runGOElite: no_plot = True
elif len(PriorColumnClusters)>0 and len(PriorRowClusters)>0 and row_method==None and column_method == None:
no_plot = True ### If trying to instantly view prior results, no dendrogram will be display, but prior GO-Elite can
else:
ax1 = fig.add_axes([ax1_x, ax1_y, ax1_w, ax1_h], frame_on=False) # frame_on may be False - this window conflicts with GO-Elite labels
except Exception:
ax1 = fig.add_axes([ax1_x, ax1_y, ax1_w, ax1_h], frame_on=False) # frame_on may be False
try: Z1 = sch.dendrogram(Y1, orientation='left',no_plot=no_plot) ### This is where plotting occurs - orientation 'right' in old matplotlib
except Exception:
row_method = 'average'
try:
Y1 = fc.linkage(D1, method=row_method, metric=row_metric)
Z1 = sch.dendrogram(Y1, orientation='right',no_plot=no_plot)
except Exception:
row_method = 'ward'
Y1 = fc.linkage(D1, method=row_method, metric=row_metric)
Z1 = sch.dendrogram(Y1, orientation='right',no_plot=no_plot)
#ind1 = sch.fcluster(Y1,0.6*D1.max(),'distance') ### get the correlations
#ind1 = sch.fcluster(Y1,0.2*D1.max(),'maxclust')
ind1 = sch.fcluster(Y1,0.7*max(Y1[:,2]),'distance') ### This is the default behavior of dendrogram
if ExportCorreleationMatrix:
Z1 = sch.dendrogram(Y2, orientation='right')
Y1 = Y2
d1 = d2
D1 = D2
ind1 = ind2
try: ax1.set_xticks([]); ax1.set_yticks([]) ### Hides ticks
except Exception: pass
time_diff = str(round(time.time()-start_time,1))
print 'Row clustering completed in %s seconds' % time_diff
else:
ind1 = ['NA']*len(row_header) ### Used for exporting the flat cluster data
# Plot distance matrix.
axm = fig.add_axes([axm_x, axm_y, axm_w, axm_h]) # axes for the data matrix
xt = x
if column_method != None:
idx2 = Z2['leaves'] ### apply the clustering for the array-dendrograms to the actual matrix data
xt = xt[:,idx2]
#ind2 = ind2[:,idx2] ### reorder the flat cluster to match the order of the leaves the dendrogram
""" Error can occur here if hopach was selected in a prior run but now running NONE """
try: ind2 = [ind2[i] for i in idx2] ### replaces the above due to numpy specific windows version issue
except Exception:
column_method=None
xt = x
ind2 = ['NA']*len(column_header) ### Used for exporting the flat cluster data
ind1 = ['NA']*len(row_header) ### Used for exporting the flat cluster data
if row_method != None:
idx1 = Z1['leaves'] ### apply the clustering for the gene-dendrograms to the actual matrix data
prior_xt = xt
xt = xt[idx1,:] # xt is transformed x
#ind1 = ind1[idx1,:] ### reorder the flat cluster to match the order of the leaves the dendrogram
try: ind1 = [ind1[i] for i in idx1] ### replaces the above due to numpy specific windows version issue
except Exception:
if 'MarkerGenes' in dataset_name:
ind1 = ['NA']*len(row_header) ### Used for exporting the flat cluster data
row_method = None
### taken from http://stackoverflow.com/questions/2982929/plotting-results-of-hierarchical-clustering-ontop-of-a-matrix-of-data-in-python/3011894#3011894
im = axm.matshow(xt, aspect='auto', origin='lower', cmap=cmap, norm=norm) ### norm=norm added to scale coloring of expression with zero = white or black
axm.set_xticks([]) ### Hides x-ticks
axm.set_yticks([])
#axm.set_axis_off() ### Hide border
#fix_verts(ax1,1)
#fix_verts(ax2,0)
### Adjust the size of the fonts for genes and arrays based on size and character length
row_fontsize = 5
column_fontsize = 5
column_text_max_len = max(map(lambda x: len(x), column_header)) ### Get the maximum length of a column annotation
if len(row_header)<75:
row_fontsize = 6.5
if len(row_header)<50:
row_fontsize = 8
if len(row_header)<25:
row_fontsize = 11
if len(column_header)<75:
column_fontsize = 6.5
if len(column_header)<50:
column_fontsize = 8
if len(column_header)<25:
column_fontsize = 11
if column_text_max_len < 15:
column_fontsize = 15
elif column_text_max_len > 30:
column_fontsize = 6.5
else:
column_fontsize = 10
try:
if len(justShowTheseIDs)>50:
column_fontsize = 7
elif len(justShowTheseIDs)>0:
column_fontsize = 10
if len(justShowTheseIDs)>0:
additional_symbols=[]
import gene_associations
try:
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
except Exception: gene_to_symbol={}; symbol_to_gene={}
JustShowTheseIDs = copy.deepcopy(justShowTheseIDs)
except Exception:
JustShowTheseIDs=[]
# Add text
new_row_header=[]
new_column_header=[]
for i in range(x.shape[0]):
if row_method != None:
new_row_header.append(row_header[idx1[i]])
else:
new_row_header.append(row_header[i])
for i in range(x.shape[1]):
if column_method != None:
new_column_header.append(column_header[idx2[i]])
else: ### When not clustering columns
new_column_header.append(column_header[i])
dataset_name = string.replace(dataset_name,'Clustering-','')### clean up the name if already a clustered file
if '-hierarchical' in dataset_name:
dataset_name = string.split(dataset_name,'-hierarchical')[0]
filename = 'Clustering-%s-hierarchical_%s_%s.pdf' % (dataset_name,column_metric,row_metric)
if 'MarkerGenes' in dataset_name:
time_stamp = timestamp() ### Don't overwrite the previous version
filename = string.replace(filename,'hierarchical',time_stamp)
elite_dir, cdt_file, SystemCode = exportFlatClusterData(root_dir + filename, root_dir, dataset_name, new_row_header,new_column_header,xt,ind1,ind2,vmax,display)
def ViewPNG(png_file_dir):
if os.name == 'nt':
try: os.startfile('"'+png_file_dir+'"')
except Exception: os.system('open "'+png_file_dir+'"')
elif 'darwin' in sys.platform: os.system('open "'+png_file_dir+'"')
elif 'linux' in sys.platform: os.system('xdg-open "'+png_file_dir+'"')
try:
try:
temp1=len(justShowTheseIDs)
if 'monocle' in justShowTheseIDs and ('guide' not in justShowTheseIDs):
import R_interface
print 'Running Monocle through R (be patient, this can take 20 minutes+)'
R_interface.performMonocleAnalysisFromHeatmap(species,cdt_file[:-3]+'txt',cdt_file[:-3]+'txt')
png_file_dir = root_dir+'/Monocle/monoclePseudotime.png'
#print png_file_dir
ViewPNG(png_file_dir)
except Exception: pass # no justShowTheseIDs
except Exception:
print '...Monocle error:'
print traceback.format_exc()
pass
cluster_elite_terms={}; ge_fontsize=11.5; top_genes=[]; proceed=True
try:
try:
if 'guide' in justShowTheseIDs: proceed = False
except Exception: pass
if proceed:
try:
cluster_elite_terms,top_genes = remoteGOElite(elite_dir,SystemCode=SystemCode)
if cluster_elite_terms['label-size']>40: ge_fontsize = 9.5
except Exception:
pass
except Exception: pass #print traceback.format_exc()
if len(cluster_elite_terms)<1:
try:
elite_dirs = string.split(elite_dir,'GO-Elite')
old_elite_dir = elite_dirs[0]+'GO-Elite'+elite_dirs[-1] ### There are actually GO-Elite/GO-Elite directories for the already clustered
old_elite_dir = string.replace(old_elite_dir,'ICGS/','')
if len(PriorColumnClusters)>0 and len(PriorRowClusters)>0 and skipClustering:
cluster_elite_terms,top_genes = importGOEliteResults(old_elite_dir)
except Exception,e:
#print traceback.format_exc()
pass
try:
if len(justShowTheseIDs)<1 and len(top_genes) > 0 and column_fontsize < 9:
column_fontsize = 10
if len(justShowTheseIDs)<1:
additional_symbols=[]
import gene_associations; from import_scripts import OBO_import
try:
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
#symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception: gene_to_symbol={}; symbol_to_gene={}
except Exception: pass
def formatpval(p):
if '-' in p: p1=p[:1]+p[-4:]
else:
p1 = '{number:.{digits}f}'.format(number=float(p), digits=3)
p1=str(p1)
#print traceback.format_exc();sys.exit()
return p1
# Add text
new_row_header=[]
new_column_header=[]
ci=0 ### index of entries in the cluster
last_cluster=1
interval = int(float(string.split(str(len(row_header)/35.0),'.')[0]))+1 ### for enrichment term labels with over 100 genes
increment=interval-2
if len(row_header)<100: increment = interval-1
label_pos=-0.03*len(column_header)-.8
alternate=1
#print label_pos
try:
if 'top' in justShowTheseIDs: justShowTheseIDs.remove('top')
if 'positive' in justShowTheseIDs: justShowTheseIDs.remove('positive')
if 'amplify' in justShowTheseIDs: justShowTheseIDs.remove('amplify')
if 'IntraCorrelatedOnly' in justShowTheseIDs: justShowTheseIDs.remove('IntraCorrelatedOnly')
if 'GuideOnlyCorrelation' in justShowTheseIDs: justShowTheseIDs.remove('GuideOnlyCorrelation')
except Exception:
pass
for i in range(x.shape[0]):
if len(row_header)<40:
radj = len(row_header)*0.009 ### row offset value to center the vertical position of the row label
elif len(row_header)<70:
radj = len(row_header)*0.007 ### row offset value to center the vertical position of the row label
else:
radj = len(row_header)*0.005
try: cluster = str(ind1[i])
except Exception: cluster = 'NA'
if cluster == 'NA':
new_index = i
try: cluster = 'cluster-'+string.split(row_header[new_index],':')[0]
except Exception: pass
if cluster != last_cluster:
ci=0
increment=0
#print cluster,i,row_header[idx1[i]]
color = 'black'
if row_method != None:
try:
if row_header[idx1[i]] in JustShowTheseIDs:
if len(row_header)>len(justShowTheseIDs):
color = 'red'
else: color = 'black'
except Exception: pass
if len(row_header)<106: ### Don't visualize gene associations when more than 100 rows
if display_label_names == False or 'ticks' in JustShowTheseIDs:
if color=='red':
axm.text(x.shape[1]-0.5, i-radj, ' '+'-',fontsize=row_fontsize, color=color, picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+'',fontsize=row_fontsize, color=color, picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+row_header[idx1[i]],fontsize=row_fontsize, color=color, picker=True)
new_row_header.append(row_header[idx1[i]])
new_index = idx1[i]
else:
try:
feature_id = row_header[i]
if ':' in feature_id:
feature_id = string.split(feature_id,':')[1]
if feature_id[-1]==' ': feature_id = feature_id[:-1]
if feature_id in JustShowTheseIDs:
color = 'red'
else: color = 'black'
except Exception: pass
if len(row_header)<106: ### Don't visualize gene associations when more than 100 rows
if display_label_names == False or 'ticks' in JustShowTheseIDs:
if color=='red':
axm.text(x.shape[1]-0.5, i-radj, ' '+'-',fontsize=row_fontsize, color=color, picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+'',fontsize=row_fontsize, color=color, picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+row_header[i],fontsize=row_fontsize, color=color, picker=True) ### When not clustering rows
new_row_header.append(row_header[i])
new_index = i ### This is different when clustering rows versus not
if len(row_header)<106:
"""
if cluster in cluster_elite_terms:
try:
term = cluster_elite_terms[cluster][ci][1]
axm.text(-1.5, i-radj, term,horizontalalignment='right',fontsize=row_fontsize)
except Exception: pass
ci+=1
"""
pass
else:
feature_id = row_header[new_index]
original_feature_id = feature_id
if ':' in feature_id:
if 'ENS' != feature_id[:3] or 'G0000' in feature_id:
feature_id = string.split(feature_id,':')[1]
if feature_id[-1]==' ': feature_id = feature_id[:-1]
else:
feature_id = string.split(feature_id,':')[0]
try: feature_id = gene_to_symbol[feature_id][0]
except Exception: pass
if (' ' in feature_id and ('ENS' in feature_id or 'G0000' in feature_id)):
feature_id = string.split(feature_id,' ')[1]
try:
if feature_id in JustShowTheseIDs or original_feature_id in JustShowTheseIDs: color = 'red'
else: color = 'black'
except Exception: pass
try:
if feature_id in justShowTheseIDs or (len(justShowTheseIDs)<1 and feature_id in top_genes) or original_feature_id in justShowTheseIDs:
if original_feature_id in justShowTheseIDs:
feature_id = original_feature_id
if display_label_names and 'ticks' not in justShowTheseIDs:
if alternate==1: buffer=1.2; alternate=2
elif alternate==2: buffer=2.4; alternate=3
elif alternate==3: buffer=3.6; alternate=4
elif alternate==4: buffer=0; alternate=1
axm.text(x.shape[1]-0.4+buffer, i-radj, feature_id,fontsize=column_fontsize, color=color,picker=True) ### When not clustering rows
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+"-",fontsize=column_fontsize, color=color,picker=True) ### When not clustering rows
elif ' ' in row_header[new_index]:
symbol = string.split(row_header[new_index], ' ')[-1]
if len(symbol)>0:
if symbol in justShowTheseIDs:
if display_label_names and 'ticks' not in justShowTheseIDs:
axm.text(x.shape[1]-0.5, i-radj, ' '+row_header[new_index],fontsize=column_fontsize, color=color,picker=True)
else:
axm.text(x.shape[1]-0.5, i-radj, ' '+"-",fontsize=column_fontsize, color=color,picker=True)
except Exception: pass
if cluster in cluster_elite_terms or 'cluster-'+cluster in cluster_elite_terms:
if 'cluster-'+cluster in cluster_elite_terms:
new_cluster_id = 'cluster-'+cluster
else:
new_cluster_id = cluster
if cluster != last_cluster:
cluster_intialized = False
try:
increment+=1
#print [increment,interval,cluster],cluster_elite_terms[cluster][ci][1];sys.exit()
#if increment == interval or (
#print increment,interval,len(row_header),cluster_intialized
if (increment == interval) or (len(row_header)>200 and increment == (interval-9) and cluster_intialized==False): ### second argument brings the label down
cluster_intialized=True
atypical_cluster = False
if ind1[i+9] == 'NA': ### This occurs for custom cluster, such MarkerFinder (not cluster numbers)
atypical_cluster = True
cluster9 = 'cluster-'+string.split(row_header[new_index+9],':')[0]
if (len(row_header)>200 and str(cluster9)!=cluster): continue
elif (len(row_header)>200 and str(ind1[i+9])!=cluster): continue ### prevents the last label in a cluster from overlapping with the first in the next cluster
pvalue,original_term = cluster_elite_terms[new_cluster_id][ci]
term = original_term
if 'GO:' in term:
term = string.split(term, '(')[0]
if ':WP' in term:
term = string.split(term, ':WP')[0]
pvalue = formatpval(str(pvalue))
term += ' p='+pvalue
if atypical_cluster == False:
term += ' (c'+str(cluster)+')'
try: cluster_elite_terms[term] = cluster_elite_terms[cluster,original_term] ### store the new term name with the associated genes
except Exception: pass
axm.text(label_pos, i-radj, term,horizontalalignment='right',fontsize=ge_fontsize, picker=True, color = 'blue')
increment=0
ci+=1
except Exception,e:
#print traceback.format_exc();sys.exit()
increment=0
last_cluster = cluster
def onpick1(event):
text = event.artist
print('onpick1 text:', text.get_text())
if 'TreeView' in text.get_text():
try: openTreeView(cdt_file)
except Exception: print 'Failed to open TreeView'
elif 'p=' not in text.get_text():
webbrowser.open('http://www.genecards.org/cgi-bin/carddisp.pl?gene='+string.replace(text.get_text(),' ',''))
else:
#"""
from visualization_scripts import TableViewer
header = ['Associated Genes']
tuple_list = []
for gene in cluster_elite_terms[text.get_text()]:
tuple_list.append([(gene)])
TableViewer.viewTable(text.get_text(),header,tuple_list) #"""
cluster_prefix = 'c'+string.split(text.get_text(),'(c')[1][:-1]+'-'
for geneSet in EliteGeneSets:
if geneSet == 'GeneOntology':
png_file_dir = elite_dir+'/GO-Elite_results/networks/'+cluster_prefix+'GO'+'.png'
elif geneSet == 'WikiPathways':
png_file_dir = elite_dir+'/GO-Elite_results/networks/'+cluster_prefix+'local'+'.png'
elif len(geneSet)>1:
png_file_dir = elite_dir+'/GO-Elite_results/networks/'+cluster_prefix+geneSet+'.png'
#try: UI.GUI(root_dir,'ViewPNG',[],png_file_dir)
#except Exception: print traceback.format_exc()
try:
alt_png_file_dir = elite_dir+'/GO-Elite_results/networks/'+cluster_prefix+eliteGeneSet+'.png'
png_file_dirs = string.split(alt_png_file_dir,'GO-Elite/')
alt_png_file_dir = png_file_dirs[0]+'GO-Elite/'+png_file_dirs[-1]
except Exception: pass
if os.name == 'nt':
try: os.startfile('"'+png_file_dir+'"')
except Exception:
try: os.system('open "'+png_file_dir+'"')
except Exception: os.startfile('"'+alt_png_file_dir+'"')
elif 'darwin' in sys.platform:
try: os.system('open "'+png_file_dir+'"')
except Exception: os.system('open "'+alt_png_file_dir+'"')
elif 'linux' in sys.platform:
try: os.system('xdg-open "'+png_file_dir+'"')
except Exception: os.system('xdg-open "'+alt_png_file_dir+'"')
#print cluster_elite_terms[text.get_text()]
fig.canvas.mpl_connect('pick_event', onpick1)
for i in range(x.shape[1]):
adji = i
### Controls the vertical position of the column (array) labels
if len(row_header)<3:
cadj = len(row_header)*-0.26 ### column offset value
elif len(row_header)<4:
cadj = len(row_header)*-0.23 ### column offset value
elif len(row_header)<6:
cadj = len(row_header)*-0.18 ### column offset value
elif len(row_header)<10:
cadj = len(row_header)*-0.08 ### column offset value
elif len(row_header)<15:
cadj = len(row_header)*-0.04 ### column offset value
elif len(row_header)<20:
cadj = len(row_header)*-0.05 ### column offset value
elif len(row_header)<22:
cadj = len(row_header)*-0.06 ### column offset value
elif len(row_header)<23:
cadj = len(row_header)*-0.08 ### column offset value
elif len(row_header)>200:
cadj = -2
else:
cadj = -0.9
#cadj = -1
if len(column_header)>15:
adji = i-0.1 ### adjust the relative position of the column label horizontally
if len(column_header)>20:
adji = i-0.2 ### adjust the relative position of the column label horizontally
if len(column_header)>25:
adji = i-0.2 ### adjust the relative position of the column label horizontally
if len(column_header)>30:
adji = i-0.25 ### adjust the relative position of the column label horizontally
if len(column_header)>35:
adji = i-0.3 ### adjust the relative position of the column label horizontally
if len(column_header)>200:
column_fontsize = 2
if column_method != None:
if len(column_header)<300: ### Don't show the headers when too many values exist
axm.text(adji, cadj, ''+column_header[idx2[i]], rotation=270, verticalalignment="top",fontsize=column_fontsize) # rotation could also be degrees
new_column_header.append(column_header[idx2[i]])
else: ### When not clustering columns
if len(column_header)<300: ### Don't show the headers when too many values exist
axm.text(adji, cadj, ''+column_header[i], rotation=270, verticalalignment="top",fontsize=column_fontsize)
new_column_header.append(column_header[i])
# Plot colside colors
# axc --> axes for column side colorbar
group_name_list=[]
ind1_clust,ind2_clust = ind1,ind2
ind1,ind2,group_name_list,cb_status = updateColorBarData(ind1,ind2,new_column_header,new_row_header,row_method)
if (column_method != None or 'column' in cb_status) and show_color_bars == True:
axc = fig.add_axes([axc_x, axc_y, axc_w, axc_h]) # axes for column side colorbar
cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF', '#CCCCE0','#000066','#FFFF00', '#FF1493'])
if use_default_colors:
cmap_c = pylab.cm.nipy_spectral
else:
#cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF','#FFFF00', '#FF1493'])
if len(unique.unique(ind2))==2: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
#cmap_c = matplotlib.colors.ListedColormap(['#7CFC00','k'])
cmap_c = matplotlib.colors.ListedColormap(['w', 'k'])
elif len(unique.unique(ind2))==3: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
#cmap_c = matplotlib.colors.ListedColormap(['r', 'y', 'b'])
elif len(unique.unique(ind2))==4: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C','#FEBC18'])
#cmap_c = matplotlib.colors.ListedColormap(['k', 'w', 'w', 'w'])
elif len(unique.unique(ind2))==5: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2))==6: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
#cmap_c = matplotlib.colors.ListedColormap(['black', '#1DA532', '#88BF47','b', 'grey','r'])
#cmap_c = matplotlib.colors.ListedColormap(['w', '#0B9B48', 'w', '#5D82C1','#4CB1E4','#71C065'])
#cmap_c = matplotlib.colors.ListedColormap(['w', 'w', 'k', 'w','w','w'])
elif len(unique.unique(ind2))==7: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
#cmap_c = matplotlib.colors.ListedColormap(['w', 'w', 'w', 'k', 'w','w','w'])
#cmap_c = matplotlib.colors.ListedColormap(['w','w', '#0B9B48', 'w', '#5D82C1','#4CB1E4','#71C065'])
#elif len(unique.unique(ind2))==9: cmap_c = matplotlib.colors.ListedColormap(['k', 'w', 'w', 'w', 'w', 'w', 'w', 'w', 'w'])
#elif len(unique.unique(ind2))==11:
#cmap_c = matplotlib.colors.ListedColormap(['w', '#DC2342', '#0B9B48', '#FDDF5E', '#E0B724', 'k', '#5D82C1', '#F79020', '#4CB1E4', '#983894', '#71C065'])
elif len(unique.unique(ind2))>0: ### cmap_c is too few colors
cmap_c = pylab.cm.nipy_spectral
dc = numpy.array(ind2, dtype=int)
dc.shape = (1,len(ind2))
im_c = axc.matshow(dc, aspect='auto', origin='lower', cmap=cmap_c)
axc.set_xticks([]) ### Hides ticks
if 'hopach' == column_method and len(group_name_list)>0:
axc.set_yticklabels(['','Groups'],fontsize=10)
else:
axc.set_yticks([])
#axc.set_frame_on(False) ### Hide border
if len(group_name_list)>0: ### Add a group color legend key
if 'hopach' == column_method: ### allows us to add the second color bar
axcd = fig.add_axes([ax2_x, ax2_y, ax2_w, color_bar_w]) # dendrogram coordinates with color_bar_w substituted - can use because dendrogram is not used
cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF', '#CCCCE0','#000066','#FFFF00', '#FF1493'])
#cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF','#FFFF00', '#FF1493'])
if use_default_colors:
cmap_c = pylab.cm.nipy_spectral
else:
if len(unique.unique(ind2_clust))==2: ### cmap_c is too few colors
#cmap_c = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
cmap_c = matplotlib.colors.ListedColormap(['w', 'k'])
elif len(unique.unique(ind2_clust))==3: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
#cmap_c = matplotlib.colors.ListedColormap(['r', 'y', 'b'])
elif len(unique.unique(ind2_clust))==4: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
#cmap_c = matplotlib.colors.ListedColormap(['black', '#1DA532', 'b','r'])
elif len(unique.unique(ind2_clust))==5: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2_clust))==6: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2_clust))==7: ### cmap_c is too few colors
cmap_c = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2_clust))>0: ### cmap_c is too few colors
cmap_c = pylab.cm.nipy_spectral
dc = numpy.array(ind2_clust, dtype=int)
dc.shape = (1,len(ind2_clust))
im_cd = axcd.matshow(dc, aspect='auto', origin='lower', cmap=cmap_c)
#axcd.text(-1,-1,'clusters')
axcd.set_yticklabels(['','Clusters'],fontsize=10)
#pylab.yticks(range(1),['HOPACH clusters'])
axcd.set_xticks([]) ### Hides ticks
#axcd.set_yticks([])
axd = fig.add_axes([axcc_x, axcc_y, axcc_w, axcc_h])
group_name_list.sort()
group_colors = map(lambda x: x[0],group_name_list)
group_names = map(lambda x: x[1],group_name_list)
cmap_d = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF', '#CCCCE0','#000066','#FFFF00', '#FF1493'])
#cmap_d = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF','#FFFF00', '#FF1493'])
if len(unique.unique(ind2))==2: ### cmap_c is too few colors
#cmap_d = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
cmap_d = matplotlib.colors.ListedColormap(['w', 'k'])
elif len(unique.unique(ind2))==3: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
#cmap_d = matplotlib.colors.ListedColormap(['r', 'y', 'b'])
elif len(unique.unique(ind2))==4: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
elif len(unique.unique(ind2))==5: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif len(unique.unique(ind2))==6: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
#cmap_d = matplotlib.colors.ListedColormap(['black', '#1DA532', '#88BF47','b', 'grey','r'])
#cmap_d = matplotlib.colors.ListedColormap(['w', '#0B9B48', 'w', '#5D82C1','#4CB1E4','#71C065'])
#cmap_d = matplotlib.colors.ListedColormap(['w', 'w', 'k', 'w', 'w','w','w'])
elif len(unique.unique(ind2))==7: ### cmap_c is too few colors
cmap_d = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
#cmap_d = matplotlib.colors.ListedColormap(['w', 'w', 'w', 'k', 'w','w','w'])
#cmap_d = matplotlib.colors.ListedColormap(['w','w', '#0B9B48', 'w', '#5D82C1','#4CB1E4','#71C065'])
#elif len(unique.unique(ind2))==10: cmap_d = matplotlib.colors.ListedColormap(['w', 'w', 'w', 'w', 'w', 'w', 'w', 'w', 'w', 'k'])
#elif len(unique.unique(ind2))==11:
#Eryth Gfi1 Gran HSCP-1 HSCP-2 IG2 MDP Meg Mono Multi-Lin Myelo
#cmap_d = matplotlib.colors.ListedColormap(['#DC2342', 'k', '#0B9B48', '#FDDF5E', '#E0B724', 'w', '#5D82C1', '#F79020', '#4CB1E4', '#983894', '#71C065'])
elif len(unique.unique(ind2))>0: ### cmap_c is too few colors
cmap_d = pylab.cm.nipy_spectral
dc = numpy.array(group_colors, dtype=int)
dc.shape = (1,len(group_colors))
im_c = axd.matshow(dc, aspect='auto', origin='lower', cmap=cmap_d)
axd.set_yticks([])
#axd.set_xticklabels(group_names, rotation=45, ha='left')
#if len(group_names)<200:
pylab.xticks(range(len(group_names)),group_names,rotation=45,ha='left')
#cmap_c = matplotlib.colors.ListedColormap(map(lambda x: GroupDB[x][-1], new_column_header))
if show_color_bars == False:
axc = fig.add_axes([axc_x, axc_y, axc_w, axc_h]) # axes for column side colorbar
axc.set_frame_on(False)
# Plot rowside colors
# axr --> axes for row side colorbar
if (row_method != None or 'row' in cb_status) and show_color_bars == True:
axr = fig.add_axes([axr_x, axr_y, axr_w, axr_h]) # axes for column side colorbar
try:
dr = numpy.array(ind1, dtype=int)
dr.shape = (len(ind1),1)
#print ind1, len(ind1)
cmap_r = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF', '#FFFF00', '#FF1493'])
if len(unique.unique(ind1))>4: ### cmap_r is too few colors
cmap_r = pylab.cm.nipy_spectral_r
if len(unique.unique(ind1))==2:
cmap_r = matplotlib.colors.ListedColormap(['w', 'k'])
im_r = axr.matshow(dr, aspect='auto', origin='lower', cmap=cmap_r)
axr.set_xticks([]) ### Hides ticks
axr.set_yticks([])
#axr.set_frame_on(False) ### Hide border
except Exception:
row_method = None
pass ### likely occurs for some reason when row_method should be None
if show_color_bars == False:
axr = fig.add_axes([axr_x, axr_y, axr_w, axr_h]) # axes for column side colorbar
axr.set_frame_on(False)
# Plot color legend
axcb = fig.add_axes([axcb_x, axcb_y, axcb_w, axcb_h], frame_on=False) # axes for colorbar
cb = matplotlib.colorbar.ColorbarBase(axcb, cmap=cmap, norm=norm, orientation='horizontal')
#axcb.set_title("colorkey",fontsize=14)
if 'LineageCorrelations' in dataset_name:
cb.set_label("Lineage Correlation Z Scores",fontsize=11)
elif 'Heatmap' in root_dir:
cb.set_label("GO-Elite Z Scores",fontsize=11)
else:
cb.set_label("Differential Expression (log2)",fontsize=10)
### Add filename label to the heatmap
if len(dataset_name)>30:fontsize = 10
else: fontsize = 12.5
fig.text(0.015, 0.970, dataset_name, fontsize = fontsize)
### Render and save the graphic
pylab.savefig(root_dir + filename,dpi=1000)
#print 'Exporting:',filename
filename = filename[:-3]+'png'
pylab.savefig(root_dir + filename, dpi=100) #,dpi=200
includeBackground=False
try:
if 'TkAgg' != matplotlib.rcParams['backend']:
includeBackground = False
except Exception: pass
if includeBackground:
fig.text(0.020, 0.070, 'Open heatmap in TreeView (click here)', fontsize = 11.5, picker=True,color = 'red', backgroundcolor='white')
else:
fig.text(0.020, 0.070, 'Open heatmap in TreeView (click here)', fontsize = 11.5, picker=True,color = 'red')
if 'Outlier' in dataset_name and 'Removed' not in dataset_name:
graphic_link.append(['Hierarchical Clustering - Outlier Genes Genes',root_dir+filename])
elif 'Relative' in dataset_name:
graphic_link.append(['Hierarchical Clustering - Significant Genes (Relative comparisons)',root_dir+filename])
elif 'LineageCorrelations' in filename:
graphic_link.append(['Hierarchical Clustering - Lineage Correlations',root_dir+filename])
elif 'MarkerGenes' in filename:
graphic_link.append(['Hierarchical Clustering - MarkerFinder',root_dir+filename])
elif 'AltExonConfirmed' in filename:
graphic_link.append(['Hierarchical Clustering - AltExonConfirmed',root_dir+filename])
elif 'AltExon' in filename:
graphic_link.append(['Hierarchical Clustering - AltExon',root_dir+filename])
elif 'alt_junction' in filename:
graphic_link.append(['Hierarchical Clustering - Variable Splice-Events',root_dir+filename])
else:
graphic_link.append(['Hierarchical Clustering - Significant Genes',root_dir+filename])
if display:
proceed=True
try:
if 'guide' in justShowTheseIDs:
proceed = False
except Exception: pass
if proceed:
print 'Exporting:',filename
try: pylab.show()
except Exception: None ### when run in headless mode
fig.clf()
#fig.close() causes segfault
#pylab.close() causes segfault
def openTreeView(filename):
import subprocess
fn = filepath("AltDatabase/TreeView/TreeView.jar")
retcode = subprocess.Popen(['java', "-Xmx500m", '-jar', fn, "-r", filename])
def remoteGOElite(elite_dir,SystemCode = None):
mod = 'Ensembl'
if SystemCode == 'Ae':
mod = 'AltExon'
pathway_permutations = 'FisherExactTest'
filter_method = 'z-score'
z_threshold = 1.96
p_val_threshold = 0.05
change_threshold = 0
if runGOElite:
resources_to_analyze = EliteGeneSets
if 'all' in resources_to_analyze:
resources_to_analyze = 'all'
returnPathways = 'no'
root = None
import GO_Elite
reload(GO_Elite)
input_files = dir_list = unique.read_directory(elite_dir) ### Are there any files to analyze?
if len(input_files)>0 and resources_to_analyze !=['']:
print '\nBeginning to run GO-Elite analysis on all results'
file_dirs = elite_dir,None,elite_dir
enrichmentAnalysisType = 'ORA'
#enrichmentAnalysisType = 'URA'
variables = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,enrichmentAnalysisType,root
try: GO_Elite.remoteAnalysis(variables,'non-UI Heatmap')
except Exception: 'GO-Elite failed for:',elite_dir
if commandLine==False:
try: UI.openDirectory(elite_dir+'/GO-Elite_results')
except Exception: None
cluster_elite_terms,top_genes = importGOEliteResults(elite_dir)
return cluster_elite_terms,top_genes
else:
return {},[]
else:
return {},[]
def importGOEliteResults(elite_dir):
global eliteGeneSet
pruned_results = elite_dir+'/GO-Elite_results/CompleteResults/ORA_pruned/pruned-results_z-score_elite.txt' ### This is the exception (not moved)
if os.path.isfile(pruned_results) == False:
pruned_results = elite_dir+'/GO-Elite_results/pruned-results_z-score_elite.txt'
firstLine=True
cluster_elite_terms={}
all_term_length=[0]
for line in open(pruned_results,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
try: symbol_index = values.index('gene symbols')
except Exception: symbol_index = None
else:
try: symbol_index = values.index('gene symbols')
except Exception: pass
try:
eliteGeneSet = string.split(values[0][1:],'-')[1][:-4]
try: cluster = str(int(float(string.split(values[0][1:],'-')[0])))
except Exception:
cluster = string.join(string.split(values[0],'-')[:-1],'-')
term = values[2]
num_genes_changed = int(values[3])
all_term_length.append(len(term))
pval = float(values[9]) ### adjusted is values[10]
#pval = float(values[10]) ### adjusted is values[10]
if num_genes_changed>2:
try: cluster_elite_terms[cluster].append([pval,term])
except Exception: cluster_elite_terms[cluster] = [[pval,term]]
if symbol_index!=None:
symbols = string.split(values[symbol_index],'|')
cluster_elite_terms[cluster,term] = symbols
except Exception,e: pass
for cluster in cluster_elite_terms:
cluster_elite_terms[cluster].sort()
cluster_elite_terms['label-size'] = max(all_term_length)
top_genes = []; count=0
ranked_genes = elite_dir+'/GO-Elite_results/CompleteResults/ORA_pruned/gene_associations/pruned-gene-ranking.txt'
for line in open(ranked_genes,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
count+=1
if len(values)>2:
if values[2]!='Symbol':
try: top_genes.append((int(values[4]),values[2]))
except Exception: pass
top_genes.sort(); top_genes.reverse()
top_genes = map(lambda x: x[1],top_genes[:21])
return cluster_elite_terms,top_genes
def mergeRotateAroundPointPage(page, page2, rotation, tx, ty):
from pyPdf import PdfFileWriter, PdfFileReader
translation = [[1, 0, 0],
[0, 1, 0],
[-tx,-ty,1]]
rotation = math.radians(rotation)
rotating = [[math.cos(rotation), math.sin(rotation),0],
[-math.sin(rotation),math.cos(rotation), 0],
[0, 0, 1]]
rtranslation = [[1, 0, 0],
[0, 1, 0],
[tx,ty,1]]
ctm = numpy.dot(translation, rotating)
ctm = numpy.dot(ctm, rtranslation)
return page.mergeTransformedPage(page2, [ctm[0][0], ctm[0][1],
ctm[1][0], ctm[1][1],
ctm[2][0], ctm[2][1]])
def mergePDFs2(pdf1,pdf2,outPdf):
from pyPdf import PdfFileWriter, PdfFileReader
input1 = PdfFileReader(file(pdf1, "rb"))
page1 = input1.getPage(0)
input2 = PdfFileReader(file(pdf2, "rb"))
page2 = input2.getPage(0)
page3 = mergeRotateAroundPointPage(page1, page2,
page1.get('/Rotate') or 0,
page2.mediaBox.getWidth()/2, page2.mediaBox.getWidth()/2)
output = PdfFileWriter()
output.addPage(page3)
outputStream = file(outPdf, "wb")
output.write(outputStream)
outputStream.close()
def mergePDFs(pdf1,pdf2,outPdf):
# http://stackoverflow.com/questions/6041244/how-to-merge-two-landscape-pdf-pages-using-pypdf
from pyPdf import PdfFileWriter, PdfFileReader
input1 = PdfFileReader(file(pdf1, "rb"))
page1 = input1.getPage(0)
page1.mediaBox.upperRight = (page1.mediaBox.getUpperRight_x(), page1.mediaBox.getUpperRight_y())
input2 = PdfFileReader(file(pdf2, "rb"))
page2 = input2.getPage(0)
page2.mediaBox.getLowerLeft_x = (page2.mediaBox.getLowerLeft_x(), page2.mediaBox.getLowerLeft_y())
# Merge
page2.mergePage(page1)
# Output
output = PdfFileWriter()
output.addPage(page1)
outputStream = file(outPdf, "wb")
output.write(outputStream)
outputStream.close()
"""
def merge_horizontal(out_filename, left_filename, right_filename):
#Merge the first page of two PDFs side-to-side
import pyPdf
# open the PDF files to be merged
with open(left_filename) as left_file, open(right_filename) as right_file, open(out_filename, 'w') as output_file:
left_pdf = pyPdf.PdfFileReader(left_file)
right_pdf = pyPdf.PdfFileReader(right_file)
output = pyPdf.PdfFileWriter()
# get the first page from each pdf
left_page = left_pdf.pages[0]
right_page = right_pdf.pages[0]
# start a new blank page with a size that can fit the merged pages side by side
page = output.addBlankPage(
width=left_page.mediaBox.getWidth() + right_page.mediaBox.getWidth(),
height=max(left_page.mediaBox.getHeight(), right_page.mediaBox.getHeight()),
)
# draw the pages on that new page
page.mergeTranslatedPage(left_page, 0, 0)
page.mergeTranslatedPage(right_page, left_page.mediaBox.getWidth(), 0)
# write to file
output.write(output_file)
"""
def inverseDist(value):
if value == 0: value = 1
return math.log(value,2)
def getGOEliteExportDir(root_dir,dataset_name):
if 'AltResults' in root_dir:
root_dir = string.split(root_dir,'AltResults')[0]
if 'ExpressionInput' in root_dir:
root_dir = string.split(root_dir,'ExpressionInput')[0]
if 'ExpressionOutput' in root_dir:
root_dir = string.split(root_dir,'ExpressionOutput')[0]
if 'DataPlots' in root_dir:
root_dir = string.replace(root_dir,'DataPlots','GO-Elite')
elite_dir = root_dir
else:
elite_dir = root_dir+'/GO-Elite'
try: os.mkdir(elite_dir)
except Exception: pass
return elite_dir+'/clustering/'+dataset_name
def systemCodeCheck(IDs):
import gene_associations
id_type_db={}
for id in IDs:
id_type = gene_associations.predictIDSourceSimple(id)
try: id_type_db[id_type]+=1
except Exception: id_type_db[id_type]=1
id_type_count=[]
for i in id_type_db:
id_type_count.append((id_type_db[i],i))
id_type_count.sort()
id_type = id_type_count[-1][-1]
return id_type
def exportFlatClusterData(filename, root_dir, dataset_name, new_row_header,new_column_header,xt,ind1,ind2,vmax,display):
""" Export the clustered results as a text file, only indicating the flat-clusters rather than the tree """
filename = string.replace(filename,'.pdf','.txt')
export_text = export.ExportFile(filename)
column_header = string.join(['UID','row_clusters-flat']+new_column_header,'\t')+'\n' ### format column-names for export
export_text.write(column_header)
column_clusters = string.join(['column_clusters-flat','']+ map(str, ind2),'\t')+'\n' ### format column-flat-clusters for export
export_text.write(column_clusters)
### The clusters, dendrogram and flat clusters are drawn bottom-up, so we need to reverse the order to match
#new_row_header = new_row_header[::-1]
#xt = xt[::-1]
try: elite_dir = getGOEliteExportDir(root_dir,dataset_name)
except Exception: elite_dir = None
elite_columns = string.join(['InputID','SystemCode'])
try: sy = systemCodeCheck(new_row_header)
except Exception: sy = None
### Export each row in the clustered data matrix xt
i=0
cluster_db={}
export_lines = []
for row in xt:
try:
id = new_row_header[i]
original_id = str(id)
if sy == '$En:Sy':
cluster = 'cluster-'+string.split(id,':')[0]
elif sy == 'S' and ':' in id:
cluster = 'cluster-'+string.split(id,':')[0]
elif sy == 'Sy' and ':' in id:
cluster = 'cluster-'+string.split(id,':')[0]
else:
cluster = 'c'+str(ind1[i])
except Exception:
pass
try:
if 'MarkerGenes' in originalFilename:
cluster = 'cluster-'+string.split(id,':')[0]
id = string.split(id,':')[1]
if ' ' in id:
id = string.split(id,' ')[0]
if 'G000' in id: sy = 'En'
else: sy = 'Sy'
except Exception: pass
try: cluster_db[cluster].append(id)
except Exception: cluster_db[cluster] = [id]
try: export_lines.append(string.join([original_id,str(ind1[i])]+map(str, row),'\t')+'\n')
except Exception:
export_lines.append(string.join([original_id,'NA']+map(str, row),'\t')+'\n')
i+=1
### Reverse the order of the file
export_lines.reverse()
for line in export_lines:
export_text.write(line)
export_text.close()
### Export GO-Elite input files
allGenes={}
for cluster in cluster_db:
export_elite = export.ExportFile(elite_dir+'/'+cluster+'.txt')
if sy==None:
export_elite.write('ID\n')
else:
export_elite.write('ID\tSystemCode\n')
for id in cluster_db[cluster]:
try:
i1,i2 = string.split(id,' ')
if i1==i2: id = i1
except Exception: pass
if sy == '$En:Sy':
id = string.split(id,':')[1]
ids = string.split(id,' ')
if 'ENS' in ids[0] or 'G0000' in ids[0]: id = ids[0]
else: id = ids[-1]
sc = 'En'
elif sy == 'Sy' and ':' in id:
id = string.split(id,':')[1]
ids = string.split(id,' ')
sc = 'Sy'
elif sy == 'En:Sy':
id = string.split(id,' ')[0]
sc = 'En'
elif sy == 'Ae':
l = string.split(id,':')
if len(l)==2:
id = string.split(id,':')[0] ### Use the Ensembl
if len(l) == 3:
id = string.split(id,':')[1] ### Use the Ensembl
sc = 'En'
if ' ' in id:
ids = string.split(id,' ')
if 'ENS' in ids[-1] or 'G0000' in ids[-1]: id = ids[-1]
else: id = ids[0]
elif sy == 'En' and '&' in id:
for i in string.split(id,'&'):
if 'G0000' in i: id = i; sc = 'En'; break
elif sy == 'Sy' and 'EFN' in id:
sc = 'En'
else:
sc = sy
if sy == 'S':
if ':' in id:
id = string.split(id,':')[-1]
sc = 'Ae'
if '&' in id:
sc = 'Ae'
if (len(id)==9 and "SRS" in id) or (len(id)==15 and "TCGA-" in id):
sc = 'En'
try: export_elite.write(id+'\t'+sc+'\n')
except Exception: export_elite.write(id+'\n') ### if no System Code known
allGenes[id]=[]
export_elite.close()
try:
if storeGeneSetName != None:
if len(storeGeneSetName)>0 and ('guide' not in justShowTheseIDs):
exportCustomGeneSet(storeGeneSetName,species,allGenes)
print 'Exported geneset to "StoredGeneSets"'
except Exception: pass
### Export as CDT file
filename = string.replace(filename,'.txt','.cdt')
if display:
try: exportJTV(filename, new_column_header, new_row_header,vmax=vmax)
except Exception: pass
export_cdt = export.ExportFile(filename)
column_header = string.join(['UNIQID','NAME','GWEIGHT']+new_column_header,'\t')+'\n' ### format column-names for export
export_cdt.write(column_header)
eweight = string.join(['EWEIGHT','','']+ ['1']*len(new_column_header),'\t')+'\n' ### format column-flat-clusters for export
export_cdt.write(eweight)
### Export each row in the clustered data matrix xt
i=0; cdt_lines=[]
for row in xt:
cdt_lines.append(string.join([new_row_header[i]]*2+['1']+map(str, row),'\t')+'\n')
i+=1
### Reverse the order of the file
cdt_lines.reverse()
for line in cdt_lines:
export_cdt.write(line)
export_cdt.close()
return elite_dir, filename, sc
def exportJTV(cdt_dir, column_header, row_header,vmax=None):
### This is a config file for TreeView
filename = string.replace(cdt_dir,'.cdt','.jtv')
export_jtv = export.ExportFile(filename)
cscale = '3'
if len(column_header)>100:
cscale = '1.5'
if len(column_header)>200:
cscale = '1.1'
if len(column_header)>300:
cscale = '0.6'
if len(column_header)>400:
cscale = '0.3'
hscale = '5'
if len(row_header)< 50:
hscale = '10'
if len(row_header)>100:
hscale = '3'
if len(row_header)>500:
hscale = '1'
if len(row_header)>1000:
hscale = '0.5'
contrast = str(float(vmax)/4)[:4] ### base the contrast on the heatmap vmax variable
"""
config = '<DocumentConfig><UrlExtractor/><ArrayUrlExtractor/><MainView><ColorExtractor>'
config+= '<ColorSet down="#00FFFF"/></ColorExtractor><ArrayDrawer/><GlobalXMap>'
config+= '<FixedMap type="Fixed" scale="'+cscale+'"/><FillMap type="Fill"/><NullMap type="Null"/>'
config+= '</GlobalXMap><GlobalYMap><FixedMap type="Fixed" scale="'+hscale+'"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></GlobalYMap><ZoomXMap><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></ZoomXMap><ZoomYMap><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></ZoomYMap><TextView><TextView><GeneSummary/></TextView><TextView>'
config+= '<GeneSummary/></TextView><TextView><GeneSummary/></TextView></TextView><ArrayNameView>'
config+= '<ArraySummary included="0"/></ArrayNameView><AtrSummary/><GtrSummary/></MainView></DocumentConfig>'
export_jtv.write(config)
"""
config = '<DocumentConfig><UrlExtractor/><ArrayUrlExtractor/><MainView><ColorExtractor>'
config+= '<ColorSet down="#00FFFF"/></ColorExtractor><ArrayDrawer/><GlobalXMap>'
config+= '<FixedMap type="Fixed" scale="'+cscale+'"/><FillMap type="Fill"/><NullMap type="Null"/>'
config+= '</GlobalXMap><GlobalYMap><FixedMap type="Fixed" scale="'+hscale+'"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></GlobalYMap><ZoomXMap><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></ZoomXMap><ZoomYMap><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></ZoomYMap><TextView><TextView><GeneSummary/></TextView><TextView>'
config+= '<GeneSummary/></TextView><TextView><GeneSummary/></TextView></TextView><ArrayNameView>'
config+= '<ArraySummary included="0"/></ArrayNameView><AtrSummary/><GtrSummary/></MainView><Views>'
config+= '<View type="Dendrogram" dock="1"><ColorExtractor contrast="'+contrast+'"><ColorSet up="#FFFF00" down="#00CCFF"/>'
config+= '</ColorExtractor><ArrayDrawer/><GlobalXMap current="Fill"><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></GlobalXMap><GlobalYMap current="Fill"><FixedMap type="Fixed"/><FillMap type="Fill"/>'
config+= '<NullMap type="Null"/></GlobalYMap><ZoomXMap><FixedMap type="Fixed"/><FillMap type="Fill"/><NullMap type="Null"/>'
config+= '</ZoomXMap><ZoomYMap current="Fixed"><FixedMap type="Fixed"/><FillMap type="Fill"/><NullMap type="Null"/></ZoomYMap>'
config+= '<TextView><TextView><GeneSummary/></TextView><TextView><GeneSummary/></TextView><TextView><GeneSummary/></TextView>'
config+= '</TextView><ArrayNameView><ArraySummary included="0"/></ArrayNameView><AtrSummary/><GtrSummary/></View></Views></DocumentConfig>'
export_jtv.write(config)
### How to create custom colors - http://matplotlib.sourceforge.net/examples/pylab_examples/custom_cmap.html
def updateColorBarData(ind1,ind2,column_header,row_header,row_method):
""" Replace the top-level cluster information with group assignments for color bar coloring (if group data present)"""
cb_status = 'original'
group_number_list=[]
group_name_list=[]
try: ### Error if GroupDB not recognized as global
if column_header[0] in GroupDB: ### Thus group assignments exist for column headers
cb_status = 'column'
for header in column_header:
group,color,color_num = GroupDB[header]
group_number_list.append(color_num) ### will replace ind2
if (color_num,group) not in group_name_list:
group_name_list.append((color_num,group))
ind2 = group_number_list
if row_header[0] in GroupDB and row_method == None: ### Thus group assignments exist for row headers
group_number_list=[]
if cb_status == 'column': cb_status = 'column-row'
else: cb_status = 'row'
for header in row_header:
group,color,color_num = GroupDB[header]
group_number_list.append(color_num) ### will replace ind2
#group_number_list.reverse()
ind1 = group_number_list
except Exception: None
return ind1,ind2,group_name_list,cb_status
def ConvertFromHex(color1,color2,color3):
c1tuple = tuple(ord(c) for c in color1.lsstrip('0x').decode('hex'))
c2tuple = tuple(ord(c) for c in color2.lsstrip('0x').decode('hex'))
c3tuple = tuple(ord(c) for c in color3.lsstrip('0x').decode('hex'))
def RedBlackSkyBlue():
cdict = {'red': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0)),
'green': ((0.0, 0.0, 0.9),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0)),
'blue': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0))
}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def RedBlackBlue():
cdict = {'red': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0)),
'green': ((0.0, 0.0, 0.0),
(1.0, 0.0, 0.0)),
'blue': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0))
}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def RedBlackGreen():
cdict = {'red': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0)),
'blue': ((0.0, 0.0, 0.0),
(1.0, 0.0, 0.0)),
'green': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0))
}
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def YellowBlackBlue():
cdict = {'red': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0)),
'green': ((0.0, 0.0, 0.8),
(0.5, 0.1, 0.0),
(1.0, 1.0, 1.0)),
'blue': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0))
}
### yellow is created by adding y = 1 to RedBlackSkyBlue green last tuple
### modulate between blue and cyan using the last y var in the first green tuple
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def BlackYellowBlue():
cdict = {'red': ((0.0, 0.0, 1.0),
(0.5, 0.1, 0.0),
(1.0, 0.0, 0.0)),
'green': ((0.0, 0.0, 0.8),
(0.5, 0.1, 0.0),
(1.0, 1.0, 1.0)),
'blue': ((0.0, 0.0, 0.0),
(0.5, 0.0, 0.1),
(1.0, 1.0, 1.0))
}
### yellow is created by adding y = 1 to RedBlackSkyBlue green last tuple
### modulate between blue and cyan using the last y var in the first green tuple
my_cmap = mc.LinearSegmentedColormap('my_colormap',cdict,256)
return my_cmap
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def filepath(filename):
fn = unique.filepath(filename)
return fn
def remoteImportData(filename,geneFilter=None,reverseOrder=True):
matrix, column_header, row_header, dataset_name, group_db = importData(filename,geneFilter=geneFilter,reverseOrder=reverseOrder)
try:
return matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters
except:
return matrix, column_header, row_header, dataset_name, group_db, [], []
def importData(filename,Normalize=False,reverseOrder=True,geneFilter=None,
zscore=False,forceClusters=False):
global priorColumnClusters
global priorRowClusters
try:
if len(priorColumnClusters)>0:
priorColumnClusters = None
priorRowClusters = None
except Exception: pass
getRowClusters=False
start_time = time.time()
fn = filepath(filename)
matrix=[]
original_matrix=[]
row_header=[]
overwriteGroupNotations=True
x=0; inputMax=0; inputMin=100
filename = string.replace(filename,'\\','/')
dataset_name = string.split(filename,'/')[-1][:-4]
if '.cdt' in filename: start = 3
else: start = 1
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
if '.cdt' in filename: t = [t[0]]+t[3:]
if t[1] == 'row_clusters-flat':
t = t = [t[0]]+t[2:]
### color samples by annotated groups if an expression file
new_headers=[]
temp_groups={}
original_headers=t[1:]
if ('exp.' in filename or 'filteredExp.' in filename or 'MarkerGene' in filename) and forceClusters==False:# and ':' not in data:
if overwriteGroupNotations:
### Use groups file annotations over any header sample separation with a ":"
for i in t:
if ':' in i: ### Don't annotate groups according to the clusters
group,i = string.split(i,':')
new_headers.append(i)
temp_groups[i] = group
else: new_headers.append(i)
filename = string.replace(filename,'-steady-state.txt','.txt')
try:
import ExpressionBuilder
try: sample_group_db = ExpressionBuilder.simplerGroupImport(filename)
except Exception: sample_group_db={}
if len(temp_groups)>0 and len(sample_group_db)==0:
sample_group_db = temp_groups
if len(new_headers)>0:
t = new_headers
new_headers = []
for v in t:
if v in sample_group_db:
v = sample_group_db[v]+':'+v
new_headers.append(v)
t = new_headers
except Exception:
#print traceback.format_exc()
pass
group_db, column_header = assignGroupColors(t[1:])
x=1
elif 'column_clusters-flat' in t:
try:
if 'NA' in t:
kill
try:
if forceClusters==False:
prior = map(lambda x: int(float(x)),t[2:])
else:
prior = map(lambda x: x,t[2:])
except Exception:
### Replace the cluster string with number
index=0
c=1; prior=[]; clusters={}
for i in t[2:]:
original_headers[index] = i+':'+original_headers[index]
if i in clusters:
c1 = clusters[i]
else:
c1 = c; clusters[i]=c1
c+=1
prior.append(c1)
index+=1
#prior=[]
if len(temp_groups)==0: ### Hence, no secondary group label combined with the sample name
if '-ReOrdered.txt' not in filename: ### Applies to cellHarmony UMAP and heatmap visualization
group_db, column_header = assignGroupColors(original_headers)
#priorColumnClusters = dict(zip(column_header, prior))
priorColumnClusters = prior
except Exception:
#print traceback.format_exc()
pass
start = 2
getRowClusters = True
priorRowClusters=[]
elif 'EWEIGHT' in t: pass
else:
gene = t[0]
if geneFilter==None:
proceed = True
elif gene in geneFilter:
proceed = True
else:
proceed = False
if proceed:
nullsPresent = False
#if ' ' not in t and '' not in t: ### Occurs for rows with missing data
try: s = map(float,t[start:])
except Exception:
nullsPresent=True
s=[]
for value in t[start:]:
try: s.append(float(value))
except Exception: s.append(0.000101)
#s = numpy.ma.masked_values(s, 0.000101)
original_matrix.append(s)
try:
if max(s)>inputMax: inputMax = max(s)
except:
continue ### empty row
if min(s)<inputMin: inputMin = min(s)
#if (abs(max(s)-min(s)))>2:
if Normalize!=False:
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
if Normalize=='row mean':
#avg = min(s)
avg = numpy.mean(s)
else:
avg = numpy.median(s)
if nullsPresent:
s=[] ### Needs to be done to zero out the values
for value in t[start:]:
try: s.append(float(value)-avg)
except Exception: s.append(0.000101)
#s = numpy.ma.masked_values(s, 0.000101)
else:
s = map(lambda x: x-avg,s) ### normalize to the mean
if ' ' in gene:
try:
g1,g2 = string.split(gene,' ')
if g1 == g2: gene = g1
except Exception: pass
if getRowClusters:
try:
#priorRowClusters[gene]=int(float(t[1]))
priorRowClusters.append(int(float(t[1])))
except Exception: pass
if zscore:
### convert to z-scores for normalization prior to PCA
avg = numpy.mean(s)
std = numpy.std(s)
if std ==0:
std = 0.1
try: s = map(lambda x: (x-avg)/std,s)
except Exception: pass
if geneFilter==None:
matrix.append(s)
row_header.append(gene)
else:
if gene in geneFilter:
matrix.append(s)
row_header.append(gene)
x+=1
if inputMax>100: ### Thus, not log values
print 'Converting values to log2...'
matrix=[]
k=0
if inputMin==0: increment = 1#0.01
else: increment = 1
for s in original_matrix:
if 'counts.' in filename:
s = map(lambda x: math.log(x+1,2),s)
else:
try: s = map(lambda x: math.log(x+increment,2),s)
except Exception:
print filename
print Normalize
print row_header[k], min(s),max(s); kill
if Normalize!=False:
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
if Normalize=='row mean':
avg = numpy.average(s)
else: avg = avg = numpy.median(s)
s = map(lambda x: x-avg,s) ### normalize to the mean
if zscore: ### The above z-score does not impact the original_matrix which is analyzed
### convert to z-scores for normalization prior to PCA
avg = numpy.mean(s)
std = numpy.std(s)
if std ==0:
std = 0.1
try: s = map(lambda x: (x-avg)/std,s)
except Exception: pass
matrix.append(s)
k+=1
del original_matrix
if zscore: print 'Converting values to normalized z-scores...'
#reverseOrder = True ### Cluster order is background (this is a temporary workaround)
if reverseOrder == True:
matrix.reverse(); row_header.reverse()
time_diff = str(round(time.time()-start_time,1))
try:
print '%d rows and %d columns imported for %s in %s seconds...' % (len(matrix),len(column_header),dataset_name,time_diff)
except Exception:
print 'No data in input file.'; force_error
### Add groups for column pre-clustered samples if there
group_db2, row_header2 = assignGroupColors(list(row_header)) ### row_header gets sorted in this function and will get permenantly screwed up if not mutated
#if '.cdt' in filename: matrix.reverse(); row_header.reverse()
for i in group_db2:
if i not in group_db: group_db[i] = group_db2[i]
##print group_db;sys.exit()
return matrix, column_header, row_header, dataset_name, group_db
def importSIF(filename):
fn = filepath(filename)
edges=[]
x=0
if '/' in filename:
dataset_name = string.split(filename,'/')[-1][:-4]
else:
dataset_name = string.split(filename,'\\')[-1][:-4]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
parent,type,child = string.split(data,'\t')
if 'AltAnalyze' in dataset_name:
### This is the order for proper directed interactions in the AltAnalyze-interaction viewer
edges.append([parent,child,type])
else:
if '(' in parent: ### for TF-target annotations
parent = string.split(parent,'(')[0]
if ':' in child:
child = string.split(child,':')[1]
if 'TF' in dataset_name or 'UserSuppliedAssociations' in dataset_name or 'WGRV' in dataset_name:
edges.append([parent,child,type]) ### Do this to indicate that the TF is regulating the target
else:
edges.append([child,parent,type])
edges = unique.unique(edges)
return edges
def assignGroupColors(t):
""" Assign a unique color to each group. Optionally used for cluster display. """
column_header=[]; group_number_db={}
groupNamesPresent=False # Some samples may have missing group names which will result in a clustering error
for i in t:
if ':' in i: groupNamesPresent = True
for i in t:
repls = {'.2txt' : '', '.2bed' : '', '.2tab' : ''}
i=reduce(lambda a, kv: a.replace(*kv), repls.iteritems(), i)
if ':' in i:
group,j = string.split(i,':')[:2]
group_number_db[group]=[]
elif groupNamesPresent:
group_number_db['UNK']=[]
i = 'UNK:'+i
column_header.append(i)
#import random
k = 0
group_db={}; color_db={}
color_list = ['r', 'b', 'y', 'g', 'w', 'k', 'm']
if len(group_number_db)>3:
color_list = []
cm = pylab.cm.get_cmap('nipy_spectral') #gist_ncar # binary
for i in range(len(group_number_db)):
color_list.append(cm(1.*i/len(group_number_db))) # color will now be an RGBA tuple
#color_list=[]
#color_template = [1,1,1,0,0,0,0.5,0.5,0.5,0.25,0.25,0.25,0.75,0.75,0.75]
t.sort() ### Ensure that all clusters have the same order of groups
for i in column_header:
repls = {'.2txt' : '', '.2bed' : '', '.2tab' : ''}
i=reduce(lambda a, kv: a.replace(*kv), repls.iteritems(), i)
if ':' in i:
group,j = string.split(i,':')[:2]
try: color,ko = color_db[group]
except Exception:
try: color_db[group] = color_list[k],k
except Exception:
### If not listed in the standard color set add a new random color
rgb = tuple(scipy.rand(3)) ### random color
#rgb = tuple(random.sample(color_template,3)) ### custom alternative method
color_list.append(rgb)
color_db[group] = color_list[k], k
color,ko = color_db[group]
k+=1
group_db[i] = group, color, ko
#column_header.append(i)
return group_db, column_header
def verifyFile(filename):
status = 'not found'
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = 'found';break
except Exception: status = 'not found'
return status
def AppendOrWrite(export_path):
export_path = filepath(export_path)
status = verifyFile(export_path)
if status == 'not found':
export_data = export.ExportFile(export_path) ### Write this new file
else:
export_data = open(export_path,'a') ### Appends to existing file
return export_path, export_data, status
def exportCustomGeneSet(geneSetName,species,allGenes):
for gene in allGenes:break
if 'ENS' not in gene:
try:
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception: symbol_to_gene={}
if species != None:
export_path, export_data, status = AppendOrWrite('AltDatabase/goelite/'+species+'/gene-mapp/Ensembl-StoredGeneSets.txt')
stored_lines=[]
for line in open(export_path,'rU').xreadlines(): stored_lines.append(line)
if status == 'not found':
export_data.write('GeneID\tEmpty\tGeneSetName\n')
for gene in allGenes:
if ' ' in gene:
a,b=string.split(gene,' ')
if 'ENS' in a: gene = a
else: gene = b
if 'ENS' not in gene and gene in symbol_to_gene:
gene = symbol_to_gene[gene][0]
line = gene+'\t\t'+geneSetName+'\n'
if line not in stored_lines:
export_data.write(line)
export_data.close()
else:
print 'Could not store since no species name provided.'
def writetSNEScores(scores,outputdir):
export_obj = export.ExportFile(outputdir)
for matrix_row in scores:
matrix_row = map(str,matrix_row)
export_obj.write(string.join(matrix_row,'\t')+'\n')
export_obj.close()
def importtSNEScores(inputdir):
#print inputdir
scores=[]
### Imports tSNE scores to allow for different visualizations of the same scatter plot
for line in open(inputdir,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
t=map(float,t)
scores.append(t)
return scores
def runUMAP(matrix, column_header,dataset_name,group_db,display=False,showLabels=False,
row_header=None,colorByGene=None,species=None,reimportModelScores=True,method="UMAP",rootDir='',finalOutputDir=''):
global root_dir
global graphic_link
graphic_link=[]
root_dir = rootDir
tSNE(matrix, column_header,dataset_name,group_db,display=False,showLabels=False,
row_header=None,colorByGene=None,species=None,reimportModelScores=True,method="UMAP")
import shutil
filename = 'Clustering-'+dataset_name+'-'+method+'.pdf'
filename = string.replace(filename,'Clustering-Clustering','Clustering')
new_file=finalOutputDir + filename
new_file=string.replace(new_file,'Clustering-','')
new_file=string.replace(new_file,'exp.','')
old_file=root_dir+filename
shutil.move(old_file,new_file)
filename = filename[:-3]+'png'
new_file=finalOutputDir + filename
new_file=string.replace(new_file,'Clustering-','')
new_file=string.replace(new_file,'exp.','')
old_file=root_dir+filename
shutil.move(old_file,new_file)
old_file=root_dir+dataset_name+'-'+method+'_scores.txt'
new_file=finalOutputDir+dataset_name+'-'+method+'_coordinates.txt'
new_file=string.replace(new_file,'exp.','')
shutil.move(old_file,new_file)
def tSNE(matrix, column_header,dataset_name,group_db,display=True,showLabels=False,
row_header=None,colorByGene=None,species=None,reimportModelScores=True,
method="tSNE",maskGroups=None):
try: prior_clusters = priorColumnClusters
except Exception: prior_clusters=[]
try:
if priorColumnClusters==None: prior_clusters=[]
except:
pass
try:
if len(prior_clusters)>0 and len(group_db)==0:
newColumnHeader=[]
i=0
for sample_name in column_header:
newColumnHeader.append(str(prior_clusters[i])+':'+sample_name)
i+=1
group_db, column_header = assignGroupColors(newColumnHeader)
except Exception,e:
print traceback.format_exc()
#print e
group_db={}
if reimportModelScores:
print 'Re-importing',method,'model scores rather than calculating from scratch',
print root_dir+dataset_name+'-'+method+'_scores.txt'
try: scores = importtSNEScores(root_dir+dataset_name+'-'+method+'_scores.txt'); print '...import finished'
except Exception:
reimportModelScores=False; print '...no existing score file found'
if reimportModelScores==False:
X=matrix.T
"""
from tsne import bh_sne
X = np.asarray(X).astype('float64')
X = X.reshape((X.shape[0], -1))
x_data = x_data.reshape((x_data.shape[0], -1))
scores = bh_sne(X)"""
#model = TSNE(n_components=2, random_state=0,init='pca',early_exaggeration=4.0,perplexity=20)
print "Performing",method
if method=="tSNE" or method=="t-SNE":
from sklearn.manifold import TSNE
model = TSNE(n_components=2)
if method=="UMAP":
try:
import umap
model=umap.UMAP(n_neighbors=50,min_dist=0.75,metric='correlation')
except:
try:
from visualization_scripts.umap_learn import umap ### Bypasses issues with Py2app importing umap (and secondarily numba/llvmlite)
model=umap.UMAP(n_neighbors=50,min_dist=0.75,metric='correlation')
except: ### requires single-threading for Windows platforms (possibly others)
from visualization_scripts.umap_learn_single import umap ### Bypasses issues with Py2app importing umap (and secondarily numba/llvmlite)
model=umap.UMAP(n_neighbors=50,min_dist=0.75,metric='correlation')
print 'UMAP run'
#model = TSNE(n_components=2,init='pca', random_state=0, verbose=1, perplexity=40, n_iter=300)
#model = TSNE(n_components=2,verbose=1, perplexity=40, n_iter=300)
#model = TSNE(n_components=2, random_state=0, n_iter=10000, early_exaggeration=10)
scores=model.fit_transform(X)
### Export the results for optional re-import later
writetSNEScores(scores,root_dir+dataset_name+'-'+method+'_scores.txt')
#pylab.scatter(scores[:,0], scores[:,1], 20, labels);
if maskGroups != None:
group_name,restricted_samples = maskGroups
dataset_name += '-'+group_name ### indicate the restricted group
### Exclude samples with high TSNE deviations
scoresT = zip(*scores)
exclude={}
try:
for vector in scoresT:
lower1th,median_val,upper99th,int_qrt_range = statistics.iqr(list(vector),k1=99.9,k2=0.1)
index=0
for i in vector:
if (i > upper99th+1) or (i<lower1th-1):
exclude[index]=None
index+=1
except Exception:
pass
print 'Not showing',len(exclude),'outlier samples.'
fig = pylab.figure()
ax = fig.add_subplot(111)
pylab.xlabel(method.upper()+'-X')
pylab.ylabel(method.upper()+'-Y')
axes = getAxesTransposed(scores,exclude=exclude) ### adds buffer space to the end of each axis and creates room for a legend
pylab.axis(axes)
marker_size = 15
if len(column_header)>20:
marker_size = 12
if len(column_header)>40:
marker_size = 10
if len(column_header)>150:
marker_size = 7
if len(column_header)>500:
marker_size = 5
if len(column_header)>1000:
marker_size = 4
if len(column_header)>2000:
marker_size = 3
if len(column_header)>4000:
marker_size = 2
if len(column_header)>6000:
marker_size = 1
### Color By Gene
if colorByGene != None and len(matrix)==0:
print 'Gene %s not found in the imported dataset... Coloring by groups.' % colorByGene
if colorByGene != None and len(matrix)>0:
gene_translation_db={}
matrix = numpy.array(matrix)
min_val = matrix.min() ### min val
if ' ' in colorByGene:
genes = string.split(colorByGene,' ')
else:
genes = [colorByGene]
genePresent=False
numberGenesPresent=[]
for gene in genes:
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
### Translate symbol to Ensembl
if len(numberGenesPresent)==0:
try:
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
for symbol in genes:
if symbol in symbol_to_gene:
gene = symbol_to_gene[symbol][0]
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
gene_translation_db[symbol]=gene
except Exception: pass
numberGenesPresent = len(numberGenesPresent)
if numberGenesPresent==1:
cm = pylab.cm.get_cmap('Reds')
else:
if numberGenesPresent==2:
cm = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
#cm = matplotlib.colors.ListedColormap(['w', 'k']) ### If you want to hide one of the groups
elif numberGenesPresent==3:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
elif numberGenesPresent==4:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
elif numberGenesPresent==5:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif numberGenesPresent==6:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif numberGenesPresent==7:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
else:
cm = pylab.cm.get_cmap('gist_rainbow')
def get_cmap(n, name='hsv'):
'''Returns a function that maps each index in 0, 1, ..., n-1 to a distinct
RGB color; the keyword argument name must be a standard mpl colormap name.'''
return pylab.cm.get_cmap(name, n)
if genePresent:
dataset_name+='-'+colorByGene
group_db={}
bestGeneAssociated={}
k=0
for gene in genes:
try:
try: i = row_header.index(gene)
except Exception: i = row_header.index(gene_translation_db[gene])
values = map(float,matrix[i])
min_val = min(values)
bin_size = (max(values)-min_val)/8
max_val = max(values)
ranges = []
iz=min_val
while iz < (max(values)-bin_size/100):
r = iz,iz+bin_size
if len(ranges)==7:
r = iz,max_val
ranges.append(r)
iz+=bin_size
color_db = {}
colors = get_cmap(len(genes))
for i in range(len(ranges)):
if i==0:
color = '#C0C0C0'
else:
if numberGenesPresent==1:
### use a single color gradient
color = cm(1.*i/len(ranges))
#color = cm(1.*(i+1)/len(ranges))
else:
if i>2:
if len(genes)<8:
color = cm(k)
else:
color = colors(k)
else:
color = '#C0C0C0'
color_db[ranges[i]] = color
i=0
for val in values:
sample = column_header[i]
for (l,u) in color_db:
range_index = ranges.index((l,u)) ### what is the ranking of this range
if val>=l and val<=u:
color = color_db[(l,u)]
color_label = [gene+'-range: '+str(l)[:4]+'-'+str(u)[:4],color,'']
group_db[sample] = color_label
try: bestGeneAssociated[sample].append([range_index,val,color_label])
except Exception: bestGeneAssociated[sample] = [[range_index,val,color_label]]
i+=1
#print min(values),min_val,bin_size,max_val
if len(genes)>1:
### Collapse and rank multiple gene results
for sample in bestGeneAssociated:
bestGeneAssociated[sample].sort()
color_label = bestGeneAssociated[sample][-1][-1]
if numberGenesPresent>1:
index = bestGeneAssociated[sample][-1][0]
if index > 2:
gene = string.split(color_label[0],'-')[0]
else:
gene = 'Null'
color_label[0] = gene
group_db[sample] = color_label
except Exception:
print [gene], 'not found in rows...'
#print traceback.format_exc()
k+=1
else:
print [colorByGene], 'not found in rows...'
pylab.title(method+' - '+dataset_name)
group_names={}
i=0
for sample_name in column_header: #scores[0]
if maskGroups != None:
base_name = sample_name
if ':' in sample_name:
base_name = string.split(base_name,':')[1]
if base_name not in restricted_samples:
exclude[i]=None ### Don't visualize this sample
if i not in exclude:
### Add the text labels for each
try:
### Get group name and color information
group_name,color,k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color
except Exception:
color = 'r'; label=None
ax.plot(scores[i][0],scores[i][1],color=color,marker='o',markersize=marker_size,label=label,markeredgewidth=0,picker=True)
#except Exception: print i, len(scores[pcB]);kill
if showLabels:
try: sample_name = ' '+string.split(sample_name,':')[1]
except Exception: pass
ax.text(scores[i][0],scores[i][1],sample_name,fontsize=11)
i+=1
group_count = []
for i in group_db:
if group_db[i][0] not in group_count:
group_count.append(group_db[i][0])
#print len(group_count)
Lfontsize = 8
if len(group_count)>20:
Lfontsize = 10
if len(group_count)>30:
Lfontsize = 8
if len(group_count)>40:
Lfontsize = 6
if len(group_count)>50:
Lfontsize = 5
i=0
box = ax.get_position()
if len(group_count) > 0: ### Make number larger to get the legend in the plot -- BUT, the axis buffer above has been disabled
# Shink current axis by 20%
ax.set_position([box.x0, box.y0, box.width * 0.8, box.height])
try: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5),fontsize = Lfontsize) ### move the legend over to the right of the plot
except Exception: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
else:
ax.set_position([box.x0, box.y0, box.width, box.height])
pylab.legend(loc="upper left", prop={'size': 10})
filename = 'Clustering-'+dataset_name+'-'+method+'.pdf'
filename = string.replace(filename,'Clustering-Clustering','Clustering')
try: pylab.savefig(root_dir + filename)
except Exception: None ### Rare error
#print 'Exporting:',filename
filename = filename[:-3]+'png'
try: pylab.savefig(root_dir + filename) #dpi=200, transparent=True
except Exception: None ### Rare error
graphic_link.append(['Principal Component Analysis',root_dir+filename])
if display:
print 'Exporting:',filename
try:
pylab.show()
except Exception:
#print traceback.format_exc()
pass### when run in headless mode
def excludeHighlyCorrelatedHits(x,row_header):
### For methylation data or other data with redundant signatures, remove these and only report the first one
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
D1 = numpy.corrcoef(x)
i=0
exclude={}
gene_correlations={}
include = []
for score_ls in D1:
k=0
for v in score_ls:
if str(v)!='nan':
if v>1.00 and k!=i:
#print row_header[i], row_header[k], v
if row_header[i] not in exclude:
exclude[row_header[k]]=[]
#if k not in exclude: include.append(row_header[k])
k+=1
#if i not in exclude: include.append(row_header[i])
i+=1
#print len(exclude),len(row_header);sys.exit()
return exclude
def PrincipalComponentAnalysis(matrix, column_header, row_header, dataset_name,
group_db, display=False, showLabels=True, algorithm='SVD', geneSetName=None,
species=None, pcA=1,pcB=2, colorByGene=None, reimportModelScores=True):
print "Performing Principal Component Analysis..."
from numpy import mean,cov,double,cumsum,dot,linalg,array,rank
try: prior_clusters = priorColumnClusters
except Exception: prior_clusters=[]
if prior_clusters == None: prior_clusters=[]
try:
if len(prior_clusters)>0 and len(group_db)==0:
newColumnHeader=[]
i=0
for sample_name in column_header:
newColumnHeader.append(str(prior_clusters[i])+':'+sample_name)
i+=1
group_db, column_header = assignGroupColors(newColumnHeader)
except Exception,e:
print traceback.format_exc()
group_db={}
pcA-=1
pcB-=1
label1=''
label2=''
""" Based in part on code from:
http://glowingpython.blogspot.com/2011/07/principal-component-analysis-with-numpy.html
Performs performs principal components analysis
(PCA) on the n-by-p data matrix A
Rows of A correspond to observations, columns to variables.
Returns :
coeff :
is a p-by-p matrix, each column containing coefficients
for one principal component.
score :
the principal component scores; that is, the representation
of A in the principal component space. Rows of SCORE
correspond to observations, columns to components.
latent :
a vector containing the eigenvalues
of the covariance matrix of A.
"""
# computing eigenvalues and eigenvectors of covariance matrix
if algorithm == 'SVD': use_svd = True
else: use_svd = False
if reimportModelScores:
print 'Re-importing PCA model scores rather than calculating from scratch',
print root_dir+dataset_name+'-PCA_scores.txt'
try:
scores = importtSNEScores(root_dir+dataset_name+'-PCA_scores.txt'); print '...import finished'
matrix = zip(*matrix)
except Exception:
reimportModelScores=False; print '...no existing score file found'
if reimportModelScores==False:
#Mdif = matrix-matrix.mean(axis=0)# subtract the mean (along columns)
#M = (matrix-mean(matrix.T,axis=1)).T # subtract the mean (along columns)
Mdif = matrix/matrix.std()
Mdif = Mdif.T
u, s, vt = svd(Mdif, 0)
fracs = s**2/np.sum(s**2)
entropy = -sum(fracs*np.log(fracs))/np.log(np.min(vt.shape))
label1 = 'PC%i (%2.1f%%)' %(pcA+1, fracs[0]*100)
label2 = 'PC%i (%2.1f%%)' %(pcB+1, fracs[1]*100)
#http://docs.scipy.org/doc/scipy/reference/sparse.html
#scipy.sparse.linalg.svds - sparse svd
#idx = numpy.argsort(vt[0,:])
#print idx;sys.exit() # Use this as your cell order or use a density analysis to get groups
#### FROM LARSSON ########
#100 most correlated Genes with PC1
#print vt
PCsToInclude = 4
correlated_db={}
allGenes={}
new_matrix = []
new_headers = []
added_indexes=[]
x = 0
#100 most correlated Genes with PC1
print 'exporting PCA loading genes to:',root_dir+'/PCA/correlated.txt'
exportData = export.ExportFile(root_dir+'/PCA/correlated.txt')
matrix = zip(*matrix) ### transpose this back to normal
try:
while x<PCsToInclude:
idx = numpy.argsort(u[:,x])
correlated = map(lambda i: row_header[i],idx[:300])
anticorrelated = map(lambda i: row_header[i],idx[-300:])
correlated_db[x] = correlated,anticorrelated
### Create a new filtered matrix of loading gene indexes
fidx = list(idx[:300])+list(idx[-300:])
for i in fidx:
if i not in added_indexes:
added_indexes.append(i)
new_headers.append(row_header[i])
new_matrix.append(matrix[i])
x+=1
#redundant_genes = excludeHighlyCorrelatedHits(numpy.array(new_matrix),new_headers)
redundant_genes = []
for x in correlated_db:
correlated,anticorrelated = correlated_db[x]
count=0
for gene in correlated:
if gene not in redundant_genes and count<100:
exportData.write(gene+'\tcorrelated-PC'+str(x+1)+'\n'); allGenes[gene]=[]
count+=1
count=0
for gene in anticorrelated:
if gene not in redundant_genes and count<100:
exportData.write(gene+'\tanticorrelated-PC'+str(x+1)+'\n'); allGenes[gene]=[]
count+=1
exportData.close()
if geneSetName != None:
if len(geneSetName)>0:
exportCustomGeneSet(geneSetName,species,allGenes)
print 'Exported geneset to "StoredGeneSets"'
except Exception:
pass
###########################
#if len(row_header)>20000:
#print '....Using eigenvectors of the real symmetric square matrix for efficiency...'
#[latent,coeff] = scipy.sparse.linalg.eigsh(cov(M))
#scores=mlab.PCA(scores)
if use_svd == False:
[latent,coeff] = linalg.eig(cov(M))
scores = dot(coeff.T,M) # projection of the data in the new space
else:
### transform u into the same structure as the original scores from linalg.eig coeff
scores = vt
writetSNEScores(scores,root_dir+dataset_name+'-PCA_scores.txt')
fig = pylab.figure()
ax = fig.add_subplot(111)
pylab.xlabel(label1)
pylab.ylabel(label2)
axes = getAxes(scores) ### adds buffer space to the end of each axis and creates room for a legend
pylab.axis(axes)
marker_size = 15
if len(column_header)>20:
marker_size = 12
if len(column_header)>40:
marker_size = 10
if len(column_header)>150:
marker_size = 7
if len(column_header)>500:
marker_size = 5
if len(column_header)>1000:
marker_size = 4
if len(column_header)>2000:
marker_size = 3
#marker_size = 9
#samples = list(column_header)
### Color By Gene
if colorByGene != None:
print 'Coloring based on feature expression.'
gene_translation_db={}
matrix = numpy.array(matrix)
min_val = matrix.min() ### min val
if ' ' in colorByGene:
genes = string.split(colorByGene,' ')
else:
genes = [colorByGene]
genePresent=False
numberGenesPresent=[]
for gene in genes:
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
### Translate symbol to Ensembl
if len(numberGenesPresent)==0:
try:
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
for symbol in genes:
if symbol in symbol_to_gene:
gene = symbol_to_gene[symbol][0]
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
gene_translation_db[symbol]=gene
except Exception: pass
numberGenesPresent = len(numberGenesPresent)
if numberGenesPresent==1:
cm = pylab.cm.get_cmap('Reds')
else:
if numberGenesPresent==2:
cm = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
#cm = matplotlib.colors.ListedColormap(['w', 'k']) ### If you want to hide one of the groups
elif numberGenesPresent==3:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
elif numberGenesPresent==4:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
elif numberGenesPresent==5:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif numberGenesPresent==6:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif numberGenesPresent==7:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
else:
cm = pylab.cm.get_cmap('gist_rainbow')
def get_cmap(n, name='hsv'):
'''Returns a function that maps each index in 0, 1, ..., n-1 to a distinct
RGB color; the keyword argument name must be a standard mpl colormap name.'''
return pylab.cm.get_cmap(name, n)
if genePresent:
dataset_name+='-'+colorByGene
group_db={}
bestGeneAssociated={}
k=0
for gene in genes:
try:
try: i = row_header.index(gene)
except Exception: i = row_header.index(gene_translation_db[gene])
values = map(float,matrix[i])
min_val = min(values)
bin_size = (max(values)-min_val)/8
max_val = max(values)
ranges = []
iz=min_val
while iz < (max(values)-bin_size/100):
r = iz,iz+bin_size
if len(ranges)==7:
r = iz,max_val
ranges.append(r)
iz+=bin_size
color_db = {}
colors = get_cmap(len(genes))
for i in range(len(ranges)):
if i==0:
color = '#C0C0C0'
else:
if numberGenesPresent==1:
### use a single color gradient
color = cm(1.*i/len(ranges))
#color = cm(1.*(i+1)/len(ranges))
else:
if i>2:
if len(genes)<8:
color = cm(k)
else:
color = colors(k)
else:
color = '#C0C0C0'
color_db[ranges[i]] = color
i=0
for val in values:
sample = column_header[i]
for (l,u) in color_db:
range_index = ranges.index((l,u)) ### what is the ranking of this range
if val>=l and val<=u:
color = color_db[(l,u)]
color_label = [gene+'-range: '+str(l)[:4]+'-'+str(u)[:4],color,'']
group_db[sample] = color_label
try: bestGeneAssociated[sample].append([range_index,val,color_label])
except Exception: bestGeneAssociated[sample] = [[range_index,val,color_label]]
i+=1
#print min(values),min_val,bin_size,max_val
if len(genes)>1:
### Collapse and rank multiple gene results
for sample in bestGeneAssociated:
bestGeneAssociated[sample].sort()
color_label = bestGeneAssociated[sample][-1][-1]
if numberGenesPresent>1:
index = bestGeneAssociated[sample][-1][0]
if index > 2:
gene = string.split(color_label[0],'-')[0]
else:
gene = 'Null'
color_label[0] = gene
group_db[sample] = color_label
except Exception:
print [gene], 'not found in rows...'
#print traceback.format_exc()
k+=1
else:
print [colorByGene], 'not found in rows...'
pylab.title('Principal Component Analysis - '+dataset_name)
group_names={}
i=0
for sample_name in column_header: #scores[0]
### Add the text labels for each
try:
### Get group name and color information
group_name,color,k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color
except Exception:
color = 'r'; label=None
try: ax.plot(scores[pcA][i],scores[1][i],color=color,marker='o',markersize=marker_size,label=label,markeredgewidth=0,picker=True)
except Exception, e: print e; print i, len(scores[pcB]);kill
if showLabels:
try: sample_name = ' '+string.split(sample_name,':')[1]
except Exception: pass
ax.text(scores[pcA][i],scores[pcB][i],sample_name,fontsize=11)
i+=1
group_count = []
for i in group_db:
if group_db[i][0] not in group_count:
group_count.append(group_db[i][0])
#print len(group_count)
Lfontsize = 8
if len(group_count)>20:
Lfontsize = 10
if len(group_count)>30:
Lfontsize = 8
if len(group_count)>40:
Lfontsize = 6
if len(group_count)>50:
Lfontsize = 5
i=0
#group_count = group_count*10 ### force the legend box out of the PCA core plot
box = ax.get_position()
if len(group_count) > 0:
# Shink current axis by 20%
ax.set_position([box.x0, box.y0, box.width * 0.8, box.height])
try: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5),fontsize = Lfontsize) ### move the legend over to the right of the plot
except Exception: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
else:
ax.set_position([box.x0, box.y0, box.width, box.height])
pylab.legend(loc="upper left", prop={'size': 10})
filename = 'Clustering-%s-PCA.pdf' % dataset_name
try: pylab.savefig(root_dir + filename)
except Exception: None ### Rare error
#print 'Exporting:',filename
filename = filename[:-3]+'png'
try: pylab.savefig(root_dir + filename) #dpi=200
except Exception: None ### Rare error
graphic_link.append(['Principal Component Analysis',root_dir+filename])
if display:
print 'Exporting:',filename
try:
pylab.show()
except Exception:
pass### when run in headless mode
fig.clf()
def ViolinPlot():
def readData(filename):
all_data = {}
headers={}
groups=[]
firstRow=True
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow=False
i=0
for x in t[1:]:
try: g,h = string.split(x,':')
except Exception: g=x; h=x
headers[i] = g
if g not in groups: groups.append(g)
i+=1
else:
#all_data.append(map(lambda x: math.log(math.pow(2,float(x))-1+0.001,2), t[1:]))
t = map(lambda x: float(x), t[1:])
i = 0
for x in t:
try: g = headers[i]
except Exception: print i;sys.exit()
try: all_data[g].append(x)
except Exception: all_data[g] = [x]
i+=1
all_data2=[]
print groups
for group in groups:
all_data2.append(all_data[group])
return all_data2
def violin_plot(ax, data, pos, bp=False):
'''
create violin plots on an axis
'''
from scipy.stats import gaussian_kde
from numpy import arange
dist = max(pos)-min(pos)
w = min(0.15*max(dist,1.0),0.5)
for d,p in zip(data,pos):
k = gaussian_kde(d) #calculates the kernel density
m = k.dataset.min() #lower bound of violin
M = k.dataset.max() #upper bound of violin
x = arange(m,M,(M-m)/100.) # support for violin
v = k.evaluate(x) #violin profile (density curve)
v = v/v.max()*w #scaling the violin to the available space
ax.fill_betweenx(x,p,v+p,facecolor='y',alpha=0.3)
ax.fill_betweenx(x,p,-v+p,facecolor='y',alpha=0.3)
if bp:
ax.boxplot(data,notch=1,positions=pos,vert=1)
def draw_all(data, output):
pos = [1,2,3]
fig = pylab.figure()
ax = fig.add_subplot(111)
violin_plot(ax, data, pos)
pylab.show()
pylab.savefig(output+'.pdf')
all_data = []
all_data = readData('/Users/saljh8/Downloads/TPM_cobound.txt')
import numpy
#all_data = map(numpy.array, zip(*all_data))
#failed_data = map(numpy.array, zip(*failed_data))
draw_all(all_data, 'alldata')
def simpleScatter(fn):
import matplotlib.patches as mpatches
values=[]
legends={}
colors={}
skip=True
scale = 100.0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if skip:
x_header, y_header, color_header,label_header, shape_header = string.split(data,'\t')
skip=False
else:
x, y, color,label,shape = string.split(data,'\t')
if color in colors:
xval,yval,label,shape = colors[color]
xval.append(float(x)); yval.append(float(y))
else:
xval = [float(x)]; yval = [float(y)]
colors[color] = xval,yval,label,shape
for color in colors:
xval,yval,label,shape = colors[color]
pylab.scatter(xval, yval, s=scale, c=color, alpha=0.75, label=label, marker=shape,edgecolor="none")
pylab.legend(loc='upper left')
pylab.title(fn)
pylab.xlabel(x_header, fontsize=15)
pylab.ylabel(y_header, fontsize=15)
marker_size = 7
#pylab.grid(True)
pylab.show()
def ica(filename):
showLabels=True
X, column_header, row_header, dataset_name, group_db = importData(filename)
X = map(numpy.array, zip(*X)) ### coverts these to tuples
column_header, row_header = row_header, column_header
ica = FastICA()
scores = ica.fit(X).transform(X) # Estimate the sources
scores /= scores.std(axis=0)
fig = pylab.figure()
ax = fig.add_subplot(111)
pylab.xlabel('ICA-X')
pylab.ylabel('ICA-Y')
pylab.title('ICA - '+dataset_name)
axes = getAxes(scores) ### adds buffer space to the end of each axis and creates room for a legend
pylab.axis(axes)
marker_size = 15
if len(column_header)>20:
marker_size = 12
if len(column_header)>40:
marker_size = 10
if len(column_header)>150:
marker_size = 7
if len(column_header)>500:
marker_size = 5
if len(column_header)>1000:
marker_size = 4
if len(column_header)>2000:
marker_size = 3
group_names={}
i=0
for sample_name in row_header: #scores[0]
### Add the text labels for each
try:
### Get group name and color information
group_name,color,k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color
except Exception:
color = 'r'; label=None
ax.plot(scores[0][i],scores[1][i],color=color,marker='o',markersize=marker_size,label=label)
if showLabels:
ax.text(scores[0][i],scores[1][i],sample_name,fontsize=8)
i+=1
pylab.title('ICA recovered signals')
pylab.show()
def plot_samples(S, axis_list=None):
pylab.scatter(S[:, 0], S[:, 1], s=20, marker='o', linewidths=0, zorder=10,
color='red', alpha=0.5)
if axis_list is not None:
colors = ['orange', 'red']
for color, axis in zip(colors, axis_list):
axis /= axis.std()
x_axis, y_axis = axis
# Trick to get legend to work
pylab.plot(0.1 * x_axis, 0.1 * y_axis, linewidth=2, color=color)
pylab.quiver(0, 0, x_axis, y_axis, zorder=11, width=2, scale=6,
color=color)
pylab.xlabel('x')
pylab.ylabel('y')
def PCA3D(matrix, column_header, row_header, dataset_name, group_db,
display=False, showLabels=True, algorithm='SVD',geneSetName=None,
species=None,colorByGene=None):
from numpy import mean,cov,double,cumsum,dot,linalg,array,rank
fig = pylab.figure()
ax = fig.add_subplot(111, projection='3d')
start = time.time()
#M = (matrix-mean(matrix.T,axis=1)).T # subtract the mean (along columns)
try: prior_clusters = priorColumnClusters
except Exception: prior_clusters=[]
if prior_clusters == None: prior_clusters=[]
try:
if len(prior_clusters)>0 and len(group_db)==0:
newColumnHeader=[]
i=0
for sample_name in column_header:
newColumnHeader.append(str(prior_clusters[i])+':'+sample_name)
i+=1
group_db, column_header = assignGroupColors(newColumnHeader)
except Exception,e:
#print e
group_db={}
if algorithm == 'SVD': use_svd = True
else: use_svd = False
Mdif = matrix/matrix.std()
Mdif = Mdif.T
u, s, vt = svd(Mdif, 0)
fracs = s**2/np.sum(s**2)
entropy = -sum(fracs*np.log(fracs))/np.log(np.min(vt.shape))
label1 = 'PC%i (%2.1f%%)' %(0+1, fracs[0]*100)
label2 = 'PC%i (%2.1f%%)' %(1+1, fracs[1]*100)
label3 = 'PC%i (%2.1f%%)' %(2+1, fracs[2]*100)
PCsToInclude = 4
correlated_db={}
allGenes={}
new_matrix = []
new_headers = []
added_indexes=[]
x = 0
#100 most correlated Genes with PC1
print 'exporting PCA loading genes to:',root_dir+'/PCA/correlated.txt'
exportData = export.ExportFile(root_dir+'/PCA/correlated.txt')
matrix = zip(*matrix) ### transpose this back to normal
try:
while x<PCsToInclude:
idx = numpy.argsort(u[:,x])
correlated = map(lambda i: row_header[i],idx[:300])
anticorrelated = map(lambda i: row_header[i],idx[-300:])
correlated_db[x] = correlated,anticorrelated
### Create a new filtered matrix of loading gene indexes
fidx = list(idx[:300])+list(idx[-300:])
for i in fidx:
if i not in added_indexes:
added_indexes.append(i)
new_headers.append(row_header[i])
new_matrix.append(matrix[i])
x+=1
#redundant_genes = excludeHighlyCorrelatedHits(numpy.array(new_matrix),new_headers)
redundant_genes = []
for x in correlated_db:
correlated,anticorrelated = correlated_db[x]
count=0
for gene in correlated:
if gene not in redundant_genes and count<100:
exportData.write(gene+'\tcorrelated-PC'+str(x+1)+'\n'); allGenes[gene]=[]
count+=1
count=0
for gene in anticorrelated:
if gene not in redundant_genes and count<100:
exportData.write(gene+'\tanticorrelated-PC'+str(x+1)+'\n'); allGenes[gene]=[]
count+=1
exportData.close()
if geneSetName != None:
if len(geneSetName)>0:
exportCustomGeneSet(geneSetName,species,allGenes)
print 'Exported geneset to "StoredGeneSets"'
except ZeroDivisionError:
pass
#numpy.Mdiff.toFile(root_dir+'/PCA/correlated.txt','\t')
if use_svd == False:
[latent,coeff] = linalg.eig(cov(M))
scores = dot(coeff.T,M) # projection of the data in the new space
else:
### transform u into the same structure as the original scores from linalg.eig coeff
scores = vt
end = time.time()
print 'PCA completed in', end-start, 'seconds.'
### Hide the axis number labels
#ax.w_xaxis.set_ticklabels([])
#ax.w_yaxis.set_ticklabels([])
#ax.w_zaxis.set_ticklabels([])
#"""
#ax.set_xticks([]) ### Hides ticks
#ax.set_yticks([])
#ax.set_zticks([])
ax.set_xlabel(label1)
ax.set_ylabel(label2)
ax.set_zlabel(label3)
#"""
#pylab.title('Principal Component Analysis\n'+dataset_name)
"""
pylab.figure()
pylab.xlabel('Principal Component 1')
pylab.ylabel('Principal Component 2')
"""
axes = getAxes(scores,PlotType='3D') ### adds buffer space to the end of each axis and creates room for a legend
pylab.axis(axes)
Lfontsize = 8
group_count = []
for i in group_db:
if group_db[i][0] not in group_count:
group_count.append(group_db[i][0])
### Color By Gene
if colorByGene != None:
gene_translation_db={}
matrix = numpy.array(matrix)
min_val = matrix.min() ### min val
if ' ' in colorByGene:
genes = string.split(colorByGene,' ')
else:
genes = [colorByGene]
genePresent=False
numberGenesPresent=[]
for gene in genes:
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
### Translate symbol to Ensembl
if len(numberGenesPresent)==0:
try:
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
for symbol in genes:
if symbol in symbol_to_gene:
gene = symbol_to_gene[symbol][0]
if gene in row_header:
numberGenesPresent.append(gene)
genePresent = True
gene_translation_db[symbol]=gene
except Exception: pass
numberGenesPresent = len(numberGenesPresent)
if numberGenesPresent==1:
cm = pylab.cm.get_cmap('Reds')
else:
if numberGenesPresent==2:
cm = matplotlib.colors.ListedColormap(['#00FF00', '#1E90FF'])
cm = matplotlib.colors.ListedColormap(['w', 'k'])
elif numberGenesPresent==3:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C'])
elif numberGenesPresent==4:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#3D3181', '#EE2C3C', '#FEBC18'])
elif numberGenesPresent==5:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#3D3181', '#FEBC18', '#EE2C3C'])
elif numberGenesPresent==6:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
elif numberGenesPresent==7:
cm = matplotlib.colors.ListedColormap(['#88BF47', '#63C6BB', '#29C3EC', '#3D3181', '#7B4976','#FEBC18', '#EE2C3C'])
else:
cm = pylab.cm.get_cmap('gist_rainbow')
if genePresent:
dataset_name+='-'+colorByGene
group_db={}
bestGeneAssociated={}
k=0
for gene in genes:
try:
try: i = row_header.index(gene)
except Exception: i = row_header.index(gene_translation_db[gene])
values = map(float,matrix[i])
min_val = min(values)
bin_size = (max(values)-min_val)/8
max_val = max(values)
ranges = []
iz=min_val
while iz < (max(values)-bin_size/100):
r = iz,iz+bin_size
if len(ranges)==7:
r = iz,max_val
ranges.append(r)
iz+=bin_size
color_db = {}
for i in range(len(ranges)):
if i==0:
color = '#C0C0C0'
else:
if numberGenesPresent==1:
### use a single color gradient
color = cm(1.*i/len(ranges))
#color = cm(1.*(i+1)/len(ranges))
else:
if i>2:
color = cm(k)
else:
color = '#C0C0C0'
color_db[ranges[i]] = color
i=0
for val in values:
sample = column_header[i]
for (l,u) in color_db:
range_index = ranges.index((l,u)) ### what is the ranking of this range
if val>=l and val<=u:
color = color_db[(l,u)]
color_label = [gene+'-range: '+str(l)[:4]+'-'+str(u)[:4],color,'']
group_db[sample] = color_label
try: bestGeneAssociated[sample].append([range_index,val,color_label])
except Exception: bestGeneAssociated[sample] = [[range_index,val,color_label]]
i+=1
#print min(values),min_val,bin_size,max_val
if len(genes)>1:
### Collapse and rank multiple gene results
for sample in bestGeneAssociated:
bestGeneAssociated[sample].sort()
color_label = bestGeneAssociated[sample][-1][-1]
if numberGenesPresent>1:
index = bestGeneAssociated[sample][-1][0]
if index > 2:
gene = string.split(color_label[0],'-')[0]
else:
gene = 'Null'
color_label[0] = gene
group_db[sample] = color_label
except Exception:
print [gene], 'not found in rows...'
#print traceback.format_exc()
k+=1
else:
print [colorByGene], 'not found in rows...'
#print len(group_count)
if len(group_count)>20:
Lfontsize = 10
if len(group_count)>30:
Lfontsize = 8
if len(group_count)>40:
Lfontsize = 6
if len(group_count)>50:
Lfontsize = 5
if len(scores[0])>150:
markersize = 7
else:
markersize = 10
i=0
group_names={}
for x in scores[0]:
### Add the text labels for each
sample_name = column_header[i]
try:
### Get group name and color information
group_name,color, k = group_db[sample_name]
if group_name not in group_names:
label = group_name ### Only add once for each group
else: label = None
group_names[group_name] = color, k
except Exception:
color = 'r'; label=None
ax.plot([scores[0][i]],[scores[1][i]],[scores[2][i]],color=color,marker='o',markersize=markersize,label=label,markeredgewidth=0,picker=True) #markeredgecolor=color
if showLabels:
#try: sample_name = ' '+string.split(sample_name,':')[1]
#except Exception: pass
ax.text(scores[0][i],scores[1][i],scores[2][i], ' '+sample_name,fontsize=9)
i+=1
# Shink current axis by 20%
box = ax.get_position()
ax.set_position([box.x0, box.y0, box.width * 0.8, box.height])
#pylab.legend(loc="upper left", prop={'size': 10})
try: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5),fontsize = Lfontsize) ### move the legend over to the right of the plot
except Exception: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
filename = 'Clustering-%s-3D-PCA.pdf' % dataset_name
pylab.savefig(root_dir + filename)
#print 'Exporting:',filename
filename = filename[:-3]+'png'
pylab.savefig(root_dir + filename) #dpi=200
graphic_link.append(['Principal Component Analysis',root_dir+filename])
if display:
print 'Exporting:',filename
try: pylab.show()
except Exception: None ### when run in headless mode
fig.clf()
def getAxes1(scores,PlotType=None):
""" Adjust these axes to account for (A) legend size (left hand upper corner)
and (B) long sample name extending to the right
"""
try:
x_range = max(scores[0])-min(scores[0])
y_range = max(scores[1])-min(scores[1])
if PlotType == '3D':
x_axis_min = min(scores[0])-(x_range/10)
x_axis_max = max(scores[0])+(x_range/10)
y_axis_min = min(scores[1])-(y_range/10)
y_axis_max = max(scores[1])+(y_range/10)
else:
x_axis_min = min(scores[0])-(x_range/10)
x_axis_max = max(scores[0])+(x_range/10)
y_axis_min = min(scores[1])-(y_range/10)
y_axis_max = max(scores[1])+(y_range/10)
except KeyError:
None
return [x_axis_min, x_axis_max, y_axis_min, y_axis_max]
def getAxes(scores,PlotType=None):
""" Adjust these axes to account for (A) legend size (left hand upper corner)
and (B) long sample name extending to the right
"""
try:
x_range = max(scores[0])-min(scores[0])
y_range = max(scores[1])-min(scores[1])
if PlotType == '3D':
x_axis_min = min(scores[0])-(x_range/1.5)
x_axis_max = max(scores[0])+(x_range/1.5)
y_axis_min = min(scores[1])-(y_range/5)
y_axis_max = max(scores[1])+(y_range/5)
else:
x_axis_min = min(scores[0])-(x_range/10)
x_axis_max = max(scores[0])+(x_range/10)
y_axis_min = min(scores[1])-(y_range/10)
y_axis_max = max(scores[1])+(y_range/10)
except KeyError:
None
return [x_axis_min, x_axis_max, y_axis_min, y_axis_max]
def getAxesTransposed(scores,exclude={}):
""" Adjust these axes to account for (A) legend size (left hand upper corner)
and (B) long sample name extending to the right
"""
scores_filtered=[]
for i in range(len(scores)):
if i not in exclude:
scores_filtered.append(scores[i])
scores = scores_filtered
scores = map(numpy.array, zip(*scores))
try:
x_range = max(scores[0])-min(scores[0])
y_range = max(scores[1])-min(scores[1])
x_axis_min = min(scores[0])-int((float(x_range)/7))
x_axis_max = max(scores[0])+int((float(x_range)/7))
y_axis_min = min(scores[1])-int(float(y_range/7))
y_axis_max = max(scores[1])+int(float(y_range/7))
except KeyError:
None
return [x_axis_min, x_axis_max, y_axis_min, y_axis_max]
def Kmeans(features, column_header, row_header):
#http://www.janeriksolem.net/2009/04/clustering-using-scipys-k-means.html
#class1 = numpy.array(numpy.random.standard_normal((100,2))) + numpy.array([5,5])
#class2 = 1.5 * numpy.array(numpy.random.standard_normal((100,2)))
features = numpy.vstack((class1,class2))
centroids,variance = scipy.cluster.vq.kmeans(features,2)
code,distance = scipy.cluster.vq.vq(features,centroids)
"""
This generates two normally distributed classes in two dimensions. To try and cluster the points, run k-means with k=2 like this.
The variance is returned but we don't really need it since the SciPy implementation computes several runs (default is 20) and selects the one with smallest variance for us. Now you can check where each data point is assigned using the vector quantization function in the SciPy package.
By checking the value of code we can see if there are any incorrect assignments. To visualize, we can plot the points and the final centroids.
"""
pylab.plot([p[0] for p in class1],[p[1] for p in class1],'*')
pylab.plot([p[0] for p in class2],[p[1] for p in class2],'r*')
pylab.plot([p[0] for p in centroids],[p[1] for p in centroids],'go')
pylab.show()
"""
def displaySimpleNetworkX():
import networkx as nx
print 'Graphing output with NetworkX'
gr = nx.Graph(rotate=90,bgcolor='white') ### commands for neworkx and pygraphviz are the same or similiar
edges = importSIF('Config/TissueFateMap.sif')
### Add nodes and edges
for (node1,node2,type) in edges:
gr.add_edge(node1,node2)
draw_networkx_edges
#gr['Myometrium']['color']='red'
# Draw as PNG
nx.draw_shell(gr) #wopi, gvcolor, wc, ccomps, tred, sccmap, fdp, circo, neato, acyclic, nop, gvpr, dot, sfdp. - fdp
pylab.savefig('LineageNetwork.png')
def displaySimpleNetwork(sif_filename,fold_db,pathway_name):
import pygraphviz as pgv
#print 'Graphing output with PygraphViz'
gr = pgv.AGraph(bgcolor='white',directed=True) ### Graph creation and setting of attributes - directed indicates arrows should be added
#gr = pgv.AGraph(rotate='90',bgcolor='lightgray')
### Set graph attributes
gr.node_attr['style']='filled'
gr.graph_attr['label']='%s Network' % pathway_name
edges = importSIF(sif_filename)
if len(edges) > 700:
print sif_filename, 'too large to visualize...'
else:
### Add nodes and edges
for (node1,node2,type) in edges:
nodes = (node1,node2)
gr.add_edge(nodes)
child, parent = nodes
edge = gr.get_edge(nodes[0],nodes[1])
if 'TF' in pathway_name or 'WGRV' in pathway_name:
node = child ### This is the regulating TF
else:
node = parent ### This is the pathway
n=gr.get_node(node)
### http://www.graphviz.org/doc/info/attrs.html
n.attr['penwidth'] = 4
n.attr['fillcolor']= '#FFFF00' ### yellow
n.attr['shape']='rectangle'
#n.attr['weight']='yellow'
#edge.attr['arrowhead'] = 'diamond' ### set the arrow type
id_color_db = WikiPathways_webservice.getHexadecimalColorRanges(fold_db,'Genes')
for gene_symbol in id_color_db:
color_code = id_color_db[gene_symbol]
try:
n=gr.get_node(gene_symbol)
n.attr['fillcolor']= '#'+string.upper(color_code) #'#FF0000'
#n.attr['rotate']=90
except Exception: None
# Draw as PNG
#gr.layout(prog='dot') #fdp (spring embedded), sfdp (OK layout), neato (compressed), circo (lots of empty space), dot (hierarchical - linear)
gr.layout(prog='neato')
output_filename = '%s.png' % sif_filename[:-4]
#print output_filename
gr.draw(output_filename)
"""
def findParentDir(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1 ### get just the parent directory
return filename[:x]
def findFilename(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1 ### get just the parent directory
return filename[x:]
def runHierarchicalClustering(matrix, row_header, column_header, dataset_name,
row_method, row_metric, column_method, column_metric,
color_gradient, display=False, contrast=None,
allowAxisCompression=True,Normalize=True):
""" Running with cosine or other distance metrics can often produce negative Z scores
during clustering, so adjustments to the clustering may be required.
=== Options Include ===
row_method = 'average'
column_method = 'single'
row_metric = 'cosine'
column_metric = 'euclidean'
color_gradient = 'red_white_blue'
color_gradient = 'red_black_sky'
color_gradient = 'red_black_blue'
color_gradient = 'red_black_green'
color_gradient = 'yellow_black_blue'
color_gradient == 'coolwarm'
color_gradient = 'seismic'
color_gradient = 'green_white_purple'
"""
try:
if allowLargeClusters: maxSize = 50000
else: maxSize = 7000
except Exception: maxSize = 7000
try:
PriorColumnClusters=priorColumnClusters
PriorRowClusters=priorRowClusters
except Exception:
PriorColumnClusters=None
PriorRowClusters=None
run = False
print 'max allowed cluster size:',maxSize
if len(matrix)>0 and (len(matrix)<maxSize or row_method == None):
#if len(matrix)>5000: row_metric = 'euclidean'
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
try:
### Default for display is False, when set to True, Pylab will render the image
heatmap(numpy.array(matrix), row_header, column_header, row_method, column_method,
row_metric, column_metric, color_gradient, dataset_name, display=display,
contrast=contrast,allowAxisCompression=allowAxisCompression,Normalize=Normalize,
PriorColumnClusters=PriorColumnClusters,PriorRowClusters=PriorRowClusters)
run = True
except Exception:
print traceback.format_exc()
try:
pylab.clf()
pylab.close() ### May result in TK associated errors later on
import gc
gc.collect()
except Exception: None
if len(matrix)<10000:
print 'Error using %s ... trying euclidean instead' % row_metric
row_metric = 'cosine'; row_method = 'average' ### cityblock
else:
print 'Error with hierarchical clustering... only clustering arrays'
row_method = None ### Skip gene clustering
try:
heatmap(numpy.array(matrix), row_header, column_header, row_method, column_method,
row_metric, column_metric, color_gradient, dataset_name, display=display,
contrast=contrast,allowAxisCompression=allowAxisCompression,Normalize=Normalize,
PriorColumnClusters=PriorColumnClusters,PriorRowClusters=PriorRowClusters)
run = True
except Exception:
print traceback.format_exc()
print 'Unable to generate cluster due to dataset incompatibilty.'
elif len(matrix)==0:
print_out = 'SKIPPING HIERARCHICAL CLUSTERING!!! - Your dataset file has no associated rows.'
print print_out
else:
print_out = 'SKIPPING HIERARCHICAL CLUSTERING!!! - Your dataset file is over the recommended size limit for clustering ('+str(maxSize)+' rows). Please cluster later using "Additional Analyses"'
print print_out
try:
pylab.clf()
pylab.close() ### May result in TK associated errors later on
import gc
gc.collect()
except Exception: None
return run
def debugTKBug():
return None
def runHCexplicit(filename, graphics, row_method, row_metric, column_method, column_metric, color_gradient,
extra_params, display=True, contrast=None, Normalize=False, JustShowTheseIDs=[],compressAxis=True):
""" Explicit method for hieararchical clustering with defaults defined by the user (see below function) """
#print [filename, graphics, row_method, row_metric, column_method, column_metric, color_gradient, contrast, Normalize]
global root_dir
global inputFilename
global originalFilename
global graphic_link
global allowLargeClusters
global GroupDB
global justShowTheseIDs
global targetGeneIDs
global normalize
global rho_cutoff
global species
global runGOElite
global EliteGeneSets
global storeGeneSetName
EliteGeneSets=[]
targetGene=[]
filterByPathways=False
runGOElite = False
justShowTheseIDs = JustShowTheseIDs
allowLargeClusters = True
if compressAxis:
allowAxisCompression = True
else:
allowAxisCompression = False
graphic_link=graphics ### Store all locations of pngs
inputFilename = filename ### Used when calling R
filterIDs = False
normalize = Normalize
try:
### Specific additional optional parameters for filtering
transpose = extra_params.Transpose()
try:
rho_cutoff = extra_params.RhoCutoff()
print 'Setting correlation cutoff to a rho of',rho_cutoff
except Exception:
rho_cutoff = 0.5 ### Always done if no rho, but only used if getGeneCorrelations == True
#print 'Setting correlation cutoff to a rho of',rho_cutoff
PathwayFilter = extra_params.PathwaySelect()
GeneSet = extra_params.GeneSet()
OntologyID = extra_params.OntologyID()
Normalize = extra_params.Normalize()
normalize = Normalize
filterIDs = True
species = extra_params.Species()
platform = extra_params.Platform()
vendor = extra_params.Vendor()
newInput = findParentDir(inputFilename)+'/GeneSetClustering/'+findFilename(inputFilename)
targetGene = extra_params.GeneSelection() ### Select a gene or ID to get the top correlating genes
getGeneCorrelations = extra_params.GetGeneCorrelations() ### Select a gene or ID to get the top correlating genes
filterByPathways = extra_params.FilterByPathways()
PathwayFilter, filterByPathways = verifyPathwayName(PathwayFilter,GeneSet,OntologyID,filterByPathways)
justShowTheseIDs_var = extra_params.JustShowTheseIDs()
if len(justShowTheseIDs_var)>0:
justShowTheseIDs = justShowTheseIDs_var
elif len(targetGene)>0:
targetGene = string.replace(targetGene,'\n',' ')
targetGene = string.replace(targetGene,'\r',' ')
justShowTheseIDs = string.split(targetGene,' ')
try:
EliteGeneSets = extra_params.ClusterGOElite()
if EliteGeneSets != ['']: runGOElite = True
except Exception:
#print traceback.format_exc()
pass
try:
storeGeneSetName = extra_params.StoreGeneSetName()
except Exception:
storeGeneSetName = ''
except Exception,e:
#print traceback.format_exc();sys.exit()
transpose = extra_params
root_dir = findParentDir(filename)
if 'ExpressionOutput/Clustering' in root_dir:
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
elif 'ExpressionOutput' in root_dir:
root_dir = string.replace(root_dir,'ExpressionOutput','DataPlots') ### Applies to clustering of LineageProfiler results
root_dir = string.replace(root_dir,'/Clustering','') ### Applies to clustering of MarkerFinder results
else:
root_dir += '/DataPlots/'
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
if row_method == 'hopach': reverseOrder = False
else: reverseOrder = True
#"""
matrix, column_header, row_header, dataset_name, group_db = importData(filename,Normalize=Normalize,reverseOrder=reverseOrder)
GroupDB = group_db
inputFilename = string.replace(inputFilename,'.cdt','.txt')
originalFilename = inputFilename
if len(justShowTheseIDs)==0:
try:
if len(priorColumnClusters)>0 and priorRowClusters>0 and row_method==None and column_method == None:
try: justShowTheseIDs = importPriorDrivers(inputFilename)
except Exception: pass #justShowTheseIDs=[]
except Exception:
#print traceback.format_exc()
pass
#print len(matrix),;print len(column_header),;print len(row_header)
if filterIDs:
transpose_update = True ### Since you can filterByPathways and getGeneCorrelations, only transpose once
if filterByPathways: ### Restrict analyses to only a single pathway/gene-set/ontology term
if isinstance(PathwayFilter, tuple) or isinstance(PathwayFilter, list):
FileName = string.join(list(PathwayFilter),' ')
FileName = string.replace(FileName,':','-')
else: FileName = PathwayFilter
if len(FileName)>40:
FileName = FileName[:40]
try: inputFilename = string.replace(newInput,'.txt','_'+FileName+'.txt') ### update the pathway reference for HOPACH
except Exception: inputFilename = string.replace(newInput,'.txt','_GeneSets.txt')
vars = filterByPathway(matrix,row_header,column_header,species,platform,vendor,GeneSet,PathwayFilter,OntologyID,transpose)
try: dataset_name += '-'+FileName
except Exception: dataset_name += '-GeneSets'
transpose_update = False
if 'amplify' in targetGene:
targetGene = string.join(vars[1],' ')+' amplify '+targetGene ### amplify the gene sets, but need the original matrix and headers (not the filtered)
else: matrix,row_header,column_header = vars
try:
alt_targetGene = string.replace(targetGene,'amplify','')
alt_targetGene = string.replace(alt_targetGene,'amplify','')
alt_targetGene = string.replace(alt_targetGene,'driver','')
alt_targetGene = string.replace(alt_targetGene,'guide','')
alt_targetGene = string.replace(alt_targetGene,'top','')
alt_targetGene = string.replace(alt_targetGene,'positive','')
alt_targetGene = string.replace(alt_targetGene,'excludeCellCycle','')
alt_targetGene = string.replace(alt_targetGene,'monocle','')
alt_targetGene = string.replace(alt_targetGene,'GuideOnlyCorrelation','')
alt_targetGene = string.replace(alt_targetGene,' ','')
except Exception:
alt_targetGene = ''
if getGeneCorrelations and targetGene != 'driver' and targetGene != 'GuideOnlyCorrelation' and \
targetGene != 'guide' and targetGene !='excludeCellCycle' and \
targetGene !='top' and targetGene != ' monocle' and \
targetGene !='positive' and len(alt_targetGene)>0: ###Restrict analyses to only genes that correlate with the target gene of interest
allowAxisCompression = False
if transpose and transpose_update == False: transpose_update = False ### If filterByPathways selected
elif transpose and transpose_update: transpose_update = True ### If filterByPathways not selected
else: transpose_update = False ### If transpose == False
if '\r' in targetGene or '\n' in targetGene:
targetGene = string.replace(targetGene, '\r',' ')
targetGene = string.replace(targetGene, '\n',' ')
if len(targetGene)>15:
inputFilename = string.replace(newInput,'.txt','-'+targetGene[:50]+'.txt') ### update the pathway reference for HOPACH
dataset_name += '-'+targetGene[:50]
else:
inputFilename = string.replace(newInput,'.txt','-'+targetGene+'.txt') ### update the pathway reference for HOPACH
dataset_name += '-'+targetGene
inputFilename = root_dir+'/'+string.replace(findFilename(inputFilename),'|',' ')
inputFilename = root_dir+'/'+string.replace(findFilename(inputFilename),':',' ') ### need to be careful of C://
dataset_name = string.replace(dataset_name,'|',' ')
dataset_name = string.replace(dataset_name,':',' ')
try:
matrix,row_header,column_header,row_method = getAllCorrelatedGenes(matrix,row_header,column_header,species,platform,vendor,targetGene,row_method,transpose_update)
except Exception:
print traceback.format_exc()
print targetGene, 'not found in input expression file. Exiting. \n\n'
badExit
targetGeneIDs = targetGene
exportTargetGeneList(targetGene,inputFilename)
else:
if transpose: ### Transpose the data matrix
print 'Transposing the data matrix'
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
#print len(matrix),;print len(column_header),;print len(row_header)
if len(column_header)>1000 or len(row_header)>1000:
print 'Performing hierarchical clustering (please be patient)...'
runHierarchicalClustering(matrix, row_header, column_header, dataset_name, row_method, row_metric,
column_method, column_metric, color_gradient, display=display,contrast=contrast,
allowAxisCompression=allowAxisCompression, Normalize=Normalize)
#"""
#graphic_link = [root_dir+'Clustering-exp.myeloid-steady-state-amplify positive Mki67 Clec4a2 Gria3 Ifitm6 Gfi1b -hierarchical_cosine_cosine.txt']
if 'guide' in targetGene:
import RNASeq
input_file = graphic_link[-1][-1][:-4]+'.txt'
if 'excludeCellCycle' in targetGene: excludeCellCycle = True
else: excludeCellCycle = False
print 'excludeCellCycle',excludeCellCycle
targetGene = RNASeq.remoteGetDriverGenes(species,platform,input_file,excludeCellCycle=excludeCellCycle,ColumnMethod=column_method)
extra_params.setGeneSelection(targetGene) ### force correlation to these
extra_params.setGeneSet('None Selected') ### silence this
graphic_link= runHCexplicit(filename, graphic_link, row_method, row_metric, column_method, column_metric, color_gradient,
extra_params, display=display, contrast=contrast, Normalize=Normalize, JustShowTheseIDs=JustShowTheseIDs,compressAxis=compressAxis)
return graphic_link
def importPriorDrivers(inputFilename):
filename = string.replace(inputFilename,'Clustering-','')
filename = string.split(filename,'-hierarchical')[0]+'-targetGenes.txt'
genes = open(filename, "rU")
genes = map(lambda x: cleanUpLine(x),genes)
return genes
def exportTargetGeneList(targetGene,inputFilename):
exclude=['positive','top','driver', 'guide', 'amplify','GuideOnlyCorrelation']
exportFile = inputFilename[:-4]+'-targetGenes.txt'
eo = export.ExportFile(root_dir+findFilename(exportFile))
targetGenes = string.split(targetGene,' ')
for gene in targetGenes:
if gene not in exclude:
try: eo.write(gene+'\n')
except Exception: print 'Error export out gene (bad ascii):', [gene]
eo.close()
def debugPylab():
pylab.figure()
pylab.close()
pylab.figure()
def verifyPathwayName(PathwayFilter,GeneSet,OntologyID,filterByPathways):
import gene_associations
### If the user supplied an Ontology ID rather than a Ontology term name, lookup the term name and return this as the PathwayFilter
if len(OntologyID)>0:
PathwayFilter = gene_associations.lookupOntologyID(GeneSet,OntologyID,type='ID')
filterByPathways = True
return PathwayFilter, filterByPathways
def filterByPathway(matrix,row_header,column_header,species,platform,vendor,GeneSet,PathwayFilter,OntologyID,transpose):
### Filter all the matrix and header entries for IDs in the selected pathway
import gene_associations
from import_scripts import OBO_import
exportData = export.ExportFile(inputFilename)
matrix2=[]; row_header2=[]
if 'Ontology' in GeneSet: directory = 'nested'
else: directory = 'gene-mapp'
print "GeneSet(s) to analyze:",PathwayFilter
if isinstance(PathwayFilter, tuple) or isinstance(PathwayFilter, list): ### see if it is one or more pathways
associated_IDs={}
for p in PathwayFilter:
associated = gene_associations.simpleGenePathwayImport(species,GeneSet,p,OntologyID,directory)
for i in associated:associated_IDs[i]=[]
else:
associated_IDs = gene_associations.simpleGenePathwayImport(species,GeneSet,PathwayFilter,OntologyID,directory)
gene_annotations = gene_associations.importGeneData(species,'Ensembl')
vendor = string.replace(vendor,'other:','') ### For other IDs
try: array_to_ens = gene_associations.filterGeneToUID(species,'Ensembl',vendor,associated_IDs)
except Exception: array_to_ens={}
if platform == "3'array":
### IDs thus won't be Ensembl - need to translate
try:
#ens_to_array = gene_associations.getGeneToUidNoExon(species,'Ensembl-'+vendor); print vendor, 'IDs imported...'
array_to_ens = gene_associations.filterGeneToUID(species,'Ensembl',vendor,associated_IDs)
except Exception:
pass
#print platform, vendor, 'not found!!! Exiting method'; badExit
#array_to_ens = gene_associations.swapKeyValues(ens_to_array)
try:
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception:
pass
i=0
original_rows={} ### Don't add the same original ID twice if it associates with different Ensembl IDs
for row_id in row_header:
original_id = row_id; symbol = row_id
if 'SampleLogFolds' in inputFilename or 'RelativeLogFolds' in inputFilename or 'AltConfirmed' in inputFilename or 'MarkerGenes' in inputFilename or 'blah' not in inputFilename:
try: row_id,symbol = string.split(row_id,' ')[:2] ### standard ID convention is ID space symbol
except Exception:
try: symbol = gene_to_symbol[row_id][0]
except Exception: None
if len(symbol)==0: symbol = row_id
if ':' in row_id:
try:
cluster,row_id = string.split(row_id,':')
updated_row_id = cluster+':'+symbol
except Exception:
pass
else:
updated_row_id = symbol
try: original_id = updated_row_id
except Exception: pass
if platform == "3'array":
try:
try: row_ids = array_to_ens[row_id]
except Exception: row_ids = symbol_to_gene[symbol]
except Exception:
row_ids = [row_id]
else:
try:
try: row_ids = array_to_ens[row_id]
except Exception: row_ids = symbol_to_gene[symbol]
except Exception:
row_ids = [row_id]
for row_id in row_ids:
if row_id in associated_IDs:
if 'SampleLogFolds' in inputFilename or 'RelativeLogFolds' in inputFilename:
if original_id != symbol:
row_id = original_id+' '+symbol
else: row_id = symbol
else:
try: row_id = gene_annotations[row_id].Symbol()
except Exception: None ### If non-Ensembl data
if original_id not in original_rows: ### Don't add the same ID twice if associated with mult. Ensembls
matrix2.append(matrix[i])
#row_header2.append(row_id)
row_header2.append(original_id)
original_rows[original_id]=None
i+=1
if transpose:
matrix2 = map(numpy.array, zip(*matrix2)) ### coverts these to tuples
column_header, row_header2 = row_header2, column_header
exportData.write(string.join(['UID']+column_header,'\t')+'\n') ### title row export
i=0
for row_id in row_header2:
exportData.write(string.join([row_id]+map(str,matrix2[i]),'\t')+'\n') ### export values
i+=1
print len(row_header2), 'filtered IDs'
exportData.close()
return matrix2,row_header2,column_header
def getAllCorrelatedGenes(matrix,row_header,column_header,species,platform,vendor,targetGene,row_method,transpose):
### Filter all the matrix and header entries for IDs in the selected targetGene
resort_by_ID_name=False
if resort_by_ID_name:
index=0; new_row_header=[]; new_matrix=[]; temp_row_header = []
for name in row_header: temp_row_header.append((name,index)); index+=1
temp_row_header.sort()
for (name,index) in temp_row_header:
new_row_header.append(name)
new_matrix.append(matrix[index])
matrix = new_matrix
row_header = new_row_header
exportData = export.ExportFile(inputFilename)
try:
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
#from import_scripts import OBO_import; symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except Exception:
print 'No Ensembl-Symbol database available for',species
if platform == "3'array":
### IDs thus won't be Ensembl - need to translate
try:
if ':' in vendor:
vendor = string.split(vendor,':')[1]
#ens_to_array = gene_associations.getGeneToUidNoExon(species,'Ensembl-'+vendor); print vendor, 'IDs imported...'
array_to_ens = gene_associations.filterGeneToUID(species,'Ensembl',vendor,{})
except Exception,e:
array_to_ens={}
for uid in array_to_ens:
for gid in array_to_ens[uid]:
if gid in gene_to_symbol:
symbol = gene_to_symbol[gid][0]
try: gene_to_symbol[uid].append(symbol)
except Exception: gene_to_symbol[uid] = [symbol]
matrix2=[]
row_header2=[]
matrix_db={} ### Used to optionally sort according to the original order
multipleGenes = False
intersecting_ids=[]
i=0
### If multiple genes entered, just display these
targetGenes=[targetGene]
if ' ' in targetGene or ',' in targetGene or '|' in targetGene or '\n' in targetGene or '\r' in targetGene:
multipleGenes = True
if ' ' in targetGene: targetGene = string.replace(targetGene,' ', ' ')
if ',' in targetGene: targetGene = string.replace(targetGene,',', ' ')
#if '|' in targetGene and 'alt_junction' not in originalFilename: targetGene = string.replace(targetGene,'|', ' ')
if '\n' in targetGene: targetGene = string.replace(targetGene,'\n', ' ')
if '\r' in targetGene: targetGene = string.replace(targetGene,'\r', ' ')
targetGenes = string.split(targetGene,' ')
if row_method != None: targetGenes.sort()
intersecting_ids = [val for val in targetGenes if val in row_header]
for row_id in row_header:
original_rowid = row_id
symbol=row_id
new_symbol = symbol
rigorous_search = True
if ':' in row_id and '|' in row_id:
rigorous_search = False
elif ':' in row_id and '|' not in row_id:
a,b = string.split(row_id,':')[:2]
if 'ENS' in a or len(a)==17:
try:
row_id = a
symbol = gene_to_symbol[row_id][0]
except Exception:
symbol =''
elif 'ENS' not in b and len(a)!=17:
row_id = b
elif 'ENS' in b:
symbol = original_rowid
row_id = a
if rigorous_search:
try: row_id,symbol = string.split(row_id,' ')[:2] ### standard ID convention is ID space symbol
except Exception:
try: symbol = gene_to_symbol[row_id][0]
except Exception:
if 'ENS' not in original_rowid:
row_id, symbol = row_id, row_id
new_symbol = symbol
if 'ENS' not in original_rowid and len(original_rowid)!=17:
if original_rowid != symbol:
symbol = original_rowid+' '+symbol
for gene in targetGenes:
if string.lower(gene) == string.lower(row_id) or string.lower(gene) == string.lower(symbol) or string.lower(original_rowid)==string.lower(gene) or string.lower(gene) == string.lower(new_symbol):
matrix2.append(matrix[i]) ### Values for the row
row_header2.append(symbol)
matrix_db[symbol]=matrix[i]
else:
if row_id in targetGenes:
matrix2.append(matrix[i])
row_header2.append(row_id)
matrix_db[row_id]=matrix[i]
i+=1
i=0
#for gene in targetGenes:
# if gene not in matrix_db: print gene
else:
i=0
original_rows={} ### Don't add the same original ID twice if it associates with different Ensembl IDs
for row_id in row_header:
original_id = row_id
symbol = 'NA'
if 'SampleLogFolds' in inputFilename or 'RelativeLogFolds' in inputFilename or 'blah' not in inputFilename:
try: row_id,symbol = string.split(row_id,' ')[:2] ### standard ID convention is ID space symbol
except Exception:
try: symbol = gene_to_symbol[row_id][0]
except Exception:
row_id, symbol = row_id, row_id
original_id = row_id
if row_id == targetGene or symbol == targetGene:
targetGeneValues = matrix[i] ### Values for the row
break
i+=1
i=0
if multipleGenes==False: limit = 50
else: limit = 140 # lower limit is 132
print 'limit:',limit
#print len(intersecting_ids),len(targetGenes), multipleGenes
if multipleGenes==False or 'amplify' in targetGene or 'correlated' in targetGene:
row_header3=[] ### Convert to symbol if possible
if multipleGenes==False:
targetGeneValue_array = [targetGeneValues]
else:
targetGeneValue_array = matrix2
if (len(row_header2)>4) or len(row_header)>15000: # and len(row_header)<20000
print 'Performing all iterative pairwise corelations...',
corr_matrix = numpyCorrelationMatrixGene(matrix,row_header,row_header2,gene_to_symbol)
print 'complete'
matrix2=[]; original_headers=row_header2; row_header2 = []
matrix2_alt=[]; row_header2_alt=[]
### If one gene entered, display the most positive and negative correlated
import markerFinder; k=0
for targetGeneValues in targetGeneValue_array:
correlated=[]
anticorrelated=[]
try: targetGeneID = original_headers[k]
except Exception: targetGeneID=''
try:
rho_results = list(corr_matrix[targetGeneID])
except Exception:
#print traceback.format_exc()
rho_results = markerFinder.simpleScipyPearson(matrix,targetGeneValues)
correlated_symbols={}
#print targetGeneID, rho_results[:100]
#print targetGeneID, rho_results[-100:];sys.exit()
for (rho,ind) in rho_results[:limit]: ### Get the top-50 correlated plus the gene of interest
proceed = True
try:
if len(rho)==2: rho = rho[0]
except: pass
if 'top' in targetGene:
if rho_results[4][0]<rho_cutoff: proceed = False
if rho>rho_cutoff and proceed: #and rho_results[3][0]>rho_cutoff:# ensures only clustered genes considered
rh = row_header[ind]
#if gene_to_symbol[rh][0] in targetGenes:correlated.append(gene_to_symbol[rh][0])
#correlated.append(gene_to_symbol[rh][0])
if len(row_header2)<100 or multipleGenes:
rh = row_header[ind]
#print rh, rho # Ly6c1, S100a8
if matrix[ind] not in matrix2:
if 'correlated' in targetGene:
if rho!=1:
matrix2.append(matrix[ind])
row_header2.append(rh)
if targetGeneValues not in matrix2: ### gene ID systems can be different between source and query
matrix2.append(targetGeneValues)
row_header2.append(targetGeneID)
try:correlated_symbols[gene_to_symbol[rh][0]]=ind
except Exception: correlated_symbols[rh]=ind
#print targetGeneValues, targetGene;sys.exit()
else:
matrix2.append(matrix[ind])
row_header2.append(rh)
try: correlated_symbols[gene_to_symbol[rh][0]]=ind
except Exception: correlated_symbols[rh]=ind
#if rho!=1: print gene_to_symbol[rh][0],'pos',targetGeneID
#sys.exit()
rho_results.reverse()
for (rho,ind) in rho_results[:limit]: ### Get the top-50 anti-correlated plus the gene of interest
try:
if len(rho)==2: rho = rho[0]
except: pass
if rho<-1*rho_cutoff and 'positive' not in targetGene:
rh = row_header[ind]
#if gene_to_symbol[rh][0] in targetGenes:anticorrelated.append(gene_to_symbol[rh][0])
#anticorrelated.append(gene_to_symbol[rh][0])
if len(row_header2)<100 or multipleGenes:
rh = row_header[ind]
if matrix[ind] not in matrix2:
if 'correlated' in targetGene:
if rho!=1:
matrix2.append(matrix[ind])
row_header2.append(rh)
if targetGeneValues not in matrix2:
matrix2.append(targetGeneValues)
row_header2.append(targetGeneID)
try: correlated_symbols[gene_to_symbol[rh][0]]=ind
except Exception: correlated_symbols[rh]=ind
#print targetGeneValues, targetGene;sys.exit()
else:
matrix2.append(matrix[ind])
row_header2.append(rh)
try: correlated_symbols[gene_to_symbol[rh][0]]=ind
except Exception: correlated_symbols[rh]=ind
#if rho!=1: print gene_to_symbol[rh][0],'neg',targetGeneID
try:
### print overlapping input genes that are correlated
if len(correlated_symbols)>0:
potentially_redundant=[]
for i in targetGenes:
if i in correlated_symbols:
if i != targetGeneID: potentially_redundant.append((i,correlated_symbols[i]))
if len(potentially_redundant)>0:
### These are intra-correlated genes based on the original filtered query
#print targetGeneID, potentially_redundant
for (rh,ind) in potentially_redundant:
matrix2_alt.append(matrix[ind])
row_header2_alt.append(rh)
rho_results.reverse()
#print targetGeneID, correlated_symbols, rho_results[:5]
except Exception:
pass
k+=1
#print targetGeneID+'\t'+str(len(correlated))+'\t'+str(len(anticorrelated))
#sys.exit()
if 'IntraCorrelatedOnly' in targetGene:
matrix2 = matrix2_alt
row_header2 = row_header2_alt
for r in row_header2:
try:
row_header3.append(gene_to_symbol[r][0])
except Exception: row_header3.append(r)
row_header2 = row_header3
#print len(row_header2),len(row_header3),len(matrix2);sys.exit()
matrix2.reverse() ### Display from top-to-bottom rather than bottom-to-top (this is how the clusters are currently ordered in the heatmap)
row_header2.reverse()
if 'amplify' not in targetGene:
row_method = None ### don't cluster the rows (row_method)
if 'amplify' not in targetGene and 'correlated' not in targetGene:
### reorder according to orignal
matrix_temp=[]
header_temp=[]
#print targetGenes
for symbol in targetGenes:
if symbol in matrix_db:
matrix_temp.append(matrix_db[symbol]); header_temp.append(symbol)
#print len(header_temp), len(matrix_db)
if len(header_temp) >= len(matrix_db): ### Hence it worked and all IDs are the same type
matrix2 = matrix_temp
row_header2 = header_temp
if transpose:
matrix2 = map(numpy.array, zip(*matrix2)) ### coverts these to tuples
column_header, row_header2 = row_header2, column_header
exclude=[]
#exclude = excludeHighlyCorrelatedHits(numpy.array(matrix2),row_header2)
exportData.write(string.join(['UID']+column_header,'\t')+'\n') ### title row export
i=0
for row_id in row_header2:
if ':' in row_id and '|' not in row_id:
a,b = string.split(row_id,':')[:2]
if 'ENS' in a:
try: row_id=string.replace(row_id,a,gene_to_symbol[a][0])
except Exception,e: pass
row_header2[i] = row_id
elif 'ENS' in row_id and ' ' in row_id and '|' not in row_id:
row_id = string.split(row_id, ' ')[1]
row_header2[i] = row_id
elif ' ' in row_id:
try: a,b = string.split(row_id, ' ')
except Exception: a = 1; b=2
if a==b:
row_id = a
if row_id not in exclude:
exportData.write(string.join([row_id]+map(str,matrix2[i]),'\t')+'\n') ### export values
i+=1
if 'amplify' not in targetGene and 'correlated' not in targetGene:
print len(row_header2), 'input gene IDs found'
else:
print len(row_header2), 'top-correlated IDs'
exportData.close()
return matrix2,row_header2,column_header,row_method
def numpyCorrelationMatrixGeneStore(x,rows,genes,gene_to_symbol):
### Decided not to use since it would require writing out the whole correlation matrix which is huge (1+GB) and time-intensive to import
start = time.time()
output_file = string.replace(originalFilename,'.txt','.corrmatrix')
status = verifyFile(output_file)
gene_correlations={}
if status == 'found':
try: symbol = gene_to_symbol[rows[i]][0]
except Exception: symbol = '$'
def splitInt(x):
rho,ind = string.split(x,'|')
return (float(rho),int(float(ind)))
for line in open(output_file,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
scores = map(lambda x: splitInt(x), t[1:])
gene_correlations[t[0]] = scores
else:
eo=export.ExportFile(output_file)
#D1 = numpy.ma.corrcoef(x)
D1 = numpy.corrcoef(x)
i=0
for score_ls in D1:
scores = []
try: symbol = gene_to_symbol[rows[i]][0]
except Exception: symbol = '$'
if rows[i] in genes or symbol in genes:
k=0
for v in score_ls:
if str(v)!='nan':
scores.append((v,k))
k+=1
scores.sort()
scores.reverse()
if len(symbol)==1: symbol = rows[i]
gene_correlations[symbol] = scores
export_values = [symbol]
for (v,k) in scores: ### re-import next time to save time
export_values.append(str(v)[:5]+'|'+str(k))
eo.write(string.join(export_values,'\t')+'\n')
i+=1
eo.close()
print len(gene_correlations)
print time.time() - start, 'seconds';sys.exit()
return gene_correlations
def numpyCorrelationMatrixGene(x,rows,genes,gene_to_symbol):
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
#D1 = numpy.ma.corrcoef(x)
D1 = numpy.corrcoef(x)
i=0
gene_correlations={}
for score_ls in D1:
scores = []
try: symbol = gene_to_symbol[rows[i]][0]
except Exception: symbol = '$'
if rows[i] in genes or symbol in genes:
k=0
for v in score_ls:
if str(v)!='nan':
scores.append((v,k))
k+=1
scores.sort()
scores.reverse()
if len(symbol)==1: symbol = rows[i]
gene_correlations[symbol] = scores
i+=1
return gene_correlations
def runHCOnly(filename,graphics,Normalize=False):
""" Simple method for hieararchical clustering with defaults defined by the function rather than the user (see above function) """
global root_dir
global graphic_link
global inputFilename
global GroupDB
global allowLargeClusters
global runGOElite
global EliteGeneSets
runGOElite = False
EliteGeneSets=[]
allowLargeClusters = False
###############
global inputFilename
global originalFilename
global GroupDB
global justShowTheseIDs
global targetGeneIDs
global normalize
global species
global storeGeneSetName
targetGene=[]
filterByPathways=False
justShowTheseIDs=[]
###############
graphic_link=graphics ### Store all locations of pngs
inputFilename = filename ### Used when calling R
root_dir = findParentDir(filename)
if 'ExpressionOutput/Clustering' in root_dir:
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
elif 'ExpressionOutput' in root_dir:
root_dir = string.replace(root_dir,'ExpressionOutput','DataPlots') ### Applies to clustering of LineageProfiler results
else:
root_dir += '/DataPlots/'
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
row_method = 'average'
column_method = 'weighted'
row_metric = 'cosine'
column_metric = 'cosine'
if 'Lineage' in filename or 'Elite' in filename:
color_gradient = 'red_white_blue'
else:
color_gradient = 'yellow_black_blue'
color_gradient = 'red_black_sky'
matrix, column_header, row_header, dataset_name, group_db = importData(filename,Normalize=Normalize)
GroupDB = group_db
runHierarchicalClustering(matrix, row_header, column_header, dataset_name,
row_method, row_metric, column_method, column_metric, color_gradient, display=False, Normalize=Normalize)
return graphic_link
def timestamp():
import datetime
today = str(datetime.date.today()); today = string.split(today,'-'); today = today[0]+''+today[1]+''+today[2]
time_stamp = string.replace(time.ctime(),':','')
time_stamp = string.replace(time_stamp,' ',' ')
time_stamp = string.split(time_stamp,' ') ###Use a time-stamp as the output dir (minus the day)
time_stamp = today+'-'+time_stamp[3]
return time_stamp
def runPCAonly(filename,graphics,transpose,showLabels=True,plotType='3D',display=True,
algorithm='SVD',geneSetName=None, species=None, zscore=True, colorByGene=None,
reimportModelScores=True, separateGenePlots=False, forceClusters=False, maskGroups=None):
global root_dir
global graphic_link
graphic_link=graphics ### Store all locations of pngs
root_dir = findParentDir(filename)
root_dir = string.replace(root_dir,'/DataPlots','')
root_dir = string.replace(root_dir,'/amplify','')
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
root_dir = string.replace(root_dir,'ExpressionInput','DataPlots')
root_dir = string.replace(root_dir,'ICGS-NMF','DataPlots')
if 'DataPlots' not in root_dir:
root_dir += '/DataPlots/'
try: os.mkdir(root_dir) ### May need to create this directory
except Exception: None
### Transpose matrix and build PCA
geneFilter=None
if (algorithm == 't-SNE' or algorithm == 'UMAP') and reimportModelScores:
dataset_name = string.split(filename,'/')[-1][:-4]
try:
### if the scores are present, we only need to import the genes of interest (save time importing large matrices)
if algorithm == 't-SNE':
importtSNEScores(root_dir+dataset_name+'-t-SNE_scores.txt')
if algorithm == 'UMAP':
importtSNEScores(root_dir+dataset_name+'-UMAP_scores.txt')
if len(colorByGene)==None:
geneFilter = [''] ### It won't import the matrix, basically
elif ' ' in colorByGene or ',' in colorByGene:
colorByGene = string.replace(colorByGene,',',' ')
geneFilter = string.split(colorByGene,' ')
else:
geneFilter = [colorByGene]
except Exception:
#print traceback.format_exc();sys.exit()
geneFilter = None ### It won't import the matrix, basically
matrix, column_header, row_header, dataset_name, group_db = importData(filename,zscore=zscore,geneFilter=geneFilter,forceClusters=forceClusters)
if transpose == False: ### We normally transpose the data, so if True, we don't transpose (I know, it's confusing)
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
if (len(column_header)>1000 or len(row_header)>1000) and algorithm != 't-SNE' and algorithm != 'UMAP':
print 'Performing Principal Component Analysis (please be patient)...'
#PrincipalComponentAnalysis(numpy.array(matrix), row_header, column_header, dataset_name, group_db, display=True)
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
if algorithm == 't-SNE' or algorithm == 'UMAP':
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
if separateGenePlots and (len(colorByGene)>0 or colorByGene==None):
for gene in geneFilter:
tSNE(numpy.array(matrix),column_header,dataset_name,group_db,display=False,
showLabels=showLabels,row_header=row_header,colorByGene=gene,species=species,
reimportModelScores=reimportModelScores,method=algorithm)
if display:
### Show the last one
tSNE(numpy.array(matrix),column_header,dataset_name,group_db,display=True,
showLabels=showLabels,row_header=row_header,colorByGene=gene,species=species,
reimportModelScores=reimportModelScores,method=algorithm)
elif maskGroups!=None:
""" Mask the samples not present in each examined group below """
import ExpressionBuilder
sample_group_db = ExpressionBuilder.simplerGroupImport(maskGroups)
##print maskGroups
#print sample_group_db;sys.exit()
group_sample_db = {}
for sample in sample_group_db:
try: group_sample_db[sample_group_db[sample]].append(sample)
except: group_sample_db[sample_group_db[sample]] = [sample]
for group in group_sample_db:
restricted_samples = group_sample_db[group]
tSNE(numpy.array(matrix),column_header,dataset_name,group_db,display=display,
showLabels=showLabels,row_header=row_header,colorByGene=colorByGene,species=species,
reimportModelScores=reimportModelScores,method=algorithm,maskGroups=(group,restricted_samples))
else:
tSNE(numpy.array(matrix),column_header,dataset_name,group_db,display=display,
showLabels=showLabels,row_header=row_header,colorByGene=colorByGene,species=species,
reimportModelScores=reimportModelScores,method=algorithm)
elif plotType == '3D':
try: PCA3D(numpy.array(matrix), row_header, column_header, dataset_name, group_db,
display=display, showLabels=showLabels, algorithm=algorithm, geneSetName=geneSetName,
species=species, colorByGene=colorByGene)
except Exception:
print traceback.format_exc()
PrincipalComponentAnalysis(numpy.array(matrix), row_header, column_header,
dataset_name, group_db, display=display, showLabels=showLabels, algorithm=algorithm,
geneSetName=geneSetName, species=species, colorByGene=colorByGene,
reimportModelScores=reimportModelScores)
else:
PrincipalComponentAnalysis(numpy.array(matrix), row_header, column_header, dataset_name,
group_db, display=display, showLabels=showLabels, algorithm=algorithm,
geneSetName=geneSetName, species=species, colorByGene=colorByGene,
reimportModelScores=reimportModelScores)
return graphic_link
def outputClusters(filenames,graphics,Normalize=False,Species=None,platform=None,vendor=None):
""" Peforms PCA and Hiearchical clustering on exported log-folds from AltAnalyze """
global root_dir
global graphic_link
global inputFilename
global GroupDB
global allowLargeClusters
global EliteGeneSets
EliteGeneSets=[]
global runGOElite
runGOElite = False
allowLargeClusters=False
graphic_link=graphics ### Store all locations of pngs
filename = filenames[0] ### This is the file to cluster with "significant" gene changes
inputFilename = filename ### Used when calling R
root_dir = findParentDir(filename)
root_dir = string.replace(root_dir,'ExpressionOutput/Clustering','DataPlots')
### Transpose matrix and build PCA
original = importData(filename,Normalize=Normalize)
matrix, column_header, row_header, dataset_name, group_db = original
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
if len(row_header)<700000 and len(column_header)<700000 and len(column_header)>2:
PrincipalComponentAnalysis(numpy.array(matrix), row_header, column_header, dataset_name, group_db)
else:
print 'SKIPPING PCA!!! - Your dataset file is over or under the recommended size limit for clustering (>7000 rows). Please cluster later using "Additional Analyses".'
row_method = 'average'
column_method = 'average'
row_metric = 'cosine'
column_metric = 'cosine'
color_gradient = 'red_white_blue'
color_gradient = 'red_black_sky'
global species
species = Species
if 'LineageCorrelations' not in filename and 'Zscores' not in filename:
EliteGeneSets=['GeneOntology']
runGOElite = True
### Generate Significant Gene HeatMap
matrix, column_header, row_header, dataset_name, group_db = original
GroupDB = group_db
runHierarchicalClustering(matrix, row_header, column_header, dataset_name, row_method, row_metric, column_method, column_metric, color_gradient, Normalize=Normalize)
### Generate Outlier and other Significant Gene HeatMap
for filename in filenames[1:]:
inputFilename = filename
matrix, column_header, row_header, dataset_name, group_db = importData(filename,Normalize=Normalize)
GroupDB = group_db
try:
runHierarchicalClustering(matrix, row_header, column_header, dataset_name, row_method, row_metric, column_method, column_metric, color_gradient, Normalize=Normalize)
except Exception: print 'Could not cluster',inputFilename,', file not found'
return graphic_link
def importEliteGeneAssociations(gene_filename):
fn = filepath(gene_filename)
x=0; fold_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#': x=0
elif x==0: x=1
else:
geneid=t[0];symbol=t[1]
fold = 0
try:
if '|' in t[6]:
fold = float(string.split(t[6])[0]) ### Sometimes there are multiple folds for a gene (multiple probesets)
except Exception:
None
try: fold=float(t[6])
except Exception: None
fold_db[symbol] = fold
return fold_db
def importPathwayLevelFolds(filename):
fn = filepath(filename)
x=0
folds_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if len(data)==0: x=0
elif x==0:
z_score_indexes = []; i=0
z_headers = []
for header in t:
if 'z_score.' in header:
z_score_indexes.append(i)
header = string.split(header,'z_score.')[1] ### Get rid of z_score.
if 'AS.' in header:
header = string.split(header,'.p')[0] ### Remove statistics details
header = 'AS.'+string.join(string.split(header,'_')[2:],'_') ### species and array type notation
else:
header = string.join(string.split(header,'-')[:-2],'-')
if '-fold' in header:
header = string.join(string.split(header,'-')[:-1],'-')
z_headers.append(header)
i+=1
headers = string.join(['Gene-Set Name']+z_headers,'\t')+'\n'
x=1
else:
term_name=t[1];geneset_type=t[2]
zscores = map(lambda x: t[x], z_score_indexes)
max_z = max(map(float, zscores)) ### If there are a lot of terms, only show the top 70
line = string.join([term_name]+zscores,'\t')+'\n'
try: zscore_db[geneset_type].append((max_z,line))
except Exception: zscore_db[geneset_type] = [(max_z,line)]
exported_files = []
for geneset_type in zscore_db:
### Create an input file for hierarchical clustering in a child directory (Heatmaps)
clusterinput_filename = findParentDir(filename)+'/Heatmaps/Clustering-Zscores-'+geneset_type+'.txt'
exported_files.append(clusterinput_filename)
export_text = export.ExportFile(clusterinput_filename)
export_text.write(headers) ### Header is the same for each file
zscore_db[geneset_type].sort()
zscore_db[geneset_type].reverse()
i=0 ### count the entries written
for (max_z,line) in zscore_db[geneset_type]:
if i<60:
export_text.write(line) ### Write z-score values and row names
i+=1
export_text.close()
return exported_files
def importOverlappingEliteScores(filename):
fn = filepath(filename)
x=0
zscore_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if len(data)==0: x=0
elif x==0:
z_score_indexes = []; i=0
z_headers = []
for header in t:
if 'z_score.' in header:
z_score_indexes.append(i)
header = string.split(header,'z_score.')[1] ### Get rid of z_score.
if 'AS.' in header:
header = string.split(header,'.p')[0] ### Remove statistics details
header = 'AS.'+string.join(string.split(header,'_')[2:],'_') ### species and array type notation
else:
header = string.join(string.split(header,'-')[:-2],'-')
if '-fold' in header:
header = string.join(string.split(header,'-')[:-1],'-')
z_headers.append(header)
i+=1
headers = string.join(['Gene-Set Name']+z_headers,'\t')+'\n'
x=1
else:
term_name=t[1];geneset_type=t[2]
zscores = map(lambda x: t[x], z_score_indexes)
max_z = max(map(float, zscores)) ### If there are a lot of terms, only show the top 70
line = string.join([term_name]+zscores,'\t')+'\n'
try: zscore_db[geneset_type].append((max_z,line))
except Exception: zscore_db[geneset_type] = [(max_z,line)]
exported_files = []
for geneset_type in zscore_db:
### Create an input file for hierarchical clustering in a child directory (Heatmaps)
clusterinput_filename = findParentDir(filename)+'/Heatmaps/Clustering-Zscores-'+geneset_type+'.txt'
exported_files.append(clusterinput_filename)
export_text = export.ExportFile(clusterinput_filename)
export_text.write(headers) ### Header is the same for each file
zscore_db[geneset_type].sort()
zscore_db[geneset_type].reverse()
i=0 ### count the entries written
for (max_z,line) in zscore_db[geneset_type]:
if i<60:
export_text.write(line) ### Write z-score values and row names
i+=1
export_text.close()
return exported_files
def buildGraphFromSIF(mod,species,sif_filename,ora_input_dir):
""" Imports a SIF and corresponding gene-association file to get fold changes for standardized gene-symbols """
global SpeciesCode; SpeciesCode = species
mod = 'Ensembl'
if sif_filename == None:
### Used for testing only
sif_filename = '/Users/nsalomonis/Desktop/dataAnalysis/collaborations/WholeGenomeRVista/Alex-Figure/GO-Elite_results/CompleteResults/ORA_pruned/up-2f_p05-WGRV.sif'
ora_input_dir = '/Users/nsalomonis/Desktop/dataAnalysis/collaborations/WholeGenomeRVista/Alex-Figure/up-stringent/up-2f_p05.txt'
#sif_filename = 'C:/Users/Nathan Salomonis/Desktop/Endothelial_Kidney/GO-Elite/GO-Elite_results/CompleteResults/ORA_pruned/GE.b_vs_a-fold2.0_rawp0.05-local.sif'
#ora_input_dir = 'C:/Users/Nathan Salomonis/Desktop/Endothelial_Kidney/GO-Elite/input/GE.b_vs_a-fold2.0_rawp0.05.txt'
gene_filename = string.replace(sif_filename,'.sif','_%s-gene-associations.txt') % mod
gene_filename = string.replace(gene_filename,'ORA_pruned','ORA_pruned/gene_associations')
pathway_name = string.split(sif_filename,'/')[-1][:-4]
output_filename = None
try: fold_db = importEliteGeneAssociations(gene_filename)
except Exception: fold_db={}
if ora_input_dir != None:
### This is an optional accessory function that adds fold changes from genes that are NOT in the GO-Elite pruned results (TFs regulating these genes)
try: fold_db = importDataSimple(ora_input_dir,species,fold_db,mod)
except Exception: None
try:
### Alternative Approaches dependening on the availability of GraphViz
#displaySimpleNetXGraph(sif_filename,fold_db,pathway_name)
output_filename = iGraphSimple(sif_filename,fold_db,pathway_name)
except Exception:
print 'igraph export failed due to an unknown error (not installed)'
print traceback.format_exc()
try: displaySimpleNetwork(sif_filename,fold_db,pathway_name)
except Exception: pass ### GraphViz problem
return output_filename
def iGraphSimple(sif_filename,fold_db,pathway_name):
""" Build a network export using iGraph and Cairo """
edges = importSIF(sif_filename)
id_color_db = WikiPathways_webservice.getHexadecimalColorRanges(fold_db,'Genes')
output_filename = iGraphDraw(edges,pathway_name,filePath=sif_filename,display=True,graph_layout='spring',colorDB=id_color_db)
return output_filename
def iGraphDraw(edges, pathway_name, labels=None, graph_layout='shell', display=False,
node_size=700, node_color='yellow', node_alpha=0.5, node_text_size=7,
edge_color='black', edge_alpha=0.5, edge_thickness=2, edges_pos=.3,
text_font='sans-serif',filePath='test',colorDB=None):
### Here node = vertex
output_filename=None
if len(edges) > 700 and 'AltAnalyze' not in pathway_name:
print findFilename(filePath), 'too large to visualize...'
elif len(edges) > 3000:
print findFilename(filePath), 'too large to visualize...'
else:
arrow_scaler = 1 ### To scale the arrow
if edges>40: arrow_scaler = .9
vars = formatiGraphEdges(edges,pathway_name,colorDB,arrow_scaler)
vertices,iGraph_edges,vertice_db,label_list,shape_list,vertex_size, color_list, vertex_label_colors, arrow_width, edge_colors = vars
if vertices>0:
import igraph
gr = igraph.Graph(vertices, directed=True)
canvas_scaler = 0.8 ### To scale the canvas size (bounding box)
if vertices<15: canvas_scaler = 0.5
elif vertices<25: canvas_scaler = .70
elif vertices>35:
canvas_scaler += len(iGraph_edges)/400.00
filePath,canvas_scaler = correctedFilePath(filePath,canvas_scaler) ### adjust for GO-Elite
#print vertices, len(iGraph_edges), pathway_name, canvas_scaler
canvas_size = (600*canvas_scaler,600*canvas_scaler)
gr.add_edges(iGraph_edges)
gr.vs["label"] = label_list
gr.vs["shape"] = shape_list
gr.vs["size"] = vertex_size
gr.vs["label_dist"] = [1.3]*vertices
gr.vs["label_size"] = [12]*vertices
gr.vs["color"]=color_list
gr.vs["label_color"]=vertex_label_colors
gr.es["color"] = edge_colors
gr.es["arrow_size"]=arrow_width
output_filename = '%s.pdf' % filePath[:-4]
output_filename = output_filename.encode('ascii','ignore') ### removes the damned unicode u proceeding the filename
layout = "kk"
visual_style = {}
#visual_style["layout"] = layout #The default is auto, which selects a layout algorithm automatically based on the size and connectedness of the graph
visual_style["margin"] = 50 ### white-space around the network (see vertex size)
visual_style["bbox"] = canvas_size
igraph.plot(gr,output_filename, **visual_style)
output_filename = '%s.png' % filePath[:-4]
output_filename = output_filename.encode('ascii','ignore') ### removes the damned unicode u proceeding the filename
if vertices <15: gr,visual_style = increasePlotSize(gr,visual_style)
igraph.plot(gr,output_filename, **visual_style)
#surface = cairo.ImageSurface(cairo.FORMAT_ARGB32, width, height)
return output_filename
def correctedFilePath(filePath,canvas_scaler):
""" Move this file to it's own network directory for GO-Elite """
if 'ORA_pruned' in filePath:
filePath = string.replace(filePath,'CompleteResults/ORA_pruned','networks')
try: os.mkdir(findParentDir(filePath))
except Exception: pass
canvas_scaler = canvas_scaler*1.3 ### These graphs tend to be more dense and difficult to read
return filePath,canvas_scaler
def increasePlotSize(gr,visual_style):
### To display the plot better, need to manually increase the size of everything
factor = 2
object_list = ["size","label_size"]
for i in object_list:
new=[]
for k in gr.vs[i]:
new.append(k*factor)
gr.vs[i] = new
new=[]
for i in gr.es["arrow_size"]:
new.append(i*factor)
new=[]
for i in visual_style["bbox"]:
new.append(i*factor)
visual_style["bbox"] = new
visual_style["margin"]=visual_style["margin"]*factor
return gr,visual_style
def getHMDBDataSimple():
### Determine which IDs are metabolites
program_type,database_dir = unique.whatProgramIsThis()
filename = database_dir+'/'+SpeciesCode+'/gene/HMDB.txt'
symbol_hmdb_db={}
x=0
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if x==0: x=1
else:
t = string.split(data,'\t')
hmdb_id = t[0]; symbol = t[1]; ProteinNames = t[-1]
symbol_hmdb_db[symbol]=hmdb_id
return symbol_hmdb_db
def formatiGraphEdges(edges,pathway_name,colorDB,arrow_scaler):
### iGraph appears to require defined vertice number and edges as numbers corresponding to these vertices
edge_db={}
edges2=[]
vertice_db={}
shape_list=[] ### node shape in order
label_list=[] ### Names of each vertix aka node
vertex_size=[]
color_list=[]
vertex_label_colors=[]
arrow_width=[] ### Indicates the presence or absence of an arrow
edge_colors=[]
k=0
try: symbol_hmdb_db = getHMDBDataSimple()
except Exception: symbol_hmdb_db={}
for (node1,node2,type) in edges:
edge_color = 'grey'
### Assign nodes to a numeric vertix ID
if 'TF' in pathway_name or 'WGRV' in pathway_name:
pathway = node1 ### This is the regulating TF
else:
pathway = node2 ### This is the pathway
if 'drugInteraction' == type: edge_color = "purple"
elif 'TBar' == type: edge_color = 'blue'
elif 'microRNAInteraction' == type: edge_color = '#53A26D'
elif 'transcription' in type: edge_color = '#FF7D7D'
if 'AltAnalyze' in pathway_name: default_node_color = 'grey'
else: default_node_color = "yellow"
if node1 in vertice_db: v1=vertice_db[node1]
else: #### Left hand node
### Only time the vertex is added to the below attribute lists
v1=k; label_list.append(node1)
rs = 1 ### relative size
if 'TF' in pathway_name or 'WGRV' in pathway_name and 'AltAnalyze' not in pathway_name:
shape_list.append('rectangle')
vertex_size.append(15)
vertex_label_colors.append('blue')
else:
if 'drugInteraction' == type:
rs = 0.75
shape_list.append('rectangle')
vertex_label_colors.append('purple')
default_node_color = "purple"
elif 'Metabolic' == type and node1 in symbol_hmdb_db:
shape_list.append('triangle-up')
vertex_label_colors.append('blue') #dark green
default_node_color = 'grey' #'#008000'
elif 'microRNAInteraction' == type:
rs = 0.75
shape_list.append('triangle-up')
vertex_label_colors.append('#008000') #dark green
default_node_color = 'grey' #'#008000'
else:
shape_list.append('circle')
vertex_label_colors.append('black')
vertex_size.append(10*rs)
vertice_db[node1]=v1; k+=1
try:
color = '#'+string.upper(colorDB[node1])
color_list.append(color) ### Hex color
except Exception:
color_list.append(default_node_color)
if node2 in vertice_db: v2=vertice_db[node2]
else: #### Right hand node
### Only time the vertex is added to the below attribute lists
v2=k; label_list.append(node2)
if 'TF' in pathway_name or 'WGRV' in pathway_name:
shape_list.append('circle')
vertex_size.append(10)
vertex_label_colors.append('black')
default_node_color = "grey"
elif 'AltAnalyze' not in pathway_name:
shape_list.append('rectangle')
vertex_size.append(15)
vertex_label_colors.append('blue')
default_node_color = "grey"
elif 'Metabolic' == type and node2 in symbol_hmdb_db:
shape_list.append('triangle-up')
vertex_label_colors.append('blue') #dark green
default_node_color = 'grey' #'#008000'
else:
shape_list.append('circle')
vertex_size.append(10)
vertex_label_colors.append('black')
default_node_color = "grey"
vertice_db[node2]=v2; k+=1
try:
color = '#'+string.upper(colorDB[node2])
color_list.append(color) ### Hex color
except Exception: color_list.append(default_node_color)
edges2.append((v1,v2))
if type == 'physical': arrow_width.append(0)
else: arrow_width.append(arrow_scaler)
try: edge_db[v1].append(v2)
except Exception: edge_db[v1]=[v2]
try: edge_db[v2].append(v1)
except Exception: edge_db[v2]=[v1]
edge_colors.append(edge_color)
vertices = len(edge_db) ### This is the number of nodes
edge_db = eliminate_redundant_dict_values(edge_db)
vertice_db2={} ### Invert
for node in vertice_db:
vertice_db2[vertice_db[node]] = node
#print len(edges2), len(edge_colors)
print vertices, 'and', len(edges2),'edges in the iGraph network.'
return vertices,edges2,vertice_db2, label_list, shape_list, vertex_size, color_list, vertex_label_colors, arrow_width, edge_colors
def eliminate_redundant_dict_values(database):
db1={}
for key in database: list = unique.unique(database[key]); list.sort(); db1[key] = list
return db1
def importDataSimple(filename,species,fold_db,mod):
""" Imports an input ID file and converts those IDs to gene symbols for analysis with folds """
import GO_Elite
from import_scripts import OBO_import
import gene_associations
fn = filepath(filename)
x=0
metabolite_codes = ['Ck','Ca','Ce','Ch','Cp']
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#': x=0
elif x==0:
si=None; symbol_present = False
try:
si= t.index('Symbol')
symbol_present = True
except: pass
x=1
else:
if x == 1:
system_code = t[1]
if system_code in metabolite_codes:
mod = 'HMDB'
system_codes,source_types,mod_types = GO_Elite.getSourceData()
try: source_data = system_codes[system_code]
except Exception:
source_data = None
if 'ENS' in t[0]: source_data = system_codes['En']
else: ### Assume the file is composed of gene symbols
source_data = system_codes['Sy']
if source_data == mod:
source_is_mod = True
elif source_data==None:
None ### Skip this
else:
source_is_mod = False
mod_source = mod+'-'+source_data+'.txt'
gene_to_source_id = gene_associations.getGeneToUid(species,('hide',mod_source))
source_to_gene = OBO_import.swapKeyValues(gene_to_source_id)
try: gene_to_symbol = gene_associations.getGeneToUid(species,('hide',mod+'-Symbol'))
except Exception: gene_to_symbol={}
try: met_to_symbol = gene_associations.importGeneData(species,'HMDB',simpleImport=True)
except Exception: met_to_symbol={}
for i in met_to_symbol: gene_to_symbol[i] = met_to_symbol[i] ### Add metabolite names
x+=1
if source_is_mod == True:
if t[0] in gene_to_symbol:
symbol = gene_to_symbol[t[0]][0]
try: fold_db[symbol] = float(t[2])
except Exception: fold_db[symbol] = 0
else:
fold_db[t[0]] = 0 ### If not found (wrong ID with the wrong system) still try to color the ID in the network as yellow
elif symbol_present:
fold_db[t[si]] = 0
try: fold_db[t[si]] = float(t[2])
except Exception:
try: fold_db[t[si]] = 0
except: fold_db[t[0]] = 0
elif t[0] in source_to_gene:
mod_ids = source_to_gene[t[0]]
try: mod_ids+=source_to_gene[t[2]] ###If the file is a SIF
except Exception:
try: mod_ids+=source_to_gene[t[1]] ###If the file is a SIF
except Exception: None
for mod_id in mod_ids:
if mod_id in gene_to_symbol:
symbol = gene_to_symbol[mod_id][0]
try: fold_db[symbol] = float(t[2]) ### If multiple Ensembl IDs in dataset, only record the last associated fold change
except Exception: fold_db[symbol] = 0
else: fold_db[t[0]] = 0
return fold_db
def clusterPathwayZscores(filename):
""" Imports a overlapping-results file and exports an input file for hierarchical clustering and clusters """
### This method is not fully written or in use yet - not sure if needed
if filename == None:
### Only used for testing
filename = '/Users/nsalomonis/Desktop/dataAnalysis/r4_Bruneau_TopHat/GO-Elite/TF-enrichment2/GO-Elite_results/overlapping-results_z-score_elite.txt'
exported_files = importOverlappingEliteScores(filename)
graphic_links=[]
for file in exported_files:
try: graphic_links = runHCOnly(file,graphic_links)
except Exception,e:
#print e
print 'Unable to generate cluster due to dataset incompatibilty.'
print 'Clustering of overlapping-results_z-score complete (see "GO-Elite_results/Heatmaps" directory)'
def clusterPathwayMeanFolds():
""" Imports the pruned-results file and exports an input file for hierarchical clustering and clusters """
filename = '/Users/nsalomonis/Desktop/User Diagnostics/Mm_spinal_cord_injury/GO-Elite/GO-Elite_results/pruned-results_z-score_elite.txt'
exported_files = importPathwayLevelFolds(filename)
def VennDiagram():
f = pylab.figure()
ax = f.gca()
rad = 1.4
c1 = Circle((-1,0),rad, alpha=.2, fc ='red',label='red')
c2 = Circle((1,0),rad, alpha=.2, fc ='blue',label='blue')
c3 = Circle((0,1),rad, alpha=.2, fc ='green',label='g')
#pylab.plot(c1,color='green',marker='o',markersize=7,label='blue')
#ax.add_patch(c1)
ax.add_patch(c2)
ax.add_patch(c3)
ax.set_xlim(-3,3)
ax.set_ylim(-3,3)
pylab.show()
def plotHistogram(filename):
matrix, column_header, row_header, dataset_name, group_db = importData(filename)
transpose=True
if transpose: ### Transpose the data matrix
print 'Transposing the data matrix'
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
pylab.figure()
for i in matrix:
pylab.hist(i, 200, normed=0, histtype='step', cumulative=-1)
#pylab.hist(matrix, 50, cumulative=-1)
pylab.show()
def stackedbarchart(filename,display=False,output=False):
header=[]
conditions = []
data_matrix=[]
for line in open(filename,'rU').xreadlines():
cd = cleanUpLine(line)
t = string.split(cd,'\t')
if len(header)==0:
header = t[4:]
exc_indexes = [0,2,4,6,8,10,12]
inc_indexes = [1,3,5,7,9,11,13]
inlc_header = map(lambda i: string.split(header[i],'_')[0],inc_indexes)
header = inlc_header
else:
condition = t[0]
data = t[4:]
conditions.append(condition+'-inclusion ')
data_matrix.append(map(lambda i: float(data[i]),inc_indexes))
conditions.append(condition+'-exclusion ')
data_matrix.append(map(lambda i: float(data[i]),exc_indexes))
data_matrix = map(numpy.array, zip(*data_matrix))
#https://www.w3resource.com/graphics/matplotlib/barchart/matplotlib-barchart-exercise-16.php
# multi-dimensional data_matrix
y_pos = np.arange(len(conditions))
fig, ax = pylab.subplots()
#fig = pylab.figure(figsize=(10,8))
#ax = fig.add_subplot(111)
#pos1 = ax.get_position() # get the original position
#pos2 = [pos1.x0 + 0.2, pos1.y0 - 0.2, pos1.width / 1.2, pos1.height / 1.2 ]
#ax.set_position(pos2) # set a new position
colors =['royalblue','salmon','grey','gold','cornflowerblue','mediumseagreen','navy']
patch_handles = []
# left alignment of data_matrix starts at zero
left = np.zeros(len(conditions))
index=0
for i, d in enumerate(data_matrix):
patch_handles.append(ax.barh(y_pos, d, 0.3,
color=colors[index], align='center',
left=left,label = header[index]))
left += d
index+=1
# search all of the bar segments and annotate
"""
for j in range(len(patch_handles)):
for i, patch in enumerate(patch_handles[j].get_children()):
bl = patch.get_xy()
x = 0.5*patch.get_width() + bl[0]
y = 0.5*patch.get_height() + bl[1]
#ax.text(x,y, "%d%%" % (percentages[i,j]), ha='center')
"""
ax.set_yticks(y_pos)
ax.set_yticklabels(conditions)
ax.set_xlabel('Events')
ax.legend(loc="best", bbox_to_anchor=(1.0, 1.0))
box = ax.get_position()
# Shink current axis by 20%
ax.set_position([box.x0+0.2, box.y0, box.width * 0.6, box.height])
try: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5),fontsize = 10) ### move the legend over to the right of the plot
except Exception: ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
ax.set_title('MultiPath-PSI Splicing Event Types')
#pylab.tight_layout(pad=0.4, w_pad=0.5, h_pad=1.0)
if output==False:
pylab.savefig(filename[:-4]+'.pdf')
pylab.savefig(filename[:-4]+'.png')
else:
pylab.savefig(output[:-4]+'.pdf')
pylab.savefig(output[:-4]+'.png')
if display:
print 'Exporting:',filename
try: pylab.show()
except Exception: None ### when run in headless mode
def barchart(filename,index1,index2,x_axis,y_axis,title,display=False,color1='gold',color2='darkviolet',output=False):
header=[]
reference_data=[]
query_data=[]
groups=[]
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if len(header)==0:
header = t
header1=header[index1]
header2=header[index2]
else:
reference_data.append(float(t[index1]))
q_value = float(t[index2])
if 'frequen' not in filename:
q_value = q_value*-1
query_data.append(q_value)
name = t[0]
if '_vs_' in name and 'event_summary' not in filename:
name = string.split(name,'_vs_')[0]
suffix=None
if '__' in name:
suffix = string.split(name,'__')[-1]
if '_' in name:
name = string.split(name,'_')[:-1]
name = string.join(name,'_')
if len(name)>20:
name = string.split(name,'_')[0]
if suffix !=None:
name+='_'+suffix
groups.append(name)
fig, ax = pylab.subplots()
pos1 = ax.get_position() # get the original position
pos2 = [pos1.x0 + 0.2, pos1.y0 + 0.1, pos1.width / 1.2, pos1.height / 1.2 ]
ax.set_position(pos2) # set a new position
ind = np.arange(len(groups)) # the x locations for the groups
width = 0.35 # the width of the bars
query_data.reverse()
reference_data.reverse()
groups.reverse()
ax.barh(ind - width/2, query_data, width, color=color2, label=header2)
ax.barh(ind + width/2, reference_data, width,color=color1, label=header1)
ax.set_xlabel(x_axis)
ax.set_ylabel(y_axis)
ax.set_yticks(ind+0.175)
ax.set_yticklabels(groups)
ax.set_title(title)
ax.legend()
if output==False:
pylab.savefig(filename[:-4]+'.pdf')
#pylab.savefig(filename[:-4]+'.png')
else:
pylab.savefig(output[:-4]+'.pdf')
#pylab.savefig(output[:-4]+'.png')
if display:
print 'Exporting:',filename
try: pylab.show()
except Exception: None ### when run in headless mode
def multipleSubPlots(filename,uids,SubPlotType='column',n=20):
#uids = [uids[-1]]+uids[:-1]
str_uids = string.join(uids,'_')
matrix, column_header, row_header, dataset_name, group_db = importData(filename,geneFilter=uids)
for uid in uids:
if uid not in row_header:
print uid,"is missing from the expression file."
fig = pylab.figure()
def ReplaceZeros(val,min_val):
if val == 0:
return min_val
else: return val
### Order the graphs based on the original gene order
new_row_header=[]
matrix2 = []
for uid in uids:
if uid in row_header:
ind = row_header.index(uid)
new_row_header.append(uid)
try: update_exp_vals = map(lambda x: ReplaceZeros(x,0.0001),matrix[ind])
except Exception: print uid, len(matrix[ind]);sys.exit()
#update_exp_vals = map(lambda x: math.pow(2,x+1),update_exp_vals) #- nonlog transform
matrix2.append(update_exp_vals)
matrix = numpy.array(matrix2)
row_header = new_row_header
#print row_header
color_list = ['r', 'b', 'y', 'g', 'w', 'k', 'm']
groups=[]
for sample in column_header:
try: group = group_db[sample][0]
except: group = '1'
if group not in groups:
groups.append(group)
fontsize=10
if len(groups)>0:
color_list = []
if len(groups)==9:
cm = matplotlib.colors.ListedColormap(['#80C241', '#118943', '#6FC8BB', '#ED1D30', '#F26E21','#8051A0', '#4684C5', '#FBD019','#3A52A4'])
elif len(groups)==3:
cm = matplotlib.colors.ListedColormap(['#4684C4','#FAD01C','#7D7D7F'])
#elif len(groups)==5: cm = matplotlib.colors.ListedColormap(['#41449B','#6182C1','#9DDAEA','#42AED0','#7F7F7F'])
else:
cm = pylab.cm.get_cmap('gist_rainbow') #gist_ncar
for i in range(len(groups)):
color_list.append(cm(1.*i/len(groups))) # color will now be an RGBA tuple
for i in range(len(matrix)):
ax = pylab.subplot(n,1,1+i)
OY = matrix[i]
pylab.xlim(0,len(OY))
pylab.subplots_adjust(right=0.85)
ind = np.arange(len(OY))
index_list = []
v_list = []
colors_list = []
if SubPlotType=='column':
index=-1
for v in OY:
index+=1
try: group = group_db[column_header[index]][0]
except: group = '1'
index_list.append(index)
v_list.append(v)
colors_list.append(color_list[groups.index(group)])
#pylab.bar(index, v,edgecolor='black',linewidth=0,color=color_list[groups.index(group)])
width = .35
#print i ,row_header[i]
print 1
barlist = pylab.bar(index_list, v_list,edgecolor='black',linewidth=0)
ci = 0
for cs in barlist:
barlist[ci].set_color(colors_list[ci])
ci+=1
if SubPlotType=='plot':
pylab.plot(x,y)
ax.text(matrix.shape[1]-0.5, i, ' '+row_header[i],fontsize=8)
fig.autofmt_xdate()
pylab.subplots_adjust(hspace = .001)
temp = tic.MaxNLocator(3)
ax.yaxis.set_major_locator(temp)
ax.set_xticks([])
#ax.title.set_visible(False)
#pylab.xticks(ind + width / 2, column_header)
#ax.set_xticklabels(column_header)
#ax.xaxis.set_ticks([-1]+range(len(OY)+1))
#xtickNames = pylab.setp(pylab.gca(), xticklabels=['']+column_header)
#pylab.setp(xtickNames, rotation=90, fontsize=10)
#pylab.show()
if len(str_uids)>50:
str_uids = str_uids[:50]
pylab.savefig(filename[:-4]+'-1'+str_uids+'.pdf')
def simpleTranspose(filename):
fn = filepath(filename)
matrix = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,' ')
matrix.append(t)
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
filename = filename[:-4]+'-transposed.txt'
ea = export.ExportFile(filename)
for i in matrix:
ea.write(string.join(i,'\t')+'\n')
ea.close()
def CorrdinateToBed(filename):
fn = filepath(filename)
matrix = []
translation={}
multiExon={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
data = string.replace(data,' ','')
t = string.split(data,'\t')
if '.gtf' in filename:
if 'chr' not in t[0]: chr = 'chr'+t[0]
else: chr = t[0]
start = t[3]; end = t[4]; strand = t[6]; annotation = t[8]
annotation = string.replace(annotation,'gene_id','')
annotation = string.replace(annotation,'transcript_id','')
annotation = string.replace(annotation,'gene_name','')
geneIDs = string.split(annotation,';')
geneID = geneIDs[0]; symbol = geneIDs[3]
else:
chr = t[4]; strand = t[5]; start = t[6]; end = t[7]
#if 'ENS' not in annotation:
t = [chr,start,end,geneID,'0',strand]
#matrix.append(t)
translation[geneID] = symbol
try: multiExon[geneID]+=1
except Exception: multiExon[geneID]=1
filename = filename[:-4]+'-new.bed'
ea = export.ExportFile(filename)
for i in translation:
#ea.write(string.join(i,'\t')+'\n')
ea.write(i+'\t'+translation[i]+'\t'+str(multiExon[i])+'\n')
ea.close()
def SimpleCorrdinateToBed(filename):
fn = filepath(filename)
matrix = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
data = string.replace(data,' ','')
t = string.split(data,'\t')
if '.bed' in filename:
print t;sys.exit()
chr = t[4]; strand = t[5]; start = t[6]; end = t[7]
if 'ENS' in t[0]:
t = [chr,start,end,t[0],'0',strand]
matrix.append(t)
filename = filename[:-4]+'-new.bed'
ea = export.ExportFile(filename)
for i in matrix:
ea.write(string.join(i,'\t')+'\n')
ea.close()
def simpleIntegrityCheck(filename):
fn = filepath(filename)
matrix = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
data = string.replace(data,' ','')
t = string.split(data,'\t')
matrix.append(t)
filename = filename[:-4]+'-new.bed'
ea = export.ExportFile(filename)
for i in matrix:
ea.write(string.join(i,'\t')+'\n')
ea.close()
def BedFileCheck(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.bed'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow = False
else:
#if len(t) != 12: print len(t);sys.exit()
ea.write(string.join(t,'\t')+'\n')
ea.close()
def simpleFilter(filename):
fn = filepath(filename)
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
matrix = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,',')
uid = t[0]
#if '=chr' in t[0]:
if 1==2:
a,b = string.split(t[0],'=')
b = string.replace(b,'_',':')
uid = a+ '='+b
matrix.append(t)
ea.write(string.join([uid]+t[1:],'\t')+'\n')
ea.close()
def test(filename):
symbols2={}
firstLine=True
fn = filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine=False
header = t
i=0; start=None; alt_start=None
value_indexes=[]
groups = {}
group = 0
for h in header:
if h == 'WikiPathways': start=i
if h == 'Select Protein Classes': alt_start=i
i+=1
if start == None: start = alt_start
for h in header:
if h>i:
group[i]
i+=1
if start == None: start = alt_start
else:
uniprot = t[0]
symbols = string.replace(t[-1],';;',';')
symbols = string.split(symbols,';')
for s in symbols:
if len(s)>0:
symbols2[string.upper(s),uniprot]=[]
for (s,u) in symbols2:
ea.write(string.join([s,u],'\t')+'\n')
ea.close()
def coincentIncedenceTest(exp_file,TFs):
fn = filepath(TFs)
tfs={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
tfs[data]=[]
comparisons={}
for tf1 in tfs:
for tf2 in tfs:
if tf1!=tf2:
temp = [tf1,tf2]
temp.sort()
comparisons[tuple(temp)]=[]
gene_data={}
firstLine=True
fn = filepath(exp_file)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if firstLine:
firstLine=False
header = string.split(data,'\t')[1:]
else:
t = string.split(data,'\t')
gene = t[0]
values = map(float,t[1:])
gene_data[gene] = values
filename = TFs[:-4]+'-all-coincident-5z.txt'
ea = export.ExportFile(filename)
comparison_db={}
for comparison in comparisons:
vals1 = gene_data[comparison[0]]
vals2 = gene_data[comparison[1]]
i=0
coincident=[]
for v1 in vals1:
v2 = vals2[i]
#print v1,v2
if v1>1 and v2>1:
coincident.append(i)
i+=1
i=0
population_db={}; coincident_db={}
for h in header:
population=string.split(h,':')[0]
if i in coincident:
try: coincident_db[population]+=1
except Exception: coincident_db[population]=1
try: population_db[population]+=1
except Exception: population_db[population]=1
i+=1
import mappfinder
final_population_percent=[]
for population in population_db:
d = population_db[population]
try: c = coincident_db[population]
except Exception: c = 0
N = float(len(header)) ### num all samples examined
R = float(len(coincident)) ### num all coincedent samples for the TFs
n = float(d) ### num all samples in cluster
r = float(c) ### num all coincident samples in cluster
try: z = mappfinder.Zscore(r,n,N,R)
except Exception: z=0
#if 'Gfi1b' in comparison and 'Gata1' in comparison: print N, R, n, r, z
final_population_percent.append([population,str(c),str(d),str(float(c)/float(d)),str(z)])
comparison_db[comparison]=final_population_percent
filtered_comparison_db={}
top_scoring_population={}
for comparison in comparison_db:
max_group=[]
for population_stat in comparison_db[comparison]:
z = float(population_stat[-1])
c = float(population_stat[1])
population = population_stat[0]
max_group.append([z,population])
max_group.sort()
z = max_group[-1][0]
pop = max_group[-1][1]
if z>(1.96)*2 and c>3:
filtered_comparison_db[comparison]=comparison_db[comparison]
top_scoring_population[comparison] = pop,z
firstLine = True
for comparison in filtered_comparison_db:
comparison_alt = string.join(list(comparison),'|')
all_percents=[]
for line in filtered_comparison_db[comparison]:
all_percents.append(line[3])
if firstLine:
all_headers=[]
for line in filtered_comparison_db[comparison]:
all_headers.append(line[0])
ea.write(string.join(['gene-pair']+all_headers+['Top Population','Top Z'],'\t')+'\n')
firstLine=False
pop,z = top_scoring_population[comparison]
ea.write(string.join([comparison_alt]+all_percents+[pop,str(z)],'\t')+'\n')
ea.close()
def getlastexon(filename):
filename2 = filename[:-4]+'-last-exon.txt'
ea = export.ExportFile(filename2)
firstLine=True
fn = filepath(filename)
last_gene = 'null'; last_exon=''
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
firstLine=False
else:
gene = t[2]
if gene != last_gene:
if ':E' in last_exon:
gene,exon = last_exon = string.split(':E')
block,region = string.split(exon,'.')
try: ea.write(last_exon+'\n')
except: pass
last_gene = gene
last_exon = t[0]
ea.close()
def replaceWithBinary(filename):
filename2 = filename[:-4]+'-binary.txt'
ea = export.ExportFile(filename2)
firstLine=True
fn = filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstLine:
ea.write(line)
firstLine=False
else:
try: values = map(float,t[1:])
except Exception: print t[1:];sys.exit()
values2=[]
for v in values:
if v == 0: values2.append('0')
else: values2.append('1')
ea.write(string.join([t[0]]+values2,'\t')+'\n')
ea.close()
def geneMethylationOutput(filename):
filename2 = filename[:-4]+'-binary.txt'
ea = export.ExportFile(filename2)
firstLine=True
fn = filepath(filename)
db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
values = (t[20],t[3]+'-methylation')
db[values]=[]
for value in db:
ea.write(string.join(list(value),'\t')+'\n')
ea.close()
def coincidentIncedence(filename,genes):
exportPairs=True
gene_data=[]
firstLine=True
fn = filepath(filename)
if exportPairs:
filename = filename[:-4]+'_'+genes[0]+'-'+genes[1]+'2.txt'
ea = export.ExportFile(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if firstLine:
firstLine=False
header = string.split(data,'\t')[1:]
else:
t = string.split(data,'\t')
gene = t[0]
if gene in genes:
values = map(float,t[1:])
gene_data.append(values)
vals1 = gene_data[0]
vals2 = gene_data[1]
i=0
coincident=[]
for v1 in vals1:
v2 = vals2[i]
#print v1,v2
if v1>1 and v2>1:
coincident.append(i)
i+=1
i=0
population_db={}; coincident_db={}
for h in header:
population=string.split(h,':')[0]
if i in coincident:
try: coincident_db[population]+=1
except Exception: coincident_db[population]=1
try: population_db[population]+=1
except Exception: population_db[population]=1
i+=1
import mappfinder
final_population_percent=[]
for population in population_db:
d = population_db[population]
try: c = coincident_db[population]
except Exception: c = 0
N = float(len(header)) ### num all samples examined
R = float(len(coincident)) ### num all coincedent samples for the TFs
n = d ### num all samples in cluster
r = c ### num all coincident samples in cluster
try: z = mappfinder.zscore(r,n,N,R)
except Exception: z = 0
final_population_percent.append([population,str(c),str(d),str(float(c)/float(d)),str(z)])
if exportPairs:
for line in final_population_percent:
ea.write(string.join(line,'\t')+'\n')
ea.close()
else:
return final_population_percent
def extractFeatures(countinp,IGH_gene_file):
import export
ExonsPresent=False
igh_genes=[]
firstLine = True
for line in open(IGH_gene_file,'rU').xreadlines():
if firstLine: firstLine=False
else:
data = cleanUpLine(line)
gene = string.split(data,'\t')[0]
igh_genes.append(gene)
if 'counts.' in countinp:
feature_file = string.replace(countinp,'counts.','IGH.')
fe = export.ExportFile(feature_file)
firstLine = True
for line in open(countinp,'rU').xreadlines():
if firstLine:
fe.write(line)
firstLine=False
else:
feature_info = string.split(line,'\t')[0]
gene = string.split(feature_info,':')[0]
if gene in igh_genes:
fe.write(line)
fe.close()
def filterForJunctions(countinp):
import export
ExonsPresent=False
igh_genes=[]
firstLine = True
count = 0
if 'counts.' in countinp:
feature_file = countinp[:-4]+'-output.txt'
fe = export.ExportFile(feature_file)
firstLine = True
for line in open(countinp,'rU').xreadlines():
if firstLine:
fe.write(line)
firstLine=False
else:
feature_info = string.split(line,'\t')[0]
junction = string.split(feature_info,'=')[0]
if '-' in junction:
fe.write(line)
count+=1
fe.close()
print count
def countIntronsExons(filename):
import export
exon_db={}
intron_db={}
firstLine = True
last_transcript=None
for line in open(filename,'rU').xreadlines():
if firstLine:
firstLine=False
else:
line = line.rstrip()
t = string.split(line,'\t')
transcript = t[-1]
chr = t[1]
strand = t[2]
start = t[3]
end = t[4]
exon_db[chr,start,end]=[]
if transcript==last_transcript:
if strand == '1':
intron_db[chr,last_end,start]=[]
else:
intron_db[chr,last_start,end]=[]
last_end = end
last_start = start
last_transcript = transcript
print len(exon_db)+1, len(intron_db)+1
def importGeneList(gene_list_file,n=20):
genesets=[]
genes=[]
for line in open(gene_list_file,'rU').xreadlines():
gene = line.rstrip()
gene = string.split(gene,'\t')[0]
genes.append(gene)
if len(genes)==n:
genesets.append(genes)
genes=[]
if len(genes)>0 and len(genes)<(n+1):
genes+=(n-len(genes))*[gene]
genesets.append(genes)
return genesets
def simpleListImport(filename):
genesets=[]
genes=[]
for line in open(filename,'rU').xreadlines():
gene = line.rstrip()
gene = string.split(gene,'\t')[0]
genes.append(gene)
return genes
def customClean(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow = False
#print len(t)
ea.write(string.join(['UID']+t,'\t')+'\n')
else:
if ';' in t[0]:
uid = string.split(t[0],';')[0]
else:
uid = t[0]
values = map(lambda x: float(x),t[1:])
values.sort()
if values[3]>=1:
ea.write(string.join([uid]+t[1:],'\t')+'\n')
ea.close()
def MakeJunctionFasta(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'.fasta'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
probeset, seq = string.split(data,'\t')[:2]
ea.write(">"+probeset+'\n')
ea.write(string.upper(seq)+'\n')
ea.close()
def ToppGeneFilter(filename):
import gene_associations; from import_scripts import OBO_import
gene_to_symbol = gene_associations.getGeneToUid('Hs',('hide','Ensembl-Symbol'))
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow = False
#print len(t)
ea.write(string.join(['Ensembl\t\tCategory'],'\t')+'\n')
else:
symbol = t[1]; category = t[3]
symbol = symbol[0]+string.lower(symbol[1:]) ### Mouse
category = category[:100]
if symbol in symbol_to_gene:
ensembl = symbol_to_gene[symbol][0]
ea.write(string.join([ensembl,symbol,category],'\t')+'\n')
ea.close()
def CountKallistoAlignedJunctions(filename):
fn = filepath(filename)
firstRow=True
#filename = filename[:-4]+'.fasta'
ea = export.ExportFile(filename)
found = False
counts=0
unique={}
ea = export.ExportFile(filename[:-4]+'-Mpo.txt')
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if 'ENS' in line and 'JUNC1201' in line:
ea.write(line)
unique[t[0]]=[]
counts+=1
print counts, len(unique)
ea.close()
def filterRandomFile(filename,col1,col2):
fn = filepath(filename)
firstRow=True
counts=0
ea = export.ExportFile(filename[:-4]+'-columns.txt')
for line in open(fn,'rU').xreadlines():
if line[0]!='#':
data = line.rstrip()
t = string.split(data,',')
#print t[col1-1]+'\t'+t[col2-1];sys.exit()
if ' ' in t[col2-1]:
t[col2-1] = string.split(t[col2-1],' ')[2]
ea.write(t[col1-1]+'\t'+t[col2-1]+'\n')
counts+=1
#print counts, len(unique)
ea.close()
def getBlockExonPositions():
fn = '/Users/saljh8/Desktop/Code/AltAnalyze/AltDatabase/EnsMart65/ensembl/Mm/Mm_Ensembl_exon.txt'
firstRow=True
filename = fn[:-4]+'.block.txt'
ea = export.ExportFile(filename)
found = False
lines=0
exon_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
gene,exonid,chromosome,strand,start,stop, a, b, c, d = string.split(data,'\t')
exonid = string.split(exonid,'.')[0]
uid = gene+':'+exonid
if lines>0:
try:
exon_db[uid,strand].append(int(start))
exon_db[uid,strand].append(int(stop))
except Exception:
exon_db[uid,strand] = [int(start)]
exon_db[uid,strand].append(int(stop))
lines+=1
print len(exon_db)
for (uid,strand) in exon_db:
exon_db[uid,strand].sort()
if strand == '-':
exon_db[uid,strand].reverse()
start = str(exon_db[uid,strand][0])
stop = str(exon_db[uid,strand][1])
coord = [start,stop]; coord.sort()
ea.write(uid+'\t'+strand+'\t'+coord[0]+'\t'+coord[1]+'\n')
ea.close()
def combineVariants(fn):
firstRow=True
filename = fn[:-4]+'.gene-level.txt'
ea = export.ExportFile(filename)
found = False
lines=0
gene_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
gene = t[9]
if lines == 0:
header = ['UID']+t[16:]
header = string.join(header,'\t')+'\n'
ea.write(header)
lines+=1
else:
var_calls = map(float,t[16:])
if gene in gene_db:
count_sum_array = gene_db[gene]
count_sum_array = [sum(value) for value in zip(*[count_sum_array,var_calls])]
gene_db[gene] = count_sum_array
else:
gene_db[gene] = var_calls
for gene in gene_db:
var_calls = gene_db[gene]
var_calls2=[]
for i in var_calls:
if i==0: var_calls2.append('0')
else: var_calls2.append('1')
ea.write(gene+'\t'+string.join(var_calls2,'\t')+'\n')
ea.close()
def compareFusions(fn):
firstRow=True
filename = fn[:-4]+'.matrix.txt'
ea = export.ExportFile(filename)
found = False
lines=0
fusion_db={}
sample_list=[]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if 'Gene_Fusion_Pair' in line:
headers = string.split(data,'\t')[1:]
try:
sample, fusion = string.split(data,'\t')
try: fusion_db[fusion].append(sample)
except Exception: fusion_db[fusion] = [sample]
if sample not in sample_list: sample_list.append(sample)
except Exception:
t = string.split(data,'\t')
fusion = t[0]
index=0
for i in t[1:]:
if i=='1':
sample = headers[index]
try: fusion_db[fusion].append(sample)
except Exception: fusion_db[fusion] = [sample]
if sample not in sample_list: sample_list.append(sample)
index+=1
fusion_db2=[]
for fusion in fusion_db:
samples = fusion_db[fusion]
samples2=[]
for s in sample_list:
if s in samples: samples2.append('1')
else: samples2.append('0')
fusion_db[fusion] = samples2
ea.write(string.join(['Fusion']+sample_list,'\t')+'\n')
for fusion in fusion_db:
print [fusion]
ea.write(fusion+'\t'+string.join(fusion_db[fusion],'\t')+'\n')
ea.close()
def customCleanSupplemental(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
line = string.split(data,', ')
gene_data=[]
for gene in line:
gene = string.replace(gene,' ','')
if '/' in gene:
genes = string.split(gene,'/')
gene_data.append(genes[0])
for i in genes[1:]:
gene_data.append(genes[0][:len(genes[1])*-1]+i)
elif '(' in gene:
genes = string.split(gene[:-1],'(')
gene_data+=genes
else:
gene_data.append(gene)
ea.write(string.join(gene_data,' ')+'\n')
ea.close()
def customCleanBinomial(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
from stats_scripts import statistics
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
headers = t
firstRow = False
ea.write(string.join(['uid']+headers,'\t')+'\n')
else:
gene = t[0]
values = map(float,t[1:])
min_val = abs(min(values))
values = map(lambda x: x+min_val,values)
values = map(str,values)
ea.write(string.join([gene]+values,'\t')+'\n')
ea.close()
class MarkerFinderInfo:
def __init__(self,gene,rho,tissue):
self.gene = gene
self.rho = rho
self.tissue = tissue
def Gene(self): return self.gene
def Rho(self): return self.rho
def Tissue(self): return self.tissue
def ReceptorLigandCellInteractions(species,lig_receptor_dir,cell_type_gene_dir):
ligand_db={}
receptor_db={}
fn = filepath(lig_receptor_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
ligand,receptor = string.split(data,'\t')
if species=='Mm':
ligand = ligand[0]+string.lower(ligand[1:])
receptor = receptor[0]+string.lower(receptor[1:])
try: ligand_db[ligand].apepnd(receptor)
except Exception: ligand_db[ligand] = [receptor]
try: receptor_db[receptor].append(ligand)
except Exception: receptor_db[receptor] = [ligand]
firstRow=True
filename = cell_type_gene_dir[:-4]+'-new.txt'
ea = export.ExportFile(filename)
found = False
cell_specific_ligands={}
cell_specific_receptor={}
fn = filepath(cell_type_gene_dir)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
gene, rho, tissue, notes, order = string.split(data,'\t')
mf = MarkerFinderInfo(gene, rho, tissue)
if gene in ligand_db:
cell_specific_ligands[gene]=mf
if gene in receptor_db:
cell_specific_receptor[gene]=mf
ligand_receptor_pairs=[]
for gene in cell_specific_ligands:
receptors = ligand_db[gene]
for receptor in receptors:
if receptor in cell_specific_receptor:
rmf = cell_specific_receptor[receptor]
lmf = cell_specific_ligands[gene]
gene_data = [gene,lmf.Tissue(),lmf.Rho(),receptor,rmf.Tissue(),rmf.Rho()]
pair = gene,receptor
if pair not in ligand_receptor_pairs:
ea.write(string.join(gene_data,'\t')+'\n')
ligand_receptor_pairs.append(pair)
for receptor in cell_specific_receptor:
ligands = receptor_db[receptor]
for gene in ligands:
if gene in cell_specific_ligands:
rmf = cell_specific_receptor[receptor]
lmf = cell_specific_ligands[gene]
gene_data = [gene,lmf.Tissue(),lmf.Rho(),receptor,rmf.Tissue(),rmf.Rho()]
pair = gene,receptor
if pair not in ligand_receptor_pairs:
ea.write(string.join(gene_data,'\t')+'\n')
ligand_receptor_pairs.append(pair)
ea.close()
def findReciprocal(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-filtered.txt'
ea = export.ExportFile(filename)
found = False
gene_ko={}; gene_oe={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
firstRow = False
headers={}
TFs={}
i=0
for v in t[1:]:
TF,direction = string.split(v,'-')
headers[i]=TF,direction,v
i+=1
if v not in TFs:
f = filename[:-4]+'-'+v+'-up.txt'
tea = export.ExportFile(f)
TFs[v+'-up']=tea
tea.write('GeneID\tEn\n')
f = filename[:-4]+'-'+v+'-down.txt'
tea = export.ExportFile(f)
TFs[v+'-down']=tea
tea.write('GeneID\tEn\n')
else:
values = map(float,t[1:])
gene = t[0]
i=0
for v in values:
TF,direction,name = headers[i]
if 'KO' in direction:
if v > 1:
gene_ko[gene,TF,1]=[]
tea = TFs[name+'-up']
tea.write(gene+'\tEn\n')
else:
gene_ko[gene,TF,-1]=[]
tea = TFs[name+'-down']
tea.write(gene+'\tEn\n')
if 'OE' in direction:
if v > 1:
gene_oe[gene,TF,1]=[]
tea = TFs[name+'-up']
tea.write(gene+'\tEn\n')
else:
gene_oe[gene,TF,-1]=[]
tea = TFs[name+'-down']
tea.write(gene+'\tEn\n')
i+=1
print len(gene_oe)
for (gene,TF,direction) in gene_oe:
alt_dir=direction*-1
if (gene,TF,alt_dir) in gene_ko:
ea.write(string.join([TF,gene,str(direction)],'\t')+'\n')
ea.close()
for TF in TFs:
TFs[TF].close()
def effectsPrioritization(filename):
fn = filepath(filename)
firstRow=True
filename = filename[:-4]+'-new.txt'
ea = export.ExportFile(filename)
from stats_scripts import statistics
found = False
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
headers = t[1:]
firstRow = False
else:
gene = t[0]
values = map(float,t[1:])
max_val = abs(max(values))
max_header = headers[values.index(max_val)]
ea.write(gene+'\t'+max_header+'\t'+str(max_val)+'\n')
ea.close()
def simpleCombine(folder):
filename = folder+'/combined/combined.txt'
ea = export.ExportFile(filename)
headers=['UID']
data_db={}
files = UI.read_directory(folder)
for file in files: #:70895507-70895600
if '.txt' in file:
fn = filepath(folder+'/'+file)
print fn
firstRow=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
for i in t[1:]:
headers.append(i+'.'+file[:-4])
firstRow = False
else:
gene = t[0]
try: data_db[gene]+=t[1:]
except Exception: data_db[gene] = t[1:]
len_db={}
ea.write(string.join(headers,'\t')+'\n')
for gene in data_db:
if len(data_db[gene])==(len(headers)-1):
values = map(float,data_db[gene])
count=0
for i in values:
if i>0.9: count+=1
if count>7:
ea.write(string.join([gene]+data_db[gene],'\t')+'\n')
len_db[len(data_db[gene])]=[]
print len(len_db)
for i in len_db:
print i
ea.close()
def simpleCombineFiles(folder):
filename = folder+'/combined/combined.txt'
ea = export.ExportFile(filename)
files = UI.read_directory(folder)
firstRow=True
for file in files:
if '.txt' in file:
fn = filepath(folder+'/'+file)
firstRow=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
t.append('Comparison')
ea.write(string.join(t,'\t')+'\n')
firstRow = False
else:
t.append(file[:-4])
ea.write(string.join(t,'\t')+'\n')
ea.close()
def evaluateMultiLinRegulatoryStructure(all_genes_TPM,MarkerFinder,SignatureGenes,state,query=None):
"""Predict multi-lineage cells and their associated coincident lineage-defining TFs"""
ICGS_State_as_Row = True
### Import all genes with TPM values for all cells
matrix, column_header, row_header, dataset_name, group_db = importData(all_genes_TPM)
group_index={}
all_indexes=[]
for sampleName in group_db:
ICGS_state = group_db[sampleName][0]
try: group_index[ICGS_state].append(column_header.index(sampleName))
except Exception: group_index[ICGS_state] = [column_header.index(sampleName)]
all_indexes.append(column_header.index(sampleName))
for ICGS_state in group_index:
group_index[ICGS_state].sort()
all_indexes.sort()
def importGeneLists(fn):
genes={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
gene,cluster = string.split(data,'\t')[0:2]
genes[gene]=cluster
return genes
def importMarkerFinderHits(fn):
genes={}
skip=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
if skip: skip=False
else:
gene,symbol,rho,ICGS_State = string.split(data,'\t')
#if ICGS_State!=state and float(rho)>0.0:
if float(rho)>0.0:
genes[gene]=float(rho),ICGS_State ### Retain all population specific genes (lax)
genes[symbol]=float(rho),ICGS_State
return genes
def importQueryDataset(fn):
matrix, column_header, row_header, dataset_name, group_db = importData(fn)
return matrix, column_header, row_header, dataset_name, group_db
signatureGenes = importGeneLists(SignatureGenes)
markerFinderGenes = importMarkerFinderHits(MarkerFinder)
#print len(signatureGenes),len(markerFinderGenes)
### Determine for each gene, its population frequency per cell state
index=0
expressedGenesPerState={}
def freqCutoff(x,cutoff):
if x>cutoff: return 1 ### minimum expression cutoff
else: return 0
for row in matrix:
ICGS_state_gene_frq={}
gene = row_header[index]
for ICGS_state in group_index:
state_values = map(lambda i: row[i],group_index[ICGS_state])
def freqCheck(x):
if x>1: return 1 ### minimum expression cutoff
else: return 0
expStateCells = sum(map(lambda x: freqCheck(x),state_values))
statePercentage = (float(expStateCells)/len(group_index[ICGS_state]))
ICGS_state_gene_frq[ICGS_state] = statePercentage
multilin_frq = ICGS_state_gene_frq[state]
datasets_values = map(lambda i: row[i],all_indexes)
all_cells_frq = sum(map(lambda x: freqCheck(x),datasets_values))/(len(datasets_values)*1.0)
all_states_frq = map(lambda x: ICGS_state_gene_frq[x],ICGS_state_gene_frq)
all_states_frq.sort() ### frequencies of all non-multilin states
rank = all_states_frq.index(multilin_frq)
states_expressed = sum(map(lambda x: freqCutoff(x,0.5),all_states_frq))/(len(all_states_frq)*1.0)
if multilin_frq > 0.25 and rank>0: #and states_expressed<0.75 #and all_cells_frq>0.75
if 'Rik' not in gene and 'Gm' not in gene:
if gene in signatureGenes:# and gene in markerFinderGenes:
if ICGS_State_as_Row:
ICGS_State = signatureGenes[gene]
if gene in markerFinderGenes:
if ICGS_State_as_Row == False:
rho, ICGS_State = markerFinderGenes[gene]
else:
rho, ICGS_Cell_State = markerFinderGenes[gene]
score = int(rho*100*multilin_frq)*(float(rank)/len(all_states_frq))
try: expressedGenesPerState[ICGS_State].append((score,gene))
except Exception: expressedGenesPerState[ICGS_State]=[(score,gene)] #(rank*multilin_frq)
index+=1
if query!=None:
matrix, column_header, row_header, dataset_name, group_db = importQueryDataset(query)
createPseudoCell=True
### The expressedGenesPerState defines genes and modules co-expressed in the multi-Lin
### Next, find the cells that are most frequent in mulitple states
representativeMarkers={}
for ICGS_State in expressedGenesPerState:
expressedGenesPerState[ICGS_State].sort()
expressedGenesPerState[ICGS_State].reverse()
if '1Multi' not in ICGS_State:
markers = expressedGenesPerState[ICGS_State][:5]
print ICGS_State,":",string.join(map(lambda x: x[1],list(markers)),', ')
if createPseudoCell:
for gene in markers:
def getBinary(x):
if x>1: return 1
else: return 0
if gene[1] in row_header: ### Only for query datasets
row_index = row_header.index(gene[1])
binaryValues = map(lambda x: getBinary(x), matrix[row_index])
#if gene[1]=='S100a8': print binaryValues;sys.exit()
try: representativeMarkers[ICGS_State].append(binaryValues)
except Exception: representativeMarkers[ICGS_State] = [binaryValues]
else:
representativeMarkers[ICGS_State]=markers[0][-1]
#int(len(markers)*.25)>5:
#print ICGS_State, markers
#sys.exit()
for ICGS_State in representativeMarkers:
if createPseudoCell:
signature_values = representativeMarkers[ICGS_State]
signature_values = [int(numpy.median(value)) for value in zip(*signature_values)]
representativeMarkers[ICGS_State] = signature_values
else:
gene = representativeMarkers[ICGS_State]
row_index = row_header.index(gene)
gene_values = matrix[row_index]
representativeMarkers[ICGS_State] = gene_values
### Determine for each gene, its population frequency per cell state
expressedStatesPerCell={}
for ICGS_State in representativeMarkers:
gene_values = representativeMarkers[ICGS_State]
index=0
for cell in column_header:
log2_tpm = gene_values[index]
if log2_tpm>=1:
try: expressedStatesPerCell[cell].append(ICGS_State)
except Exception: expressedStatesPerCell[cell] = [ICGS_State]
index+=1
cell_mutlilin_ranking=[]
for cell in expressedStatesPerCell:
lineageCount = expressedStatesPerCell[cell]
cell_mutlilin_ranking.append((len(lineageCount),cell))
cell_mutlilin_ranking.sort()
cell_mutlilin_ranking.reverse()
for cell in cell_mutlilin_ranking:
print cell[0], cell[1], string.join(expressedStatesPerCell[cell[1]],'|')
def compareGenomicLocationAndICGSClusters():
species = 'Mm'
array_type = 'RNASeq'
from build_scripts import EnsemblImport
gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,array_type,'key_by_array')
markerfinder = '/Users/saljh8/Desktop/Old Mac/Desktop/Grimes/Kallisto/ExpressionOutput/MarkerFinder/AllCorrelationsAnnotated-ProteinCodingOnly.txt'
eo = export.ExportFile(markerfinder[:-4]+'-bidirectional_promoters.txt')
firstRow=True
chr_cellTypeSpecific={}
for line in open(markerfinder,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
symbol = t[1]
ensembl = t[0]
try: rho = float(t[6])
except Exception: pass
cellType = t[7]
if firstRow:
firstRow = False
else:
if ensembl in gene_location_db and rho>0.2:
chr,strand,start,end = gene_location_db[ensembl]
start = int(start)
end = int(end)
#region = start[:-5]
try:
db = chr_cellTypeSpecific[chr,cellType]
try: db[strand].append([start,end,symbol,ensembl])
except Exception: db[strand] = [[start,end,symbol,ensembl]]
except Exception:
db={}
db[strand] = [[start,end,symbol,ensembl]]
chr_cellTypeSpecific[chr,cellType] = db
bidirectional={}
eo.write(string.join(['CellType','Chr','Ensembl1','Symbol1','Start1','End1','Strand1','Ensembl2','Symbol2','Start2','End2','Strand2'],'\t')+'\n')
for (chr,cellType) in chr_cellTypeSpecific:
db = chr_cellTypeSpecific[chr,cellType]
if len(db)>1: ### hence two strands
for (start,end,symbol,ens) in db['+']:
for (start2,end2,symbol2,ens2) in db['-']:
if abs(start-end2)<100000 and start>end2:
eo.write(string.join([cellType,chr,ens,symbol,str(start),str(end),'+',ens2,symbol2,str(end2),str(start2),'-'],'\t')+'\n')
try: bidirectional[chr,cellType].append([start,end,symbol,ens,start2,end2,symbol2,ens2])
except Exception: bidirectional[chr,cellType] = [[start,end,symbol,ens,start2,end2,symbol2,ens2]]
eo.close()
def filterCountsFile(filename):
fn = filepath(filename)
firstRow=True
def countif(value,cutoff=9):
if float(value)>cutoff: return 1
else: return 0
header = True
unique_genes = {}
ea = export.ExportFile(filename[:-4]+'-filtered.txt')
for line in open(fn,'rU').xreadlines():
if header:
header = False
ea.write(line)
else:
data = line.rstrip()
t = string.split(data,'\t')
gene = string.split(t[0],':')[0]
unique_genes[gene]=[]
expressedSamples = map(countif,t[1:])
if sum(expressedSamples)>2:
ea.write(line)
ea.close()
print len(unique_genes),'unique genes.'
def filterPSIValues(filename):
fn = filepath(filename)
firstRow=True
header = True
rows=0
filtered=0
new_file = filename[:-4]+'-75p.txt'
new_file_clust = new_file[:-4]+'-clustID.txt'
ea = export.ExportFile(new_file)
eac = export.ExportFile(new_file_clust)
added=[]
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
header = False
t = [t[1]]+t[8:]
header_length = len(t)-1
minimum_values_present = int(0.75*int(header_length))
not_detected = header_length-minimum_values_present
new_line = string.join(t,'\t')+'\n'
ea.write(new_line)
else:
cID = t[5]
t = [t[1]]+t[8:]
missing_values_at_the_end = (header_length+1)-len(t)
missing = missing_values_at_the_end+t.count('')
if missing<not_detected:
#if cID not in added:
added.append(cID)
new_line = string.join(t,'\t')+'\n'
ea.write(new_line)
eac.write(t[0]+'\t'+cID+'\n')
filtered+=1
rows+=1
print rows, filtered
ea.close()
eac.close()
#removeRedundantCluster(new_file,new_file_clust)
def removeRedundantCluster(filename,clusterID_file):
from scipy import stats
import ExpressionBuilder
sort_col=0
export_count=0
### Sort the filtered PSI model by gene name
ExpressionBuilder.exportSorted(filename, sort_col, excludeHeader=True)
new_file = filename[:-4]+'-unique.txt'
ea = export.ExportFile(new_file)
event_clusterID_db={}
for line in open(clusterID_file,'rU').xreadlines():
data = line.rstrip()
eventID,clusterID = string.split(data,'\t')
event_clusterID_db[eventID]=clusterID
def compareEvents(events_to_compare,export_count):
### This is where we compare the events and write out the unique entries
if len(events_to_compare)==1:
ea.write(events_to_compare[0][-1])
export_count+=1
else:
exclude={}
compared={}
for event1 in events_to_compare:
if event1[0] not in exclude:
ea.write(event1[-1])
exclude[event1[0]]=[]
export_count+=1
for event2 in events_to_compare:
if event2[0] not in exclude:
if event1[0] != event2[0] and (event1[0],event2[0]) not in compared:
uid1,values1,line1 = event1
uid2,values2,line2 = event2
coefr=numpy.ma.corrcoef(values1,values2)
#rho,p = stats.pearsonr(values1,values2)
rho = coefr[0][1]
if rho>0.6 or rho<-0.6:
exclude[event2[0]]=[]
compared[event1[0],event2[0]]=[]
compared[event2[0],event1[0]]=[]
for event in events_to_compare:
if event[0] not in exclude:
ea.write(event[-1]) ### write out the line
exclude.append(event[0])
export_count+=1
return export_count
header = True
rows=0
filtered=0
prior_cID = 0
events_to_compare=[]
for line in open(filename,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
ea.write(line)
header_row = t
header=False
else:
uid = t[0]
cID = event_clusterID_db[uid]
empty_offset = len(header_row)-len(t)
t+=['']*empty_offset
values = ['0.000101' if x=='' else x for x in t[1:]]
values = map(float,values)
values = numpy.ma.masked_values(values,0.000101)
if prior_cID==0: prior_cID = cID ### Occurs for the first entry
if cID == prior_cID:
### Replace empty values with 0
events_to_compare.append((uid,values,line))
else:
export_count = compareEvents(events_to_compare,export_count)
events_to_compare=[(uid,values,line)]
prior_cID = cID
if len(events_to_compare)>0: ### If the laster cluster set not written out yet
export_count = compareEvents(events_to_compare,export_count)
ea.close()
print export_count,'Non-redundant splice-events exported'
def convertToGOElite(folder):
files = UI.read_directory(folder)
for file in files:
if '.txt' in file:
gene_count=0; up_count=0; down_count=0
new_filename = string.split(file[3:],"_")[0]+'.txt'
ea = export.ExportFile(folder+'/GO-Elite/'+new_filename)
fn = folder+'/'+file
ea.write('GeneID\tSystemCode\n')
firstLine = True
for line in open(fn,'rU').xreadlines():
if firstLine:
firstLine= False
continue
data = line.rstrip()
t = string.split(data,'\t')
if ':' in t[0]:
ea.write(string.split(t[0],':')[0]+'\tSy\n')
else:
gene_count+=1
if '-' in t[2]: down_count+=1
else: up_count+=1
ea.close()
print file,'\t',gene_count,'\t',up_count,'\t',down_count
def geneExpressionSummary(folder):
import collections
event_db = collections.OrderedDict()
groups_list=['']
files = UI.read_directory(folder)
for file in files:
if '.txt' in file and 'GE.' in file:
ls=[]
event_db[file[:-4]]=ls
groups_list.append(file[:-4])
fn = folder+'/'+file
firstLine = True
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
fold_index = t.index('LogFold')
firstLine= False
continue
uid = t[0]
if float(t[fold_index])>0:
fold_dir = 1
else:
fold_dir = -1
ls.append((uid,fold_dir))
for file in event_db:
print file,'\t',len(event_db[file])
def compareEventLists(folder):
import collections
event_db = collections.OrderedDict()
groups_list=['']
files = UI.read_directory(folder)
file_headers = {}
for file in files:
if '.txt' in file and 'PSI.' in file:
ls={}
event_db[file[:-4]]=ls
groups_list.append(file[:-4])
fn = folder+'/'+file
firstLine = True
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
file_headers[file[:-4]] = t ### Store the headers
cid = t.index('ClusterID')
try: event_index = t.index('Event-Direction')
except:
try: event_index = t.index('Inclusion-Junction') ### legacy
except: print file, 'Event-Direction error';sys.exit()
firstLine= False
continue
uid = t[0]
uid = string.split(uid,'|')[0]
#uid = t[cid]
if 'U2AF1-l' in file or 'U2AF1-E' in file:
if t[2] == "inclusion":
ls[(uid,t[event_index])]=t ### Keep the event data for output
else:
ls[(uid,t[event_index])]=t ### Keep the event data for output
def convertEvents(events):
opposite_events=[]
for (event,direction) in events:
if direction == 'exclusion':
direction = 'inclusion'
else:
direction = 'exclusion'
opposite_events.append((event,direction))
return opposite_events
ea1 = export.ExportFile(folder+'/overlaps-same-direction.txt')
ea2 = export.ExportFile(folder+'/overlaps-opposite-direction.txt')
ea3 = export.ExportFile(folder+'/concordance.txt')
#ea4 = export.ExportFile(folder+'/overlap-same-direction-events.txt')
ea1.write(string.join(groups_list,'\t')+'\n')
ea2.write(string.join(groups_list,'\t')+'\n')
ea3.write(string.join(groups_list,'\t')+'\n')
comparison_db={}
best_hits={}
for comparison1 in event_db:
events1 = event_db[comparison1]
hits1=[comparison1]
hits2=[comparison1]
hits3=[comparison1]
best_hits[comparison1]=[]
for comparison2 in event_db:
events2 = event_db[comparison2]
events3 = convertEvents(events2)
overlapping_events = list(set(events1).intersection(events2))
overlap = len(overlapping_events)
inverse_overlap = len(set(events1).intersection(events3)) ### Get opposite events
### Calculate ratios based on the size of the smaller set
min_events1 = min([len(events1),len(events2)])
min_events2 = min([len(events1),len(events3)])
denom = overlap+inverse_overlap
if denom == 0: denom = 0.00001
#comparison_db[comparison1,comparison2]=overlap
if min_events1 == 0: min_events1 = 1
if (overlap+inverse_overlap)<20:
hits1.append('0.5')
hits2.append('0.5')
hits3.append('0.5|0.5')
else:
hits1.append(str((1.00*overlap)/min_events1))
hits2.append(str((1.00*inverse_overlap)/min_events1))
hits3.append(str(1.00*overlap/denom)+'|'+str(1.00*inverse_overlap/denom)+':'+str(overlap+inverse_overlap))
if 'Leu' not in comparison2:
comp_name = string.split(comparison2,'_vs')[0]
best_hits[comparison1].append([abs(1.00*overlap/denom),'cor',comp_name])
best_hits[comparison1].append([abs(1.00*inverse_overlap/denom),'anti',comp_name])
if comparison1 != comparison2:
if len(overlapping_events)>0:
#ea4.write(string.join(['UID',comparison1]+file_headers[comparison1]+[comparison2]+file_headers[comparison2],'\t')+'\n')
pass
overlapping_events.sort()
for event in overlapping_events:
vals = string.join([event[0],comparison1]+event_db[comparison1][event]+[comparison2]+event_db[comparison2][event],'\t')
#ea4.write(vals+'\n')
pass
ea1.write(string.join(hits1,'\t')+'\n')
ea2.write(string.join(hits2,'\t')+'\n')
ea3.write(string.join(hits3,'\t')+'\n')
ea1.close()
ea2.close()
ea3.close()
#ea4.close()
for comparison in best_hits:
best_hits[comparison].sort()
best_hits[comparison].reverse()
hits = best_hits[comparison][:10]
hits2=[]
for (score,dir,comp) in hits:
h = str(score)[:4]+'|'+dir+'|'+comp
hits2.append(h)
print comparison,'\t',string.join(hits2,', ')
def convertGroupsToBinaryMatrix(groups_file,sample_order,cellHarmony=False):
eo = export.ExportFile(groups_file[:-4]+'-matrix.txt')
print groups_file[:-4]+'-matrix.txt'
firstRow=True
samples = []
### Import a file with the sample names in the groups file in the correct order
for line in open(sample_order,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if 'row_clusters-flat' in t:
samples=[]
samples1 = t[2:]
for name in samples1:
if ':' in name:
group,name = string.split(name,':')
samples.append(name)
if cellHarmony==False:
break
elif 'column_clusters-flat' in t and cellHarmony:
clusters = t[2:]
elif groups_file == sample_order:
samples.append(t[0])
elif firstRow:
samples = t[1:]
firstRow=False
### Import a groups file
import collections
sample_groups = collections.OrderedDict()
for line in open(groups_file,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
sample, groupNum, groupName = t[:3]
if cellHarmony == False: ### JUST USE THE PROVIDED GROUPS FOR SAMPLES FOUND IN BOTH FILES
if sample in samples:
si=samples.index(sample) ### Index of the sample
try: sample_groups[groupName][si] = '1' ### set that sample to 1
except Exception:
sample_groups[groupName] = ['0']*len(samples)
sample_groups[groupName][si] = '1' ### set that sample to 1
else: ### JUST GRAB THE GROUP NAMES FOR THE SAMPLE GROUPS NOT THE SAMPES
sample_groups[groupNum]=groupName
if cellHarmony:
i=0
for sample in samples1:
cluster = clusters[i]
group_name = sample_groups[cluster]
eo.write(sample+'\t'+cluster+'\t'+group_name+'\n')
i+=1
eo.close()
else:
eo.write(string.join(['GroupName']+samples,'\t')+'\n')
for group in sample_groups:
eo.write(string.join([group]+sample_groups[group],'\t')+'\n')
eo.close()
def returnIntronJunctionRatio(counts_file,species = 'Mm'):
eo = export.ExportFile(counts_file[:-4]+'-intron-ratios.txt')
### Import a groups file
header=True
prior_gene=[]
exon_junction_values=[]
intron_junction_values=[]
eoi = export.ExportFile(counts_file[:-4]+'-intron-ratios-gene.txt')
rows=0
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
def logratio(list):
try: return list[0]/list[1]
except Exception: return 0
for line in open(counts_file,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
junctionID = t[0]
if header:
eoi.write(line)
samples = t[1:]
#zero_ref =[0]*len(samples)
global_intron_ratios={}
i=0
for val in samples:
global_intron_ratios[i]=[]
i+=1
header = False
continue
else:
uid,coords = string.split(junctionID,'=')
genes = string.split(uid,':') ### can indicate trans-splicing
if len(genes)>2: trans_splicing = True
else: trans_splicing = False
coords = string.split(coords,':')[1]
coords = string.split(coords,'-')
coords = map(int,coords)
coord_diff = abs(coords[1]-coords[0])
#ENSMUSG00000027770:I23.1-E24.1=chr3:62470748-62470747
gene = string.split(junctionID,':')[0]
rows+=1
if rows == 1:
prior_gene = gene
if gene != prior_gene:
#print gene
### merge all of the gene level counts for all samples
if len(intron_junction_values)==0:
#global_intron_ratios = [sum(value) for value in zip(*[global_intron_ratios,zero_ref])]
pass
else:
intron_junction_values_original = list(intron_junction_values)
exon_junction_values_original = list(exon_junction_values)
intron_junction_values = [sum(i) for i in zip(*intron_junction_values)]
exon_junction_values = [sum(i) for i in zip(*exon_junction_values)]
intron_ratios = [logratio(value) for value in zip(*[intron_junction_values,exon_junction_values])]
#if sum(intron_ratios)>3:
intron_ratios2=[]
if prior_gene in gene_to_symbol:
symbol = gene_to_symbol[prior_gene][0]
else:
symbol = prior_gene
i=0
#"""
if symbol == 'Pi4ka':
print samples[482:487]
for x in exon_junction_values_original:
print x[482:487]
print exon_junction_values[482:487]
print intron_ratios[482:487]
#"""
for val in intron_ratios:
if exon_junction_values[i]>9:
if val>0:
### stringent requirement - make sure it's not just a few reads
if intron_junction_values[i]>9:
intron_ratios2.append(val)
else:
intron_ratios2.append(0)
else:
intron_ratios2.append(0)
else:
"""
if val>0:
print val
print intron_junction_values
print exon_junction_values;sys.exit()"""
intron_ratios2.append('')
i+=1
eoi.write(string.join([symbol]+map(str,intron_ratios2),'\t')+'\n')
i = 0
for val in intron_ratios:
if exon_junction_values[i]!=0: ### Only consider values with a non-zero denominator
global_intron_ratios[i].append(intron_ratios[i])
i+=1
exon_junction_values = []
intron_junction_values = []
prior_gene = gene
values = map(float,t[1:])
if 'I' in junctionID and '_' not in junctionID and coord_diff==1 and trans_splicing == False:
intron_junction_values.append(values)
exon_junction_values.append(values)
elif trans_splicing == False:
exon_junction_values.append(values)
print rows, 'processed'
import numpy
i=0; global_intron_ratios_values=[]
for val in samples:
global_intron_ratios_values.append(100*numpy.mean(global_intron_ratios[i])) ### list of lists
i+=1
eo.write(string.join(['UID']+samples,'\t')+'\n')
eo.write(string.join(['Global-Intron-Retention-Ratio']+map(str,global_intron_ratios_values),'\t')+'\n')
eo.close()
eoi.close()
def convertSymbolLog(input_file,ensembl_symbol):
gene_symbol_db={}
for line in open(ensembl_symbol,'rU').xreadlines():
data = cleanUpLine(line)
ensembl,symbol = string.split(data,'\t')
gene_symbol_db[ensembl]=symbol
convert = False
eo = export.ExportFile(input_file[:-4]+'-log2.txt')
header=0
added_symbols=[]
not_found=[]
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
gene = values[0]
if header == 0:
#eo.write(line)
data = cleanUpLine(line)
headers = []
values = string.split(data,'\t')
for v in values:
if "exp." in v:
headers.append(string.split(v,'.exp.')[0])
else:
headers.append(v)
eo.write(string.join(headers,'\t')+'\n')
header +=1
if gene in gene_symbol_db:
symbol = gene_symbol_db[gene]
if symbol not in added_symbols:
added_symbols.append(symbol)
values = map(lambda x: math.log(float(x)+1,2),values[1:])
if max(values)> 0.5:
values = map(lambda x: str(x)[:5],values)
eo.write(string.join([symbol]+values,'\t')+'\n')
elif convert==False and header>1:
values = map(lambda x: math.log(float(x)+1,2),values[1:])
if max(values)> 0.5:
values = map(lambda x: str(x)[:5],values)
eo.write(string.join([gene]+values,'\t')+'\n')
else:
not_found.append(gene)
print len(not_found),not_found[:10]
eo.close()
def convertXenaBrowserIsoformDataToStandardRatios(input_file):
eo = open(input_file[:-4]+'-log2.txt','w')
header=0
count=0
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
uid = string.split(values[0],'.')[0]
isoform = values[0]
if header == 0:
eo.write(line)
header +=1
else:
values = map(lambda x: math.pow(2,float(x)),values[1:]) # convert list out of log space
values = map(lambda x: math.log(float(x)+1,2),values) # convert to value+1 log2
def percentExp(x):
if x>1: return 1
else: return 0
counts = map(lambda x: percentExp(x),values) # find how many values > 1
if sum(counts)/(len(values)*1.000)>0.1: # only write out genes with >10% of values > 1
values = map(str,values)
values = string.join([uid]+values,'\t')
eo.write(values+'\n')
count+=1
eo.close()
print count,'genes written'
def outputForGOElite(folds_dir):
matrix, column_header, row_header, dataset_name, group_db = importData(folds_dir,Normalize=False)
matrix = zip(*matrix) ### transpose
ci=0
root_dir = findParentDir(folds_dir)
for group_data in matrix:
group_name = column_header[ci]
eo = export.ExportFile(root_dir+'/folds/'+group_name+'.txt')
gi=0
eo.write('geneID'+'\tSy\t'+'log2-fold'+'\n')
for fold in group_data:
gene = row_header[gi]
if fold>0:
eo.write(gene+'\tSy\t'+str(fold)+'\n')
gi+=1
eo.close()
ci+=1
def transposeMatrix(input_file):
arrays=[]
eo = export.ExportFile(input_file[:-4]+'-transposed.txt')
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
arrays.append(values)
t_arrays = zip(*arrays)
for t in t_arrays:
eo.write(string.join(t,'\t')+'\n')
eo.close()
def simpleStatsSummary(input_file):
cluster_counts={}
header=True
for line in open(input_file,'rU').xreadlines():
data = cleanUpLine(line)
if header:
header = False
else:
sample,cluster,counts = string.split(data,'\t')
try: cluster_counts[cluster].append(float(counts))
except Exception: cluster_counts[cluster]=[float(counts)]
for cluster in cluster_counts:
avg = statistics.avg(cluster_counts[cluster])
stdev = statistics.stdev(cluster_counts[cluster])
print cluster+'\t'+str(avg)+'\t'+str(stdev)
def latteralMerge(file1, file2):
import collections
cluster_db = collections.OrderedDict()
eo = export.ExportFile(file2[:-4]+'combined.txt')
for line in open(file1,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
cluster_db[t[0]]=t
for line in open(file2,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if t[0] in cluster_db:
t1=cluster_db[t[0]]
eo.write(string.join(t1+t[2:],'\t')+'\n')
eo.close()
def removeMarkerFinderDoublets(heatmap_file,diff=1):
matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters = remoteImportData(heatmap_file)
priorRowClusters.reverse()
if len(priorColumnClusters)==0:
for c in column_header:
cluster = string.split(c,':')[0]
priorColumnClusters.append(cluster)
for r in row_header:
cluster = string.split(r,':')[0]
priorRowClusters.append(cluster)
import collections
cluster_db = collections.OrderedDict()
i=0
for cluster in priorRowClusters:
try: cluster_db[cluster].append(matrix[i])
except: cluster_db[cluster] = [matrix[i]]
i+=1
transposed_data_matrix=[]
clusters=[]
for cluster in cluster_db:
cluster_cell_means = numpy.mean(cluster_db[cluster],axis=0)
cluster_db[cluster] = cluster_cell_means
transposed_data_matrix.append(cluster_cell_means)
if cluster not in clusters:
clusters.append(cluster)
transposed_data_matrix = zip(*transposed_data_matrix)
i=0
cell_max_scores=[]
cell_max_score_db = collections.OrderedDict()
for cell_scores in transposed_data_matrix:
cluster = priorColumnClusters[i]
cell = column_header[i]
ci = clusters.index(cluster)
#print ci, cell, cluster, cell_scores;sys.exit()
cell_state_score = cell_scores[ci] ### This is the score for that cell for it's assigned MarkerFinder cluster
alternate_state_scores=[]
for score in cell_scores:
if score != cell_state_score:
alternate_state_scores.append(score)
alt_max_score = max(alternate_state_scores)
alt_sum_score = sum(alternate_state_scores)
cell_max_scores.append([cell_state_score,alt_max_score,alt_sum_score]) ### max and secondary max score - max for the cell-state should be greater than secondary max
try: cell_max_score_db[cluster].append(([cell_state_score,alt_max_score,alt_sum_score]))
except: cell_max_score_db[cluster] = [[cell_state_score,alt_max_score,alt_sum_score]]
i+=1
for cluster in cell_max_score_db:
cluster_cell_means = numpy.median(cell_max_score_db[cluster],axis=0)
cell_max_score_db[cluster] = cluster_cell_means ### This is the cell-state mean score for all cells in that cluster and the alternative max mean score (difference gives you the threshold for detecting double)
i=0
print len(cell_max_scores)
keep=['row_clusters-flat']
keep_alt=['row_clusters-flat']
remove = ['row_clusters-flat']
remove_alt = ['row_clusters-flat']
min_val = 1000
for (cell_score,alt_score,alt_sum) in cell_max_scores:
cluster = priorColumnClusters[i]
cell = column_header[i]
ref_max, ref_alt, ref_sum = cell_max_score_db[cluster]
ci = clusters.index(cluster)
ref_diff= math.pow(2,(ref_max-ref_alt))*diff #1.1
ref_alt = math.pow(2,(ref_alt))
cell_diff = math.pow(2,(cell_score-alt_score))
cell_score = math.pow(2,cell_score)
if cell_diff<min_val: min_val = cell_diff
if cell_diff>ref_diff and cell_diff>diff: #cell_score cutoff removes some, but cell_diff is more crucial
#if alt_sum<cell_score:
assignment=0 #1.2
keep.append(cell)
try: keep_alt.append(string.split(cell,':')[1]) ### if prefix added
except Exception:
keep_alt.append(cell)
else:
remove.append(cell)
try: remove_alt.append(string.split(cell,':')[1])
except Exception: remove_alt.append(cell)
assignment=1
#print assignment
i+=1
print min_val
print len(keep), len(remove)
from import_scripts import sampleIndexSelection
input_file=heatmap_file
output_file = heatmap_file[:-4]+'-Singlets.txt'
try: sampleIndexSelection.filterFile(input_file,output_file,keep)
except: sampleIndexSelection.filterFile(input_file,output_file,keep_alt)
output_file = heatmap_file[:-4]+'-Multiplets.txt'
try: sampleIndexSelection.filterFile(input_file,output_file,remove)
except: sampleIndexSelection.filterFile(input_file,output_file,remove_alt)
def exportTFcorrelations(filename,TF_file,threshold,anticorrelation=False):
eo = export.ExportFile(filename[:-4]+'-TF-correlations.txt')
TFs = simpleListImport(TF_file)
x, column_header, row_header, dataset_name, group_db = importData(filename)
### For methylation data or other data with redundant signatures, remove these and only report the first one
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
D1 = numpy.corrcoef(x)
i=0
correlation_pairs=[]
for score_ls in D1:
k=0
for v in score_ls:
if str(v)!='nan':
if k!=i:
#print row_header[i], row_header[k], v
if row_header[i] in TFs or row_header[k] in TFs:
#correlation_pairs.append([row_header[i],row_header[k],v])
if anticorrelation:
if v<(-1*threshold):
eo.write(row_header[i]+'\t'+row_header[k]+'\t'+str(v)+'\n')
elif v<(-1*threshold) or v>threshold:
eo.write(row_header[i]+'\t'+row_header[k]+'\t'+str(v)+'\n')
k+=1
i+=1
eo.close()
def TFisoformImport(filename):
isoform_db={}
for line in open(filename,'rU').xreadlines():
data = line.rstrip()
trans, prot, gene, symbol, uid, uid2, uid3 = string.split(data,'\t')
isoform_db[trans]=symbol,prot
return isoform_db
def exportIntraTFIsoformCorrelations(filename,TF_file,threshold,anticorrelation=False):
eo = export.ExportFile(filename[:-4]+'-TF-correlations.txt')
isoform_db = TFisoformImport(TF_file)
x, column_header, row_header, dataset_name, group_db = importData(filename)
### For methylation data or other data with redundant signatures, remove these and only report the first one
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
D1 = numpy.corrcoef(x)
i=0
correlation_pairs=[]
for score_ls in D1:
k=0
for v in score_ls:
if str(v)!='nan':
if k!=i:
#print row_header[i], row_header[k], v
if row_header[i] in isoform_db or row_header[k] in isoform_db:
try:
gene1,prot1 = isoform_db[row_header[i]]
gene2,prot2 = isoform_db[row_header[k]]
#correlation_pairs.append([row_header[i],row_header[k],v])
if gene1==gene2:
if anticorrelation:
if v<(-1*threshold):
eo.write(row_header[i]+'\t'+row_header[k]+'\t'+str(v)+'\n')
elif v<(-1*threshold) or v>threshold:
eo.write(row_header[i]+'\t'+row_header[k]+'\t'+str(v)+'\n')
except:
pass
k+=1
i+=1
eo.close()
def PSIfilterAndImpute(folder):
### Filter a PSI file and impute missing values based on neighbors
files = UI.read_directory(folder)
for file in files:
filename = folder+'/'+file
if '.txt' in file:
eo = export.ExportFile(filename[:-4]+'-impute.txt')
header = True
count=0
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
t0=values[1]
tl=values[-1]
vs = values[1:]
if header:
header=False
eo.write(line)
elif len(vs) == len(vs)-vs.count(''):
sum_val = sum(map(float,vs))/len(vs)
if sum_val!=1 and sum_val!=0: ### No variance in row
eo.write(line)
count+=1
elif (len(vs)-vs.count(''))>len(vs)-3:
new_values=[]; i=0
for v in vs:
if v=='':
if i==0: ### if the first element is null
try: new_values.append((float(vs[i+1])+float(tl))/2)
except: new_values.append(None) ### If two nulls occur in a row
elif i==len(vs)-1: ### if the last element is null
try: new_values.append((float(vs[i-1])+float(t0))/2)
except: new_values.append(None) ### If two nulls occur in a row
else: ### if the another element is null
try: new_values.append((float(vs[i-1])+float(vs[i+1]))/2)
except: new_values.append(None) ### If two nulls occur in a row
else:
new_values.append(v)
i+=1
if None not in new_values:
sum_val = sum(map(float,new_values))/len(new_values)
if sum_val!=1 and sum_val!=0: ### No variance in row
eo.write(string.join([values[0]]+map(str,new_values),'\t')+'\n')
count+=1
eo.close()
print count, '\t',fileg
def summarizePSIresults(folder, TF_file):
TFs = simpleListImport(TF_file)
### Import PSI results and report number of impacted TFs
files = UI.read_directory(folder)
eo = export.ExportFile(folder+'/TF_events.txt')
all_TFs=[]
for file in files:
TFs_in_file=[]
filename = folder+'/'+file
if '.txt' in file and 'PSI.' in file:
header = True
count=0
header=True
for line in open(filename,'rU').xreadlines():
if header:
header = False
else:
data = cleanUpLine(line)
t = string.split(data,'\t')
symbol = string.split(t[0],':')[0]
dPSI = abs(float(t[-5]))
#if symbol == 'CACFD1':
if symbol in TFs and symbol not in TFs_in_file and dPSI>0.2:
eo.write(string.join(t+[file],'\t')+'\n')
TFs_in_file.append(symbol)
if symbol not in all_TFs:
all_TFs.append(symbol)
count+=1
print file, count, len(all_TFs),string.join(TFs_in_file,',')
eo.close()
def convertPSICoordinatesToBED(folder):
### Import PSI results and report number of impacted TFs
files = UI.read_directory(folder)
eo = export.ExportFile(folder+'/combined.bed')
all_TFs=[]
for file in files:
TFs_in_file=[]
filename = folder+'/'+file
if '.txt' in file:
header = True
count=0
header=True
for line in open(filename,'rU').xreadlines():
if header:
header = False
else:
data = cleanUpLine(line)
t = string.split(data,'\t')
symbol = string.split(t[0],':')[0]
try: coordinates = t[7]
except: print t;sys.exit()
j1,j2 = string.split(coordinates,'|')
c1a,c1b = map(int,string.split(j1.split(':')[1],'-'))
strand='+'
if c1a>c1b:
c1a,c1b = c1b,c1a
strand='-'
c2a,c2b = map(int,string.split(j2.split(':')[1],'-'))
if c2a>c2b:
c2a,c2b = c2b,c2a
chr = string.split(coordinates,':')[0]
uid = string.replace(t[0],':','__')
eo.write(string.join([chr,str(c1a),str(c1b),uid+'--'+file,strand,str(c1a),str(c1b),'0'],'\t')+'\n')
eo.write(string.join([chr,str(c2a),str(c2b),uid+'--'+file,strand,str(c2a),str(c2b),'0'],'\t')+'\n')
eo.close()
def convertPSICoordinatesToBED(Mm_Ba_coordinates,Ba_events):
equivalencies={'Heart':['Heart'],
'Kidney':['Kidney-cortex','Kidney-medulla'],
'WFAT':['White-adipose-pericardial','White-adipose-mesenteric','White-adipose-subcutaneous','Omental-fat'],
'BFAT':['White-adipose-pericardial','White-adipose-mesenteric','White-adipose-subcutaneous','Omental-fat'],
'Lung':['Lungs'],
'Cere':['Cerebellum','Ventromedial-hypothalamus','Habenula','Pons','Pineal-gland','Visual-cortex','Lateral-globus-pallidus',
'Paraventricular-nuclei','Arcuate-nucleus','Suprachiasmatic-nuclei','Putamen','Optic-nerve-head', 'Medial-globus-pallidus',
'Amygdala','Prefontal-cortex','Dorsomedial-hypothalamus'],
'BS':['Cerebellum','Ventromedial-hypothalamus','Habenula','Pons','Pineal-gland','Visual-cortex','Lateral-globus-pallidus',
'Paraventricular-nuclei','Arcuate-nucleus','Suprachiasmatic-nuclei','Putamen','Optic-nerve-head', 'Medial-globus-pallidus',
'Amygdala','Prefontal-cortex','Dorsomedial-hypothalamus'],
'Hypo':['Cerebellum','Ventromedial-hypothalamus','Habenula','Pons','Pineal-gland','Visual-cortex','Lateral-globus-pallidus',
'Paraventricular-nuclei','Arcuate-nucleus','Suprachiasmatic-nuclei','Putamen','Optic-nerve-head', 'Medial-globus-pallidus',
'Amygdala','Prefontal-cortex','Dorsomedial-hypothalamus'],
'Adrenal':['Adrenal-cortex','Adrenal-medulla'],
'SM':['Muscle-gastrocnemian','Muscle-abdominal'],
'Liver':['Liver'],
}
eo = export.ExportFile(Mm_Ba_coordinates[:-4]+'-matched.txt')
eo2 = export.ExportFile(Mm_Ba_coordinates[:-4]+'-matrix.txt')
mouse_events={}
baboon_events={}
baboon_corridinates={}
### This mouse circadian events file has been lifted over to baboon coordinates
countX=0
for line in open(Mm_Ba_coordinates,'rU').xreadlines():
#Xpo6:ENSMUSG00000000131:E25.5-E28.1|ENSMUSG00000000131:E27.1-E28.1
data = cleanUpLine(line)
values = string.split(data,'\t')
chr,c1,c2,event,strand,null,null,null=values
event = string.replace(event,'__',':')
event,tissue = event.split('--')
junctions = string.split(event,':')[1:]
junctions = string.join(junctions,':')
junctions = string.split(junctions,'|')
junctions.sort() ### make a unique event
junctions = string.join(junctions,'|')
symbol = string.split(event,':')[0]
event = symbol+':'+junctions
#if countX==10: sys.exit()
countX+=1
tissue = string.replace(tissue,'_event_annot_file.txt','')
tissue = string.replace(tissue,'PSI.','')
tissue = string.replace(tissue,'_Mm','')
if strand == '-':
junction = chr+':'+c2+'-'+c1
else:
junction = chr+':'+c1+'-'+c2
try: mouse_events[junction].append([event,tissue])
except: mouse_events[junction] = [[event,tissue]]
for line in open(Ba_events,'rU').xreadlines():
data = cleanUpLine(line)
values = string.split(data,'\t')
event,tissue_num,tissues,coordinates=values
junctions = string.split(event,':')[1:]
junctions = string.join(junctions,':')
junctions = string.split(junctions,'|')
junctions.sort() ### make a unique event
junctions = string.join(junctions,'|')
symbol = string.split(event,':')[0]
event = symbol+':'+junctions
baboon_corridinates[event]=coordinates
try: j1,j2 = string.split(coordinates,"|")
except: continue
tissues = tissues.split('|')
try: baboon_events[j1].append([event,tissues])
except: baboon_events[j1] = [[event,tissues]]
try: baboon_events[j2].append([event,tissues])
except: baboon_events[j2] = [[event,tissues]]
print len(mouse_events), len(baboon_events)
common=0
matched_events={}
matched_mm_events={}
tissue_matrix={}
mm_single_tissue_counts={}
ba_single_tissue_counts={}
for junction in mouse_events:
if junction in baboon_events:
common+=1
mm_events={}
### Aggregate tissues for mouse
for (mm_event,mm_tissue) in mouse_events[junction]:
try: mm_events[mm_event].append(mm_tissue)
except: mm_events[mm_event] = [mm_tissue]
for mm_event in mm_events:
mm_tissues = mm_events[mm_event]
mm_tissues = unique.unique(mm_tissues)
for ba_event,ba_tissues in baboon_events[junction]:
ba_tissues = unique.unique(ba_tissues)
matched_events[mm_event,ba_event] = mm_tissues,ba_tissues
matched_mm_events[mm_event]=[]
def matchingTissues(mouse,baboon):
m_matches=[]
b_matches=[]
for m in mouse:
for b in baboon:
if m in equivalencies:
if b in equivalencies[m]:
m_matches.append(m)
b_matches.append(b)
if len(m_matches)==0:
return ''
else:
m_matches = string.join(unique.unique(m_matches),', ')
b_matches = string.join(unique.unique(b_matches),', ')
return m_matches+':'+b_matches
for (mm_event,ba_event) in matched_events:
mm_tissues,ba_tissues = matched_events[mm_event,ba_event]
matching_tissues=matchingTissues(mm_tissues,ba_tissues)
eo.write(string.join([mm_event,ba_event,string.join(mm_tissues,'|'),string.join(ba_tissues,'|'),str(len(mm_tissues)),str(len(ba_tissues)),matching_tissues],'\t')+'\n')
for mt in mm_tissues:
for bt in ba_tissues:
#if mt=='Heart' and bt=='Thyroid': print mm_event+'\t'+ba_event
try: tissue_matrix[mt,bt]+=1
except: tissue_matrix[mt,bt]=1
try: mm_single_tissue_counts[mt]+=1
except: mm_single_tissue_counts[mt]=1
try: ba_single_tissue_counts[bt]+=1
except: ba_single_tissue_counts[bt]=1
print mm_single_tissue_counts['Heart']
print ba_single_tissue_counts['Thyroid']
print tissue_matrix['Heart','Heart']
tissue_matrix_table=[]
ba_tissues=['Tissues']
for bt in ba_single_tissue_counts:
ba_tissues.append(bt)
eo2.write(string.join(ba_tissues,'\t')+'\n')
for mt in mm_single_tissue_counts:
table=[]
for bt in ba_single_tissue_counts:
if bt=='Thyroid' and mt=='Heart':
print tissue_matrix[mt,bt]
print tissue_matrix[mt,bt]/(1.00*ba_single_tissue_counts[bt])
try: table.append(str(tissue_matrix[mt,bt]/(1.00*ba_single_tissue_counts[bt])))
except: table.append('0')
eo2.write(string.join([mt]+table,'\t')+'\n')
print common, len(matched_events), len(matched_mm_events)
eo.close()
eo2.close()
if __name__ == '__main__':
TF_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/NCI-R01/CCSB_TFIso_Clones.txt'
PSI_dir = '/Volumes/salomonis2/NCI-R01/TCGA-BREAST-CANCER/TCGA-files-Ens91/bams/AltResults/AlternativeOutput/OncoSPlice-All-Samples-filtered-names/SubtypeAnalyses-Results/round1/Events-dPSI_0.1_adjp/'
#convertPSICoordinatesToBED(PSI_dir);sys.exit()
summarizePSIresults(PSI_dir,TF_file);sys.exit()
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Anukana/Breast-Cancer/TF-isoform/TF_ratio_correlation-analysis/tcga_rsem_isopct_filtered-filtered.2-filtered.txt'
TF_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Anukana/Breast-Cancer/TF-isoform/Ensembl-isoform-key-CCSB.txt'
exportIntraTFIsoformCorrelations(filename,TF_file,0.3,anticorrelation=True);sys.exit()
input_file= '/Volumes/salomonis2/NCI-R01/TCGA-BREAST-CANCER/Anukana/UO1analysis/xenabrowserFiles/tcga_rsem_isoform_tpm_filtered.txt'
#convertXenaBrowserIsoformDataToStandardRatios(input_file);sys.exit()
Mm_Ba_coordinates = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Krithika/Baboon-Mouse/mm10-circadian_liftOverTo_baboon.txt'
Ba_events = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Krithika/Baboon-Mouse/Baboon_metacycle-significant-AS-coordinates.txt'
#convertPSICoordinatesToBED(Mm_Ba_coordinates,Ba_events);sys.exit()
#PSIfilterAndImpute('/Volumes/salomonis2/LabFiles/krithika_circadian/GSE98965-Papio_Anubis/files/grp-files/Filtered-Psi-groups-files'); sys.exit()
filename='/Users/saljh8/Desktop/DemoData/Venetoclax/D4/cellHarmony-rawp-stringent/gene_summary.txt'
filename = '/Volumes/salomonis2/LabFiles/Nathan/10x-PBMC-CD34+/AML-p27-pre-post/pre/cellHarmony-latest/gene_summary-p27.txt'
filename = '/Volumes/salomonis2/LabFiles/Dan-Schnell/To_cellHarmony/MIToSham/Input/cellHarmony/cell-frequency-stats.txt'
index1=2;index2=3; x_axis='Number of Differentially Expressed Genes'; y_axis = 'Comparisons'; title='Hippocampus - Number of Differentially Expressed Genes'
#OutputFile = export.findParentDir(filename)
#OutputFile = export.findParentDir(OutputFile[:-1])+'/test.pdf'
#exportTFcorrelations('/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/SuperPan/ExpressionInput/exp.Cdt1-2139-genes.txt','/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Marie.Dominique/TF-to-gene/228-tfs.txt',0.1);sys.exit()
#stackedbarchart(filename,display=True,output=OutputFile);sys.exit()
index1=2;index2=3; x_axis='Number of DEGs'; y_axis = 'Reference clusters'; title='cellHarmony Differentially Expressed Genes'
index1=-2;index2=-1; x_axis='Cell-State Percentage'; y_axis = 'Reference clusters'; title='Assigned Cell Frequencies'
#barchart(filename,index1,index2,x_axis,y_axis,title,display=True)
#barchart(filename,index1,index2,x_axis,y_axis,title,display=True,color1='IndianRed',color2='SkyBlue');sys.exit()
diff=0.7
print 'diff:',diff
#latteralMerge(file1, file2);sys.exit()
#removeMarkerFinderDoublets('/Volumes/salomonis2/Nancy_ratner/2mo-NF/exp.Figure_SX-ICGS-MarkerFinder.filt.txt',diff=diff);sys.exit()
#outputForGOElite('/Users/saljh8/Desktop/R412X/completed/centroids.WT.R412X.median.txt');sys.exit()
#simpleStatsSummary('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/HCA/Mean-Comparisons/ExpressionInput/MergedFiles.Counts.UMI.txt');sys.exit()
a = '/Users/saljh8/Downloads/groups.CellTypes-Predicted-Label-Transfer-For-Nuclei-matrix.txt'
b = '/Volumes/salomonis2/Immune-10x-data-Human-Atlas/Bone-Marrow/Stuart/Browser/ExpressionInput/HS-compatible_symbols.txt'
#b = '/data/salomonis2/GSE107727_RAW-10X-Mm/filtered-counts/ExpressionInput/Mm_compatible_symbols.txt'
#a = '/Volumes/salomonis2/Immune-10x-data-Human-Atlas/Bone-Marrow/Stuart/Browser/head.txt'
##transposeMatrix(a);sys.exit()
#convertSymbolLog(a,b);sys.exit()
#returnIntronJunctionRatio('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Fluidigm_scRNA-Seq/12.09.2107/counts.WT-R412X.txt');sys.exit()
#geneExpressionSummary('/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/ExpressionInput/DEGs-LogFold_1.0_rawp');sys.exit()
b = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/C202SC19040013/raw_data/ModGMP_PMLRARTG/ModGMP_PMLRARTG/outs/filtered_feature_bc_matrix/AltAnalyze-Outliers-removed/ICGS-NMF-euclidean-ref/groups.ICGS.txt'
a = '/Volumes/salomonis2/Grimes/RNA/scRNA-Seq/10x-Genomics/C202SC19040013/raw_data/ModGMP_PMLRARTG/ModGMP_PMLRARTG/outs/filtered_feature_bc_matrix/AltAnalyze-Outliers-removed/ICGS-NMF-euclidean-ref/exp.ICGS.txt'
#convertGroupsToBinaryMatrix(b,a,cellHarmony=False);sys.exit()
a = '/Users/saljh8/Desktop/temp/groups.TNBC.txt'
b = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/tests/clusters.txt'
#simpleCombineFiles('/Users/saljh8/Desktop/dataAnalysis/Collaborative/Jose/NewTranscriptome/CombinedDataset/ExpressionInput/Events-LogFold_0.58_rawp')
#removeRedundantCluster(a,b);sys.exit()
a = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/PSI/SpliceICGS.R1.Depleted.12.27.17/all-depleted-and-KD'
#a = '/Users/saljh8/Desktop/Ashish/all/Events-dPSI_0.1_rawp-0.01/'
compareEventLists(a);sys.exit()
#filterPSIValues('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/PSI/CORNEL-AML/PSI/exp.Cornell-Bulk.txt');sys.exit()
#compareGenomicLocationAndICGSClusters();sys.exit()
#ViolinPlot();sys.exit()
#simpleScatter('/Users/saljh8/Downloads/CMdiff_paper/calcium_data-KO4.txt');sys.exit()
query_dataset = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/exp.GSE81682_HTSeq-cellHarmony-filtered.txt'
all_tpm = '/Users/saljh8/Desktop/demo/BoneMarrow/ExpressionInput/exp.BoneMarrow-scRNASeq.txt'
markerfinder = '/Users/saljh8/Desktop/demo/BoneMarrow/ExpressionOutput1/MarkerFinder/AllGenes_correlations-ReplicateBasedOriginal.txt'
signature_genes = '/Users/saljh8/Desktop/Grimes/KashishNormalization/test/Panorama.txt'
state = 'Multi-Lin'
#evaluateMultiLinRegulatoryStructure(all_tpm,markerfinder,signature_genes,state);sys.exit()
query_dataset = None
all_tpm = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/MultiLin/Gottgens_HarmonizeReference.txt'
signature_genes = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/MultiLin/Gottgens_HarmonizeReference.txt'
markerfinder = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
state = 'Eryth_Multi-Lin'
#evaluateMultiLinRegulatoryStructure(all_tpm,markerfinder,signature_genes,state,query = query_dataset);sys.exit()
#simpleCombine("/Volumes/My Passport/Ari-10X/input");sys.exit()
#effectsPrioritization('/Users/saljh8/Documents/1-dataAnalysis/RBM20-collaboration/RBM20-BAG3_splicing/Missing Values-Splicing/Effects.txt');sys.exit()
#customCleanBinomial('/Volumes/salomonis2-1/Lab backup/Theresa-Microbiome-DropSeq/NegBinomial/ExpressionInput/exp.Instesinal_microbiome2.txt');sys.exit()
#findReciprocal('/Volumes/HomeBackup/CCHMC/Jared-KO/BatchCorrectedFiltered/exp.CM-KO-steady-state.txt');sys.exit()
#ReceptorLigandCellInteractions('Mm','/Users/saljh8/Downloads/ncomms8866-s3.txt','/Users/saljh8/Downloads/Round3-MarkerFinder_All-Genes.txt');sys.exit()
#compareFusions('/Volumes/salomonis2-2/CPMC_Melanoma-GBM/Third-batch-files/Complete_analysis/temp/Combined_Fusion_GBM.txt');sys.exit()
#combineVariants('/Volumes/salomonis2/CPMC_Melanoma-GBM/Third-batch-files/Complete_analysis/Variant_results/GBM/Variants_HighModerate-GBM_selected.txt');sys.exit()
#customCleanSupplemental('/Users/saljh8/Desktop/dataAnalysis/CPMC/TCGA_MM/MM_genes_published.txt');sys.exit()
#customClean('/Users/saljh8/Desktop/dataAnalysis/Driscoll/R3/2000_run1708A_normalized.txt');sys.exit()
#simpleFilter('/Volumes/SEQ-DATA 1/all_10.5_mapped_norm_GC.csv');sys.exit()
#filterRandomFile('/Users/saljh8/Downloads/HuGene-1_1-st-v1.na36.hg19.transcript2.csv',1,8);sys.exit()
filename = '/Users/saljh8/Desktop/Grimes/GEC14078/MergedFiles.txt'
#CountKallistoAlignedJunctions(filename);sys.exit()
filename = '/Users/saljh8/Desktop/Code/AltAnalyze/AltDatabase/EnsMart72/Mm/junction1/junction_critical-junction-seq.txt'
#MakeJunctionFasta(filename);sys.exit()
filename = '/Users/saljh8/Downloads/CoexpressionAtlas.txt'
#ToppGeneFilter(filename); sys.exit()
#countIntronsExons(filename);sys.exit()
#filterForJunctions(filename);sys.exit()
#filename = '/Users/saljh8/Desktop/Grimes/GEC14074/ExpressionOutput/LineageCorrelations-test-protein_coding-zscores.txt'
#runHCOnly(filename,[]); sys.exit()
folder = '/Users/saljh8/Desktop/Code/AltAnalyze/AltDatabase/EnsMart72/ensembl/Hs'
try:
files = UI.read_directory(folder)
for file in files: #:70895507-70895600
if '.bed' in file:
#BedFileCheck(folder+'/'+file)
pass
except Exception: pass
#sys.exit()
#runPCAonly(filename,[],False,showLabels=False,plotType='2D');sys.exit()
countinp = '/Volumes/salomonis2/SinghLab/20150715_single_GCBCell/bams/ExpressionInput/counts.Bcells.txt'
IGH_gene_file = '/Volumes/salomonis2/SinghLab/20150715_single_GCBCell/bams/ExpressionInput/IGH_genes.txt'
#extractFeatures(countinp,IGH_gene_file);sys.exit()
import UI
#geneMethylationOutput(filename);sys.exit()
#ica(filename);sys.exit()
#replaceWithBinary('/Users/saljh8/Downloads/Neg_Bi_wholegenome.txt');sys.exit()
#simpleFilter('/Volumes/SEQ-DATA/AML-TCGA/ExpressionInput/counts.LAML1.txt');sys.exit()
filename = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/genes.tpm_tracking-ordered.txt'
#filename = '/Users/saljh8/Desktop/Grimes/KashishNormalization/6-5-2015/ExpressionInput/amplify/exp.All-wt-output.txt'
#getlastexon(filename);sys.exit()
TFs = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/TF-by-gene_matrix/all-TFs2.txt'
folder = '/Users/saljh8/Downloads/BLASTX2_Gecko.tab'
genes = ['Gfi1', 'Irf8'] #'Cebpe', 'Mecom', 'Vwf', 'Itga2b', 'Meis1', 'Gata2','Ctsg','Elane', 'Klf4','Gata1']
#genes = ['Gata1','Gfi1b']
#coincentIncedenceTest(filename,TFs);sys.exit()
#coincidentIncedence(filename,genes);sys.exit()
#test(folder);sys.exit()
#files = UI.read_directory(folder)
#for file in files: SimpleCorrdinateToBed(folder+'/'+file)
#filename = '/Users/saljh8/Desktop/bed/RREs0.5_exons_unique.txt'
#simpleIntegrityCheck(filename);sys.exit()
gene_list = ['S100a8','Chd7','Ets1','Chd7','S100a8']
gene_list_file = '/Users/saljh8/Desktop/demo/Amit/ExpressionInput/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/Grimes/Comb-plots/AML_genes-interest.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Grimes/Mm_Sara-single-cell-AML/alt/AdditionalHOPACH/ExpressionInput/AML_combplots.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Grimes/MDS-array/Comb-plot genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Grimes/All-Fluidigm/ExpressionInput/comb_plot3.txt'
gene_list_file = '/Users/saljh8/Desktop/Grimes/MultiLin-Code/MultiLin-TFs.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/ExpressionInput/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/10X-DropSeq-comparison/Final-Classifications/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/TFs/Myelo_TFs2.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/R412X/customGenes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/ExpressionInput/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/R412X/genes.txt'
gene_list_file = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/HCA/BM1-8_CD34+/ExpressionInput/MixedLinPrimingGenes.txt'
gene_list_file = '/Users/saljh8/Dropbox/Manuscripts/MBNL1/genes.txt'
genesets = importGeneList(gene_list_file,n=1)
filename = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/comb-plots/exp.IG2_GG1-extended-output.txt'
filename = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/comb-plots/genes.tpm_tracking-ordered.txt'
filename = '/Users/saljh8/Desktop/demo/Amit/ExpressedCells/GO-Elite_results/3k_selected_LineageGenes-CombPlotInput2.txt'
filename = '/Users/saljh8/Desktop/Grimes/Comb-plots/exp.AML_single-cell-output.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Grimes/Mm_Sara-single-cell-AML/alt/AdditionalHOPACH/ExpressionInput/exp.AML.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Grimes/MDS-array/comb-plot/input.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Grimes/All-Fluidigm/ExpressionInput/exp.Lsk_panorama.txt'
filename = '/Users/saljh8/Desktop/demo/BoneMarrow/ExpressionInput/exp.BoneMarrow-scRNASeq.txt'
filename = '/Users/saljh8/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/exp.GSE81682_HTSeq-cellHarmony-filtered.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Harinder/scRNASeq_Mm-Plasma/PCA-loading/ExpressionInput/exp.PCA-Symbol.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/10X-DropSeq-comparison/Final-Classifications/cellHarmony/MF-analysis/ExpressionInput/exp.Fluidigm-log2-NearestNeighbor-800.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/10X-DropSeq-comparison/Final-Classifications/cellHarmony/MF-analysis/ExpressionInput/exp.10X-log2-NearestNeighbor.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/10X-DropSeq-comparison/DropSeq/MultiLinDetect/ExpressionInput/DataPlots/exp.DropSeq-2k-log2.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/R412X/exp.allcells-v2.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/HCA/BM1-8_CD34+/ExpressionInput/exp.CD34+.v5-log2.txt'
filename = '/Users/saljh8/Dropbox/Manuscripts/MBNL1/exp.MBNL1-all.txt'
#filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/CITE-Seq-MF-indexed/ExpressionInput/exp.cellHarmony.v3.txt'
#filename = '/Volumes/salomonis2/Theodosia-Kalfa/Combined-10X-CPTT/ExpressionInput/exp.MergedFiles-ICGS.txt'
#filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/Ly6g/combined-ICGS-Final/R412X/exp.cellHarmony-WT-R412X-relative.txt'
#filename = '/Users/saljh8/Desktop/Old Mac/Desktop/Grimes/Kallisto/Ly6g/CodingOnly/Guide3-Kallisto-Coding-NatureAugmented/SubClustering/Nov-27-Final-version/ExpressionInput/exp.wt-panorama.txt'
#filename = '/Volumes/salomonis2/Harinder-singh/Run2421-10X/10X_IRF4_Lo/outs/filtered_gene_bc_matrices/ExpressionInput/exp.10X_IRF4_Lo_matrix_CPTT-ICGS.txt'
#filename = '/Users/saljh8/Desktop/Old Mac/Desktop/Grimes/Kallisto/Ly6g/CodingOnly/Guide3-Kallisto-Coding-NatureAugmented/SubClustering/Nov-27-Final-version/R412X/exp.R412X-RSEM-order.txt'
print genesets
for gene_list in genesets:
multipleSubPlots(filename,gene_list,SubPlotType='column',n=1)
sys.exit()
plotHistogram(filename);sys.exit()
filename = '/Users/saljh8/Desktop/Grimes/Expression_final_files/ExpressionInput/amplify-wt/DataPlots/Clustering-exp.myeloid-steady-state-PCA-all_wt_myeloid_SingleCell-Klhl7 Dusp7 Slc25a33 H6pd Bcorl1 Sdpr Ypel3 251000-hierarchical_cosine_cosine.cdt'
openTreeView(filename);sys.exit()
pdf1 = "/Users/saljh8/Desktop/Grimes/1.pdf"
pdf2 = "/Users/saljh8/Desktop/Grimes/2.pdf"
outPdf = "/Users/saljh8/Desktop/Grimes/3.pdf"
merge_horizontal(outPdf, pdf1, pdf2);sys.exit()
mergePDFs(pdf1,pdf2,outPdf);sys.exit()
filename = '/Volumes/SEQ-DATA/CardiacRNASeq/BedFiles/ExpressionOutput/Clustering/SampleLogFolds-CardiacRNASeq.txt'
ica(filename);sys.exit()
features = 5
matrix, column_header, row_header, dataset_name, group_db = importData(filename)
Kmeans(features, column_header, row_header); sys.exit()
#graphViz();sys.exit()
filename = '/Users/saljh8/Desktop/delete.txt'
filenames = [filename]
outputClusters(filenames,[]); sys.exit()
#runPCAonly(filename,[],False);sys.exit()
#VennDiagram(); sys.exit()
#buildGraphFromSIF('Ensembl','Mm',None,None); sys.exit()
#clusterPathwayZscores(None); sys.exit()
pruned_folder = '/Users/nsalomonis/Desktop/CBD/LogTransformed/GO-Elite/GO-Elite_results/CompleteResults/ORA_pruned/'
input_ora_folder = '/Users/nsalomonis/Desktop/CBD/LogTransformed/GO-Elite/input/'
files = UI.read_directory(pruned_folder)
for file in files:
if '.sif' in file:
input_file = string.join(string.split(file,'-')[:-1],'-')+'.txt'
sif_file = pruned_folder+file
input_file = input_ora_folder+input_file
buildGraphFromSIF('Ensembl','Hs',sif_file,input_file)
sys.exit()
filenames = [filename]
outputClusters(filenames,[])
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/QC.py
|
QC.py
|
import base64
import csv
import getpass
try:
import lxml
except Exception:
print 'library lxml not supported. WikiPathways and LineageProfiler visualization will not work. Please install with pip install lxml.'
from lxml import etree as ET
from lxml import _elementpath
import re
try: import requests
except Exception:
print 'library requests not supported. WikiPathways and LineageProfiler visualization will not work. Please install with pip install requests.'
import sys
class WikipathwaysApiClient(object):
"""Returns :class:`WikipathwaysApiClient` object.
:param identifier: WikiPathways ID for the new :class:`WikipathwaysApiClient` object.
"""
def __invert_dict(dictionary):
return dict((v, k) for k, v in dictionary.iteritems())
def __get_bridgedb_datasets(self):
if hasattr(self, 'bridgedb_datasets'):
bridgedb_datasets = self.bridgedb_datasets
else:
bridgedb_datasets_request = requests.get('https://raw.githubusercontent.com/bridgedb/BridgeDb/master/org.bridgedb.bio/resources/org/bridgedb/bio/datasources.txt')
bridgedb_datasets_string = bridgedb_datasets_request.text
bridgedb_datasets_csv = csv.reader(bridgedb_datasets_string.split('\n'), delimiter='\t')
bridgedb_datasets_parsed = [];
for bridgedb_dataset_csv in bridgedb_datasets_csv:
if bridgedb_dataset_csv:
bridgedb_dataset_parsed = {}
bridgedb_dataset_parsed['system_code'] = bridgedb_dataset_csv[1]
bridgedb_dataset_parsed['miriam'] = bridgedb_dataset_csv[8]
bridgedb_datasets_parsed.append(bridgedb_dataset_parsed)
self.bridgedb_datasets = bridgedb_datasets_parsed
return self.bridgedb_datasets
def __parse_identifiers_iri(self, iri):
iri_components = iri.split('identifiers.org')
iri_path = iri_components[len(iri_components) - 1]
iri_path_components = iri_path.split('/')
preferred_prefix = iri_path_components[1]
identifier = iri_path_components[2]
bridgedb_datasets = self.__get_bridgedb_datasets()
for bridgedb_dataset in bridgedb_datasets:
if 'urn:miriam:' + preferred_prefix == bridgedb_dataset['miriam']:
system_code = bridgedb_dataset['system_code']
return {
'preferred_prefix': preferred_prefix,
'identifier': identifier,
'system_code': system_code
}
api_to_standard_term_mappings = {
'id': 'identifier',
'ids': 'identifiers',
'pwId': 'identifier',
'revision': 'version',
'graphId': 'element_identifiers',
'color': 'colors',
'fileType': 'file_format',
'species': 'organism',
'url': 'web_page',
'codes': 'system_codes'
}
filename_extension_to_media_type_mappings = {
'svg': 'image/svg+xml',
'png': 'image/png',
'pdf': 'application/pdf',
'gpml': 'application/gpml+xml',
'txt': 'text/vnd.genelist+tab-separated-values',
'pwf': 'text/vnd.eu.gene+plain',
'owl': 'application/vnd.biopax.owl+xml',
}
filename_extensions = filename_extension_to_media_type_mappings.keys()
media_types = filename_extension_to_media_type_mappings.values()
media_type_to_filename_extension_mappings = __invert_dict(filename_extension_to_media_type_mappings)
english_name_to_iri_mappings = {
'African malaria mosquito': 'http://identifiers.org/taxonomy/7165',
'beet': 'http://identifiers.org/taxonomy/161934',
'thale cress': 'http://identifiers.org/taxonomy/3702',
'cattle': 'http://identifiers.org/taxonomy/9913',
'roundworm': 'http://identifiers.org/taxonomy/6239',
'dog': 'http://identifiers.org/taxonomy/9615',
'sea vase': 'http://identifiers.org/taxonomy/7719',
'zebrafish': 'http://identifiers.org/taxonomy/7955',
'fruit fly': 'http://identifiers.org/taxonomy/7227',
'Escherichia coli': 'http://identifiers.org/taxonomy/562',
'horse': 'http://identifiers.org/taxonomy/9796',
'chicken': 'http://identifiers.org/taxonomy/9031',
'soybean': 'http://identifiers.org/taxonomy/3847',
'human': 'http://identifiers.org/taxonomy/9606',
'barley': 'http://identifiers.org/taxonomy/4513',
'Rhesus monkey': 'http://identifiers.org/taxonomy/9544',
'mouse': 'http://identifiers.org/taxonomy/10090',
'platypus': 'http://identifiers.org/taxonomy/9258',
'long-grained rice': 'http://identifiers.org/taxonomy/39946',
'rice': 'http://identifiers.org/taxonomy/4530',
'black cottonwood': 'http://identifiers.org/taxonomy/3694',
'chimpanzee': 'http://identifiers.org/taxonomy/9598',
'Norway rat': 'http://identifiers.org/taxonomy/10116',
'baker\'s yeast': 'http://identifiers.org/taxonomy/4932',
'tomato': 'http://identifiers.org/taxonomy/4081',
'pig': 'http://identifiers.org/taxonomy/9823',
'wine grape': 'http://identifiers.org/taxonomy/29760',
'western clawed frog': 'http://identifiers.org/taxonomy/8364',
'maize': 'http://identifiers.org/taxonomy/4577'
}
iri_to_english_name_mappings = __invert_dict(english_name_to_iri_mappings)
latin_name_to_iri_mappings = {
'Anopheles gambiae': 'http://identifiers.org/taxonomy/7165',
'Arabidopsis thaliana': 'http://identifiers.org/taxonomy/3702',
'Aspergillus niger': 'http://identifiers.org/taxonomy/5061',
'Bacillus subtilis': 'http://identifiers.org/taxonomy/1423',
'Beta vulgaris': 'http://identifiers.org/taxonomy/161934',
'Bos taurus': 'http://identifiers.org/taxonomy/9913',
'Caenorhabditis elegans': 'http://identifiers.org/taxonomy/6239',
'Canis familiaris': 'http://identifiers.org/taxonomy/9615',
'Ciona intestinalis': 'http://identifiers.org/taxonomy/7719',
'Ruminiclostridium thermocellum': 'http://identifiers.org/taxonomy/1515',
'Clostridium thermocellum': 'http://identifiers.org/taxonomy/1515',
'Danio rerio': 'http://identifiers.org/taxonomy/7955',
'Drosophila melanogaster': 'http://identifiers.org/taxonomy/7227',
'Escherichia coli': 'http://identifiers.org/taxonomy/562',
'Equus caballus': 'http://identifiers.org/taxonomy/9796',
'Gallus gallus': 'http://identifiers.org/taxonomy/9031',
'Gibberella zeae': 'http://identifiers.org/taxonomy/5518',
'Glycine max': 'http://identifiers.org/taxonomy/3847',
'Homo sapiens': 'http://identifiers.org/taxonomy/9606',
'Hordeum vulgare': 'http://identifiers.org/taxonomy/4513',
'Macaca mulatta': 'http://identifiers.org/taxonomy/9544',
'Mus musculus': 'http://identifiers.org/taxonomy/10090',
'Mycobacterium tuberculosis': 'http://identifiers.org/taxonomy/1773',
'Ornithorhynchus anatinus': 'http://identifiers.org/taxonomy/9258',
'Oryza indica': 'http://identifiers.org/taxonomy/39946',
'Oryza sativa': 'http://identifiers.org/taxonomy/4530',
'Oryza sativa Indica Group': 'http://identifiers.org/taxonomy/39946',
'Populus trichocarpa': 'http://identifiers.org/taxonomy/3694',
'Pan troglodytes': 'http://identifiers.org/taxonomy/9598',
'Rattus norvegicus': 'http://identifiers.org/taxonomy/10116',
'Saccharomyces cerevisiae': 'http://identifiers.org/taxonomy/4932',
'Solanum lycopersicum': 'http://identifiers.org/taxonomy/4081',
'Sus scrofa': 'http://identifiers.org/taxonomy/9823',
'Vitis vinifera': 'http://identifiers.org/taxonomy/29760',
'Xenopus tropicalis': 'http://identifiers.org/taxonomy/8364',
'Zea mays': 'http://identifiers.org/taxonomy/4577'
}
def __init__(self, base_iri=None):
if base_iri is None:
base_iri = 'http://webservice.wikipathways.org/'
self.base_iri = base_iri
# define namespaces
self.NAMESPACES = {'ns1':'http://www.wso2.org/php/xsd','ns2':'http://www.wikipathways.org/webservice/'}
def __convert_standard_terms_to_api_terms(self, input_params):
terms_to_convert = self.api_to_standard_term_mappings
standard_terms = terms_to_convert.values()
api_terms = terms_to_convert.keys()
request_params = {}
for key, value in input_params.iteritems():
if (key in standard_terms):
def get_api_term(candidate_api_term):
return self.api_to_standard_term_mappings[candidate_api_term] == key
api_term = filter(get_api_term, api_terms)[0]
request_params[api_term] = input_params[key]
else:
request_params[key] = input_params[key]
return request_params
def __convert_api_terms_to_standard_terms(self, input_object):
terms_to_convert = self.api_to_standard_term_mappings
standard_terms = terms_to_convert.values()
api_terms = terms_to_convert.keys()
output_object = {}
for key, value in input_object.iteritems():
if (key in api_terms):
api_term = terms_to_convert[key]
output_object[api_term] = input_object[key]
else:
output_object[key] = input_object[key]
return output_object
def __convert_organism_to_dict(self, organism):
if hasattr(self, 'organism_dicts'):
organism_dicts = self.organism_dicts
else:
organism_dicts = self.organism_dicts = self.list_organisms()
for organism_dict in organism_dicts:
if isinstance(organism, basestring):
if organism_dict['@id'] == organism:
return organism_dict
elif organism_dict['name']['la'] == organism:
return organism_dict
elif organism_dict['name'].get('en') and organism_dict['name']['en'] == organism:
return organism_dict
elif organism.get('@id') and organism['@id'] == organism_dict['@id']:
return organism_dict
def __enrich_pathway(self, pathway):
pathway['@id'] = 'http://identifiers.org/wikipathways/' + pathway['identifier']
if pathway.get('organism') and isinstance(pathway['organism'], basestring):
pathway['organism'] = self.__convert_organism_to_dict(pathway['organism'])
return pathway
def create_pathway(self):
###
# author: msk ([email protected])
###
# login
pswd = getpass.getpass('Password:')
auth = {'name' : username , 'pass' : pswd}
r_login = requests.get(self.base_iri + 'login', params=auth)
dom = ET.fromstring(r_login.text)
authentication = ''
for node in dom.findall('ns1:auth', namespaces):
authentication = node.text
# read gpml file
f = open(gpml_file, 'r')
gpml = f.read()
# create pathway
update_params = {'auth' : username+'-'+authentication, 'gpml': gpml}
re = requests.post(self.base_iri + 'createPathway', params=update_params)
#print re.text
def get_colored_pathway(self, identifier, element_identifiers, colors, version = '0', file_format = 'svg'):
"""Sends a GET request. Returns file as string.
Args:
identifier (str): WikiPathways ID.
element_identifiers (list of str): means of identifying one or more elements in a pathway,
for example, specify GPML GraphIds as ["ffffff90","ffffffe5"].
colors (list of str): one or more hexadecimal number(s), representing the colors to use for
the corresponding element_identifier (if the length of the colors list is equal to the
length of the element_identifiers list) or the single color to use for all element_identifiers
(if the colors list is not equal in length to the element_identifiers list).
Example: ["#0000FF","#0000FF"].
version (str, optional): The version of the pathway. Defaults to '0', which means latest.
file_format (str): IANA media type (http://www.iana.org/assignments/media-types/media-types.xhtml)
or filename extension desired for response. Defaults to 'svg'. Examples:
Media types:
* 'image/svg+xml'
* 'image/png'
* 'application/pdf'
Filename extensions:
* 'svg'
* 'png'
* 'pdf'
"""
# API does not yet support content-type negotiation, so we need to convert
# filename extension to be used as a query parameter.
if file_format in self.media_types:
file_format = self.media_type_to_filename_extension_mappings[file_format]
# HTML/CSS defaults use a pound sign before the HEX number, e.g. #FFFFFF.
# But the API does not use this, so to make it easier for users, we are
# accepting the pound sign in the input args and stripping it here.
input_colors = colors
colors = []
non_letter_number_pattern = re.compile('[^a-zA-Z0-9]+')
for input_color in input_colors:
color = non_letter_number_pattern.sub('', input_color)
colors.append(color)
input_params = {
'identifier': identifier,
'version': version,
'element_identifiers': element_identifiers,
'colors': colors,
'file_format': file_format
}
request_params = self.__convert_standard_terms_to_api_terms(input_params)
response = requests.get(self.base_iri + 'getColoredPathway', params=request_params)
dom = ET.fromstring(response.text)
node = dom.find('ns1:data', self.NAMESPACES)
file = base64.b64decode(node.text) ### decode this file
return file
def get_pathway_as(self, identifier, version = '0', file_format = 'gpml'):
"""
Sends a GET request. Returns an LXML object for any XML media type
and a string for anything else
Args:
identifier (str): WikiPathways ID.
version (str, optional): The version of the pathway. Defaults to '0', which means latest.
file_format (str): IANA media type (http://www.iana.org/assignments/media-types/media-types.xhtml)
or filename extension desired for response. Defaults to 'gpml'.
Examples:
Media types:
* 'application/gpml+xml'
* 'text/vnd.genelist+tab-separated-values'
* 'text/vnd.eu.gene+plain'
* 'application/vnd.biopax.owl+xml'
* 'image/svg+xml'
* 'image/png'
* 'application/pdf'
Filename extensions:
* 'gpml'
* 'txt'
* 'pwf'
* 'owl'
* 'svg'
* 'png'
* 'pdf'
"""
# API does not yet support content-type negotiation, so we need to convert
# filename extension to be used as a query parameter.
if file_format in self.media_types:
file_format = self.media_type_to_filename_extension_mappings[file_format]
input_params = {
'identifier': identifier,
'version': version,
'file_format': file_format
}
request_params = self.__convert_standard_terms_to_api_terms(input_params)
response = requests.get(self.base_iri + 'getPathwayAs', params=request_params)
#print [response.text];sys.exit()
dom = ET.fromstring(response.text)
node = dom.find('ns1:data', self.NAMESPACES)
response_string = base64.b64decode(node.text) ### decode this file
if request_params['fileType'] == 'gpml' or request_params['fileType'] == 'owl' or request_params['fileType'] == 'svg':
#response = ET.fromstring(response_string)
response = response_string
else:
response = response_string
return response
def get_pathway_info(self, identifier):
"""Sends a GET request. Returns pathway metadata as dict.
Args:
identifier (str): WikiPathways ID.
"""
request_params = {'pwId' : identifier}
response = requests.get(self.base_iri + 'getPathwayInfo', params=request_params)
dom = ET.fromstring(response.text)
pathway_using_api_terms = {}
for node in dom.findall('ns1:pathwayInfo', self.NAMESPACES):
for attribute in node:
pathway_using_api_terms[ET.QName(attribute).localname] = attribute.text
pathway = self.__convert_api_terms_to_standard_terms(pathway_using_api_terms)
pathway = self.__enrich_pathway(pathway)
return pathway
def find_pathways_by_text(self, query, organism = None):
"""Sends a GET request. Returns pathways as list of dicts.
Args:
query (str): Text to search for.
organism (str or dict, optional): Limit to organism with given name
(Latin or English) or @id (from http://identifiers.org/taxonomy/)
"""
input_params = {}
input_params['query'] = query
if organism:
input_params['organism'] = self.__convert_organism_to_dict(organism)['name']['la']
request_params = self.__convert_standard_terms_to_api_terms(input_params)
response = requests.get(self.base_iri + 'findPathwaysByText', params=request_params)
dom = ET.fromstring(response.text)
pathways = []
for node in dom.findall('ns1:result', self.NAMESPACES):
pathway_using_api_terms = {}
for child in node:
pathway_using_api_terms[ET.QName(child).localname] = child.text
pathway = self.__convert_api_terms_to_standard_terms(pathway_using_api_terms)
pathway = self.__enrich_pathway(pathway)
pathways.append(pathway)
return pathways
def find_pathways_by_xref(self, **kwargs):
"""Sends a GET request. Returns pathways as a list of dicts.
Required: either just @id or both system_codes and identifiers.
Args:
@id (list of str): One or more identifiers.org IRIs, like ['http://identifiers.org/ncbigene/7040'].
system_codes (list of str): One or more BridgeDb system codes.
identifiers (list of str): One or more entity reference identifiers.
"""
if kwargs.get('@id'):
if not isinstance(kwargs['@id'], list):
kwargs['@id'] = [kwargs['@id']]
system_codes = []
identifiers = []
for iri in kwargs['@id']:
identifiers_iri_components = self.__parse_identifiers_iri(iri)
system_codes.append(identifiers_iri_components['system_code'])
identifiers.append(identifiers_iri_components['identifier'])
input_params = {
'system_codes': system_codes,
'identifiers': identifiers
}
else:
input_params = kwargs
request_params = self.__convert_standard_terms_to_api_terms(input_params)
response = requests.get(self.base_iri + 'findPathwaysByXref', params=request_params)
dom = ET.fromstring(response.text)
pathways = []
for resultNode in dom.findall('ns1:result', self.NAMESPACES):
pathway_using_api_terms = {}
pathway_using_api_terms['fields'] = []
for childNode in resultNode:
if ET.QName(childNode).localname != 'fields':
pathway_using_api_terms[ET.QName(childNode).localname] = childNode.text
elif ET.QName(childNode).localname == 'fields':
field = {}
for fieldChildNode in childNode:
#TODO standardize names & values from fieldChildNode.text
field[ET.QName(fieldChildNode).localname] = fieldChildNode.text
pathway_using_api_terms['fields'].append(field)
pathway = self.__convert_api_terms_to_standard_terms(pathway_using_api_terms)
pathway = self.__enrich_pathway(pathway)
pathways.append(pathway)
return pathways
def list_organisms(self):
"""Sends a GET request. Returns :list:`organisms` object, each an organism as a dict,
with the IRI, Latin name and English name (when available).
"""
response = requests.get(self.base_iri + 'listOrganisms')
dom = ET.fromstring(response.text)
organisms = []
for node in dom:
try:
organism = {}
organism['@context'] = [
{
'name': {
'@id': 'biopax:name',
'@container': '@language'
},
'Organism': 'http://identifiers.org/snomedct/410607006'
}
]
organism['@type'] = 'Organism'
organism['name'] = {}
organism['name']['la'] = latin_name = node.text
organism['@id'] = self.latin_name_to_iri_mappings[latin_name]
english_name = self.iri_to_english_name_mappings.get(organism['@id'])
if english_name != None:
organism['name']['en'] = english_name
organisms.append(organism)
except Exception:
pass
return organisms
# list pathways
def list_pathways(self, organism):
request_params = {'organism': organism}
response = requests.get(self.base_iri + 'listPathways', params=request_params)
dom = ET.fromstring(response.text)
pathways = []
for pathway_node in dom.findall('ns1:pathways', self.NAMESPACES):
pathway_using_api_terms = {}
for child_node in pathway_node:
pathway_using_api_terms[ET.QName(child_node).localname] = child_node.text
pathway = self.__convert_api_terms_to_standard_terms(pathway_using_api_terms)
pathway = self.__enrich_pathway(pathway)
pathways.append(pathway)
return pathways
if __name__ == '__main__':
client = WikipathwaysApiClient()
wp_id_data = client.get_pathway_as(file_format = 'gpml',identifier = 'WP254', version = 0)
with open('WP205.gpml', 'a') as file_out:
file_out.write(wp_id_data)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/wikipathways_api_client.py
|
wikipathways_api_client.py
|
import numpy as np
import pylab as pl
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from misopy import index_gff as ifg
import subprocess
import multiprocessing
import time
import unique
import traceback
samplelis=[]
samp=[]
sample_read={}
PSIJunctions=[]
new_lis=[]
added_gff_entries={}
class PositionData:
def __init__(self,position_str):
self.chr,interval = string.split(position_str,'__') #chr16__18810389-18807423
self.start,self.end = string.split(interval,'-')
def Chr(self): return self.chr
def Start(self): return int(self.start)
def End(self): return int(self.end)
def reimportFeatures(featureFile):
""" Import the exon and gene coordinates """
gene_event_db={}
featureFile = unique.filepath(featureFile)
head=0
for line in open(featureFile,'rU').xreadlines():
#for k in range(len(strand['AltAnalyze_ID'])):
if head ==0: head=1
else:
line = line.rstrip('\n')
event=string.split(line,'\t')[0] #example event: ENSMUSG00000025915:E17.2-E17.5=chr1:9885753-9886047
event = string.replace(event,':','__')
event_split=string.split(event,'__')
for i in range(len(event_split)):
if "ENS" in event_split[i] or '00000' in event_split[i]:
if '-' in event_split[i]:
ji=string.split(event_split[i],'-')
gene=ji[1]
else:
gene=event_split[i]
featureID,position = string.split(event,'=') ### store the feature (exon or junction) position and ID separately
pd = PositionData(position)
if gene in gene_event_db:
feature_db = gene_event_db[gene]
feature_db[featureID] = pd
else:
feature_db = {featureID:pd}
gene_event_db[gene]=feature_db
return gene_event_db
def writegene(chromosome,junction_start,junction_end,strand,uid):
#junction_start = str(int(junction_start)-1000)
#junction_end = str(int(junction_end)+2000)
temp = [int(junction_start),int(junction_end)]
temp.sort()
junction_start,junction_end = str(temp[0]),str(temp[1])
if 'M' not in chromosome:
gff_export_obj.write(chromosome+'\t'+'SE'+'\t'+'gene'+'\t'+junction_start+'\t'+junction_end+'\t'+'.'+'\t'+strand+'\t'+'.'+'\t'+'ID='+uid+';'+'Name='+uid+';'+'\n')
gff_export_obj.write(chromosome+'\t'+'SE'+'\t'+'mRNA'+'\t'+junction_start+'\t'+junction_end+'\t'+'.'+'\t'+strand+'\t'+'.'+'\t'+'ID='+uid+'.STRAND;'+'Parent='+uid+';'+'\n')
def manualWriteExon(chromosome,junction_start,junction_end,strand,uid):
if 'M' not in chromosome:
if strand== '-': i1=-5; i2=5
else: i1=5; i2=-5
gff_export_obj.write(chromosome+'\t'+'SE'+'\t'+'exon'+'\t'+junction_start+'\t'+str(int(float(junction_start)))+'\t'+'.'+'\t'+strand+'\t'+'.'+'\t'+'ID='+uid+';'+'Parent='+uid+'.STRAND;'+'\n')
gff_export_obj.write(chromosome+'\t'+'SE'+'\t'+'exon'+'\t'+junction_end+'\t'+str(int(float(junction_end)))+'\t'+'.'+'\t'+strand+'\t'+'.'+'\t'+'ID='+uid+';'+'Parent='+uid+'.STRAND;'+'\n')
def importReciprocalJunctions(PSIFileDir,PSIJunctions):
### Also include other predicted splicing events from ASPIRE or LinearRegression
alt_dir = string.split(PSIFileDir,'AlternativeOutput')[0]+'AlternativeOutput'
files = unique.read_directory(alt_dir)
added=0
already_added=0
for file in files:
if 'ASPIRE-exon-inclusion-results' in file or 'linearregres-exon-inclusion-results' in file:
alt_exon_path = alt_dir+'/'+file
header=True
for line in open(alt_exon_path,'rU').xreadlines():
line = line.rstrip(os.linesep)
if header: header=False
else:
t=string.split(line,'\t')
inclusion_junction = string.replace(t[8],':','__')
exclusion_junction = string.replace(t[10],':','__')
pair = inclusion_junction,exclusion_junction
if pair in PSIJunctions:
already_added+=1
else:
PSIJunctions.append(pair)
added+=1
return PSIJunctions
def importPSIJunctions(fname):
All_PSI_Reciprocol_Junctions=[]
fname = unique.filepath(fname)
header=True
for line in open(fname,'rU').xreadlines():
line = line.rstrip(os.linesep)
if header: header = False
else:
t=string.split(line,'\t')
junction1 = t[2]
junction2 = t[3]
try:
### Re-order these to have the exclusion be listed first
j1a,j1b = string.split(t[2],'-')
j2a,j2b = string.split(t[3],'-')
j1a = string.split(j1a,':')[1]
j2a = string.split(j2a,':')[1]
j1a = int(float(string.split(j1a,'.')[0][1:]))
j1b = int(float(string.split(j1b,'.')[0][1:]))
j2a = int(float(string.split(j2a,'.')[0][1:]))
j2b = int(float(string.split(j2b,'.')[0][1:]))
#print [j1a,j2a,j1b,j2b], t[2], t[3]
event1 = string.replace(junction2,":","__") ### first listed junction
event2 = string.replace(junction2,":","__") ### second listed junction
if j1a>j2a or j1b<j2b:
event_pair = event1,event2
else:
event_pair=event2,event1
except Exception:
#print traceback.format_exc();sys.exit()
event_pair=event1,event2
if '-' not in event1:
event_pair = event2,event1
All_PSI_Reciprocol_Junctions.append(event_pair)
return All_PSI_Reciprocol_Junctions
def checkForNovelJunctions(exon):
### if a novel junction, make this a non-novel for indexing
exon_alt = string.replace(exon,'__',':') #revert back to :
if '_' in exon_alt:
exon = string.split(exon_alt,'_')[0] ### Only report the non-novel prefix exonID
exon = string.replace(exon,':','__')
if 'I' in exon:
exon = string.replace(exon,'I','E')
### We should also increment the exon ID by 1, to skip the intron
### e.g., ENSMUSG00000000934__I6.1 => ENSMUSG00000000934__E7.1
try:
gene,block_region = string.split(exon,'__E')
block,region = string.split(block_region,'.')
exon = gene+'__E'+str(int(block)+1)+'.'+region
except Exception:
block,region = string.split(exon[1:],'.')
exon = 'E'+str(int(block)+1)+'.'+region
if 'U0' in exon:
exon = string.replace(exon,'U0','E1')
return exon
def exportToGff(incl_junction,excl_junction,feature_positions,gene):
""" Write the exon and gene coordinates to strand gff file for any alternatively regulated and flanking exons """
proceed = False
if '-' in excl_junction: selected_event=excl_junction ### should be the exclusion junction
else: selected_event=incl_junction
### For the first and last exon, determine their positions then later loop through the exons in between
gene_prefix = gene+'__'
pds = feature_positions[selected_event]
e1,e2 = string.split(selected_event,'-')
e1 = checkForNovelJunctions(e1)
e2 = checkForNovelJunctions(e2)
chr = pds.Chr()
if pds.Start() > pds.End(): strand = '-'
else: strand = '+'
try: pd1 = feature_positions[e1]
except Exception:
e1 = string.split(e1,'.')[0]+'.1' ### occurs with IDS such as ENSMUSG00000002107__E26.27, where .27 is not in the database (not sure why)
try: pd1 = feature_positions[e1]
except Exception: ### Occurs with gene entry, e.g., ENSMUSG00000028180__E1.1-E100.1
pd1 = PositionData(chr+'__'+str(pds.Start())+'-'+str(pds.Start())) #chr3:157544964-157545122
selected_event = gene ### Again, set this for gene entry coordinates
try: pd2 = feature_positions[gene_prefix+e2]
except Exception:
e2 = string.split(e2,'.')[0]+'.1' ### occurs with IDS such as ENSMUSG00000002107__E26.27, where .27 is not in the database (not sure why)
try: pd2 = feature_positions[gene_prefix+e2]
except Exception: ### Occurs with gene entry, e.g., ENSMUSG00000028180__E1.1-E100.1
pd2 = PositionData(chr+'__'+str(pds.End())+'-'+str(pds.End())) #chr3:157544964-157545122
selected_event = gene ### Again, set this for gene entry coordinates
#print pd.Start(),pd.End(), pd1.Start(),pd1.End(), pd2.Start(), pd2.End()
first_start = pd1.Start()
last_end = pd2.End()
### Now, loop through all gene features and only select the ones that are in between the spliced exons
for exonID in feature_positions:
proceed = False
if '-' not in exonID: ### Hence strand junction
geneID, block_region = string.split(exonID,'__')
exon_region = string.replace(block_region,'E','')
if ('I' not in exon_region) and (';' not in exon_region) and ('-' not in exon_region):
pd = feature_positions[exonID]
exon_start = pd.Start()
exon_end = pd.End()
if(exon_start >= first_start and exon_end <= last_end):
proceed = True
if(exon_start <= first_start and exon_end >= last_end):
proceed = True
if proceed:
export_string = chr+'\t'+'SE'+'\t'+'exon'+'\t'+str(exon_start)+'\t'+str(exon_end)+'\t'+'.'+'\t'+strand+'\t'+'.'+'\t'+'ID='+exonID+';'+'Parent='+selected_event+'.STRAND;'+'\n'
if 'M' not in chr:
key = exonID,selected_event
#if key not in added_gff_entries:
gff_export_obj.write(export_string)
added_gff_entries[key]=[]
if strand == '-':
junction_exon_start=int(pd2.Start())-300 ### increase the window size
junction_exon_end=int(pd1.End())+300 ### increase the window size
else:
junction_exon_start=int(pd1.Start())-300 ### increase the window size
junction_exon_end=int(pd2.End())+300 ### increase the window size
return chr,str(junction_exon_start),str(junction_exon_end),strand,selected_event
def Indexing(counts,PSIFileDir,output):
""" Indexing is the process of creating strand machine readable binary for SashimiPlot """
gene_feature_db=reimportFeatures(counts) ### import the exon and gene coordinates
PSIJunctions=importPSIJunctions(PSIFileDir) ### Retreive all PSI junction pairs
PSIJunctions=importReciprocalJunctions(PSIFileDir,PSIJunctions) ### Include other Junctions from ASPIRE/LinRegress
exported = False
for (junction1,junction2) in PSIJunctions:
#if 'ENSMUSG00000066007:E5.1-ENSMUSG00000063245:E3.1' in PSIJunctions[event_pair]:
geneID = string.split(junction1,'__')[0]
if geneID in gene_feature_db:
try:
chr,junction_start,junction_end,strand,selected_event=exportToGff(junction1,junction2,gene_feature_db[geneID],geneID)
exported=True
except Exception: pass ### Not handling trans-splicing well
if exported:
### For each exon entry exported for this event, we need a gene entry with the boundary exon positions exported (returned from the above function)
try: writegene(chr,junction_start,junction_end,strand,selected_event) #('chrX', '38600355', '38606652', '+', 'ENSMUSG00000000355__E1.2-E5.1')
except Exception: pass
### Export coordinate information for the entire gene
for geneID in gene_feature_db:
feature_db = gene_feature_db[geneID]
for event in feature_db:
if 'E1.1-E100.1' in event:
pd = feature_db[event]
chr = pd.Chr()
start,stop = pd.Start(), pd.End()
if start>stop: strand = '-'
else: strand = '+'
try:
chr,junction_start,junction_end,strand,selected_event=exportToGff('null',event,gene_feature_db[geneID],geneID)
writegene(chr,junction_start,junction_end,strand,selected_event) #('chrX', '38600355', '38606652', '+', 'ENSMUSG00000000355__E1.2-E5.1')
except Exception:
pass
time.sleep(2)
gff_export_obj.close()
newout=findParentDir(output)+'sashimi_index/'
try: ifg.index_gff(output,newout)
except Exception:
print traceback.format_exc();sys.exit()
print('error in indexing')
def findParentDir(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1
return filename[:x]
def remoteIndexing(species,fl):
""" Begin building strand gff and index files for SashimiPlot based on the AltAnalyze database
exon, junction and gene annotations """
global gff_export_obj
try:
### When fl is strand dataset information object
countsFileDir = fl.CountsFile() ### Counts file containing exon and junction positions
root_dir = fl.RootDir() ### Root folder location
except Exception:
### STRAND proper object may not be supplied with this information. Use the root directory alone to infer these
root_dir = fl
search_dir = root_dir+'/ExpressionInput'
files = unique.read_directory(search_dir) ### all files in ExpressionInput
for file in files:
if 'counts.' in file and 'steady-state.txt' not in file:
countsFileDir = search_dir+'/'+file ### counts file with exon positions
PSIFileDir = root_dir+'/AltResults/AlternativeOutput/'+species+'_RNASeq_top_alt_junctions-PSI.txt'
OutputDir=findParentDir(PSIFileDir)
output=OutputDir+"events_sashimi.gff"
gff_export_obj=open(output,'w')
### Sometimes only junctions are in the count file so create strand new file with detected junctions and all exons
### This information and associated featrues is extracted from the counts file
featuresEvaluated = extractFeatures(species,countsFileDir)
### Compile and export the coordinates to gff format and index these coordinates for fast retreival by Miso
Indexing(featuresEvaluated,PSIFileDir,output)
def extractFeatures(species,countsFileDir):
import export
ExonsPresent=False
lastgene = None
lastend = None
genes_detected={}
count=0
first_last_exons = {} ### Make strand fake junction comprised of the first and last exon
if 'counts.' in countsFileDir:
### The feature_file contains only ExonID or Gene IDs and associated coordinates
feature_file = string.replace(countsFileDir,'counts.','features.')
fe = export.ExportFile(feature_file)
firstLine = True
for line in open(countsFileDir,'rU').xreadlines():
if firstLine: firstLine=False
else:
feature_info = string.split(line,'\t')[0]
fe.write(feature_info+'\n')
junction_annotation = string.split(feature_info,'=')[0]
if '-' in junction_annotation:
geneid = string.split(junction_annotation,':')[0]
genes_detected[geneid]=[]
if ExonsPresent == False:
exon = string.split(feature_info,'=')[0]
if '-' not in exon:
ExonsPresent = True
### Add exon-info if necessary
exons_file = unique.filepath('AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt')
firstLine = True
for line in open(exons_file,'rU').xreadlines():
if firstLine: firstLine=False
else:
line = line.rstrip('\n')
t = string.split(line,'\t')
gene,exon,chr,strand,start,end = t[:6]
if gene!=lastgene:
if len(genes_detected)==0 or gene in genes_detected: ### restrict to detected genes
first_last_exons[gene,strand] = [(chr,start)]
if len(genes_detected)==0 or lastgene in genes_detected: ### restrict to detected genes
try: first_last_exons[lastgene,laststrand].append(lastend)
except Exception:
pass ### occurs for the first gene
if ExonsPresent == False:
fe.write(gene+':'+exon+'='+chr+':'+start+'-'+end+'\n')
lastgene = gene; lastend = end; laststrand = strand
if len(genes_detected)==0 or lastgene in genes_detected:
first_last_exons[lastgene,laststrand].append(lastend)
### Add strand fake junction for the whole gene
for (gene,strand) in first_last_exons:
(chr,start),end = first_last_exons[gene,strand]
if strand == '-':
start,end = end,start # Need to encode strand in this annotation, do this by strand orienting the positions
fe.write(gene+':E1.1-E100.1'+'='+chr+':'+start+'-'+end+'\n')
fe.close()
return feature_file ### return the location of the exon and gene coordinates file
def obtainTopGeneResults():
pass
if __name__ == '__main__':
Species = 'Hs'
root = '/Volumes/salomonis2-1/Ichi_data/bams/Insertion_analysis/'
#root = '/Users/saljh8/Desktop/Grimes/GEC14074/'
remoteIndexing(Species,root)
sys.exit()
#"""
countsFileDir = os.path.abspath(os.path.expanduser(sys.argv[1]))
PSIFileDir = os.path.abspath(os.path.expanduser(sys.argv[2]))
OutputDir=findParentDir(PSIFileDir)
output=OutputDir+"events_sashimi.gff"
gff_export_obj=open(output,'w')
Indexing(countsFileDir,PSIFileDir,output)
#"""
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/SashimiIndex.py
|
SashimiIndex.py
|
from __future__ import print_function
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy as np
import pylab as pl
import sys,string
import os
import os.path
import scipy
import operator
from collections import OrderedDict
from collections import defaultdict
from operator import itemgetter
from visualization_scripts import clustering; reload(clustering)
import export
def Classify(filename,Mutlabels={},dire="",flag=True):
count=0
start=1
orderdict=OrderedDict()
countdict=OrderedDict()
countlst=[]
Y=[]
head=0
rownames=[]
colnames=[]
q=[]
Z=[]
if dire!="":
output_dir = dire+'Results'
export.createExportFolder(output_dir)
if flag:
output_file=output_dir+"/Consolidated-Increasing"+".txt"
else:
output_file=output_dir+"/Consolidated-Decreasing"+".txt"
else:
output_file=filename[:-4]+"-ordered.txt"
export_object = open(output_file,'w')
for line in open(filename,'rU').xreadlines():
if head >0:
val=[]
counter2=0
val2=[]
me=0.0
line=line.rstrip('\r\n')
q= string.split(line,'\t')
# rownames.append(q[0])
if q[0]=="":
continue
orderdict[q[0]]=[q[0],]
for i in range(start,len(q)):
try:
val2.append(float(q[i]))
try:
orderdict[q[0]].append(float(q[i]))
except Exception:
orderdict[q[0]]=[float(q[i]),]
try:
countdict[i].append(float(q[i]))
except Exception:
countdict[i]=[float(q[i]),]
except Exception:
continue
count+=1
else:
#export_object.write(line)
head=1
line=line.rstrip('\r\n')
q= string.split(line,'\t')
header=q
continue
for i in countdict:
countlst.append(sum(countdict[i]))
#print countlst
B=sorted(range(len(countlst)),key=lambda x:countlst[x],reverse=flag)
C=sorted(range(len(countlst)),key=lambda x:B[x])
qu=0
for i in orderdict.keys():
Y.append(orderdict[i])
qu+=1
#print Y
for i in range(0,len(C)):
jk= C.index(i)+1
#print jk
#print Y[jk]
Y=sorted(Y,key=itemgetter(jk))
#orderdict=OrderedDict(sorted(orderdict,key=itemgetter(jk)))
#colnames.append(header[C.index(i)+1])
Y=np.array(Y)
Y=zip(*Y)
Y=np.array(Y)
Z.append(Y[0,:])
for i in range(0,len(C)):
jk= C.index(i)+1
Z.append(Y[jk,:])
Z=np.array(Z)
q= Z.shape
export_object.write("uid")
for i in range(q[1]):
export_object.write("\t"+Z[0][i])
export_object.write("\n")
for ij in range(1,q[0]):
jk= C.index(ij-1)+1
if header[jk] in Mutlabels:
export_object.write(Mutlabels[header[jk]])
else:
export_object.write(header[jk])
for jq in range(0,q[1]):
export_object.write("\t"+str(Z[ij][jq]))
export_object.write("\n")
export_object.close()
graphic_links=[]
row_method = None
column_method=None
column_metric='cosine'
row_metric='cosine'
color_gradient = 'yellow_black_blue'
transpose=False
graphic_links = clustering.runHCexplicit(output_file,graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=False)
if __name__ == '__main__':
import getopt
group=[]
grplst=[]
name=[]
matrix={}
compared_groups={}
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['Guidedir='])
for opt, arg in options:
if opt == '--Guidedir':
Guidedir=arg
Classify(Guidedir)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/Orderedheatmap.py
|
Orderedheatmap.py
|
import platform
useDefaultBackend=False
if platform.system()=='Darwin':
if platform.mac_ver()[0] == '10.14.6':
useDefaultBackend=True
try:
import warnings
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
import matplotlib
if useDefaultBackend == False:
matplotlib.rcParams['backend'] = 'TkAgg'
import matplotlib.pyplot as pylab
matplotlib.rcParams['axes.linewidth'] = 0.5
matplotlib.rcParams['pdf.fonttype'] = 42
matplotlib.rcParams['font.family'] = 'sans-serif'
matplotlib.rcParams['font.sans-serif'] = 'Arial'
except Exception:
None
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from matplotlib import pyplot as pylab
from matplotlib.patches import Circle, Ellipse
from itertools import chain
from collections import Iterable
import UI
import time, datetime
today = str(datetime.date.today()); today = string.split(today,'-'); today = today[0]+''+today[1]+''+today[2]
time_stamp = string.replace(time.ctime(),':',''); time_stamp = string.replace(time_stamp,' ',' ')
time_stamp = string.split(time_stamp,' '); time_stamp = today+'-'+time_stamp[3]
venn_export = 'Standard_VennDiagram-'+time_stamp
venn_export_weighted = 'Weighted_VennDiagram-'+time_stamp
#--------------------------------------------------------------------
alignment = {'horizontalalignment':'center', 'verticalalignment':'baseline'}
#--------------------------------------------------------------------
def venn(data, names=None, fill="number", show_names=True, show_plot=True, outputDir=False, **kwds):
"""
data: a list
names: names of groups in data
fill = ["number"|"logic"|"both"], fill with number, logic label, or both
show_names = [True|False]
show_plot = [True|False]
"""
if data is None:
raise Exception("No data!")
if len(data) == 2:
venn2(data, names, fill, show_names, show_plot, outputDir, **kwds)
elif len(data) == 3:
venn3(data, names, fill, show_names, show_plot, outputDir, **kwds)
elif len(data) == 4:
venn4(data, names, fill, show_names, show_plot, outputDir, **kwds)
else:
print len(data), 'files submitted, must be less than 4 and greater than 1...'
#raise Exception("currently only 2-4 sets venn diagrams are supported")
#--------------------------------------------------------------------
def get_labels(data, fill="number"):
"""
to get a dict of labels for groups in data
input
data: data to get label for
fill = ["number"|"logic"|"both"], fill with number, logic label, or both
return
labels: a dict of labels for different sets
example:
In [12]: get_labels([range(10), range(5,15), range(3,8)], fill="both")
Out[12]:
{'001': '001: 0',
'010': '010: 5',
'011': '011: 0',
'100': '100: 3',
'101': '101: 2',
'110': '110: 2',
'111': '111: 3'}
"""
N = len(data)
sets_data = [set(data[i]) for i in range(N)] # sets for separate groups
s_all = set(chain(*data)) # union of all sets
# bin(3) --> '0b11', so bin(3).split('0b')[-1] will remove "0b"
set_collections = {}
for n in range(1, 2**N):
key = bin(n).split('0b')[-1].zfill(N)
value = s_all
sets_for_intersection = [sets_data[i] for i in range(N) if key[i] == '1']
sets_for_difference = [sets_data[i] for i in range(N) if key[i] == '0']
for s in sets_for_intersection:
value = value & s
for s in sets_for_difference:
value = value - s
set_collections[key] = value
"""for i in set_collections:
if len(set_collections[i])<100:
print set_collections[i]"""
labels={}
if fill == "number":
for k in set_collections:
#labels[k] = len(set_collections[k])
labels[k] = set_collections[k]
elif fill == "logic":
for k in set_collections: labels[k] = k
elif fill == "both":
for k in set_collections: labels[k] = ("%s: %d" % (k, len(set_collections[k])))
#labels = {k: ("%s: %d" % (k, len(set_collections[k]))) for k in set_collections}
else: # invalid value
raise Exception("invalid value for fill")
return labels
#--------------------------------------------------------------------
def venn2(data=None, names=None, fill="number", show_names=True, show_plot=True, outputDir=False, **kwds):
global coordinates
coordinates={}
if (data is None) or len(data) != 2:
raise Exception("length of data should be 2!")
if (names is None) or (len(names) != 2):
names = ("set 1", "set 2")
labels = get_labels(data, fill=fill)
# set figure size
if 'figsize' in kwds and len(kwds['figsize']) == 2:
# if 'figsize' is in kwds, and it is a list or tuple with length of 2
figsize = kwds['figsize']
else: # default figure size
figsize = (11, 8)
fig = pylab.figure(figsize=figsize)
ax = fig.gca(); ax.set_aspect("equal")
ax.set_xticks([]); ax.set_yticks([]);
ax.set_xlim(0, 11); ax.set_ylim(0, 8)
# r: radius of the circles
# (x1, y1), (x2, y2): center of circles
r, x1, y1, x2, y2 = 2.0, 3.0, 4.0, 5.0, 4.0
# set colors for different Circles or ellipses
if 'colors' in kwds and isinstance(kwds['colors'], Iterable) and len(kwds['colors']) >= 2:
colors = kwds['colors']
else:
colors = ['red', 'green']
c1 = Circle((x1,y1), radius=r, alpha=0.5, color=colors[0])
c2 = Circle((x2,y2), radius=r, alpha=0.5, color=colors[1])
ax.add_patch(c1)
ax.add_patch(c2)
## draw text
#1
pylab.text(round(x1-r/2), round(y1), len(labels['10']),fontsize=16, picker=True, **alignment); coordinates[round(x1-r/2), round(y1)]=labels['10']
pylab.text(round(x2+r/2), round(y2), len(labels['01']),fontsize=16, picker=True, **alignment); coordinates[round(x2+r/2), round(y2)]=labels['01']
# 2
pylab.text(round((x1+x2)/2), round(y1), len(labels['11']),fontsize=16, picker=True, **alignment); coordinates[round((x1+x2)/2), round(y1)]=labels['11']
# names of different groups
if show_names:
pylab.text(x1, y1-1.2*r, names[0], fontsize=16, **alignment)
pylab.text(x2, y2-1.2*r, names[1], fontsize=16, **alignment)
leg = ax.legend(names, loc='best', fancybox=True)
leg.get_frame().set_alpha(0.5)
fig.canvas.mpl_connect('pick_event', onpick)
try:
if outputDir!=False:
filename = outputDir+'/%s.pdf' % venn_export
pylab.savefig(filename)
filename = outputDir+'/%s.png' % venn_export
pylab.savefig(filename, dpi=100) #,dpi=200
except Exception:
print 'Image file not saved...'
if show_plot:
pylab.show()
try:
import gc
fig.clf()
pylab.close()
gc.collect()
except Exception:
pass
#--------------------------------------------------------------------
def venn3(data=None, names=None, fill="number", show_names=True, show_plot=True, outputDir=False, **kwds):
global coordinates
coordinates={}
if (data is None) or len(data) != 3:
raise Exception("length of data should be 3!")
if (names is None) or (len(names) != 3):
names = ("set 1", "set 2", "set 3")
labels = get_labels(data, fill=fill)
# set figure size
if 'figsize' in kwds and len(kwds['figsize']) == 2:
# if 'figsize' is in kwds, and it is a list or tuple with length of 2
figsize = kwds['figsize']
else: # default figure size
figsize = (11, 8.3)
fig = pylab.figure(figsize=figsize) # set figure size
ax = fig.gca()
ax.set_aspect("equal") # set aspect ratio to 1
ax.set_xticks([]); ax.set_yticks([]);
ax.set_xlim(0, 11); ax.set_ylim(0, 8.3)
# r: radius of the circles
# (x1, y1), (x2, y2), (x3, y3): center of circles
r, x1, y1, x2, y2 = 2.0, 3.0, 3.0, 5.0, 3.0
x3, y3 = (x1+x2)/2.0, y1 + 3**0.5/2*r
# set colors for different Circles or ellipses
if 'colors' in kwds and isinstance(kwds['colors'], Iterable) and len(kwds['colors']) >= 3:
colors = kwds['colors']
else:
colors = ['red', 'green', 'blue']
c1 = Circle((x1,y1), radius=r, alpha=0.5, color=colors[0])
c2 = Circle((x2,y2), radius=r, alpha=0.5, color=colors[1])
c3 = Circle((x3,y3), radius=r, alpha=0.5, color=colors[2])
for c in (c1, c2, c3):
ax.add_patch(c)
## draw text
# 1
pylab.text(x1-r/2, round(y1-r/2), len(labels['100']),fontsize=16, picker=True, **alignment); coordinates[x1-r/2, round(y1-r/2)]=labels['100']
pylab.text(x2+r/2, round(y2-r/2), len(labels['010']), fontsize=16, picker=True, **alignment); coordinates[x2+r/2, round(y2-r/2)]=labels['010']
pylab.text((x1+x2)/2, round(y3+r/2), len(labels['001']), fontsize=16, picker=True, **alignment); coordinates[(x1+x2)/2, round(y3+r/2)]=labels['001']
# 2
pylab.text((x1+x2)/2, round(y1-r/2), len(labels['110']),fontsize=16,picker=True, **alignment); coordinates[(x1+x2)/2, round(y1-r/2)]=labels['110']
pylab.text(x1, round(y1+2*r/3), len(labels['101']),fontsize=16,picker=True, **alignment); coordinates[x1, round(y1+2*r/3)]=labels['101']
pylab.text(x2, round(y2+2*r/3), len(labels['011']),fontsize=16,picker=True, **alignment); coordinates[x2, round(y2+2*r/3)]=labels['011']
# 3
pylab.text((x1+x2)/2, round(y1+r/3),len(labels['111']), fontsize=16,picker=True, **alignment); coordinates[(x1+x2)/2, round(y1+r/3)]=labels['111']
# names of different groups
if show_names:
pylab.text(x1-r, y1-r, names[0], fontsize=16, **alignment)
pylab.text(x2+r, y2-r, names[1], fontsize=16, **alignment)
pylab.text(x3, y3+1.2*r, names[2], fontsize=16, **alignment)
leg = ax.legend(names, loc='best', fancybox=True)
leg.get_frame().set_alpha(0.5)
fig.canvas.mpl_connect('pick_event', onpick)
try:
if outputDir!=False:
filename = outputDir+'/%s.pdf' % venn_export
pylab.savefig(filename)
filename = outputDir+'/%s.png' % venn_export
pylab.savefig(filename, dpi=100) #,dpi=200
except Exception:
print 'Image file not saved...'
if show_plot:
pylab.show()
try:
import gc
fig.clf()
pylab.close()
gc.collect()
except Exception:
pass
#--------------------------------------------------------------------
def venn4(data=None, names=None, fill="number", show_names=True, show_plot=True, outputDir=False, **kwds):
global coordinates
coordinates={}
if (data is None) or len(data) != 4:
raise Exception("length of data should be 4!")
if (names is None) or (len(names) != 4):
names = ("set 1", "set 2", "set 3", "set 4")
labels = get_labels(data, fill=fill)
# set figure size
if 'figsize' in kwds and len(kwds['figsize']) == 2:
# if 'figsize' is in kwds, and it is a list or tuple with length of 2
figsize = kwds['figsize']
else: # default figure size
figsize = (11, 10)
# set colors for different Circles or ellipses
if 'colors' in kwds and isinstance(kwds['colors'], Iterable) and len(kwds['colors']) >= 4:
colors = kwds['colors']
else:
colors = ['r', 'g', 'b', 'c']
# draw ellipse, the coordinates are hard coded in the rest of the function
fig = pylab.figure(figsize=figsize) # set figure size
ax = fig.gca()
patches = []
width, height = 170, 110 # width and height of the ellipses
patches.append(Ellipse((170, 170), width, height, -45, color=colors[0], alpha=0.5, label=names[0])) ### some disconnect with the colors and the labels (see above) so had to re-order here
patches.append(Ellipse((200, 200), width, height, -45, color=colors[2], alpha=0.5, label=names[2]))
patches.append(Ellipse((200, 200), width, height, -135, color=colors[3], alpha=0.5, label=names[3]))
patches.append(Ellipse((230, 170), width, height, -135, color=colors[1], alpha=0.5, label=names[1]))
for e in patches:
ax.add_patch(e)
ax.set_xlim(80, 320); ax.set_ylim(80, 320)
ax.set_xticks([]); ax.set_yticks([]);
ax.set_aspect("equal")
### draw text
# 1
pylab.text(120, 200, len(labels['1000']),fontsize=16, picker=True, **alignment);coordinates[120, 200]=labels['1000']
pylab.text(280, 200, len(labels['0100']),fontsize=16, picker=True, **alignment);coordinates[280, 200]=labels['0100']
pylab.text(155, 250, len(labels['0010']),fontsize=16, picker=True, **alignment);coordinates[155, 250]=labels['0010']
pylab.text(245, 250, len(labels['0001']),fontsize=16, picker=True, **alignment);coordinates[245, 250]=labels['0001']
# 2
pylab.text(200, 115, len(labels['1100']),fontsize=16, picker=True, **alignment);coordinates[200, 115]=labels['1100']
pylab.text(140, 225, len(labels['1010']),fontsize=16, picker=True, **alignment);coordinates[140, 225]=labels['1010']
pylab.text(145, 155, len(labels['1001']),fontsize=16, picker=True, **alignment);coordinates[145, 155]=labels['1001']
pylab.text(255, 155, len(labels['0110']),fontsize=16, picker=True, **alignment);coordinates[255, 155]=labels['0110']
pylab.text(260, 225, len(labels['0101']),fontsize=16, picker=True, **alignment);coordinates[260, 225]=labels['0101']
pylab.text(200, 240, len(labels['0011']),fontsize=16, picker=True, **alignment);coordinates[200, 240]=labels['0011']
# 3
pylab.text(235, 205, len(labels['0111']),fontsize=16, picker=True, **alignment);coordinates[235, 205]=labels['0111']
pylab.text(165, 205, len(labels['1011']),fontsize=16, picker=True, **alignment);coordinates[165, 205]=labels['1011']
pylab.text(225, 135, len(labels['1101']),fontsize=16, picker=True, **alignment);coordinates[225, 135]=labels['1101']
pylab.text(175, 135, len(labels['1110']),fontsize=16, picker=True, **alignment);coordinates[175, 135]=labels['1110']
# 4
pylab.text(200, 175, len(labels['1111']),fontsize=16, picker=True, **alignment);coordinates[200, 175]=labels['1111']
# names of different groups
if show_names:
pylab.text(110, 110, names[0], fontsize=16, **alignment)
pylab.text(290, 110, names[1], fontsize=16, **alignment)
pylab.text(130, 275, names[2], fontsize=16, **alignment)
pylab.text(270, 275, names[3], fontsize=16, **alignment)
leg = ax.legend(loc='best', fancybox=True)
leg.get_frame().set_alpha(0.5)
fig.canvas.mpl_connect('pick_event', onpick)
try:
if outputDir!=False:
filename = outputDir+'/%s.pdf' % venn_export
pylab.savefig(filename)
filename = outputDir+'/%s.png' % venn_export
pylab.savefig(filename, dpi=100) #,dpi=200
except Exception:
print 'Image file not saved...'
if show_plot:
pylab.show()
try:
import gc
fig.clf()
pylab.close()
gc.collect()
except Exception:
pass
#--------------------------------------------------------------------
def onpick(event):
text = event.artist
x = int(event.mouseevent.xdata)
y = int(event.mouseevent.ydata)
#print [text.get_text()]
if (x,y) in coordinates:
#print text.get_text()
from visualization_scripts import TableViewer
header = ['Associated Genes']
tuple_list = []
for gene in coordinates[(x,y)]:
tuple_list.append([(gene)])
TableViewer.viewTable(text.get_text(),header,tuple_list) #"""
def test():
""" a test function to show basic usage of venn()"""
# venn3()
venn([range(10), range(5,15), range(3,8)], ["aaaa", "bbbb", "cccc"], fill="both", show_names=False)
# venn2()
venn([range(10), range(5,15)])
venn([range(10), range(5,15)], ["aaaa", "bbbb"], fill="logic", show_names=False)
# venn4()
venn([range(10), range(5,15), range(3,8), range(4,9)], ["aaaa", "bbbb", "cccc", "dddd"], figsize=(12,12))
######### Added for AltAnalyze #########
def test2():
""" a test function to show basic usage of venn()"""
venn([['a','b','c','d','e'], ['a','b','c','d','e'], ['a','b','c','d','e']], ["a", "b", "c"], fill="number", show_names=True)
def compareDirectoryFiles(import_dir):
file_list = UI.read_directory(import_dir)
compareImportedTables(file_list,import_dir,importDir=import_dir)
def compareInputFiles(file_list,outputDir,display=True):
compareImportedTables(file_list,outputDir,display=display)
def compareImportedTables(file_list,outputDir,importDir=False,considerNumericDirection=False,display=True):
### added for AltAnalyze
print 'Creating Venn Diagram from input files...'
import UI
import export
file_id_db={}
file_list2=[]
for file in file_list:
x=0
if '.txt' in file:
if importDir !=False: ### When all files in a directory are analyzed
fn=UI.filepath(import_dir+'/'+file)
else:
fn = file
file = export.findFilename(fn) ### Only report the actual filename
file_list2.append(file)
for line in open(fn,'rU').xreadlines():
if x == 0:
data_type = examineFields(line)
x+=1
else:
data = UI.cleanUpLine(line)
t = string.split(data,'\t')
uid = t[0]
if string.lower(uid) == 'uid':
continue
valid = True
if data_type != 'first':
if data_type == 'comparison':
score = float(string.split(t[6],'|')[0])
if 'yes' not in t[5]:
valid = False ### not replicated independently
if data_type == 'reciprocal':
uid = t[8]+'-'+t[10]
score = float(t[1])
if data_type == 'single':
uid = t[6]
score = float(t[1])
else:
try:
score = float(t[1]) #t[2]
except Exception: score = None
if score != None and considerNumericDirection: ### change the UID so that it only matches if the same direction
if score>0:
uid+='+' ### encode the ID with a negative sign
else:
uid+='-' ### encode the ID with a negative sign
#if score>0:
if valid:
try: file_id_db[file].append(uid)
except Exception: file_id_db[file] = [uid]
id_lists=[]
new_file_list=[]
for file in file_list2: ### Use the sorted names
if file in file_id_db:
uids = file_id_db[file]
id_lists.append(uids)
new_file_list.append(file)
#print file, len(new_file_list), len(uids)
if len(file_id_db):
if len(new_file_list)==2 or len(new_file_list)==3:
SimpleMatplotVenn(new_file_list,id_lists,outputDir=outputDir,display=False) ### display both below
venn(id_lists, new_file_list, fill="number", show_names=False, outputDir=outputDir, show_plot=display)
def examineFields(data):
if 'independent confirmation' in data:
data_type = 'comparison'
elif 'norm-p2' in data:
data_type = 'reciprocal'
elif 'functional_prediction' in data:
data_type = 'single'
else:
data_type = 'first' ### first column
return data_type
def SimpleMatplotVenn(names,data,outputDir=False,display=True):
""" Uses http://pypi.python.org/pypi/matplotlib-venn (code combined into one module) to export
simple or complex, overlapp weighted venn diagrams as an alternative to the default methods in
this module """
import numpy as np
pylab.figure(figsize=(11,7),facecolor='w')
vd = get_labels(data, fill="number")
set_labels=[]
for i in names:
set_labels.append(string.replace(i,'.txt',''))
if len(set_labels)==2:
from matplotlib_venn import venn2, venn2_circles
set_colors = ('r', 'g')
subsets = (vd['10'], vd['01'], vd['11'])
v = venn2(subsets=subsets, set_labels = set_labels, set_colors=set_colors)
c = venn2_circles(subsets=subsets, alpha=0.5, linewidth=1.5, linestyle='dashed')
if len(set_labels)==3:
from matplotlib_venn import venn3, venn3_circles
set_colors = ('r', 'g', 'b')
subsets = (vd['100'], vd['010'], vd['110'], vd['001'], vd['101'], vd['011'], vd['111'])
v = venn3(subsets=subsets, set_labels = set_labels,set_colors=set_colors)
c = venn3_circles(subsets=subsets, alpha=0.5, linewidth=1.5, linestyle='dashed')
pylab.title("Overlap Weighted Venn Diagram",fontsize=24)
try:
if outputDir!=False:
filename = outputDir+'/%s.pdf' % venn_export_weighted
pylab.savefig(filename)
filename = outputDir+'/%s.png' % venn_export_weighted
pylab.savefig(filename, dpi=100) #,dpi=200
except Exception:
print 'Image file not saved...'
if display:
pylab.show()
try:
import gc
fig.clf()
pylab.close()
gc.collect()
except Exception:
pass
######### End Added for AltAnalyze #########
if __name__ == '__main__':
names = ['a','b','c']
venn_data = {}
venn_data['100']=1
venn_data['010']=2
venn_data['110']=3
venn_data['001']=4
venn_data['101']=5
venn_data['011']=6
venn_data['111']=7
#SimpleMatplotVenn(names,venn_data); sys.exit()
import_dir = '/Users/nsalomonis/Desktop/code/python/combine-lists/input'
import_dir = '/Users/nsalomonis/Downloads/GSE31327_RAW/GO-Elite/input/compare'
import_dir = '/Users/nsalomonis/Desktop/dataAnalysis/r4_Bruneau_TopHat/AltResults/AlternativeOutput/ASPIRE-comp'
import_dir = '/Users/nsalomonis/Desktop/dataAnalysis/r4_Bruneau_TopHat/AltResults/AlternativeOutput/SI-comp'
import_dir = '/Users/nsalomonis/Desktop/dataAnalysis/r4_Bruneau_TopHat/AltResults/AlternativeOutput/stringent-comp'
import_dir = '/Users/nsalomonis/Desktop/dataAnalysis/collaborations/Faith/MarkerClustering/NC-type-markers'
compareDirectoryFiles(import_dir)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/VennDiagram.py
|
VennDiagram.py
|
_VERSION = '1.3'
def setversion(version):
if version != _VERSION:
raise ValueError, 'Dynamic versioning not available'
def setalphaversions(*alpha_versions):
if alpha_versions != ():
raise ValueError, 'Dynamic versioning not available'
def version(alpha = 0):
if alpha:
return ()
else:
return _VERSION
def installedversions(alpha = 0):
if alpha:
return ()
else:
return (_VERSION,)
######################################################################
### File: PmwBase.py
# Pmw megawidget base classes.
# This module provides a foundation for building megawidgets. It
# contains the MegaArchetype class which manages component widgets and
# configuration options. Also provided are the MegaToplevel and
# MegaWidget classes, derived from the MegaArchetype class. The
# MegaToplevel class contains a Tkinter Toplevel widget to act as the
# container of the megawidget. This is used as the base class of all
# megawidgets that are contained in their own top level window, such
# as a Dialog window. The MegaWidget class contains a Tkinter Frame
# to act as the container of the megawidget. This is used as the base
# class of all other megawidgets, such as a ComboBox or ButtonBox.
#
# Megawidgets are built by creating a class that inherits from either
# the MegaToplevel or MegaWidget class.
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import traceback
import types
import Tkinter
# Special values used in index() methods of several megawidgets.
END = ['end']
SELECT = ['select']
DEFAULT = ['default']
# Constant used to indicate that an option can only be set by a call
# to the constructor.
INITOPT = ['initopt']
_DEFAULT_OPTION_VALUE = ['default_option_value']
_useTkOptionDb = 0
# Symbolic constants for the indexes into an optionInfo list.
_OPT_DEFAULT = 0
_OPT_VALUE = 1
_OPT_FUNCTION = 2
# Stacks
_busyStack = []
# Stack which tracks nested calls to show/hidebusycursor (called
# either directly or from activate()/deactivate()). Each element
# is a dictionary containing:
# 'newBusyWindows' : List of windows which had busy_hold called
# on them during a call to showbusycursor().
# The corresponding call to hidebusycursor()
# will call busy_release on these windows.
# 'busyFocus' : The blt _Busy window which showbusycursor()
# set the focus to.
# 'previousFocus' : The focus as it was when showbusycursor()
# was called. The corresponding call to
# hidebusycursor() will restore this focus if
# the focus has not been changed from busyFocus.
_grabStack = []
# Stack of grabbed windows. It tracks calls to push/popgrab()
# (called either directly or from activate()/deactivate()). The
# window on the top of the stack is the window currently with the
# grab. Each element is a dictionary containing:
# 'grabWindow' : The window grabbed by pushgrab(). The
# corresponding call to popgrab() will release
# the grab on this window and restore the grab
# on the next window in the stack (if there is one).
# 'globalMode' : True if the grabWindow was grabbed with a
# global grab, false if the grab was local
# and 'nograb' if no grab was performed.
# 'previousFocus' : The focus as it was when pushgrab()
# was called. The corresponding call to
# popgrab() will restore this focus.
# 'deactivateFunction' :
# The function to call (usually grabWindow.deactivate) if
# popgrab() is called (usually from a deactivate() method)
# on a window which is not at the top of the stack (that is,
# does not have the grab or focus). For example, if a modal
# dialog is deleted by the window manager or deactivated by
# a timer. In this case, all dialogs above and including
# this one are deactivated, starting at the top of the
# stack.
# Note that when dealing with focus windows, the name of the Tk
# widget is used, since it may be the '_Busy' window, which has no
# python instance associated with it.
#=============================================================================
# Functions used to forward methods from a class to a component.
# Fill in a flattened method resolution dictionary for a class (attributes are
# filtered out). Flattening honours the MI method resolution rules
# (depth-first search of bases in order). The dictionary has method names
# for keys and functions for values.
def __methodDict(cls, dict):
# the strategy is to traverse the class in the _reverse_ of the normal
# order, and overwrite any duplicates.
baseList = list(cls.__bases__)
baseList.reverse()
# do bases in reverse order, so first base overrides last base
for super in baseList:
__methodDict(super, dict)
# do my methods last to override base classes
for key, value in cls.__dict__.items():
# ignore class attributes
if type(value) == types.FunctionType:
dict[key] = value
def __methods(cls):
# Return all method names for a class.
# Return all method names for a class (attributes are filtered
# out). Base classes are searched recursively.
dict = {}
__methodDict(cls, dict)
return dict.keys()
# Function body to resolve a forwarding given the target method name and the
# attribute name. The resulting lambda requires only self, but will forward
# any other parameters.
__stringBody = (
'def %(method)s(this, *args, **kw): return ' +
'apply(this.%(attribute)s.%(method)s, args, kw)')
# Get a unique id
__counter = 0
def __unique():
global __counter
__counter = __counter + 1
return str(__counter)
# Function body to resolve a forwarding given the target method name and the
# index of the resolution function. The resulting lambda requires only self,
# but will forward any other parameters. The target instance is identified
# by invoking the resolution function.
__funcBody = (
'def %(method)s(this, *args, **kw): return ' +
'apply(this.%(forwardFunc)s().%(method)s, args, kw)')
def forwardmethods(fromClass, toClass, toPart, exclude = ()):
# Forward all methods from one class to another.
# Forwarders will be created in fromClass to forward method
# invocations to toClass. The methods to be forwarded are
# identified by flattening the interface of toClass, and excluding
# methods identified in the exclude list. Methods already defined
# in fromClass, or special methods with one or more leading or
# trailing underscores will not be forwarded.
# For a given object of class fromClass, the corresponding toClass
# object is identified using toPart. This can either be a String
# denoting an attribute of fromClass objects, or a function taking
# a fromClass object and returning a toClass object.
# Example:
# class MyClass:
# ...
# def __init__(self):
# ...
# self.__target = TargetClass()
# ...
# def findtarget(self):
# return self.__target
# forwardmethods(MyClass, TargetClass, '__target', ['dangerous1', 'dangerous2'])
# # ...or...
# forwardmethods(MyClass, TargetClass, MyClass.findtarget,
# ['dangerous1', 'dangerous2'])
# In both cases, all TargetClass methods will be forwarded from
# MyClass except for dangerous1, dangerous2, special methods like
# __str__, and pre-existing methods like findtarget.
# Allow an attribute name (String) or a function to determine the instance
if type(toPart) != types.StringType:
# check that it is something like a function
if callable(toPart):
# If a method is passed, use the function within it
if hasattr(toPart, 'im_func'):
toPart = toPart.im_func
# After this is set up, forwarders in this class will use
# the forwarding function. The forwarding function name is
# guaranteed to be unique, so that it can't be hidden by subclasses
forwardName = '__fwdfunc__' + __unique()
fromClass.__dict__[forwardName] = toPart
# It's not a valid type
else:
raise TypeError, 'toPart must be attribute name, function or method'
# get the full set of candidate methods
dict = {}
__methodDict(toClass, dict)
# discard special methods
for ex in dict.keys():
if ex[:1] == '_' or ex[-1:] == '_':
del dict[ex]
# discard dangerous methods supplied by the caller
for ex in exclude:
if dict.has_key(ex):
del dict[ex]
# discard methods already defined in fromClass
for ex in __methods(fromClass):
if dict.has_key(ex):
del dict[ex]
for method, func in dict.items():
d = {'method': method, 'func': func}
if type(toPart) == types.StringType:
execString = \
__stringBody % {'method' : method, 'attribute' : toPart}
else:
execString = \
__funcBody % {'forwardFunc' : forwardName, 'method' : method}
exec execString in d
# this creates a method
fromClass.__dict__[method] = d[method]
#=============================================================================
def setgeometryanddeiconify(window, geom):
# To avoid flashes on X and to position the window correctly on NT
# (caused by Tk bugs).
if os.name == 'nt' or \
(os.name == 'posix' and sys.platform[:6] == 'cygwin'):
# Require overrideredirect trick to stop window frame
# appearing momentarily.
redirect = window.overrideredirect()
if not redirect:
window.overrideredirect(1)
window.deiconify()
if geom is not None:
window.geometry(geom)
# Call update_idletasks to ensure NT moves the window to the
# correct position it is raised.
window.update_idletasks()
window.tkraise()
if not redirect:
window.overrideredirect(0)
else:
if geom is not None:
window.geometry(geom)
# Problem!? Which way around should the following two calls
# go? If deiconify() is called first then I get complaints
# from people using the enlightenment or sawfish window
# managers that when a dialog is activated it takes about 2
# seconds for the contents of the window to appear. But if
# tkraise() is called first then I get complaints from people
# using the twm window manager that when a dialog is activated
# it appears in the top right corner of the screen and also
# takes about 2 seconds to appear.
#window.tkraise()
# Call update_idletasks to ensure certain window managers (eg:
# enlightenment and sawfish) do not cause Tk to delay for
# about two seconds before displaying window.
#window.update_idletasks()
#window.deiconify()
window.deiconify()
if window.overrideredirect():
# The window is not under the control of the window manager
# and so we need to raise it ourselves.
window.tkraise()
#=============================================================================
class MegaArchetype:
# Megawidget abstract root class.
# This class provides methods which are inherited by classes
# implementing useful bases (this class doesn't provide a
# container widget inside which the megawidget can be built).
def __init__(self, parent = None, hullClass = None):
# Mapping from each megawidget option to a list of information
# about the option
# - default value
# - current value
# - function to call when the option is initialised in the
# call to initialiseoptions() in the constructor or
# modified via configure(). If this is INITOPT, the
# option is an initialisation option (an option that can
# be set by the call to the constructor but can not be
# used with configure).
# This mapping is not initialised here, but in the call to
# defineoptions() which precedes construction of this base class.
#
# self._optionInfo = {}
# Mapping from each component name to a tuple of information
# about the component.
# - component widget instance
# - configure function of widget instance
# - the class of the widget (Frame, EntryField, etc)
# - cget function of widget instance
# - the name of the component group of this component, if any
self.__componentInfo = {}
# Mapping from alias names to the names of components or
# sub-components.
self.__componentAliases = {}
# Contains information about the keywords provided to the
# constructor. It is a mapping from the keyword to a tuple
# containing:
# - value of keyword
# - a boolean indicating if the keyword has been used.
# A keyword is used if, during the construction of a megawidget,
# - it is defined in a call to defineoptions() or addoptions(), or
# - it references, by name, a component of the megawidget, or
# - it references, by group, at least one component
# At the end of megawidget construction, a call is made to
# initialiseoptions() which reports an error if there are
# unused options given to the constructor.
#
# After megawidget construction, the dictionary contains
# keywords which refer to a dynamic component group, so that
# these components can be created after megawidget
# construction and still use the group options given to the
# constructor.
#
# self._constructorKeywords = {}
# List of dynamic component groups. If a group is included in
# this list, then it not an error if a keyword argument for
# the group is given to the constructor or to configure(), but
# no components with this group have been created.
# self._dynamicGroups = ()
if hullClass is None:
self._hull = None
else:
if parent is None:
parent = Tkinter._default_root
# Create the hull.
self._hull = self.createcomponent('hull',
(), None,
hullClass, (parent,))
_hullToMegaWidget[self._hull] = self
if _useTkOptionDb:
# Now that a widget has been created, query the Tk
# option database to get the default values for the
# options which have not been set in the call to the
# constructor. This assumes that defineoptions() is
# called before the __init__().
option_get = self.option_get
_VALUE = _OPT_VALUE
_DEFAULT = _OPT_DEFAULT
for name, info in self._optionInfo.items():
value = info[_VALUE]
if value is _DEFAULT_OPTION_VALUE:
resourceClass = string.upper(name[0]) + name[1:]
value = option_get(name, resourceClass)
if value != '':
try:
# Convert the string to int/float/tuple, etc
value = eval(value, {'__builtins__': {}})
except:
pass
info[_VALUE] = value
else:
info[_VALUE] = info[_DEFAULT]
def destroy(self):
# Clean up optionInfo in case it contains circular references
# in the function field, such as self._settitle in class
# MegaToplevel.
self._optionInfo = {}
if self._hull is not None:
del _hullToMegaWidget[self._hull]
self._hull.destroy()
#======================================================================
# Methods used (mainly) during the construction of the megawidget.
def defineoptions(self, keywords, optionDefs, dynamicGroups = ()):
# Create options, providing the default value and the method
# to call when the value is changed. If any option created by
# base classes has the same name as one in <optionDefs>, the
# base class's value and function will be overriden.
# This should be called before the constructor of the base
# class, so that default values defined in the derived class
# override those in the base class.
if not hasattr(self, '_constructorKeywords'):
# First time defineoptions has been called.
tmp = {}
for option, value in keywords.items():
tmp[option] = [value, 0]
self._constructorKeywords = tmp
self._optionInfo = {}
self._initialiseoptions_counter = 0
self._initialiseoptions_counter = self._initialiseoptions_counter + 1
if not hasattr(self, '_dynamicGroups'):
self._dynamicGroups = ()
self._dynamicGroups = self._dynamicGroups + tuple(dynamicGroups)
self.addoptions(optionDefs)
def addoptions(self, optionDefs):
# Add additional options, providing the default value and the
# method to call when the value is changed. See
# "defineoptions" for more details
# optimisations:
optionInfo = self._optionInfo
optionInfo_has_key = optionInfo.has_key
keywords = self._constructorKeywords
keywords_has_key = keywords.has_key
FUNCTION = _OPT_FUNCTION
for name, default, function in optionDefs:
if '_' not in name:
# The option will already exist if it has been defined
# in a derived class. In this case, do not override the
# default value of the option or the callback function
# if it is not None.
if not optionInfo_has_key(name):
if keywords_has_key(name):
value = keywords[name][0]
optionInfo[name] = [default, value, function]
del keywords[name]
else:
if _useTkOptionDb:
optionInfo[name] = \
[default, _DEFAULT_OPTION_VALUE, function]
else:
optionInfo[name] = [default, default, function]
elif optionInfo[name][FUNCTION] is None:
optionInfo[name][FUNCTION] = function
else:
# This option is of the form "component_option". If this is
# not already defined in self._constructorKeywords add it.
# This allows a derived class to override the default value
# of an option of a component of a base class.
if not keywords_has_key(name):
keywords[name] = [default, 0]
def createcomponent(self, componentName, componentAliases,
componentGroup, widgetClass, *widgetArgs, **kw):
# Create a component (during construction or later).
if self.__componentInfo.has_key(componentName):
raise ValueError, 'Component "%s" already exists' % componentName
if '_' in componentName:
raise ValueError, \
'Component name "%s" must not contain "_"' % componentName
if hasattr(self, '_constructorKeywords'):
keywords = self._constructorKeywords
else:
keywords = {}
for alias, component in componentAliases:
# Create aliases to the component and its sub-components.
index = string.find(component, '_')
if index < 0:
self.__componentAliases[alias] = (component, None)
else:
mainComponent = component[:index]
subComponent = component[(index + 1):]
self.__componentAliases[alias] = (mainComponent, subComponent)
# Remove aliases from the constructor keyword arguments by
# replacing any keyword arguments that begin with *alias*
# with corresponding keys beginning with *component*.
alias = alias + '_'
aliasLen = len(alias)
for option in keywords.keys():
if len(option) > aliasLen and option[:aliasLen] == alias:
newkey = component + '_' + option[aliasLen:]
keywords[newkey] = keywords[option]
del keywords[option]
componentPrefix = componentName + '_'
nameLen = len(componentPrefix)
for option in keywords.keys():
if len(option) > nameLen and option[:nameLen] == componentPrefix:
# The keyword argument refers to this component, so add
# this to the options to use when constructing the widget.
kw[option[nameLen:]] = keywords[option][0]
del keywords[option]
else:
# Check if this keyword argument refers to the group
# of this component. If so, add this to the options
# to use when constructing the widget. Mark the
# keyword argument as being used, but do not remove it
# since it may be required when creating another
# component.
index = string.find(option, '_')
if index >= 0 and componentGroup == option[:index]:
rest = option[(index + 1):]
kw[rest] = keywords[option][0]
keywords[option][1] = 1
if kw.has_key('pyclass'):
widgetClass = kw['pyclass']
del kw['pyclass']
if widgetClass is None:
return None
if len(widgetArgs) == 1 and type(widgetArgs[0]) == types.TupleType:
# Arguments to the constructor can be specified as either
# multiple trailing arguments to createcomponent() or as a
# single tuple argument.
widgetArgs = widgetArgs[0]
widget = apply(widgetClass, widgetArgs, kw)
componentClass = widget.__class__.__name__
self.__componentInfo[componentName] = (widget, widget.configure,
componentClass, widget.cget, componentGroup)
return widget
def destroycomponent(self, name):
# Remove a megawidget component.
# This command is for use by megawidget designers to destroy a
# megawidget component.
self.__componentInfo[name][0].destroy()
del self.__componentInfo[name]
def createlabel(self, parent, childCols = 1, childRows = 1):
labelpos = self['labelpos']
labelmargin = self['labelmargin']
if labelpos is None:
return
label = self.createcomponent('label',
(), None,
Tkinter.Label, (parent,))
if labelpos[0] in 'ns':
# vertical layout
if labelpos[0] == 'n':
row = 0
margin = 1
else:
row = childRows + 3
margin = row - 1
label.grid(column=2, row=row, columnspan=childCols, sticky=labelpos)
parent.grid_rowconfigure(margin, minsize=labelmargin)
else:
# horizontal layout
if labelpos[0] == 'w':
col = 0
margin = 1
else:
col = childCols + 3
margin = col - 1
label.grid(column=col, row=2, rowspan=childRows, sticky=labelpos)
parent.grid_columnconfigure(margin, minsize=labelmargin)
def initialiseoptions(self, dummy = None):
self._initialiseoptions_counter = self._initialiseoptions_counter - 1
if self._initialiseoptions_counter == 0:
unusedOptions = []
keywords = self._constructorKeywords
for name in keywords.keys():
used = keywords[name][1]
if not used:
# This keyword argument has not been used. If it
# does not refer to a dynamic group, mark it as
# unused.
index = string.find(name, '_')
if index < 0 or name[:index] not in self._dynamicGroups:
unusedOptions.append(name)
if len(unusedOptions) > 0:
if len(unusedOptions) == 1:
text = 'Unknown option "'
else:
text = 'Unknown options "'
raise KeyError, text + string.join(unusedOptions, ', ') + \
'" for ' + self.__class__.__name__
# Call the configuration callback function for every option.
FUNCTION = _OPT_FUNCTION
for info in self._optionInfo.values():
func = info[FUNCTION]
if func is not None and func is not INITOPT:
func()
#======================================================================
# Method used to configure the megawidget.
def configure(self, option=None, **kw):
# Query or configure the megawidget options.
#
# If not empty, *kw* is a dictionary giving new
# values for some of the options of this megawidget or its
# components. For options defined for this megawidget, set
# the value of the option to the new value and call the
# configuration callback function, if any. For options of the
# form <component>_<option>, where <component> is a component
# of this megawidget, call the configure method of the
# component giving it the new value of the option. The
# <component> part may be an alias or a component group name.
#
# If *option* is None, return all megawidget configuration
# options and settings. Options are returned as standard 5
# element tuples
#
# If *option* is a string, return the 5 element tuple for the
# given configuration option.
# First, deal with the option queries.
if len(kw) == 0:
# This configure call is querying the values of one or all options.
# Return 5-tuples:
# (optionName, resourceName, resourceClass, default, value)
if option is None:
rtn = {}
for option, config in self._optionInfo.items():
resourceClass = string.upper(option[0]) + option[1:]
rtn[option] = (option, option, resourceClass,
config[_OPT_DEFAULT], config[_OPT_VALUE])
return rtn
else:
config = self._optionInfo[option]
resourceClass = string.upper(option[0]) + option[1:]
return (option, option, resourceClass, config[_OPT_DEFAULT],
config[_OPT_VALUE])
# optimisations:
optionInfo = self._optionInfo
optionInfo_has_key = optionInfo.has_key
componentInfo = self.__componentInfo
componentInfo_has_key = componentInfo.has_key
componentAliases = self.__componentAliases
componentAliases_has_key = componentAliases.has_key
VALUE = _OPT_VALUE
FUNCTION = _OPT_FUNCTION
# This will contain a list of options in *kw* which
# are known to this megawidget.
directOptions = []
# This will contain information about the options in
# *kw* of the form <component>_<option>, where
# <component> is a component of this megawidget. It is a
# dictionary whose keys are the configure method of each
# component and whose values are a dictionary of options and
# values for the component.
indirectOptions = {}
indirectOptions_has_key = indirectOptions.has_key
for option, value in kw.items():
if optionInfo_has_key(option):
# This is one of the options of this megawidget.
# Make sure it is not an initialisation option.
if optionInfo[option][FUNCTION] is INITOPT:
raise KeyError, \
'Cannot configure initialisation option "' \
+ option + '" for ' + self.__class__.__name__
optionInfo[option][VALUE] = value
directOptions.append(option)
else:
index = string.find(option, '_')
if index >= 0:
# This option may be of the form <component>_<option>.
component = option[:index]
componentOption = option[(index + 1):]
# Expand component alias
if componentAliases_has_key(component):
component, subComponent = componentAliases[component]
if subComponent is not None:
componentOption = subComponent + '_' \
+ componentOption
# Expand option string to write on error
option = component + '_' + componentOption
if componentInfo_has_key(component):
# Configure the named component
componentConfigFuncs = [componentInfo[component][1]]
else:
# Check if this is a group name and configure all
# components in the group.
componentConfigFuncs = []
for info in componentInfo.values():
if info[4] == component:
componentConfigFuncs.append(info[1])
if len(componentConfigFuncs) == 0 and \
component not in self._dynamicGroups:
raise KeyError, 'Unknown option "' + option + \
'" for ' + self.__class__.__name__
# Add the configure method(s) (may be more than
# one if this is configuring a component group)
# and option/value to dictionary.
for componentConfigFunc in componentConfigFuncs:
if not indirectOptions_has_key(componentConfigFunc):
indirectOptions[componentConfigFunc] = {}
indirectOptions[componentConfigFunc][componentOption] \
= value
else:
raise KeyError, 'Unknown option "' + option + \
'" for ' + self.__class__.__name__
# Call the configure methods for any components.
map(apply, indirectOptions.keys(),
((),) * len(indirectOptions), indirectOptions.values())
# Call the configuration callback function for each option.
for option in directOptions:
info = optionInfo[option]
func = info[_OPT_FUNCTION]
if func is not None:
func()
def __setitem__(self, key, value):
apply(self.configure, (), {key: value})
#======================================================================
# Methods used to query the megawidget.
def component(self, name):
# Return a component widget of the megawidget given the
# component's name
# This allows the user of a megawidget to access and configure
# widget components directly.
# Find the main component and any subcomponents
index = string.find(name, '_')
if index < 0:
component = name
remainingComponents = None
else:
component = name[:index]
remainingComponents = name[(index + 1):]
# Expand component alias
if self.__componentAliases.has_key(component):
component, subComponent = self.__componentAliases[component]
if subComponent is not None:
if remainingComponents is None:
remainingComponents = subComponent
else:
remainingComponents = subComponent + '_' \
+ remainingComponents
widget = self.__componentInfo[component][0]
if remainingComponents is None:
return widget
else:
return widget.component(remainingComponents)
def interior(self):
return self._hull
def hulldestroyed(self):
return not _hullToMegaWidget.has_key(self._hull)
def __str__(self):
return str(self._hull)
def cget(self, option):
# Get current configuration setting.
# Return the value of an option, for example myWidget['font'].
if self._optionInfo.has_key(option):
return self._optionInfo[option][_OPT_VALUE]
else:
index = string.find(option, '_')
if index >= 0:
component = option[:index]
componentOption = option[(index + 1):]
# Expand component alias
if self.__componentAliases.has_key(component):
component, subComponent = self.__componentAliases[component]
if subComponent is not None:
componentOption = subComponent + '_' + componentOption
# Expand option string to write on error
option = component + '_' + componentOption
if self.__componentInfo.has_key(component):
# Call cget on the component.
componentCget = self.__componentInfo[component][3]
return componentCget(componentOption)
else:
# If this is a group name, call cget for one of
# the components in the group.
for info in self.__componentInfo.values():
if info[4] == component:
componentCget = info[3]
return componentCget(componentOption)
raise KeyError, 'Unknown option "' + option + \
'" for ' + self.__class__.__name__
__getitem__ = cget
def isinitoption(self, option):
return self._optionInfo[option][_OPT_FUNCTION] is INITOPT
def options(self):
options = []
if hasattr(self, '_optionInfo'):
for option, info in self._optionInfo.items():
isinit = info[_OPT_FUNCTION] is INITOPT
default = info[_OPT_DEFAULT]
options.append((option, default, isinit))
options.sort()
return options
def components(self):
# Return a list of all components.
# This list includes the 'hull' component and all widget subcomponents
names = self.__componentInfo.keys()
names.sort()
return names
def componentaliases(self):
# Return a list of all component aliases.
componentAliases = self.__componentAliases
names = componentAliases.keys()
names.sort()
rtn = []
for alias in names:
(mainComponent, subComponent) = componentAliases[alias]
if subComponent is None:
rtn.append((alias, mainComponent))
else:
rtn.append((alias, mainComponent + '_' + subComponent))
return rtn
def componentgroup(self, name):
return self.__componentInfo[name][4]
#=============================================================================
# The grab functions are mainly called by the activate() and
# deactivate() methods.
#
# Use pushgrab() to add a new window to the grab stack. This
# releases the grab by the window currently on top of the stack (if
# there is one) and gives the grab and focus to the new widget.
#
# To remove the grab from the window on top of the grab stack, call
# popgrab().
#
# Use releasegrabs() to release the grab and clear the grab stack.
def pushgrab(grabWindow, globalMode, deactivateFunction):
prevFocus = grabWindow.tk.call('focus')
grabInfo = {
'grabWindow' : grabWindow,
'globalMode' : globalMode,
'previousFocus' : prevFocus,
'deactivateFunction' : deactivateFunction,
}
_grabStack.append(grabInfo)
_grabtop()
grabWindow.focus_set()
def popgrab(window):
# Return the grab to the next window in the grab stack, if any.
# If this window is not at the top of the grab stack, then it has
# just been deleted by the window manager or deactivated by a
# timer. Call the deactivate method for the modal dialog above
# this one on the stack.
if _grabStack[-1]['grabWindow'] != window:
for index in range(len(_grabStack)):
if _grabStack[index]['grabWindow'] == window:
_grabStack[index + 1]['deactivateFunction']()
break
grabInfo = _grabStack[-1]
del _grabStack[-1]
topWidget = grabInfo['grabWindow']
prevFocus = grabInfo['previousFocus']
globalMode = grabInfo['globalMode']
if globalMode != 'nograb':
topWidget.grab_release()
if len(_grabStack) > 0:
_grabtop()
if prevFocus != '':
try:
topWidget.tk.call('focus', prevFocus)
except Tkinter.TclError:
# Previous focus widget has been deleted. Set focus
# to root window.
Tkinter._default_root.focus_set()
else:
# Make sure that focus does not remain on the released widget.
if len(_grabStack) > 0:
topWidget = _grabStack[-1]['grabWindow']
topWidget.focus_set()
else:
Tkinter._default_root.focus_set()
def grabstacktopwindow():
if len(_grabStack) == 0:
return None
else:
return _grabStack[-1]['grabWindow']
def releasegrabs():
# Release grab and clear the grab stack.
current = Tkinter._default_root.grab_current()
if current is not None:
current.grab_release()
_grabStack[:] = []
def _grabtop():
grabInfo = _grabStack[-1]
topWidget = grabInfo['grabWindow']
globalMode = grabInfo['globalMode']
if globalMode == 'nograb':
return
while 1:
try:
if globalMode:
topWidget.grab_set_global()
else:
topWidget.grab_set()
break
except Tkinter.TclError:
# Another application has grab. Keep trying until
# grab can succeed.
topWidget.after(100)
#=============================================================================
class MegaToplevel(MegaArchetype):
def __init__(self, parent = None, **kw):
# Define the options for this megawidget.
optiondefs = (
('activatecommand', None, None),
('deactivatecommand', None, None),
('master', None, None),
('title', None, self._settitle),
('hull_class', self.__class__.__name__, None),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaArchetype.__init__(self, parent, Tkinter.Toplevel)
# Initialise instance.
# Set WM_DELETE_WINDOW protocol, deleting any old callback, so
# memory does not leak.
if hasattr(self._hull, '_Pmw_WM_DELETE_name'):
self._hull.tk.deletecommand(self._hull._Pmw_WM_DELETE_name)
self._hull._Pmw_WM_DELETE_name = \
self.register(self._userDeleteWindow, needcleanup = 0)
self.protocol('WM_DELETE_WINDOW', self._hull._Pmw_WM_DELETE_name)
# Initialise instance variables.
self._firstShowing = 1
# Used by show() to ensure window retains previous position on screen.
# The IntVar() variable to wait on during a modal dialog.
self._wait = None
self._active = 0
self._userDeleteFunc = self.destroy
self._userModalDeleteFunc = self.deactivate
# Check keywords and initialise options.
self.initialiseoptions()
def _settitle(self):
title = self['title']
if title is not None:
self.title(title)
def userdeletefunc(self, func=None):
if func:
self._userDeleteFunc = func
else:
return self._userDeleteFunc
def usermodaldeletefunc(self, func=None):
if func:
self._userModalDeleteFunc = func
else:
return self._userModalDeleteFunc
def _userDeleteWindow(self):
if self.active():
self._userModalDeleteFunc()
else:
self._userDeleteFunc()
def destroy(self):
# Allow this to be called more than once.
if _hullToMegaWidget.has_key(self._hull):
self.deactivate()
# Remove circular references, so that object can get cleaned up.
del self._userDeleteFunc
del self._userModalDeleteFunc
MegaArchetype.destroy(self)
def show(self, master = None):
if self.state() != 'normal':
if self._firstShowing:
# Just let the window manager determine the window
# position for the first time.
geom = None
else:
# Position the window at the same place it was last time.
geom = self._sameposition()
setgeometryanddeiconify(self, geom)
if self._firstShowing:
self._firstShowing = 0
else:
if self.transient() == '':
self.tkraise()
# Do this last, otherwise get flashing on NT:
if master is not None:
if master == 'parent':
parent = self.winfo_parent()
# winfo_parent() should return the parent widget, but the
# the current version of Tkinter returns a string.
if type(parent) == types.StringType:
parent = self._hull._nametowidget(parent)
master = parent.winfo_toplevel()
self.transient(master)
self.focus()
def _centreonscreen(self):
# Centre the window on the screen. (Actually halfway across
# and one third down.)
parent = self.winfo_parent()
if type(parent) == types.StringType:
parent = self._hull._nametowidget(parent)
# Find size of window.
self.update_idletasks()
width = self.winfo_width()
height = self.winfo_height()
if width == 1 and height == 1:
# If the window has not yet been displayed, its size is
# reported as 1x1, so use requested size.
width = self.winfo_reqwidth()
height = self.winfo_reqheight()
# Place in centre of screen:
x = (self.winfo_screenwidth() - width) / 2 - parent.winfo_vrootx()
y = (self.winfo_screenheight() - height) / 3 - parent.winfo_vrooty()
if x < 0:
x = 0
if y < 0:
y = 0
return '+%d+%d' % (x, y)
def _sameposition(self):
# Position the window at the same place it was last time.
geometry = self.geometry()
index = string.find(geometry, '+')
if index >= 0:
return geometry[index:]
else:
return None
def activate(self, globalMode = 0, geometry = 'centerscreenfirst'):
if self._active:
raise ValueError, 'Window is already active'
if self.state() == 'normal':
self.withdraw()
self._active = 1
showbusycursor()
if self._wait is None:
self._wait = Tkinter.IntVar()
self._wait.set(0)
if geometry == 'centerscreenalways':
geom = self._centreonscreen()
elif geometry == 'centerscreenfirst':
if self._firstShowing:
# Centre the window the first time it is displayed.
geom = self._centreonscreen()
else:
# Position the window at the same place it was last time.
geom = self._sameposition()
elif geometry[:5] == 'first':
if self._firstShowing:
geom = geometry[5:]
else:
# Position the window at the same place it was last time.
geom = self._sameposition()
else:
geom = geometry
self._firstShowing = 0
setgeometryanddeiconify(self, geom)
# Do this last, otherwise get flashing on NT:
master = self['master']
if master is not None:
if master == 'parent':
parent = self.winfo_parent()
# winfo_parent() should return the parent widget, but the
# the current version of Tkinter returns a string.
if type(parent) == types.StringType:
parent = self._hull._nametowidget(parent)
master = parent.winfo_toplevel()
self.transient(master)
pushgrab(self._hull, globalMode, self.deactivate)
command = self['activatecommand']
if callable(command):
command()
self.wait_variable(self._wait)
return self._result
def deactivate(self, result=None):
if not self._active:
return
self._active = 0
# Restore the focus before withdrawing the window, since
# otherwise the window manager may take the focus away so we
# can't redirect it. Also, return the grab to the next active
# window in the stack, if any.
popgrab(self._hull)
command = self['deactivatecommand']
if callable(command):
command()
self.withdraw()
hidebusycursor(forceFocusRestore = 1)
self._result = result
self._wait.set(1)
def active(self):
return self._active
forwardmethods(MegaToplevel, Tkinter.Toplevel, '_hull')
#=============================================================================
class MegaWidget(MegaArchetype):
def __init__(self, parent = None, **kw):
# Define the options for this megawidget.
optiondefs = (
('hull_class', self.__class__.__name__, None),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaArchetype.__init__(self, parent, Tkinter.Frame)
# Check keywords and initialise options.
self.initialiseoptions()
forwardmethods(MegaWidget, Tkinter.Frame, '_hull')
#=============================================================================
# Public functions
#-----------------
_traceTk = 0
def tracetk(root = None, on = 1, withStackTrace = 0, file=None):
global _withStackTrace
global _traceTkFile
global _traceTk
if root is None:
root = Tkinter._default_root
_withStackTrace = withStackTrace
_traceTk = on
if on:
if hasattr(root.tk, '__class__'):
# Tracing already on
return
if file is None:
_traceTkFile = sys.stderr
else:
_traceTkFile = file
tk = _TraceTk(root.tk)
else:
if not hasattr(root.tk, '__class__'):
# Tracing already off
return
tk = root.tk.getTclInterp()
_setTkInterps(root, tk)
def showbusycursor():
_addRootToToplevelBusyInfo()
root = Tkinter._default_root
busyInfo = {
'newBusyWindows' : [],
'previousFocus' : None,
'busyFocus' : None,
}
_busyStack.append(busyInfo)
if _disableKeyboardWhileBusy:
# Remember the focus as it is now, before it is changed.
busyInfo['previousFocus'] = root.tk.call('focus')
if not _havebltbusy(root):
# No busy command, so don't call busy hold on any windows.
return
for (window, winInfo) in _toplevelBusyInfo.items():
if (window.state() != 'withdrawn' and not winInfo['isBusy']
and not winInfo['excludeFromBusy']):
busyInfo['newBusyWindows'].append(window)
winInfo['isBusy'] = 1
_busy_hold(window, winInfo['busyCursorName'])
# Make sure that no events for the busy window get
# through to Tkinter, otherwise it will crash in
# _nametowidget with a 'KeyError: _Busy' if there is
# a binding on the toplevel window.
window.tk.call('bindtags', winInfo['busyWindow'], 'Pmw_Dummy_Tag')
if _disableKeyboardWhileBusy:
# Remember previous focus widget for this toplevel window
# and set focus to the busy window, which will ignore all
# keyboard events.
winInfo['windowFocus'] = \
window.tk.call('focus', '-lastfor', window._w)
window.tk.call('focus', winInfo['busyWindow'])
busyInfo['busyFocus'] = winInfo['busyWindow']
if len(busyInfo['newBusyWindows']) > 0:
if os.name == 'nt':
# NT needs an "update" before it will change the cursor.
window.update()
else:
window.update_idletasks()
def hidebusycursor(forceFocusRestore = 0):
# Remember the focus as it is now, before it is changed.
root = Tkinter._default_root
if _disableKeyboardWhileBusy:
currentFocus = root.tk.call('focus')
# Pop the busy info off the stack.
busyInfo = _busyStack[-1]
del _busyStack[-1]
for window in busyInfo['newBusyWindows']:
# If this window has not been deleted, release the busy cursor.
if _toplevelBusyInfo.has_key(window):
winInfo = _toplevelBusyInfo[window]
winInfo['isBusy'] = 0
_busy_release(window)
if _disableKeyboardWhileBusy:
# Restore previous focus window for this toplevel window,
# but only if is still set to the busy window (it may have
# been changed).
windowFocusNow = window.tk.call('focus', '-lastfor', window._w)
if windowFocusNow == winInfo['busyWindow']:
try:
window.tk.call('focus', winInfo['windowFocus'])
except Tkinter.TclError:
# Previous focus widget has been deleted. Set focus
# to toplevel window instead (can't leave focus on
# busy window).
window.focus_set()
if _disableKeyboardWhileBusy:
# Restore the focus, depending on whether the focus had changed
# between the calls to showbusycursor and hidebusycursor.
if forceFocusRestore or busyInfo['busyFocus'] == currentFocus:
# The focus had not changed, so restore it to as it was before
# the call to showbusycursor,
previousFocus = busyInfo['previousFocus']
if previousFocus is not None:
try:
root.tk.call('focus', previousFocus)
except Tkinter.TclError:
# Previous focus widget has been deleted; forget it.
pass
else:
# The focus had changed, so restore it to what it had been
# changed to before the call to hidebusycursor.
root.tk.call('focus', currentFocus)
def clearbusycursor():
while len(_busyStack) > 0:
hidebusycursor()
def setbusycursorattributes(window, **kw):
_addRootToToplevelBusyInfo()
for name, value in kw.items():
if name == 'exclude':
_toplevelBusyInfo[window]['excludeFromBusy'] = value
elif name == 'cursorName':
_toplevelBusyInfo[window]['busyCursorName'] = value
else:
raise KeyError, 'Unknown busycursor attribute "' + name + '"'
def _addRootToToplevelBusyInfo():
# Include the Tk root window in the list of toplevels. This must
# not be called before Tkinter has had a chance to be initialised by
# the application.
root = Tkinter._default_root
if root == None:
root = Tkinter.Tk()
if not _toplevelBusyInfo.has_key(root):
_addToplevelBusyInfo(root)
def busycallback(command, updateFunction = None):
if not callable(command):
raise ValueError, \
'cannot register non-command busy callback %s %s' % \
(repr(command), type(command))
wrapper = _BusyWrapper(command, updateFunction)
return wrapper.callback
_errorReportFile = None
_errorWindow = None
def reporterrorstofile(file = None):
global _errorReportFile
_errorReportFile = file
def displayerror(text):
global _errorWindow
if _errorReportFile is not None:
_errorReportFile.write(text + '\n')
else:
# Print error on standard error as well as to error window.
# Useful if error window fails to be displayed, for example
# when exception is triggered in a <Destroy> binding for root
# window.
sys.stderr.write(text + '\n')
if _errorWindow is None:
# The error window has not yet been created.
_errorWindow = _ErrorWindow()
_errorWindow.showerror(text)
_root = None
_disableKeyboardWhileBusy = 1
def initialise(
root = None,
size = None,
fontScheme = None,
useTkOptionDb = 0,
noBltBusy = 0,
disableKeyboardWhileBusy = None,
):
# Remember if show/hidebusycursor should ignore keyboard events.
global _disableKeyboardWhileBusy
if disableKeyboardWhileBusy is not None:
_disableKeyboardWhileBusy = disableKeyboardWhileBusy
# Do not use blt busy command if noBltBusy is set. Otherwise,
# use blt busy if it is available.
global _haveBltBusy
if noBltBusy:
_haveBltBusy = 0
# Save flag specifying whether the Tk option database should be
# queried when setting megawidget option default values.
global _useTkOptionDb
_useTkOptionDb = useTkOptionDb
# If we haven't been given a root window, use the default or
# create one.
if root is None:
if Tkinter._default_root is None:
root = Tkinter.Tk()
else:
root = Tkinter._default_root
# If this call is initialising a different Tk interpreter than the
# last call, then re-initialise all global variables. Assume the
# last interpreter has been destroyed - ie: Pmw does not (yet)
# support multiple simultaneous interpreters.
global _root
if _root is not None and _root != root:
global _busyStack
global _errorWindow
global _grabStack
global _hullToMegaWidget
global _toplevelBusyInfo
_busyStack = []
_errorWindow = None
_grabStack = []
_hullToMegaWidget = {}
_toplevelBusyInfo = {}
_root = root
# Trap Tkinter Toplevel constructors so that a list of Toplevels
# can be maintained.
Tkinter.Toplevel.title = __TkinterToplevelTitle
# Trap Tkinter widget destruction so that megawidgets can be
# destroyed when their hull widget is destoyed and the list of
# Toplevels can be pruned.
Tkinter.Toplevel.destroy = __TkinterToplevelDestroy
Tkinter.Widget.destroy = __TkinterWidgetDestroy
# Modify Tkinter's CallWrapper class to improve the display of
# errors which occur in callbacks.
Tkinter.CallWrapper = __TkinterCallWrapper
# Make sure we get to know when the window manager deletes the
# root window. Only do this if the protocol has not yet been set.
# This is required if there is a modal dialog displayed and the
# window manager deletes the root window. Otherwise the
# application will not exit, even though there are no windows.
if root.protocol('WM_DELETE_WINDOW') == '':
root.protocol('WM_DELETE_WINDOW', root.destroy)
# Set the base font size for the application and set the
# Tk option database font resources.
_font_initialise(root, size, fontScheme)
return root
def alignlabels(widgets, sticky = None):
if len(widgets) == 0:
return
widgets[0].update_idletasks()
# Determine the size of the maximum length label string.
maxLabelWidth = 0
for iwid in widgets:
labelWidth = iwid.grid_bbox(0, 1)[2]
if labelWidth > maxLabelWidth:
maxLabelWidth = labelWidth
# Adjust the margins for the labels such that the child sites and
# labels line up.
for iwid in widgets:
if sticky is not None:
iwid.component('label').grid(sticky=sticky)
iwid.grid_columnconfigure(0, minsize = maxLabelWidth)
#=============================================================================
# Private routines
#-----------------
_callToTkReturned = 1
_recursionCounter = 1
class _TraceTk:
def __init__(self, tclInterp):
self.tclInterp = tclInterp
def getTclInterp(self):
return self.tclInterp
# Calling from python into Tk.
def call(self, *args, **kw):
global _callToTkReturned
global _recursionCounter
_callToTkReturned = 0
if len(args) == 1 and type(args[0]) == types.TupleType:
argStr = str(args[0])
else:
argStr = str(args)
_traceTkFile.write('CALL TK> %d:%s%s' %
(_recursionCounter, ' ' * _recursionCounter, argStr))
_recursionCounter = _recursionCounter + 1
try:
result = apply(self.tclInterp.call, args, kw)
except Tkinter.TclError, errorString:
_callToTkReturned = 1
_recursionCounter = _recursionCounter - 1
_traceTkFile.write('\nTK ERROR> %d:%s-> %s\n' %
(_recursionCounter, ' ' * _recursionCounter,
repr(errorString)))
if _withStackTrace:
_traceTkFile.write('CALL TK> stack:\n')
traceback.print_stack()
raise Tkinter.TclError, errorString
_recursionCounter = _recursionCounter - 1
if _callToTkReturned:
_traceTkFile.write('CALL RTN> %d:%s-> %s' %
(_recursionCounter, ' ' * _recursionCounter, repr(result)))
else:
_callToTkReturned = 1
if result:
_traceTkFile.write(' -> %s' % repr(result))
_traceTkFile.write('\n')
if _withStackTrace:
_traceTkFile.write('CALL TK> stack:\n')
traceback.print_stack()
_traceTkFile.flush()
return result
def __getattr__(self, key):
return getattr(self.tclInterp, key)
def _setTkInterps(window, tk):
window.tk = tk
for child in window.children.values():
_setTkInterps(child, tk)
#=============================================================================
# Functions to display a busy cursor. Keep a list of all toplevels
# and display the busy cursor over them. The list will contain the Tk
# root toplevel window as well as all other toplevel windows.
# Also keep a list of the widget which last had focus for each
# toplevel.
# Map from toplevel windows to
# {'isBusy', 'windowFocus', 'busyWindow',
# 'excludeFromBusy', 'busyCursorName'}
_toplevelBusyInfo = {}
# Pmw needs to know all toplevel windows, so that it can call blt busy
# on them. This is a hack so we get notified when a Tk topevel is
# created. Ideally, the __init__ 'method' should be overridden, but
# it is a 'read-only special attribute'. Luckily, title() is always
# called from the Tkinter Toplevel constructor.
def _addToplevelBusyInfo(window):
if window._w == '.':
busyWindow = '._Busy'
else:
busyWindow = window._w + '._Busy'
_toplevelBusyInfo[window] = {
'isBusy' : 0,
'windowFocus' : None,
'busyWindow' : busyWindow,
'excludeFromBusy' : 0,
'busyCursorName' : None,
}
def __TkinterToplevelTitle(self, *args):
# If this is being called from the constructor, include this
# Toplevel in the list of toplevels and set the initial
# WM_DELETE_WINDOW protocol to destroy() so that we get to know
# about it.
if not _toplevelBusyInfo.has_key(self):
_addToplevelBusyInfo(self)
self._Pmw_WM_DELETE_name = self.register(self.destroy, None, 0)
self.protocol('WM_DELETE_WINDOW', self._Pmw_WM_DELETE_name)
return apply(Tkinter.Wm.title, (self,) + args)
_haveBltBusy = None
def _havebltbusy(window):
global _busy_hold, _busy_release, _haveBltBusy
if _haveBltBusy is None:
import PmwBlt
_haveBltBusy = PmwBlt.havebltbusy(window)
_busy_hold = PmwBlt.busy_hold
if os.name == 'nt':
# There is a bug in Blt 2.4i on NT where the busy window
# does not follow changes in the children of a window.
# Using forget works around the problem.
_busy_release = PmwBlt.busy_forget
else:
_busy_release = PmwBlt.busy_release
return _haveBltBusy
class _BusyWrapper:
def __init__(self, command, updateFunction):
self._command = command
self._updateFunction = updateFunction
def callback(self, *args):
showbusycursor()
rtn = apply(self._command, args)
# Call update before hiding the busy windows to clear any
# events that may have occurred over the busy windows.
if callable(self._updateFunction):
self._updateFunction()
hidebusycursor()
return rtn
#=============================================================================
def drawarrow(canvas, color, direction, tag, baseOffset = 0.25, edgeOffset = 0.15):
canvas.delete(tag)
bw = (string.atoi(canvas['borderwidth']) +
string.atoi(canvas['highlightthickness']))
width = string.atoi(canvas['width'])
height = string.atoi(canvas['height'])
if direction in ('up', 'down'):
majorDimension = height
minorDimension = width
else:
majorDimension = width
minorDimension = height
offset = round(baseOffset * majorDimension)
if direction in ('down', 'right'):
base = bw + offset
apex = bw + majorDimension - offset
else:
base = bw + majorDimension - offset
apex = bw + offset
if minorDimension > 3 and minorDimension % 2 == 0:
minorDimension = minorDimension - 1
half = int(minorDimension * (1 - 2 * edgeOffset)) / 2
low = round(bw + edgeOffset * minorDimension)
middle = low + half
high = low + 2 * half
if direction in ('up', 'down'):
coords = (low, base, high, base, middle, apex)
else:
coords = (base, low, base, high, apex, middle)
kw = {'fill' : color, 'outline' : color, 'tag' : tag}
apply(canvas.create_polygon, coords, kw)
#=============================================================================
# Modify the Tkinter destroy methods so that it notifies us when a Tk
# toplevel or frame is destroyed.
# A map from the 'hull' component of a megawidget to the megawidget.
# This is used to clean up a megawidget when its hull is destroyed.
_hullToMegaWidget = {}
def __TkinterToplevelDestroy(tkWidget):
if _hullToMegaWidget.has_key(tkWidget):
mega = _hullToMegaWidget[tkWidget]
try:
mega.destroy()
except:
_reporterror(mega.destroy, ())
else:
# Delete the busy info structure for this toplevel (if the
# window was created before initialise() was called, it
# will not have any.
if _toplevelBusyInfo.has_key(tkWidget):
del _toplevelBusyInfo[tkWidget]
if hasattr(tkWidget, '_Pmw_WM_DELETE_name'):
tkWidget.tk.deletecommand(tkWidget._Pmw_WM_DELETE_name)
del tkWidget._Pmw_WM_DELETE_name
Tkinter.BaseWidget.destroy(tkWidget)
def __TkinterWidgetDestroy(tkWidget):
if _hullToMegaWidget.has_key(tkWidget):
mega = _hullToMegaWidget[tkWidget]
try:
mega.destroy()
except:
_reporterror(mega.destroy, ())
else:
Tkinter.BaseWidget.destroy(tkWidget)
#=============================================================================
# Add code to Tkinter to improve the display of errors which occur in
# callbacks.
class __TkinterCallWrapper:
def __init__(self, func, subst, widget):
self.func = func
self.subst = subst
self.widget = widget
# Calling back from Tk into python.
def __call__(self, *args):
try:
if self.subst:
args = apply(self.subst, args)
if _traceTk:
if not _callToTkReturned:
_traceTkFile.write('\n')
if hasattr(self.func, 'im_class'):
name = self.func.im_class.__name__ + '.' + \
self.func.__name__
else:
name = self.func.__name__
if len(args) == 1 and hasattr(args[0], 'type'):
# The argument to the callback is an event.
eventName = _eventTypeToName[string.atoi(args[0].type)]
if eventName in ('KeyPress', 'KeyRelease',):
argStr = '(%s %s Event: %s)' % \
(eventName, args[0].keysym, args[0].widget)
else:
argStr = '(%s Event, %s)' % (eventName, args[0].widget)
else:
argStr = str(args)
_traceTkFile.write('CALLBACK> %d:%s%s%s\n' %
(_recursionCounter, ' ' * _recursionCounter, name, argStr))
_traceTkFile.flush()
return apply(self.func, args)
except SystemExit, msg:
raise SystemExit, msg
except:
_reporterror(self.func, args)
_eventTypeToName = {
2 : 'KeyPress', 15 : 'VisibilityNotify', 28 : 'PropertyNotify',
3 : 'KeyRelease', 16 : 'CreateNotify', 29 : 'SelectionClear',
4 : 'ButtonPress', 17 : 'DestroyNotify', 30 : 'SelectionRequest',
5 : 'ButtonRelease', 18 : 'UnmapNotify', 31 : 'SelectionNotify',
6 : 'MotionNotify', 19 : 'MapNotify', 32 : 'ColormapNotify',
7 : 'EnterNotify', 20 : 'MapRequest', 33 : 'ClientMessage',
8 : 'LeaveNotify', 21 : 'ReparentNotify', 34 : 'MappingNotify',
9 : 'FocusIn', 22 : 'ConfigureNotify', 35 : 'VirtualEvents',
10 : 'FocusOut', 23 : 'ConfigureRequest', 36 : 'ActivateNotify',
11 : 'KeymapNotify', 24 : 'GravityNotify', 37 : 'DeactivateNotify',
12 : 'Expose', 25 : 'ResizeRequest', 38 : 'MouseWheelEvent',
13 : 'GraphicsExpose', 26 : 'CirculateNotify',
14 : 'NoExpose', 27 : 'CirculateRequest',
}
def _reporterror(func, args):
# Fetch current exception values.
exc_type, exc_value, exc_traceback = sys.exc_info()
# Give basic information about the callback exception.
if type(exc_type) == types.ClassType:
# Handle python 1.5 class exceptions.
exc_type = exc_type.__name__
msg = str(exc_type) + ' Exception in Tk callback\n'
msg = msg + ' Function: %s (type: %s)\n' % (repr(func), type(func))
msg = msg + ' Args: %s\n' % str(args)
if type(args) == types.TupleType and len(args) > 0 and \
hasattr(args[0], 'type'):
eventArg = 1
else:
eventArg = 0
# If the argument to the callback is an event, add the event type.
if eventArg:
eventNum = string.atoi(args[0].type)
if eventNum in _eventTypeToName.keys():
msg = msg + ' Event type: %s (type num: %d)\n' % \
(_eventTypeToName[eventNum], eventNum)
else:
msg = msg + ' Unknown event type (type num: %d)\n' % eventNum
# Add the traceback.
msg = msg + 'Traceback (innermost last):\n'
for tr in traceback.extract_tb(exc_traceback):
msg = msg + ' File "%s", line %s, in %s\n' % (tr[0], tr[1], tr[2])
msg = msg + ' %s\n' % tr[3]
msg = msg + '%s: %s\n' % (exc_type, exc_value)
# If the argument to the callback is an event, add the event contents.
if eventArg:
msg = msg + '\n================================================\n'
msg = msg + ' Event contents:\n'
keys = args[0].__dict__.keys()
keys.sort()
for key in keys:
msg = msg + ' %s: %s\n' % (key, args[0].__dict__[key])
clearbusycursor()
try:
displayerror(msg)
except:
pass
class _ErrorWindow:
def __init__(self):
self._errorQueue = []
self._errorCount = 0
self._open = 0
self._firstShowing = 1
# Create the toplevel window
self._top = Tkinter.Toplevel()
self._top.protocol('WM_DELETE_WINDOW', self._hide)
self._top.title('Error in background function')
self._top.iconname('Background error')
# Create the text widget and scrollbar in a frame
upperframe = Tkinter.Frame(self._top)
scrollbar = Tkinter.Scrollbar(upperframe, orient='vertical')
scrollbar.pack(side = 'right', fill = 'y')
self._text = Tkinter.Text(upperframe, yscrollcommand=scrollbar.set)
self._text.pack(fill = 'both', expand = 1)
scrollbar.configure(command=self._text.yview)
# Create the buttons and label in a frame
lowerframe = Tkinter.Frame(self._top)
ignore = Tkinter.Button(lowerframe,
text = 'Ignore remaining errors', command = self._hide)
ignore.pack(side='left')
self._nextError = Tkinter.Button(lowerframe,
text = 'Show next error', command = self._next)
self._nextError.pack(side='left')
self._label = Tkinter.Label(lowerframe, relief='ridge')
self._label.pack(side='left', fill='x', expand=1)
# Pack the lower frame first so that it does not disappear
# when the window is resized.
lowerframe.pack(side = 'bottom', fill = 'x')
upperframe.pack(side = 'bottom', fill = 'both', expand = 1)
def showerror(self, text):
if self._open:
self._errorQueue.append(text)
else:
self._display(text)
self._open = 1
# Display the error window in the same place it was before.
if self._top.state() == 'normal':
# If update_idletasks is not called here, the window may
# be placed partially off the screen. Also, if it is not
# called and many errors are generated quickly in
# succession, the error window may not display errors
# until the last one is generated and the interpreter
# becomes idle.
# XXX: remove this, since it causes omppython to go into an
# infinite loop if an error occurs in an omp callback.
# self._top.update_idletasks()
pass
else:
if self._firstShowing:
geom = None
else:
geometry = self._top.geometry()
index = string.find(geometry, '+')
if index >= 0:
geom = geometry[index:]
else:
geom = None
setgeometryanddeiconify(self._top, geom)
if self._firstShowing:
self._firstShowing = 0
else:
self._top.tkraise()
self._top.focus()
self._updateButtons()
# Release any grab, so that buttons in the error window work.
releasegrabs()
def _hide(self):
self._errorCount = self._errorCount + len(self._errorQueue)
self._errorQueue = []
self._top.withdraw()
self._open = 0
def _next(self):
# Display the next error in the queue.
text = self._errorQueue[0]
del self._errorQueue[0]
self._display(text)
self._updateButtons()
def _display(self, text):
self._errorCount = self._errorCount + 1
text = 'Error: %d\n%s' % (self._errorCount, text)
self._text.delete('1.0', 'end')
self._text.insert('end', text)
def _updateButtons(self):
numQueued = len(self._errorQueue)
if numQueued > 0:
self._label.configure(text='%d more errors' % numQueued)
self._nextError.configure(state='normal')
else:
self._label.configure(text='No more errors')
self._nextError.configure(state='disabled')
_bltImported = 1
_bltbusyOK = 0
######################################################################
### File: PmwDialog.py
# Based on iwidgets2.2.0/dialog.itk and iwidgets2.2.0/dialogshell.itk code.
# Convention:
# Each dialog window should have one of these as the rightmost button:
# Close Close a window which only displays information.
# Cancel Close a window which may be used to change the state of
# the application.
import sys
import types
import Tkinter
# A Toplevel with a ButtonBox and child site.
class Dialog(MegaToplevel):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('buttonbox_hull_borderwidth', 1, None),
('buttonbox_hull_relief', 'raised', None),
('buttonboxpos', 's', INITOPT),
('buttons', ('OK',), self._buttons),
('command', None, None),
('dialogchildsite_borderwidth', 1, None),
('dialogchildsite_relief', 'raised', None),
('defaultbutton', None, self._defaultButton),
('master', 'parent', None),
('separatorwidth', 0, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaToplevel.__init__(self, parent)
# Create the components.
oldInterior = MegaToplevel.interior(self)
# Set up pack options according to the position of the button box.
pos = self['buttonboxpos']
if pos not in 'nsew':
raise ValueError, \
'bad buttonboxpos option "%s": should be n, s, e, or w' \
% pos
if pos in 'ns':
orient = 'horizontal'
fill = 'x'
if pos == 'n':
side = 'top'
else:
side = 'bottom'
else:
orient = 'vertical'
fill = 'y'
if pos == 'w':
side = 'left'
else:
side = 'right'
# Create the button box.
self._buttonBox = self.createcomponent('buttonbox',
(), None,
ButtonBox, (oldInterior,), orient = orient)
self._buttonBox.pack(side = side, fill = fill)
# Create the separating line.
width = self['separatorwidth']
if width > 0:
self._separator = self.createcomponent('separator',
(), None,
Tkinter.Frame, (oldInterior,), relief = 'sunken',
height = width, width = width, borderwidth = width / 2)
self._separator.pack(side = side, fill = fill)
# Create the child site.
self.__dialogChildSite = self.createcomponent('dialogchildsite',
(), None,
Tkinter.Frame, (oldInterior,))
self.__dialogChildSite.pack(side=side, fill='both', expand=1)
self.oldButtons = ()
self.oldDefault = None
self.bind('<Return>', self._invokeDefault)
self.userdeletefunc(self._doCommand)
self.usermodaldeletefunc(self._doCommand)
# Check keywords and initialise options.
self.initialiseoptions()
def interior(self):
return self.__dialogChildSite
def invoke(self, index = DEFAULT):
return self._buttonBox.invoke(index)
def _invokeDefault(self, event):
try:
self._buttonBox.index(DEFAULT)
except ValueError:
return
self._buttonBox.invoke()
def _doCommand(self, name = None):
if name is not None and self.active() and \
grabstacktopwindow() != self.component('hull'):
# This is a modal dialog but is not on the top of the grab
# stack (ie: should not have the grab), so ignore this
# event. This seems to be a bug in Tk and may occur in
# nested modal dialogs.
#
# An example is the PromptDialog demonstration. To
# trigger the problem, start the demo, then move the mouse
# to the main window, hit <TAB> and then <TAB> again. The
# highlight border of the "Show prompt dialog" button
# should now be displayed. Now hit <SPACE>, <RETURN>,
# <RETURN> rapidly several times. Eventually, hitting the
# return key invokes the password dialog "OK" button even
# though the confirm dialog is active (and therefore
# should have the keyboard focus). Observed under Solaris
# 2.5.1, python 1.5.2 and Tk8.0.
# TODO: Give focus to the window on top of the grabstack.
return
command = self['command']
if callable(command):
return command(name)
else:
if self.active():
self.deactivate(name)
else:
self.withdraw()
def _buttons(self):
buttons = self['buttons']
if type(buttons) != types.TupleType and type(buttons) != types.ListType:
raise ValueError, \
'bad buttons option "%s": should be a tuple' % str(buttons)
if self.oldButtons == buttons:
return
self.oldButtons = buttons
for index in range(self._buttonBox.numbuttons()):
self._buttonBox.delete(0)
for name in buttons:
self._buttonBox.add(name,
command=lambda self=self, name=name: self._doCommand(name))
if len(buttons) > 0:
defaultbutton = self['defaultbutton']
if defaultbutton is None:
self._buttonBox.setdefault(None)
else:
try:
self._buttonBox.index(defaultbutton)
except ValueError:
pass
else:
self._buttonBox.setdefault(defaultbutton)
self._buttonBox.alignbuttons()
def _defaultButton(self):
defaultbutton = self['defaultbutton']
if self.oldDefault == defaultbutton:
return
self.oldDefault = defaultbutton
if len(self['buttons']) > 0:
if defaultbutton is None:
self._buttonBox.setdefault(None)
else:
try:
self._buttonBox.index(defaultbutton)
except ValueError:
pass
else:
self._buttonBox.setdefault(defaultbutton)
######################################################################
### File: PmwTimeFuncs.py
# Functions for dealing with dates and times.
import re
import string
def timestringtoseconds(text, separator = ':'):
inputList = string.split(string.strip(text), separator)
if len(inputList) != 3:
raise ValueError, 'invalid value: ' + text
sign = 1
if len(inputList[0]) > 0 and inputList[0][0] in ('+', '-'):
if inputList[0][0] == '-':
sign = -1
inputList[0] = inputList[0][1:]
if re.search('[^0-9]', string.join(inputList, '')) is not None:
raise ValueError, 'invalid value: ' + text
hour = string.atoi(inputList[0])
minute = string.atoi(inputList[1])
second = string.atoi(inputList[2])
if minute >= 60 or second >= 60:
raise ValueError, 'invalid value: ' + text
return sign * (hour * 60 * 60 + minute * 60 + second)
_year_pivot = 50
_century = 2000
def setyearpivot(pivot, century = None):
global _year_pivot
global _century
oldvalues = (_year_pivot, _century)
_year_pivot = pivot
if century is not None:
_century = century
return oldvalues
def datestringtojdn(text, format = 'ymd', separator = '/'):
inputList = string.split(string.strip(text), separator)
if len(inputList) != 3:
raise ValueError, 'invalid value: ' + text
if re.search('[^0-9]', string.join(inputList, '')) is not None:
raise ValueError, 'invalid value: ' + text
formatList = list(format)
day = string.atoi(inputList[formatList.index('d')])
month = string.atoi(inputList[formatList.index('m')])
year = string.atoi(inputList[formatList.index('y')])
if _year_pivot is not None:
if year >= 0 and year < 100:
if year <= _year_pivot:
year = year + _century
else:
year = year + _century - 100
jdn = ymdtojdn(year, month, day)
if jdntoymd(jdn) != (year, month, day):
raise ValueError, 'invalid value: ' + text
return jdn
def _cdiv(a, b):
# Return a / b as calculated by most C language implementations,
# assuming both a and b are integers.
if a * b > 0:
return a / b
else:
return -(abs(a) / abs(b))
def ymdtojdn(year, month, day, julian = -1, papal = 1):
# set Julian flag if auto set
if julian < 0:
if papal: # Pope Gregory XIII's decree
lastJulianDate = 15821004L # last day to use Julian calendar
else: # British-American usage
lastJulianDate = 17520902L # last day to use Julian calendar
julian = ((year * 100L) + month) * 100 + day <= lastJulianDate
if year < 0:
# Adjust BC year
year = year + 1
if julian:
return 367L * year - _cdiv(7 * (year + 5001L + _cdiv((month - 9), 7)), 4) + \
_cdiv(275 * month, 9) + day + 1729777L
else:
return (day - 32076L) + \
_cdiv(1461L * (year + 4800L + _cdiv((month - 14), 12)), 4) + \
_cdiv(367 * (month - 2 - _cdiv((month - 14), 12) * 12), 12) - \
_cdiv((3 * _cdiv((year + 4900L + _cdiv((month - 14), 12)), 100)), 4) + \
1 # correction by rdg
def jdntoymd(jdn, julian = -1, papal = 1):
# set Julian flag if auto set
if julian < 0:
if papal: # Pope Gregory XIII's decree
lastJulianJdn = 2299160L # last jdn to use Julian calendar
else: # British-American usage
lastJulianJdn = 2361221L # last jdn to use Julian calendar
julian = (jdn <= lastJulianJdn);
x = jdn + 68569L
if julian:
x = x + 38
daysPer400Years = 146100L
fudgedDaysPer4000Years = 1461000L + 1
else:
daysPer400Years = 146097L
fudgedDaysPer4000Years = 1460970L + 31
z = _cdiv(4 * x, daysPer400Years)
x = x - _cdiv((daysPer400Years * z + 3), 4)
y = _cdiv(4000 * (x + 1), fudgedDaysPer4000Years)
x = x - _cdiv(1461 * y, 4) + 31
m = _cdiv(80 * x, 2447)
d = x - _cdiv(2447 * m, 80)
x = _cdiv(m, 11)
m = m + 2 - 12 * x
y = 100 * (z - 49) + y + x
# Convert from longs to integers.
yy = int(y)
mm = int(m)
dd = int(d)
if yy <= 0:
# Adjust BC years.
yy = yy - 1
return (yy, mm, dd)
def stringtoreal(text, separator = '.'):
if separator != '.':
if string.find(text, '.') >= 0:
raise ValueError, 'invalid value: ' + text
index = string.find(text, separator)
if index >= 0:
text = text[:index] + '.' + text[index + 1:]
return string.atof(text)
######################################################################
### File: PmwBalloon.py
import os
import string
import Tkinter
class Balloon(MegaToplevel):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('initwait', 500, None), # milliseconds
('label_background', 'lightyellow', None),
('label_foreground', 'black', None),
('label_justify', 'left', None),
('master', 'parent', None),
('relmouse', 'none', self._relmouse),
('state', 'both', self._state),
('statuscommand', None, None),
('xoffset', 20, None), # pixels
('yoffset', 1, None), # pixels
('hull_highlightthickness', 1, None),
('hull_highlightbackground', 'black', None),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaToplevel.__init__(self, parent)
self.withdraw()
self.overrideredirect(1)
# Create the components.
interior = self.interior()
self._label = self.createcomponent('label',
(), None,
Tkinter.Label, (interior,))
self._label.pack()
# The default hull configuration options give a black border
# around the balloon, but avoids a black 'flash' when the
# balloon is deiconified, before the text appears.
if not kw.has_key('hull_background'):
self.configure(hull_background = \
str(self._label.cget('background')))
# Initialise instance variables.
self._timer = None
# The widget or item that is currently triggering the balloon.
# It is None if the balloon is not being displayed. It is a
# one-tuple if the balloon is being displayed in response to a
# widget binding (value is the widget). It is a two-tuple if
# the balloon is being displayed in response to a canvas or
# text item binding (value is the widget and the item).
self._currentTrigger = None
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
if self._timer is not None:
self.after_cancel(self._timer)
self._timer = None
MegaToplevel.destroy(self)
def bind(self, widget, balloonHelp, statusHelp = None):
# If a previous bind for this widget exists, remove it.
self.unbind(widget)
if balloonHelp is None and statusHelp is None:
return
if statusHelp is None:
statusHelp = balloonHelp
enterId = widget.bind('<Enter>',
lambda event, self = self, w = widget,
sHelp = statusHelp, bHelp = balloonHelp:
self._enter(event, w, sHelp, bHelp, 0))
# Set Motion binding so that if the pointer remains at rest
# within the widget until the status line removes the help and
# then the pointer moves again, then redisplay the help in the
# status line.
# Note: The Motion binding only works for basic widgets, and
# the hull of megawidgets but not for other megawidget components.
motionId = widget.bind('<Motion>',
lambda event = None, self = self, statusHelp = statusHelp:
self.showstatus(statusHelp))
leaveId = widget.bind('<Leave>', self._leave)
buttonId = widget.bind('<ButtonPress>', self._buttonpress)
# Set Destroy binding so that the balloon can be withdrawn and
# the timer can be cancelled if the widget is destroyed.
destroyId = widget.bind('<Destroy>', self._destroy)
# Use the None item in the widget's private Pmw dictionary to
# store the widget's bind callbacks, for later clean up.
if not hasattr(widget, '_Pmw_BalloonBindIds'):
widget._Pmw_BalloonBindIds = {}
widget._Pmw_BalloonBindIds[None] = \
(enterId, motionId, leaveId, buttonId, destroyId)
def unbind(self, widget):
if hasattr(widget, '_Pmw_BalloonBindIds'):
if widget._Pmw_BalloonBindIds.has_key(None):
(enterId, motionId, leaveId, buttonId, destroyId) = \
widget._Pmw_BalloonBindIds[None]
# Need to pass in old bindings, so that Tkinter can
# delete the commands. Otherwise, memory is leaked.
widget.unbind('<Enter>', enterId)
widget.unbind('<Motion>', motionId)
widget.unbind('<Leave>', leaveId)
widget.unbind('<ButtonPress>', buttonId)
widget.unbind('<Destroy>', destroyId)
del widget._Pmw_BalloonBindIds[None]
if self._currentTrigger is not None and len(self._currentTrigger) == 1:
# The balloon is currently being displayed and the current
# trigger is a widget.
triggerWidget = self._currentTrigger[0]
if triggerWidget == widget:
if self._timer is not None:
self.after_cancel(self._timer)
self._timer = None
self.withdraw()
self.clearstatus()
self._currentTrigger = None
def tagbind(self, widget, tagOrItem, balloonHelp, statusHelp = None):
# If a previous bind for this widget's tagOrItem exists, remove it.
self.tagunbind(widget, tagOrItem)
if balloonHelp is None and statusHelp is None:
return
if statusHelp is None:
statusHelp = balloonHelp
enterId = widget.tag_bind(tagOrItem, '<Enter>',
lambda event, self = self, w = widget,
sHelp = statusHelp, bHelp = balloonHelp:
self._enter(event, w, sHelp, bHelp, 1))
motionId = widget.tag_bind(tagOrItem, '<Motion>',
lambda event = None, self = self, statusHelp = statusHelp:
self.showstatus(statusHelp))
leaveId = widget.tag_bind(tagOrItem, '<Leave>', self._leave)
buttonId = widget.tag_bind(tagOrItem, '<ButtonPress>', self._buttonpress)
# Use the tagOrItem item in the widget's private Pmw dictionary to
# store the tagOrItem's bind callbacks, for later clean up.
if not hasattr(widget, '_Pmw_BalloonBindIds'):
widget._Pmw_BalloonBindIds = {}
widget._Pmw_BalloonBindIds[tagOrItem] = \
(enterId, motionId, leaveId, buttonId)
def tagunbind(self, widget, tagOrItem):
if hasattr(widget, '_Pmw_BalloonBindIds'):
if widget._Pmw_BalloonBindIds.has_key(tagOrItem):
(enterId, motionId, leaveId, buttonId) = \
widget._Pmw_BalloonBindIds[tagOrItem]
widget.tag_unbind(tagOrItem, '<Enter>', enterId)
widget.tag_unbind(tagOrItem, '<Motion>', motionId)
widget.tag_unbind(tagOrItem, '<Leave>', leaveId)
widget.tag_unbind(tagOrItem, '<ButtonPress>', buttonId)
del widget._Pmw_BalloonBindIds[tagOrItem]
if self._currentTrigger is None:
# The balloon is not currently being displayed.
return
if len(self._currentTrigger) == 1:
# The current trigger is a widget.
return
if len(self._currentTrigger) == 2:
# The current trigger is a canvas item.
(triggerWidget, triggerItem) = self._currentTrigger
if triggerWidget == widget and triggerItem == tagOrItem:
if self._timer is not None:
self.after_cancel(self._timer)
self._timer = None
self.withdraw()
self.clearstatus()
self._currentTrigger = None
else: # The current trigger is a text item.
(triggerWidget, x, y) = self._currentTrigger
if triggerWidget == widget:
currentPos = widget.index('@%d,%d' % (x, y))
currentTags = widget.tag_names(currentPos)
if tagOrItem in currentTags:
if self._timer is not None:
self.after_cancel(self._timer)
self._timer = None
self.withdraw()
self.clearstatus()
self._currentTrigger = None
def showstatus(self, statusHelp):
if self['state'] in ('status', 'both'):
cmd = self['statuscommand']
if callable(cmd):
cmd(statusHelp)
def clearstatus(self):
self.showstatus(None)
def _state(self):
if self['state'] not in ('both', 'balloon', 'status', 'none'):
raise ValueError, 'bad state option ' + repr(self['state']) + \
': should be one of \'both\', \'balloon\', ' + \
'\'status\' or \'none\''
def _relmouse(self):
if self['relmouse'] not in ('both', 'x', 'y', 'none'):
raise ValueError, 'bad relmouse option ' + repr(self['relmouse'])+ \
': should be one of \'both\', \'x\', ' + '\'y\' or \'none\''
def _enter(self, event, widget, statusHelp, balloonHelp, isItem):
# Do not display balloon if mouse button is pressed. This
# will only occur if the button was pressed inside a widget,
# then the mouse moved out of and then back into the widget,
# with the button still held down. The number 0x1f00 is the
# button mask for the 5 possible buttons in X.
buttonPressed = (event.state & 0x1f00) != 0
if not buttonPressed and balloonHelp is not None and \
self['state'] in ('balloon', 'both'):
if self._timer is not None:
self.after_cancel(self._timer)
self._timer = None
self._timer = self.after(self['initwait'],
lambda self = self, widget = widget, help = balloonHelp,
isItem = isItem:
self._showBalloon(widget, help, isItem))
if isItem:
if hasattr(widget, 'canvasx'):
# The widget is a canvas.
item = widget.find_withtag('current')
if len(item) > 0:
item = item[0]
else:
item = None
self._currentTrigger = (widget, item)
else:
# The widget is a text widget.
self._currentTrigger = (widget, event.x, event.y)
else:
self._currentTrigger = (widget,)
self.showstatus(statusHelp)
def _leave(self, event):
if self._timer is not None:
self.after_cancel(self._timer)
self._timer = None
self.withdraw()
self.clearstatus()
self._currentTrigger = None
def _destroy(self, event):
# Only withdraw the balloon and cancel the timer if the widget
# being destroyed is the widget that triggered the balloon.
# Note that in a Tkinter Destroy event, the widget field is a
# string and not a widget as usual.
if self._currentTrigger is None:
# The balloon is not currently being displayed
return
if len(self._currentTrigger) == 1:
# The current trigger is a widget (not an item)
triggerWidget = self._currentTrigger[0]
if str(triggerWidget) == event.widget:
if self._timer is not None:
self.after_cancel(self._timer)
self._timer = None
self.withdraw()
self.clearstatus()
self._currentTrigger = None
def _buttonpress(self, event):
if self._timer is not None:
self.after_cancel(self._timer)
self._timer = None
self.withdraw()
self._currentTrigger = None
def _showBalloon(self, widget, balloonHelp, isItem):
self._label.configure(text = balloonHelp)
# First, display the balloon offscreen to get dimensions.
screenWidth = self.winfo_screenwidth()
screenHeight = self.winfo_screenheight()
self.geometry('+%d+0' % (screenWidth + 1))
self.update_idletasks()
if isItem:
# Get the bounding box of the current item.
bbox = widget.bbox('current')
if bbox is None:
# The item that triggered the balloon has disappeared,
# perhaps by a user's timer event that occured between
# the <Enter> event and the 'initwait' timer calling
# this method.
return
# The widget is either a text or canvas. The meaning of
# the values returned by the bbox method is different for
# each, so use the existence of the 'canvasx' method to
# distinguish between them.
if hasattr(widget, 'canvasx'):
# The widget is a canvas. Place balloon under canvas
# item. The positions returned by bbox are relative
# to the entire canvas, not just the visible part, so
# need to convert to window coordinates.
leftrel = bbox[0] - widget.canvasx(0)
toprel = bbox[1] - widget.canvasy(0)
bottomrel = bbox[3] - widget.canvasy(0)
else:
# The widget is a text widget. Place balloon under
# the character closest to the mouse. The positions
# returned by bbox are relative to the text widget
# window (ie the visible part of the text only).
leftrel = bbox[0]
toprel = bbox[1]
bottomrel = bbox[1] + bbox[3]
else:
leftrel = 0
toprel = 0
bottomrel = widget.winfo_height()
xpointer, ypointer = widget.winfo_pointerxy() # -1 if off screen
if xpointer >= 0 and self['relmouse'] in ('both', 'x'):
x = xpointer
else:
x = leftrel + widget.winfo_rootx()
x = x + self['xoffset']
if ypointer >= 0 and self['relmouse'] in ('both', 'y'):
y = ypointer
else:
y = bottomrel + widget.winfo_rooty()
y = y + self['yoffset']
edges = (string.atoi(str(self.cget('hull_highlightthickness'))) +
string.atoi(str(self.cget('hull_borderwidth')))) * 2
if x + self._label.winfo_reqwidth() + edges > screenWidth:
x = screenWidth - self._label.winfo_reqwidth() - edges
if y + self._label.winfo_reqheight() + edges > screenHeight:
if ypointer >= 0 and self['relmouse'] in ('both', 'y'):
y = ypointer
else:
y = toprel + widget.winfo_rooty()
y = y - self._label.winfo_reqheight() - self['yoffset'] - edges
setgeometryanddeiconify(self, '+%d+%d' % (x, y))
######################################################################
### File: PmwButtonBox.py
# Based on iwidgets2.2.0/buttonbox.itk code.
import types
import Tkinter
class ButtonBox(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('orient', 'horizontal', INITOPT),
('padx', 3, INITOPT),
('pady', 3, INITOPT),
)
self.defineoptions(kw, optiondefs, dynamicGroups = ('Button',))
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
if self['labelpos'] is None:
self._buttonBoxFrame = self._hull
columnOrRow = 0
else:
self._buttonBoxFrame = self.createcomponent('frame',
(), None,
Tkinter.Frame, (interior,))
self._buttonBoxFrame.grid(column=2, row=2, sticky='nsew')
columnOrRow = 2
self.createlabel(interior)
orient = self['orient']
if orient == 'horizontal':
interior.grid_columnconfigure(columnOrRow, weight = 1)
elif orient == 'vertical':
interior.grid_rowconfigure(columnOrRow, weight = 1)
else:
raise ValueError, 'bad orient option ' + repr(orient) + \
': must be either \'horizontal\' or \'vertical\''
# Initialise instance variables.
# List of tuples describing the buttons:
# - name
# - button widget
self._buttonList = []
# The index of the default button.
self._defaultButton = None
self._timerId = None
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
if self._timerId:
self.after_cancel(self._timerId)
self._timerId = None
MegaWidget.destroy(self)
def numbuttons(self):
return len(self._buttonList)
def index(self, index, forInsert = 0):
listLength = len(self._buttonList)
if type(index) == types.IntType:
if forInsert and index <= listLength:
return index
elif not forInsert and index < listLength:
return index
else:
raise ValueError, 'index "%s" is out of range' % index
elif index is END:
if forInsert:
return listLength
elif listLength > 0:
return listLength - 1
else:
raise ValueError, 'ButtonBox has no buttons'
elif index is DEFAULT:
if self._defaultButton is not None:
return self._defaultButton
raise ValueError, 'ButtonBox has no default'
else:
names = map(lambda t: t[0], self._buttonList)
if index in names:
return names.index(index)
validValues = 'a name, a number, END or DEFAULT'
raise ValueError, \
'bad index "%s": must be %s' % (index, validValues)
def insert(self, componentName, beforeComponent = 0, **kw):
if componentName in self.components():
raise ValueError, 'button "%s" already exists' % componentName
if not kw.has_key('text'):
kw['text'] = componentName
kw['default'] = 'normal'
button = apply(self.createcomponent, (componentName,
(), 'Button',
Tkinter.Button, (self._buttonBoxFrame,)), kw)
index = self.index(beforeComponent, 1)
horizontal = self['orient'] == 'horizontal'
numButtons = len(self._buttonList)
# Shift buttons up one position.
for i in range(numButtons - 1, index - 1, -1):
widget = self._buttonList[i][1]
pos = i * 2 + 3
if horizontal:
widget.grid(column = pos, row = 0)
else:
widget.grid(column = 0, row = pos)
# Display the new button.
if horizontal:
button.grid(column = index * 2 + 1, row = 0, sticky = 'ew',
padx = self['padx'], pady = self['pady'])
self._buttonBoxFrame.grid_columnconfigure(
numButtons * 2 + 2, weight = 1)
else:
button.grid(column = 0, row = index * 2 + 1, sticky = 'ew',
padx = self['padx'], pady = self['pady'])
self._buttonBoxFrame.grid_rowconfigure(
numButtons * 2 + 2, weight = 1)
self._buttonList.insert(index, (componentName, button))
return button
def add(self, componentName, **kw):
return apply(self.insert, (componentName, len(self._buttonList)), kw)
def delete(self, index):
index = self.index(index)
(name, widget) = self._buttonList[index]
widget.grid_forget()
self.destroycomponent(name)
numButtons = len(self._buttonList)
# Shift buttons down one position.
horizontal = self['orient'] == 'horizontal'
for i in range(index + 1, numButtons):
widget = self._buttonList[i][1]
pos = i * 2 - 1
if horizontal:
widget.grid(column = pos, row = 0)
else:
widget.grid(column = 0, row = pos)
if horizontal:
self._buttonBoxFrame.grid_columnconfigure(numButtons * 2 - 1,
minsize = 0)
self._buttonBoxFrame.grid_columnconfigure(numButtons * 2, weight = 0)
else:
self._buttonBoxFrame.grid_rowconfigure(numButtons * 2, weight = 0)
del self._buttonList[index]
def setdefault(self, index):
# Turn off the default ring around the current default button.
if self._defaultButton is not None:
button = self._buttonList[self._defaultButton][1]
button.configure(default = 'normal')
self._defaultButton = None
# Turn on the default ring around the new default button.
if index is not None:
index = self.index(index)
self._defaultButton = index
button = self._buttonList[index][1]
button.configure(default = 'active')
def invoke(self, index = DEFAULT, noFlash = 0):
# Invoke the callback associated with the *index* button. If
# *noFlash* is not set, flash the button to indicate to the
# user that something happened.
button = self._buttonList[self.index(index)][1]
if not noFlash:
state = button.cget('state')
relief = button.cget('relief')
button.configure(state = 'active', relief = 'sunken')
self.update_idletasks()
self.after(100)
button.configure(state = state, relief = relief)
return button.invoke()
def button(self, buttonIndex):
return self._buttonList[self.index(buttonIndex)][1]
def alignbuttons(self, when = 'later'):
if when == 'later':
if not self._timerId:
self._timerId = self.after_idle(self.alignbuttons, 'now')
return
self.update_idletasks()
self._timerId = None
# Determine the width of the maximum length button.
max = 0
horizontal = (self['orient'] == 'horizontal')
for index in range(len(self._buttonList)):
gridIndex = index * 2 + 1
if horizontal:
width = self._buttonBoxFrame.grid_bbox(gridIndex, 0)[2]
else:
width = self._buttonBoxFrame.grid_bbox(0, gridIndex)[2]
if width > max:
max = width
# Set the width of all the buttons to be the same.
if horizontal:
for index in range(len(self._buttonList)):
self._buttonBoxFrame.grid_columnconfigure(index * 2 + 1,
minsize = max)
else:
self._buttonBoxFrame.grid_columnconfigure(0, minsize = max)
######################################################################
### File: PmwEntryField.py
# Based on iwidgets2.2.0/entryfield.itk code.
import re
import string
import types
import Tkinter
# Possible return values of validation functions.
OK = 1
ERROR = 0
PARTIAL = -1
class EntryField(MegaWidget):
_classBindingsDefinedFor = 0
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('command', None, None),
('errorbackground', 'pink', None),
('invalidcommand', self.bell, None),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('modifiedcommand', None, None),
('sticky', 'ew', INITOPT),
('validate', None, self._validate),
('extravalidators', {}, None),
('value', '', INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
self._entryFieldEntry = self.createcomponent('entry',
(), None,
Tkinter.Entry, (interior,))
self._entryFieldEntry.grid(column=2, row=2, sticky=self['sticky'])
if self['value'] != '':
self.__setEntry(self['value'])
interior.grid_columnconfigure(2, weight=1)
interior.grid_rowconfigure(2, weight=1)
self.createlabel(interior)
# Initialise instance variables.
self.normalBackground = None
self._previousText = None
# Initialise instance.
_registerEntryField(self._entryFieldEntry, self)
# Establish the special class bindings if not already done.
# Also create bindings if the Tkinter default interpreter has
# changed. Use Tkinter._default_root to create class
# bindings, so that a reference to root is created by
# bind_class rather than a reference to self, which would
# prevent object cleanup.
if EntryField._classBindingsDefinedFor != Tkinter._default_root:
tagList = self._entryFieldEntry.bindtags()
root = Tkinter._default_root
allSequences = {}
for tag in tagList:
sequences = root.bind_class(tag)
if type(sequences) is types.StringType:
# In old versions of Tkinter, bind_class returns a string
sequences = root.tk.splitlist(sequences)
for sequence in sequences:
allSequences[sequence] = None
for sequence in allSequences.keys():
root.bind_class('EntryFieldPre', sequence, _preProcess)
root.bind_class('EntryFieldPost', sequence, _postProcess)
EntryField._classBindingsDefinedFor = root
self._entryFieldEntry.bindtags(('EntryFieldPre',) +
self._entryFieldEntry.bindtags() + ('EntryFieldPost',))
self._entryFieldEntry.bind('<Return>', self._executeCommand)
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
_deregisterEntryField(self._entryFieldEntry)
MegaWidget.destroy(self)
def _getValidatorFunc(self, validator, index):
# Search the extra and standard validator lists for the
# given 'validator'. If 'validator' is an alias, then
# continue the search using the alias. Make sure that
# self-referencial aliases do not cause infinite loops.
extraValidators = self['extravalidators']
traversedValidators = []
while 1:
traversedValidators.append(validator)
if extraValidators.has_key(validator):
validator = extraValidators[validator][index]
elif _standardValidators.has_key(validator):
validator = _standardValidators[validator][index]
else:
return validator
if validator in traversedValidators:
return validator
def _validate(self):
dict = {
'validator' : None,
'min' : None,
'max' : None,
'minstrict' : 1,
'maxstrict' : 1,
}
opt = self['validate']
if type(opt) is types.DictionaryType:
dict.update(opt)
else:
dict['validator'] = opt
# Look up validator maps and replace 'validator' field with
# the corresponding function.
validator = dict['validator']
valFunction = self._getValidatorFunc(validator, 0)
self._checkValidateFunction(valFunction, 'validate', validator)
dict['validator'] = valFunction
# Look up validator maps and replace 'stringtovalue' field
# with the corresponding function.
if dict.has_key('stringtovalue'):
stringtovalue = dict['stringtovalue']
strFunction = self._getValidatorFunc(stringtovalue, 1)
self._checkValidateFunction(
strFunction, 'stringtovalue', stringtovalue)
else:
strFunction = self._getValidatorFunc(validator, 1)
if strFunction == validator:
strFunction = len
dict['stringtovalue'] = strFunction
self._validationInfo = dict
args = dict.copy()
del args['validator']
del args['min']
del args['max']
del args['minstrict']
del args['maxstrict']
del args['stringtovalue']
self._validationArgs = args
self._previousText = None
if type(dict['min']) == types.StringType and strFunction is not None:
dict['min'] = apply(strFunction, (dict['min'],), args)
if type(dict['max']) == types.StringType and strFunction is not None:
dict['max'] = apply(strFunction, (dict['max'],), args)
self._checkValidity()
def _checkValidateFunction(self, function, option, validator):
# Raise an error if 'function' is not a function or None.
if function is not None and not callable(function):
extraValidators = self['extravalidators']
extra = extraValidators.keys()
extra.sort()
extra = tuple(extra)
standard = _standardValidators.keys()
standard.sort()
standard = tuple(standard)
msg = 'bad %s value "%s": must be a function or one of ' \
'the standard validators %s or extra validators %s'
raise ValueError, msg % (option, validator, standard, extra)
def _executeCommand(self, event = None):
cmd = self['command']
if callable(cmd):
if event is None:
# Return result of command for invoke() method.
return cmd()
else:
cmd()
def _preProcess(self):
self._previousText = self._entryFieldEntry.get()
self._previousICursor = self._entryFieldEntry.index('insert')
self._previousXview = self._entryFieldEntry.index('@0')
if self._entryFieldEntry.selection_present():
self._previousSel= (self._entryFieldEntry.index('sel.first'),
self._entryFieldEntry.index('sel.last'))
else:
self._previousSel = None
def _postProcess(self):
# No need to check if text has not changed.
previousText = self._previousText
if previousText == self._entryFieldEntry.get():
return self.valid()
valid = self._checkValidity()
if self.hulldestroyed():
# The invalidcommand called by _checkValidity() destroyed us.
return valid
cmd = self['modifiedcommand']
if callable(cmd) and previousText != self._entryFieldEntry.get():
cmd()
return valid
def checkentry(self):
# If there is a variable specified by the entry_textvariable
# option, checkentry() should be called after the set() method
# of the variable is called.
self._previousText = None
return self._postProcess()
def _getValidity(self):
text = self._entryFieldEntry.get()
dict = self._validationInfo
args = self._validationArgs
if dict['validator'] is not None:
status = apply(dict['validator'], (text,), args)
if status != OK:
return status
# Check for out of (min, max) range.
if dict['stringtovalue'] is not None:
min = dict['min']
max = dict['max']
if min is None and max is None:
return OK
val = apply(dict['stringtovalue'], (text,), args)
if min is not None and val < min:
if dict['minstrict']:
return ERROR
else:
return PARTIAL
if max is not None and val > max:
if dict['maxstrict']:
return ERROR
else:
return PARTIAL
return OK
def _checkValidity(self):
valid = self._getValidity()
oldValidity = valid
if valid == ERROR:
# The entry is invalid.
cmd = self['invalidcommand']
if callable(cmd):
cmd()
if self.hulldestroyed():
# The invalidcommand destroyed us.
return oldValidity
# Restore the entry to its previous value.
if self._previousText is not None:
self.__setEntry(self._previousText)
self._entryFieldEntry.icursor(self._previousICursor)
self._entryFieldEntry.xview(self._previousXview)
if self._previousSel is not None:
self._entryFieldEntry.selection_range(self._previousSel[0],
self._previousSel[1])
# Check if the saved text is valid as well.
valid = self._getValidity()
self._valid = valid
if self.hulldestroyed():
# The validator or stringtovalue commands called by
# _checkValidity() destroyed us.
return oldValidity
if valid == OK:
if self.normalBackground is not None:
self._entryFieldEntry.configure(
background = self.normalBackground)
self.normalBackground = None
else:
if self.normalBackground is None:
self.normalBackground = self._entryFieldEntry.cget('background')
self._entryFieldEntry.configure(
background = self['errorbackground'])
return oldValidity
def invoke(self):
return self._executeCommand()
def valid(self):
return self._valid == OK
def clear(self):
self.setentry('')
def __setEntry(self, text):
oldState = str(self._entryFieldEntry.cget('state'))
if oldState != 'normal':
self._entryFieldEntry.configure(state='normal')
self._entryFieldEntry.delete(0, 'end')
self._entryFieldEntry.insert(0, text)
if oldState != 'normal':
self._entryFieldEntry.configure(state=oldState)
def setentry(self, text):
self._preProcess()
self.__setEntry(text)
return self._postProcess()
def getvalue(self):
return self._entryFieldEntry.get()
def setvalue(self, text):
return self.setentry(text)
forwardmethods(EntryField, Tkinter.Entry, '_entryFieldEntry')
# ======================================================================
# Entry field validation functions
_numericregex = re.compile('^[0-9]*$')
_alphabeticregex = re.compile('^[a-z]*$', re.IGNORECASE)
_alphanumericregex = re.compile('^[0-9a-z]*$', re.IGNORECASE)
def numericvalidator(text):
if text == '':
return PARTIAL
else:
if _numericregex.match(text) is None:
return ERROR
else:
return OK
def integervalidator(text):
if text in ('', '-', '+'):
return PARTIAL
try:
string.atol(text)
return OK
except ValueError:
return ERROR
def alphabeticvalidator(text):
if _alphabeticregex.match(text) is None:
return ERROR
else:
return OK
def alphanumericvalidator(text):
if _alphanumericregex.match(text) is None:
return ERROR
else:
return OK
def hexadecimalvalidator(text):
if text in ('', '0x', '0X', '+', '+0x', '+0X', '-', '-0x', '-0X'):
return PARTIAL
try:
string.atol(text, 16)
return OK
except ValueError:
return ERROR
def realvalidator(text, separator = '.'):
if separator != '.':
if string.find(text, '.') >= 0:
return ERROR
index = string.find(text, separator)
if index >= 0:
text = text[:index] + '.' + text[index + 1:]
try:
string.atof(text)
return OK
except ValueError:
# Check if the string could be made valid by appending a digit
# eg ('-', '+', '.', '-.', '+.', '1.23e', '1E-').
if len(text) == 0:
return PARTIAL
if text[-1] in string.digits:
return ERROR
try:
string.atof(text + '0')
return PARTIAL
except ValueError:
return ERROR
def timevalidator(text, separator = ':'):
try:
timestringtoseconds(text, separator)
return OK
except ValueError:
if len(text) > 0 and text[0] in ('+', '-'):
text = text[1:]
if re.search('[^0-9' + separator + ']', text) is not None:
return ERROR
return PARTIAL
def datevalidator(text, format = 'ymd', separator = '/'):
try:
datestringtojdn(text, format, separator)
return OK
except ValueError:
if re.search('[^0-9' + separator + ']', text) is not None:
return ERROR
return PARTIAL
_standardValidators = {
'numeric' : (numericvalidator, string.atol),
'integer' : (integervalidator, string.atol),
'hexadecimal' : (hexadecimalvalidator, lambda s: string.atol(s, 16)),
'real' : (realvalidator, stringtoreal),
'alphabetic' : (alphabeticvalidator, len),
'alphanumeric' : (alphanumericvalidator, len),
'time' : (timevalidator, timestringtoseconds),
'date' : (datevalidator, datestringtojdn),
}
_entryCache = {}
def _registerEntryField(entry, entryField):
# Register an EntryField widget for an Entry widget
_entryCache[entry] = entryField
def _deregisterEntryField(entry):
# Deregister an Entry widget
del _entryCache[entry]
def _preProcess(event):
# Forward preprocess events for an Entry to it's EntryField
_entryCache[event.widget]._preProcess()
def _postProcess(event):
# Forward postprocess events for an Entry to it's EntryField
# The function specified by the 'command' option may have destroyed
# the megawidget in a binding earlier in bindtags, so need to check.
if _entryCache.has_key(event.widget):
_entryCache[event.widget]._postProcess()
######################################################################
### File: PmwGroup.py
import string
import Tkinter
def aligngrouptags(groups):
# Adjust the y position of the tags in /groups/ so that they all
# have the height of the highest tag.
maxTagHeight = 0
for group in groups:
if group._tag is None:
height = (string.atoi(str(group._ring.cget('borderwidth'))) +
string.atoi(str(group._ring.cget('highlightthickness'))))
else:
height = group._tag.winfo_reqheight()
if maxTagHeight < height:
maxTagHeight = height
for group in groups:
ringBorder = (string.atoi(str(group._ring.cget('borderwidth'))) +
string.atoi(str(group._ring.cget('highlightthickness'))))
topBorder = maxTagHeight / 2 - ringBorder / 2
group._hull.grid_rowconfigure(0, minsize = topBorder)
group._ring.grid_rowconfigure(0,
minsize = maxTagHeight - topBorder - ringBorder)
if group._tag is not None:
group._tag.place(y = maxTagHeight / 2)
class Group( MegaWidget ):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('collapsedsize', 6, INITOPT),
('ring_borderwidth', 2, None),
('ring_relief', 'groove', None),
('tagindent', 10, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = MegaWidget.interior(self)
self._ring = self.createcomponent(
'ring',
(), None,
Tkinter.Frame, (interior,),
)
self._groupChildSite = self.createcomponent(
'groupchildsite',
(), None,
Tkinter.Frame, (self._ring,)
)
self._tag = self.createcomponent(
'tag',
(), None,
Tkinter.Label, (interior,),
)
ringBorder = (string.atoi(str(self._ring.cget('borderwidth'))) +
string.atoi(str(self._ring.cget('highlightthickness'))))
if self._tag is None:
tagHeight = ringBorder
else:
tagHeight = self._tag.winfo_reqheight()
self._tag.place(
x = ringBorder + self['tagindent'],
y = tagHeight / 2,
anchor = 'w')
topBorder = tagHeight / 2 - ringBorder / 2
self._ring.grid(column = 0, row = 1, sticky = 'nsew')
interior.grid_columnconfigure(0, weight = 1)
interior.grid_rowconfigure(1, weight = 1)
interior.grid_rowconfigure(0, minsize = topBorder)
self._groupChildSite.grid(column = 0, row = 1, sticky = 'nsew')
self._ring.grid_columnconfigure(0, weight = 1)
self._ring.grid_rowconfigure(1, weight = 1)
self._ring.grid_rowconfigure(0,
minsize = tagHeight - topBorder - ringBorder)
self.showing = 1
# Check keywords and initialise options.
self.initialiseoptions()
def toggle(self):
if self.showing:
self.collapse()
else:
self.expand()
self.showing = not self.showing
def expand(self):
self._groupChildSite.grid(column = 0, row = 1, sticky = 'nsew')
def collapse(self):
self._groupChildSite.grid_forget()
if self._tag is None:
tagHeight = 0
else:
tagHeight = self._tag.winfo_reqheight()
self._ring.configure(height=(tagHeight / 2) + self['collapsedsize'])
def interior(self):
return self._groupChildSite
######################################################################
### File: PmwLabeledWidget.py
import Tkinter
class LabeledWidget(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('sticky', 'nsew', INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = MegaWidget.interior(self)
self._labelChildSite = self.createcomponent('labelchildsite',
(), None,
Tkinter.Frame, (interior,))
self._labelChildSite.grid(column=2, row=2, sticky=self['sticky'])
interior.grid_columnconfigure(2, weight=1)
interior.grid_rowconfigure(2, weight=1)
self.createlabel(interior)
# Check keywords and initialise options.
self.initialiseoptions()
def interior(self):
return self._labelChildSite
######################################################################
### File: PmwMainMenuBar.py
# Main menubar
import string
import types
import Tkinter
class MainMenuBar(MegaArchetype):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('balloon', None, None),
('hotkeys', 1, INITOPT),
('hull_tearoff', 0, None),
)
self.defineoptions(kw, optiondefs, dynamicGroups = ('Menu',))
# Initialise the base class (after defining the options).
MegaArchetype.__init__(self, parent, Tkinter.Menu)
self._menuInfo = {}
self._menuInfo[None] = (None, [])
# Map from a menu name to a tuple of information about the menu.
# The first item in the tuple is the name of the parent menu (for
# toplevel menus this is None). The second item in the tuple is
# a list of status help messages for each item in the menu.
# The key for the information for the main menubar is None.
self._menu = self.interior()
self._menu.bind('<Leave>', self._resetHelpmessage)
self._menu.bind('<Motion>',
lambda event=None, self=self: self._menuHelp(event, None))
# Check keywords and initialise options.
self.initialiseoptions()
def deletemenuitems(self, menuName, start, end = None):
self.component(menuName).delete(start, end)
if end is None:
del self._menuInfo[menuName][1][start]
else:
self._menuInfo[menuName][1][start:end+1] = []
def deletemenu(self, menuName):
"""Delete should be called for cascaded menus before main menus.
"""
parentName = self._menuInfo[menuName][0]
del self._menuInfo[menuName]
if parentName is None:
parentMenu = self._menu
else:
parentMenu = self.component(parentName)
menu = self.component(menuName)
menuId = str(menu)
for item in range(parentMenu.index('end') + 1):
if parentMenu.type(item) == 'cascade':
itemMenu = str(parentMenu.entrycget(item, 'menu'))
if itemMenu == menuId:
parentMenu.delete(item)
del self._menuInfo[parentName][1][item]
break
self.destroycomponent(menuName)
def disableall(self):
for index in range(len(self._menuInfo[None][1])):
self.entryconfigure(index, state = 'disabled')
def enableall(self):
for index in range(len(self._menuInfo[None][1])):
self.entryconfigure(index, state = 'normal')
def addmenu(self, menuName, balloonHelp, statusHelp = None,
traverseSpec = None, **kw):
if statusHelp is None:
statusHelp = balloonHelp
self._addmenu(None, menuName, balloonHelp, statusHelp,
traverseSpec, kw)
def addcascademenu(self, parentMenuName, menuName, statusHelp='',
traverseSpec = None, **kw):
self._addmenu(parentMenuName, menuName, None, statusHelp,
traverseSpec, kw)
def _addmenu(self, parentMenuName, menuName, balloonHelp, statusHelp,
traverseSpec, kw):
if (menuName) in self.components():
raise ValueError, 'menu "%s" already exists' % menuName
menukw = {}
if kw.has_key('tearoff'):
menukw['tearoff'] = kw['tearoff']
del kw['tearoff']
else:
menukw['tearoff'] = 0
if kw.has_key('name'):
menukw['name'] = kw['name']
del kw['name']
if not kw.has_key('label'):
kw['label'] = menuName
self._addHotkeyToOptions(parentMenuName, kw, traverseSpec)
if parentMenuName is None:
parentMenu = self._menu
balloon = self['balloon']
# Bug in Tk: balloon help not implemented
# if balloon is not None:
# balloon.mainmenubind(parentMenu, balloonHelp, statusHelp)
else:
parentMenu = self.component(parentMenuName)
apply(parentMenu.add_cascade, (), kw)
menu = apply(self.createcomponent, (menuName,
(), 'Menu',
Tkinter.Menu, (parentMenu,)), menukw)
parentMenu.entryconfigure('end', menu = menu)
self._menuInfo[parentMenuName][1].append(statusHelp)
self._menuInfo[menuName] = (parentMenuName, [])
menu.bind('<Leave>', self._resetHelpmessage)
menu.bind('<Motion>',
lambda event=None, self=self, menuName=menuName:
self._menuHelp(event, menuName))
def addmenuitem(self, menuName, itemType, statusHelp = '',
traverseSpec = None, **kw):
menu = self.component(menuName)
if itemType != 'separator':
self._addHotkeyToOptions(menuName, kw, traverseSpec)
if itemType == 'command':
command = menu.add_command
elif itemType == 'separator':
command = menu.add_separator
elif itemType == 'checkbutton':
command = menu.add_checkbutton
elif itemType == 'radiobutton':
command = menu.add_radiobutton
elif itemType == 'cascade':
command = menu.add_cascade
else:
raise ValueError, 'unknown menuitem type "%s"' % itemType
self._menuInfo[menuName][1].append(statusHelp)
apply(command, (), kw)
def _addHotkeyToOptions(self, menuName, kw, traverseSpec):
if (not self['hotkeys'] or kw.has_key('underline') or
not kw.has_key('label')):
return
if type(traverseSpec) == types.IntType:
kw['underline'] = traverseSpec
return
if menuName is None:
menu = self._menu
else:
menu = self.component(menuName)
hotkeyList = []
end = menu.index('end')
if end is not None:
for item in range(end + 1):
if menu.type(item) not in ('separator', 'tearoff'):
underline = \
string.atoi(str(menu.entrycget(item, 'underline')))
if underline != -1:
label = str(menu.entrycget(item, 'label'))
if underline < len(label):
hotkey = string.lower(label[underline])
if hotkey not in hotkeyList:
hotkeyList.append(hotkey)
name = kw['label']
if type(traverseSpec) == types.StringType:
lowerLetter = string.lower(traverseSpec)
if traverseSpec in name and lowerLetter not in hotkeyList:
kw['underline'] = string.index(name, traverseSpec)
else:
targets = string.digits + string.letters
lowerName = string.lower(name)
for letter_index in range(len(name)):
letter = lowerName[letter_index]
if letter in targets and letter not in hotkeyList:
kw['underline'] = letter_index
break
def _menuHelp(self, event, menuName):
if menuName is None:
menu = self._menu
index = menu.index('@%d'% event.x)
else:
menu = self.component(menuName)
index = menu.index('@%d'% event.y)
balloon = self['balloon']
if balloon is not None:
if index is None:
balloon.showstatus('')
else:
if str(menu.cget('tearoff')) == '1':
index = index - 1
if index >= 0:
help = self._menuInfo[menuName][1][index]
balloon.showstatus(help)
def _resetHelpmessage(self, event=None):
balloon = self['balloon']
if balloon is not None:
balloon.clearstatus()
forwardmethods(MainMenuBar, Tkinter.Menu, '_hull')
######################################################################
### File: PmwMenuBar.py
# Manager widget for menus.
import string
import types
import Tkinter
class MenuBar(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('balloon', None, None),
('hotkeys', 1, INITOPT),
('padx', 0, INITOPT),
)
self.defineoptions(kw, optiondefs, dynamicGroups = ('Menu', 'Button'))
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
self._menuInfo = {}
# Map from a menu name to a tuple of information about the menu.
# The first item in the tuple is the name of the parent menu (for
# toplevel menus this is None). The second item in the tuple is
# a list of status help messages for each item in the menu.
# The third item in the tuple is the id of the binding used
# to detect mouse motion to display status help.
# Information for the toplevel menubuttons is not stored here.
self._mydeletecommand = self.component('hull').tk.deletecommand
# Cache this method for use later.
# Check keywords and initialise options.
self.initialiseoptions()
def deletemenuitems(self, menuName, start, end = None):
self.component(menuName + '-menu').delete(start, end)
if end is None:
del self._menuInfo[menuName][1][start]
else:
self._menuInfo[menuName][1][start:end+1] = []
def deletemenu(self, menuName):
"""Delete should be called for cascaded menus before main menus.
"""
# Clean up binding for this menu.
parentName = self._menuInfo[menuName][0]
bindId = self._menuInfo[menuName][2]
_bindtag = 'PmwMenuBar' + str(self) + menuName
self.unbind_class(_bindtag, '<Motion>')
self._mydeletecommand(bindId) # unbind_class does not clean up
del self._menuInfo[menuName]
if parentName is None:
self.destroycomponent(menuName + '-button')
else:
parentMenu = self.component(parentName + '-menu')
menu = self.component(menuName + '-menu')
menuId = str(menu)
for item in range(parentMenu.index('end') + 1):
if parentMenu.type(item) == 'cascade':
itemMenu = str(parentMenu.entrycget(item, 'menu'))
if itemMenu == menuId:
parentMenu.delete(item)
del self._menuInfo[parentName][1][item]
break
self.destroycomponent(menuName + '-menu')
def disableall(self):
for menuName in self._menuInfo.keys():
if self._menuInfo[menuName][0] is None:
menubutton = self.component(menuName + '-button')
menubutton.configure(state = 'disabled')
def enableall(self):
for menuName in self._menuInfo.keys():
if self._menuInfo[menuName][0] is None:
menubutton = self.component(menuName + '-button')
menubutton.configure(state = 'normal')
def addmenu(self, menuName, balloonHelp, statusHelp = None,
side = 'left', traverseSpec = None, **kw):
self._addmenu(None, menuName, balloonHelp, statusHelp,
traverseSpec, side, 'text', kw)
def addcascademenu(self, parentMenuName, menuName, statusHelp = '',
traverseSpec = None, **kw):
self._addmenu(parentMenuName, menuName, None, statusHelp,
traverseSpec, None, 'label', kw)
def _addmenu(self, parentMenuName, menuName, balloonHelp, statusHelp,
traverseSpec, side, textKey, kw):
if (menuName + '-menu') in self.components():
raise ValueError, 'menu "%s" already exists' % menuName
menukw = {}
if kw.has_key('tearoff'):
menukw['tearoff'] = kw['tearoff']
del kw['tearoff']
else:
menukw['tearoff'] = 0
if not kw.has_key(textKey):
kw[textKey] = menuName
self._addHotkeyToOptions(parentMenuName, kw, textKey, traverseSpec)
if parentMenuName is None:
button = apply(self.createcomponent, (menuName + '-button',
(), 'Button',
Tkinter.Menubutton, (self.interior(),)), kw)
button.pack(side=side, padx = self['padx'])
balloon = self['balloon']
if balloon is not None:
balloon.bind(button, balloonHelp, statusHelp)
parentMenu = button
else:
parentMenu = self.component(parentMenuName + '-menu')
apply(parentMenu.add_cascade, (), kw)
self._menuInfo[parentMenuName][1].append(statusHelp)
menu = apply(self.createcomponent, (menuName + '-menu',
(), 'Menu',
Tkinter.Menu, (parentMenu,)), menukw)
if parentMenuName is None:
button.configure(menu = menu)
else:
parentMenu.entryconfigure('end', menu = menu)
# Need to put this binding after the class bindings so that
# menu.index() does not lag behind.
_bindtag = 'PmwMenuBar' + str(self) + menuName
bindId = self.bind_class(_bindtag, '<Motion>',
lambda event=None, self=self, menuName=menuName:
self._menuHelp(menuName))
menu.bindtags(menu.bindtags() + (_bindtag,))
menu.bind('<Leave>', self._resetHelpmessage)
self._menuInfo[menuName] = (parentMenuName, [], bindId)
def addmenuitem(self, menuName, itemType, statusHelp = '',
traverseSpec = None, **kw):
menu = self.component(menuName + '-menu')
if itemType != 'separator':
self._addHotkeyToOptions(menuName, kw, 'label', traverseSpec)
if itemType == 'command':
command = menu.add_command
elif itemType == 'separator':
command = menu.add_separator
elif itemType == 'checkbutton':
command = menu.add_checkbutton
elif itemType == 'radiobutton':
command = menu.add_radiobutton
elif itemType == 'cascade':
command = menu.add_cascade
else:
raise ValueError, 'unknown menuitem type "%s"' % itemType
self._menuInfo[menuName][1].append(statusHelp)
apply(command, (), kw)
def _addHotkeyToOptions(self, menuName, kw, textKey, traverseSpec):
if (not self['hotkeys'] or kw.has_key('underline') or
not kw.has_key(textKey)):
return
if type(traverseSpec) == types.IntType:
kw['underline'] = traverseSpec
return
hotkeyList = []
if menuName is None:
for menuName in self._menuInfo.keys():
if self._menuInfo[menuName][0] is None:
menubutton = self.component(menuName + '-button')
underline = string.atoi(str(menubutton.cget('underline')))
if underline != -1:
label = str(menubutton.cget(textKey))
if underline < len(label):
hotkey = string.lower(label[underline])
if hotkey not in hotkeyList:
hotkeyList.append(hotkey)
else:
menu = self.component(menuName + '-menu')
end = menu.index('end')
if end is not None:
for item in range(end + 1):
if menu.type(item) not in ('separator', 'tearoff'):
underline = string.atoi(
str(menu.entrycget(item, 'underline')))
if underline != -1:
label = str(menu.entrycget(item, textKey))
if underline < len(label):
hotkey = string.lower(label[underline])
if hotkey not in hotkeyList:
hotkeyList.append(hotkey)
name = kw[textKey]
if type(traverseSpec) == types.StringType:
lowerLetter = string.lower(traverseSpec)
if traverseSpec in name and lowerLetter not in hotkeyList:
kw['underline'] = string.index(name, traverseSpec)
else:
targets = string.digits + string.letters
lowerName = string.lower(name)
for letter_index in range(len(name)):
letter = lowerName[letter_index]
if letter in targets and letter not in hotkeyList:
kw['underline'] = letter_index
break
def _menuHelp(self, menuName):
menu = self.component(menuName + '-menu')
index = menu.index('active')
balloon = self['balloon']
if balloon is not None:
if index is None:
balloon.showstatus('')
else:
if str(menu.cget('tearoff')) == '1':
index = index - 1
if index >= 0:
help = self._menuInfo[menuName][1][index]
balloon.showstatus(help)
def _resetHelpmessage(self, event=None):
balloon = self['balloon']
if balloon is not None:
balloon.clearstatus()
######################################################################
### File: PmwMessageBar.py
# Class to display messages in an information line.
import string
import Tkinter
class MessageBar(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
defaultMessageTypes = {
# (priority, showtime, bells, logmessage)
'systemerror' : (5, 10, 2, 1),
'usererror' : (4, 5, 1, 0),
'busy' : (3, 0, 0, 0),
'systemevent' : (2, 5, 0, 0),
'userevent' : (2, 5, 0, 0),
'help' : (1, 5, 0, 0),
'state' : (0, 0, 0, 0),
}
optiondefs = (
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('messagetypes', defaultMessageTypes, INITOPT),
('silent', 0, None),
('sticky', 'ew', INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
self._messageBarEntry = self.createcomponent('entry',
(), None,
Tkinter.Entry, (interior,))
# Can't always use 'disabled', since this greys out text in Tk 8.4.2
try:
self._messageBarEntry.configure(state = 'readonly')
except Tkinter.TclError:
self._messageBarEntry.configure(state = 'disabled')
self._messageBarEntry.grid(column=2, row=2, sticky=self['sticky'])
interior.grid_columnconfigure(2, weight=1)
interior.grid_rowconfigure(2, weight=1)
self.createlabel(interior)
# Initialise instance variables.
self._numPriorities = 0
for info in self['messagetypes'].values():
if self._numPriorities < info[0]:
self._numPriorities = info[0]
self._numPriorities = self._numPriorities + 1
self._timer = [None] * self._numPriorities
self._messagetext = [''] * self._numPriorities
self._activemessage = [0] * self._numPriorities
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
for timerId in self._timer:
if timerId is not None:
self.after_cancel(timerId)
self._timer = [None] * self._numPriorities
MegaWidget.destroy(self)
def message(self, type, text):
# Display a message in the message bar.
(priority, showtime, bells, logmessage) = self['messagetypes'][type]
if not self['silent']:
for i in range(bells):
if i != 0:
self.after(100)
self.bell()
self._activemessage[priority] = 1
if text is None:
text = ''
self._messagetext[priority] = string.replace(text, '\n', ' ')
self._redisplayInfoMessage()
if logmessage:
# Should log this text to a text widget.
pass
if showtime > 0:
if self._timer[priority] is not None:
self.after_cancel(self._timer[priority])
# Define a callback to clear this message after a time.
def _clearmessage(self=self, priority=priority):
self._clearActivemessage(priority)
mseconds = int(showtime * 1000)
self._timer[priority] = self.after(mseconds, _clearmessage)
def helpmessage(self, text):
if text is None:
self.resetmessages('help')
else:
self.message('help', text)
def resetmessages(self, type):
priority = self['messagetypes'][type][0]
self._clearActivemessage(priority)
for messagetype, info in self['messagetypes'].items():
thisPriority = info[0]
showtime = info[1]
if thisPriority < priority and showtime != 0:
self._clearActivemessage(thisPriority)
def _clearActivemessage(self, priority):
self._activemessage[priority] = 0
if self._timer[priority] is not None:
self.after_cancel(self._timer[priority])
self._timer[priority] = None
self._redisplayInfoMessage()
def _redisplayInfoMessage(self):
text = ''
for priority in range(self._numPriorities - 1, -1, -1):
if self._activemessage[priority]:
text = self._messagetext[priority]
break
self._messageBarEntry.configure(state = 'normal')
self._messageBarEntry.delete(0, 'end')
self._messageBarEntry.insert('end', text)
# Can't always use 'disabled', since this greys out text in Tk 8.4.2
try:
self._messageBarEntry.configure(state = 'readonly')
except Tkinter.TclError:
self._messageBarEntry.configure(state = 'disabled')
forwardmethods(MessageBar, Tkinter.Entry, '_messageBarEntry')
######################################################################
### File: PmwMessageDialog.py
# Based on iwidgets2.2.0/messagedialog.itk code.
import Tkinter
class MessageDialog(Dialog):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderx', 20, INITOPT),
('bordery', 20, INITOPT),
('iconmargin', 20, INITOPT),
('iconpos', None, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
Dialog.__init__(self, parent)
# Create the components.
interior = self.interior()
self._message = self.createcomponent('message',
(), None,
Tkinter.Label, (interior,))
iconpos = self['iconpos']
iconmargin = self['iconmargin']
borderx = self['borderx']
bordery = self['bordery']
border_right = 2
border_bottom = 2
if iconpos is None:
self._message.grid(column = 1, row = 1)
else:
self._icon = self.createcomponent('icon',
(), None,
Tkinter.Label, (interior,))
if iconpos not in 'nsew':
raise ValueError, \
'bad iconpos option "%s": should be n, s, e, or w' \
% iconpos
if iconpos in 'nw':
icon = 1
message = 3
else:
icon = 3
message = 1
if iconpos in 'ns':
# vertical layout
self._icon.grid(column = 1, row = icon)
self._message.grid(column = 1, row = message)
interior.grid_rowconfigure(2, minsize = iconmargin)
border_bottom = 4
else:
# horizontal layout
self._icon.grid(column = icon, row = 1)
self._message.grid(column = message, row = 1)
interior.grid_columnconfigure(2, minsize = iconmargin)
border_right = 4
interior.grid_columnconfigure(0, minsize = borderx)
interior.grid_rowconfigure(0, minsize = bordery)
interior.grid_columnconfigure(border_right, minsize = borderx)
interior.grid_rowconfigure(border_bottom, minsize = bordery)
# Check keywords and initialise options.
self.initialiseoptions()
######################################################################
### File: PmwNoteBook.py
import string
import types
import Tkinter
class NoteBook(MegaArchetype):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('hull_highlightthickness', 0, None),
('hull_borderwidth', 0, None),
('arrownavigation', 1, INITOPT),
('borderwidth', 2, INITOPT),
('createcommand', None, None),
('lowercommand', None, None),
('pagemargin', 4, INITOPT),
('raisecommand', None, None),
('tabpos', 'n', INITOPT),
)
self.defineoptions(kw, optiondefs, dynamicGroups = ('Page', 'Tab'))
# Initialise the base class (after defining the options).
MegaArchetype.__init__(self, parent, Tkinter.Canvas)
self.bind('<Map>', self._handleMap)
self.bind('<Configure>', self._handleConfigure)
tabpos = self['tabpos']
if tabpos is not None and tabpos != 'n':
raise ValueError, \
'bad tabpos option %s: should be n or None' % repr(tabpos)
self._withTabs = (tabpos is not None)
self._pageMargin = self['pagemargin']
self._borderWidth = self['borderwidth']
# Use a dictionary as a set of bits indicating what needs to
# be redisplayed the next time _layout() is called. If
# dictionary contains 'topPage' key, the value is the new top
# page to be displayed. None indicates that all pages have
# been deleted and that _layout() should draw a border under where
# the tabs should be.
self._pending = {}
self._pending['size'] = 1
self._pending['borderColor'] = 1
self._pending['topPage'] = None
if self._withTabs:
self._pending['tabs'] = 1
self._canvasSize = None # This gets set by <Configure> events
# Set initial height of space for tabs
if self._withTabs:
self.tabBottom = 35
else:
self.tabBottom = 0
self._lightBorderColor, self._darkBorderColor = \
Color.bordercolors(self, self['hull_background'])
self._pageNames = [] # List of page names
# Map from page name to page info. Each item is itself a
# dictionary containing the following items:
# page the Tkinter.Frame widget for the page
# created set to true the first time the page is raised
# tabbutton the Tkinter.Button widget for the button (if any)
# tabreqwidth requested width of the tab
# tabreqheight requested height of the tab
# tabitems the canvas items for the button: the button
# window item, the lightshadow and the darkshadow
# left the left and right canvas coordinates of the tab
# right
self._pageAttrs = {}
# Name of page currently on top (actually displayed, using
# create_window, not pending). Ignored if current top page
# has been deleted or new top page is pending. None indicates
# no pages in notebook.
self._topPageName = None
# Canvas items used:
# Per tab:
# top and left shadow
# right shadow
# button
# Per notebook:
# page
# top page
# left shadow
# bottom and right shadow
# top (one or two items)
# Canvas tags used:
# lighttag - top and left shadows of tabs and page
# darktag - bottom and right shadows of tabs and page
# (if no tabs then these are reversed)
# (used to color the borders by recolorborders)
# Create page border shadows.
if self._withTabs:
self._pageLeftBorder = self.create_polygon(0, 0, 0, 0, 0, 0,
fill = self._lightBorderColor, tags = 'lighttag')
self._pageBottomRightBorder = self.create_polygon(0, 0, 0, 0, 0, 0,
fill = self._darkBorderColor, tags = 'darktag')
self._pageTop1Border = self.create_polygon(0, 0, 0, 0, 0, 0,
fill = self._darkBorderColor, tags = 'lighttag')
self._pageTop2Border = self.create_polygon(0, 0, 0, 0, 0, 0,
fill = self._darkBorderColor, tags = 'lighttag')
else:
self._pageLeftBorder = self.create_polygon(0, 0, 0, 0, 0, 0,
fill = self._darkBorderColor, tags = 'darktag')
self._pageBottomRightBorder = self.create_polygon(0, 0, 0, 0, 0, 0,
fill = self._lightBorderColor, tags = 'lighttag')
self._pageTopBorder = self.create_polygon(0, 0, 0, 0, 0, 0,
fill = self._darkBorderColor, tags = 'darktag')
# Check keywords and initialise options.
self.initialiseoptions()
def insert(self, pageName, before = 0, **kw):
if self._pageAttrs.has_key(pageName):
msg = 'Page "%s" already exists.' % pageName
raise ValueError, msg
# Do this early to catch bad <before> spec before creating any items.
beforeIndex = self.index(before, 1)
pageOptions = {}
if self._withTabs:
# Default tab button options.
tabOptions = {
'text' : pageName,
'borderwidth' : 0,
}
# Divide the keyword options into the 'page_' and 'tab_' options.
for key in kw.keys():
if key[:5] == 'page_':
pageOptions[key[5:]] = kw[key]
del kw[key]
elif self._withTabs and key[:4] == 'tab_':
tabOptions[key[4:]] = kw[key]
del kw[key]
else:
raise KeyError, 'Unknown option "' + key + '"'
# Create the frame to contain the page.
page = apply(self.createcomponent, (pageName,
(), 'Page',
Tkinter.Frame, self._hull), pageOptions)
attributes = {}
attributes['page'] = page
attributes['created'] = 0
if self._withTabs:
# Create the button for the tab.
def raiseThisPage(self = self, pageName = pageName):
self.selectpage(pageName)
tabOptions['command'] = raiseThisPage
tab = apply(self.createcomponent, (pageName + '-tab',
(), 'Tab',
Tkinter.Button, self._hull), tabOptions)
if self['arrownavigation']:
# Allow the use of the arrow keys for Tab navigation:
def next(event, self = self, pageName = pageName):
self.nextpage(pageName)
def prev(event, self = self, pageName = pageName):
self.previouspage(pageName)
tab.bind('<Left>', prev)
tab.bind('<Right>', next)
attributes['tabbutton'] = tab
attributes['tabreqwidth'] = tab.winfo_reqwidth()
attributes['tabreqheight'] = tab.winfo_reqheight()
# Create the canvas item to manage the tab's button and the items
# for the tab's shadow.
windowitem = self.create_window(0, 0, window = tab, anchor = 'nw')
lightshadow = self.create_polygon(0, 0, 0, 0, 0, 0,
tags = 'lighttag', fill = self._lightBorderColor)
darkshadow = self.create_polygon(0, 0, 0, 0, 0, 0,
tags = 'darktag', fill = self._darkBorderColor)
attributes['tabitems'] = (windowitem, lightshadow, darkshadow)
self._pending['tabs'] = 1
self._pageAttrs[pageName] = attributes
self._pageNames.insert(beforeIndex, pageName)
# If this is the first page added, make it the new top page
# and call the create and raise callbacks.
if self.getcurselection() is None:
self._pending['topPage'] = pageName
self._raiseNewTop(pageName)
self._layout()
return page
def add(self, pageName, **kw):
return apply(self.insert, (pageName, len(self._pageNames)), kw)
def delete(self, *pageNames):
newTopPage = 0
for page in pageNames:
pageIndex = self.index(page)
pageName = self._pageNames[pageIndex]
pageInfo = self._pageAttrs[pageName]
if self.getcurselection() == pageName:
if len(self._pageNames) == 1:
newTopPage = 0
self._pending['topPage'] = None
elif pageIndex == len(self._pageNames) - 1:
newTopPage = 1
self._pending['topPage'] = self._pageNames[pageIndex - 1]
else:
newTopPage = 1
self._pending['topPage'] = self._pageNames[pageIndex + 1]
if self._topPageName == pageName:
self._hull.delete(self._topPageItem)
self._topPageName = None
if self._withTabs:
self.destroycomponent(pageName + '-tab')
apply(self._hull.delete, pageInfo['tabitems'])
self.destroycomponent(pageName)
del self._pageAttrs[pageName]
del self._pageNames[pageIndex]
# If the old top page was deleted and there are still pages
# left in the notebook, call the create and raise callbacks.
if newTopPage:
pageName = self._pending['topPage']
self._raiseNewTop(pageName)
if self._withTabs:
self._pending['tabs'] = 1
self._layout()
def page(self, pageIndex):
pageName = self._pageNames[self.index(pageIndex)]
return self._pageAttrs[pageName]['page']
def pagenames(self):
return list(self._pageNames)
def getcurselection(self):
if self._pending.has_key('topPage'):
return self._pending['topPage']
else:
return self._topPageName
def tab(self, pageIndex):
if self._withTabs:
pageName = self._pageNames[self.index(pageIndex)]
return self._pageAttrs[pageName]['tabbutton']
else:
return None
def index(self, index, forInsert = 0):
listLength = len(self._pageNames)
if type(index) == types.IntType:
if forInsert and index <= listLength:
return index
elif not forInsert and index < listLength:
return index
else:
raise ValueError, 'index "%s" is out of range' % index
elif index is END:
if forInsert:
return listLength
elif listLength > 0:
return listLength - 1
else:
raise ValueError, 'NoteBook has no pages'
elif index is SELECT:
if listLength == 0:
raise ValueError, 'NoteBook has no pages'
return self._pageNames.index(self.getcurselection())
else:
if index in self._pageNames:
return self._pageNames.index(index)
validValues = 'a name, a number, END or SELECT'
raise ValueError, \
'bad index "%s": must be %s' % (index, validValues)
def selectpage(self, page):
pageName = self._pageNames[self.index(page)]
oldTopPage = self.getcurselection()
if pageName != oldTopPage:
self._pending['topPage'] = pageName
if oldTopPage == self._topPageName:
self._hull.delete(self._topPageItem)
cmd = self['lowercommand']
if cmd is not None:
cmd(oldTopPage)
self._raiseNewTop(pageName)
self._layout()
# Set focus to the tab of new top page:
if self._withTabs and self['arrownavigation']:
self._pageAttrs[pageName]['tabbutton'].focus_set()
def previouspage(self, pageIndex = None):
if pageIndex is None:
curpage = self.index(SELECT)
else:
curpage = self.index(pageIndex)
if curpage > 0:
self.selectpage(curpage - 1)
def nextpage(self, pageIndex = None):
if pageIndex is None:
curpage = self.index(SELECT)
else:
curpage = self.index(pageIndex)
if curpage < len(self._pageNames) - 1:
self.selectpage(curpage + 1)
def setnaturalsize(self, pageNames = None):
self.update_idletasks()
maxPageWidth = 1
maxPageHeight = 1
if pageNames is None:
pageNames = self.pagenames()
for pageName in pageNames:
pageInfo = self._pageAttrs[pageName]
page = pageInfo['page']
w = page.winfo_reqwidth()
h = page.winfo_reqheight()
if maxPageWidth < w:
maxPageWidth = w
if maxPageHeight < h:
maxPageHeight = h
pageBorder = self._borderWidth + self._pageMargin
width = maxPageWidth + pageBorder * 2
height = maxPageHeight + pageBorder * 2
if self._withTabs:
maxTabHeight = 0
for pageInfo in self._pageAttrs.values():
if maxTabHeight < pageInfo['tabreqheight']:
maxTabHeight = pageInfo['tabreqheight']
height = height + maxTabHeight + self._borderWidth * 1.5
# Note that, since the hull is a canvas, the width and height
# options specify the geometry *inside* the borderwidth and
# highlightthickness.
self.configure(hull_width = width, hull_height = height)
def recolorborders(self):
self._pending['borderColor'] = 1
self._layout()
def _handleMap(self, event):
self._layout()
def _handleConfigure(self, event):
self._canvasSize = (event.width, event.height)
self._pending['size'] = 1
self._layout()
def _raiseNewTop(self, pageName):
if not self._pageAttrs[pageName]['created']:
self._pageAttrs[pageName]['created'] = 1
cmd = self['createcommand']
if cmd is not None:
cmd(pageName)
cmd = self['raisecommand']
if cmd is not None:
cmd(pageName)
# This is the vertical layout of the notebook, from top (assuming
# tabpos is 'n'):
# hull highlightthickness (top)
# hull borderwidth (top)
# borderwidth (top border of tabs)
# borderwidth * 0.5 (space for bevel)
# tab button (maximum of requested height of all tab buttons)
# borderwidth (border between tabs and page)
# pagemargin (top)
# the page itself
# pagemargin (bottom)
# borderwidth (border below page)
# hull borderwidth (bottom)
# hull highlightthickness (bottom)
#
# canvasBorder is sum of top two elements.
# tabBottom is sum of top five elements.
#
# Horizontal layout (and also vertical layout when tabpos is None):
# hull highlightthickness
# hull borderwidth
# borderwidth
# pagemargin
# the page itself
# pagemargin
# borderwidth
# hull borderwidth
# hull highlightthickness
#
def _layout(self):
if not self.winfo_ismapped() or self._canvasSize is None:
# Don't layout if the window is not displayed, or we
# haven't yet received a <Configure> event.
return
hullWidth, hullHeight = self._canvasSize
borderWidth = self._borderWidth
canvasBorder = string.atoi(self._hull['borderwidth']) + \
string.atoi(self._hull['highlightthickness'])
if not self._withTabs:
self.tabBottom = canvasBorder
oldTabBottom = self.tabBottom
if self._pending.has_key('borderColor'):
self._lightBorderColor, self._darkBorderColor = \
Color.bordercolors(self, self['hull_background'])
# Draw all the tabs.
if self._withTabs and (self._pending.has_key('tabs') or
self._pending.has_key('size')):
# Find total requested width and maximum requested height
# of tabs.
sumTabReqWidth = 0
maxTabHeight = 0
for pageInfo in self._pageAttrs.values():
sumTabReqWidth = sumTabReqWidth + pageInfo['tabreqwidth']
if maxTabHeight < pageInfo['tabreqheight']:
maxTabHeight = pageInfo['tabreqheight']
if maxTabHeight != 0:
# Add the top tab border plus a bit for the angled corners
self.tabBottom = canvasBorder + maxTabHeight + borderWidth * 1.5
# Prepare for drawing the border around each tab button.
tabTop = canvasBorder
tabTop2 = tabTop + borderWidth
tabTop3 = tabTop + borderWidth * 1.5
tabBottom2 = self.tabBottom
tabBottom = self.tabBottom + borderWidth
numTabs = len(self._pageNames)
availableWidth = hullWidth - 2 * canvasBorder - \
numTabs * 2 * borderWidth
x = canvasBorder
cumTabReqWidth = 0
cumTabWidth = 0
# Position all the tabs.
for pageName in self._pageNames:
pageInfo = self._pageAttrs[pageName]
(windowitem, lightshadow, darkshadow) = pageInfo['tabitems']
if sumTabReqWidth <= availableWidth:
tabwidth = pageInfo['tabreqwidth']
else:
# This ugly calculation ensures that, when the
# notebook is not wide enough for the requested
# widths of the tabs, the total width given to
# the tabs exactly equals the available width,
# without rounding errors.
cumTabReqWidth = cumTabReqWidth + pageInfo['tabreqwidth']
tmp = (2*cumTabReqWidth*availableWidth + sumTabReqWidth) \
/ (2 * sumTabReqWidth)
tabwidth = tmp - cumTabWidth
cumTabWidth = tmp
# Position the tab's button canvas item.
self.coords(windowitem, x + borderWidth, tabTop3)
self.itemconfigure(windowitem,
width = tabwidth, height = maxTabHeight)
# Make a beautiful border around the tab.
left = x
left2 = left + borderWidth
left3 = left + borderWidth * 1.5
right = left + tabwidth + 2 * borderWidth
right2 = left + tabwidth + borderWidth
right3 = left + tabwidth + borderWidth * 0.5
self.coords(lightshadow,
left, tabBottom2, left, tabTop2, left2, tabTop,
right2, tabTop, right3, tabTop2, left3, tabTop2,
left2, tabTop3, left2, tabBottom,
)
self.coords(darkshadow,
right2, tabTop, right, tabTop2, right, tabBottom2,
right2, tabBottom, right2, tabTop3, right3, tabTop2,
)
pageInfo['left'] = left
pageInfo['right'] = right
x = x + tabwidth + 2 * borderWidth
# Redraw shadow under tabs so that it appears that tab for old
# top page is lowered and that tab for new top page is raised.
if self._withTabs and (self._pending.has_key('topPage') or
self._pending.has_key('tabs') or self._pending.has_key('size')):
if self.getcurselection() is None:
# No pages, so draw line across top of page area.
self.coords(self._pageTop1Border,
canvasBorder, self.tabBottom,
hullWidth - canvasBorder, self.tabBottom,
hullWidth - canvasBorder - borderWidth,
self.tabBottom + borderWidth,
borderWidth + canvasBorder, self.tabBottom + borderWidth,
)
# Ignore second top border.
self.coords(self._pageTop2Border, 0, 0, 0, 0, 0, 0)
else:
# Draw two lines, one on each side of the tab for the
# top page, so that the tab appears to be raised.
pageInfo = self._pageAttrs[self.getcurselection()]
left = pageInfo['left']
right = pageInfo['right']
self.coords(self._pageTop1Border,
canvasBorder, self.tabBottom,
left, self.tabBottom,
left + borderWidth, self.tabBottom + borderWidth,
canvasBorder + borderWidth, self.tabBottom + borderWidth,
)
self.coords(self._pageTop2Border,
right, self.tabBottom,
hullWidth - canvasBorder, self.tabBottom,
hullWidth - canvasBorder - borderWidth,
self.tabBottom + borderWidth,
right - borderWidth, self.tabBottom + borderWidth,
)
# Prevent bottom of dark border of tabs appearing over
# page top border.
self.tag_raise(self._pageTop1Border)
self.tag_raise(self._pageTop2Border)
# Position the page border shadows.
if self._pending.has_key('size') or oldTabBottom != self.tabBottom:
self.coords(self._pageLeftBorder,
canvasBorder, self.tabBottom,
borderWidth + canvasBorder,
self.tabBottom + borderWidth,
borderWidth + canvasBorder,
hullHeight - canvasBorder - borderWidth,
canvasBorder, hullHeight - canvasBorder,
)
self.coords(self._pageBottomRightBorder,
hullWidth - canvasBorder, self.tabBottom,
hullWidth - canvasBorder, hullHeight - canvasBorder,
canvasBorder, hullHeight - canvasBorder,
borderWidth + canvasBorder,
hullHeight - canvasBorder - borderWidth,
hullWidth - canvasBorder - borderWidth,
hullHeight - canvasBorder - borderWidth,
hullWidth - canvasBorder - borderWidth,
self.tabBottom + borderWidth,
)
if not self._withTabs:
self.coords(self._pageTopBorder,
canvasBorder, self.tabBottom,
hullWidth - canvasBorder, self.tabBottom,
hullWidth - canvasBorder - borderWidth,
self.tabBottom + borderWidth,
borderWidth + canvasBorder, self.tabBottom + borderWidth,
)
# Color borders.
if self._pending.has_key('borderColor'):
self.itemconfigure('lighttag', fill = self._lightBorderColor)
self.itemconfigure('darktag', fill = self._darkBorderColor)
newTopPage = self._pending.get('topPage')
pageBorder = borderWidth + self._pageMargin
# Raise new top page.
if newTopPage is not None:
self._topPageName = newTopPage
self._topPageItem = self.create_window(
pageBorder + canvasBorder, self.tabBottom + pageBorder,
window = self._pageAttrs[newTopPage]['page'],
anchor = 'nw',
)
# Change position of top page if tab height has changed.
if self._topPageName is not None and oldTabBottom != self.tabBottom:
self.coords(self._topPageItem,
pageBorder + canvasBorder, self.tabBottom + pageBorder)
# Change size of top page if,
# 1) there is a new top page.
# 2) canvas size has changed, but not if there is no top
# page (eg: initially or when all pages deleted).
# 3) tab height has changed, due to difference in the height of a tab
if (newTopPage is not None or \
self._pending.has_key('size') and self._topPageName is not None
or oldTabBottom != self.tabBottom):
self.itemconfigure(self._topPageItem,
width = hullWidth - 2 * canvasBorder - pageBorder * 2,
height = hullHeight - 2 * canvasBorder - pageBorder * 2 -
(self.tabBottom - canvasBorder),
)
self._pending = {}
# Need to do forwarding to get the pack, grid, etc methods.
# Unfortunately this means that all the other canvas methods are also
# forwarded.
forwardmethods(NoteBook, Tkinter.Canvas, '_hull')
######################################################################
### File: PmwOptionMenu.py
import types
import Tkinter
import sys
class OptionMenu(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('command', None, None),
('items', (), INITOPT),
('initialitem', None, INITOPT),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('sticky', 'ew', INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
self._menubutton = self.createcomponent('menubutton',
(), None,
Tkinter.Menubutton, (interior,),
borderwidth = 2,
indicatoron = 1,
relief = 'raised',
anchor = 'c',
highlightthickness = 2,
direction = 'flush',
takefocus = 1,
)
self._menubutton.grid(column = 2, row = 2, sticky = self['sticky'])
self._menu = self.createcomponent('menu',
(), None,
Tkinter.Menu, (self._menubutton,),
tearoff=0
)
self._menubutton.configure(menu = self._menu)
interior.grid_columnconfigure(2, weight = 1)
interior.grid_rowconfigure(2, weight = 1)
# Create the label.
self.createlabel(interior)
# Add the items specified by the initialisation option.
self._itemList = []
self.setitems(self['items'], self['initialitem'])
# Check keywords and initialise options.
self.initialiseoptions()
def setitems(self, items, index = None):
# python version check
# python versions >= 2.5.4 automatically clean commands
# and manually cleaning them causes errors when deleting items
if sys.version_info[0] * 100 + sys.version_info[1] * 10 + \
sys.version_info[2] < 254:
# Clean up old items and callback commands.
for oldIndex in range(len(self._itemList)):
tclCommandName = str(self._menu.entrycget(oldIndex, 'command'))
if tclCommandName != '':
self._menu.deletecommand(tclCommandName)
self._menu.delete(0, 'end')
self._itemList = list(items)
# Set the items in the menu component.
for item in items:
self._menu.add_command(label = item,
command = lambda self = self, item = item: self._invoke(item))
# Set the currently selected value.
if index is None:
var = str(self._menubutton.cget('textvariable'))
if var != '':
# None means do not change text variable.
return
if len(items) == 0:
text = ''
elif str(self._menubutton.cget('text')) in items:
# Do not change selection if it is still valid
return
else:
text = items[0]
else:
index = self.index(index)
text = self._itemList[index]
self.setvalue(text)
def getcurselection(self):
var = str(self._menubutton.cget('textvariable'))
if var == '':
return str(self._menubutton.cget('text'))
else:
return self._menu.tk.globalgetvar(var)
def getvalue(self):
return self.getcurselection()
def setvalue(self, text):
var = str(self._menubutton.cget('textvariable'))
if var == '':
self._menubutton.configure(text = text)
else:
self._menu.tk.globalsetvar(var, text)
def index(self, index):
listLength = len(self._itemList)
if type(index) == types.IntType:
if index < listLength:
return index
else:
raise ValueError, 'index "%s" is out of range' % index
elif index is END:
if listLength > 0:
return listLength - 1
else:
raise ValueError, 'OptionMenu has no items'
else:
if index is SELECT:
if listLength > 0:
index = self.getcurselection()
else:
raise ValueError, 'OptionMenu has no items'
if index in self._itemList:
return self._itemList.index(index)
raise ValueError, \
'bad index "%s": must be a ' \
'name, a number, END or SELECT' % (index,)
def invoke(self, index = SELECT):
index = self.index(index)
text = self._itemList[index]
return self._invoke(text)
def _invoke(self, text):
self.setvalue(text)
command = self['command']
if callable(command):
return command(text)
######################################################################
### File: PmwPanedWidget.py
# PanedWidget
# a frame which may contain several resizable sub-frames
import string
import sys
import types
import Tkinter
class PanedWidget(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('command', None, None),
('orient', 'vertical', INITOPT),
('separatorrelief', 'sunken', INITOPT),
('separatorthickness', 2, INITOPT),
('handlesize', 8, INITOPT),
('hull_width', 400, None),
('hull_height', 400, None),
)
self.defineoptions(kw, optiondefs,
dynamicGroups = ('Frame', 'Separator', 'Handle'))
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
self.bind('<Configure>', self._handleConfigure)
if self['orient'] not in ('horizontal', 'vertical'):
raise ValueError, 'bad orient option ' + repr(self['orient']) + \
': must be either \'horizontal\' or \'vertical\''
self._separatorThickness = self['separatorthickness']
self._handleSize = self['handlesize']
self._paneNames = [] # List of pane names
self._paneAttrs = {} # Map from pane name to pane info
self._timerId = None
self._frame = {}
self._separator = []
self._button = []
self._totalSize = 0
self._movePending = 0
self._relsize = {}
self._relmin = {}
self._relmax = {}
self._size = {}
self._min = {}
self._max = {}
self._rootp = None
self._curSize = None
self._beforeLimit = None
self._afterLimit = None
self._buttonIsDown = 0
self._majorSize = 100
self._minorSize = 100
# Check keywords and initialise options.
self.initialiseoptions()
def insert(self, name, before = 0, **kw):
# Parse <kw> for options.
self._initPaneOptions(name)
self._parsePaneOptions(name, kw)
insertPos = self._nameToIndex(before)
atEnd = (insertPos == len(self._paneNames))
# Add the frame.
self._paneNames[insertPos:insertPos] = [name]
self._frame[name] = self.createcomponent(name,
(), 'Frame',
Tkinter.Frame, (self.interior(),))
# Add separator, if necessary.
if len(self._paneNames) > 1:
self._addSeparator()
else:
self._separator.append(None)
self._button.append(None)
# Add the new frame and adjust the PanedWidget
if atEnd:
size = self._size[name]
if size > 0 or self._relsize[name] is not None:
if self['orient'] == 'vertical':
self._frame[name].place(x=0, relwidth=1,
height=size, y=self._totalSize)
else:
self._frame[name].place(y=0, relheight=1,
width=size, x=self._totalSize)
else:
if self['orient'] == 'vertical':
self._frame[name].place(x=0, relwidth=1,
y=self._totalSize)
else:
self._frame[name].place(y=0, relheight=1,
x=self._totalSize)
else:
self._updateSizes()
self._totalSize = self._totalSize + self._size[name]
return self._frame[name]
def add(self, name, **kw):
return apply(self.insert, (name, len(self._paneNames)), kw)
def delete(self, name):
deletePos = self._nameToIndex(name)
name = self._paneNames[deletePos]
self.destroycomponent(name)
del self._paneNames[deletePos]
del self._frame[name]
del self._size[name]
del self._min[name]
del self._max[name]
del self._relsize[name]
del self._relmin[name]
del self._relmax[name]
last = len(self._paneNames)
del self._separator[last]
del self._button[last]
if last > 0:
self.destroycomponent(self._sepName(last))
self.destroycomponent(self._buttonName(last))
self._plotHandles()
def setnaturalsize(self):
self.update_idletasks()
totalWidth = 0
totalHeight = 0
maxWidth = 0
maxHeight = 0
for name in self._paneNames:
frame = self._frame[name]
w = frame.winfo_reqwidth()
h = frame.winfo_reqheight()
totalWidth = totalWidth + w
totalHeight = totalHeight + h
if maxWidth < w:
maxWidth = w
if maxHeight < h:
maxHeight = h
# Note that, since the hull is a frame, the width and height
# options specify the geometry *outside* the borderwidth and
# highlightthickness.
bw = string.atoi(str(self.cget('hull_borderwidth')))
hl = string.atoi(str(self.cget('hull_highlightthickness')))
extra = (bw + hl) * 2
if str(self.cget('orient')) == 'horizontal':
totalWidth = totalWidth + extra
maxHeight = maxHeight + extra
self.configure(hull_width = totalWidth, hull_height = maxHeight)
else:
totalHeight = (totalHeight + extra +
(len(self._paneNames) - 1) * self._separatorThickness)
maxWidth = maxWidth + extra
self.configure(hull_width = maxWidth, hull_height = totalHeight)
def move(self, name, newPos, newPosOffset = 0):
# see if we can spare ourselves some work
numPanes = len(self._paneNames)
if numPanes < 2:
return
newPos = self._nameToIndex(newPos) + newPosOffset
if newPos < 0 or newPos >=numPanes:
return
deletePos = self._nameToIndex(name)
if deletePos == newPos:
# inserting over ourself is a no-op
return
# delete name from old position in list
name = self._paneNames[deletePos]
del self._paneNames[deletePos]
# place in new position
self._paneNames[newPos:newPos] = [name]
# force everything to redraw
self._plotHandles()
self._updateSizes()
def _nameToIndex(self, nameOrIndex):
try:
pos = self._paneNames.index(nameOrIndex)
except ValueError:
pos = nameOrIndex
return pos
def _initPaneOptions(self, name):
# Set defaults.
self._size[name] = 0
self._relsize[name] = None
self._min[name] = 0
self._relmin[name] = None
self._max[name] = 100000
self._relmax[name] = None
def _parsePaneOptions(self, name, args):
# Parse <args> for options.
for arg, value in args.items():
if type(value) == types.FloatType:
relvalue = value
value = self._absSize(relvalue)
else:
relvalue = None
if arg == 'size':
self._size[name], self._relsize[name] = value, relvalue
elif arg == 'min':
self._min[name], self._relmin[name] = value, relvalue
elif arg == 'max':
self._max[name], self._relmax[name] = value, relvalue
else:
raise ValueError, 'keyword must be "size", "min", or "max"'
def _absSize(self, relvalue):
return int(round(relvalue * self._majorSize))
def _sepName(self, n):
return 'separator-%d' % n
def _buttonName(self, n):
return 'handle-%d' % n
def _addSeparator(self):
n = len(self._paneNames) - 1
downFunc = lambda event, s = self, num=n: s._btnDown(event, num)
upFunc = lambda event, s = self, num=n: s._btnUp(event, num)
moveFunc = lambda event, s = self, num=n: s._btnMove(event, num)
# Create the line dividing the panes.
sep = self.createcomponent(self._sepName(n),
(), 'Separator',
Tkinter.Frame, (self.interior(),),
borderwidth = 1,
relief = self['separatorrelief'])
self._separator.append(sep)
sep.bind('<ButtonPress-1>', downFunc)
sep.bind('<Any-ButtonRelease-1>', upFunc)
sep.bind('<B1-Motion>', moveFunc)
if self['orient'] == 'vertical':
cursor = 'sb_v_double_arrow'
sep.configure(height = self._separatorThickness,
width = 10000, cursor = cursor)
else:
cursor = 'sb_h_double_arrow'
sep.configure(width = self._separatorThickness,
height = 10000, cursor = cursor)
self._totalSize = self._totalSize + self._separatorThickness
# Create the handle on the dividing line.
handle = self.createcomponent(self._buttonName(n),
(), 'Handle',
Tkinter.Frame, (self.interior(),),
relief = 'raised',
borderwidth = 1,
width = self._handleSize,
height = self._handleSize,
cursor = cursor,
)
self._button.append(handle)
handle.bind('<ButtonPress-1>', downFunc)
handle.bind('<Any-ButtonRelease-1>', upFunc)
handle.bind('<B1-Motion>', moveFunc)
self._plotHandles()
for i in range(1, len(self._paneNames)):
self._separator[i].tkraise()
for i in range(1, len(self._paneNames)):
self._button[i].tkraise()
def _btnUp(self, event, item):
self._buttonIsDown = 0
self._updateSizes()
try:
self._button[item].configure(relief='raised')
except:
pass
def _btnDown(self, event, item):
self._button[item].configure(relief='sunken')
self._getMotionLimit(item)
self._buttonIsDown = 1
self._movePending = 0
def _handleConfigure(self, event = None):
self._getNaturalSizes()
if self._totalSize == 0:
return
iterRange = list(self._paneNames)
iterRange.reverse()
if self._majorSize > self._totalSize:
n = self._majorSize - self._totalSize
self._iterate(iterRange, self._grow, n)
elif self._majorSize < self._totalSize:
n = self._totalSize - self._majorSize
self._iterate(iterRange, self._shrink, n)
self._plotHandles()
self._updateSizes()
def _getNaturalSizes(self):
# Must call this in order to get correct winfo_width, winfo_height
self.update_idletasks()
self._totalSize = 0
if self['orient'] == 'vertical':
self._majorSize = self.winfo_height()
self._minorSize = self.winfo_width()
majorspec = Tkinter.Frame.winfo_reqheight
else:
self._majorSize = self.winfo_width()
self._minorSize = self.winfo_height()
majorspec = Tkinter.Frame.winfo_reqwidth
bw = string.atoi(str(self.cget('hull_borderwidth')))
hl = string.atoi(str(self.cget('hull_highlightthickness')))
extra = (bw + hl) * 2
self._majorSize = self._majorSize - extra
self._minorSize = self._minorSize - extra
if self._majorSize < 0:
self._majorSize = 0
if self._minorSize < 0:
self._minorSize = 0
for name in self._paneNames:
# adjust the absolute sizes first...
if self._relsize[name] is None:
#special case
if self._size[name] == 0:
self._size[name] = apply(majorspec, (self._frame[name],))
self._setrel(name)
else:
self._size[name] = self._absSize(self._relsize[name])
if self._relmin[name] is not None:
self._min[name] = self._absSize(self._relmin[name])
if self._relmax[name] is not None:
self._max[name] = self._absSize(self._relmax[name])
# now adjust sizes
if self._size[name] < self._min[name]:
self._size[name] = self._min[name]
self._setrel(name)
if self._size[name] > self._max[name]:
self._size[name] = self._max[name]
self._setrel(name)
self._totalSize = self._totalSize + self._size[name]
# adjust for separators
self._totalSize = (self._totalSize +
(len(self._paneNames) - 1) * self._separatorThickness)
def _setrel(self, name):
if self._relsize[name] is not None:
if self._majorSize != 0:
self._relsize[name] = round(self._size[name]) / self._majorSize
def _iterate(self, names, proc, n):
for i in names:
n = apply(proc, (i, n))
if n == 0:
break
def _grow(self, name, n):
canGrow = self._max[name] - self._size[name]
if canGrow > n:
self._size[name] = self._size[name] + n
self._setrel(name)
return 0
elif canGrow > 0:
self._size[name] = self._max[name]
self._setrel(name)
n = n - canGrow
return n
def _shrink(self, name, n):
canShrink = self._size[name] - self._min[name]
if canShrink > n:
self._size[name] = self._size[name] - n
self._setrel(name)
return 0
elif canShrink > 0:
self._size[name] = self._min[name]
self._setrel(name)
n = n - canShrink
return n
def _updateSizes(self):
totalSize = 0
for name in self._paneNames:
size = self._size[name]
if self['orient'] == 'vertical':
self._frame[name].place(x = 0, relwidth = 1,
y = totalSize,
height = size)
else:
self._frame[name].place(y = 0, relheight = 1,
x = totalSize,
width = size)
totalSize = totalSize + size + self._separatorThickness
# Invoke the callback command
cmd = self['command']
if callable(cmd):
cmd(map(lambda x, s = self: s._size[x], self._paneNames))
def _plotHandles(self):
if len(self._paneNames) == 0:
return
if self['orient'] == 'vertical':
btnp = self._minorSize - 13
else:
h = self._minorSize
if h > 18:
btnp = 9
else:
btnp = h - 9
firstPane = self._paneNames[0]
totalSize = self._size[firstPane]
first = 1
last = len(self._paneNames) - 1
# loop from first to last, inclusive
for i in range(1, last + 1):
handlepos = totalSize - 3
prevSize = self._size[self._paneNames[i - 1]]
nextSize = self._size[self._paneNames[i]]
offset1 = 0
if i == first:
if prevSize < 4:
offset1 = 4 - prevSize
else:
if prevSize < 8:
offset1 = (8 - prevSize) / 2
offset2 = 0
if i == last:
if nextSize < 4:
offset2 = nextSize - 4
else:
if nextSize < 8:
offset2 = (nextSize - 8) / 2
handlepos = handlepos + offset1
if self['orient'] == 'vertical':
height = 8 - offset1 + offset2
if height > 1:
self._button[i].configure(height = height)
self._button[i].place(x = btnp, y = handlepos)
else:
self._button[i].place_forget()
self._separator[i].place(x = 0, y = totalSize,
relwidth = 1)
else:
width = 8 - offset1 + offset2
if width > 1:
self._button[i].configure(width = width)
self._button[i].place(y = btnp, x = handlepos)
else:
self._button[i].place_forget()
self._separator[i].place(y = 0, x = totalSize,
relheight = 1)
totalSize = totalSize + nextSize + self._separatorThickness
def pane(self, name):
return self._frame[self._paneNames[self._nameToIndex(name)]]
# Return the name of all panes
def panes(self):
return list(self._paneNames)
def configurepane(self, name, **kw):
name = self._paneNames[self._nameToIndex(name)]
self._parsePaneOptions(name, kw)
self._handleConfigure()
def updatelayout(self):
self._handleConfigure()
def _getMotionLimit(self, item):
curBefore = (item - 1) * self._separatorThickness
minBefore, maxBefore = curBefore, curBefore
for name in self._paneNames[:item]:
curBefore = curBefore + self._size[name]
minBefore = minBefore + self._min[name]
maxBefore = maxBefore + self._max[name]
curAfter = (len(self._paneNames) - item) * self._separatorThickness
minAfter, maxAfter = curAfter, curAfter
for name in self._paneNames[item:]:
curAfter = curAfter + self._size[name]
minAfter = minAfter + self._min[name]
maxAfter = maxAfter + self._max[name]
beforeToGo = min(curBefore - minBefore, maxAfter - curAfter)
afterToGo = min(curAfter - minAfter, maxBefore - curBefore)
self._beforeLimit = curBefore - beforeToGo
self._afterLimit = curBefore + afterToGo
self._curSize = curBefore
self._plotHandles()
# Compress the motion so that update is quick even on slow machines
#
# theRootp = root position (either rootx or rooty)
def _btnMove(self, event, item):
self._rootp = event
if self._movePending == 0:
self._timerId = self.after_idle(
lambda s = self, i = item: s._btnMoveCompressed(i))
self._movePending = 1
def destroy(self):
if self._timerId is not None:
self.after_cancel(self._timerId)
self._timerId = None
MegaWidget.destroy(self)
def _btnMoveCompressed(self, item):
if not self._buttonIsDown:
return
if self['orient'] == 'vertical':
p = self._rootp.y_root - self.winfo_rooty()
else:
p = self._rootp.x_root - self.winfo_rootx()
if p == self._curSize:
self._movePending = 0
return
if p < self._beforeLimit:
p = self._beforeLimit
if p >= self._afterLimit:
p = self._afterLimit
self._calculateChange(item, p)
self.update_idletasks()
self._movePending = 0
# Calculate the change in response to mouse motions
def _calculateChange(self, item, p):
if p < self._curSize:
self._moveBefore(item, p)
elif p > self._curSize:
self._moveAfter(item, p)
self._plotHandles()
def _moveBefore(self, item, p):
n = self._curSize - p
# Shrink the frames before
iterRange = list(self._paneNames[:item])
iterRange.reverse()
self._iterate(iterRange, self._shrink, n)
# Adjust the frames after
iterRange = self._paneNames[item:]
self._iterate(iterRange, self._grow, n)
self._curSize = p
def _moveAfter(self, item, p):
n = p - self._curSize
# Shrink the frames after
iterRange = self._paneNames[item:]
self._iterate(iterRange, self._shrink, n)
# Adjust the frames before
iterRange = list(self._paneNames[:item])
iterRange.reverse()
self._iterate(iterRange, self._grow, n)
self._curSize = p
######################################################################
### File: PmwPromptDialog.py
# Based on iwidgets2.2.0/promptdialog.itk code.
# A Dialog with an entryfield
class PromptDialog(Dialog):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderx', 20, INITOPT),
('bordery', 20, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
Dialog.__init__(self, parent)
# Create the components.
interior = self.interior()
aliases = (
('entry', 'entryfield_entry'),
('label', 'entryfield_label'),
)
self._promptDialogEntry = self.createcomponent('entryfield',
aliases, None,
EntryField, (interior,))
self._promptDialogEntry.pack(fill='x', expand=1,
padx = self['borderx'], pady = self['bordery'])
if not kw.has_key('activatecommand'):
# Whenever this dialog is activated, set the focus to the
# EntryField's entry widget.
tkentry = self.component('entry')
self.configure(activatecommand = tkentry.focus_set)
# Check keywords and initialise options.
self.initialiseoptions()
# Supply aliases to some of the entry component methods.
def insertentry(self, index, text):
self._promptDialogEntry.insert(index, text)
def deleteentry(self, first, last=None):
self._promptDialogEntry.delete(first, last)
def indexentry(self, index):
return self._promptDialogEntry.index(index)
forwardmethods(PromptDialog, EntryField, '_promptDialogEntry')
######################################################################
### File: PmwRadioSelect.py
import types
import Tkinter
class RadioSelect(MegaWidget):
# A collection of several buttons. In single mode, only one
# button may be selected. In multiple mode, any number of buttons
# may be selected.
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('buttontype', 'button', INITOPT),
('command', None, None),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('orient', 'horizontal', INITOPT),
('padx', 5, INITOPT),
('pady', 5, INITOPT),
('selectmode', 'single', INITOPT),
)
self.defineoptions(kw, optiondefs, dynamicGroups = ('Button',))
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
if self['labelpos'] is None:
self._radioSelectFrame = self._hull
else:
self._radioSelectFrame = self.createcomponent('frame',
(), None,
Tkinter.Frame, (interior,))
self._radioSelectFrame.grid(column=2, row=2, sticky='nsew')
interior.grid_columnconfigure(2, weight=1)
interior.grid_rowconfigure(2, weight=1)
self.createlabel(interior)
# Initialise instance variables.
self._buttonList = []
if self['selectmode'] == 'single':
self._singleSelect = 1
elif self['selectmode'] == 'multiple':
self._singleSelect = 0
else:
raise ValueError, 'bad selectmode option "' + \
self['selectmode'] + '": should be single or multiple'
if self['buttontype'] == 'button':
self.buttonClass = Tkinter.Button
elif self['buttontype'] == 'radiobutton':
self._singleSelect = 1
self.var = Tkinter.StringVar()
self.buttonClass = Tkinter.Radiobutton
elif self['buttontype'] == 'checkbutton':
self._singleSelect = 0
self.buttonClass = Tkinter.Checkbutton
else:
raise ValueError, 'bad buttontype option "' + \
self['buttontype'] + \
'": should be button, radiobutton or checkbutton'
if self._singleSelect:
self.selection = None
else:
self.selection = []
if self['orient'] not in ('horizontal', 'vertical'):
raise ValueError, 'bad orient option ' + repr(self['orient']) + \
': must be either \'horizontal\' or \'vertical\''
# Check keywords and initialise options.
self.initialiseoptions()
def getcurselection(self):
if self._singleSelect:
return self.selection
else:
return tuple(self.selection)
def getvalue(self):
return self.getcurselection()
def setvalue(self, textOrList):
if self._singleSelect:
self.__setSingleValue(textOrList)
else:
# Multiple selections
oldselection = self.selection
self.selection = textOrList
for button in self._buttonList:
if button in oldselection:
if button not in self.selection:
# button is currently selected but should not be
widget = self.component(button)
if self['buttontype'] == 'checkbutton':
widget.deselect()
else: # Button
widget.configure(relief='raised')
else:
if button in self.selection:
# button is not currently selected but should be
widget = self.component(button)
if self['buttontype'] == 'checkbutton':
widget.select()
else: # Button
widget.configure(relief='sunken')
def numbuttons(self):
return len(self._buttonList)
def index(self, index):
# Return the integer index of the button with the given index.
listLength = len(self._buttonList)
if type(index) == types.IntType:
if index < listLength:
return index
else:
raise ValueError, 'index "%s" is out of range' % index
elif index is END:
if listLength > 0:
return listLength - 1
else:
raise ValueError, 'RadioSelect has no buttons'
else:
for count in range(listLength):
name = self._buttonList[count]
if index == name:
return count
validValues = 'a name, a number or END'
raise ValueError, \
'bad index "%s": must be %s' % (index, validValues)
def button(self, buttonIndex):
name = self._buttonList[self.index(buttonIndex)]
return self.component(name)
def add(self, componentName, **kw):
if componentName in self._buttonList:
raise ValueError, 'button "%s" already exists' % componentName
kw['command'] = \
lambda self=self, name=componentName: self.invoke(name)
if not kw.has_key('text'):
kw['text'] = componentName
if self['buttontype'] == 'radiobutton':
if not kw.has_key('anchor'):
kw['anchor'] = 'w'
if not kw.has_key('variable'):
kw['variable'] = self.var
if not kw.has_key('value'):
kw['value'] = kw['text']
elif self['buttontype'] == 'checkbutton':
if not kw.has_key('anchor'):
kw['anchor'] = 'w'
button = apply(self.createcomponent, (componentName,
(), 'Button',
self.buttonClass, (self._radioSelectFrame,)), kw)
if self['orient'] == 'horizontal':
self._radioSelectFrame.grid_rowconfigure(0, weight=1)
col = len(self._buttonList)
button.grid(column=col, row=0, padx = self['padx'],
pady = self['pady'], sticky='nsew')
self._radioSelectFrame.grid_columnconfigure(col, weight=1)
else:
self._radioSelectFrame.grid_columnconfigure(0, weight=1)
row = len(self._buttonList)
button.grid(column=0, row=row, padx = self['padx'],
pady = self['pady'], sticky='ew')
self._radioSelectFrame.grid_rowconfigure(row, weight=1)
self._buttonList.append(componentName)
return button
def deleteall(self):
for name in self._buttonList:
self.destroycomponent(name)
self._buttonList = []
if self._singleSelect:
self.selection = None
else:
self.selection = []
def __setSingleValue(self, value):
self.selection = value
if self['buttontype'] == 'radiobutton':
widget = self.component(value)
widget.select()
else: # Button
for button in self._buttonList:
widget = self.component(button)
if button == value:
widget.configure(relief='sunken')
else:
widget.configure(relief='raised')
def invoke(self, index):
index = self.index(index)
name = self._buttonList[index]
if self._singleSelect:
self.__setSingleValue(name)
command = self['command']
if callable(command):
return command(name)
else:
# Multiple selections
widget = self.component(name)
if name in self.selection:
if self['buttontype'] == 'checkbutton':
widget.deselect()
else:
widget.configure(relief='raised')
self.selection.remove(name)
state = 0
else:
if self['buttontype'] == 'checkbutton':
widget.select()
else:
widget.configure(relief='sunken')
self.selection.append(name)
state = 1
command = self['command']
if callable(command):
return command(name, state)
######################################################################
### File: PmwScrolledCanvas.py
import Tkinter
class ScrolledCanvas(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderframe', 0, INITOPT),
('canvasmargin', 0, INITOPT),
('hscrollmode', 'dynamic', self._hscrollMode),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('scrollmargin', 2, INITOPT),
('usehullsize', 0, INITOPT),
('vscrollmode', 'dynamic', self._vscrollMode),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
self.origInterior = MegaWidget.interior(self)
if self['usehullsize']:
self.origInterior.grid_propagate(0)
if self['borderframe']:
# Create a frame widget to act as the border of the canvas.
self._borderframe = self.createcomponent('borderframe',
(), None,
Tkinter.Frame, (self.origInterior,),
relief = 'sunken',
borderwidth = 2,
)
self._borderframe.grid(row = 2, column = 2, sticky = 'news')
# Create the canvas widget.
self._canvas = self.createcomponent('canvas',
(), None,
Tkinter.Canvas, (self._borderframe,),
highlightthickness = 0,
borderwidth = 0,
)
self._canvas.pack(fill = 'both', expand = 1)
else:
# Create the canvas widget.
self._canvas = self.createcomponent('canvas',
(), None,
Tkinter.Canvas, (self.origInterior,),
relief = 'sunken',
borderwidth = 2,
)
self._canvas.grid(row = 2, column = 2, sticky = 'news')
self.origInterior.grid_rowconfigure(2, weight = 1, minsize = 0)
self.origInterior.grid_columnconfigure(2, weight = 1, minsize = 0)
# Create the horizontal scrollbar
self._horizScrollbar = self.createcomponent('horizscrollbar',
(), 'Scrollbar',
Tkinter.Scrollbar, (self.origInterior,),
orient='horizontal',
command=self._canvas.xview
)
# Create the vertical scrollbar
self._vertScrollbar = self.createcomponent('vertscrollbar',
(), 'Scrollbar',
Tkinter.Scrollbar, (self.origInterior,),
orient='vertical',
command=self._canvas.yview
)
self.createlabel(self.origInterior, childCols = 3, childRows = 3)
# Initialise instance variables.
self._horizScrollbarOn = 0
self._vertScrollbarOn = 0
self.scrollTimer = None
self._scrollRecurse = 0
self._horizScrollbarNeeded = 0
self._vertScrollbarNeeded = 0
self.setregionTimer = None
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
if self.scrollTimer is not None:
self.after_cancel(self.scrollTimer)
self.scrollTimer = None
if self.setregionTimer is not None:
self.after_cancel(self.setregionTimer)
self.setregionTimer = None
MegaWidget.destroy(self)
# ======================================================================
# Public methods.
def interior(self):
return self._canvas
def resizescrollregion(self):
if self.setregionTimer is None:
self.setregionTimer = self.after_idle(self._setRegion)
# ======================================================================
# Configuration methods.
def _hscrollMode(self):
# The horizontal scroll mode has been configured.
mode = self['hscrollmode']
if mode == 'static':
if not self._horizScrollbarOn:
self._toggleHorizScrollbar()
elif mode == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
elif mode == 'none':
if self._horizScrollbarOn:
self._toggleHorizScrollbar()
else:
message = 'bad hscrollmode option "%s": should be static, dynamic, or none' % mode
raise ValueError, message
self._configureScrollCommands()
def _vscrollMode(self):
# The vertical scroll mode has been configured.
mode = self['vscrollmode']
if mode == 'static':
if not self._vertScrollbarOn:
self._toggleVertScrollbar()
elif mode == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
elif mode == 'none':
if self._vertScrollbarOn:
self._toggleVertScrollbar()
else:
message = 'bad vscrollmode option "%s": should be static, dynamic, or none' % mode
raise ValueError, message
self._configureScrollCommands()
# ======================================================================
# Private methods.
def _configureScrollCommands(self):
# If both scrollmodes are not dynamic we can save a lot of
# time by not having to create an idle job to handle the
# scroll commands.
# Clean up previous scroll commands to prevent memory leak.
tclCommandName = str(self._canvas.cget('xscrollcommand'))
if tclCommandName != '':
self._canvas.deletecommand(tclCommandName)
tclCommandName = str(self._canvas.cget('yscrollcommand'))
if tclCommandName != '':
self._canvas.deletecommand(tclCommandName)
if self['hscrollmode'] == self['vscrollmode'] == 'dynamic':
self._canvas.configure(
xscrollcommand=self._scrollBothLater,
yscrollcommand=self._scrollBothLater
)
else:
self._canvas.configure(
xscrollcommand=self._scrollXNow,
yscrollcommand=self._scrollYNow
)
def _scrollXNow(self, first, last):
self._horizScrollbar.set(first, last)
self._horizScrollbarNeeded = ((first, last) != ('0', '1'))
if self['hscrollmode'] == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
def _scrollYNow(self, first, last):
self._vertScrollbar.set(first, last)
self._vertScrollbarNeeded = ((first, last) != ('0', '1'))
if self['vscrollmode'] == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
def _scrollBothLater(self, first, last):
# Called by the canvas to set the horizontal or vertical
# scrollbar when it has scrolled or changed scrollregion.
if self.scrollTimer is None:
self.scrollTimer = self.after_idle(self._scrollBothNow)
def _scrollBothNow(self):
# This performs the function of _scrollXNow and _scrollYNow.
# If one is changed, the other should be updated to match.
self.scrollTimer = None
# Call update_idletasks to make sure that the containing frame
# has been resized before we attempt to set the scrollbars.
# Otherwise the scrollbars may be mapped/unmapped continuously.
self._scrollRecurse = self._scrollRecurse + 1
self.update_idletasks()
self._scrollRecurse = self._scrollRecurse - 1
if self._scrollRecurse != 0:
return
xview = self._canvas.xview()
yview = self._canvas.yview()
self._horizScrollbar.set(xview[0], xview[1])
self._vertScrollbar.set(yview[0], yview[1])
self._horizScrollbarNeeded = (xview != (0.0, 1.0))
self._vertScrollbarNeeded = (yview != (0.0, 1.0))
# If both horizontal and vertical scrollmodes are dynamic and
# currently only one scrollbar is mapped and both should be
# toggled, then unmap the mapped scrollbar. This prevents a
# continuous mapping and unmapping of the scrollbars.
if (self['hscrollmode'] == self['vscrollmode'] == 'dynamic' and
self._horizScrollbarNeeded != self._horizScrollbarOn and
self._vertScrollbarNeeded != self._vertScrollbarOn and
self._vertScrollbarOn != self._horizScrollbarOn):
if self._horizScrollbarOn:
self._toggleHorizScrollbar()
else:
self._toggleVertScrollbar()
return
if self['hscrollmode'] == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
if self['vscrollmode'] == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
def _toggleHorizScrollbar(self):
self._horizScrollbarOn = not self._horizScrollbarOn
interior = self.origInterior
if self._horizScrollbarOn:
self._horizScrollbar.grid(row = 4, column = 2, sticky = 'news')
interior.grid_rowconfigure(3, minsize = self['scrollmargin'])
else:
self._horizScrollbar.grid_forget()
interior.grid_rowconfigure(3, minsize = 0)
def _toggleVertScrollbar(self):
self._vertScrollbarOn = not self._vertScrollbarOn
interior = self.origInterior
if self._vertScrollbarOn:
self._vertScrollbar.grid(row = 2, column = 4, sticky = 'news')
interior.grid_columnconfigure(3, minsize = self['scrollmargin'])
else:
self._vertScrollbar.grid_forget()
interior.grid_columnconfigure(3, minsize = 0)
def _setRegion(self):
self.setregionTimer = None
region = self._canvas.bbox('all')
if region is not None:
canvasmargin = self['canvasmargin']
region = (region[0] - canvasmargin, region[1] - canvasmargin,
region[2] + canvasmargin, region[3] + canvasmargin)
self._canvas.configure(scrollregion = region)
# Need to explicitly forward this to override the stupid
# (grid_)bbox method inherited from Tkinter.Frame.Grid.
def bbox(self, *args):
return apply(self._canvas.bbox, args)
forwardmethods(ScrolledCanvas, Tkinter.Canvas, '_canvas')
######################################################################
### File: PmwScrolledField.py
import Tkinter
class ScrolledField(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('sticky', 'ew', INITOPT),
('text', '', self._text),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
self._scrolledFieldEntry = self.createcomponent('entry',
(), None,
Tkinter.Entry, (interior,))
# Can't always use 'disabled', since this greys out text in Tk 8.4.2
try:
self._scrolledFieldEntry.configure(state = 'readonly')
except Tkinter.TclError:
self._scrolledFieldEntry.configure(state = 'disabled')
self._scrolledFieldEntry.grid(column=2, row=2, sticky=self['sticky'])
interior.grid_columnconfigure(2, weight=1)
interior.grid_rowconfigure(2, weight=1)
self.createlabel(interior)
# Check keywords and initialise options.
self.initialiseoptions()
def _text(self):
text = self['text']
self._scrolledFieldEntry.configure(state = 'normal')
self._scrolledFieldEntry.delete(0, 'end')
self._scrolledFieldEntry.insert('end', text)
# Can't always use 'disabled', since this greys out text in Tk 8.4.2
try:
self._scrolledFieldEntry.configure(state = 'readonly')
except Tkinter.TclError:
self._scrolledFieldEntry.configure(state = 'disabled')
forwardmethods(ScrolledField, Tkinter.Entry, '_scrolledFieldEntry')
######################################################################
### File: PmwScrolledFrame.py
import string
import types
import Tkinter
class ScrolledFrame(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderframe', 1, INITOPT),
('horizflex', 'fixed', self._horizflex),
('horizfraction', 0.05, INITOPT),
('hscrollmode', 'dynamic', self._hscrollMode),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('scrollmargin', 2, INITOPT),
('usehullsize', 0, INITOPT),
('vertflex', 'fixed', self._vertflex),
('vertfraction', 0.05, INITOPT),
('vscrollmode', 'dynamic', self._vscrollMode),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
self.origInterior = MegaWidget.interior(self)
if self['usehullsize']:
self.origInterior.grid_propagate(0)
if self['borderframe']:
# Create a frame widget to act as the border of the clipper.
self._borderframe = self.createcomponent('borderframe',
(), None,
Tkinter.Frame, (self.origInterior,),
relief = 'sunken',
borderwidth = 2,
)
self._borderframe.grid(row = 2, column = 2, sticky = 'news')
# Create the clipping window.
self._clipper = self.createcomponent('clipper',
(), None,
Tkinter.Frame, (self._borderframe,),
width = 400,
height = 300,
highlightthickness = 0,
borderwidth = 0,
)
self._clipper.pack(fill = 'both', expand = 1)
else:
# Create the clipping window.
self._clipper = self.createcomponent('clipper',
(), None,
Tkinter.Frame, (self.origInterior,),
width = 400,
height = 300,
relief = 'sunken',
borderwidth = 2,
)
self._clipper.grid(row = 2, column = 2, sticky = 'news')
self.origInterior.grid_rowconfigure(2, weight = 1, minsize = 0)
self.origInterior.grid_columnconfigure(2, weight = 1, minsize = 0)
# Create the horizontal scrollbar
self._horizScrollbar = self.createcomponent('horizscrollbar',
(), 'Scrollbar',
Tkinter.Scrollbar, (self.origInterior,),
orient='horizontal',
command=self.xview
)
# Create the vertical scrollbar
self._vertScrollbar = self.createcomponent('vertscrollbar',
(), 'Scrollbar',
Tkinter.Scrollbar, (self.origInterior,),
orient='vertical',
command=self.yview
)
self.createlabel(self.origInterior, childCols = 3, childRows = 3)
# Initialise instance variables.
self._horizScrollbarOn = 0
self._vertScrollbarOn = 0
self.scrollTimer = None
self._scrollRecurse = 0
self._horizScrollbarNeeded = 0
self._vertScrollbarNeeded = 0
self.startX = 0
self.startY = 0
self._flexoptions = ('fixed', 'expand', 'shrink', 'elastic')
# Create a frame in the clipper to contain the widgets to be
# scrolled.
self._frame = self.createcomponent('frame',
(), None,
Tkinter.Frame, (self._clipper,)
)
# Whenever the clipping window or scrolled frame change size,
# update the scrollbars.
self._frame.bind('<Configure>', self._reposition)
self._clipper.bind('<Configure>', self._reposition)
# Work around a bug in Tk where the value returned by the
# scrollbar get() method is (0.0, 0.0, 0.0, 0.0) rather than
# the expected 2-tuple. This occurs if xview() is called soon
# after the ScrolledFrame has been created.
self._horizScrollbar.set(0.0, 1.0)
self._vertScrollbar.set(0.0, 1.0)
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
if self.scrollTimer is not None:
self.after_cancel(self.scrollTimer)
self.scrollTimer = None
MegaWidget.destroy(self)
# ======================================================================
# Public methods.
def interior(self):
return self._frame
# Set timer to call real reposition method, so that it is not
# called multiple times when many things are reconfigured at the
# same time.
def reposition(self):
if self.scrollTimer is None:
self.scrollTimer = self.after_idle(self._scrollBothNow)
# Called when the user clicks in the horizontal scrollbar.
# Calculates new position of frame then calls reposition() to
# update the frame and the scrollbar.
def xview(self, mode = None, value = None, units = None):
if type(value) == types.StringType:
value = string.atof(value)
if mode is None:
return self._horizScrollbar.get()
elif mode == 'moveto':
frameWidth = self._frame.winfo_reqwidth()
self.startX = value * float(frameWidth)
else: # mode == 'scroll'
clipperWidth = self._clipper.winfo_width()
if units == 'units':
jump = int(clipperWidth * self['horizfraction'])
else:
jump = clipperWidth
self.startX = self.startX + value * jump
self.reposition()
# Called when the user clicks in the vertical scrollbar.
# Calculates new position of frame then calls reposition() to
# update the frame and the scrollbar.
def yview(self, mode = None, value = None, units = None):
if type(value) == types.StringType:
value = string.atof(value)
if mode is None:
return self._vertScrollbar.get()
elif mode == 'moveto':
frameHeight = self._frame.winfo_reqheight()
self.startY = value * float(frameHeight)
else: # mode == 'scroll'
clipperHeight = self._clipper.winfo_height()
if units == 'units':
jump = int(clipperHeight * self['vertfraction'])
else:
jump = clipperHeight
self.startY = self.startY + value * jump
self.reposition()
# ======================================================================
# Configuration methods.
def _hscrollMode(self):
# The horizontal scroll mode has been configured.
mode = self['hscrollmode']
if mode == 'static':
if not self._horizScrollbarOn:
self._toggleHorizScrollbar()
elif mode == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
elif mode == 'none':
if self._horizScrollbarOn:
self._toggleHorizScrollbar()
else:
message = 'bad hscrollmode option "%s": should be static, dynamic, or none' % mode
raise ValueError, message
def _vscrollMode(self):
# The vertical scroll mode has been configured.
mode = self['vscrollmode']
if mode == 'static':
if not self._vertScrollbarOn:
self._toggleVertScrollbar()
elif mode == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
elif mode == 'none':
if self._vertScrollbarOn:
self._toggleVertScrollbar()
else:
message = 'bad vscrollmode option "%s": should be static, dynamic, or none' % mode
raise ValueError, message
def _horizflex(self):
# The horizontal flex mode has been configured.
flex = self['horizflex']
if flex not in self._flexoptions:
message = 'bad horizflex option "%s": should be one of %s' % \
(flex, str(self._flexoptions))
raise ValueError, message
self.reposition()
def _vertflex(self):
# The vertical flex mode has been configured.
flex = self['vertflex']
if flex not in self._flexoptions:
message = 'bad vertflex option "%s": should be one of %s' % \
(flex, str(self._flexoptions))
raise ValueError, message
self.reposition()
# ======================================================================
# Private methods.
def _reposition(self, event):
self.reposition()
def _getxview(self):
# Horizontal dimension.
clipperWidth = self._clipper.winfo_width()
frameWidth = self._frame.winfo_reqwidth()
if frameWidth <= clipperWidth:
# The scrolled frame is smaller than the clipping window.
self.startX = 0
endScrollX = 1.0
if self['horizflex'] in ('expand', 'elastic'):
relwidth = 1
else:
relwidth = ''
else:
# The scrolled frame is larger than the clipping window.
if self['horizflex'] in ('shrink', 'elastic'):
self.startX = 0
endScrollX = 1.0
relwidth = 1
else:
if self.startX + clipperWidth > frameWidth:
self.startX = frameWidth - clipperWidth
endScrollX = 1.0
else:
if self.startX < 0:
self.startX = 0
endScrollX = (self.startX + clipperWidth) / float(frameWidth)
relwidth = ''
# Position frame relative to clipper.
self._frame.place(x = -self.startX, relwidth = relwidth)
return (self.startX / float(frameWidth), endScrollX)
def _getyview(self):
# Vertical dimension.
clipperHeight = self._clipper.winfo_height()
frameHeight = self._frame.winfo_reqheight()
if frameHeight <= clipperHeight:
# The scrolled frame is smaller than the clipping window.
self.startY = 0
endScrollY = 1.0
if self['vertflex'] in ('expand', 'elastic'):
relheight = 1
else:
relheight = ''
else:
# The scrolled frame is larger than the clipping window.
if self['vertflex'] in ('shrink', 'elastic'):
self.startY = 0
endScrollY = 1.0
relheight = 1
else:
if self.startY + clipperHeight > frameHeight:
self.startY = frameHeight - clipperHeight
endScrollY = 1.0
else:
if self.startY < 0:
self.startY = 0
endScrollY = (self.startY + clipperHeight) / float(frameHeight)
relheight = ''
# Position frame relative to clipper.
self._frame.place(y = -self.startY, relheight = relheight)
return (self.startY / float(frameHeight), endScrollY)
# According to the relative geometries of the frame and the
# clipper, reposition the frame within the clipper and reset the
# scrollbars.
def _scrollBothNow(self):
self.scrollTimer = None
# Call update_idletasks to make sure that the containing frame
# has been resized before we attempt to set the scrollbars.
# Otherwise the scrollbars may be mapped/unmapped continuously.
self._scrollRecurse = self._scrollRecurse + 1
self.update_idletasks()
self._scrollRecurse = self._scrollRecurse - 1
if self._scrollRecurse != 0:
return
xview = self._getxview()
yview = self._getyview()
self._horizScrollbar.set(xview[0], xview[1])
self._vertScrollbar.set(yview[0], yview[1])
self._horizScrollbarNeeded = (xview != (0.0, 1.0))
self._vertScrollbarNeeded = (yview != (0.0, 1.0))
# If both horizontal and vertical scrollmodes are dynamic and
# currently only one scrollbar is mapped and both should be
# toggled, then unmap the mapped scrollbar. This prevents a
# continuous mapping and unmapping of the scrollbars.
if (self['hscrollmode'] == self['vscrollmode'] == 'dynamic' and
self._horizScrollbarNeeded != self._horizScrollbarOn and
self._vertScrollbarNeeded != self._vertScrollbarOn and
self._vertScrollbarOn != self._horizScrollbarOn):
if self._horizScrollbarOn:
self._toggleHorizScrollbar()
else:
self._toggleVertScrollbar()
return
if self['hscrollmode'] == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
if self['vscrollmode'] == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
def _toggleHorizScrollbar(self):
self._horizScrollbarOn = not self._horizScrollbarOn
interior = self.origInterior
if self._horizScrollbarOn:
self._horizScrollbar.grid(row = 4, column = 2, sticky = 'news')
interior.grid_rowconfigure(3, minsize = self['scrollmargin'])
else:
self._horizScrollbar.grid_forget()
interior.grid_rowconfigure(3, minsize = 0)
def _toggleVertScrollbar(self):
self._vertScrollbarOn = not self._vertScrollbarOn
interior = self.origInterior
if self._vertScrollbarOn:
self._vertScrollbar.grid(row = 2, column = 4, sticky = 'news')
interior.grid_columnconfigure(3, minsize = self['scrollmargin'])
else:
self._vertScrollbar.grid_forget()
interior.grid_columnconfigure(3, minsize = 0)
######################################################################
### File: PmwScrolledListBox.py
# Based on iwidgets2.2.0/scrolledlistbox.itk code.
import types
import Tkinter
class ScrolledListBox(MegaWidget):
_classBindingsDefinedFor = 0
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('dblclickcommand', None, None),
('hscrollmode', 'dynamic', self._hscrollMode),
('items', (), INITOPT),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('scrollmargin', 2, INITOPT),
('selectioncommand', None, None),
('usehullsize', 0, INITOPT),
('vscrollmode', 'dynamic', self._vscrollMode),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
if self['usehullsize']:
interior.grid_propagate(0)
# Create the listbox widget.
self._listbox = self.createcomponent('listbox',
(), None,
Tkinter.Listbox, (interior,))
self._listbox.grid(row = 2, column = 2, sticky = 'news')
interior.grid_rowconfigure(2, weight = 1, minsize = 0)
interior.grid_columnconfigure(2, weight = 1, minsize = 0)
# Create the horizontal scrollbar
self._horizScrollbar = self.createcomponent('horizscrollbar',
(), 'Scrollbar',
Tkinter.Scrollbar, (interior,),
orient='horizontal',
command=self._listbox.xview)
# Create the vertical scrollbar
self._vertScrollbar = self.createcomponent('vertscrollbar',
(), 'Scrollbar',
Tkinter.Scrollbar, (interior,),
orient='vertical',
command=self._listbox.yview)
self.createlabel(interior, childCols = 3, childRows = 3)
# Add the items specified by the initialisation option.
items = self['items']
if type(items) != types.TupleType:
items = tuple(items)
if len(items) > 0:
apply(self._listbox.insert, ('end',) + items)
_registerScrolledList(self._listbox, self)
# Establish the special class bindings if not already done.
# Also create bindings if the Tkinter default interpreter has
# changed. Use Tkinter._default_root to create class
# bindings, so that a reference to root is created by
# bind_class rather than a reference to self, which would
# prevent object cleanup.
theTag = 'ScrolledListBoxTag'
if ScrolledListBox._classBindingsDefinedFor != Tkinter._default_root:
root = Tkinter._default_root
def doubleEvent(event):
_handleEvent(event, 'double')
def keyEvent(event):
_handleEvent(event, 'key')
def releaseEvent(event):
_handleEvent(event, 'release')
# Bind space and return keys and button 1 to the selectioncommand.
root.bind_class(theTag, '<Key-space>', keyEvent)
root.bind_class(theTag, '<Key-Return>', keyEvent)
root.bind_class(theTag, '<ButtonRelease-1>', releaseEvent)
# Bind double button 1 click to the dblclickcommand.
root.bind_class(theTag, '<Double-ButtonRelease-1>', doubleEvent)
ScrolledListBox._classBindingsDefinedFor = root
bindtags = self._listbox.bindtags()
self._listbox.bindtags(bindtags + (theTag,))
# Initialise instance variables.
self._horizScrollbarOn = 0
self._vertScrollbarOn = 0
self.scrollTimer = None
self._scrollRecurse = 0
self._horizScrollbarNeeded = 0
self._vertScrollbarNeeded = 0
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
if self.scrollTimer is not None:
self.after_cancel(self.scrollTimer)
self.scrollTimer = None
_deregisterScrolledList(self._listbox)
MegaWidget.destroy(self)
# ======================================================================
# Public methods.
def clear(self):
self.setlist(())
def getcurselection(self):
rtn = []
for sel in self.curselection():
rtn.append(self._listbox.get(sel))
return tuple(rtn)
def getvalue(self):
return self.getcurselection()
def setvalue(self, textOrList):
self._listbox.selection_clear(0, 'end')
listitems = list(self._listbox.get(0, 'end'))
if type(textOrList) == types.StringType:
if textOrList in listitems:
self._listbox.selection_set(listitems.index(textOrList))
else:
raise ValueError, 'no such item "%s"' % textOrList
else:
for item in textOrList:
if item in listitems:
self._listbox.selection_set(listitems.index(item))
else:
raise ValueError, 'no such item "%s"' % item
def setlist(self, items):
self._listbox.delete(0, 'end')
if len(items) > 0:
if type(items) != types.TupleType:
items = tuple(items)
apply(self._listbox.insert, (0,) + items)
# Override Tkinter.Listbox get method, so that if it is called with
# no arguments, return all list elements (consistent with other widgets).
def get(self, first=None, last=None):
if first is None:
return self._listbox.get(0, 'end')
else:
return self._listbox.get(first, last)
# ======================================================================
# Configuration methods.
def _hscrollMode(self):
'''The horizontal scroll mode has been configured.'''
mode = self['hscrollmode']
if mode == 'static':
if not self._horizScrollbarOn:
self._toggleHorizScrollbar()
elif mode == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
elif mode == 'none':
if self._horizScrollbarOn:
self._toggleHorizScrollbar()
else:
message = 'bad hscrollmode option "%s": should be static, dynamic, or none' % mode
raise ValueError, message
self._configureScrollCommands()
def _vscrollMode(self):
'''The vertical scroll mode has been configured.'''
mode = self['vscrollmode']
if mode == 'static':
if not self._vertScrollbarOn:
self._toggleVertScrollbar()
elif mode == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
elif mode == 'none':
if self._vertScrollbarOn:
self._toggleVertScrollbar()
else:
message = 'bad vscrollmode option "%s": should be static, dynamic, or none' % mode
raise ValueError, message
self._configureScrollCommands()
# ======================================================================
# Private methods.
def _configureScrollCommands(self):
# If both scrollmodes are not dynamic we can save a lot of
# time by not having to create an idle job to handle the
# scroll commands.
# Clean up previous scroll commands to prevent memory leak.
tclCommandName = str(self._listbox.cget('xscrollcommand'))
if tclCommandName != '':
self._listbox.deletecommand(tclCommandName)
tclCommandName = str(self._listbox.cget('yscrollcommand'))
if tclCommandName != '':
self._listbox.deletecommand(tclCommandName)
if self['hscrollmode'] == self['vscrollmode'] == 'dynamic':
self._listbox.configure(
xscrollcommand=self._scrollBothLater,
yscrollcommand=self._scrollBothLater
)
else:
self._listbox.configure(
xscrollcommand=self._scrollXNow,
yscrollcommand=self._scrollYNow
)
def _scrollXNow(self, first, last):
self._horizScrollbar.set(first, last)
self._horizScrollbarNeeded = ((first, last) != ('0', '1'))
if self['hscrollmode'] == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
def _scrollYNow(self, first, last):
self._vertScrollbar.set(first, last)
self._vertScrollbarNeeded = ((first, last) != ('0', '1'))
if self['vscrollmode'] == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
def _scrollBothLater(self, first, last):
'''Called by the listbox to set the horizontal or vertical
scrollbar when it has scrolled or changed size or contents.
'''
if self.scrollTimer is None:
self.scrollTimer = self.after_idle(self._scrollBothNow)
def _scrollBothNow(self):
'''This performs the function of _scrollXNow and _scrollYNow.
If one is changed, the other should be updated to match.
'''
self.scrollTimer = None
# Call update_idletasks to make sure that the containing frame
# has been resized before we attempt to set the scrollbars.
# Otherwise the scrollbars may be mapped/unmapped continuously.
self._scrollRecurse = self._scrollRecurse + 1
self.update_idletasks()
self._scrollRecurse = self._scrollRecurse - 1
if self._scrollRecurse != 0:
return
xview = self._listbox.xview()
yview = self._listbox.yview()
self._horizScrollbar.set(xview[0], xview[1])
self._vertScrollbar.set(yview[0], yview[1])
self._horizScrollbarNeeded = (xview != (0.0, 1.0))
self._vertScrollbarNeeded = (yview != (0.0, 1.0))
# If both horizontal and vertical scrollmodes are dynamic and
# currently only one scrollbar is mapped and both should be
# toggled, then unmap the mapped scrollbar. This prevents a
# continuous mapping and unmapping of the scrollbars.
if (self['hscrollmode'] == self['vscrollmode'] == 'dynamic' and
self._horizScrollbarNeeded != self._horizScrollbarOn and
self._vertScrollbarNeeded != self._vertScrollbarOn and
self._vertScrollbarOn != self._horizScrollbarOn):
if self._horizScrollbarOn:
self._toggleHorizScrollbar()
else:
self._toggleVertScrollbar()
return
if self['hscrollmode'] == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
if self['vscrollmode'] == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
def _toggleHorizScrollbar(self):
self._horizScrollbarOn = not self._horizScrollbarOn
interior = self.interior()
if self._horizScrollbarOn:
self._horizScrollbar.grid(row = 4, column = 2, sticky = 'news')
interior.grid_rowconfigure(3, minsize = self['scrollmargin'])
else:
self._horizScrollbar.grid_forget()
interior.grid_rowconfigure(3, minsize = 0)
def _toggleVertScrollbar(self):
self._vertScrollbarOn = not self._vertScrollbarOn
interior = self.interior()
if self._vertScrollbarOn:
self._vertScrollbar.grid(row = 2, column = 4, sticky = 'news')
interior.grid_columnconfigure(3, minsize = self['scrollmargin'])
else:
self._vertScrollbar.grid_forget()
interior.grid_columnconfigure(3, minsize = 0)
def _handleEvent(self, event, eventType):
if eventType == 'double':
command = self['dblclickcommand']
elif eventType == 'key':
command = self['selectioncommand']
else: #eventType == 'release'
# Do not execute the command if the mouse was released
# outside the listbox.
if (event.x < 0 or self._listbox.winfo_width() <= event.x or
event.y < 0 or self._listbox.winfo_height() <= event.y):
return
command = self['selectioncommand']
if callable(command):
command()
# Need to explicitly forward this to override the stupid
# (grid_)size method inherited from Tkinter.Frame.Grid.
def size(self):
return self._listbox.size()
# Need to explicitly forward this to override the stupid
# (grid_)bbox method inherited from Tkinter.Frame.Grid.
def bbox(self, index):
return self._listbox.bbox(index)
forwardmethods(ScrolledListBox, Tkinter.Listbox, '_listbox')
# ======================================================================
_listboxCache = {}
def _registerScrolledList(listbox, scrolledList):
# Register an ScrolledList widget for a Listbox widget
_listboxCache[listbox] = scrolledList
def _deregisterScrolledList(listbox):
# Deregister a Listbox widget
del _listboxCache[listbox]
def _handleEvent(event, eventType):
# Forward events for a Listbox to it's ScrolledListBox
# A binding earlier in the bindtags list may have destroyed the
# megawidget, so need to check.
if _listboxCache.has_key(event.widget):
_listboxCache[event.widget]._handleEvent(event, eventType)
######################################################################
### File: PmwScrolledText.py
# Based on iwidgets2.2.0/scrolledtext.itk code.
import Tkinter
class ScrolledText(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderframe', 0, INITOPT),
('columnheader', 0, INITOPT),
('hscrollmode', 'dynamic', self._hscrollMode),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('rowcolumnheader',0, INITOPT),
('rowheader', 0, INITOPT),
('scrollmargin', 2, INITOPT),
('usehullsize', 0, INITOPT),
('vscrollmode', 'dynamic', self._vscrollMode),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
if self['usehullsize']:
interior.grid_propagate(0)
if self['borderframe']:
# Create a frame widget to act as the border of the text
# widget. Later, pack the text widget so that it fills
# the frame. This avoids a problem in Tk, where window
# items in a text widget may overlap the border of the
# text widget.
self._borderframe = self.createcomponent('borderframe',
(), None,
Tkinter.Frame, (interior,),
relief = 'sunken',
borderwidth = 2,
)
self._borderframe.grid(row = 4, column = 4, sticky = 'news')
# Create the text widget.
self._textbox = self.createcomponent('text',
(), None,
Tkinter.Text, (self._borderframe,),
highlightthickness = 0,
borderwidth = 0,
)
self._textbox.pack(fill = 'both', expand = 1)
bw = self._borderframe.cget('borderwidth'),
ht = self._borderframe.cget('highlightthickness'),
else:
# Create the text widget.
self._textbox = self.createcomponent('text',
(), None,
Tkinter.Text, (interior,),
)
self._textbox.grid(row = 4, column = 4, sticky = 'news')
bw = self._textbox.cget('borderwidth'),
ht = self._textbox.cget('highlightthickness'),
# Create the header text widgets
if self['columnheader']:
self._columnheader = self.createcomponent('columnheader',
(), 'Header',
Tkinter.Text, (interior,),
height=1,
wrap='none',
borderwidth = bw,
highlightthickness = ht,
)
self._columnheader.grid(row = 2, column = 4, sticky = 'ew')
self._columnheader.configure(
xscrollcommand = self._columnheaderscrolled)
if self['rowheader']:
self._rowheader = self.createcomponent('rowheader',
(), 'Header',
Tkinter.Text, (interior,),
wrap='none',
borderwidth = bw,
highlightthickness = ht,
)
self._rowheader.grid(row = 4, column = 2, sticky = 'ns')
self._rowheader.configure(
yscrollcommand = self._rowheaderscrolled)
if self['rowcolumnheader']:
self._rowcolumnheader = self.createcomponent('rowcolumnheader',
(), 'Header',
Tkinter.Text, (interior,),
height=1,
wrap='none',
borderwidth = bw,
highlightthickness = ht,
)
self._rowcolumnheader.grid(row = 2, column = 2, sticky = 'nsew')
interior.grid_rowconfigure(4, weight = 1, minsize = 0)
interior.grid_columnconfigure(4, weight = 1, minsize = 0)
# Create the horizontal scrollbar
self._horizScrollbar = self.createcomponent('horizscrollbar',
(), 'Scrollbar',
Tkinter.Scrollbar, (interior,),
orient='horizontal',
command=self._textbox.xview
)
# Create the vertical scrollbar
self._vertScrollbar = self.createcomponent('vertscrollbar',
(), 'Scrollbar',
Tkinter.Scrollbar, (interior,),
orient='vertical',
command=self._textbox.yview
)
self.createlabel(interior, childCols = 5, childRows = 5)
# Initialise instance variables.
self._horizScrollbarOn = 0
self._vertScrollbarOn = 0
self.scrollTimer = None
self._scrollRecurse = 0
self._horizScrollbarNeeded = 0
self._vertScrollbarNeeded = 0
self._textWidth = None
# These four variables avoid an infinite loop caused by the
# row or column header's scrollcommand causing the main text
# widget's scrollcommand to be called and vice versa.
self._textboxLastX = None
self._textboxLastY = None
self._columnheaderLastX = None
self._rowheaderLastY = None
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
if self.scrollTimer is not None:
self.after_cancel(self.scrollTimer)
self.scrollTimer = None
MegaWidget.destroy(self)
# ======================================================================
# Public methods.
def clear(self):
self.settext('')
def importfile(self, fileName, where = 'end'):
file = open(fileName, 'r')
self._textbox.insert(where, file.read())
file.close()
def exportfile(self, fileName):
file = open(fileName, 'w')
file.write(self._textbox.get('1.0', 'end'))
file.close()
def settext(self, text):
disabled = (str(self._textbox.cget('state')) == 'disabled')
if disabled:
self._textbox.configure(state='normal')
self._textbox.delete('0.0', 'end')
self._textbox.insert('end', text)
if disabled:
self._textbox.configure(state='disabled')
# Override Tkinter.Text get method, so that if it is called with
# no arguments, return all text (consistent with other widgets).
def get(self, first=None, last=None):
if first is None:
return self._textbox.get('1.0', 'end')
else:
return self._textbox.get(first, last)
def getvalue(self):
return self.get()
def setvalue(self, text):
return self.settext(text)
def appendtext(self, text):
oldTop, oldBottom = self._textbox.yview()
disabled = (str(self._textbox.cget('state')) == 'disabled')
if disabled:
self._textbox.configure(state='normal')
self._textbox.insert('end', text)
if disabled:
self._textbox.configure(state='disabled')
if oldBottom == 1.0:
self._textbox.yview('moveto', 1.0)
# ======================================================================
# Configuration methods.
def _hscrollMode(self):
# The horizontal scroll mode has been configured.
mode = self['hscrollmode']
if mode == 'static':
if not self._horizScrollbarOn:
self._toggleHorizScrollbar()
elif mode == 'dynamic':
if self._horizScrollbarNeeded != self._horizScrollbarOn:
self._toggleHorizScrollbar()
elif mode == 'none':
if self._horizScrollbarOn:
self._toggleHorizScrollbar()
else:
message = 'bad hscrollmode option "%s": should be static, dynamic, or none' % mode
raise ValueError, message
self._configureScrollCommands()
def _vscrollMode(self):
# The vertical scroll mode has been configured.
mode = self['vscrollmode']
if mode == 'static':
if not self._vertScrollbarOn:
self._toggleVertScrollbar()
elif mode == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
elif mode == 'none':
if self._vertScrollbarOn:
self._toggleVertScrollbar()
else:
message = 'bad vscrollmode option "%s": should be static, dynamic, or none' % mode
raise ValueError, message
self._configureScrollCommands()
# ======================================================================
# Private methods.
def _configureScrollCommands(self):
# If both scrollmodes are not dynamic we can save a lot of
# time by not having to create an idle job to handle the
# scroll commands.
# Clean up previous scroll commands to prevent memory leak.
tclCommandName = str(self._textbox.cget('xscrollcommand'))
if tclCommandName != '':
self._textbox.deletecommand(tclCommandName)
tclCommandName = str(self._textbox.cget('yscrollcommand'))
if tclCommandName != '':
self._textbox.deletecommand(tclCommandName)
if self['hscrollmode'] == self['vscrollmode'] == 'dynamic':
self._textbox.configure(
xscrollcommand=self._scrollBothLater,
yscrollcommand=self._scrollBothLater
)
else:
self._textbox.configure(
xscrollcommand=self._scrollXNow,
yscrollcommand=self._scrollYNow
)
def _scrollXNow(self, first, last):
self._horizScrollbar.set(first, last)
self._horizScrollbarNeeded = ((first, last) != ('0', '1'))
# This code is the same as in _scrollBothNow. Keep it that way.
if self['hscrollmode'] == 'dynamic':
currentWidth = self._textbox.winfo_width()
if self._horizScrollbarNeeded != self._horizScrollbarOn:
if self._horizScrollbarNeeded or \
self._textWidth != currentWidth:
self._toggleHorizScrollbar()
self._textWidth = currentWidth
if self['columnheader']:
if self._columnheaderLastX != first:
self._columnheaderLastX = first
self._columnheader.xview('moveto', first)
def _scrollYNow(self, first, last):
if first == '0' and last == '0':
return
self._vertScrollbar.set(first, last)
self._vertScrollbarNeeded = ((first, last) != ('0', '1'))
if self['vscrollmode'] == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
if self['rowheader']:
if self._rowheaderLastY != first:
self._rowheaderLastY = first
self._rowheader.yview('moveto', first)
def _scrollBothLater(self, first, last):
# Called by the text widget to set the horizontal or vertical
# scrollbar when it has scrolled or changed size or contents.
if self.scrollTimer is None:
self.scrollTimer = self.after_idle(self._scrollBothNow)
def _scrollBothNow(self):
# This performs the function of _scrollXNow and _scrollYNow.
# If one is changed, the other should be updated to match.
self.scrollTimer = None
# Call update_idletasks to make sure that the containing frame
# has been resized before we attempt to set the scrollbars.
# Otherwise the scrollbars may be mapped/unmapped continuously.
self._scrollRecurse = self._scrollRecurse + 1
self.update_idletasks()
self._scrollRecurse = self._scrollRecurse - 1
if self._scrollRecurse != 0:
return
xview = self._textbox.xview()
yview = self._textbox.yview()
# The text widget returns a yview of (0.0, 0.0) just after it
# has been created. Ignore this.
if yview == (0.0, 0.0):
return
if self['columnheader']:
if self._columnheaderLastX != xview[0]:
self._columnheaderLastX = xview[0]
self._columnheader.xview('moveto', xview[0])
if self['rowheader']:
if self._rowheaderLastY != yview[0]:
self._rowheaderLastY = yview[0]
self._rowheader.yview('moveto', yview[0])
self._horizScrollbar.set(xview[0], xview[1])
self._vertScrollbar.set(yview[0], yview[1])
self._horizScrollbarNeeded = (xview != (0.0, 1.0))
self._vertScrollbarNeeded = (yview != (0.0, 1.0))
# If both horizontal and vertical scrollmodes are dynamic and
# currently only one scrollbar is mapped and both should be
# toggled, then unmap the mapped scrollbar. This prevents a
# continuous mapping and unmapping of the scrollbars.
if (self['hscrollmode'] == self['vscrollmode'] == 'dynamic' and
self._horizScrollbarNeeded != self._horizScrollbarOn and
self._vertScrollbarNeeded != self._vertScrollbarOn and
self._vertScrollbarOn != self._horizScrollbarOn):
if self._horizScrollbarOn:
self._toggleHorizScrollbar()
else:
self._toggleVertScrollbar()
return
if self['hscrollmode'] == 'dynamic':
# The following test is done to prevent continuous
# mapping and unmapping of the horizontal scrollbar.
# This may occur when some event (scrolling, resizing
# or text changes) modifies the displayed text such
# that the bottom line in the window is the longest
# line displayed. If this causes the horizontal
# scrollbar to be mapped, the scrollbar may "cover up"
# the bottom line, which would mean that the scrollbar
# is no longer required. If the scrollbar is then
# unmapped, the bottom line will then become visible
# again, which would cause the scrollbar to be mapped
# again, and so on...
#
# The idea is that, if the width of the text widget
# has not changed and the scrollbar is currently
# mapped, then do not unmap the scrollbar even if it
# is no longer required. This means that, during
# normal scrolling of the text, once the horizontal
# scrollbar has been mapped it will not be unmapped
# (until the width of the text widget changes).
currentWidth = self._textbox.winfo_width()
if self._horizScrollbarNeeded != self._horizScrollbarOn:
if self._horizScrollbarNeeded or \
self._textWidth != currentWidth:
self._toggleHorizScrollbar()
self._textWidth = currentWidth
if self['vscrollmode'] == 'dynamic':
if self._vertScrollbarNeeded != self._vertScrollbarOn:
self._toggleVertScrollbar()
def _columnheaderscrolled(self, first, last):
if self._textboxLastX != first:
self._textboxLastX = first
self._textbox.xview('moveto', first)
def _rowheaderscrolled(self, first, last):
if self._textboxLastY != first:
self._textboxLastY = first
self._textbox.yview('moveto', first)
def _toggleHorizScrollbar(self):
self._horizScrollbarOn = not self._horizScrollbarOn
interior = self.interior()
if self._horizScrollbarOn:
self._horizScrollbar.grid(row = 6, column = 4, sticky = 'news')
interior.grid_rowconfigure(5, minsize = self['scrollmargin'])
else:
self._horizScrollbar.grid_forget()
interior.grid_rowconfigure(5, minsize = 0)
def _toggleVertScrollbar(self):
self._vertScrollbarOn = not self._vertScrollbarOn
interior = self.interior()
if self._vertScrollbarOn:
self._vertScrollbar.grid(row = 4, column = 6, sticky = 'news')
interior.grid_columnconfigure(5, minsize = self['scrollmargin'])
else:
self._vertScrollbar.grid_forget()
interior.grid_columnconfigure(5, minsize = 0)
# Need to explicitly forward this to override the stupid
# (grid_)bbox method inherited from Tkinter.Frame.Grid.
def bbox(self, index):
return self._textbox.bbox(index)
forwardmethods(ScrolledText, Tkinter.Text, '_textbox')
######################################################################
### File: PmwHistoryText.py
_ORIGINAL = 0
_MODIFIED = 1
_DISPLAY = 2
class HistoryText(ScrolledText):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('compressany', 1, None),
('compresstail', 1, None),
('historycommand', None, None),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
ScrolledText.__init__(self, parent)
# Initialise instance variables.
self._list = []
self._currIndex = 0
self._pastIndex = None
self._lastIndex = 0 # pointer to end of history list
# Check keywords and initialise options.
self.initialiseoptions()
def addhistory(self):
text = self.get()
if text[-1] == '\n':
text = text[:-1]
if len(self._list) == 0:
# This is the first history entry. Add it.
self._list.append([text, text, _MODIFIED])
return
currentEntry = self._list[self._currIndex]
if text == currentEntry[_ORIGINAL]:
# The current history entry has not been modified. Check if
# we need to add it again.
if self['compresstail'] and self._currIndex == self._lastIndex:
return
if self['compressany']:
return
# Undo any changes for the current history entry, since they
# will now be available in the new entry.
currentEntry[_MODIFIED] = currentEntry[_ORIGINAL]
historycommand = self['historycommand']
if self._currIndex == self._lastIndex:
# The last history entry is currently being displayed,
# so disable the special meaning of the 'Next' button.
self._pastIndex = None
nextState = 'disabled'
else:
# A previous history entry is currently being displayed,
# so allow the 'Next' button to go to the entry after this one.
self._pastIndex = self._currIndex
nextState = 'normal'
if callable(historycommand):
historycommand('normal', nextState)
# Create the new history entry.
self._list.append([text, text, _MODIFIED])
# Move the pointer into the history entry list to the end.
self._lastIndex = self._lastIndex + 1
self._currIndex = self._lastIndex
def next(self):
if self._currIndex == self._lastIndex and self._pastIndex is None:
self.bell()
else:
self._modifyDisplay('next')
def prev(self):
self._pastIndex = None
if self._currIndex == 0:
self.bell()
else:
self._modifyDisplay('prev')
def undo(self):
if len(self._list) != 0:
self._modifyDisplay('undo')
def redo(self):
if len(self._list) != 0:
self._modifyDisplay('redo')
def gethistory(self):
return self._list
def _modifyDisplay(self, command):
# Modify the display to show either the next or previous
# history entry (next, prev) or the original or modified
# version of the current history entry (undo, redo).
# Save the currently displayed text.
currentText = self.get()
if currentText[-1] == '\n':
currentText = currentText[:-1]
currentEntry = self._list[self._currIndex]
if currentEntry[_DISPLAY] == _MODIFIED:
currentEntry[_MODIFIED] = currentText
elif currentEntry[_ORIGINAL] != currentText:
currentEntry[_MODIFIED] = currentText
if command in ('next', 'prev'):
currentEntry[_DISPLAY] = _MODIFIED
if command in ('next', 'prev'):
prevstate = 'normal'
nextstate = 'normal'
if command == 'next':
if self._pastIndex is not None:
self._currIndex = self._pastIndex
self._pastIndex = None
self._currIndex = self._currIndex + 1
if self._currIndex == self._lastIndex:
nextstate = 'disabled'
elif command == 'prev':
self._currIndex = self._currIndex - 1
if self._currIndex == 0:
prevstate = 'disabled'
historycommand = self['historycommand']
if callable(historycommand):
historycommand(prevstate, nextstate)
currentEntry = self._list[self._currIndex]
else:
if command == 'undo':
currentEntry[_DISPLAY] = _ORIGINAL
elif command == 'redo':
currentEntry[_DISPLAY] = _MODIFIED
# Display the new text.
self.delete('1.0', 'end')
self.insert('end', currentEntry[currentEntry[_DISPLAY]])
######################################################################
### File: PmwSelectionDialog.py
# Not Based on iwidgets version.
class SelectionDialog(Dialog):
# Dialog window with selection list.
# Dialog window displaying a list and requesting the user to
# select one.
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderx', 10, INITOPT),
('bordery', 10, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
Dialog.__init__(self, parent)
# Create the components.
interior = self.interior()
aliases = (
('listbox', 'scrolledlist_listbox'),
('label', 'scrolledlist_label'),
)
self._list = self.createcomponent('scrolledlist',
aliases, None,
ScrolledListBox, (interior,),
dblclickcommand = self.invoke)
self._list.pack(side='top', expand='true', fill='both',
padx = self['borderx'], pady = self['bordery'])
if not kw.has_key('activatecommand'):
# Whenever this dialog is activated, set the focus to the
# ScrolledListBox's listbox widget.
listbox = self.component('listbox')
self.configure(activatecommand = listbox.focus_set)
# Check keywords and initialise options.
self.initialiseoptions()
# Need to explicitly forward this to override the stupid
# (grid_)size method inherited from Tkinter.Toplevel.Grid.
def size(self):
return self.component('listbox').size()
# Need to explicitly forward this to override the stupid
# (grid_)bbox method inherited from Tkinter.Toplevel.Grid.
def bbox(self, index):
return self.component('listbox').size(index)
forwardmethods(SelectionDialog, ScrolledListBox, '_list')
######################################################################
### File: PmwTextDialog.py
# A Dialog with a ScrolledText widget.
class TextDialog(Dialog):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderx', 10, INITOPT),
('bordery', 10, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
Dialog.__init__(self, parent)
# Create the components.
interior = self.interior()
aliases = (
('text', 'scrolledtext_text'),
('label', 'scrolledtext_label'),
)
self._text = self.createcomponent('scrolledtext',
aliases, None,
ScrolledText, (interior,))
self._text.pack(side='top', expand=1, fill='both',
padx = self['borderx'], pady = self['bordery'])
# Check keywords and initialise options.
self.initialiseoptions()
# Need to explicitly forward this to override the stupid
# (grid_)bbox method inherited from Tkinter.Toplevel.Grid.
def bbox(self, index):
return self._text.bbox(index)
forwardmethods(TextDialog, ScrolledText, '_text')
######################################################################
### File: PmwTimeCounter.py
# Authors: Joe VanAndel and Greg McFarlane
import string
import sys
import time
import Tkinter
class TimeCounter(MegaWidget):
"""Up-down counter
A TimeCounter is a single-line entry widget with Up and Down arrows
which increment and decrement the Time value in the entry.
"""
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('autorepeat', 1, None),
('buttonaspect', 1.0, INITOPT),
('command', None, None),
('initwait', 300, None),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('max', None, self._max),
('min', None, self._min),
('padx', 0, INITOPT),
('pady', 0, INITOPT),
('repeatrate', 50, None),
('value', None, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
self.arrowDirection = {}
self._flag = 'stopped'
self._timerId = None
self._createComponents(kw)
value = self['value']
if value is None:
now = time.time()
value = time.strftime('%H:%M:%S', time.localtime(now))
self.setvalue(value)
# Check keywords and initialise options.
self.initialiseoptions()
def _createComponents(self, kw):
# Create the components.
interior = self.interior()
# If there is no label, put the arrows and the entry directly
# into the interior, otherwise create a frame for them. In
# either case the border around the arrows and the entry will
# be raised (but not around the label).
if self['labelpos'] is None:
frame = interior
if not kw.has_key('hull_relief'):
frame.configure(relief = 'raised')
if not kw.has_key('hull_borderwidth'):
frame.configure(borderwidth = 1)
else:
frame = self.createcomponent('frame',
(), None,
Tkinter.Frame, (interior,),
relief = 'raised', borderwidth = 1)
frame.grid(column=2, row=2, sticky='nsew')
interior.grid_columnconfigure(2, weight=1)
interior.grid_rowconfigure(2, weight=1)
# Create the down arrow buttons.
# Create the hour down arrow.
self._downHourArrowBtn = self.createcomponent('downhourarrow',
(), 'Arrow',
Tkinter.Canvas, (frame,),
width = 16, height = 16, relief = 'raised', borderwidth = 2)
self.arrowDirection[self._downHourArrowBtn] = 'down'
self._downHourArrowBtn.grid(column = 0, row = 2)
# Create the minute down arrow.
self._downMinuteArrowBtn = self.createcomponent('downminutearrow',
(), 'Arrow',
Tkinter.Canvas, (frame,),
width = 16, height = 16, relief = 'raised', borderwidth = 2)
self.arrowDirection[self._downMinuteArrowBtn] = 'down'
self._downMinuteArrowBtn.grid(column = 1, row = 2)
# Create the second down arrow.
self._downSecondArrowBtn = self.createcomponent('downsecondarrow',
(), 'Arrow',
Tkinter.Canvas, (frame,),
width = 16, height = 16, relief = 'raised', borderwidth = 2)
self.arrowDirection[self._downSecondArrowBtn] = 'down'
self._downSecondArrowBtn.grid(column = 2, row = 2)
# Create the entry fields.
# Create the hour entry field.
self._hourCounterEntry = self.createcomponent('hourentryfield',
(('hourentry', 'hourentryfield_entry'),), None,
EntryField, (frame,), validate='integer', entry_width = 2)
self._hourCounterEntry.grid(column = 0, row = 1, sticky = 'news')
# Create the minute entry field.
self._minuteCounterEntry = self.createcomponent('minuteentryfield',
(('minuteentry', 'minuteentryfield_entry'),), None,
EntryField, (frame,), validate='integer', entry_width = 2)
self._minuteCounterEntry.grid(column = 1, row = 1, sticky = 'news')
# Create the second entry field.
self._secondCounterEntry = self.createcomponent('secondentryfield',
(('secondentry', 'secondentryfield_entry'),), None,
EntryField, (frame,), validate='integer', entry_width = 2)
self._secondCounterEntry.grid(column = 2, row = 1, sticky = 'news')
# Create the up arrow buttons.
# Create the hour up arrow.
self._upHourArrowBtn = self.createcomponent('uphourarrow',
(), 'Arrow',
Tkinter.Canvas, (frame,),
width = 16, height = 16, relief = 'raised', borderwidth = 2)
self.arrowDirection[self._upHourArrowBtn] = 'up'
self._upHourArrowBtn.grid(column = 0, row = 0)
# Create the minute up arrow.
self._upMinuteArrowBtn = self.createcomponent('upminutearrow',
(), 'Arrow',
Tkinter.Canvas, (frame,),
width = 16, height = 16, relief = 'raised', borderwidth = 2)
self.arrowDirection[self._upMinuteArrowBtn] = 'up'
self._upMinuteArrowBtn.grid(column = 1, row = 0)
# Create the second up arrow.
self._upSecondArrowBtn = self.createcomponent('upsecondarrow',
(), 'Arrow',
Tkinter.Canvas, (frame,),
width = 16, height = 16, relief = 'raised', borderwidth = 2)
self.arrowDirection[self._upSecondArrowBtn] = 'up'
self._upSecondArrowBtn.grid(column = 2, row = 0)
# Make it resize nicely.
padx = self['padx']
pady = self['pady']
for col in range(3):
frame.grid_columnconfigure(col, weight = 1, pad = padx)
frame.grid_rowconfigure(0, pad = pady)
frame.grid_rowconfigure(2, pad = pady)
frame.grid_rowconfigure(1, weight = 1)
# Create the label.
self.createlabel(interior)
# Set bindings.
# Up hour
self._upHourArrowBtn.bind('<Configure>',
lambda event, s=self,button=self._upHourArrowBtn:
s._drawArrow(button, 'up'))
self._upHourArrowBtn.bind('<1>',
lambda event, s=self,button=self._upHourArrowBtn:
s._countUp(button, 3600))
self._upHourArrowBtn.bind('<Any-ButtonRelease-1>',
lambda event, s=self, button=self._upHourArrowBtn:
s._stopUpDown(button))
# Up minute
self._upMinuteArrowBtn.bind('<Configure>',
lambda event, s=self,button=self._upMinuteArrowBtn:
s._drawArrow(button, 'up'))
self._upMinuteArrowBtn.bind('<1>',
lambda event, s=self,button=self._upMinuteArrowBtn:
s._countUp(button, 60))
self._upMinuteArrowBtn.bind('<Any-ButtonRelease-1>',
lambda event, s=self, button=self._upMinuteArrowBtn:
s._stopUpDown(button))
# Up second
self._upSecondArrowBtn.bind('<Configure>',
lambda event, s=self,button=self._upSecondArrowBtn:
s._drawArrow(button, 'up'))
self._upSecondArrowBtn.bind('<1>',
lambda event, s=self,button=self._upSecondArrowBtn:
s._countUp(button, 1))
self._upSecondArrowBtn.bind('<Any-ButtonRelease-1>',
lambda event, s=self, button=self._upSecondArrowBtn:
s._stopUpDown(button))
# Down hour
self._downHourArrowBtn.bind('<Configure>',
lambda event, s=self,button=self._downHourArrowBtn:
s._drawArrow(button, 'down'))
self._downHourArrowBtn.bind('<1>',
lambda event, s=self,button=self._downHourArrowBtn:
s._countDown(button, 3600))
self._downHourArrowBtn.bind('<Any-ButtonRelease-1>',
lambda event, s=self, button=self._downHourArrowBtn:
s._stopUpDown(button))
# Down minute
self._downMinuteArrowBtn.bind('<Configure>',
lambda event, s=self,button=self._downMinuteArrowBtn:
s._drawArrow(button, 'down'))
self._downMinuteArrowBtn.bind('<1>',
lambda event, s=self,button=self._downMinuteArrowBtn:
s._countDown(button, 60))
self._downMinuteArrowBtn.bind('<Any-ButtonRelease-1>',
lambda event, s=self, button=self._downMinuteArrowBtn:
s._stopUpDown(button))
# Down second
self._downSecondArrowBtn.bind('<Configure>',
lambda event, s=self,button=self._downSecondArrowBtn:
s._drawArrow(button, 'down'))
self._downSecondArrowBtn.bind('<1>',
lambda event, s=self, button=self._downSecondArrowBtn:
s._countDown(button,1))
self._downSecondArrowBtn.bind('<Any-ButtonRelease-1>',
lambda event, s=self, button=self._downSecondArrowBtn:
s._stopUpDown(button))
self._hourCounterEntry.component('entry').bind(
'<Return>', self._invoke)
self._minuteCounterEntry.component('entry').bind(
'<Return>', self._invoke)
self._secondCounterEntry.component('entry').bind(
'<Return>', self._invoke)
self._hourCounterEntry.bind('<Configure>', self._resizeArrow)
self._minuteCounterEntry.bind('<Configure>', self._resizeArrow)
self._secondCounterEntry.bind('<Configure>', self._resizeArrow)
def _drawArrow(self, arrow, direction):
drawarrow(arrow, self['hourentry_foreground'], direction, 'arrow')
def _resizeArrow(self, event = None):
for btn in (self._upHourArrowBtn, self._upMinuteArrowBtn,
self._upSecondArrowBtn,
self._downHourArrowBtn,
self._downMinuteArrowBtn, self._downSecondArrowBtn):
bw = (string.atoi(btn['borderwidth']) +
string.atoi(btn['highlightthickness']))
newHeight = self._hourCounterEntry.winfo_reqheight() - 2 * bw
newWidth = int(newHeight * self['buttonaspect'])
btn.configure(width=newWidth, height=newHeight)
self._drawArrow(btn, self.arrowDirection[btn])
def _min(self):
min = self['min']
if min is None:
self._minVal = 0
else:
self._minVal = timestringtoseconds(min)
def _max(self):
max = self['max']
if max is None:
self._maxVal = None
else:
self._maxVal = timestringtoseconds(max)
def getvalue(self):
return self.getstring()
def setvalue(self, text):
list = string.split(text, ':')
if len(list) != 3:
raise ValueError, 'invalid value: ' + text
self._hour = string.atoi(list[0])
self._minute = string.atoi(list[1])
self._second = string.atoi(list[2])
self._setHMS()
def getstring(self):
return '%02d:%02d:%02d' % (self._hour, self._minute, self._second)
def getint(self):
return self._hour * 3600 + self._minute * 60 + self._second
def _countUp(self, button, increment):
self._relief = self._upHourArrowBtn.cget('relief')
button.configure(relief='sunken')
self._count(1, 'start', increment)
def _countDown(self, button, increment):
self._relief = self._downHourArrowBtn.cget('relief')
button.configure(relief='sunken')
self._count(-1, 'start', increment)
def increment(self, seconds = 1):
self._count(1, 'force', seconds)
def decrement(self, seconds = 1):
self._count(-1, 'force', seconds)
def _count(self, factor, newFlag = None, increment = 1):
if newFlag != 'force':
if newFlag is not None:
self._flag = newFlag
if self._flag == 'stopped':
return
value = (string.atoi(self._hourCounterEntry.get()) *3600) + \
(string.atoi(self._minuteCounterEntry.get()) *60) + \
string.atoi(self._secondCounterEntry.get()) + \
factor * increment
min = self._minVal
max = self._maxVal
if value < min:
value = min
if max is not None and value > max:
value = max
self._hour = value /3600
self._minute = (value - (self._hour*3600)) / 60
self._second = value - (self._hour*3600) - (self._minute*60)
self._setHMS()
if newFlag != 'force':
if self['autorepeat']:
if self._flag == 'start':
delay = self['initwait']
self._flag = 'running'
else:
delay = self['repeatrate']
self._timerId = self.after(
delay, lambda self=self, factor=factor,increment=increment:
self._count(factor,'running', increment))
def _setHMS(self):
self._hourCounterEntry.setentry('%02d' % self._hour)
self._minuteCounterEntry.setentry('%02d' % self._minute)
self._secondCounterEntry.setentry('%02d' % self._second)
def _stopUpDown(self, button):
if self._timerId is not None:
self.after_cancel(self._timerId)
self._timerId = None
button.configure(relief=self._relief)
self._flag = 'stopped'
def _invoke(self, event):
cmd = self['command']
if callable(cmd):
cmd()
def invoke(self):
cmd = self['command']
if callable(cmd):
return cmd()
def destroy(self):
if self._timerId is not None:
self.after_cancel(self._timerId)
self._timerId = None
MegaWidget.destroy(self)
######################################################################
### File: PmwAboutDialog.py
class AboutDialog(MessageDialog):
# Window to display version and contact information.
# Class members containing resettable 'default' values:
_version = ''
_copyright = ''
_contact = ''
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('applicationname', '', INITOPT),
('iconpos', 'w', None),
('icon_bitmap', 'info', None),
('buttons', ('Close',), None),
('defaultbutton', 0, None),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MessageDialog.__init__(self, parent)
applicationname = self['applicationname']
if not kw.has_key('title'):
self.configure(title = 'About ' + applicationname)
if not kw.has_key('message_text'):
text = applicationname + '\n\n'
if AboutDialog._version != '':
text = text + 'Version ' + AboutDialog._version + '\n'
if AboutDialog._copyright != '':
text = text + AboutDialog._copyright + '\n\n'
if AboutDialog._contact != '':
text = text + AboutDialog._contact
self.configure(message_text=text)
# Check keywords and initialise options.
self.initialiseoptions()
def aboutversion(value):
AboutDialog._version = value
def aboutcopyright(value):
AboutDialog._copyright = value
def aboutcontact(value):
AboutDialog._contact = value
######################################################################
### File: PmwComboBox.py
# Based on iwidgets2.2.0/combobox.itk code.
import os
import string
import types
import Tkinter
class ComboBox(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('autoclear', 0, INITOPT),
('buttonaspect', 1.0, INITOPT),
('dropdown', 1, INITOPT),
('fliparrow', 0, INITOPT),
('history', 1, INITOPT),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('listheight', 200, INITOPT),
('selectioncommand', None, None),
('sticky', 'ew', INITOPT),
('unique', 1, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Create the components.
interior = self.interior()
self._entryfield = self.createcomponent('entryfield',
(('entry', 'entryfield_entry'),), None,
EntryField, (interior,))
self._entryfield.grid(column=2, row=2, sticky=self['sticky'])
interior.grid_columnconfigure(2, weight = 1)
self._entryWidget = self._entryfield.component('entry')
if self['dropdown']:
self._isPosted = 0
interior.grid_rowconfigure(2, weight = 1)
# Create the arrow button.
self._arrowBtn = self.createcomponent('arrowbutton',
(), None,
Tkinter.Canvas, (interior,), borderwidth = 2,
relief = 'raised',
width = 16, height = 16)
if 'n' in self['sticky']:
sticky = 'n'
else:
sticky = ''
if 's' in self['sticky']:
sticky = sticky + 's'
self._arrowBtn.grid(column=3, row=2, sticky = sticky)
self._arrowRelief = self._arrowBtn.cget('relief')
# Create the label.
self.createlabel(interior, childCols=2)
# Create the dropdown window.
self._popup = self.createcomponent('popup',
(), None,
Tkinter.Toplevel, (interior,))
self._popup.withdraw()
self._popup.overrideredirect(1)
# Create the scrolled listbox inside the dropdown window.
self._list = self.createcomponent('scrolledlist',
(('listbox', 'scrolledlist_listbox'),), None,
ScrolledListBox, (self._popup,),
hull_borderwidth = 2,
hull_relief = 'raised',
hull_height = self['listheight'],
usehullsize = 1,
listbox_exportselection = 0)
self._list.pack(expand=1, fill='both')
self.__listbox = self._list.component('listbox')
# Bind events to the arrow button.
self._arrowBtn.bind('<1>', self._postList)
self._arrowBtn.bind('<Configure>', self._drawArrow)
self._arrowBtn.bind('<3>', self._next)
self._arrowBtn.bind('<Shift-3>', self._previous)
self._arrowBtn.bind('<Down>', self._next)
self._arrowBtn.bind('<Up>', self._previous)
self._arrowBtn.bind('<Control-n>', self._next)
self._arrowBtn.bind('<Control-p>', self._previous)
self._arrowBtn.bind('<Shift-Down>', self._postList)
self._arrowBtn.bind('<Shift-Up>', self._postList)
self._arrowBtn.bind('<F34>', self._postList)
self._arrowBtn.bind('<F28>', self._postList)
self._arrowBtn.bind('<space>', self._postList)
# Bind events to the dropdown window.
self._popup.bind('<Escape>', self._unpostList)
self._popup.bind('<space>', self._selectUnpost)
self._popup.bind('<Return>', self._selectUnpost)
self._popup.bind('<ButtonRelease-1>', self._dropdownBtnRelease)
self._popup.bind('<ButtonPress-1>', self._unpostOnNextRelease)
# Bind events to the Tk listbox.
self.__listbox.bind('<Enter>', self._unpostOnNextRelease)
# Bind events to the Tk entry widget.
self._entryWidget.bind('<Configure>', self._resizeArrow)
self._entryWidget.bind('<Shift-Down>', self._postList)
self._entryWidget.bind('<Shift-Up>', self._postList)
self._entryWidget.bind('<F34>', self._postList)
self._entryWidget.bind('<F28>', self._postList)
# Need to unpost the popup if the entryfield is unmapped (eg:
# its toplevel window is withdrawn) while the popup list is
# displayed.
self._entryWidget.bind('<Unmap>', self._unpostList)
else:
# Create the scrolled listbox below the entry field.
self._list = self.createcomponent('scrolledlist',
(('listbox', 'scrolledlist_listbox'),), None,
ScrolledListBox, (interior,),
selectioncommand = self._selectCmd)
self._list.grid(column=2, row=3, sticky='nsew')
self.__listbox = self._list.component('listbox')
# The scrolled listbox should expand vertically.
interior.grid_rowconfigure(3, weight = 1)
# Create the label.
self.createlabel(interior, childRows=2)
self._entryWidget.bind('<Down>', self._next)
self._entryWidget.bind('<Up>', self._previous)
self._entryWidget.bind('<Control-n>', self._next)
self._entryWidget.bind('<Control-p>', self._previous)
self.__listbox.bind('<Control-n>', self._next)
self.__listbox.bind('<Control-p>', self._previous)
if self['history']:
self._entryfield.configure(command=self._addHistory)
# Check keywords and initialise options.
self.initialiseoptions()
def destroy(self):
if self['dropdown'] and self._isPosted:
popgrab(self._popup)
MegaWidget.destroy(self)
#======================================================================
# Public methods
def get(self, first = None, last=None):
if first is None:
return self._entryWidget.get()
else:
return self._list.get(first, last)
def invoke(self):
if self['dropdown']:
self._postList()
else:
return self._selectCmd()
def selectitem(self, index, setentry=1):
if isinstance(index, basestring):
text = index
items = self._list.get(0, 'end')
if text in items:
index = list(items).index(text)
else:
raise IndexError, 'index "%s" not found' % text
elif setentry:
text = self._list.get(0, 'end')[index]
self._list.select_clear(0, 'end')
self._list.select_set(index, index)
self._list.activate(index)
self.see(index)
if setentry:
self._entryfield.setentry(text)
# Need to explicitly forward this to override the stupid
# (grid_)size method inherited from Tkinter.Frame.Grid.
def size(self):
return self._list.size()
# Need to explicitly forward this to override the stupid
# (grid_)bbox method inherited from Tkinter.Frame.Grid.
def bbox(self, index):
return self._list.bbox(index)
def clear(self):
self._entryfield.clear()
self._list.clear()
#======================================================================
# Private methods for both dropdown and simple comboboxes.
def _addHistory(self):
input = self._entryWidget.get()
if input != '':
index = None
if self['unique']:
# If item is already in list, select it and return.
items = self._list.get(0, 'end')
if input in items:
index = list(items).index(input)
if index is None:
index = self._list.index('end')
self._list.insert('end', input)
self.selectitem(index)
if self['autoclear']:
self._entryWidget.delete(0, 'end')
# Execute the selectioncommand on the new entry.
self._selectCmd()
def _next(self, event):
size = self.size()
if size <= 1:
return
cursels = self.curselection()
if len(cursels) == 0:
index = 0
else:
index = string.atoi(cursels[0])
if index == size - 1:
index = 0
else:
index = index + 1
self.selectitem(index)
def _previous(self, event):
size = self.size()
if size <= 1:
return
cursels = self.curselection()
if len(cursels) == 0:
index = size - 1
else:
index = string.atoi(cursels[0])
if index == 0:
index = size - 1
else:
index = index - 1
self.selectitem(index)
def _selectCmd(self, event=None):
sels = self.getcurselection()
if len(sels) == 0:
item = None
else:
item = sels[0]
self._entryfield.setentry(item)
cmd = self['selectioncommand']
if callable(cmd):
if event is None:
# Return result of selectioncommand for invoke() method.
return cmd(item)
else:
cmd(item)
#======================================================================
# Private methods for dropdown combobox.
def _drawArrow(self, event=None, sunken=0):
arrow = self._arrowBtn
if sunken:
self._arrowRelief = arrow.cget('relief')
arrow.configure(relief = 'sunken')
else:
arrow.configure(relief = self._arrowRelief)
if self._isPosted and self['fliparrow']:
direction = 'up'
else:
direction = 'down'
drawarrow(arrow, self['entry_foreground'], direction, 'arrow')
def _postList(self, event = None):
self._isPosted = 1
self._drawArrow(sunken=1)
# Make sure that the arrow is displayed sunken.
self.update_idletasks()
x = self._entryfield.winfo_rootx()
y = self._entryfield.winfo_rooty() + \
self._entryfield.winfo_height()
w = self._entryfield.winfo_width() + self._arrowBtn.winfo_width()
h = self.__listbox.winfo_height()
sh = self.winfo_screenheight()
if y + h > sh and y > sh / 2:
y = self._entryfield.winfo_rooty() - h
self._list.configure(hull_width=w)
setgeometryanddeiconify(self._popup, '+%d+%d' % (x, y))
# Grab the popup, so that all events are delivered to it, and
# set focus to the listbox, to make keyboard navigation
# easier.
pushgrab(self._popup, 1, self._unpostList)
self.__listbox.focus_set()
self._drawArrow()
# Ignore the first release of the mouse button after posting the
# dropdown list, unless the mouse enters the dropdown list.
self._ignoreRelease = 1
def _dropdownBtnRelease(self, event):
if (event.widget == self._list.component('vertscrollbar') or
event.widget == self._list.component('horizscrollbar')):
return
if self._ignoreRelease:
self._unpostOnNextRelease()
return
self._unpostList()
if (event.x >= 0 and event.x < self.__listbox.winfo_width() and
event.y >= 0 and event.y < self.__listbox.winfo_height()):
self._selectCmd()
def _unpostOnNextRelease(self, event = None):
self._ignoreRelease = 0
def _resizeArrow(self, event):
bw = (string.atoi(self._arrowBtn['borderwidth']) +
string.atoi(self._arrowBtn['highlightthickness']))
newHeight = self._entryfield.winfo_reqheight() - 2 * bw
newWidth = int(newHeight * self['buttonaspect'])
self._arrowBtn.configure(width=newWidth, height=newHeight)
self._drawArrow()
def _unpostList(self, event=None):
if not self._isPosted:
# It is possible to get events on an unposted popup. For
# example, by repeatedly pressing the space key to post
# and unpost the popup. The <space> event may be
# delivered to the popup window even though
# popgrab() has set the focus away from the
# popup window. (Bug in Tk?)
return
# Restore the focus before withdrawing the window, since
# otherwise the window manager may take the focus away so we
# can't redirect it. Also, return the grab to the next active
# window in the stack, if any.
popgrab(self._popup)
self._popup.withdraw()
self._isPosted = 0
self._drawArrow()
def _selectUnpost(self, event):
self._unpostList()
self._selectCmd()
forwardmethods(ComboBox, ScrolledListBox, '_list')
forwardmethods(ComboBox, EntryField, '_entryfield')
######################################################################
### File: PmwComboBoxDialog.py
# Not Based on iwidgets version.
class ComboBoxDialog(Dialog):
# Dialog window with simple combobox.
# Dialog window displaying a list and entry field and requesting
# the user to make a selection or enter a value
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderx', 10, INITOPT),
('bordery', 10, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
Dialog.__init__(self, parent)
# Create the components.
interior = self.interior()
aliases = (
('listbox', 'combobox_listbox'),
('scrolledlist', 'combobox_scrolledlist'),
('entry', 'combobox_entry'),
('label', 'combobox_label'),
)
self._combobox = self.createcomponent('combobox',
aliases, None,
ComboBox, (interior,),
scrolledlist_dblclickcommand = self.invoke,
dropdown = 0,
)
self._combobox.pack(side='top', expand='true', fill='both',
padx = self['borderx'], pady = self['bordery'])
if not kw.has_key('activatecommand'):
# Whenever this dialog is activated, set the focus to the
# ComboBox's listbox widget.
listbox = self.component('listbox')
self.configure(activatecommand = listbox.focus_set)
# Check keywords and initialise options.
self.initialiseoptions()
# Need to explicitly forward this to override the stupid
# (grid_)size method inherited from Tkinter.Toplevel.Grid.
def size(self):
return self._combobox.size()
# Need to explicitly forward this to override the stupid
# (grid_)bbox method inherited from Tkinter.Toplevel.Grid.
def bbox(self, index):
return self._combobox.bbox(index)
forwardmethods(ComboBoxDialog, ComboBox, '_combobox')
######################################################################
### File: PmwCounter.py
import string
import sys
import types
import Tkinter
class Counter(MegaWidget):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('autorepeat', 1, None),
('buttonaspect', 1.0, INITOPT),
('datatype', 'numeric', self._datatype),
('increment', 1, None),
('initwait', 300, None),
('labelmargin', 0, INITOPT),
('labelpos', None, INITOPT),
('orient', 'horizontal', INITOPT),
('padx', 0, INITOPT),
('pady', 0, INITOPT),
('repeatrate', 50, None),
('sticky', 'ew', INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
MegaWidget.__init__(self, parent)
# Initialise instance variables.
self._timerId = None
self._normalRelief = None
# Create the components.
interior = self.interior()
# If there is no label, put the arrows and the entry directly
# into the interior, otherwise create a frame for them. In
# either case the border around the arrows and the entry will
# be raised (but not around the label).
if self['labelpos'] is None:
frame = interior
if not kw.has_key('hull_relief'):
frame.configure(relief = 'raised')
if not kw.has_key('hull_borderwidth'):
frame.configure(borderwidth = 1)
else:
frame = self.createcomponent('frame',
(), None,
Tkinter.Frame, (interior,),
relief = 'raised', borderwidth = 1)
frame.grid(column=2, row=2, sticky=self['sticky'])
interior.grid_columnconfigure(2, weight=1)
interior.grid_rowconfigure(2, weight=1)
# Create the down arrow.
self._downArrowBtn = self.createcomponent('downarrow',
(), 'Arrow',
Tkinter.Canvas, (frame,),
width = 16, height = 16, relief = 'raised', borderwidth = 2)
# Create the entry field.
self._counterEntry = self.createcomponent('entryfield',
(('entry', 'entryfield_entry'),), None,
EntryField, (frame,))
# Create the up arrow.
self._upArrowBtn = self.createcomponent('uparrow',
(), 'Arrow',
Tkinter.Canvas, (frame,),
width = 16, height = 16, relief = 'raised', borderwidth = 2)
padx = self['padx']
pady = self['pady']
orient = self['orient']
if orient == 'horizontal':
self._downArrowBtn.grid(column = 0, row = 0)
self._counterEntry.grid(column = 1, row = 0,
sticky = self['sticky'])
self._upArrowBtn.grid(column = 2, row = 0)
frame.grid_columnconfigure(1, weight = 1)
frame.grid_rowconfigure(0, weight = 1)
if Tkinter.TkVersion >= 4.2:
frame.grid_columnconfigure(0, pad = padx)
frame.grid_columnconfigure(2, pad = padx)
frame.grid_rowconfigure(0, pad = pady)
elif orient == 'vertical':
self._upArrowBtn.grid(column = 0, row = 0, sticky = 's')
self._counterEntry.grid(column = 0, row = 1,
sticky = self['sticky'])
self._downArrowBtn.grid(column = 0, row = 2, sticky = 'n')
frame.grid_columnconfigure(0, weight = 1)
frame.grid_rowconfigure(0, weight = 1)
frame.grid_rowconfigure(2, weight = 1)
if Tkinter.TkVersion >= 4.2:
frame.grid_rowconfigure(0, pad = pady)
frame.grid_rowconfigure(2, pad = pady)
frame.grid_columnconfigure(0, pad = padx)
else:
raise ValueError, 'bad orient option ' + repr(orient) + \
': must be either \'horizontal\' or \'vertical\''
self.createlabel(interior)
self._upArrowBtn.bind('<Configure>', self._drawUpArrow)
self._upArrowBtn.bind('<1>', self._countUp)
self._upArrowBtn.bind('<Any-ButtonRelease-1>', self._stopCounting)
self._downArrowBtn.bind('<Configure>', self._drawDownArrow)
self._downArrowBtn.bind('<1>', self._countDown)
self._downArrowBtn.bind('<Any-ButtonRelease-1>', self._stopCounting)
self._counterEntry.bind('<Configure>', self._resizeArrow)
entry = self._counterEntry.component('entry')
entry.bind('<Down>', lambda event, s = self: s._key_decrement(event))
entry.bind('<Up>', lambda event, s = self: s._key_increment(event))
# Need to cancel the timer if an arrow button is unmapped (eg:
# its toplevel window is withdrawn) while the mouse button is
# held down. The canvas will not get the ButtonRelease event
# if it is not mapped, since the implicit grab is cancelled.
self._upArrowBtn.bind('<Unmap>', self._stopCounting)
self._downArrowBtn.bind('<Unmap>', self._stopCounting)
# Check keywords and initialise options.
self.initialiseoptions()
def _resizeArrow(self, event):
for btn in (self._upArrowBtn, self._downArrowBtn):
bw = (string.atoi(btn['borderwidth']) +
string.atoi(btn['highlightthickness']))
newHeight = self._counterEntry.winfo_reqheight() - 2 * bw
newWidth = int(newHeight * self['buttonaspect'])
btn.configure(width=newWidth, height=newHeight)
self._drawArrow(btn)
def _drawUpArrow(self, event):
self._drawArrow(self._upArrowBtn)
def _drawDownArrow(self, event):
self._drawArrow(self._downArrowBtn)
def _drawArrow(self, arrow):
if self['orient'] == 'vertical':
if arrow == self._upArrowBtn:
direction = 'up'
else:
direction = 'down'
else:
if arrow == self._upArrowBtn:
direction = 'right'
else:
direction = 'left'
drawarrow(arrow, self['entry_foreground'], direction, 'arrow')
def _stopCounting(self, event = None):
if self._timerId is not None:
self.after_cancel(self._timerId)
self._timerId = None
if self._normalRelief is not None:
button, relief = self._normalRelief
button.configure(relief=relief)
self._normalRelief = None
def _countUp(self, event):
self._normalRelief = (self._upArrowBtn, self._upArrowBtn.cget('relief'))
self._upArrowBtn.configure(relief='sunken')
# Force arrow down (it may come up immediately, if increment fails).
self._upArrowBtn.update_idletasks()
self._count(1, 1)
def _countDown(self, event):
self._normalRelief = (self._downArrowBtn, self._downArrowBtn.cget('relief'))
self._downArrowBtn.configure(relief='sunken')
# Force arrow down (it may come up immediately, if increment fails).
self._downArrowBtn.update_idletasks()
self._count(-1, 1)
def increment(self):
self._forceCount(1)
def decrement(self):
self._forceCount(-1)
def _key_increment(self, event):
self._forceCount(1)
self.update_idletasks()
def _key_decrement(self, event):
self._forceCount(-1)
self.update_idletasks()
def _datatype(self):
datatype = self['datatype']
if type(datatype) is types.DictionaryType:
self._counterArgs = datatype.copy()
if self._counterArgs.has_key('counter'):
datatype = self._counterArgs['counter']
del self._counterArgs['counter']
else:
datatype = 'numeric'
else:
self._counterArgs = {}
if _counterCommands.has_key(datatype):
self._counterCommand = _counterCommands[datatype]
elif callable(datatype):
self._counterCommand = datatype
else:
validValues = _counterCommands.keys()
validValues.sort()
raise ValueError, ('bad datatype value "%s": must be a' +
' function or one of %s') % (datatype, validValues)
def _forceCount(self, factor):
if not self.valid():
self.bell()
return
text = self._counterEntry.get()
try:
value = apply(self._counterCommand,
(text, factor, self['increment']), self._counterArgs)
except ValueError:
self.bell()
return
previousICursor = self._counterEntry.index('insert')
if self._counterEntry.setentry(value) == OK:
self._counterEntry.xview('end')
self._counterEntry.icursor(previousICursor)
def _count(self, factor, first):
if not self.valid():
self.bell()
return
self._timerId = None
origtext = self._counterEntry.get()
try:
value = apply(self._counterCommand,
(origtext, factor, self['increment']), self._counterArgs)
except ValueError:
# If text is invalid, stop counting.
self._stopCounting()
self.bell()
return
# If incrementing produces an invalid value, restore previous
# text and stop counting.
previousICursor = self._counterEntry.index('insert')
valid = self._counterEntry.setentry(value)
if valid != OK:
self._stopCounting()
self._counterEntry.setentry(origtext)
if valid == PARTIAL:
self.bell()
return
self._counterEntry.xview('end')
self._counterEntry.icursor(previousICursor)
if self['autorepeat']:
if first:
delay = self['initwait']
else:
delay = self['repeatrate']
self._timerId = self.after(delay,
lambda self=self, factor=factor: self._count(factor, 0))
def destroy(self):
self._stopCounting()
MegaWidget.destroy(self)
forwardmethods(Counter, EntryField, '_counterEntry')
def _changeNumber(text, factor, increment):
value = string.atol(text)
if factor > 0:
value = (value / increment) * increment + increment
else:
value = ((value - 1) / increment) * increment
# Get rid of the 'L' at the end of longs (in python up to 1.5.2).
rtn = str(value)
if rtn[-1] == 'L':
return rtn[:-1]
else:
return rtn
def _changeReal(text, factor, increment, separator = '.'):
value = stringtoreal(text, separator)
div = value / increment
# Compare reals using str() to avoid problems caused by binary
# numbers being only approximations to decimal numbers.
# For example, if value is -0.3 and increment is 0.1, then
# int(value/increment) = -2, not -3 as one would expect.
if str(div)[-2:] == '.0':
# value is an even multiple of increment.
div = round(div) + factor
else:
# value is not an even multiple of increment.
div = int(div) * 1.0
if value < 0:
div = div - 1
if factor > 0:
div = (div + 1)
value = div * increment
text = str(value)
if separator != '.':
index = string.find(text, '.')
if index >= 0:
text = text[:index] + separator + text[index + 1:]
return text
def _changeDate(value, factor, increment, format = 'ymd',
separator = '/', yyyy = 0):
jdn = datestringtojdn(value, format, separator) + factor * increment
y, m, d = jdntoymd(jdn)
result = ''
for index in range(3):
if index > 0:
result = result + separator
f = format[index]
if f == 'y':
if yyyy:
result = result + '%02d' % y
else:
result = result + '%02d' % (y % 100)
elif f == 'm':
result = result + '%02d' % m
elif f == 'd':
result = result + '%02d' % d
return result
_SECSPERDAY = 24 * 60 * 60
def _changeTime(value, factor, increment, separator = ':', time24 = 0):
unixTime = timestringtoseconds(value, separator)
if factor > 0:
chunks = unixTime / increment + 1
else:
chunks = (unixTime - 1) / increment
unixTime = chunks * increment
if time24:
while unixTime < 0:
unixTime = unixTime + _SECSPERDAY
while unixTime >= _SECSPERDAY:
unixTime = unixTime - _SECSPERDAY
if unixTime < 0:
unixTime = -unixTime
sign = '-'
else:
sign = ''
secs = unixTime % 60
unixTime = unixTime / 60
mins = unixTime % 60
hours = unixTime / 60
return '%s%02d%s%02d%s%02d' % (sign, hours, separator, mins, separator, secs)
# hexadecimal, alphabetic, alphanumeric not implemented
_counterCommands = {
'numeric' : _changeNumber, # } integer
'integer' : _changeNumber, # } these two use the same function
'real' : _changeReal, # real number
'time' : _changeTime,
'date' : _changeDate,
}
######################################################################
### File: PmwCounterDialog.py
# A Dialog with a counter
class CounterDialog(Dialog):
def __init__(self, parent = None, **kw):
# Define the megawidget options.
optiondefs = (
('borderx', 20, INITOPT),
('bordery', 20, INITOPT),
)
self.defineoptions(kw, optiondefs)
# Initialise the base class (after defining the options).
Dialog.__init__(self, parent)
# Create the components.
interior = self.interior()
# Create the counter.
aliases = (
('entryfield', 'counter_entryfield'),
('entry', 'counter_entryfield_entry'),
('label', 'counter_label')
)
self._cdCounter = self.createcomponent('counter',
aliases, None,
Counter, (interior,))
self._cdCounter.pack(fill='x', expand=1,
padx = self['borderx'], pady = self['bordery'])
if not kw.has_key('activatecommand'):
# Whenever this dialog is activated, set the focus to the
# Counter's entry widget.
tkentry = self.component('entry')
self.configure(activatecommand = tkentry.focus_set)
# Check keywords and initialise options.
self.initialiseoptions()
# Supply aliases to some of the entry component methods.
def insertentry(self, index, text):
self._cdCounter.insert(index, text)
def deleteentry(self, first, last=None):
self._cdCounter.delete(first, last)
def indexentry(self, index):
return self._cdCounter.index(index)
forwardmethods(CounterDialog, Counter, '_cdCounter')
######################################################################
### File: PmwLogicalFont.py
import os
import string
def _font_initialise(root, size=None, fontScheme = None):
global _fontSize
if size is not None:
_fontSize = size
if fontScheme in ('pmw1', 'pmw2'):
if os.name == 'posix':
defaultFont = logicalfont('Helvetica')
menuFont = logicalfont('Helvetica', weight='bold', slant='italic')
scaleFont = logicalfont('Helvetica', slant='italic')
root.option_add('*Font', defaultFont, 'userDefault')
root.option_add('*Menu*Font', menuFont, 'userDefault')
root.option_add('*Menubutton*Font', menuFont, 'userDefault')
root.option_add('*Scale.*Font', scaleFont, 'userDefault')
if fontScheme == 'pmw1':
balloonFont = logicalfont('Helvetica', -6, pixel = '12')
else: # fontScheme == 'pmw2'
balloonFont = logicalfont('Helvetica', -2)
root.option_add('*Balloon.*Font', balloonFont, 'userDefault')
else:
defaultFont = logicalfont('Helvetica')
root.option_add('*Font', defaultFont, 'userDefault')
elif fontScheme == 'default':
defaultFont = ('Helvetica', '-%d' % (_fontSize,), 'bold')
entryFont = ('Helvetica', '-%d' % (_fontSize,))
textFont = ('Courier', '-%d' % (_fontSize,))
root.option_add('*Font', defaultFont, 'userDefault')
root.option_add('*Entry*Font', entryFont, 'userDefault')
root.option_add('*Text*Font', textFont, 'userDefault')
def logicalfont(name='Helvetica', sizeIncr = 0, **kw):
if not _fontInfo.has_key(name):
raise ValueError, 'font %s does not exist' % name
rtn = []
for field in _fontFields:
if kw.has_key(field):
logicalValue = kw[field]
elif _fontInfo[name].has_key(field):
logicalValue = _fontInfo[name][field]
else:
logicalValue = '*'
if _propertyAliases[name].has_key((field, logicalValue)):
realValue = _propertyAliases[name][(field, logicalValue)]
elif _propertyAliases[name].has_key((field, None)):
realValue = _propertyAliases[name][(field, None)]
elif _propertyAliases[None].has_key((field, logicalValue)):
realValue = _propertyAliases[None][(field, logicalValue)]
elif _propertyAliases[None].has_key((field, None)):
realValue = _propertyAliases[None][(field, None)]
else:
realValue = logicalValue
if field == 'size':
if realValue == '*':
realValue = _fontSize
realValue = str((realValue + sizeIncr) * 10)
rtn.append(realValue)
return string.join(rtn, '-')
def logicalfontnames():
return _fontInfo.keys()
if os.name == 'nt':
_fontSize = 16
else:
_fontSize = 14
_fontFields = (
'registry', 'foundry', 'family', 'weight', 'slant', 'width', 'style',
'pixel', 'size', 'xres', 'yres', 'spacing', 'avgwidth', 'charset', 'encoding')
# <_propertyAliases> defines other names for which property values may
# be known by. This is required because italics in adobe-helvetica
# are specified by 'o', while other fonts use 'i'.
_propertyAliases = {}
_propertyAliases[None] = {
('slant', 'italic') : 'i',
('slant', 'normal') : 'r',
('weight', 'light') : 'normal',
('width', 'wide') : 'normal',
('width', 'condensed') : 'normal',
}
# <_fontInfo> describes a 'logical' font, giving the default values of
# some of its properties.
_fontInfo = {}
_fontInfo['Helvetica'] = {
'foundry' : 'adobe',
'family' : 'helvetica',
'registry' : '',
'charset' : 'iso8859',
'encoding' : '1',
'spacing' : 'p',
'slant' : 'normal',
'width' : 'normal',
'weight' : 'normal',
}
_propertyAliases['Helvetica'] = {
('slant', 'italic') : 'o',
('weight', 'normal') : 'medium',
('weight', 'light') : 'medium',
}
_fontInfo['Times'] = {
'foundry' : 'adobe',
'family' : 'times',
'registry' : '',
'charset' : 'iso8859',
'encoding' : '1',
'spacing' : 'p',
'slant' : 'normal',
'width' : 'normal',
'weight' : 'normal',
}
_propertyAliases['Times'] = {
('weight', 'normal') : 'medium',
('weight', 'light') : 'medium',
}
_fontInfo['Fixed'] = {
'foundry' : 'misc',
'family' : 'fixed',
'registry' : '',
'charset' : 'iso8859',
'encoding' : '1',
'spacing' : 'c',
'slant' : 'normal',
'width' : 'normal',
'weight' : 'normal',
}
_propertyAliases['Fixed'] = {
('weight', 'normal') : 'medium',
('weight', 'light') : 'medium',
('style', None) : '',
('width', 'condensed') : 'semicondensed',
}
_fontInfo['Courier'] = {
'foundry' : 'adobe',
'family' : 'courier',
'registry' : '',
'charset' : 'iso8859',
'encoding' : '1',
'spacing' : 'm',
'slant' : 'normal',
'width' : 'normal',
'weight' : 'normal',
}
_propertyAliases['Courier'] = {
('weight', 'normal') : 'medium',
('weight', 'light') : 'medium',
('style', None) : '',
}
_fontInfo['Typewriter'] = {
'foundry' : 'b&h',
'family' : 'lucidatypewriter',
'registry' : '',
'charset' : 'iso8859',
'encoding' : '1',
'spacing' : 'm',
'slant' : 'normal',
'width' : 'normal',
'weight' : 'normal',
}
_propertyAliases['Typewriter'] = {
('weight', 'normal') : 'medium',
('weight', 'light') : 'medium',
}
if os.name == 'nt':
# For some reason 'fixed' fonts on NT aren't.
_fontInfo['Fixed'] = _fontInfo['Courier']
_propertyAliases['Fixed'] = _propertyAliases['Courier']
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/PmwFreeze.py
|
PmwFreeze.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from scipy.optimize import brentq
import numpy as np
tol = 1e-10
def circle_intersection_area(r, R, d):
'''
Formula from: http://mathworld.wolfram.com/Circle-CircleIntersection.html
Does not make sense for negative r, R or d
>>> circle_intersection_area(0.0, 0.0, 0.0)
0.0
>>> circle_intersection_area(1.0, 1.0, 0.0)
3.1415...
>>> circle_intersection_area(1.0, 1.0, 1.0)
1.2283...
'''
if np.abs(d) < tol:
minR = np.min([r, R])
return np.pi * minR**2
if np.abs(r - 0) < tol or np.abs(R - 0) < tol:
return 0.0
d2, r2, R2 = float(d**2), float(r**2), float(R**2)
arg = (d2 + r2 - R2) / 2 / d / r
arg = np.max([np.min([arg, 1.0]), -1.0]) # Even with valid arguments, the above computation may result in things like -1.001
A = r2 * np.arccos(arg)
arg = (d2 + R2 - r2) / 2 / d / R
arg = np.max([np.min([arg, 1.0]), -1.0])
B = R2 * np.arccos(arg)
arg = (-d + r + R) * (d + r - R) * (d - r + R) * (d + r + R)
arg = np.max([arg, 0])
C = -0.5 * np.sqrt(arg)
return A + B + C
def circle_line_intersection(center, r, a, b):
'''
Computes two intersection points between the circle centered at <center> and radius <r> and a line given by two points a and b.
If no intersection exists, or if a==b, None is returned. If one intersection exists, it is repeated in the answer.
>>> circle_line_intersection(np.array([0.0, 0.0]), 1, np.array([-1.0, 0.0]), np.array([1.0, 0.0]))
array([[ 1., 0.],
[-1., 0.]])
>>> np.round(circle_line_intersection(np.array([1.0, 1.0]), np.sqrt(2), np.array([-1.0, 1.0]), np.array([1.0, -1.0])), 6)
array([[ 0., 0.],
[ 0., 0.]])
'''
s = b - a
# Quadratic eqn coefs
A = np.linalg.norm(s)**2
if abs(A) < tol:
return None
B = 2 * np.dot(a - center, s)
C = np.linalg.norm(a - center)**2 - r**2
disc = B**2 - 4 * A * C
if disc < 0.0:
return None
t1 = (-B + np.sqrt(disc)) / 2.0 / A
t2 = (-B - np.sqrt(disc)) / 2.0 / A
return np.array([a + t1 * s, a + t2 * s])
def find_distance_by_area(r, R, a, numeric_correction=0.0001):
'''
Solves circle_intersection_area(r, R, d) == a for d numerically (analytical solution seems to be too ugly to pursue).
Assumes that a < pi * min(r, R)**2, will fail otherwise.
The numeric correction parameter is used whenever the computed distance is exactly (R - r) (i.e. one circle must be inside another).
In this case the result returned is (R-r+correction). This helps later when we position the circles and need to ensure they intersect.
>>> find_distance_by_area(1, 1, 0, 0.0)
2.0
>>> round(find_distance_by_area(1, 1, 3.1415, 0.0), 4)
0.0
>>> d = find_distance_by_area(2, 3, 4, 0.0)
>>> d
3.37...
>>> round(circle_intersection_area(2, 3, d), 10)
4.0
>>> find_distance_by_area(1, 2, np.pi)
1.0001
'''
if r > R:
r, R = R, r
if np.abs(a) < tol:
return float(r + R)
if np.abs(min([r, R])**2 * np.pi - a) < tol:
return np.abs(R - r + numeric_correction)
return brentq(lambda x: circle_intersection_area(r, R, x) - a, R - r, R + r)
def circle_circle_intersection(C_a, r_a, C_b, r_b):
'''
Finds the coordinates of the intersection points of two circles A and B.
Circle center coordinates C_a and C_b, should be given as tuples (or 1x2 arrays).
Returns a 2x2 array result with result[0] being the first intersection point (to the right of the vector C_a -> C_b)
and result[1] being the second intersection point.
If there is a single intersection point, it is repeated in output.
If there are no intersection points or an infinite number of those, None is returned.
>>> circle_circle_intersection([0, 0], 1, [1, 0], 1) # Two intersection points
array([[ 0.5 , -0.866...],
[ 0.5 , 0.866...]])
>>> circle_circle_intersection([0, 0], 1, [2, 0], 1) # Single intersection point (circles touch from outside)
array([[ 1., 0.],
[ 1., 0.]])
>>> circle_circle_intersection([0, 0], 1, [0.5, 0], 0.5) # Single intersection point (circles touch from inside)
array([[ 1., 0.],
[ 1., 0.]])
>>> circle_circle_intersection([0, 0], 1, [0, 0], 1) is None # Infinite number of intersections (circles coincide)
True
>>> circle_circle_intersection([0, 0], 1, [0, 0.1], 0.8) is None # No intersections (one circle inside another)
True
>>> circle_circle_intersection([0, 0], 1, [2.1, 0], 1) is None # No intersections (one circle outside another)
True
'''
C_a, C_b = np.array(C_a, float), np.array(C_b, float)
v_ab = C_b - C_a
d_ab = np.linalg.norm(v_ab)
if np.abs(d_ab) < tol: # No intersection points
return None
cos_gamma = (d_ab**2 + r_a**2 - r_b**2) / 2.0 / d_ab / r_a
if abs(cos_gamma) > 1.0:
return None
sin_gamma = np.sqrt(1 - cos_gamma**2)
u = v_ab / d_ab
v = np.array([-u[1], u[0]])
pt1 = C_a + r_a * cos_gamma * u - r_a * sin_gamma * v
pt2 = C_a + r_a * cos_gamma * u + r_a * sin_gamma * v
return np.array([pt1, pt2])
def vector_angle_in_degrees(v):
'''
Given a vector, returns its elevation angle in degrees (-180..180).
>>> vector_angle_in_degrees([1, 0])
0.0
>>> vector_angle_in_degrees([1, 1])
45.0
>>> vector_angle_in_degrees([0, 1])
90.0
>>> vector_angle_in_degrees([-1, 1])
135.0
>>> vector_angle_in_degrees([-1, 0])
180.0
>>> vector_angle_in_degrees([-1, -1])
-135.0
>>> vector_angle_in_degrees([0, -1])
-90.0
>>> vector_angle_in_degrees([1, -1])
-45.0
'''
return np.arctan2(v[1], v[0]) * 180 / np.pi
def normalize_by_center_of_mass(coords, radii):
'''
Given coordinates of circle centers and radii, as two arrays,
returns new coordinates array, computed such that the center of mass of the
three circles is (0, 0).
>>> normalize_by_center_of_mass(np.array([[0.0, 0.0], [2.0, 0.0], [1.0, 3.0]]), np.array([1.0, 1.0, 1.0]))
array([[-1., -1.],
[ 1., -1.],
[ 0., 2.]])
>>> normalize_by_center_of_mass(np.array([[0.0, 0.0], [2.0, 0.0], [1.0, 2.0]]), np.array([1.0, 1.0, np.sqrt(2.0)]))
array([[-1., -1.],
[ 1., -1.],
[ 0., 1.]])
'''
# Now find the center of mass.
radii = radii**2
sum_r = np.sum(radii)
if sum_r < tol:
return coords
else:
return coords - np.dot(radii, coords) / np.sum(radii)
#### End __math.py
#### Begin __venn2.py
'''
Venn diagram plotting routines.
Two-circle venn plotter.
Copyright 2012, Konstantin Tretyakov.
http://kt.era.ee/
Licensed under MIT license.
'''
import numpy as np
from matplotlib.patches import Circle
from matplotlib.colors import ColorConverter
from matplotlib.pyplot import gca
#from _math import *
#from _venn3 import make_venn3_region_patch, prepare_venn3_axes, mix_colors
def compute_venn2_areas(diagram_areas, normalize_to=1.0):
'''
The list of venn areas is given as 3 values, corresponding to venn diagram areas in the following order:
(Ab, aB, AB) (i.e. last element corresponds to the size of intersection A&B&C).
The return value is a list of areas (A, B, AB), such that the total area is normalized
to normalize_to. If total area was 0, returns
(1.0, 1.0, 0.0)/2.0
Assumes all input values are nonnegative (to be more precise, all areas are passed through and abs() function)
>>> compute_venn2_areas((1, 1, 0))
(0.5, 0.5, 0.0)
>>> compute_venn2_areas((0, 0, 0))
(0.5, 0.5, 0.0)
>>> compute_venn2_areas((1, 1, 1), normalize_to=3)
(2.0, 2.0, 1.0)
>>> compute_venn2_areas((1, 2, 3), normalize_to=6)
(4.0, 5.0, 3.0)
'''
# Normalize input values to sum to 1
areas = np.array(np.abs(diagram_areas), float)
total_area = np.sum(areas)
if np.abs(total_area) < tol:
return (0.5, 0.5, 0.0)
else:
areas = areas / total_area * normalize_to
return (areas[0] + areas[2], areas[1] + areas[2], areas[2])
def solve_venn2_circles(venn_areas):
'''
Given the list of "venn areas" (as output from compute_venn2_areas, i.e. [A, B, AB]),
finds the positions and radii of the two circles.
The return value is a tuple (coords, radii), where coords is a 2x2 array of coordinates and
radii is a 2x1 array of circle radii.
Assumes the input values to be nonnegative and not all zero.
In particular, the first two values must be positive.
>>> c, r = solve_venn2_circles((1, 1, 0))
>>> np.round(r, 3)
array([ 0.564, 0.564])
>>> c, r = solve_venn2_circles(compute_venn2_areas((1, 2, 3)))
>>> np.round(r, 3)
array([ 0.461, 0.515])
'''
(A_a, A_b, A_ab) = map(float, venn_areas)
r_a, r_b = np.sqrt(A_a / np.pi), np.sqrt(A_b / np.pi)
radii = np.array([r_a, r_b])
if A_ab > tol:
# Nonzero intersection
coords = np.zeros((2, 2))
coords[1][0] = find_distance_by_area(radii[0], radii[1], A_ab)
else:
# Zero intersection
coords = np.zeros((2, 2))
coords[1][0] = radii[0] + radii[1] + np.mean(radii) * 1.1
coords = normalize_by_center_of_mass(coords, radii)
return (coords, radii)
def compute_venn2_regions(centers, radii):
'''
See compute_venn3_regions for explanations.
>>> centers, radii = solve_venn2_circles((1, 1, 0.5))
>>> regions = compute_venn2_regions(centers, radii)
'''
intersection = circle_circle_intersection(centers[0], radii[0], centers[1], radii[1])
if intersection is None:
# Two circular regions
regions = [("CIRCLE", (centers[a], radii[a], True), centers[a]) for a in [0, 1]] + [None]
else:
# Three curved regions
regions = []
for (a, b) in [(0, 1), (1, 0)]:
# Make region a¬ b: [(AB, A-), (BA, B+)]
points = np.array([intersection[a], intersection[b]])
arcs = [(centers[a], radii[a], False), (centers[b], radii[b], True)]
if centers[a][0] < centers[b][0]:
# We are to the left
label_pos_x = (centers[a][0] - radii[a] + centers[b][0] - radii[b]) / 2.0
else:
# We are to the right
label_pos_x = (centers[a][0] + radii[a] + centers[b][0] + radii[b]) / 2.0
label_pos = np.array([label_pos_x, centers[a][1]])
regions.append((points, arcs, label_pos))
# Make region a&b: [(AB, A+), (BA, B+)]
(a, b) = (0, 1)
points = np.array([intersection[a], intersection[b]])
arcs = [(centers[a], radii[a], True), (centers[b], radii[b], True)]
label_pos_x = (centers[a][0] + radii[a] + centers[b][0] - radii[b]) / 2.0
label_pos = np.array([label_pos_x, centers[a][1]])
regions.append((points, arcs, label_pos))
return regions
def compute_venn2_colors(set_colors):
'''
Given two base colors, computes combinations of colors corresponding to all regions of the venn diagram.
returns a list of 3 elements, providing colors for regions (10, 01, 11).
>>> compute_venn2_colors(('r', 'g'))
(array([ 1., 0., 0.]), array([ 0. , 0.5, 0. ]), array([ 0.7 , 0.35, 0. ]))
'''
ccv = ColorConverter()
base_colors = [np.array(ccv.to_rgb(c)) for c in set_colors]
return (base_colors[0], base_colors[1], mix_colors(base_colors[0], base_colors[1]))
def venn2_circles(subsets, normalize_to=1.0, alpha=1.0, color='black', linestyle='solid', linewidth=2.0, **kwargs):
'''
Plots only the two circles for the corresponding Venn diagram.
Useful for debugging or enhancing the basic venn diagram.
parameters sets and normalize_to are the same as in venn2()
kwargs are passed as-is to matplotlib.patches.Circle.
returns a list of three Circle patches.
>>> c = venn2_circles((1, 2, 3))
>>> c = venn2_circles({'10': 1, '01': 2, '11': 3}) # Same effect
'''
complete_subets = subsets
subsets_abrev = []
for i in subsets:
subsets_abrev.append(len(i))
subsets = tuple(subsets_abrev)
if isinstance(subsets, dict):
subsets = [subsets.get(t, 0) for t in ['10', '01', '11']]
areas = compute_venn2_areas(subsets, normalize_to)
centers, radii = solve_venn2_circles(areas)
ax = gca()
prepare_venn3_axes(ax, centers, radii)
result = []
for (c, r) in zip(centers, radii):
circle = Circle(c, r, alpha=alpha, edgecolor=color, facecolor='none', linestyle=linestyle, linewidth=linewidth, **kwargs)
ax.add_patch(circle)
result.append(circle)
return result
class Venn2:
'''
A container for a set of patches and patch labels and set labels, which make up the rendered venn diagram.
'''
id2idx = {'10': 0, '01': 1, '11': 2, 'A': 0, 'B': 1}
def __init__(self, patches, subset_labels, set_labels):
self.patches = patches
self.subset_labels = subset_labels
self.set_labels = set_labels
def get_patch_by_id(self, id):
'''Returns a patch by a "region id". A region id is a string '10', '01' or '11'.'''
return self.patches[self.id2idx[id]]
def get_label_by_id(self, id):
'''
Returns a subset label by a "region id". A region id is a string '10', '01' or '11'.
Alternatively, if the string 'A' or 'B' is given, the label of the
corresponding set is returned (or None).'''
if len(id) == 1:
return self.set_labels[self.id2idx[id]] if self.set_labels is not None else None
else:
return self.subset_labels[self.id2idx[id]]
def venn2(subsets, set_labels=('A', 'B'), set_colors=('r', 'g'), alpha=0.4, normalize_to=1.0):
'''Plots a 2-set area-weighted Venn diagram.
The subsets parameter is either a dict or a list.
- If it is a dict, it must map regions to their sizes.
The regions are identified via two-letter binary codes ('10', '01', and '11'), hence a valid set could look like:
{'01': 10, '01': 20, '11': 40}. Unmentioned codes are considered to map to 0.
- If it is a list, it must have 3 elements, denoting the sizes of the regions in the following order:
(10, 10, 11)
Set labels parameter is a list of two strings - set labels. Set it to None to disable set labels.
The set_colors parameter should be a list of two elements, specifying the "base colors" of the two circles.
The color of circle intersection will be computed based on those.
The normalize_to parameter specifies the total (on-axes) area of the circles to be drawn. Sometimes tuning it (together
with the overall fiture size) may be useful to fit the text labels better.
The return value is a Venn2 object, that keeps references to the Text and Patch objects used on the plot.
>>> from matplotlib_venn import *
>>> v = venn2(subsets={'10': 1, '01': 1, '11': 1}, set_labels = ('A', 'B'))
>>> c = venn2_circles(subsets=(1, 1, 1), linestyle='dashed')
>>> v.get_patch_by_id('10').set_alpha(1.0)
>>> v.get_patch_by_id('10').set_color('white')
>>> v.get_label_by_id('10').set_text('Unknown')
>>> v.get_label_by_id('A').set_text('Set A')
'''
complete_subets = subsets
subsets_abrev = []
for i in subsets:
subsets_abrev.append(len(i))
subsets = subsets_abrev
if isinstance(subsets, dict):
subsets = [subsets.get(t, 0) for t in ['10', '01', '11']]
areas = compute_venn2_areas(subsets, normalize_to)
centers, radii = solve_venn2_circles(areas)
if (areas[0] < tol or areas[1] < tol):
raise Exception("Both circles in the diagram must have positive areas.")
centers, radii = solve_venn2_circles(areas)
regions = compute_venn2_regions(centers, radii)
colors = compute_venn2_colors(set_colors)
ax = gca()
prepare_venn3_axes(ax, centers, radii)
# Create and add patches and text
patches = [make_venn3_region_patch(r) for r in regions]
for (p, c) in zip(patches, colors):
if p is not None:
p.set_facecolor(c)
p.set_edgecolor('none')
p.set_alpha(alpha)
ax.add_patch(p)
texts = [ax.text(r[2][0], r[2][1], str(s), va='center', ha='center', size = 17) if r is not None else None for (r, s) in zip(regions, subsets)]
# Position labels
if set_labels is not None:
padding = np.mean([r * 0.1 for r in radii])
label_positions = [centers[0] + np.array([0.0, - radii[0] - padding]),
centers[1] + np.array([0.0, - radii[1] - padding])]
labels = [ax.text(pos[0], pos[1], txt, size=20, ha='right', va='top') for (pos, txt) in zip(label_positions, set_labels)]
labels[1].set_ha('left')
else:
labels = None
return Venn2(patches, texts, labels)
#### End __venn2.py
#### Begin __venn3.py
'''
Venn diagram plotting routines.
Three-circle venn plotter.
Copyright 2012, Konstantin Tretyakov.
http://kt.era.ee/
Licensed under MIT license.
'''
import numpy as np
import warnings
from matplotlib.patches import Circle, PathPatch
from matplotlib.path import Path
from matplotlib.colors import ColorConverter
from matplotlib.pyplot import gca
#from _math import *
def compute_venn3_areas(diagram_areas, normalize_to=1.0):
'''
The list of venn areas is given as 7 values, corresponding to venn diagram areas in the following order:
(Abc, aBc, ABc, abC, AbC, aBC, ABC)
(i.e. last element corresponds to the size of intersection A&B&C).
The return value is a list of areas (A_a, A_b, A_c, A_ab, A_bc, A_ac, A_abc),
such that the total area of all circles is normalized to normalize_to. If total area was 0, returns
(1.0, 1.0, 1.0, 0.0, 0.0, 0.0, 0.0)/3.0
Assumes all input values are nonnegative (to be more precise, all areas are passed through and abs() function)
>>> compute_venn3_areas((1, 1, 0, 1, 0, 0, 0))
(0.33..., 0.33..., 0.33..., 0.0, 0.0, 0.0, 0.0)
>>> compute_venn3_areas((0, 0, 0, 0, 0, 0, 0))
(0.33..., 0.33..., 0.33..., 0.0, 0.0, 0.0, 0.0)
>>> compute_venn3_areas((1, 1, 1, 1, 1, 1, 1), normalize_to=7)
(4.0, 4.0, 4.0, 2.0, 2.0, 2.0, 1.0)
>>> compute_venn3_areas((1, 2, 3, 4, 5, 6, 7), normalize_to=56/2)
(16.0, 18.0, 22.0, 10.0, 13.0, 12.0, 7.0)
'''
# Normalize input values to sum to 1
areas = np.array(np.abs(diagram_areas), float)
total_area = np.sum(areas)
if np.abs(total_area) < tol:
return (1.0 / 3.0, 1.0 / 3.0, 1.0 / 3.0, 0.0, 0.0, 0.0, 0.0)
else:
areas = areas / total_area * normalize_to
A_a = areas[0] + areas[2] + areas[4] + areas[6]
A_b = areas[1] + areas[2] + areas[5] + areas[6]
A_c = areas[3] + areas[4] + areas[5] + areas[6]
# Areas of the three intersections (ab, ac, bc)
A_ab, A_ac, A_bc = areas[2] + areas[6], areas[4] + areas[6], areas[5] + areas[6]
return (A_a, A_b, A_c, A_ab, A_bc, A_ac, areas[6])
def solve_venn3_circles(venn_areas):
'''
Given the list of "venn areas" (as output from compute_venn3_areas, i.e. [A, B, C, AB, BC, AC, ABC]),
finds the positions and radii of the three circles.
The return value is a tuple (coords, radii), where coords is a 3x2 array of coordinates and
radii is a 3x1 array of circle radii.
Assumes the input values to be nonnegative and not all zero.
In particular, the first three values must all be positive.
The overall match is only approximate (to be precise, what is matched are the areas of the circles and the
three pairwise intersections).
>>> c, r = solve_venn3_circles((1, 1, 1, 0, 0, 0, 0))
>>> np.round(r, 3)
array([ 0.564, 0.564, 0.564])
>>> c, r = solve_venn3_circles(compute_venn3_areas((1, 2, 40, 30, 4, 40, 4)))
>>> np.round(r, 3)
array([ 0.359, 0.476, 0.453])
'''
(A_a, A_b, A_c, A_ab, A_bc, A_ac, A_abc) = map(float, venn_areas)
r_a, r_b, r_c = np.sqrt(A_a / np.pi), np.sqrt(A_b / np.pi), np.sqrt(A_c / np.pi)
intersection_areas = [A_ab, A_bc, A_ac]
radii = np.array([r_a, r_b, r_c])
# Hypothetical distances between circle centers that assure
# that their pairwise intersection areas match the requirements.
dists = [find_distance_by_area(radii[i], radii[j], intersection_areas[i]) for (i, j) in [(0, 1), (1, 2), (2, 0)]]
# How many intersections have nonzero area?
num_nonzero = sum(np.array([A_ab, A_bc, A_ac]) > tol)
# Handle four separate cases:
# 1. All pairwise areas nonzero
# 2. Two pairwise areas nonzero
# 3. One pairwise area nonzero
# 4. All pairwise areas zero.
if num_nonzero == 3:
# The "generic" case, simply use dists to position circles at the vertices of a triangle.
# Before we need to ensure that resulting circles can be at all positioned on a triangle,
# use an ad-hoc fix.
for i in range(3):
i, j, k = (i, (i + 1) % 3, (i + 2) % 3)
if dists[i] > dists[j] + dists[k]:
dists[i] = 0.8 * (dists[j] + dists[k])
warnings.warn("Bad circle positioning")
coords = position_venn3_circles_generic(radii, dists)
elif num_nonzero == 2:
# One pair of circles is not intersecting.
# In this case we can position all three circles in a line
# The two circles that have no intersection will be on either sides.
for i in range(3):
if intersection_areas[i] < tol:
(left, right, middle) = (i, (i + 1) % 3, (i + 2) % 3)
coords = np.zeros((3, 2))
coords[middle][0] = dists[middle]
coords[right][0] = dists[middle] + dists[right]
# We want to avoid the situation where left & right still intersect
if coords[left][0] + radii[left] > coords[right][0] - radii[right]:
mid = (coords[left][0] + radii[left] + coords[right][0] - radii[right]) / 2.0
coords[left][0] = mid - radii[left] - 1e-5
coords[right][0] = mid + radii[right] + 1e-5
break
elif num_nonzero == 1:
# Only one pair of circles is intersecting, and one circle is independent.
# Position all on a line first two intersecting, then the free one.
for i in range(3):
if intersection_areas[i] > tol:
(left, right, side) = (i, (i + 1) % 3, (i + 2) % 3)
coords = np.zeros((3, 2))
coords[right][0] = dists[left]
coords[side][0] = dists[left] + radii[right] + radii[side] * 1.1 # Pad by 10%
break
else:
# All circles are non-touching. Put them all in a sequence
coords = np.zeros((3, 2))
coords[1][0] = radii[0] + radii[1] * 1.1
coords[2][0] = radii[0] + radii[1] * 1.1 + radii[1] + radii[2] * 1.1
coords = normalize_by_center_of_mass(coords, radii)
return (coords, radii)
def position_venn3_circles_generic(radii, dists):
'''
Given radii = (r_a, r_b, r_c) and distances between the circles = (d_ab, d_bc, d_ac),
finds the coordinates of the centers for the three circles so that they form a proper triangle.
The current positioning method puts the center of A and B on a horizontal line y==0,
and C just below.
Returns a 3x2 array with circle center coordinates in rows.
>>> position_venn3_circles_generic((1, 1, 1), (0, 0, 0))
array([[ 0., 0.],
[ 0., 0.],
[ 0., -0.]])
>>> position_venn3_circles_generic((1, 1, 1), (2, 2, 2))
array([[ 0. , 0. ],
[ 2. , 0. ],
[ 1. , -1.73205081]])
'''
(d_ab, d_bc, d_ac) = dists
(r_a, r_b, r_c) = radii
coords = np.array([[0, 0], [d_ab, 0], [0, 0]], float)
C_x = (d_ac**2 - d_bc**2 + d_ab**2) / 2.0 / d_ab if np.abs(d_ab) > tol else 0.0
C_y = -np.sqrt(d_ac**2 - C_x**2)
coords[2, :] = C_x, C_y
return coords
def compute_venn3_regions(centers, radii):
'''
Given the 3x2 matrix with circle center coordinates, and a 3-element list (or array) with circle radii [as returned from solve_venn3_circles],
returns the 7 regions, comprising the venn diagram.
Each region is given as [array([pt_1, pt_2, pt_3]), (arc_1, arc_2, arc_3), label_pos] where each pt_i gives the coordinates of a point,
and each arc_i is in turn a triple (circle_center, circle_radius, direction), and label_pos is the recommended center point for
positioning region label.
The region is the poly-curve constructed by moving from pt_1 to pt_2 along arc_1, then to pt_3 along arc_2 and back to pt_1 along arc_3.
Arc direction==True denotes positive (CCW) direction.
There is also a special case, where the region is given as
["CIRCLE", (center, radius, True), label_pos], which corresponds to a completely circular region.
Regions are returned in order (Abc, aBc, ABc, abC, AbC, aBC, ABC)
>>> centers, radii = solve_venn3_circles((1, 1, 1, 1, 1, 1, 1))
>>> regions = compute_venn3_regions(centers, radii)
'''
# First compute all pairwise circle intersections
intersections = [circle_circle_intersection(centers[i], radii[i], centers[j], radii[j]) for (i, j) in [(0, 1), (1, 2), (2, 0)]]
regions = []
# Regions [Abc, aBc, abC]
for i in range(3):
(a, b, c) = (i, (i + 1) % 3, (i + 2) % 3)
if intersections[a] is not None and intersections[c] is not None:
# Current circle intersects both of the other circles.
if intersections[b] is not None:
# .. and the two other circles intersect, this is either the "normal" situation
# or it can also be a case of bad placement
if np.linalg.norm(intersections[b][0] - centers[a]) < radii[a]:
# In the "normal" situation we use the scheme [(BA, B+), (BC, C+), (AC, A-)]
points = np.array([intersections[a][1], intersections[b][0], intersections[c][1]])
arcs = [(centers[b], radii[b], True), (centers[c], radii[c], True), (centers[a], radii[a], False)]
# Ad-hoc label positioning
pt_a = intersections[b][0]
pt_b = intersections[b][1]
pt_c = circle_line_intersection(centers[a], radii[a], pt_a, pt_b)
if pt_c is None:
label_pos = circle_circle_intersection(centers[b], radii[b] + 0.1 * radii[a], centers[c], radii[c] + 0.1 * radii[c])[0]
else:
label_pos = 0.5 * (pt_c[1] + pt_a)
else:
# This is the "bad" situation (basically one disc covers two touching disks)
# We use the scheme [(BA, B+), (AB, A-)] if (AC is inside B) and
# [(CA, C+), (AC, A-)] otherwise
if np.linalg.norm(intersections[c][0] - centers[b]) < radii[b]:
points = np.array([intersections[a][1], intersections[a][0]])
arcs = [(centers[b], radii[b], True), (centers[a], radii[a], False)]
else:
points = np.array([intersections[c][0], intersections[c][1]])
arcs = [(centers[c], radii[c], True), (centers[a], radii[a], False)]
label_pos = centers[a]
else:
# .. and the two other circles do not intersect. This means we are in the "middle" of a OoO placement.
# The patch is then a [(AB, B-), (BA, A+), (AC, C-), (CA, A+)]
points = np.array([intersections[a][0], intersections[a][1], intersections[c][1], intersections[c][0]])
arcs = [(centers[b], radii[b], False), (centers[a], radii[a], True), (centers[c], radii[c], False), (centers[a], radii[a], True)]
# Label will be between the b and c circles
leftc, rightc = (b, c) if centers[b][0] < centers[c][0] else (c, b)
label_x = ((centers[leftc][0] + radii[leftc]) + (centers[rightc][0] - radii[rightc])) / 2.0
label_y = centers[a][1] + radii[a] / 2.0
label_pos = np.array([label_x, label_y])
elif intersections[a] is None and intersections[c] is None:
# Current circle is completely separate from others
points = "CIRCLE"
arcs = (centers[a], radii[a], True)
label_pos = centers[a]
else:
# Current circle intersects one of the other circles
other_circle = b if intersections[a] is not None else c
other_circle_intersection = a if intersections[a] is not None else c
i1, i2 = (0, 1) if intersections[a] is not None else (1, 0)
# The patch is a [(AX, A-), (XA, X+)]
points = np.array([intersections[other_circle_intersection][i1], intersections[other_circle_intersection][i2]])
arcs = [(centers[a], radii[a], False), (centers[other_circle], radii[other_circle], True)]
if centers[a][0] < centers[other_circle][0]:
# We are to the left
label_pos_x = (centers[a][0] - radii[a] + centers[other_circle][0] - radii[other_circle]) / 2.0
else:
# We are to the right
label_pos_x = (centers[a][0] + radii[a] + centers[other_circle][0] + radii[other_circle]) / 2.0
label_pos = np.array([label_pos_x, centers[a][1]])
regions.append((points, arcs, label_pos))
(a, b, c) = (0, 1, 2)
# Regions [aBC, AbC, ABc]
for i in range(3):
(a, b, c) = (i, (i + 1) % 3, (i + 2) % 3)
if intersections[b] is None: # No region there
regions.append(None)
continue
has_middle_region = np.linalg.norm(intersections[b][0] - centers[a]) < radii[a]
if has_middle_region:
# This is the "normal" situation (i.e. all three circles have a common area)
# We then use the scheme [(CB, C+), (CA, A-), (AB, B+)]
points = np.array([intersections[b][1], intersections[c][0], intersections[a][0]])
arcs = [(centers[c], radii[c], True), (centers[a], radii[a], False), (centers[b], radii[b], True)]
# Ad-hoc label positioning
pt_a = intersections[b][1]
dir_to_a = pt_a - centers[a]
dir_to_a = dir_to_a / np.linalg.norm(dir_to_a)
pt_b = centers[a] + dir_to_a * radii[a]
label_pos = 0.5 * (pt_a + pt_b)
else:
# This is the situation, where there is no common area
# Then the corresponding area is made by scheme [(CB, C+), (BC, B+), None]
points = np.array([intersections[b][1], intersections[b][0]])
arcs = [(centers[c], radii[c], True), (centers[b], radii[b], True)]
label_pos = 0.5 * (intersections[b][1] + intersections[b][0])
regions.append((points, arcs, label_pos))
# Central region made by scheme [(BC, B+), (AB, A+), (CA, C+)]
(a, b, c) = (0, 1, 2)
if intersections[a] is None or intersections[b] is None or intersections[c] is None:
# No middle region
regions.append(None)
else:
points = np.array([intersections[b][0], intersections[a][0], intersections[c][0]])
label_pos = np.mean(points, 0) # Middle of the central region
arcs = [(centers[b], radii[b], True), (centers[a], radii[a], True), (centers[c], radii[c], True)]
has_middle_region = np.linalg.norm(intersections[b][0] - centers[a]) < radii[a]
if has_middle_region:
regions.append((points, arcs, label_pos))
else:
regions.append(([], [], label_pos))
# (Abc, aBc, ABc, abC, AbC, aBC, ABC)
return (regions[0], regions[1], regions[5], regions[2], regions[4], regions[3], regions[6])
def make_venn3_region_patch(region):
'''
Given a venn3 region (as returned from compute_venn3_regions) produces a Patch object,
depicting the region as a curve.
>>> centers, radii = solve_venn3_circles((1, 1, 1, 1, 1, 1, 1))
>>> regions = compute_venn3_regions(centers, radii)
>>> patches = [make_venn3_region_patch(r) for r in regions]
'''
if region is None or len(region[0]) == 0:
return None
if region[0] == "CIRCLE":
return Circle(region[1][0], region[1][1])
pts, arcs, label_pos = region
path = [pts[0]]
for i in range(len(pts)):
j = (i + 1) % len(pts)
(center, radius, direction) = arcs[i]
fromangle = vector_angle_in_degrees(pts[i] - center)
toangle = vector_angle_in_degrees(pts[j] - center)
if direction:
vertices = Path.arc(fromangle, toangle).vertices
else:
vertices = Path.arc(toangle, fromangle).vertices
vertices = vertices[np.arange(len(vertices) - 1, -1, -1)]
vertices = vertices * radius + center
path = path + list(vertices[1:])
codes = [1] + [4] * (len(path) - 1)
return PathPatch(Path(path, codes))
def mix_colors(col1, col2, col3=None):
'''
Mixes two colors to compute a "mixed" color (for purposes of computing
colors of the intersection regions based on the colors of the sets.
Note that we do not simply compute averages of given colors as those seem
too dark for some default configurations. Thus, we lighten the combination up a bit.
Inputs are (up to) three RGB triples of floats 0.0-1.0 given as numpy arrays.
>>> mix_colors(np.array([1.0, 0., 0.]), np.array([1.0, 0., 0.])) # doctest: +NORMALIZE_WHITESPACE
array([ 1., 0., 0.])
>>> mix_colors(np.array([1.0, 1., 0.]), np.array([1.0, 0.9, 0.]), np.array([1.0, 0.8, 0.1])) # doctest: +NORMALIZE_WHITESPACE
array([ 1. , 1. , 0.04])
'''
if col3 is None:
mix_color = 0.7 * (col1 + col2)
else:
mix_color = 0.4 * (col1 + col2 + col3)
mix_color = np.min([mix_color, [1.0, 1.0, 1.0]], 0)
return mix_color
def compute_venn3_colors(set_colors):
'''
Given three base colors, computes combinations of colors corresponding to all regions of the venn diagram.
returns a list of 7 elements, providing colors for regions (100, 010, 110, 001, 101, 011, 111).
>>> compute_venn3_colors(['r', 'g', 'b'])
(array([ 1., 0., 0.]),..., array([ 0.4, 0.2, 0.4]))
'''
ccv = ColorConverter()
base_colors = [np.array(ccv.to_rgb(c)) for c in set_colors]
return (base_colors[0], base_colors[1], mix_colors(base_colors[0], base_colors[1]), base_colors[2],
mix_colors(base_colors[0], base_colors[2]), mix_colors(base_colors[1], base_colors[2]), mix_colors(base_colors[0], base_colors[1], base_colors[2]))
def prepare_venn3_axes(ax, centers, radii):
'''
Sets properties of the axis object to suit venn plotting. I.e. hides ticks, makes proper xlim/ylim.
'''
ax.set_aspect('equal')
ax.set_xticks([])
ax.set_yticks([])
min_x = min([centers[i][0] - radii[i] for i in range(len(radii))])
max_x = max([centers[i][0] + radii[i] for i in range(len(radii))])
min_y = min([centers[i][1] - radii[i] for i in range(len(radii))])
max_y = max([centers[i][1] + radii[i] for i in range(len(radii))])
ax.set_xlim([min_x - 0.1, max_x + 0.1])
ax.set_ylim([min_y - 0.1, max_y + 0.1])
ax.set_axis_off()
def venn3_circles(subsets, normalize_to=1.0, alpha=1.0, color='black', linestyle='solid', linewidth=2.0, **kwargs):
'''
Plots only the three circles for the corresponding Venn diagram.
Useful for debugging or enhancing the basic venn diagram.
parameters sets and normalize_to are the same as in venn3()
kwargs are passed as-is to matplotlib.patches.Circle.
returns a list of three Circle patches.
>>> plot = venn3_circles({'001': 10, '100': 20, '010': 21, '110': 13, '011': 14})
'''
complete_subets = subsets
subsets_abrev = []
for i in subsets:
subsets_abrev.append(len(i))
subsets = tuple(subsets_abrev)
# Prepare parameters
if isinstance(subsets, dict):
subsets = [subsets.get(t, 0) for t in ['100', '010', '110', '001', '101', '011', '111']]
areas = compute_venn3_areas(subsets, normalize_to)
centers, radii = solve_venn3_circles(areas)
ax = gca()
prepare_venn3_axes(ax, centers, radii)
result = []
for (c, r) in zip(centers, radii):
circle = Circle(c, r, alpha=alpha, edgecolor=color, facecolor='none', linestyle=linestyle, linewidth=linewidth, **kwargs)
ax.add_patch(circle)
result.append(circle)
return result
class Venn3:
'''
A container for a set of patches and patch labels and set labels, which make up the rendered venn diagram.
'''
id2idx = {'100': 0, '010': 1, '110': 2, '001': 3, '101': 4, '011': 5, '111': 6, 'A': 0, 'B': 1, 'C': 2}
def __init__(self, patches, subset_labels, set_labels):
self.patches = patches
self.subset_labels = subset_labels
self.set_labels = set_labels
def get_patch_by_id(self, id):
'''Returns a patch by a "region id". A region id is a string like 001, 011, 010, etc.'''
return self.patches[self.id2idx[id]]
def get_label_by_id(self, id):
'''
Returns a subset label by a "region id". A region id is a string like 001, 011, 010, etc.
Alternatively, if you provide either of 'A', 'B' or 'C', you will obtain the label of the
corresponding set (or None).'''
if len(id) == 1:
return self.set_labels[self.id2idx[id]] if self.set_labels is not None else None
else:
return self.subset_labels[self.id2idx[id]]
def venn3(subsets, set_labels=('A', 'B', 'C'), set_colors=('r', 'g', 'b'), alpha=0.4, normalize_to=1.0):
global coordinates
coordinates={}
'''Plots a 3-set area-weighted Venn diagram.
The subsets parameter is either a dict or a list.
- If it is a dict, it must map regions to their sizes.
The regions are identified via three-letter binary codes ('100', '010', etc), hence a valid set could look like:
{'001': 10, '010': 20, '110':30, ...}. Unmentioned codes are considered to map to 0.
- If it is a list, it must have 7 elements, denoting the sizes of the regions in the following order:
(100, 010, 110, 001, 101, 011, 111).
Set labels parameter is a list of three strings - set labels. Set it to None to disable set labels.
The set_colors parameter should be a list of three elements, specifying the "base colors" of the three circles.
The colors of circle intersections will be computed based on those.
The normalize_to parameter specifies the total (on-axes) area of the circles to be drawn. Sometimes tuning it (together
with the overall fiture size) may be useful to fit the text labels better.
The return value is a Venn3 object, that keeps references to the Text and Patch objects used on the plot.
>>> from matplotlib_venn import *
>>> v = venn3(subsets=(1, 1, 1, 1, 1, 1, 1), set_labels = ('A', 'B', 'C'))
>>> c = venn3_circles(subsets=(1, 1, 1, 1, 1, 1, 1), linestyle='dashed')
>>> v.get_patch_by_id('100').set_alpha(1.0)
>>> v.get_patch_by_id('100').set_color('white')
>>> v.get_label_by_id('100').set_text('Unknown')
>>> v.get_label_by_id('C').set_text('Set C')
'''
# Prepare parameters
complete_subets = subsets
subsets_abrev = []
for i in subsets:
subsets_abrev.append(len(i))
subsets = tuple(subsets_abrev)
if isinstance(subsets, dict):
subsets = [subsets.get(t, 0) for t in ['100', '010', '110', '001', '101', '011', '111']]
areas = compute_venn3_areas(subsets, normalize_to)
if (areas[0] < tol or areas[1] < tol or areas[2] < tol):
raise Exception("All three circles in the diagram must have positive areas. Use venn2 or just a circle to draw diagrams with two or one circle.")
centers, radii = solve_venn3_circles(areas)
regions = compute_venn3_regions(centers, radii)
colors = compute_venn3_colors(set_colors)
ax = gca()
prepare_venn3_axes(ax, centers, radii)
# Create and add patches and text
patches = [make_venn3_region_patch(r) for r in regions]
for (p, c) in zip(patches, colors):
if p is not None:
p.set_facecolor(c)
p.set_edgecolor('none')
p.set_alpha(alpha)
ax.add_patch(p)
subset_labels = [ax.text(r[2][0], r[2][1], str(s), va='center', ha='center', size=17) if r is not None else None for (r, s) in zip(regions, subsets)]
#null = [coordinates[120, 200]=labels['1000'] if r is not None else None for (r, s) in zip(regions, complete_subets)]
# Position labels
if set_labels is not None:
# There are two situations, when set C is not on the same line with sets A and B, and when the three are on the same line.
if abs(centers[2][1] - centers[0][1]) > tol:
# Three circles NOT on the same line
label_positions = [centers[0] + np.array([-radii[0] / 2, radii[0]]),
centers[1] + np.array([radii[1] / 2, radii[1]]),
centers[2] + np.array([0.0, -radii[2] * 1.1])]
labels = [ax.text(pos[0], pos[1], txt, size=20) for (pos, txt) in zip(label_positions, set_labels)]
labels[0].set_horizontalalignment('right')
labels[1].set_horizontalalignment('left')
labels[2].set_verticalalignment('top')
labels[2].set_horizontalalignment('center')
else:
padding = np.mean([r * 0.1 for r in radii])
# Three circles on the same line
label_positions = [centers[0] + np.array([0.0, - radii[0] - padding]),
centers[1] + np.array([0.0, - radii[1] - padding]),
centers[2] + np.array([0.0, - radii[2] - padding])]
labels = [ax.text(pos[0], pos[1], txt, size='large', ha='center', va='top') for (pos, txt) in zip(label_positions, set_labels)]
else:
labels = None
return Venn3(patches, subset_labels, labels)
#### End __venn3.py
if __name__ == '__main__':
from matplotlib import pyplot as plt
import numpy as np
#from matplotlib_venn import venn3, venn3_circles
plt.figure(figsize=(4,4))
v = venn3(subsets=(1, 1, 1, 1, 1, 1, 1), set_labels = ('A', 'B', 'C'))
v.get_patch_by_id('100').set_alpha(1.0)
v.get_patch_by_id('100').set_color('white')
v.get_label_by_id('100').set_text('Unknown')
v.get_label_by_id('A').set_text('Set "A"')
c = venn3_circles(subsets=(1, 1, 1, 1, 1, 1, 1), linestyle='dashed')
c[0].set_lw(1.0)
c[0].set_ls('dotted')
plt.title("Sample Venn diagram")
plt.annotate('Unknown set', xy=v.get_label_by_id('100').get_position() - np.array([0, 0.05]), xytext=(-70,-70),
ha='center', textcoords='offset points', bbox=dict(boxstyle='round,pad=0.5', fc='gray', alpha=0.1),
arrowprops=dict(arrowstyle='->', connectionstyle='arc3,rad=0.5',color='gray'))
plt.show()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/matplotlib_venn.py
|
matplotlib_venn.py
|
from __future__ import print_function
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy
import export
import traceback
import collections as c
import unique
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>0 and '--' in command_args: commandLine=True
else: commandLine=False
try:
import math
import warnings
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
import matplotlib
#try: matplotlib.use('TkAgg')
#except Exception: pass
#if commandLine==False:
#try: matplotlib.rcParams['backend'] = 'TkAgg'
#except Exception: pass
try:
import matplotlib.pyplot as pylab
import matplotlib.colors as mc
import matplotlib.mlab as mlab
import matplotlib.ticker as tic
from matplotlib.patches import Circle
from mpl_toolkits.mplot3d import Axes3D
matplotlib.rcParams['axes.linewidth'] = 0.5
matplotlib.rcParams['pdf.fonttype'] = 42
matplotlib.rcParams['font.family'] = 'sans-serif'
matplotlib.rcParams['font.sans-serif'] = 'Arial'
from matplotlib.patches import Rectangle
import matplotlib.patches
import matplotlib.backend_bases as event_plot
from mpldatacursor import datacursor
from matplotlib.widgets import Slider as matplotSlider
except Exception:
print(traceback.format_exc())
print('Matplotlib support not enabled')
except Exception:
print(traceback.format_exc())
#os.chdir("/Users/saljh8/Desktop/Code/AltAnalyze/Config/ExonViewFiles")
class EnsemblRegionClass:
def __init__(self,start,stop,ensembl_exon,exon_region,strand):
self.start = start
self.stop = stop
self.ensembl_exon = ensembl_exon
self.exon_region = exon_region
self.strand = strand
def Start(self): return int(self.start)
def Stop(self): return int(self.stop)
def EnsemblExon(self): return self.ensembl_exon
def EnsemblRegion(self): return self.exon_region
def ExonBlock(self):
return string.split(self.EnsemblRegion(),'.')[0]
def Strand(self): return self.strand
def Length(self): return abs(int(self.Start())-int(self.Stop()))
def setChr(self, chr): self.chr = chr
def Chr(self): return self.chr
class SplicingIndexClass:
def __init__(self, reg_call, splicing_index, p_val, midas):
self.reg_call = reg_call
self.splicing_index = splicing_index
self.p_val = p_val
self.midas = midas
def RegCall(self): return self.reg_call
def SplicingIndex(self): return self.splicing_index
def PVal(self): return self.p_val
def Midas(self): return self.midas
class MicroRNAClass:
def __init__(self, exon_name, description, basepairs, algorithms):
self.exon_name = exon_name
self.description = description
self.basepairs = basepairs
self.algorithms = algorithms
def ExonBlock(self): return self.exon_name
def Description(self): return self.description
def BP(self): return self.basepairs
def Algorithms(self): return self.algorithms
def ProteinCentricIsoformView(Selected_Gene):
Transcript_List = []
Transcript_db = {}
Exon_db = {}
for line in open(Transcript_Annotations_File, "rU").xreadlines():
line = line.rstrip()
line = line.split("\t")
if(line[0] == Selected_Gene):
transcriptID = line[-1]
exonID = line[5]
start = line[3]
stop = line[4]
strand = line[2]
chr = line[1]
if 'chr' not in chr:
chr = 'chr'+chr
exon_data = EnsemblRegionClass(start,stop,exonID,None,strand)
exon_data.setChr(chr)
Transcript_List.append((transcriptID, exonID))
try:
Transcript_db[transcriptID].append(exon_data)
except Exception:
Transcript_db[transcriptID]=[exon_data]
try:
Exon_db[exonID].append(transcriptID)
except Exception:
Exon_db[exonID]=[transcriptID]
Transcript_Protein_db = {}
Protein_Transcript_db = {}
Protein_List = []
count = 0
for line in open(Prt_Trans_File, "rU").xreadlines():
if(count == 0):
count = 1
continue
line = line.rstrip()
line = line.split("\t")
if(len(line) != 3):
continue
geneID = line[0]
transcriptID = line[1]
proteinID = line[2]
if Selected_Gene == geneID:
Transcript_Protein_db[transcriptID] = proteinID
Protein_Transcript_db[proteinID] = transcriptID
Protein_List.append(proteinID)
#MicroRNA File
microRNA_db = {}
for line in open(microRNA_File, "rU").xreadlines():
line = line.rstrip()
line = line.split("\t")
try:
gene_and_exon_id = line[0].split(":")
current_gene_id = gene_and_exon_id[0]
current_exon_id = gene_and_exon_id[1]
except Exception:
continue
#print([current_gene_id,current_exon_id,Selected_Gene]);break
current_description = line[1]
current_base_pairs = line[2]
algorithms = line[3]
if(current_gene_id == Selected_Gene):
m = MicroRNAClass(current_exon_id, current_description, current_base_pairs, algorithms)
try:
if(len(microRNA_db[current_exon_id]) > 6):
continue
microRNA_db[current_exon_id].append(m)
#print("ADDED!")
except:
microRNA_db[current_exon_id] = [m]
Transcript_ExonRegion_db={}
geneExonRegion_db={}
exon_coord_db={}
exonRegion_db={}
AllBlocks = [("E", []), ("I", [])]
# Store the exon region positions and later link them to the Ensembl exons
for line in open(ExonRegion_File, "rU").xreadlines():
line = line.rstrip()
line = line.split("\t")
geneID = line[0]
exon_region = line[1]
chr = line[2]
exonID = line[1]
strand = line[3]
start = line[4]
stop = line[5]
er = EnsemblRegionClass(start,stop,exonID,exon_region,strand)
if(geneID == Selected_Gene):
Block_Num = exon_region[1:]
I_E_id = exon_region[0]
if(I_E_id == "E"):
AllBlocks[0][1].append(Block_Num)
if(I_E_id == "I"):
AllBlocks[1][1].append(Block_Num)
continue
exon_added = False
#Exon_List = line[7].split("|")
exon_coord_db[chr,int(start),'start'] = exon_region
exon_coord_db[chr,int(stop),'stop'] = exon_region
exonRegion_db[Selected_Gene,exon_region] = er
#print chr,start,'start'
probeset_to_ExonID={}
if platform != 'RNASeq':
for line in open(unique.filepath('AltDatabase/'+species+'/'+string.lower(platform)+'/'+species+'_Ensembl_probesets.txt'), "rU").xreadlines():
line = line.rstrip()
line = line.split("\t")
gene = line[2]
if gene == Selected_Gene:
probeset = line[0]
exon_region = line[12]
if '.' not in exon_region:
exon_region = string.replace(exon_region,'-','.')
probeset_to_ExonID[probeset] = exon_region
ETC_List = []
for line in open(SplicingIndex_File, "rU").xreadlines():
line = line.rstrip()
line = line.split("\t")
if ':' in line[0]:
GeneLine = line[0].split(":")
FeatureID = GeneLine[1]
else:
FeatureID = line[0]
Gene = line[1]
regcall = line[2]
spl_index = line[3]
pval = line[4]
midas = line[5]
S_I_data = SplicingIndexClass(regcall, spl_index, pval, midas)
if(Gene == Selected_Gene):
if platform != 'RNASeq':
if FeatureID in probeset_to_ExonID:
FeatureID = probeset_to_ExonID[FeatureID]
#print(FeatureID)
ETC_List.append((FeatureID, S_I_data))
else:
try:
FeatureID = FeatureID.split("_")
FeatureID = FeatureID[0]
ETC_List.append((FeatureID, S_I_data))
except:
pass
ETC_dict = {}
# Link the exon regions to the Ensembl exons
for transcriptID in Transcript_db:
for exon_data in Transcript_db[transcriptID]:
start = exon_data.Start()
stop = exon_data.Stop()
chr = exon_data.Chr()
strand = exon_data.Strand()
try:
start_exon_region = exon_coord_db[chr,start,'start']
stop_exon_region = exon_coord_db[chr,stop,'stop']
proceed = True
except Exception: ### Not clear why this error occurs. Erroring region was found to be an intron region start position (I7.2 ENSMUSG00000020385)
proceed = False
if proceed:
if '-' in strand:
stop_exon_region,start_exon_region = start_exon_region,stop_exon_region
regions = [start_exon_region]
block,start_region = start_exon_region.split('.')
start_region = int(float(start_region))
block,stop_region = stop_exon_region.split('.')
stop_region = int(float(stop_region))
region = start_region+1
while region<stop_region:
er = block+'.'+str(region)
regions.append(er)
region+=1
if stop_region != start_region:
regions.append(stop_exon_region)
for region in regions:
er = exonRegion_db[Selected_Gene,region]
try:
Transcript_ExonRegion_db[transcriptID].append(er)
except:
Transcript_ExonRegion_db[transcriptID] = [er]
exon_virtualToRealPos= c.OrderedDict()
junction_transcript_db = {}
for transcriptID in Transcript_ExonRegion_db:
#print('transcripts:',transcriptID)
position=0
Buffer=15
for exon_object in Transcript_ExonRegion_db[transcriptID]:
if position!=0:
if last_exon != exon_object.ExonBlock():
#print last_exon_region+'-'+exon_object.EnsemblRegion(),position,Buffer
junctionID = last_exon_region+'-'+exon_object.EnsemblRegion()
try: junction_transcript_db[transcriptID].append((position,position+Buffer, junctionID)) ### virtual junction positions
except: junction_transcript_db[transcriptID] = [(position,position+Buffer, junctionID)]
position+=Buffer
virtualStart = position
virtualStop = virtualStart + exon_object.Length()
position = virtualStop
try:
exon_virtualToRealPos[transcriptID].append(([virtualStart,virtualStop],[exon_object.Start(), exon_object.Stop()],exon_object))
except Exception:
exon_virtualToRealPos[transcriptID]=[([virtualStart,virtualStop],[exon_object.Start(), exon_object.Stop()],exon_object)]
#print transcriptID,exon_object.ExonBlock(),exon_object.EnsemblExon(),exon_object.EnsemblRegion(),exon_object.Start(),exon_object.Stop(),virtualStart,virtualStop,"\n"
last_exon = exon_object.ExonBlock()
last_exon_region = exon_object.EnsemblRegion()
for i in ETC_List:
Region = i[0]
S_I = i[1]
Region = Region.split("-")
if(len(Region) > 1):
#Delete underscores from values.
R_Start = Region[0]
R_End = Region[1]
R_Start = R_Start.split("_")
R_End = R_End.split("_")
R_Start = R_Start[0]
R_End = R_End[0]
R_Final = R_Start + "-" + R_End
R_Type = R_Final[0]
#print(R_Final)
ETC_dict[R_Final] = S_I
else:
Region = Region[0]
Region = Region.split("_")
Region = Region[0]
Region_type = Region[0]
ETC_dict[Region] = S_I
#if(Region_type == "E"):
# for entry in AllBlocks[0][1]:
# if(Region[1:] == entry):
# ETC_dict[("E" + entry)] = S_I
#if(Region_type == "I"):
# for entry in AllBlocks[1][1]:
# if(Region[1:] == entry):
# ETC_dict[("I" + entry)] = S_I
#for a in ETC_dict:
# print(ETC_dict[a].RegCall(), a)
#for i in junction_transcript_db:
# print i, junction_transcript_db[i], "\n"
Protein_Pos_Db = {}
last_protein=None
stored_stop=None
for line in open(Prt_Boundaries_File, "rU").xreadlines():
line = line.rstrip()
line = line.split("\t")
proteinID = line[0]
if(proteinID in Protein_List):
Stop = int(line[-1])
Start = int(line[-2])
if(proteinID != last_protein):
if stored_stop !=None:
#print proteinID,stored_start,stored_stop
Protein_Pos_Db[last_protein] = [[stored_start,stored_stop,None]]
stored_start = int(Start)
if(proteinID == last_protein):
stored_stop = int(Stop)
last_protein = str(proteinID)
Protein_Pos_Db[last_protein] = [(stored_start,stored_stop,None)]
Protein_virtualPos = RealToVirtual(Protein_Pos_Db, exon_virtualToRealPos, Protein_Transcript_db,Transcript_ExonRegion_db)
Domain_Pos_Db={}
domainAnnotation_db={}
#"""
for line in open(Prt_Regions_File, "rU").xreadlines():
line = line.rstrip()
line = line.split("\t")
proteinID = line[0]
if proteinID in Protein_Pos_Db:
domain_start = int(float(line[3]))
domain_stop = int(float(line[4]))
domainID = line[-2]
domainName = line[-1]
try:
Domain_Pos_Db[proteinID].append((domain_start,domain_stop,domainID))
except:
Domain_Pos_Db[proteinID] = [(domain_start,domain_stop,domainID)]
domainAnnotation_db[domainID] = domainName
#"""
for line in open(UniPrt_Regions_File, "rU").xreadlines():
line = line.rstrip()
line = line.split("\t")
proteinID = line[0]
if proteinID in Protein_Pos_Db:
domain_start = int(float(line[3]))
domain_stop = int(float(line[4]))
domainID = line[-1]
domainName = line[-1]
try:
Domain_Pos_Db[proteinID].append((domain_start,domain_stop,domainID))
except:
Domain_Pos_Db[proteinID] = [(domain_start,domain_stop,domainID)]
domainAnnotation_db[domainID] = domainName
#print('--',domainName,domain_start,domain_stop)
# Do the same for domain coordinates
Domain_virtualPos = RealToVirtual(Domain_Pos_Db, exon_virtualToRealPos, Protein_Transcript_db,Transcript_ExonRegion_db)
return_val = ((junction_transcript_db, Protein_virtualPos, Domain_virtualPos, Transcript_db, exon_virtualToRealPos, ETC_dict, microRNA_db, domainAnnotation_db))
return return_val
def RealToVirtual(Protein_Pos_Db, exon_virtualToRealPos, Protein_Transcript_db,Transcript_ExonRegion_db):
Transcript_to_Protein_Coords = {}
for proteinID in Protein_Pos_Db:
transcript = Protein_Transcript_db[proteinID]
strand = Transcript_ExonRegion_db[transcript][0].Strand()
e_coords = exon_virtualToRealPos[transcript]
if proteinID in Protein_Pos_Db:
for q in Protein_Pos_Db[proteinID]:
#print("Protein: ", proteinID)
p_start = q[0]
p_stop = q[1]
annotation = q[2]
if '-' not in strand:
p_start +=1
p_stop -= 1 ### seems to be off by 1
virtual_p_start = None
virtual_p_stop = None
#print("E", len(e_coords))
#print("Protein: ", proteinID)
for i in range(len(e_coords)):
e_virtual_start, e_virtual_stop = e_coords[i][0]
#print("Sub-E", e_virtual_start, e_virtual_stop)
e_real_start,e_real_stop = e_coords[i][1]
#print e_real_start,e_real_stop
e = [e_real_start,e_real_stop]
e.sort()
p = [p_start,p_stop]
p.sort()
coord = e+p
coord.sort()
if (p_start<e[1] and p_start>e[0]) or p_start==e[1] or p_start==e[0]:
if '-' in strand:
offset = e_real_stop-p_start
else:
offset = p_start-e_real_start
virtual_p_start = offset+e_virtual_start
#print("Final_Val", proteinID, virtual_p_start)
if (p_stop<e[1] and p_stop>e[0]) or p_stop==e[1] or p_stop==e[0]:
if '-' in strand:
offset = e_real_stop-p_stop
else:
offset = p_stop-e_real_start
virtual_p_stop = offset+e_virtual_start
if annotation != None:
#print("Entered", proteinID, virtual_p_start)
try:
Transcript_to_Protein_Coords[transcript].append((proteinID, annotation, virtual_p_start, virtual_p_stop, e_coords[0][0][0],e_coords[-1][0][1]))
except Exception:
Transcript_to_Protein_Coords[transcript] = [(proteinID, annotation, virtual_p_start, virtual_p_stop, e_coords[0][0][0],e_coords[-1][0][1])]
else:
#print("Entered_2", proteinID, virtual_p_start)
Transcript_to_Protein_Coords[transcript] = proteinID, virtual_p_start, virtual_p_stop, e_coords[0][0][0],e_coords[-1][0][1]
#print transcript, proteinID, virtual_p_start, virtual_p_stop, p_start,p_stop, e_coords[0][0][0],e_coords[-1][0][1],annotation
return Transcript_to_Protein_Coords
def searchDirectory(directory,var,secondary=None):
directory = unique.filepath(directory)
files = unique.read_directory(directory)
for file in files:
if var in file:
if secondary== None:
return directory+'/'+file
break
elif secondary in file:
return directory+'/'+file
break
### if all else fails
return directory+'/'+file
def getPlatform(filename):
prefix = string.split(export.findFilename(filename),'.')[0]
array_type = string.split(prefix,'_')[1]
if array_type != 'RNASeq':
array_type = string.lower(array_type)
return array_type
def remoteGene(gene,Species,root_dir,comparison_file):
global Transcript_Annotations_File
global ExonRegion_File
global Selected_Gene
global Prt_Trans_File
global Prt_Regions_File
global Prt_Boundaries_File
global SplicingIndex_File
global UniPrt_Regions_File
global microRNA_File
global domainAnnotation_db
global platform
global species
Selected_Gene = str(gene)
species = Species
comparison_name = string.split(export.findFilename(comparison_file),'.')[0]
ExonRegion_File = unique.filepath("AltDatabase/ensembl/"+species+"/"+species+"_Ensembl_exon.txt")
Transcript_Annotations_File = unique.filepath("AltDatabase/ensembl/"+species+"/"+species+"_Ensembl_transcript-annotations.txt")
Prt_Trans_File = searchDirectory("AltDatabase/ensembl/"+species+"/",'Ensembl_Protein')
Prt_Regions_File = searchDirectory("AltDatabase/ensembl/"+species+"/",'ProteinFeatures')
Prt_Boundaries_File = searchDirectory("AltDatabase/ensembl/"+species+"/",'ProteinCoordinates')
UniPrt_Regions_File = searchDirectory("AltDatabase/uniprot/"+species+"/",'FeatureCoordinate')
SplicingIndex_File = searchDirectory(root_dir+'/AltResults/ProcessedSpliceData/','splicing-index',secondary=comparison_name)
platform = getPlatform(SplicingIndex_File)
microRNA_File = searchDirectory("AltDatabase/"+species+"/"+platform,'microRNAs_multiple')
#print(SplicingIndex_File)
total_val = ProteinCentricIsoformView(Selected_Gene)
junctions = total_val[0]
p_boundaries = total_val[1]
p_domains = total_val[2]
transcript_db = total_val[3]
exon_db = total_val[4]
splice_db = total_val[5]
microRNA_db = total_val[6]
domainAnnotation_db = total_val[7]
#for i in exon_db:
# print("THE", i, exon_db[i], "\n")
#for i in microRNA_db:
# m_test = microRNA_db[i]
# print(len(m_test))
# for q in m_test:
# print("microRNA", q.ExonBlock(), q.Description(), q.BP(), "\n")
#for i in exon_db["ENST00000349238"]:
# print(i[2].EnsemblRegion())
domain_color_list = []
for i in p_domains:
ploy = p_domains[i]
for a in ploy:
domain_color_list.append(a[1])
domain_color_list = list(set(domain_color_list))
domain_color_key = {}
c_color1 = [0.8, 0.6, 0.1]
c_color2 = [0.1, 0.6, 0.8]
c_color3 = [0.6, 0.1, 0.8]
c_color4 = [0.95, 0.6, 0.3]
c_color5 = [0.3, 0.6, 0.95]
c_color6 = [0.6, 0.3, 0.95]
FLAG = 1
for item in domain_color_list:
if(FLAG == 1):
domain_color_key[item] = c_color1
FLAG = FLAG + 1
continue
if(FLAG == 2):
domain_color_key[item] = c_color2
FLAG = FLAG + 1
continue
if(FLAG == 3):
domain_color_key[item] = c_color3
FLAG = FLAG + 1
continue
if(FLAG == 4):
domain_color_key[item] = c_color4
FLAG = FLAG + 1
continue
if(FLAG == 5):
domain_color_key[item] = c_color5
FLAG = FLAG + 1
continue
if(FLAG == 6):
domain_color_key[item] = c_color6
FLAG = 1
continue
#for i in domain_color_key:
#print(i, domain_color_key[i], "\n")
Y = 100
Transcript_to_Y = {}
for transcript in transcript_db:
Transcript_to_Y[transcript] = Y
Y = Y + 300
import traceback
def onpick(event):
#ind = event.ind
print(event.artist.get_label())
#for i in domainAnnotation_db: print(i,len(domainAnnotation_db));break
fig = pylab.figure()
ylim = Y + 200
currentAxis = pylab.gca()
#ax = pylab.axes()
ax = fig.add_subplot(111)
X_Pos_List = []
CoordsBank = []
for transcript in transcript_db:
try:
Junc_List = junctions[transcript]
y_pos = Transcript_to_Y[transcript]
Gene_List = exon_db[transcript]
color_flag = 1
for entry in Gene_List:
G_start = entry[0][0]
G_end = entry[0][1]
Exon_Object = entry[2]
try:
LabelClass = splice_db[Exon_Object.EnsemblRegion()]
ExonName = Exon_Object.EnsemblExon()
RegCall = LabelClass.RegCall()
SplicingIndex = LabelClass.SplicingIndex()
PVal = LabelClass.PVal()
Midas = LabelClass.Midas()
Label = "\n" + "Exon: " + str(ExonName) + "\n" + "RegCall: " + str(RegCall) + "\n" + "Splicing Index: " + str(SplicingIndex) + "\n" + "P-Value: " + str(PVal) + "\n" + "Midas Value: " + str(Midas) + "\n"
Label = string.replace(Label,"\n"," ")
if(RegCall == "UC"):
color_choice = "Grey"
else:
S_Int = float(SplicingIndex)
if(S_Int > 0):
#color_choice = (0.7, 0.7, 0.99)
color_choice = 'blue'
if(S_Int < 0):
#color_choice = (0.8, 0.4, 0.4)
color_choice = 'red'
except:
#print(traceback.format_exc());sys.exit()
Label = ""
color_choice = "Grey"
#print("Start", G_start, "end", G_end, "Region", entry[2].EnsemblRegion())
if((color_flag % 2) == 0):
currentAxis.add_patch(Rectangle((G_start, y_pos), (G_end - G_start), 50, color = color_choice, label = (entry[2].EnsemblRegion() + Label), picker = True))
y_end = y_pos + 50
try: CoordsBank.append((G_start, G_end, y_pos, y_end, 'Exon: '+entry[2].EnsemblRegion()+' '+ 'SI: '+str(SplicingIndex)[:4]+' Pval: '+str(Midas)[:4]))
except Exception:
CoordsBank.append((G_start, G_end, y_pos, y_end, 'Exon: '+entry[2].EnsemblRegion()))
#print(entry[2].EnsemblRegion(),y_pos,y_end)
if((color_flag % 2) != 0):
currentAxis.add_patch(Rectangle((G_start, y_pos), (G_end - G_start), 50, color = color_choice, label = (entry[2].EnsemblRegion() + Label), picker = True))
y_end = y_pos + 50
try: CoordsBank.append((G_start, G_end, y_pos, y_end, 'Exon: '+entry[2].EnsemblRegion()+' '+ 'SI: '+str(SplicingIndex)[:4]+' p-value: '+str(Midas)[:4]))
except Exception:
CoordsBank.append((G_start, G_end, y_pos, y_end, 'Exon: '+entry[2].EnsemblRegion()))
#print(entry[2].EnsemblRegion(),y_pos,y_end)
color_flag = color_flag + 1
if(entry[2].EnsemblRegion() in microRNA_db):
microRNA_object = microRNA_db[entry[2].EnsemblRegion()]
mr_label = "MICRORNA MATCHES" + "\n"
for class_object in microRNA_object:
mr_exonname = class_object.ExonBlock()
mr_desc = class_object.Description() + " " + class_object.Algorithms()
#print(mr_desc)
mr_label = mr_label + mr_desc + "\n"
currentAxis.add_patch(Rectangle((G_start, (y_pos - 75)), (G_end - G_start), 40, color = "Green", label = (mr_label), picker = True))
y_start = y_pos - 75
y_end = y_pos - 35
CoordsBank.append((G_start, G_end, y_start, y_end, mr_desc))
for entry in Junc_List:
junctionID = entry[-1]
try:
LabelClass = splice_db[entry[2]]
RegCall = LabelClass.RegCall()
SplicingIndex = LabelClass.SplicingIndex()
PVal = LabelClass.PVal()
Midas = LabelClass.Midas()
Label = "\n" + "RegCall: " + str(RegCall) + "\n" + "Splicing Index: " + str(SplicingIndex) + "\n" + "P-Value: " + str(PVal) + "\n" + "Midas Value: " + str(Midas) + "\n"
if(float(SplicingIndex) > 0):
color_junc = "blue"
if(float(SplicingIndex) < 0):
color_junc = "red"
if(RegCall == "UC"):
color_junc = "grey"
except:
Label = ""
color_junc = "grey"
currentAxis.add_patch(Rectangle((entry[0], y_pos), (entry[1] - entry[0]), 50, color = "White", label = (str(entry[2]) + Label), picker = True))
ax.arrow(entry[0], (y_pos+50), 8, 40, label = (str(entry[2]) + Label), color = color_junc, picker = True)
ax.arrow((entry[0] + 8), (y_pos+90), 11, -40, label = (str(entry[2]) + Label), color = color_junc, picker = True)
y_start = y_pos
y_end = y_pos + 30
#print(junctionID,y_start,y_end)
CoordsBank.append((G_start, G_end, y_start, y_end, junctionID))
try:
P_Bound_List = p_boundaries[transcript]
E_Start = P_Bound_List[-2]
E_End = P_Bound_List[-1]
P_Start = P_Bound_List[1]
P_End = P_Bound_List[2]
#print("Boundaries: ", P_Start, P_End)
X_Pos_List.append(int(E_End))
#currentAxis.add_patch(Rectangle((E_Start, y_pos), E_End, 50, color = "Blue"))
try:
currentAxis.add_patch(Rectangle((P_Start, (y_pos + 120)), (P_End - P_Start), 10))
except:
pass
p_label_list = ["DEF"]
#CoordsBank.append((P_Start, P_End, y_pos, P_End - P_Start, transcript)) ### Added by NS - needs work
try: P_Domain_List = p_domains[transcript]
except Exception: P_Domain_List=[]
for entry in P_Domain_List:
#print("Domain", entry)
color_domain_choice = domain_color_key[entry[1]]
domain_annotation = domainAnnotation_db[entry[1]]
#domain_annotation = string.replace(domain_annotation,'REGION-','')
p_label = (str(entry[0]) + " " + str(domain_annotation))
#print(entry[0], entry[2], entry[3], P_Start, P_End, domain_annotation, )
Repeat_Flag = 0
for i in p_label_list:
if(p_label == i):
Repeat_Flag = 1
if(Repeat_Flag == 1):
continue
p_label_list.append(p_label)
currentAxis.add_patch(Rectangle((entry[2], y_pos + 100), (entry[3] - entry[2]), 50, color = color_domain_choice, label= p_label, picker = True))
y_start = y_pos + 100
y_end = y_pos + 150
CoordsBank.append((entry[2], entry[3], y_start, y_end, p_label))
except Exception:
pass
#print(traceback.format_exc())
except:
#print(traceback.format_exc())
pass
pylab.ylim([0.0, ylim])
try:
max_x = max(X_Pos_List)
except:
max_x = 5000
try:
pylab.xlim([0.0, max_x])
except:
pylab.xlim([0.0, 3000])
fig.canvas.mpl_connect('pick_event', onpick)
def format_coord(x, y):
for m in CoordsBank:
if(x >= m[0] and x <= m[1] and y >= m[2] and y <= m[3]):
string_display = m[4]
return string_display
string_display = " "
return string_display
ax.format_coord = format_coord
#datacursor(hover=True, formatter='{label}'.format, bbox=dict(fc='yellow', alpha=1), arrowprops=None)
pylab.show()
if __name__ == "__main__":
#Selected_Gene = sys.argv[1]
Selected_Gene = 'ENSG00000132906'
Species = 'Hs'
root_dir = '/Volumes/salomonis2/Leucegene_project/STAR_TOP-SRSF2-U2AF1-like/'
comparison_file = '/Volumes/salomonis2/Leucegene_project/STAR_TOP-SRSF2-U2AF1-like/AltResults/RawSpliceData/Hs/splicing-index/Hs_RNASeq_U2AF1-like_vs_SRSF2-like.ExpCutoff-5.0_average.txt'
remoteGene(Selected_Gene,Species,root_dir,comparison_file)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/ExPlot.py
|
ExPlot.py
|
from __future__ import print_function
import numpy as np
import numba
import os
from utils import (
tau_rand,
make_heap,
heap_push,
unchecked_heap_push,
smallest_flagged,
rejection_sample,
build_candidates,
new_build_candidates,
deheap_sort,
)
from rp_tree import search_flat_tree
def make_nn_descent(dist, dist_args):
"""Create a numba accelerated version of nearest neighbor descent
specialised for the given distance metric and metric arguments. Numba
doesn't support higher order functions directly, but we can instead JIT
compile the version of NN-descent for any given metric.
Parameters
----------
dist: function
A numba JITd distance function which, given two arrays computes a
dissimilarity between them.
dist_args: tuple
Any extra arguments that need to be passed to the distance function
beyond the two arrays to be compared.
Returns
-------
A numba JITd function for nearest neighbor descent computation that is
specialised to the given metric.
"""
@numba.njit()
def nn_descent(
data,
n_neighbors,
rng_state,
max_candidates=50,
n_iters=10,
delta=0.001,
rho=0.5,
rp_tree_init=True,
leaf_array=None,
verbose=False,
):
n_vertices = data.shape[0]
current_graph = make_heap(data.shape[0], n_neighbors)
for i in range(data.shape[0]):
indices = rejection_sample(n_neighbors, data.shape[0], rng_state)
for j in range(indices.shape[0]):
d = dist(data[i], data[indices[j]], *dist_args)
heap_push(current_graph, i, d, indices[j], 1)
heap_push(current_graph, indices[j], d, i, 1)
if rp_tree_init:
for n in range(leaf_array.shape[0]):
for i in range(leaf_array.shape[1]):
if leaf_array[n, i] < 0:
break
for j in range(i + 1, leaf_array.shape[1]):
if leaf_array[n, j] < 0:
break
d = dist(
data[leaf_array[n, i]], data[leaf_array[n, j]], *dist_args
)
heap_push(
current_graph, leaf_array[n, i], d, leaf_array[n, j], 1
)
heap_push(
current_graph, leaf_array[n, j], d, leaf_array[n, i], 1
)
for n in range(n_iters):
if verbose:
print("\t", n, " / ", n_iters)
candidate_neighbors = build_candidates(
current_graph, n_vertices, n_neighbors, max_candidates, rng_state
)
c = 0
for i in range(n_vertices):
for j in range(max_candidates):
p = int(candidate_neighbors[0, i, j])
if p < 0 or tau_rand(rng_state) < rho:
continue
for k in range(max_candidates):
q = int(candidate_neighbors[0, i, k])
if (
q < 0
or not candidate_neighbors[2, i, j]
and not candidate_neighbors[2, i, k]
):
continue
d = dist(data[p], data[q], *dist_args)
c += heap_push(current_graph, p, d, q, 1)
c += heap_push(current_graph, q, d, p, 1)
if c <= delta * n_neighbors * data.shape[0]:
break
return deheap_sort(current_graph)
return nn_descent
def make_initialisations(dist, dist_args):
@numba.njit()
def init_from_random(n_neighbors, data, query_points, heap, rng_state):
for i in range(query_points.shape[0]):
indices = rejection_sample(n_neighbors, data.shape[0], rng_state)
for j in range(indices.shape[0]):
if indices[j] < 0:
continue
d = dist(data[indices[j]], query_points[i], *dist_args)
heap_push(heap, i, d, indices[j], 1)
return
@numba.njit()
def init_from_tree(tree, data, query_points, heap, rng_state):
for i in range(query_points.shape[0]):
indices = search_flat_tree(
query_points[i],
tree.hyperplanes,
tree.offsets,
tree.children,
tree.indices,
rng_state,
)
for j in range(indices.shape[0]):
if indices[j] < 0:
continue
d = dist(data[indices[j]], query_points[i], *dist_args)
heap_push(heap, i, d, indices[j], 1)
return
return init_from_random, init_from_tree
def initialise_search(
forest, data, query_points, n_neighbors, init_from_random, init_from_tree, rng_state
):
results = make_heap(query_points.shape[0], n_neighbors)
init_from_random(n_neighbors, data, query_points, results, rng_state)
if forest is not None:
for tree in forest:
init_from_tree(tree, data, query_points, results, rng_state)
return results
def make_initialized_nnd_search(dist, dist_args):
@numba.njit()
def initialized_nnd_search(data, indptr, indices, initialization, query_points):
for i in numba.prange(query_points.shape[0]):
tried = set(initialization[0, i])
while True:
# Find smallest flagged vertex
vertex = smallest_flagged(initialization, i)
if vertex == -1:
break
candidates = indices[indptr[vertex] : indptr[vertex + 1]]
for j in range(candidates.shape[0]):
if (
candidates[j] == vertex
or candidates[j] == -1
or candidates[j] in tried
):
continue
d = dist(data[candidates[j]], query_points[i], *dist_args)
unchecked_heap_push(initialization, i, d, candidates[j], 1)
tried.add(candidates[j])
return initialization
return initialized_nnd_search
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn_single/nndescent.py
|
nndescent.py
|
import numpy as np
import numba
_mock_identity = np.eye(2, dtype=np.float64)
_mock_ones = np.ones(2, dtype=np.float64)
@numba.njit(fastmath=True)
def euclidean(x, y):
"""Standard euclidean distance.
..math::
D(x, y) = \sqrt{\sum_i (x_i - y_i)^2}
"""
result = 0.0
for i in range(x.shape[0]):
result += (x[i] - y[i]) ** 2
return np.sqrt(result)
@numba.njit()
def standardised_euclidean(x, y, sigma=_mock_ones):
"""Euclidean distance standardised against a vector of standard
deviations per coordinate.
..math::
D(x, y) = \sqrt{\sum_i \frac{(x_i - y_i)**2}{v_i}}
"""
result = 0.0
for i in range(x.shape[0]):
result += ((x[i] - y[i]) ** 2) / sigma[i]
return np.sqrt(result)
@numba.njit()
def manhattan(x, y):
"""Manhatten, taxicab, or l1 distance.
..math::
D(x, y) = \sum_i |x_i - y_i|
"""
result = 0.0
for i in range(x.shape[0]):
result += np.abs(x[i] - y[i])
return result
@numba.njit()
def chebyshev(x, y):
"""Chebyshev or l-infinity distance.
..math::
D(x, y) = \max_i |x_i - y_i|
"""
result = 0.0
for i in range(x.shape[0]):
result = max(result, np.abs(x[i] - y[i]))
return result
@numba.njit()
def minkowski(x, y, p=2):
"""Minkowski distance.
..math::
D(x, y) = \left(\sum_i |x_i - y_i|^p\right)^{\frac{1}{p}}
This is a general distance. For p=1 it is equivalent to
manhattan distance, for p=2 it is Euclidean distance, and
for p=infinity it is Chebyshev distance. In general it is better
to use the more specialised functions for those distances.
"""
result = 0.0
for i in range(x.shape[0]):
result += (np.abs(x[i] - y[i])) ** p
return result ** (1.0 / p)
@numba.njit()
def weighted_minkowski(x, y, w=_mock_ones, p=2):
"""A weighted version of Minkowski distance.
..math::
D(x, y) = \left(\sum_i w_i |x_i - y_i|^p\right)^{\frac{1}{p}}
If weights w_i are inverse standard deviations of data in each dimension
then this represented a standardised Minkowski distance (and is
equivalent to standardised Euclidean distance for p=1).
"""
result = 0.0
for i in range(x.shape[0]):
result += (w[i] * np.abs(x[i] - y[i])) ** p
return result ** (1.0 / p)
@numba.njit()
def mahalanobis(x, y, vinv=_mock_identity):
result = 0.0
diff = np.empty(x.shape[0], dtype=np.float64)
for i in range(x.shape[0]):
diff[i] = x[i] - y[i]
for i in range(x.shape[0]):
tmp = 0.0
for j in range(x.shape[0]):
tmp += vinv[i, j] * diff[j]
result += tmp * diff[i]
return np.sqrt(result)
@numba.njit()
def hamming(x, y):
result = 0.0
for i in range(x.shape[0]):
if x[i] != y[i]:
result += 1.0
return float(result) / x.shape[0]
@numba.njit()
def canberra(x, y):
result = 0.0
for i in range(x.shape[0]):
denominator = np.abs(x[i]) + np.abs(y[i])
if denominator > 0:
result += np.abs(x[i] - y[i]) / denominator
return result
@numba.njit()
def bray_curtis(x, y):
numerator = 0.0
denominator = 0.0
for i in range(x.shape[0]):
numerator += np.abs(x[i] - y[i])
denominator += np.abs(x[i] + y[i])
if denominator > 0.0:
return float(numerator) / denominator
else:
return 0.0
@numba.njit()
def jaccard(x, y):
num_non_zero = 0.0
num_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_non_zero += x_true or y_true
num_equal += x_true and y_true
if num_non_zero == 0.0:
return 0.0
else:
return float(num_non_zero - num_equal) / num_non_zero
@numba.njit()
def matching(x, y):
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_not_equal += x_true != y_true
return float(num_not_equal) / x.shape[0]
@numba.njit()
def dice(x, y):
num_true_true = 0.0
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
num_not_equal += x_true != y_true
if num_not_equal == 0.0:
return 0.0
else:
return num_not_equal / (2.0 * num_true_true + num_not_equal)
@numba.njit()
def kulsinski(x, y):
num_true_true = 0.0
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
num_not_equal += x_true != y_true
if num_not_equal == 0:
return 0.0
else:
return float(num_not_equal - num_true_true + x.shape[0]) / (
num_not_equal + x.shape[0]
)
@numba.njit()
def rogers_tanimoto(x, y):
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_not_equal += x_true != y_true
return (2.0 * num_not_equal) / (x.shape[0] + num_not_equal)
@numba.njit()
def russellrao(x, y):
num_true_true = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
if num_true_true == np.sum(x != 0) and num_true_true == np.sum(y != 0):
return 0.0
else:
return float(x.shape[0] - num_true_true) / (x.shape[0])
@numba.njit()
def sokal_michener(x, y):
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_not_equal += x_true != y_true
return (2.0 * num_not_equal) / (x.shape[0] + num_not_equal)
@numba.njit()
def sokal_sneath(x, y):
num_true_true = 0.0
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
num_not_equal += x_true != y_true
if num_not_equal == 0.0:
return 0.0
else:
return num_not_equal / (0.5 * num_true_true + num_not_equal)
@numba.njit()
def haversine(x, y):
if x.shape[0] != 2:
raise ValueError("haversine is only defined for 2 dimensional data")
sin_lat = np.sin(0.5 * (x[0] - y[0]))
sin_long = np.sin(0.5 * (x[1] - y[1]))
result = np.sqrt(sin_lat ** 2 + np.cos(x[0]) * np.cos(y[0]) * sin_long ** 2)
return 2.0 * np.arcsin(result)
@numba.njit()
def yule(x, y):
num_true_true = 0.0
num_true_false = 0.0
num_false_true = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
num_true_false += x_true and (not y_true)
num_false_true += (not x_true) and y_true
num_false_false = x.shape[0] - num_true_true - num_true_false - num_false_true
if num_true_false == 0.0 or num_false_true == 0.0:
return 0.0
else:
return (2.0 * num_true_false * num_false_true) / (
num_true_true * num_false_false + num_true_false * num_false_true
)
@numba.njit()
def cosine(x, y):
result = 0.0
norm_x = 0.0
norm_y = 0.0
for i in range(x.shape[0]):
result += x[i] * y[i]
norm_x += x[i] ** 2
norm_y += y[i] ** 2
if norm_x == 0.0 and norm_y == 0.0:
return 0.0
elif norm_x == 0.0 or norm_y == 0.0:
return 1.0
else:
return 1.0 - (result / np.sqrt(norm_x * norm_y))
@numba.njit()
def correlation(x, y):
mu_x = 0.0
mu_y = 0.0
norm_x = 0.0
norm_y = 0.0
dot_product = 0.0
for i in range(x.shape[0]):
mu_x += x[i]
mu_y += y[i]
mu_x /= x.shape[0]
mu_y /= x.shape[0]
for i in range(x.shape[0]):
shifted_x = x[i] - mu_x
shifted_y = y[i] - mu_y
norm_x += shifted_x ** 2
norm_y += shifted_y ** 2
dot_product += shifted_x * shifted_y
if norm_x == 0.0 and norm_y == 0.0:
return 0.0
elif dot_product == 0.0:
return 1.0
else:
return 1.0 - (dot_product / np.sqrt(norm_x * norm_y))
named_distances = {
# general minkowski distances
"euclidean": euclidean,
"l2": euclidean,
"manhattan": manhattan,
"taxicab": manhattan,
"l1": manhattan,
"chebyshev": chebyshev,
"linfinity": chebyshev,
"linfty": chebyshev,
"linf": chebyshev,
"minkowski": minkowski,
# Standardised/weighted distances
"seuclidean": standardised_euclidean,
"standardised_euclidean": standardised_euclidean,
"wminkowski": weighted_minkowski,
"weighted_minkowski": weighted_minkowski,
"mahalanobis": mahalanobis,
# Other distances
"canberra": canberra,
"cosine": cosine,
"correlation": correlation,
"haversine": haversine,
"braycurtis": bray_curtis,
# Binary distances
"hamming": hamming,
"jaccard": jaccard,
"dice": dice,
"matching": matching,
"kulsinski": kulsinski,
"rogerstanimoto": rogers_tanimoto,
"russellrao": russellrao,
"sokalsneath": sokal_sneath,
"sokalmichener": sokal_michener,
"yule": yule,
}
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn_single/distances.py
|
distances.py
|
import numpy as np
import numba
from sklearn.neighbors import KDTree
from umap_learn.distances import named_distances
@numba.njit()
def trustworthiness_vector_bulk(
indices_source, indices_embedded, max_k
): # pragma: no cover
n_samples = indices_embedded.shape[0]
trustworthiness = np.zeros(max_k + 1, dtype=np.float64)
for i in range(n_samples):
for j in range(max_k):
rank = 0
while indices_source[i, rank] != indices_embedded[i, j]:
rank += 1
for k in range(j + 1, max_k + 1):
if rank > k:
trustworthiness[k] += rank - k
for k in range(1, max_k + 1):
trustworthiness[k] = 1.0 - trustworthiness[k] * (
2.0 / (n_samples * k * (2.0 * n_samples - 3.0 * k - 1.0))
)
return trustworthiness
def make_trustworthiness_calculator(metric): # pragma: no cover
@numba.njit()
def trustworthiness_vector_lowmem(source, indices_embedded, max_k):
n_samples = indices_embedded.shape[0]
trustworthiness = np.zeros(max_k + 1, dtype=np.float64)
dist_vector = np.zeros(n_samples, dtype=np.float64)
for i in range(n_samples):
for j in numba.prange(n_samples):
dist_vector[j] = metric(source[i], source[j])
indices_source = np.argsort(dist_vector)
for j in range(max_k):
rank = 0
while indices_source[rank] != indices_embedded[i, j]:
rank += 1
for k in range(j + 1, max_k + 1):
if rank > k:
trustworthiness[k] += rank - k
for k in range(1, max_k + 1):
trustworthiness[k] = 1.0 - trustworthiness[k] * (
2.0 / (n_samples * k * (2.0 * n_samples - 3.0 * k - 1.0))
)
trustworthiness[0] = 1.0
return trustworthiness
return trustworthiness_vector_lowmem
@numba.jit()
def trustworthiness_vector(
source, embedding, max_k, metric="euclidean"
): # pragma: no cover
tree = KDTree(embedding, metric=metric)
indices_embedded = tree.query(embedding, k=max_k, return_distance=False)
# Drop the actual point itself
indices_embedded = indices_embedded[:, 1:]
dist = named_distances[metric]
vec_calculator = make_trustworthiness_calculator(dist)
result = vec_calculator(source, indices_embedded, max_k)
return result
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn_single/validation.py
|
validation.py
|
from __future__ import print_function
import numpy as np
import numba
from utils import (
tau_rand_int,
tau_rand,
norm,
make_heap,
heap_push,
rejection_sample,
build_candidates,
deheap_sort,
)
import locale
locale.setlocale(locale.LC_NUMERIC, "C")
# Just reproduce a simpler version of numpy unique (not numba supported yet)
@numba.njit()
def arr_unique(arr):
aux = np.sort(arr)
flag = np.concatenate((np.ones(1, dtype=np.bool_), aux[1:] != aux[:-1]))
return aux[flag]
# Just reproduce a simpler version of numpy union1d (not numba supported yet)
@numba.njit()
def arr_union(ar1, ar2):
if ar1.shape[0] == 0:
return ar2
elif ar2.shape[0] == 0:
return ar1
else:
return arr_unique(np.concatenate((ar1, ar2)))
# Just reproduce a simpler version of numpy intersect1d (not numba supported
# yet)
@numba.njit()
def arr_intersect(ar1, ar2):
aux = np.concatenate((ar1, ar2))
aux.sort()
return aux[:-1][aux[1:] == aux[:-1]]
@numba.njit()
def sparse_sum(ind1, data1, ind2, data2):
result_ind = arr_union(ind1, ind2)
result_data = np.zeros(result_ind.shape[0], dtype=np.float32)
i1 = 0
i2 = 0
nnz = 0
# pass through both index lists
while i1 < ind1.shape[0] and i2 < ind2.shape[0]:
j1 = ind1[i1]
j2 = ind2[i2]
if j1 == j2:
val = data1[i1] + data2[i2]
if val != 0:
result_ind[nnz] = j1
result_data[nnz] = val
nnz += 1
i1 += 1
i2 += 1
elif j1 < j2:
val = data1[i1]
if val != 0:
result_ind[nnz] = j1
result_data[nnz] = val
nnz += 1
i1 += 1
else:
val = data2[i2]
if val != 0:
result_ind[nnz] = j2
result_data[nnz] = val
nnz += 1
i2 += 1
# pass over the tails
while i1 < ind1.shape[0]:
val = data1[i1]
if val != 0:
result_ind[nnz] = i1
result_data[nnz] = val
nnz += 1
i1 += 1
while i2 < ind2.shape[0]:
val = data2[i2]
if val != 0:
result_ind[nnz] = i2
result_data[nnz] = val
nnz += 1
i2 += 1
# truncate to the correct length in case there were zeros created
result_ind = result_ind[:nnz]
result_data = result_data[:nnz]
return result_ind, result_data
@numba.njit()
def sparse_diff(ind1, data1, ind2, data2):
return sparse_sum(ind1, data1, ind2, -data2)
@numba.njit()
def sparse_mul(ind1, data1, ind2, data2):
result_ind = arr_intersect(ind1, ind2)
result_data = np.zeros(result_ind.shape[0], dtype=np.float32)
i1 = 0
i2 = 0
nnz = 0
# pass through both index lists
while i1 < ind1.shape[0] and i2 < ind2.shape[0]:
j1 = ind1[i1]
j2 = ind2[i2]
if j1 == j2:
val = data1[i1] * data2[i2]
if val != 0:
result_ind[nnz] = j1
result_data[nnz] = val
nnz += 1
i1 += 1
i2 += 1
elif j1 < j2:
i1 += 1
else:
i2 += 1
# truncate to the correct length in case there were zeros created
result_ind = result_ind[:nnz]
result_data = result_data[:nnz]
return result_ind, result_data
def make_sparse_nn_descent(sparse_dist, dist_args):
"""Create a numba accelerated version of nearest neighbor descent
specialised for the given distance metric and metric arguments on sparse
matrix data provided in CSR ind, indptr and data format. Numba
doesn't support higher order functions directly, but we can instead JIT
compile the version of NN-descent for any given metric.
Parameters
----------
sparse_dist: function
A numba JITd distance function which, given four arrays (two sets of
indices and data) computes a dissimilarity between them.
dist_args: tuple
Any extra arguments that need to be passed to the distance function
beyond the two arrays to be compared.
Returns
-------
A numba JITd function for nearest neighbor descent computation that is
specialised to the given metric.
"""
@numba.njit()
def nn_descent(
inds,
indptr,
data,
n_vertices,
n_neighbors,
rng_state,
max_candidates=50,
n_iters=10,
delta=0.001,
rho=0.5,
rp_tree_init=True,
leaf_array=None,
verbose=False,
):
current_graph = make_heap(n_vertices, n_neighbors)
for i in range(n_vertices):
indices = rejection_sample(n_neighbors, n_vertices, rng_state)
for j in range(indices.shape[0]):
from_inds = inds[indptr[i] : indptr[i + 1]]
from_data = data[indptr[i] : indptr[i + 1]]
to_inds = inds[indptr[indices[j]] : indptr[indices[j] + 1]]
to_data = data[indptr[indices[j]] : indptr[indices[j] + 1]]
d = sparse_dist(from_inds, from_data, to_inds, to_data, *dist_args)
heap_push(current_graph, i, d, indices[j], 1)
heap_push(current_graph, indices[j], d, i, 1)
if rp_tree_init:
for n in range(leaf_array.shape[0]):
for i in range(leaf_array.shape[1]):
if leaf_array[n, i] < 0:
break
for j in range(i + 1, leaf_array.shape[1]):
if leaf_array[n, j] < 0:
break
from_inds = inds[
indptr[leaf_array[n, i]] : indptr[leaf_array[n, i] + 1]
]
from_data = data[
indptr[leaf_array[n, i]] : indptr[leaf_array[n, i] + 1]
]
to_inds = inds[
indptr[leaf_array[n, j]] : indptr[leaf_array[n, j] + 1]
]
to_data = data[
indptr[leaf_array[n, j]] : indptr[leaf_array[n, j] + 1]
]
d = sparse_dist(
from_inds, from_data, to_inds, to_data, *dist_args
)
heap_push(
current_graph, leaf_array[n, i], d, leaf_array[n, j], 1
)
heap_push(
current_graph, leaf_array[n, j], d, leaf_array[n, i], 1
)
for n in range(n_iters):
if verbose:
print("\t", n, " / ", n_iters)
candidate_neighbors = build_candidates(
current_graph, n_vertices, n_neighbors, max_candidates, rng_state
)
c = 0
for i in range(n_vertices):
for j in range(max_candidates):
p = int(candidate_neighbors[0, i, j])
if p < 0 or tau_rand(rng_state) < rho:
continue
for k in range(max_candidates):
q = int(candidate_neighbors[0, i, k])
if (
q < 0
or not candidate_neighbors[2, i, j]
and not candidate_neighbors[2, i, k]
):
continue
from_inds = inds[indptr[p] : indptr[p + 1]]
from_data = data[indptr[p] : indptr[p + 1]]
to_inds = inds[indptr[q] : indptr[q + 1]]
to_data = data[indptr[q] : indptr[q + 1]]
d = sparse_dist(
from_inds, from_data, to_inds, to_data, *dist_args
)
c += heap_push(current_graph, p, d, q, 1)
c += heap_push(current_graph, q, d, p, 1)
if c <= delta * n_neighbors * n_vertices:
break
return deheap_sort(current_graph)
return nn_descent
@numba.njit()
def general_sset_intersection(
indptr1,
indices1,
data1,
indptr2,
indices2,
data2,
result_row,
result_col,
result_val,
mix_weight=0.5,
):
left_min = max(data1.min() / 2.0, 1.0e-8)
right_min = max(data2.min() / 2.0, 1.0e-8)
for idx in range(result_row.shape[0]):
i = result_row[idx]
j = result_col[idx]
left_val = left_min
for k in range(indptr1[i], indptr1[i + 1]):
if indices1[k] == j:
left_val = data1[k]
right_val = right_min
for k in range(indptr2[i], indptr2[i + 1]):
if indices2[k] == j:
right_val = data2[k]
if left_val > left_min or right_val > right_min:
if mix_weight < 0.5:
result_val[idx] = left_val * pow(
right_val, mix_weight / (1.0 - mix_weight)
)
else:
result_val[idx] = (
pow(left_val, (1.0 - mix_weight) / mix_weight) * right_val
)
return
@numba.njit()
def sparse_euclidean(ind1, data1, ind2, data2):
aux_inds, aux_data = sparse_diff(ind1, data1, ind2, data2)
result = 0.0
for i in range(aux_data.shape[0]):
result += aux_data[i] ** 2
return np.sqrt(result)
@numba.njit()
def sparse_manhattan(ind1, data1, ind2, data2):
aux_inds, aux_data = sparse_diff(ind1, data1, ind2, data2)
result = 0.0
for i in range(aux_data.shape[0]):
result += np.abs(aux_data[i])
return result
@numba.njit()
def sparse_chebyshev(ind1, data1, ind2, data2):
aux_inds, aux_data = sparse_diff(ind1, data1, ind2, data2)
result = 0.0
for i in range(aux_data.shape[0]):
result = max(result, np.abs(aux_data[i]))
return result
@numba.njit()
def sparse_minkowski(ind1, data1, ind2, data2, p=2.0):
aux_inds, aux_data = sparse_diff(ind1, data1, ind2, data2)
result = 0.0
for i in range(aux_data.shape[0]):
result += np.abs(aux_data[i]) ** p
return result ** (1.0 / p)
@numba.njit()
def sparse_hamming(ind1, data1, ind2, data2, n_features):
num_not_equal = sparse_diff(ind1, data1, ind2, data2)[0].shape[0]
return float(num_not_equal) / n_features
@numba.njit()
def sparse_canberra(ind1, data1, ind2, data2):
abs_data1 = np.abs(data1)
abs_data2 = np.abs(data2)
denom_inds, denom_data = sparse_sum(ind1, abs_data1, ind2, abs_data2)
denom_data = 1.0 / denom_data
numer_inds, numer_data = sparse_diff(ind1, data1, ind2, data2)
numer_data = np.abs(numer_data)
val_inds, val_data = sparse_mul(numer_inds, numer_data, denom_inds, denom_data)
return np.sum(val_data)
@numba.njit()
def sparse_bray_curtis(ind1, data1, ind2, data2): # pragma: no cover
abs_data1 = np.abs(data1)
abs_data2 = np.abs(data2)
denom_inds, denom_data = sparse_sum(ind1, abs_data1, ind2, abs_data2)
if denom_data.shape[0] == 0:
return 0.0
denominator = np.sum(denom_data)
numer_inds, numer_data = sparse_diff(ind1, data1, ind2, data2)
numer_data = np.abs(numer_data)
numerator = np.sum(numer_data)
return float(numerator) / denominator
@numba.njit()
def sparse_jaccard(ind1, data1, ind2, data2):
num_non_zero = arr_union(ind1, ind2).shape[0]
num_equal = arr_intersect(ind1, ind2).shape[0]
if num_non_zero == 0:
return 0.0
else:
return float(num_non_zero - num_equal) / num_non_zero
@numba.njit()
def sparse_matching(ind1, data1, ind2, data2, n_features):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
return float(num_not_equal) / n_features
@numba.njit()
def sparse_dice(ind1, data1, ind2, data2):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
if num_not_equal == 0.0:
return 0.0
else:
return num_not_equal / (2.0 * num_true_true + num_not_equal)
@numba.njit()
def sparse_kulsinski(ind1, data1, ind2, data2, n_features):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
if num_not_equal == 0:
return 0.0
else:
return float(num_not_equal - num_true_true + n_features) / (
num_not_equal + n_features
)
@numba.njit()
def sparse_rogers_tanimoto(ind1, data1, ind2, data2, n_features):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
return (2.0 * num_not_equal) / (n_features + num_not_equal)
@numba.njit()
def sparse_russellrao(ind1, data1, ind2, data2, n_features):
if ind1.shape[0] == ind2.shape[0] and np.all(ind1 == ind2):
return 0.0
num_true_true = arr_intersect(ind1, ind2).shape[0]
if num_true_true == np.sum(data1 != 0) and num_true_true == np.sum(data2 != 0):
return 0.0
else:
return float(n_features - num_true_true) / (n_features)
@numba.njit()
def sparse_sokal_michener(ind1, data1, ind2, data2, n_features):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
return (2.0 * num_not_equal) / (n_features + num_not_equal)
@numba.njit()
def sparse_sokal_sneath(ind1, data1, ind2, data2):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
if num_not_equal == 0.0:
return 0.0
else:
return num_not_equal / (0.5 * num_true_true + num_not_equal)
@numba.njit()
def sparse_cosine(ind1, data1, ind2, data2):
aux_inds, aux_data = sparse_mul(ind1, data1, ind2, data2)
result = 0.0
norm1 = norm(data1)
norm2 = norm(data2)
for i in range(aux_data.shape[0]):
result += aux_data[i]
if norm1 == 0.0 and norm2 == 0.0:
return 0.0
elif norm1 == 0.0 or norm2 == 0.0:
return 1.0
else:
return 1.0 - (result / (norm1 * norm2))
@numba.njit()
def sparse_correlation(ind1, data1, ind2, data2, n_features):
mu_x = 0.0
mu_y = 0.0
dot_product = 0.0
if ind1.shape[0] == 0 and ind2.shape[0] == 0:
return 0.0
elif ind1.shape[0] == 0 or ind2.shape[0] == 0:
return 1.0
for i in range(data1.shape[0]):
mu_x += data1[i]
for i in range(data2.shape[0]):
mu_y += data2[i]
mu_x /= n_features
mu_y /= n_features
shifted_data1 = np.empty(data1.shape[0], dtype=np.float32)
shifted_data2 = np.empty(data2.shape[0], dtype=np.float32)
for i in range(data1.shape[0]):
shifted_data1[i] = data1[i] - mu_x
for i in range(data2.shape[0]):
shifted_data2[i] = data2[i] - mu_y
norm1 = np.sqrt(
(norm(shifted_data1) ** 2) + (n_features - ind1.shape[0]) * (mu_x ** 2)
)
norm2 = np.sqrt(
(norm(shifted_data2) ** 2) + (n_features - ind2.shape[0]) * (mu_y ** 2)
)
dot_prod_inds, dot_prod_data = sparse_mul(ind1, shifted_data1, ind2, shifted_data2)
common_indices = set(dot_prod_inds)
for i in range(dot_prod_data.shape[0]):
dot_product += dot_prod_data[i]
for i in range(ind1.shape[0]):
if ind1[i] not in common_indices:
dot_product -= shifted_data1[i] * (mu_y)
for i in range(ind2.shape[0]):
if ind2[i] not in common_indices:
dot_product -= shifted_data2[i] * (mu_x)
all_indices = arr_union(ind1, ind2)
dot_product += mu_x * mu_y * (n_features - all_indices.shape[0])
if norm1 == 0.0 and norm2 == 0.0:
return 0.0
elif dot_product == 0.0:
return 1.0
else:
return 1.0 - (dot_product / (norm1 * norm2))
sparse_named_distances = {
# general minkowski distances
"euclidean": sparse_euclidean,
"manhattan": sparse_manhattan,
"l1": sparse_manhattan,
"taxicab": sparse_manhattan,
"chebyshev": sparse_chebyshev,
"linf": sparse_chebyshev,
"linfty": sparse_chebyshev,
"linfinity": sparse_chebyshev,
"minkowski": sparse_minkowski,
# Other distances
"canberra": sparse_canberra,
# 'braycurtis': sparse_bray_curtis,
# Binary distances
"hamming": sparse_hamming,
"jaccard": sparse_jaccard,
"dice": sparse_dice,
"matching": sparse_matching,
"kulsinski": sparse_kulsinski,
"rogerstanimoto": sparse_rogers_tanimoto,
"russellrao": sparse_russellrao,
"sokalmichener": sparse_sokal_michener,
"sokalsneath": sparse_sokal_sneath,
"cosine": sparse_cosine,
"correlation": sparse_correlation,
}
sparse_need_n_features = (
"hamming",
"matching",
"kulsinski",
"rogerstanimoto",
"russellrao",
"sokalmichener",
"correlation",
)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn_single/sparse.py
|
sparse.py
|
from __future__ import print_function
from collections import deque, namedtuple
from warnings import warn
import numpy as np
import numba
from sparse import sparse_mul, sparse_diff, sparse_sum
from utils import tau_rand_int, norm
import scipy.sparse
import locale
locale.setlocale(locale.LC_NUMERIC, "C")
RandomProjectionTreeNode = namedtuple(
"RandomProjectionTreeNode",
["indices", "is_leaf", "hyperplane", "offset", "left_child", "right_child"],
)
FlatTree = namedtuple("FlatTree", ["hyperplanes", "offsets", "children", "indices"])
@numba.njit(fastmath=True)
def angular_random_projection_split(data, indices, rng_state):
"""Given a set of ``indices`` for data points from ``data``, create
a random hyperplane to split the data, returning two arrays indices
that fall on either side of the hyperplane. This is the basis for a
random projection tree, which simply uses this splitting recursively.
This particular split uses cosine distance to determine the hyperplane
and which side each data sample falls on.
Parameters
----------
data: array of shape (n_samples, n_features)
The original data to be split
indices: array of shape (tree_node_size,)
The indices of the elements in the ``data`` array that are to
be split in the current operation.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
indices_left: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
indices_right: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
"""
dim = data.shape[1]
# Select two random points, set the hyperplane between them
left_index = tau_rand_int(rng_state) % indices.shape[0]
right_index = tau_rand_int(rng_state) % indices.shape[0]
right_index += left_index == right_index
right_index = right_index % indices.shape[0]
left = indices[left_index]
right = indices[right_index]
left_norm = norm(data[left])
right_norm = norm(data[right])
if left_norm == 0.0:
left_norm = 1.0
if right_norm == 0.0:
right_norm = 1.0
# Compute the normal vector to the hyperplane (the vector between
# the two points)
hyperplane_vector = np.empty(dim, dtype=np.float32)
for d in range(dim):
hyperplane_vector[d] = (data[left, d] / left_norm) - (
data[right, d] / right_norm
)
hyperplane_norm = norm(hyperplane_vector)
if hyperplane_norm == 0.0:
hyperplane_norm = 1.0
for d in range(dim):
hyperplane_vector[d] = hyperplane_vector[d] / hyperplane_norm
# For each point compute the margin (project into normal vector)
# If we are on lower side of the hyperplane put in one pile, otherwise
# put it in the other pile (if we hit hyperplane on the nose, flip a coin)
n_left = 0
n_right = 0
side = np.empty(indices.shape[0], np.int8)
for i in range(indices.shape[0]):
margin = 0.0
for d in range(dim):
margin += hyperplane_vector[d] * data[indices[i], d]
if margin == 0:
side[i] = tau_rand_int(rng_state) % 2
if side[i] == 0:
n_left += 1
else:
n_right += 1
elif margin > 0:
side[i] = 0
n_left += 1
else:
side[i] = 1
n_right += 1
# Now that we have the counts allocate arrays
indices_left = np.empty(n_left, dtype=np.int64)
indices_right = np.empty(n_right, dtype=np.int64)
# Populate the arrays with indices according to which side they fell on
n_left = 0
n_right = 0
for i in range(side.shape[0]):
if side[i] == 0:
indices_left[n_left] = indices[i]
n_left += 1
else:
indices_right[n_right] = indices[i]
n_right += 1
return indices_left, indices_right, hyperplane_vector, None
@numba.njit(fastmath=True)
def euclidean_random_projection_split(data, indices, rng_state):
"""Given a set of ``indices`` for data points from ``data``, create
a random hyperplane to split the data, returning two arrays indices
that fall on either side of the hyperplane. This is the basis for a
random projection tree, which simply uses this splitting recursively.
This particular split uses euclidean distance to determine the hyperplane
and which side each data sample falls on.
Parameters
----------
data: array of shape (n_samples, n_features)
The original data to be split
indices: array of shape (tree_node_size,)
The indices of the elements in the ``data`` array that are to
be split in the current operation.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
indices_left: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
indices_right: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
"""
dim = data.shape[1]
# Select two random points, set the hyperplane between them
left_index = tau_rand_int(rng_state) % indices.shape[0]
right_index = tau_rand_int(rng_state) % indices.shape[0]
right_index += left_index == right_index
right_index = right_index % indices.shape[0]
left = indices[left_index]
right = indices[right_index]
# Compute the normal vector to the hyperplane (the vector between
# the two points) and the offset from the origin
hyperplane_offset = 0.0
hyperplane_vector = np.empty(dim, dtype=np.float32)
for d in range(dim):
hyperplane_vector[d] = data[left, d] - data[right, d]
hyperplane_offset -= (
hyperplane_vector[d] * (data[left, d] + data[right, d]) / 2.0
)
# For each point compute the margin (project into normal vector, add offset)
# If we are on lower side of the hyperplane put in one pile, otherwise
# put it in the other pile (if we hit hyperplane on the nose, flip a coin)
n_left = 0
n_right = 0
side = np.empty(indices.shape[0], np.int8)
for i in range(indices.shape[0]):
margin = hyperplane_offset
for d in range(dim):
margin += hyperplane_vector[d] * data[indices[i], d]
if margin == 0:
side[i] = tau_rand_int(rng_state) % 2
if side[i] == 0:
n_left += 1
else:
n_right += 1
elif margin > 0:
side[i] = 0
n_left += 1
else:
side[i] = 1
n_right += 1
# Now that we have the counts allocate arrays
indices_left = np.empty(n_left, dtype=np.int64)
indices_right = np.empty(n_right, dtype=np.int64)
# Populate the arrays with indices according to which side they fell on
n_left = 0
n_right = 0
for i in range(side.shape[0]):
if side[i] == 0:
indices_left[n_left] = indices[i]
n_left += 1
else:
indices_right[n_right] = indices[i]
n_right += 1
return indices_left, indices_right, hyperplane_vector, hyperplane_offset
@numba.njit(fastmath=True)
def sparse_angular_random_projection_split(inds, indptr, data, indices, rng_state):
"""Given a set of ``indices`` for data points from a sparse data set
presented in csr sparse format as inds, indptr and data, create
a random hyperplane to split the data, returning two arrays indices
that fall on either side of the hyperplane. This is the basis for a
random projection tree, which simply uses this splitting recursively.
This particular split uses cosine distance to determine the hyperplane
and which side each data sample falls on.
Parameters
----------
inds: array
CSR format index array of the matrix
indptr: array
CSR format index pointer array of the matrix
data: array
CSR format data array of the matrix
indices: array of shape (tree_node_size,)
The indices of the elements in the ``data`` array that are to
be split in the current operation.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
indices_left: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
indices_right: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
"""
# Select two random points, set the hyperplane between them
left_index = tau_rand_int(rng_state) % indices.shape[0]
right_index = tau_rand_int(rng_state) % indices.shape[0]
right_index += left_index == right_index
right_index = right_index % indices.shape[0]
left = indices[left_index]
right = indices[right_index]
left_inds = inds[indptr[left] : indptr[left + 1]]
left_data = data[indptr[left] : indptr[left + 1]]
right_inds = inds[indptr[right] : indptr[right + 1]]
right_data = data[indptr[right] : indptr[right + 1]]
left_norm = norm(left_data)
right_norm = norm(right_data)
# Compute the normal vector to the hyperplane (the vector between
# the two points)
normalized_left_data = left_data / left_norm
normalized_right_data = right_data / right_norm
hyperplane_inds, hyperplane_data = sparse_diff(
left_inds, normalized_left_data, right_inds, normalized_right_data
)
hyperplane_norm = norm(hyperplane_data)
for d in range(hyperplane_data.shape[0]):
hyperplane_data[d] = hyperplane_data[d] / hyperplane_norm
# For each point compute the margin (project into normal vector)
# If we are on lower side of the hyperplane put in one pile, otherwise
# put it in the other pile (if we hit hyperplane on the nose, flip a coin)
n_left = 0
n_right = 0
side = np.empty(indices.shape[0], np.int8)
for i in range(indices.shape[0]):
margin = 0.0
i_inds = inds[indptr[indices[i]] : indptr[indices[i] + 1]]
i_data = data[indptr[indices[i]] : indptr[indices[i] + 1]]
mul_inds, mul_data = sparse_mul(
hyperplane_inds, hyperplane_data, i_inds, i_data
)
for d in range(mul_data.shape[0]):
margin += mul_data[d]
if margin == 0:
side[i] = tau_rand_int(rng_state) % 2
if side[i] == 0:
n_left += 1
else:
n_right += 1
elif margin > 0:
side[i] = 0
n_left += 1
else:
side[i] = 1
n_right += 1
# Now that we have the counts allocate arrays
indices_left = np.empty(n_left, dtype=np.int64)
indices_right = np.empty(n_right, dtype=np.int64)
# Populate the arrays with indices according to which side they fell on
n_left = 0
n_right = 0
for i in range(side.shape[0]):
if side[i] == 0:
indices_left[n_left] = indices[i]
n_left += 1
else:
indices_right[n_right] = indices[i]
n_right += 1
hyperplane = np.vstack((hyperplane_inds, hyperplane_data))
return indices_left, indices_right, hyperplane, None
@numba.njit(fastmath=True)
def sparse_euclidean_random_projection_split(inds, indptr, data, indices, rng_state):
"""Given a set of ``indices`` for data points from a sparse data set
presented in csr sparse format as inds, indptr and data, create
a random hyperplane to split the data, returning two arrays indices
that fall on either side of the hyperplane. This is the basis for a
random projection tree, which simply uses this splitting recursively.
This particular split uses cosine distance to determine the hyperplane
and which side each data sample falls on.
Parameters
----------
inds: array
CSR format index array of the matrix
indptr: array
CSR format index pointer array of the matrix
data: array
CSR format data array of the matrix
indices: array of shape (tree_node_size,)
The indices of the elements in the ``data`` array that are to
be split in the current operation.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
indices_left: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
indices_right: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
"""
# Select two random points, set the hyperplane between them
left_index = tau_rand_int(rng_state) % indices.shape[0]
right_index = tau_rand_int(rng_state) % indices.shape[0]
right_index += left_index == right_index
right_index = right_index % indices.shape[0]
left = indices[left_index]
right = indices[right_index]
left_inds = inds[indptr[left] : indptr[left + 1]]
left_data = data[indptr[left] : indptr[left + 1]]
right_inds = inds[indptr[right] : indptr[right + 1]]
right_data = data[indptr[right] : indptr[right + 1]]
# Compute the normal vector to the hyperplane (the vector between
# the two points) and the offset from the origin
hyperplane_offset = 0.0
hyperplane_inds, hyperplane_data = sparse_diff(
left_inds, left_data, right_inds, right_data
)
offset_inds, offset_data = sparse_sum(left_inds, left_data, right_inds, right_data)
offset_data = offset_data / 2.0
offset_inds, offset_data = sparse_mul(
hyperplane_inds, hyperplane_data, offset_inds, offset_data
)
for d in range(offset_data.shape[0]):
hyperplane_offset -= offset_data[d]
# For each point compute the margin (project into normal vector, add offset)
# If we are on lower side of the hyperplane put in one pile, otherwise
# put it in the other pile (if we hit hyperplane on the nose, flip a coin)
n_left = 0
n_right = 0
side = np.empty(indices.shape[0], np.int8)
for i in range(indices.shape[0]):
margin = hyperplane_offset
i_inds = inds[indptr[indices[i]] : indptr[indices[i] + 1]]
i_data = data[indptr[indices[i]] : indptr[indices[i] + 1]]
mul_inds, mul_data = sparse_mul(
hyperplane_inds, hyperplane_data, i_inds, i_data
)
for d in range(mul_data.shape[0]):
margin += mul_data[d]
if margin == 0:
side[i] = tau_rand_int(rng_state) % 2
if side[i] == 0:
n_left += 1
else:
n_right += 1
elif margin > 0:
side[i] = 0
n_left += 1
else:
side[i] = 1
n_right += 1
# Now that we have the counts allocate arrays
indices_left = np.empty(n_left, dtype=np.int64)
indices_right = np.empty(n_right, dtype=np.int64)
# Populate the arrays with indices according to which side they fell on
n_left = 0
n_right = 0
for i in range(side.shape[0]):
if side[i] == 0:
indices_left[n_left] = indices[i]
n_left += 1
else:
indices_right[n_right] = indices[i]
n_right += 1
hyperplane = np.vstack((hyperplane_inds, hyperplane_data))
return indices_left, indices_right, hyperplane, hyperplane_offset
@numba.jit()
def make_euclidean_tree(data, indices, rng_state, leaf_size=30):
if indices.shape[0] > leaf_size:
left_indices, right_indices, hyperplane, offset = euclidean_random_projection_split(
data, indices, rng_state
)
left_node = make_euclidean_tree(data, left_indices, rng_state, leaf_size)
right_node = make_euclidean_tree(data, right_indices, rng_state, leaf_size)
node = RandomProjectionTreeNode(
None, False, hyperplane, offset, left_node, right_node
)
else:
node = RandomProjectionTreeNode(indices, True, None, None, None, None)
return node
@numba.jit()
def make_angular_tree(data, indices, rng_state, leaf_size=30):
if indices.shape[0] > leaf_size:
left_indices, right_indices, hyperplane, offset = angular_random_projection_split(
data, indices, rng_state
)
left_node = make_angular_tree(data, left_indices, rng_state, leaf_size)
right_node = make_angular_tree(data, right_indices, rng_state, leaf_size)
node = RandomProjectionTreeNode(
None, False, hyperplane, offset, left_node, right_node
)
else:
node = RandomProjectionTreeNode(indices, True, None, None, None, None)
return node
@numba.jit()
def make_sparse_euclidean_tree(inds, indptr, data, indices, rng_state, leaf_size=30):
if indices.shape[0] > leaf_size:
left_indices, right_indices, hyperplane, offset = sparse_euclidean_random_projection_split(
inds, indptr, data, indices, rng_state
)
left_node = make_sparse_euclidean_tree(
inds, indptr, data, left_indices, rng_state, leaf_size
)
right_node = make_sparse_euclidean_tree(
inds, indptr, data, right_indices, rng_state, leaf_size
)
node = RandomProjectionTreeNode(
None, False, hyperplane, offset, left_node, right_node
)
else:
node = RandomProjectionTreeNode(indices, True, None, None, None, None)
return node
@numba.jit()
def make_sparse_angular_tree(inds, indptr, data, indices, rng_state, leaf_size=30):
if indices.shape[0] > leaf_size:
left_indices, right_indices, hyperplane, offset = sparse_angular_random_projection_split(
inds, indptr, data, indices, rng_state
)
left_node = make_sparse_angular_tree(
inds, indptr, data, left_indices, rng_state, leaf_size
)
right_node = make_sparse_angular_tree(
inds, indptr, data, right_indices, rng_state, leaf_size
)
node = RandomProjectionTreeNode(
None, False, hyperplane, offset, left_node, right_node
)
else:
node = RandomProjectionTreeNode(indices, True, None, None, None, None)
return node
def make_tree(data, rng_state, leaf_size=30, angular=False):
"""Construct a random projection tree based on ``data`` with leaves
of size at most ``leaf_size``.
Parameters
----------
data: array of shape (n_samples, n_features)
The original data to be split
rng_state: array of int64, shape (3,)
The internal state of the rng
leaf_size: int (optional, default 30)
The maximum size of any leaf node in the tree. Any node in the tree
with more than ``leaf_size`` will be split further to create child
nodes.
angular: bool (optional, default False)
Whether to use cosine/angular distance to create splits in the tree,
or euclidean distance.
Returns
-------
node: RandomProjectionTreeNode
A random projection tree node which links to its child nodes. This
provides the full tree below the returned node.
"""
is_sparse = scipy.sparse.isspmatrix_csr(data)
indices = np.arange(data.shape[0])
# Make a tree recursively until we get below the leaf size
if is_sparse:
inds = data.indices
indptr = data.indptr
spdata = data.data
if angular:
return make_sparse_angular_tree(
inds, indptr, spdata, indices, rng_state, leaf_size
)
else:
return make_sparse_euclidean_tree(
inds, indptr, spdata, indices, rng_state, leaf_size
)
else:
if angular:
return make_angular_tree(data, indices, rng_state, leaf_size)
else:
return make_euclidean_tree(data, indices, rng_state, leaf_size)
def num_nodes(tree):
"""Determine the number of nodes in a tree"""
if tree.is_leaf:
return 1
else:
return 1 + num_nodes(tree.left_child) + num_nodes(tree.right_child)
def num_leaves(tree):
"""Determine the number of leaves in a tree"""
if tree.is_leaf:
return 1
else:
return num_leaves(tree.left_child) + num_leaves(tree.right_child)
def max_sparse_hyperplane_size(tree):
"""Determine the most number on non zeros in a hyperplane entry"""
if tree.is_leaf:
return 0
else:
return max(
tree.hyperplane.shape[1],
max_sparse_hyperplane_size(tree.left_child),
max_sparse_hyperplane_size(tree.right_child),
)
def recursive_flatten(
tree, hyperplanes, offsets, children, indices, node_num, leaf_num
):
if tree.is_leaf:
children[node_num, 0] = -leaf_num
indices[leaf_num, : tree.indices.shape[0]] = tree.indices
leaf_num += 1
return node_num, leaf_num
else:
if len(tree.hyperplane.shape) > 1:
# spare case
hyperplanes[node_num][:, : tree.hyperplane.shape[1]] = tree.hyperplane
else:
hyperplanes[node_num] = tree.hyperplane
offsets[node_num] = tree.offset
children[node_num, 0] = node_num + 1
old_node_num = node_num
node_num, leaf_num = recursive_flatten(
tree.left_child,
hyperplanes,
offsets,
children,
indices,
node_num + 1,
leaf_num,
)
children[old_node_num, 1] = node_num + 1
node_num, leaf_num = recursive_flatten(
tree.right_child,
hyperplanes,
offsets,
children,
indices,
node_num + 1,
leaf_num,
)
return node_num, leaf_num
def flatten_tree(tree, leaf_size):
n_nodes = num_nodes(tree)
n_leaves = num_leaves(tree)
if len(tree.hyperplane.shape) > 1:
# sparse case
max_hyperplane_nnz = max_sparse_hyperplane_size(tree)
hyperplanes = np.zeros(
(n_nodes, tree.hyperplane.shape[0], max_hyperplane_nnz), dtype=np.float32
)
else:
hyperplanes = np.zeros((n_nodes, tree.hyperplane.shape[0]), dtype=np.float32)
offsets = np.zeros(n_nodes, dtype=np.float32)
children = -1 * np.ones((n_nodes, 2), dtype=np.int64)
indices = -1 * np.ones((n_leaves, leaf_size), dtype=np.int64)
recursive_flatten(tree, hyperplanes, offsets, children, indices, 0, 0)
return FlatTree(hyperplanes, offsets, children, indices)
@numba.njit()
def select_side(hyperplane, offset, point, rng_state):
margin = offset
for d in range(point.shape[0]):
margin += hyperplane[d] * point[d]
if margin == 0:
side = tau_rand_int(rng_state) % 2
if side == 0:
return 0
else:
return 1
elif margin > 0:
return 0
else:
return 1
@numba.njit()
def search_flat_tree(point, hyperplanes, offsets, children, indices, rng_state):
node = 0
while children[node, 0] > 0:
side = select_side(hyperplanes[node], offsets[node], point, rng_state)
if side == 0:
node = children[node, 0]
else:
node = children[node, 1]
return indices[-children[node, 0]]
def make_forest(data, n_neighbors, n_trees, rng_state, angular=False):
"""Build a random projection forest with ``n_trees``.
Parameters
----------
data
n_neighbors
n_trees
rng_state
angular
Returns
-------
forest: list
A list of random projection trees.
"""
result = []
leaf_size = max(10, n_neighbors)
try:
result = [
flatten_tree(make_tree(data, rng_state, leaf_size, angular), leaf_size)
for i in range(n_trees)
]
except (RuntimeError, RecursionError):
warn(
"Random Projection forest initialisation failed due to recursion"
"limit being reached. Something is a little strange with your "
"data, and this may take longer than normal to compute."
)
return result
def rptree_leaf_array(rp_forest):
"""Generate an array of sets of candidate nearest neighbors by
constructing a random projection forest and taking the leaves of all the
trees. Any given tree has leaves that are a set of potential nearest
neighbors. Given enough trees the set of all such leaves gives a good
likelihood of getting a good set of nearest neighbors in composite. Since
such a random projection forest is inexpensive to compute, this can be a
useful means of seeding other nearest neighbor algorithms.
Parameters
----------
data: array of shape (n_samples, n_features)
The data for which to generate nearest neighbor approximations.
n_neighbors: int
The number of nearest neighbors to attempt to approximate.
rng_state: array of int64, shape (3,)
The internal state of the rng
n_trees: int (optional, default 10)
The number of trees to build in the forest construction.
angular: bool (optional, default False)
Whether to use angular/cosine distance for random projection tree
construction.
Returns
-------
leaf_array: array of shape (n_leaves, max(10, n_neighbors))
Each row of leaf array is a list of indices found in a given leaf.
Since not all leaves are the same size the arrays are padded out with -1
to ensure we can return a single ndarray.
"""
if len(rp_forest) > 0:
leaf_array = np.vstack([tree.indices for tree in rp_forest])
else:
leaf_array = np.array([[-1]])
return leaf_array
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn_single/rp_tree.py
|
rp_tree.py
|
from __future__ import print_function
from warnings import warn
from scipy.optimize import curve_fit
from sklearn.base import BaseEstimator
from sklearn.utils import check_random_state, check_array
from sklearn.metrics import pairwise_distances
from sklearn.preprocessing import normalize
from sklearn.neighbors import KDTree
from sklearn.externals import joblib
import numpy as np
import scipy.sparse
import scipy.sparse.csgraph
import numba
import distances as dist
from utils import tau_rand_int, deheap_sort, submatrix
from rp_tree import rptree_leaf_array, make_forest
from nndescent import (
make_nn_descent,
make_initialisations,
make_initialized_nnd_search,
initialise_search,
)
from spectral import spectral_layout
import locale
locale.setlocale(locale.LC_NUMERIC, "C")
INT32_MIN = np.iinfo(np.int32).min + 1
INT32_MAX = np.iinfo(np.int32).max - 1
SMOOTH_K_TOLERANCE = 1e-5
MIN_K_DIST_SCALE = 1e-3
NPY_INFINITY = np.inf
@numba.njit(fastmath=True)
def smooth_knn_dist(distances, k, n_iter=64, local_connectivity=1.0, bandwidth=1.0):
"""Compute a continuous version of the distance to the kth nearest
neighbor. That is, this is similar to knn-distance but allows continuous
k values rather than requiring an integral k. In esscence we are simply
computing the distance such that the cardinality of fuzzy set we generate
is k.
Parameters
----------
distances: array of shape (n_samples, n_neighbors)
Distances to nearest neighbors for each samples. Each row should be a
sorted list of distances to a given samples nearest neighbors.
k: float
The number of nearest neighbors to approximate for.
n_iter: int (optional, default 64)
We need to binary search for the correct distance value. This is the
max number of iterations to use in such a search.
local_connectivity: int (optional, default 1)
The local connectivity required -- i.e. the number of nearest
neighbors that should be assumed to be connected at a local level.
The higher this value the more connected the manifold becomes
locally. In practice this should be not more than the local intrinsic
dimension of the manifold.
bandwidth: float (optional, default 1)
The target bandwidth of the kernel, larger values will produce
larger return values.
Returns
-------
knn_dist: array of shape (n_samples,)
The distance to kth nearest neighbor, as suitably approximated.
nn_dist: array of shape (n_samples,)
The distance to the 1st nearest neighbor for each point.
"""
target = np.log2(k) * bandwidth
rho = np.zeros(distances.shape[0])
result = np.zeros(distances.shape[0])
for i in range(distances.shape[0]):
lo = 0.0
hi = NPY_INFINITY
mid = 1.0
# TODO: This is very inefficient, but will do for now. FIXME
ith_distances = distances[i]
non_zero_dists = ith_distances[ith_distances > 0.0]
if non_zero_dists.shape[0] >= local_connectivity:
index = int(np.floor(local_connectivity))
interpolation = local_connectivity - index
if index > 0:
rho[i] = non_zero_dists[index - 1]
if interpolation > SMOOTH_K_TOLERANCE:
rho[i] += interpolation * (non_zero_dists[index] - non_zero_dists[index - 1])
else:
rho[i] = interpolation * non_zero_dists[0]
elif non_zero_dists.shape[0] > 0:
rho[i] = np.max(non_zero_dists)
for n in range(n_iter):
psum = 0.0
for j in range(1, distances.shape[1]):
d = distances[i, j] - rho[i]
if d > 0:
psum += np.exp(-(d / mid))
else:
psum += 1.0
if np.fabs(psum - target) < SMOOTH_K_TOLERANCE:
break
if psum > target:
hi = mid
mid = (lo + hi) / 2.0
else:
lo = mid
if hi == NPY_INFINITY:
mid *= 2
else:
mid = (lo + hi) / 2.0
result[i] = mid
# TODO: This is very inefficient, but will do for now. FIXME
if rho[i] > 0.0:
if result[i] < MIN_K_DIST_SCALE * np.mean(ith_distances):
result[i] = MIN_K_DIST_SCALE * np.mean(ith_distances)
else:
if result[i] < MIN_K_DIST_SCALE * np.mean(distances):
result[i] = MIN_K_DIST_SCALE * np.mean(distances)
return result, rho
def nearest_neighbors(
X, n_neighbors, metric, metric_kwds, angular, random_state, verbose=False
):
"""Compute the ``n_neighbors`` nearest points for each data point in ``X``
under ``metric``. This may be exact, but more likely is approximated via
nearest neighbor descent.
Parameters
----------
X: array of shape (n_samples, n_features)
The input data to compute the k-neighbor graph of.
n_neighbors: int
The number of nearest neighbors to compute for each sample in ``X``.
metric: string or callable
The metric to use for the computation.
metric_kwds: dict
Any arguments to pass to the metric computation function.
angular: bool
Whether to use angular rp trees in NN approximation.
random_state: np.random state
The random state to use for approximate NN computations.
verbose: bool
Whether to print status data during the computation.
Returns
-------
knn_indices: array of shape (n_samples, n_neighbors)
The indices on the ``n_neighbors`` closest points in the dataset.
knn_dists: array of shape (n_samples, n_neighbors)
The distances to the ``n_neighbors`` closest points in the dataset.
"""
if metric == "precomputed":
# Note that this does not support sparse distance matrices yet ...
# Compute indices of n nearest neighbors
knn_indices = np.argsort(X)[:, :n_neighbors]
# Compute the nearest neighbor distances
# (equivalent to np.sort(X)[:,:n_neighbors])
knn_dists = X[np.arange(X.shape[0])[:, None], knn_indices].copy()
rp_forest = []
else:
if callable(metric):
distance_func = metric
elif metric in dist.named_distances:
distance_func = dist.named_distances[metric]
else:
raise ValueError("Metric is neither callable, " + "nor a recognised string")
if metric in ("cosine", "correlation", "dice", "jaccard"):
angular = True
rng_state = random_state.randint(INT32_MIN, INT32_MAX, 3).astype(np.int64)
if scipy.sparse.isspmatrix_csr(X):
if metric in sparse.sparse_named_distances:
distance_func = sparse.sparse_named_distances[metric]
if metric in sparse.sparse_need_n_features:
metric_kwds["n_features"] = X.shape[1]
else:
raise ValueError(
"Metric {} not supported for sparse " + "data".format(metric)
)
metric_nn_descent = sparse.make_sparse_nn_descent(
distance_func, tuple(metric_kwds.values())
)
# TODO: Hacked values for now
n_trees = 5 + int(round((X.shape[0]) ** 0.5 / 20.0))
n_iters = max(5, int(round(np.log2(X.shape[0]))))
rp_forest = make_forest(X, n_neighbors, n_trees, rng_state, angular)
leaf_array = rptree_leaf_array(rp_forest)
knn_indices, knn_dists = metric_nn_descent(
X.indices,
X.indptr,
X.data,
X.shape[0],
n_neighbors,
rng_state,
max_candidates=60,
rp_tree_init=True,
leaf_array=leaf_array,
n_iters=n_iters,
verbose=verbose,
)
else:
metric_nn_descent = make_nn_descent(
distance_func, tuple(metric_kwds.values())
)
# TODO: Hacked values for now
n_trees = 5 + int(round((X.shape[0]) ** 0.5 / 20.0))
n_iters = max(5, int(round(np.log2(X.shape[0]))))
rp_forest = make_forest(X, n_neighbors, n_trees, rng_state, angular)
leaf_array = rptree_leaf_array(rp_forest)
knn_indices, knn_dists = metric_nn_descent(
X,
n_neighbors,
rng_state,
max_candidates=60,
rp_tree_init=True,
leaf_array=leaf_array,
n_iters=n_iters,
verbose=verbose,
)
if np.any(knn_indices < 0):
warn(
"Failed to correctly find n_neighbors for some samples."
"Results may be less than ideal. Try re-running with"
"different parameters."
)
return knn_indices, knn_dists, rp_forest
@numba.njit(fastmath=True)
def compute_membership_strengths(knn_indices, knn_dists, sigmas, rhos):
"""Construct the membership strength data for the 1-skeleton of each local
fuzzy simplicial set -- this is formed as a sparse matrix where each row is
a local fuzzy simplicial set, with a membership strength for the
1-simplex to each other data point.
Parameters
----------
knn_indices: array of shape (n_samples, n_neighbors)
The indices on the ``n_neighbors`` closest points in the dataset.
knn_dists: array of shape (n_samples, n_neighbors)
The distances to the ``n_neighbors`` closest points in the dataset.
sigmas: array of shape(n_samples)
The normalization factor derived from the metric tensor approximation.
rhos: array of shape(n_samples)
The local connectivity adjustment.
Returns
-------
rows: array of shape (n_samples * n_neighbors)
Row data for the resulting sparse matrix (coo format)
cols: array of shape (n_samples * n_neighbors)
Column data for the resulting sparse matrix (coo format)
vals: array of shape (n_samples * n_neighbors)
Entries for the resulting sparse matrix (coo format)
"""
n_samples = knn_indices.shape[0]
n_neighbors = knn_indices.shape[1]
rows = np.zeros((n_samples * n_neighbors), dtype=np.int64)
cols = np.zeros((n_samples * n_neighbors), dtype=np.int64)
vals = np.zeros((n_samples * n_neighbors), dtype=np.float64)
for i in range(n_samples):
for j in range(n_neighbors):
if knn_indices[i, j] == -1:
continue # We didn't get the full knn for i
if knn_indices[i, j] == i:
val = 0.0
elif knn_dists[i, j] - rhos[i] <= 0.0:
val = 1.0
else:
val = np.exp(-((knn_dists[i, j] - rhos[i]) / (sigmas[i])))
rows[i * n_neighbors + j] = i
cols[i * n_neighbors + j] = knn_indices[i, j]
vals[i * n_neighbors + j] = val
return rows, cols, vals
@numba.jit()
def fuzzy_simplicial_set(
X,
n_neighbors,
random_state,
metric,
metric_kwds={},
knn_indices=None,
knn_dists=None,
angular=False,
set_op_mix_ratio=1.0,
local_connectivity=1.0,
verbose=False,
):
"""Given a set of data X, a neighborhood size, and a measure of distance
compute the fuzzy simplicial set (here represented as a fuzzy graph in
the form of a sparse matrix) associated to the data. This is done by
locally approximating geodesic distance at each point, creating a fuzzy
simplicial set for each such point, and then combining all the local
fuzzy simplicial sets into a global one via a fuzzy union.
Parameters
----------
X: array of shape (n_samples, n_features)
The data to be modelled as a fuzzy simplicial set.
n_neighbors: int
The number of neighbors to use to approximate geodesic distance.
Larger numbers induce more global estimates of the manifold that can
miss finer detail, while smaller values will focus on fine manifold
structure to the detriment of the larger picture.
random_state: numpy RandomState or equivalent
A state capable being used as a numpy random state.
metric: string or function (optional, default 'euclidean')
The metric to use to compute distances in high dimensional space.
If a string is passed it must match a valid predefined metric. If
a general metric is required a function that takes two 1d arrays and
returns a float can be provided. For performance purposes it is
required that this be a numba jit'd function. Valid string metrics
include:
* euclidean (or l2)
* manhattan (or l1)
* cityblock
* braycurtis
* canberra
* chebyshev
* correlation
* cosine
* dice
* hamming
* jaccard
* kulsinski
* mahalanobis
* matching
* minkowski
* rogerstanimoto
* russellrao
* seuclidean
* sokalmichener
* sokalsneath
* sqeuclidean
* yule
* wminkowski
Metrics that take arguments (such as minkowski, mahalanobis etc.)
can have arguments passed via the metric_kwds dictionary. At this
time care must be taken and dictionary elements must be ordered
appropriately; this will hopefully be fixed in the future.
metric_kwds: dict (optional, default {})
Arguments to pass on to the metric, such as the ``p`` value for
Minkowski distance.
knn_indices: array of shape (n_samples, n_neighbors) (optional)
If the k-nearest neighbors of each point has already been calculated
you can pass them in here to save computation time. This should be
an array with the indices of the k-nearest neighbors as a row for
each data point.
knn_dists: array of shape (n_samples, n_neighbors) (optional)
If the k-nearest neighbors of each point has already been calculated
you can pass them in here to save computation time. This should be
an array with the distances of the k-nearest neighbors as a row for
each data point.
angular: bool (optional, default False)
Whether to use angular/cosine distance for the random projection
forest for seeding NN-descent to determine approximate nearest
neighbors.
set_op_mix_ratio: float (optional, default 1.0)
Interpolate between (fuzzy) union and intersection as the set operation
used to combine local fuzzy simplicial sets to obtain a global fuzzy
simplicial sets. Both fuzzy set operations use the product t-norm.
The value of this parameter should be between 0.0 and 1.0; a value of
1.0 will use a pure fuzzy union, while 0.0 will use a pure fuzzy
intersection.
local_connectivity: int (optional, default 1)
The local connectivity required -- i.e. the number of nearest
neighbors that should be assumed to be connected at a local level.
The higher this value the more connected the manifold becomes
locally. In practice this should be not more than the local intrinsic
dimension of the manifold.
verbose: bool (optional, default False)
Whether to report information on the current progress of the algorithm.
Returns
-------
fuzzy_simplicial_set: coo_matrix
A fuzzy simplicial set represented as a sparse matrix. The (i,
j) entry of the matrix represents the membership strength of the
1-simplex between the ith and jth sample points.
"""
if knn_indices is None or knn_dists is None:
knn_indices, knn_dists, _ = nearest_neighbors(
X, n_neighbors, metric, metric_kwds, angular, random_state, verbose=verbose
)
sigmas, rhos = smooth_knn_dist(
knn_dists, n_neighbors, local_connectivity=local_connectivity
)
rows, cols, vals = compute_membership_strengths(
knn_indices, knn_dists, sigmas, rhos
)
result = scipy.sparse.coo_matrix(
(vals, (rows, cols)), shape=(X.shape[0], X.shape[0])
)
result.eliminate_zeros()
transpose = result.transpose()
prod_matrix = result.multiply(transpose)
result = (
set_op_mix_ratio * (result + transpose - prod_matrix)
+ (1.0 - set_op_mix_ratio) * prod_matrix
)
result.eliminate_zeros()
return result
@numba.jit()
def fast_intersection(rows, cols, values, target, unknown_dist=1.0, far_dist=5.0):
"""Under the assumption of categorical distance for the intersecting
simplicial set perform a fast intersection.
Parameters
----------
rows: array
An array of the row of each non-zero in the sparse matrix
representation.
cols: array
An array of the column of each non-zero in the sparse matrix
representation.
values: array
An array of the value of each non-zero in the sparse matrix
representation.
target: array of shape (n_samples)
The categorical labels to use in the intersection.
unknown_dist: float (optional, default 1.0)
The distance an unknown label (-1) is assumed to be from any point.
far_dist float (optional, default 5.0)
The distance between unmatched labels.
Returns
-------
None
"""
for nz in range(rows.shape[0]):
i = rows[nz]
j = cols[nz]
if target[i] == -1 or target[j] == -1:
values[nz] *= np.exp(-unknown_dist)
elif target[i] != target[j]:
values[nz] *= np.exp(-far_dist)
return
@numba.jit()
def reset_local_connectivity(simplicial_set):
"""Reset the local connectivity requirement -- each data sample should
have complete confidence in at least one 1-simplex in the simplicial set.
We can enforce this by locally rescaling confidences, and then remerging the
different local simplicial sets together.
Parameters
----------
simplicial_set: sparse matrix
The simplicial set for which to recalculate with respect to local
connectivity.
Returns
-------
simplicial_set: sparse_matrix
The recalculated simplicial set, now with the local connectivity
assumption restored.
"""
simplicial_set = normalize(simplicial_set, norm="max")
transpose = simplicial_set.transpose()
prod_matrix = simplicial_set.multiply(transpose)
simplicial_set = simplicial_set + transpose - prod_matrix
simplicial_set.eliminate_zeros()
return simplicial_set
@numba.jit()
def categorical_simplicial_set_intersection(
simplicial_set, target, unknown_dist=1.0, far_dist=5.0
):
"""Combine a fuzzy simplicial set with another fuzzy simplicial set
generated from categorical data using categorical distances. The target
data is assumed to be categorical label data (a vector of labels),
and this will update the fuzzy simplicial set to respect that label data.
TODO: optional category cardinality based weighting of distance
Parameters
----------
simplicial_set: sparse matrix
The input fuzzy simplicial set.
target: array of shape (n_samples)
The categorical labels to use in the intersection.
unknown_dist: float (optional, default 1.0)
The distance an unknown label (-1) is assumed to be from any point.
far_dist float (optional, default 5.0)
The distance between unmatched labels.
Returns
-------
simplicial_set: sparse matrix
The resulting intersected fuzzy simplicial set.
"""
simplicial_set = simplicial_set.tocoo()
fast_intersection(
simplicial_set.row,
simplicial_set.col,
simplicial_set.data,
target,
unknown_dist,
far_dist,
)
simplicial_set.eliminate_zeros()
return reset_local_connectivity(simplicial_set)
@numba.jit()
def general_simplicial_set_intersection(simplicial_set1, simplicial_set2, weight):
result = (simplicial_set1 + simplicial_set2).tocoo()
left = simplicial_set1.tocsr()
right = simplicial_set2.tocsr()
sparse.general_sset_intersection(
left.indptr,
left.indices,
left.data,
right.indptr,
right.indices,
right.data,
result.row,
result.col,
result.data,
weight,
)
return result
@numba.jit()
def make_epochs_per_sample(weights, n_epochs):
"""Given a set of weights and number of epochs generate the number of
epochs per sample for each weight.
Parameters
----------
weights: array of shape (n_1_simplices)
The weights ofhow much we wish to sample each 1-simplex.
n_epochs: int
The total number of epochs we want to train for.
Returns
-------
An array of number of epochs per sample, one for each 1-simplex.
"""
result = -1.0 * np.ones(weights.shape[0], dtype=np.float64)
n_samples = n_epochs * (weights / weights.max())
result[n_samples > 0] = float(n_epochs) / n_samples[n_samples > 0]
return result
@numba.njit()
def clip(val):
"""Standard clamping of a value into a fixed range (in this case -4.0 to
4.0)
Parameters
----------
val: float
The value to be clamped.
Returns
-------
The clamped value, now fixed to be in the range -4.0 to 4.0.
"""
if val > 4.0:
return 4.0
elif val < -4.0:
return -4.0
else:
return val
@numba.njit("f4(f4[:],f4[:])", fastmath=True)
def rdist(x, y):
"""Reduced Euclidean distance.
Parameters
----------
x: array of shape (embedding_dim,)
y: array of shape (embedding_dim,)
Returns
-------
The squared euclidean distance between x and y
"""
result = 0.0
for i in range(x.shape[0]):
result += (x[i] - y[i]) ** 2
return result
@numba.njit(fastmath=True)
def optimize_layout(
head_embedding,
tail_embedding,
head,
tail,
n_epochs,
n_vertices,
epochs_per_sample,
a,
b,
rng_state,
gamma=1.0,
initial_alpha=1.0,
negative_sample_rate=5.0,
verbose=False,
):
"""Improve an embedding using stochastic gradient descent to minimize the
fuzzy set cross entropy between the 1-skeletons of the high dimensional
and low dimensional fuzzy simplicial sets. In practice this is done by
sampling edges based on their membership strength (with the (1-p) terms
coming from negative sampling similar to word2vec).
Parameters
----------
head_embedding: array of shape (n_samples, n_components)
The initial embedding to be improved by SGD.
tail_embedding: array of shape (source_samples, n_components)
The reference embedding of embedded points. If not embedding new
previously unseen points with respect to an existing embedding this
is simply the head_embedding (again); otherwise it provides the
existing embedding to embed with respect to.
head: array of shape (n_1_simplices)
The indices of the heads of 1-simplices with non-zero membership.
tail: array of shape (n_1_simplices)
The indices of the tails of 1-simplices with non-zero membership.
n_epochs: int
The number of training epochs to use in optimization.
n_vertices: int
The number of vertices (0-simplices) in the dataset.
epochs_per_samples: array of shape (n_1_simplices)
A float value of the number of epochs per 1-simplex. 1-simplices with
weaker membership strength will have more epochs between being sampled.
a: float
Parameter of differentiable approximation of right adjoint functor
b: float
Parameter of differentiable approximation of right adjoint functor
rng_state: array of int64, shape (3,)
The internal state of the rng
gamma: float (optional, default 1.0)
Weight to apply to negative samples.
initial_alpha: float (optional, default 1.0)
Initial learning rate for the SGD.
negative_sample_rate: int (optional, default 5)
Number of negative samples to use per positive sample.
verbose: bool (optional, default False)
Whether to report information on the current progress of the algorithm.
Returns
-------
embedding: array of shape (n_samples, n_components)
The optimized embedding.
"""
dim = head_embedding.shape[1]
move_other = head_embedding.shape[0] == tail_embedding.shape[0]
alpha = initial_alpha
epochs_per_negative_sample = epochs_per_sample / negative_sample_rate
epoch_of_next_negative_sample = epochs_per_negative_sample.copy()
epoch_of_next_sample = epochs_per_sample.copy()
for n in range(n_epochs):
for i in range(epochs_per_sample.shape[0]):
if epoch_of_next_sample[i] <= n:
j = head[i]
k = tail[i]
current = head_embedding[j]
other = tail_embedding[k]
dist_squared = rdist(current, other)
if dist_squared > 0.0:
grad_coeff = -2.0 * a * b * pow(dist_squared, b - 1.0)
grad_coeff /= a * pow(dist_squared, b) + 1.0
else:
grad_coeff = 0.0
for d in range(dim):
grad_d = clip(grad_coeff * (current[d] - other[d]))
current[d] += grad_d * alpha
if move_other:
other[d] += -grad_d * alpha
epoch_of_next_sample[i] += epochs_per_sample[i]
n_neg_samples = int(
(n - epoch_of_next_negative_sample[i])
/ epochs_per_negative_sample[i]
)
for p in range(n_neg_samples):
k = tau_rand_int(rng_state) % n_vertices
other = tail_embedding[k]
dist_squared = rdist(current, other)
if dist_squared > 0.0:
grad_coeff = 2.0 * gamma * b
grad_coeff /= (0.001 + dist_squared) * (
a * pow(dist_squared, b) + 1
)
else:
grad_coeff = 0.0
for d in range(dim):
if grad_coeff > 0.0:
grad_d = clip(grad_coeff * (current[d] - other[d]))
else:
grad_d = 4.0
current[d] += grad_d * alpha
epoch_of_next_negative_sample[i] += (
n_neg_samples * epochs_per_negative_sample[i]
)
alpha = initial_alpha * (1.0 - (float(n) / float(n_epochs)))
if verbose and n % int(n_epochs / 10) == 0:
print("\tcompleted ", n, " / ", n_epochs, "epochs")
return head_embedding
def simplicial_set_embedding(
data,
graph,
n_components,
initial_alpha,
a,
b,
gamma,
negative_sample_rate,
n_epochs,
init,
random_state,
metric,
metric_kwds,
verbose,
):
"""Perform a fuzzy simplicial set embedding, using a specified
initialisation method and then minimizing the fuzzy set cross entropy
between the 1-skeletons of the high and low dimensional fuzzy simplicial
sets.
Parameters
----------
data: array of shape (n_samples, n_features)
The source data to be embedded by UMAP.
graph: sparse matrix
The 1-skeleton of the high dimensional fuzzy simplicial set as
represented by a graph for which we require a sparse matrix for the
(weighted) adjacency matrix.
n_components: int
The dimensionality of the euclidean space into which to embed the data.
initial_alpha: float
Initial learning rate for the SGD.
a: float
Parameter of differentiable approximation of right adjoint functor
b: float
Parameter of differentiable approximation of right adjoint functor
gamma: float
Weight to apply to negative samples.
negative_sample_rate: int (optional, default 5)
The number of negative samples to select per positive sample
in the optimization process. Increasing this value will result
in greater repulsive force being applied, greater optimization
cost, but slightly more accuracy.
n_epochs: int (optional, default 0)
The number of training epochs to be used in optimizing the
low dimensional embedding. Larger values result in more accurate
embeddings. If 0 is specified a value will be selected based on
the size of the input dataset (200 for large datasets, 500 for small).
init: string
How to initialize the low dimensional embedding. Options are:
* 'spectral': use a spectral embedding of the fuzzy 1-skeleton
* 'random': assign initial embedding positions at random.
* A numpy array of initial embedding positions.
random_state: numpy RandomState or equivalent
A state capable being used as a numpy random state.
metric: string
The metric used to measure distance in high dimensional space; used if
multiple connected components need to be layed out.
metric_kwds: dict
Key word arguments to be passed to the metric function; used if
multiple connected components need to be layed out.
verbose: bool (optional, default False)
Whether to report information on the current progress of the algorithm.
Returns
-------
embedding: array of shape (n_samples, n_components)
The optimized of ``graph`` into an ``n_components`` dimensional
euclidean space.
"""
graph = graph.tocoo()
graph.sum_duplicates()
n_vertices = graph.shape[1]
if n_epochs <= 0:
# For smaller datasets we can use more epochs
if graph.shape[0] <= 10000:
n_epochs = 500
else:
n_epochs = 200
graph.data[graph.data < (graph.data.max() / float(n_epochs))] = 0.0
graph.eliminate_zeros()
if isinstance(init, str) and init == "random":
embedding = random_state.uniform(
low=-10.0, high=10.0, size=(graph.shape[0], n_components)
).astype(np.float32)
elif isinstance(init, str) and init == "spectral":
# We add a little noise to avoid local minima for optimization to come
initialisation = spectral_layout(
data,
graph,
n_components,
random_state,
metric=metric,
metric_kwds=metric_kwds,
)
expansion = 10.0 / initialisation.max()
embedding = (initialisation * expansion).astype(
np.float32
) + random_state.normal(
scale=0.0001, size=[graph.shape[0], n_components]
).astype(
np.float32
)
else:
init_data = np.array(init)
if len(init_data.shape) == 2:
if np.unique(init_data, axis=0).shape[0] < init_data.shape[0]:
tree = KDTree(init_data)
dist, ind = tree.query(init_data, k=2)
nndist = np.mean(dist[:, 1])
embedding = init_data + np.random.normal(
scale=0.001 * nndist, size=init_data.shape
).astype(np.float32)
else:
embedding = init_data
epochs_per_sample = make_epochs_per_sample(graph.data, n_epochs)
head = graph.row
tail = graph.col
rng_state = random_state.randint(INT32_MIN, INT32_MAX, 3).astype(np.int64)
embedding = optimize_layout(
embedding,
embedding,
head,
tail,
n_epochs,
n_vertices,
epochs_per_sample,
a,
b,
rng_state,
gamma,
initial_alpha,
negative_sample_rate,
verbose=verbose,
)
return embedding
@numba.njit()
def init_transform(indices, weights, embedding):
"""Given indices and weights and an original embeddings
initialize the positions of new points relative to the
indices and weights (of their neighbors in the source data).
Parameters
----------
indices: array of shape (n_new_samples, n_neighbors)
The indices of the neighbors of each new sample
weights: array of shape (n_new_samples, n_neighbors)
The membership strengths of associated 1-simplices
for each of the new samples.
embedding: array of shape (n_samples, dim)
The original embedding of the source data.
Returns
-------
new_embedding: array of shape (n_new_samples, dim)
An initial embedding of the new sample points.
"""
result = np.zeros((indices.shape[0], embedding.shape[1]), dtype=np.float32)
for i in range(indices.shape[0]):
for j in range(indices.shape[1]):
for d in range(embedding.shape[1]):
result[i, d] += weights[i, j] * embedding[indices[i, j], d]
return result
def find_ab_params(spread, min_dist):
"""Fit a, b params for the differentiable curve used in lower
dimensional fuzzy simplicial complex construction. We want the
smooth curve (from a pre-defined family with simple gradient) that
best matches an offset exponential decay.
"""
def curve(x, a, b):
return 1.0 / (1.0 + a * x ** (2 * b))
xv = np.linspace(0, spread * 3, 300)
yv = np.zeros(xv.shape)
yv[xv < min_dist] = 1.0
yv[xv >= min_dist] = np.exp(-(xv[xv >= min_dist] - min_dist) / spread)
params, covar = curve_fit(curve, xv, yv)
return params[0], params[1]
class UMAP(BaseEstimator):
"""Uniform Manifold Approximation and Projection
Finds a low dimensional embedding of the data that approximates
an underlying manifold.
Parameters
----------
n_neighbors: float (optional, default 15)
The size of local neighborhood (in terms of number of neighboring
sample points) used for manifold approximation. Larger values
result in more global views of the manifold, while smaller
values result in more local data being preserved. In general
values should be in the range 2 to 100.
n_components: int (optional, default 2)
The dimension of the space to embed into. This defaults to 2 to
provide easy visualization, but can reasonably be set to any
integer value in the range 2 to 100.
metric: string or function (optional, default 'euclidean')
The metric to use to compute distances in high dimensional space.
If a string is passed it must match a valid predefined metric. If
a general metric is required a function that takes two 1d arrays and
returns a float can be provided. For performance purposes it is
required that this be a numba jit'd function. Valid string metrics
include:
* euclidean
* manhattan
* chebyshev
* minkowski
* canberra
* braycurtis
* mahalanobis
* wminkowski
* seuclidean
* cosine
* correlation
* haversine
* hamming
* jaccard
* dice
* russelrao
* kulsinski
* rogerstanimoto
* sokalmichener
* sokalsneath
* yule
Metrics that take arguments (such as minkowski, mahalanobis etc.)
can have arguments passed via the metric_kwds dictionary. At this
time care must be taken and dictionary elements must be ordered
appropriately; this will hopefully be fixed in the future.
n_epochs: int (optional, default None)
The number of training epochs to be used in optimizing the
low dimensional embedding. Larger values result in more accurate
embeddings. If None is specified a value will be selected based on
the size of the input dataset (200 for large datasets, 500 for small).
learning_rate: float (optional, default 1.0)
The initial learning rate for the embedding optimization.
init: string (optional, default 'spectral')
How to initialize the low dimensional embedding. Options are:
* 'spectral': use a spectral embedding of the fuzzy 1-skeleton
* 'random': assign initial embedding positions at random.
* A numpy array of initial embedding positions.
min_dist: float (optional, default 0.1)
The effective minimum distance between embedded points. Smaller values
will result in a more clustered/clumped embedding where nearby points
on the manifold are drawn closer together, while larger values will
result on a more even dispersal of points. The value should be set
relative to the ``spread`` value, which determines the scale at which
embedded points will be spread out.
spread: float (optional, default 1.0)
The effective scale of embedded points. In combination with ``min_dist``
this determines how clustered/clumped the embedded points are.
set_op_mix_ratio: float (optional, default 1.0)
Interpolate between (fuzzy) union and intersection as the set operation
used to combine local fuzzy simplicial sets to obtain a global fuzzy
simplicial sets. Both fuzzy set operations use the product t-norm.
The value of this parameter should be between 0.0 and 1.0; a value of
1.0 will use a pure fuzzy union, while 0.0 will use a pure fuzzy
intersection.
local_connectivity: int (optional, default 1)
The local connectivity required -- i.e. the number of nearest
neighbors that should be assumed to be connected at a local level.
The higher this value the more connected the manifold becomes
locally. In practice this should be not more than the local intrinsic
dimension of the manifold.
repulsion_strength: float (optional, default 1.0)
Weighting applied to negative samples in low dimensional embedding
optimization. Values higher than one will result in greater weight
being given to negative samples.
negative_sample_rate: int (optional, default 5)
The number of negative samples to select per positive sample
in the optimization process. Increasing this value will result
in greater repulsive force being applied, greater optimization
cost, but slightly more accuracy.
transform_queue_size: float (optional, default 4.0)
For transform operations (embedding new points using a trained model_
this will control how aggressively to search for nearest neighbors.
Larger values will result in slower performance but more accurate
nearest neighbor evaluation.
a: float (optional, default None)
More specific parameters controlling the embedding. If None these
values are set automatically as determined by ``min_dist`` and
``spread``.
b: float (optional, default None)
More specific parameters controlling the embedding. If None these
values are set automatically as determined by ``min_dist`` and
``spread``.
random_state: int, RandomState instance or None, optional (default: None)
If int, random_state is the seed used by the random number generator;
If RandomState instance, random_state is the random number generator;
If None, the random number generator is the RandomState instance used
by `np.random`.
metric_kwds: dict (optional, default None)
Arguments to pass on to the metric, such as the ``p`` value for
Minkowski distance. If None then no arguments are passed on.
angular_rp_forest: bool (optional, default False)
Whether to use an angular random projection forest to initialise
the approximate nearest neighbor search. This can be faster, but is
mostly on useful for metric that use an angular style distance such
as cosine, correlation etc. In the case of those metrics angular forests
will be chosen automatically.
target_n_neighbors: int (optional, default -1)
The number of nearest neighbors to use to construct the target simplcial
set. If set to -1 use the ``n_neighbors`` value.
target_metric: string or callable (optional, default 'categorical')
The metric used to measure distance for a target array is using supervised
dimension reduction. By default this is 'categorical' which will measure
distance in terms of whether categories match or are different. Furthermore,
if semi-supervised is required target values of -1 will be trated as
unlabelled under the 'categorical' metric. If the target array takes
continuous values (e.g. for a regression problem) then metric of 'l1'
or 'l2' is probably more appropriate.
target_metric_kwds: dict (optional, default None)
Keyword argument to pass to the target metric when performing
supervised dimension reduction. If None then no arguments are passed on.
target_weight: float (optional, default 0.5)
weighting factor between data topology and target topology. A value of
0.0 weights entirely on data, a value of 1.0 weights entirely on target.
The default of 0.5 balances the weighting equally between data and target.
transform_seed: int (optional, default 42)
Random seed used for the stochastic aspects of the transform operation.
This ensures consistency in transform operations.
verbose: bool (optional, default False)
Controls verbosity of logging.
"""
def __init__(
self,
n_neighbors=15,
n_components=2,
metric="euclidean",
n_epochs=None,
learning_rate=1.0,
init="spectral",
min_dist=0.1,
spread=1.0,
set_op_mix_ratio=1.0,
local_connectivity=1.0,
repulsion_strength=1.0,
negative_sample_rate=5,
transform_queue_size=4.0,
a=None,
b=None,
random_state=None,
metric_kwds=None,
angular_rp_forest=False,
target_n_neighbors=-1,
target_metric="categorical",
target_metric_kwds=None,
target_weight=0.5,
transform_seed=42,
verbose=False,
):
self.n_neighbors = n_neighbors
self.metric = metric
self.metric_kwds = metric_kwds
self.n_epochs = n_epochs
self.init = init
self.n_components = n_components
self.repulsion_strength = repulsion_strength
self.learning_rate = learning_rate
self.spread = spread
self.min_dist = min_dist
self.set_op_mix_ratio = set_op_mix_ratio
self.local_connectivity = local_connectivity
self.negative_sample_rate = negative_sample_rate
self.random_state = random_state
self.angular_rp_forest = angular_rp_forest
self.transform_queue_size = transform_queue_size
self.target_n_neighbors = target_n_neighbors
self.target_metric = target_metric
self.target_metric_kwds = target_metric_kwds
self.target_weight = target_weight
self.transform_seed = transform_seed
self.verbose = verbose
self.a = a
self.b = b
def _validate_parameters(self):
if self.set_op_mix_ratio < 0.0 or self.set_op_mix_ratio > 1.0:
raise ValueError("set_op_mix_ratio must be between 0.0 and 1.0")
if self.repulsion_strength < 0.0:
raise ValueError("repulsion_strength cannot be negative")
if self.min_dist > self.spread:
raise ValueError("min_dist must be less than or equal to spread")
if self.min_dist < 0.0:
raise ValueError("min_dist must be greater than 0.0")
if not isinstance(self.init, str) and not isinstance(self.init, np.ndarray):
raise ValueError("init must be a string or ndarray")
if isinstance(self.init, str) and self.init not in ("spectral", "random"):
raise ValueError('string init values must be "spectral" or "random"')
if (
isinstance(self.init, np.ndarray)
and self.init.shape[1] != self.n_components
):
raise ValueError("init ndarray must match n_components value")
if not isinstance(self.metric, str) and not callable(self.metric):
raise ValueError("metric must be string or callable")
if self.negative_sample_rate < 0:
raise ValueError("negative sample rate must be positive")
if self._initial_alpha < 0.0:
raise ValueError("learning_rate must be positive")
if self.n_neighbors < 2:
raise ValueError("n_neighbors must be greater than 2")
if self.target_n_neighbors < 2 and self.target_n_neighbors != -1:
raise ValueError("target_n_neighbors must be greater than 2")
if not isinstance(self.n_components, int):
raise ValueError("n_components must be an int")
if self.n_components < 1:
raise ValueError("n_components must be greater than 0")
if self.n_epochs is not None and (
self.n_epochs <= 10 or not isinstance(self.n_epochs, int)
):
raise ValueError("n_epochs must be a positive integer "
"larger than 10")
def fit(self, X, y=None):
"""Fit X into an embedded space.
Optionally use y for supervised dimension reduction.
Parameters
----------
X : array, shape (n_samples, n_features) or (n_samples, n_samples)
If the metric is 'precomputed' X must be a square distance
matrix. Otherwise it contains a sample per row. If the method
is 'exact', X may be a sparse matrix of type 'csr', 'csc'
or 'coo'.
y : array, shape (n_samples)
A target array for supervised dimension reduction. How this is
handled is determined by parameters UMAP was instantiated with.
The relevant attributes are ``target_metric`` and
``target_metric_kwds``.
"""
X = check_array(X, dtype=np.float32, accept_sparse="csr")
self._raw_data = X
# Handle all the optional arguments, setting default
if self.a is None or self.b is None:
self._a, self._b = find_ab_params(self.spread, self.min_dist)
else:
self._a = self.a
self._b = self.b
if self.metric_kwds is not None:
self._metric_kwds = self.metric_kwds
else:
self._metric_kwds = {}
if self.target_metric_kwds is not None:
self._target_metric_kwds = self.target_metric_kwds
else:
self._target_metric_kwds = {}
if isinstance(self.init, np.ndarray):
init = check_array(self.init, dtype=np.float32, accept_sparse=False)
else:
init = self.init
self._initial_alpha = self.learning_rate
self._validate_parameters()
if self.verbose:
print(str(self))
# Error check n_neighbors based on data size
if X.shape[0] <= self.n_neighbors:
if X.shape[0] == 1:
self.embedding_ = np.zeros((1, self.n_components)) # needed to sklearn comparability
return self
warn(
"n_neighbors is larger than the dataset size; truncating to "
"X.shape[0] - 1"
)
self._n_neighbors = X.shape[0] - 1
else:
self._n_neighbors = self.n_neighbors
if scipy.sparse.isspmatrix_csr(X):
if not X.has_sorted_indices:
X.sort_indices()
self._sparse_data = True
else:
self._sparse_data = False
random_state = check_random_state(self.random_state)
if self.verbose:
print("Construct fuzzy simplicial set")
# Handle small cases efficiently by computing all distances
if X.shape[0] < 4096:
self._small_data = True
dmat = pairwise_distances(X, metric=self.metric, **self._metric_kwds)
self.graph_ = fuzzy_simplicial_set(
dmat,
self._n_neighbors,
random_state,
"precomputed",
self._metric_kwds,
None,
None,
self.angular_rp_forest,
self.set_op_mix_ratio,
self.local_connectivity,
self.verbose,
)
else:
self._small_data = False
# Standard case
(self._knn_indices, self._knn_dists, self._rp_forest) = nearest_neighbors(
X,
self._n_neighbors,
self.metric,
self._metric_kwds,
self.angular_rp_forest,
random_state,
self.verbose,
)
self.graph_ = fuzzy_simplicial_set(
X,
self.n_neighbors,
random_state,
self.metric,
self._metric_kwds,
self._knn_indices,
self._knn_dists,
self.angular_rp_forest,
self.set_op_mix_ratio,
self.local_connectivity,
self.verbose,
)
self._search_graph = scipy.sparse.lil_matrix(
(X.shape[0], X.shape[0]), dtype=np.int8
)
self._search_graph.rows = self._knn_indices
self._search_graph.data = (self._knn_dists != 0).astype(np.int8)
self._search_graph = self._search_graph.maximum(
self._search_graph.transpose()
).tocsr()
if callable(self.metric):
self._distance_func = self.metric
elif self.metric in dist.named_distances:
self._distance_func = dist.named_distances[self.metric]
elif self.metric == 'precomputed':
warn('Using precomputed metric; transform will be unavailable for new data')
else:
raise ValueError(
"Metric is neither callable, " + "nor a recognised string"
)
if self.metric != 'precomputed':
self._dist_args = tuple(self._metric_kwds.values())
self._random_init, self._tree_init = make_initialisations(
self._distance_func, self._dist_args
)
self._search = make_initialized_nnd_search(
self._distance_func, self._dist_args
)
if y is not None:
y_ = check_array(y, ensure_2d=False)
if self.target_metric == "categorical":
if self.target_weight < 1.0:
far_dist = 2.5 * (1.0 / (1.0 - self.target_weight))
else:
far_dist = 1.0e12
self.graph_ = categorical_simplicial_set_intersection(
self.graph_, y_, far_dist=far_dist
)
else:
if self.target_n_neighbors == -1:
target_n_neighbors = self._n_neighbors
else:
target_n_neighbors = self.target_n_neighbors
# Handle the small case as precomputed as before
if y.shape[0] < 4096:
ydmat = pairwise_distances(y_[np.newaxis, :].T,
metric=self.target_metric,
**self._target_metric_kwds)
target_graph = fuzzy_simplicial_set(
ydmat,
target_n_neighbors,
random_state,
"precomputed",
self._target_metric_kwds,
None,
None,
False,
1.0,
1.0,
False
)
else:
# Standard case
target_graph = fuzzy_simplicial_set(
y_[np.newaxis, :].T,
target_n_neighbors,
random_state,
self.target_metric,
self._target_metric_kwds,
None,
None,
False,
1.0,
1.0,
False,
)
# product = self.graph_.multiply(target_graph)
# # self.graph_ = 0.99 * product + 0.01 * (self.graph_ +
# # target_graph -
# # product)
# self.graph_ = product
self.graph_ = general_simplicial_set_intersection(
self.graph_, target_graph, self.target_weight
)
self.graph_ = reset_local_connectivity(self.graph_)
if self.n_epochs is None:
n_epochs = 0
else:
n_epochs = self.n_epochs
if self.verbose:
print("Construct embedding")
self.embedding_ = simplicial_set_embedding(
self._raw_data,
self.graph_,
self.n_components,
self._initial_alpha,
self._a,
self._b,
self.repulsion_strength,
self.negative_sample_rate,
n_epochs,
init,
random_state,
self.metric,
self._metric_kwds,
self.verbose,
)
self._input_hash = joblib.hash(self._raw_data)
return self
def fit_transform(self, X, y=None):
"""Fit X into an embedded space and return that transformed
output.
Parameters
----------
X : array, shape (n_samples, n_features) or (n_samples, n_samples)
If the metric is 'precomputed' X must be a square distance
matrix. Otherwise it contains a sample per row.
y : array, shape (n_samples)
A target array for supervised dimension reduction. How this is
handled is determined by parameters UMAP was instantiated with.
The relevant attributes are ``target_metric`` and
``target_metric_kwds``.
Returns
-------
X_new : array, shape (n_samples, n_components)
Embedding of the training data in low-dimensional space.
"""
self.fit(X, y)
return self.embedding_
def transform(self, X):
"""Transform X into the existing embedded space and return that
transformed output.
Parameters
----------
X : array, shape (n_samples, n_features)
New data to be transformed.
Returns
-------
X_new : array, shape (n_samples, n_components)
Embedding of the new data in low-dimensional space.
"""
# If we fit just a single instance then error
if self.embedding_.shape[0] == 1:
raise ValueError('Transform unavailable when model was fit with'
'only a single data sample.')
# If we just have the original input then short circuit things
X = check_array(X, dtype=np.float32, accept_sparse="csr")
x_hash = joblib.hash(X)
if x_hash == self._input_hash:
return self.embedding_
if self._sparse_data:
raise ValueError("Transform not available for sparse input.")
elif self.metric == 'precomputed':
raise ValueError("Transform of new data not available for "
"precomputed metric.")
X = check_array(X, dtype=np.float32, order="C")
random_state = check_random_state(self.transform_seed)
rng_state = random_state.randint(INT32_MIN, INT32_MAX, 3).astype(np.int64)
if self._small_data:
dmat = pairwise_distances(
X, self._raw_data, metric=self.metric, **self._metric_kwds
)
indices = np.argpartition(dmat,
self._n_neighbors)[:, :self._n_neighbors]
dmat_shortened = submatrix(dmat, indices, self._n_neighbors)
indices_sorted = np.argsort(dmat_shortened)
indices = submatrix(indices, indices_sorted, self._n_neighbors)
dists = submatrix(dmat_shortened, indices_sorted,
self._n_neighbors)
else:
init = initialise_search(
self._rp_forest,
self._raw_data,
X,
int(self._n_neighbors * self.transform_queue_size),
self._random_init,
self._tree_init,
rng_state,
)
result = self._search(
self._raw_data,
self._search_graph.indptr,
self._search_graph.indices,
init,
X,
)
indices, dists = deheap_sort(result)
indices = indices[:, : self._n_neighbors]
dists = dists[:, : self._n_neighbors]
adjusted_local_connectivity = max(0, self.local_connectivity - 1.0)
sigmas, rhos = smooth_knn_dist(
dists, self._n_neighbors, local_connectivity=adjusted_local_connectivity
)
rows, cols, vals = compute_membership_strengths(indices, dists, sigmas, rhos)
graph = scipy.sparse.coo_matrix(
(vals, (rows, cols)), shape=(X.shape[0], self._raw_data.shape[0])
)
# This was a very specially constructed graph with constant degree.
# That lets us do fancy unpacking by reshaping the csr matrix indices
# and data. Doing so relies on the constant degree assumption!
csr_graph = normalize(graph.tocsr(), norm="l1")
inds = csr_graph.indices.reshape(X.shape[0], self._n_neighbors)
weights = csr_graph.data.reshape(X.shape[0], self._n_neighbors)
embedding = init_transform(inds, weights, self.embedding_)
if self.n_epochs is None:
# For smaller datasets we can use more epochs
if graph.shape[0] <= 10000:
n_epochs = 100
else:
n_epochs = 30
else:
n_epochs = self.n_epochs // 3.0
graph.data[graph.data < (graph.data.max() / float(n_epochs))] = 0.0
graph.eliminate_zeros()
epochs_per_sample = make_epochs_per_sample(graph.data, n_epochs)
head = graph.row
tail = graph.col
embedding = optimize_layout(
embedding,
self.embedding_,
head,
tail,
n_epochs,
graph.shape[1],
epochs_per_sample,
self._a,
self._b,
rng_state,
self.repulsion_strength,
self._initial_alpha,
self.negative_sample_rate,
verbose=self.verbose,
)
return embedding
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn_single/umap.py
|
umap.py
|
import numpy as np
import numba
import os
@numba.njit("i4(i8[:])")
def tau_rand_int(state):
"""A fast (pseudo)-random number generator.
Parameters
----------
state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
A (pseudo)-random int32 value
"""
state[0] = (((state[0] & 4294967294) << 12) & 0xffffffff) ^ (
(((state[0] << 13) & 0xffffffff) ^ state[0]) >> 19
)
state[1] = (((state[1] & 4294967288) << 4) & 0xffffffff) ^ (
(((state[1] << 2) & 0xffffffff) ^ state[1]) >> 25
)
state[2] = (((state[2] & 4294967280) << 17) & 0xffffffff) ^ (
(((state[2] << 3) & 0xffffffff) ^ state[2]) >> 11
)
return state[0] ^ state[1] ^ state[2]
@numba.njit("f4(i8[:])")
def tau_rand(state):
"""A fast (pseudo)-random number generator for floats in the range [0,1]
Parameters
----------
state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
A (pseudo)-random float32 in the interval [0, 1]
"""
integer = tau_rand_int(state)
return float(integer) / 0x7fffffff
@numba.njit()
def norm(vec):
"""Compute the (standard l2) norm of a vector.
Parameters
----------
vec: array of shape (dim,)
Returns
-------
The l2 norm of vec.
"""
result = 0.0
for i in range(vec.shape[0]):
result += vec[i] ** 2
return np.sqrt(result)
@numba.njit()
def rejection_sample(n_samples, pool_size, rng_state):
"""Generate n_samples many integers from 0 to pool_size such that no
integer is selected twice. The duplication constraint is achieved via
rejection sampling.
Parameters
----------
n_samples: int
The number of random samples to select from the pool
pool_size: int
The size of the total pool of candidates to sample from
rng_state: array of int64, shape (3,)
Internal state of the random number generator
Returns
-------
sample: array of shape(n_samples,)
The ``n_samples`` randomly selected elements from the pool.
"""
result = np.empty(n_samples, dtype=np.int64)
for i in range(n_samples):
reject_sample = True
while reject_sample:
j = tau_rand_int(rng_state) % pool_size
for k in range(i):
if j == result[k]:
break
else:
reject_sample = False
result[i] = j
return result
@numba.njit("f8[:, :, :](i8,i8)")
def make_heap(n_points, size):
"""Constructor for the numba enabled heap objects. The heaps are used
for approximate nearest neighbor search, maintaining a list of potential
neighbors sorted by their distance. We also flag if potential neighbors
are newly added to the list or not. Internally this is stored as
a single ndarray; the first axis determines whether we are looking at the
array of candidate indices, the array of distances, or the flag array for
whether elements are new or not. Each of these arrays are of shape
(``n_points``, ``size``)
Parameters
----------
n_points: int
The number of data points to track in the heap.
size: int
The number of items to keep on the heap for each data point.
Returns
-------
heap: An ndarray suitable for passing to other numba enabled heap functions.
"""
result = np.zeros((3, int(n_points), int(size)), dtype=np.float64)
result[0] = -1
result[1] = np.infty
result[2] = 0
return result
@numba.jit("i8(f8[:,:,:],i8,f8,i8,i8)")
def heap_push(heap, row, weight, index, flag):
"""Push a new element onto the heap. The heap stores potential neighbors
for each data point. The ``row`` parameter determines which data point we
are addressing, the ``weight`` determines the distance (for heap sorting),
the ``index`` is the element to add, and the flag determines whether this
is to be considered a new addition.
Parameters
----------
heap: ndarray generated by ``make_heap``
The heap object to push into
row: int
Which actual heap within the heap object to push to
weight: float
The priority value of the element to push onto the heap
index: int
The actual value to be pushed
flag: int
Whether to flag the newly added element or not.
Returns
-------
success: The number of new elements successfully pushed into the heap.
"""
row = int(row)
indices = heap[0, row]
weights = heap[1, row]
is_new = heap[2, row]
if weight >= weights[0]:
return 0
# break if we already have this element.
for i in range(indices.shape[0]):
if index == indices[i]:
return 0
# insert val at position zero
weights[0] = weight
indices[0] = index
is_new[0] = flag
# descend the heap, swapping values until the max heap criterion is met
i = 0
while True:
ic1 = 2 * i + 1
ic2 = ic1 + 1
if ic1 >= heap.shape[2]:
break
elif ic2 >= heap.shape[2]:
if weights[ic1] > weight:
i_swap = ic1
else:
break
elif weights[ic1] >= weights[ic2]:
if weight < weights[ic1]:
i_swap = ic1
else:
break
else:
if weight < weights[ic2]:
i_swap = ic2
else:
break
weights[i] = weights[i_swap]
indices[i] = indices[i_swap]
is_new[i] = is_new[i_swap]
i = i_swap
weights[i] = weight
indices[i] = index
is_new[i] = flag
return 1
@numba.jit("i8(f8[:,:,:],i8,f8,i8,i8)")
def unchecked_heap_push(heap, row, weight, index, flag):
"""Push a new element onto the heap. The heap stores potential neighbors
for each data point. The ``row`` parameter determines which data point we
are addressing, the ``weight`` determines the distance (for heap sorting),
the ``index`` is the element to add, and the flag determines whether this
is to be considered a new addition.
Parameters
----------
heap: ndarray generated by ``make_heap``
The heap object to push into
row: int
Which actual heap within the heap object to push to
weight: float
The priority value of the element to push onto the heap
index: int
The actual value to be pushed
flag: int
Whether to flag the newly added element or not.
Returns
-------
success: The number of new elements successfully pushed into the heap.
"""
indices = heap[0, row]
weights = heap[1, row]
is_new = heap[2, row]
if weight >= weights[0]:
return 0
# insert val at position zero
weights[0] = weight
indices[0] = index
is_new[0] = flag
# descend the heap, swapping values until the max heap criterion is met
i = 0
while True:
ic1 = 2 * i + 1
ic2 = ic1 + 1
if ic1 >= heap.shape[2]:
break
elif ic2 >= heap.shape[2]:
if weights[ic1] > weight:
i_swap = ic1
else:
break
elif weights[ic1] >= weights[ic2]:
if weight < weights[ic1]:
i_swap = ic1
else:
break
else:
if weight < weights[ic2]:
i_swap = ic2
else:
break
weights[i] = weights[i_swap]
indices[i] = indices[i_swap]
is_new[i] = is_new[i_swap]
i = i_swap
weights[i] = weight
indices[i] = index
is_new[i] = flag
return 1
@numba.njit()
def siftdown(heap1, heap2, elt):
"""Restore the heap property for a heap with an out of place element
at position ``elt``. This works with a heap pair where heap1 carries
the weights and heap2 holds the corresponding elements."""
while elt * 2 + 1 < heap1.shape[0]:
left_child = elt * 2 + 1
right_child = left_child + 1
swap = elt
if heap1[swap] < heap1[left_child]:
swap = left_child
if right_child < heap1.shape[0] and heap1[swap] < heap1[right_child]:
swap = right_child
if swap == elt:
break
else:
heap1[elt], heap1[swap] = heap1[swap], heap1[elt]
heap2[elt], heap2[swap] = heap2[swap], heap2[elt]
elt = swap
@numba.njit()
def deheap_sort(heap):
"""Given an array of heaps (of indices and weights), unpack the heap
out to give and array of sorted lists of indices and weights by increasing
weight. This is effectively just the second half of heap sort (the first
half not being required since we already have the data in a heap).
Parameters
----------
heap : array of shape (3, n_samples, n_neighbors)
The heap to turn into sorted lists.
Returns
-------
indices, weights: arrays of shape (n_samples, n_neighbors)
The indices and weights sorted by increasing weight.
"""
indices = heap[0]
weights = heap[1]
for i in range(indices.shape[0]):
ind_heap = indices[i]
dist_heap = weights[i]
for j in range(ind_heap.shape[0] - 1):
ind_heap[0], ind_heap[ind_heap.shape[0] - j - 1] = (
ind_heap[ind_heap.shape[0] - j - 1],
ind_heap[0],
)
dist_heap[0], dist_heap[dist_heap.shape[0] - j - 1] = (
dist_heap[dist_heap.shape[0] - j - 1],
dist_heap[0],
)
siftdown(
dist_heap[: dist_heap.shape[0] - j - 1],
ind_heap[: ind_heap.shape[0] - j - 1],
0,
)
return indices.astype(np.int64), weights
@numba.njit("i8(f8[:, :, :],i8)")
def smallest_flagged(heap, row):
"""Search the heap for the smallest element that is
still flagged.
Parameters
----------
heap: array of shape (3, n_samples, n_neighbors)
The heaps to search
row: int
Which of the heaps to search
Returns
-------
index: int
The index of the smallest flagged element
of the ``row``th heap, or -1 if no flagged
elements remain in the heap.
"""
ind = heap[0, row]
dist = heap[1, row]
flag = heap[2, row]
min_dist = np.inf
result_index = -1
for i in range(ind.shape[0]):
if flag[i] == 1 and dist[i] < min_dist:
min_dist = dist[i]
result_index = i
if result_index >= 0:
flag[result_index] = 0.0
return int(ind[result_index])
else:
return -1
#@numba.njit()
@numba.njit()
def build_candidates(current_graph, n_vertices, n_neighbors, max_candidates, rng_state):
"""Build a heap of candidate neighbors for nearest neighbor descent. For
each vertex the candidate neighbors are any current neighbors, and any
vertices that have the vertex as one of their nearest neighbors.
Parameters
----------
current_graph: heap
The current state of the graph for nearest neighbor descent.
n_vertices: int
The total number of vertices in the graph.
n_neighbors: int
The number of neighbor edges per node in the current graph.
max_candidates: int
The maximum number of new candidate neighbors.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
candidate_neighbors: A heap with an array of (randomly sorted) candidate
neighbors for each vertex in the graph.
"""
candidate_neighbors = make_heap(n_vertices, max_candidates)
for i in range(n_vertices):
for j in range(n_neighbors):
if current_graph[0, i, j] < 0:
continue
idx = current_graph[0, i, j]
isn = current_graph[2, i, j]
d = tau_rand(rng_state)
heap_push(candidate_neighbors, i, d, idx, isn)
heap_push(candidate_neighbors, idx, d, i, isn)
current_graph[2, i, j] = 0
return candidate_neighbors
@numba.njit()
def new_build_candidates(
current_graph, n_vertices, n_neighbors, max_candidates, rng_state, rho=0.5
): # pragma: no cover
"""Build a heap of candidate neighbors for nearest neighbor descent. For
each vertex the candidate neighbors are any current neighbors, and any
vertices that have the vertex as one of their nearest neighbors.
Parameters
----------
current_graph: heap
The current state of the graph for nearest neighbor descent.
n_vertices: int
The total number of vertices in the graph.
n_neighbors: int
The number of neighbor edges per node in the current graph.
max_candidates: int
The maximum number of new candidate neighbors.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
candidate_neighbors: A heap with an array of (randomly sorted) candidate
neighbors for each vertex in the graph.
"""
new_candidate_neighbors = make_heap(n_vertices, max_candidates)
old_candidate_neighbors = make_heap(n_vertices, max_candidates)
for i in numba.prange(n_vertices):
for j in range(n_neighbors):
if current_graph[0, i, j] < 0:
continue
idx = current_graph[0, i, j]
isn = current_graph[2, i, j]
d = tau_rand(rng_state)
if tau_rand(rng_state) < rho:
c = 0
if isn:
c += heap_push(new_candidate_neighbors, i, d, idx, isn)
c += heap_push(new_candidate_neighbors, idx, d, i, isn)
else:
heap_push(old_candidate_neighbors, i, d, idx, isn)
heap_push(old_candidate_neighbors, idx, d, i, isn)
if c > 0:
current_graph[2, i, j] = 0
return new_candidate_neighbors, old_candidate_neighbors
@numba.njit()
def submatrix(dmat, indices_col, n_neighbors):
"""Return a submatrix given an orginal matrix and the indices to keep.
Parameters
----------
mat: array, shape (n_samples, n_samples)
Original matrix.
indices_col: array, shape (n_samples, n_neighbors)
Indices to keep. Each row consists of the indices of the columns.
n_neighbors: int
Number of neighbors.
Returns
-------
submat: array, shape (n_samples, n_neighbors)
The corresponding submatrix.
"""
n_samples_transform, n_samples_fit = dmat.shape
submat = np.zeros((n_samples_transform, n_neighbors), dtype=dmat.dtype)
for i in numba.prange(n_samples_transform):
for j in numba.prange(n_neighbors):
submat[i, j] = dmat[i, indices_col[i, j]]
return submat
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn_single/utils.py
|
utils.py
|
import numpy as np
import scipy.sparse
import scipy.sparse.csgraph
from sklearn.manifold import SpectralEmbedding
from sklearn.metrics import pairwise_distances
from warnings import warn
def component_layout(
data, n_components, component_labels, dim, metric="euclidean", metric_kwds={}
):
"""Provide a layout relating the separate connected components. This is done
by taking the centroid of each component and then performing a spectral embedding
of the centroids.
Parameters
----------
data: array of shape (n_samples, n_features)
The source data -- required so we can generate centroids for each
connected component of the graph.
n_components: int
The number of distinct components to be layed out.
component_labels: array of shape (n_samples)
For each vertex in the graph the label of the component to
which the vertex belongs.
dim: int
The chosen embedding dimension.
metric: string or callable (optional, default 'euclidean')
The metric used to measure distances among the source data points.
metric_kwds: dict (optional, default {})
Keyword arguments to be passed to the metric function.
Returns
-------
component_embedding: array of shape (n_components, dim)
The ``dim``-dimensional embedding of the ``n_components``-many
connected components.
"""
component_centroids = np.empty((n_components, data.shape[1]), dtype=np.float64)
for label in range(n_components):
component_centroids[label] = data[component_labels == label].mean(axis=0)
distance_matrix = pairwise_distances(
component_centroids, metric=metric, **metric_kwds
)
affinity_matrix = np.exp(-distance_matrix ** 2)
component_embedding = SpectralEmbedding(
n_components=dim, affinity="precomputed"
).fit_transform(affinity_matrix)
component_embedding /= component_embedding.max()
return component_embedding
def multi_component_layout(
data,
graph,
n_components,
component_labels,
dim,
random_state,
metric="euclidean",
metric_kwds={},
):
"""Specialised layout algorithm for dealing with graphs with many connected components.
This will first fid relative positions for the components by spectrally embedding
their centroids, then spectrally embed each individual connected component positioning
them according to the centroid embeddings. This provides a decent embedding of each
component while placing the components in good relative positions to one another.
Parameters
----------
data: array of shape (n_samples, n_features)
The source data -- required so we can generate centroids for each
connected component of the graph.
graph: sparse matrix
The adjacency matrix of the graph to be emebdded.
n_components: int
The number of distinct components to be layed out.
component_labels: array of shape (n_samples)
For each vertex in the graph the label of the component to
which the vertex belongs.
dim: int
The chosen embedding dimension.
metric: string or callable (optional, default 'euclidean')
The metric used to measure distances among the source data points.
metric_kwds: dict (optional, default {})
Keyword arguments to be passed to the metric function.
Returns
-------
embedding: array of shape (n_samples, dim)
The initial embedding of ``graph``.
"""
result = np.empty((graph.shape[0], dim), dtype=np.float32)
if n_components > 2 * dim:
meta_embedding = component_layout(
data,
n_components,
component_labels,
dim,
metric=metric,
metric_kwds=metric_kwds,
)
else:
k = int(np.ceil(n_components / 2.0))
base = np.hstack([np.eye(k), np.zeros((k, dim - k))])
meta_embedding = np.vstack([base, -base])[:n_components]
for label in range(n_components):
component_graph = graph.tocsr()[component_labels == label, :].tocsc()
component_graph = component_graph[:, component_labels == label].tocoo()
distances = pairwise_distances([meta_embedding[label]], meta_embedding)
data_range = distances[distances > 0.0].min() / 2.0
if component_graph.shape[0] < 2 * dim:
result[component_labels == label] = (
random_state.uniform(
low=-data_range,
high=data_range,
size=(component_graph.shape[0], dim),
)
+ meta_embedding[label]
)
continue
diag_data = np.asarray(component_graph.sum(axis=0))
# standard Laplacian
# D = scipy.sparse.spdiags(diag_data, 0, graph.shape[0], graph.shape[0])
# L = D - graph
# Normalized Laplacian
I = scipy.sparse.identity(component_graph.shape[0], dtype=np.float64)
D = scipy.sparse.spdiags(
1.0 / np.sqrt(diag_data),
0,
component_graph.shape[0],
component_graph.shape[0],
)
L = I - D * component_graph * D
k = dim + 1
num_lanczos_vectors = max(2 * k + 1, int(np.sqrt(component_graph.shape[0])))
try:
eigenvalues, eigenvectors = scipy.sparse.linalg.eigsh(
L,
k,
which="SM",
ncv=num_lanczos_vectors,
tol=1e-4,
v0=np.ones(L.shape[0]),
maxiter=graph.shape[0] * 5,
)
order = np.argsort(eigenvalues)[1:k]
component_embedding = eigenvectors[:, order]
expansion = data_range / np.max(np.abs(component_embedding))
component_embedding *= expansion
result[component_labels == label] = (
component_embedding + meta_embedding[label]
)
except scipy.sparse.linalg.ArpackError:
warn(
"WARNING: spectral initialisation failed! The eigenvector solver\n"
"failed. This is likely due to too small an eigengap. Consider\n"
"adding some noise or jitter to your data.\n\n"
"Falling back to random initialisation!"
)
result[component_labels == label] = (
random_state.uniform(
low=-data_range,
high=data_range,
size=(component_graph.shape[0], dim),
)
+ meta_embedding[label]
)
return result
def spectral_layout(data, graph, dim, random_state, metric="euclidean", metric_kwds={}):
"""Given a graph compute the spectral embedding of the graph. This is
simply the eigenvectors of the laplacian of the graph. Here we use the
normalized laplacian.
Parameters
----------
data: array of shape (n_samples, n_features)
The source data
graph: sparse matrix
The (weighted) adjacency matrix of the graph as a sparse matrix.
dim: int
The dimension of the space into which to embed.
random_state: numpy RandomState or equivalent
A state capable being used as a numpy random state.
Returns
-------
embedding: array of shape (n_vertices, dim)
The spectral embedding of the graph.
"""
n_samples = graph.shape[0]
n_components, labels = scipy.sparse.csgraph.connected_components(graph)
if n_components > 1:
warn(
"Embedding a total of {} separate connected components using meta-embedding (experimental)".format(
n_components
)
)
return multi_component_layout(
data,
graph,
n_components,
labels,
dim,
random_state,
metric=metric,
metric_kwds=metric_kwds,
)
diag_data = np.asarray(graph.sum(axis=0))
# standard Laplacian
# D = scipy.sparse.spdiags(diag_data, 0, graph.shape[0], graph.shape[0])
# L = D - graph
# Normalized Laplacian
I = scipy.sparse.identity(graph.shape[0], dtype=np.float64)
D = scipy.sparse.spdiags(
1.0 / np.sqrt(diag_data), 0, graph.shape[0], graph.shape[0]
)
L = I - D * graph * D
k = dim + 1
num_lanczos_vectors = max(2 * k + 1, int(np.sqrt(graph.shape[0])))
try:
if L.shape[0] < 2000000:
eigenvalues, eigenvectors = scipy.sparse.linalg.eigsh(
L,
k,
which="SM",
ncv=num_lanczos_vectors,
tol=1e-4,
v0=np.ones(L.shape[0]),
maxiter=graph.shape[0] * 5,
)
else:
eigenvalues, eigenvectors = scipy.sparse.linalg.lobpcg(
L,
random_state.normal(size=(L.shape[0], k)),
largest=False,
tol=1e-8
)
order = np.argsort(eigenvalues)[1:k]
return eigenvectors[:, order]
except scipy.sparse.linalg.ArpackError:
warn(
"WARNING: spectral initialisation failed! The eigenvector solver\n"
"failed. This is likely due to too small an eigengap. Consider\n"
"adding some noise or jitter to your data.\n\n"
"Falling back to random initialisation!"
)
return random_state.uniform(low=-10.0, high=10.0, size=(graph.shape[0], dim))
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn_single/spectral.py
|
spectral.py
|
from __future__ import print_function
import numpy as np
import numba
from utils import (
tau_rand,
make_heap,
heap_push,
unchecked_heap_push,
smallest_flagged,
rejection_sample,
build_candidates,
new_build_candidates,
deheap_sort,
)
from rp_tree import search_flat_tree
def make_nn_descent(dist, dist_args):
"""Create a numba accelerated version of nearest neighbor descent
specialised for the given distance metric and metric arguments. Numba
doesn't support higher order functions directly, but we can instead JIT
compile the version of NN-descent for any given metric.
Parameters
----------
dist: function
A numba JITd distance function which, given two arrays computes a
dissimilarity between them.
dist_args: tuple
Any extra arguments that need to be passed to the distance function
beyond the two arrays to be compared.
Returns
-------
A numba JITd function for nearest neighbor descent computation that is
specialised to the given metric.
"""
@numba.njit(parallel=True)
def nn_descent(
data,
n_neighbors,
rng_state,
max_candidates=50,
n_iters=10,
delta=0.001,
rho=0.5,
rp_tree_init=True,
leaf_array=None,
verbose=False,
):
n_vertices = data.shape[0]
current_graph = make_heap(data.shape[0], n_neighbors)
for i in range(data.shape[0]):
indices = rejection_sample(n_neighbors, data.shape[0], rng_state)
for j in range(indices.shape[0]):
d = dist(data[i], data[indices[j]], *dist_args)
heap_push(current_graph, i, d, indices[j], 1)
heap_push(current_graph, indices[j], d, i, 1)
if rp_tree_init:
for n in range(leaf_array.shape[0]):
for i in range(leaf_array.shape[1]):
if leaf_array[n, i] < 0:
break
for j in range(i + 1, leaf_array.shape[1]):
if leaf_array[n, j] < 0:
break
d = dist(
data[leaf_array[n, i]], data[leaf_array[n, j]], *dist_args
)
heap_push(
current_graph, leaf_array[n, i], d, leaf_array[n, j], 1
)
heap_push(
current_graph, leaf_array[n, j], d, leaf_array[n, i], 1
)
for n in range(n_iters):
if verbose:
print("\t", n, " / ", n_iters)
candidate_neighbors = build_candidates(
current_graph, n_vertices, n_neighbors, max_candidates, rng_state
)
c = 0
for i in range(n_vertices):
for j in range(max_candidates):
p = int(candidate_neighbors[0, i, j])
if p < 0 or tau_rand(rng_state) < rho:
continue
for k in range(max_candidates):
q = int(candidate_neighbors[0, i, k])
if (
q < 0
or not candidate_neighbors[2, i, j]
and not candidate_neighbors[2, i, k]
):
continue
d = dist(data[p], data[q], *dist_args)
c += heap_push(current_graph, p, d, q, 1)
c += heap_push(current_graph, q, d, p, 1)
if c <= delta * n_neighbors * data.shape[0]:
break
return deheap_sort(current_graph)
return nn_descent
def make_initialisations(dist, dist_args):
@numba.njit(parallel=True)
def init_from_random(n_neighbors, data, query_points, heap, rng_state):
for i in range(query_points.shape[0]):
indices = rejection_sample(n_neighbors, data.shape[0], rng_state)
for j in range(indices.shape[0]):
if indices[j] < 0:
continue
d = dist(data[indices[j]], query_points[i], *dist_args)
heap_push(heap, i, d, indices[j], 1)
return
@numba.njit(parallel=True)
def init_from_tree(tree, data, query_points, heap, rng_state):
for i in range(query_points.shape[0]):
indices = search_flat_tree(
query_points[i],
tree.hyperplanes,
tree.offsets,
tree.children,
tree.indices,
rng_state,
)
for j in range(indices.shape[0]):
if indices[j] < 0:
continue
d = dist(data[indices[j]], query_points[i], *dist_args)
heap_push(heap, i, d, indices[j], 1)
return
return init_from_random, init_from_tree
def initialise_search(
forest, data, query_points, n_neighbors, init_from_random, init_from_tree, rng_state
):
results = make_heap(query_points.shape[0], n_neighbors)
init_from_random(n_neighbors, data, query_points, results, rng_state)
if forest is not None:
for tree in forest:
init_from_tree(tree, data, query_points, results, rng_state)
return results
def make_initialized_nnd_search(dist, dist_args):
@numba.njit(parallel=True)
def initialized_nnd_search(data, indptr, indices, initialization, query_points):
for i in numba.prange(query_points.shape[0]):
tried = set(initialization[0, i])
while True:
# Find smallest flagged vertex
vertex = smallest_flagged(initialization, i)
if vertex == -1:
break
candidates = indices[indptr[vertex] : indptr[vertex + 1]]
for j in range(candidates.shape[0]):
if (
candidates[j] == vertex
or candidates[j] == -1
or candidates[j] in tried
):
continue
d = dist(data[candidates[j]], query_points[i], *dist_args)
unchecked_heap_push(initialization, i, d, candidates[j], 1)
tried.add(candidates[j])
return initialization
return initialized_nnd_search
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn/nndescent.py
|
nndescent.py
|
import numpy as np
import numba
_mock_identity = np.eye(2, dtype=np.float64)
_mock_ones = np.ones(2, dtype=np.float64)
@numba.njit(fastmath=True)
def euclidean(x, y):
"""Standard euclidean distance.
..math::
D(x, y) = \sqrt{\sum_i (x_i - y_i)^2}
"""
result = 0.0
for i in range(x.shape[0]):
result += (x[i] - y[i]) ** 2
return np.sqrt(result)
@numba.njit()
def standardised_euclidean(x, y, sigma=_mock_ones):
"""Euclidean distance standardised against a vector of standard
deviations per coordinate.
..math::
D(x, y) = \sqrt{\sum_i \frac{(x_i - y_i)**2}{v_i}}
"""
result = 0.0
for i in range(x.shape[0]):
result += ((x[i] - y[i]) ** 2) / sigma[i]
return np.sqrt(result)
@numba.njit()
def manhattan(x, y):
"""Manhatten, taxicab, or l1 distance.
..math::
D(x, y) = \sum_i |x_i - y_i|
"""
result = 0.0
for i in range(x.shape[0]):
result += np.abs(x[i] - y[i])
return result
@numba.njit()
def chebyshev(x, y):
"""Chebyshev or l-infinity distance.
..math::
D(x, y) = \max_i |x_i - y_i|
"""
result = 0.0
for i in range(x.shape[0]):
result = max(result, np.abs(x[i] - y[i]))
return result
@numba.njit()
def minkowski(x, y, p=2):
"""Minkowski distance.
..math::
D(x, y) = \left(\sum_i |x_i - y_i|^p\right)^{\frac{1}{p}}
This is a general distance. For p=1 it is equivalent to
manhattan distance, for p=2 it is Euclidean distance, and
for p=infinity it is Chebyshev distance. In general it is better
to use the more specialised functions for those distances.
"""
result = 0.0
for i in range(x.shape[0]):
result += (np.abs(x[i] - y[i])) ** p
return result ** (1.0 / p)
@numba.njit()
def weighted_minkowski(x, y, w=_mock_ones, p=2):
"""A weighted version of Minkowski distance.
..math::
D(x, y) = \left(\sum_i w_i |x_i - y_i|^p\right)^{\frac{1}{p}}
If weights w_i are inverse standard deviations of data in each dimension
then this represented a standardised Minkowski distance (and is
equivalent to standardised Euclidean distance for p=1).
"""
result = 0.0
for i in range(x.shape[0]):
result += (w[i] * np.abs(x[i] - y[i])) ** p
return result ** (1.0 / p)
@numba.njit()
def mahalanobis(x, y, vinv=_mock_identity):
result = 0.0
diff = np.empty(x.shape[0], dtype=np.float64)
for i in range(x.shape[0]):
diff[i] = x[i] - y[i]
for i in range(x.shape[0]):
tmp = 0.0
for j in range(x.shape[0]):
tmp += vinv[i, j] * diff[j]
result += tmp * diff[i]
return np.sqrt(result)
@numba.njit()
def hamming(x, y):
result = 0.0
for i in range(x.shape[0]):
if x[i] != y[i]:
result += 1.0
return float(result) / x.shape[0]
@numba.njit()
def canberra(x, y):
result = 0.0
for i in range(x.shape[0]):
denominator = np.abs(x[i]) + np.abs(y[i])
if denominator > 0:
result += np.abs(x[i] - y[i]) / denominator
return result
@numba.njit()
def bray_curtis(x, y):
numerator = 0.0
denominator = 0.0
for i in range(x.shape[0]):
numerator += np.abs(x[i] - y[i])
denominator += np.abs(x[i] + y[i])
if denominator > 0.0:
return float(numerator) / denominator
else:
return 0.0
@numba.njit()
def jaccard(x, y):
num_non_zero = 0.0
num_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_non_zero += x_true or y_true
num_equal += x_true and y_true
if num_non_zero == 0.0:
return 0.0
else:
return float(num_non_zero - num_equal) / num_non_zero
@numba.njit()
def matching(x, y):
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_not_equal += x_true != y_true
return float(num_not_equal) / x.shape[0]
@numba.njit()
def dice(x, y):
num_true_true = 0.0
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
num_not_equal += x_true != y_true
if num_not_equal == 0.0:
return 0.0
else:
return num_not_equal / (2.0 * num_true_true + num_not_equal)
@numba.njit()
def kulsinski(x, y):
num_true_true = 0.0
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
num_not_equal += x_true != y_true
if num_not_equal == 0:
return 0.0
else:
return float(num_not_equal - num_true_true + x.shape[0]) / (
num_not_equal + x.shape[0]
)
@numba.njit()
def rogers_tanimoto(x, y):
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_not_equal += x_true != y_true
return (2.0 * num_not_equal) / (x.shape[0] + num_not_equal)
@numba.njit()
def russellrao(x, y):
num_true_true = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
if num_true_true == np.sum(x != 0) and num_true_true == np.sum(y != 0):
return 0.0
else:
return float(x.shape[0] - num_true_true) / (x.shape[0])
@numba.njit()
def sokal_michener(x, y):
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_not_equal += x_true != y_true
return (2.0 * num_not_equal) / (x.shape[0] + num_not_equal)
@numba.njit()
def sokal_sneath(x, y):
num_true_true = 0.0
num_not_equal = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
num_not_equal += x_true != y_true
if num_not_equal == 0.0:
return 0.0
else:
return num_not_equal / (0.5 * num_true_true + num_not_equal)
@numba.njit()
def haversine(x, y):
if x.shape[0] != 2:
raise ValueError("haversine is only defined for 2 dimensional data")
sin_lat = np.sin(0.5 * (x[0] - y[0]))
sin_long = np.sin(0.5 * (x[1] - y[1]))
result = np.sqrt(sin_lat ** 2 + np.cos(x[0]) * np.cos(y[0]) * sin_long ** 2)
return 2.0 * np.arcsin(result)
@numba.njit()
def yule(x, y):
num_true_true = 0.0
num_true_false = 0.0
num_false_true = 0.0
for i in range(x.shape[0]):
x_true = x[i] != 0
y_true = y[i] != 0
num_true_true += x_true and y_true
num_true_false += x_true and (not y_true)
num_false_true += (not x_true) and y_true
num_false_false = x.shape[0] - num_true_true - num_true_false - num_false_true
if num_true_false == 0.0 or num_false_true == 0.0:
return 0.0
else:
return (2.0 * num_true_false * num_false_true) / (
num_true_true * num_false_false + num_true_false * num_false_true
)
@numba.njit()
def cosine(x, y):
result = 0.0
norm_x = 0.0
norm_y = 0.0
for i in range(x.shape[0]):
result += x[i] * y[i]
norm_x += x[i] ** 2
norm_y += y[i] ** 2
if norm_x == 0.0 and norm_y == 0.0:
return 0.0
elif norm_x == 0.0 or norm_y == 0.0:
return 1.0
else:
return 1.0 - (result / np.sqrt(norm_x * norm_y))
@numba.njit()
def correlation(x, y):
mu_x = 0.0
mu_y = 0.0
norm_x = 0.0
norm_y = 0.0
dot_product = 0.0
for i in range(x.shape[0]):
mu_x += x[i]
mu_y += y[i]
mu_x /= x.shape[0]
mu_y /= x.shape[0]
for i in range(x.shape[0]):
shifted_x = x[i] - mu_x
shifted_y = y[i] - mu_y
norm_x += shifted_x ** 2
norm_y += shifted_y ** 2
dot_product += shifted_x * shifted_y
if norm_x == 0.0 and norm_y == 0.0:
return 0.0
elif dot_product == 0.0:
return 1.0
else:
return 1.0 - (dot_product / np.sqrt(norm_x * norm_y))
named_distances = {
# general minkowski distances
"euclidean": euclidean,
"l2": euclidean,
"manhattan": manhattan,
"taxicab": manhattan,
"l1": manhattan,
"chebyshev": chebyshev,
"linfinity": chebyshev,
"linfty": chebyshev,
"linf": chebyshev,
"minkowski": minkowski,
# Standardised/weighted distances
"seuclidean": standardised_euclidean,
"standardised_euclidean": standardised_euclidean,
"wminkowski": weighted_minkowski,
"weighted_minkowski": weighted_minkowski,
"mahalanobis": mahalanobis,
# Other distances
"canberra": canberra,
"cosine": cosine,
"correlation": correlation,
"haversine": haversine,
"braycurtis": bray_curtis,
# Binary distances
"hamming": hamming,
"jaccard": jaccard,
"dice": dice,
"matching": matching,
"kulsinski": kulsinski,
"rogerstanimoto": rogers_tanimoto,
"russellrao": russellrao,
"sokalsneath": sokal_sneath,
"sokalmichener": sokal_michener,
"yule": yule,
}
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn/distances.py
|
distances.py
|
import numpy as np
import numba
from sklearn.neighbors import KDTree
from umap_learn.distances import named_distances
@numba.njit()
def trustworthiness_vector_bulk(
indices_source, indices_embedded, max_k
): # pragma: no cover
n_samples = indices_embedded.shape[0]
trustworthiness = np.zeros(max_k + 1, dtype=np.float64)
for i in range(n_samples):
for j in range(max_k):
rank = 0
while indices_source[i, rank] != indices_embedded[i, j]:
rank += 1
for k in range(j + 1, max_k + 1):
if rank > k:
trustworthiness[k] += rank - k
for k in range(1, max_k + 1):
trustworthiness[k] = 1.0 - trustworthiness[k] * (
2.0 / (n_samples * k * (2.0 * n_samples - 3.0 * k - 1.0))
)
return trustworthiness
def make_trustworthiness_calculator(metric): # pragma: no cover
@numba.njit(parallel=True)
def trustworthiness_vector_lowmem(source, indices_embedded, max_k):
n_samples = indices_embedded.shape[0]
trustworthiness = np.zeros(max_k + 1, dtype=np.float64)
dist_vector = np.zeros(n_samples, dtype=np.float64)
for i in range(n_samples):
for j in numba.prange(n_samples):
dist_vector[j] = metric(source[i], source[j])
indices_source = np.argsort(dist_vector)
for j in range(max_k):
rank = 0
while indices_source[rank] != indices_embedded[i, j]:
rank += 1
for k in range(j + 1, max_k + 1):
if rank > k:
trustworthiness[k] += rank - k
for k in range(1, max_k + 1):
trustworthiness[k] = 1.0 - trustworthiness[k] * (
2.0 / (n_samples * k * (2.0 * n_samples - 3.0 * k - 1.0))
)
trustworthiness[0] = 1.0
return trustworthiness
return trustworthiness_vector_lowmem
@numba.jit()
def trustworthiness_vector(
source, embedding, max_k, metric="euclidean"
): # pragma: no cover
tree = KDTree(embedding, metric=metric)
indices_embedded = tree.query(embedding, k=max_k, return_distance=False)
# Drop the actual point itself
indices_embedded = indices_embedded[:, 1:]
dist = named_distances[metric]
vec_calculator = make_trustworthiness_calculator(dist)
result = vec_calculator(source, indices_embedded, max_k)
return result
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn/validation.py
|
validation.py
|
from __future__ import print_function
import numpy as np
import numba
from utils import (
tau_rand_int,
tau_rand,
norm,
make_heap,
heap_push,
rejection_sample,
build_candidates,
deheap_sort,
)
import locale
locale.setlocale(locale.LC_NUMERIC, "C")
# Just reproduce a simpler version of numpy unique (not numba supported yet)
@numba.njit()
def arr_unique(arr):
aux = np.sort(arr)
flag = np.concatenate((np.ones(1, dtype=np.bool_), aux[1:] != aux[:-1]))
return aux[flag]
# Just reproduce a simpler version of numpy union1d (not numba supported yet)
@numba.njit()
def arr_union(ar1, ar2):
if ar1.shape[0] == 0:
return ar2
elif ar2.shape[0] == 0:
return ar1
else:
return arr_unique(np.concatenate((ar1, ar2)))
# Just reproduce a simpler version of numpy intersect1d (not numba supported
# yet)
@numba.njit()
def arr_intersect(ar1, ar2):
aux = np.concatenate((ar1, ar2))
aux.sort()
return aux[:-1][aux[1:] == aux[:-1]]
@numba.njit()
def sparse_sum(ind1, data1, ind2, data2):
result_ind = arr_union(ind1, ind2)
result_data = np.zeros(result_ind.shape[0], dtype=np.float32)
i1 = 0
i2 = 0
nnz = 0
# pass through both index lists
while i1 < ind1.shape[0] and i2 < ind2.shape[0]:
j1 = ind1[i1]
j2 = ind2[i2]
if j1 == j2:
val = data1[i1] + data2[i2]
if val != 0:
result_ind[nnz] = j1
result_data[nnz] = val
nnz += 1
i1 += 1
i2 += 1
elif j1 < j2:
val = data1[i1]
if val != 0:
result_ind[nnz] = j1
result_data[nnz] = val
nnz += 1
i1 += 1
else:
val = data2[i2]
if val != 0:
result_ind[nnz] = j2
result_data[nnz] = val
nnz += 1
i2 += 1
# pass over the tails
while i1 < ind1.shape[0]:
val = data1[i1]
if val != 0:
result_ind[nnz] = i1
result_data[nnz] = val
nnz += 1
i1 += 1
while i2 < ind2.shape[0]:
val = data2[i2]
if val != 0:
result_ind[nnz] = i2
result_data[nnz] = val
nnz += 1
i2 += 1
# truncate to the correct length in case there were zeros created
result_ind = result_ind[:nnz]
result_data = result_data[:nnz]
return result_ind, result_data
@numba.njit()
def sparse_diff(ind1, data1, ind2, data2):
return sparse_sum(ind1, data1, ind2, -data2)
@numba.njit()
def sparse_mul(ind1, data1, ind2, data2):
result_ind = arr_intersect(ind1, ind2)
result_data = np.zeros(result_ind.shape[0], dtype=np.float32)
i1 = 0
i2 = 0
nnz = 0
# pass through both index lists
while i1 < ind1.shape[0] and i2 < ind2.shape[0]:
j1 = ind1[i1]
j2 = ind2[i2]
if j1 == j2:
val = data1[i1] * data2[i2]
if val != 0:
result_ind[nnz] = j1
result_data[nnz] = val
nnz += 1
i1 += 1
i2 += 1
elif j1 < j2:
i1 += 1
else:
i2 += 1
# truncate to the correct length in case there were zeros created
result_ind = result_ind[:nnz]
result_data = result_data[:nnz]
return result_ind, result_data
def make_sparse_nn_descent(sparse_dist, dist_args):
"""Create a numba accelerated version of nearest neighbor descent
specialised for the given distance metric and metric arguments on sparse
matrix data provided in CSR ind, indptr and data format. Numba
doesn't support higher order functions directly, but we can instead JIT
compile the version of NN-descent for any given metric.
Parameters
----------
sparse_dist: function
A numba JITd distance function which, given four arrays (two sets of
indices and data) computes a dissimilarity between them.
dist_args: tuple
Any extra arguments that need to be passed to the distance function
beyond the two arrays to be compared.
Returns
-------
A numba JITd function for nearest neighbor descent computation that is
specialised to the given metric.
"""
@numba.njit(parallel=True)
def nn_descent(
inds,
indptr,
data,
n_vertices,
n_neighbors,
rng_state,
max_candidates=50,
n_iters=10,
delta=0.001,
rho=0.5,
rp_tree_init=True,
leaf_array=None,
verbose=False,
):
current_graph = make_heap(n_vertices, n_neighbors)
for i in range(n_vertices):
indices = rejection_sample(n_neighbors, n_vertices, rng_state)
for j in range(indices.shape[0]):
from_inds = inds[indptr[i] : indptr[i + 1]]
from_data = data[indptr[i] : indptr[i + 1]]
to_inds = inds[indptr[indices[j]] : indptr[indices[j] + 1]]
to_data = data[indptr[indices[j]] : indptr[indices[j] + 1]]
d = sparse_dist(from_inds, from_data, to_inds, to_data, *dist_args)
heap_push(current_graph, i, d, indices[j], 1)
heap_push(current_graph, indices[j], d, i, 1)
if rp_tree_init:
for n in range(leaf_array.shape[0]):
for i in range(leaf_array.shape[1]):
if leaf_array[n, i] < 0:
break
for j in range(i + 1, leaf_array.shape[1]):
if leaf_array[n, j] < 0:
break
from_inds = inds[
indptr[leaf_array[n, i]] : indptr[leaf_array[n, i] + 1]
]
from_data = data[
indptr[leaf_array[n, i]] : indptr[leaf_array[n, i] + 1]
]
to_inds = inds[
indptr[leaf_array[n, j]] : indptr[leaf_array[n, j] + 1]
]
to_data = data[
indptr[leaf_array[n, j]] : indptr[leaf_array[n, j] + 1]
]
d = sparse_dist(
from_inds, from_data, to_inds, to_data, *dist_args
)
heap_push(
current_graph, leaf_array[n, i], d, leaf_array[n, j], 1
)
heap_push(
current_graph, leaf_array[n, j], d, leaf_array[n, i], 1
)
for n in range(n_iters):
if verbose:
print("\t", n, " / ", n_iters)
candidate_neighbors = build_candidates(
current_graph, n_vertices, n_neighbors, max_candidates, rng_state
)
c = 0
for i in range(n_vertices):
for j in range(max_candidates):
p = int(candidate_neighbors[0, i, j])
if p < 0 or tau_rand(rng_state) < rho:
continue
for k in range(max_candidates):
q = int(candidate_neighbors[0, i, k])
if (
q < 0
or not candidate_neighbors[2, i, j]
and not candidate_neighbors[2, i, k]
):
continue
from_inds = inds[indptr[p] : indptr[p + 1]]
from_data = data[indptr[p] : indptr[p + 1]]
to_inds = inds[indptr[q] : indptr[q + 1]]
to_data = data[indptr[q] : indptr[q + 1]]
d = sparse_dist(
from_inds, from_data, to_inds, to_data, *dist_args
)
c += heap_push(current_graph, p, d, q, 1)
c += heap_push(current_graph, q, d, p, 1)
if c <= delta * n_neighbors * n_vertices:
break
return deheap_sort(current_graph)
return nn_descent
@numba.njit()
def general_sset_intersection(
indptr1,
indices1,
data1,
indptr2,
indices2,
data2,
result_row,
result_col,
result_val,
mix_weight=0.5,
):
left_min = max(data1.min() / 2.0, 1.0e-8)
right_min = max(data2.min() / 2.0, 1.0e-8)
for idx in range(result_row.shape[0]):
i = result_row[idx]
j = result_col[idx]
left_val = left_min
for k in range(indptr1[i], indptr1[i + 1]):
if indices1[k] == j:
left_val = data1[k]
right_val = right_min
for k in range(indptr2[i], indptr2[i + 1]):
if indices2[k] == j:
right_val = data2[k]
if left_val > left_min or right_val > right_min:
if mix_weight < 0.5:
result_val[idx] = left_val * pow(
right_val, mix_weight / (1.0 - mix_weight)
)
else:
result_val[idx] = (
pow(left_val, (1.0 - mix_weight) / mix_weight) * right_val
)
return
@numba.njit()
def sparse_euclidean(ind1, data1, ind2, data2):
aux_inds, aux_data = sparse_diff(ind1, data1, ind2, data2)
result = 0.0
for i in range(aux_data.shape[0]):
result += aux_data[i] ** 2
return np.sqrt(result)
@numba.njit()
def sparse_manhattan(ind1, data1, ind2, data2):
aux_inds, aux_data = sparse_diff(ind1, data1, ind2, data2)
result = 0.0
for i in range(aux_data.shape[0]):
result += np.abs(aux_data[i])
return result
@numba.njit()
def sparse_chebyshev(ind1, data1, ind2, data2):
aux_inds, aux_data = sparse_diff(ind1, data1, ind2, data2)
result = 0.0
for i in range(aux_data.shape[0]):
result = max(result, np.abs(aux_data[i]))
return result
@numba.njit()
def sparse_minkowski(ind1, data1, ind2, data2, p=2.0):
aux_inds, aux_data = sparse_diff(ind1, data1, ind2, data2)
result = 0.0
for i in range(aux_data.shape[0]):
result += np.abs(aux_data[i]) ** p
return result ** (1.0 / p)
@numba.njit()
def sparse_hamming(ind1, data1, ind2, data2, n_features):
num_not_equal = sparse_diff(ind1, data1, ind2, data2)[0].shape[0]
return float(num_not_equal) / n_features
@numba.njit()
def sparse_canberra(ind1, data1, ind2, data2):
abs_data1 = np.abs(data1)
abs_data2 = np.abs(data2)
denom_inds, denom_data = sparse_sum(ind1, abs_data1, ind2, abs_data2)
denom_data = 1.0 / denom_data
numer_inds, numer_data = sparse_diff(ind1, data1, ind2, data2)
numer_data = np.abs(numer_data)
val_inds, val_data = sparse_mul(numer_inds, numer_data, denom_inds, denom_data)
return np.sum(val_data)
@numba.njit()
def sparse_bray_curtis(ind1, data1, ind2, data2): # pragma: no cover
abs_data1 = np.abs(data1)
abs_data2 = np.abs(data2)
denom_inds, denom_data = sparse_sum(ind1, abs_data1, ind2, abs_data2)
if denom_data.shape[0] == 0:
return 0.0
denominator = np.sum(denom_data)
numer_inds, numer_data = sparse_diff(ind1, data1, ind2, data2)
numer_data = np.abs(numer_data)
numerator = np.sum(numer_data)
return float(numerator) / denominator
@numba.njit()
def sparse_jaccard(ind1, data1, ind2, data2):
num_non_zero = arr_union(ind1, ind2).shape[0]
num_equal = arr_intersect(ind1, ind2).shape[0]
if num_non_zero == 0:
return 0.0
else:
return float(num_non_zero - num_equal) / num_non_zero
@numba.njit()
def sparse_matching(ind1, data1, ind2, data2, n_features):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
return float(num_not_equal) / n_features
@numba.njit()
def sparse_dice(ind1, data1, ind2, data2):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
if num_not_equal == 0.0:
return 0.0
else:
return num_not_equal / (2.0 * num_true_true + num_not_equal)
@numba.njit()
def sparse_kulsinski(ind1, data1, ind2, data2, n_features):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
if num_not_equal == 0:
return 0.0
else:
return float(num_not_equal - num_true_true + n_features) / (
num_not_equal + n_features
)
@numba.njit()
def sparse_rogers_tanimoto(ind1, data1, ind2, data2, n_features):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
return (2.0 * num_not_equal) / (n_features + num_not_equal)
@numba.njit()
def sparse_russellrao(ind1, data1, ind2, data2, n_features):
if ind1.shape[0] == ind2.shape[0] and np.all(ind1 == ind2):
return 0.0
num_true_true = arr_intersect(ind1, ind2).shape[0]
if num_true_true == np.sum(data1 != 0) and num_true_true == np.sum(data2 != 0):
return 0.0
else:
return float(n_features - num_true_true) / (n_features)
@numba.njit()
def sparse_sokal_michener(ind1, data1, ind2, data2, n_features):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
return (2.0 * num_not_equal) / (n_features + num_not_equal)
@numba.njit()
def sparse_sokal_sneath(ind1, data1, ind2, data2):
num_true_true = arr_intersect(ind1, ind2).shape[0]
num_non_zero = arr_union(ind1, ind2).shape[0]
num_not_equal = num_non_zero - num_true_true
if num_not_equal == 0.0:
return 0.0
else:
return num_not_equal / (0.5 * num_true_true + num_not_equal)
@numba.njit()
def sparse_cosine(ind1, data1, ind2, data2):
aux_inds, aux_data = sparse_mul(ind1, data1, ind2, data2)
result = 0.0
norm1 = norm(data1)
norm2 = norm(data2)
for i in range(aux_data.shape[0]):
result += aux_data[i]
if norm1 == 0.0 and norm2 == 0.0:
return 0.0
elif norm1 == 0.0 or norm2 == 0.0:
return 1.0
else:
return 1.0 - (result / (norm1 * norm2))
@numba.njit()
def sparse_correlation(ind1, data1, ind2, data2, n_features):
mu_x = 0.0
mu_y = 0.0
dot_product = 0.0
if ind1.shape[0] == 0 and ind2.shape[0] == 0:
return 0.0
elif ind1.shape[0] == 0 or ind2.shape[0] == 0:
return 1.0
for i in range(data1.shape[0]):
mu_x += data1[i]
for i in range(data2.shape[0]):
mu_y += data2[i]
mu_x /= n_features
mu_y /= n_features
shifted_data1 = np.empty(data1.shape[0], dtype=np.float32)
shifted_data2 = np.empty(data2.shape[0], dtype=np.float32)
for i in range(data1.shape[0]):
shifted_data1[i] = data1[i] - mu_x
for i in range(data2.shape[0]):
shifted_data2[i] = data2[i] - mu_y
norm1 = np.sqrt(
(norm(shifted_data1) ** 2) + (n_features - ind1.shape[0]) * (mu_x ** 2)
)
norm2 = np.sqrt(
(norm(shifted_data2) ** 2) + (n_features - ind2.shape[0]) * (mu_y ** 2)
)
dot_prod_inds, dot_prod_data = sparse_mul(ind1, shifted_data1, ind2, shifted_data2)
common_indices = set(dot_prod_inds)
for i in range(dot_prod_data.shape[0]):
dot_product += dot_prod_data[i]
for i in range(ind1.shape[0]):
if ind1[i] not in common_indices:
dot_product -= shifted_data1[i] * (mu_y)
for i in range(ind2.shape[0]):
if ind2[i] not in common_indices:
dot_product -= shifted_data2[i] * (mu_x)
all_indices = arr_union(ind1, ind2)
dot_product += mu_x * mu_y * (n_features - all_indices.shape[0])
if norm1 == 0.0 and norm2 == 0.0:
return 0.0
elif dot_product == 0.0:
return 1.0
else:
return 1.0 - (dot_product / (norm1 * norm2))
sparse_named_distances = {
# general minkowski distances
"euclidean": sparse_euclidean,
"manhattan": sparse_manhattan,
"l1": sparse_manhattan,
"taxicab": sparse_manhattan,
"chebyshev": sparse_chebyshev,
"linf": sparse_chebyshev,
"linfty": sparse_chebyshev,
"linfinity": sparse_chebyshev,
"minkowski": sparse_minkowski,
# Other distances
"canberra": sparse_canberra,
# 'braycurtis': sparse_bray_curtis,
# Binary distances
"hamming": sparse_hamming,
"jaccard": sparse_jaccard,
"dice": sparse_dice,
"matching": sparse_matching,
"kulsinski": sparse_kulsinski,
"rogerstanimoto": sparse_rogers_tanimoto,
"russellrao": sparse_russellrao,
"sokalmichener": sparse_sokal_michener,
"sokalsneath": sparse_sokal_sneath,
"cosine": sparse_cosine,
"correlation": sparse_correlation,
}
sparse_need_n_features = (
"hamming",
"matching",
"kulsinski",
"rogerstanimoto",
"russellrao",
"sokalmichener",
"correlation",
)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn/sparse.py
|
sparse.py
|
from __future__ import print_function
from collections import deque, namedtuple
from warnings import warn
import numpy as np
import numba
from sparse import sparse_mul, sparse_diff, sparse_sum
from utils import tau_rand_int, norm
import scipy.sparse
import locale
locale.setlocale(locale.LC_NUMERIC, "C")
RandomProjectionTreeNode = namedtuple(
"RandomProjectionTreeNode",
["indices", "is_leaf", "hyperplane", "offset", "left_child", "right_child"],
)
FlatTree = namedtuple("FlatTree", ["hyperplanes", "offsets", "children", "indices"])
@numba.njit(fastmath=True)
def angular_random_projection_split(data, indices, rng_state):
"""Given a set of ``indices`` for data points from ``data``, create
a random hyperplane to split the data, returning two arrays indices
that fall on either side of the hyperplane. This is the basis for a
random projection tree, which simply uses this splitting recursively.
This particular split uses cosine distance to determine the hyperplane
and which side each data sample falls on.
Parameters
----------
data: array of shape (n_samples, n_features)
The original data to be split
indices: array of shape (tree_node_size,)
The indices of the elements in the ``data`` array that are to
be split in the current operation.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
indices_left: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
indices_right: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
"""
dim = data.shape[1]
# Select two random points, set the hyperplane between them
left_index = tau_rand_int(rng_state) % indices.shape[0]
right_index = tau_rand_int(rng_state) % indices.shape[0]
right_index += left_index == right_index
right_index = right_index % indices.shape[0]
left = indices[left_index]
right = indices[right_index]
left_norm = norm(data[left])
right_norm = norm(data[right])
if left_norm == 0.0:
left_norm = 1.0
if right_norm == 0.0:
right_norm = 1.0
# Compute the normal vector to the hyperplane (the vector between
# the two points)
hyperplane_vector = np.empty(dim, dtype=np.float32)
for d in range(dim):
hyperplane_vector[d] = (data[left, d] / left_norm) - (
data[right, d] / right_norm
)
hyperplane_norm = norm(hyperplane_vector)
if hyperplane_norm == 0.0:
hyperplane_norm = 1.0
for d in range(dim):
hyperplane_vector[d] = hyperplane_vector[d] / hyperplane_norm
# For each point compute the margin (project into normal vector)
# If we are on lower side of the hyperplane put in one pile, otherwise
# put it in the other pile (if we hit hyperplane on the nose, flip a coin)
n_left = 0
n_right = 0
side = np.empty(indices.shape[0], np.int8)
for i in range(indices.shape[0]):
margin = 0.0
for d in range(dim):
margin += hyperplane_vector[d] * data[indices[i], d]
if margin == 0:
side[i] = tau_rand_int(rng_state) % 2
if side[i] == 0:
n_left += 1
else:
n_right += 1
elif margin > 0:
side[i] = 0
n_left += 1
else:
side[i] = 1
n_right += 1
# Now that we have the counts allocate arrays
indices_left = np.empty(n_left, dtype=np.int64)
indices_right = np.empty(n_right, dtype=np.int64)
# Populate the arrays with indices according to which side they fell on
n_left = 0
n_right = 0
for i in range(side.shape[0]):
if side[i] == 0:
indices_left[n_left] = indices[i]
n_left += 1
else:
indices_right[n_right] = indices[i]
n_right += 1
return indices_left, indices_right, hyperplane_vector, None
@numba.njit(fastmath=True)
def euclidean_random_projection_split(data, indices, rng_state):
"""Given a set of ``indices`` for data points from ``data``, create
a random hyperplane to split the data, returning two arrays indices
that fall on either side of the hyperplane. This is the basis for a
random projection tree, which simply uses this splitting recursively.
This particular split uses euclidean distance to determine the hyperplane
and which side each data sample falls on.
Parameters
----------
data: array of shape (n_samples, n_features)
The original data to be split
indices: array of shape (tree_node_size,)
The indices of the elements in the ``data`` array that are to
be split in the current operation.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
indices_left: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
indices_right: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
"""
dim = data.shape[1]
# Select two random points, set the hyperplane between them
left_index = tau_rand_int(rng_state) % indices.shape[0]
right_index = tau_rand_int(rng_state) % indices.shape[0]
right_index += left_index == right_index
right_index = right_index % indices.shape[0]
left = indices[left_index]
right = indices[right_index]
# Compute the normal vector to the hyperplane (the vector between
# the two points) and the offset from the origin
hyperplane_offset = 0.0
hyperplane_vector = np.empty(dim, dtype=np.float32)
for d in range(dim):
hyperplane_vector[d] = data[left, d] - data[right, d]
hyperplane_offset -= (
hyperplane_vector[d] * (data[left, d] + data[right, d]) / 2.0
)
# For each point compute the margin (project into normal vector, add offset)
# If we are on lower side of the hyperplane put in one pile, otherwise
# put it in the other pile (if we hit hyperplane on the nose, flip a coin)
n_left = 0
n_right = 0
side = np.empty(indices.shape[0], np.int8)
for i in range(indices.shape[0]):
margin = hyperplane_offset
for d in range(dim):
margin += hyperplane_vector[d] * data[indices[i], d]
if margin == 0:
side[i] = tau_rand_int(rng_state) % 2
if side[i] == 0:
n_left += 1
else:
n_right += 1
elif margin > 0:
side[i] = 0
n_left += 1
else:
side[i] = 1
n_right += 1
# Now that we have the counts allocate arrays
indices_left = np.empty(n_left, dtype=np.int64)
indices_right = np.empty(n_right, dtype=np.int64)
# Populate the arrays with indices according to which side they fell on
n_left = 0
n_right = 0
for i in range(side.shape[0]):
if side[i] == 0:
indices_left[n_left] = indices[i]
n_left += 1
else:
indices_right[n_right] = indices[i]
n_right += 1
return indices_left, indices_right, hyperplane_vector, hyperplane_offset
@numba.njit(fastmath=True)
def sparse_angular_random_projection_split(inds, indptr, data, indices, rng_state):
"""Given a set of ``indices`` for data points from a sparse data set
presented in csr sparse format as inds, indptr and data, create
a random hyperplane to split the data, returning two arrays indices
that fall on either side of the hyperplane. This is the basis for a
random projection tree, which simply uses this splitting recursively.
This particular split uses cosine distance to determine the hyperplane
and which side each data sample falls on.
Parameters
----------
inds: array
CSR format index array of the matrix
indptr: array
CSR format index pointer array of the matrix
data: array
CSR format data array of the matrix
indices: array of shape (tree_node_size,)
The indices of the elements in the ``data`` array that are to
be split in the current operation.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
indices_left: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
indices_right: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
"""
# Select two random points, set the hyperplane between them
left_index = tau_rand_int(rng_state) % indices.shape[0]
right_index = tau_rand_int(rng_state) % indices.shape[0]
right_index += left_index == right_index
right_index = right_index % indices.shape[0]
left = indices[left_index]
right = indices[right_index]
left_inds = inds[indptr[left] : indptr[left + 1]]
left_data = data[indptr[left] : indptr[left + 1]]
right_inds = inds[indptr[right] : indptr[right + 1]]
right_data = data[indptr[right] : indptr[right + 1]]
left_norm = norm(left_data)
right_norm = norm(right_data)
# Compute the normal vector to the hyperplane (the vector between
# the two points)
normalized_left_data = left_data / left_norm
normalized_right_data = right_data / right_norm
hyperplane_inds, hyperplane_data = sparse_diff(
left_inds, normalized_left_data, right_inds, normalized_right_data
)
hyperplane_norm = norm(hyperplane_data)
for d in range(hyperplane_data.shape[0]):
hyperplane_data[d] = hyperplane_data[d] / hyperplane_norm
# For each point compute the margin (project into normal vector)
# If we are on lower side of the hyperplane put in one pile, otherwise
# put it in the other pile (if we hit hyperplane on the nose, flip a coin)
n_left = 0
n_right = 0
side = np.empty(indices.shape[0], np.int8)
for i in range(indices.shape[0]):
margin = 0.0
i_inds = inds[indptr[indices[i]] : indptr[indices[i] + 1]]
i_data = data[indptr[indices[i]] : indptr[indices[i] + 1]]
mul_inds, mul_data = sparse_mul(
hyperplane_inds, hyperplane_data, i_inds, i_data
)
for d in range(mul_data.shape[0]):
margin += mul_data[d]
if margin == 0:
side[i] = tau_rand_int(rng_state) % 2
if side[i] == 0:
n_left += 1
else:
n_right += 1
elif margin > 0:
side[i] = 0
n_left += 1
else:
side[i] = 1
n_right += 1
# Now that we have the counts allocate arrays
indices_left = np.empty(n_left, dtype=np.int64)
indices_right = np.empty(n_right, dtype=np.int64)
# Populate the arrays with indices according to which side they fell on
n_left = 0
n_right = 0
for i in range(side.shape[0]):
if side[i] == 0:
indices_left[n_left] = indices[i]
n_left += 1
else:
indices_right[n_right] = indices[i]
n_right += 1
hyperplane = np.vstack((hyperplane_inds, hyperplane_data))
return indices_left, indices_right, hyperplane, None
@numba.njit(fastmath=True)
def sparse_euclidean_random_projection_split(inds, indptr, data, indices, rng_state):
"""Given a set of ``indices`` for data points from a sparse data set
presented in csr sparse format as inds, indptr and data, create
a random hyperplane to split the data, returning two arrays indices
that fall on either side of the hyperplane. This is the basis for a
random projection tree, which simply uses this splitting recursively.
This particular split uses cosine distance to determine the hyperplane
and which side each data sample falls on.
Parameters
----------
inds: array
CSR format index array of the matrix
indptr: array
CSR format index pointer array of the matrix
data: array
CSR format data array of the matrix
indices: array of shape (tree_node_size,)
The indices of the elements in the ``data`` array that are to
be split in the current operation.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
indices_left: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
indices_right: array
The elements of ``indices`` that fall on the "left" side of the
random hyperplane.
"""
# Select two random points, set the hyperplane between them
left_index = tau_rand_int(rng_state) % indices.shape[0]
right_index = tau_rand_int(rng_state) % indices.shape[0]
right_index += left_index == right_index
right_index = right_index % indices.shape[0]
left = indices[left_index]
right = indices[right_index]
left_inds = inds[indptr[left] : indptr[left + 1]]
left_data = data[indptr[left] : indptr[left + 1]]
right_inds = inds[indptr[right] : indptr[right + 1]]
right_data = data[indptr[right] : indptr[right + 1]]
# Compute the normal vector to the hyperplane (the vector between
# the two points) and the offset from the origin
hyperplane_offset = 0.0
hyperplane_inds, hyperplane_data = sparse_diff(
left_inds, left_data, right_inds, right_data
)
offset_inds, offset_data = sparse_sum(left_inds, left_data, right_inds, right_data)
offset_data = offset_data / 2.0
offset_inds, offset_data = sparse_mul(
hyperplane_inds, hyperplane_data, offset_inds, offset_data
)
for d in range(offset_data.shape[0]):
hyperplane_offset -= offset_data[d]
# For each point compute the margin (project into normal vector, add offset)
# If we are on lower side of the hyperplane put in one pile, otherwise
# put it in the other pile (if we hit hyperplane on the nose, flip a coin)
n_left = 0
n_right = 0
side = np.empty(indices.shape[0], np.int8)
for i in range(indices.shape[0]):
margin = hyperplane_offset
i_inds = inds[indptr[indices[i]] : indptr[indices[i] + 1]]
i_data = data[indptr[indices[i]] : indptr[indices[i] + 1]]
mul_inds, mul_data = sparse_mul(
hyperplane_inds, hyperplane_data, i_inds, i_data
)
for d in range(mul_data.shape[0]):
margin += mul_data[d]
if margin == 0:
side[i] = tau_rand_int(rng_state) % 2
if side[i] == 0:
n_left += 1
else:
n_right += 1
elif margin > 0:
side[i] = 0
n_left += 1
else:
side[i] = 1
n_right += 1
# Now that we have the counts allocate arrays
indices_left = np.empty(n_left, dtype=np.int64)
indices_right = np.empty(n_right, dtype=np.int64)
# Populate the arrays with indices according to which side they fell on
n_left = 0
n_right = 0
for i in range(side.shape[0]):
if side[i] == 0:
indices_left[n_left] = indices[i]
n_left += 1
else:
indices_right[n_right] = indices[i]
n_right += 1
hyperplane = np.vstack((hyperplane_inds, hyperplane_data))
return indices_left, indices_right, hyperplane, hyperplane_offset
@numba.jit()
def make_euclidean_tree(data, indices, rng_state, leaf_size=30):
if indices.shape[0] > leaf_size:
left_indices, right_indices, hyperplane, offset = euclidean_random_projection_split(
data, indices, rng_state
)
left_node = make_euclidean_tree(data, left_indices, rng_state, leaf_size)
right_node = make_euclidean_tree(data, right_indices, rng_state, leaf_size)
node = RandomProjectionTreeNode(
None, False, hyperplane, offset, left_node, right_node
)
else:
node = RandomProjectionTreeNode(indices, True, None, None, None, None)
return node
@numba.jit()
def make_angular_tree(data, indices, rng_state, leaf_size=30):
if indices.shape[0] > leaf_size:
left_indices, right_indices, hyperplane, offset = angular_random_projection_split(
data, indices, rng_state
)
left_node = make_angular_tree(data, left_indices, rng_state, leaf_size)
right_node = make_angular_tree(data, right_indices, rng_state, leaf_size)
node = RandomProjectionTreeNode(
None, False, hyperplane, offset, left_node, right_node
)
else:
node = RandomProjectionTreeNode(indices, True, None, None, None, None)
return node
@numba.jit()
def make_sparse_euclidean_tree(inds, indptr, data, indices, rng_state, leaf_size=30):
if indices.shape[0] > leaf_size:
left_indices, right_indices, hyperplane, offset = sparse_euclidean_random_projection_split(
inds, indptr, data, indices, rng_state
)
left_node = make_sparse_euclidean_tree(
inds, indptr, data, left_indices, rng_state, leaf_size
)
right_node = make_sparse_euclidean_tree(
inds, indptr, data, right_indices, rng_state, leaf_size
)
node = RandomProjectionTreeNode(
None, False, hyperplane, offset, left_node, right_node
)
else:
node = RandomProjectionTreeNode(indices, True, None, None, None, None)
return node
@numba.jit()
def make_sparse_angular_tree(inds, indptr, data, indices, rng_state, leaf_size=30):
if indices.shape[0] > leaf_size:
left_indices, right_indices, hyperplane, offset = sparse_angular_random_projection_split(
inds, indptr, data, indices, rng_state
)
left_node = make_sparse_angular_tree(
inds, indptr, data, left_indices, rng_state, leaf_size
)
right_node = make_sparse_angular_tree(
inds, indptr, data, right_indices, rng_state, leaf_size
)
node = RandomProjectionTreeNode(
None, False, hyperplane, offset, left_node, right_node
)
else:
node = RandomProjectionTreeNode(indices, True, None, None, None, None)
return node
def make_tree(data, rng_state, leaf_size=30, angular=False):
"""Construct a random projection tree based on ``data`` with leaves
of size at most ``leaf_size``.
Parameters
----------
data: array of shape (n_samples, n_features)
The original data to be split
rng_state: array of int64, shape (3,)
The internal state of the rng
leaf_size: int (optional, default 30)
The maximum size of any leaf node in the tree. Any node in the tree
with more than ``leaf_size`` will be split further to create child
nodes.
angular: bool (optional, default False)
Whether to use cosine/angular distance to create splits in the tree,
or euclidean distance.
Returns
-------
node: RandomProjectionTreeNode
A random projection tree node which links to its child nodes. This
provides the full tree below the returned node.
"""
is_sparse = scipy.sparse.isspmatrix_csr(data)
indices = np.arange(data.shape[0])
# Make a tree recursively until we get below the leaf size
if is_sparse:
inds = data.indices
indptr = data.indptr
spdata = data.data
if angular:
return make_sparse_angular_tree(
inds, indptr, spdata, indices, rng_state, leaf_size
)
else:
return make_sparse_euclidean_tree(
inds, indptr, spdata, indices, rng_state, leaf_size
)
else:
if angular:
return make_angular_tree(data, indices, rng_state, leaf_size)
else:
return make_euclidean_tree(data, indices, rng_state, leaf_size)
def num_nodes(tree):
"""Determine the number of nodes in a tree"""
if tree.is_leaf:
return 1
else:
return 1 + num_nodes(tree.left_child) + num_nodes(tree.right_child)
def num_leaves(tree):
"""Determine the number of leaves in a tree"""
if tree.is_leaf:
return 1
else:
return num_leaves(tree.left_child) + num_leaves(tree.right_child)
def max_sparse_hyperplane_size(tree):
"""Determine the most number on non zeros in a hyperplane entry"""
if tree.is_leaf:
return 0
else:
return max(
tree.hyperplane.shape[1],
max_sparse_hyperplane_size(tree.left_child),
max_sparse_hyperplane_size(tree.right_child),
)
def recursive_flatten(
tree, hyperplanes, offsets, children, indices, node_num, leaf_num
):
if tree.is_leaf:
children[node_num, 0] = -leaf_num
indices[leaf_num, : tree.indices.shape[0]] = tree.indices
leaf_num += 1
return node_num, leaf_num
else:
if len(tree.hyperplane.shape) > 1:
# spare case
hyperplanes[node_num][:, : tree.hyperplane.shape[1]] = tree.hyperplane
else:
hyperplanes[node_num] = tree.hyperplane
offsets[node_num] = tree.offset
children[node_num, 0] = node_num + 1
old_node_num = node_num
node_num, leaf_num = recursive_flatten(
tree.left_child,
hyperplanes,
offsets,
children,
indices,
node_num + 1,
leaf_num,
)
children[old_node_num, 1] = node_num + 1
node_num, leaf_num = recursive_flatten(
tree.right_child,
hyperplanes,
offsets,
children,
indices,
node_num + 1,
leaf_num,
)
return node_num, leaf_num
def flatten_tree(tree, leaf_size):
n_nodes = num_nodes(tree)
n_leaves = num_leaves(tree)
if len(tree.hyperplane.shape) > 1:
# sparse case
max_hyperplane_nnz = max_sparse_hyperplane_size(tree)
hyperplanes = np.zeros(
(n_nodes, tree.hyperplane.shape[0], max_hyperplane_nnz), dtype=np.float32
)
else:
hyperplanes = np.zeros((n_nodes, tree.hyperplane.shape[0]), dtype=np.float32)
offsets = np.zeros(n_nodes, dtype=np.float32)
children = -1 * np.ones((n_nodes, 2), dtype=np.int64)
indices = -1 * np.ones((n_leaves, leaf_size), dtype=np.int64)
recursive_flatten(tree, hyperplanes, offsets, children, indices, 0, 0)
return FlatTree(hyperplanes, offsets, children, indices)
@numba.njit()
def select_side(hyperplane, offset, point, rng_state):
margin = offset
for d in range(point.shape[0]):
margin += hyperplane[d] * point[d]
if margin == 0:
side = tau_rand_int(rng_state) % 2
if side == 0:
return 0
else:
return 1
elif margin > 0:
return 0
else:
return 1
@numba.njit()
def search_flat_tree(point, hyperplanes, offsets, children, indices, rng_state):
node = 0
while children[node, 0] > 0:
side = select_side(hyperplanes[node], offsets[node], point, rng_state)
if side == 0:
node = children[node, 0]
else:
node = children[node, 1]
return indices[-children[node, 0]]
def make_forest(data, n_neighbors, n_trees, rng_state, angular=False):
"""Build a random projection forest with ``n_trees``.
Parameters
----------
data
n_neighbors
n_trees
rng_state
angular
Returns
-------
forest: list
A list of random projection trees.
"""
result = []
leaf_size = max(10, n_neighbors)
try:
result = [
flatten_tree(make_tree(data, rng_state, leaf_size, angular), leaf_size)
for i in range(n_trees)
]
except (RuntimeError, RecursionError):
warn(
"Random Projection forest initialisation failed due to recursion"
"limit being reached. Something is a little strange with your "
"data, and this may take longer than normal to compute."
)
return result
def rptree_leaf_array(rp_forest):
"""Generate an array of sets of candidate nearest neighbors by
constructing a random projection forest and taking the leaves of all the
trees. Any given tree has leaves that are a set of potential nearest
neighbors. Given enough trees the set of all such leaves gives a good
likelihood of getting a good set of nearest neighbors in composite. Since
such a random projection forest is inexpensive to compute, this can be a
useful means of seeding other nearest neighbor algorithms.
Parameters
----------
data: array of shape (n_samples, n_features)
The data for which to generate nearest neighbor approximations.
n_neighbors: int
The number of nearest neighbors to attempt to approximate.
rng_state: array of int64, shape (3,)
The internal state of the rng
n_trees: int (optional, default 10)
The number of trees to build in the forest construction.
angular: bool (optional, default False)
Whether to use angular/cosine distance for random projection tree
construction.
Returns
-------
leaf_array: array of shape (n_leaves, max(10, n_neighbors))
Each row of leaf array is a list of indices found in a given leaf.
Since not all leaves are the same size the arrays are padded out with -1
to ensure we can return a single ndarray.
"""
if len(rp_forest) > 0:
leaf_array = np.vstack([tree.indices for tree in rp_forest])
else:
leaf_array = np.array([[-1]])
return leaf_array
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn/rp_tree.py
|
rp_tree.py
|
from __future__ import print_function
from warnings import warn
from scipy.optimize import curve_fit
from sklearn.base import BaseEstimator
from sklearn.utils import check_random_state, check_array
from sklearn.metrics import pairwise_distances
from sklearn.preprocessing import normalize
from sklearn.neighbors import KDTree
from sklearn.externals import joblib
import numpy as np
import scipy.sparse
import scipy.sparse.csgraph
import numba
import distances as dist
from utils import tau_rand_int, deheap_sort, submatrix
from rp_tree import rptree_leaf_array, make_forest
from nndescent import (
make_nn_descent,
make_initialisations,
make_initialized_nnd_search,
initialise_search,
)
from spectral import spectral_layout
import locale
locale.setlocale(locale.LC_NUMERIC, "C")
INT32_MIN = np.iinfo(np.int32).min + 1
INT32_MAX = np.iinfo(np.int32).max - 1
SMOOTH_K_TOLERANCE = 1e-5
MIN_K_DIST_SCALE = 1e-3
NPY_INFINITY = np.inf
@numba.njit(parallel=True, fastmath=True)
def smooth_knn_dist(distances, k, n_iter=64, local_connectivity=1.0, bandwidth=1.0):
"""Compute a continuous version of the distance to the kth nearest
neighbor. That is, this is similar to knn-distance but allows continuous
k values rather than requiring an integral k. In esscence we are simply
computing the distance such that the cardinality of fuzzy set we generate
is k.
Parameters
----------
distances: array of shape (n_samples, n_neighbors)
Distances to nearest neighbors for each samples. Each row should be a
sorted list of distances to a given samples nearest neighbors.
k: float
The number of nearest neighbors to approximate for.
n_iter: int (optional, default 64)
We need to binary search for the correct distance value. This is the
max number of iterations to use in such a search.
local_connectivity: int (optional, default 1)
The local connectivity required -- i.e. the number of nearest
neighbors that should be assumed to be connected at a local level.
The higher this value the more connected the manifold becomes
locally. In practice this should be not more than the local intrinsic
dimension of the manifold.
bandwidth: float (optional, default 1)
The target bandwidth of the kernel, larger values will produce
larger return values.
Returns
-------
knn_dist: array of shape (n_samples,)
The distance to kth nearest neighbor, as suitably approximated.
nn_dist: array of shape (n_samples,)
The distance to the 1st nearest neighbor for each point.
"""
target = np.log2(k) * bandwidth
rho = np.zeros(distances.shape[0])
result = np.zeros(distances.shape[0])
for i in range(distances.shape[0]):
lo = 0.0
hi = NPY_INFINITY
mid = 1.0
# TODO: This is very inefficient, but will do for now. FIXME
ith_distances = distances[i]
non_zero_dists = ith_distances[ith_distances > 0.0]
if non_zero_dists.shape[0] >= local_connectivity:
index = int(np.floor(local_connectivity))
interpolation = local_connectivity - index
if index > 0:
rho[i] = non_zero_dists[index - 1]
if interpolation > SMOOTH_K_TOLERANCE:
rho[i] += interpolation * (non_zero_dists[index] - non_zero_dists[index - 1])
else:
rho[i] = interpolation * non_zero_dists[0]
elif non_zero_dists.shape[0] > 0:
rho[i] = np.max(non_zero_dists)
for n in range(n_iter):
psum = 0.0
for j in range(1, distances.shape[1]):
d = distances[i, j] - rho[i]
if d > 0:
psum += np.exp(-(d / mid))
else:
psum += 1.0
if np.fabs(psum - target) < SMOOTH_K_TOLERANCE:
break
if psum > target:
hi = mid
mid = (lo + hi) / 2.0
else:
lo = mid
if hi == NPY_INFINITY:
mid *= 2
else:
mid = (lo + hi) / 2.0
result[i] = mid
# TODO: This is very inefficient, but will do for now. FIXME
if rho[i] > 0.0:
if result[i] < MIN_K_DIST_SCALE * np.mean(ith_distances):
result[i] = MIN_K_DIST_SCALE * np.mean(ith_distances)
else:
if result[i] < MIN_K_DIST_SCALE * np.mean(distances):
result[i] = MIN_K_DIST_SCALE * np.mean(distances)
return result, rho
def nearest_neighbors(
X, n_neighbors, metric, metric_kwds, angular, random_state, verbose=False
):
"""Compute the ``n_neighbors`` nearest points for each data point in ``X``
under ``metric``. This may be exact, but more likely is approximated via
nearest neighbor descent.
Parameters
----------
X: array of shape (n_samples, n_features)
The input data to compute the k-neighbor graph of.
n_neighbors: int
The number of nearest neighbors to compute for each sample in ``X``.
metric: string or callable
The metric to use for the computation.
metric_kwds: dict
Any arguments to pass to the metric computation function.
angular: bool
Whether to use angular rp trees in NN approximation.
random_state: np.random state
The random state to use for approximate NN computations.
verbose: bool
Whether to print status data during the computation.
Returns
-------
knn_indices: array of shape (n_samples, n_neighbors)
The indices on the ``n_neighbors`` closest points in the dataset.
knn_dists: array of shape (n_samples, n_neighbors)
The distances to the ``n_neighbors`` closest points in the dataset.
"""
if metric == "precomputed":
# Note that this does not support sparse distance matrices yet ...
# Compute indices of n nearest neighbors
knn_indices = np.argsort(X)[:, :n_neighbors]
# Compute the nearest neighbor distances
# (equivalent to np.sort(X)[:,:n_neighbors])
knn_dists = X[np.arange(X.shape[0])[:, None], knn_indices].copy()
rp_forest = []
else:
if callable(metric):
distance_func = metric
elif metric in dist.named_distances:
distance_func = dist.named_distances[metric]
else:
raise ValueError("Metric is neither callable, " + "nor a recognised string")
if metric in ("cosine", "correlation", "dice", "jaccard"):
angular = True
rng_state = random_state.randint(INT32_MIN, INT32_MAX, 3).astype(np.int64)
if scipy.sparse.isspmatrix_csr(X):
if metric in sparse.sparse_named_distances:
distance_func = sparse.sparse_named_distances[metric]
if metric in sparse.sparse_need_n_features:
metric_kwds["n_features"] = X.shape[1]
else:
raise ValueError(
"Metric {} not supported for sparse " + "data".format(metric)
)
metric_nn_descent = sparse.make_sparse_nn_descent(
distance_func, tuple(metric_kwds.values())
)
# TODO: Hacked values for now
n_trees = 5 + int(round((X.shape[0]) ** 0.5 / 20.0))
n_iters = max(5, int(round(np.log2(X.shape[0]))))
rp_forest = make_forest(X, n_neighbors, n_trees, rng_state, angular)
leaf_array = rptree_leaf_array(rp_forest)
knn_indices, knn_dists = metric_nn_descent(
X.indices,
X.indptr,
X.data,
X.shape[0],
n_neighbors,
rng_state,
max_candidates=60,
rp_tree_init=True,
leaf_array=leaf_array,
n_iters=n_iters,
verbose=verbose,
)
else:
metric_nn_descent = make_nn_descent(
distance_func, tuple(metric_kwds.values())
)
# TODO: Hacked values for now
n_trees = 5 + int(round((X.shape[0]) ** 0.5 / 20.0))
n_iters = max(5, int(round(np.log2(X.shape[0]))))
rp_forest = make_forest(X, n_neighbors, n_trees, rng_state, angular)
leaf_array = rptree_leaf_array(rp_forest)
knn_indices, knn_dists = metric_nn_descent(
X,
n_neighbors,
rng_state,
max_candidates=60,
rp_tree_init=True,
leaf_array=leaf_array,
n_iters=n_iters,
verbose=verbose,
)
if np.any(knn_indices < 0):
warn(
"Failed to correctly find n_neighbors for some samples."
"Results may be less than ideal. Try re-running with"
"different parameters."
)
return knn_indices, knn_dists, rp_forest
@numba.njit(parallel=True, fastmath=True)
def compute_membership_strengths(knn_indices, knn_dists, sigmas, rhos):
"""Construct the membership strength data for the 1-skeleton of each local
fuzzy simplicial set -- this is formed as a sparse matrix where each row is
a local fuzzy simplicial set, with a membership strength for the
1-simplex to each other data point.
Parameters
----------
knn_indices: array of shape (n_samples, n_neighbors)
The indices on the ``n_neighbors`` closest points in the dataset.
knn_dists: array of shape (n_samples, n_neighbors)
The distances to the ``n_neighbors`` closest points in the dataset.
sigmas: array of shape(n_samples)
The normalization factor derived from the metric tensor approximation.
rhos: array of shape(n_samples)
The local connectivity adjustment.
Returns
-------
rows: array of shape (n_samples * n_neighbors)
Row data for the resulting sparse matrix (coo format)
cols: array of shape (n_samples * n_neighbors)
Column data for the resulting sparse matrix (coo format)
vals: array of shape (n_samples * n_neighbors)
Entries for the resulting sparse matrix (coo format)
"""
n_samples = knn_indices.shape[0]
n_neighbors = knn_indices.shape[1]
rows = np.zeros((n_samples * n_neighbors), dtype=np.int64)
cols = np.zeros((n_samples * n_neighbors), dtype=np.int64)
vals = np.zeros((n_samples * n_neighbors), dtype=np.float64)
for i in range(n_samples):
for j in range(n_neighbors):
if knn_indices[i, j] == -1:
continue # We didn't get the full knn for i
if knn_indices[i, j] == i:
val = 0.0
elif knn_dists[i, j] - rhos[i] <= 0.0:
val = 1.0
else:
val = np.exp(-((knn_dists[i, j] - rhos[i]) / (sigmas[i])))
rows[i * n_neighbors + j] = i
cols[i * n_neighbors + j] = knn_indices[i, j]
vals[i * n_neighbors + j] = val
return rows, cols, vals
@numba.jit()
def fuzzy_simplicial_set(
X,
n_neighbors,
random_state,
metric,
metric_kwds={},
knn_indices=None,
knn_dists=None,
angular=False,
set_op_mix_ratio=1.0,
local_connectivity=1.0,
verbose=False,
):
"""Given a set of data X, a neighborhood size, and a measure of distance
compute the fuzzy simplicial set (here represented as a fuzzy graph in
the form of a sparse matrix) associated to the data. This is done by
locally approximating geodesic distance at each point, creating a fuzzy
simplicial set for each such point, and then combining all the local
fuzzy simplicial sets into a global one via a fuzzy union.
Parameters
----------
X: array of shape (n_samples, n_features)
The data to be modelled as a fuzzy simplicial set.
n_neighbors: int
The number of neighbors to use to approximate geodesic distance.
Larger numbers induce more global estimates of the manifold that can
miss finer detail, while smaller values will focus on fine manifold
structure to the detriment of the larger picture.
random_state: numpy RandomState or equivalent
A state capable being used as a numpy random state.
metric: string or function (optional, default 'euclidean')
The metric to use to compute distances in high dimensional space.
If a string is passed it must match a valid predefined metric. If
a general metric is required a function that takes two 1d arrays and
returns a float can be provided. For performance purposes it is
required that this be a numba jit'd function. Valid string metrics
include:
* euclidean (or l2)
* manhattan (or l1)
* cityblock
* braycurtis
* canberra
* chebyshev
* correlation
* cosine
* dice
* hamming
* jaccard
* kulsinski
* mahalanobis
* matching
* minkowski
* rogerstanimoto
* russellrao
* seuclidean
* sokalmichener
* sokalsneath
* sqeuclidean
* yule
* wminkowski
Metrics that take arguments (such as minkowski, mahalanobis etc.)
can have arguments passed via the metric_kwds dictionary. At this
time care must be taken and dictionary elements must be ordered
appropriately; this will hopefully be fixed in the future.
metric_kwds: dict (optional, default {})
Arguments to pass on to the metric, such as the ``p`` value for
Minkowski distance.
knn_indices: array of shape (n_samples, n_neighbors) (optional)
If the k-nearest neighbors of each point has already been calculated
you can pass them in here to save computation time. This should be
an array with the indices of the k-nearest neighbors as a row for
each data point.
knn_dists: array of shape (n_samples, n_neighbors) (optional)
If the k-nearest neighbors of each point has already been calculated
you can pass them in here to save computation time. This should be
an array with the distances of the k-nearest neighbors as a row for
each data point.
angular: bool (optional, default False)
Whether to use angular/cosine distance for the random projection
forest for seeding NN-descent to determine approximate nearest
neighbors.
set_op_mix_ratio: float (optional, default 1.0)
Interpolate between (fuzzy) union and intersection as the set operation
used to combine local fuzzy simplicial sets to obtain a global fuzzy
simplicial sets. Both fuzzy set operations use the product t-norm.
The value of this parameter should be between 0.0 and 1.0; a value of
1.0 will use a pure fuzzy union, while 0.0 will use a pure fuzzy
intersection.
local_connectivity: int (optional, default 1)
The local connectivity required -- i.e. the number of nearest
neighbors that should be assumed to be connected at a local level.
The higher this value the more connected the manifold becomes
locally. In practice this should be not more than the local intrinsic
dimension of the manifold.
verbose: bool (optional, default False)
Whether to report information on the current progress of the algorithm.
Returns
-------
fuzzy_simplicial_set: coo_matrix
A fuzzy simplicial set represented as a sparse matrix. The (i,
j) entry of the matrix represents the membership strength of the
1-simplex between the ith and jth sample points.
"""
if knn_indices is None or knn_dists is None:
knn_indices, knn_dists, _ = nearest_neighbors(
X, n_neighbors, metric, metric_kwds, angular, random_state, verbose=verbose
)
sigmas, rhos = smooth_knn_dist(
knn_dists, n_neighbors, local_connectivity=local_connectivity
)
rows, cols, vals = compute_membership_strengths(
knn_indices, knn_dists, sigmas, rhos
)
result = scipy.sparse.coo_matrix(
(vals, (rows, cols)), shape=(X.shape[0], X.shape[0])
)
result.eliminate_zeros()
transpose = result.transpose()
prod_matrix = result.multiply(transpose)
result = (
set_op_mix_ratio * (result + transpose - prod_matrix)
+ (1.0 - set_op_mix_ratio) * prod_matrix
)
result.eliminate_zeros()
return result
@numba.jit()
def fast_intersection(rows, cols, values, target, unknown_dist=1.0, far_dist=5.0):
"""Under the assumption of categorical distance for the intersecting
simplicial set perform a fast intersection.
Parameters
----------
rows: array
An array of the row of each non-zero in the sparse matrix
representation.
cols: array
An array of the column of each non-zero in the sparse matrix
representation.
values: array
An array of the value of each non-zero in the sparse matrix
representation.
target: array of shape (n_samples)
The categorical labels to use in the intersection.
unknown_dist: float (optional, default 1.0)
The distance an unknown label (-1) is assumed to be from any point.
far_dist float (optional, default 5.0)
The distance between unmatched labels.
Returns
-------
None
"""
for nz in range(rows.shape[0]):
i = rows[nz]
j = cols[nz]
if target[i] == -1 or target[j] == -1:
values[nz] *= np.exp(-unknown_dist)
elif target[i] != target[j]:
values[nz] *= np.exp(-far_dist)
return
@numba.jit()
def reset_local_connectivity(simplicial_set):
"""Reset the local connectivity requirement -- each data sample should
have complete confidence in at least one 1-simplex in the simplicial set.
We can enforce this by locally rescaling confidences, and then remerging the
different local simplicial sets together.
Parameters
----------
simplicial_set: sparse matrix
The simplicial set for which to recalculate with respect to local
connectivity.
Returns
-------
simplicial_set: sparse_matrix
The recalculated simplicial set, now with the local connectivity
assumption restored.
"""
simplicial_set = normalize(simplicial_set, norm="max")
transpose = simplicial_set.transpose()
prod_matrix = simplicial_set.multiply(transpose)
simplicial_set = simplicial_set + transpose - prod_matrix
simplicial_set.eliminate_zeros()
return simplicial_set
@numba.jit()
def categorical_simplicial_set_intersection(
simplicial_set, target, unknown_dist=1.0, far_dist=5.0
):
"""Combine a fuzzy simplicial set with another fuzzy simplicial set
generated from categorical data using categorical distances. The target
data is assumed to be categorical label data (a vector of labels),
and this will update the fuzzy simplicial set to respect that label data.
TODO: optional category cardinality based weighting of distance
Parameters
----------
simplicial_set: sparse matrix
The input fuzzy simplicial set.
target: array of shape (n_samples)
The categorical labels to use in the intersection.
unknown_dist: float (optional, default 1.0)
The distance an unknown label (-1) is assumed to be from any point.
far_dist float (optional, default 5.0)
The distance between unmatched labels.
Returns
-------
simplicial_set: sparse matrix
The resulting intersected fuzzy simplicial set.
"""
simplicial_set = simplicial_set.tocoo()
fast_intersection(
simplicial_set.row,
simplicial_set.col,
simplicial_set.data,
target,
unknown_dist,
far_dist,
)
simplicial_set.eliminate_zeros()
return reset_local_connectivity(simplicial_set)
@numba.jit()
def general_simplicial_set_intersection(simplicial_set1, simplicial_set2, weight):
result = (simplicial_set1 + simplicial_set2).tocoo()
left = simplicial_set1.tocsr()
right = simplicial_set2.tocsr()
sparse.general_sset_intersection(
left.indptr,
left.indices,
left.data,
right.indptr,
right.indices,
right.data,
result.row,
result.col,
result.data,
weight,
)
return result
@numba.jit()
def make_epochs_per_sample(weights, n_epochs):
"""Given a set of weights and number of epochs generate the number of
epochs per sample for each weight.
Parameters
----------
weights: array of shape (n_1_simplices)
The weights ofhow much we wish to sample each 1-simplex.
n_epochs: int
The total number of epochs we want to train for.
Returns
-------
An array of number of epochs per sample, one for each 1-simplex.
"""
result = -1.0 * np.ones(weights.shape[0], dtype=np.float64)
n_samples = n_epochs * (weights / weights.max())
result[n_samples > 0] = float(n_epochs) / n_samples[n_samples > 0]
return result
@numba.njit()
def clip(val):
"""Standard clamping of a value into a fixed range (in this case -4.0 to
4.0)
Parameters
----------
val: float
The value to be clamped.
Returns
-------
The clamped value, now fixed to be in the range -4.0 to 4.0.
"""
if val > 4.0:
return 4.0
elif val < -4.0:
return -4.0
else:
return val
@numba.njit("f4(f4[:],f4[:])", fastmath=True)
def rdist(x, y):
"""Reduced Euclidean distance.
Parameters
----------
x: array of shape (embedding_dim,)
y: array of shape (embedding_dim,)
Returns
-------
The squared euclidean distance between x and y
"""
result = 0.0
for i in range(x.shape[0]):
result += (x[i] - y[i]) ** 2
return result
@numba.njit(fastmath=True, parallel=True)
def optimize_layout(
head_embedding,
tail_embedding,
head,
tail,
n_epochs,
n_vertices,
epochs_per_sample,
a,
b,
rng_state,
gamma=1.0,
initial_alpha=1.0,
negative_sample_rate=5.0,
verbose=False,
):
"""Improve an embedding using stochastic gradient descent to minimize the
fuzzy set cross entropy between the 1-skeletons of the high dimensional
and low dimensional fuzzy simplicial sets. In practice this is done by
sampling edges based on their membership strength (with the (1-p) terms
coming from negative sampling similar to word2vec).
Parameters
----------
head_embedding: array of shape (n_samples, n_components)
The initial embedding to be improved by SGD.
tail_embedding: array of shape (source_samples, n_components)
The reference embedding of embedded points. If not embedding new
previously unseen points with respect to an existing embedding this
is simply the head_embedding (again); otherwise it provides the
existing embedding to embed with respect to.
head: array of shape (n_1_simplices)
The indices of the heads of 1-simplices with non-zero membership.
tail: array of shape (n_1_simplices)
The indices of the tails of 1-simplices with non-zero membership.
n_epochs: int
The number of training epochs to use in optimization.
n_vertices: int
The number of vertices (0-simplices) in the dataset.
epochs_per_samples: array of shape (n_1_simplices)
A float value of the number of epochs per 1-simplex. 1-simplices with
weaker membership strength will have more epochs between being sampled.
a: float
Parameter of differentiable approximation of right adjoint functor
b: float
Parameter of differentiable approximation of right adjoint functor
rng_state: array of int64, shape (3,)
The internal state of the rng
gamma: float (optional, default 1.0)
Weight to apply to negative samples.
initial_alpha: float (optional, default 1.0)
Initial learning rate for the SGD.
negative_sample_rate: int (optional, default 5)
Number of negative samples to use per positive sample.
verbose: bool (optional, default False)
Whether to report information on the current progress of the algorithm.
Returns
-------
embedding: array of shape (n_samples, n_components)
The optimized embedding.
"""
dim = head_embedding.shape[1]
move_other = head_embedding.shape[0] == tail_embedding.shape[0]
alpha = initial_alpha
epochs_per_negative_sample = epochs_per_sample / negative_sample_rate
epoch_of_next_negative_sample = epochs_per_negative_sample.copy()
epoch_of_next_sample = epochs_per_sample.copy()
for n in range(n_epochs):
for i in range(epochs_per_sample.shape[0]):
if epoch_of_next_sample[i] <= n:
j = head[i]
k = tail[i]
current = head_embedding[j]
other = tail_embedding[k]
dist_squared = rdist(current, other)
if dist_squared > 0.0:
grad_coeff = -2.0 * a * b * pow(dist_squared, b - 1.0)
grad_coeff /= a * pow(dist_squared, b) + 1.0
else:
grad_coeff = 0.0
for d in range(dim):
grad_d = clip(grad_coeff * (current[d] - other[d]))
current[d] += grad_d * alpha
if move_other:
other[d] += -grad_d * alpha
epoch_of_next_sample[i] += epochs_per_sample[i]
n_neg_samples = int(
(n - epoch_of_next_negative_sample[i])
/ epochs_per_negative_sample[i]
)
for p in range(n_neg_samples):
k = tau_rand_int(rng_state) % n_vertices
other = tail_embedding[k]
dist_squared = rdist(current, other)
if dist_squared > 0.0:
grad_coeff = 2.0 * gamma * b
grad_coeff /= (0.001 + dist_squared) * (
a * pow(dist_squared, b) + 1
)
else:
grad_coeff = 0.0
for d in range(dim):
if grad_coeff > 0.0:
grad_d = clip(grad_coeff * (current[d] - other[d]))
else:
grad_d = 4.0
current[d] += grad_d * alpha
epoch_of_next_negative_sample[i] += (
n_neg_samples * epochs_per_negative_sample[i]
)
alpha = initial_alpha * (1.0 - (float(n) / float(n_epochs)))
if verbose and n % int(n_epochs / 10) == 0:
print("\tcompleted ", n, " / ", n_epochs, "epochs")
return head_embedding
def simplicial_set_embedding(
data,
graph,
n_components,
initial_alpha,
a,
b,
gamma,
negative_sample_rate,
n_epochs,
init,
random_state,
metric,
metric_kwds,
verbose,
):
"""Perform a fuzzy simplicial set embedding, using a specified
initialisation method and then minimizing the fuzzy set cross entropy
between the 1-skeletons of the high and low dimensional fuzzy simplicial
sets.
Parameters
----------
data: array of shape (n_samples, n_features)
The source data to be embedded by UMAP.
graph: sparse matrix
The 1-skeleton of the high dimensional fuzzy simplicial set as
represented by a graph for which we require a sparse matrix for the
(weighted) adjacency matrix.
n_components: int
The dimensionality of the euclidean space into which to embed the data.
initial_alpha: float
Initial learning rate for the SGD.
a: float
Parameter of differentiable approximation of right adjoint functor
b: float
Parameter of differentiable approximation of right adjoint functor
gamma: float
Weight to apply to negative samples.
negative_sample_rate: int (optional, default 5)
The number of negative samples to select per positive sample
in the optimization process. Increasing this value will result
in greater repulsive force being applied, greater optimization
cost, but slightly more accuracy.
n_epochs: int (optional, default 0)
The number of training epochs to be used in optimizing the
low dimensional embedding. Larger values result in more accurate
embeddings. If 0 is specified a value will be selected based on
the size of the input dataset (200 for large datasets, 500 for small).
init: string
How to initialize the low dimensional embedding. Options are:
* 'spectral': use a spectral embedding of the fuzzy 1-skeleton
* 'random': assign initial embedding positions at random.
* A numpy array of initial embedding positions.
random_state: numpy RandomState or equivalent
A state capable being used as a numpy random state.
metric: string
The metric used to measure distance in high dimensional space; used if
multiple connected components need to be layed out.
metric_kwds: dict
Key word arguments to be passed to the metric function; used if
multiple connected components need to be layed out.
verbose: bool (optional, default False)
Whether to report information on the current progress of the algorithm.
Returns
-------
embedding: array of shape (n_samples, n_components)
The optimized of ``graph`` into an ``n_components`` dimensional
euclidean space.
"""
graph = graph.tocoo()
graph.sum_duplicates()
n_vertices = graph.shape[1]
if n_epochs <= 0:
# For smaller datasets we can use more epochs
if graph.shape[0] <= 10000:
n_epochs = 500
else:
n_epochs = 200
graph.data[graph.data < (graph.data.max() / float(n_epochs))] = 0.0
graph.eliminate_zeros()
if isinstance(init, str) and init == "random":
embedding = random_state.uniform(
low=-10.0, high=10.0, size=(graph.shape[0], n_components)
).astype(np.float32)
elif isinstance(init, str) and init == "spectral":
# We add a little noise to avoid local minima for optimization to come
initialisation = spectral_layout(
data,
graph,
n_components,
random_state,
metric=metric,
metric_kwds=metric_kwds,
)
expansion = 10.0 / initialisation.max()
embedding = (initialisation * expansion).astype(
np.float32
) + random_state.normal(
scale=0.0001, size=[graph.shape[0], n_components]
).astype(
np.float32
)
else:
init_data = np.array(init)
if len(init_data.shape) == 2:
if np.unique(init_data, axis=0).shape[0] < init_data.shape[0]:
tree = KDTree(init_data)
dist, ind = tree.query(init_data, k=2)
nndist = np.mean(dist[:, 1])
embedding = init_data + np.random.normal(
scale=0.001 * nndist, size=init_data.shape
).astype(np.float32)
else:
embedding = init_data
epochs_per_sample = make_epochs_per_sample(graph.data, n_epochs)
head = graph.row
tail = graph.col
rng_state = random_state.randint(INT32_MIN, INT32_MAX, 3).astype(np.int64)
embedding = optimize_layout(
embedding,
embedding,
head,
tail,
n_epochs,
n_vertices,
epochs_per_sample,
a,
b,
rng_state,
gamma,
initial_alpha,
negative_sample_rate,
verbose=verbose,
)
return embedding
@numba.njit()
def init_transform(indices, weights, embedding):
"""Given indices and weights and an original embeddings
initialize the positions of new points relative to the
indices and weights (of their neighbors in the source data).
Parameters
----------
indices: array of shape (n_new_samples, n_neighbors)
The indices of the neighbors of each new sample
weights: array of shape (n_new_samples, n_neighbors)
The membership strengths of associated 1-simplices
for each of the new samples.
embedding: array of shape (n_samples, dim)
The original embedding of the source data.
Returns
-------
new_embedding: array of shape (n_new_samples, dim)
An initial embedding of the new sample points.
"""
result = np.zeros((indices.shape[0], embedding.shape[1]), dtype=np.float32)
for i in range(indices.shape[0]):
for j in range(indices.shape[1]):
for d in range(embedding.shape[1]):
result[i, d] += weights[i, j] * embedding[indices[i, j], d]
return result
def find_ab_params(spread, min_dist):
"""Fit a, b params for the differentiable curve used in lower
dimensional fuzzy simplicial complex construction. We want the
smooth curve (from a pre-defined family with simple gradient) that
best matches an offset exponential decay.
"""
def curve(x, a, b):
return 1.0 / (1.0 + a * x ** (2 * b))
xv = np.linspace(0, spread * 3, 300)
yv = np.zeros(xv.shape)
yv[xv < min_dist] = 1.0
yv[xv >= min_dist] = np.exp(-(xv[xv >= min_dist] - min_dist) / spread)
params, covar = curve_fit(curve, xv, yv)
return params[0], params[1]
class UMAP(BaseEstimator):
"""Uniform Manifold Approximation and Projection
Finds a low dimensional embedding of the data that approximates
an underlying manifold.
Parameters
----------
n_neighbors: float (optional, default 15)
The size of local neighborhood (in terms of number of neighboring
sample points) used for manifold approximation. Larger values
result in more global views of the manifold, while smaller
values result in more local data being preserved. In general
values should be in the range 2 to 100.
n_components: int (optional, default 2)
The dimension of the space to embed into. This defaults to 2 to
provide easy visualization, but can reasonably be set to any
integer value in the range 2 to 100.
metric: string or function (optional, default 'euclidean')
The metric to use to compute distances in high dimensional space.
If a string is passed it must match a valid predefined metric. If
a general metric is required a function that takes two 1d arrays and
returns a float can be provided. For performance purposes it is
required that this be a numba jit'd function. Valid string metrics
include:
* euclidean
* manhattan
* chebyshev
* minkowski
* canberra
* braycurtis
* mahalanobis
* wminkowski
* seuclidean
* cosine
* correlation
* haversine
* hamming
* jaccard
* dice
* russelrao
* kulsinski
* rogerstanimoto
* sokalmichener
* sokalsneath
* yule
Metrics that take arguments (such as minkowski, mahalanobis etc.)
can have arguments passed via the metric_kwds dictionary. At this
time care must be taken and dictionary elements must be ordered
appropriately; this will hopefully be fixed in the future.
n_epochs: int (optional, default None)
The number of training epochs to be used in optimizing the
low dimensional embedding. Larger values result in more accurate
embeddings. If None is specified a value will be selected based on
the size of the input dataset (200 for large datasets, 500 for small).
learning_rate: float (optional, default 1.0)
The initial learning rate for the embedding optimization.
init: string (optional, default 'spectral')
How to initialize the low dimensional embedding. Options are:
* 'spectral': use a spectral embedding of the fuzzy 1-skeleton
* 'random': assign initial embedding positions at random.
* A numpy array of initial embedding positions.
min_dist: float (optional, default 0.1)
The effective minimum distance between embedded points. Smaller values
will result in a more clustered/clumped embedding where nearby points
on the manifold are drawn closer together, while larger values will
result on a more even dispersal of points. The value should be set
relative to the ``spread`` value, which determines the scale at which
embedded points will be spread out.
spread: float (optional, default 1.0)
The effective scale of embedded points. In combination with ``min_dist``
this determines how clustered/clumped the embedded points are.
set_op_mix_ratio: float (optional, default 1.0)
Interpolate between (fuzzy) union and intersection as the set operation
used to combine local fuzzy simplicial sets to obtain a global fuzzy
simplicial sets. Both fuzzy set operations use the product t-norm.
The value of this parameter should be between 0.0 and 1.0; a value of
1.0 will use a pure fuzzy union, while 0.0 will use a pure fuzzy
intersection.
local_connectivity: int (optional, default 1)
The local connectivity required -- i.e. the number of nearest
neighbors that should be assumed to be connected at a local level.
The higher this value the more connected the manifold becomes
locally. In practice this should be not more than the local intrinsic
dimension of the manifold.
repulsion_strength: float (optional, default 1.0)
Weighting applied to negative samples in low dimensional embedding
optimization. Values higher than one will result in greater weight
being given to negative samples.
negative_sample_rate: int (optional, default 5)
The number of negative samples to select per positive sample
in the optimization process. Increasing this value will result
in greater repulsive force being applied, greater optimization
cost, but slightly more accuracy.
transform_queue_size: float (optional, default 4.0)
For transform operations (embedding new points using a trained model_
this will control how aggressively to search for nearest neighbors.
Larger values will result in slower performance but more accurate
nearest neighbor evaluation.
a: float (optional, default None)
More specific parameters controlling the embedding. If None these
values are set automatically as determined by ``min_dist`` and
``spread``.
b: float (optional, default None)
More specific parameters controlling the embedding. If None these
values are set automatically as determined by ``min_dist`` and
``spread``.
random_state: int, RandomState instance or None, optional (default: None)
If int, random_state is the seed used by the random number generator;
If RandomState instance, random_state is the random number generator;
If None, the random number generator is the RandomState instance used
by `np.random`.
metric_kwds: dict (optional, default None)
Arguments to pass on to the metric, such as the ``p`` value for
Minkowski distance. If None then no arguments are passed on.
angular_rp_forest: bool (optional, default False)
Whether to use an angular random projection forest to initialise
the approximate nearest neighbor search. This can be faster, but is
mostly on useful for metric that use an angular style distance such
as cosine, correlation etc. In the case of those metrics angular forests
will be chosen automatically.
target_n_neighbors: int (optional, default -1)
The number of nearest neighbors to use to construct the target simplcial
set. If set to -1 use the ``n_neighbors`` value.
target_metric: string or callable (optional, default 'categorical')
The metric used to measure distance for a target array is using supervised
dimension reduction. By default this is 'categorical' which will measure
distance in terms of whether categories match or are different. Furthermore,
if semi-supervised is required target values of -1 will be trated as
unlabelled under the 'categorical' metric. If the target array takes
continuous values (e.g. for a regression problem) then metric of 'l1'
or 'l2' is probably more appropriate.
target_metric_kwds: dict (optional, default None)
Keyword argument to pass to the target metric when performing
supervised dimension reduction. If None then no arguments are passed on.
target_weight: float (optional, default 0.5)
weighting factor between data topology and target topology. A value of
0.0 weights entirely on data, a value of 1.0 weights entirely on target.
The default of 0.5 balances the weighting equally between data and target.
transform_seed: int (optional, default 42)
Random seed used for the stochastic aspects of the transform operation.
This ensures consistency in transform operations.
verbose: bool (optional, default False)
Controls verbosity of logging.
"""
def __init__(
self,
n_neighbors=15,
n_components=2,
metric="euclidean",
n_epochs=None,
learning_rate=1.0,
init="spectral",
min_dist=0.1,
spread=1.0,
set_op_mix_ratio=1.0,
local_connectivity=1.0,
repulsion_strength=1.0,
negative_sample_rate=5,
transform_queue_size=4.0,
a=None,
b=None,
random_state=None,
metric_kwds=None,
angular_rp_forest=False,
target_n_neighbors=-1,
target_metric="categorical",
target_metric_kwds=None,
target_weight=0.5,
transform_seed=42,
verbose=False,
):
self.n_neighbors = n_neighbors
self.metric = metric
self.metric_kwds = metric_kwds
self.n_epochs = n_epochs
self.init = init
self.n_components = n_components
self.repulsion_strength = repulsion_strength
self.learning_rate = learning_rate
self.spread = spread
self.min_dist = min_dist
self.set_op_mix_ratio = set_op_mix_ratio
self.local_connectivity = local_connectivity
self.negative_sample_rate = negative_sample_rate
self.random_state = random_state
self.angular_rp_forest = angular_rp_forest
self.transform_queue_size = transform_queue_size
self.target_n_neighbors = target_n_neighbors
self.target_metric = target_metric
self.target_metric_kwds = target_metric_kwds
self.target_weight = target_weight
self.transform_seed = transform_seed
self.verbose = verbose
self.a = a
self.b = b
def _validate_parameters(self):
if self.set_op_mix_ratio < 0.0 or self.set_op_mix_ratio > 1.0:
raise ValueError("set_op_mix_ratio must be between 0.0 and 1.0")
if self.repulsion_strength < 0.0:
raise ValueError("repulsion_strength cannot be negative")
if self.min_dist > self.spread:
raise ValueError("min_dist must be less than or equal to spread")
if self.min_dist < 0.0:
raise ValueError("min_dist must be greater than 0.0")
if not isinstance(self.init, str) and not isinstance(self.init, np.ndarray):
raise ValueError("init must be a string or ndarray")
if isinstance(self.init, str) and self.init not in ("spectral", "random"):
raise ValueError('string init values must be "spectral" or "random"')
if (
isinstance(self.init, np.ndarray)
and self.init.shape[1] != self.n_components
):
raise ValueError("init ndarray must match n_components value")
if not isinstance(self.metric, str) and not callable(self.metric):
raise ValueError("metric must be string or callable")
if self.negative_sample_rate < 0:
raise ValueError("negative sample rate must be positive")
if self._initial_alpha < 0.0:
raise ValueError("learning_rate must be positive")
if self.n_neighbors < 2:
raise ValueError("n_neighbors must be greater than 2")
if self.target_n_neighbors < 2 and self.target_n_neighbors != -1:
raise ValueError("target_n_neighbors must be greater than 2")
if not isinstance(self.n_components, int):
raise ValueError("n_components must be an int")
if self.n_components < 1:
raise ValueError("n_components must be greater than 0")
if self.n_epochs is not None and (
self.n_epochs <= 10 or not isinstance(self.n_epochs, int)
):
raise ValueError("n_epochs must be a positive integer "
"larger than 10")
def fit(self, X, y=None):
"""Fit X into an embedded space.
Optionally use y for supervised dimension reduction.
Parameters
----------
X : array, shape (n_samples, n_features) or (n_samples, n_samples)
If the metric is 'precomputed' X must be a square distance
matrix. Otherwise it contains a sample per row. If the method
is 'exact', X may be a sparse matrix of type 'csr', 'csc'
or 'coo'.
y : array, shape (n_samples)
A target array for supervised dimension reduction. How this is
handled is determined by parameters UMAP was instantiated with.
The relevant attributes are ``target_metric`` and
``target_metric_kwds``.
"""
X = check_array(X, dtype=np.float32, accept_sparse="csr")
self._raw_data = X
# Handle all the optional arguments, setting default
if self.a is None or self.b is None:
self._a, self._b = find_ab_params(self.spread, self.min_dist)
else:
self._a = self.a
self._b = self.b
if self.metric_kwds is not None:
self._metric_kwds = self.metric_kwds
else:
self._metric_kwds = {}
if self.target_metric_kwds is not None:
self._target_metric_kwds = self.target_metric_kwds
else:
self._target_metric_kwds = {}
if isinstance(self.init, np.ndarray):
init = check_array(self.init, dtype=np.float32, accept_sparse=False)
else:
init = self.init
self._initial_alpha = self.learning_rate
self._validate_parameters()
if self.verbose:
print(str(self))
# Error check n_neighbors based on data size
if X.shape[0] <= self.n_neighbors:
if X.shape[0] == 1:
self.embedding_ = np.zeros((1, self.n_components)) # needed to sklearn comparability
return self
warn(
"n_neighbors is larger than the dataset size; truncating to "
"X.shape[0] - 1"
)
self._n_neighbors = X.shape[0] - 1
else:
self._n_neighbors = self.n_neighbors
if scipy.sparse.isspmatrix_csr(X):
if not X.has_sorted_indices:
X.sort_indices()
self._sparse_data = True
else:
self._sparse_data = False
random_state = check_random_state(self.random_state)
if self.verbose:
print("Construct fuzzy simplicial set")
# Handle small cases efficiently by computing all distances
if X.shape[0] < 4096:
self._small_data = True
dmat = pairwise_distances(X, metric=self.metric, **self._metric_kwds)
self.graph_ = fuzzy_simplicial_set(
dmat,
self._n_neighbors,
random_state,
"precomputed",
self._metric_kwds,
None,
None,
self.angular_rp_forest,
self.set_op_mix_ratio,
self.local_connectivity,
self.verbose,
)
else:
self._small_data = False
# Standard case
(self._knn_indices, self._knn_dists, self._rp_forest) = nearest_neighbors(
X,
self._n_neighbors,
self.metric,
self._metric_kwds,
self.angular_rp_forest,
random_state,
self.verbose,
)
self.graph_ = fuzzy_simplicial_set(
X,
self.n_neighbors,
random_state,
self.metric,
self._metric_kwds,
self._knn_indices,
self._knn_dists,
self.angular_rp_forest,
self.set_op_mix_ratio,
self.local_connectivity,
self.verbose,
)
self._search_graph = scipy.sparse.lil_matrix(
(X.shape[0], X.shape[0]), dtype=np.int8
)
self._search_graph.rows = self._knn_indices
self._search_graph.data = (self._knn_dists != 0).astype(np.int8)
self._search_graph = self._search_graph.maximum(
self._search_graph.transpose()
).tocsr()
if callable(self.metric):
self._distance_func = self.metric
elif self.metric in dist.named_distances:
self._distance_func = dist.named_distances[self.metric]
elif self.metric == 'precomputed':
warn('Using precomputed metric; transform will be unavailable for new data')
else:
raise ValueError(
"Metric is neither callable, " + "nor a recognised string"
)
if self.metric != 'precomputed':
self._dist_args = tuple(self._metric_kwds.values())
self._random_init, self._tree_init = make_initialisations(
self._distance_func, self._dist_args
)
self._search = make_initialized_nnd_search(
self._distance_func, self._dist_args
)
if y is not None:
y_ = check_array(y, ensure_2d=False)
if self.target_metric == "categorical":
if self.target_weight < 1.0:
far_dist = 2.5 * (1.0 / (1.0 - self.target_weight))
else:
far_dist = 1.0e12
self.graph_ = categorical_simplicial_set_intersection(
self.graph_, y_, far_dist=far_dist
)
else:
if self.target_n_neighbors == -1:
target_n_neighbors = self._n_neighbors
else:
target_n_neighbors = self.target_n_neighbors
# Handle the small case as precomputed as before
if y.shape[0] < 4096:
ydmat = pairwise_distances(y_[np.newaxis, :].T,
metric=self.target_metric,
**self._target_metric_kwds)
target_graph = fuzzy_simplicial_set(
ydmat,
target_n_neighbors,
random_state,
"precomputed",
self._target_metric_kwds,
None,
None,
False,
1.0,
1.0,
False
)
else:
# Standard case
target_graph = fuzzy_simplicial_set(
y_[np.newaxis, :].T,
target_n_neighbors,
random_state,
self.target_metric,
self._target_metric_kwds,
None,
None,
False,
1.0,
1.0,
False,
)
# product = self.graph_.multiply(target_graph)
# # self.graph_ = 0.99 * product + 0.01 * (self.graph_ +
# # target_graph -
# # product)
# self.graph_ = product
self.graph_ = general_simplicial_set_intersection(
self.graph_, target_graph, self.target_weight
)
self.graph_ = reset_local_connectivity(self.graph_)
if self.n_epochs is None:
n_epochs = 0
else:
n_epochs = self.n_epochs
if self.verbose:
print("Construct embedding")
self.embedding_ = simplicial_set_embedding(
self._raw_data,
self.graph_,
self.n_components,
self._initial_alpha,
self._a,
self._b,
self.repulsion_strength,
self.negative_sample_rate,
n_epochs,
init,
random_state,
self.metric,
self._metric_kwds,
self.verbose,
)
self._input_hash = joblib.hash(self._raw_data)
return self
def fit_transform(self, X, y=None):
"""Fit X into an embedded space and return that transformed
output.
Parameters
----------
X : array, shape (n_samples, n_features) or (n_samples, n_samples)
If the metric is 'precomputed' X must be a square distance
matrix. Otherwise it contains a sample per row.
y : array, shape (n_samples)
A target array for supervised dimension reduction. How this is
handled is determined by parameters UMAP was instantiated with.
The relevant attributes are ``target_metric`` and
``target_metric_kwds``.
Returns
-------
X_new : array, shape (n_samples, n_components)
Embedding of the training data in low-dimensional space.
"""
self.fit(X, y)
return self.embedding_
def transform(self, X):
"""Transform X into the existing embedded space and return that
transformed output.
Parameters
----------
X : array, shape (n_samples, n_features)
New data to be transformed.
Returns
-------
X_new : array, shape (n_samples, n_components)
Embedding of the new data in low-dimensional space.
"""
# If we fit just a single instance then error
if self.embedding_.shape[0] == 1:
raise ValueError('Transform unavailable when model was fit with'
'only a single data sample.')
# If we just have the original input then short circuit things
X = check_array(X, dtype=np.float32, accept_sparse="csr")
x_hash = joblib.hash(X)
if x_hash == self._input_hash:
return self.embedding_
if self._sparse_data:
raise ValueError("Transform not available for sparse input.")
elif self.metric == 'precomputed':
raise ValueError("Transform of new data not available for "
"precomputed metric.")
X = check_array(X, dtype=np.float32, order="C")
random_state = check_random_state(self.transform_seed)
rng_state = random_state.randint(INT32_MIN, INT32_MAX, 3).astype(np.int64)
if self._small_data:
dmat = pairwise_distances(
X, self._raw_data, metric=self.metric, **self._metric_kwds
)
indices = np.argpartition(dmat,
self._n_neighbors)[:, :self._n_neighbors]
dmat_shortened = submatrix(dmat, indices, self._n_neighbors)
indices_sorted = np.argsort(dmat_shortened)
indices = submatrix(indices, indices_sorted, self._n_neighbors)
dists = submatrix(dmat_shortened, indices_sorted,
self._n_neighbors)
else:
init = initialise_search(
self._rp_forest,
self._raw_data,
X,
int(self._n_neighbors * self.transform_queue_size),
self._random_init,
self._tree_init,
rng_state,
)
result = self._search(
self._raw_data,
self._search_graph.indptr,
self._search_graph.indices,
init,
X,
)
indices, dists = deheap_sort(result)
indices = indices[:, : self._n_neighbors]
dists = dists[:, : self._n_neighbors]
adjusted_local_connectivity = max(0, self.local_connectivity - 1.0)
sigmas, rhos = smooth_knn_dist(
dists, self._n_neighbors, local_connectivity=adjusted_local_connectivity
)
rows, cols, vals = compute_membership_strengths(indices, dists, sigmas, rhos)
graph = scipy.sparse.coo_matrix(
(vals, (rows, cols)), shape=(X.shape[0], self._raw_data.shape[0])
)
# This was a very specially constructed graph with constant degree.
# That lets us do fancy unpacking by reshaping the csr matrix indices
# and data. Doing so relies on the constant degree assumption!
csr_graph = normalize(graph.tocsr(), norm="l1")
inds = csr_graph.indices.reshape(X.shape[0], self._n_neighbors)
weights = csr_graph.data.reshape(X.shape[0], self._n_neighbors)
embedding = init_transform(inds, weights, self.embedding_)
if self.n_epochs is None:
# For smaller datasets we can use more epochs
if graph.shape[0] <= 10000:
n_epochs = 100
else:
n_epochs = 30
else:
n_epochs = self.n_epochs // 3.0
graph.data[graph.data < (graph.data.max() / float(n_epochs))] = 0.0
graph.eliminate_zeros()
epochs_per_sample = make_epochs_per_sample(graph.data, n_epochs)
head = graph.row
tail = graph.col
embedding = optimize_layout(
embedding,
self.embedding_,
head,
tail,
n_epochs,
graph.shape[1],
epochs_per_sample,
self._a,
self._b,
rng_state,
self.repulsion_strength,
self._initial_alpha,
self.negative_sample_rate,
verbose=self.verbose,
)
return embedding
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn/umap.py
|
umap.py
|
import numpy as np
import numba
@numba.njit("i4(i8[:])")
def tau_rand_int(state):
"""A fast (pseudo)-random number generator.
Parameters
----------
state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
A (pseudo)-random int32 value
"""
state[0] = (((state[0] & 4294967294) << 12) & 0xffffffff) ^ (
(((state[0] << 13) & 0xffffffff) ^ state[0]) >> 19
)
state[1] = (((state[1] & 4294967288) << 4) & 0xffffffff) ^ (
(((state[1] << 2) & 0xffffffff) ^ state[1]) >> 25
)
state[2] = (((state[2] & 4294967280) << 17) & 0xffffffff) ^ (
(((state[2] << 3) & 0xffffffff) ^ state[2]) >> 11
)
return state[0] ^ state[1] ^ state[2]
@numba.njit("f4(i8[:])")
def tau_rand(state):
"""A fast (pseudo)-random number generator for floats in the range [0,1]
Parameters
----------
state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
A (pseudo)-random float32 in the interval [0, 1]
"""
integer = tau_rand_int(state)
return float(integer) / 0x7fffffff
@numba.njit()
def norm(vec):
"""Compute the (standard l2) norm of a vector.
Parameters
----------
vec: array of shape (dim,)
Returns
-------
The l2 norm of vec.
"""
result = 0.0
for i in range(vec.shape[0]):
result += vec[i] ** 2
return np.sqrt(result)
@numba.njit()
def rejection_sample(n_samples, pool_size, rng_state):
"""Generate n_samples many integers from 0 to pool_size such that no
integer is selected twice. The duplication constraint is achieved via
rejection sampling.
Parameters
----------
n_samples: int
The number of random samples to select from the pool
pool_size: int
The size of the total pool of candidates to sample from
rng_state: array of int64, shape (3,)
Internal state of the random number generator
Returns
-------
sample: array of shape(n_samples,)
The ``n_samples`` randomly selected elements from the pool.
"""
result = np.empty(n_samples, dtype=np.int64)
for i in range(n_samples):
reject_sample = True
while reject_sample:
j = tau_rand_int(rng_state) % pool_size
for k in range(i):
if j == result[k]:
break
else:
reject_sample = False
result[i] = j
return result
@numba.njit("f8[:, :, :](i8,i8)")
def make_heap(n_points, size):
"""Constructor for the numba enabled heap objects. The heaps are used
for approximate nearest neighbor search, maintaining a list of potential
neighbors sorted by their distance. We also flag if potential neighbors
are newly added to the list or not. Internally this is stored as
a single ndarray; the first axis determines whether we are looking at the
array of candidate indices, the array of distances, or the flag array for
whether elements are new or not. Each of these arrays are of shape
(``n_points``, ``size``)
Parameters
----------
n_points: int
The number of data points to track in the heap.
size: int
The number of items to keep on the heap for each data point.
Returns
-------
heap: An ndarray suitable for passing to other numba enabled heap functions.
"""
result = np.zeros((3, int(n_points), int(size)), dtype=np.float64)
result[0] = -1
result[1] = np.infty
result[2] = 0
return result
@numba.jit("i8(f8[:,:,:],i8,f8,i8,i8)")
def heap_push(heap, row, weight, index, flag):
"""Push a new element onto the heap. The heap stores potential neighbors
for each data point. The ``row`` parameter determines which data point we
are addressing, the ``weight`` determines the distance (for heap sorting),
the ``index`` is the element to add, and the flag determines whether this
is to be considered a new addition.
Parameters
----------
heap: ndarray generated by ``make_heap``
The heap object to push into
row: int
Which actual heap within the heap object to push to
weight: float
The priority value of the element to push onto the heap
index: int
The actual value to be pushed
flag: int
Whether to flag the newly added element or not.
Returns
-------
success: The number of new elements successfully pushed into the heap.
"""
row = int(row)
indices = heap[0, row]
weights = heap[1, row]
is_new = heap[2, row]
if weight >= weights[0]:
return 0
# break if we already have this element.
for i in range(indices.shape[0]):
if index == indices[i]:
return 0
# insert val at position zero
weights[0] = weight
indices[0] = index
is_new[0] = flag
# descend the heap, swapping values until the max heap criterion is met
i = 0
while True:
ic1 = 2 * i + 1
ic2 = ic1 + 1
if ic1 >= heap.shape[2]:
break
elif ic2 >= heap.shape[2]:
if weights[ic1] > weight:
i_swap = ic1
else:
break
elif weights[ic1] >= weights[ic2]:
if weight < weights[ic1]:
i_swap = ic1
else:
break
else:
if weight < weights[ic2]:
i_swap = ic2
else:
break
weights[i] = weights[i_swap]
indices[i] = indices[i_swap]
is_new[i] = is_new[i_swap]
i = i_swap
weights[i] = weight
indices[i] = index
is_new[i] = flag
return 1
@numba.jit("i8(f8[:,:,:],i8,f8,i8,i8)")
def unchecked_heap_push(heap, row, weight, index, flag):
"""Push a new element onto the heap. The heap stores potential neighbors
for each data point. The ``row`` parameter determines which data point we
are addressing, the ``weight`` determines the distance (for heap sorting),
the ``index`` is the element to add, and the flag determines whether this
is to be considered a new addition.
Parameters
----------
heap: ndarray generated by ``make_heap``
The heap object to push into
row: int
Which actual heap within the heap object to push to
weight: float
The priority value of the element to push onto the heap
index: int
The actual value to be pushed
flag: int
Whether to flag the newly added element or not.
Returns
-------
success: The number of new elements successfully pushed into the heap.
"""
indices = heap[0, row]
weights = heap[1, row]
is_new = heap[2, row]
if weight >= weights[0]:
return 0
# insert val at position zero
weights[0] = weight
indices[0] = index
is_new[0] = flag
# descend the heap, swapping values until the max heap criterion is met
i = 0
while True:
ic1 = 2 * i + 1
ic2 = ic1 + 1
if ic1 >= heap.shape[2]:
break
elif ic2 >= heap.shape[2]:
if weights[ic1] > weight:
i_swap = ic1
else:
break
elif weights[ic1] >= weights[ic2]:
if weight < weights[ic1]:
i_swap = ic1
else:
break
else:
if weight < weights[ic2]:
i_swap = ic2
else:
break
weights[i] = weights[i_swap]
indices[i] = indices[i_swap]
is_new[i] = is_new[i_swap]
i = i_swap
weights[i] = weight
indices[i] = index
is_new[i] = flag
return 1
@numba.njit()
def siftdown(heap1, heap2, elt):
"""Restore the heap property for a heap with an out of place element
at position ``elt``. This works with a heap pair where heap1 carries
the weights and heap2 holds the corresponding elements."""
while elt * 2 + 1 < heap1.shape[0]:
left_child = elt * 2 + 1
right_child = left_child + 1
swap = elt
if heap1[swap] < heap1[left_child]:
swap = left_child
if right_child < heap1.shape[0] and heap1[swap] < heap1[right_child]:
swap = right_child
if swap == elt:
break
else:
heap1[elt], heap1[swap] = heap1[swap], heap1[elt]
heap2[elt], heap2[swap] = heap2[swap], heap2[elt]
elt = swap
@numba.njit()
def deheap_sort(heap):
"""Given an array of heaps (of indices and weights), unpack the heap
out to give and array of sorted lists of indices and weights by increasing
weight. This is effectively just the second half of heap sort (the first
half not being required since we already have the data in a heap).
Parameters
----------
heap : array of shape (3, n_samples, n_neighbors)
The heap to turn into sorted lists.
Returns
-------
indices, weights: arrays of shape (n_samples, n_neighbors)
The indices and weights sorted by increasing weight.
"""
indices = heap[0]
weights = heap[1]
for i in range(indices.shape[0]):
ind_heap = indices[i]
dist_heap = weights[i]
for j in range(ind_heap.shape[0] - 1):
ind_heap[0], ind_heap[ind_heap.shape[0] - j - 1] = (
ind_heap[ind_heap.shape[0] - j - 1],
ind_heap[0],
)
dist_heap[0], dist_heap[dist_heap.shape[0] - j - 1] = (
dist_heap[dist_heap.shape[0] - j - 1],
dist_heap[0],
)
siftdown(
dist_heap[: dist_heap.shape[0] - j - 1],
ind_heap[: ind_heap.shape[0] - j - 1],
0,
)
return indices.astype(np.int64), weights
@numba.njit("i8(f8[:, :, :],i8)")
def smallest_flagged(heap, row):
"""Search the heap for the smallest element that is
still flagged.
Parameters
----------
heap: array of shape (3, n_samples, n_neighbors)
The heaps to search
row: int
Which of the heaps to search
Returns
-------
index: int
The index of the smallest flagged element
of the ``row``th heap, or -1 if no flagged
elements remain in the heap.
"""
ind = heap[0, row]
dist = heap[1, row]
flag = heap[2, row]
min_dist = np.inf
result_index = -1
for i in range(ind.shape[0]):
if flag[i] == 1 and dist[i] < min_dist:
min_dist = dist[i]
result_index = i
if result_index >= 0:
flag[result_index] = 0.0
return int(ind[result_index])
else:
return -1
@numba.njit(parallel=True)
def build_candidates(current_graph, n_vertices, n_neighbors, max_candidates, rng_state):
"""Build a heap of candidate neighbors for nearest neighbor descent. For
each vertex the candidate neighbors are any current neighbors, and any
vertices that have the vertex as one of their nearest neighbors.
Parameters
----------
current_graph: heap
The current state of the graph for nearest neighbor descent.
n_vertices: int
The total number of vertices in the graph.
n_neighbors: int
The number of neighbor edges per node in the current graph.
max_candidates: int
The maximum number of new candidate neighbors.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
candidate_neighbors: A heap with an array of (randomly sorted) candidate
neighbors for each vertex in the graph.
"""
candidate_neighbors = make_heap(n_vertices, max_candidates)
for i in range(n_vertices):
for j in range(n_neighbors):
if current_graph[0, i, j] < 0:
continue
idx = current_graph[0, i, j]
isn = current_graph[2, i, j]
d = tau_rand(rng_state)
heap_push(candidate_neighbors, i, d, idx, isn)
heap_push(candidate_neighbors, idx, d, i, isn)
current_graph[2, i, j] = 0
return candidate_neighbors
@numba.njit(parallel=True)
def new_build_candidates(
current_graph, n_vertices, n_neighbors, max_candidates, rng_state, rho=0.5
): # pragma: no cover
"""Build a heap of candidate neighbors for nearest neighbor descent. For
each vertex the candidate neighbors are any current neighbors, and any
vertices that have the vertex as one of their nearest neighbors.
Parameters
----------
current_graph: heap
The current state of the graph for nearest neighbor descent.
n_vertices: int
The total number of vertices in the graph.
n_neighbors: int
The number of neighbor edges per node in the current graph.
max_candidates: int
The maximum number of new candidate neighbors.
rng_state: array of int64, shape (3,)
The internal state of the rng
Returns
-------
candidate_neighbors: A heap with an array of (randomly sorted) candidate
neighbors for each vertex in the graph.
"""
new_candidate_neighbors = make_heap(n_vertices, max_candidates)
old_candidate_neighbors = make_heap(n_vertices, max_candidates)
for i in numba.prange(n_vertices):
for j in range(n_neighbors):
if current_graph[0, i, j] < 0:
continue
idx = current_graph[0, i, j]
isn = current_graph[2, i, j]
d = tau_rand(rng_state)
if tau_rand(rng_state) < rho:
c = 0
if isn:
c += heap_push(new_candidate_neighbors, i, d, idx, isn)
c += heap_push(new_candidate_neighbors, idx, d, i, isn)
else:
heap_push(old_candidate_neighbors, i, d, idx, isn)
heap_push(old_candidate_neighbors, idx, d, i, isn)
if c > 0:
current_graph[2, i, j] = 0
return new_candidate_neighbors, old_candidate_neighbors
@numba.njit(parallel=True)
def submatrix(dmat, indices_col, n_neighbors):
"""Return a submatrix given an orginal matrix and the indices to keep.
Parameters
----------
mat: array, shape (n_samples, n_samples)
Original matrix.
indices_col: array, shape (n_samples, n_neighbors)
Indices to keep. Each row consists of the indices of the columns.
n_neighbors: int
Number of neighbors.
Returns
-------
submat: array, shape (n_samples, n_neighbors)
The corresponding submatrix.
"""
n_samples_transform, n_samples_fit = dmat.shape
submat = np.zeros((n_samples_transform, n_neighbors), dtype=dmat.dtype)
for i in numba.prange(n_samples_transform):
for j in numba.prange(n_neighbors):
submat[i, j] = dmat[i, indices_col[i, j]]
return submat
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn/utils.py
|
utils.py
|
import numpy as np
import scipy.sparse
import scipy.sparse.csgraph
from sklearn.manifold import SpectralEmbedding
from sklearn.metrics import pairwise_distances
from warnings import warn
def component_layout(
data, n_components, component_labels, dim, metric="euclidean", metric_kwds={}
):
"""Provide a layout relating the separate connected components. This is done
by taking the centroid of each component and then performing a spectral embedding
of the centroids.
Parameters
----------
data: array of shape (n_samples, n_features)
The source data -- required so we can generate centroids for each
connected component of the graph.
n_components: int
The number of distinct components to be layed out.
component_labels: array of shape (n_samples)
For each vertex in the graph the label of the component to
which the vertex belongs.
dim: int
The chosen embedding dimension.
metric: string or callable (optional, default 'euclidean')
The metric used to measure distances among the source data points.
metric_kwds: dict (optional, default {})
Keyword arguments to be passed to the metric function.
Returns
-------
component_embedding: array of shape (n_components, dim)
The ``dim``-dimensional embedding of the ``n_components``-many
connected components.
"""
component_centroids = np.empty((n_components, data.shape[1]), dtype=np.float64)
for label in range(n_components):
component_centroids[label] = data[component_labels == label].mean(axis=0)
distance_matrix = pairwise_distances(
component_centroids, metric=metric, **metric_kwds
)
affinity_matrix = np.exp(-distance_matrix ** 2)
component_embedding = SpectralEmbedding(
n_components=dim, affinity="precomputed"
).fit_transform(affinity_matrix)
component_embedding /= component_embedding.max()
return component_embedding
def multi_component_layout(
data,
graph,
n_components,
component_labels,
dim,
random_state,
metric="euclidean",
metric_kwds={},
):
"""Specialised layout algorithm for dealing with graphs with many connected components.
This will first fid relative positions for the components by spectrally embedding
their centroids, then spectrally embed each individual connected component positioning
them according to the centroid embeddings. This provides a decent embedding of each
component while placing the components in good relative positions to one another.
Parameters
----------
data: array of shape (n_samples, n_features)
The source data -- required so we can generate centroids for each
connected component of the graph.
graph: sparse matrix
The adjacency matrix of the graph to be emebdded.
n_components: int
The number of distinct components to be layed out.
component_labels: array of shape (n_samples)
For each vertex in the graph the label of the component to
which the vertex belongs.
dim: int
The chosen embedding dimension.
metric: string or callable (optional, default 'euclidean')
The metric used to measure distances among the source data points.
metric_kwds: dict (optional, default {})
Keyword arguments to be passed to the metric function.
Returns
-------
embedding: array of shape (n_samples, dim)
The initial embedding of ``graph``.
"""
result = np.empty((graph.shape[0], dim), dtype=np.float32)
if n_components > 2 * dim:
meta_embedding = component_layout(
data,
n_components,
component_labels,
dim,
metric=metric,
metric_kwds=metric_kwds,
)
else:
k = int(np.ceil(n_components / 2.0))
base = np.hstack([np.eye(k), np.zeros((k, dim - k))])
meta_embedding = np.vstack([base, -base])[:n_components]
for label in range(n_components):
component_graph = graph.tocsr()[component_labels == label, :].tocsc()
component_graph = component_graph[:, component_labels == label].tocoo()
distances = pairwise_distances([meta_embedding[label]], meta_embedding)
data_range = distances[distances > 0.0].min() / 2.0
if component_graph.shape[0] < 2 * dim:
result[component_labels == label] = (
random_state.uniform(
low=-data_range,
high=data_range,
size=(component_graph.shape[0], dim),
)
+ meta_embedding[label]
)
continue
diag_data = np.asarray(component_graph.sum(axis=0))
# standard Laplacian
# D = scipy.sparse.spdiags(diag_data, 0, graph.shape[0], graph.shape[0])
# L = D - graph
# Normalized Laplacian
I = scipy.sparse.identity(component_graph.shape[0], dtype=np.float64)
D = scipy.sparse.spdiags(
1.0 / np.sqrt(diag_data),
0,
component_graph.shape[0],
component_graph.shape[0],
)
L = I - D * component_graph * D
k = dim + 1
num_lanczos_vectors = max(2 * k + 1, int(np.sqrt(component_graph.shape[0])))
try:
eigenvalues, eigenvectors = scipy.sparse.linalg.eigsh(
L,
k,
which="SM",
ncv=num_lanczos_vectors,
tol=1e-4,
v0=np.ones(L.shape[0]),
maxiter=graph.shape[0] * 5,
)
order = np.argsort(eigenvalues)[1:k]
component_embedding = eigenvectors[:, order]
expansion = data_range / np.max(np.abs(component_embedding))
component_embedding *= expansion
result[component_labels == label] = (
component_embedding + meta_embedding[label]
)
except scipy.sparse.linalg.ArpackError:
warn(
"WARNING: spectral initialisation failed! The eigenvector solver\n"
"failed. This is likely due to too small an eigengap. Consider\n"
"adding some noise or jitter to your data.\n\n"
"Falling back to random initialisation!"
)
result[component_labels == label] = (
random_state.uniform(
low=-data_range,
high=data_range,
size=(component_graph.shape[0], dim),
)
+ meta_embedding[label]
)
return result
def spectral_layout(data, graph, dim, random_state, metric="euclidean", metric_kwds={}):
"""Given a graph compute the spectral embedding of the graph. This is
simply the eigenvectors of the laplacian of the graph. Here we use the
normalized laplacian.
Parameters
----------
data: array of shape (n_samples, n_features)
The source data
graph: sparse matrix
The (weighted) adjacency matrix of the graph as a sparse matrix.
dim: int
The dimension of the space into which to embed.
random_state: numpy RandomState or equivalent
A state capable being used as a numpy random state.
Returns
-------
embedding: array of shape (n_vertices, dim)
The spectral embedding of the graph.
"""
n_samples = graph.shape[0]
n_components, labels = scipy.sparse.csgraph.connected_components(graph)
if n_components > 1:
warn(
"Embedding a total of {} separate connected components using meta-embedding (experimental)".format(
n_components
)
)
return multi_component_layout(
data,
graph,
n_components,
labels,
dim,
random_state,
metric=metric,
metric_kwds=metric_kwds,
)
diag_data = np.asarray(graph.sum(axis=0))
# standard Laplacian
# D = scipy.sparse.spdiags(diag_data, 0, graph.shape[0], graph.shape[0])
# L = D - graph
# Normalized Laplacian
I = scipy.sparse.identity(graph.shape[0], dtype=np.float64)
D = scipy.sparse.spdiags(
1.0 / np.sqrt(diag_data), 0, graph.shape[0], graph.shape[0]
)
L = I - D * graph * D
k = dim + 1
num_lanczos_vectors = max(2 * k + 1, int(np.sqrt(graph.shape[0])))
try:
if L.shape[0] < 2000000:
eigenvalues, eigenvectors = scipy.sparse.linalg.eigsh(
L,
k,
which="SM",
ncv=num_lanczos_vectors,
tol=1e-4,
v0=np.ones(L.shape[0]),
maxiter=graph.shape[0] * 5,
)
else:
eigenvalues, eigenvectors = scipy.sparse.linalg.lobpcg(
L,
random_state.normal(size=(L.shape[0], k)),
largest=False,
tol=1e-8
)
order = np.argsort(eigenvalues)[1:k]
return eigenvectors[:, order]
except scipy.sparse.linalg.ArpackError:
warn(
"WARNING: spectral initialisation failed! The eigenvector solver\n"
"failed. This is likely due to too small an eigengap. Consider\n"
"adding some noise or jitter to your data.\n\n"
"Falling back to random initialisation!"
)
return random_state.uniform(low=-10.0, high=10.0, size=(graph.shape[0], dim))
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/visualization_scripts/umap_learn/spectral.py
|
spectral.py
|
import warnings
warnings.filterwarnings("ignore")
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning)
import pandas ### Some weird issue with dateutils being detected twice (suppress the warning)
import pandas as pd
import patsy
import sys
import numpy.linalg as la
import numpy as np
import sys, string
import os.path
import export
import time
# Downloaded on 12-12-18 from: https://github.com/brentp/combat.py
def adjust_nums(numerical_covariates, drop_idxs):
# if we dropped some values, have to adjust those with a larger index.
if numerical_covariates is None: return drop_idxs
return [nc - sum(nc < di for di in drop_idxs) for nc in numerical_covariates]
def design_mat(mod, numerical_covariates, batch_levels):
# require levels to make sure they are in the same order as we use in the
# rest of the script.
design = patsy.dmatrix("~ 0 + C(batch, levels=%s)" % str(batch_levels),
mod, return_type="dataframe")
mod = mod.drop(["batch"], axis=1)
numerical_covariates = list(numerical_covariates)
sys.stderr.write("found %i batches\n" % design.shape[1])
other_cols = [c for i, c in enumerate(mod.columns)
if not i in numerical_covariates]
factor_matrix = mod[other_cols]
design = pd.concat((design, factor_matrix), axis=1)
if numerical_covariates is not None:
sys.stderr.write("found %i numerical covariates...\n"
% len(numerical_covariates))
for i, nC in enumerate(numerical_covariates):
cname = mod.columns[nC]
sys.stderr.write("\t{0}\n".format(cname))
design[cname] = mod[mod.columns[nC]]
sys.stderr.write("found %i categorical variables:" % len(other_cols))
sys.stderr.write("\t" + ", ".join(other_cols) + '\n')
return design
def combat(data, batch, model=None, numerical_covariates=None):
"""Correct for batch effects in a dataset
Parameters
----------
data : pandas.DataFrame
A (n_features, n_samples) dataframe of the expression or methylation
data to batch correct
batch : pandas.Series
A column corresponding to the batches in the data, with index same as
the columns that appear in ``data``
model : patsy.design_info.DesignMatrix, optional
A model matrix describing metadata on the samples which could be
causing batch effects. If not provided, then will attempt to coarsely
correct just from the information provided in ``batch``
numerical_covariates : list-like
List of covariates in the model which are numerical, rather than
categorical
Returns
-------
corrected : pandas.DataFrame
A (n_features, n_samples) dataframe of the batch-corrected data
"""
if isinstance(numerical_covariates, str):
numerical_covariates = [numerical_covariates]
if numerical_covariates is None:
numerical_covariates = []
if model is not None and isinstance(model, pd.DataFrame):
model["batch"] = list(batch)
else:
model = pd.DataFrame({'batch': batch})
batch_items = model.groupby("batch").groups.items()
batch_levels = [k for k, v in batch_items]
batch_info = [v for k, v in batch_items]
n_batch = len(batch_info)
n_batches = np.array([len(v) for v in batch_info])
n_array = float(sum(n_batches))
# drop intercept
drop_cols = [cname for cname, inter in ((model == 1).all()).iteritems() if inter == True]
drop_idxs = [list(model.columns).index(cdrop) for cdrop in drop_cols]
model = model[[c for c in model.columns if not c in drop_cols]]
numerical_covariates = [list(model.columns).index(c) if isinstance(c, str) else c
for c in numerical_covariates if not c in drop_cols]
design = design_mat(model, numerical_covariates, batch_levels)
sys.stderr.write("Standardizing Data across genes.\n")
B_hat = np.dot(np.dot(la.inv(np.dot(design.T, design)), design.T), data.T)
grand_mean = np.dot((n_batches / n_array).T, B_hat[:n_batch,:])
var_pooled = np.dot(((data - np.dot(design, B_hat).T)**2), np.ones((int(n_array), 1)) / int(n_array))
stand_mean = np.dot(grand_mean.T.reshape((len(grand_mean), 1)), np.ones((1, int(n_array))))
tmp = np.array(design.copy())
tmp[:,:n_batch] = 0
stand_mean += np.dot(tmp, B_hat).T
s_data = ((data - stand_mean) / np.dot(np.sqrt(var_pooled), np.ones((1, int(n_array)))))
sys.stderr.write("Fitting L/S model and finding priors\n")
batch_design = design[design.columns[:n_batch]]
gamma_hat = np.dot(np.dot(la.inv(np.dot(batch_design.T, batch_design)), batch_design.T), s_data.T)
delta_hat = []
for i, batch_idxs in enumerate(batch_info):
#batches = [list(model.columns).index(b) for b in batches]
delta_hat.append(s_data[batch_idxs].var(axis=1))
gamma_bar = gamma_hat.mean(axis=1)
t2 = gamma_hat.var(axis=1)
a_prior = list(map(aprior, delta_hat))
b_prior = list(map(bprior, delta_hat))
sys.stderr.write("Finding parametric adjustments\n")
gamma_star, delta_star = [], []
for i, batch_idxs in enumerate(batch_info):
#print '18 20 22 28 29 31 32 33 35 40 46'
#print batch_info[batch_id]
temp = it_sol(s_data[batch_idxs], gamma_hat[i],
delta_hat[i], gamma_bar[i], t2[i], a_prior[i], b_prior[i])
gamma_star.append(temp[0])
delta_star.append(temp[1])
sys.stdout.write("Adjusting data\n")
bayesdata = s_data
gamma_star = np.array(gamma_star)
delta_star = np.array(delta_star)
for j, batch_idxs in enumerate(batch_info):
dsq = np.sqrt(delta_star[j,:])
dsq = dsq.reshape((len(dsq), 1))
denom = np.dot(dsq, np.ones((1, n_batches[j])))
numer = np.array(bayesdata[batch_idxs] - np.dot(batch_design.loc[batch_idxs], gamma_star).T)
bayesdata[batch_idxs] = numer / denom
vpsq = np.sqrt(var_pooled).reshape((len(var_pooled), 1))
bayesdata = bayesdata * np.dot(vpsq, np.ones((1, int(n_array)))) + stand_mean
return bayesdata
def it_sol(sdat, g_hat, d_hat, g_bar, t2, a, b, conv=0.0001):
n = (1 - np.isnan(sdat)).sum(axis=1)
g_old = g_hat.copy()
d_old = d_hat.copy()
change = 1
count = 0
while change > conv:
#print g_hat.shape, g_bar.shape, t2.shape
g_new = postmean(g_hat, g_bar, n, d_old, t2)
sum2 = ((sdat - np.dot(g_new.values.reshape((g_new.shape[0], 1)), np.ones((1, sdat.shape[1])))) ** 2).sum(axis=1)
d_new = postvar(sum2, n, a, b)
change = max((abs(g_new - g_old) / g_old).max(), (abs(d_new - d_old) / d_old).max())
g_old = g_new #.copy()
d_old = d_new #.copy()
count = count + 1
adjust = (g_new, d_new)
return adjust
def aprior(gamma_hat):
m = gamma_hat.mean()
s2 = gamma_hat.var()
return (2 * s2 +m**2) / s2
def bprior(gamma_hat):
m = gamma_hat.mean()
s2 = gamma_hat.var()
return (m*s2+m**3)/s2
def postmean(g_hat, g_bar, n, d_star, t2):
return (t2*n*g_hat+d_star * g_bar) / (t2*n+d_star)
def postvar(sum2, n, a, b):
return (0.5 * sum2 + b) / (n / 2.0 + a - 1.0)
######## Begin AltAnalyze Functions ########
def formatPhenoFile(fl):
expr_group_dir = fl.GroupsFile()
expr_batch_dir = string.replace(expr_group_dir,'groups.','batch.')
pheno_dir = string.replace(expr_group_dir,'groups.','pheno.')
import gene_associations
group_db = gene_associations.importGeneric(expr_group_dir)
bath_db = gene_associations.importGeneric(expr_batch_dir)
pheno_obj = export.ExportFile(pheno_dir)
pheno_obj.write('sample\tgroup\tbatch\n')
for sample in group_db:
group_name = group_db[sample][-1]
batch_number = bath_db[sample][0]
pheno_obj.write(string.join([sample,group_name,batch_number],'\t')+'\n')
pheno_obj.close()
return pheno_dir
def runPyCombat(fl):
""" This method was added specifically for AltAnalyze version 2.0.8 (not in the original GitHub code) """
print 'Running Combat...',
expr_input_dir = fl.ExpFile()
pheno_dir = formatPhenoFile(fl)
moved_exp_dir = export.findParentDir(expr_input_dir)+'Non-Combat/'+export.findFilename(expr_input_dir)
try:
export.copyFile(expr_input_dir, moved_exp_dir)
print 'Moved original expression file to:'
print '\t'+moved_exp_dir
### now overwrite the origin excluding the commented rows
export.cleanFile(expr_input_dir,removeExtra='#') ### remove comments from the original file
except Exception: None
pheno = pd.read_table(pheno_dir, index_col=0)
dat = pd.read_table(expr_input_dir, index_col=0)
mod = patsy.dmatrix("group", pheno, return_type="dataframe")
t = time.time()
#print dat, pheno.batch, mod;sys.exit()
ebat = combat(dat, pheno.batch, mod)
print "...Combat completed in %.2f seconds" % (time.time() - t)
print 'Original expression file over-written with batch effect removal results...'
ebat.to_csv(expr_input_dir, sep="\t")
if __name__ == "__main__":
expfile = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Heping/4-runs/ExpressionInput/exp.singh-results-317.txt'
groupsfile = string.replace(expfile,'exp.','groups.')
import UI
fl = UI.ExpressionFileLocationData(expfile,'',groupsfile,'')
runPyCombat(fl);sys.exit()
# NOTE: run this first to get the bladder batch stuff written to files.
# NOTE: run this first to get the bladder batch stuff written to files.
"""
source("http://bioconductor.org/biocLite.R")
biocLite("sva")
library("sva")
options(stringsAsFactors=FALSE)
library(bladderbatch)
data(bladderdata)
pheno = pData(bladderEset)
# add fake age variable for numeric
pheno$age = c(1:7, rep(1:10, 5))
write.table(data.frame(cel=rownames(pheno), pheno), row.names=F, quote=F, sep="\t", file="bladder-pheno.txt")
edata = exprs(bladderEset)
write.table(edata, row.names=T, quote=F, sep="\t", file="bladder-expr.txt")
# use dataframe instead of matrix
mod = model.matrix(~as.factor(cancer) + age, data=pheno)
t = Sys.time()
cdata = ComBat(dat=edata, batch=as.factor(pheno$batch), mod=mod, numCov=match("age", colnames(mod)))
print(Sys.time() - t)
print(cdata[1:5, 1:5])
write.table(cdata, row.names=True, quote=F, sep="\t", file="r-batch.txt")
"""
pheno = pd.read_table('bladder-pheno.txt', index_col=0)
dat = pd.read_table('bladder-expr.txt', index_col=0)
mod = patsy.dmatrix("~ age + cancer", pheno, return_type="dataframe")
import time
t = time.time()
ebat = combat(dat, pheno['batch'], mod, "age")
sys.stdout.write("%.2f seconds\n" % (time.time() - t))
sys.stdout.write(str(ebat.iloc[:5, :5]))
ebat.to_csv("py-batch.txt", sep="\t")
ebat = combat(dat, pheno['batch'], None)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/combat.py
|
combat.py
|
import numpy as np
import sys,string
import os
import os.path
from collections import defaultdict
try:from stats_scripts import statistics
except Exception: import statistics
import export; reload(export)
import re
from stats_scripts import fishers_exact_test
import traceback
import warnings
import math
import export
def importDenominator(denom_dir):
denominator_events={}
firstRow=True
for line in open(denom_dir,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstRow:
uid_index = t.index('UID')
firstRow=False
else:
uid = t[uid_index]
denominator_events[uid]=[]
return denominator_events
def importEvents(folder,denom={}):
### For example, knockdown signatures
import collections
unique_events = {}
import UI
files = UI.read_directory(folder)
comparison_events={}
for file in files:
if '.txt' in file and 'PSI.' in file:
fn = folder+'/'+file
firstLine = True
comparison = file[:-4]
comparison_events[comparison,'inclusion']=[]
comparison_events[comparison,'exclusion']=[]
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
try: event_index = t.index('Event-Direction')
except:
try: event_index = t.index('Inclusion-Junction') ### legacy
except: print file, 'Event-Direction error';sys.exit()
firstLine= False
continue
event = t[0]
#event = string.split(event,'|')[0]
unique_events[event]=[]
event_dictionary = comparison_events[comparison,t[event_index]]
if len(denom)>0:
if event in denom:
event_dictionary.append(event)
else:
event_dictionary.append(event)
return unique_events,comparison_events
def performMutualEnrichment(unique_inp_events,event_inp_dictionary,unique_ref_events,event_ref_dictionary):
N = len(unique_inp_events)
N = 88000
for (comparison,direction) in event_inp_dictionary:
if direction == 'inclusion': alt_direction = 'exclusion'
else: alt_direction = 'inclusion'
comparison_events1 = event_inp_dictionary[(comparison,direction)]
comparison_events2 = event_inp_dictionary[(comparison,alt_direction)]
for (reference_comp,ref_direction) in event_ref_dictionary:
if direction == ref_direction and direction == 'inclusion':
if ref_direction == 'inclusion': alt_ref_direction = 'exclusion'
else: alt_ref_direction = 'inclusion'
ref_events1 = event_ref_dictionary[(reference_comp,ref_direction)]
ref_events2 = event_ref_dictionary[(reference_comp,alt_ref_direction)]
concordant1 = len(list(set(comparison_events1) & set(ref_events1)))
concordant2 = len(list(set(comparison_events2) & set(ref_events2)))
r1 = concordant1+concordant2
n = len(ref_events1)+len(ref_events2)
R = len(comparison_events1)+len(comparison_events2)
disconcordant1 = len(list(set(comparison_events1) & set(ref_events2)))
disconcordant2 = len(list(set(comparison_events2) & set(ref_events1)))
r2 = disconcordant1+disconcordant2
#n = r1+r2
try: z_concordant = Zscore(r1,n,N,R)
except ZeroDivisionError: z_concordant = 0.0000
try: z_discordant = Zscore(r2,n,N,R)
except ZeroDivisionError: z_discordant = 0.0000
try: null_z = Zscore(0,n,N,R)
except ZeroDivisionError: null_z = 0.000
### Calculate a Fischer's Exact P-value
import mappfinder
pval1 = mappfinder.FishersExactTest(r1,n,R,N)
pval2 = mappfinder.FishersExactTest(r2,n,R,N)
### Store these data in an object
#zsd = mappfinder.ZScoreData(signature,r,n,z,null_z,n)
#zsd.SetP(pval)
print comparison+'\t'+reference_comp+'\t'+ref_direction+'\t'+str(z_concordant)+'\t'+str(z_discordant)+'\t'+str(r2)+'\t'+str(n)+'\t'+str(pval1)+'\t'+str(pval2)
def Zscore(r,n,N,R):
"""where N is the total number of events measured:
R is the total number of events meeting the criterion:
n is the total number of events in this specific reference gene-set:
r is the number of events meeting the criterion in the examined reference gene-set: """
N=float(N) ### This bring all other values into float space
z = (r - n*(R/N))/math.sqrt(n*(R/N)*(1-(R/N))*(1-((n-1)/(N-1))))
return z
if __name__ == '__main__':
import getopt
mutdict=defaultdict(list)
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['i=','r=','d='])
for opt, arg in options:
if opt == '--i': input_directory=arg
elif opt == '--r':reference_directory=arg
elif opt == '--d': denominator_directory=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
denominator_events = importDenominator(denominator_directory)
unique_ref_events,event_ref_dictionary = importEvents(reference_directory,denom=denominator_events)
#unique_inp_events,event_inp_dictionary = importEvents(input_directory,denom=unique_ref_events)
unique_inp_events,event_inp_dictionary = importEvents(input_directory)
performMutualEnrichment(unique_inp_events,event_inp_dictionary,unique_ref_events,event_ref_dictionary)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/SpliceMutualEnrich.py
|
SpliceMutualEnrich.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys,string,os,copy
import export
import unique
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>0 and '--' in command_args: commandLine=True
else: commandLine=False
display_label_names = True
import traceback
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def importViralBarcodeReferences(barcode1,barcode2):
### Derive all possible viral barcode combination sequences truncated to 38nt (not ideal but necessary with our read length)
b1_ls=[]
spacer='TGGT'
for line in open(barcode1,'rU').xreadlines():
b1 = cleanUpLine(line)
b1_ls.append(b1)
reference_48mers={}
for line in open(barcode2,'rU').xreadlines():
b2 = cleanUpLine(line)
for b1 in b1_ls:
#reference_48mers[b1+spacer+b2[:20]]=[]
reference_48mers[b1+spacer+b2]=[]
return reference_48mers
def processBarcodes(viral_barcode_file,cell_cluster_file,reference_48mers):
eo = export.ExportFile(viral_barcode_file[:-4]+'-cleaned.txt')
parent = export.findParentDir(viral_barcode_file)
eom = export.ExportFile(parent+'/MultiLin-cells.txt')
### Import a file with the sample names in the groups file in the correct order
viral_barcodes={}
repair={}
short={}
cluster_header=[]
cell_clusters={}
for line in open(cell_cluster_file,'rU').xreadlines():
data = cleanUpLine(line)
cell, cluster, cluster_name = string.split(data,'\t')
cell_clusters[cell]=cluster_name
if cluster_name not in cluster_header:
cluster_header.append(cluster_name)
cells_with_virus={}
for line in open(viral_barcode_file,'rU').xreadlines():
data = cleanUpLine(line)
cellular, viral = string.split(data,'\t')
if cellular in cell_clusters:
try:
if viral not in cells_with_virus[cellular]:
cells_with_virus[cellular].append(viral)
except Exception: cells_with_virus[cellular]=[viral]
if len(viral)<48:
#if len(viral)<38:
if viral not in repair:
repair[viral]=[cellular]
else:
if cellular not in repair[viral]:
repair[viral].append(cellular)
else:
#short[viral[:35]]=viral
try:
if cellular not in viral_barcodes[viral]:
viral_barcodes[viral].append(cellular)
except Exception: viral_barcodes[viral] = [cellular]
### Repair the short sequences
for viral_short in repair:
cellular_barcodes = repair[viral_short]
if viral_short[:35] in short:
viral = short[viral_short[:35]]
for cellular in cellular_barcodes:
try:
if cellular not in viral_barcodes[viral]:
viral_barcodes[viral].append(cellular)
except Exception: viral_barcodes[viral] = [cellular]
print len(viral_barcodes),'unique viral barcodes present'
#print cells_with_virus['ACGCCGATCTGTTGAG']
#print cells_with_virus['CAGAATCCAAACTGCT']
#sys.exit()
valid_barcodes = 0
for viral in viral_barcodes:
if viral in reference_48mers:
valid_barcodes+=1
print valid_barcodes, 'unique valid viral barcodes present'
#"""
### If the viral barcodes have frequent errors - associate the error with the reference in a cell-specific manner
### Only one virus for cell should be present unless it is a doublet
print len(cells_with_virus), 'cells with viral barcodes'
doublet_cell={}
mismatch_to_match={}
cells_with_valid_barcodes=0
viral_barcodes_overide={}
cellular_barcodes_overide={}
for cellular in cells_with_virus:
cell_5prime={}
cell_3prime={}
ref_sequences=[]
if len(cells_with_virus[cellular])>1:
for i in cells_with_virus[cellular]:
try: cell_5prime[i[:10]].append(i)
except Exception: cell_5prime[i[:10]]=[i]
try: cell_3prime[i[-10:]].append(i)
except Exception: cell_3prime[i[-10:]]=[i]
if i in reference_48mers:
ref_sequences.append(i)
if len(ref_sequences)>0:
cells_with_valid_barcodes+=1 ### Determine how many cells have valid viral barcodes
cell_5prime_ls=[]
cell_3prime_ls=[]
for i in cell_5prime:
cell_5prime_ls.append([len(cell_5prime[i]),i])
for i in cell_3prime:
cell_3prime_ls.append([len(cell_3prime[i]),i])
cell_5prime_ls.sort(); cell_3prime_ls.sort()
for seq in ref_sequences:
if cell_5prime_ls[-1][1] in seq and cell_3prime_ls[-1][1] in seq:
ref_seq = seq
try: viral_barcodes_overide[ref_seq].append(cellular)
except: viral_barcodes_overide[ref_seq]=[cellular]
cellular_barcodes_overide[cellular]=[ref_seq]
for y in cell_5prime[cell_5prime_ls[-1][1]]:
mismatch_to_match[y] = ref_seq
for y in cell_3prime[cell_3prime_ls[-1][1]]:
mismatch_to_match[y] = ref_seq
else:
for i in cells_with_virus[cellular]:
if i in reference_48mers:
cells_with_valid_barcodes+=1 ### Determine how many cells have valid viral barcodes
try: viral_barcodes_overide[i].append(cellular)
except: viral_barcodes_overide[i]=[cellular]
viral_barcodes = viral_barcodes_overide
cells_with_virus = cellular_barcodes_overide
### Update the viral_barcodes dictionary
viral_barcodes2={}; cells_with_virus2={}
for v in viral_barcodes:
cell_barcodes = viral_barcodes[v]
proceed = False
if v in mismatch_to_match:
v = mismatch_to_match[v]
proceed = True
elif v in reference_48mers:
proceed = True
if proceed:
if v in viral_barcodes2:
for c in cell_barcodes:
if c not in viral_barcodes2:
viral_barcodes2[v].append(c)
else:
viral_barcodes2[v] = cell_barcodes
print cells_with_valid_barcodes, 'cells with valid viral barcodes.'
viral_barcodes = viral_barcodes2
### Update the cells_with_virus dictionary
for v in viral_barcodes:
cell_barcodes = viral_barcodes[v]
for c in cell_barcodes:
if c in cells_with_virus2:
if v not in cells_with_virus2[c]:
cells_with_virus2[c].append(v)
else:
cells_with_virus2[c]=[v]
cells_with_virus = cells_with_virus2
for c in cells_with_virus:
if len(cells_with_virus[c])>1:
doublet_cell[c]=[]
print len(doublet_cell),'doublets'
#print cells_with_virus['ACGCCGATCTGTTGAG']
#print cells_with_virus['CAGAATCCAAACTGCT']
#sys.exit()
print len(cells_with_virus),'updated cells with virus'
print len(viral_barcodes),'updated unique viral barcodes'
#"""
#reference_48mers={}
multi_cell_mapping=0
unique_cells={}
multiMappingFinal={}
import collections
import unique
event_db = collections.OrderedDict()
for cluster in cluster_header:
event_db[cluster]='0'
k_value = 1
import unique
cluster_hits_counts={}
cluster_pairs={}
custom=[]
cells_per_pattern={}
for viral in viral_barcodes:
clusters=[]
k=len(unique.unique(viral_barcodes[viral]))
if k>k_value:
proceed=True
if len(reference_48mers)>0:
if viral in reference_48mers:
proceed = True
else: proceed = False
if proceed:
viral_cluster_db = copy.deepcopy(event_db) ### copy this
multi_cell_mapping+=1
cell_tracker=[]
multilin=[]
all_cells=[]
for cell in viral_barcodes[viral]:
#if cell not in doublet_cell:
cell_tracker.append(cell)
try: unique_cells[cell].append(viral)
except: unique_cells[cell] = [viral]
if cell in cell_clusters:
cluster = cell_clusters[cell]
if 'Multi-Lin' == cluster:
multilin.append(cell)
all_cells.append(cell)
viral_cluster_db[cluster]='1'
clusters.append(cluster)
c1= unique.unique(clusters)
c2 = string.join(c1,'|')
try: cells_per_pattern[c2]+=all_cells
except: cells_per_pattern[c2]=all_cells
#if c1 == ['Multi-Lin c4-Mast']:
#if c1 == ['MultiLin','MEP','Myelo-1'] or c1 == ['MultiLin','MEP','Myelo-2'] or c1 == ['MultiLin','MEP','Myelo-4']:
#if 'Multi-Lin c4-Mast' in c1 and ('ERP-primed' not in c1 and 'MEP' not in c1 and 'MKP-primed' not in c1 and 'MKP' not in c1 and 'ERP' not in c1) and 'Monocyte' not in c1 and 'e-Mono' not in c1 and ('Gran' in c1 or 'Myelo-1' in c1 or 'Myelo-2' in c1 and 'Myelo-3' in c1 and 'Myelo-4' in c1):
#if 'Multi-Lin' in c1 and ('e-Mono' in c1 or 'Monocyte' in c1) and ('ERP-primed' in c1 or 'MEP' in c1 or 'MKP-primed' in c1 or 'MKP' in c1) and ('Gran' in c1 or 'Myelo-4' in c1 or 'Myelo-1' in c1 or 'Myelo-2' in c1 or 'Myelo-3' in c1):
if 'Multi-Lin' in c1:
for cell in multilin:
eom.write(string.join(c1,'|')+'\t'+cell+'\t'+viral+'\n')
custom+=viral_barcodes[viral]
#print 'custom:',custom
multiMappingFinal[viral]=viral_cluster_db
### Count the number of cluster pairs to make a weighted network
for c1 in clusters:
for c2 in clusters:
if c1 != c2:
try:
cx = cluster_pairs[c1]
try: cx[c2]+=1
except: cx[c2]=1
except:
cx={}
cx[c2]=1
cluster_pairs[c1] = cx
clusters = string.join(unique.unique(clusters),'|')
try: cluster_hits_counts[clusters]+=1
except Exception: cluster_hits_counts[clusters]=1
#sys.exit()
#print custom
for cluster in cluster_pairs:
cluster_counts=[]
cx = cluster_pairs[cluster]
for c2 in cx:
count=cx[c2]
cluster_counts.append([count,c2])
cluster_counts.sort()
cluster_counts.reverse()
#print cluster, cluster_counts
print len(multiMappingFinal)
final_ranked_cluster_hits=[]
for clusters in cluster_hits_counts:
final_ranked_cluster_hits.append([cluster_hits_counts[clusters],clusters])
final_ranked_cluster_hits.sort()
final_ranked_cluster_hits.reverse()
for (counts,clusters) in final_ranked_cluster_hits:
try:
print str(counts)+'\t'+clusters+'\t'+str(len(unique.unique(cells_per_pattern[clusters])))
#print cells_per_pattern[clusters];sys.exit()
except: print str(counts)+'\t'+clusters
eo.write(string.join(['UID']+cluster_header,'\t')+'\n')
for viral_barcode in multiMappingFinal:
cluster_db = multiMappingFinal[viral_barcode]
hits=[]
for cluster in cluster_db:
hits.append(cluster_db[cluster])
eo.write(string.join([viral_barcode]+hits,'\t')+'\n')
eo.close()
eo = export.ExportFile(viral_barcode_file[:-4]+'-cells-'+str(k_value)+'.txt')
for cell in unique_cells:
#eo.write(cell+'\t1\t1\t'+str(len(unique_cells[cell]))+'\t'+string.join(unique_cells[cell],'|')+'\n')
eo.write(cell+'\t1\t1\t\n')
eo.close()
#print multi_cell_mapping
#print len(unique_cells)
if __name__ == '__main__':
cellBarcodes = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/Viral-tracking/3-prime/cellular-viral_und-det.txt'
cellClusters = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/Viral-tracking/3-prime/groups.cellHarmony-Celexa5prime-Merged.txt'
#cellClusters = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/Viral-tracking/3-prime/groups.cellHarmony-Celexa5prime-MultiMerge.txt'
#cellClusters = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/Viral-tracking/3-prime/groups.cellHarmony-Celexa5prime-Baso.txt'
barcode1 = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/Viral-tracking/3-prime/14mer.txt'
barcode2 = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/Viral-tracking/3-prime/30mer.txt'
references = importViralBarcodeReferences(barcode1,barcode2)
#references={}
processBarcodes(cellBarcodes,cellClusters,references);sys.exit()
import getopt
filter_rows=False
filter_file=None
genome = 'hg19'
dataset_name = '10X_filtered'
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Insufficient options provided";sys.exit()
#Filtering samples in a datasets
#python 10XProcessing.py --i /Users/test/10X/outs/filtered_gene_bc_matrices/ --g hg19 --n My10XExperiment
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['i=','g=','n='])
#print sys.argv[1:]
for opt, arg in options:
if opt == '--i': matrices_dir=arg
elif opt == '--g': genome=arg
elif opt == '--n': dataset_name=arg
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/quantifyBarcodes.py
|
quantifyBarcodes.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from scipy import sparse, io
import numpy
import LineageProfilerIterate
import cluster_corr
from import_scripts import ChromiumProcessing
import traceback
""" cellHarmony with Louvain Clustering Funcitons """
def manage_louvain_alignment(species,platform,query_exp_file,exp_output,
customMarkers=False,useMulti=False,fl=None,customLabels=None):
""" Align an query expression dataset (txt, mtx, h5) to a reference expression dataset
(txt, mtx, h5) or ICGS results file (marker genes and cell clusters). In the absence of
an ICGS results file, a customMarkers gene file (list of genes) and customLabels two
column text files with barcodes (left) and group labels (right) should be supplied"""
if customLabels==None or customLabels == '':
try: customLabels = fl.Labels()
except: pass
try: returnCentroids = fl.ReturnCentroids()
except Exception: returnCentroids = 'community'
if customLabels == '':
customLabels = None
if customLabels !=None:
customLabels = cluster_corr.read_labels_dictionary(customLabels)
try:
reference_exp_file = fl.reference_exp_file()
except:
reference_exp_file = False
sparse_ref, full_ref_dense, peformDiffExpAnalysis = pre_process_files(reference_exp_file,species,fl,'reference',customMarkers)
sparse_query, full_query_dense, peformDiffExpAnalysis = pre_process_files(query_exp_file,fl,species,'query',customMarkers)
if sparse_ref or sparse_query:
### One file is h5 or mtx
if sparse_ref:
ref = reference_exp_file
reference_exp_file = full_ref_dense
reference = full_ref_dense
try: ### Should always replace the h5 file with dense matrix
fl.set_reference_exp_file(full_ref_dense)
except: pass
if sparse_query:
query = query_exp_file
query_exp_file = full_query_dense
#if 'ICGS' in customMarkers or 'MarkerGene' in customMarkers:
""" When performing cellHarmony, build an ICGS expression reference with log2 TPM values rather than fold """
print 'Attempting to convert ICGS folds to ICGS expression values as a reference first...'
try: customMarkers = LineageProfilerIterate.convertICGSClustersToExpression(customMarkers,query_exp_file,returnCentroids=False,species=species,fl=fl)
except:
print "Using the supplied reference file only (not importing raw expression)...Proceeding without differential expression analsyes..."
peformDiffExpAnalysis = False
try: fl.setPeformDiffExpAnalysis(peformDiffExpAnalysis)
except: pass
print traceback.format_exc()
reference = customMarkers ### Not sparse
gene_list = None
if species != None:
gene_list = cluster_corr.read_gene_list(customMarkers)
export_directory = os.path.abspath(os.path.join(query_exp_file, os.pardir))
dataset_name = string.replace(string.split(query_exp_file,'/')[-1][:-4],'exp.','')
try: os.mkdir(export_directory+'/CellClassification/')
except: pass
output_classification_file = export_directory+'/CellClassification/'+dataset_name+'-CellClassification.txt'
if sparse_ref and sparse_query:
### Use the h5 files for alignment
pass
else:
ref = reference
query = query_exp_file
louvain_results = cluster_corr.find_nearest_cells(ref,
query,
gene_list=gene_list,
num_neighbors=10,
num_trees=100,
louvain_level=0,
min_cluster_correlation=-1,
genome=species)
cluster_corr.write_results_to_file(louvain_results, output_classification_file, labels=customLabels)
try:
LineageProfilerIterate.harmonizeClassifiedSamples(species, reference, query_exp_file, output_classification_file,fl=fl)
except:
print '\nFAILED TO COMPLETE THE FULL CELLHARMONY ANALYSIS (SEE LOG FILE)...'
print traceback.format_exc()
return True
def pre_process_files(exp_file,species,fl,type,customMarkers):
""" If a matrix or h5 file, produce the full matrix if performing a full analysis """
ICGS=True
with open(customMarkers, 'rU') as f:
for line in f:
if len(line.split('\t', 1))>10:
ICGS=True
break
try:
peformDiffExpAnalysis=fl.PeformDiffExpAnalysis()
except:
peformDiffExpAnalysis = True
sparse_file = False
file_path = False
if exp_file != False and exp_file !=None:
if '.h5' in exp_file or '.mtx' in exp_file:
sparse_file = True
if ICGS: ### Hence, cellHarmony can visualize the data as combined heatmaps
#if not os.path.exists(output_file):
print 'Pre-Processing matrix file'
file_path = ChromiumProcessing.import10XSparseMatrix(exp_file,species,'cellHarmony-'+type)
return sparse_file, file_path, peformDiffExpAnalysis
if __name__ == '__main__':
from argparse import ArgumentParser
parser = ArgumentParser(description="find the cells in reference_h5 that are most similar to the cells in query_h5")
parser.add_argument("reference_h5", help="a CellRanger h5 file")
parser.add_argument("query_h5", help="a CellRanger h5 file")
parser.add_argument("output", help="the result file to write")
parser.add_argument("-g", "--genes", default=None, help="an ICGS file with the genes to use")
parser.add_argument("-s", "--genome", default=None, help="genome aligned to")
parser.add_argument("-k", "--num_neighbors", type=int, default=10,
help="number of nearest neighbors to use in clustering, default: %(default)s")
parser.add_argument("-t", "--num_trees", type=int, default=100,
help="number of trees to use in random forest for approximating nearest neighbors, default: %(default)s")
parser.add_argument("-l", "--louvain", type=int, default=0,
help="what level to cut the clustering dendrogram. 0 is the most granular, -1 the least. Default: %(default)s")
parser.add_argument("-m", "--min_correlation", type=float, default=-1,
help="the lowest correlation permissible between clusters. Any clusters in query that don't correlate to ref at least this well will be skipped. Default: %(default)s")
parser.add_argument("-d", "--diff_expression", type=float, default=True,
help="perform differential expression analyses. Default: %(default)s")
parser.add_argument("-b", "--labels", type=str, default=None, help = "a tab-delimited text file with two columns (reference cell barcode and cluster name)")
args = parser.parse_args()
genome = None
if args.genes != None:
genome = args.genome
if args.labels != None:
labels = cluster_corr.read_labels_dictionary(args.labels)
platform = 'RNASeq'
manage_louvain_alignment(genome,platform,args.query_h5,None,
customMarkers=args.reference_h5,useMulti=False,fl=None,customLabels=labels)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/cellHarmony.py
|
cellHarmony.py
|
from __future__ import print_function
from cell_collection import CellCollection
from annoy import AnnoyIndex
import community # python-louvain package, imported as community
import networkx
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) # import parent dir dependencies
import numpy as np
import time
def read_gene_list(filename):
"""
Reads the gene list from a file
"""
gene_list = []
with open(filename, 'rU') as f:
for line in f:
gene = line.split('\t', 1)[0]
if ' ' in gene:
gene = string.split(gene.rstrip(),' ')[0]
if ':' in gene:
gene_list.append((gene.rstrip().split(':'))[1])
else:
gene_list.append(gene.rstrip())
return gene_list
def read_labels_dictionary(filename):
"""
Reads the labels assigned by the user to each barcode (first and last columns)
"""
label_dictionary = {}
with open(filename, 'rU') as f:
header_lines = 2
for line in f:
barcode = line.split('\t', 1)[0]
label = line.split('\t', 1)[-1]
label_dictionary[barcode]=label.rstrip()
return label_dictionary
def data_check(ref_h5_filename, query_h5_filename):
""" If h5 files, are both h5? """
if '.h5' in ref_h5_filename and '.h5' not in query_h5_filename:
return False
else:
return True
def find_nearest_cells(ref_h5_filename, query_h5_filename, gene_list=None, genome=None,
num_neighbors=10, num_trees=100, louvain_level=-1, min_cluster_correlation=-1):
"""
For every cell in query_h5_filename, identifies the most similar cell in ref_h5_filename.
For parameter definitions, see partition_h5_file() and find_closest_cluster(), this is a convenience
function that calls them (among others)
"""
### Do the two input file formats match?
matching = data_check(ref_h5_filename, query_h5_filename)
startT = time.time()
if '.mtx' not in ref_h5_filename and '.h5' not in ref_h5_filename:
""" Assumes partial overlapping gene lists present """
gene_list = find_shared_genes(ref_h5_filename,genome=genome,gene_list=gene_list)
gene_list = find_shared_genes(query_h5_filename,genome=genome,gene_list=gene_list)
ref_partition = partition_h5_file(ref_h5_filename, gene_list=gene_list, num_neighbors=num_neighbors,
num_trees=num_trees, louvain_level=louvain_level,genome=genome)
query_partition = partition_h5_file(query_h5_filename, gene_list=gene_list, num_neighbors=num_neighbors,
num_trees=num_trees, louvain_level=louvain_level,genome=genome)
best_match = find_closest_cluster(query_partition, ref_partition, min_correlation=min_cluster_correlation)
q_result = {}
r_result = {}
for query_part_id, ref_part_id in best_match:
ref = ref_partition[ref_part_id]
query = query_partition[query_part_id]
for idx in range(query.num_cells()):
q_barcode = query.get_barcode(idx)
best_bc, best_cor = ref.find_best_correlated(query.get_cell_expression_vector(idx))
q_result[q_barcode] = {'barcode': best_bc,
'correlation': best_cor,
'query_partition': query_part_id,
'ref_partition': ref_part_id}
### Prepare the reference partition data for export
for ref_part_id in ref_partition:
ref = ref_partition[ref_part_id]
for idx in range(ref.num_cells()):
r_barcode = ref.get_barcode(idx)
r_result[r_barcode] = {'barcode': r_barcode,
'correlation': 'NA',
'query_partition': 'NA',
'ref_partition': ref_part_id}
print('cellHarmony-community alignment complete in %s seconds' % str(time.time()-startT))
return q_result, r_result
def write_results_to_file(results, filename, labels=None):
def add_labels(barcode):
alt_barcode = string.replace(barcode,'.Reference','')
if barcode in labels:
return labels[barcode]
if alt_barcode in labels:
return labels[alt_barcode]
if ':' in barcode:
return labels[barcode.split(':')[1]]
else:
return 'NA'
if labels == None:
with open(filename, 'w') as f:
print("\t".join( ("Query Barcode", "Ref Barcode", "Correlation", "Query Partition", "Ref Partition") ), file=f)
for q in results.keys():
print("\t".join( (q,
results[q]['barcode'],
str(results[q]['correlation']),
str(results[q]['query_partition']),
str(results[q]['ref_partition'])) ), file=f)
else:
with open(filename, 'w') as f:
print("\t".join( ("Query Barcode", "Ref Barcode", "Correlation", "Query Partition", "Ref Partition", "Label") ), file=f)
for q in results.keys():
print("\t".join( (q,
results[q]['barcode'],
str(results[q]['correlation']),
str(results[q]['query_partition']),
str(results[q]['ref_partition']),
add_labels(results[q]['barcode'])) ), file=f)
def nearest_neighbors(collection, num_neighbors=10, n_trees=100):
"""
Finds the num_neighbors nearest neighbors to each cell in the sparse matrix
Return result is a dictionary of lists, where the key is an index into the cells,
and the value is the neighbors of that cell
"""
nn_idx = AnnoyIndex(collection.num_genes())
# Add the elements in reverse order because Annoy allocates the memory based on
# the value of the element added - so adding in increasing order will trigger
# lots of allocations
for i in range(collection.num_cells()-1, -1, -1):
nn_idx.add_item(i, collection.get_cell_expression_vector(i))
nn_idx.build(n_trees)
return { i: nn_idx.get_nns_by_item(i, num_neighbors) for i in range(collection.num_cells()) }
def identify_clusters(graph, louvain_level=-1):
"""
Identifies clusters in the given NetworkX Graph by Louvain partitioning.
The parameter louvain_level controls the degree of partitioning. 0 is the most granular
partition, and granularity decreases as louvain_level increases. Since the number of
levels can't be known a priori, negative values "count down" from the max - ie, -1
means to use the maximum possible value and thus get the largest clusters
"""
dendrogram = community.generate_dendrogram(graph)
if louvain_level < 0:
louvain_level = max(0, len(dendrogram) + louvain_level)
if louvain_level >= len(dendrogram):
#print("Warning [identify_clusters]: louvain_level set to {}, max allowable is {}. Resetting".format(louvain_level, len(dendrogram)-1), file=sys.stderr)
louvain_level = len(dendrogram) - 1
#print("Cutting the Louvain dendrogram at level {}".format(louvain_level), file=sys.stderr)
return community.partition_at_level(dendrogram, louvain_level)
def find_shared_genes(h5_filename,genome=None,gene_list=None):
"""
Selects genes shared by the reference, query and gene_list
for filtering genes
"""
if gene_list !=None:
if '.h5' in h5_filename:
genes = CellCollection.from_cellranger_h5(h5_filename,returnGenes=True)
elif 'txt' in h5_filename:
try:
genes = CellCollection.from_tsvfile_alt(h5_filename,genome,returnGenes=True,gene_list=gene_list)
except:
genes = CellCollection.from_tsvfile(h5_filename,genome,returnGenes=True,gene_list=gene_list)
else:
genes = CellCollection.from_cellranger_mtx(h5_filename,genome,returnGenes=True)
gene_list = list(set(genes) & set(gene_list))
return gene_list
def partition_h5_file(h5_filename, gene_list=None, num_neighbors=10, num_trees=100,
louvain_level=-1,genome=None):
"""
Reads a CellRanger h5 file and partitions it by clustering on the k-nearest neighbor graph
Keyword arguments:
gene_list - restricts the analysis to the specified list of gene symbols. Default is to not restrict
num_neighbors - the number of nearest neighbors to compute for each cell
num_trees - the number of trees used in the random forest that approximates the nearest neighbor calculation
louvain_level - the level of the Louvain clustering dendrogram to cut at. Level 0 is the lowest (most granular)
level, and higher levels get less granular. The highest level is considered the "best" set
of clusters, but the number of levels is not known a priori. Hence, negative values will
count down from the highest level, so -1 will always be the "best" clustering, regardless of
the actual number of levels in the dendrogram
Return Result: A dictionary, where the keys are partition ids and the values are the CellCollection for that partition
"""
if '.h5' in h5_filename:
collection = CellCollection.from_cellranger_h5(h5_filename)
data_type = 'h5'
elif 'txt' in h5_filename:
try:
collection = CellCollection.from_tsvfile_alt(h5_filename,genome,gene_list=gene_list)
except:
collection = CellCollection.from_tsvfile(h5_filename,genome)
data_type = 'txt'
else:
collection = CellCollection.from_cellranger_mtx(h5_filename,genome)
data_type = 'mtx'
if gene_list != None:
collection.filter_genes_by_symbol(gene_list,data_type)
neighbor_dict = nearest_neighbors(collection, num_neighbors=num_neighbors, n_trees=num_trees)
cluster_definition = identify_clusters(networkx.from_dict_of_lists(neighbor_dict), louvain_level=louvain_level)
return collection.partition(cluster_definition)
def compute_centroids(collection_dict):
"""Returns (centroid matrix, ID list) for the given dictionary of CellCollections"""
centroids = np.concatenate( [ p.centroid() for p in collection_dict.values() ], axis=1 )
return centroids, collection_dict.keys()
def find_closest_cluster(query, ref, min_correlation=-1):
"""
For each collection in query, identifies the collection in ref that is most similar
query and ref are both dictionaries of CellCollections, keyed by a "partition id"
Returns a list containing the best matches for each collection in query that meet the
min_correlation threshold. Each member of the list is itself a list containing the
id of the query collection and the id of its best match in ref
"""
query_centroids, query_ids = compute_centroids(query)
ref_centroids, ref_ids = compute_centroids(ref)
print('number of reference partions %d, number of query partions %d' % (len(ref_ids),len(query_ids)))
all_correlations = np.corrcoef(np.concatenate((ref_centroids, query_centroids), axis=1), rowvar=False)
# At this point, we have the correlations of everything vs everything. We only care about query vs ref
# Extract the top-right corner of the matrix
nref = len(ref)
corr = np.hsplit(np.vsplit(all_correlations, (nref, ))[0], (nref,))[1]
best_match = zip(range(corr.shape[1]), np.argmax(corr, 0))
# At this point, best_match is: 1) using indices into the array rather than ids,
# and 2) not restricted by the threshold. Fix before returning
return ( (query_ids[q], ref_ids[r]) for q, r in best_match if corr[r,q] >= min_correlation )
if __name__ == "__main__":
# genes = read_gene_list('data/FinalMarkerHeatmap_all.txt')
# results = find_nearest_cells('data/reference-filtered_gene_bc_matrices_h5.h5',
# 'data/query-filtered_gene_bc_matrices_h5.h5',
# gene_list=genes, louvain_level=-1)
# write_results_to_file(results, 'temp.txt')
from argparse import ArgumentParser
parser = ArgumentParser(description="find the cells in reference_h5 that are most similar to the cells in query_h5")
parser.add_argument("reference_h5", help="a CellRanger h5 file")
parser.add_argument("query_h5", help="a CellRanger h5 file")
parser.add_argument("output", help="the result file to write")
parser.add_argument("-g", "--genes", default=None, help="an ICGS file with the genes to use")
parser.add_argument("-s", "--genome", default=None, help="genome aligned to")
parser.add_argument("-k", "--num_neighbors", type=int, default=10,
help="number of nearest neighbors to use in clustering, default: %(default)s")
parser.add_argument("-t", "--num_trees", type=int, default=100,
help="number of trees to use in random forest for approximating nearest neighbors, default: %(default)s")
parser.add_argument("-l", "--louvain", type=int, default=0,
help="what level to cut the clustering dendrogram. 0 is the most granular, -1 the least. Default: %(default)s")
parser.add_argument("-m", "--min_correlation", type=float, default=-1,
help="the lowest correlation permissible between clusters. Any clusters in query that don't correlate to ref at least this well will be skipped. Default: %(default)s")
parser.add_argument("-b", "--labels", type=str, default=None, help = "a tab-delimited text file with two columns (reference cell barcode and cluster name)")
args = parser.parse_args()
gene_list = None
genome = None
labels = None
if args.genes != None:
gene_list = read_gene_list(args.genes)
if args.labels != None:
labels = read_labels_dictionary(args.labels)
if args.genome != None:
genome = args.genome
q_results, r_results = find_nearest_cells(args.reference_h5,
args.query_h5,
gene_list=gene_list,
num_neighbors=args.num_neighbors,
num_trees=args.num_trees,
louvain_level=args.louvain,
min_cluster_correlation=args.min_correlation,
genome=genome)
write_results_to_file(q_results, args.output,labels=labels)
write_results_to_file(r_results, args.output[:-4]+'-refererence.txt',labels=labels)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/cluster_corr.py
|
cluster_corr.py
|
from __future__ import print_function
try:
import h5py
from h5py import defs, utils, h5ac, _proxy # for py2app
except:
print ('Missing the h5py library (hdf5 support)...')
import gzip
import scipy.io
from scipy import sparse, stats, io
import numpy as np
import sys, string, os, csv, math
import time
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
def index_items(universe, itemset):
"""
Returns a list of indices to the items in universe that match items in itemset
"""
return [ idx for idx, item in enumerate(universe) if item in itemset ]
class CellCollection:
"""
Encapsulates a cohort of cells, ie from a CellRanger run
Expression values are stored in a sparse matrix, and barcodes/gene identifiers are
maintained in parallel arrays. Construct by calling CellCollection.from_file(), or one
of the other specialized static constructors
"""
@staticmethod
def from_cellranger_h5(h5_filename, genome=None, returnGenes=False):
"""
Creates a CellCollection from the contents of an H5 file created by CellRanger.
The meaning of the genome parameter differs depending on the version of CellRanger that created the h5.
For CellRanger version 2, the genome parameters specifies the matrix to load. If genome is None, the
single matrix present will be loaded (using genome==None when multiple genomes are present in the file
is an error and will cause an exception).
For CellRanger version 3, genome is now specified as an attribute of the features (typically genes).
In this version, specifying a genome will filter the matrix to only include features from that genome.
Whether a genome is specified or not, non-gene features will be removed
"""
start = time.time()
coll = CellCollection()
f = h5py.File(h5_filename, 'r')
if 'matrix' in f:
# CellRanger v3
coll._barcodes = f['matrix']['barcodes']
coll._gene_ids = f['matrix']['features']['id']
coll._gene_names = f['matrix']['features']['name']
if returnGenes:
""" Do not import the matrix at this point """
return list(coll._gene_names)
coll._matrix = sparse.csc_matrix((f['matrix']['data'], f['matrix']['indices'], f['matrix']['indptr']), shape=f['matrix']['shape'])
indices = np.flatnonzero(np.array(f['matrix']['features']['genome']) != '') if \
genome == None else \
np.flatnonzero(np.array(f['matrix']['features']['genome']) == genome)
coll._filter_genes_by_index(indices.tolist())
else:
# CellRanger v2
if genome == None:
possible_genomes = f.keys()
if len(possible_genomes) != 1:
raise Exception("{} contains multiple genomes ({}). Explicitly select one".format(h5_filename, ", ".join(possible_genomes)))
genome = possible_genomes[0]
#print("Auto-selecting genome {}".format(genome), file=sys.stderr)
coll._gene_names = f[genome]['gene_names']
if returnGenes:
""" Do not import the matrix at this point """
return list(coll._gene_names)
coll._matrix = sparse.csc_matrix((f[genome]['data'], f[genome]['indices'], f[genome]['indptr']))
coll._barcodes = f[genome]['barcodes']
coll._gene_ids = f[genome]['genes']
print('sparse matrix data imported from h5 file in %s seconds' % str(time.time()-start))
return coll
@staticmethod
def from_cellranger_mtx(mtx_directory, genome=None, returnGenes=False):
"""
Creates a CellCollection from a sparse matrix (.mtx and associated files) exported by CellRanger
Recognize directories from CellRanger version 2 (files: matrix.mtx, genes.tsv, barcodes.tsv) and
CellRanger v3 (files: matrix.mtx.gz, features.tsv.gz, barcodes.tsv.gz)
"""
start = time.time()
coll = CellCollection()
cellranger_version = 2
if '.mtx' in mtx_directory:
mtx_file = mtx_directory ### Hence an mtx file was directly supplied
mtx_directory = os.path.abspath(os.path.join(mtx_file, os.pardir))
else:
mtx_file = os.path.join(mtx_directory, "matrix.mtx")
if not os.path.exists(mtx_file):
cellranger_version = 3
mtx_file = mtx_file + ".gz"
if not os.path.exists(mtx_file):
raise Exception("Directory {} does not contain a recognizable matrix file".format(mtx_directory))
if '.gz' in mtx_file:
cellranger_version = 3
sparse_matrix = io.mmread(mtx_file)
coll._matrix = sparse_matrix.tocsc()
coll._gene_ids = np.empty((coll._matrix.shape[0], ), np.object)
coll._gene_names = np.empty((coll._matrix.shape[0], ), np.object)
if cellranger_version == 2:
with open(os.path.join(mtx_directory, "genes.tsv"), "rU") as f:
idx = 0
for line in f:
i, n = line.rstrip().split("\t")
coll._gene_ids[idx] = i
coll._gene_names[idx] = n
idx += 1
with open(os.path.join(mtx_directory, "barcodes.tsv"), "rU") as f:
coll._barcodes = np.array( [ line.rstrip() for line in f ] )
else:
with gzip.open(os.path.join(mtx_directory, "features.tsv.gz"), "rt") as f:
idx = 0
indices = []
for line in f:
i, n, t = line.rstrip().split("\t")
coll._gene_ids[idx] = i
coll._gene_names[idx] = n
if t == 'Gene Expression':
indices.append(idx)
idx += 1
coll._filter_genes_by_index(indices)
with gzip.open(os.path.join(mtx_directory, "barcodes.tsv.gz"), "rt") as f:
coll._barcodes = np.array( [ line.rstrip() for line in f ] )
if returnGenes:
""" Do not import the matrix at this point """
return list(coll._gene_names)
print('sparse matrix data imported from mtx file in %s seconds' % str(time.time()-start))
return coll
@staticmethod
def from_tsvfile_alt(tsv_file, genome=None, returnGenes=False, gene_list=None):
"""
Creates a CellCollection from the contents of a tab-separated text file.
"""
startT = time.time()
coll = CellCollection()
UseDense=False
header=True
skip=False
for line in open(tsv_file,'rU').xreadlines():
if header:
delimiter = ',' # CSV file
start = 1
if 'row_clusters' in line:
start=2 # An extra column and row are present from the ICGS file
skip=True
if '\t' in line:
delimiter = '\t' # TSV file
barcodes = string.split(line.rstrip(),delimiter)[start:]
if ':' in line:
barcodes = map(lambda x:x.split(':')[1],barcodes)
coll._barcodes=barcodes
coll._gene_names=[]
data_array=[]
header=False
elif skip:
skip=False # Igore the second row in the file that has cluster info
else:
values = line.rstrip().split(delimiter)
gene = values[0]
if ' ' in gene:
gene = string.split(gene,' ')[0]
if ':' in gene:
gene = (gene.rstrip().split(':'))[1]
if gene_list!=None:
if gene not in gene_list:
continue
coll._gene_names.append(gene)
""" If the data (always log2) is a float, increment by 0.5 to round up """
if returnGenes==False:
if UseDense:
data_array.append(map(float,values[start:]))
else:
#data_array.append(map(lambda x: round(math.pow(2,float(x))),values[start:]))
data_array.append(map(float,values[start:]))
if returnGenes:
""" Do not import the matrix at this point """
return list(coll._gene_names)
if UseDense:
coll._matrix = np.array(data_array)
else:
""" Convert to a sparse matrix """
coll._matrix = sparse.csc_matrix(np.array(data_array))
coll._barcodes = np.array(coll._barcodes)
coll._gene_names = np.array(coll._gene_names)
coll._gene_ids = coll._gene_names
print('sparse matrix data imported from TSV file in %s seconds' % str(time.time()-startT))
#print (len(coll._gene_ids),len(coll._barcodes))
return coll
@staticmethod
def from_tsvfile(tsv_filename, genome=None, returnGenes=False, gene_list=None):
"""
Generates a CellCollection from a (dense) tab-separated file, where cells are in
columns and
"""
start = time.time()
coll = CellCollection()
with open(tsv_filename, "rU") as f:
try:
line = next(f)
except StopIteration:
raise Exception("TSV file {} is empty".format(tsv_filename))
### Check formatting
skip=False
if '\t' in line:
delimiter = '\t' # TSV file
else:
delimiter = ','
col_start = 1
if 'row_clusters' in line:
col_start=2 # An extra column and row are present from the ICGS file
skip=True
### Check formatting end
coll._barcodes = np.array(line.rstrip().split(delimiter)[col_start:])
sparse_matrix = sparse.lil_matrix((50000, len(coll._barcodes)), dtype=np.float_)
coll._gene_names = np.empty((sparse_matrix.shape[0], ), np.object)
row = 0
for line in f:
if row==0 and skip:
skip = False
continue
vals = line.rstrip().split(delimiter)
coll._gene_names[row] = vals[0]
if returnGenes==False:
for i in range(col_start, len(vals)):
if vals[i] != "0":
sparse_matrix[row, i-col_start] = float(vals[i])
if row == sparse_matrix.shape[0]-1:
sparse_matrix.resize(sparse_matrix.shape + (10000, 0))
coll._gene_names.resize(coll._gene_names.shape + (10000, 0))
row += 1
coll._gene_names.resize((row, ))
if returnGenes:
""" Do not import the matrix at this point """
return list(coll._gene_names)
sparse_matrix.resize((row, len(coll._barcodes)))
coll._matrix = sparse_matrix.tocsc()
coll._gene_ids = coll._gene_names
#print('matrix shape: {}'.format(coll._matrix.shape))
print('sparse matrix data imported from TSV file in %s seconds' % str(time.time()-start))
return coll
def __init__(self):
self._matrix = sparse.csc_matrix((0,0), dtype=np.int8)
self._barcodes = ()
self._gene_names = ()
self._gene_ids = ()
def __getattr__(self, name):
"""
Methods/attributes not explicitly defined in the CellCollection are passed down
to the matrix
"""
return getattr(self._matrix, name)
def num_genes(self):
return len(self._gene_ids)
def num_cells(self):
return len(self._barcodes)
def get_barcode(self, cell_index):
return self._barcodes[cell_index]
def get_cell_expression_vector(self, cell_index):
"""
Returns a (standard, non-sparse) sequence of expression values for a given cell
"""
#try:
return self._matrix.getcol(cell_index).todense()
#except:
# return self._matrix[:,cell_index] # ith column for existing dense matrix
def centroid(self):
"""
Returns the centroid of this collection as a (standard, non-sparse) sequence.
The centroid is defined as the mean expression of each gene
"""
return self._matrix.mean(axis=1)
def partition(self, partition_dict):
"""
Returns a dictionary of CellCollections, each a distinct subset (by cell) of self.
partition_dict is a dictionary of cell index => set id, as generated by
the python-louvain methods
"""
partitions = {}
for k, v in partition_dict.items():
if v not in partitions: partitions[v] = []
partitions[v].append(k)
result = {}
for part_id in partitions.keys():
result[part_id] = self.subset_by_cell_index(partitions[part_id])
return result
def find_best_correlated(self, query):
"""
Identifies the cell in this collection that has the highest Pearson's correlation
with query (a sequence of expression values in the same order as in this collection)
Returns the pair of (barcode, r^2 value) for the best match in ref
"""
best_cor = -2
best_bc = "<None>"
for idx in range(self.num_cells()):
r = self.get_cell_expression_vector(idx)
cor = stats.pearsonr(query, r)[0][0] # pearsonr returns the pair (r^2, p-val), and for some reason the r^2 is a list
if cor > best_cor:
best_cor = cor
best_bc = self.get_barcode(idx)
return best_bc, best_cor
def filter_by_cell_index(self, cell_index):
self._matrix = self._matrix[:, cell_index]
self._barcodes = self._barcodes[cell_index]
def subset_by_cell_index(self, cell_index):
"""
Returns a new CellCollection containing only chosen cells from self
"""
cc = CellCollection()
cc._gene_ids = self._gene_ids
cc._gene_names = self._gene_names
cc._matrix = self._matrix[:, cell_index]
cc._barcodes = self._barcodes[cell_index]
return cc
def filter_barcodes(self, barcode_list):
"""
Reduces the CellCollection in-place to only contain the barcodes requested
"""
barcode_subset = set(barcode_list)
#print("Selecting {} barcodes".format(len(barcode_subset)), file=sys.stderr)
barcode_index = index_items(self._barcodes, barcode_subset)
self.filter_by_cell_index(barcode_index)
def subset_barcodes(self, barcode_list):
barcode_subset = set(barcode_list)
barcode_index = index_items(self._barcodes, barcode_subset)
return self.subset_by_cell_index(barcode_index)
def _filter_genes_by_index(self, gene_index):
#print(gene_index);sys.exit()
self._matrix = self._matrix[gene_index, :]
self._gene_ids = self._gene_ids[gene_index]
self._gene_names = self._gene_names[gene_index]
#mat_array_original = self._matrix.toarray()
#print(len(mat_array_original))
def filter_genes_by_symbol(self, symbol_list, data_type):
"""
Reduces the CellCollection in-place to only contain the genes requested.
Note that gene symbols could be non-unique, and thus more genes may remain in the
filtered collection than were requested. The order of the genes in the h5 may also
differ and the same genes may not be present in the different sets
"""
gene_subset = set(symbol_list)
#print("Selecting {} genes".format(len(gene_subset)), file=sys.stderr)
gene_index=[]
gene_names = list(self._gene_names)
if data_type == 'txt':
### below code is problematic for h5 and probably sparse matrix files
for gene in gene_subset:
if gene in gene_names:
gene_index.append(gene_names.index(gene))
else:
gene_index = index_items(self._gene_names, gene_subset) # will output genes in the full dataset order
self._filter_genes_by_index(gene_index)
def filter_genes_by_id(self, id_list):
"""
Reduces the CellCollection in-place to only contain the genes requested.
"""
gene_subset = set(id_list)
#print("Selecting {} genes".format(len(gene_subset)), file=sys.stderr)
gene_index = index_items(self._gene_ids, gene_subset)
self._filter_genes_by_index(gene_index)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/cell_collection.py
|
cell_collection.py
|
import sys,string,os,shutil
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from scipy import sparse, io
import numpy
import LineageProfilerIterate
import cluster_corr
import export
from import_scripts import ChromiumProcessing
import traceback
""" cellHarmony without alignment """
def cellHarmony(species,platform,query_exp_file,exp_output,
customMarkers=False,useMulti=False,fl=None,customLabels=None):
""" Prepare pre-aligned result files in a pre-defined format for cellHarmony post-aligment
differential and visualization analyses """
customLabels = fl.Labels()
reference_exp_file = customMarkers ### pre-formatted from Seurat or other outputs
export_directory = os.path.abspath(os.path.join(query_exp_file, os.pardir))
if 'ExpressionInput' in query_exp_file:
### Change to the root directory above ExpressionINput
export_directory = os.path.abspath(os.path.join(export_directory, os.pardir))
dataset_name = string.replace(string.split(query_exp_file,'/')[-1][:-4],'exp.','')
try: os.mkdir(export_directory+'/cellHarmony/')
except: pass
try: os.mkdir(export_directory+'/cellHarmony/CellClassification/')
except: pass
try: os.mkdir(export_directory+'/cellHarmony/OtherFiles/')
except: pass
### Get the query and reference cells, dataset names
refererence_cells, query_cells, reference_dataset, query_dataset = importCelltoClusterAnnotations(customLabels) ### Get the reference and query cells in their respective order
### copy and re-name the input expression file to the output cellHarmony directory
if len(reference_dataset)>0 and len(query_dataset)>0:
target_exp_dir = export_directory+'/cellHarmony/exp.'+reference_dataset+'__'+query_dataset+'-AllCells.txt'
else:
target_exp_dir = export_directory+'/cellHarmony/exp.cellHarmony-reference__Query-AllCells.txt'
reference_dataset = 'cellHarmony-reference'
shutil.copy(query_exp_file,target_exp_dir)
### filter and export the heatmap with just reference cells
cell_cluster_order = simpleHeaderImport(reference_exp_file)
filtered_reference_cells=[]
filtered_query_cells_db={}
filtered_query_cells=[]
representative_refcluster_cell = {}
for cell_id in cell_cluster_order:
if cell_id in refererence_cells:
filtered_reference_cells.append(cell_id)
cluster_label = refererence_cells[cell_id].Label()
### Identifies where to place each query cell
try: representative_refcluster_cell[cluster_label].append(cell_id)
except: representative_refcluster_cell[cluster_label] = [cell_id]
elif cell_id in query_cells:
filtered_query_cells_db[cell_id]=query_cells[cell_id]
filtered_query_cells.append(cell_id)
#reference_output_file = export.findParentDir(reference_exp_file)+'/'+reference_dataset+'.txt'
reference_output_file = export_directory+'/cellHarmony/OtherFiles/'+reference_dataset+'.txt'
reference_output_file2 = export_directory+'/cellHarmony/exp.'+reference_dataset+'__'+query_dataset+'-Reference.txt'
query_output_file =export_directory+'/'+query_dataset+'.txt'
### Write out separate refernece and query files
from import_scripts import sampleIndexSelection
sampleIndexSelection.filterFile(reference_exp_file,reference_output_file,['row_clusters-flat']+filtered_reference_cells,force=True)
sampleIndexSelection.filterFile(target_exp_dir,query_output_file,filtered_query_cells,force=True)
shutil.copy(reference_output_file,reference_output_file2)
### export the CellClassification file
output_classification_file = export_directory+'/cellHarmony/CellClassification/CellClassification.txt'
exportCellClassifications(output_classification_file,filtered_query_cells_db,filtered_query_cells,representative_refcluster_cell)
labels_file = export_directory+'/labels.txt'
exportLabels(labels_file,filtered_reference_cells,refererence_cells)
fl.setLabels(labels_file)
print 'Files formatted for cellHarmony... running differential expression analyses'
try:
print reference_output_file
print query_output_file
print output_classification_file
LineageProfilerIterate.harmonizeClassifiedSamples(species, reference_output_file, query_output_file, output_classification_file,fl=fl)
except:
print '\nFAILED TO COMPLETE THE FULL CELLHARMONY ANALYSIS (SEE LOG FILE)...'
print traceback.format_exc()
return True
def exportCellClassifications(output_file,query_cells,filtered_query_cells,representative_refcluster_cell):
""" Match the Louvain cellHarmony export format for the classification file """
header = 'Query Barcode\tRef Barcode\tCorrelation\tQuery Partition\tRef Partition\tLabel\n'
o = open(output_file,'w')
o.write(header)
for query_barcode in filtered_query_cells:
CI = query_cells[query_barcode]
cluster_number = CI.ClusterNumber()
label = CI.Label()
ref_barcode = representative_refcluster_cell[label][-1]
values = [query_barcode,ref_barcode,'1.0',cluster_number,cluster_number,label]
o.write(string.join(values,'\t')+'\n')
o.close()
def exportLabels(labels_file,filtered_reference_cells,refererence_cells):
l = open(labels_file,'w')
for cell_id in filtered_reference_cells:
CI = refererence_cells[cell_id]
cluster_number = CI.ClusterNumber()
label = CI.Label()
values = [cell_id,cluster_number,label]
l.write(string.join(values,'\t')+'\n')
l.close()
def simpleHeaderImport(filename):
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
if '\t' in data:
t = string.split(data,'\t')
else:
t = string.split(data,',')
header = t[2:]
header2 = []
for h in header:
if ":" in h:
h = string.split(h,':')[-1]
header2.append(h)
break
return header2
class CellInfo:
def __init__(self,cell_id, cluster_number, dataset_name, dataset_type, label):
self.cell_id = cell_id; self.cluster_number = cluster_number; self.dataset_name = dataset_name
self.dataset_type = dataset_type; self.label = label
def CellID(self): return self.cell_id
def ClusterNumber(self): return self.cluster_number
def DatasetName(self): return self.dataset_name
def DataSetType(self): return self.dataset_type
def Label(self): return self.label
def __repr__(self):
return self.CellID()+'|'+self.Label()+'|'+self.DataSetType()
def importCelltoClusterAnnotations(filename):
firstRow = True
refererence_cells={}
query_cells={}
for line in open(filename,'rU').xreadlines():
data = cleanUpLine(line)
if '\t' in data:
t = string.split(data,'\t')
else:
t = string.split(data,',')
if firstRow:
ci = t.index('cell_id')
cn = t.index('cluster_number')
try: cm = t.index('cluster_name')
except: cm = False
dn = t.index('dataset_name')
dt = t.index('dataset_type')
firstRow = False
else:
cell_id = t[ci]
cluster_number = t[cn]
dataset_name = t[dn]
dataset_type = t[dt]
if cm != False:
cluster_name = t[cm]
label = cluster_name + '_c'+cluster_number
else:
label = 'c'+cluster_number
if string.lower(dataset_type)[0] == 'r':
dataset_type = 'Reference'
reference_dataset = dataset_name
CI = CellInfo(cell_id, cluster_number, dataset_name, dataset_type, label)
refererence_cells[cell_id]=CI
else:
dataset_type = 'Query'
query_dataset = dataset_name
CI = CellInfo(cell_id, cluster_number, dataset_name, dataset_type, label)
query_cells[cell_id]=CI
return refererence_cells, query_cells, reference_dataset, query_dataset
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
if __name__ == '__main__':
platform = 'RNASeq'
cellHarmony(genome,platform,args.query_h5,None,
customMarkers=args.reference_h5,useMulti=False,fl=None,customLabels=labels)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/preAligned.py
|
preAligned.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import scipy, numpy
import statistics
from visualization_scripts import clustering
def evaluateMultiLinRegulatoryStructure(all_genes_TPM,MarkerFinder,SignatureGenes,state=None,query=None):
all_indexes, group_index, expressionData = loopThroughEachState(all_genes_TPM)
if state!=None:
states = [state] ### For example, we only want to look in annotated Multi-Lin's
else:
states = group_index
state_scores=[]
for state in states:
print '\n',state, 'running now.'
score = evaluateStateRegulatoryStructure(expressionData,all_indexes,group_index,MarkerFinder,SignatureGenes,state,query=query)
state_scores.append([score,state])
print state, score
state_scores.sort()
state_scores.reverse()
print state_scores
def loopThroughEachState(all_genes_TPM):
### Import all genes with TPM values for all cells
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(all_genes_TPM)
group_index={}
all_indexes=[]
for sampleName in group_db:
ICGS_state = group_db[sampleName][0]
try: group_index[ICGS_state].append(column_header.index(sampleName))
except Exception: group_index[ICGS_state] = [column_header.index(sampleName)]
all_indexes.append(column_header.index(sampleName))
for ICGS_state in group_index:
group_index[ICGS_state].sort()
all_indexes.sort()
expressionData = matrix, column_header, row_header, dataset_name, group_db
return all_indexes, group_index, expressionData
def evaluateStateRegulatoryStructure(expressionData, all_indexes,group_index,MarkerFinder,SignatureGenes,state,query=None):
"""Predict multi-lineage cells and their associated coincident lineage-defining TFs"""
useProbablityOfExpression=False
ICGS_State_as_Row = False
matrix, column_header, row_header, dataset_name, group_db = expressionData
def importGeneLists(fn):
genes={}
for line in open(fn,'rU').xreadlines():
data = clustering.cleanUpLine(line)
gene,cluster = string.split(data,'\t')[0:2]
genes[gene]=cluster
return genes
def importMarkerFinderHits(fn):
genes={}
genes_to_symbol={}
ICGS_State_ranked={}
skip=True
for line in open(fn,'rU').xreadlines():
data = clustering.cleanUpLine(line)
if skip: skip=False
else:
try:
gene,symbol,rho,ICGS_State = string.split(data,'\t')
except Exception:
gene,symbol,rho,rho_p,ICGS_State = string.split(data,'\t')
genes_to_symbol[gene]=symbol
#if ICGS_State!=state and float(rho)>0.0:
if float(rho)>0.3:
try: ICGS_State_ranked[ICGS_State].append([float(rho),gene,symbol])
except Exception: ICGS_State_ranked[ICGS_State] = [[float(rho),gene,symbol]]
for ICGS_State in ICGS_State_ranked:
ICGS_State_ranked[ICGS_State].sort()
ICGS_State_ranked[ICGS_State].reverse()
#print ICGS_State, ICGS_State_ranked[ICGS_State][:50]
for (rho,gene,symbol) in ICGS_State_ranked[ICGS_State][:50]:
genes[gene]=rho,ICGS_State ### Retain all population specific genes (lax)
genes[symbol]=rho,ICGS_State
return genes, genes_to_symbol
def importQueryDataset(fn):
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(fn)
return matrix, column_header, row_header, dataset_name, group_db
signatureGenes = importGeneLists(SignatureGenes)
markerFinderGenes, genes_to_symbol = importMarkerFinderHits(MarkerFinder)
#print len(signatureGenes),len(markerFinderGenes)
### Determine for each gene, its population frequency per cell state
index=0
expressedGenesPerState={}
stateAssociatedMarkers={}
def freqCutoff(x,cutoff):
if x>cutoff: return 1 ### minimum expression cutoff
else: return 0
for row in matrix:
ICGS_state_gene_frq={}
gene = row_header[index]
for ICGS_state in group_index:
state_values = map(lambda i: row[i],group_index[ICGS_state])
def freqCheck(x):
if x>1: return 1 ### minimum expression cutoff
else: return 0
expStateCells = sum(map(lambda x: freqCheck(x),state_values))
statePercentage = (float(expStateCells)/len(group_index[ICGS_state]))
ICGS_state_gene_frq[ICGS_state] = statePercentage
datasets_values = map(lambda i: row[i],all_indexes)
all_cells_frq = sum(map(lambda x: freqCheck(x),datasets_values))/(len(datasets_values)*1.0)
all_states_frq = map(lambda x: ICGS_state_gene_frq[x],ICGS_state_gene_frq)
all_states_frq.sort() ### frequencies of all non-multilin states
states_expressed = sum(map(lambda x: freqCutoff(x,0.5),all_states_frq))/(len(all_states_frq)*1.0)
for State in ICGS_state_gene_frq:
state_frq = ICGS_state_gene_frq[State]
rank = all_states_frq.index(state_frq)
if state_frq > 0.25 and rank>0: #and states_expressed<0.75 #and all_cells_frq>0.75
if 'Rik' not in gene and 'Gm' not in gene and '-' not in gene:
if gene in markerFinderGenes:# and gene in markerFinderGenes:
if ICGS_State_as_Row:
ICGS_State = signatureGenes[gene]
if gene in markerFinderGenes:
if ICGS_State_as_Row == False:
rho, ICGS_State = markerFinderGenes[gene]
else:
rho, ICGS_Cell_State = markerFinderGenes[gene] #ICGS_Cell_State
#try: gene = genes_to_symbol[gene]
#except: gene = gene
score = int(rho*100*state_frq)*(float(rank)/len(all_states_frq))
try: expressedGenesPerState[ICGS_State].append((score,gene))
except Exception: expressedGenesPerState[ICGS_State]=[(score,gene)] #(rank*multilin_frq)
try: stateAssociatedMarkers[gene,ICGS_State].append(State)
except Exception: stateAssociatedMarkers[gene,ICGS_State] = [State]
index+=1
if query!=None:
matrix, column_header, row_header, dataset_name, group_db = importQueryDataset(query)
markers_to_exclude=[]
expressedGenesPerState2={}
for (gene,ICGS_State) in stateAssociatedMarkers:
if len(stateAssociatedMarkers[(gene,ICGS_State)])<2: # or len(stateAssociatedMarkers[(gene,ICGS_State)])>len(ICGS_state_gene_frq)/2.0:
markers_to_exclude.append(gene)
else:
print ICGS_State, gene, stateAssociatedMarkers[(gene,ICGS_State)]
for ICGS_State in expressedGenesPerState:
for (score,gene) in expressedGenesPerState[ICGS_State]:
if gene not in markers_to_exclude:
try: expressedGenesPerState2[ICGS_State].append((score,gene))
except Exception: expressedGenesPerState2[ICGS_State] = [(score,gene)]
expressedGenesPerState = expressedGenesPerState2
createPseudoCell=True
### The expressedGenesPerState defines genes and modules co-expressed in the multi-Lin
### Next, find the cells that are most frequent in mulitple states
representativeMarkers={}
for ICGS_State in expressedGenesPerState:
expressedGenesPerState[ICGS_State].sort()
expressedGenesPerState[ICGS_State].reverse()
if '1Multi' not in ICGS_State:
markers = expressedGenesPerState[ICGS_State]#[:5]
markers_unique = list(set(map(lambda x: x[1],list(markers))))
print ICGS_State,":",string.join(markers_unique,', ')
if createPseudoCell:
for gene in markers:
def getBinary(x):
if x>1: return 1
else: return 0
if gene[1] in row_header: ### Only for query datasets
row_index = row_header.index(gene[1])
if useProbablityOfExpression:
pvalues = calculateGeneExpressProbilities(matrix[row_index]) ### probability of expression
values = pvalues
else:
binaryValues = map(lambda x: getBinary(x), matrix[row_index])
values = binaryValues
#values = matrix[row_index]
#if gene[1]=='S100a8': print binaryValues;sys.exit()
try: representativeMarkers[ICGS_State].append(values)
except Exception: representativeMarkers[ICGS_State] = [values]
else:
representativeMarkers[ICGS_State]=markers[0][-1]
#int(len(markers)*.25)>5:
#print ICGS_State, markers
#sys.exit()
for ICGS_State in representativeMarkers:
if createPseudoCell:
signature_values = representativeMarkers[ICGS_State]
if useProbablityOfExpression:
signature_values = [numpy.sum(value) for value in zip(*signature_values)]
else:
signature_values = [float(numpy.mean(value)) for value in zip(*signature_values)]
representativeMarkers[ICGS_State] = signature_values
else:
gene = representativeMarkers[ICGS_State]
row_index = row_header.index(gene)
gene_values = matrix[row_index]
representativeMarkers[ICGS_State] = gene_values
### Determine for each gene, its population frequency per cell state
expressedStatesPerCell={}
multilin_probability={}
import export
print 'Writing results matrix to:',MarkerFinder[:-4]+'-cellStateScores.txt'
eo = export.ExportFile(MarkerFinder[:-4]+'-cellStateScores.txt')
eo.write(string.join(['UID']+column_header,'\t')+'\n')
print 'a'
print len(representativeMarkers)
for ICGS_State in representativeMarkers:
gene_values = representativeMarkers[ICGS_State]
index=0
scoreMatrix=[]
HitsCount=0
for cell in column_header:
value = gene_values[index]
"""
expressedLiklihood = '0'
if (value<0.05 and useProbablityOfExpression==True) or (value==1 and useProbablityOfExpression==False):
try: expressedStatesPerCell[cell].append(ICGS_State)
except Exception: expressedStatesPerCell[cell] = [ICGS_State]
expressedLiklihood = '1'
HitsCount+=1
if useProbablityOfExpression:
try: multilin_probability[cell].append(value)
except Exception: multilin_probability[cell] = [value]
"""
index+=1
HitsCount+=1
scoreMatrix.append(str(value))
if HitsCount>1:
#print ICGS_State,HitsCount
eo.write(string.join([ICGS_State]+scoreMatrix,'\t')+'\n')
eo.close()
sys.exit()
def multiply(values):
p = 1
for i in values:
if i>0:
p = p*i
else:
p = p*1.e-16
return p
cell_mutlilin_ranking=[]
for cell in expressedStatesPerCell:
#if 'Multi-Lin:Gmp.R3.10' in cell: sys.exit()
if useProbablityOfExpression:
p = numpy.mean(multilin_probability[cell]) ### mean state probability
lineageCount = expressedStatesPerCell[cell]
if useProbablityOfExpression:
cell_mutlilin_ranking.append((p,len(lineageCount),cell))
else:
cell_mutlilin_ranking.append((len(lineageCount),cell))
cell_mutlilin_ranking.sort()
if useProbablityOfExpression == False:
cell_mutlilin_ranking.reverse()
scores = []
state_scores={}
cellsPerState={} ### Denominator for z-score analysis
for cell in cell_mutlilin_ranking:
score = cell[0]
scores.append(score)
cell_state = string.split(cell[-1],':')[0]
try: cellsPerState[cell_state]+=1
except Exception: cellsPerState[cell_state]=1
try: state_scores[cell_state].append(float(score))
except Exception: state_scores[cell_state] = [float(score)]
scoreMean = numpy.mean(scores)
scoreSD = numpy.std(scores)
oneSD = scoreMean+scoreSD
twoSD = scoreMean+scoreSD+scoreSD
oneStandDeviationAway={}
twoStandDeviationsAway={}
oneStandDeviationAwayTotal=0
twoStandDeviationsAwayTotal=0
print 'Mean:',scoreMean
print 'STDev:',scoreSD
state_scores2=[]
for cell_state in state_scores:
state_scores2.append((numpy.mean(state_scores[cell_state]),cell_state))
i=0
for cell in cell_mutlilin_ranking:
score,cellName = cell
CellState,CellName = string.split(cellName,':')
if score>=oneSD:
try: oneStandDeviationAway[CellState]+=1
except Exception: oneStandDeviationAway[CellState]=1
oneStandDeviationAwayTotal+=1
if score>=twoSD:
try: twoStandDeviationsAway[CellState]+=1
except Exception: twoStandDeviationsAway[CellState]=1
twoStandDeviationsAwayTotal+=1
print cell, string.join(expressedStatesPerCell[cell[-1]],'|')
i+=1
state_scores2
state_scores2.sort()
state_scores2.reverse()
twoStandDeviationsAway = oneStandDeviationAway
twoStandDeviationsAwayTotal = oneStandDeviationAwayTotal
print '\n\n'
import statistics
zscores = []
for CellState in twoStandDeviationsAway:
#print CellState
highMetaScoreCells = twoStandDeviationsAway[CellState]
totalCellsPerState = cellsPerState[CellState]
r = highMetaScoreCells
n = twoStandDeviationsAwayTotal
R = totalCellsPerState
N = len(column_header)
z = statistics.zscore(r,n,N,R)
scores = [z, CellState,statistics.p_value(z)]
zscores.append(scores)
zscores.sort()
zscores.reverse()
for scores in zscores:
scores = string.join(map(str,scores),'\t')
print scores
"""
for i in state_scores2:
print str(i[0])+'\t'+str(i[1])"""
sys.exit()
return numpy.mean(state_scores)
def calculateGeneExpressProbilities(values, useZ=False):
### First calculate z-scores - scipy.stats.mstats.zscore for the entire matrix
avg = numpy.mean(values)
std = numpy.std(values)
if std ==0:
std = 0.1
if useZ:
values = map(lambda x: (x-avg)/std,values)
else:
values = map(lambda x: x*2,values)
p_values = 1 - scipy.special.ndtr(values)
return p_values
if __name__ == '__main__':
#query_dataset = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/exp.GSE81682_HTSeq-cellHarmony-filtered.txt'
all_tpm = '/Users/saljh8/Downloads/test1/exp.cellHarmony.txt'
markerfinder = '/Users/saljh8/Downloads/test1/AllGenes_correlations-ReplicateBased.txt'
signature_genes = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/CITE-Seq_mLSK-60ADT/Merged/ExpressionInput/MF.txt'
state = 'DC'
#all_tpm = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/ExpressionInput/exp.Guide3-cellHarmony-revised.txt'
#markerfinder = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
#signature_genes = '/Users/saljh8/Desktop/Old Mac/Desktop/Grimes/KashishNormalization/test/Panorama.txt'
query_dataset = None
query_dataset = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/exp.NaturePan-PreGM-CD150-.txt'
query_dataset = None
"""
#all_tpm = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/MultiLin/exp.Gottgens_HarmonizeReference.txt'
all_tpm = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/MultiLin/ExpressionInput/exp.Gottgens_HarmonizeReference.txt'
#signature_genes = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/MultiLin/ExpressionInput/Gottgens_HarmonizeReference.txt'
signature_genes = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/MultiLin/Gottgens_HarmonizeReference.txt'
#markerfinder = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
markerfinder = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/MultiLin/ExpressionInput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
state = 'Eryth_Multi-Lin'
"""
state = None
import getopt
options, remainder = getopt.getopt(sys.argv[1:],'', ['q=','expdir=','m=','ICGS=','state='])
#print sys.argv[1:]
for opt, arg in options:
if opt == '--q': query_dataset=arg
elif opt == '--expdir': all_tpm=arg
elif opt == '--m': markerfinder=arg
elif opt == '--ICGS': signature_genes=arg
elif opt == '--state': state=arg
#state = None
#evaluateMultiLinRegulatoryStructure(all_tpm,markerfinder,signature_genes,state);sys.exit()
evaluateMultiLinRegulatoryStructure(all_tpm,markerfinder,signature_genes,state,query = query_dataset);sys.exit()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/multiLineagePredict.py
|
multiLineagePredict.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy as np
import os.path
from collections import defaultdict
import export
#import statistics
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def filterRows(input_file,output_file,filterDB=None,logData=False):
orderlst={}
counter=[]
export_object = export.ExportFile(output_file)
firstLine = True
Flag=0;
print len(filterDB)
#for i in filterDB:
for line in open(input_file,'rU').xreadlines():
#for i in filterDB:
flag1=0
data = cleanUpLine(line)
values = string.split(data,'\t')
if firstLine:
firstLine = False
k=values.index('UID')
if Flag==0:
export_object.write(line)
else:
if values[k] in filterDB:
counter=[index for index, value in enumerate(filterDB) if value == values[k]]
#print counter
for it in range(0,len(counter)):
orderlst[counter[it]]=line
#export_object.write(line)
#firstLine=True
# Flag=1;
#else:
# max_val = max(map(float,values[1:]))
#min_val = min(map(float,values[1:]))
#if max_val>0.1:
# export_object.write(line)
try:
for i in range(0,len(orderlst)):
export_object.write(orderlst[i])
except Exception:
print i,filterDB[i]
export_object.close()
print 'Filtered rows printed to:',output_file
def FilterFile(Guidefile,PSI,turn=0):
if 'Clustering' in Guidefile:
count=1
else:
count=0
val=[]
head=0
for line in open(Guidefile,'rU').xreadlines():
if head >count:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
val.append(q[0])
else:
head+=1
continue
dire = export.findParentDir(export.findParentDir(Guidefile)[:-1])
output_dir = dire+'SubtypeAnalyses-Results'
if os.path.exists(output_dir)==False:
export.createExportFolder(output_dir)
#output_file = output_dir+'/round'+str(turn)+'/'+export.findFilename(PSI)+'-filtered.txt'
output_file = output_dir+'/round'+str(turn)+'/'+export.findFilename(PSI)[:-4]+'-filtered.txt'
filterRows(PSI,output_file,filterDB=val)
return output_file
if __name__ == '__main__':
import getopt
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['PSIfile=','PSIEvent='])
for opt, arg in options:
if opt == '--PSIfile': PSIfile=arg
elif opt == '--PSIEvent':PSIEvent=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
#mutfile="/Users/meenakshi/Desktop/Leucegene-data1/Mutation_Annotations.txt"
#Guidefile="/Users/meenakshi/Documents/leucegene/ICGS/Round2_cor_0.6_280default/Clustering-exp.round2_insignificantU2like-Guide1 DDX5&ENSG00000108654&E3.4-E3.9__ENSG0000010-hierarchical_cosine_correlation.txt"
inputfile=FilterFile(PSIfile,PSIEvent)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/filterEventAnnotation.py
|
filterEventAnnotation.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy as np
import pylab as pl
import os.path
import scipy
from import_scripts import sampleIndexSelection
import matplotlib.pyplot as plt
import export
from sklearn import datasets, linear_model
from sklearn.preprocessing import StandardScaler
from sklearn.neighbors import KNeighborsClassifier
from sklearn.svm import SVC
from sklearn.tree import DecisionTreeClassifier
from sklearn.ensemble import RandomForestClassifier, AdaBoostClassifier
from sklearn.metrics.pairwise import euclidean_distances
from sklearn.metrics.pairwise import pairwise_distances
from visualization_scripts import Orderedheatmap
#from sklearn import cross_validation
from sklearn import svm
from sklearn.multiclass import OneVsOneClassifier
from sklearn.multiclass import OneVsRestClassifier
from sklearn.svm import LinearSVC
from sklearn import linear_model
import operator
from collections import OrderedDict
from collections import defaultdict
upd_guides=[]
#upd_guides.append("uid")
def FindTopUniqueEvents(Guidefile,psi,Guidedir):
head=0
guidekeys=[]
exportnam=os.path.join(Guidedir,"SplicingeventCount1.txt")
export_class=open(exportnam,"w")
#commonkeys=[]
tempkeys={}
global upd_guides
global train
omitcluster=0
unique_clusters={}
for line in open(Guidefile,'rU').xreadlines():
if head==0:
head=1
continue
else:
line1=line.rstrip('\r\n')
q= string.split(line1,'\t')
if abs(float(q[8]))>0.15:
try:
tempkeys[q[2]].append([q[0],float(q[10]),q[11]])
except KeyError:
tempkeys[q[2]]=[[q[0],float(q[10]),q[11]],]
for i in tempkeys:
if len(tempkeys[i])>1:
#print tempkeys[i]
tempkeys[i].sort(key=operator.itemgetter(1),reverse=False)
#print tempkeys[i][0]
try:
unique_clusters[0].append(tempkeys[i][0])
except KeyError:
unique_clusters[0]=[tempkeys[i][0],]
else:
try:
unique_clusters[0].append(tempkeys[i][0])
except KeyError:
unique_clusters[0]=[tempkeys[i][0],]
unique_clusters[0].sort(key=operator.itemgetter(1))
if len(unique_clusters[0])>100:
guidekeys=unique_clusters[0]
for i in range(0,len(guidekeys)):
#upd_guides[i]=[upd_guides[i][3],upd_guides[i][4]]
upd_guides.append(guidekeys[i][0])
else:
omitcluster=1
export_class.write(psi+"\t"+str(len(unique_clusters[0]))+"\n")
return omitcluster
#return upd_guides,train
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def filterRows(input_file,output_file,filterDB=None,logData=False,firstLine=True):
orderlst={}
counter=[]
export_object = open(output_file,'w')
row_count=0
for line in open(input_file,'rU').xreadlines():
#for i in filterDB:
flag1=0
row_count+=1
data = cleanUpLine(line)
line = data+'\n'
values = string.split(data,'\t')
if row_count == 1:
initial_length = len(values)
if len(values)!=initial_length:
print values
if firstLine:
firstLine = False
export_object.write(line)
else:
if values[0] in filterDB:
counter=[index for index, value in enumerate(filterDB) if value == values[0]]
for it in range(0,len(counter)):
orderlst[counter[it]]=line
for i in range(0,len(orderlst)):
try: export_object.write(orderlst[i])
except Exception:
print i,filterDB[i]
continue
export_object.close()
print 'Filtered rows printed to:',output_file
def filterRows_data(input_file,output_file,filterDB=None,logData=False):
filteredevents=[]
tempevents=[]
orderlst={}
counter=[]
export_object = open(output_file,'w')
firstLine = True
Flag=0;
for i in filterDB:
event=string.split(i,"|")[0]
tempevents.append(event)
for line in open(input_file,'rU').xreadlines():
#for i in filterDB:
flag1=0
data = cleanUpLine(line)
values = string.split(data,'\t')
event=string.split(values[0],"|")[0]
if firstLine:
firstLine = False
if Flag==0:
export_object.write(line)
else:
if event in tempevents:
counter=[index for index, value in enumerate(tempevents) if value == event]
#print counter
filteredevents.append(event)
for it in range(0,len(counter)):
orderlst[counter[it]]=line
if logData:
line = string.join([values[0]]+map(str,(map(lambda x: math.log(float(x)+1,2),values[1:]))),'\t')+'\n'
for i in range(0,len(tempevents)):
if i in orderlst:
export_object.write(orderlst[i])
if "\n" not in orderlst[i]:
#print i
export_object.write("\n")
export_object.close()
tempevents2=[]
#print 'Filtered rows printed to:',output_file
for i in range(len(tempevents)):
if tempevents[i] in filteredevents:
tempevents2.append(tempevents[i])
# print len(tempevents2)
return tempevents2
def findParentDir(filename):
filename = string.replace(filename,'//','/')
filename = string.replace(filename,'\\','/')
x = string.find(filename[::-1],'/')*-1
return filename[:x]
def Classify(header,Xobs,output_file,grplst,name,turn,platform,output_dir,root_dir):
count=0
start=1
Y=[]
head=0
for line in open(output_file,'rU').xreadlines():
if head >count:
val=[]
counter2=0
val2=[]
me=0.0
line=line.rstrip('\r\n')
q= string.split(line,'\t')
for i in range(start,len(q)):
try:
val2.append(float(q[i]))
except Exception:
continue
me=np.median(val2)
for i in range(start,len(q)):
try:
val.append(float(q[i]))
except Exception:
val.append(float(me))
#if q[1]==prev:
Y.append(val)
else:
head+=1
continue
Xobs=zip(*Xobs)
Xobs=np.array(Xobs)
Xobs=zip(*Xobs)
Xobs=np.array(Xobs)
X=grplst
X=zip(*X)
X=np.array(X)
#print X
Y=zip(*Y)
Y=np.array(Y)
#np.savetxt("/Volumes/MyPassport/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/PSI/ExpressionProfiles/DataPlots/complete_KNN.txt",q)
#if platform=="PSI":
#else:
output_dir = output_dir+'/SVMOutputs'
output_dir2 = root_dir+'/ICGS-NMF'
if os.path.exists(output_dir)==False:
export.createExportFolder(output_dir)
if os.path.exists(output_dir2)==False:
export.createExportFolder(output_dir2)
#exportnam=output_dir+'/round'+str(turn)+'SVC_test_50cor.txt'
#export_class=open(exportnam,"w")
exportnam1=output_dir+'/round'+str(turn)+'SVC_decision_func.txt'
export_class1=open(exportnam1,"w")
if platform=="PSI":
exportnam2=output_dir+'/round'+str(turn)+'SVC_Results.txt'
export_class2=open(exportnam2,"w")
else:
exportnam2=output_dir2+'/FinalGroups.txt'
export_class2=open(exportnam2,"w")
exportnam3=output_dir+'/round'+str(turn)+'SVC_Results_max.txt'
export_class3=open(exportnam3,"w")
#export_class2.write("uid"+"\t"+"group"+"\t"+"class"+"\n")
regr = LinearSVC()
regr.fit(Xobs,X[:,0])
q=regr.predict(Y)
#print q
count=1
ordersamp={}
order=[]
for i in q:
gr=string.split(name[int(i)-1],"_")[0]
gr=gr.replace("V","")
#export_class2.write(header[count]+"\t"+str(i)+"\t"+name[int(i)-1]+"\n")
# export_class2.write(header[count]+"\t"+str(i)+"\t"+gr+"\n")
ordersamp[header[count]]=[name[int(i)-1],str(i)]
count+=1
#print len(X[:,0])
if len(X[:,0])>2:
prob_=regr.fit(Xobs,X[:,0]).decision_function(Y)
#k=list(prob_)
export_class1.write("uid")
#export_class2.write("uid")
export_class3.write("uid")
for ni in name:
export_class1.write("\t"+"R"+str(turn)+"-"+ni)
#export_class2.write("\t"+"R"+str(turn)+"-"+ni)
export_class3.write("\t"+"R"+str(turn)+"-"+ni)
export_class1.write("\n")
#export_class2.write("\n")
export_class3.write("\n")
#print prob_
for iq in range(0,len(header)-1):
export_class1.write(header[iq+1])
#export_class2.write(header[iq+1])
export_class3.write(header[iq+1])
for jq in range(0,len(name)):
export_class1.write("\t"+str(prob_[iq][jq]))
if prob_[iq][jq]==max(prob_[iq,:]):
#print ordersamp[header[iq+1]],name[jq]
if ordersamp[header[iq+1]][0]==name[jq]:
order.append([header[iq+1],name[jq],prob_[iq][jq],ordersamp[header[iq+1]][1]])
export_class3.write("\t"+str(1))
else:
export_class3.write("\t"+str(0))
export_class1.write("\n")
#export_class2.write("\n")
export_class3.write("\n")
export_class1.close()
export_class3.close()
else:
if platform=="PSI":
prob_=regr.fit(Xobs,X[:,0]).decision_function(Y)
#k=list(prob_)
export_class1.write("uid"+"\t")
export_class2.write("uid"+"\t")
export_class1.write("group")
export_class2.write("round"+str(turn)+"-V1"+"\t"+"round"+str(turn)+"-V2"+"\n")
#for ni in name:
# export_class1.write("\t"+ni)
# export_class2.write("\t"+ni)
export_class1.write("\n")
export_class2.write("\n")
#print prob_
#export_class1.write(header[1])
#export_class2.write(header[1])
for iq in range(0,len(header)-1):
export_class1.write(header[iq+1])
export_class2.write(header[iq+1])
#for jq in range(0,len(X[:,0])):
export_class1.write("\t"+str(prob_[iq]))
if prob_[iq]>0.5:
export_class2.write("\t"+str(1)+"\t"+str(0))
else:
if prob_[iq]<-0.5:
export_class2.write("\t"+str(0)+"\t"+str(1))
else:
export_class2.write("\t"+str(0)+"\t"+str(0))
export_class1.write("\n")
export_class2.write("\n")
else:
prob_=regr.fit(Xobs,X[:,0]).decision_function(Y)
#k=list(prob_)
export_class1.write("uid")
#export_class2.write("uid")
export_class3.write("uid")
for ni in name:
export_class1.write("\t"+"R"+str(turn)+"-"+ni)
#export_class2.write("\t"+"R"+str(turn)+"-"+ni)
export_class3.write("\t"+"R"+str(turn)+"-"+ni)
export_class1.write("\n")
#export_class2.write("\n")
export_class3.write("\n")
#print prob_
for iq in range(0,len(header)-1):
export_class1.write(header[iq+1])
#export_class2.write(header[iq+1])
export_class3.write(header[iq+1])
# for jq in range(0,len(name)):
export_class1.write("\t"+str(prob_[iq]))
if prob_[iq]>0.0:
#print ordersamp[header[iq+1]],name[jq]
if ordersamp[header[iq+1]][0]==name[jq]:
order.append([header[iq+1],name[jq],prob_[iq],ordersamp[header[iq+1]][1]])
export_class3.write("\t"+str(1))
else:
export_class3.write("\t"+str(0))
export_class1.write("\n")
#export_class2.write("\n")
export_class3.write("\n")
export_class1.close()
export_class3.close()
order = sorted(order, key = operator.itemgetter(2),reverse=True)
order = sorted(order, key = operator.itemgetter(1))
for i in range(len(order)):
#export_class2.write(order[i][0]+"\t"+order[i][3]+"\t"+order[i][1]+"\n")
gr=string.split(order[i][1],"_")[0]
gr=gr.replace("V","")
#export_class2.write(header[count]+"\t"+str(i)+"\t"+name[int(i)-1]+"\n")
export_class2.write(order[i][0]+"\t"+order[i][3]+"\t"+gr+"\n")
export_class2.close()
if platform=="PSI":
Orderedheatmap.Classify(exportnam2)
else:
Orderedheatmap.Classify(exportnam3)
def header_file(fname, delimiter=None):
head=0
header=[]
new_head=[]
with open(fname, 'rU') as fin:
for line in fin:
if head==0:
line = line.rstrip(os.linesep)
header=string.split(line,'\t')
for i in header:
if ":" in i:
i=string.split(i,":")[1]
new_head.append(i)
head=1
else:break
return new_head
def avg(array):
total = sum(map(float, array))
average = total/len(array)
return average
def Findgroups(NMF_annot,name):
head=0
groups=[]
counter=1
line=[]
for exp1 in open(NMF_annot,"rU").xreadlines():
lin=exp1.rstrip('\r\n')
lin=string.split(lin,"\t")
mapping={}
if head==0:
head=1
continue
else:
line.append(lin)
for j in range(0,len(name)):
#print name[j]
for i in range(len(line)):
lin=line[i]
key=lin[0]
key1=key+"_vs"
key2="vs_"+key+".txt"
if key1 in name[j] or key2 in name[j]:
tot=0
for q in range(1,len(lin)):
if lin[q]=='1':
tot=tot+1
groups.append(counter)
counter=counter+1
groups=np.asarray(groups)
return groups
def TrainDataGeneration(output_file,NMF_annot,name,scaling=False,exports=False,rootDir='',calculatecentroid=True):
head=0
groups=[1,2]
centroid_heatmap_input=''
matrix=defaultdict(list)
compared_groups={}
for exp1 in open(NMF_annot,"rU").xreadlines():
lin=exp1.rstrip('\r\n')
lin=string.split(lin,"\t")
mapping={}
if head==0:
header=lin
head=1
continue
else:
for i in range(1,len(lin)):
if lin[i]=='1':
try:mapping[1].append(header[i])
except Exception: mapping[1]=[header[i]]
else:
try:mapping[0].append(header[i])
except Exception: mapping[0]=[header[i]]
head2=0
#print len(mapping[1]),len(mapping[0])
#print lin[0]
eventname=[]
for exp2 in open(output_file,"rU").xreadlines():
lin2=exp2.rstrip('\r\n')
lin2=string.split(lin2,"\t")
if head2==0:
group_db={}
index=0
try:
if len(mapping[1])>0 and len(mapping[0])>0:
for i in lin2[1:]:
if i in mapping[1]:
try: group_db[1].append(index)
except Exception: group_db[1] = [index]
else:
try: group_db[2].append(index)
except Exception: group_db[2] = [index]
index+=1
except Exception:
break
#print len(group_db[1])
head2=1
continue
else:
key = lin2[0]
lin2=lin2[1:]
grouped_floats=[]
associated_groups=[]
### string values
gvalues_list=[]
for i in group_db[1]:
try:
x=float(lin2[i])
gvalues_list.append(x)
except Exception:
#try: gvalues_list.append('') ### Thus are missing values
#except Exception: pass
pass
if calculatecentroid:
try:
matrix[lin[0]].append(avg(gvalues_list))
eventname.append(key)
except Exception:
matrix[lin[0]].append(float(0))
eventname.append(key)
else:
#matrix[lin[0]].append(gvalues_list)
try:
matrix[lin[0]].append(gvalues_list)
eventname.append(key)
except Exception:
matrix[lin[0]].append(float(0))
eventname.append(key)
#export_class=open(exportnam,"w")
#export_class.write('uid')
#for i in range(len(eventname)):
#export_class.write('\t'+eventname[i])
#export_class.write('\n')
keylist=[]
matri=[]
train=[]
for j in range(0,len(name)):
mediod=[]
for key in matrix:
"""
temp_ls=[]
for ls in matrix[key]:
temp_ls.append(np.median(ls))
print key,sum(temp_ls)
if sum(temp_ls)==0 and scaling==False:
continue
"""
if exports:
key1=string.split(key,"-")[1]
key2=string.split(key,"-")[1]
else:
key1=key+"_vs"
key2="vs_"+key+".txt"
if key1 in name[j] or key2 in name[j]:
keylist.append(key)
#print name[j]
if calculatecentroid:
train.append(matrix[key])
else:
#mediod.append(matrix[key])
matri=zip(*matrix[key])
#print len(matri)
k=0
if calculatecentroid==False:
matri=np.array(matri)
#print matri.shape
n=matri.shape[0]
D=pairwise_distances(matri,metric='euclidean').tolist()
D=np.array(D)
dist=np.mean(D,0)
for i in np.argsort(dist):
if k<1:
train.append(np.array(matri[i]))
k=k+1
#Xst=10000000
#temp=[]
#for i in range(n):
# Xd=np.array(matri[i])
# Xsd=Xd.reshape(1, -1)
#
# Xxd=pairwise_distances(Xsd,matri,metric='euclidean')
# #print Xxd
# dist=np.mean(Xxd)
# #print dist
# if dist<Xst:
# Xst=dist
# temp=Xd
#train.append(temp)
if exports:
train1=zip(*train)
centroid_heatmap_input=rootDir+'NMF-SVM/centroids/exp.MF.txt'
centroid_heatmap_groups=rootDir+'NMF-SVM/centroids/groups.MF.txt'
centroid_heatmap_obj = export.ExportFile(centroid_heatmap_input)
centroid_heatmap_groups_obj = export.ExportFile(centroid_heatmap_groups)
centroid_heatmap_obj.write("uid")
train1=zip(*train)
count=1
for k in range(len(keylist)):
centroid_heatmap_obj.write("\t"+keylist[k])
centroid_heatmap_groups_obj.write(keylist[k]+"\t"+str(k)+"\t"+str(k)+"\n")
count=count+1
centroid_heatmap_obj.write("\n")
train=zip(*train)
for i in range(len(train)):
ls=[]
ls.append(eventname[i])
for j in range(len(train[i])):
ls.append(train[i][j])
s=sum(ls[1:])
#print eventname[i], s
if s==0 and scaling == False:
pass
else:
centroid_heatmap_obj.write(string.join(map(str,ls),'\t')+'\n')
centroid_heatmap_obj.close()
centroid_heatmap_groups_obj.close()
#for i in range(len(matrix[key])):
#export_class.write('\t'+str(matrix[key][i]))
#export_class.write('\n')
train=zip(*train)
scaling=False
train=np.array(train)
if scaling==True:
trainlst=[None]*len(train[0])
for i in range(len(train)):
for j in range(len(train[i])):
try:trainlst[j]=trainlst[j]+train[i][j]
except Exception:trainlst[j]=train[i][j]
trainlst=zip(*trainlst)
train=np.array(trainlst)
return train, centroid_heatmap_input
if __name__ == '__main__':
import getopt
group=[]
grplst=[]
name=[]
matrix={}
compared_groups={}
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['Guidedir=','PSIdir=','PSI=','NMF_annot='])
for opt, arg in options:
if opt == '--Guidedir': Guidedir=arg
elif opt =='--PSIdir':PSIdir=arg
elif opt =='--PSI':PSI=arg
elif opt =='--NMF_annot':NMF_annot=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
#commonkeys=[]
counter=1
#filename="/Users/meenakshi/Documents/leucegene/ICGS/Clustering-exp.Hs_RNASeq_top_alt_junctions367-Leucegene-75p_no149-Guide1 TRAK1&ENSG00000182606&I1.1_42075542-E2.1__E-hierarchical_cosine_correlation.txt"
#PSIfile="/Users/meenakshi/Documents/leucegene/ExpressionInput/exp.Hs_RNASeq_top_alt_junctions-PSI_EventAnnotation-367-Leucegene-75p-unique-filtered-filtered.txt"
#keylabel="/Users/meenakshi/Documents/leucegene/ExpressionInput/exp.round2_glmfilteredKmeans_label.txt"
for filename in os.listdir(Guidedir):
if filename.startswith("PSI."):
Guidefile=os.path.join(Guidedir, filename)
psi=string.replace(filename,"PSI.","")
PSIfile=os.path.join(PSIdir, psi)
print Guidefile,PSIfile
#output_file=PSIfile[:-4]+"-filtered.txt"
#sampleIndexSelection.filterFile(PSIfile,output_file,header)
omitcluster=FindTopUniqueEvents(Guidefile,psi,Guidedir)
print omitcluster
if omitcluster==0:
group.append(counter)
name.append(psi)
counter+=1
output_file=PSI[:-4]+"-filtered.txt"
print len(upd_guides)
filterRows(PSI,output_file,filterDB=upd_guides,logData=False)
header=header_file(output_file)
train=TrainDataGeneration(output_file,NMF_annot,name)
grplst.append(group)
print grplst
Classify(header,train,output_file,grplst,name)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/ExpandSampleClusters.py
|
ExpandSampleClusters.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import export
import unique
import traceback
""" Intersecting Coordinate Files """
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def importLookupTable(fn):
""" Import a gRNA to valid tag lookup table """
lookup_table = []
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
gRNA,tag = t
lookup_table.append((gRNA,tag))
return lookup_table
def importCountMatrix(fn,mask=False):
""" Import a count matrix """
classification = {}
firstRow = True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if firstRow:
headers = t[1:]
firstRow = False
else:
barcode = t[0]
values = map(int,t[1:])
if mask:
sum_counts = sum(values[2:])
else:
sum_counts = sum(values)
def threshold(val):
if val>0.3: return 1
else: return 0
if sum_counts>0:
ratios = map(lambda x: (1.000*x)/sum_counts, values)
if mask:
original_ratios = ratios
ratios = ratios[2:] ### mask the first two controls which are saturating
else:
original_ratios = ratios
hits = map(lambda x: threshold(x), ratios)
hits = sum(hits)
if sum_counts>20 and hits == 1:
index=0
for ratio in ratios:
if ratio>0.3:
header = headers[index]
index+=1
classification[barcode] = header
print len(classification),fn
return classification
def exportGuideToTags(lookup_table,gRNA_barcode,tag_barcode,output):
export_object = open(output,'w')
for barcode in gRNA_barcode:
gRNA = gRNA_barcode[barcode]
if barcode in tag_barcode:
tag = tag_barcode[barcode]
if (gRNA,tag) in lookup_table:
uid = tag+'__'+gRNA
export_object.write(barcode+'\t'+uid+'\t'+uid+'\n')
export_object.close()
if __name__ == '__main__':
################ Comand-line arguments ################
import getopt
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print 'WARNING!!!! Too commands supplied.'
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['species=','gRNA=', 'tag=', 'lookup=','output='])
#print sys.argv[1:]
for opt, arg in options:
if opt == '--gRNA':
gRNA = arg
elif opt == '--tag':
tag = arg
elif opt == '--lookup':
lookup = arg
elif opt == '--output':
output = arg
lookup_table = importLookupTable(lookup)
gRNA_barcode = importCountMatrix(gRNA)
tag_barcode = importCountMatrix(tag)
exportGuideToTags(lookup_table,gRNA_barcode,tag_barcode,output)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/perturbSeqAnalysis.py
|
perturbSeqAnalysis.py
|
import sys,string,os,re
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
#import altanalyze.unique as unique
import unique
import export
import math
from stats_scripts import statistics
import traceback
import collections
import junctionGraph
"""
This script takes existing formatted metadata (C4 PCBC approved fields) and filters them to determine unique and non-unique
donors for a specific covariate and derives comparison and group relationships to extract from an existing expression file
"""
def BatchCheck(sample_id,nonpreferential_batchs,preferential_samples,platform):
priority=0
for batch in nonpreferential_batchs:
if batch in sample_id:
priority=1
if platform == 'RNASeq' or platform == 'PSI':
if sample_id not in preferential_samples: priority = 1
elif sample_id in preferential_samples: priority = 0
return priority
class MetaDataQuery:
def __init__(self, field_name, field_type, field_values):
self.field_name = field_name
self.field_type = field_type
self.field_values = field_values
def FieldName(self): return self.field_name
def FieldType(self): return self.field_type
def FieldValues(self): ### can be a list of values
if self.field_type == 'NumFilter':
cutoff = float(self.field_values[1:])
if self.field_values[0]=='>':
direction = '>'
else:
direction = '<'
return cutoff, direction
else:
ls_results = string.split(self.field_values,',')
return ls_results
def __repr__(self): return self.FieldName(), self.FieldName(), self.FieldType(),FieldValues()
def importMetaDataDescriptions(field_description_file):
###Import the looks ups to indicate which fields to consider for metadata filtering
metadata_filters={}
for line in open(field_description_file,'rU').xreadlines():
data = line.rstrip()
data = string.replace(data,'"','')
values = string.split(data,'\t')
if len(values)>1: ### Hence this field should be considered for filtering
field_name = values[0]
field_type = values[1]
field_values = values[2]
mdq = MetaDataQuery(field_name,field_type,field_values)
metadata_filters[field_name] = mdq
return metadata_filters
def prepareComparisonData(metadata_file,metadata_filters,groups_db,comps_db):
"""Import the metadata and include/exclude fields/samples based on the user inputs"""
firstLine = True
samplesToRetain=[]
samplesToRemove=[]
uniqueDB={}
indexesToFilterOn={}
covariateSamples={}
for line in open(metadata_file,'rU').xreadlines():
data = line.rstrip()
data = string.replace(data,'"','')
values = string.split(data,'\t')
if len(values)==1: continue
if firstLine:
headers = values
covariateIndex=None
index=0
for h in headers:
if h in metadata_filters:
md = metadata_filters[h]
if md.FieldType()=='UID':
uid_index = index
else:
indexesToFilterOn[index] = md
index+=1
firstLine = False
else:
try: covariateType = values[covariateIndex]
except Exception: covariateType = None
try: sampleID = values[uid_index] ### Must always be present
except: continue ### Typically caused by rows with no values or variable blank values
if '.bed' in sampleID:
sampleID = string.replace(sampleID,'.bed','')
for index in indexesToFilterOn:
sample_value = values[index]
md = indexesToFilterOn[index]
if md.FieldType() == 'Unique' and 'TRUE' in md.FieldValues():
uniqueID_sample_name = values[index]
### For example, unique donor ID (remove duplicate samples)
try: uniqueDB[uniqueID_sample_name].append(sampleID)
except Exception: uniqueDB[uniqueID_sample_name] = [sampleID]
elif md.FieldType() == 'Restrict':
if sample_value in md.FieldValues():
samplesToRetain.append(sampleID)
else:
#if 'M' not in md.FieldValues() and 'Training' not in md.FieldValues() and 'Bone marrow' not in md.FieldValues():
#print md.FieldValues(), md.FieldType(), sample_value, sampleID;sys.exit()
samplesToRemove.append(sampleID)
#if sampleID == 'SRS874639':
#print md.FieldValues(), md.FieldType(), [sample_value], sampleID;sys.exit()
elif md.FieldType() == 'Exclude':
if sample_value in md.FieldValues():
samplesToRemove.append(sampleID)
elif md.FieldType() == 'Covariate':
""" This is the field we are deriving our groups and comps from """
### If the covariateSamples key already present
if sample_value in md.FieldValues():
### Hence, this sample is TRUE to the field name (e.g., diseaseX)
sample_type = md.FieldName()
else:
sample_type = 'Others'
if md.FieldName() in covariateSamples:
groups = covariateSamples[md.FieldName()]
else:
groups={}
if sample_type in groups:
groups[sample_type].append(sampleID)
else:
groups[sample_type] = [sampleID]
covariateSamples[md.FieldName()]=groups
elif md.FieldType() == 'NumFilter':
cutoff,direction = md.FieldValues()
try:
if direction==">":
if float(sample_value)>cutoff:
samplesToRetain.append(sampleID)
else:
samplesToRemove.append(sampleID)
if direction=="<":
if float(sample_value)<cutoff:
samplesToRetain.append(sampleID)
else:
samplesToRemove.append(sampleID)
except Exception: ### Sample value not a float
samplesToRemove.append(sampleID)
if len(samplesToRetain)==0 and len(samplesToRemove) ==0:
for sample_type in groups:
samplesToRetain+=groups[sample_type]
#print len(list(set(samplesToRetain)))
#print len(list(set(samplesToRemove)));sys.exit()
for unique_donor_id in uniqueDB:
if len(uniqueDB[unique_donor_id])>1:
### This method needs to be updated with a prioritization schema for duplicates, based on metadata
for sample in uniqueDB[unique_donor_id][1:]:
samplesToRemove.append(sample)
samplesToRetainFinal=[]
for sample in samplesToRetain:
if sample not in samplesToRemove:
samplesToRetainFinal.append(sample)
for field in covariateSamples:
groups = covariateSamples[field]
for group_id in groups:
updated_samples = []
#print field, group_id, len(groups[group_id]),
for sample in groups[group_id]:
if sample in samplesToRetainFinal:
updated_samples.append(sample)
#print len(updated_samples)
groups[group_id] = updated_samples
if len(covariateSamples[field])>0:
groups_db[field] = covariateSamples[field]
### Determine comparisons
for field in groups_db:
group_names=[]
comps=[]
groups = groups_db[field]
for group_id in groups:
if len(groups[group_id])>1: ### Ensure sufficient samples to compare
if group_id not in group_names:
group_names.append(group_id)
if len(group_names) == 2:
if 'Others'in group_names:
comps.append(tuple(group_names))
if 'Others'not in group_names:
comps.append(tuple(group_names))
else:
for group1 in group_names:
for group2 in group_names:
if group1!=group2:
if (group2,group1) not in comps:
comps.append((group1,group2))
comps_db[field]=comps
print len(list(set(samplesToRetainFinal))), 'samples considered for analysis.', len(list(set(samplesToRemove))), 'removed.'
return groups_db,comps_db
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def importGroupsComps(groups_file):
initial_groups={}
group_id_lookup={}
groups_db={}
comps_db={}
for line in open(groups_file,'rU').xreadlines():
data = cleanUpLine(line)
try: sample,ID,group_name = string.split(data,'\t')
except:
print [data]
print traceback.format_exc();sys.exit()
sample = string.replace(sample,'.bed','')
group_id_lookup[ID]=group_name
try: initial_groups[group_name].append(sample)
except Exception: initial_groups[group_name] = [sample]
groups_db[1] = initial_groups
comps=[]
comps_file = string.replace(groups_file,'groups.','comps.')
try:
for line in open(comps_file,'rU').xreadlines():
data = cleanUpLine(line)
g1,g2 = string.split(data,'\t')
try:
g1_name = group_id_lookup[g1]
g2_name = group_id_lookup[g2]
comps.append((g1_name,g2_name))
except Exception:
""" The group ID in the comps file does not exist in the groups file """
print g1, "or", g2, "groups do not exist in the groups file, but do in the comps."
groups_db[1] = initial_groups
comps_db[1] = comps
except:
print traceback.format_exc()
print '\nNo comparison file identified... proceeding with all pairwise comparisons'
for group_name1 in initial_groups:
for group_name2 in initial_groups:
groups=[group_name1,group_name2]
groups.sort()
if group_name1 != group_name2:
if groups not in comps:
comps.append(groups)
groups_db[1] = initial_groups
comps_db[1] = comps
return groups_db,comps_db
class PSIData:
def __init__(self, clusterID, altexons, event_annotation, protein_predictions, coordinates):
self.clusterID = clusterID
self.altexons = altexons
self.event_annotation = event_annotation
self.protein_predictions = protein_predictions
self.coordinates = coordinates
def setUpdatedClusterID(self,updated_clusterID):
self.updatedClusterID = updated_clusterID ### update this object
def ClusterID(self): return self.clusterID
def UpdatedClusterID(self): return self.updatedClusterID
def AltExons(self): return self.altexons
def EventAnnotation(self): return self.event_annotation
def ProteinPredictions(self,dPSI):
if dPSI<0:
pp = string.replace(self.protein_predictions,'(+)','(--)')
pp = string.replace(pp,'(-)','(+)')
pp = string.replace(pp,'(--)','(-)')
return pp
else:
return self.protein_predictions
def Coordinates(self): return self.coordinates
def Inclusion(self):
""" Determines if the first junction is the inclusion or the second """
junction1,junction2=string.split(self.Coordinates(),'|')
j1s,j1e = string.split(string.split(junction1,':')[1],'-')
j2s,j2e = string.split(string.split(junction2,':')[1],'-')
j1_dist = abs(int(j1s)-int(j1e))
j2_dist = abs(int(j2s)-int(j2e))
if j1_dist>j2_dist:
self.junction_type = 'exclusion'
else:
self.junction_type = 'inclusion'
return self.junction_type
def InclusionJunction(self):
if self.junction_type == 'inclusion':
return 'True'
else:
return 'False'
def RelativeInclusion(self,dPSI):
try: junction_type = self.junction_type
except Exception: junction_type = self.Inclusion()
if junction_type == 'exclusion' and dPSI<0:
return 'inclusion'
elif junction_type == 'inclusion' and dPSI<0:
return 'exclusion'
else:
return junction_type
def performDifferentialExpressionAnalysis(species,platform,input_file,groups_db,comps_db,
CovariateQuery,splicingEventTypes={},suppressPrintOuts=False):
### filter the expression file for the samples of interest and immediately calculate comparison statistics
firstLine = True
group_index_db={}
pval_summary_db={}
group_avg_exp_db={}
export_object_db={}
psi_annotations={}
rootdir=export.findParentDir(input_file)
#print rootdir;sys.exit()
if platform != 'PSI':
try:
import ExpressionBuilder
expressionDataFormat,increment,convertNonLogToLog = ExpressionBuilder.checkExpressionFileFormat(input_file)
except:
increment = 0
convertNonLogToLog=True
else:
convertNonLogToLog=True
print 'convertNonLogToLog:',convertNonLogToLog
try:
gene_to_symbol,system_code = getAnnotations(species,platform)
from import_scripts import OBO_import
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except: gene_to_symbol={}; system_code=''
for groups in comps_db:
group1, group2 = groups
pval_summary_db[groups] = {} ### setup this data structure for later
filename='ExpressionProfiles/'+string.join(groups,'_vs_')+'.txt'
eo = export.ExportFile(rootdir+filename) ### create and store an export object for the comparison (for raw expression)
export_object_db[groups] = eo
compared_ids={}
row_count=0
header_db={}
headers_compared={}
for line in open(input_file,'rU').xreadlines():
row_count+=1
if '.bed' in line:
line = string.replace(line,'.bed','')
data = line.rstrip()
data = string.replace(data,'"','')
values = string.split(data,'\t')
if firstLine:
header = values
for group in groups_db:
samplesToEvaluate = groups_db[group]
try: sample_index_list = map(lambda x: values.index(x), samplesToEvaluate)
except Exception: ### For datasets with mising samples (removed due to other QC issues)
sample_index_list=[]
filteredSamples=[]
for x in samplesToEvaluate:
try:
sample_index_list.append(values.index(x))
filteredSamples.append(x)
except Exception: pass
samplesToEvaluate = filteredSamples
groups_db[group] = samplesToEvaluate
group_index_db[group] = sample_index_list
group_avg_exp_db[group] = {}
### Write out headers for grouped expression values
for (group1,group2) in comps_db:
eo = export_object_db[(group1,group2)]
g1_headers = groups_db[group1]
g2_headers = groups_db[group2]
g1_headers = map(lambda x: group1+':'+x,g1_headers)
g2_headers = map(lambda x: group2+':'+x,g2_headers)
eo.write(string.join(['UID']+g1_headers+g2_headers,'\t')+'\n')
#print (group1,group2),len(g1_headers),len(g2_headers)
header_db[(group1,group2)] = g1_headers, g2_headers
index=0
uid_header=0
for i in header:
if i=='UID': uid_header=index
if i=='AltExons': alt_exon_header=index
if i=='ProteinPredictions': protein_prediction=index
if i=='ClusterID': cluster_id_index=index
if i=='Coordinates': coordinates_index=index
if i=='EventAnnotation': event_annotation_index=index
index+=1
firstLine = False
else:
uid = values[uid_header]
if platform == 'RNASeq':
try: uid = symbol_to_gene[uid][0]
except Exception: pass
elif platform == 'PSI':
clusterID = values[cluster_id_index]
altexons = values[alt_exon_header]
protein_predictions = values[protein_prediction]
coordinates = values[coordinates_index]
try: event_annotation = values[event_annotation_index]
except:
continue ### Occurs with a filtered PSI value and no event annotation and anything follwoing
ps = PSIData(clusterID, altexons, event_annotation, protein_predictions, coordinates)
psi_annotations[uid]=ps
group_expression_values={}
original_group={}
for group in group_index_db:
sample_index_list = group_index_db[group]
if platform != 'PSI' and platform != 'methylation' and '' not in values and 'NA' not in values and len(values)==len(header):
try: filtered_values = map(lambda x: float(values[x]), sample_index_list) ### simple and fast way to reorganize the samples
except ValueError:
### Strings rather than values present - skip this row
continue
else: ### for splice-event comparisons where there a missing values at the end of the row
if len(header) != len(values):
diff = len(header)-len(values)
values+=diff*['']
initial_filtered=[] ### the blanks can cause problems here so we loop through each entry and catch exceptions
unfiltered=[]
for x in sample_index_list:
initial_filtered.append(values[x])
filtered_values=[]
for x in initial_filtered:
if x != '' and x!= 'NA':
filtered_values.append(float(x))
unfiltered.append(x)
#if uid == 'ENSG00000105321:E3.2-E4.2 ENSG00000105321:E2.3-E4.2' and 'inner cell mass' in group:
#print filtered_values;sys.exit()
if platform == 'PSI' or platform == 'methylation':
original_group[group]=unfiltered
else:
original_group[group]=filtered_values
if platform == 'RNASeq':# or platform == 'miRSeq':
if convertNonLogToLog:
filtered_values = map(lambda x: math.log(x+increment,2),filtered_values) ### increment and log2 adjusted
group_expression_values[group] = filtered_values
for groups in comps_db:
group1,group2 = groups
g1_headers,g2_headers=header_db[groups]
header_samples = (tuple(g1_headers),tuple(g2_headers))
if header_samples not in headers_compared:
headers_compared[header_samples]=[]
if suppressPrintOuts==False:
print len(g1_headers),len(g2_headers),groups
try:
data_list1 = group_expression_values[group1]
except KeyError:
### This error is linked to the above Strings rather than values present error
continue
try:
data_list2 = group_expression_values[group2]
except:
continue
combined = data_list1+data_list2
if g1_headers==0 or g2_headers==0:
continue ### no samples matching the criterion
try: diff = max(combined)-min(combined) ### Is there any variance?
except Exception:
### No PSI values for this splice-event and these groups
continue
if diff==0:
continue
elif (100.00*len(data_list1))/len(g1_headers)>=PercentExp and (100.00*len(data_list2))/len(g2_headers)>=PercentExp:
if len(data_list1)<minSampleNumber or len(data_list2)<minSampleNumber:
continue ### Don't analyze
compared_ids[uid]=[]
pass
else:
continue ### Don't analyze
#print len(g1_headers), len(g2_headers);sys.exit()
if (len(data_list1)>1 and len(data_list2)>1) or (len(g1_headers)==1 or len(g2_headers)==1): ### For splicing data
### Just a One-Way ANOVA at first - moderation happens later !!!!
try:
p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic)
except Exception: p = 1
avg1 = statistics.avg(data_list1)
avg2 = statistics.avg(data_list2)
log_fold = avg1-avg2
if platform == 'RNASeq':# or platform == 'miRSeq':
max_avg = math.pow(2,max([avg1,avg2]))-1
else: max_avg = 10000
valid = True
try:
if max_avg<minRPKM:
log_fold = 'Insufficient Expression'
except Exception: pass
gs = statistics.GroupStats(log_fold,None,p)
gs.setAdditionalStats(data_list1,data_list2) ### Assuming equal variance
pval_db = pval_summary_db[groups] ### for calculated adjusted statistics
pval_db[uid] = gs ### store the statistics here
if len(restricted_gene_denominator)>0:
if uid not in restricted_gene_denominator:
proceed = False
### store a new instance
gsg = statistics.GroupStats(log_fold,None,p)
gsg.setAdditionalStats(data_list1,data_list2) ### Assuming equal variance
global_adjp_db[CovariateQuery,groups,uid] = gsg ### for global adjustment across comparisons
group_avg_exp_db[group1][uid] = avg1 ### store the group expression values
group_avg_exp_db[group2][uid] = avg2 ### store the group expression values
if 'Insufficient Expression2' != log_fold:
#if abs(log_fold)>logfold_threshold:
eo = export_object_db[groups]
ls1 = map(str,original_group[group1])
ls2 = map(str,original_group[group2])
eo.write(string.join([uid]+ls1+ls2,'\t')+'\n')
for groups in export_object_db:
export_object_db[groups].close()
### Calculate adjusted p-values for all pairwise comparisons
global_count=0
ensembls_found=False
try: to = export.ExportFile(rootdir+'/top50/MultiPath-PSI.txt')
except: pass
for groups in pval_summary_db:
psi_results = collections.OrderedDict()
group1,group2 = groups
group_comp_name = string.join(groups,'_vs_')
if platform == 'PSI':
filename=CovariateQuery+'/PSI.'+group_comp_name+'.txt'
else:
filename=CovariateQuery+'/GE.'+group_comp_name+'.txt'
eo = export.ExportFile(rootdir+'/'+filename)
#do = export.ExportFile(rootdir+'/Downregulated/'+filename)
#uo = export.ExportFile(rootdir+'/Upregulated/'+filename)
try:
so = export.ExportFile(rootdir+'PValues/'+CovariateQuery+'-'+string.join(groups,'_vs_')+'.txt')
except IOError:
### Occurs when the filename is too long - so we shorten this (not ideal)
groups2 = list(groups)
groups2[-1] = 'controls'
try:
so = export.ExportFile(rootdir+'PValues/'+CovariateQuery+'-'+string.join(groups2,'_vs_')+'.txt')
except IOError:
groups2[0] = groups2[0][:30] ### restrict to 30 characters
so = export.ExportFile(rootdir+'PValues/'+CovariateQuery+'-'+string.join(groups2,'_vs_')+'.txt')
header = 'GeneID\tSystemCode\tLogFold\trawp\tadjp\tSymbol\tavg-%s\tavg-%s\n' % (group1,group2)
if platform == 'PSI':
header = 'UID\tInclusion-Junction\tEvent-Direction\tClusterID\tUpdatedClusterID\tAltExons\tEventAnnotation\tCoordinates\tProteinPredictions\tdPSI\trawp\tadjp\tavg-%s\tavg-%s\n' % (group1,group2)
eo.write(header)
#do.write(header)
#uo.write(header)
so.write('Gene\tPval\n')
pval_db = pval_summary_db[groups]
if 'moderated' in probability_statistic: # and pval_threshold<1:
try: statistics.moderateTestStats(pval_db,probability_statistic) ### Moderates the original reported test p-value prior to adjusting
except Exception:
print 'Moderated test failed... using student t-test instead',group1,group2
#print traceback.format_exc()
else:
'Skipping moderated t-test...'
statistics.adjustPermuteStats(pval_db) ### sets the adjusted p-values for objects
pval_sort=[]
for uid in pval_db:
gs = pval_db[uid]
pval_sort.append((gs.Pval(),uid))
pval_sort.sort()
ranked_count = 0
for (pval,uid) in pval_sort:
if ranked_count<11 and global_count<51:
try: to.write(uid+'\n')
except: pass
gs = pval_db[uid]
ranked_count+=1
global_count+=1
group1_avg = str(group_avg_exp_db[group1][uid])
group2_avg = str(group_avg_exp_db[group2][uid])
if use_adjusted_p:
pval = float(gs.AdjP())
else:
pval = gs.Pval()
if platform == 'miRSeq':
symbol=[]
altIDs = getAltID(uid)
for id in altIDs:
if id in gene_to_symbol:
symbol.append(gene_to_symbol[id][0])
symbol.append(id)
symbol = string.join(symbol,'|')
elif uid in gene_to_symbol:
symbols = unique.unique(gene_to_symbol[uid])
symbol = string.join(symbols,'|')
elif 'ENS' in uid and ':' in uid:
ens_gene = string.split(uid,':')[0]
try: symbol = gene_to_symbol[ens_gene][0]
except Exception: symbol=''
else:
symbol = ''
proceed = True
### Remove genes not a predetermined list (optional)
if len(restricted_gene_denominator)>0:
if uid not in restricted_gene_denominator:
if symbol not in restricted_gene_denominator:
proceed = False
if 'Insufficient Expression' != gs.LogFold() and proceed:
#pval_threshold=1; logfold_threshold=0
if abs(gs.LogFold())>logfold_threshold and pval<=pval_threshold:
#print uid, groups, abs(gs.LogFold()), logfold_threshold;sys.exit()
if platform == 'PSI':
psi_results[uid]=gs,group1_avg,group2_avg
### Write these out once all entries for that gene have been parsed
else:
if gs.LogFold()>0: fold = 'upregulated'
else: fold = 'downregulated'
try: splicingEventTypes[group_comp_name].append(fold)
except Exception: splicingEventTypes[group_comp_name] = [fold]
if 'ENS' in uid and ensembls_found==False:
ensembls_found = True
else:
if 'ENS' not in uid:
system_code = 'Sy'; symbol = uid
if 'ENS' in uid:
system_code = 'En'
values = string.join([uid,system_code,str(gs.LogFold()),str(gs.Pval()),str(gs.AdjP()),symbol,group1_avg,group2_avg],'\t')+'\n'
eo.write(values)
proceed = True
if proceed:
if gs.LogFold()>0:
#uo.write(values)
pass
if gs.LogFold()<0:
#do.write(values)
pass
so.write(uid+'\t'+str(gs.Pval())+'\n')
if platform == 'PSI':
### Although we could have easily written the results above, we need to update the cluster ID here
### based on the significant results for this comparison only.
gene_db={}
for uid in psi_results:
ps=psi_annotations[uid]
geneID = string.split(uid,':')[1]
if geneID in gene_db:
event_coordinates = gene_db[geneID]
event_coordinates[uid]=ps.Coordinates()
else:
event_coordinates={}
event_coordinates[uid]=ps.Coordinates()
gene_db[geneID] = event_coordinates
event_cluster_db=junctionGraph.createFeaturesFromEvents(gene_db)
for uid in psi_results:
updated_cluster_id=event_cluster_db[uid]
ps=psi_annotations[uid]
ps.setUpdatedClusterID(updated_cluster_id)
gs,group1_avg,group2_avg=psi_results[uid]
event_direction = ps.RelativeInclusion(gs.LogFold())
incl_junction = ps.InclusionJunction()
values = [uid,incl_junction,event_direction,ps.ClusterID(),ps.UpdatedClusterID(),ps.AltExons(),ps.EventAnnotation(),ps.Coordinates(),
ps.ProteinPredictions(gs.LogFold()),str(gs.LogFold()),str(gs.Pval()),str(gs.AdjP()),group1_avg,group2_avg]
values = string.join(values,'\t')+'\n'
eo.write(values)
try: splicingEventTypes[group_comp_name].append((ps.ClusterID(),event_direction,ps.EventAnnotation()))
except Exception: splicingEventTypes[group_comp_name] = [(ps.ClusterID(),event_direction,ps.EventAnnotation())]
eo.close()
#do.close()
#uo.close()
so.close()
to.close()
print len(compared_ids),'unique IDs compared (denominator).'
return rootdir, splicingEventTypes
def outputGeneExpressionSummaries(rootdir,DEGs):
eo = export.ExportFile(rootdir+'/gene_summary.txt')
header = ['ComparisonName','RegulatedGenes','Upregulated','Downregulated']
eo.write(string.join(header,'\t')+'\n')
for comparison in DEGs:
folds={}
genes = DEGs[comparison]
for fold in genes:
try: folds[fold]+=1
except Exception: folds[fold]=1
try: up = folds['upregulated']
except: up = 0
try: down = folds['downregulated']
except: down = 0
eo.write(string.join([comparison,str(len(genes)),str(up),str(down)],'\t')+'\n')
eo.close()
def outputSplicingSummaries(rootdir,splicingEventTypes):
events_file = rootdir+'/event_summary.txt'
eo = export.ExportFile(events_file)
header = ['ComparisonName','UniqueJunctionClusters','InclusionEvents','ExclusionEvents']
header+= ["alt-3'_exclusion","alt-3'_inclusion","alt-5'_exclusion","alt-5'_inclusion"]
header+= ["alt-C-term_exclusion","alt-C-term_inclusion","altPromoter_exclusion","altPromoter_inclusion"]
header+= ["cassette-exon_exclusion","cassette-exon_inclusion","intron-retention_exclusion"]
header+= ["intron-retention_inclusion","trans-splicing_exclusion","trans-splicing_inclusion"]
header = string.join(header,'\t')+'\n'
eo.write(header)
for comparison in splicingEventTypes:
uniqueEvents={}
for (clusterID,inclusionType,eventAnnotation) in splicingEventTypes[comparison]:
try: uniqueEvents[clusterID].append((eventAnnotation,inclusionType))
except Exception: uniqueEvents[clusterID] =[(eventAnnotation,inclusionType)]
eventAnnotations={}
inclusionEvents={}
for clusterID in uniqueEvents:
events = uniqueEvents[clusterID]
events = list(set(events)) ### get unique (multiple distinct events can occur together)
events.sort()
### Try to ignore events for the same clustering without an eventAnnotation
for (eventAnnotation,inclusionType) in events:
if len(eventAnnotation)>1:
if '|' in eventAnnotation: ### Occurs rarely - alt-3'|cassette-exon
annotations = string.split(eventAnnotation,'|')
else:
annotations = [eventAnnotation]
for annotation in annotations:
try: eventAnnotations[eventAnnotation+'_'+inclusionType]+=1
except Exception: eventAnnotations[eventAnnotation+'_'+inclusionType]=1
try: inclusionEvents[inclusionType]+=1
except Exception: inclusionEvents[inclusionType]=1
try: a3e = str(eventAnnotations["alt-3'_exclusion"])
except Exception: a3e = "0"
try: a3i = str(eventAnnotations["alt-3'_inclusion"])
except Exception: a3i = "0"
try: a5e = str(eventAnnotations["alt-5'_exclusion"])
except Exception: a5e = "0"
try: a5i = str(eventAnnotations["alt-5'_inclusion"])
except Exception: a5i = "0"
try: aCe = str(eventAnnotations["alt-C-term_exclusion"])
except Exception: aCe = "0"
try: aCi = str(eventAnnotations["alt-C-term_inclusion"])
except Exception: aCi = "0"
try: ae = str(eventAnnotations["altPromoter_exclusion"])
except Exception: ae = "0"
try: ai = str(eventAnnotations["altPromoter_inclusion"])
except Exception: ai = "0"
try: ce = str(eventAnnotations["cassette-exon_exclusion"])
except Exception: ce = "0"
try: ci = str(eventAnnotations["cassette-exon_inclusion"])
except Exception: ci = "0"
try: ie = str(eventAnnotations["intron-retention_exclusion"])
except Exception: ie = "0"
try: ii = str(eventAnnotations["intron-retention_inclusion"])
except Exception: ii = "0"
try: te = str(eventAnnotations["trans-splicing_exclusion"])
except Exception: te = "0"
try: ti = str(eventAnnotations["trans-splicing_inclusion"])
except Exception: ti = "0"
try: incEvents = str(inclusionEvents['inclusion'])
except Exception: incEvents = "0"
try: exclEvents = str(inclusionEvents['exclusion'])
except Exception: exclEvents = "0"
values = [comparison,str(len(uniqueEvents)),incEvents,
exclEvents,a3e,a3i,a5e,a5i,
aCe,aCi,ae,ai,ce,ci,ie,ii,te,ti]
values = string.join(values,'\t')+'\n'
eo.write(values)
eo.close()
graphics = []
try:
from visualization_scripts import clustering
parent_dir = export.findParentDir(events_file)
parent_dir = export.findParentDir(parent_dir[:-1])
OutputFile1 = parent_dir+'/SpliceEvent-Types.png'
clustering.stackedbarchart(events_file,display=False,output=OutputFile1)
OutputFile2 = parent_dir+'/SignificantEvents.png'
index1=2;index2=3; x_axis='Number of Alternative Events'; y_axis = 'Comparisons'; title='MultiPath-PSI Alternative Splicing Events'
clustering.barchart(events_file,index1,index2,x_axis,y_axis,title,display=False,color1='Orange',color2='SkyBlue',output=OutputFile2)
graphics.append(['SpliceEvent-Types',OutputFile1])
graphics.append(['Significant MultiPath-PSI Events',OutputFile2])
except Exception:
print traceback.format_exc()
return graphics
def getAltID(uid):
altID = string.replace(uid,'hsa-mir-','MIR')
altID = string.replace(altID,'hsa-miR-','MIR')
altID = string.replace(altID,'3p','')
altID = string.replace(altID,'5p','')
altID = string.upper(string.replace(altID,'hsa-let-','LET'))
altID = string.replace(altID,'-','')
altIDs = string.split(altID,'_')
altIDs+=string.split(uid,'_')
altIDs = unique.unique(altIDs)
return altIDs
def getAnnotations(species,platform):
import gene_associations
if platform == 'RNASeq' or platform == 'PSI' or platform == 'miRSeq':
try: gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
except Exception: gene_to_symbol={}
system_code = 'En'
if platform == 'miRSeq':
from import_scripts import OBO_import
gene_to_symbol = OBO_import.swapKeyValues(gene_to_symbol)
if platform == 'methylation':
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
gene_to_symbol = importMethylationAnnotations(species,gene_to_symbol)
system_code = 'Ilm'
return gene_to_symbol, system_code
def importMethylationAnnotations(species,gene_to_symbol):
filename = 'AltDatabase/ucsc/'+species+'/illumina_genes.txt'
from import_scripts import OBO_import
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
firstLine=True
probe_gene_db={}
for line in open(OBO_import.filepath(filename),'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
geneIndex = values.index('UCSC_RefGene_Name')
locationIndex = values.index('UCSC_RefGene_Group')
firstLine = False
else:
probeID = values[0]
try: genes = string.split(values[geneIndex],';')
except Exception: genes=[]
try:
locations = unique.unique(string.split(values[locationIndex],';'))
locations = string.join(locations,';')
except Exception:
locations = ''
for symbol in genes:
if len(symbol)>0:
if symbol in symbol_to_gene:
for geneID in symbol_to_gene[symbol]:
try: probe_gene_db[probeID].append(geneID)
except Exception: probe_gene_db[probeID] = [geneID]
probe_gene_db[probeID].append(symbol)
probe_gene_db[probeID].append(locations)
return probe_gene_db
def getDatasetSamples(expression_file,sample_metadata,cellLines):
### Required as samples may exist in the metadata but were excluded due to QC
for line in open(expression_file,'rU').xreadlines():
if '.bed' in line:
line = string.replace(line,'.bed','')
data = line.rstrip()
headers = string.split(data,'\t')[1:]
break
supported_cellLines={}
for s in headers:
if s in sample_metadata:
metadata = sample_metadata[s]
if len(metadata.CellLine())>0:
try:
if s == cellLines[metadata.CellLine()]: ### Make sure the correct samples is being matched
supported_cellLines[metadata.CellLine()]=None
except Exception: pass
return supported_cellLines
def importExpressionData(species,platform,expression_file,cell_line_db,common_lines):
### Imports the omics data, filters/orders samples, transforms (if necessary), keys by a common geneID
filtered_exp_data={}
try: gene_to_symbol,system_code = getAnnotations(species,platform)
except ZeroDivisionError: gene_to_symbol={}; system_code=''
firstLine=True
for line in open(expression_file,'rU').xreadlines():
if '.bed' in line:
line = string.replace(line,'.bed','')
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
samplesToEvaluate = map(lambda x: cell_line_db[x], common_lines)
sample_index_list = map(lambda x: values.index(x), samplesToEvaluate)
header = values
#print len(samplesToEvaluate),platform, samplesToEvaluate
firstLine = False
else:
try: filtered_values = map(lambda x: float(values[x]), sample_index_list) ### simple and fast way to reorganize the samples
except Exception: ### for splice-event comparisons
if len(header) != len(values):
diff = len(header)-len(values)
values+=diff*['']
initial_filtered=[] ### the blanks can cause problems here so we loop through each entry and catch exceptions
initial_filtered = map(lambda x: values[x], sample_index_list)
filtered_values=[]
for x in initial_filtered:
if x != '': filtered_values.append(float(x))
if platform == 'RNASeq':# or platform == 'miRSeq':
filtered_values = map(lambda x: math.log(x+1,2),filtered_values) ### increment and log2 adjusted
uid = values[0]
geneIDs = []
if platform == 'miRSeq':
altID = string.replace(uid,'hsa-mir-','MIR')
altID = string.replace(altID,'hsa-miR-','MIR')
altID = string.replace(altID,'3p','')
altID = string.replace(altID,'5p','')
altID = string.upper(string.replace(altID,'hsa-let-','MIRLET'))
altID = string.replace(altID,'-','')
altIDs = string.split(altID,'_')
altIDs+=string.split(uid,'_')
altIDs = unique.unique(altIDs)
for id in altIDs:
if id in gene_to_symbol:
geneIDs.append((gene_to_symbol[id][0],uid))
original_uid = uid
if platform == 'methylation' and ':' in uid:
uid=string.split(uid,':')[1]
if 'ENS' in uid and '.' in uid and ':' not in uid:
uid = string.split(uid,'.')[0] ### for cufflinks
if 'ENS' in uid:
if uid in gene_to_symbol:
symbol = gene_to_symbol[uid][0]
else:
symbol = ''
geneIDs = [(uid,symbol)]
elif uid in gene_to_symbol:
if 'uid'== 'cg21028156':
print gene_to_symbol[uid]
for g in gene_to_symbol[uid]:
if 'ENS' in g:
if platform == 'methylation' and ':' in original_uid:
uid = original_uid
geneIDs.append((g,uid))
for (geneID,uid) in geneIDs:
try: filtered_exp_data[geneID].append((uid,filtered_values))
except Exception: filtered_exp_data[geneID] = [(uid,filtered_values)]
print len(filtered_exp_data)
return filtered_exp_data, samplesToEvaluate
def combineAndCompareMatrices(input_file,filtered_exp_data1,filtered_exp_data2,platform1,platform2,samplesToEvaluate):
### Get the matching genes and identify anti-correlated rows (same sample order) to export to a merged file
rootdir=export.findParentDir(input_file)
compared_uid_pairs={}
count=0
import warnings
from scipy import stats
p1_samples = map(lambda x: platform1+':'+x, samplesToEvaluate)
p2_samples = map(lambda x: platform2+':'+x, samplesToEvaluate)
exportFile = rootdir+'/MergedOmicsTables/'+platform1+'-'+platform2+'.txt'
if uniqueDonors:
exportFile = rootdir+'/MergedOmicsTables/'+platform1+'-'+platform2+'-UniqueDonors.txt'
co = export.ExportFile(exportFile)
co.write(string.join(['GeneID',platform1+'-UID','Pearson-rho']+p1_samples+[platform2+'-UID']+p2_samples,'\t')+'\n')
correlated_geneIDs={}
for geneID in filtered_exp_data1:
if geneID in filtered_exp_data2:
rows1 = filtered_exp_data1[geneID]
rows2 = filtered_exp_data2[geneID]
for (uid1,row1) in rows1:
"""
if platform1 == 'RNASeq' and platform2 == 'methylation':
try: row1 = map(lambda x: math.pow(2,x)-1,row1)
except Exception: print uid1,row1;sys.exit()
"""
for (uid2,row2) in rows2:
try: null=compared_uid_pairs[(uid1,uid2)] ### already compared
except Exception:
with warnings.catch_warnings():
warnings.filterwarnings("ignore") ### hides import warnings
try: rho,p = stats.pearsonr(row1,row2)
except Exception: print 'The rows are not of equal length, likely due to missing values in that row:',uid1,uid2;sys.exit()
compared_uid_pairs[(uid1,uid2)]=None
if rho < -0.5:
values = [geneID,uid1,rho]+row1+[uid2]+row2
values = string.join(map(str, values),'\t')+'\n'
correlated_geneIDs[geneID]=None
co.write(values)
count+=1
co.close()
print 'Writing out %d entries to %s:' % (count,exportFile)
correlated_geneIDs_ls=[]
for i in correlated_geneIDs:
correlated_geneIDs_ls.append(i)
print len(correlated_geneIDs_ls)
def getFiles(sub_dir,directories=True):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
for entry in dir_list:
if directories:
if '.' not in entry: dir_list2.append(entry)
else:
if '.' in entry: dir_list2.append(entry)
return dir_list2
class GeneData:
def __init__(self,geneID, systemCode, logFold, rawp, adjp, symbol, avg1, avg2):
self.geneID = geneID; self.systemCode = systemCode; self.logFold = logFold; self.rawp = rawp; self.adjp = adjp
self.symbol = symbol; self.avg1 = avg1; self.avg2 = avg2
def GeneID(self): return self.geneID
def LogFold(self): return self.logFold
def Rawp(self): return self.rawp
def Adjp(self): return self.adjp
def Symbol(self): return self.symbol
def Avg1(self): return self.avg1
def Avg2(self): return self.avg2
def SystemCode(self): return self.systemCode
def __repr__(self): return self.GeneID()
def importResultsSummary(filepath,comparison,gene_associations_db):
#'GeneID\tSystemCode\tLogFold\trawp\tadjp\tSymbol\tavg-%s\tavg-%s\n
firstLine=True
for line in open(filepath,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
else:
geneID, systemCode, logFold, rawp, adjp, symbol, avg1, avg2 = values
gd = GeneData(geneID, systemCode, logFold, rawp, adjp, symbol, avg1, avg2)
if comparison in gene_associations_db:
gene_db = gene_associations_db[comparison]
gene_db[geneID]=gd
else:
gene_db = {}
gene_db[geneID]=gd
gene_associations_db[comparison]=gene_db
return gene_associations_db
def compareGOEliteEnrichmentProfiles(expressionDir,eliteDir):
up_elite_miRNAs={}
down_elite_miRNAs={}
folders = getFiles(expressionDir)
for folder in folders:
subdirs = getFiles(expressionDir+'/'+folder)
for sub_dir in subdirs:
subdir = expressionDir+'/'+folder + '/'+ sub_dir
elite_dirs = getFiles(subdir) ### Are there any files to analyze?
if 'GO-Elite_results' in elite_dirs:
elitedir = subdir + '/GO-Elite_results/pruned-results_z-score_elite.txt'
if 'Down' in folder:
try: down_elite_miRNAs = getMIRAssociations(elitedir,sub_dir,down_elite_miRNAs)
except Exception: pass
else:
try: up_elite_miRNAs = getMIRAssociations(elitedir,sub_dir,up_elite_miRNAs)
except Exception: pass
if '.txt' not in eliteDir:
if 'CombinedResults' not in eliteDir:
eliteDir += '/CombinedResults/allTopGenes.txt'
else: eliteDir += '/allTopGenes.txt'
firstLine=True
for line in open(eliteDir,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
else:
comparison, gene, symbol, up_rawp, ng_adjp, up_logfold,ng_logofold, ng_avg1, ng_avg2 = values[:9]
comparison = string.replace(comparison,'.txt','')
miRNAs = string.split(gene,'_')+string.split(symbol,'|')
log_fold = float(up_logfold)
if log_fold>0:
if comparison in down_elite_miRNAs:
for miRNA in miRNAs:
miRNA = string.lower(miRNA)
if miRNA in down_elite_miRNAs[comparison]:
if 'lab' not in comparison and 'CD34+ cells_vs_mononuclear' not in comparison:
print miRNA, comparison, 'down'
else:
if comparison in up_elite_miRNAs:
for miRNA in miRNAs:
miRNA = string.lower(miRNA)
if miRNA in up_elite_miRNAs[comparison]:
if 'lab' not in comparison and 'CD34+ cells_vs_mononuclear' not in comparison:
print miRNA, comparison, 'up'
def importRestrictedSetOfGenesToQuery(filepath):
### Applied to predetermined expressed genes matching some criterion (e.g., FPKM > 5 and 20% expression in EBs)
restricted_gene_denominator_db={}
firstLine=True
for line in open(filepath,'rU').xreadlines():
data = line.rstrip()
gene = string.split(data,'\t')[0]
if firstLine:
firstLine=False
else:
restricted_gene_denominator_db[gene]=[]
return restricted_gene_denominator_db
def getMIRAssociations(filepath,diffstate_comparison,elite_miRNAs):
firstLine=True
for line in open(filepath,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
else:
try:
#if 'Combined' in values[0]: ### Restrict comparison to these
regulated_geneset_name = values[0] # GE.group1_vs_group2-microRNATargets.txt
regulated_geneset_name = string.split(regulated_geneset_name,'-')[0]
#regulated_geneset_name = string.replace(regulated_geneset_name,'-UniqueDonors','')
#regulated_geneset_name = string.replace(regulated_geneset_name,'-Combined','')
miRNA = string.lower(values[2])
miRNA = string.replace(miRNA,'*','')
miRNA2 = string.replace(miRNA,'hsa-mir-','MIR')
miRNA2 = string.replace(miRNA2,'hsa-let-','LET')
miRNA3 = string.replace(miRNA2,'-5p','')
miRNA3 = string.replace(miRNA3,'-3p','')
miRNAs = [miRNA,miRNA2,miRNA3]
for miRNA in miRNAs:
try: elite_miRNAs[diffstate_comparison+':'+regulated_geneset_name].append(string.lower(miRNA))
except Exception: elite_miRNAs[diffstate_comparison+':'+regulated_geneset_name] = [string.lower(miRNA)]
except Exception:
pass
return elite_miRNAs
def runGOEliteAnalysis(species,resultsDirectory):
mod = 'Ensembl'
pathway_permutations = 'FisherExactTest'
filter_method = 'z-score'
z_threshold = 1.96
p_val_threshold = 0.05
change_threshold = 2
resources_to_analyze = ['microRNATargets','pictar','miRanda','mirbase','RNAhybrid','TargetScan','microRNATargets_All']
returnPathways = 'no'
root = None
import GO_Elite
print '\nBeginning to run GO-Elite analysis on all results'
folders = getFiles(resultsDirectory)
for folder in folders:
subdirs = getFiles(resultsDirectory+'/'+folder)
for subdir in subdirs:
subdir = resultsDirectory+'/'+folder + '/'+ subdir
file_dirs = subdir,None,subdir
input_files = getFiles(subdir,directories=False) ### Are there any files to analyze?
if len(input_files)>0:
variables = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,root
try: GO_Elite.remoteAnalysis(variables,'non-UI')
except Exception: 'GO-Elite failed for:',subdir
def identifyCommonGenes(resultsDirectory):
""" Compares results from parallel statistical analyses for unique and non-unique genetic donor workflows """
uniqueDonorGenes = {}
nonUniqueDonorGenes={}
folders = getFiles(resultsDirectory)
for folder in folders:
files = getFiles(resultsDirectory+'/'+folder,directories=False)
for file in files:
if '.txt' in file and 'GE.'== file[:3]:
filepath = resultsDirectory+'/'+folder+'/'+file
comparison = folder+':'+string.replace(file,'-UniqueDonors.txt','.txt')
if 'UniqueDonors.txt' in filepath:
uniqueDonorGenes = importResultsSummary(filepath,comparison,uniqueDonorGenes)
else:
nonUniqueDonorGenes = importResultsSummary(filepath,comparison,nonUniqueDonorGenes)
#nonUniqueDonorGenes = uniqueDonorGenes
from build_scripts import EnsemblImport
try: gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,platform,'key_by_array')
except Exception: gene_location_db={}
includeGlobalAdjustedPvals = False
if len(global_adjp_db)>0: ### When all comparisons are run together
#global_adjp_db[CovariateQuery,uniqueDonors,groups,geneID] = gs
if 'moderated' in probability_statistic:
try: statistics.moderateTestStats(global_adjp_db,probability_statistic) ### Moderates the original reported test p-value prior to adjusting
except Exception: print 'Moderated test failed... using student t-test instead'
statistics.adjustPermuteStats(global_adjp_db) ### sets the adjusted p-values for objects
includeGlobalAdjustedPvals = True
output_dir = resultsDirectory+'/CombinedResults/allTopGenes.txt'
eo = export.ExportFile(output_dir)
header = 'Comparison\tGeneID\tSymbol\tUniqueDonor-rawp\tNonUnique-adjp\tUniqueDonor-LogFold\tNonUnique-LogFold\tNonUnique-Avg1\tNonUnique-Avg2'
if includeGlobalAdjustedPvals:
header+='\tGlobalAdjustedP'
eo.write(header+'\n')
topComparisonAssociations={}
for comparison in uniqueDonorGenes:
if comparison in nonUniqueDonorGenes:
CovariateQuery,groups = string.split(comparison[:-4],':')
groups = tuple(string.split(groups[3:],'_vs_'))
comparison_dir = string.replace(comparison,':','/')[:-4]
do = export.ExportFile(resultsDirectory+'/Downregulated/'+comparison_dir+'-Combined.txt')
uo = export.ExportFile(resultsDirectory+'/Upregulated/'+comparison_dir+'-Combined.txt')
header = 'GeneID\tSy\tFoldChange\trawp\n'
uo.write(header)
do.write(header)
unique_gene_db = uniqueDonorGenes[comparison]
nonunique_gene_db = nonUniqueDonorGenes[comparison]
for gene in unique_gene_db: ### loop through the gene dictionary
if gene in nonunique_gene_db: ### common genes between unique and non-unique donors
ug = unique_gene_db[gene]
ng = nonunique_gene_db[gene]
values = [comparison,gene, ug.Symbol(),ug.Rawp(),ng.Adjp(),ug.LogFold(),ng.LogFold(),ng.Avg1(),ng.Avg2()]
if includeGlobalAdjustedPvals:
try:
gs = global_adjp_db[CovariateQuery,groups,gene]
ng_adjp = float(gs.AdjP())
values+=[str(ng_adjp)]
if platform == 'miRSeq' or platform == 'PSI' and use_adjusted_p == False:
ng_adjp = float(ug.Rawp())
except Exception:
if platform == 'miRSeq' or platform == 'PSI' and use_adjusted_p == False:
ng_adjp = float(ug.Rawp())
else:
ng_adjp = float(ug.Rawp())
values = string.join(values,'\t')+'\n'
eo.write(values)
if ng_adjp<pval_threshold:
try: topComparisonAssociations[gene].append((float(ug.Rawp()),values))
except Exception: topComparisonAssociations[gene] = [(float(ug.Rawp()),values)]
values = [ug.GeneID(), ug.SystemCode(), ug.LogFold(), ug.Rawp()]
values = string.join(values,'\t')+'\n'
try: chr = gene_location_db[ug.GeneID()][0]
except Exception: chr = ''
proceed = True
if 'Gender' in comparison:
if 'Y' in chr: proceed = False
if proceed:
if float(ug.LogFold())>0:
uo.write(values)
else:
do.write(values)
do.close()
uo.close()
eo.close()
print 'Matching Unique-Donor and NonUnique Donor results written to:',output_dir
### Write out the comparison for each gene with the most significant result (best associations)
output_dir = resultsDirectory+'/CombinedResults/eliteTopGenes.txt'
eo = export.ExportFile(output_dir)
eo.write('Comparison\tGeneID\tSymbol\tUniqueDonor-rawp\tNonUnique-adjp\tUniqueDonor-LogFold\tNonUnique-LogFold\tNonUnique-Avg1\tNonUnique-Avg2\n')
for gene in topComparisonAssociations:
topComparisonAssociations[gene].sort()
eo.write(topComparisonAssociations[gene][0][1])
eo.close()
print 'The most significant comparisons for each gene reported to:',output_dir
def getCommonCellLines(cellLines1,cellLines2,exp_cellLines1,exp_cellLines2,uniqueDonor_db,uniqueDonors,donor_db):
common_lines = list(cellLines1.viewkeys() & cellLines2.viewkeys() & exp_cellLines1.viewkeys() & exp_cellLines2.viewkeys())
common_lines.sort()
exclude = ['SC11-009','SC11-008'] ### selected the line with the greatest XIST for this donor
if uniqueDonors:
common_lines2=[]; donor_added=[]
for donorID in donor_db:
donor_db[donorID].sort()
for (priority,cellLine) in donor_db[donorID]: ### Prioritize based on sample QC and donor preference
if cellLine in common_lines and cellLine not in exclude:
if donorID not in donor_added and 'H9' not in cellLine:
common_lines2.append(cellLine)
donor_added.append(donorID)
common_lines = common_lines2
print 'Common Approved Lines:',common_lines
return common_lines
def downloadSynapseFile(synid,output_dir):
import synapseclient
import os,sys,string,shutil
syn = synapseclient.Synapse()
syn.login()
matrix = syn.get(synid, downloadLocation=output_dir, ifcollision="keep.local")
return matrix.path
def buildAdditionalMirTargetGeneSets():
miR_association_file = 'AltDatabase/ensembl/Hs/Hs_microRNA-Ensembl.txt'
output_dir = 'AltDatabase/EnsMart72/goelite/Hs/gene-mapp/Ensembl-'
eo = export.ExportFile(output_dir+'microRNATargets_All.txt')
header = 'GeneID\tSystemCode\tmiRNA\n'
eo.write(header)
miRNA_source_db={}
for line in open(unique.filepath(miR_association_file),'rU').xreadlines():
data = line.rstrip()
miRNA,ensembl,source = string.split(data,'\t')
output = ensembl+'\t\t'+miRNA+'\n'
eo.write(output)
sources = string.split(source,'|')
for source in sources:
try: miRNA_source_db[source].append(output)
except KeyError: miRNA_source_db[source]=[output]
eo.close()
for source in miRNA_source_db:
eo = export.ExportFile(output_dir+source+'.txt')
eo.write(header)
for line in miRNA_source_db[source]:
eo.write(line)
eo.close()
def returnSynFileLocations(file1,file2,output_dir):
if 'syn' in file1:
try: file1 = downloadSynapseFile(file1,output_dir)
except Exception:
print 'Is the destination file %s already open?' % file1;sys.exit()
if 'syn' in file2:
try: file2 = downloadSynapseFile(file2,output_dir)
except Exception:
print 'Is the destination file %s already open?' % file2;sys.exit()
return file1,file2
def synapseStore(file_dirs,root,parent_syn,executed_urls,used):
for file in file_dirs:
file_dir = root+'/'+file
file = synapseclient.File(file_dir, parent=parent_syn)
file = syn.store(file,executed=executed_urls,used=used)
def synapseStoreFolder(dir_path,parent_syn):
data_folder = synapseclient.Folder(dir_path, parent=parent_syn)
data_folder = syn.store(data_folder)
sub_parent = data_folder.id
return sub_parent
def synapseDirectoryUpload(expressionDir, parent_syn, executed_urls, used):
root = string.split(expressionDir,'/')[-1]
sub_parent = synapseStoreFolder(root,parent_syn)
folders = getFiles(expressionDir)
for folder in folders:
### Create the folder in Synapse
dir_path_level1 = expressionDir+'/'+folder
sub_parent1 = synapseStoreFolder(folder,sub_parent)
f2 = getFiles(dir_path_level1)
files = getFiles(dir_path_level1,False)
synapseStore(files,dir_path_level1,sub_parent1,executed_urls,used)
for folder in f2:
dir_path_level2 = dir_path_level1+'/'+folder
sub_parent2 = synapseStoreFolder(folder,sub_parent1)
f3 = getFiles(dir_path_level2)
files = getFiles(dir_path_level2,False)
synapseStore(files,dir_path_level2,sub_parent2,executed_urls,used)
for folder in f3:
dir_path_level3 = dir_path_level2+'/'+folder
sub_parent3 = synapseStoreFolder(folder,sub_parent2)
files = getFiles(dir_path_level3,False)
### These are the GO-Elite result files (not folders)
synapseStore(files,dir_path_level3,sub_parent3,executed_urls,used)
def exportUpDownGenes(results_dir):
files = os.listdir(results_dir)
for file in files:
filename = results_dir+'/'+file
output_dir = results_dir+'/regulated/'+file
firstLine=True
if '.txt' in filename and 'GE.' in filename:
ou = export.ExportFile(output_dir[:-4]+'-up.txt')
od = export.ExportFile(output_dir[:-4]+'-down.txt')
for line in open(filename,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
ou.write(line)
od.write(line)
lfi = values.index('LogFold')
else:
if float(values[lfi]) >0:
ou.write(line)
else:
od.write(line)
ou.close()
od.close()
def exportGeneSetsFromCombined(filename):
firstLine=True
synapse_format = True
simple_format = False
reverse = True
comparison_to_gene={}
rootdir = export.findParentDir(filename)
file = export.findFilename(filename)
for line in open(filename,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
else:
comparison, gene, symbol, up_rawp, ng_adjp, up_logfold,ng_logofold, ng_avg1, ng_avg2 = values[:9]
comparison = string.replace(comparison,'GE.','')
prefix = string.split(comparison,':')[0]
state = string.split(prefix,'-')[0]
if state=='NA': state = 'All'
comparison = file[:-4]+'-'+string.split(comparison,':')[1]
comparison = string.replace(comparison,'allTopGenes-','')[:-4]
c1,c2 = string.split(comparison,'_vs_')
c1 = re.sub('[^0-9a-zA-Z]+', '', c1)
c2 = re.sub('[^0-9a-zA-Z]+', '', c2)
comparison = c1+'_vs_'+c2
#comparison = string.replace(comparison,':','-')
log_fold = float(up_logfold)
#print c1,c2,reverse,log_fold
if c1>c2:
comparison = c2+'_vs_'+c1
log_fold = log_fold *-1
if log_fold<0:
if synapse_format:
comparison+='__down'
else:
comparison = c2+'_vs_'+c1 ### reverse the regulation direction
else:
if synapse_format:
comparison+='__up'
if synapse_format:
if len(symbol) == 0: symbol = gene
gene = symbol
#comparison = string.replace(comparison,'_vs_','_')
comparison = string.replace(comparison,'NA','NotApplicable')
#comparison = string.replace(comparison,'MESO-5','MESO-EARLY')
comparison = string.replace(comparison,' ','_')
#if state == 'MESO': state = 'MESOEARLY'
if 'ENS' in gene:
SystemCode = 'En'
if ':' in gene:
gene = string.split(gene,':')[0]
genes = [gene]
elif 'cg' in gene:
SystemCode = 'En'
genes = []
for i in string.split(symbol,'|'):
if 'ENS' in i:
genes.append(i)
else:
SystemCode = 'Sy'
genes = [gene]
for g in genes:
if synapse_format:
if simple_format:
try: comparison_to_gene[comparison].append([g,log_fold])
except Exception: comparison_to_gene[comparison] = [[g,log_fold]]
else:
try: comparison_to_gene[state+'__'+comparison].append([g,log_fold])
except Exception: comparison_to_gene[state+'__'+comparison] = [[g,log_fold]]
elif simple_format:
try: comparison_to_gene[comparison].append([g,log_fold])
except Exception: comparison_to_gene[comparison] = [[g,log_fold]]
else:
try: comparison_to_gene[state+'-'+comparison].append([g,log_fold])
except Exception: comparison_to_gene[state+'-'+comparison] = [[g,log_fold]]
aro = export.ExportFile(rootdir+'/Regulated/combined.txt')
aro.write('Gene\tLogFold\tComparison\n')
for comparison in comparison_to_gene:
ro = export.ExportFile(rootdir+'/Regulated/'+comparison+'.txt')
ro.write('Gene\tSystemCode\n')
for (gene,logfold) in comparison_to_gene[comparison]:
ro.write(gene+'\t'+SystemCode+'\n')
aro.write(gene+'\t'+str(logfold)+'\t'+string.replace(comparison,'.txt','')+'\n')
ro.close()
aro.close()
def remoteAnalysis(species,expression_file,groups_file,platform='PSI',log_fold_cutoff=0.1,
use_adjusted_pval=True,pvalThreshold=0.05,use_custom_output_dir='',
suppressPrintOuts=False):
print "Performing a differential expression analysis (be patient)..."
global pval_threshold
global PercentExp
global restricted_gene_denominator
global global_adjp_db
global probability_statistic
global use_adjusted_p
global logfold_threshold
global minSampleNumber
minSampleNumber = 2
logfold_threshold = log_fold_cutoff
use_adjusted_p = use_adjusted_pval
global_adjp_db={}
restricted_gene_denominator={}
probability_statistic = 'moderated t-test'
PercentExp = 0.5
if platform == 'PSI':
CovariateQuery = 'Events'
else:
CovariateQuery = 'DEGs'
pval_threshold = float(pvalThreshold)
metadata_files = [groups_file]
meta_description_file = None
if platform == 'PSI' or platform == 'methylation':
if log_fold_cutoff==None:
logfold_threshold=0.1 ### equivalent to a 0.15 dPSI or 0.15 beta differences
else:
logfold_threshold= log_fold_cutoff
if platform == 'PSI':
print 'Using a dPSI of:',logfold_threshold
if platform == 'methylation':
use_adjusted_p = True
if platform == 'miRSeq':
use_adjusted_p = False
logfold_threshold=math.log(1,2)
print 'Filtering on adjusted p-value:',use_adjusted_p
if platform != 'PSI':
from build_scripts import EnsemblImport
try: gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,platform,'key_by_array')
except Exception: gene_location_db={}
if meta_description_file !=None:
if '.txt' in meta_description_file:
meta_description_files = [meta_description_file]
else:
meta_description_files=[]
files = os.listdir(meta_description_file)
for file in files:
if '.txt' in file:
meta_description_files.append(meta_description_file+'/'+file)
splicingEventTypes={}
all_groups_db={}
all_comps_db={}
if 'groups.' in metadata_files[0]:
all_groups_db, all_comps_db = importGroupsComps(metadata_files[0])
else:
for meta_description_file in meta_description_files:
metadata_filters = importMetaDataDescriptions(meta_description_file)
all_groups_db, all_comps_db = prepareComparisonData(metadata_files[0],metadata_filters,all_groups_db, all_comps_db)
for i in all_groups_db:
#print i
for k in all_groups_db[i]:
if suppressPrintOuts == False:
print ' ',k,'\t',len(all_groups_db[i][k])
#print all_comps_db
if platform == 'PSI':
result_type = 'dPSI'
else:
result_type = 'LogFold'
if len(use_custom_output_dir)>0:
CovariateQuery = use_custom_output_dir
elif use_adjusted_p:
CovariateQuery += '-'+result_type+'_'+str(logfold_threshold)[:4]+'_adjp'
else:
CovariateQuery += '-'+result_type+'_'+str(logfold_threshold)+'_rawp'
for specificCovariate in all_groups_db:
comps_db = all_comps_db[specificCovariate]
groups_db = all_groups_db[specificCovariate]
rootdir,splicingEventTypes = performDifferentialExpressionAnalysis(species,platform,expression_file,
groups_db,comps_db,CovariateQuery,splicingEventTypes,suppressPrintOuts=suppressPrintOuts)
if platform == 'PSI':
graphics = outputSplicingSummaries(rootdir+'/'+CovariateQuery,splicingEventTypes)
else:
graphics=[]
outputGeneExpressionSummaries(rootdir+'/'+CovariateQuery,splicingEventTypes)
#except: pass
return graphics
def compareDomainComposition(folder):
### Compare domain composition
import collections
event_db = collections.OrderedDict()
background_db = collections.OrderedDict()
groups_list=['']
import UI
files = UI.read_directory(folder)
for file in files:
if '.txt' in file and 'PSI.' in file:
db={}
event_db[file[:-4]]=db
all_unique_events = {}
background_db[file[:-4]]=all_unique_events
groups_list.append(file[:-4])
fn = folder+'/'+file
firstLine = True
output_file = fn[:-4]+'-ProteinImpact.txt'
output_file = string.replace(output_file,'PSI.','Impact.')
eo = export.ExportFile(output_file)
eo.write('GeneSymbol\tSy\tImpact\tImpactDescription\tUID\n')
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
#(+)alt-coding, (+)AA:232(ENSP00000230685)->296(ENSP00000009530)|(-)MHC_II-assoc_invar_chain-IPR022339, (+)DISULFID, (+)DOMAIN-Thyroglobulin type-1, (+)HELIX, (+)MHC_II-assoc_invar_chain-IPR022339, (+)SITE-Breakpoint for translocation to form a CD74-ROS1 fusion protein, (+)STRAND, (+)TOPO_DOM-Extracellular, (+)TURN, (+)Thyroglobulin_1-IPR000716
if firstLine:
protein_index = t.index('ProteinPredictions')
clusterID_index = t.index('UpdatedClusterID')
event_index = t.index('EventAnnotation')
firstLine= False
continue
uid = t[0]
symbol = string.split(uid,':')[0]
clusterID=t[clusterID_index]
all_unique_events[clusterID]=None
if len(t[protein_index])>0:
protein_prediction = t[protein_index]
protein_predictions = string.split(protein_prediction,',')
if 'Promoter' in t[event_index]:
continue
for annotation in protein_predictions:
if 'AA:' in annotation:
if annotation[0]==' ':
direction = annotation[2]
diff = string.split(annotation[7:],'|')[0]
else:
direction = annotation[1]
diff = string.split(annotation[6:],'|')[0]
try: diff1,diff2 = string.split(diff,'->')
except:
diff1,diff2 = string.split(diff,'+>')
try: diff1 = int(string.split(diff1,'(')[0])
except: print diff,diff1,diff2,[annotation];sys.exit()
diff2 = int(string.split(diff2,'(')[0])
coding_diff = (diff1)/float(diff2)
if coding_diff < 0.333:
type = 'Truncation'
elif coding_diff < 0.75:
type = 'Protein Length'
else:
type = None
if '|' in annotation:
domain_difference = True
else:
domain_difference = False
if type==None and domain_difference == False:
pass
else:
if direction == '+':
direction = 'decreased' ### seems counter intuitive but we are specificall looking at decreased protein length
""" specifically, if a (+)200->1000, this means in the experiment the protein is longer but truncation is decreased """
elif direction == '-':
direction = 'increased'
if direction != '~':
if type == 'Truncation':
score = 2
elif domain_difference:
score = 1
elif type == 'Protein Length':
score = 0.5
if direction == 'decreased':
score = score*-1
if type != None:
updated_type = type
if domain_difference:
updated_type += '|domain-impact'
else:
updated_type = ''
if domain_difference:
updated_type += 'domain-impact'
eo.write(string.join([symbol,'Sy',str(score),updated_type,uid],'\t')+'\n')
db[clusterID]=type,domain_difference,direction
continue
eo = export.ExportFile(folder+'/Protein-Level-Impact-Summary.txt')
file_list = []
for file in event_db:
file_list.append(file)
domain_difference_db={}
impact_type_db={}
impacted_events = event_db[file]
for clusterID in impacted_events:
type,domain_difference,direction = impacted_events[clusterID]
### Create the entries in the dictionary so these can be consistently populated below
if domain_difference!=False:
domain_difference_db[direction,domain_difference]=[]
if type !=None:
impact_type_db[direction,type]=[]
for file in event_db:
impacted_events = event_db[file]
unique_events = len(background_db[file])
file_domain_difference_db={}
file_impact_type_db={}
for clusterID in impacted_events:
type,domain_difference,direction = impacted_events[clusterID]
try:
if domain_difference!=False:
file_domain_difference_db[direction,domain_difference]+=1
if type!=None:
file_impact_type_db[direction,type]+=1
except:
if domain_difference!=False:
file_domain_difference_db[direction,domain_difference]=1
if type!=None:
file_impact_type_db[direction,type]=1
for (direction,type) in impact_type_db:
try: impact_type_db[direction,type].append(str(file_impact_type_db[direction,type]/float(unique_events)))
except: impact_type_db[direction,type].append('0')
for (direction,domain_difference) in domain_difference_db:
try: domain_difference_db[direction,domain_difference].append(str(file_domain_difference_db[direction,domain_difference]/float(unique_events)))
except: domain_difference_db[direction,domain_difference].append('0')
eo.write(string.join(['UID']+file_list,'\t')+'\n')
for (direction,domain_difference) in domain_difference_db:
out = [direction+' Domain Impact']+domain_difference_db[direction,domain_difference]
out = string.join(map(str,out),'\t')+'\n'
eo.write(out)
for (direction,type) in impact_type_db:
out = [direction+' '+type]+impact_type_db[direction,type]
out = string.join(map(str,out),'\t')+'\n'
#print file, out, unique_events;sys.exit()
eo.write(out)
eo.close()
sys.exit()
if __name__ == '__main__':
species = 'Hs';
expression_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Kumar/July-26-2017/Hs_RNASeq_top_alt_junctions-PSI_EventAnnotation.txt'
groups_file = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Kumar/July-26-2017/groups.KD.txt'
computed_results_dir = '/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/Leucegene/July-2017/PSI/SpliceICGS.R1.Depleted.12.27.17/all-depleted-and-KD'
#exportUpDownGenes('/Users/saljh8/Desktop/dataAnalysis/SalomonisLab/cellHarmony-evaluation/HCA-alignment/DEGs');sys.exit()
#remoteAnalysis(species,expression_file,groups_file,platform='PSI',log_fold_cutoff=0.1,use_adjusted_pval=True,pvalThreshold=0.05);sys.exit()
#compareDomainComposition(computed_results_dir)
################ Comand-line arguments ################
#buildAdditionalMirTargetGeneSets();sys.exit()
filename = '/Users/saljh8/Desktop/PCBC_MetaData_Comparisons/eXpress/CombinedResults/allTopGenes.txt' #DiffStateComps Reprogramming
#exportGeneSetsFromCombined(filename);sys.exit()
platform='RNASeq'
species='Hs'
probability_statistic = 'moderated t-test'
#probability_statistic = 'Kolmogorov Smirnov'
#probability_statistic = 'unpaired t-test'
#probability_statistic = 'paired t-test'
minRPKM=-1000
PercentExp = 75
minSampleNumber = 2
logfold_threshold=math.log(2,2)
pval_threshold=0.05
use_adjusted_p = False
expression_files=[]
platforms=[]
metadata_files=[]
gender_restricted = None
runGOElite=False
compareEnrichmentProfiles=False
restrictCovariateTerm=None
compDiffState=None
runAgain=False
output_dir=None
include_only=''
meta_description_file=None
sampleSetQuery = 'NA'
used=[]
executed_urls=[]
restricted_gene_denominator={}
global_adjp_db={}
splicingEventTypes={}
CovariateQuery = None
log_fold_cutoff=None
print sys.argv[1:]
import getopt
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print 'Supply the argument --i location'
### python metaDataAnalysis.py --i /Volumes/SEQ-DATA\ 1/PCBC/RNASeq/July2014/MetaData/RNASeq_MetaData_July2014.txt --key "CellType" --value "Cell Type of Origin"
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['m=','i=','d=','c=','u=','p=','s=','f=',
'g=','e=','ce=','rc=','cd=','o=','md=','in=','target=','parent=','urls=','used=','mf=',
'adjp=','dPSI=','pval=','percentExp=','percentExp=', 'fold=', 'species=', 'platform=', 'expdir='])
for opt, arg in options:
if opt == '--m': metadata_files.append(arg)
if opt == '--o':
if output_dir==None:
output_dir = arg
else:
output_dir = [output_dir,arg]
if opt == '--i' or opt == '--expdir': expression_files.append(arg)
if opt == '--e': runGOElite=True
if opt == '--f' or opt == '--fold':
try: logfold_threshold = math.log(float(arg),2)
except Exception: logfold_threshold = 0
if opt == '--ce': compareEnrichmentProfiles = True
if opt == '--d': sampleSetQuery=arg
if opt == '--c': CovariateQuery=arg
if opt == '--p' or opt == '--platform': platforms.append(arg)
if opt == '--g': gender_restricted=arg
if opt == '--s' or opt == '--species': species=arg
if opt == '--rc': restrictCovariateTerm=arg
if opt == '--cd': compDiffState=arg
if opt == '--md': mirDataDir=arg
if opt == '--in': include_only=arg
if opt == '--pval': pval_threshold = float(arg)
if opt == '--target': target_dir=arg
if opt == '--mf': meta_description_file=arg
if opt == '--parent': parent_syn=arg
if opt == '--urls': executed_urls.append(arg) ### options are: all, junction, exon, reference
if opt == '--used': used.append(arg)
if opt == '--percentExp': PercentExp = int(arg)
if opt == '--adjp':
if string.lower(arg) == 'yes' or string.lower(arg) == 'true':
use_adjusted_p = True
if opt == '--dPSI':
log_fold_cutoff = float(arg)
if opt == '--u':
if string.lower(arg) == 'yes' or string.lower(arg) == 'true':
uniqueDonors=True
use_adjusted_p = False
else:
uniqueDonors = False
if string.lower(arg) == 'both':
runAgain = True
if len(used)>0:
###Upload existing results folder to Synapse
import synapseclient
import os,sys,string,shutil,getopt
syn = synapseclient.Synapse()
syn.login()
synapseDirectoryUpload(target_dir, parent_syn, executed_urls, used)
sys.exit()
elif compareEnrichmentProfiles:
#print expression_files
compareGOEliteEnrichmentProfiles(expression_files[0],expression_files[1])
elif runGOElite:
runGOEliteAnalysis(species,expression_files[0])
elif (len(expression_files)==1 and '.txt' in expression_files[0]) or (len(expression_files)==1 and 'syn' in expression_files[0]):
### Perform a covariate based analysis on the lone input expression file
metadata_file = metadata_files[0]
if 'syn' in metadata_file:
try: metadata_file = downloadSynapseFile(metadata_file,output_dir)
except Exception:
print 'Is the destination file %s already open?' % metadata_file;sys.exit()
expression_file = expression_files[0]
if 'syn' in expression_file:
try:expression_file = downloadSynapseFile(expression_file,output_dir)
except Exception:
print 'Is the destination file %s already open?' % expression_file;sys.exit()
if 'syn' in include_only:
try:include_only = downloadSynapseFile(include_only,output_dir)
except Exception:
print 'Is the destination file %s already open?' % include_only;sys.exit()
if len(platforms)>0: platform = platforms[0]
if platform == 'PSI' or platform == 'methylation':
if log_fold_cutoff==None:
logfold_threshold=0.1 ### equivalent to a 0.15 dPSI or 0.15 beta differences
else:
logfold_threshold= log_fold_cutoff
if platform == 'PSI':
print 'Using a dPSI of:',logfold_threshold
if platform == 'methylation':
use_adjusted_p = True
if platform == 'miRSeq':
use_adjusted_p = False
logfold_threshold=math.log(1,2)
print 'Filtering on adjusted p-value:',use_adjusted_p
if platform == 'PSI' and CovariateQuery == None:
CovariateQuery = 'Events'
else:
CovariateQuery = 'DEGs'
if platform != 'PSI':
from build_scripts import EnsemblImport
try: gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,platform,'key_by_array')
except Exception: gene_location_db={}
if len(include_only)>0:
restricted_gene_denominator = importRestrictedSetOfGenesToQuery(include_only)
if meta_description_file !=None:
if '.txt' in meta_description_file:
meta_description_files = [meta_description_file]
else:
meta_description_files=[]
files = os.listdir(meta_description_file)
for file in files:
if '.txt' in file:
meta_description_files.append(meta_description_file+'/'+file)
splicingEventTypes={}
all_groups_db={}
all_comps_db={}
if 'groups.' in metadata_files[0]:
all_groups_db, all_comps_db = importGroupsComps(metadata_files[0])
else:
for meta_description_file in meta_description_files:
metadata_filters = importMetaDataDescriptions(meta_description_file)
all_groups_db, all_comps_db = prepareComparisonData(metadata_files[0],metadata_filters,all_groups_db, all_comps_db)
for i in all_groups_db:
print i
for k in all_groups_db[i]: print ' ',k,'\t',len(all_groups_db[i][k])
print all_comps_db
if platform == 'PSI':
result_type = 'dPSI'
else:
result_type = 'LogFold'
if use_adjusted_p:
CovariateQuery += '-'+result_type+'_'+str(logfold_threshold)[:4]+'_adjp'
else:
CovariateQuery += '-'+result_type+'_'+str(logfold_threshold)+'_rawp'
for specificCovariate in all_groups_db:
comps_db = all_comps_db[specificCovariate]
groups_db = all_groups_db[specificCovariate]
rootdir,splicingEventTypes = performDifferentialExpressionAnalysis(species,platform,expression_file,groups_db,comps_db,CovariateQuery,splicingEventTypes)
if platform == 'PSI':
graphics = outputSplicingSummaries(rootdir+'/'+CovariateQuery,splicingEventTypes)
else:
outputGeneExpressionSummaries(rootdir+'/'+CovariateQuery,splicingEventTypes)
#except: pass
sys.exit()
for CovariateQuery in covariate_set:
for sampleSetQuery in Sample_set:
print 'Analyzing the covariate:',CovariateQuery, 'and diffState:',sampleSetQuery, 'unique donor analysis:',uniqueDonors
if 'XIST' in CovariateQuery: gender_restricted='female'
genderRestricted = gender_restricted
try:
sample_metadata,groups_db,comps_db = prepareComparisonData(metadata_file,metadata_filters,sampleSetQuery,CovariateQuery,uniqueDonors,genderRestricted,platform=platform,compDiffState=compDiffState,restrictCovariateTerm=restrictCovariateTerm)
performDifferentialExpressionAnalysis(species,platform,expression_file,sample_metadata,groups_db,comps_db,sampleSetQuery+'-'+CovariateQuery,uniqueDonors)
except Exception:
print traceback.format_exc()
if runAgain:
uniqueDonors=True
use_adjusted_p = False
print 'Analyzing the covariate:',CovariateQuery, 'and diffState:',sampleSetQuery, 'unique donor analysis:',uniqueDonors
try:
sample_metadata,groups_db,comps_db = prepareComparisonData(metadata_file,sampleSetQuery,CovariateQuery,uniqueDonors,genderRestricted,platform=platform,compDiffState=compDiffState,restrictCovariateTerm=restrictCovariateTerm)
performDifferentialExpressionAnalysis(species,platform,expression_file,sample_metadata,groups_db,comps_db,sampleSetQuery+'-'+CovariateQuery,uniqueDonors)
except Exception: pass
uniqueDonors=False; use_adjusted_p = True
if platform == 'miRSeq' or platform == 'PSI': use_adjusted_p = False
if runAgain:
root_exp_dir=export.findParentDir(expression_file)
identifyCommonGenes(root_exp_dir)
runGOEliteAnalysis(species,root_exp_dir)
try: compareGOEliteEnrichmentProfiles(root_exp_dir,mirDataDir)
except Exception: pass
elif '.txt' not in expression_files[0] and 'syn' not in expression_files[0]:
### Compare unique and non-unique results to get overlapps (single high-confidence result file)
### The parent directory that contain results from the above analysis serve as input
resultsDirectory = expression_files[0]
identifyCommonGenes(resultsDirectory)
else:
### Perform a correlation analysis between two omics technologies
expression_file1,expression_file2 = expression_files
platform1,platform2 = platforms
metadata_file1,metadata_file2 = metadata_files
output_dir1, output_dir2 = output_dir
print expression_files, output_dir1
print metadata_files, output_dir2
expression_file1,metadata_file1 = returnSynFileLocations(expression_file1,metadata_file1,output_dir1)
metadata_file2,expression_file2 = returnSynFileLocations(metadata_file2,expression_file2,output_dir2)
print expression_file1, expression_file2
print metadata_file1, metadata_file2
cellLines1,sample_metadata1,uniqueDonor_db,donor_db = prepareComparisonData(metadata_file1,sampleSetQuery,None,uniqueDonors,gender_restricted,platform=platform1)
cellLines2,sample_metadata2,uniqueDonor_db,donor_db = prepareComparisonData(metadata_file2,sampleSetQuery,None,uniqueDonors,gender_restricted,platform=platform2)
exp_cellLines1 = getDatasetSamples(expression_file1,sample_metadata1,cellLines1)
exp_cellLines2 = getDatasetSamples(expression_file2,sample_metadata2,cellLines2)
common_lines = getCommonCellLines(cellLines1,cellLines2,exp_cellLines1,exp_cellLines2,uniqueDonor_db,uniqueDonors,donor_db)
filtered_exp_data1, samplesToEvaluate = importExpressionData(species,platform1,expression_file1,cellLines1,common_lines)
filtered_exp_data2, samplesToEvaluate = importExpressionData(species,platform2,expression_file2,cellLines2,common_lines)
combineAndCompareMatrices(expression_file1,filtered_exp_data1,filtered_exp_data2,platform1,platform2,samplesToEvaluate) ### export results to two different files and a combined file
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/metaDataAnalysis.py
|
metaDataAnalysis.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import math
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from stats_scripts import statistics
import os.path
import unique
import UI
import export
import time
import traceback
import RNASeq
import ExpressionBuilder
def normalizeDataset(filename,output = None, normalization='quantile',platform="3'array"):
""" Perform Quantile Normalization on an input expression dataset """
if output==None:
output = filename
moved_exp_dir = export.findParentDir(filename)+'Non-Normalized/'+export.findFilename(filename)
try:
export.copyFile(filename, moved_exp_dir)
print 'Moved original expression file to:'
print '\t'+moved_exp_dir
except Exception: None
if normalization == 'Quantile' or normalization == 'quantile':
print "Importing data..."
sample_expression_db = importExpressionValues(filename)
print "Performing quantile normalization..."
sample_expression_db = RNASeq.quantileNormalizationSimple(sample_expression_db)
exportExpressionData(output,sample_expression_db)
elif normalization == 'group':
performGroupNormalization(moved_exp_dir,filename,platform)
print 'Exported expression input file to:',output
def importGroups(fn):
try: group_db=collections.OrderedDict()
except Exception:
try:
import ordereddict
group_db=ordereddict.OrderedDict()
except Exception: group_db={}
for line in open(fn,'rU').xreadlines():
data = ExpressionBuilder.cleanUpLine(line)
sample_filename,group_number,group_name = string.split(data,'\t')
try: group_db[group_name].append(sample_filename)
except Exception: group_db[group_name] = [sample_filename]
return group_db
def performGroupNormalization(filename,export_dir,platform):
expressionDataFormat,increment,convertNonLogToLog = ExpressionBuilder.checkExpressionFileFormat(filename)
groups_dir = string.replace(export_dir,'exp.','batch.')
fn=unique.filepath(filename); row_number=0; exp_db={}; relative_headers_exported = False
group_db = importGroups(groups_dir)
export_data = export.ExportFile(export_dir)
for line in open(fn,'rU').xreadlines():
data = ExpressionBuilder.cleanUpLine(line)
t = string.split(data,'\t')
if data[0]=='#' and row_number==0: row_number = 0
elif row_number==0:
sample_list = t[1:]
new_sample_list = []
for group in group_db:
group_samples = group_db[group]
try:
sample_index_list = map(lambda x: sample_list.index(x), group_samples)
group_db[group] = sample_index_list
new_sample_list+=group_samples
except Exception:
missing=[]
for x in sample_list:
if x not in t[1:]: missing.append(x)
print 'missing:',missing
print t
print sample_list
print filename, groups_dir
print 'Unknown Error!!! Skipping cluster input file build (check column and row formats for conflicts)'; forceExit
title = string.join([t[0]]+new_sample_list,'\t')+'\n' ### output the new sample order (group file order)
export_data.write(title)
row_number=1
else:
gene = t[0]
if expressionDataFormat == 'non-log' and (convertNonLogToLog or platform == 'RNASeq'):
### Convert to log2 RPKM values - or counts
try: all_values = map(lambda x: math.log(float(x)+increment,2), t[1:])
except Exception:
all_values = ExpressionBuilder.logTransformWithNAs(t[1:],increment)
else:
try: all_values = map(float,t[1:])
except Exception:
all_values = ExpressionBuilder.logTransformWithNAs(t[1:],increment)
row_number+=1 ### Keep track of the first gene as to write out column headers for the relative outputs
gene_log_folds = []
for group in group_db:
sample_index_list = group_db[group]
### Calculate log-fold values relative to the mean of all sample expression values
try: values = map(lambda x: all_values[x], sample_index_list) ### simple and fast way to reorganize the samples
except Exception:
print len(values), sample_index_list;kill
try: avg = statistics.avg(values)
except Exception:
values2=[]
for v in values:
try: values2.append(float(v))
except Exception: pass
values = values2
try: avg = statistics.avg(values)
except Exception:
if len(values)>0: avg = values[0]
else: avg = 0
try: log_folds = map(lambda x: (x-avg), values)
except Exception:
log_folds=[]
for x in values:
try: log_folds.append(x-avg)
except Exception: log_folds.append('')
gene_log_folds+=log_folds
gene_log_folds = map(lambda x: str(x),gene_log_folds)
export_data.write(string.join([gene]+gene_log_folds,'\t')+'\n')
export_data.close()
def calculateRatios(db1,db2):
ratio_db={}
for array in db1:
exp_ratios={}
exp_db = db1[array]
for probe_name in exp_db:
exp_ratios[probe_name] = str(float(exp_db[probe_name])-float(db2[array][probe_name])) ### log2 ratio
ratio_db[array]=exp_ratios
return ratio_db
def importExpressionValues(filename):
""" Imports tab-delimited expression values"""
header = True
sample_expression_db={}
fn=unique.filepath(filename)
for line in open(fn,'rU').xreadlines():
data = UI.cleanUpLine(line)
if header:
sample_names = string.split(data,'\t')
header = False
else:
exp_values = string.split(data,'\t')
gene = exp_values[0]
index=1
for value in exp_values[1:]:
sample_name = sample_names[index]
if sample_name in sample_expression_db:
gene_expression_db = sample_expression_db[sample_name]
gene_expression_db[gene] = value
else:
gene_expression_db={}
gene_expression_db[gene] = value
sample_expression_db[sample_name] = gene_expression_db
index+=1
return sample_expression_db
def exportExpressionData(filename,sample_db):
export_text = export.ExportFile(filename)
all_genes_db = {}
sample_list=[]
for sample in sample_db:
sample_list.append(sample)
gene_db = sample_db[sample]
for geneid in gene_db:
all_genes_db[geneid]=[]
sample_list.sort() ### Organize these alphabetically rather than randomly
column_header = string.join(['ProbeName']+sample_list,'\t')+'\n' ### format column-names for export
export_text.write(column_header)
for geneid in all_genes_db:
values=[]
for sample in sample_list:
try: values.append(sample_db[sample][geneid]) ### protein_expression
except Exception: values.append(0)
export_text.write(string.join([geneid]+map(str, values),'\t')+'\n')
export_text.close()
if __name__ == '__main__':
filename = "/Volumes/salomonis1/projects/Beena2/fastq/ExpressionInput/exp.AF.txt'"
normalizeDataset(filename,normalization='Group')
#normalizeDataset(filename)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/NormalizeDataset.py
|
NormalizeDataset.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
try:
from stats_scripts import statistics
except Exception:
import statistics
import math
import os.path
import unique
import update
import copy
import time
import export
try: from build_scripts import EnsemblImport; reload(EnsemblImport)
except Exception: import EnsemblImport
try: from build_scripts import JunctionArrayEnsemblRules
except Exception: pass ### occurs with circular imports
try: from build_scripts import JunctionArray; reload(JunctionArray)
except Exception: pass ### occurs with circular imports
try: from build_scripts import ExonArrayEnsemblRules
except Exception: pass ### occurs with circular imports
import multiprocessing
import logging
import traceback
import warnings
import bisect
try: from visualization_scripts import clustering; reload(clustering)
except Exception: import clustering_new as clustering;
try:
import scipy
import scipy.cluster.hierarchy as sch
import scipy.spatial.distance as dist
except Exception: pass
try: import numpy
except Exception: pass
LegacyMode = True
try:
from scipy import average as Average
from scipy import stats
except Exception:
from statistics import avg as Average
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir)
return dir_list
def makeUnique(item):
db1={}; list1=[]; k=0
for i in item:
try: db1[i]=[]
except TypeError: db1[tuple(i)]=[]; k=1
for i in db1:
if k==0: list1.append(i)
else: list1.append(list(i))
list1.sort()
return list1
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
######### Below code deals with building the AltDatabase #########
def collapseNoveExonBoundaries(novel_exon_coordinates,dataset_dir):
""" Merge exon predictions based on junction measurments from TopHat. The predicted exons are
bound by the identified splice site and the consensus length of reads in that sample"""
dataset_dir = string.replace(dataset_dir,'exp.','ExpressionInput/novel.')
export_data,status = AppendOrWrite(dataset_dir) ### Export all novel exons
if status == 'not found':
export_data.write('GeneID\tStrand\tExonID\tCoordinates\n')
novel_gene_exon_db={}
for (chr,coord) in novel_exon_coordinates:
key = (chr,coord)
ji,side,coord2 = novel_exon_coordinates[(chr,coord)]
try:
if side == 'left': ### left corresponds to the position of coord
intron = string.split(string.split(ji.ExonRegionID(),'-')[1][:2],'.')[0]
else:
intron = string.split(string.split(ji.ExonRegionID(),'-'),'.')[0]
ls = [coord,coord2]
ls.sort() ### The order of this is variable
if ji.Strand() == '-':
coord2,coord = ls
else: coord,coord2 = ls
if 'I' in intron and ji.Novel() == 'side':
#if 'ENSG00000221983' == ji.GeneID():
try: novel_gene_exon_db[ji.GeneID(),ji.Strand(),intron].append((coord,coord2,ji,key,side))
except Exception: novel_gene_exon_db[ji.GeneID(),ji.Strand(),intron] = [(coord,coord2,ji,key,side)]
except Exception: pass
outdatedExons={} ### merging novel exons, delete one of the two original
for key in novel_gene_exon_db:
firstNovel=True ### First putative novel exon coordinates examined for that gene
novel_gene_exon_db[key].sort()
if key[1]=='-':
novel_gene_exon_db[key].reverse()
for (c1,c2,ji,k,s) in novel_gene_exon_db[key]:
if firstNovel==False:
#print [c1,l2] #abs(c1-l2);sys.exit()
### see if the difference between the start position of the second exon is less than 300 nt away from the end of the last
if abs(c2-l1) < 300 and os!=s: ### 80% of human exons are less than 200nt - PMID: 15217358
proceed = True
#if key[1]=='-':
if c2 in k:
novel_exon_coordinates[k] = ji,s,l1
outdatedExons[ok]=None ### merged out entry
elif l1 in ok:
novel_exon_coordinates[ok] = li,os,c2
outdatedExons[k]=None ### merged out entry
else:
proceed = False ### Hence, the two splice-site ends are pointing to two distinct versus one common exons
"""
if c2 == 18683670 or l1 == 18683670:
print key,abs(c2-l1), c1, c2, l1, l2, li.ExonRegionID(), ji.ExonRegionID();
print k,novel_exon_coordinates[k]
print ok,novel_exon_coordinates[ok]
"""
if proceed:
values = string.join([ji.GeneID(),ji.Strand(),key[2],ji.Chr()+':'+str(l1)+'-'+str(c2)],'\t')+'\n'
export_data.write(values)
### For negative strand genes, c1 is larger than c2 but is the 5' begining of the exon
l1,l2,li,ok,os = c1,c2,ji,k,s ### record the last entry
firstNovel=False
for key in outdatedExons: ### Delete the non-merged entry
del novel_exon_coordinates[key]
export_data.close()
return novel_exon_coordinates
def exportNovelExonToBedCoordinates(species,novel_exon_coordinates,chr_status,searchChr=None):
### Export the novel exon coordinates based on those in the junction BED file to examine the differential expression of the predicted novel exon
#bamToBed -i accepted_hits.bam -split| coverageBed -a stdin -b /home/databases/hESC_differentiation_exons.bed > day20_7B__exons-novel.bed
bed_export_path = filepath('AltDatabase/'+species+'/RNASeq/chr/'+species + '_Ensembl_exons'+searchChr+'.bed')
bed_data = open(bed_export_path,'w') ### Appends to existing file
for (chr,coord) in novel_exon_coordinates:
ji,side,coord2 = novel_exon_coordinates[(chr,coord)]
if side == 'left': start,stop = coord,coord2
if side == 'right': start,stop = coord2,coord
try: gene = ji.GeneID()
except Exception: gene = 'NA'
if gene == None: gene = 'NA'
if gene == None: gene = 'NA'
if gene != 'NA': ### Including these has no benefit for AltAnalyze (just slows down alignment and piles up memory)
if ji.Strand() == '-': stop,start=start,stop
if chr_status == False:
chr = string.replace(chr,'chr','') ### This will thus match up to the BAM files
a = [start,stop]; a.sort(); start,stop = a
bed_values = [chr,str(start),str(stop),gene,'0',str(ji.Strand())]
bed_values = cleanUpLine(string.join(bed_values,'\t'))+'\n'
bed_data.write(bed_values)
bed_data.close()
return bed_export_path
def moveBAMtoBEDFile(species,dataset_name,root_dir):
bed_export_path = filepath('AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_exons.bed')
dataset_name = string.replace(dataset_name,'exp.','')
new_fn = root_dir+'/BAMtoBED/'+species + '_'+dataset_name+'_exons.bed'
new_fn = string.replace(new_fn,'.txt','')
print 'Writing exon-level coordinates to BED file:'
print new_fn
catFiles(bed_export_path,'chr') ### concatenate the files ot the main AltDatabase directory then move
export.customFileMove(bed_export_path,new_fn)
return new_fn
def reformatExonFile(species,type,chr_status):
if type == 'exon':
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt'
export_path = 'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_exons.txt'
### Used by BEDTools to get counts per specific AltAnalyze exon region (should augment with de novo regions identified from junction analyses)
bed_export_path = 'AltDatabase/'+species+'/RNASeq/chr/'+species + '_Ensembl_exons.bed'
bed_data = export.ExportFile(bed_export_path)
else:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_junction.txt'
export_path = 'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_junctions.txt'
print 'Writing',export_path
export_data = export.ExportFile(export_path)
fn=filepath(filename); x=0
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0:
x+=1
export_title = ['AltAnalyzeID','exon_id','ensembl_gene_id','transcript_cluster_id','chromosome','strand','probeset_start','probeset_stop']
export_title +=['affy_class','constitutive_probeset','ens_exon_ids','ens_constitutive_status','exon_region','exon-region-start(s)','exon-region-stop(s)','splice_events','splice_junctions']
export_title = string.join(export_title,'\t')+'\n'; export_data.write(export_title)
else:
try: gene, exonid, chr, strand, start, stop, constitutive_call, ens_exon_ids, splice_events, splice_junctions = t
except Exception: print t;kill
if chr == 'chrM': chr = 'chrMT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if chr == 'M': chr = 'MT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention,
if constitutive_call == 'yes': ens_constitutive_status = '1'
else: ens_constitutive_status = '0'
export_values = [gene+':'+exonid, exonid, gene, '', chr, strand, start, stop, 'known', constitutive_call, ens_exon_ids, ens_constitutive_status]
export_values+= [exonid, start, stop, splice_events, splice_junctions]
export_values = string.join(export_values,'\t')+'\n'; export_data.write(export_values)
if type == 'exon':
if chr_status == False:
chr = string.replace(chr,'chr','') ### This will thus match up to the BAM files
bed_values = [chr,start,stop,gene+':'+exonid+'_'+ens_exon_ids,'0',strand]
bed_values = string.join(bed_values,'\t')+'\n'; bed_data.write(bed_values)
export_data.close()
if type == 'exon': bed_data.close()
def importExonAnnotations(species,type,search_chr):
if 'exon' in type:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt'
else:
filename = 'AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_junction.txt'
fn=filepath(filename); x=0; exon_annotation_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: x=1
else:
gene, exonid, chr, strand, start, stop, constitutive_call, ens_exon_ids, splice_events, splice_junctions = t; proceed = 'yes'
if chr == 'chrM': chr = 'chrMT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if chr == 'M': chr = 'MT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if len(search_chr)>0:
if chr != search_chr: proceed = 'no'
if proceed == 'yes':
if type == 'exon': start = int(start); stop = int(stop)
ea = EnsemblImport.ExonAnnotationsSimple(chr, strand, start, stop, gene, ens_exon_ids, constitutive_call, exonid, splice_events, splice_junctions)
if type == 'junction_coordinates':
exon1_start,exon1_stop = string.split(start,'|')
exon2_start,exon2_stop = string.split(stop,'|')
if strand == '-':
exon1_stop,exon1_start = exon1_start,exon1_stop
exon2_stop,exon2_start = exon2_start,exon2_stop
#if gene == 'ENSMUSG00000027340': print chr,int(exon1_stop),int(exon2_start)
exon_annotation_db[chr,int(exon1_stop),int(exon2_start)]=ea
elif type == 'distal-exon':
exon_annotation_db[gene] = exonid
else:
try: exon_annotation_db[gene].append(ea)
except KeyError: exon_annotation_db[gene]=[ea]
return exon_annotation_db
def exportKnownJunctionComparisons(species):
gene_junction_db = JunctionArrayEnsemblRules.importEnsemblUCSCAltJunctions(species,'standard')
gene_intronjunction_db = JunctionArrayEnsemblRules.importEnsemblUCSCAltJunctions(species,'_intronic')
for i in gene_intronjunction_db: gene_junction_db[i]=[]
gene_junction_db2={}
for (gene,critical_exon,incl_junction,excl_junction) in gene_junction_db:
critical_exons = string.split(critical_exon,'|')
for critical_exon in critical_exons:
try: gene_junction_db2[gene,incl_junction,excl_junction].append(critical_exon)
except Exception: gene_junction_db2[gene,incl_junction,excl_junction] = [critical_exon]
gene_junction_db = gene_junction_db2; gene_junction_db2=[]
junction_export = 'AltDatabase/' + species + '/RNASeq/'+ species + '_junction_comps.txt'
fn=filepath(junction_export); data = open(fn,'w')
print "Exporting",junction_export
title = 'gene'+'\t'+'critical_exon'+'\t'+'exclusion_junction_region'+'\t'+'inclusion_junction_region'+'\t'+'exclusion_probeset'+'\t'+'inclusion_probeset'+'\t'+'data_source'+'\n'
data.write(title); temp_list=[]
for (gene,incl_junction,excl_junction) in gene_junction_db:
critical_exons = unique.unique(gene_junction_db[(gene,incl_junction,excl_junction)])
critical_exon = string.join(critical_exons,'|')
temp_list.append(string.join([gene,critical_exon,excl_junction,incl_junction,gene+':'+excl_junction,gene+':'+incl_junction,'AltAnalyze'],'\t')+'\n')
temp_list = unique.unique(temp_list)
for i in temp_list: data.write(i)
data.close()
def getExonAndJunctionSequences(species):
export_exon_filename = 'AltDatabase/'+species+'/RNASeq/'+species+'_Ensembl_exons.txt'
ensembl_exon_db = ExonArrayEnsemblRules.reimportEnsemblProbesetsForSeqExtraction(export_exon_filename,'null',{})
### Import just the probeset region for mRNA alignment analysis
analysis_type = ('region_only','get_sequence'); array_type = 'RNASeq'
dir = 'AltDatabase/'+species+'/SequenceData/chr/'+species; gene_seq_filename = dir+'_gene-seq-2000_flank.fa'
ensembl_exon_db = EnsemblImport.import_sequence_data(gene_seq_filename,ensembl_exon_db,species,analysis_type)
critical_exon_file = 'AltDatabase/'+species+'/'+ array_type + '/' + array_type+'_critical-exon-seq.txt'
getCriticalJunctionSequences(critical_exon_file,species,ensembl_exon_db)
"""
### Import the full Ensembl exon sequence (not just the probeset region) for miRNA binding site analysis
analysis_type = 'get_sequence'; array_type = 'RNASeq'
dir = 'AltDatabase/'+species+'/SequenceData/chr/'+species; gene_seq_filename = dir+'_gene-seq-2000_flank.fa'
ensembl_exon_db = EnsemblImport.import_sequence_data(gene_seq_filename,ensembl_exon_db,species,analysis_type)
"""
critical_exon_file = 'AltDatabase/'+species+'/'+ array_type + '/' + array_type+'_critical-exon-seq.txt'
updateCriticalExonSequences(critical_exon_file, ensembl_exon_db)
def updateCriticalExonSequences(filename,ensembl_exon_db):
exon_seq_db_filename = filename[:-4]+'_updated.txt'
exonseq_data = export.ExportFile(exon_seq_db_filename)
critical_exon_seq_db={}; null_count={}
for gene in ensembl_exon_db:
gene_exon_data={}
for probe_data in ensembl_exon_db[gene]:
exon_id,((probe_start,probe_stop,probeset_id,exon_class,transcript_clust),ed) = probe_data
try: gene_exon_data[probeset_id] = ed.ExonSeq()
except Exception: null_count[gene]=[] ### Occurs for non-chromosomal DNA (could also download this sequence though)
if len(gene_exon_data)>0: critical_exon_seq_db[gene] = gene_exon_data
print len(null_count),'genes not assigned sequenced (e.g.,non-chromosomal)'
ensembl_exon_db=[]
### Export exon sequences
for gene in critical_exon_seq_db:
gene_exon_data = critical_exon_seq_db[gene]
for probeset in gene_exon_data:
critical_exon_seq = gene_exon_data[probeset]
values = [probeset,'',critical_exon_seq]
values = string.join(values,'\t')+'\n'
exonseq_data.write(values)
exonseq_data.close()
print exon_seq_db_filename, 'exported....'
def getCriticalJunctionSequences(filename,species,ensembl_exon_db):
### Assemble and export junction sequences
junction_seq_db_filename = string.replace(filename,'exon-seq','junction-seq')
junctionseq_data = export.ExportFile(junction_seq_db_filename)
critical_exon_seq_db={}; null_count={}
for gene in ensembl_exon_db:
gene_exon_data={}
for probe_data in ensembl_exon_db[gene]:
exon_id,((probe_start,probe_stop,probeset_id,exon_class,transcript_clust),ed) = probe_data
try: gene_exon_data[probeset_id] = ed.ExonSeq()
except Exception: null_count[gene]=[] ### Occurs for non-chromosomal DNA (could also download this sequence though)
if len(gene_exon_data)>0: critical_exon_seq_db[gene] = gene_exon_data
print len(null_count),'genes not assigned sequenced (e.g.,non-chromosomal)'
ensembl_exon_db=[]
junction_annotation_db = importExonAnnotations(species,'junction',[])
for gene in junction_annotation_db:
if gene in critical_exon_seq_db:
gene_exon_data = critical_exon_seq_db[gene]
for jd in junction_annotation_db[gene]:
exon1,exon2=string.split(jd.ExonRegionIDs(),'-')
p1=gene+':'+exon1
p2=gene+':'+exon2
p1_seq=gene_exon_data[p1][-15:]
p2_seq=gene_exon_data[p2][:15]
junction_seq = p1_seq+'|'+p2_seq
junctionseq_data.write(gene+':'+jd.ExonRegionIDs()+'\t'+junction_seq+'\t\n')
junctionseq_data.close()
print junction_seq_db_filename, 'exported....'
def getEnsemblAssociations(species,data_type,test_status,force):
### Get UCSC associations (download databases if necessary)
from build_scripts import UCSCImport
mRNA_Type = 'mrna'; run_from_scratch = 'yes'
export_all_associations = 'no' ### YES only for protein prediction analysis
update.buildUCSCAnnoationFiles(species,mRNA_Type,export_all_associations,run_from_scratch,force)
null = EnsemblImport.getEnsemblAssociations(species,data_type,test_status); null=[]
reformatExonFile(species,'exon',True); reformatExonFile(species,'junction',True)
exportKnownJunctionComparisons(species)
getExonAndJunctionSequences(species)
######### Below code deals with user read alignment as opposed to building the AltDatabase #########
class ExonInfo:
def __init__(self,start,unique_id,annotation):
self.start = start; self.unique_id = unique_id; self.annotation = annotation
def ReadStart(self): return self.start
def UniqueID(self): return self.unique_id
def Annotation(self): return self.annotation
def setExonRegionData(self,rd): self.rd = rd
def ExonRegionData(self): return self.rd
def setExonRegionID(self,region_id): self.region_id = region_id
def ExonRegionID(self): return self.region_id
def setAlignmentRegion(self,region_type): self.region_type = region_type
def AlignmentRegion(self): return self.region_type
def __repr__(self): return "ExonData values"
class JunctionData:
def __init__(self,chr,strand,exon1_stop,exon2_start,junction_id,biotype):
self.chr = chr; self.strand = strand; self._chr = chr
self.exon1_stop = exon1_stop; self.exon2_start = exon2_start
self.junction_id = junction_id; self.biotype = biotype
#self.reads = reads; self.condition = condition
self.left_exon = None; self.right_exon = None; self.jd = None; self.gene_id = None
self.trans_splicing = None
self.splice_events=''
self.splice_junctions=''
self.seq_length=''
self.uid = None
def Chr(self): return self.chr
def Strand(self): return self.strand
def Exon1Stop(self): return self.exon1_stop
def Exon2Start(self): return self.exon2_start
def setExon1Stop(self,exon1_stop): self.exon1_stop = exon1_stop
def setExon2Start(self,exon2_start): self.exon2_start = exon2_start
def setSeqLength(self,seq_length): self.seq_length = seq_length
def SeqLength(self): return self.seq_length
def BioType(self): return self.biotype
def checkExonPosition(self,exon_pos):
if exon_pos == self.Exon1Stop(): return 'left'
else: return 'right'
### These are used to report novel exon boundaries
def setExon1Start(self,exon1_start): self.exon1_start = exon1_start
def setExon2Stop(self,exon2_stop): self.exon2_stop = exon2_stop
def Exon1Start(self): return self.exon1_start
def Exon2Stop(self): return self.exon2_stop
def Reads(self): return self.reads
def JunctionID(self): return self.junction_id
def Condition(self): return self.condition
def setExonAnnotations(self,jd):
self.jd = jd
self.splice_events = jd.AssociatedSplicingEvent()
self.splice_junctions = jd.AssociatedSplicingJunctions()
self.exon_region = jd.ExonRegionIDs()
self.exonid = jd.ExonID()
self.gene_id = jd.GeneID()
self.uid = jd.GeneID()+':'+jd.ExonRegionIDs()
def ExonAnnotations(self): return self.jd
def setLeftExonAnnotations(self,ld): self.gene_id,self.left_exon = ld
def LeftExonAnnotations(self): return self.left_exon
def setRightExonAnnotations(self,rd): self.secondary_geneid,self.right_exon = rd
def RightExonAnnotations(self): return self.right_exon
def setGeneID(self,geneid): self.gene_id = geneid
def GeneID(self): return self.gene_id
def setSecondaryGeneID(self,secondary_geneid): self.secondary_geneid = secondary_geneid
def SecondaryGeneID(self): return self.secondary_geneid
def setTransSplicing(self): self.trans_splicing = 'yes'
def TransSplicing(self): return self.trans_splicing
def SpliceSitesFound(self):
if self.jd != None: sites_found = 'both'
elif self.left_exon != None and self.right_exon != None: sites_found = 'both'
elif self.left_exon != None: sites_found = 'left'
elif self.right_exon != None: sites_found = 'right'
else: sites_found = None
return sites_found
def setConstitutive(self,constitutive): self.constitutive = constitutive
def Constitutive(self): return self.constitutive
def setAssociatedSplicingEvent(self,splice_events): self.splice_events = splice_events
def AssociatedSplicingEvent(self): return self.splice_events
def setAssociatedSplicingJunctions(self,splice_junctions): self.splice_junctions = splice_junctions
def AssociatedSplicingJunctions(self): return self.splice_junctions
def setExonID(self,exonid): self.exonid = exonid
def ExonID(self): return self.exonid
def setExonRegionID(self,exon_region): self.exon_region = exon_region
def ExonRegionID(self): return self.exon_region
def setUniqueID(self,uid): self.uid = uid
def UniqueID(self): return self.uid
def setLeftExonRegionData(self,li): self.li = li
def LeftExonRegionData(self): return self.li
def setRightExonRegionData(self,ri): self.ri = ri
def RightExonRegionData(self): return self.ri
def setNovel(self, side): self.side = side
def Novel(self): return self.side
def __repr__(self): return "JunctionData values"
def checkBEDFileFormat(bed_dir,root_dir):
""" This method checks to see if the BED files (junction or exon) have 'chr' proceeding the chr number.
It also checks to see if some files have two underscores and one has none or if double underscores are missing from all."""
dir_list = read_directory(bed_dir)
x=0
break_now = False
chr_present = False
condition_db={}
for filename in dir_list:
fn=filepath(bed_dir+filename)
#if ('.bed' in fn or '.BED' in fn): delim = 'r'
delim = 'rU'
if '.tab' in string.lower(filename) or '.bed' in string.lower(filename) or '.junction_quantification.txt' in string.lower(filename):
condition_db[filename]=[]
for line in open(fn,delim).xreadlines(): ### changed rU to r to remove \r effectively, rather than read as end-lines
if line[0] == '#': x=0 ### BioScope
elif x == 0: x=1 ###skip the first line
elif x < 10: ### Only check the first 10 lines
if 'chr' in line: ### Need to look at multiple input formats (chr could be in t[0] or t[1])
chr_present = True
x+=1
else:
break_now = True
break
if break_now == True:
break
### Check to see if exon.bed and junction.bed file names are propper or faulty (which will result in downstream errors)
double_underscores=[]
no_doubles=[]
for condition in condition_db:
if '__' in condition:
double_underscores.append(condition)
else:
no_doubles.append(condition)
exon_beds=[]
junctions_beds=[]
if len(double_underscores)>0 and len(no_doubles)>0:
### Hence, a problem is likely due to inconsistent naming
print 'The input files appear to have inconsistent naming. If both exon and junction sample data are present, make sure they are named propperly.'
print 'For example: cancer1__exon.bed, cancer1__junction.bed (double underscore required to match these samples up)!'
print 'Exiting AltAnalyze'; forceError
elif len(no_doubles)>0:
for condition in no_doubles:
condition = string.lower(condition)
if 'exon' in condition:
exon_beds.append(condition)
if 'junction' in condition:
junctions_beds.append(condition)
if len(exon_beds)>0 and len(junctions_beds)>0:
print 'The input files appear to have inconsistent naming. If both exon and junction sample data are present, make sure they are named propperly.'
print 'For example: cancer1__exon.bed, cancer1__junction.bed (double underscore required to match these samples up)!'
print 'Exiting AltAnalyze'; forceError
return chr_present
def getStrandMappingData(species):
splicesite_db={}
refExonCoordinateFile = unique.filepath('AltDatabase/ensembl/'+species+'/'+species+'_Ensembl_exon.txt')
firstLine=True
for line in open(refExonCoordinateFile,'rU').xreadlines():
if firstLine: firstLine=False
else:
line = line.rstrip('\n')
t = string.split(line,'\t'); #'gene', 'exon-id', 'chromosome', 'strand', 'exon-region-start(s)', 'exon-region-stop(s)', 'constitutive_call', 'ens_exon_ids', 'splice_events', 'splice_junctions'
geneID, exon, chr, strand, start, stop = t[:6]
splicesite_db[chr,int(start)]=strand
splicesite_db[chr,int(stop)]=strand
return splicesite_db
def importBEDFile(bed_dir,root_dir,species,normalize_feature_exp,getReads=False,searchChr=None,getBiotype=None,testImport=False,filteredJunctions=None):
dir_list = read_directory(bed_dir)
begin_time = time.time()
if 'chr' not in searchChr:
searchChr = 'chr'+searchChr
condition_count_db={}; neg_count=0; pos_count=0; junction_db={}; biotypes={}; algorithms={}; exon_len_db={}; splicesite_db={}
if testImport == 'yes': print "Reading user RNA-seq input data files"
for filename in dir_list:
count_db={}; rows=0
fn=filepath(bed_dir+filename)
condition = export.findFilename(fn)
if '__' in condition:
### Allow multiple junction files per sample to be combined (e.g. canonical and non-canonical junction alignments)
condition=string.split(condition,'__')[0]+filename[-4:]
if ('.bed' in fn or '.BED' in fn or '.tab' in fn or '.TAB' in fn or '.junction_quantification.txt' in fn) and '._' not in condition:
if ('.bed' in fn or '.BED' in fn): delim = 'r'
else: delim = 'rU'
### The below code removes .txt if still in the filename along with .tab or .bed
if '.tab' in fn: condition = string.replace(condition,'.txt','.tab')
elif '.bed' in fn: condition = string.replace(condition,'.txt','.bed')
if '.TAB' in fn: condition = string.replace(condition,'.txt','.TAB')
elif '.BED' in fn: condition = string.replace(condition,'.txt','.BED')
if testImport == 'yes': print "Reading the bed file", [fn], condition
### If the BED was manually created on a Mac, will neeed 'rU' - test this
for line in open(fn,delim).xreadlines(): break
if len(line)>500: delim = 'rU'
for line in open(fn,delim).xreadlines(): ### changed rU to r to remove \r effectively, rather than read as end-lines
data = cleanUpLine(line)
t = string.split(data,'\t')
rows+=1
if rows==1 or '#' == data[0]:
format_description = data
algorithm = 'Unknown'
if 'TopHat' in format_description: algorithm = 'TopHat'
elif 'HMMSplicer' in format_description: algorithm = 'HMMSplicer'
elif 'SpliceMap junctions' in format_description: algorithm = 'SpliceMap'
elif t[0] == 'E1': algorithm = 'BioScope-junction'
elif '# filterOrphanedMates=' in data or 'alignmentFilteringMode=' in data or '#number_of_mapped_reads=' in data:
algorithm = 'BioScope-exon'
elif '.junction_quantification.txt' in fn:
algorithm = 'TCGA format'
if 'barcode' in t: junction_position = 1
else: junction_position = 0
elif '.tab' in fn and len(t)==9:
try: start = float(t[1]) ### expect this to be a numerical coordinate
except Exception: continue
algorithm = 'STAR'
strand = '-' ### If no strand exists
rows=2 ### allows this first row to be processed
if len(splicesite_db)==0: ### get strand to pos info
splicesite_db = getStrandMappingData(species)
if testImport == 'yes': print condition, algorithm
if rows>1:
try:
if ':' in t[0]:
chr = string.split(t[0],':')[0]
else: chr = t[0]
if 'chr' not in chr:
chr = 'chr'+chr
if searchChr == chr or ('BioScope' in algorithm and searchChr == t[1]): proceed = True
elif searchChr == 'chrMT' and ('BioScope' not in algorithm):
if 'M' in chr and len(chr)<6: proceed = True ### If you don't have the length, any random thing with an M will get included
else: proceed = False
else: proceed = False
except IndexError:
print 'The input file:\n',filename
print 'is not formated as expected (format='+algorithm+').'
print 'search chromosome:',searchChr
print t; force_bad_exit
if proceed:
proceed = False
if '.tab' in fn or '.TAB' in fn:
### Applies to non-BED format Junction and Exon inputs (BioScope)
if 'BioScope' in algorithm:
if algorithm == 'BioScope-exon': ### Not BED format
chr,source,data_type,start,end,reads,strand,null,gene_info=t[:9]
if 'chr' not in chr: chr = 'chr'+chr
if data_type == 'exon': ### Can also be CDS
gene_info,test,rpkm_info,null = string.split(gene_info,';')
symbol = string.split(gene_info,' ')[-1]
#refseq = string.split(transcript_info,' ')[-1]
rpkm = string.split(rpkm_info,' ')[-1]
#if normalize_feature_exp == 'RPKM': reads = rpkm ### The RPKM should be adjusted +1 counts, so don't use this
biotype = 'exon'; biotypes[biotype]=[]
exon1_stop,exon2_start = int(start),int(end); junction_id=''
### Adjust exon positions - not ideal but necessary. Needed as a result of exon regions overlapping by 1nt (due to build process)
exon1_stop+=1; exon2_start-=1
#if float(reads)>4 or getReads:
proceed = True ### Added in version 2.0.9 to remove rare novel isoforms
seq_length = abs(exon1_stop-exon2_start)
if algorithm == 'BioScope-junction':
chr = t[1]; strand = t[2]; exon1_stop = int(t[4]); exon2_start = int(t[8]); count_paired = t[17]; count_single = t[19]; score=t[21]
if 'chr' not in chr: chr = 'chr'+chr
try: exon1_start = int(t[3]); exon2_stop = int(t[9])
except Exception: pass ### If missing, these are not assigned
reads = str(int(float(count_paired))+int(float(count_single))) ### Users will either have paired or single read (this uses either)
biotype = 'junction'; biotypes[biotype]=[]; junction_id=''
if float(reads)>4 or getReads: proceed = True ### Added in version 2.0.9 to remove rare novel isoforms
seq_length = abs(float(exon1_stop-exon2_start))
if 'STAR' in algorithm:
chr = t[0]; exon1_stop = int(t[1])-1; exon2_start = int(t[2])+1; strand=''
if 'chr' not in chr: chr = 'chr'+chr
reads = str(int(t[7])+int(t[6]))
biotype = 'junction'; biotypes[biotype]=[]; junction_id=''
if float(reads)>4 or getReads: proceed = True ### Added in version 2.0.9 to remove rare novel isoforms
if (chr,exon1_stop) in splicesite_db:
strand = splicesite_db[chr,exon1_stop]
elif (chr,exon2_start) in splicesite_db:
strand = splicesite_db[chr,exon2_start]
#else: proceed = False
seq_length = abs(float(exon1_stop-exon2_start))
if strand == '-': ### switch the orientation of the positions
exon1_stop,exon2_start=exon2_start,exon1_stop
exon1_start = exon1_stop; exon2_stop = exon2_start
#if 9996685==exon1_stop and 10002682==exon2_stop:
#print chr, strand, reads, exon1_stop, exon2_start,proceed;sys.exit()
else:
try:
if algorithm == 'TCGA format':
coordinates = string.split(t[junction_position],',')
try: chr,pos1,strand = string.split(coordinates[0],':')
except Exception: print t;sys.exit()
chr,pos2,strand = string.split(coordinates[1],':')
if 'chr' not in chr: chr = 'chr'+chr
pos2 = str(int(pos2)-1) ### This is the bed format conversion with exons of 0 length
exon1_start, exon2_stop = pos1, pos2
reads = t[junction_position+1]
junction_id = t[junction_position]
exon1_len=0; exon2_len=0
else:
### Applies to BED format Junction input
chr, exon1_start, exon2_stop, junction_id, reads, strand, null, null, null, null, lengths, null = t
if 'chr' not in chr: chr = 'chr'+chr
exon1_len,exon2_len=string.split(lengths,',')[:2]; exon1_len = int(exon1_len); exon2_len = int(exon2_len)
exon1_start = int(exon1_start); exon2_stop = int(exon2_stop)
biotype = 'junction'; biotypes[biotype]=[]
if strand == '-':
exon1_stop = exon1_start+exon1_len; exon2_start=exon2_stop-exon2_len+1
### Exons have the opposite order
a = exon1_start,exon1_stop; b = exon2_start,exon2_stop
exon1_stop,exon1_start = b; exon2_stop,exon2_start = a
else:
exon1_stop = exon1_start+exon1_len; exon2_start=exon2_stop-exon2_len+1
if float(reads)>4 or getReads: proceed = True
if algorithm == 'HMMSplicer':
if '|junc=' in junction_id: reads = string.split(junction_id,'|junc=')[-1]
else: proceed = False
if algorithm == 'SpliceMap':
if ')' in junction_id and len(junction_id)>1: reads = string.split(junction_id,')')[0][1:]
else: proceed = False
seq_length = abs(float(exon1_stop-exon2_start)) ### Junction distance
except Exception,e:
#print traceback.format_exc();sys.exit()
### Applies to BED format exon input (BEDTools export)
# bamToBed -i accepted_hits.bam -split| coverageBed -a stdin -b /home/nsalomonis/databases/Mm_Ensembl_exons.bed > day0_8B__exons.bed
try: chr, start, end, exon_id, null, strand, reads, bp_coverage, bp_total, percent_coverage = t
except Exception:
print 'The file',fn,'does not appear to be propperly formatted as input.'
print t; force_exception
if 'chr' not in chr: chr = 'chr'+chr
algorithm = 'TopHat-exon'; biotype = 'exon'; biotypes[biotype]=[]
exon1_stop,exon2_start = int(start),int(end); junction_id=exon_id; seq_length = float(bp_total)
if seq_length == 0:
seq_length = abs(float(exon1_stop-exon2_start))
### Adjust exon positions - not ideal but necessary. Needed as a result of exon regions overlapping by 1nt (due to build process)
exon1_stop+=1; exon2_start-=1
#if float(reads)>4 or getReads: ### Added in version 2.0.9 to remove rare novel isoforms
proceed = True
#else: proceed = False
if proceed:
if 'chr' not in chr:
chr = 'chr'+chr ### Add the chromosome prefix
if chr == 'chrM': chr = 'chrMT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if chr == 'M': chr = 'MT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if strand == '+': pos_count+=1
else: neg_count+=1
if getReads and seq_length>0:
if getBiotype == biotype:
if biotype == 'junction':
### We filtered for junctions>4 reads before, now we include all reads for expressed junctions
if (chr,exon1_stop,exon2_start) in filteredJunctions:
count_db[chr,exon1_stop,exon2_start] = reads
try: exon_len_db[chr,exon1_stop,exon2_start] = seq_length
except Exception: exon_len_db[chr,exon1_stop,exon2_start] = []
else:
count_db[chr,exon1_stop,exon2_start] = reads
try: exon_len_db[chr,exon1_stop,exon2_start] = seq_length
except Exception: exon_len_db[chr,exon1_stop,exon2_start] = []
elif seq_length>0:
if (chr,exon1_stop,exon2_start) not in junction_db:
ji = JunctionData(chr,strand,exon1_stop,exon2_start,junction_id,biotype)
junction_db[chr,exon1_stop,exon2_start] = ji
try: ji.setSeqLength(seq_length) ### If RPKM imported or calculated
except Exception: null=[]
try: ji.setExon1Start(exon1_start);ji.setExon2Stop(exon2_stop)
except Exception: null=[]
key = chr,exon1_stop,exon2_start
algorithms[algorithm]=[]
if getReads:
if condition in condition_count_db:
### combine the data from the different files for the same sample junction alignments
count_db1 = condition_count_db[condition]
for key in count_db:
if key not in count_db1: count_db1[key] = count_db[key]
else:
combined_counts = int(count_db1[key])+int(count_db[key])
count_db1[key] = str(combined_counts)
condition_count_db[condition]=count_db1
else:
try: condition_count_db[condition] = count_db
except Exception: null=[] ### Occurs for other text files in the directory that are not used for the analysis
end_time = time.time()
if testImport == 'yes': print 'Read coordinates imported in',int(end_time-begin_time),'seconds'
if getReads:
#print len(exon_len_db), getBiotype, 'read counts present for',algorithm
return condition_count_db,exon_len_db,biotypes,algorithms
else:
if testImport == 'yes':
if 'exon' not in biotypes and 'BioScope' not in algorithm:
print len(junction_db),'junctions present in',algorithm,'format BED files.' # ('+str(pos_count),str(neg_count)+' by strand).'
elif 'exon' in biotypes and 'BioScope' not in algorithm:
print len(junction_db),'sequence identifiers present in input files.'
else: print len(junction_db),'sequence identifiers present in BioScope input files.'
return junction_db,biotypes,algorithms
def importExonCoordinates(probeCoordinateFile,search_chr,getBiotype):
probe_coordinate_db={}
junction_db={}
biotypes={}
x=0
fn=filepath(probeCoordinateFile)
for line in open(fn,'rU').xreadlines(): ### changed rU to r to remove \r effectively, rather than read as end-lines
data = cleanUpLine(line)
if x==0: x=1
else:
t = string.split(data,'\t')
probe_id = t[0]; probeset_id=t[1]; chr=t[2]; strand=t[3]; start=t[4]; end=t[5]
exon1_stop,exon2_start = int(start),int(end)
seq_length = abs(float(exon1_stop-exon2_start))
if 'chr' not in chr:
chr = 'chr'+chr ### Add the chromosome prefix
if chr == 'chrM': chr = 'chrMT' ### MT is the Ensembl convention whereas M is the Affymetrix and UCSC convention
if search_chr == chr or search_chr == None:
try: biotype = t[6]
except Exception:
if seq_length>25:biotype = 'junction'
else: biotype = 'exon'
if strand == '-':
exon1_stop,exon2_start = exon2_start, exon1_stop ### this is their actual 5' -> 3' orientation
if biotype == 'junction':
exon1_start,exon2_stop = exon1_stop,exon2_start
else:
exon1_stop+=1; exon2_start-=1
biotypes[biotype]=[]
if getBiotype == biotype or getBiotype == None:
ji = JunctionData(chr,strand,exon1_stop,exon2_start,probe_id,biotype)
junction_db[chr,exon1_stop,exon2_start] = ji
try: ji.setSeqLength(seq_length) ### If RPKM imported or calculated
except Exception: null=[]
try: ji.setExon1Start(exon1_start);ji.setExon2Stop(exon2_stop)
except Exception: null=[]
probe_coordinate_db[probe_id] = chr,exon1_stop,exon2_start ### Import the expression data for the correct chromosomes with these IDs
return probe_coordinate_db, junction_db, biotypes
def importExpressionMatrix(exp_dir,root_dir,species,fl,getReads,search_chr=None,getBiotype=None):
""" Non-RNA-Seq expression data (typically Affymetrix microarray) import and mapping to an external probe-coordinate database """
begin_time = time.time()
condition_count_db={}; neg_count=0; pos_count=0; algorithms={}; exon_len_db={}
probe_coordinate_db, junction_db, biotypes = importExonCoordinates(fl.ExonMapFile(),search_chr,getBiotype)
x=0
fn=filepath(exp_dir)[:-1]
condition = export.findFilename(fn)
### If the BED was manually created on a Mac, will neeed 'rU' - test this
for line in open(fn,'rU').xreadlines(): ### changed rU to r to remove \r effectively, rather than read as end-lines
data = cleanUpLine(line)
t = string.split(data,'\t')
if '#' == data[0]: None
elif x==0:
if 'block' in t:
start_index = 7
else:
start_index = 1
headers = t[start_index:]
x=1
else:
proceed = 'yes' ### restrict by chromosome with minimum line parsing (unless we want counts instead)
probe_id=t[0]
if probe_id in probe_coordinate_db:
key = probe_coordinate_db[probe_id]
if getReads == 'no':
pass
else:
expression_data = t[start_index:]
i=0
for sample in headers:
if sample in condition_count_db:
count_db = condition_count_db[sample]
count_db[key] = expression_data[i]
exon_len_db[key]=[]
else:
count_db={}
count_db[key] = expression_data[i]
condition_count_db[sample] = count_db
exon_len_db[key]=[]
i+=1
algorithms['ProbeData']=[]
end_time = time.time()
if testImport == 'yes': print 'Probe data imported in',int(end_time-begin_time),'seconds'
if getReads == 'yes':
return condition_count_db,exon_len_db,biotypes,algorithms
else:
return junction_db,biotypes,algorithms
def adjustCounts(condition_count_db,exon_len_db):
for key in exon_len_db:
try:
null=exon_len_db[key]
for condition in condition_count_db:
count_db = condition_count_db[condition]
try: read_count = float(count_db[key])+1 ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
except KeyError: read_count = 1 ###Was zero, but needs to be one for more realistic log2 fold calculations
count_db[key] = str(read_count) ### Replace original counts with adjusted counts
except Exception: null=[]
return condition_count_db
def calculateRPKM(condition_count_db,exon_len_db,biotype_to_examine):
"""Determines the total number of reads in a sample and then calculates RPMK relative to a pre-determined junction length (60).
60 was choosen, based on Illumina single-end read lengths of 35 (5 nt allowed overhand on either side of the junction)"""
### Get the total number of mapped reads
mapped_reads={}
for condition in condition_count_db:
mapped_reads[condition]=0
count_db = condition_count_db[condition]
for key in count_db:
read_count = count_db[key]
mapped_reads[condition]+=float(read_count)
### Use the average_total_reads when no counts reported such that 0 counts are comparable
average_total_reads = 0
for i in mapped_reads:
average_total_reads+=mapped_reads[i]
if testImport == 'yes':
print 'condition:',i,'total reads:',mapped_reads[i]
average_total_reads = average_total_reads/len(condition_count_db)
if testImport == 'yes':
print 'average_total_reads:',average_total_reads
k=0
c=math.pow(10.0,9.0)
for key in exon_len_db:
try:
for condition in condition_count_db:
total_mapped_reads = mapped_reads[condition]
try: read_count = float(condition_count_db[condition][key])+1 ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
except KeyError: read_count = 1 ###Was zero, but needs to be one for more realistic log2 fold calculations
if biotype_to_examine == 'junction': region_length = 60.0
else:
try: region_length = exon_len_db[key]
except Exception: continue ### This should only occur during testing (when restricting to one or few chromosomes)
if read_count == 1: ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
rpkm = c*(float(read_count)/(float(average_total_reads)*region_length))
try:
if region_length == 0:
region_length = abs(int(key[2]-key[1]))
rpkm = c*(read_count/(float(total_mapped_reads)*region_length))
except Exception:
print condition, key
print 'Error Encountered... Exon or Junction of zero length encoutered... RPKM failed... Exiting AltAnalyze.'
print 'This error may be due to inconsistent file naming. If both exon and junction sample data is present, make sure they are named propperly.'
print 'For example: cancer1__exon.bed, cancer1__junction.bed (double underscore required to match these samples up)!'
print [read_count,total_mapped_reads,region_length];k=1; forceError
condition_count_db[condition][key] = str(rpkm) ### Replace original counts with RPMK
except Exception:
if k == 1: kill
null=[]
return condition_count_db
def calculateGeneLevelStatistics(steady_state_export,species,expressed_gene_exon_db,normalize_feature_exp,array_names,fl,excludeLowExp=True,exportRPKMs=False):
global UserOptions; UserOptions = fl
exp_file = string.replace(steady_state_export,'-steady-state','')
if normalize_feature_exp == 'RPKM':
exp_dbase, all_exp_features, array_count = importRawCountData(exp_file,expressed_gene_exon_db,excludeLowExp=excludeLowExp)
steady_state_db = obtainGeneCounts(expressed_gene_exon_db,species,exp_dbase,array_count,normalize_feature_exp,excludeLowExp=excludeLowExp); exp_dbase=[]
exportGeneCounts(steady_state_export,array_names,steady_state_db)
steady_state_db = calculateGeneRPKM(steady_state_db)
if exportRPKMs:
exportGeneCounts(steady_state_export,array_names,steady_state_db,dataType='RPKMs')
else:
exp_dbase, all_exp_features, array_count = importNormalizedCountData(exp_file,expressed_gene_exon_db)
steady_state_db = obtainGeneCounts(expressed_gene_exon_db,species,exp_dbase,array_count,normalize_feature_exp); exp_dbase=[]
exportGeneCounts(steady_state_export,array_names,steady_state_db)
return steady_state_db, all_exp_features
def exportGeneCounts(steady_state_export,headers,gene_count_db,dataType='counts'):
### In addition to RPKM gene-level data, export gene level counts and lengths (should be able to calculate gene RPKMs from this file)
if dataType=='counts':
export_path = string.replace(steady_state_export,'exp.','counts.')
else:
export_path = steady_state_export
export_data = export.ExportFile(export_path)
title = string.join(['Ensembl']+headers,'\t')+'\n'
export_data.write(title)
for gene in gene_count_db:
sample_counts=[]
for count_data in gene_count_db[gene]:
try: read_count,region_length = count_data
except Exception: read_count = count_data
sample_counts.append(str(read_count))
sample_counts = string.join([gene]+sample_counts,'\t')+'\n'
export_data.write(sample_counts)
export_data.close()
def importGeneCounts(filename,import_type):
### Import non-normalized original counts and return the max value
counts_filename = string.replace(filename,'exp.','counts.')
status = verifyFile(counts_filename)
if status == 'not found': ### Occurs for non-normalized counts
counts_filename = filename
fn=filepath(counts_filename); x=0; count_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: array_names = t[1:]; x=1
else:
gene = t[0]
if import_type == 'max':
count_db[gene] = str(max(map(float,t[1:])))
else:
count_db[gene] = map(float,t[1:])
return count_db,array_names
def calculateGeneRPKM(gene_count_db):
"""Determines the total number of reads in a sample and then calculates RPMK relative to a pre-determined junction length (60).
60 was choosen, based on Illumina single-end read lengths of 35 (5 nt allowed overhand on either side of the junction)"""
### Get the total number of mapped reads (relative to all gene aligned rather than genome aligned exon reads)
mapped_reads={}
for gene in gene_count_db:
index=0
for (read_count,total_len) in gene_count_db[gene]:
try: mapped_reads[index]+=float(read_count)
except Exception: mapped_reads[index]=float(read_count)
index+=1
### Use the average_total_reads when no counts reported such that 0 counts are comparable
average_total_reads = 0
for i in mapped_reads: average_total_reads+=mapped_reads[i]
average_total_reads = average_total_reads/(index+1) ###
c=math.pow(10.0,9.0)
for gene in gene_count_db:
index=0; rpkms = []
for (read_count,region_length) in gene_count_db[gene]:
total_mapped_reads = mapped_reads[index]
#print [read_count],[region_length],[total_mapped_reads]
#if gene == 'ENSMUSG00000028186': print [read_count, index, total_mapped_reads,average_total_reads,region_length]
if read_count == 0: read_count=1; rpkm = c*(float(read_count)/(float(average_total_reads)*region_length)) ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
else:
try: rpkm = c*(float(read_count+1)/(float(total_mapped_reads)*region_length)) ### read count is incremented +1 (see next line)
except Exception: read_count=1; rpkm = c*(float(read_count)/(float(average_total_reads)*region_length)) ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
#if gene == 'ENSMUSG00000028186': print rpkm,read_count,index,total_mapped_reads,average_total_reads,region_length
#if gene == 'ENSMUSG00000026049': print gene_count_db[gene], mapped_reads[index], rpkm
rpkms.append(rpkm)
index+=1
gene_count_db[gene] = rpkms ### Replace original counts with RPMK
return gene_count_db
def deleteOldAnnotations(species,root_dir,dataset_name):
db_dir = root_dir+'AltDatabase/'+species
try:
status = export.deleteFolder(db_dir)
if status == 'success':
print "...Previous experiment database deleted"
except Exception: null=[]
count_dir = root_dir+'ExpressionInput/Counts'
try: status = export.deleteFolder(count_dir)
except Exception: pass
if 'exp.' not in dataset_name: dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name: dataset_name+='.txt'
export_path = root_dir+'ExpressionInput/'+dataset_name
try: os.remove(filepath(export_path))
except Exception: null=[]
try: os.remove(filepath(string.replace(export_path,'exp.','counts.')))
except Exception: null=[]
try: os.remove(filepath(string.replace(export_path,'exp.','novel.')))
except Exception: null=[]
from copy_reg import pickle
from types import MethodType
def _pickle_method(method):
func_name = method.im_func.__name__
obj = method.im_self
cls = method.im_class
return _unpickle_method, (func_name, obj, cls)
def _unpickle_method(func_name, obj, cls):
for cls in cls.mro():
try:
func = cls.__dict__[func_name]
except KeyError:
pass
else:
break
return func.__get__(obj, cls)
def call_it(instance, name, args=(), kwargs=None):
"indirect caller for instance methods and multiprocessing"
if kwargs is None:
kwargs = {}
return getattr(instance, name)(*args, **kwargs)
def alignExonsAndJunctionsToEnsembl(species,exp_file_location_db,dataset_name,Multi=None):
fl = exp_file_location_db[dataset_name]
try: multiThreading = fl.multiThreading()
except Exception: multiThreading = True
print 'multiThreading:',multiThreading
normalize_feature_exp = fl.FeatureNormalization()
testImport='no'
if 'demo_data' in fl.ExpFile():
### If the input files are in the AltAnalyze test directory, only analyze select chromosomes
print 'Running AltAnalyze in TEST MODE... restricting to select chromosomes only!!!!!'
testImport='yes'
rnaseq_begin_time = time.time()
p = AlignExonsAndJunctionsToEnsembl(species,exp_file_location_db,dataset_name,testImport)
chromosomes = p.getChromosomes()
### The following files need to be produced from chromosome specific sets later
countsFile = p.countsFile()
exonFile = p.exonFile()
junctionFile = p.junctionFile()
junctionCompFile = p.junctionCompFile()
novelJunctionAnnotations = p.novelJunctionAnnotations()
#chromosomes = ['chrMT']
#p('chrY'); p('chr1'); p('chr2')
#chromosomes = ['chr8','chr17']
multiprocessing_pipe = True
if 'exp.' not in dataset_name:
dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name:
dataset_name+='.txt'
try:
mlp=Multi
pool_size = mlp.cpu_count()
print 'Using %d processes' % pool_size
if multiprocessing_pipe and multiThreading:
### This is like pool, but less efficient (needed to get print outs)
s = pool_size; b=0
chr_blocks=[]
while s<len(chromosomes):
chr_blocks.append(chromosomes[b:s])
b+=pool_size; s+=pool_size
chr_blocks.append(chromosomes[b:s])
queue = mlp.Queue()
results=[]
#parent_conn, child_conn=multiprocessing.Pipe()
for chromosomes in chr_blocks:
procs=list()
#print 'Block size:',len(chromosomes)
for search_chr in chromosomes:
proc = mlp.Process(target=p, args=(queue,search_chr)) ### passing sys.stdout unfortunately doesn't work to pass the Tk string
procs.append(proc)
proc.start()
for _ in procs:
val = queue.get()
if p.AnalysisMode() == 'GUI': print '*',
results.append(val)
for proc in procs:
proc.join()
elif multiThreading:
pool = mlp.Pool(processes=pool_size)
chr_vars=[]
for search_chr in chromosomes:
chr_vars.append(([],search_chr)) ### As an alternative for the pipe version above, pass an empty list rather than queue
results = pool.map(p, chr_vars) ### worker jobs initiated in tandem
try:pool.close(); pool.join(); pool = None
except Exception: pass
else:
forceThreadingError
print 'Read exon and junction mapping complete'
except Exception,e:
#print e
print 'Proceeding with single-processor version align...'
try: proc.close; proc.join; proc = None
except Exception: pass
try: pool.close(); pool.join(); pool = None
except Exception: pass
results=[] ### For single-thread compatible versions of Python
for search_chr in chromosomes:
result = p([],search_chr)
results.append(result)
results_organized=[]
for result_set in results:
if len(result_set[0])>0: ### Sometimes chromsomes are missing
biotypes = result_set[0]
results_organized.append(list(result_set[1:]))
pooled_results = [sum(value) for value in zip(*results_organized)] # combine these counts
pooled_results = [biotypes]+pooled_results
p.setCountsOverview(pooled_results) # store as retreivable objects
catFiles(countsFile,'Counts')
catFiles(junctionFile,'junctions')
catFiles(exonFile,'exons')
catFiles(junctionCompFile,'comps')
catFiles(novelJunctionAnnotations,'denovo')
if normalize_feature_exp == 'RPKM':
fastRPKMCalculate(countsFile)
rnaseq_end_time = time.time()
print '...RNA-seq import completed in',int(rnaseq_end_time-rnaseq_begin_time),'seconds\n'
biotypes = p.outputResults()
return biotypes
def alignCoordinatesToGeneExternal(species,coordinates_to_annotate):
chr_strand_gene_dbs,location_gene_db,chromosomes,gene_location_db = getChromosomeStrandCoordinates(species,'no')
read_aligned_to_gene=0
for (chr,strand) in coordinates_to_annotate:
if (chr,strand) in chr_strand_gene_dbs:
chr_gene_locations = chr_strand_gene_dbs[chr,strand]
chr_reads = coordinates_to_annotate[chr,strand]
chr_gene_locations.sort(); chr_reads.sort()
### Set GeneID for each coordinate object (primary and seconardary GeneIDs)
read_aligned_to_gene=geneAlign(chr,chr_gene_locations,location_gene_db,chr_reads,'no',read_aligned_to_gene)
### Gene objects will be updated
def catFiles(outFileDir,folder):
""" Concatenate all the chromosomal files but retain only the first header """
root_dir = export.findParentDir(outFileDir)+folder+'/'
dir_list = read_directory(root_dir)
firstFile=True
with open(filepath(outFileDir), 'w') as outfile:
for fname in dir_list:
chr_file = root_dir+fname
header=True
with open(filepath(chr_file)) as infile:
for line in infile:
if header:
header=False
if firstFile:
outfile.write(line)
firstFile=False
else: outfile.write(line)
export.deleteFolder(root_dir)
def error(msg, *args):
return multiprocessing.get_logger().error(msg, *args)
class AlignExonsAndJunctionsToEnsembl:
def setCountsOverview(self, overview):
self.biotypes_store, self.known_count, self.novel_junction_count, self.trans_splicing_reads, self.junctions_without_exon_gene_alignments, self.exons_without_gene_alignment_count = overview
def getChromosomes(self):
chr_list=list()
for c in self.chromosomes:
### Sort chromosome by int number
ci=string.replace(c,'chr','')
try: ci = int(ci)
except Exception: pass
chr_list.append((ci,c))
chr_list.sort()
chr_list2=list()
for (i,c) in chr_list: chr_list2.append(c) ### sorted
return chr_list2
def countsFile(self):
return string.replace(self.expfile,'exp.','counts.')
def junctionFile(self):
junction_file = self.root_dir+'AltDatabase/'+self.species+'/RNASeq/'+self.species + '_Ensembl_junctions.txt'
return junction_file
def exonFile(self):
exon_file = self.root_dir+'AltDatabase/'+self.species+'/RNASeq/'+self.species + '_Ensembl_exons.txt'
return exon_file
def junctionCompFile(self):
junction_comp_file = self.root_dir+'AltDatabase/'+self.species+'/RNASeq/'+self.species + '_junction_comps_updated.txt'
return junction_comp_file
def novelJunctionAnnotations(self):
junction_annotation_file = self.root_dir+'AltDatabase/ensembl/'+self.species+'/'+self.species + '_alternative_junctions_de-novo.txt'
return junction_annotation_file
def AnalysisMode(self): return self.analysisMode
def __init__(self,species,exp_file_location_db,dataset_name,testImport):
self.species = species; self.dataset_name = dataset_name
self.testImport = testImport
fl = exp_file_location_db[dataset_name]
bed_dir=fl.BEDFileDir()
root_dir=fl.RootDir()
#self.stdout = fl.STDOUT()
try: platformType = fl.PlatformType()
except Exception: platformType = 'RNASeq'
try: analysisMode = fl.AnalysisMode()
except Exception: analysisMode = 'GUI'
### This occurs when run using the BAMtoBED pipeline in the GUI
if 'exp.' not in dataset_name:
dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name:
dataset_name+='.txt'
self.dataset_name = dataset_name
### Import experimentally identified junction splice-sites
normalize_feature_exp = fl.FeatureNormalization()
if platformType == 'RNASeq':
chr_status = checkBEDFileFormat(bed_dir,root_dir) ### If false, need to remove 'chr' from the search_chr
else:
chr_status = True
#self.fl = fl # Can not pass this object in pool or it breaks
self.platformType = platformType
self.analysisMode = analysisMode
self.root_dir = root_dir
self.normalize_feature_exp = normalize_feature_exp
self.bed_dir = bed_dir
self.chr_status = chr_status
self.exonBedBuildStatus = fl.ExonBedBuildStatus()
self.expfile = root_dir+'ExpressionInput/'+dataset_name
if testImport == 'yes':
print 'Chromosome annotation detected =',chr_status
#if self.exonBedBuildStatus == 'yes':
reformatExonFile(species,'exon',chr_status) ### exports BED format exons for exon expression extraction
"""
Strategies to reduce memory in RNASeq:
1) (done)Delete old AltDatabase-local version if it exists before starting
2) (done)Check to see if a file exists before writing it and if so append rather than create
3) (done)Get counts last and normalize last in for exons and junctions separately.
4) (done)Delete objects explicitly before importing any new data (define a new function that just does this).
5) (done)Get all chromosomes first then parse exon and junction coordinate data on a per known chromosome basis.
6) (done)Prior to deleting all junction/exon object info for each chromsome, save the coordinate(key)-to-annotation information for the read count export file."""
### Delete any existing annotation databases that currently exist (redundant with below)
deleteOldAnnotations(species,root_dir,dataset_name)
###Define variables to report once reads for all chromosomes have been aligned
#global self.known_count; global self.novel_junction_count; global self.one_found; global self.not_found; global self.both_found; global self.trans_splicing_reads
#global self.junctions_without_exon_gene_alignments; global self.exons_without_gene_alignment_count; global self.junction_simple_db; global self.chr_strand_gene_dbs
self.known_count=0; self.novel_junction_count=0; self.one_found=0; self.not_found=0; self.both_found=0; self.trans_splicing_reads=0
self.junctions_without_exon_gene_alignments=0; self.exons_without_gene_alignment_count=0; self.junction_simple_db={}
###Begin Chromosome specific read to exon alignments
self.chr_strand_gene_dbs,self.location_gene_db,chromosomes,self.gene_location_db = getChromosomeStrandCoordinates(species,testImport)
self.chromosomes = chromosomes
print "Processing exon/junction coordinates sequentially by chromosome"
print "Note: this step is time intensive (can be hours) and no print statements may post for a while"
def outputResults(self):
exportDatasetLinkedGenes(self.species,self.gene_location_db,self.root_dir) ### Include an entry for gene IDs to include constitutive expression for RPKM normalized data
chr_gene_locations=[]; self.location_gene_db=[]; self.chr_strand_gene_dbs=[]
#print 'user coordinates imported/processed'
#print 'Importing read counts from coordinate data...'
biotypes = self.biotypes_store
### Output summary statistics
if self.normalize_feature_exp != 'none':
print self.normalize_feature_exp, 'normalization complete'
if 'junction' in biotypes:
print 'Imported Junction Statistics:'
print ' ',self.known_count, 'junctions found in Ensembl/UCSC and',self.novel_junction_count,'are novel'
print ' ',self.trans_splicing_reads,'trans-splicing junctions found (two aligning Ensembl genes)'
print ' ',self.junctions_without_exon_gene_alignments, 'junctions where neither splice-site aligned to a gene'
if (float(self.known_count)*10)<float(self.novel_junction_count):
print '\nWARNING!!!!! Few junctions aligned to known exons. Ensure that the AltAnalyze Ensembl database\nversion matches the genome build aligned to!\n'
if 'exon' in biotypes:
print 'Imported Exon Statistics:'
print ' ',self.exons_without_gene_alignment_count, 'exons where neither aligned to a gene'
print 'User databases and read counts written to:', self.root_dir[:-1]+'ExpressionInput'
### END CHROMOSOME SPECIFIC ANALYSES
if self.exonBedBuildStatus == 'yes':
bedfile = moveBAMtoBEDFile(self.species,self.dataset_name,self.root_dir)
print 'Exon BED file updated with novel exon predictions from junction file'
return bedfile; sys.exit()
clearObjectsFromMemory(self.junction_simple_db); self.junction_simple_db=[]
return biotypes
def test(self, search_chr):
print search_chr
def __call__(self, queue, search_chr):
try:
#sys.stdout = self.stdout
platformType = self.platformType
testImport = self.testImport
species = self.species
dataset_name = self.dataset_name
platformType = self.platformType
analysisMode = self.analysisMode
root_dir = self.root_dir
normalize_feature_exp = self.normalize_feature_exp
bed_dir = self.bed_dir
chr_status = self.chr_status
junction_annotations={}
if chr_status == False:
searchchr = string.replace(search_chr,'chr','')
else:
searchchr = search_chr
if platformType == 'RNASeq':
junction_db,biotypes,algorithms = importBEDFile(bed_dir,root_dir,species,normalize_feature_exp,searchChr=searchchr,testImport=testImport)
else:
normalize_feature_exp = 'quantile'
junction_db,biotypes,algorithms = importExpressionMatrix(bed_dir,root_dir,species,fl,'no',search_chr=searchchr)
self.biotypes_store = biotypes
if len(junction_db)>0:
### Determine which kind of data is being imported, junctions, exons or both
unmapped_exon_db={}
if 'junction' in biotypes:
### Get all known junction splice-sites
ens_junction_coord_db = importExonAnnotations(species,'junction_coordinates',search_chr)
if testImport == 'yes':
print len(ens_junction_coord_db),'Ensembl/UCSC junctions imported'
### Identify known junctions sites found in the experimental dataset (perfect match)
novel_junction_db={}; novel_exon_db={}
for key in junction_db:
ji=junction_db[key]
if ji.BioType()=='junction':
if key in ens_junction_coord_db:
jd=ens_junction_coord_db[key]
ji.setExonAnnotations(jd)
self.known_count+=1
else:
novel_junction_db[key]=junction_db[key]; self.novel_junction_count+=1
#if 75953254 in key: print key; sys.exit()
else:
unmapped_exon_db[key]=junction_db[key]
ens_exon_db = importExonAnnotations(species,'exon',search_chr)
if 'junction' in biotypes:
if testImport == 'yes':
print self.known_count, 'junctions found in Ensembl/UCSC and',len(novel_junction_db),'are novel.'
### Separate each junction into a 5' and 3' splice site (exon1_coord_db and exon2_coord_db)
exon1_coord_db={}; exon2_coord_db={}
for (chr,exon1_stop,exon2_start) in ens_junction_coord_db:
jd = ens_junction_coord_db[(chr,exon1_stop,exon2_start)]
exon1_coord_db[chr,exon1_stop] = jd.GeneID(),string.split(jd.ExonRegionIDs(),'-')[0]
exon2_coord_db[chr,exon2_start] = jd.GeneID(),string.split(jd.ExonRegionIDs(),'-')[1]
clearObjectsFromMemory(ens_junction_coord_db); ens_junction_coord_db=[] ### Clear object from memory
### Get and re-format individual exon info
exon_region_db={}
#if 'exon' not in biotypes:
for gene in ens_exon_db:
for rd in ens_exon_db[gene]:
exon_region_db[gene,rd.ExonRegionIDs()]=rd
### Add the exon annotations from the known junctions to the exons to export dictionary
exons_to_export={}
for key in junction_db:
ji=junction_db[key]
if ji.ExonAnnotations() != None:
jd = ji.ExonAnnotations()
exon1, exon2 = string.split(jd.ExonRegionIDs(),'-')
key1 = jd.GeneID(),exon1; key2 = jd.GeneID(),exon2
exons_to_export[key1] = exon_region_db[key1]
exons_to_export[key2] = exon_region_db[key2]
### For novel experimental junctions, identify those with at least one matching known 5' or 3' site
exons_not_identified = {}; novel_exon_coordinates={}
for (chr,exon1_stop,exon2_start) in novel_junction_db:
ji = novel_junction_db[(chr,exon1_stop,exon2_start)]
coord = [exon1_stop,exon2_start]; coord.sort()
if (chr,exon1_stop) in exon1_coord_db and (chr,exon2_start) in exon2_coord_db:
### Assign exon annotations to junctions where both splice-sites are known in Ensembl/UCSC
### Store the exon objects, genes and regions (le is a tuple of gene and exon region ID)
### Do this later for the below un-assigned exons
le=exon1_coord_db[(chr,exon1_stop)]; ji.setLeftExonAnnotations(le); ji.setLeftExonRegionData(exon_region_db[le])
re=exon2_coord_db[(chr,exon2_start)]; ji.setRightExonAnnotations(re); ji.setRightExonRegionData(exon_region_db[re])
if le[0] != re[0]: ### Indicates Trans-splicing (e.g., chr7:52,677,568-52,711,750 mouse mm9)
ji.setTransSplicing(); #print exon1_stop,le,exon2_start,re,ji.Chr(),ji.Strand()
self.both_found+=1; #print 'five',(chr,exon1_stop,exon2_start),exon1_coord_db[(chr,exon1_stop)]
else:
if (chr,exon1_stop) in exon1_coord_db: ### hence, exon1_stop is known, so report the coordinates of exon2 as novel
le=exon1_coord_db[(chr,exon1_stop)]; ji.setLeftExonAnnotations(le)
self.one_found+=1; #print 'three',(chr,exon1_stop,exon2_start),exon1_coord_db[(chr,exon1_stop)]
novel_exon_coordinates[ji.Chr(),exon2_start] = ji,'left',ji.Exon2Stop() ### Employ this strategy to avoid duplicate exons with differing lengths (mainly an issue if analyzing only exons results)
ji.setNovel('side')
elif (chr,exon2_start) in exon2_coord_db: ### hence, exon2_start is known, so report the coordinates of exon1 as novel
re=exon2_coord_db[(chr,exon2_start)]; ji.setRightExonAnnotations(re) ### In very rare cases, a gene can be assigned here, even though the splice-site is on the opposite strand (not worthwhile filtering out)
self.one_found+=1; #print 'three',(chr,exon1_stop,exon2_start),exon1_coord_db[(chr,exon1_stop)]
novel_exon_coordinates[ji.Chr(),exon1_stop] = ji,'right',ji.Exon1Start()
ji.setNovel('side')
else:
self.not_found+=1; #if self.not_found < 10: print (chr,exon1_stop,exon2_start)
novel_exon_coordinates[ji.Chr(),exon1_stop] = ji,'right',ji.Exon1Start()
novel_exon_coordinates[ji.Chr(),exon2_start] = ji,'left',ji.Exon2Stop()
ji.setNovel('both')
### We examine reads where one splice-site aligns to a known but the other not, to determine if trans-splicing occurs
try: exons_not_identified[chr,ji.Strand()].append((coord,ji))
except KeyError: exons_not_identified[chr,ji.Strand()] = [(coord,ji)]
"""
if fl.ExonBedBuildStatus() == 'no':
exportNovelJunctions(species,novel_junction_db,condition_count_db,root_dir,dataset_name,'junction') ### Includes known exons
"""
#print self.both_found, ' where both and', self.one_found, 'where one splice-site are known out of',self.both_found+self.one_found+self.not_found
#print 'Novel junctions where both splice-sites are known:',self.both_found
#print 'Novel junctions where one splice-site is known:',self.one_found
#print 'Novel junctions where the splice-sites are not known:',self.not_found
clearObjectsFromMemory(exon_region_db); exon_region_db=[] ### Clear memory of this object
read_aligned_to_gene=0
for (chr,strand) in exons_not_identified:
if (chr,strand) in self.chr_strand_gene_dbs:
chr_gene_locations = self.chr_strand_gene_dbs[chr,strand]
chr_reads = exons_not_identified[chr,strand]
chr_gene_locations.sort(); chr_reads.sort()
### Set GeneID for each coordinate object (primary and seconardary GeneIDs)
read_aligned_to_gene=geneAlign(chr,chr_gene_locations,self.location_gene_db,chr_reads,'no',read_aligned_to_gene)
#print read_aligned_to_gene, 'novel junctions aligned to Ensembl genes out of',self.one_found+self.not_found
clearObjectsFromMemory(exons_not_identified); exons_not_identified=[] ## Clear memory of this object
for key in novel_junction_db:
(chr,exon1_stop,exon2_start) = key
ji=novel_junction_db[key]
if ji.GeneID() == None:
try:
if ji.SecondaryGeneID() != None:
### Occurs if mapping is to the 5'UTR of a gene for the left splice-site (novel alternative promoter)
ji.setGeneID(ji.SecondaryGeneID()); ji.setSecondaryGeneID(''); #print key, ji.GeneID(), ji.Strand(), ji.SecondaryGeneID()
except Exception: null=[]
if ji.GeneID() != None:
geneid = ji.GeneID()
proceed = 'no'
if ji.SpliceSitesFound() == None: proceed = 'yes'; coordinates = [exon1_stop,exon2_start]
elif ji.SpliceSitesFound() == 'left': proceed = 'yes'; coordinates = [exon1_stop,exon2_start]
elif ji.SpliceSitesFound() == 'right': proceed = 'yes'; coordinates = [exon1_stop,exon2_start]
if proceed == 'yes':
for coordinate in coordinates:
if ji.TransSplicing() == 'yes':
#print ji.Chr(),ji.GeneID(), ji.SecondaryGeneID(), ji.Exon1Stop(), ji.Exon2Start()
self.trans_splicing_reads+=1
if ji.checkExonPosition(coordinate) == 'right': geneid = ji.SecondaryGeneID()
if abs(exon2_start-exon1_stop)==1: eventType = 'novel-exon-intron' ### Indicates intron-exon boundary (intron retention)
else: eventType = 'novel'
exon_data = (coordinate,ji.Chr()+'-'+str(coordinate),eventType)
try: novel_exon_db[geneid].append(exon_data)
except KeyError: novel_exon_db[geneid] = [exon_data]
else:
### write these out
self.junctions_without_exon_gene_alignments+=1
### Remove redundant exon entries and store objects
for key in novel_exon_db:
exon_data_objects=[]
exon_data_list = unique.unique(novel_exon_db[key])
exon_data_list.sort()
for e in exon_data_list:
ed = ExonInfo(e[0],e[1],e[2])
exon_data_objects.append(ed)
novel_exon_db[key] = exon_data_objects
#print self.trans_splicing_reads,'trans-splicing junctions found (two aligning Ensembl genes).'
#print self.junctions_without_exon_gene_alignments, 'junctions where neither splice-site aligned to a gene'
#if 'X' in search_chr: print len(ens_exon_db),len(ens_exon_db['ENSMUSG00000044424'])
alignReadsToExons(novel_exon_db,ens_exon_db,testImport=testImport)
### Link exon annotations up with novel junctions
junction_region_db,exons_to_export = annotateNovelJunctions(novel_junction_db,novel_exon_db,exons_to_export)
### Add the exon region data from known Ensembl/UCSC matched junctions to junction_region_db for recipricol junction analysis
for key in junction_db:
ji=junction_db[key]; jd = ji.ExonAnnotations()
try:
uid = jd.GeneID()+':'+jd.ExonRegionIDs(); ji.setUniqueID(uid)
try: junction_region_db[jd.GeneID()].append((formatID(uid),jd.ExonRegionIDs()))
except KeyError: junction_region_db[jd.GeneID()] = [(formatID(uid),jd.ExonRegionIDs())]
except AttributeError: null=[] ### Occurs since not all entries in the dictionary are perfect junction matches
try: novel_exon_coordinates = collapseNoveExonBoundaries(novel_exon_coordinates,root_dir+dataset_name) ### Joins inferred novel exon-IDs (5' and 3' splice sites) from adjacent and close junction predictions
except Exception: pass ### No errors encountered before
#if self.exonBedBuildStatus == 'yes':
### Append to the exported BED format exon coordinate file
bedfile = exportNovelExonToBedCoordinates(species,novel_exon_coordinates,chr_status,searchChr=searchchr)
### Identify reciprocol junctions and retrieve splice-event annotations for exons and inclusion junctions
junction_annotations,critical_exon_annotations = JunctionArray.inferJunctionComps(species,('RNASeq',junction_region_db,root_dir),searchChr=searchchr)
clearObjectsFromMemory(junction_region_db); junction_region_db=[]
### Reformat these dictionaries to combine annotations from multiple reciprocol junctions
junction_annotations = combineExonAnnotations(junction_annotations)
critical_exon_annotations = combineExonAnnotations(critical_exon_annotations)
if 'exon' in biotypes:
if testImport == 'yes':
print len(unmapped_exon_db),'exon genomic locations imported.'
### Create a new dictionary keyed by chromosome and strand
exons_not_aligned={}
for (chr,exon1_stop,exon2_start) in unmapped_exon_db:
ji = unmapped_exon_db[(chr,exon1_stop,exon2_start)]
coord = [exon1_stop,exon2_start]; coord.sort()
try: exons_not_aligned[chr,ji.Strand()].append((coord,ji))
except KeyError: exons_not_aligned[chr,ji.Strand()] = [(coord,ji)]
read_aligned_to_gene=0
for (chr,strand) in exons_not_aligned:
if (chr,strand) in self.chr_strand_gene_dbs:
chr_gene_locations = self.chr_strand_gene_dbs[chr,strand]
chr_reads = exons_not_aligned[chr,strand]
chr_gene_locations.sort(); chr_reads.sort()
read_aligned_to_gene=geneAlign(chr,chr_gene_locations,self.location_gene_db,chr_reads,'no',read_aligned_to_gene)
#print read_aligned_to_gene, 'exons aligned to Ensembl genes out of',self.one_found+self.not_found
align_exon_db={}; exons_without_gene_alignments={}; multigene_exon=0
for key in unmapped_exon_db:
(chr,exon1_stop,exon2_start) = key
ji=unmapped_exon_db[key]
if ji.GeneID() == None:
try:
if ji.SecondaryGeneID() != None:
### Occurs if mapping outside known exon boundaries for one side of the exon
ji.setGeneID(ji.SecondaryGeneID()); ji.setSecondaryGeneID(''); #print key, ji.GeneID(), ji.Strand(), ji.SecondaryGeneID()
except Exception: null=[]
else:
if 'ENS' in ji.JunctionID():
if ji.GeneID() not in ji.JunctionID(): ### Hence, there were probably two overlapping Ensembl genes and the wrong was assigned based on the initial annotations
original_geneid = string.split(ji.JunctionID(),':')[0]
if original_geneid in ens_exon_db: ji.setGeneID(original_geneid) #check if in ens_exon_db (since chromosome specific)
if ji.GeneID() != None:
geneid = ji.GeneID()
coordinates = [exon1_stop,exon2_start]
for coordinate in coordinates:
if ji.TransSplicing() != 'yes': ### This shouldn't occur for exons
exon_data = (coordinate,ji.Chr()+'-'+str(coordinate),'novel')
try: align_exon_db[geneid].append(exon_data)
except KeyError: align_exon_db[geneid] = [exon_data]
else:
multigene_exon+=1 ### Shouldn't occur due to a fix in the gene-alignment method which will find the correct gene on the 2nd interation
else: exons_without_gene_alignments[key]=ji; self.exons_without_gene_alignment_count+=1
### Remove redundant exon entries and store objects (this step may be unnecessary)
for key in align_exon_db:
exon_data_objects=[]
exon_data_list = unique.unique(align_exon_db[key])
exon_data_list.sort()
for e in exon_data_list:
ed = ExonInfo(e[0],e[1],e[2])
exon_data_objects.append(ed)
align_exon_db[key] = exon_data_objects
#print self.exons_without_gene_alignment_count, 'exons where neither aligned to a gene'
#if self.exons_without_gene_alignment_count>3000: print 'NOTE: Poor mapping of these exons may be due to an older build of\nEnsembl than the current version. Update BAMtoBED mappings to correct.'
begin_time = time.time()
alignReadsToExons(align_exon_db,ens_exon_db)
end_time = time.time()
if testImport == 'yes':
print 'Exon sequences aligned to exon regions in',int(end_time-begin_time),'seconds'
### Combine the start and end region alignments into a single exon annotation entry
combineDetectedExons(unmapped_exon_db,align_exon_db,novel_exon_db)
clearObjectsFromMemory(unmapped_exon_db); clearObjectsFromMemory(align_exon_db); clearObjectsFromMemory(novel_exon_db)
unmapped_exon_db=[]; align_exon_db=[]; novel_exon_db=[]
"""
if fl.ExonBedBuildStatus() == 'no':
exportNovelJunctions(species,exons_without_gene_alignments,condition_count_db,root_dir,dataset_name,'exon') ### Includes known exons
"""
clearObjectsFromMemory(exons_without_gene_alignments); exons_without_gene_alignments=[]
### Export both exon and junction annotations
if 'junction' in biotypes:
### Export the novel user exon annotations
exportDatasetLinkedExons(species,exons_to_export,critical_exon_annotations,root_dir,testImport=testImport,searchChr=searchchr)
### Export the novel user exon-junction annotations (original junction_db objects updated by above processing)
exportDatasetLinkedJunctions(species,junction_db,junction_annotations,root_dir,testImport=testImport,searchChr=searchchr)
### Clear memory once results are exported (don't want to delete actively used objects)
if 'junction' in biotypes:
clearObjectsFromMemory(exons_to_export); clearObjectsFromMemory(critical_exon_annotations)
clearObjectsFromMemory(novel_junction_db); novel_junction_db=[]
clearObjectsFromMemory(novel_exon_coordinates); novel_exon_coordinates=[]
exons_to_export=[]; critical_exon_annotations=[]
clearObjectsFromMemory(exon1_coord_db); clearObjectsFromMemory(exon2_coord_db)
exon1_coord_db=[]; exon2_coord_db=[]
if 'exon' in biotypes:
clearObjectsFromMemory(exons_not_aligned); exons_not_aligned=[]
clearObjectsFromMemory(ens_exon_db); ens_exon_db=[]
### Add chromsome specific junction_db data to a simple whole genome dictionary
for key in junction_db:
ji = junction_db[key]
if ji.GeneID()!=None and ji.UniqueID()!=None: self.junction_simple_db[key]=ji.UniqueID()
#returnLargeGlobalVars()
clearObjectsFromMemory(junction_db); clearObjectsFromMemory(junction_annotations)
junction_db=[]; junction_annotations=[]; chr_reads=[]
for biotype in biotypes:
### Import Read Counts (do this last to conserve memory)
if platformType == 'RNASeq':
condition_count_db,exon_len_db,biotypes2,algorithms = importBEDFile(bed_dir,root_dir,species,normalize_feature_exp,getReads=True,searchChr=searchchr,getBiotype=biotype,testImport=testImport,filteredJunctions=self.junction_simple_db)
else:
condition_count_db,exon_len_db,biotypes2,algorithms = importExpressionMatrix(bed_dir,root_dir,species,fl,'yes',getBiotype=biotype)
###First export original counts, rather than quantile normalized or RPKM
self.exportJunctionCounts(species,self.junction_simple_db,exon_len_db,condition_count_db,root_dir,dataset_name,biotype,'counts',searchChr=searchchr)
clearObjectsFromMemory(condition_count_db); clearObjectsFromMemory(exon_len_db); condition_count_db=[]; exon_len_db=[]
if analysisMode == 'commandline':
print 'finished parsing data for chromosome:',search_chr ### Unix platforms are not displaying the progress in real-time
else:
pass #print "*",
try: queue.put([self.biotypes_store, self.known_count, self.novel_junction_count, self.trans_splicing_reads, self.junctions_without_exon_gene_alignments, self.exons_without_gene_alignment_count])
except Exception:
### If queue is not a multiprocessing object
queue = [self.biotypes_store, self.known_count, self.novel_junction_count, self.trans_splicing_reads, self.junctions_without_exon_gene_alignments, self.exons_without_gene_alignment_count]
return queue
except Exception:
print traceback.format_exc()
error(traceback.format_exc())
multiprocessing.log_to_stderr().setLevel(logging.DEBUG)
raise
def exportJunctionCounts(self,species,junction_simple_db,exon_len_db,condition_count_db,root_dir,dataset_name,biotype,count_type,searchChr=None):
if 'exp.' not in dataset_name: dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name: dataset_name+='.txt'
export_path = root_dir+'ExpressionInput/'+dataset_name
if count_type == 'counts':
export_path = string.replace(export_path,'exp.','counts.') ### separately export counts
if searchChr !=None:
export_path = string.replace(export_path,'ExpressionInput','ExpressionInput/Counts')
export_path = string.replace(export_path,'.txt','.'+searchChr+'.txt')
self.countsFile = export_path
if self.testImport == 'yes':
print 'Writing',export_path
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
title = ['AltAnalyze_ID']
for condition in condition_count_db: title.append(condition)
export_data.write(string.join(title,'\t')+'\n')
for key in self.junction_simple_db:
chr,exon1_stop,exon2_start = key
if biotype == 'junction':
coordinates = chr+':'+str(exon1_stop)+'-'+str(exon2_start)
elif biotype == 'exon':
coordinates = chr+':'+str(exon1_stop-1)+'-'+str(exon2_start+1)
try:
null=exon_len_db[key]
if count_type == 'counts': values = [self.junction_simple_db[key]+'='+coordinates]
else: values = [self.junction_simple_db[key]]
for condition in condition_count_db: ###Memory crash here
count_db = condition_count_db[condition]
try: read_count = count_db[key]
except KeyError: read_count = '0'
values.append(read_count)
export_data.write(string.join(values,'\t')+'\n')
except Exception: null=[]
export_data.close()
def countsDir(self):
return self.countsFile
def calculateRPKMsFromGeneCounts(filename,species,AdjustExpression):
""" Manual way of calculating gene RPKMs from gene counts only """
gene_lengths = getGeneExonLengths(species)
fastRPKMCalculate(filename,GeneLengths=gene_lengths,AdjustExpression=AdjustExpression)
def fastRPKMCalculate(counts_file,GeneLengths=None,AdjustExpression=True):
export_path = string.replace(counts_file,'counts.','exp.')
export_data = export.ExportFile(export_path) ### Write this new file
fn=filepath(counts_file); header=True
exon_sum_array=[]; junction_sum_array=[]
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
samples = t[1:]
header=False
exon_sum_array=[0]*len(samples)
junction_sum_array=[0]*len(samples)
else:
try: values = map(float,t[1:])
except Exception:
print traceback.format_exc()
print t
badCountsLine
### get the total reads/sample
if '-' in string.split(t[0],'=')[0]:
junction_sum_array = [sum(value) for value in zip(*[junction_sum_array,values])]
else:
exon_sum_array = [sum(value) for value in zip(*[exon_sum_array,values])]
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides warnings associated with Scipy for n=1 sample comparisons
jatr=Average(junction_sum_array) # Average of the total maped reads
eatr=Average(exon_sum_array) # Average of the total maped reads
if AdjustExpression:
offset = 1
else:
offset = 0
header=True
c=math.pow(10.0,9.0)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
export_data.write(line) ### Write header
header=False
else:
try:
exon_id,coordinates = string.split(t[0],'=')
coordinates = string.split(coordinates,':')[1]
coordinates = string.split(coordinates,'-')
l=abs(int(coordinates[1])-int(coordinates[0])) ### read-length
except Exception: ### Manual way of calculating gene RPKMs from gene counts only
exon_id = t[0]
try: l = GeneLengths[exon_id]
except Exception: continue #Occurs when Ensembl genes supplied from an external analysis
try: read_counts = map(lambda x: int(x)+offset, t[1:])
except Exception: read_counts = map(lambda x: int(float(x))+offset, t[1:])
if '-' in exon_id:
count_stats = zip(read_counts,junction_sum_array)
atr = jatr
l=60
else:
count_stats = zip(read_counts,exon_sum_array)
atr = eatr
values=[]
#rpkm = map(lambda (r,t): c*(r/(t*l)), count_stats) ### Efficent way to convert to rpkm, but doesn't work for 0 counts
for (r,t) in count_stats:
if r == 1: ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
t = atr
try:
rpkm = str(c*(r/(t*l)))
#print c,r,t,l,exon_id,rpkm;sys.exit()
values.append(rpkm)
except Exception,e:
print e
print t[0]
print 'Error Encountered... Exon or Junction of zero length encoutered... RPKM failed... Exiting AltAnalyze.'
print 'This error may be due to inconsistent file naming. If both exon and junction sample data is present, make sure they are named propperly.'
print 'For example: cancer1__exon.bed, cancer1__junction.bed (double underscore required to match these samples up)!'
print [r,t,l];k=1; forceError
values = string.join([exon_id]+values,'\t')+'\n'
export_data.write(values)
export_data.close()
def mergeCountFiles(counts_file1,counts_file2):
### Used internally to merge count files that are very large and too time-consuming to recreate (regenerate them)
export_path = string.replace(counts_file2,'counts.','temp-counts.')
export_data = export.ExportFile(export_path) ### Write this new file
fn=filepath(counts_file1); header=True
count_db={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
samples = t[1:]
header=False
si = samples.index('H9.102.2.5.bed')+1
else:
try: value = t[si]
except Exception: print t; sys.exit()
### get the total reads/sample
count_db[t[0]] = value
fn=filepath(counts_file2); header=True
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if header:
samples = t[1:]
header=False
si = samples.index('H9.102.2.5.bed')+1
export_data.write(line)
else:
try: t[si] = count_db[t[0]]
except Exception: pass ### keep the current value
export_data.write(string.join(t,'\t')+'\n')
export_data.close()
def getGeneExonLengths(species):
gene_lengths={}
filename = 'AltDatabase/'+species+'/RNASeq/'+species+'_Ensembl_exons.txt'
fn=filepath(filename)
firstLine=True
for line in open(fn,'rU').xreadlines():
line = line.rstrip('\n')
if firstLine:
firstLine=False
else:
t = string.split(line,'\t')
geneID = t[2]; start = int(t[6]); end = int(t[7]); exonID = t[1]
if 'E' in exonID:
try: gene_lengths[geneID]+=abs(end-start)
except Exception: gene_lengths[geneID]=abs(end-start)
return gene_lengths
def importRawCountData(filename,expressed_gene_exon_db,excludeLowExp=True):
""" Identifies exons or junctions to evaluate gene-level expression. This function, as it is currently written:
1) examines the RPKM and original read counts associated with all exons
2) removes exons/junctions that do not meet their respective RPKM AND read count cutoffs
3) returns ONLY those exons and genes deemed expressed, whether constitutive selected or all exons
"""
### Get expression values for exon/junctions to analyze
seq_ids_to_import={}
for gene in expressed_gene_exon_db:
for exonid in expressed_gene_exon_db[gene]: seq_ids_to_import[exonid]=[]
### Define thresholds
exon_exp_threshold = UserOptions.ExonExpThreshold()
junction_exp_threshold = UserOptions.JunctionExpThreshold()
exon_rpkm_threshold = UserOptions.ExonRPKMThreshold()
gene_rpkm_threshold = UserOptions.RPKMThreshold()
gene_exp_threshold = UserOptions.GeneExpThreshold()
### Import RPKM normalized expression values
fn=filepath(filename); x=0; rpkm_dbase={}
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: array_names = t[1:]; x=1
else:
exon_id=t[0]
max_count=max(map(float,t[1:]))
if max_count>=exon_rpkm_threshold or excludeLowExp==False: rpkm_dbase[exon_id]=[] ### Only retain exons/junctions meeting the RPKM threshold
### Import non-normalized original counts
counts_filename = string.replace(filename,'exp.','counts.')
fn=filepath(counts_filename); x=0; exp_dbase={}
all_exp_features={} ### Don't filter for only gene-expression reporting
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: array_names = t[1:]; x=1
else:
exon_id,coordinates = string.split(t[0],'=')
coordinates = string.split(coordinates,':')[1]
coordinates = string.split(coordinates,'-')
length=abs(int(coordinates[1])-int(coordinates[0]))
max_count=max(map(float,t[1:])); proceed = 'no'
if '-' in exon_id:
length = 60.0
if max_count>=junction_exp_threshold or excludeLowExp==False:
### Only considered when exon data is not present in the analysis
proceed = 'yes'
elif max_count>=exon_exp_threshold or excludeLowExp==False: proceed = 'yes'
if proceed == 'yes' and exon_id in rpkm_dbase: ### Ensures that the maximum sample (not group) user defined count threshold is achieved at the exon or junction-level
all_exp_features[exon_id]=None
if exon_id in seq_ids_to_import:### Forces an error if not in the steady-state pre-determined set (CS or all-exons) - INCLUDE HERE TO FILTER ALL FEATURES
exp_dbase[exon_id] = t[1:],length ### Include sequence length for normalization
for exon in exp_dbase: array_count = len(exp_dbase[exon][0]); break
try:null=array_count
except Exception:
print 'No exons or junctions considered expressed (based user thresholds). Exiting analysis.'; force_exit
return exp_dbase, all_exp_features, array_count
def importNormalizedCountData(filename,expressed_gene_exon_db):
### Get expression values for exon/junctions to analyze
seq_ids_to_import={}
for gene in expressed_gene_exon_db:
for exonid in expressed_gene_exon_db[gene]: seq_ids_to_import[exonid]=[]
### Define thresholds
exon_exp_threshold = UserOptions.ExonExpThreshold()
junction_exp_threshold = UserOptions.JunctionExpThreshold()
exon_rpkm_threshold = UserOptions.ExonRPKMThreshold()
gene_rpkm_threshold = UserOptions.RPKMThreshold()
gene_exp_threshold = UserOptions.GeneExpThreshold()
### Import non-normalized original counts
fn=filepath(filename); x=0; exp_dbase={}
all_exp_features={} ### Don't filter for only gene-expression reporting
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: array_names = t[1:]; x=1
else:
exon_id=t[0]; proceed = 'no'
max_count=max(map(float,t[1:]))
if '-' in exon_id:
if max_count>=junction_exp_threshold: proceed = 'yes'
elif max_count>=exon_exp_threshold: proceed = 'yes'
if proceed == 'yes': ### Ensures that the maximum sample (not group) user defined count threshold is achieved at the exon or junction-level
all_exp_features[exon_id]=None
if exon_id in seq_ids_to_import: ### If a "constitutive" or exon-level feature (filter missing prior to 2.0.8 - bug)
exp_dbase[exon_id] = t[1:],0 ### Add the zero just to comply with the raw count input format (indicates exon length)
for exon in exp_dbase: array_count = len(exp_dbase[exon][0]); break
return exp_dbase, all_exp_features, array_count
def obtainGeneCounts(expressed_gene_exon_db,species,exp_dbase,array_count,normalize_feature_exp,excludeLowExp=True):
###Calculate avg expression for each sample for each exon (using constitutive or all exon values)
if excludeLowExp == False:
gene_lengths = getGeneExonLengths(species)
steady_state_db={}
for gene in expressed_gene_exon_db:
x = 0; gene_sum=0
exon_list = expressed_gene_exon_db[gene]
while x < array_count:
exp_list=[]; len_list=[]
for exon in exon_list:
try:
exp_val = exp_dbase[exon][0][x]
if normalize_feature_exp == 'RPKM':
### Decided to include all exons, expressed or not to prevent including lowly expressed exons that are long, that can bias the expression call
#if float(exp_val) != 0: ### Here, we use the original raw count data, whereas above is the adjusted quantile or raw count data
exp_list.append(exp_val); len_list.append(exp_dbase[exon][1]) ### This is for RNASeq -> don't include undetected exons - made in v.204
else: exp_list.append(exp_val) #elif float(exp_val) != 1:
except KeyError: null =[] ###occurs if the expression exon list is missing some of these exons
try:
if len(exp_list)==0:
for exon in exon_list:
try:
exp_list.append(exp_dbase[exon][0][x]); len_list.append(exp_dbase[exon][1])
#kill
except KeyError: null=[] ### Gene entries will cause this error, since they are in the database but not in the count file
if normalize_feature_exp == 'RPKM':
sum_const_exp=sum(map(float,exp_list)); gene_sum+=sum_const_exp
sum_length=sum(len_list) ### can have different lengths for each sample, since only expressed exons are considered
if excludeLowExp == False:
sum_length = gene_lengths[gene] ### Uses the all annotated exon lengths
### Add only one avg-expression value for each array, this loop
try: steady_state_db[gene].append((sum_const_exp,sum_length))
except KeyError: steady_state_db[gene] = [(sum_const_exp,sum_length)]
else:
avg_const_exp=Average(exp_list)
if avg_const_exp != 1: gene_sum+=avg_const_exp
### Add only one avg-expression value for each array, this loop
try: steady_state_db[gene].append(avg_const_exp)
except KeyError: steady_state_db[gene] = [avg_const_exp]
except Exception: null=[] ### Occurs when processing a truncated dataset (for testing usually) - no values for the gene should be included
x += 1
if gene_sum==0:
try:
del steady_state_db[gene] ### Hence, no genes showed evidence of expression (most critical for RNA-Seq)
except Exception: null=[] ### Error occurs when a gene is added to the database from self.location_gene_db, but is not expressed
return steady_state_db
def returnLargeGlobalVars():
### Prints all large global variables retained in memory (taking up space)
all = [var for var in globals() if (var[:2], var[-2:]) != ("__", "__")]
for var in all:
try:
if len(globals()[var])>1:
print var, len(globals()[var])
except Exception: null=[]
def clearObjectsFromMemory(db_to_clear):
db_keys={}
for key in db_to_clear: db_keys[key]=[]
for key in db_keys:
try: del db_to_clear[key]
except Exception:
try:
for i in key: del i ### For lists of tuples
except Exception: del key ### For plain lists
def verifyFile(filename):
status = 'not found'
try:
fn=filepath(filename)
for line in open(fn,'rU').xreadlines(): status = 'found';break
except Exception: status = 'not found'
return status
def AppendOrWrite(export_path):
export_path = filepath(export_path)
status = verifyFile(export_path)
if status == 'not found':
export_data = export.ExportFile(export_path) ### Write this new file
else:
export_data = open(export_path,'a') ### Appends to existing file
return export_data, status
def quantileNormalizationSimple(condition_count_db):
### Basic quantile normalization method (average ranked expression values)
### Get all junction or exon entries
key_db={}
for condition in condition_count_db:
count_db = condition_count_db[condition]
for key in count_db: key_db[key]=[]
condition_unnormalized_db={}
for key in key_db:
### Only look at the specific biotype of interest for each normalization
for condition in condition_count_db:
count_db = condition_count_db[condition]
try:
count = float(count_db[key])+1 ###This adjustment allows us to obtain more realist folds where 0 is compared and use log2
count_db[key] = [] ### Set equal to null as a temporary measure to save memory
except KeyError: count = 1.00 ###Was zero, but needs to be one for more realistic log2 fold calculations
### store the minimal information to recover the original count and ID data prior to quantile normalization
try: condition_unnormalized_db[condition].append([count,key])
except Exception: condition_unnormalized_db[condition]=[[count,key]]
quantile_normalize_db={}; key_db={}
for condition in condition_unnormalized_db:
condition_unnormalized_db[condition].sort() ### Sort lists by count number
rank=0 ### thus, the ID is the rank order of counts
for (count,key) in condition_unnormalized_db[condition]:
try: quantile_normalize_db[rank].append(count)
except KeyError: quantile_normalize_db[rank] = [count]
rank+=1
### Get the average value for each index
for rank in quantile_normalize_db:
quantile_normalize_db[rank] = Average(quantile_normalize_db[rank])
for condition in condition_unnormalized_db:
rank=0
count_db = condition_count_db[condition]
for (count,key) in condition_unnormalized_db[condition]:
avg_count = quantile_normalize_db[rank]
rank+=1
count_db[key] = str(avg_count) ### re-set this value to the normalized value
try:
clearObjectsFromMemory(condition_unnormalized_db); condition_unnormalized_db = []
clearObjectsFromMemory(quantile_normalize_db); quantile_normalize_db = []
except Exception: None
return condition_count_db
def combineExonAnnotations(db):
for i in db:
list1=[]; list2=[]
for (junctions,splice_event) in db[i]:
list1.append(junctions); list2.append(splice_event)
junctions = EnsemblImport.combineAnnotations(list1)
splice_event = EnsemblImport.combineAnnotations(list2)
db[i] = junctions,splice_event
return db
def formatID(id):
### JunctionArray methods handle IDs with ":" different than those that lack this
return string.replace(id,':','@')
def filterChromosomes(chromosome_names):
### If transcriptome only aligned to Ensembl reference, many chromosomes are not real
updated_chromosomes=[]
chr_count=0
for chr in chromosome_names:
if 'chr' in chr and len(chr)<7:
chr_count+=1
updated_chromosomes.append(chr)
if chr_count>1:
return updated_chromosomes
else:
return chromosome_names
def getChromosomeStrandCoordinates(species,testImport):
### For novel junctions with no known-splice site, map to genes
gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,'RNASeq','key_by_array')
chr_strand_gene_db = {}; location_gene_db = {}; chromosome_names={}; all_chromosomes={}
for gene in gene_location_db:
chr,strand,start,end = gene_location_db[gene]
location_gene_db[chr,int(start),int(end)] = gene,strand
try: chr_strand_gene_db[chr,strand].append((int(start),int(end)))
except KeyError: chr_strand_gene_db[chr,strand] = [(int(start),int(end))]
if testImport == 'yes':
if chr=='chr1': chromosome_names[chr]=[]
#if chr=='chr19': chromosome_names[chr]=[] ### Gene rich chromosome
#if chr=='chrMT': chromosome_names[chr]=[] ### Gene rich chromosome
elif len(chr)<7: chromosome_names[chr]=[]
all_chromosomes[chr]=[]
#chromosome_names = filterChromosomes(chromosome_names)
### Some organisms aren't organized into classical chromosomes (why I don't know)
if len(chromosome_names)<10 and len(all_chromosomes)>9 and testImport=='no': chromosome_names = all_chromosomes
return chr_strand_gene_db,location_gene_db,chromosome_names,gene_location_db
def exportDatasetLinkedExons(species,exons_to_export,critical_exon_annotations,root_dir,testImport=None,searchChr=None):
export_path = root_dir+'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_exons.txt'
if searchChr != None:
export_path = string.replace(export_path,'RNASeq/'+species,'RNASeq/exons/'+species)
export_path = string.replace(export_path,'.txt','.'+searchChr+'.txt')
if testImport == 'yes': print 'Writing',export_path
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
export_title = ['AltAnalyzeID','exon_id','ensembl_gene_id','transcript_cluster_id','chromosome','strand','probeset_start','probeset_stop']
export_title +=['class','constitutive_probeset','ens_exon_ids','ens_constitutive_status','exon_region','exon-region-start(s)','exon-region-stop(s)','splice_events','splice_junctions']
export_title = string.join(export_title,'\t')+'\n'; export_data.write(export_title)
### We stored these in a dictionary to make sure each exon is written only once and so we can organize by gene
exons_to_export_list=[]
for key in exons_to_export:
ed = exons_to_export[key]
exons_to_export_list.append((key,ed))
exons_to_export_list.sort()
for (key,ed) in exons_to_export_list:
constitutive_call = 'no'; ens_constitutive_status = '0'
try:
red = ed.ExonRegionData()
exon_region = ed.ExonRegionID()
start = str(ed.ReadStart()); stop = start
if '-' not in exon_region and '_' not in exon_region: annotation = 'known'
else: annotation = 'novel'
except Exception:
red = ed ### For annotated exons, no difference in the annotations
exon_region = ed.ExonRegionIDs()
start = str(red.ExonStart()); stop = str(red.ExonStop())
constitutive_call = red.Constitutive()
if constitutive_call == 'yes': ens_constitutive_status = '1'
annotation = 'known'
uid = red.GeneID()+':'+exon_region
splice_events = red.AssociatedSplicingEvent(); splice_junctions = red.AssociatedSplicingJunctions()
if uid in critical_exon_annotations:
splice_junctions,splice_events = critical_exon_annotations[uid]
export_values = [uid, exon_region, red.GeneID(), '', red.Chr(), red.Strand(), start, stop, annotation, constitutive_call, red.ExonID(), ens_constitutive_status]
export_values+= [exon_region, str(red.ExonStart()), str(red.ExonStop()), splice_events, splice_junctions]
export_values = string.join(export_values,'\t')+'\n'; export_data.write(export_values)
export_data.close()
def exportNovelJunctions(species,novel_junction_db,condition_count_db,root_dir,dataset_name,biotype):
if 'exp.' not in dataset_name: dataset_name = 'exp.'+dataset_name
if '.txt' not in dataset_name: dataset_name+='.txt'
dataset_name = string.replace(dataset_name,'exp','novel')
dataset_name = string.replace(dataset_name,'.txt','.'+biotype+'.txt')
export_path = root_dir+'ExpressionInput/'+dataset_name
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
title = ['chr','strand','start','stop','start Ensembl','end Ensembl','known start', 'known end']
for condition in condition_count_db: title.append(condition)
export_data.write(string.join(title,'\t')+'\n')
for key in novel_junction_db:
ji = novel_junction_db[key]
try: gene1 = str(ji.GeneID())
except Exception: gene1=''
try: gene2 = str(ji.SecondaryGeneID())
except Exception: gene2 = 'None'
try: le = str(ji.LeftExonAnnotations())
except Exception: le = ''
try: re = str(ji.RightExonAnnotations())
except Exception: re = ''
if biotype == 'junction':
values = [ji.Chr(), ji.Strand(), str(ji.Exon1Stop()), str(ji.Exon2Start())]
elif biotype == 'exon':
values = [ji.Chr(), ji.Strand(), str(ji.Exon1Stop()-1), str(ji.Exon2Start()+1)] ### correct for initial adjustment
values += [gene1,gene2,le,re]
for condition in condition_count_db:
count_db = condition_count_db[condition]
try: read_count = count_db[key]
except KeyError: read_count = '0'
values.append(read_count)
export_data.write(string.join(values,'\t')+'\n')
export_data.close()
def exportDatasetLinkedGenes(species,gene_location_db,root_dir):
"""Include an entry for gene IDs to include constitutive expression for RPKM normalized data"""
export_path = root_dir+'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_junctions.txt'
export_data,status = AppendOrWrite(export_path)
for gene in gene_location_db:
chr,strand,start,end = gene_location_db[gene]
export_values = [gene, 'E0.1',gene, '', chr, strand, str(start), str(end), 'known', 'yes', gene, '1']
export_values+= ['E0.1', str(start), str(end), '', '']
export_values = string.join(export_values,'\t')+'\n'; export_data.write(export_values)
export_data.close()
def exportDatasetLinkedJunctions(species,junction_db,junction_annotations,root_dir,testImport=False,searchChr=None):
export_path = root_dir+'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_junctions.txt'
if searchChr != None:
export_path = string.replace(export_path,'RNASeq/'+species,'RNASeq/junctions/'+species)
export_path = string.replace(export_path,'.txt','.'+searchChr+'.txt')
if testImport == 'yes': print 'Writing',export_path
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
export_title = ['AltAnalyzeID','exon_id','ensembl_gene_id','transcript_cluster_id','chromosome','strand','probeset_start','probeset_stop']
export_title +=['class','constitutive_probeset','ens_exon_ids','ens_constitutive_status','exon_region','exon-region-start(s)','exon-region-stop(s)','splice_events','splice_junctions']
export_title = string.join(export_title,'\t')+'\n'; export_data.write(export_title)
for key in junction_db:
(chr,exon1_stop,exon2_start) = key
ji=junction_db[key]
#print key, ji.UniqueID(), ji.GeneID()
if ji.GeneID()!=None and ji.UniqueID()!=None:
if ji.UniqueID() in junction_annotations: ### Obtained from JunctionArray.inferJunctionComps()
junctions,splice_events = junction_annotations[ji.UniqueID()]
if ji.TransSplicing() == 'yes':
if len(splice_events)>0: splice_events+= '|trans-splicing'
else: splice_events = 'trans-splicing'
ji.setAssociatedSplicingEvent(splice_events); ji.setAssociatedSplicingJunctions(junctions)
elif ji.TransSplicing() == 'yes':
ji.setAssociatedSplicingEvent('trans-splicing')
try:
try: constitutive_call = ji.Constitutive()
except Exception:
jd = ji.ExonAnnotations()
constitutive_call = jd.Constitutive()
if constitutive_call == 'yes': ens_constitutive_status = '1'
else: ens_constitutive_status = '0'
annotation = 'known'
except Exception:
constitutive_call = 'no'; ens_constitutive_status = '0'; annotation = 'novel'
if 'I' in ji.ExonRegionID() or 'U' in ji.ExonRegionID() or '_' in ji.ExonRegionID():
annotation = 'novel' ### Not previously indicated well (as I remember) for exon-level reads - so do this
export_values = [ji.UniqueID(), ji.ExonRegionID(), ji.GeneID(), '', ji.Chr(), ji.Strand(), str(ji.Exon1Stop()), str(ji.Exon2Start()), annotation, constitutive_call, ji.ExonID(), ens_constitutive_status]
export_values+= [ji.ExonRegionID(), str(ji.Exon1Stop()), str(ji.Exon2Start()), ji.AssociatedSplicingEvent(), ji.AssociatedSplicingJunctions()]
export_values = string.join(export_values,'\t')+'\n'; export_data.write(export_values)
export_data.close()
def combineDetectedExons(unmapped_exon_db,align_exon_db,novel_exon_db):
### Used for exon alignments (both start position and end position aligned to exon/intron/UTR regions)
### Reformat align_exon_db to easily lookup exon data
aligned_exon_lookup_db={}
for gene in align_exon_db:
for ed in align_exon_db[gene]:
aligned_exon_lookup_db[gene,ed.ReadStart()]=ed
#if gene == 'ENSMUSG00000064181': print ed.ReadStart(),ed.ExonRegionID()
### Reformat novel_exon_db to easily lookup exon data - created from junction analysis (rename above exons to match novel junctions)
novel_exon_lookup_db={}
for gene in novel_exon_db:
for ed in novel_exon_db[gene]:
try:
### Only store exons that are found in the novel exon file
null = aligned_exon_lookup_db[gene,ed.ReadStart()+1] ### offset introduced on import
novel_exon_lookup_db[gene,ed.ReadStart()+1]=ed
except Exception: null=[]
try:
### Only store exons that are found in the novel exon file
null = aligned_exon_lookup_db[gene,ed.ReadStart()-1] ### offset introduced on import
novel_exon_lookup_db[gene,ed.ReadStart()-1]=ed
except Exception: null=[]
### Lookup the propper exon region ID and gene ID to format the unique ID and export coordinates
x = 0
for key in unmapped_exon_db:
(chr,exon1_stop,exon2_start) = key
ji=unmapped_exon_db[key]
proceed = 'no'
if ji.GeneID() != None:
e1 = (ji.GeneID(),exon1_stop)
e2 = (ji.GeneID(),exon2_start)
exon_info=[]; override_annotation = None; found=[]
try: null = aligned_exon_lookup_db[e1]; found.append(1)
except Exception: null=[]
try: null = aligned_exon_lookup_db[e2]; found.append(2)
except Exception: null=[]
try: null = novel_exon_lookup_db[e1]; override_annotation = 1
except Exception:
try: null = novel_exon_lookup_db[e2]; override_annotation = 2
except Exception: null=[]
if len(found)>0:
### Below is not the simplist way to do this, but should be the fastest
if 1 in found: exon_info.append(aligned_exon_lookup_db[e1])
if 2 in found: exon_info.append(aligned_exon_lookup_db[e2])
if len(exon_info) == 2: ed1,ed2 = exon_info
else:
ed1 = exon_info[0]; ed2 = ed1; x+=1 ### if only one splice site aligned to a gene region (shouldn't occur)
if x == 2: null=[]; #print 'SOME EXONS FOUND WITH ONLY ONE ALIGNING POSITION...',key,ji.GeneID(),ed1.ExonRegionID(),e1,e2
try: red1 = ed1.ExonRegionData(); red2 = ed2.ExonRegionData()
except Exception:
"""
print [ji.GeneID(), ji.Chr(), key]
print e1, e2
try: print ed1.ExonRegionData()
except Exception: 'ed1 failed'
try: print ed2.ExonRegionData()
except Exception: 'ed2 failed'
"""
continue
region1 = ed1.ExonRegionID(); region2 = ed2.ExonRegionID()
#print region1,region2,ji.GeneID(),ji.Chr(),ji.Strand()
try: splice_junctions = EnsemblImport.combineAnnotations([red1.AssociatedSplicingJunctions(),red2.AssociatedSplicingJunctions()])
except Exception: print red1, red2;sys.exit()
splice_events = EnsemblImport.combineAnnotations([red1.AssociatedSplicingEvent(),red2.AssociatedSplicingEvent()])
ji.setAssociatedSplicingJunctions(splice_junctions)
ji.setAssociatedSplicingEvent(splice_events)
ens_exon_ids = EnsemblImport.combineAnnotations([red1.ExonID(),red2.ExonID()])
ji.setExonID(ens_exon_ids)
if red1.Constitutive() == 'yes' or red2.Constitutive() == 'yes': constitutive_call = 'yes'
else: constitutive_call = 'no'
ji.setConstitutive(constitutive_call)
report_both_regions = 'no'
try:
### If the annotations are from a BED file produced by AltAnalyze, novel alternative splice sites may be present
### if the below variable is not created, then this exon may over-ride the annotated exon region (e.g., E15.1 is over-written by E15.1_1234;E15.1_1256)
if 'ENS' in ji.JunctionID() and ':' not in ji.JunctionID(): report_both_regions = 'yes'
except Exception: null=[]
try:
### If the annotations are from a BED file produced by AltAnalyze, it is possible for to a known exon to share a splice-site coordinate
### with a novel junction exon. This will cause both to have the same override_annotation. Prevent this with the below 2nd override
if 'ENS' in ji.JunctionID() and ':' in ji.JunctionID(): override_annotation = None
except Exception: null=[]
if override_annotation != None:
if '_' in region1: region1 = string.split(region1,'_')[0]+'_'+str(int(string.split(region1,'_')[-1])-1)
if '_' in region2: region2 = string.split(region2,'_')[0]+'_'+str(int(string.split(region2,'_')[-1])+1)
if override_annotation == 1: region_id = region1 ### This forces a TopHat exon to be named for the splice-site position
else: region_id = region2
else:
if report_both_regions == 'no':
### Don't include specific start and end coordinates if inside a known exon
if ed1.AlignmentRegion() == 'exon': region1 = string.split(region1,'_')[0]
if ed2.AlignmentRegion() == 'exon': region2 = string.split(region2,'_')[0]
if ed1.AlignmentRegion() == 'full-intron' and ed2.AlignmentRegion() == 'full-intron':
region1 = string.split(region1,'_')[0]; region2 = string.split(region2,'_')[0]
### Below adjustmements need to compenstate for adjustments made upon import
if '_' in region1: region1 = string.split(region1,'_')[0]+'_'+str(int(string.split(region1,'_')[-1])-1)
if '_' in region2: region2 = string.split(region2,'_')[0]+'_'+str(int(string.split(region2,'_')[-1])+1)
ji.setExon1Stop(ji.Exon1Stop()-1); ji.setExon2Start(ji.Exon2Start()+1)
if override_annotation != None: null=[] ### It is already assigned above
elif region1 == region2: region_id = region1
elif ji.Strand() == '+': region_id = region1+';'+region2
else: region_id = region2+';'+region1 ### start and stop or genomically assigned
uid = ji.GeneID()+':'+region_id
#try: exon_region_db[ji.GeneID()].append((formatID(uid),region_id))
#except KeyError: exon_region_db[ji.GeneID()]=[(formatID(uid),region_id)]
ji.setExonRegionID(region_id)
ji.setUniqueID(uid) ### hgu133
### Export format for new exons to add to the existing critical exon database (those in exon_region_db are combined with analyzed junctions)
#exons_to_export[ji.GeneID(),region_id] = ji
else:
#print key, ji.GeneID(), ji.JunctionID(); sys.exit()
null=[] ### Occurs because two genes are overlapping
#return exons_to_export
def annotateNovelJunctions(novel_junction_db,novel_exon_db,exons_to_export):
### Reformat novel_exon_db to easily lookup exon data
novel_exon_lookup_db={}
for gene in novel_exon_db:
for ed in novel_exon_db[gene]:
novel_exon_lookup_db[gene,ed.ReadStart()]=ed
### Lookup the propper exon region ID and gene ID to format the unique ID and export coordinates
junction_region_db={}
unknown_gene_junctions={}
for key in novel_junction_db:
(chr,exon1_stop,exon2_start) = key
ji=novel_junction_db[key]
proceed = 'no'
if ji.GeneID() != None:
if ji.SpliceSitesFound() != 'both':
e1 = (ji.GeneID(),exon1_stop)
if ji.TransSplicing() == 'yes':
e2 = (ji.SecondaryGeneID(),exon2_start)
else: e2 = (ji.GeneID(),exon2_start)
if e1 in novel_exon_lookup_db and e2 in novel_exon_lookup_db:
proceed = 'yes'
try: ed1 = novel_exon_lookup_db[e1]; red1 = ed1.ExonRegionData(); gene1 = e1[0]
except Exception:
print chr, key, e1; kill
ed2 = novel_exon_lookup_db[e2]; red2 = ed2.ExonRegionData(); gene2 = e2[0]
### If the splice-site was a match to a known junciton splice site, use it instead of that identified by exon-region location overlapp
if ji.LeftExonAnnotations() != None: region1 = ji.LeftExonAnnotations()
else: region1 = ed1.ExonRegionID(); exons_to_export[gene1,region1] = ed1
if ji.RightExonAnnotations() != None: region2 = ji.RightExonAnnotations()
else: region2 = ed2.ExonRegionID(); exons_to_export[gene2,region2] = ed2
#print region1,region2,ji.GeneID(),ji.Chr(),ji.Strand(), ji.LeftExonAnnotations(), ji.RightExonAnnotations()
else:
proceed = 'yes'
region1 = ji.LeftExonAnnotations()
region2 = ji.RightExonAnnotations()
red1 = ji.LeftExonRegionData()
red2 = ji.RightExonRegionData()
### Store the individual exons for export
gene1 = ji.GeneID()
if ji.TransSplicing() == 'yes': gene2 = ji.SecondaryGeneID()
else: gene2 = ji.GeneID()
exons_to_export[gene1,region1] = red1
exons_to_export[gene2,region2] = red2
if proceed == 'yes':
try: splice_junctions = EnsemblImport.combineAnnotations([red1.AssociatedSplicingJunctions(),red2.AssociatedSplicingJunctions()])
except Exception: print red1, red2;sys.exit()
splice_events = EnsemblImport.combineAnnotations([red1.AssociatedSplicingEvent(),red2.AssociatedSplicingEvent()])
ji.setAssociatedSplicingJunctions(splice_junctions)
ji.setAssociatedSplicingEvent(splice_events)
ens_exon_ids = EnsemblImport.combineAnnotations([red1.ExonID(),red2.ExonID()])
ji.setExonID(ens_exon_ids)
if ji.TransSplicing() == 'yes':
uid = ji.GeneID()+':'+region1+'-'+ji.SecondaryGeneID()+':'+region2
region_id = uid
### When trans-splicing occurs, add the data twice to junction_region_db for the two different genes
### in JunctionArray.inferJunctionComps, establish two separate gene junctions with a unique ID for the non-gene exon
try: junction_region_db[ji.GeneID()].append((formatID(uid),region1+'-'+'U1000.1_'+str(ji.Exon2Start())))
except KeyError: junction_region_db[ji.GeneID()]=[(formatID(uid),region1+'-'+'U1000.1_'+str(ji.Exon2Start()))]
try: junction_region_db[ji.SecondaryGeneID()].append((formatID(uid),'U0.1_'+str(ji.Exon1Stop())+'-'+region2))
except KeyError: junction_region_db[ji.SecondaryGeneID()]=[(formatID(uid),'U0.1_'+str(ji.Exon1Stop())+'-'+region2)]
else:
uid = ji.GeneID()+':'+region1+'-'+region2
region_id = region1+'-'+region2
try: junction_region_db[ji.GeneID()].append((formatID(uid),region_id))
except KeyError: junction_region_db[ji.GeneID()]=[(formatID(uid),region_id)]
ji.setExonRegionID(region_id)
ji.setUniqueID(uid)
else:
unknown_gene_junctions[key]=[]
return junction_region_db,exons_to_export
def alignReadsToExons(novel_exon_db,ens_exon_db,testImport=False):
### Simple method for aligning a single coordinate to an exon/intron region of an already matched gene
examined_exons=0; aligned_exons=0
for gene in ens_exon_db: #novel_exon_db
try:
region_numbers=[]; region_starts=[]; region_stops=[]
for ed in novel_exon_db[gene]:
examined_exons+=1; aligned_status=0; index=-1
for rd in ens_exon_db[gene]:
index+=1 ### keep track of exon/intron we are in
region_numbers.append(int(string.split(rd.ExonRegionIDs()[1:],'.')[0]))
if rd.Strand() == '-': region_starts.append(rd.ExonStop()); region_stops.append(rd.ExonStart())
else: region_starts.append(rd.ExonStart()); region_stops.append(rd.ExonStop())
#print [rd.ExonStart(),rd.ExonStop(), rd.Strand()]
#print [ed.ReadStart(),rd.ExonStart(),rd.ExonStop()]
if ed.ReadStart()>=rd.ExonStart() and ed.ReadStart()<=rd.ExonStop():
ed.setAlignmentRegion('exon')
if 'I' in rd.ExonRegionIDs(): ### In an annotated intron
ed.setAlignmentRegion('intron')
ord = rd; updated = None
try: ### If the splice site is a novel 3' splice site then annotate as the 3' exon (less than 50nt away)
nrd = ens_exon_db[gene][index+1]
if (abs(ed.ReadStart()-nrd.ExonStart())<3) or (abs(ed.ReadStart()-nrd.ExonStop())<3):
ed.setAlignmentRegion('full-intron') ### this is the start/end of intron coordinates
elif (abs(ed.ReadStart()-nrd.ExonStart())<50) or (abs(ed.ReadStart()-nrd.ExonStop())<50): rd = nrd; updated = 1
except Exception: null=[]
try:
prd = ens_exon_db[gene][index-1]
if (abs(ed.ReadStart()-prd.ExonStart())<3) or (abs(ed.ReadStart()-prd.ExonStop())<3):
ed.setAlignmentRegion('full-intron')### this is the start/end of intron coordinates
elif (abs(ed.ReadStart()-prd.ExonStart())<50) or (abs(ed.ReadStart()-prd.ExonStop())<50):
if updated==1: rd = ord; ###Hence the intron is too small to descriminate between alt5' and alt3' exons
else: rd = prd
except Exception: null=[]
ed.setExonRegionData(rd); aligned_exons+=1; aligned_status=1
if rd.ExonStop()==ed.ReadStart():
ed.setExonRegionID(rd.ExonRegionIDs())
elif rd.ExonStart()==ed.ReadStart():
ed.setExonRegionID(rd.ExonRegionIDs())
elif 'exon-intron' in ed.Annotation(): ### intron retention
ed.setExonRegionID(rd.ExonRegionIDs()) ### Hence there is a 1nt difference between read
else:
ed.setExonRegionID(rd.ExonRegionIDs()+'_'+str(ed.ReadStart()))
break
if aligned_status == 0: ### non-exon/intron alinging sequences
region_numbers.sort(); region_starts.sort(); region_stops.sort()
if (rd.Strand() == '+' and ed.ReadStart()>=rd.ExonStop()) or (rd.Strand() == '-' and rd.ExonStop()>=ed.ReadStart()):
### Applicable to 3'UTR (or other trans-splicing) aligning
utr_id = 'U'+str(region_numbers[-1])+'.1_'+str(ed.ReadStart())
ud = EnsemblImport.ExonAnnotationsSimple(rd.Chr(),rd.Strand(),region_stops[-1],region_stops[-1],gene,'','no',utr_id,'','')
ed.setExonRegionID(utr_id)
else:
### Applicable to 5'UTR (or other trans-splicing) aligning
utr_id = 'U0.1'+'_'+str(ed.ReadStart())
ud = EnsemblImport.ExonAnnotationsSimple(rd.Chr(),rd.Strand(),region_starts[0],region_starts[0],gene,'','no',utr_id,'','')
ed.setExonRegionID(utr_id)
ed.setExonRegionData(ud)
ed.setAlignmentRegion('UTR')
except Exception: null=[]
if testImport == 'yes': print aligned_exons, 'splice sites aligned to exon region out of', examined_exons
def geneAlign(chr,chr_gene_locations,location_gene_db,chr_reads,switch_coord,read_aligned_to_gene):
""" This function aligns the start or end position for each feature (junction or exon) to a gene, in two
steps by calling this function twice. In the second interation, the coordinates are reversed """
index = 0 ### Don't examine genes already looked at
genes_assigned = 0; trans_splicing=[]
for (coord,ji) in chr_reads: ### junction coordinates or exon coordinates with gene object
if index >5: index -=5 ### It is possible for some genes to overlap, so set back the index of genomically ranked genes each time
gene_id_obtained = 'no'
if switch_coord == 'no': rs,re=coord ### reverse the coordinates for the second iteration
else: re,rs=coord ### first-interation coordinates (start and end)
while index < len(chr_gene_locations):
cs,ce = chr_gene_locations[index]
#print [re,rs,cs,ce, ji.Chromosome()];sys.exit()
### Determine if the first listed coordinate lies within the gene
if cs <= rs and ce >= rs:
### Yes, it does
gene,strand = location_gene_db[chr,cs,ce]
if switch_coord == 'yes': ### Only applies to coordinates, where the end-position didn't lie in the same gene as the start-position
if cs <= re and ce >= re:
### This occurs when the first iteration detects a partial overlap, but the gene containing both coordinates is downstream
### Hence, not trans-splicing
ji.setGeneID(gene)
break
first_geneid = ji.GeneID() ### see what gene was assigned in the first iteration (start position only)
#print ['trans',coord, first_geneid, gene] ### Note: in rare cases, an exon can overlap with two genes (bad Ensembl annotations?)
ji.setTransSplicing()
side = ji.checkExonPosition(rs)
if side == 'left':
ji.setGeneID(gene)
ji.setSecondaryGeneID(first_geneid)
else:
ji.setSecondaryGeneID(gene)
#if ji.GeneID() == None: print 'B',coord, ji.GeneID(), secondaryGeneID()
#print ji.GeneID(), ji.SecondaryGeneID();kill
genes_assigned+=1; gene_id_obtained = 'yes'
### Check to see if this gene represents a multi-gene spanning region (overlaps with multiple gene loci)
try:
### This code was used to check and see if the gene is multi-spanning. Appears that the < sign is wrong > anyways, never go to the next gene unless the next read has passed it
#cs2,ce2 = chr_gene_locations[index+1]
#if cs2 < ce: index+=1 ### Continue analysis (if above is correct, the gene will have already been assigned)
#else: break
break
except Exception: break
else:
### First iteration, store the identified gene ID (only looking at the start position)
ji.setGeneID(gene); gene_id_obtained = 'yes'
#print gene, rs, re, cs, ce
### Check the end position, to ensure it is also lies within the gene region
if cs <= re and ce >= re:
genes_assigned+=1
else:
### Hence, the end lies outside the gene region
trans_splicing.append((coord,ji))
### Check to see if this gene represents a multi-gene spanning region (overlaps with multiple gene loci)
try:
### This code was used to check and see if the gene is multi-spanning. Appears that the < sign is wrong > anyways, never go to the next gene unless the next read has passed it
#cs2,ce2 = chr_gene_locations[index+1]
#if cs2 < ce: index+=1 ### Continue analysis (if above is correct, the gene will have already been assigned)
#else: break
break
except Exception: break
else:
if rs < ce and re < ce: break
elif switch_coord == 'no' and cs <= re and ce >= re:
### This can occur if the left junction splice site is in an exon and the other is the UTR as opposed to another gene
gene,strand = location_gene_db[chr,cs,ce]
ji.setSecondaryGeneID(gene); gene_id_obtained = 'yes'
#print gene, coord, ji.Strand(), ji.GeneID()
index+=1
if gene_id_obtained == 'no':
### These often appear to be genes predicted by tBLASTn at UCSC but not by Ensembl (e.g., chr17:27,089,652-27,092,318 mouse mm9)
null=[]
#ji.setGeneID(None) ### This is not necessary, since if one exon does not align to a gene it is still a valid alignment
#print chr,coord
read_aligned_to_gene += genes_assigned
#print genes_assigned, chr, 'Gene IDs assigned out of', len(chr_reads)
#print len(trans_splicing),'reads with evidence of trans-splicing'
### For any coordinate-pair where the end-position doesn't lie within the same gene as the start, re-run for those to see which gene they are in
if switch_coord == 'no' and len(trans_splicing)>0:
read_aligned_to_gene = geneAlign(chr,chr_gene_locations,location_gene_db,trans_splicing,'yes',read_aligned_to_gene)
return read_aligned_to_gene
def getNovelExonCoordinates(species,root_dir):
""" Currently, any novel exon determined during initial RNA-Seq read annotation with defined start and end coordinates, only has
the exon-end coordinate, not start, in it's name. However, the start and stop are indicated in the counts.Experiment.txt file.
To get this, we parse that file and only store exons with an I or U in them and then correct for this in the matching function below """
exp_dir = root_dir+'/ExpressionInput/'
dir_list = read_directory(exp_dir)
counts_file = None
for file in dir_list:
if 'counts.' in file and 'steady' not in file:
counts_file = file
### Example
#ENSG00000137076:I17.1_35718353=chr9:35718353-35718403 (novel exon coordinates - just sorted, not necessarily in the correct order)
#ENSG00000137076:E17.1-I17.1_35718403=chr9:35718809-35718403 (5' supporting junction)
#ENSG00000137076:I17.1_35718353-E18.1=chr9:35718353-35717783 (3' supporting junction)
#here, once we see that I17.1_35718353 is the exon ID, we know we need to get the function with -I17.1_35718403 (always the second value)
if counts_file!=None:
fn=filepath(exp_dir+counts_file)
print 'Reading counts file'
novel_exon_db = parseCountFile(fn,'exons',{}) ### Get novel exons
print 'Reading counts file'
novel_exon_db = parseCountFile(fn,'junctions',novel_exon_db) ### Get novel exons
return novel_exon_db
def getMaxCounts(fn,cutoff,filterExport=False,filterExportDir=False):
firstLine=True
expressed_uids={}
if filterExport != False:
eo=export.ExportFile(filterExportDir)
for line in open(fn,'rU').xreadlines():
Line = cleanUpLine(line)
t = string.split(Line,'\t')
key = t[0]
if firstLine:
firstLine = False
if filterExport != False:
eo.write(line)
else:
if filterExport != False:
if key in filterExport:
eo.write(line)
else:
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
try: maxExp = max(map(lambda x: float(x), t[1:])); #print maxExp;sys.exit()
except Exception:
#print t[1:];sys.exit()
if 'NA' in t[1:]:
tn = [0 if x=='NA' else x for x in t[1:]] ### Replace NAs
maxExp = max(map(lambda x: float(x), tn))
elif '' in t[1:]:
tn = [0 if x=='' else x for x in t[1:]] ### Replace blanks
maxExp = max(map(lambda x: float(x), tn))
else:
maxExp=cutoff+1
#gene = string.split(uid,':')[0]
if maxExp > cutoff:
expressed_uids[uid] = []
return expressed_uids
def importBiologicalRelationships(species):
### Combine non-coding Ensembl gene annotations with UniProt functional annotations
import ExpressionBuilder
custom_annotation_dbase={}
try: coding_db = ExpressionBuilder.importTranscriptBiotypeAnnotations(species)
except Exception: coding_db = {}
try: gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
except Exception: gene_to_symbol_db = {}
for gene in coding_db:
#coding_type = string.split(coding_db[gene][-1],'|')
coding_type = coding_db[gene][-1]
if 'protein_coding' in coding_type:
coding_type = 'protein_coding'
else:
coding_type = 'ncRNA'
if gene in gene_to_symbol_db:
symbol = string.lower(gene_to_symbol_db[gene][0])
### The below genes cause issues with many single cell datasets in terms of being highly correlated
if 'rpl'==symbol[:3] or 'rps'==symbol[:3] or 'mt-'==symbol[:3] or '.' in symbol or 'gm'==symbol[:2]:
coding_type = 'ncRNA'
try: gene_db = custom_annotation_dbase[coding_type]; gene_db[gene]=[]
except Exception: custom_annotation_dbase[coding_type] = {gene:[]}
filename = 'AltDatabase/uniprot/'+species+'/custom_annotations.txt'
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
ens_gene,compartment,custom_class = t[:3]
if 'GPCR' in custom_class:
custom_class = ['GPCR']
else:
custom_class = string.split(custom_class,'|')
custom_class = string.split(compartment,'|')+custom_class
for cc in custom_class:
try: gene_db = custom_annotation_dbase[cc]; gene_db[ens_gene]=[]
except Exception: custom_annotation_dbase[cc] = {ens_gene:[]}
#custom_annotation_dbase={}
try:
filename = 'AltDatabase/goelite/'+species+'/gene-mapp/Ensembl-BioMarkers.txt'
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
gene,null,celltype = t[:3]
try: gene_db = custom_annotation_dbase['BioMarker']; gene_db[gene]=[]
except Exception: custom_annotation_dbase['BioMarker'] = {gene:[]}
print len(custom_annotation_dbase), 'gene classes imported'
except Exception: pass
return custom_annotation_dbase
def importGeneSets(geneSetType,filterType=None,geneAnnotations=None):
gene_db={}
if 'Ontology' in geneSetType:
filename = 'AltDatabase/goelite/'+species+'/nested/Ensembl_to_Nested-GO.txt'
ontology=True
else:
filename = 'AltDatabase/goelite/'+species+'/gene-mapp/Ensembl-'+geneSetType+'.txt'
ontology=False
fn=filepath(filename)
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if ontology:
gene,category = t
else: gene,null,category = t[:3]
if filterType==None:
try: gene_db[gene].append(category)
except Exception: gene_db[gene] = [category]
elif filterType in category:
if gene in geneAnnotations:
gene = geneAnnotations[gene][0]
gene_db[gene]=[]
return gene_db
def singleCellRNASeqWorkflow(Species, platform, expFile, mlp, exp_threshold=5, rpkm_threshold=5, drivers=False, parameters = None, reportOnly=False):
global species
global rho_cutoff
species = Species
removeOutliers = False
if parameters != None:
rpkm_threshold = parameters.ExpressionCutoff()
exp_threshold = parameters.CountsCutoff()
rho_cutoff = parameters.RhoCutoff()
restrictBy = parameters.RestrictBy()
try: removeOutliers = parameters.RemoveOutliers()
except Exception: pass
if platform == 'exons' or platform == 'PSI':
rpkm_threshold=0
exp_threshold=0
else:
rho_cutoff = 0.4
restrictBy = 'protein_coding'
onlyIncludeDrivers=True
if platform != 'exons' and platform != 'PSI':
platform = checkExpressionFileFormat(expFile,platform)
if platform != 'RNASeq':
if rpkm_threshold>1.9999:
rpkm_threshold = math.log(rpkm_threshold,2) ### log2 transform
if removeOutliers:
### Remove samples with low relative number of genes expressed
try:
import shutil
print '***Removing outlier samples***'
from import_scripts import sampleIndexSelection
reload(sampleIndexSelection)
output_file = expFile[:-4]+'-OutliersRemoved.txt'
sampleIndexSelection.statisticallyFilterFile(expFile,output_file,rpkm_threshold)
if 'exp.' in expFile:
### move the original groups and comps files
groups_file = string.replace(expFile,'exp.','groups.')
groups_file = string.replace(groups_file,'-steady-state','')
groups_filtered_file = groups_file[:-4]+'-OutliersRemoved.txt'
#comps_file = string.replace(groups_file,'groups.','comps.')
#comps_filtered_file = string.replace(groups_filtered_file,'groups.','comps.')
#counts_file = string.replace(expFile,'exp.','counts.')
#counts_filtered_file = string.replace(output_file,'exp.','counts.')
try: shutil.copyfile(groups_file,groups_filtered_file) ### if present copy over
except Exception: pass
try: shutil.copyfile(comps_file,comps_filtered_file) ### if present copy over
except Exception: pass
#try: shutil.copyfile(counts_file,counts_filtered_file) ### if present copy over
#except Exception: pass
expFile = output_file
print ''
except Exception:
print '***Filtering FAILED***'
print traceback.format_exc()
expressed_uids_rpkm = getMaxCounts(expFile,rpkm_threshold)
try: expressed_uids_counts = getMaxCounts(string.replace(expFile,'exp.','counts.'),exp_threshold)
except Exception: expressed_uids_counts=expressed_uids_rpkm
if len(expressed_uids_counts) > 0:
try: expressed_uids = expressed_uids_rpkm.viewkeys() & expressed_uids_counts.viewkeys() ### common
except Exception: expressed_uids = getOverlappingKeys(expressed_uids_rpkm,expressed_uids_counts)
else:
expressed_uids = expressed_uids_rpkm
print 'Genes filtered by counts:',len(expressed_uids_counts)
print 'Genes filtered by expression:',len(expressed_uids_rpkm),len(expressed_uids)
#expressed_uids = filterByProteinAnnotation(species,expressed_uids)
print len(expressed_uids), 'expressed genes by RPKM/TPM (%d) and counts (%d)' % (rpkm_threshold,exp_threshold)
#"""
from import_scripts import OBO_import; import ExpressionBuilder
gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
try: biological_categories = importBiologicalRelationships(species)
except Exception:
restrictBy = None
biological_categories={}
print 'Missing annotation file in:','AltDatabase/uniprot/'+species+'/custom_annotations.txt !!!!!'
if restrictBy !=None:
print 'Attempting to restrict analysis to protein coding genes only (flag --RestrictBy protein_coding)'
genes = biological_categories['protein_coding']
genes_temp=dict(genes)
for gene in genes_temp:
if gene in gene_to_symbol_db:
genes[gene_to_symbol_db[gene][0]]=[] ### add symbols
genes_temp={}
else:
genes = {}
for i in expressed_uids: genes[i]=[]
"""
genes.update(biological_categories['BioMarker'])
genes.update(biological_categories['transcription regulator'])
genes.update(biological_categories['splicing regulator'])
genes.update(biological_categories['kinase'])
genes.update(biological_categories['GPCR'])
"""
expressed_uids_db={}; guide_genes={}
for id in expressed_uids: expressed_uids_db[id]=[]
if platform == 'exons' or platform == 'PSI': ### For splicing-index value filtering
expressed_uids=[]
for uid in expressed_uids_db:
geneID = string.split(uid,':')[0]
geneID = string.split(geneID,' ')[-1]
if geneID in genes: expressed_uids.append(uid)
else:
try: expressed_uids = genes.viewkeys() & expressed_uids_db.viewkeys() ### common
except Exception: expressed_uids = getOverlappingKeys(genes,expressed_uids_db)
#print len(expressed_uids)
expressed_uids_db2={}
for id in expressed_uids: expressed_uids_db2[id]=[]
if drivers != False:
guide_genes = getDrivers(drivers)
if onlyIncludeDrivers:
try: expressed_uids = guide_genes.viewkeys() & expressed_uids_db2.viewkeys() ### common
except Exception: expressed_uids = getOverlappingKeys(guide_genes,expressed_uids_db2)
if len(expressed_uids)<10:
print 'NOTE: The input IDs do not sufficiently map to annotated protein coding genes...',
print 'skipping protein coding annotation filtering.'
expressed_uids=[]
for uid in expressed_uids_db:
expressed_uids.append(uid)
print len(expressed_uids), 'expressed IDs being further analyzed'
#sys.exit()
print_out = findCommonExpressionProfiles(expFile,species,platform,expressed_uids,guide_genes,mlp,parameters=parameters,reportOnly=reportOnly)
return print_out
def getOverlappingKeys(db1,db2):
db3=[]
for key in db1:
if key in db2:
db3.append(key)
return db3
def getDrivers(filename):
fn = filepath(filename)
firstLine=True
drivers={}
for line in open(fn,'rU').xreadlines():
line = line.rstrip()
t = string.split(line,'\t')
if firstLine: firstLine = False
else:
gene = t[0]
drivers[gene]=[]
print 'Imported %d guide genes' % len(drivers)
return drivers
def filterByProteinAnnotation(species,expressed_uids):
import ExpressionBuilder
custom_annotation_dbase = ExpressionBuilder.importTranscriptBiotypeAnnotations(species)
expressed_uids_protein=[]
for gene in expressed_uids:
if gene in custom_annotation_dbase:
compartment,custom_class = custom_annotation_dbase[gene]
if 'protein_coding' in custom_class:
expressed_uids_protein.append(gene)
if len(expressed_uids_protein)>10:
return expressed_uids_protein
else:
return expressed_uids
def CoeffVar(expFile,platform,expressed_uids,fold=2,samplesDiffering=2,guideGenes=[]):
firstLine=True
expressed_values={}
expressed_values_filtered={}
cv_list=[]
for line in open(expFile,'rU').xreadlines():
key = string.split(line,'\t')[0]
t = string.split(line,'\t')
if firstLine:
headers = line
firstLine = False
else:
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
values = map(lambda x: float(x), t[1:])
#gene = string.split(uid,':')[0]
if uid in expressed_uids:
vs = list(values); vs.sort()
cv = statistics.stdev(values)/statistics.avg(values)
if samplesDiffering<1: samplesDiffering=1
if platform == 'RNASeq':
if (vs[-1*samplesDiffering]/vs[samplesDiffering])>fold: ### Ensures that atleast 4 samples are significantly different in the set
expressed_values[uid] = values
cv_list.append((cv,uid))
else:
if (vs[-1*samplesDiffering]-vs[samplesDiffering])>fold: ### Ensures that atleast 4 samples are significantly different in the set
expressed_values[uid] = values
cv_list.append((cv,uid))
if uid in guideGenes:
expressed_values[uid] = values
cv_list.append((10000,uid)) ### Very high CV
cv_list.sort()
cv_list.reverse()
x=0
for (cv,uid) in cv_list:
x+=1
"""
if uid == 'ENSMUSG00000003882':
print x, 'ilr7'
"""
for (cv,uid) in cv_list[:5000]:
expressed_values_filtered[uid] = expressed_values[uid]
return expressed_values_filtered, fold, samplesDiffering, headers
def determinePattern(vs):
max_vs = max(vs)
min_vs = min(vs)
lower_max = max_vs - (max_vs*0.01)
upper_min = abs(max_vs)*0.01
s = bisect.bisect_right(vs,upper_min) ### starting low 15% index position
e = bisect.bisect_left(vs,lower_max) ### ending upper 85% index position
#print vs
#print max_vs, min_vs
#print lower_max, upper_min
#print s, e
avg = statistics.avg(vs[s:e+1])
m = bisect.bisect_left(vs,avg)
ratio = vs[m]/vs[((e-s)/2)+s-2] ### If the ratio is close to 1, a sigmoidal or linear pattern likely exists
print ratio
#sys.exit()
return ratio
def checkExpressionFileFormat(expFile,platform):
firstLine=True
inputMax=0; inputMin=10000
expressed_values={}
for line in open(expFile,'rU').xreadlines():
key = string.split(line,'\t')[0]
t = string.split(line,'\t')
if firstLine:
headers = line
firstLine = False
else:
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
try: values = map(lambda x: float(x), t[1:])
except Exception:
values=[]
for value in t[1:]:
try: values.append(float(value))
except Exception:pass
try:
if max(values)>inputMax: inputMax = max(values)
except Exception:
pass
if inputMax>100: ### Thus, not log values
platform = 'RNASeq'
else:
platform = "3'array"
return platform
def optimizeNumberOfGenesForDiscovery(expFile,platform,expressed_uids,fold=2,samplesDiffering=2,guideGenes=[]):
firstLine=True
expressed_values={}
for line in open(expFile,'rU').xreadlines():
key = string.split(line,'\t')[0]
t = string.split(line,'\t')
if firstLine:
headers = line
firstLine = False
else:
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
try: values = map(lambda x: float(x), t[1:])
except Exception:
values = t[1:]
if 'NA' in values:
values = [0 if x=='NA' else x for x in values] ### Replace NAs
values = map(lambda x: float(x), values)
else:
values=[]
for value in t[1:]:
try: values.append(float(value))
except Exception: values.append(-9999)
values = numpy.ma.masked_values(values, -9999.)
#gene = string.split(uid,':')[0]
#if uid == 'ENSMUSG00000041515': print 'IRF8'
if uid in expressed_uids:
#slope_exp_ratio = determinePattern(vs)
#if slope_exp_ratio<2 and slope_exp_ratio>0.5:
if platform == 'RNASeq':
try: values = map(lambda x: math.log(x+1,2),values)
except Exception:
if 'NA' in values:
values = [0 if x=='NA' else x for x in values] ### Replace NAs
values = map(lambda x: math.log(x+1,2),values)
elif '' in values:
values = [0 if x=='' else x for x in values] ### Replace NAs
values = map(lambda x: math.log(x+1,2),values)
vs = list(values); vs.sort()
if (vs[-1*samplesDiffering]-vs[samplesDiffering-1])>math.log(fold,2): ### Ensures that atleast 4 samples are significantly different in the set
expressed_values[uid] = values
else:
vs = list(values); vs.sort()
if (vs[-1*samplesDiffering]-vs[samplesDiffering-1])>math.log(fold,2): ### Ensures that atleast 4 samples are significantly different in the set
expressed_values[uid] = values
if uid in guideGenes:
expressed_values[uid] = values
#if uid == 'ENSMUSG00000062825': print (vs[-1*samplesDiffering]-vs[samplesDiffering]),math.log(fold,2);sys.exit()
print len(expressed_uids),'genes examined and', len(expressed_values),'genes expressed for a fold cutoff of', fold
if len(expressed_uids)==0 or len(expressed_values)==0:
print options_result_in_no_genes
elif len(expressed_uids) < 50 and len(expressed_values)>0:
return expressed_values, fold, samplesDiffering, headers
elif len(expressed_values)>14000:
if platform == 'exons' or platform == 'PSI':
fold+=0.1
else:
fold+=1
samplesDiffering+=1
expressed_values, fold, samplesDiffering, headers = optimizeNumberOfGenesForDiscovery(expFile,platform,expressed_uids,fold=fold,samplesDiffering=samplesDiffering,guideGenes=guideGenes)
elif fold == 1.2 and samplesDiffering == 1:
return expressed_values, fold, samplesDiffering, headers
elif len(expressed_values)<50:
fold-=0.2
samplesDiffering-=1
if samplesDiffering<1: samplesDiffering = 1
if fold < 1.1: fold = 1.2
expressed_values, fold, samplesDiffering, headers = optimizeNumberOfGenesForDiscovery(expFile,platform,expressed_uids,fold=fold,samplesDiffering=samplesDiffering,guideGenes=guideGenes)
else:
return expressed_values, fold, samplesDiffering, headers
return expressed_values, fold, samplesDiffering, headers
def intraCorrelation(expressed_values,mlp):
if mlp.cpu_count() < 3:
processors = mlp.cpu_count()
else: processors = 8
pool = mlp.Pool(processes=processors)
si = (len(expressed_values)/processors)
s = si; b=0
db_ls=[]
if len(expressed_values)<10: forceError ### will si to be zero and an infanite loop
while s<len(expressed_values):
db_ls.append(dict(expressed_values.items()[b:s]))
b+=si; s+=si
db_ls.append(dict(expressed_values.items()[b:s]))
### Create an instance of MultiZscoreWorker (store the variables to save memory)
workerMulti = MultiCorrelatePatterns(expressed_values)
results = pool.map(workerMulti,db_ls)
#for i in db_ls: workerMulti(i)
pool.close(); pool.join(); pool = None
correlated_genes={}
for a in results:
for k in a: correlated_genes[k] = a[k]
return correlated_genes
def findCommonExpressionProfiles(expFile,species,platform,expressed_uids,guide_genes,mlp,fold=2,samplesDiffering=2,parameters=None,reportOnly=False):
use_CV=False
row_metric = 'correlation'; row_method = 'average'
column_metric = 'cosine'; column_method = 'hopach'
original_column_metric = column_metric
original_column_method = column_method
color_gradient = 'yellow_black_blue'; transpose = False; graphic_links=[]
if parameters != None:
try: excludeGuides = parameters.ExcludeGuides() ### Remove signatures
except Exception: excludeGuides = None
fold = parameters.FoldDiff()
samplesDiffering = parameters.SamplesDiffering()
amplifyGenes = parameters.amplifyGenes()
if 'Guide' in parameters.GeneSelection():
amplifyGenes = False ### This occurs when running ICGS with the BOTH option, in which Guide3 genes are retained - ignore these
parameters.setGeneSelection('')
parameters.setClusterGOElite('')
excludeCellCycle = parameters.ExcludeCellCycle()
from visualization_scripts import clustering
row_metric = 'correlation'; row_method = 'average'
column_metric = parameters.ColumnMetric(); column_method = parameters.ColumnMethod()
original_column_metric = column_metric
original_column_method = column_method
color_gradient = 'yellow_black_blue'; graphic_links=[]
if platform == 'exons' or platform =='PSI': color_gradient = 'yellow_black_blue'
guide_genes = parameters.JustShowTheseIDs()
cell_cycle_id_list = []
else:
amplifyGenes = False
excludeCellCycle = False
if platform != 'exons'and platform !='PSI':
platform = checkExpressionFileFormat(expFile,platform)
else:
if LegacyMode: pass
else:
fold = math.pow(2,0.5)
fold = 1.25
#"""
if use_CV:
expressed_values, fold, samplesDiffering, headers = CoeffVar(expFile,platform,expressed_uids,fold=2,samplesDiffering=2,guideGenes=guide_genes)
else:
print 'Finding an optimal number of genes based on differing thresholds to include for clustering...'
#fold=1; samplesDiffering=1
expressed_values, fold, samplesDiffering, headers = optimizeNumberOfGenesForDiscovery(expFile,platform,expressed_uids,fold=fold,samplesDiffering=samplesDiffering,guideGenes=guide_genes) #fold=2,samplesDiffering=2
print 'Evaluating',len(expressed_values),'genes, differentially expressed',fold,'fold for at least',samplesDiffering*2,'samples'
#sys.exit()
from import_scripts import OBO_import; import ExpressionBuilder
gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol_db)
areYouSure=False
if (excludeCellCycle == 'strict' or excludeCellCycle == True) and areYouSure:
cc_param = copy.deepcopy(parameters)
cc_param.setPathwaySelect('cell cycle')
cc_param.setGeneSet('GeneOntology')
cc_param.setGeneSelection('amplify')
transpose = cc_param
filtered_file = export.findParentDir(expFile)+'/amplify/'+export.findFilename(expFile)
writeFilteredFile(filtered_file,platform,headers,{},expressed_values,[])
if len(expressed_values)<1000:
row_method = 'hopach'; row_metric = 'correlation'
if column_method != 'hopach': row_method = 'average' ### needed due to PC errors
if len(headers)>7000: ### For very ultra-large datasets
column_method = 'average'
cc_graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
cell_cycle_id_list = genericRowIDImport(string.replace(cc_graphic_links[0][-1],'.png','.txt'))
expressed_values2 = {}
for id in expressed_values:
try: symbolID = gene_to_symbol_db[id][0]
except Exception: symbolID = id
if id not in cell_cycle_id_list and symbolID not in cell_cycle_id_list:
expressed_values2[id]=expressed_values[id]
print len(expressed_values)-len(expressed_values2),'cell-cycle associated genes removed for cluster discovery'
expressed_values = expressed_values2
print 'amplifyGenes:',amplifyGenes
### Write out filtered list to amplify and to filtered.YourExperiment.txt
filtered_file = export.findParentDir(expFile)+'/amplify/'+export.findFilename(expFile)
groups_file = string.replace(expFile,'exp.','groups.')
groups_filtered_file = string.replace(filtered_file,'exp.','groups.')
groups_file = string.replace(groups_file,'-steady-state','')
groups_filtered_file = string.replace(groups_filtered_file,'-steady-state','')
try: export.customFileCopy(groups_file,groups_filtered_file) ### if present copy over
except Exception: pass
writeFilteredFile(filtered_file,platform,headers,{},expressed_values,[])
filtered_file_new = string.replace(expFile,'exp.','filteredExp.')
try: export.customFileCopy(filtered_file,filtered_file_new) ### if present copy over
except Exception: pass
if reportOnly:
print_out = '%d genes, differentially expressed %d fold for at least %d samples' % (len(expressed_values), fold, samplesDiffering*2)
return print_out
if len(expressed_values)<1400 and column_method == 'hopach':
row_method = 'hopach'; row_metric = 'correlation'
else:
row_method = 'weighted'; row_metric = 'cosine'
if amplifyGenes:
transpose = parameters
try:
if len(parameters.GeneSelection())>0:
parameters.setGeneSelection(parameters.GeneSelection()+' amplify')
print 'Finding correlated genes to the input geneset(s)...'
else:
print 'Finding intra-correlated genes from the input geneset(s)...'
parameters.setGeneSelection(parameters.GeneSelection()+' IntraCorrelatedOnly amplify')
except Exception:
parameters.setGeneSelection(parameters.GeneSelection()+' IntraCorrelatedOnly amplify')
print 'Finding intra-correlated genes from the input geneset(s)...'
if column_method != 'hopach': row_method = 'average' ### needed due to PC errors
graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
#return graphic_links
from visualization_scripts import clustering
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(graphic_links[-1][-1][:-4]+'.txt')
headers = ['UID']+column_header
expressed_values2={}
for i in row_header: ### Filter the expressed values for the intra-correlated queried gene set and replace
try: expressed_values2[i]=expressed_values[i]
except Exception:
try:
e = symbol_to_gene[i][0]
expressed_values2[e]=expressed_values[e]
except Exception:
pass
expressed_values = expressed_values2
print 'Looking for common gene expression profiles for class assignment...',
begin_time = time.time()
useNumpyCorr=True
negative_rho = rho_cutoff*-1
#results_file = string.replace(expFile[:-4]+'-CORRELATED-FEATURES.txt','exp.','/SamplePrediction/')
#eo = export.ExportFile(results_file[:-4]+'-genes.txt')
if useNumpyCorr:
row_ids=[]
x = []
for id in expressed_values:
row_ids.append(id)
x.append(expressed_values[id])
#if id== 'Bcl2l11': print expressed_values[id];sys.exit()
D1 = numpy.corrcoef(x)
print 'initial correlations obtained'
i=0
correlated_genes={}
if 'exons' == platform or 'PSI' == platform:
for score_ls in D1:
proceed = True
correlated = []
geneID = row_ids[i]
refgene = string.split(geneID,':')[0]
k=0
if excludeGuides!=None:
if geneID in excludeGuides: ### skip this main event
proceed=False
continue
for v in score_ls:
if v>rho_cutoff:# or v<negative_rho:
if refgene not in row_ids[k]:
correlated.append((v,row_ids[k]))
if excludeGuides!=None:
if row_ids[k] in excludeGuides: ### skip this main event
proceed=False
break
k+=1
correlated.sort()
if LegacyMode == False:
correlated.reverse()
if proceed:
correlated = map(lambda x:x[1],correlated)
correlated_genes[geneID] = correlated
i+=1
else:
for score_ls in D1:
correlated = []
geneID = row_ids[i]
k=0; temp=[]
for v in score_ls:
if v>rho_cutoff:# or v<negative_rho:
#scores.append((v,row_ids[k]))
correlated.append((v,row_ids[k]))
#temp.append((geneID,row_ids[k],str(v)))
k+=1
correlated.sort()
if LegacyMode == False:
correlated.reverse()
correlated = map(lambda x:x[1],correlated)
if len(correlated)>0:
correlated_genes[geneID] = correlated
#for (a,b,c) in temp: eo.write(a+'\t'+b+'\t'+c+'\n')
i+=1
else:
### Find common patterns now
performAllPairwiseComparisons = True
if performAllPairwiseComparisons:
correlated_genes = intraCorrelation(expressed_values,mlp)
print len(correlated_genes), 'highly correlated genes found for downstream clustering.'
else: correlated_genes={}
atleast_10={}
if len(correlated_genes)<70: connections = 0
elif len(correlated_genes)<110: connections = 4
else: connections = 5
numb_corr=[]
for i in correlated_genes:
if len(correlated_genes[i])>connections:
numb_corr.append([len(correlated_genes[i]),i])
atleast_10[i]=correlated_genes[i] ### if atleast 10 genes apart of this pattern
x=0
for k in correlated_genes[i]:
if x<30: ### cap it at 30
try: atleast_10[k]=correlated_genes[k] ### add all correlated keys and values
except Exception: pass
x+=1
if len(atleast_10)<30:
print 'Initial correlated set too small, getting anything correlated'
for i in correlated_genes:
if len(correlated_genes[i])>0:
numb_corr.append([len(correlated_genes[i]),i])
try: atleast_10[i]=correlated_genes[i] ### if atleast 10 genes apart of this pattern
except Exception: pass
for k in correlated_genes[i]:
try: atleast_10[k]=correlated_genes[k] ### add all correlated keys and values
except Exception: pass
if len(atleast_10) == 0:
atleast_10 = expressed_values
#eo.close()
print len(atleast_10), 'genes correlated to multiple other members (initial filtering)'
### go through the list from the most linked to the least linked genes, only reported the most linked partners
removeOutlierDrivenCorrelations=True
exclude_corr=[]
numb_corr.sort(); numb_corr.reverse()
numb_corr2=[]
#print len(numb_corr)
if removeOutlierDrivenCorrelations and samplesDiffering != 1:
for key in numb_corr: ### key gene
associations,gene = key
temp_corr_matrix_db={}; rows=[]; temp_corr_matrix=[]
gene_exp_vals = list(expressed_values[gene]) ### copy the list
max_index = gene_exp_vals.index(max(gene_exp_vals))
del gene_exp_vals[max_index]
#temp_corr_matrix.append(exp_vals); rows.append(gene)
#if 'ENSG00000016082' in correlated_genes[gene] or 'ENSG00000016082' == gene: print gene_to_symbol_db[gene],associations
if gene not in exclude_corr:
#print len(correlated_genes[gene])
for k in correlated_genes[gene]:
exp_vals = list(expressed_values[k]) ### copy the list
#print exp_vals
del exp_vals[max_index]
#temp_corr_matrix.append(exp_vals); rows.append(gene)
#print exp_vals,'\n'
temp_corr_matrix_db[k]=exp_vals
temp_corr_matrix.append(exp_vals); rows.append(gene)
correlated_hits = pearsonCorrelations(gene_exp_vals,temp_corr_matrix_db)
try: avg_corr = numpyCorrelationMatrix(temp_corr_matrix,rows,gene)
except Exception: avg_corr = 0
#if gene_to_symbol_db[gene][0] == 'ISL1' or gene_to_symbol_db[gene][0] == 'CD10' or gene_to_symbol_db[gene][0] == 'POU3F2':
if len(correlated_hits)>0:
if LegacyMode:
if (float(len(correlated_hits))+1)/len(correlated_genes[gene])<0.5 or avg_corr<rho_cutoff: ### compare to the below
pass
else:
numb_corr2.append([len(correlated_hits),gene])
else:
if (float(len(correlated_hits))+1)/len(correlated_genes[gene])<0.5 or avg_corr<(rho_cutoff-0.1):
#exclude_corr.append(key)
#if gene == 'XXX': print len(correlated_hits),len(correlated_genes[gene]), avg_corr, rho_cutoff-0.1
pass
else:
numb_corr2.append([len(correlated_hits),gene])
#print (float(len(correlated_hits))+1)/len(correlated_genes[gene]), len(correlated_genes[gene]), key
numb_corr = numb_corr2
numb_corr.sort(); numb_corr.reverse()
#print len(numb_corr)
exclude_corr={}; new_filtered_set={}
limit=0
for key in numb_corr: ### key gene
associations,gene = key
#if 'ENSG00000016082' in correlated_genes[gene] or 'ENSG00000016082' == gene: print gene_to_symbol_db[gene],associations
if gene not in exclude_corr:
for k in correlated_genes[gene]:
exclude_corr[k]=[]
new_filtered_set[k]=[]
new_filtered_set[gene]=[]
limit+=1
#print key
#if limit==1: break
atleast_10 = new_filtered_set
addMultipleDrivers=True
if len(guide_genes)>0 and addMultipleDrivers: ### Artificially weight the correlated genes with known biological driverse
for gene in guide_genes:
y=1
while y<2:
if y==1:
try: atleast_10[gene]=expressed_values[gene]
except Exception: break
else:
try: atleast_10[gene+'-'+str(y)]=expressed_values[gene]
except Exception: break
expressed_values[gene+'-'+str(y)]=expressed_values[gene] ### Add this new ID to the database
#print gene+'-'+str(y)
y+=1
#atleast_10 = expressed_values
results_file = string.replace(expFile[:-4]+'-CORRELATED-FEATURES.txt','exp.','/SamplePrediction/')
writeFilteredFile(results_file,platform,headers,gene_to_symbol_db,expressed_values,atleast_10)
print len(atleast_10),'final correlated genes'
end_time = time.time()
print 'Initial clustering completed in',int(end_time-begin_time),'seconds'
results_file = string.replace(expFile[:-4]+'-CORRELATED-FEATURES.txt','exp.','/SamplePrediction/')
if len(atleast_10)<1200 and column_method == 'hopach':
row_method = 'hopach'; row_metric = 'correlation'
else:
if LegacyMode:
row_method = 'average'; row_metric = 'euclidean'
else:
row_method = 'weighted'; row_metric = 'cosine'
#print row_method, row_metric
correlateByArrayDirectly = False
if correlateByArrayDirectly:
from visualization_scripts import clustering
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(results_file)
new_column_header = map(lambda x: int(x[5:]),column_header)
matrix = [new_column_header]+matrix
matrix = zip(*matrix) ### transpose
exp_sample_db={}
for sample_data in matrix:
exp_sample_db[sample_data[0]] = sample_data[1:]
correlated_arrays = intraCorrelation(exp_sample_db,mpl)
print len(correlated_arrays), 'highly correlated arrays from gene subsets.'
mimum_corr_arrays={}
for i in correlated_arrays:
if len(correlated_arrays[i])>1:
linked_lists=correlated_arrays[i]+[i]
for k in correlated_arrays[i]:
linked_lists+=correlated_arrays[k]
linked_lists = unique.unique(linked_lists)
linked_lists.sort()
# print len(linked_lists), linked_lists
else:
try:
from visualization_scripts import clustering
if platform == 'exons': color_gradient = 'yellow_black_blue'
transpose = False
if column_method != 'hopach': row_method = 'average' ### needed due to PC errors (possibly outside of LegacyMode)
graphic_links = clustering.runHCexplicit(results_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
if len(graphic_links)==0:
graphic_links = clustering.runHCexplicit(results_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
cluster_file = string.replace(graphic_links[0][1],'.png','.txt')
except Exception: pass
#exportGroupsFromClusters(cluster_file,expFile,platform)
#"""
#filtered_file = export.findParentDir(expFile)+'/amplify/'+export.findFilename(expFile)
#graphic_links = [(1,'/Users/saljh8/Desktop/Grimes/KashishNormalization/test/ExpressionInput/SamplePrediction/DataPlots/Clustering-CombinedSingleCell_March_15_2015-CORRELATED-FEATURES-hierarchical_cosine_euclidean.txt')]
try: graphic_links,new_results_file = correlateClusteredGenes(platform,graphic_links[-1][-1][:-4]+'.txt',numSamplesClustered=samplesDiffering,excludeCellCycle=excludeCellCycle,graphics=graphic_links,ColumnMethod=column_method)
except Exception: print traceback.format_exc()
row_metric = 'correlation'; row_method = 'hopach'
#column_metric = 'cosine'
#if LegacyMode: column_method = 'hopach'
cellCycleRemove1=[]; cellCycleRemove2=[]
try:
newDriverGenes1, cellCycleRemove1 = correlateClusteredGenes(platform,graphic_links[-1][-1][:-4]+'.txt',stringency='strict',numSamplesClustered=samplesDiffering,excludeCellCycle=excludeCellCycle,ColumnMethod=column_method)
newDriverGenes1_str = 'Guide1 '+string.join(newDriverGenes1.keys(),' ')+' amplify positive'
parameters.setGeneSelection(newDriverGenes1_str) ### force correlation to these targetGenes
parameters.setGeneSet('None Selected') ### silence this
parameters.setPathwaySelect('None Selected')
if column_method != 'hopach': row_method = 'average' ### needed due to PC errors
graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, parameters, display=False, Normalize=True)
newDriverGenes2, cellCycleRemove2 = correlateClusteredGenes(platform,graphic_links[-1][-1][:-4]+'.txt',stringency='strict',numSamplesClustered=samplesDiffering,excludeCellCycle=excludeCellCycle,ColumnMethod=column_method)
newDriverGenes2_str = 'Guide2 '+string.join(newDriverGenes2.keys(),' ')+' amplify positive'
parameters.setGeneSelection(newDriverGenes2_str) ### force correlation to these targetGenes
parameters.setGeneSet('None Selected') ### silence this
parameters.setPathwaySelect('None Selected')
graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, parameters, display=False, Normalize=True)
newDriverGenes3 = unique.unique(newDriverGenes1.keys()+newDriverGenes2.keys())
cellCycleRemove=cellCycleRemove1+cellCycleRemove2 ### It is possible for a cell cycle guide-gene to be reported in both guide1 and 2, but only as cell cycle associated in one of them
newDriverGenes3_filtered=[]
for i in newDriverGenes3:
if not i in cellCycleRemove:
newDriverGenes3_filtered.append(i)
newDriverGenes3_str = 'Guide3 '+string.join(newDriverGenes3_filtered,' ')+' amplify positive'
parameters.setGeneSelection(newDriverGenes3_str)
try:
parameters.setClusterGOElite('BioMarkers')
"""
if species == 'Mm' or species == 'Hs' or species == 'Rn':
parameters.setClusterGOElite('BioMarkers')
else:
parameters.setClusterGOElite('GeneOntology')
"""
except Exception, e:
print e
graphic_links = clustering.runHCexplicit(filtered_file, graphic_links, row_method, row_metric, column_method, column_metric, color_gradient, parameters, display=False, Normalize=True)
except Exception:
print traceback.format_exc()
try: copyICGSfiles(expFile,graphic_links)
except Exception: pass
return graphic_links
def copyICGSfiles(expFile,graphic_links):
if 'ExpressionInput' in expFile:
root_dir = string.split(expFile,'ExpressionInput')[0]
else:
root_dir = string.split(expFile,'AltResults')[0]
import shutil
destination_folder = root_dir+'/ICGS'
try: os.mkdir(destination_folder)
except Exception: pass
for (order,png) in graphic_links:
file = export.findFilename(png)
txt = string.replace(file,'.png','.txt')
pdf = string.replace(file,'.png','.pdf')
dest_png = destination_folder+'/'+file
dest_txt = destination_folder+'/'+txt
dest_pdf = destination_folder+'/'+pdf
shutil.copy(png, dest_png)
shutil.copy(png[:-4]+'.txt', dest_txt)
shutil.copy(png[:-4]+'.pdf', dest_pdf)
def pearsonCorrelations(ref_gene_exp,exp_value_db):
correlated=[]
for gene in exp_value_db:
rho,p = stats.pearsonr(ref_gene_exp,exp_value_db[gene])
if rho>rho_cutoff or rho<(rho_cutoff*-1):
if rho!= 1:
correlated.append(gene)
#print len(exp_value_db),len(correlated);sys.exit()
return correlated
def numpyCorrelationMatrix(x,rows,gene):
D1 = numpy.corrcoef(x)
gene_correlations={}
i=0
scores = []
for score_ls in D1:
for v in score_ls:
scores.append(v)
return numpy.average(scores)
def numpyCorrelationMatrixCount(x,rows,cutoff=0.4,geneTypeReport=None):
### Find which genes are most correlated
D1 = numpy.corrcoef(x)
gene_correlation_counts={}
i=0
for score_ls in D1:
correlated_genes=[]
geneID = rows[i]
k=0; genes_to_report=[]
for rho in score_ls:
if rho>cutoff:
correlated_genes.append(rows[k])
if rows[k] in geneTypeReport:
genes_to_report.append(rows[k])
k+=1
gene_correlation_counts[geneID]=len(correlated_genes),genes_to_report
i+=1
return gene_correlation_counts
def numpyCorrelationMatrixGene(x,rows,gene):
D1 = numpy.corrcoef(x)
gene_correlations={}
i=0
for score_ls in D1:
scores = []
geneID = rows[i]
k=0
for v in score_ls:
scores.append((v,rows[k]))
k+=1
scores.sort()
gene_correlations[geneID] = scores
i+=1
correlated_genes={}
rho_values = map(lambda (r,g): r,gene_correlations[gene])
genes = map(lambda (r,g): g,gene_correlations[gene])
s1 = bisect.bisect_right(rho_values,rho_cutoff)
s2 = bisect.bisect_left(rho_values,-1*rho_cutoff)
correlated = genes[:s2] ### for the right bisect, remove self correlations with -1
correlated = genes[s1:] ### for the left bisect, remove self correlations with -1
#print len(rows), len(correlated);sys.exit()
return len(correlated)/len(rows)
def numpyCorrelationMatrixGeneAlt(x,rows,genes,gene_to_symbol,rho_cutoff):
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
D1 = numpy.ma.corrcoef(x)
i=0
gene_correlations={}
for score_ls in D1:
scores = []
try: symbol = gene_to_symbol[rows[i]][0]
except Exception: symbol = '$'
if rows[i] in genes or symbol in genes:
k=0
for v in score_ls:
if str(v)!='nan':
if v > rho_cutoff:
uid = rows[k]
if uid in gene_to_symbol: uid = gene_to_symbol[uid][0]
scores.append((v,uid))
k+=1
scores.sort()
scores.reverse()
scores = map(lambda x: x[1], scores[:140]) ### grab the top 140 correlated gene symbols only
if len(symbol)==1: symbol = rows[i]
gene_correlations[symbol] = scores
i+=1
return gene_correlations
def genericRowIDImport(filename):
id_list=[]
for line in open(filename,'rU').xreadlines():
uid = string.split(line,'\t')[0]
if ' ' in uid:
for id in string.split(uid,' '):
id_list.append(id)
else:
id_list.append(uid)
return id_list
def writeFilteredFile(results_file,platform,headers,gene_to_symbol_db,expressed_values,atleast_10,excludeGenes=[]):
eo = export.ExportFile(results_file)
try: headers = string.replace(headers,'row_clusters-flat','UID')
except Exception:
headers = string.join(headers,'\t')+'\n'
headers = string.replace(headers,'row_clusters-flat','UID')
eo.write(headers)
keep=[]; sort_genes=False
e=0
if len(atleast_10)==0:
atleast_10 = expressed_values
sort_genes = True
for i in atleast_10:
if i in gene_to_symbol_db:
symbol = gene_to_symbol_db[i][0]
else: symbol = i
if i not in excludeGenes and symbol not in excludeGenes:
if i not in keep:
keep.append((symbol,i))
if sort_genes:
keep.sort(); keep.reverse()
for (symbol,i) in keep:
"""
if platform == 'RNASeq':
values = map(lambda x: logTransform(x), expressed_values[i])
else:
"""
values = map(str,expressed_values[i])
eo.write(string.join([symbol]+values,'\t')+'\n')
e+=1
eo.close()
def remoteGetDriverGenes(Species,platform,results_file,numSamplesClustered=3,excludeCellCycle=False,ColumnMethod='hopach'):
global species
species = Species
guideGenes, cellCycleRemove = correlateClusteredGenes(platform,results_file,stringency='strict',excludeCellCycle=excludeCellCycle,ColumnMethod=ColumnMethod)
guideGenes = string.join(guideGenes.keys(),' ')+' amplify positive'
return guideGenes
def correlateClusteredGenes(platform,results_file,stringency='medium',numSamplesClustered=3,
excludeCellCycle=False,graphics=[],ColumnMethod='hopach',rhoCutOff=0.2, transpose=False,
includeMoreCells=False):
if numSamplesClustered<1: numSamplesClustered=1
### Get all highly variably but low complexity differences, typically one or two samples that are really different
if stringency == 'medium':
new_results_file = string.replace(results_file,'.txt','-filtered.txt')
new_results_file = string.replace(new_results_file,'.cdt','-filtered.txt')
eo = export.ExportFile(new_results_file)
medVarHighComplexity=[]; medVarLowComplexity=[]; highVarHighComplexity=[]; highVarLowComplexity=[]
if transpose==False or includeMoreCells:
medVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=3,hits_to_report=6,transpose=transpose)
medVarHighComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.1,hits_cutoff=3,hits_to_report=6,transpose=transpose) #hits_cutoff=6
highVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.5,hits_cutoff=1,hits_to_report=4,transpose=transpose)
highVarHighComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.2,hits_cutoff=1,hits_to_report=6,filter=True,numSamplesClustered=numSamplesClustered,transpose=transpose)
else:
highVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.5,hits_cutoff=1,hits_to_report=4,transpose=transpose)
#combined_results = dict(medVarLowComplexity.items() + medVarLowComplexity.items() + highVarLowComplexity.items() + highVarHighComplexity.items())
combined_results={}
for i in medVarLowComplexity: combined_results[i]=[]
for i in medVarHighComplexity: combined_results[i]=[]
for i in highVarLowComplexity: combined_results[i]=[]
for i in highVarHighComplexity: combined_results[i]=[]
#combined_results = highVarHighComplexity
if stringency == 'strict':
medVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=4,hits_to_report=50,filter=True,numSamplesClustered=numSamplesClustered)
medVarHighComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.1,hits_cutoff=4,hits_to_report=50,filter=True,numSamplesClustered=numSamplesClustered) #hits_cutoff=6
highVarLowComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.5,hits_cutoff=3,hits_to_report=50,filter=True,numSamplesClustered=numSamplesClustered)
highVarHighComplexity, column_header = correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=3,hits_to_report=50,filter=True,numSamplesClustered=numSamplesClustered)
#combined_results = dict(medVarLowComplexity.items() + medVarLowComplexity.items() + highVarLowComplexity.items() + highVarHighComplexity.items())
combined_results={}
for i in medVarLowComplexity: combined_results[i]=[]
for i in medVarHighComplexity: combined_results[i]=[]
for i in highVarLowComplexity: combined_results[i]=[]
for i in highVarHighComplexity: combined_results[i]=[]
guideGenes, addition_cell_cycle_associated = correlateClusteredGenesParameters(results_file,rho_cutoff=rhoCutOff,hits_cutoff=0,hits_to_report=1,geneFilter=combined_results,excludeCellCycle=excludeCellCycle)
if guideGenes == 'TooFewBlocks':
guideGenes, addition_cell_cycle_associated = correlateClusteredGenesParameters(results_file,rho_cutoff=rhoCutOff+0.1,hits_cutoff=0,hits_to_report=1,geneFilter=combined_results,excludeCellCycle=excludeCellCycle)
if guideGenes == 'TooFewBlocks':
guideGenes, addition_cell_cycle_associated = correlateClusteredGenesParameters(results_file,rho_cutoff=rhoCutOff+0.2,hits_cutoff=0,hits_to_report=1,geneFilter=combined_results,excludeCellCycle=excludeCellCycle,forceOutput=True)
if len(guideGenes)>200:
print 'Too many guides selected (>200)... performing more stringent filtering...'
guideGenes, addition_cell_cycle_associated = correlateClusteredGenesParameters(results_file,rho_cutoff=0.1,hits_cutoff=0,hits_to_report=1,geneFilter=combined_results,excludeCellCycle=excludeCellCycle,restrictTFs=True)
return guideGenes, addition_cell_cycle_associated
#B4galt6, Prom1
for tuple_ls in combined_results:
data_length = len(tuple_ls);break
if data_length == len(column_header):
eo.write(string.join(column_header,'\t')+'\n')
else:
eo.write(string.join(['UID']+column_header,'\t')+'\n')
#combined_results = highVarHighComplexity
for tuple_ls in combined_results:
eo.write(string.join(list(tuple_ls),'\t')+'\n')
eo.close()
cluster = True
if cluster == True and transpose==False:
from visualization_scripts import clustering
if ColumnMethod == 'hopach':
row_method = 'hopach'
column_method = 'hopach'
else:
column_method = ColumnMethod
row_method = 'average'
row_metric = 'correlation'
column_metric = 'cosine'
color_gradient = 'yellow_black_blue'
if platform == 'exons': color_gradient = 'yellow_black_blue'
transpose = False
try:
len(guide_genes)
except Exception:
guide_genes = []
graphics = clustering.runHCexplicit(new_results_file, graphics, row_method, row_metric, column_method, column_metric, color_gradient, transpose, display=False, Normalize=True, JustShowTheseIDs=guide_genes)
cluster_file = string.replace(graphics[0][1],'.png','.txt')
#exportGroupsFromClusters(cluster_file,expFile,platform)
return graphics, new_results_file
def exportReDefinedClusterBlocks(results_file,block_db,rho_cutoff):
### Re-import the matrix to get the column cluster IDs
matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters = clustering.remoteImportData(results_file)
new_block_db = {}
centroid_blocks=[]
centroids = []
for block in block_db:
if len(block_db[block])>3:
new_block_db[block] = block_db[block] ### Keep track of the row_header indexes associated with each blcok
data = map(lambda x: matrix[x],block_db[block])
### Compute an expression centroid from the block (cluster)
centroid = [float(sum(col))/len(col) for col in zip(*data)]
centroids.append(centroid)
centroid_blocks.append(block)
### Compare block centroids
#change this code
D1 = numpy.corrcoef(centroids)
i=0
correlated_blocks=[]
for score_ls in D1:
scores = []
block = centroid_blocks[i]
k=0
for v in score_ls:
#absolute correlations
if str(v)!='nan' and v>0.5:
if block !=centroid_blocks[k]:
blocks = [block,centroid_blocks[k]]
blocks.sort()
if blocks not in correlated_blocks:
correlated_blocks.append(blocks)
k+=1
i+=1
newBlock=0
existing=[]
updated_blocks={}
correlated_blocks.sort()
#print correlated_blocks
### Build a tree of related blocks (based on the code in junctionGraph)
for (block1,block2) in correlated_blocks:
if block1 not in existing and block2 not in existing:
newBlock=newBlock+1
updated_blocks[newBlock]=[block1,]
updated_blocks[newBlock].append(block2)
existing.append(block1)
existing.append(block2)
elif block1 in existing and block2 not in existing:
for i in updated_blocks:
if block1 in updated_blocks[i]:
updated_blocks[i].append(block2)
existing.append(block2)
elif block2 in existing and block1 not in existing:
for i in updated_blocks:
if block2 in updated_blocks[i]:
updated_blocks[i].append(block1)
existing.append(block1)
elif block1 in existing and block2 in existing:
for i in updated_blocks:
if block1 in updated_blocks[i]:
b1=i
if block2 in updated_blocks[i]:
b2=i
if b1!=b2:
for b in updated_blocks[b2]:
if b not in updated_blocks[b1]:
updated_blocks[b1].append(b)
del updated_blocks[b2]
### Add blocks not correlated to other blocks (not in correlated_blocks)
#print len(existing),len(centroid_blocks)
#print updated_blocks
for block in centroid_blocks:
if block not in existing:
newBlock+=1
updated_blocks[newBlock]=[block]
import collections
row_order = collections.OrderedDict()
for newBlock in updated_blocks:
events_in_block=0
for block in updated_blocks[newBlock]:
for i in new_block_db[block]:
events_in_block+=1
if events_in_block>6:
for block in updated_blocks[newBlock]:
for i in new_block_db[block]:
row_order[i] = newBlock ### i is a row_header index - row_header[i] is a UID
#if newBlock==3:
#if row_header[i]=='TAF2&ENSG00000064313&E9.1-I9.1_120807184__ENSG00000064313&E9.1-E10.1':
#print row_header[i]
#print updated_blocks
### Non-clustered block results - Typically not used by good to refer back to when testing
original_block_order = collections.OrderedDict()
for block in new_block_db:
for i in new_block_db[block]:
original_block_order[i]=block
#row_order = original_block_order
### Export the results
row_header.reverse() ### Reverse order is the default
priorColumnClusters = map(str,priorColumnClusters)
new_results_file = results_file[:-4]+'-BlockIDs.txt'
eo = export.ExportFile(new_results_file)
eo.write(string.join(['UID','row_clusters-flat']+column_header,'\t')+'\n')
eo.write(string.join(['column_clusters-flat','']+priorColumnClusters,'\t')+'\n')
for i in row_order:
cluster_number = str(row_order[i])
uid = row_header[i]
values = map(str,matrix[i])
eo.write(string.join([uid,cluster_number]+values,'\t')+'\n')
eo.close()
print 'Filtered, grouped expression clusters exported to:',new_results_file
def correlateClusteredGenesParameters(results_file,rho_cutoff=0.3,hits_cutoff=4,hits_to_report=5,
filter=False,geneFilter=None,numSamplesClustered=3,excludeCellCycle=False,restrictTFs=False,
forceOutput=False,ReDefinedClusterBlocks=False,transpose=False):
try: from visualization_scripts import clustering; reload(clustering)
except Exception: import clustering
addition_cell_cycle_associated=[]
if geneFilter != None:
geneFilter_db={}
for i in geneFilter:
geneFilter_db[i[0]]=[]
geneFilter=geneFilter_db
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(results_file,geneFilter=geneFilter)
if transpose: ### If performing reduce cluster heterogeneity on cells rather than on genes
#print 'Transposing matrix'
matrix = map(numpy.array, zip(*matrix)) ### coverts these to tuples
column_header, row_header = row_header, column_header
Platform = None
for i in row_header:
if 'ENS' in i and '-' in i and ':' in i: Platform = 'exons'
#print hits_to_report
if hits_to_report == 1:
### Select the best gene using correlation counts and TFs
try:
from import_scripts import OBO_import; import ExpressionBuilder
gene_to_symbol_db = ExpressionBuilder.importGeneAnnotations(species)
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol_db)
try: TFs = importGeneSets('Biotypes',filterType='transcription regulator',geneAnnotations=gene_to_symbol_db)
except Exception: TFs = importGeneSets('BioTypes',filterType='transcription regulator',geneAnnotations=gene_to_symbol_db)
if excludeCellCycle == True or excludeCellCycle == 'strict':
cell_cycle = importGeneSets('KEGG',filterType='Cell cycle:',geneAnnotations=gene_to_symbol_db)
cell_cycle_go = importGeneSets('GeneOntology',filterType='GO:0022402',geneAnnotations=gene_to_symbol_db)
for i in cell_cycle_go:
cell_cycle[i]=[]
print len(cell_cycle),'cell cycle genes being considered.'
else:
cell_cycle={}
except Exception:
print traceback.format_exc()
symbol_to_gene={}; TFs={}; cell_cycle={}
gene_corr_counts = numpyCorrelationMatrixCount(matrix,row_header,cutoff=0.4,geneTypeReport=TFs)
#try: column_header = map(lambda x: string.split(x,':')[1],column_header[1:])
#except Exception: column_header = column_header[1:]
i=0
block=0
if ReDefinedClusterBlocks:
import collections
block_db=collections.OrderedDict() ### seems benign but could alter legacy results
else:
block_db={}
for row in matrix:
list1=[]
list2=[]
for iq in range(0,len(row)):
if row[iq]!=0.0 and matrix[i-1][iq]!=0.0:
list1.append(row[iq])
list2.append(matrix[i-1][iq])
if i!=0:
#need to change here
rho,p = stats.pearsonr(list1,list2) ### correlate to the last ordered row
#if row_header[i] == 'Pax6': print [block],row_header[i-1],rho,rho_cutoff
"""
try:
if row_header[i] in guide_genes: print row_header[i], rho
if row_header[i-1] in guide_genes: print row_header[i-1], rho
if row_header[i+1] in guide_genes: print row_header[i+1], rho
except Exception:
pass
"""
#if hits_to_report == 1: print [block],row_header[i], row_header[i-1],rho,rho_cutoff
#print rho
if rho>0.95:
pass ### don't store this
elif rho>rho_cutoff:
try:
block_db[block].append(i) ### store the row index
except Exception:
block_db[block] = [i] ### store the row index
else:
block+=1
block_db[block] = [i] ### store the row index
else:
block_db[block] = [i] ### store the row index
i+=1
if ReDefinedClusterBlocks:
### Produces a filtered-down and centroid organized heatmap text file
exportReDefinedClusterBlocks(results_file,block_db,rho_cutoff)
if hits_to_report == 1:
if len(block_db)<4 and forceOutput==False:
return 'TooFewBlocks', None
guideGenes={}
### Select the top TFs or non-TFs with the most gene correlations
for b in block_db:
corr_counts_gene = []; cell_cycle_count=[]
#print len(block_db), b, map(lambda i: row_header[i],block_db[b])
for (gene,i) in map(lambda i: (row_header[i],i),block_db[b]):
corr_counts_gene.append((len(gene_corr_counts[gene][1]),gene_corr_counts[gene][0],gene))
if gene in cell_cycle:
cell_cycle_count.append(gene)
corr_counts_gene.sort(); tfs=[]
#print b, corr_counts_gene, '***',len(cell_cycle_count)
if (len(cell_cycle_count)>1) or (len(corr_counts_gene)<4 and (len(cell_cycle_count)>0)): pass
else:
tf_count=0
for (r,t, gene) in corr_counts_gene:
if gene in TFs:
if gene not in cell_cycle:
if restrictTFs==True and tf_count==0: pass
else:
guideGenes[gene]=[]
tf_count+=1
if len(tfs)==0:
gene = corr_counts_gene[-1][-1]
if gene in cell_cycle and LegacyMode: pass
else:
guideGenes[gene]=[]
#block_db[b]= [corr_counts_gene[-1][-1]] ### save just the selected gene indexes
### Additional filter to remove guides that will bring in cell cycle genes (the more guides the more likely)
if excludeCellCycle == 'strict':
#print 'guides',len(guideGenes)
guideCorrelated = numpyCorrelationMatrixGeneAlt(matrix,row_header,guideGenes,gene_to_symbol_db,rho_cutoff)
guideGenes={}
for gene in guideCorrelated:
cell_cycle_count=[]
for corr_gene in guideCorrelated[gene]:
if corr_gene in cell_cycle: cell_cycle_count.append(corr_gene)
#print gene, len(cell_cycle_count),len(guideCorrelated[gene])
if (float(len(cell_cycle_count))/len(guideCorrelated[gene]))>.15 or (len(guideCorrelated[gene])<4 and (len(cell_cycle_count)>0)):
print gene, cell_cycle_count
addition_cell_cycle_associated.append(gene)
pass
else:
guideGenes[gene]=[]
print 'additional Cell Cycle guide genes removed:',addition_cell_cycle_associated
print len(guideGenes), 'novel guide genes discovered:', guideGenes.keys()
return guideGenes,addition_cell_cycle_associated
def greaterThan(x,results_file,numSamplesClustered):
if 'alt_junctions' not in results_file and Platform == None:
if x>(numSamplesClustered-1): return 1
else: return 0
else:
return 1
max_block_size=0
### Sometimes the hits_cutoff is too stringent so take the largest size instead
for block in block_db:
indexes = len(block_db[block])
if indexes>max_block_size: max_block_size=indexes
max_block_size-=1
retained_ids={}; final_rows = {}
for block in block_db:
indexes = block_db[block]
#print [block], len(indexes),hits_cutoff,max_block_size
if len(indexes)>hits_cutoff or len(indexes)>max_block_size: ###Increasing this helps get rid of homogenous clusters of little significance
#if statistics.avg(matrix[indexes[0]][1:]) < -2: print statistics.avg(matrix[indexes[0]][1:]), len(indexes)
gene_names = map(lambda i: row_header[i], indexes)
#if 'Pax6' in gene_names or 'WNT8A' in gene_names: print '******',hits_to_report, gene_names
indexes = indexes[:hits_to_report]
if filter:
new_indexes = []
for index in indexes:
vs = list(matrix[index])
a = map(lambda x: greaterThan(x,results_file,numSamplesClustered),vs)
b=[1]*numSamplesClustered
c = [(i, i+len(b)) for i in range(len(a)) if a[i:i+len(b)] == b]
if len(c)>0: #http://stackoverflow.com/questions/10459493/find-indexes-of-sequence-in-list-in-python
new_indexes.append(index)
"""
vs.sort()
try:
if abs(vs[-5]-vs[5])>6: new_indexes.append(index)
except Exception:
if abs(vs[-1]-vs[1])>6: new_indexes.append(index)"""
indexes = new_indexes
#if block == 1: print map(lambda i:row_header[i],indexes)
#print indexes;sys.exit()
for ls in map(lambda i: [row_header[i]]+map(str,(matrix[i])), indexes):
final_rows[tuple(ls)]=[]
for i in indexes:
retained_ids[row_header[i]]=[]
if len(final_rows)==0:
for block in block_db:
indexes = block_db[block]
if len(indexes)>hits_cutoff or len(indexes)>max_block_size:
indexes = indexes[:hits_to_report]
for ls in map(lambda i: [row_header[i]]+map(str,(matrix[i])), indexes):
final_rows[tuple(ls)]=[]
if len(final_rows)==0:
for block in block_db:
indexes = block_db[block]
for ls in map(lambda i: [row_header[i]]+map(str,(matrix[i])), indexes):
final_rows[tuple(ls)]=[]
#print 'block length:',len(block_db), 'genes retained:',len(retained_ids)
return final_rows, column_header
def exportGroupsFromClusters(cluster_file,expFile,platform,suffix=None):
lineNum=1
for line in open(cluster_file,'rU').xreadlines():
line = line[:-1]
t = string.split(line,'\t')
if lineNum==1: names = t[2:]; lineNum+=1
elif lineNum==2: clusters = t[2:]; lineNum+=1
else: break
unique_clusters=[] ### Export groups
new_groups_dir = string.replace(expFile,'exp.','groups.')
new_comps_dir = string.replace(expFile,'exp.','comps.')
if suffix != None:
new_groups_dir = new_groups_dir[:-4]+'-'+suffix+'.txt' ###Usually end in ICGS
new_comps_dir = new_comps_dir[:-4]+'-'+suffix+'.txt'
out_obj = export.ExportFile(new_groups_dir)
for name in names:
cluster = clusters[names.index(name)]
if platform == 'RNASeq':
if 'junction_quantification' not in name and '.bed' not in name:
name = name+'.bed'
elif 'junction_quantification.txt' not in name and '.txt' not in name and '.bed' not in name:
name = name+'.txt'
if ':' in name:
name = string.split(name,':')[1]
out_obj.write(name+'\t'+cluster+'\t'+cluster+'\n')
if cluster not in unique_clusters: unique_clusters.append(cluster)
out_obj.close()
comps=[] #Export comps
out_obj = export.ExportFile(new_comps_dir)
""" ### All possible pairwise group comparisons
for c1 in unique_clusters:
for c2 in unique_clusters:
temp=[int(c2),int(c1)]; temp.sort(); temp.reverse()
if c1 != c2 and temp not in comps:
out_obj.write(str(temp[0])+'\t'+str(temp[1])+'\n')
comps.append(temp)
"""
### Simple method comparing each subsequent ordered cluster (HOPACH orders based on relative similarity)
last_cluster = None
for c1 in unique_clusters:
if last_cluster !=None:
out_obj.write(c1+'\t'+last_cluster+'\n')
last_cluster=c1
out_obj.close()
return new_groups_dir
def logTransform(value):
try: v = math.log(value,2)
except Exception: v = math.log(0.001,2)
return str(v)
class MultiCorrelatePatterns():
def __init__(self,expressed_values):
self.expressed_values = expressed_values
def __call__(self,features_to_correlate):
from scipy import stats
correlated_genes={}
for uid in features_to_correlate:
ref_values = self.expressed_values[uid]
for uid2 in self.expressed_values:
values = self.expressed_values[uid2]
rho,p = stats.pearsonr(values,ref_values)
if rho>rho_cutoff or rho<-1*rho_cutoff:
if uid!=uid2 and rho != 1.0:
try: correlated_genes[uid].append(uid2)
except Exception: correlated_genes[uid] = [uid]
return correlated_genes
def parseCountFile(fn,parseFeature,search_exon_db):
novel_exon_db={}; firstLine=True
unique_genes={}
for line in open(fn,'rU').xreadlines():
key = string.split(line,'\t')[0]
#t = string.split(line,'\t')
if firstLine: firstLine = False
else:
#uid, coordinates = string.split(key,'=')
#values = map(lambda x: float(x), t[1:])
#gene = string.split(uid,':')[0]
#if max(values)>5: unique_genes[gene] = []
if '_' in key: ### Only look at novel exons
#ENSG00000112695:I2.1_75953139=chr6:75953139-75953254
uid, coordinates = string.split(key,'=')
gene = string.split(uid,':')[0]
if parseFeature == 'exons':
if '-' not in uid:
chr,coordinates = string.split(coordinates,':') ### Exclude the chromosome
coord1,coord2 = string.split(coordinates,'-')
intron = string.split(uid,'_')[0]
intron = string.split(intron,':')[1]
first = intron+'_'+coord1
second = intron+'_'+coord2
proceed = True
if first in uid: search_uid = second ### if the first ID is already the one looked for, store the second with the exon ID
elif second in uid: search_uid = first
else:
proceed = False
#print uid, first, second; sys.exit()
#example: ENSG00000160785:E2.15_156170151;E2.16_156170178=chr1:156170151-156170178
if proceed:
try: novel_exon_db[gene].append((uid,search_uid))
except Exception: novel_exon_db[gene] = [(uid,search_uid)]
elif '-' in uid and 'I' in uid: ### get junctions
if gene in search_exon_db:
for (u,search_uid) in search_exon_db[gene]:
#if gene == 'ENSG00000137076': print u,search_uid,uid
if search_uid in uid:
novel_exon_db[uid] = u ### Relate the currently examined novel exon ID to the junction not current associated
#if gene == 'ENSG00000137076': print u, uid
#print uid;sys.exit()
#print len(unique_genes); sys.exit()
return novel_exon_db
def getJunctionType(species,fn):
root_dir = string.split(fn,'ExpressionInput')[0]
fn = filepath(root_dir+'AltDatabase/'+species+'/RNASeq/'+species + '_Ensembl_junctions.txt')
firstLine=True
junction_type_db={}; type_db={}
for line in open(fn,'rU').xreadlines():
t = string.split(line,'\t')
if firstLine: firstLine = False
else:
id=t[0]; junction_type = t[8]
if '-' in id:
if 'trans-splicing' in line:
junction_type = 'trans-splicing'
junction_type_db[id] = junction_type
try: type_db[junction_type]+=1
except Exception: type_db[junction_type]=1
print 'Breakdown of event types'
for type in type_db:
print type, type_db[type]
return junction_type_db
def maxCount(ls):
c=0
for i in ls:
if i>0.5: c+=1
return c
def getHighExpNovelExons(species,fn):
""" Idea - if the ranking of exons based on expression changes from one condition to another, alternative splicing is occuring """
junction_type_db = getJunctionType(species,fn)
### Possible issue detected with novel exon reads: ['ENSG00000121577'] ['119364543'] cardiac
exon_max_exp_db={}; uid_key_db={}; firstLine=True
novel_intronic_junctions = {}
novel_intronic_exons = {}
cutoff = 0.2
read_threshold = 0.5
expressed_junction_types={}
features_to_export={}
exon_coord_db={}
for line in open(fn,'rU').xreadlines():
t = string.split(line,'\t')
if firstLine: firstLine = False
else:
key=t[0]
#ENSG00000112695:I2.1_75953139=chr6:75953139-75953254
try: uid, coordinates = string.split(key,'=')
except Exception: uid = key
gene = string.split(uid,':')[0]
values = map(lambda x: float(x), t[1:])
max_read_counts = max(values)
try: exon_max_exp_db[gene].append((max_read_counts,uid))
except Exception: exon_max_exp_db[gene] = [(max_read_counts,uid)]
uid_key_db[uid] = key ### retain the coordinate info
if '-' in uid and (':E' in uid or '-E' in uid):
junction_type = junction_type_db[uid]
if max_read_counts>read_threshold:
samples_expressed = maxCount(values)
if samples_expressed>2:
try: expressed_junction_types[junction_type]+=1
except Exception: expressed_junction_types[junction_type]=1
if junction_type == 'trans-splicing' and'_' not in uid:
try: expressed_junction_types['known transplicing']+=1
except Exception: expressed_junction_types['known transplicing']=1
elif junction_type == 'novel' and '_' not in uid:
try: expressed_junction_types['novel but known sites']+=1
except Exception: expressed_junction_types['novel but known sites']=1
elif junction_type == 'novel' and 'I' not in uid:
try: expressed_junction_types['novel but within 50nt of a known sites']+=1
except Exception: expressed_junction_types['novel but within 50nt of a known sites']=1
elif 'I' in uid and '_' in uid and junction_type!='trans-splicing':
#print uid;sys.exit()
try: expressed_junction_types['novel intronic junctions']+=1
except Exception: expressed_junction_types['novel intronic junctions']=1
coord = string.split(uid,'_')[-1]
if '-' in coord:
coord = string.split(coord,'-')[0]
try: novel_intronic_junctions[gene]=[coord]
except Exception: novel_intronic_junctions[gene].append(coord)
elif ('I' in uid or 'U' in uid) and '_' in uid and max_read_counts>read_threshold:
if '-' not in uid:
samples_expressed = maxCount(values)
if samples_expressed>2:
try: expressed_junction_types['novel intronic exon']+=1
except Exception: expressed_junction_types['novel intronic exon']=1
coord = string.split(uid,'_')[-1]
#print uid, coord;sys.exit()
#if 'ENSG00000269897' in uid: print [gene,coord]
try: novel_intronic_exons[gene].append(coord)
except Exception: novel_intronic_exons[gene]=[coord]
exon_coord_db[gene,coord]=uid
print 'Expressed (count>%s for at least 3 samples) junctions' % read_threshold
for junction_type in expressed_junction_types:
print junction_type, expressed_junction_types[junction_type]
expressed_junction_types={}
#print len(novel_intronic_junctions)
#print len(novel_intronic_exons)
for gene in novel_intronic_junctions:
if gene in novel_intronic_exons:
for coord in novel_intronic_junctions[gene]:
if coord in novel_intronic_exons[gene]:
try: expressed_junction_types['confirmed novel intronic exons']+=1
except Exception: expressed_junction_types['confirmed novel intronic exons']=1
uid = exon_coord_db[gene,coord]
features_to_export[uid]=[]
#else: print [gene], novel_intronic_junctions[gene]; sys.exit()
for junction_type in expressed_junction_types:
print junction_type, expressed_junction_types[junction_type]
out_file = string.replace(fn,'.txt','-highExp.txt')
print 'Exporting the highest expressed exons to:', out_file
out_obj = export.ExportFile(out_file)
### Compare the relative expression of junctions and exons separately for each gene (junctions are more comparable)
for gene in exon_max_exp_db:
junction_set=[]; exon_set=[]; junction_exp=[]; exon_exp=[]
exon_max_exp_db[gene].sort()
exon_max_exp_db[gene].reverse()
for (exp,uid) in exon_max_exp_db[gene]:
if '-' in uid: junction_set.append((exp,uid)); junction_exp.append(exp)
else: exon_set.append((exp,uid)); exon_exp.append(exp)
if len(junction_set)>0:
maxJunctionExp = junction_set[0][0]
try: lower25th,median_val,upper75th,int_qrt_range = statistics.iqr(junction_exp)
except Exception: print junction_exp;sys.exit()
if int_qrt_range>0:
maxJunctionExp = int_qrt_range
junction_percent_exp = map(lambda x: (x[1],expThreshold(x[0]/maxJunctionExp,cutoff)), junction_set)
high_exp_junctions = []
for (uid,p) in junction_percent_exp: ### ID and percentage of expression
if p!='NA':
if uid in features_to_export: ### novel exons only right now
out_obj.write(uid_key_db[uid]+'\t'+p+'\n') ### write out the original ID with coordinates
if len(exon_set)>0:
maxExonExp = exon_set[0][0]
lower25th,median_val,upper75th,int_qrt_range = statistics.iqr(exon_exp)
if int_qrt_range>0:
maxExonExp = int_qrt_range
exon_percent_exp = map(lambda x: (x[1],expThreshold(x[0]/maxExonExp,cutoff)), exon_set)
high_exp_exons = []
for (uid,p) in exon_percent_exp: ### ID and percentage of expression
if p!='NA':
if uid in features_to_export:
out_obj.write(uid_key_db[uid]+'\t'+p+'\n')
out_obj.close()
def expThreshold(ratio,cutoff):
#print [ratio,cutoff]
if ratio>cutoff: return str(ratio)
else: return 'NA'
def compareExonAndJunctionResults(species,array_type,summary_results_db,root_dir):
results_dir = root_dir +'AltResults/AlternativeOutput/'
dir_list = read_directory(results_dir)
filtered_dir_db={}
#"""
try: novel_exon_junction_db = getNovelExonCoordinates(species,root_dir)
except Exception:
#print traceback.format_exc()
print 'No counts file found.'
novel_exon_junction_db={} ### only relevant to RNA-Seq analyses
for comparison_file in summary_results_db:
for results_file in dir_list:
if (comparison_file in results_file and '-exon-inclusion-results.txt' in results_file) and ('comparison' not in results_file):
try: filtered_dir_db[comparison_file].append(results_file)
except Exception: filtered_dir_db[comparison_file] = [results_file]
try: os.remove(string.split(results_dir,'AltResults')[0]+'AltResults/Clustering/Combined-junction-exon-evidence.txt')
except Exception: pass
for comparison_file in filtered_dir_db:
alt_result_files = filtered_dir_db[comparison_file]
#print alt_result_files, comparison_file
importAltAnalyzeExonResults(alt_result_files,novel_exon_junction_db,results_dir)
#"""
### Build combined clusters of high-confidence exons
graphics2=[]; graphics=[]
import ExpressionBuilder
try:
input_dir = string.split(results_dir,'AltResults')[0]+'GO-Elite/AltExonConfirmed/'
cluster_file, rows_in_file = ExpressionBuilder.buildAltExonClusterInputs(input_dir,species,array_type,dataType='AltExonConfirmed')
if rows_in_file > 5000: useHOPACH = False
else: useHOPACH = True
if rows_in_file < 12000:
graphics = ExpressionBuilder.exportHeatmap(cluster_file,useHOPACH=useHOPACH)
except Exception: pass
try:
input_dir = string.split(results_dir,'AltResults')[0]+'GO-Elite/AltExon/'
cluster_file, rows_in_file = ExpressionBuilder.buildAltExonClusterInputs(input_dir,species,array_type,dataType='AltExon')
if rows_in_file > 5000: useHOPACH = False
else: useHOPACH = True
if rows_in_file < 12000:
graphics2 = ExpressionBuilder.exportHeatmap(cluster_file,useHOPACH=useHOPACH)
except Exception: pass
return graphics+graphics2
class SplicingData:
def __init__(self,score,symbol,description,exonid,probesets,direction,splicing_event,external_exon,genomic_loc,gene_exp,protein_annot,domain_inferred,domain_overlap,method,dataset):
self.score = score; self.dataset = dataset
self.symbol = symbol;
self.description=description;self.exonid=exonid;self.probesets=probesets;self.direction=direction
self.splicing_event=splicing_event;self.external_exon=external_exon;self.genomic_loc=genomic_loc;
self.gene_exp=gene_exp;self.protein_annot=protein_annot;self.domain_inferred=domain_inferred
self.domain_overlap=domain_overlap;self.method=method
def Score(self): return self.score
def setScore(self,score): self.score = score
def GeneExpression(self): return self.gene_exp
def Dataset(self): return self.dataset
def Symbol(self): return self.symbol
def Description(self): return self.description
def ExonID(self): return self.exonid
def appendExonID(self,exonid): self.exonid+='|'+exonid
def Probesets(self): return self.probesets
def ProbesetDisplay(self):
if len(self.Probesets()[1])>0:
return string.join(self.Probesets(),'-')
else:
return self.Probesets()[0]
def ProbesetsSorted(self):
### Don't sort the original list
a = [self.probesets[0],self.probesets[1]]
a.sort()
return a
def Direction(self): return self.direction
def setDirection(self,direction): self.direction = direction
def SplicingEvent(self): return self.splicing_event
def ProteinAnnotation(self): return self.protein_annot
def DomainInferred(self): return self.domain_inferred
def DomainOverlap(self): return self.domain_overlap
def Method(self): return self.method
def setEvidence(self,evidence): self.evidence = evidence
def Evidence(self): return self.evidence
def GenomicLocation(self): return self.genomic_loc
def setExonExpStatus(self, exon_expressed): self.exon_expressed = exon_expressed
def ExonExpStatus(self): return self.exon_expressed
def importAltAnalyzeExonResults(dir_list,novel_exon_junction_db,results_dir):
regulated_critical_exons={}; converted_db={}
includeExonJunctionComps=True ### Allow ASPIRE comparisons with the inclusion feature as an exon to count for additive reciprocal evidence
print "Reading AltAnalyze results file"
root_dir = string.split(results_dir,'AltResults')[0]
for filename in dir_list:
x=0; regulated_critical_exon_temp={}
fn=filepath(results_dir+filename)
new_filename = string.join(string.split(filename,'-')[:-5],'-')
if '_vs_' in filename and '_vs_' in new_filename: export_filename = new_filename
else: export_filename = string.join(string.split(filename,'-')[:-5],'-')
export_path = results_dir+export_filename+'-comparison-evidence.txt'
try: os.remove(filepath(export_path)) ### If we don't do this, the old results get added to the new
except Exception: null=[]
if 'AltMouse' in filename:
altmouse_ensembl_db = importAltMouseEnsembl()
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if x==0: x=1; #print t[12],t[13],t[22],t[23]
else:
converted = False ### Indicates both junction sides were regulated
geneid = t[0]; exonid = t[4]; probeset1 = t[6]; probeset2 = ''; score = t[1][:4]; symbol = t[2]; description = t[3]; regions = t[-4]; direction = t[5]
genomic_loc = t[-1]; splicing_event = t[-3]; external_exon = t[-6]; gene_exp_fold = t[-8]; protein_annot = t[14]; domain_inferred = t[15]; domain_overlap = t[17]
expressed_exon = 'NA'
if 'RNASeq' in filename: expressed_exon = 'no' ### Set by default
if ':' in geneid: geneid = string.split(geneid,':')[0] ### User reported that gene:gene was appearing and not sure exactly where or why but added this to address it
if 'FIRMA' in fn: method = 'FIRMA'
elif 'splicing-index' in fn: method = 'splicing-index'
if 'ASPIRE' in filename or 'linearregres' in filename:
f1=float(t[12]); f2=float(t[13]); probeset1 = t[8]; probeset2 = t[10]; direction = t[6]; exonid2 = t[5]; splicing_event = t[-4]
protein_annot = t[19]; domain_inferred = t[20]; domain_overlap = t[24]; method = 'linearregres'; regions = t[-5]
exon1_exp=float(t[-15]); exon2_exp=float(t[-14]); fold1=float(t[12]); fold2=float(t[13])
if fold1<0: fold1 = 1 ### don't factor in negative changes
if fold2<0: fold2 = 1 ### don't factor in negative changes
"""
if 'RNASeq' not in filename:
exon1_exp = math.pow(2,exon1_exp)
exon2_exp = math.log(2,exon2_exp)
m1 = exon1_exp*fold1
m2 = exon2_exp*fold2
max_exp = max([m1,m2])
min_exp = min([m1,m2])
percent_exon_expression = str(min_exp/max_exp)
"""
if 'ASPIRE' in filename: method = 'ASPIRE'; score = t[1][:5]
if '-' not in exonid and includeExonJunctionComps == False:
exonid=None ### Occurs when the inclusion just in an exon (possibly won't indicate confirmation so exclude)
else: exonid = exonid+' vs. '+exonid2
if 'AltMouse' in filename:
try: geneid = altmouse_ensembl_db[geneid]
except Exception: geneid = geneid
if 'RNASeq' not in filename and 'junction' not in filename: regions = string.replace(regions,'-','.')
else:
if 'RNASeq' in filename and '-' not in exonid:
fold = float(t[10]); exon_exp = float(t[18]); gene_exp = float(t[19])
if fold < 0: fold = -1.0/fold
GE_fold = float(gene_exp_fold)
if GE_fold < 0: GE_fold = -1.0/float(gene_exp_fold)
exon_psi1 = abs(exon_exp)/(abs(gene_exp))
exon_psi2 = (abs(exon_exp)*fold)/(abs(gene_exp)*GE_fold)
max_incl_exon_exp = max([exon_psi1,exon_psi2])
#if max_incl_exon_exp>0.20: expressed_exon = 'yes'
expressed_exon = max_incl_exon_exp
#if 'I2.1_75953139' in probeset1:
#print [exon_exp,gene_exp,exon_exp*fold,gene_exp*GE_fold]
#print exon_psi1, exon_psi2;sys.exit()
probesets = [probeset1,probeset2]
if (method == 'splicing-index' or method == 'FIRMA') and ('-' in exonid) or exonid == None:
pass #exclude junction IDs
else:
regions = string.replace(regions,';','|')
regions = string.replace(regions,'-','|')
regions = string.split(regions,'|')
for region in regions:
if len(region) == 0:
try: region = t[17]+t[18] ### For junction introns where no region ID exists
except Exception: null=[]
if ':' in region: region = string.split(region,':')[-1] ### User reported that gene:gene was appearing and not sure exactly where or why but added this to address it
if probeset1 in novel_exon_junction_db:
uid = novel_exon_junction_db[probeset1] ### convert the uid (alternative exon) to the annotated ID for the novel exon
converted_db[uid] = probeset1
else:
uid = geneid+':'+region
ss = SplicingData(score,symbol,description,exonid,probesets,direction,splicing_event,external_exon,genomic_loc,gene_exp_fold,protein_annot,domain_inferred,domain_overlap,method,filename)
ss.setExonExpStatus(str(expressed_exon))
try: regulated_critical_exon_temp[uid].append(ss)
except Exception: regulated_critical_exon_temp[uid] = [ss]
#print filename, len(regulated_critical_exon_temp)
for uid in regulated_critical_exon_temp:
report=None
if len(regulated_critical_exon_temp[uid])>1:
### We are only reporting one here and that's OK, since we are only reporting the top scores... won't include all inclusion junctions.
scores=[]
for ss in regulated_critical_exon_temp[uid]: scores.append((float(ss.Score()),ss))
scores.sort()
if (scores[0][0]*scores[-1][0])<0:
ss1 = scores[0][1]; ss2 = scores[-1][1]
if ss1.ProbesetsSorted() == ss2.ProbesetsSorted(): ss1.setDirection('mutual') ### same exons, hence, mutually exclusive event (or similiar)
else: ss1.setDirection('both') ### opposite directions in the same comparison-file, hence, conflicting data
report=[ss1]
else:
if abs(scores[0][0])>abs(scores[-1][0]): report=[scores[0][1]]
else: report=[scores[-1][1]]
else:
report=regulated_critical_exon_temp[uid]
### Combine data from different analysis files
try: regulated_critical_exons[uid]+=report
except Exception: regulated_critical_exons[uid]=report
"""if 'ENSG00000204120' in uid:
print uid,
for i in regulated_critical_exon_temp[uid]:
print i.Probesets(),
print ''
"""
try: report[0].setEvidence(len(regulated_critical_exon_temp[uid])) ###set the number of exons demonstrating regulation of this exons
except Exception: null=[]
clearObjectsFromMemory(regulated_critical_exon_temp)
export_data,status = AppendOrWrite(export_path)
if status == 'not found':
header = string.join(['uid','source-IDs','symbol','description','exonids','independent confirmation','score','regulation direction','alternative exon annotations','associated isoforms','inferred regulated domains','overlapping domains','method','supporting evidence score','novel exon: high-confidence','percent exon expression of gene','differential gene-expression','genomic location'],'\t')+'\n'
export_data.write(header)
combined_export_path = string.split(results_dir,'AltResults')[0]+'AltResults/Clustering/Combined-junction-exon-evidence.txt'
combined_export_data, status= AppendOrWrite(combined_export_path)
if status == 'not found':
header = string.join(['uid','source-IDs','symbol','description','exonids','independent confirmation','score','regulation direction','alternative exon annotations','associated isoforms','inferred regulated domains','overlapping domains','method','supporting evidence score','novel exon: high-confidence','percent exon expression of gene','differential gene-expression','genomic location','comparison'],'\t')+'\n'
combined_export_data.write(header)
print len(regulated_critical_exons), 'regulated exon IDs imported.\n'
print 'writing:',export_path; n=0
# print [len(converted_db)]
### Check for alternative 3' or alternative 5' exon regions that were not matched to the right reciprocal junctions (occurs because only one of the exon regions is called alternative)
regulated_critical_exons_copy={}
for uid in regulated_critical_exons:
regulated_critical_exons_copy[uid]=regulated_critical_exons[uid]
u=0
### This is most applicable to RNA-Seq since the junction IDs correspond to the Exon Regions not the probeset Exon IDs
for uid in regulated_critical_exons_copy: ### Look through the copied version since we can't delete entries while iterating through
ls = regulated_critical_exons_copy[uid]
u+=1
#if u<20: print uid
for jd in ls:
if jd.Method() != 'splicing-index' and jd.Method() != 'FIRMA':
try: ### Applicable to RNA-Seq
gene,exonsEx = string.split(jd.Probesets()[1],':') ### Exclusion probeset will have the exon not annotated as the critical exon (although it should be as well)
gene,exonsIn = string.split(jd.Probesets()[0],':')
except Exception:
gene, ce = string.split(uid,':')
exonsIn, exonsEx = string.split(jd.ExonID(),'vs.')
if gene !=None:
critical_exon = None
five_prime,three_prime = string.split(exonsEx,'-')
try: five_primeIn,three_primeIn = string.split(exonsIn,'-')
except Exception: five_primeIn = exonsIn; three_primeIn = exonsIn ### Only should occur during testing when a exon rather than junction ID is considered
#if gene == 'ENSG00000133083': print five_prime,three_prime, five_primeIn,three_primeIn
if five_primeIn == five_prime: ### Hence, the exclusion 3' exon should be added
critical_exon = gene+':'+three_prime
exonid = three_prime
elif three_primeIn == three_prime: ### Hence, the exclusion 3' exon should be added
critical_exon = gene+':'+five_prime
exonid = five_prime
else:
if ('5' in jd.SplicingEvent()) or ('five' in jd.SplicingEvent()):
critical_exon = gene+':'+five_prime
exonid = five_prime
elif ('3' in jd.SplicingEvent()) or ('three' in jd.SplicingEvent()):
critical_exon = gene+':'+three_prime
exonid = three_prime
elif ('alt-N-term' in jd.SplicingEvent()) or ('altPromoter' in jd.SplicingEvent()):
critical_exon = gene+':'+five_prime
exonid = five_prime
elif ('alt-C-term' in jd.SplicingEvent()):
critical_exon = gene+':'+three_prime
exonid = three_prime
#print critical_exon, uid, jd.ExonID(),jd.SplicingEvent(); sys.exit()
if critical_exon != None:
if critical_exon in regulated_critical_exons:
#print uid, critical_exon; sys.exit()
if len(regulated_critical_exons[critical_exon]) == 1:
if len(ls)==1 and uid in regulated_critical_exons: ### Can be deleted by this method
if 'vs.' not in regulated_critical_exons[critical_exon][0].ExonID() and 'vs.' not in regulated_critical_exons[critical_exon][0].ExonID():
regulated_critical_exons[uid].append(regulated_critical_exons[critical_exon][0])
del regulated_critical_exons[critical_exon]
elif uid in regulated_critical_exons: ###If two entries already exit
ed = regulated_critical_exons[uid][1]
ed2 = regulated_critical_exons[critical_exon][0]
if 'vs.' not in ed.ExonID() and 'vs.' not in ed2.ExonID():
if ed.Direction() != ed2.Direction(): ### should be opposite directions
ed.appendExonID(exonid)
ed.setEvidence(ed.Evidence()+1)
ed.setScore(ed.Score()+'|'+ed2.Score())
del regulated_critical_exons[critical_exon]
firstEntry=True
for uid in regulated_critical_exons:
if uid in converted_db:
converted = True
else: converted = False
#if 'ENSG00000133083' in uid: print [uid]
exon_level_confirmation = 'no'
ls = regulated_critical_exons[uid]
jd = regulated_critical_exons[uid][0] ### We are only reporting one here and that's OK, since we are only reporting the top scores... won't include all inclusion junctions.
if len(ls)>1:
methods = []; scores = []; direction = []; exonids = []; probesets = []; evidence = 0; genomic_location = []
junctionids=[]
junction_data_found = 'no'; exon_data_found = 'no'
for jd in ls:
if jd.Method() == 'ASPIRE' or jd.Method() == 'linearregres':
junction_data_found = 'yes'
methods.append(jd.Method())
scores.append(jd.Score())
direction.append(jd.Direction())
exonids.append(jd.ExonID())
junctionids.append(jd.ExonID())
probesets.append(jd.ProbesetDisplay())
evidence+=jd.Evidence()
genomic_location.append(jd.GenomicLocation())
### Prefferentially obtain isoform annotations from the reciprocal analysis which is likely more accurate
isoform_annotations = [jd.ProteinAnnotation(), jd.DomainInferred(), jd.DomainOverlap()]
for ed in ls:
if ed.Method() == 'splicing-index' or ed.Method() == 'FIRMA':
exon_data_found = 'yes' ### pick one of them
methods.append(ed.Method())
scores.append(ed.Score())
direction.append(ed.Direction())
exonids.append(ed.ExonID())
probesets.append(ed.ProbesetDisplay())
evidence+=ed.Evidence()
genomic_location.append(ed.GenomicLocation())
#isoform_annotations = [ed.ProteinAnnotation(), ed.DomainInferred(), ed.DomainOverlap()]
if junction_data_found == 'yes' and exon_data_found == 'yes':
exon_level_confirmation = 'yes'
for junctions in junctionids:
if 'vs.' in junctions:
j1 = string.split(junctions,' vs. ')[0] ### inclusion exon or junction
if '-' not in j1: ### not a junction, hence, may not be sufficient to use for confirmation (see below)
if 'I' in j1: ### intron feature
if '_' in j1: ### novel predicted exon
exon_level_confirmation = 'no'
else:
exon_level_confirmation = 'yes'
else:
if '_' in j1:
exon_level_confirmation = 'no'
else:
exon_level_confirmation = 'partial'
method = string.join(methods,'|')
unique_direction = unique.unique(direction)
genomic_location = unique.unique(genomic_location)
if len(unique_direction) == 1: direction = unique_direction[0]
else: direction = string.join(direction,'|')
score = string.join(scores,'|')
probesets = string.join(probesets,'|')
exonids_unique = unique.unique(exonids)
if len(exonids_unique) == 1: exonids = exonids_unique[0]
else: exonids = string.join(exonids,'|')
if len(genomic_location) == 1: genomic_location = genomic_location[0]
else: genomic_location = string.join(genomic_location,'|')
evidence = str(evidence)
if 'mutual' in direction: direction = 'mutual'
if len(ls) == 1:
probesets = jd.ProbesetDisplay()
direction = jd.Direction()
score = jd.Score()
method = jd.Method()
exonids = jd.ExonID()
evidence = jd.Evidence()
genomic_location = jd.GenomicLocation()
isoform_annotations = [jd.ProteinAnnotation(), jd.DomainInferred(), jd.DomainOverlap()]
try:
#if int(evidence)>4 and 'I' in uid: novel_exon = 'yes' ### high-evidence novel exon
#else: novel_exon = 'no'
if converted == True:
novel_exon = 'yes'
splicing_event = 'cassette-exon'
else:
novel_exon = 'no'
splicing_event = jd.SplicingEvent()
values = [uid, probesets, jd.Symbol(), jd.Description(), exonids, exon_level_confirmation, score, direction, splicing_event]
values += isoform_annotations+[method, str(evidence),novel_exon,jd.ExonExpStatus(),jd.GeneExpression(),genomic_location]
values = string.join(values,'\t')+'\n'
#if 'yes' in exon_level_confirmation:
export_data.write(values); n+=1
if exon_level_confirmation != 'no' and ('|' not in direction):
geneID = string.split(uid,':')[0]
try: relative_exon_exp = float(jd.ExonExpStatus())
except Exception: relative_exon_exp = 1
if firstEntry:
### Also export high-confidence predictions for GO-Elite
elite_export_path = string.split(results_dir,'AltResults')[0]+'GO-Elite/AltExonConfirmed/'+export_filename+'-junction-exon-evidence.txt'
elite_export_data = export.ExportFile(elite_export_path)
elite_export_data.write('GeneID\tEn\tExonID\tScores\tGenomicLocation\n')
firstEntry = False
if relative_exon_exp>0.10:
elite_export_data.write(string.join([geneID,'En',uid,score,genomic_location],'\t')+'\n')
#if 'DNA' in isoform_annotations[-1]:
if '2moter' not in jd.SplicingEvent() and '2lt-N' not in jd.SplicingEvent():
values = [uid, probesets, jd.Symbol(), jd.Description(), exonids, exon_level_confirmation, score, direction, splicing_event]
values += isoform_annotations+[method, str(evidence),novel_exon,jd.ExonExpStatus(),jd.GeneExpression(),genomic_location,export_filename]
values = string.join(values,'\t')+'\n'
combined_export_data.write(values)
except Exception, e:
#print traceback.format_exc();sys.exit()
pass ### Unknown error - not evaluated in 2.0.8 - isoform_annotations not referenced
print n,'exon IDs written to file.'
export_data.close()
try: elite_export_data.close()
except Exception: pass
clearObjectsFromMemory(regulated_critical_exons)
clearObjectsFromMemory(regulated_critical_exons_copy)
#print '!!!!Within comparison evidence'
#returnLargeGlobalVars()
def FeatureCounts(bed_ref, bam_file):
output = bam_file[:-4]+'__FeatureCounts.bed'
import subprocess
#if '/bin' in kallisto_dir: kallisto_file = kallisto_dir +'/apt-probeset-summarize' ### if the user selects an APT directory
kallisto_dir= 'AltDatabase/subreads/'
if os.name == 'nt':
featurecounts_file = kallisto_dir + 'PC/featureCounts.exe'; plat = 'Windows'
elif 'darwin' in sys.platform:
featurecounts_file = kallisto_dir + 'Mac/featureCounts'; plat = 'MacOSX'
elif 'linux' in sys.platform:
featurecounts_file = kallisto_dir + '/Linux/featureCounts'; plat = 'linux'
print 'Using',featurecounts_file
featurecounts_file = filepath(featurecounts_file)
featurecounts_root = string.split(featurecounts_file,'bin/featureCounts')[0]
featurecounts_file = filepath(featurecounts_file)
print [featurecounts_file,"-a", "-F", "SAF",bed_ref, "-o", output, bam_file]
retcode = subprocess.call([featurecounts_file,"-a",bed_ref, "-F", "SAF", "-o", output, bam_file])
def runKallisto(species,dataset_name,root_dir,fastq_folder,returnSampleNames=False,customFASTA=None):
#print 'Running Kallisto...please be patient'
import subprocess
kallisto_dir_objects = os.listdir(unique.filepath('AltDatabase/kallisto'))
### Determine version
version = '0.42.1'
for subdir in kallisto_dir_objects:
if subdir.count('.')>1: version = subdir
kallisto_dir= 'AltDatabase/kallisto/'+version+'/'
if os.name == 'nt':
kallisto_file = kallisto_dir + 'PC/bin/kallisto.exe'; plat = 'Windows'
elif 'darwin' in sys.platform:
kallisto_file = kallisto_dir + 'Mac/bin/kallisto'; plat = 'MacOSX'
elif 'linux' in sys.platform:
kallisto_file = kallisto_dir + '/Linux/bin/kallisto'; plat = 'linux'
print 'Using',kallisto_file
kallisto_file = filepath(kallisto_file)
kallisto_root = string.split(kallisto_file,'bin/kallisto')[0]
fn = filepath(kallisto_file)
output_dir=root_dir+'/ExpressionInput/kallisto/'
try: os.mkdir(root_dir+'/ExpressionInput')
except Exception: pass
try: os.mkdir(root_dir+'/ExpressionInput/kallisto')
except Exception: pass
fastq_folder += '/'
dir_list = read_directory(fastq_folder)
fastq_paths = []
for file in dir_list:
file_lower = string.lower(file)
if 'fastq' in file_lower and '._' not in file[:4]: ### Hidden files
fastq_paths.append(fastq_folder+file)
fastq_paths,paired = findPairs(fastq_paths)
### Check to see if Kallisto files already exist and use these if so (could be problematic but allows for outside quantification)
kallisto_tsv_paths=[]
dir_list = read_directory(output_dir)
for folder in dir_list:
kallisto_outdir = output_dir+folder+'/abundance.tsv'
status = os.path.isfile(kallisto_outdir)
if status:
kallisto_tsv_paths.append(fastq_folder+file)
if returnSampleNames:
return fastq_paths
### Store/retreive the Kallisto index in the Ensembl specific SequenceData location
kallisto_index_root = 'AltDatabase/'+species+'/SequenceData/'
try: os.mkdir(filepath(kallisto_index_root))
except Exception: pass
indexFile = filepath(kallisto_index_root+species)
indexStatus = os.path.isfile(indexFile)
if indexStatus == False or customFASTA!=None:
try: fasta_file = getFASTAFile(species)
except Exception: fasta_file = None
index_file = filepath(kallisto_index_root+species)
if fasta_file==None and customFASTA==None:
###download Ensembl fasta file to the above directory
from build_scripts import EnsemblSQL
ensembl_version = string.replace(unique.getCurrentGeneDatabaseVersion(),'EnsMart','')
EnsemblSQL.getEnsemblTranscriptSequences(ensembl_version,species,restrictTo='cDNA')
fasta_file = getFASTAFile(species)
elif customFASTA!=None: ### Custom FASTA file supplied by the user
fasta_file = customFASTA
indexFile = filepath(kallisto_index_root+species+'-custom')
try: os.remove(indexFile) ### erase any pre-existing custom index
except Exception: pass
if fasta_file!=None:
print 'Building kallisto index file...'
try: retcode = subprocess.call([kallisto_file, "index","-i", indexFile, fasta_file])
except Exception:
print traceback.format_exc()
### If installed on the local OS
retcode = subprocess.call(['kallisto', "index","-i", indexFile, fasta_file])
if customFASTA!=None:
reimportExistingKallistoOutput = False
elif len(kallisto_tsv_paths) == len(fastq_paths):
reimportExistingKallistoOutput = True
elif len(kallisto_tsv_paths) > len(fastq_paths):
reimportExistingKallistoOutput = True ### If working with a directory of kallisto results
else:
reimportExistingKallistoOutput = False
if reimportExistingKallistoOutput:
print 'NOTE: Re-import PREVIOUSLY GENERATED kallisto output:',reimportExistingKallistoOutput
print '...To force re-analysis of FASTQ files, delete the folder "kallisto" in "ExpressionInput"'
### Just get the existing Kallisto output folders
fastq_paths = read_directory(output_dir)
kallisto_folders=[]
try:
import collections
expMatrix = collections.OrderedDict()
countMatrix = collections.OrderedDict()
countSampleMatrix = collections.OrderedDict()
sample_total_counts = collections.OrderedDict()
except Exception:
try:
import ordereddict
expMatrix = ordereddict.OrderedDict()
countMatrix = ordereddict.OrderedDict()
countSampleMatrix = ordereddict.OrderedDict()
sample_total_counts = ordereddict.OrderedDict()
except Exception:
expMatrix={}
countMatrix={}
countSampleMatrix={}
sample_total_counts={}
headers=['UID']
for n in fastq_paths:
output_path = output_dir+n
kallisto_folders.append(output_path)
if reimportExistingKallistoOutput == False:
begin_time = time.time()
print 'Running kallisto on:',n,
p=fastq_paths[n]
b=[" > "+n+'.sam']
#"""
if paired == 'paired':
try:
#retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path,"--pseudobam"]+p+b)
retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path]+p)
except Exception:
print traceback.format_exc()
retcode = subprocess.call(['kallisto', "quant","-i", indexFile, "-o", output_path]+p)
else:
if os.name == 'nt':
try:
try: retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path,"--single","-l","200"]+p)
except Exception: retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path,"--single","-l","200","-s","20"]+p)
except Exception:
try: retcode = subprocess.call(['kallisto', "quant","-i", indexFile, "-o", output_path,"--single","-l","200"]+p)
except Exception:
retcode = subprocess.call(['kallisto', "quant","-i", indexFile, "-o", output_path,"--single","-l","200","-s","20"]+p)
else:
try: retcode = subprocess.call([kallisto_file, "quant","-i", indexFile, "-o", output_path,"--single","-l","200","-s","20"]+p)
except Exception:
retcode = subprocess.call(['kallisto', "quant","-i", indexFile, "-o", output_path,"--single","-l","200","-s","20"]+p)
if retcode == 0: print 'completed in', int(time.time()-begin_time), 'seconds'
else: print 'kallisto failed due to an unknown error (report to altanalyze.org help).'
#"""
input_path = output_path+'/abundance.txt'
try:
try: expMatrix,countMatrix,countSampleMatrix=importTPMs(n,input_path,expMatrix,countMatrix,countSampleMatrix)
except Exception:
input_path = output_path+'/abundance.tsv'
expMatrix,countMatrix,countSampleMatrix=importTPMs(n,input_path,expMatrix,countMatrix,countSampleMatrix)
headers.append(n)
sample_total_counts = importTotalReadCounts(n,output_path+'/run_info.json',sample_total_counts)
except Exception:
print traceback.format_exc();sys.exit()
print n, 'TPM expression import failed'
if paired == 'paired':
print '\n...Make sure the paired-end samples were correctly assigned:'
for i in fastq_paths:
print 'Common name:',i,
for x in fastq_paths[i]:
print export.findParentDir(x),
print '\n'
### Summarize alignment information
for sample in countSampleMatrix:
try: estCounts = int(float(countSampleMatrix[sample]))
except Exception: estCounts='NA'
try: totalCounts = sample_total_counts[sample]
except Exception: totalCounts = 'NA'
try: aligned = str(100*estCounts/float(totalCounts))
except Exception: aligned = 'NA'
try: aligned = string.split(aligned,'.')[0]+'.'+string.split(aligned,'.')[1][:2]
except Exception: aligned = 'NA'
countSampleMatrix[sample] = [str(estCounts),totalCounts,aligned]
dataset_name = string.replace(dataset_name,'exp.','')
to = export.ExportFile(root_dir+'/ExpressionInput/transcript.'+dataset_name+'.txt')
ico = export.ExportFile(root_dir+'/ExpressionInput/isoCounts.'+dataset_name+'.txt')
go = export.ExportFile(root_dir+'/ExpressionInput/exp.'+dataset_name+'.txt')
co = export.ExportFile(root_dir+'/ExpressionInput/counts.'+dataset_name+'.txt')
so = export.ExportFile(root_dir+'/ExpressionInput/summary.'+dataset_name+'.txt')
exportMatrix(to,headers,expMatrix) ### Export transcript expression matrix
exportMatrix(ico,headers,countMatrix) ### Export transcript count matrix
try:
geneMatrix = calculateGeneTPMs(species,expMatrix) ### calculate combined gene level TPMs
countsGeneMatrix = calculateGeneTPMs(species,countMatrix) ### calculate combined gene level TPMs
exportMatrix(go,headers,geneMatrix) ### export gene expression matrix
exportMatrix(co,headers,countsGeneMatrix) ### export gene expression matrix
except Exception:
print 'AltAnalyze was unable to summarize gene TPMs from transcripts, proceeding with transcripts.'
export.copyFile(root_dir+'/ExpressionInput/transcript.'+dataset_name+'.txt',root_dir+'/ExpressionInput/exp.'+dataset_name+'.txt')
exportMatrix(so,['SampleID','Estimated Counts','Total Fragments','Percent Aligned'],countSampleMatrix) ### export gene expression matrix
def calculateGeneTPMs(species,expMatrix):
import gene_associations
try:
gene_to_transcript_db = gene_associations.getGeneToUid(species,('hide','Ensembl-EnsTranscript'))
if len(gene_to_transcript_db)<10:
raise ValueError('Ensembl-EnsTranscript file missing, forcing download of this file')
except Exception:
try:
print 'Missing transcript-to-gene associations... downloading from Ensembl.'
from build_scripts import EnsemblSQL
db_version = unique.getCurrentGeneDatabaseVersion()
EnsemblSQL.getGeneTranscriptOnly(species,'Basic',db_version,'yes')
gene_to_transcript_db = gene_associations.getGeneToUid(species,('hide','Ensembl-EnsTranscript'))
except Exception:
from build_scripts import GeneSetDownloader
print 'Ensembl-EnsTranscripts required for gene conversion... downloading from the web...'
GeneSetDownloader.remoteDownloadEnsemblTranscriptAssocations(species)
gene_to_transcript_db = gene_associations.getGeneToUid(species,('hide','Ensembl-EnsTranscript'))
if len(gene_to_transcript_db)<10:
print 'NOTE: No valid Ensembl-EnsTranscripts available, proceeding with the analysis of transcripts rather than genes...'
from import_scripts import OBO_import
transcript_to_gene_db = OBO_import.swapKeyValues(gene_to_transcript_db)
gene_matrix = {}
present_gene_transcripts={}
for transcript in expMatrix:
if '.' in transcript:
transcript_alt = string.split(transcript,'.')[0]
else:
transcript_alt = transcript
if transcript_alt in transcript_to_gene_db:
gene = transcript_to_gene_db[transcript_alt][0]
try: present_gene_transcripts[gene].append(transcript)
except Exception: present_gene_transcripts[gene] = [transcript]
else: pass ### could keep track of the missing transcripts
for gene in present_gene_transcripts:
gene_values = []
for transcript in present_gene_transcripts[gene]:
gene_values.append(map(float,expMatrix[transcript]))
gene_tpms = [sum(value) for value in zip(*gene_values)] ### sum of all transcript tmp's per sample
gene_tpms = map(str,gene_tpms)
gene_matrix[gene] = gene_tpms
if len(gene_matrix)>0:
return gene_matrix
else:
print "NOTE: No valid transcript-gene associations available... proceeding with Transcript IDs rather than gene."
return expMatrix
def exportMatrix(eo,headers,matrix):
eo.write(string.join(headers,'\t')+'\n')
for gene in matrix:
eo.write(string.join([gene]+matrix[gene],'\t')+'\n')
eo.close()
def importTPMs(sample,input_path,expMatrix,countMatrix,countSampleMatrix):
firstLine=True
for line in open(input_path,'rU').xreadlines():
data = cleanUpLine(line)
if firstLine:
firstLine=False
header = string.split(data,'\t')
else:
target_id,length,eff_length,est_counts,tpm = string.split(data,'\t')
try: float(est_counts);
except Exception: ### nan instead of float found due to lack of alignment
est_counts = '0.0'
tpm = '0.0'
if '.' in target_id:
target_id = string.split(target_id,'.')[0] ### Ensembl isoform IDs in more recent Ensembl builds
try: expMatrix[target_id].append(tpm)
except Exception: expMatrix[target_id]=[tpm]
try: countSampleMatrix[sample]+=float(est_counts)
except Exception: countSampleMatrix[sample]=float(est_counts)
try: countMatrix[target_id].append(est_counts)
except Exception: countMatrix[target_id]=[est_counts]
return expMatrix,countMatrix,countSampleMatrix
def importTotalReadCounts(sample,input_path,sample_total_counts):
### Import from Kallisto Json file
for line in open(input_path,'rU').xreadlines():
data = cleanUpLine(line)
if "n_processed: " in data:
total = string.split(data,"n_processed: ")[1]
total = string.split(total,',')[0]
sample_total_counts[sample]=total
return sample_total_counts
def findPairs(fastq_paths):
#fastq_paths = ['/Volumes/test/run0718_lane12_read1_index701=Kopan_RBP_02_14999.fastq.gz','/Volumes/run0718_lane12_read2_index701=Kopan_RBP_02_14999.fastq.gz']
import export
read_notation=0
under_suffix_notation=0
suffix_notation=0
equal_notation=0
suffix_db={}
for i in fastq_paths:
if 'read1' in i or 'read2' in i or 'pair1' in i or 'pair2' or 'R1' in i or 'R2' in i:
read_notation+=1
f = export.findFilename(i)
if 'fastq' in f:
name = string.split(f,'fastq')[0]
elif 'FASTQ' in f:
name = string.split(f,'FASTQ')[0]
elif 'fq' in f:
name = string.split(f,'fq')[0]
if '_1.' in name or '_2.' in name:
under_suffix_notation+=1
elif '1.' in name or '2.' in name:
suffix_notation+=1
suffix_db[name[-2:]]=[]
if '=' in name:
equal_notation+=1
if read_notation==0 and suffix_notation==0 and under_suffix_notation==0:
new_names={}
for i in fastq_paths:
if '/' in i or '\\' in i:
n = export.findFilename(i)
if '=' in n:
n = string.split(n,'=')[1]
new_names[n] = [i]
### likely single-end samples
return new_names, 'single'
else:
new_names={}
paired = 'paired'
if equal_notation==len(fastq_paths):
for i in fastq_paths:
name = string.split(i,'=')[-1]
name = string.replace(name,'.fastq.gz','')
name = string.replace(name,'.fastq','')
name = string.replace(name,'.FASTQ.gz','')
name = string.replace(name,'.FASTQ','')
name = string.replace(name,'.fq.gz','')
name = string.replace(name,'.fq','')
if '/' in name or '\\' in name:
name = export.findFilename(name)
if '=' in name:
name = string.split(name,'=')[1]
try: new_names[name].append(i)
except Exception: new_names[name]=[i]
else:
for i in fastq_paths:
if suffix_notation == len(fastq_paths) and len(suffix_db)==2: ### requires that files end in both .1 and .2
pairs = ['1.','2.']
else:
pairs = ['-read1','-read2','-pair1','-pair2','_read1','_read2','_pair1','_pair2','read1','read2','pair1','pair2','_1.','_2.','_R1','_R2','-R1','-R2','R1','R2']
n=str(i)
n = string.replace(n,'fastq.gz','')
n = string.replace(n,'fastq','')
for p in pairs: n = string.replace(n,p,'')
if '/' in n or '\\' in n:
n = export.findFilename(n)
if '=' in n:
n = string.split(n,'=')[1]
if n[-1]=='.':
n = n[:-1] ###remove the last decimal
try: new_names[n].append(i)
except Exception: new_names[n]=[i]
for i in new_names:
if len(new_names[i])>1:
pass
else:
paired = 'single'
new_names = checkForMultipleLanes(new_names)
return new_names, paired
def checkForMultipleLanes(new_names):
""" This function further aggregates samples run across multiple flowcells """
read_count = 0
lane_count = 0
updated_names={}
for sample in new_names:
reads = new_names[sample]
count=0
for read in reads:
read_count+=1
if '_L00' in read and '_001':
### assumes no more than 9 lanes/sample
count+=1
if len(reads) == count: ### Multiple lanes run per sample
lane_count+=count
if lane_count==read_count:
for sample in new_names:
sample_v1 = string.replace(sample,'_001','')
sample_v1 = string.split(sample_v1,'_L00')
if len(sample_v1[-1])==1: ### lane number
sample_v1 = sample_v1[0]
if sample_v1 in updated_names:
updated_names[sample_v1]+=new_names[sample]
else:
updated_names[sample_v1]=new_names[sample]
if len(updated_names)==0:
updated_names = new_names
return updated_names
def getFASTAFile(species):
fasta_file=None
fasta_folder = 'AltDatabase/'+species+'/SequenceData/'
dir_list = read_directory(filepath(fasta_folder))
for file in dir_list:
if '.fa' in file: fasta_file = filepath(fasta_folder+file)
return fasta_file
if __name__ == '__main__':
import getopt
samplesDiffering = 3
column_method = 'hopach'
species = 'Hs'
excludeCellCycle = False
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['Guidefile='])
for opt, arg in options:
if opt == '--Guidefile': Guidefile=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
platform = 'RNASeq'; graphic_links=[('','/Volumes/HomeBackup/CCHMC/PBMC-10X/ExpressionInput/SamplePrediction/DataPlots/Clustering-33k_CPTT_matrix-CORRELATED-FEATURES-iterFilt-hierarchical_cosine_cosine.txt')]
"""
graphic_links,new_results_file = correlateClusteredGenes(platform,graphic_links[-1][-1][:-4]+'.txt',
numSamplesClustered=samplesDiffering,excludeCellCycle=excludeCellCycle,graphics=graphic_links,
ColumnMethod=column_method, transpose=True, includeMoreCells=True)
"""
#results_file='/Users/meenakshi/DataPlots/Clustering-exp.round2_correlation0.4_cor0.4-Guide2 TMBIM4&ENSG00000228144&E8.1-I5.1__ENSG00000-hierarchical_euclidean_correlation.txt'
#Guidefile='/Volumes/Pass/NewLeucegene_3samplesdel/ICGS/Clustering-exp.splicing-Guide3 CLEC7A ENSG00000172243 E2.1-E3.1 ENSG000001-hierarchical_euclidean_correlation.txt'
#Guidefile='/Volumes/Pass/NewLeucegene_3samplesdel/ICGS/Clustering-exp.splicing-filteredcor_depleted-Guide3 TMBIM4 ENSG00000228144 E8.1-I5.1 ENSG000002-hierarchical_euclidean_correlation.txt'
#Guidefile=' /Volumes/Pass/complete_splice/ExpressionInput/amplify/DataPlots/Clustering-exp.input-Guide3 ZNF275 ENSG00000063587 E2.1-E4.3 ENSG000000-hierarchical_euclidean_correlation.txt'
correlateClusteredGenesParameters(Guidefile,rho_cutoff=0.4,hits_cutoff=4,hits_to_report=50,ReDefinedClusterBlocks=True,filter=True)
sys.exit()
correlateClusteredGenes('exons',results_file,stringency='strict',rhoCutOff=0.6);sys.exit()
#sys.exit()
species='Hs'; platform = "3'array"; vendor = "3'array"
#FeatureCounts('/Users/saljh8/Downloads/subread-1.5.2-MaxOSX-x86_64/annotation/mm10_AltAnalyze.txt', '/Users/saljh8/Desktop/Grimes/GEC14074/Grimes_092914_Cell12.bam')
#sys.exit()
import UI; import multiprocessing as mlp
gsp = UI.GeneSelectionParameters(species,platform,vendor)
gsp.setGeneSet('None Selected')
gsp.setPathwaySelect('')
gsp.setGeneSelection('')
gsp.setJustShowTheseIDs('')
gsp.setNormalize('median')
gsp.setSampleDiscoveryParameters(1,50,4,4,
True,'gene','protein_coding',False,'cosine','hopach',0.4)
#expFile = '/Users/saljh8/Desktop/Grimes/KashishNormalization/test/Original/ExpressionInput/exp.CombinedSingleCell_March_15_2015.txt'
expFile = '/Volumes/My Passport/salomonis2/SRP042161_GBM-single-cell/bams/ExpressionInput/exp.GBM_scRNA-Seq-steady-state.txt'
#singleCellRNASeqWorkflow('Hs', "RNASeq", expFile, mlp, parameters=gsp);sys.exit()
filename = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/Trumpp-HSC-2017/counts.rawTrumpp.txt'
filename = '/Volumes/salomonis2/Erica-data/GSE98451/counts.GSE98451_uterus_single_cell_RNA-Seq_counts-Ensembl.txt'
#fastRPKMCalculate(filename);sys.exit()
#calculateRPKMsFromGeneCounts(filename,'Mm',AdjustExpression=False);sys.exit()
#copyICGSfiles('','');sys.exit()
runKallisto('Hs','test','tests/demo_data/FASTQ','tests/demo_data/FASTQ');sys.exit()
import multiprocessing as mlp
import UI
species='Mm'; platform = "3'array"; vendor = 'Ensembl'
gsp = UI.GeneSelectionParameters(species,platform,vendor)
gsp.setGeneSet('None Selected')
gsp.setPathwaySelect('')
gsp.setGeneSelection('')
gsp.setJustShowTheseIDs('')
gsp.setNormalize('median')
gsp.setSampleDiscoveryParameters(0,0,1.5,3,
False,'PSI','protein_coding',False,'cosine','hopach',0.35)
#gsp.setSampleDiscoveryParameters(1,1,4,3, True,'Gene','protein_coding',False,'cosine','hopach',0.5)
filename = '/Volumes/SEQ-DATA/AML_junction/AltResults/AlternativeOutput/Hs_RNASeq_top_alt_junctions-PSI-clust.txt'
#fastRPKMCalculate(filename);sys.exit()
results_file = '/Volumes/SEQ-DATA/Grimes/14018_gmp-pro/ExpressionInput/DataPlots/400 fold for at least 4 samples/Clustering-myeloblast-steady-state-correlated-features-hierarchical_euclidean_cosine-hopach.txt'
guideGeneFile = '/Volumes/SEQ-DATA/Grimes/14018_gmp-pro/ExpressionInput/drivingTFs-symbol.txt'
expFile = '/Users/saljh8/Desktop/Grimes/KashishNormalization/3-25-2015/ExpressionInput/exp.CombinedSingleCell_March_15_2015.txt'
expFile = '/Users/saljh8/Desktop/dataAnalysis/Mm_Kiddney_tubual/ExpressionInput/exp.E15.5_Adult_IRI Data-output.txt'
expFile = '/Users/saljh8/Desktop/PCBC_MetaData_Comparisons/temp/C4Meth450-filtered-SC-3_regulated.txt'
expFile = '/Volumes/SEQ-DATA/Grimeslab/TopHat/AltResults/AlternativeOutput/Mm_RNASeq_top_alt_junctions-PSI-clust-filter.txt'
expFile = '/Users/saljh8/Documents/Leucegene_TargetPSIFiles/exp.TArget_psi_noif_uncorr_03-50missing-12high.txt'
expFile = '/Volumes/BOZEMAN2015/Hs_RNASeq_top_alt_junctions-PSI-clust-filter.txt'
singleCellRNASeqWorkflow('Hs', "exons", expFile, mlp, exp_threshold=0, rpkm_threshold=0, parameters=gsp);sys.exit()
#expFile = '/Users/saljh8/Desktop/Grimes/AltSplice/Gmp-cluster-filter.txt'
#singleCellRNASeqWorkflow('Mm', "exons", expFile, mlp, exp_threshold=0, rpkm_threshold=0, parameters=gsp);sys.exit()
#expFile = '/Users/saljh8/Downloads/methylation/ExpressionInput/exp.female-steady-state.txt'
#singleCellRNASeqWorkflow('Hs', 'RNASeq', expFile, mlp, exp_threshold=50, rpkm_threshold=5) # drivers=guideGeneFile)
#sys.exit()
#correlateClusteredGenes(results_file);sys.exit()
#reformatExonFile('Hs','exon',True);sys.exit()
filename = '/Volumes/Time Machine Backups/dataAnalysis/PCBC_Sep2013/C4-reference/ExpressionInput/counts.C4.txt'
#fastRPKMCalculate(filename);sys.exit()
file1 = '/Volumes/My Passport/dataAnalysis/CardiacRNASeq/BedFiles/ExpressionInput/exp.CardiacRNASeq.txt'
file2 = '/Volumes/Time Machine Backups/dataAnalysis/PCBC_Sep2013/C4-reference/ReferenceComps/ExpressionInput/counts.C4.txt'
#getHighExpNovelExons('Hs',file1);sys.exit()
#mergeCountFiles(file1,file2); sys.exit()
import UI
test_status = 'yes'
data_type = 'ncRNA'
data_type = 'mRNA'
array_type = 'RNASeq'
array_type = 'junction'
species = 'Hs' ### edit this
summary_results_db = {}
root_dir = '/Volumes/Time Machine Backups/dataAnalysis/Human Blood/Exon/Multiple Sclerosis/Untreated_MS-analysis/'
#root_dir = '/Volumes/Time Machine Backups/dataAnalysis/Human Blood/Exon/Multiple Sclerosis/2-3rds_training-untreated/'
root_dir = '/Volumes/SEQ-DATA/Grimes/14018_gmp-pro/400-original/'
#root_dir = '/Volumes/My Passport/dataAnalysis/PCBC_Dec2013/All/bedFiles/'
root_dir = '/Users/saljh8/Desktop/dataAnalysis/HTA2.0 Files/'
#summary_results_db['Hs_Junction_d14_vs_d7.p5_average-ASPIRE-exon-inclusion-results.txt'] = [] ### edit this
#summary_results_db['Hs_Junction_d14_vs_d7.p5_average-splicing-index-exon-inclusion-results.txt'] = [] ### edit this
results_dir = root_dir +'AltResults/AlternativeOutput/'
dir_list = read_directory(results_dir)
for i in dir_list:
if '_average' in i:
comparison, end = string.split(i,'_average')
if '-exon-inclusion-results.txt' in i: summary_results_db[comparison]=[]
compareExonAndJunctionResults(species,array_type,summary_results_db,root_dir); sys.exit()
fl = UI.ExpressionFileLocationData('','','',''); fl.setCELFileDir(loc); fl.setRootDir(loc)
exp_file_location_db={}; exp_file_location_db['test']=fl
alignJunctionsToEnsembl(species,exp_file_location_db,'test'); sys.exit()
getEnsemblAssociations(species,data_type,test_status,'yes'); sys.exit()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/RNASeq_blockIdentification.py
|
RNASeq_blockIdentification.py
|
import os, sys, string, getopt
import math, numpy
from scipy import stats
import warnings
import time
def importFile(filename,convertToFloat=False):
db={}
firstRow=True
dataset_max=0
for line in open(filename,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
uid = t[0]
if firstRow: samples = t[1:]; firstRow=False
if len(t)>1:
values = t[1:]
if convertToFloat:
try:
values = map(float,values)
max_val = max(values)
if max_val > dataset_max:
dataset_max = max_val
except:
continue ### Header row
else:
values = t[1] ### Just store the gene symbol
else:
values = uid
db[uid]=values
if convertToFloat:
if dataset_max>100: ### Data is not log2
print 'Converting gene expression data to log2 values'
for uid in db:
db[uid] = map(lambda x: math.log(x+1,2), db[uid])
print 'Imported %d gene expression rows' % len(db)
return db, samples
else:
print 'Imported %d splicing factors' % len(db)
return db
def importPSIData(PSI_dir,samples):
"""Import PSI data from either EventAnnotation or PSI value file"""
firstRow=True
PSI_data_db={}
for line in open(PSI_dir,'rU').xreadlines():
data = line.rstrip()
PSI_data = string.split(data,'\t')
if firstRow:
data = string.replace(data,'.bed','')
PSI_data = string.split(data,'\t')
header_row = PSI_data
if 'ProteinPredictions' in PSI_data:
data_index = PSI_data.index('EventAnnotation')+1
uid_index = PSI_data.index('UID')
else:
uid_index = 0
data_index = 1
psi_samples = PSI_data[data_index:]
if psi_samples != samples:
print 'Error: The gene expression sample order does not match the PSI. Exiting';sys.exit()
else:
print 'Confirmed: The sample order of the gene expression and splicing files match.'
firstRow=False
else:
if len(PSI_data) != len(header_row):
empty_offset = len(header_row)-len(PSI_data)
PSI_data+=['']*empty_offset
junctionID = PSI_data[uid_index]
PSI_data = PSI_data[data_index:]
try:
values = map(lambda x: float(x), PSI_data)
except Exception:
values=[]
for value in PSI_data:
try: values.append(float(value))
except:
values.append(0.000101) ### Missing value
values = numpy.ma.masked_values(values,0.000101)
PSI_data_db[junctionID]=values
print 'Imported %d splicing event rows' % len(PSI_data_db)
return PSI_data_db
def findcorrelations(SF_dir, PSI_dir, exp_dir, output_dir, PearsonCutoff):
print ''
### Import the list of splicing factors or other genes of interest
genesToExamine = importFile(SF_dir)
### Import the tab-delimited gene expression matrix
geneExpression_db, samples = importFile(exp_dir,convertToFloat=True)
### Import the PSI data
PSI_data_db = importPSIData(PSI_dir,samples)
### Create an export directory
results_dir = output_dir+'/SFCorrelations_rho-'+str(PearsonCutoff)
try: os.mkdir(results_dir)
except: pass
eo=open(output_dir+'/SFCorrelations/SF_correlations.txt','w')
eo.write('Splicing Factor'+'\t'+'Events Count'+'\n')
counter=0
gene_correlation_time = []
for gene in genesToExamine:
gene_name = genesToExamine[gene]
if gene in geneExpression_db:
start_time = time.time()
### Hence, the gene is a splicing factor
expression_values = geneExpression_db[gene]
Corrsflist=[]
count=0
for junctionID in PSI_data_db:
psi_values = PSI_data_db[junctionID]
if 0.000101 in psi_values:
coefr=numpy.ma.corrcoef(expression_values,psi_values)
rho = coefr[0][1]
else:
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning)
rho,p = stats.pearsonr(expression_values,psi_values)
if abs(rho)>PearsonCutoff:
count+=1
Corrsflist.append([junctionID,rho])
gene_correlation_time.append(time.time()-start_time)
eo.write(gene_name+'\t'+str(count)+'\n')
filename=results_dir+"/"+gene_name+"_"+str(count)+".txt"
if count>20:
eg=open(filename,"w")
eg.write("SplicingEvent\tSystemCode\tPearsonRho\n")
for (junctionID,rho) in Corrsflist:
eg.write(junctionID+"\t"+"Ae\t"+str(rho)+"\n")
eg.close()
counter+=1
print '*',
print '\n...Correlations obtained on average of %d seconds/gene' % numpy.mean(gene_correlation_time)
def performEventEnrichment(output_dir,eventDir,species):
"""Import significant splicing events from metaDataAnalysis.py comparisons and test for their
statistical enrichmet relative to the Splicing Factor correlated events."""
import collections
import mappfinder
event_db = collections.OrderedDict()
import UI
### Import the splice-ICGS significant splicing events per signature
files = UI.read_directory(eventDir)
for file in files:
if '.txt' in file and 'PSI.' in file:
ls=[]
event_db[file[:-4]]=ls ### This list is subsequently updated below
fn = eventDir+'/'+file
firstLine = True
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
event_index = t.index('Event-Direction')
firstLine= False
continue
uid = t[0]
if 'U2AF1-like' in file:
if t[1] == "inclusion":
ls.append(uid) #ls.append((uid,t[event_index]))
else:
ls.append(uid) #ls.append((uid,t[event_index]))
### Import the splicing-factor correlated splicing events to identify associated signatures
splicing_factor_correlated_scores={}
gene_to_symbol=None
files = UI.read_directory(output_dir)
for file in files:
if '.txt' in file and '_' in file:
R_ls=[]
if 'ENS' in file:
splicing_factor = file[:-4]
if gene_to_symbol==None: ### Import only once
import gene_associations
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
sf = 'ENS'+string.split(splicing_factor,'ENS')[1]
splicing_factor = string.split(sf,'_')[0]
if splicing_factor in gene_to_symbol:
splicing_factor = gene_to_symbol[splicing_factor][0]
else:
splicing_factor = string.split(file[:-4],'_')[0]
fn = output_dir+'/'+file
firstLine = True
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
event = t[0]
R_ls.append(event)
R=len(R_ls)
N=80000
for signature in event_db:
n_ls=event_db[signature]
n = len(n_ls)
r_ls=set(R_ls).intersection(n_ls)
r = len(r_ls)
### Calculate a Z-score
try: z = Zscore(r,n,N,R)
except ZeroDivisionError: z = 0.0000
### Calculate a Z-score assuming zero matching entries
try: null_z = Zscore(0,n,N,R)
except ZeroDivisionError: null_z = 0.000
### Calculate a Fischer's Exact P-value
pval = mappfinder.FishersExactTest(r,n,R,N)
### Store these data in an object
zsd = mappfinder.ZScoreData(signature,r,n,z,null_z,n)
zsd.SetP(pval)
zsd.setAssociatedIDs(r_ls)
#print splicing_factor,'\t', signature,'\t', z, pval;sys.exit()
if splicing_factor in splicing_factor_correlated_scores:
signature_db = splicing_factor_correlated_scores[splicing_factor]
signature_db[signature]=zsd ### Necessary format for the permutation function
else:
signature_db={signature:zsd}
splicing_factor_correlated_scores[splicing_factor] = signature_db
results_dir = output_dir+'/SFEnrichmentResults'
result_file = results_dir+'/SF-correlated_SignatureScores.txt'
try: os.mkdir(results_dir)
except: pass
eo=open(result_file,'w')
eo.write(string.join(['Splicing Factor','Signature', 'Number Changed', 'Number Measured', 'Z-score','FisherExactP','AdjustedP'],'\t')+'\n') #'Events'
### Perform a permutation analysis to get BH adjusted p-values
for splicing_factor in splicing_factor_correlated_scores:
sorted_results=[]
signature_db = splicing_factor_correlated_scores[splicing_factor]
### Updates the adjusted p-value instances
mappfinder.adjustPermuteStats(signature_db)
for signature in signature_db:
zsd = signature_db[signature]
if float(zsd.ZScore())>1.96 and float(zsd.Changed())>2 and float(zsd.PermuteP())<0.05:
enriched_SFs={}
results = [splicing_factor,signature, zsd.Changed(), zsd.Measured(), zsd.ZScore(), zsd.PermuteP(), zsd.AdjP()] #string.join(zsd.AssociatedIDs(),'|')
sorted_results.append([float(zsd.PermuteP()),results])
sorted_results.sort() ### Sort by p-value
for (p,values) in sorted_results:
eo.write(string.join(values,'\t')+'\n')
if len(sorted_results)==0:
eo.write(string.join([splicing_factor,'NONE','NONE','NONE','NONE','NONE','NONE'],'\t')+'\n')
eo.close()
def Zscore(r,n,N,R):
"""where N is the total number of events measured:
R is the total number of events meeting the criterion:
n is the total number of events in this specific reference gene-set:
r is the number of events meeting the criterion in the examined reference gene-set: """
N=float(N) ### This bring all other values into float space
z = (r - n*(R/N))/math.sqrt(n*(R/N)*(1-(R/N))*(1-((n-1)/(N-1))))
return z
if __name__ == '__main__':
try:
import multiprocessing as mlp
mlp.freeze_support()
except Exception:
mpl = None
################ Default Variables ################
species = 'Hs'
platform = "RNASeq"
useMulti = False
output_dir = None
eventDir = None
PSI_dir = None
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Please designate a tab-delimited input expression file in the command-line"
print 'Example: SpliceEnricher.py --PSI "/Data/PSI_data.txt" --geneExp "/Data/GeneExp_data.txt" --geneList "/Data/SplicingFactors.txt" --rho 0.5'
else:
try:
options, remainder = getopt.getopt(sys.argv[1:],'', ['PSI=','species=','o=','platform=','useMulti=',
'geneExp=','geneList=','rho=','eventDir='])
except Exception,e:
print "Error",e
for opt, arg in options:
if opt == '--PSI': PSI_dir=arg
elif opt == '--geneExp': exp_dir=arg
elif opt == '--geneList': SF_dir=arg
elif opt == '--species': species=arg
elif opt == '--o': output_dir=arg
elif opt == '--platform': platform=arg
elif opt == '--rho': PearsonCutoff=float(arg)
elif opt == '--eventDir': eventDir=arg
if output_dir==None:
output_dir = string.replace(PSI_dir,'\\','/')
output_dir = string.join(string.split(output_dir,'/')[:-1],'/')
if PSI_dir !=None:
findcorrelations(SF_dir, PSI_dir, exp_dir, output_dir, PearsonCutoff)
if eventDir !=None:
performEventEnrichment(output_dir,eventDir,species)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/SpliceEnricher.py
|
SpliceEnricher.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import os,string
import numpy as np
from sklearn.preprocessing import scale
from numpy import linalg as LA
import scipy
def estimateK(inputfile):
header=[]
X=[]
head=0
counter=0
hgv={}
hgvgenes=[]
diclst={}
for line in open(inputfile,'rU').xreadlines():
if head==0:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
header=q
head=1
continue
else:
val=[]
line=line.rstrip('\r\n')
q= string.split(line,'\t')
#header.append(q[0])
for i in range(1,len(q)):
try:
val.append(float(q[i]))
except Exception:
continue
counter+=1
# break
X.append(val)
#X=zip(*X)
X=np.array(X)
n=float(X.shape[0])
p=float(X.shape[1])
print n
print p
X=scale(X)
Xt=np.transpose(X)
muTW=float((np.sqrt(n-1))+float(np.sqrt(p)))**2.0
sigmaTW=(float(np.sqrt(n - 1.0)) + float(np.sqrt(p))) * (1.0/float(np.sqrt(n - 1)) + 1.0/float(np.sqrt(p)))**(1.0/3.0)
sigmaHat=np.dot(Xt,X)
bd = 3.273 * sigmaTW + muTW
print bd
w,v = LA.eig(sigmaHat)
w=w.tolist()
k=0
for i in range(len(w)):
try:
if w[i]>bd:
k=k+1
except Exception:
if w[i].real>bd:
k=k+1
print k
return k
inputfile="/Volumes/Pass/Immune-complete/ExpressionInput/OncoInputs/NMFInput-Round1.txt"
estimateK(inputfile)
#inputfile="/Volumes/Pass/Singlecellbest/Pollen_upd/ExpressionInput/SamplePrediction/input-CORRELATED-FEATURES.txt"
#estimateK(inputfile)
#inputfile="/Volumes/Pass/Singlecellbest/Usoskin_upd/ExpressionInput/SamplePrediction/input-CORRELATED-FEATURES.txt"
#estimateK(inputfile)
#inputfile="/Volumes/Pass/Singlecellbest/Zeisel_upd/ExpressionInput/SamplePrediction/input-CORRELATED-FEATURES.txt"
#estimateK(inputfile)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/estimateK.py
|
estimateK.py
|
import os, sys, string
def createFeatures(junction_coordinates):
features={}
for junction_id in junction_coordinates:
location = junction_coordinates[junction_id]
pos1,pos2 = string.split(string.split(location,':')[-1],'-')
pos = [int(pos1),int(pos2)]
pos.sort()
features[junction_id] = pos
return features
def createFeaturesFromEvents(gene_db):
event_cluster_db={}
cluster_name='NewClu_1'
cluster_id=1
count=1
for gene in gene_db:
features={}
junction_to_event={}
event_coordinates = gene_db[gene]
for event in event_coordinates:
junction1, junction2 = string.split(event,'|')
junction1 = string.join(string.split(junction1,':')[1:],':')
coordinates1,coordinates2 = string.split(event_coordinates[event],'|')
pos1,pos2 = string.split(string.split(coordinates1,':')[-1],'-')
pos = [int(pos1),int(pos2)]
pos.sort(); features[junction1] = pos
pos1,pos2 = string.split(string.split(coordinates2,':')[-1],'-')
pos = [int(pos1),int(pos2)]
pos.sort(); features[junction2] = pos
try: junction_to_event[junction1].append(event)
except Exception: junction_to_event[junction1]=[event]
try: junction_to_event[junction2].append(event)
except Exception: junction_to_event[junction2]=[event]
cluster_junction,cluster_name,cluster_id,count = filterByLocalJunctionExp(features,cluster_name,cluster_id,count)
for junction in cluster_junction:
events = junction_to_event[junction]
for event in events:
event_cluster_db[event]=cluster_junction[junction]
return event_cluster_db
def filterByLocalJunctionExp(features,cluster_name,cluster_id,count):
junctions_to_compare={}
overlapping_junctions_exp={}
ovelapping_pos={}
existing=[]
overlapping_junctions_test={}
for feature in features:
pos1,pos2 = features[feature]
for f2 in features:
flag=False
if f2!=feature:
alt_pos1,alt_pos2 = features[f2]
positions = [pos1,pos2,alt_pos1,alt_pos2]
positions.sort()
diff = positions.index(pos2)-positions.index(pos1)
if diff!=1: ### Hence the two junctions are overlapping
flag=True
else:
diff = positions.index(alt_pos2)-positions.index(alt_pos1)
if diff!=1:
flag=True ### Hence the two junctions are overlapping
if flag==True:
if feature not in existing and f2 not in existing:
count=count+1
overlapping_junctions_test[count]=[feature,]
overlapping_junctions_test[count].append(f2)
existing.append(feature)
existing.append(f2)
elif feature in existing and f2 not in existing:
for i in overlapping_junctions_test:
if feature in overlapping_junctions_test[i]:
overlapping_junctions_test[i].append(f2)
existing.append(f2)
elif f2 in existing and feature not in existing:
for i in overlapping_junctions_test:
if f2 in overlapping_junctions_test[i]:
overlapping_junctions_test[i].append(feature)
existing.append(feature)
elif feature in existing and f2 in existing:
for i in overlapping_junctions_test:
if feature in overlapping_junctions_test[i]:
loc1=i
if f2 in overlapping_junctions_test[i]:
loc2=i
if loc1!=loc2:
for jun in overlapping_junctions_test[loc2]:
if jun not in overlapping_junctions_test[loc1]:
overlapping_junctions_test[loc1].append(jun)
del overlapping_junctions_test[loc2]
cluster_junction={}
#Finding clusters and corresponding junctions
for count in overlapping_junctions_test:
for feature in overlapping_junctions_test[count]:
cluster_junction[feature]=cluster_name
#print feature,cluster_name
pass
cluster_id+=1
cluster_name=string.join('NewClu_'+str(cluster_id))
cluster_name=cluster_name.replace(" ","")
return cluster_junction,cluster_name,cluster_id,count
if __name__ == '__main__':
uids = {1:'chr8:134583936-134558169',
2:'chr8:134583936-134574921',
3:'chr8:134558017-134519564',
4:'chr8:134558017-134511432',
5:'chr8:134511378-134478333',
6:'chr8:134478137-134477336',
7:'chr8:134475657-134472180',
8:'chr8:134474118-134472180',
9:'chr8:134583936-134579563',
10:'chr8:134511378-134488843',
11:'chr8:134487962-134478333',
12:'chr8:134488521-134488317',
13:'chr8:134478137-134477200',
14:'chr8:134475657-134474237'}
cluster_name='NewClu_1'
cluster_id=1
count=1
features = createFeatures(uids)
cluster_junction,cluster_name,cluster_id,count = filterByLocalJunctionExp(features,cluster_name,cluster_id,count)
cluster_junction,cluster_name,cluster_id,count = filterByLocalJunctionExp(features,cluster_name,cluster_id,count)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/junctionGraph.py
|
junctionGraph.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import math
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from stats_scripts import statistics
import export
import os.path
import unique
import time
import AltAnalyze
def filepath(filename):
fn = unique.filepath(filename)
return fn
def read_directory(sub_dir):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
###Code to prevent folder names from being included
for entry in dir_list:
if entry[-4:] == ".txt" or entry[-4:] == ".csv": dir_list2.append(entry)
return dir_list2
def eliminate_redundant_dict_values(database):
db1={}
for key in database:
list = unique.unique(database[key])
list.sort()
db1[key] = list
return db1
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
########### Begin Analyses ###########
def importSplicingAnnotations(species,array_type,avg_all_for_ss):
if array_type == 'exon' or array_type == 'gene': probeset_type = 'full'
else: probeset_type = 'all'
exon_db,constitutive_probeset_db = AltAnalyze.importSplicingAnnotations(array_type,species,probeset_type,avg_all_for_ss,root_dir)
return exon_db,constitutive_probeset_db
def import_altmerge(filename,array_type):
global exon_db
fn=filepath(filename)
altmerge_constituitive ={}; constituitive_gene={}; constitutive_original={}
exon_db={} #use this as a general annotation database
count = 0; x = 0
original_probesets_add = 0
if array_type == 'AltMouse':
for line in open(fn,'rU').xreadlines():
probeset_data = cleanUpLine(line) #remove endline
probeset,affygene,exons,transcript_num,transcripts,probe_type_call,ensembl,block_exon_ids,block_structure,comparison_info = string.split(probeset_data,'\t')
###note: currently exclude comparison_info since not applicable for existing analyses
if x == 0: x = 1
else:
probe_data = affygene,exons,ensembl,block_exon_ids,block_structure
if exons[-1] == '|': exons = exons[0:-1]
if affygene[-1] == '|': affygene = affygene[0:-1]
exon_db[probeset] = affygene
if probe_type_call == 'gene': #looked through the probe annotations and the gene seems to be the most consistent constituitive feature
altmerge_constituitive[probeset] = affygene
else:
for line in open(fn,'rU').xreadlines():
probeset_data = cleanUpLine(line) #remove endline
if x == 0: x = 1
else:
probeset_id, exon_id, ensembl_gene_id, transcript_cluster_id, chromosome, strand, probeset_start, probeset_stop, affy_class, constitutitive_probeset, ens_exon_ids, ens_const_exons, exon_region, exon_region_start, exon_region_stop, splicing_event, splice_junctions = string.split(probeset_data,'\t')
#probeset_id, exon_id, ensembl_gene_id, transcript_cluster_id, chromosome, strand, probeset_start, probeset_stop, affy_class, constitutitive_probeset, ens_exon_ids, exon_annotations = string.split(line,'\t')
probe_data = ensembl_gene_id
exon_db[probeset_id] = probe_data
original_constitutitive_probeset_call = constitutitive_probeset
if len(splicing_event)>0: constitutitive_probeset = 'no'
if constitutitive_probeset == 'yes':
altmerge_constituitive[probeset_id] = ensembl_gene_id
constituitive_gene[ensembl_gene_id]=[]
if original_constitutitive_probeset_call == 'yes':
try: constitutive_original[ensembl_gene_id].append(probeset_id)
except KeyError: constitutive_original[ensembl_gene_id] = [probeset_id]
###If no constitutive probesets for a gene as a result of additional filtering (removing all probesets associated with a splice event), add these back
for gene in constitutive_original:
if gene not in constituitive_gene:
original_probesets_add +=1
for probeset in constitutive_original[gene]: altmerge_constituitive[probeset] = gene
if array_type == 'RNASeq': id_name = 'junction IDs'
else: id_name = 'array IDs'
print original_probesets_add, 'genes not viewed as constitutive as a result of filtering ',id_name,' based on splicing evidence, added back'
return exon_db, altmerge_constituitive
def parse_input_data(filename,data_type):
fn=filepath(filename); first_line = 1; array_group_name_db = {}; z=0; array_group_db = {}; output_file = []
#print "Reading",filename
secondary_data_type = export.getParentDir(filename) ### e.g., expression or counts
for line in open(fn,'rU').xreadlines():
data = cleanUpLine(line); t = string.split(data,'\t'); probeset = t[0]; z+=1
if first_line == 1:
first_line = 0 #makes this value null for the next loop of actual array data
###Below ocucrs if the data is raw opposed to precomputed
if data_type == 'export':
if array_type == 'exon': folder = 'ExonArray'+'/'+species + '/'
elif array_type == 'gene': folder = 'GeneArray'+'/'+species + '/'
elif array_type == 'junction': folder = 'JunctionArray'+'/'+species + '/'
elif array_type == 'RNASeq': folder = 'RNASeq'+'/'+species + '/'
else: folder = array_type + '/'
parent_path = root_dir+'AltExpression/'+folder
if array_type == 'RNASeq':
output_file = altanalzye_input[0:-4] + '.ExpCutoff-' + str(original_exp_threshold) +'_'+ filter_method+'.txt'
else:
output_file = altanalzye_input[0:-4] + '.p' + str(int(100*p)) +'_'+ filter_method+'.txt'
output_file_dir = parent_path+output_file
print "...Exporting",output_file_dir
export_data = export.createExportFile(output_file_dir,root_dir+'AltExpression/'+folder)
fn=filepath(output_file_dir); export_data = open(fn,'w');
export_data.write(line)
if ':' in t[1]:
array_group_list = []; x=0 ###gives us an original index value for each entry in the group
for entry in t[1:]:
array_group,array_name = string.split(entry,':')
try:
array_group_db[array_group].append(x)
array_group_name_db[array_group].append(array_name)
except KeyError:
array_group_db[array_group] = [x]
array_group_name_db[array_group] = [array_name]
### below only occurs with a new group addition
array_group_list.append(array_group) #use this to generate comparisons in the below linked function
x += 1
#print '##### array_group_list',array_group_list
elif len(probeset)>0 and data_type != 'export':
###Use the index values from above to assign each expression value to a new database
temp_group_array={}; array_index_list = [] ###Use this list for permutation analysis
for group in array_group_db:
#array_index_list.append(array_group_db[group])
group_values = []
for array_index in array_group_db[group]:
try: exp_val = float(t[array_index+1])
except IndexError: print t, z,'\n',array_index,'\n',group, probeset;kill
group_values.append(exp_val)
avg_stat = statistics.avg(group_values)
if data_type == 'expression':
###If non-log array data
if exp_data_format == 'non-log':
### This works better for RNASeq as opposed to log transforming and then filtering which is more stringent and different than the filtering in ExonArray().
if array_type == 'RNASeq':
if normalization_method == 'RPKM' and secondary_data_type == 'expression':
if ':I' in probeset: k=1 ### Don't require an RPKM threshold for intron IDs (these will likely never meet this unless small or fully retained and highly expressed)
elif ':' not in probeset:
if avg_stat>=gene_rpkm_threshold: k=1
else: k=0
elif avg_stat>=exon_rpkm_threshold: k=1
elif '-' in probeset: k=1 ### Don't consider RPKM for junctions, just counts
else: k=0
#if 'ENSMUSG00000045991:E2.2' in probeset: print [probeset, normalization_method, secondary_data_type, gene_rpkm_threshold, avg_stat, k]
else: ### Otherwise, we are looking at count data
if '-' in probeset: ### junction meeting minimum read-count number
if avg_stat>=junction_exp_threshold: k=1 ### junction_exp_threshold is the same as nonlog_exp_threshold
else: k=0
elif ':' not in probeset:
if avg_stat>=gene_exp_threshold: k=1
else: k=0
else: ### exon or intron meeting minimum read-count number
if avg_stat>=exon_exp_threshold: k=1
else: k=0
#if 'ENSMUSG00000045991:E2.2' in probeset: print [probeset, normalization_method, secondary_data_type, exon_exp_threshold, junction_exp_threshold, avg_stat, k]
else:
if avg_stat>=nonlog_exp_threshold: k=1
else: k=0
elif avg_stat>=log_expression_threshold: k=1
else: k=0
if normalization_method == 'RPKM' and secondary_data_type == 'expression': ### Treat as dabp p-value
try: pvalue_status_db[probeset].append(k)
except KeyError: pvalue_status_db[probeset] = [k]
else:
try: expression_status_db[probeset].append(k)
except KeyError: expression_status_db[probeset] = [k]
#if probeset == '3209315': print [group],k,len(group_values),array_group_list
if data_type == 'p-value':
if avg_stat<=p: k=1
else: k=0
#if 'G7216513_a_at' in probeset: print k, avg_stat
try: pvalue_status_db[probeset].append(k)
except KeyError: pvalue_status_db[probeset] = [k]
elif data_type == 'export':
if exp_data_format == 'non-log':
### This code was added in version 1.16 in conjunction with a switch from logstatus to
### non-log in AltAnalyze to prevent "Process AltAnalyze Filtered" associated errors
exp_values = t[1:]; exp_values_log2=[]
for exp_val in exp_values:
exp_values_log2.append(str(math.log(float(exp_val),2))) ### exp_val+=1 was removed in 2.0.5
line = string.join([probeset]+exp_values_log2,'\t')+'\n'
try: null = export_db[probeset]; export_data.write(line)
except KeyError: null = [] ### occurs if not a probeset to include in the filtered results export file
if data_type == 'export': export_data.close()
return output_file
def expr_analysis(filename,filename2,altmerge_constituitive,exon_db,analyze_dabg):
"""import list of expression values for arrayids and calculates statistics"""
constitutive_keep={}; keep_probesets={}; keep_genes={}
global expression_status_db; global pvalue_status_db; global export_db
expression_status_db={}; pvalue_status_db={}; export_db={}
if normalization_method == 'RPKM':
parse_input_data(filename,'expression') ### Parse as a DABG p-value file
expression_file = filename
expression_file = string.replace(expression_file,'\\','/')
filename = string.replace(expression_file,'/expression/','/counts/') ### Set this to the counts. file
parse_input_data(filename,'expression') ### Parse expression file
if analyze_dabg == 'yes':
parse_input_data(filename2,'p-value') ### Parse DABG p-value file
if normalization_method == 'RPKM': filename = expression_file ### Set this back to the exp. file
count=0; probesets_not_found=0
for probeset in expression_status_db:
proceed = 'no'; count+=1
if probeset in pvalue_status_db: proceed = 'yes' ### Indicates there are both expression and dabg files with the same probe sets
elif normalization_method == 'RPKM': proceed = 'no' ### Indicates the rpkm expression file exon/junction does not meet the required expression threshold
elif analyze_dabg == 'no': proceed = 'yes' ### Indicates there is only an expression file and no dabg
if proceed == 'yes':
try: exp_stats = expression_status_db[probeset]; exp_stat1,exp_stat2 = exp_stats[:2]
except Exception:
print 'probeset:',probeset, 'count:',count
print "expression values (should only be 2):",expression_status_db[probeset]
print 'expression file:', filename
print 'dabg file:', filename2
print "length of expression_status_db", len(expression_status_db)
print "UNEXPECTED ERROR ENCOUNTERED - REPORT THIS ERROR TO THE ALTANALYZE HELP DESK"
forceBadExit
if analyze_dabg == 'yes' or normalization_method == 'RPKM': p_stats = pvalue_status_db[probeset]; p_stat1,p_stat2 = p_stats[:2]
else: p_stat1=1; p_stat2=1 ### Automatically assigned an "expressed" call.
try:
ed = exon_db[probeset] ### This is where the exception is
try: affygene = ed.GeneID()
except Exception: affygene = exon_db[probeset]
if exp_stat1 == 1 and p_stat1 == 1: k = 1 ### Thus it is "expressed"
else: k = 0
if exp_stat2 == 1 and p_stat2 == 1: b = 1 ### Thus it is "expressed"
else: b = 0
#if 'ENSMUSG00000045991:E2.2' in probeset: print b,k,affygene,(probeset,exp_stat1,p_stat1, exp_stat2, p_stat2),pvalue_status_db[probeset]
if probeset in altmerge_constituitive:
if b == 1 or k == 1:
keep_probesets[probeset] = [] ### If either have an "expressed" call, keep... but evaluate constitutive below
try: keep_genes[affygene].append(probeset)
except KeyError: keep_genes[affygene] = [probeset]
if b==1 and k==1:
""" This will only keep a gene in the analysis if at least one probeset is 'expressed' in both conditions. Although this can
result in "constitutive probesets with expression in only one group, it solves the problem of treating all core as "constitutive"."""
constitutive_keep[affygene] = []
else:
if b == 0 and k == 0: null = []
else:
keep_probesets[probeset] = []
try: keep_genes[affygene].append(probeset)
except KeyError: keep_genes[affygene] = [probeset]
except Exception: probesets_not_found+=0
if probesets_not_found!=0:
print probesets_not_found, 'AltAnalyze IDs missing from database! Possible version difference relative to inputs.'
for gene in constitutive_keep:
probeset_list = keep_genes[gene]
for probeset in probeset_list: export_db[probeset]=[]
output_file = parse_input_data(filename,'export') ### Parse expression file
expression_status_db={}; pvalue_status_db={}; export_db={}
return output_file
def combine_profiles(profile_list):
profile_group_sizes={}
for db in profile_list:
for key in db: profile_group_sizes[key] = len(db[key])
break
new_profile_db={}
for key in profile_group_sizes:
x = profile_group_sizes[key] ###number of elements in list for key
new_val_list=[]; i = 0
while i<x:
temp_val_list=[]
for db in profile_list:
if key in db: val = db[key][i]; temp_val_list.append(val)
i+=1; val_avg = statistics.avg(temp_val_list); new_val_list.append(val_avg)
new_profile_db[key] = new_val_list
return new_profile_db
########### Misc. Functions ###########
def eliminate_redundant_dict_values(database):
db1={}
for key in database:
list = unique.unique(database[key])
list.sort()
db1[key] = list
return db1
def remoteRun(fl,Species,Array_type,expression_threshold,filter_method_type,p_val,express_data_format,altanalyze_file_list,avg_all_for_ss):
start_time = time.time()
global p; global filter_method; global exp_data_format; global array_type; global species; global root_dir; global original_exp_threshold
global normalization_method; global exon_exp_threshold; global gene_rpkm_threshold; global junction_exp_threshold
global exon_rpkm_threshold; global gene_exp_threshold
original_exp_threshold = expression_threshold
aspire_output_list=[]; aspire_output_gene_list=[]
filter_method = filter_method_type
altanalyze_files = altanalyze_file_list
p = p_val; species = Species; array_type = Array_type
exp_data_format = express_data_format
### Define global variables from the object fl
try: normalization_method = fl.FeatureNormalization()
except Exception: normalization_method = 'NA'
try: exon_exp_threshold = fl.ExonExpThreshold()
except Exception: exon_exp_threshold = 0
try: gene_rpkm_threshold = fl.RPKMThreshold()
except Exception: gene_rpkm_threshold = 0
root_dir = fl.RootDir()
try: junction_exp_threshold = fl.JunctionExpThreshold()
except Exception: junction_exp_threshold = 0
try: exon_rpkm_threshold = fl.ExonRPKMThreshold()
except Exception: exon_rpkm_threshold = 0
try: gene_exp_threshold = fl.GeneExpThreshold()
except Exception: gene_exp_threshold = 0
if 'exon' in array_type: array_type = 'exon' ###In AnalayzeExpressionDataset module, this is named 'exon-array'
global log_expression_threshold; global nonlog_exp_threshold; nonlog_exp_threshold = expression_threshold
try: log_expression_threshold = math.log(expression_threshold,2)
except Exception: log_expression_threshold = 0 ###Occurs if expression_threshold == 0
import_dir = root_dir+'AltExpression/pre-filtered/expression/'; import_dir_dabg = root_dir+'AltExpression/pre-filtered/dabg/'
try: dir_list = read_directory(import_dir) #send a sub_directory to a function to identify all files in a directory
except Exception: dir_list=[]
try: dir_list2 = read_directory(import_dir_dabg)
except Exception: dir_list2=[]
if len(altanalyze_files) == 0: altanalyze_files = dir_list ###if no filenames input
if array_type == 'RNASeq':
altmerge_db = root_dir+'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_junctions.txt'
elif array_type != 'AltMouse': altmerge_db = 'AltDatabase/'+species+'/'+array_type+'/'+species+'_Ensembl_probesets.txt'
else: altmerge_db = "AltDatabase/"+species+"/"+array_type+"/MASTER-probeset-transcript.txt"
###Import probe-level associations
if array_type != 'AltMouse':
exon_db,altmerge_constituitive = importSplicingAnnotations(species,array_type,avg_all_for_ss)
else:
exon_db,altmerge_constituitive = import_altmerge(altmerge_db,array_type) ### Prior to version 2.0, this function was distinct from that in AltAnalyze(), so replaced it for consistency
global altanalzye_input; altanalyze_output=[]
if len(dir_list)>0:
for altanalzye_input in dir_list: #loop through each file in the directory to output results
if altanalzye_input in altanalyze_files:
if altanalzye_input in dir_list2: analyze_dabg = 'yes'
else: analyze_dabg = 'no'
ind_start_time = time.time()
array_db = import_dir + "/"+ altanalzye_input
dabg_db = import_dir_dabg + "/"+ altanalzye_input
#array_db = array_db[1:] #not sure why, but the '\' needs to be there while reading initally but not while accessing the file late
#dabg_db = dabg_db[1:]
dataset_name = altanalzye_input[0:-4] + '-'
print "Begining to filter",dataset_name[0:-1]
#print "Array type is:",array_type
#print "Species is:", species
#print "Expression format is:",exp_data_format
#print "DABG p-value cut-off is:",p
#print "Filter method is:",filter_method
#print "Log2 expression cut-off is:",log_expression_threshold
###Import expression data and stats
try:
output_file = expr_analysis(array_db,dabg_db,altmerge_constituitive,exon_db,analyze_dabg) #filter the expression data based on fold and p-value OR expression threshold
altanalyze_output.append(output_file)
except KeyError: print "Impropper array type (",dataset_name[0:-1],") for",array_type,species,'. Skipping array.'
ind_end_time = time.time(); time_diff = int(ind_end_time-ind_start_time)
#print dataset_name,"filtering finished in %d seconds" % time_diff
end_time = time.time(); time_diff = int(end_time-start_time)
#print "Filtering complete for all files in %d seconds" % time_diff
AltAnalyze.clearObjectsFromMemory(exon_db)
exon_db={}; altmerge_constituitive={}; constitutive_probeset_db={}
else: print "No expression files to filter found..."
return altanalyze_output
if __name__ == '__main__':
m = 'Mm'; h = 'Hs'
Species = h
x = 'AltMouse'; y = 'exon'
Array_type = y
l = 'log'; n = 'non-log'
express_data_format = l
p_val = 0.75
p_val = 0.05
filter_method_type = 'average'
expression_threshold = 30
avg_all_for_ss = 'yes'
import UI
loc = '/Users/saljh8/Desktop/dataAnalysis/AltAnalyze/CP-GSE13297_RAW/'
fl = UI.ExpressionFileLocationData('','','',''); fl.setRootDir(loc)
altanalyze_file_list = ['Hs_Exon_CP_vs_wt.txt']
remoteRun(fl,Species,Array_type,expression_threshold,filter_method_type,p_val,express_data_format,altanalyze_file_list,avg_all_for_ss)
sys.exit()
print "Filter Data For:"
print "1) Human 1.0 ST exon data\n2) AltMouse"
inp = sys.stdin.readline(); inp = inp.strip()
if inp == "1": species = h; p = 0.05; array_type = y; expression_threshold = 70
elif inp == "2": species = m; p = 0.75; expression_threshold = 0
altanalyze_files = []
remoteRun(species,array_type,expression_threshold,filter_method,p,exp_data_format,altanalyze_files)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/FilterDabg.py
|
FilterDabg.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
from sklearn.metrics.cluster import adjusted_rand_score
import numpy as np
def ari(truelabel,predlabel):
lab={}
truelab=[]
predlab=[]
for line in open(truelabel,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
lab[t[0]]=[int(t[1]),]
for line in open(predlabel,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
try:lab[t[0]].append(int(t[1]))
except Exception: print "Sample missing true label"
for key in lab:
try:
predlab.append(lab[key][1])
truelab.append(lab[key][0])
except Exception:
print "Sample missing predicted label"
continue
print len(truelab)
truelab=np.array(truelab)
predlab=np.array(predlab)
ari=adjusted_rand_score(truelab,predlab)
return ari
#truelabel="/Volumes/Pass/Archive_Zeisel/SVMOutputs/groups.round1SVC_Results_max.txt"
#predlabel="/Volumes/Pass/Singlecellbest/Zeisel_upd/SVMOutputs/round1SVC_Results.txt"
#predlabel="/Volumes/Pass/Singlecellbest/Zeisel_upd/SVMOutputs/round1SVC_Results.txt"
#truelabel="/Volumes/Pass/Singlecellbest/Pollen_upd/SVMOutputs/groups.round1SVC_Results_max.txt"
#predlabel="/Volumes/Pass/Singlecellbest/Pollen_upd/SVMOutputs/round1SVC_Results.txt"
#predlabel="/Volumes/Pass/Data/Pollen_cluster.txt"
#predlabel="/Users/meenakshi/Usoskin_Sc3_test.txt"
#truelabel="/Volumes/Pass/Singlecellbest/Usoskin_upd/SVMOutputs/groups.round1SVC_Results_max.txt"
#predlabel="/Users/meenakshi/Downloads/k-11-Usoskin.txt"
#predlabel="/Users/meenakshi/Documents/ZeiselCluster.txt"
#truelabel="/Users/meenakshi/Desktop/groups.Pollen.txt"
#predlabel="/Users/meenakshi/Downloads/SC3_pollen.txt"
#predlabel="/Users/meenakshi/groups-filtered.txt"
truelabel="/Users/meenakshi/Documents/Singlecelldata/groups.CD34.v5_test.txt"
#predlabel="/Volumes/Pass/HCA_ICGS/ICGS_complete/SVMOutputs/round1SVC_Results_old30.txt"
#truelabel="/Volumes/Pass/HCA_ICGS/ICGS_complete/SVMOutputs/round1SVC_Results.txt"
#truelabel="/Volumes/Pass/HCA_Umap/SVMOutputs/groups.round1SVC_Results_max3.txt"
#truelabel="/Users/meenakshi/cluster-filtered.txt"
#predlabel="/Volumes/Pass/Immune-complete/SVMOutputs/round1SVC_Results_HGV_UMAP_PR_0.2_0.2.txt"
#truelabel="/Volumes/Pass/paperResults/tsne_in/Known_groups.txt"
predlabel="/Volumes/Pass/ICGS2_testrun/ICGS-NMF/FinalGrous_Merged.txt"
#truelabel="/Volumes/Pass/Final_scicgs/groups.round1SVC_Results-orig.txt"
#predlabel="/Volumes/Pass/HCA_Umap/SVMOutputs/round1SVC_Results.txt"
#predlabel="/Volumes/Pass/Deng/SVMOutputs/round1SVC_Results.txt"
#truelabel="/Volumes/Pass/Deng/groups.Deng.txt"
#truelabel="/Users/meenakshi/Documents/paperResults/tsne_in/donor_all.txt"
#predlabel="/Users/meenakshi/Documents/paperResults/tsne_in/round1SVC_Results_HGV_UMAP_PR_0.2_0.2.txt"
arival=ari(truelabel,predlabel)
print str(arival)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/ARI.py
|
ARI.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy as np
import os.path
from numpy import corrcoef, sum, log, arange
from scipy.stats.stats import pearsonr
import traceback
tandem = dict()
dem=dict()
new=dict()
samplelis=[]
key_length=[]
list_g=[]
lis1=[]
correc=dict()
lis2=[]
def create_corr_matrix(lines,tfs, gene_labels):
maskedArray = False
header_row = True
#print len(lines),len(tfs)
for l in range(len(lines)):
line = lines[l]
line = line.rstrip()
t=string.split(line,'\t')
t1=t[0]
if header_row:
header_row = False
columns = len(t[1:])
if t1 in tfs:
t=t[1:]
if columns-len(t)>0:
t += ['']*(columns-len(t)) ### Add NAs for empty columns
if '' in t:
maskedArray = True
t = ['0.000101' if i=='' else i for i in t]
t = map(float,t)
t = np.ma.masked_values(t,0.000101)
for k in range(len(lines)):
if k ==0:
continue
list1=[]
list2=[]
ind=[]
linek = lines[k]
linek = linek.rstrip()
p=string.split(linek,'\t')
p=p[1:]
if columns-len(p)>0:
p += ['']*(columns-len(p)) ### Add NAs for empty columns
if '' in p:
p = ['0.000101' if i=='' else i for i in p]
p = map(float,p)
p = np.ma.masked_values(p,0.000101)
for i in range(len(t)-1):
if(t[i]!='' and p[i]!=''):
ind.append(i)
else:
continue
for i in range(len(ind)-1):
list1.append(float(t[ind[i]]))
list2.append(float(p[ind[i]]))
if len(list1)==0 or len(list2)==0:
print l;sys.exit()
correc[t1,gene_labels[k-1]]=0
continue
else:
if (max(list1)-min(list1))>0 and (max(list2)-min(list2)):
if maskedArray:
coefr=np.ma.corrcoef(list1,list2)
coef = coefr[0][1]
else:
coefr=pearsonr(list1,list2)
coef=coefr[0]
correc[t1,gene_labels[k-1]]=coef
return correc, maskedArray
def strip_first_col(fname, delimiter=None):
with open(fname, 'r') as fin:
for line in fin:
try:
yield line.split(delimiter, 1)[1]
except IndexError:
continue
def genelist(fname,Filter=None):
head=0
genes=[]
for line in open(fname,'rU').xreadlines():
line = line.rstrip(os.linesep)
t=string.split(line,'\t')
gene=t[0]
if Filter!=None:
if gene in Filter:
genes.append(gene)
else:
genes.append(gene)
return genes
def sample(fname):
head=0
for line in open(fname,'rU').xreadlines():
line = line.rstrip(os.linesep)
if head ==0:
t=string.split(line,'\t')
#print t
for p in range(9,len(t)):
samplelis.append(t[p])
head=1
else:
break;
return samplelis
def create_corr_files(correc,filename, tfs, gene_labels,maskedArray=False):
export_corrmat=open(filename[:-4]+'-corr.txt','w')
#export_corrmat=open('correlation_matrix_up.txt','w')
temp = ['UID']
for li in range(len(gene_labels)):
temp.append(gene_labels[li])
export_corrmat.write(string.join(temp,'\t')+'\n')
for i in range(len(tfs)):
export_corrmat.write(tfs[i]+'\t')
for li in range(len(gene_labels)):
try:
export_corrmat.write(str(correc[tfs[i],gene_labels[li]])+'\t')
except Exception:
print traceback.format_exc()
#export_corrmat.write('NA')
#else:
# export_corrmat.write(str(0)+'\t')
export_corrmat.write('\n')
#export_corrmat.write('\n')
export_corrmat.close()
def strip_first_col(fname, delimiter=None):
with open(fname, 'r') as fin:
for line in fin:
try:
yield line.split(delimiter, 1)[1]
except IndexError:
continue
def runTFCorrelationAnalysis(query_exp_file,query_tf_file):
query_data = open(query_exp_file,'rU')
lines = query_data.readlines()
print "Number or rows in file:",len(lines)
query_data.close()
genes=genelist(query_exp_file)
genes = genes[1:]
tfs=genelist(query_tf_file,Filter=genes) ### Require that the TF is in the gene list
correc,maskedArray=create_corr_matrix(lines,tfs,genes)
create_corr_files(correc,query_exp_file,tfs,genes,maskedArray=maskedArray)
if __name__ == '__main__':
import getopt
filter_rows=False
filter_file=None
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Insufficient arguments";sys.exit()
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['i=','t='])
#print sys.argv[1:]
for opt, arg in options:
if opt == '--i': query_exp_file=arg
elif opt == '--t': query_tf_file=arg
runTFCorrelationAnalysis(query_exp_file,query_tf_file)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/TFCorrelationPlot.py
|
TFCorrelationPlot.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy as np
import scipy as sp
import sys, pickle, pdb
import scipy.stats as st
import scipy.interpolate
def estimate(pv, m = None, verbose = False, lowmem = False, pi0 = None):
"""
Estimates q-values from p-values
Args
=====
m: number of tests. If not specified m = pv.size
verbose: print verbose messages? (default False)
lowmem: use memory-efficient in-place algorithm
pi0: if None, it's estimated as suggested in Storey and Tibshirani, 2003.
For most GWAS this is not necessary, since pi0 is extremely likely to be
1
"""
assert(pv.min() >= 0 and pv.max() <= 1), "p-values should be between 0 and 1"
original_shape = pv.shape
pv = pv.ravel() # flattens the array in place, more efficient than flatten()
if m == None:
m = float(len(pv))
else:
# the user has supplied an m
m *= 1.0
# if the number of hypotheses is small, just set pi0 to 1
if len(pv) < 100 and pi0 == None:
pi0 = 1.0
elif pi0 != None:
pi0 = pi0
else:
# evaluate pi0 for different lambdas
pi0 = []
lam = sp.arange(0, 0.90, 0.01)
counts = sp.array([(pv > i).sum() for i in sp.arange(0, 0.9, 0.01)])
for l in range(len(lam)):
pi0.append(counts[l]/(m*(1-lam[l])))
pi0 = sp.array(pi0)
# fit natural cubic spline
tck = sp.interpolate.splrep(lam, pi0, k = 3)
pi0 = sp.interpolate.splev(lam[-1], tck)
if pi0 > 1:
if verbose:
print("got pi0 > 1 (%.3f) while estimating qvalues, setting it to 1" % pi0)
pi0 = 1.0
assert(pi0 >= 0 and pi0 <= 1), "pi0 is not between 0 and 1: %f" % pi0
if lowmem:
# low memory version, only uses 1 pv and 1 qv matrices
qv = sp.zeros((len(pv),))
last_pv = pv.argmax()
qv[last_pv] = (pi0*pv[last_pv]*m)/float(m)
pv[last_pv] = -sp.inf
prev_qv = last_pv
for i in xrange(int(len(pv))-2, -1, -1):
cur_max = pv.argmax()
qv_i = (pi0*m*pv[cur_max]/float(i+1))
pv[cur_max] = -sp.inf
qv_i1 = prev_qv
qv[cur_max] = min(qv_i, qv_i1)
prev_qv = qv[cur_max]
else:
p_ordered = sp.argsort(pv)
pv = pv[p_ordered]
qv = pi0 * m/len(pv) * pv
qv[-1] = min(qv[-1],1.0)
for i in xrange(len(pv)-2, -1, -1):
qv[i] = min(pi0*m*pv[i]/(i+1.0), qv[i+1])
# reorder qvalues
qv_temp = qv.copy()
qv = sp.zeros_like(qv)
qv[p_ordered] = qv_temp
# reshape qvalues
qv = qv.reshape(original_shape)
return qv
if __name__ == '__main__':
pv = np.random.uniform(0.0, 1.0, size = (1000,))
pv.sort()
print pv[:10],pv[-10:]
pv = estimate(pv)
pv = map(float,pv)
print pv[:10],pv[-10:]
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/qvalue.py
|
qvalue.py
|
import numpy as np
import sys,string
import os
import os.path
from collections import defaultdict
try:from stats_scripts import statistics
except Exception: import statistics
import export; reload(export)
import re
from stats_scripts import fishers_exact_test
import traceback
import warnings
import math
import export
def importDenominator(denom_dir):
denominator_events={}
firstRow=True
for line in open(denom_dir,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstRow:
uid_index = t.index('UID')
firstRow=False
else:
uid = t[uid_index]
denominator_events[uid]=[]
return denominator_events
def importEvents(folder,denom={}):
### For example, knockdown signatures
import collections
unique_events = {}
import UI
files = UI.read_directory(folder)
comparison_events={}
for file in files:
if '.txt' in file and 'PSI.' in file and '._PSI' not in file:
fn = folder+'/'+file
firstLine = True
comparison = file[:-4]
comparison_events[comparison,'inclusion']=[]
comparison_events[comparison,'exclusion']=[]
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if firstLine:
try: event_index = t.index('Event-Direction')
except:
try: event_index = t.index('Inclusion-Junction') ### legacy
except: print file, 'Event-Direction error';sys.exit()
firstLine= False
continue
event = t[0]
#event = string.split(event,'|')[0]
unique_events[event]=[]
event_dictionary = comparison_events[comparison,t[event_index]]
if len(denom)>0:
if event in denom:
event_dictionary.append(event)
else:
event_dictionary.append(event)
return unique_events,comparison_events
def performMutualEnrichment(unique_inp_events,event_inp_dictionary,unique_ref_events,event_ref_dictionary):
N = len(unique_inp_events)
N = 88000
for (comparison,direction) in event_inp_dictionary:
if direction == 'inclusion': alt_direction = 'exclusion'
else: alt_direction = 'inclusion'
comparison_events1 = event_inp_dictionary[(comparison,direction)]
comparison_events2 = event_inp_dictionary[(comparison,alt_direction)]
for (reference_comp,ref_direction) in event_ref_dictionary:
if direction == ref_direction and direction == 'inclusion':
if ref_direction == 'inclusion': alt_ref_direction = 'exclusion'
else: alt_ref_direction = 'inclusion'
ref_events1 = event_ref_dictionary[(reference_comp,ref_direction)]
ref_events2 = event_ref_dictionary[(reference_comp,alt_ref_direction)]
concordant1 = len(list(set(comparison_events1) & set(ref_events1)))
concordant2 = len(list(set(comparison_events2) & set(ref_events2)))
r1 = concordant1+concordant2
n = len(ref_events1)+len(ref_events2)
R = len(comparison_events1)+len(comparison_events2)
disconcordant1 = len(list(set(comparison_events1) & set(ref_events2)))
disconcordant2 = len(list(set(comparison_events2) & set(ref_events1)))
r2 = disconcordant1+disconcordant2
if r1>r2:
r1=r1-r2
try: z_concordant = Zscore(r1,n,N,R)
except ZeroDivisionError: z_concordant = 0.0000
z_discordant = 0.0000
else:
r1=r2-r1
try: z_discordant = Zscore(r1,n,N,R)
except ZeroDivisionError: z_discordant = 0.0000
z_concordant=0
#try: z_concordant = Zscore(r1,n,N,R)
#except ZeroDivisionError: z_concordant = 0.0000
#
#try: z_discordant = Zscore(r2,n,N,R)
#except ZeroDivisionError: z_discordant = 0.0000
try: null_z = Zscore(0,n,N,R)
except ZeroDivisionError: null_z = 0.000
### Calculate a Fischer's Exact P-value
import mappfinder
#pval1 = mappfinder.FishersExactTest(r1,n,R,N)
#pval2 = mappfinder.FishersExactTest(r2,n,R,N)
table = [[int(concordant1),int(disconcordant1)], [int(disconcordant2),int(concordant2)]]
print table
try: ### Scipy version - cuts down rutime by ~1/3rd the time
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
oddsratio, pvalue = stats.fisher_exact(table)
# print pvalue
except Exception:
ft = fishers_exact_test.FishersExactTest(table)
# print ft.two_tail_p()
pvalue=ft.two_tail_p()
#if pvalue2< pvalue:
# pvalue=pvalue2
### Store these data in an object
#zsd = mappfinder.ZScoreData(signature,r,n,z,null_z,n)
#zsd.SetP(pval)
print comparison+'\t'+reference_comp+'\t'+ref_direction+'\t'+str(z_concordant)+'\t'+str(z_discordant)+'\t'+str(r2)+'\t'+str(n)+'\t'+str(pvalue)
def Zscore(r,n,N,R):
"""where N is the total number of events measured:
R is the total number of events meeting the criterion:
n is the total number of events in this specific reference gene-set:
r is the number of events meeting the criterion in the examined reference gene-set: """
N=float(N) ### This bring all other values into float space
z = (r - n*(R/N))/math.sqrt(n*(R/N)*(1-(R/N))*(1-((n-1)/(N-1))))
return z
if __name__ == '__main__':
import getopt
mutdict=defaultdict(list)
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['i=','r=','d='])
for opt, arg in options:
if opt == '--i': input_directory=arg
elif opt == '--r':reference_directory=arg
elif opt == '--d': denominator_directory=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
denominator_events = importDenominator(denominator_directory)
unique_ref_events,event_ref_dictionary = importEvents(reference_directory,denom=denominator_events)
#unique_inp_events,event_inp_dictionary = importEvents(input_directory,denom=unique_ref_events)
unique_inp_events,event_inp_dictionary = importEvents(input_directory)
performMutualEnrichment(unique_inp_events,event_inp_dictionary,unique_ref_events,event_ref_dictionary)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/SpliceMutualEnrich2.py
|
SpliceMutualEnrich2.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy as np
import pylab as pl
import os.path
from collections import defaultdict
from sklearn.cluster import KMeans
import export
def strip_first_col(fname, delimiter=None):
with open(fname, 'r') as fin:
for line in fin:
try:
yield line.split(delimiter, 1)[1]
except IndexError:
continue
def header_file(fname, delimiter=None):
head=0
header=[]
with open(fname, 'rU') as fin:
for line in fin:
if head==0:
line = line.rstrip(os.linesep)
header=string.split(line,'\t')
head=1
else:break
return header
def KmeansAnalysis(filename,header,InputFile,turn):
X=defaultdict(list)
prev=""
head=0
for line in open(filename,'rU').xreadlines():
if head >1:
val=[]
line=line.rstrip('\r\n')
q= string.split(line,'\t')
for i in range(2,len(q)):
val.append(float(q[i]))
if q[1]==prev:
X[prev].append(val)
else:
prev=q[1]
X[prev].append(val)
else:
head+=1
continue
for key in X:
print key
X[key]=np.array(X[key])
print X[key].shape
mat=[]
dire= export.findParentDir(export.findParentDir(InputFile)[:-1])
output_dir = dire+'SVMOutputs'
if os.path.exists(output_dir)==False:
export.createExportFolder(output_dir)
exportname=output_dir+'/round'+str(turn)+'Kmeans_result.txt'
#exportname=filename[:-4]+key+'.txt'
export_results=open(exportname,"w")
mat=zip(*X[key])
mat=np.array(mat)
print mat.shape
kmeans = KMeans(n_clusters=2, random_state=0).fit(mat)
y=kmeans.labels_
#cent=kmeans.cluster_centers_
y=y.tolist()
total=len(y)
cent_1=y.count(0)
cent_2=y.count(1)
print cent_1,cent_2
export_results.write("uid"+"\t"+"group"+"\n")
if cent_1<cent_2:
count=2
for j in y:
if j==0:
export_results.write(header[count]+"\t"+"1"+"\n")
else:
export_results.write(header[count]+"\t"+"0"+"\n")
count+=1
else:
count=2
for j in y:
if j==1:
export_results.write(header[count]+"\t"+"1"+"\n")
else:
export_results.write(header[count]+"\t"+"0"+"\n")
count+=1
if __name__ == '__main__':
import getopt
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['Guidefile='])
for opt, arg in options:
if opt == '--Guidefile': Guidefile=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
#Guidefile="/Users/meenakshi/Documents/leucegene/ICGS/Round2_cor_0.6_280default/Clustering-exp.round2_insignificantU2like-Guide1 DDX5&ENSG00000108654&E3.4-E3.9__ENSG0000010-hierarchical_cosine_correlation.txt"
header=header_file(Guidefile)
KmeansAnalysis(Guidefile)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/Kmeans.py
|
Kmeans.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import re
import unique
import export
import math
from stats_scripts import statistics
import traceback
"""
This script takes existing formatted metadata (C4 PCBC approved fields) and filters them to determine unique and non-unique
donors for a specific covariate and derives comparison and group relationships to extract from an existing expression file
"""
def BatchCheck(sample_id,nonpreferential_batchs,preferential_samples,platform):
priority=0
for batch in nonpreferential_batchs:
if batch in sample_id:
priority=1
if platform == 'RNASeq' or platform == 'exon':
if sample_id not in preferential_samples: priority = 1
elif sample_id in preferential_samples: priority = 0
return priority
class MetaData:
def __init__(self,cellLine,donor_id,sex,diffState,quality,coi,vector,genes,lab,public,priority):
self.donor_id = donor_id; self.sex = sex; self.diffState = diffState; self.quality = quality; self.coi = coi
self.cellLine = cellLine; self.vector = vector; self.genes = genes ; self.lab = lab; self.public = public
self.priority = priority
def CellLine(self): return self.cellLine
def Gender(self): return self.sex
def DiffState(self): return self.diffState
def Quality(self): return self.quality
def COI(self): return self.coi
def Vector(self): return self.vector
def Genes(self): return self.genes
def Lab(self): return self.lab
def Public(self): return self.public
def Priority(self): return self.priority
def __repr__(self): return self.CellLine()
def prepareComparisonData(input_file,diffStateQuery,CovariateQuery,uniqueDonors,genderRestricted,platform=None,compDiffState=None,restrictCovariateTerm=None):
removeConfoundedCovariates=True
firstLine = True
notation_db={}; donor_sex_db={}
failed_QC = ['FAIL','bad','EXCLUDE']
nonpreferential_batchs = ['.144.7.','.144.6.','.219.2','.219.5','H9'] ### used when removing non-unique donors
preferential_samples = ['SC11-004A.133.1.7', 'SC11-008B.149.3.14', 'SC11-010A.149.5.16', 'SC11-010B.149.5.18', 'SC11-012A.149.5.19', 'SC11-012B.149.5.20', 'SC11-013A.149.5.21', 'SC11-013B.149.5.22', 'SC12-002A.154.1.4', 'SC12-005A.144.7.19', 'SC12-005B.144.7.21', 'SC12-007.181.7.1', 'SC13-043.420.12.3', 'SC13-044.219.2.9', 'SC13-045.219.5.10', 'SC14-066.558.12.18', 'SC14-067.558.12.19', 'SC14-069.569.12.25', 'IPS18-4-1.102.2.2', 'IPS18-4-2.102.2.4', 'SC11-005B.119.5.9', 'SC11-006A.119.1.4', 'SC11-006B.119.1.10', 'SC11-007A.119.3.5', 'SC11-007B.119.3.1', 'SC11-014A.133.1.13', 'SC11-014B.133.2.4', 'SC11-015A.133.1.14', 'SC11-015B.133.2.5', 'SC11-016A.133.1.8', 'SC11-016B.133.2.15', 'SC11-017A.144.6.16', 'SC11-017B.154.1.2', 'SC11-018A.144.6.18', 'SC11-018B.154.1.3', 'SC12-006A.144.7.22', 'SC12-006B.144.7.23', 'SC12-019.181.7.2', 'SC12-020.181.7.3', 'SC12-022A.172.5.8', 'SC12-024.219.2.7', 'SC12-025A.172.5.9', 'SC12-028.181.7.5', 'SC12-029.181.7.6', 'SC12-030.181.7.4', 'SC12-031.181.7.7', 'SC12-034.182.1.7', 'SC12-035.569.12.16', 'SC12-036.182.2.20', 'SC12-037.182.1.12', 'SC12-038.420.12.1', 'SC13-049.219.2.8']
preferential_samples += ['SC11-010BEB.144.6.6', 'SC13-045EB.219.6.11', 'SC12-005EB.585.2.13', 'SC11-013EB.558.12.7', 'SC13-043BEB.419.12.13', 'SC14-067EB.558.12.1', 'SC13-044EB.219.6.10', 'SC11-012BEB.144.6.7', 'SC14-066EB.585.2.14'] # variable XIST 'SC11-009A.133.3.5', 'SC11-009A.149.5.15'
preferential_samples += ['H9.119.3.7']
unique_covariate_samples={}
covariate_samples={}
sample_metadata={}
cellLineIDs={}
uniqueDonor_db={}
donor_db={}
allCovariateSamples={}
blood = ['CD34+ cells','mononuclear']
integrating = ['lentivirus','retrovirus']
nonintegrating = ['plasmid','RNA','Sendai Virus']
for line in open(input_file,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
if 'Cell_Line_Type' in data:
values = string.replace(data,'_',' ')
values = string.replace(values,'"','')
values = string.split(values,'\t')
headers = values
covariateIndex=None
mergeBlood = False
index=0
for h in headers:
if 'CellType' in h or 'Diffname short' in h: cellTypeIndex = index ### diff-state
#if 'uid' in h: uidIndex = index
if 'Sample' == h or 'Decorated Name' == h: uidIndex = index
if 'CellLine' in h or 'C4 Cell Line ID' in h: cellLineIndex = index
if 'Pass QC' in h: qualityIndex = index
if 'Cell Type of Origin' in h: coiIndex = index
if 'Reprogramming Vector Type' in h: vectorIndex = index
if 'Reprogramming Gene Combination' in h: geneIndex = index
if 'Gender' in h: sex_index = index
if 'originating lab' in h or 'Originating Lab' in h: labIndex = index
if 'High Confidence Donor ID (HCDID)' in h or 'Donor ID' == h:
unique_donor_index = index
if 'ublic' in h: publicIndex = index
if 'C4 Karyotype Result' in h: karyotypeIndex = index
if 'Donor Life Stage' in h: donorStageIndex = index
if 'Other Conditions During Reprogramming' in h: otherConditionIndex = index
if 'Small Molecules' in h: smallMoleculeIndex = index
if CovariateQuery == h: covariateIndex = index
if 'UID' == h: uidIndexAlt = index
index+=1
firstLine = False
if CovariateQuery == 'Cell Type of Origin Combined' and covariateIndex==None:
covariateIndex = headers.index('Cell Type of Origin')
mergeBlood = True
if CovariateQuery == 'originating lab' and covariateIndex==None:
covariateIndex = headers.index('Originating Lab ID')
else:
try: sample_id = values[uidIndex]
except Exception:
uidIndex = uidIndexAlt
sample_id = values[uidIndex]
cellLine = values[cellLineIndex]
try: donor_id = values[unique_donor_index]
except Exception:
#print values
continue
#print len(values), unique_donor_index;sys.exit()
if donor_id == '': donor_id = cellLine
sex = values[sex_index]
diffState = values[cellTypeIndex]
try: quality = values[qualityIndex]
except Exception: quality = ''
COI = values[coiIndex]
vector = values[vectorIndex]
genes = values[geneIndex]
lab = values[labIndex]
public = values[publicIndex]
karyotype = values[karyotypeIndex]
priority = BatchCheck(sample_id,nonpreferential_batchs,preferential_samples,platform)
md = MetaData(cellLine,donor_id,sex,diffState,quality,COI,vector,genes,lab,public,priority)
sample_metadata[sample_id] = md ### store all relevant metadata for future access (e.g., comparing omics types)
try: covariateType = values[covariateIndex]
except Exception: covariateType = None
if CovariateQuery == 'integrating vectors': ### Build this covariate type interactively
if values[vectorIndex] in integrating:
covariateType = 'integrating'
elif values[vectorIndex] in nonintegrating:
covariateType = 'non-integrating'
else:
covariateType = 'NA'
#print covariateType, values[vectorIndex]
#print covariateType, CovariateQuery, diffState, diffStateQuery, public, sample_id;sys.exit()
if covariateType!=None:
try: covariateType = values[covariateIndex] ### e.g., COI
except Exception: pass
if mergeBlood:
if covariateType in blood: covariateType = 'blood'
if covariateType == 'N/A': covariateType = 'NA'
elif covariateType == '': covariateType = 'NA'
elif '/' in covariateType: covariateType = string.replace(covariateType,'/','-')
if (diffState==diffStateQuery or diffState==compDiffState or diffStateQuery=='NA') and (public == 'yes' or public == 'TRUE') and '_PE' not in sample_id and karyotype != 'abnormal':
if quality not in failed_QC:
proceed=True
if genderRestricted!=None:
if genderRestricted == sex: proceed = True
else: proceed = False
if restrictCovariateTerm != None:
if restrictCovariateTerm == covariateType:
proceed = True
if diffState==compDiffState:
covariateType = compDiffState +' '+covariateType ### Make this a unique name
else:
covariateType = diffStateQuery +' '+covariateType ### Make this a unique name
else:
proceed = False
if proceed:
try:
donor_db = unique_covariate_samples[covariateType]
try: donor_db[donor_id].append((priority,sample_id))
except Exception: donor_db[donor_id] = [(priority,sample_id)]
except Exception:
### get unique donor samples sorted by priority
donor_db={donor_id:[(priority,sample_id)]}
unique_covariate_samples[covariateType] = donor_db
try: ### do the same for non-unique data
covariate_samples[covariateType].append(sample_id)
except Exception:
covariate_samples[covariateType] = [sample_id]
else:
if len(cellLine)>0 and diffState==diffStateQuery and public == 'yes' and '_PE' not in sample_id and quality not in failed_QC:
proceed=True
if genderRestricted!=None:
if genderRestricted == sex: proceed = True
else: proceed = False
if proceed:
try: cellLineIDs[cellLine].append((priority,sample_id))
except Exception: cellLineIDs[cellLine] = [(priority,sample_id)]
try: uniqueDonor_db[donor_id].append(cellLine)
except Exception: uniqueDonor_db[donor_id] = [cellLine]
try: donor_db[donor_id].append((priority,cellLine))
except Exception: donor_db[donor_id] = [(priority,cellLine)]
### Now, do an exhaustive search to exclude covariateType that are confounded by another covariate such that either could attribute to the effect
### Only do this relative to the CovariateQuery being compared
if removeConfoundedCovariates:
index=0
for value in values:
header = headers[index]
if len(value)>0:
try: allCovariateSamples[header+':'+value].append(sample_id)
except Exception: allCovariateSamples[header+':'+value] = [sample_id]
index+=1
if len(covariate_samples)==0:
cellLinesPerUniqueDonor={}
for cellLine in cellLineIDs:
cellLineIDs[cellLine].sort()
cellLineIDs[cellLine] = cellLineIDs[cellLine][0][1]
for donor_id in uniqueDonor_db:
for cellLine in uniqueDonor_db[donor_id]:
cellLinesPerUniqueDonor[cellLine]=donor_id
return cellLineIDs,sample_metadata,cellLinesPerUniqueDonor, donor_db
if uniqueDonors:
### Select a single representative sample for each donor
allUniqueDonors={}
for covariateType in unique_covariate_samples:
unique_donors=[]
donor_db = unique_covariate_samples[covariateType]
#print covariateType, donor_db
for donor_id in donor_db:
donor_db[donor_id].sort() ### should now be consistent between different types of covariate analyses in terms of ranking
#print donor_db[donor_id]
uniqueDonorSample = donor_db[donor_id][0][1]
unique_donors.append(uniqueDonorSample)
allUniqueDonors[uniqueDonorSample]=None
covariate_samples[covariateType] = unique_donors
### Check to see which covariates completely conflict
conflictingCovariates={}
for variable in allCovariateSamples:
samples=[]
if uniqueDonors:
for sample in allCovariateSamples[variable]:
if sample in allUniqueDonors: samples.append(sample)
else:
samples = allCovariateSamples[variable]
samples.sort()
try: conflictingCovariates[tuple(samples)].append(variable)
except Exception: conflictingCovariates[tuple(samples)] = [variable]
conflicting=[]
for samples in conflictingCovariates:
if len(conflictingCovariates[samples])>1:
if variable not in conflicting:
if 'Run' not in variable and 'uid' not in variable and 'Sample' not in variable:
if 'other conditions during reprogramming:N/A' not in variable:
conflicting.append(variable)
if len(conflicting)>0:
print 'There were conflicting covariates present:',conflicting
covariates_to_consider=[]
for covariateType in covariate_samples:
if len(covariate_samples[covariateType])>1: ### Thus at least two samples to compare
covariates_to_consider.append(covariateType)
comps_db={}
for covariateType in covariates_to_consider:
covariatePairs=[]
for covariateType2 in covariates_to_consider:
if covariateType!=covariateType2:
covariatePairs = [covariateType,covariateType2]
covariatePairs.sort()
comps_db[tuple(covariatePairs)]=None
groups_db={}
for covariateType in covariates_to_consider:
groups_db[covariateType] = covariate_samples[covariateType]
### print out the associated unique donor samples
for i in covariate_samples[covariateType]: print covariateType,i
#sys.exit()
return sample_metadata,groups_db,comps_db
def performDifferentialExpressionAnalysis(species,platform,input_file,sample_metadata,groups_db,comps_db,CovariateQuery,uniqueDonors):
### filter the expression file for the samples of interest and immediately calculate comparison statistics
firstLine = True
group_index_db={}
pval_summary_db={}
group_avg_exp_db={}
export_object_db={}
rootdir=export.findParentDir(input_file)
#print rootdir;sys.exit()
try:
gene_to_symbol,system_code = getAnnotations(species,platform)
from import_scripts import OBO_import
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
except ZeroDivisionError: gene_to_symbol={}; system_code=''
for groups in comps_db:
group1, group2 = groups
pval_summary_db[groups] = {} ### setup this data structure for later
filename='ExpressionProfiles/'+string.join(groups,'_vs_')+'.txt'
eo = export.ExportFile(rootdir+filename) ### create and store an export object for the comparison (for raw expression)
export_object_db[groups] = eo
for line in open(input_file,'rU').xreadlines():
if '.bed' in line:
line = string.replace(line,'.bed','')
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
header = values
for group in groups_db:
samplesToEvaluate = groups_db[group]
try: sample_index_list = map(lambda x: values.index(x), samplesToEvaluate)
except Exception: ### For datasets with mising samples (removed due to other QC issues)
sample_index_list=[]
filteredSamples=[]
for x in samplesToEvaluate:
try:
sample_index_list.append(values.index(x))
filteredSamples.append(x)
except Exception: pass
samplesToEvaluate = filteredSamples
groups_db[group] = samplesToEvaluate
#print group, sample_index_list
group_index_db[group] = sample_index_list
group_avg_exp_db[group] = {}
### Write out headers for grouped expression values
for (group1,group2) in comps_db:
eo = export_object_db[(group1,group2)]
g1_headers = groups_db[group1]
g2_headers = groups_db[group2]
g1_headers = map(lambda x: group1+':'+x,g1_headers)
g2_headers = map(lambda x: group2+':'+x,g2_headers)
eo.write(string.join(['GeneID']+g1_headers+g2_headers,'\t')+'\n')
firstLine = False
else:
geneID = values[0]
if ',' in geneID:
geneID = string.replace(geneID,',','_')
if 'ENS' in geneID and '.' in geneID and ':' not in geneID:
geneID = string.split(geneID,'.')[0] ### for cufflinks
elif platform == 'RNASeq':
try: geneID = symbol_to_gene[geneID][0]
except Exception: pass
group_expression_values={}
original_group={}
for group in group_index_db:
sample_index_list = group_index_db[group]
if platform != 'exon':
filtered_values = map(lambda x: float(values[x]), sample_index_list) ### simple and fast way to reorganize the samples
else: ### for splice-event comparisons
if len(header) != len(values):
diff = len(header)-len(values)
values+=diff*['']
initial_filtered=[] ### the blanks can cause problems here so we loop through each entry and catch exceptions
unfiltered=[]
for x in sample_index_list:
initial_filtered.append(values[x])
filtered_values=[]
for x in initial_filtered:
if x != '':
filtered_values.append(float(x))
unfiltered.append(x)
#if geneID == 'ENSG00000105321:E3.2-E4.2 ENSG00000105321:E2.3-E4.2' and 'inner cell mass' in group:
#print filtered_values;sys.exit()
if platform == 'exon':
original_group[group]=unfiltered
else:
original_group[group]=filtered_values
if platform == 'RNASeq':# or platform == 'miRSeq':
filtered_values = map(lambda x: math.log(x+1,2),filtered_values) ### increment and log2 adjusted
group_expression_values[group] = filtered_values
for groups in comps_db:
group1,group2 = groups
data_list1 = group_expression_values[group1]
data_list2 = group_expression_values[group2]
if len(data_list1)>1 and len(data_list2)>1: ### For splicing data
p = statistics.runComparisonStatistic(data_list1,data_list2,probability_statistic) ### this is going to just be a oneway anova first
avg1 = statistics.avg(data_list1)
avg2 = statistics.avg(data_list2)
log_fold = avg1-avg2
if platform == 'RNASeq':# or platform == 'miRSeq':
max_avg = math.pow(2,max([avg1,avg2]))-1
else: max_avg = 10000
#if platform == 'miRSeq': max_avg = 10000
valid = True
if max_avg<minRPKM:
log_fold = 'Insufficient Expression'
gs = statistics.GroupStats(log_fold,None,p)
gs.setAdditionalStats(data_list1,data_list2) ### Assuming equal variance
pval_db = pval_summary_db[groups] ### for calculated adjusted statistics
pval_db[geneID] = gs ### store the statistics here
proceed = True
if len(restricted_gene_denominator)>0:
if geneID not in restricted_gene_denominator:
proceed = False
if uniqueDonors == False and proceed:
### store a new instance
gsg = statistics.GroupStats(log_fold,None,p)
gsg.setAdditionalStats(data_list1,data_list2) ### Assuming equal variance
global_adjp_db[CovariateQuery,groups,geneID] = gsg ### for global adjustment across comparisons
#if 'lentivirus' in group1 and geneID == 'ENSG00000184470':
#print groups,log_fold, p, avg1,avg2, max_avg, original_group[group1],original_group[group2]
if geneID == 'ENSG00000140416:I1.2-E6.4 ENSG00000140416:E3.6-E6.4' and 'male' in groups and 'female' in groups:
print groups,log_fold, p, avg1,avg2, max_avg, original_group[group1],original_group[group2],data_list1,data_list2
group_avg_exp_db[group1][geneID] = avg1 ### store the group expression values
group_avg_exp_db[group2][geneID] = avg2 ### store the group expression values
#if geneID == 'ENSG00000213973': print log_fold
if 'Insufficient Expression2' != log_fold:
if geneID == 'hsa-mir-512-1_hsa-miR-512-3p' and 'CD34+ cells' in groups and 'mononuclear' in groups:
ls1 = map(str,original_group[group1])
ls2 = map(str,original_group[group2])
#print groups, log_fold, logfold_threshold, p, ls1, ls2, avg1, avg2
#print data_list1, data_list2
if abs(log_fold)>logfold_threshold and p<pval_threshold:
print 'yes'
else: print 'no'
#sys.exit()
pass
#if abs(log_fold)>logfold_threshold:
eo = export_object_db[groups]
ls1 = map(str,original_group[group1])
ls2 = map(str,original_group[group2])
eo.write(string.join([geneID]+ls1+ls2,'\t')+'\n')
for groups in export_object_db:
export_object_db[groups].close()
### Calculate adjusted p-values for all pairwise comparisons
for groups in pval_summary_db:
group1,group2 = groups
if uniqueDonors:
filename=CovariateQuery+'/GE.'+string.join(groups,'_vs_')+'-UniqueDonors.txt'
else:
filename=CovariateQuery+'/GE.'+string.join(groups,'_vs_')+'.txt'
eo = export.ExportFile(rootdir+'/'+filename)
do = export.ExportFile(rootdir+'/Downregulated/'+filename)
uo = export.ExportFile(rootdir+'/Upregulated/'+filename)
so = export.ExportFile(rootdir+'PValues/'+CovariateQuery+'-'+string.join(groups,'_vs_')+'.txt')
header = 'GeneID\tSystemCode\tLogFold\trawp\tadjp\tSymbol\tavg-%s\tavg-%s\n' % (group1,group2)
eo.write(header)
do.write(header)
uo.write(header)
so.write('Gene\tPval\n')
pval_db = pval_summary_db[groups]
if 'moderated' in probability_statistic:
try: statistics.moderateTestStats(pval_db,probability_statistic) ### Moderates the original reported test p-value prior to adjusting
except Exception: print 'Moderated test failed... using student t-test instead'
statistics.adjustPermuteStats(pval_db) ### sets the adjusted p-values for objects
for geneID in pval_db:
gs = pval_db[geneID]
group1_avg = str(group_avg_exp_db[group1][geneID])
group2_avg = str(group_avg_exp_db[group2][geneID])
if use_adjusted_p:
pval = float(gs.AdjP())
else:
pval = gs.Pval()
if platform == 'miRSeq':
symbol=[]
altID = string.replace(geneID,'hsa-mir-','MIR')
altID = string.replace(altID,'hsa-miR-','MIR')
altID = string.replace(altID,'3p','')
altID = string.replace(altID,'5p','')
altID = string.upper(string.replace(altID,'hsa-let-','LET'))
altID = string.replace(altID,'-','')
altIDs = string.split(altID,'_')
altIDs+=string.split(geneID,'_')
altIDs = unique.unique(altIDs)
for id in altIDs:
if id in gene_to_symbol:
symbol.append(gene_to_symbol[id][0])
symbol.append(id)
symbol = string.join(symbol,'|')
elif geneID in gene_to_symbol:
symbols = unique.unique(gene_to_symbol[geneID])
symbol = string.join(symbols,'|')
elif 'ENS' in geneID and ':' in geneID:
ens_gene = string.split(geneID,':')[0]
try: symbol = gene_to_symbol[ens_gene][0]
except Exception: symbol=''
else:
symbol = ''
proceed = True
### Remove genes not a predetermined list (optional)
if len(restricted_gene_denominator)>0:
if geneID not in restricted_gene_denominator:
if symbol not in restricted_gene_denominator:
proceed = False
if geneID == 'ENSG00000105321:E3.2-E4.2 ENSG00000105321:E2.3-E4.2' and 'fibroblast' in groups and 'inner cell mass' in groups:
print groups, gs.LogFold(), logfold_threshold, pval, pval_threshold, gs.Pval(), gs.AdjP(), symbol#;sys.exit()
if 'Insufficient Expression' != gs.LogFold() and proceed:
#if geneID == 'hsa-mir-512-1_hsa-miR-512-3p' and 'CD34+ cells' in groups and 'mononuclear' in groups:
#print groups, gs.LogFold()
if abs(gs.LogFold())>logfold_threshold and pval<pval_threshold:
values = string.join([geneID,system_code,str(gs.LogFold()),str(gs.Pval()),str(gs.AdjP()),symbol,group1_avg,group2_avg],'\t')+'\n'
eo.write(values)
try: chr = gene_location_db[ug.GeneID()][0]
except Exception: chr = ''
proceed = True
if 'Gender' in filename:
if 'Y' in chr: proceed = False
if proceed:
if gs.LogFold()>0:
uo.write(values)
if gs.LogFold()<0:
do.write(values)
so.write(geneID+'\t'+str(gs.Pval())+'\n')
eo.close()
do.close()
uo.close()
so.close()
def getAnnotations(species,platform):
import gene_associations
if platform == 'RNASeq' or platform == 'exon' or platform == 'miRSeq':
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
system_code = 'En'
if platform == 'miRSeq':
from import_scripts import OBO_import
gene_to_symbol = OBO_import.swapKeyValues(gene_to_symbol)
if platform == 'methylation':
gene_to_symbol = gene_associations.getGeneToUid(species,('hide','Ensembl-Symbol'))
gene_to_symbol = importMethylationAnnotations(species,gene_to_symbol)
system_code = 'Ilm'
return gene_to_symbol, system_code
def importMethylationAnnotations(species,gene_to_symbol):
filename = 'AltDatabase/ucsc/'+species+'/illumina_genes.txt'
from import_scripts import OBO_import
symbol_to_gene = OBO_import.swapKeyValues(gene_to_symbol)
firstLine=True
probe_gene_db={}
for line in open(OBO_import.filepath(filename),'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
geneIndex = values.index('UCSC_RefGene_Name')
locationIndex = values.index('UCSC_RefGene_Group')
firstLine = False
else:
probeID = values[0]
try: genes = string.split(values[geneIndex],';')
except Exception: genes=[]
try:
locations = unique.unique(string.split(values[locationIndex],';'))
locations = string.join(locations,';')
except Exception:
locations = ''
for symbol in genes:
if len(symbol)>0:
if symbol in symbol_to_gene:
for geneID in symbol_to_gene[symbol]:
try: probe_gene_db[probeID].append(geneID)
except Exception: probe_gene_db[probeID] = [geneID]
probe_gene_db[probeID].append(symbol)
probe_gene_db[probeID].append(locations)
return probe_gene_db
def getDatasetSamples(expression_file,sample_metadata,cellLines):
### Required as samples may exist in the metadata but were excluded due to QC
for line in open(expression_file,'rU').xreadlines():
if '.bed' in line:
line = string.replace(line,'.bed','')
data = line.rstrip()
headers = string.split(data,'\t')[1:]
break
supported_cellLines={}
for s in headers:
if s in sample_metadata:
metadata = sample_metadata[s]
if len(metadata.CellLine())>0:
try:
if s == cellLines[metadata.CellLine()]: ### Make sure the correct samples is being matched
supported_cellLines[metadata.CellLine()]=None
except Exception: pass
return supported_cellLines
def importExpressionData(species,platform,expression_file,cell_line_db,common_lines):
### Imports the omics data, filters/orders samples, transforms (if necessary), keys by a common geneID
filtered_exp_data={}
try: gene_to_symbol,system_code = getAnnotations(species,platform)
except ZeroDivisionError: gene_to_symbol={}; system_code=''
firstLine=True
for line in open(expression_file,'rU').xreadlines():
if '.bed' in line:
line = string.replace(line,'.bed','')
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
samplesToEvaluate = map(lambda x: cell_line_db[x], common_lines)
sample_index_list = map(lambda x: values.index(x), samplesToEvaluate)
header = values
#print len(samplesToEvaluate),platform, samplesToEvaluate
firstLine = False
else:
try: filtered_values = map(lambda x: float(values[x]), sample_index_list) ### simple and fast way to reorganize the samples
except Exception: ### for splice-event comparisons
if len(header) != len(values):
diff = len(header)-len(values)
values+=diff*['']
initial_filtered=[] ### the blanks can cause problems here so we loop through each entry and catch exceptions
initial_filtered = map(lambda x: values[x], sample_index_list)
filtered_values=[]
for x in initial_filtered:
if x != '': filtered_values.append(float(x))
if platform == 'RNASeq':# or platform == 'miRSeq':
filtered_values = map(lambda x: math.log(x+1,2),filtered_values) ### increment and log2 adjusted
uid = values[0]
geneIDs = []
if platform == 'miRSeq':
altID = string.replace(uid,'hsa-mir-','MIR')
altID = string.replace(altID,'hsa-miR-','MIR')
altID = string.replace(altID,'3p','')
altID = string.replace(altID,'5p','')
altID = string.upper(string.replace(altID,'hsa-let-','MIRLET'))
altID = string.replace(altID,'-','')
altIDs = string.split(altID,'_')
altIDs+=string.split(uid,'_')
altIDs = unique.unique(altIDs)
for id in altIDs:
if id in gene_to_symbol:
geneIDs.append((gene_to_symbol[id][0],uid))
original_uid = uid
if platform == 'methylation' and ':' in uid:
uid=string.split(uid,':')[1]
if 'ENS' in uid and '.' in uid and ':' not in uid:
uid = string.split(uid,'.')[0] ### for cufflinks
if 'ENS' in uid:
if uid in gene_to_symbol:
symbol = gene_to_symbol[uid][0]
else:
symbol = ''
geneIDs = [(uid,symbol)]
elif uid in gene_to_symbol:
if 'uid'== 'cg21028156':
print gene_to_symbol[uid]
for g in gene_to_symbol[uid]:
if 'ENS' in g:
if platform == 'methylation' and ':' in original_uid:
uid = original_uid
geneIDs.append((g,uid))
for (geneID,uid) in geneIDs:
try: filtered_exp_data[geneID].append((uid,filtered_values))
except Exception: filtered_exp_data[geneID] = [(uid,filtered_values)]
print len(filtered_exp_data)
return filtered_exp_data, samplesToEvaluate
def combineAndCompareMatrices(input_file,filtered_exp_data1,filtered_exp_data2,platform1,platform2,samplesToEvaluate):
### Get the matching genes and identify anti-correlated rows (same sample order) to export to a merged file
rootdir=export.findParentDir(input_file)
compared_uid_pairs={}
count=0
import warnings
from scipy import stats
p1_samples = map(lambda x: platform1+':'+x, samplesToEvaluate)
p2_samples = map(lambda x: platform2+':'+x, samplesToEvaluate)
exportFile = rootdir+'/MergedOmicsTables/'+platform1+'-'+platform2+'.txt'
if uniqueDonors:
exportFile = rootdir+'/MergedOmicsTables/'+platform1+'-'+platform2+'-UniqueDonors.txt'
co = export.ExportFile(exportFile)
co.write(string.join(['GeneID',platform1+'-UID','Pearson-rho']+p1_samples+[platform2+'-UID']+p2_samples,'\t')+'\n')
correlated_geneIDs={}
for geneID in filtered_exp_data1:
if geneID in filtered_exp_data2:
rows1 = filtered_exp_data1[geneID]
rows2 = filtered_exp_data2[geneID]
for (uid1,row1) in rows1:
"""
if platform1 == 'RNASeq' and platform2 == 'methylation':
try: row1 = map(lambda x: math.pow(2,x)-1,row1)
except Exception: print uid1,row1;sys.exit()
"""
for (uid2,row2) in rows2:
try: null=compared_uid_pairs[(uid1,uid2)] ### already compared
except Exception:
with warnings.catch_warnings():
warnings.filterwarnings("ignore") ### hides import warnings
try: rho,p = stats.pearsonr(row1,row2)
except Exception: print 'The rows are not of equal length, likely due to missing values in that row:',uid1,uid2;sys.exit()
compared_uid_pairs[(uid1,uid2)]=None
if rho < -0.5:
values = [geneID,uid1,rho]+row1+[uid2]+row2
values = string.join(map(str, values),'\t')+'\n'
correlated_geneIDs[geneID]=None
co.write(values)
count+=1
co.close()
print 'Writing out %d entries to %s:' % (count,exportFile)
correlated_geneIDs_ls=[]
for i in correlated_geneIDs:
correlated_geneIDs_ls.append(i)
print len(correlated_geneIDs_ls)
def getFiles(sub_dir,directories=True):
dir_list = unique.read_directory(sub_dir); dir_list2 = []
for entry in dir_list:
if directories:
if '.' not in entry: dir_list2.append(entry)
else:
if '.' in entry: dir_list2.append(entry)
return dir_list2
class GeneData:
def __init__(self,geneID, systemCode, logFold, rawp, adjp, symbol, avg1, avg2):
self.geneID = geneID; self.systemCode = systemCode; self.logFold = logFold; self.rawp = rawp; self.adjp = adjp
self.symbol = symbol; self.avg1 = avg1; self.avg2 = avg2
def GeneID(self): return self.geneID
def LogFold(self): return self.logFold
def Rawp(self): return self.rawp
def Adjp(self): return self.adjp
def Symbol(self): return self.symbol
def Avg1(self): return self.avg1
def Avg2(self): return self.avg2
def SystemCode(self): return self.systemCode
def __repr__(self): return self.GeneID()
def importResultsSummary(filepath,comparison,gene_associations_db):
#'GeneID\tSystemCode\tLogFold\trawp\tadjp\tSymbol\tavg-%s\tavg-%s\n
firstLine=True
for line in open(filepath,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
else:
geneID, systemCode, logFold, rawp, adjp, symbol, avg1, avg2 = values
gd = GeneData(geneID, systemCode, logFold, rawp, adjp, symbol, avg1, avg2)
if comparison in gene_associations_db:
gene_db = gene_associations_db[comparison]
gene_db[geneID]=gd
else:
gene_db = {}
gene_db[geneID]=gd
gene_associations_db[comparison]=gene_db
return gene_associations_db
def compareGOEliteEnrichmentProfiles(expressionDir,eliteDir):
up_elite_miRNAs={}
down_elite_miRNAs={}
folders = getFiles(expressionDir)
for folder in folders:
subdirs = getFiles(expressionDir+'/'+folder)
for sub_dir in subdirs:
subdir = expressionDir+'/'+folder + '/'+ sub_dir
elite_dirs = getFiles(subdir) ### Are there any files to analyze?
if 'GO-Elite_results' in elite_dirs:
elitedir = subdir + '/GO-Elite_results/pruned-results_z-score_elite.txt'
if 'Down' in folder:
try: down_elite_miRNAs = getMIRAssociations(elitedir,sub_dir,down_elite_miRNAs)
except Exception: pass
else:
try: up_elite_miRNAs = getMIRAssociations(elitedir,sub_dir,up_elite_miRNAs)
except Exception: pass
if '.txt' not in eliteDir:
if 'CombinedResults' not in eliteDir:
eliteDir += '/CombinedResults/allTopGenes.txt'
else: eliteDir += '/allTopGenes.txt'
firstLine=True
for line in open(eliteDir,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
else:
comparison, gene, symbol, up_rawp, ng_adjp, up_logfold,ng_logofold, ng_avg1, ng_avg2 = values[:9]
comparison = string.replace(comparison,'.txt','')
miRNAs = string.split(gene,'_')+string.split(symbol,'|')
log_fold = float(up_logfold)
if log_fold>0:
if comparison in down_elite_miRNAs:
for miRNA in miRNAs:
miRNA = string.lower(miRNA)
if miRNA in down_elite_miRNAs[comparison]:
if 'lab' not in comparison and 'CD34+ cells_vs_mononuclear' not in comparison:
print miRNA, comparison, 'down'
else:
if comparison in up_elite_miRNAs:
for miRNA in miRNAs:
miRNA = string.lower(miRNA)
if miRNA in up_elite_miRNAs[comparison]:
if 'lab' not in comparison and 'CD34+ cells_vs_mononuclear' not in comparison:
print miRNA, comparison, 'up'
def importRestrictedSetOfGenesToQuery(filepath):
### Applied to predetermined expressed genes matching some criterion (e.g., FPKM > 5 and 20% expression in EBs)
restricted_gene_denominator_db={}
firstLine=True
for line in open(filepath,'rU').xreadlines():
data = line.rstrip()
gene = string.split(data,'\t')[0]
if firstLine:
firstLine=False
else:
restricted_gene_denominator_db[gene]=[]
return restricted_gene_denominator_db
def getMIRAssociations(filepath,diffstate_comparison,elite_miRNAs):
firstLine=True
for line in open(filepath,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
else:
try:
#if 'Combined' in values[0]: ### Restrict comparison to these
regulated_geneset_name = values[0] # GE.group1_vs_group2-microRNATargets.txt
regulated_geneset_name = string.split(regulated_geneset_name,'-')[0]
#regulated_geneset_name = string.replace(regulated_geneset_name,'-UniqueDonors','')
#regulated_geneset_name = string.replace(regulated_geneset_name,'-Combined','')
miRNA = string.lower(values[2])
miRNA = string.replace(miRNA,'*','')
miRNA2 = string.replace(miRNA,'hsa-mir-','MIR')
miRNA2 = string.replace(miRNA2,'hsa-let-','LET')
miRNA3 = string.replace(miRNA2,'-5p','')
miRNA3 = string.replace(miRNA3,'-3p','')
miRNAs = [miRNA,miRNA2,miRNA3]
for miRNA in miRNAs:
try: elite_miRNAs[diffstate_comparison+':'+regulated_geneset_name].append(string.lower(miRNA))
except Exception: elite_miRNAs[diffstate_comparison+':'+regulated_geneset_name] = [string.lower(miRNA)]
except Exception:
pass
return elite_miRNAs
def runGOEliteAnalysis(species,resultsDirectory):
mod = 'Ensembl'
pathway_permutations = 'FisherExactTest'
filter_method = 'z-score'
z_threshold = 1.96
p_val_threshold = 0.05
change_threshold = 2
resources_to_analyze = ['microRNATargets','pictar','miRanda','mirbase','RNAhybrid','TargetScan','microRNATargets_All']
returnPathways = 'no'
root = None
import GO_Elite
print '\nBeginning to run GO-Elite analysis on all results'
folders = getFiles(resultsDirectory)
for folder in folders:
subdirs = getFiles(resultsDirectory+'/'+folder)
for subdir in subdirs:
subdir = resultsDirectory+'/'+folder + '/'+ subdir
file_dirs = subdir,None,subdir
input_files = getFiles(subdir,directories=False) ### Are there any files to analyze?
if len(input_files)>0:
variables = species,mod,pathway_permutations,filter_method,z_threshold,p_val_threshold,change_threshold,resources_to_analyze,returnPathways,file_dirs,root
try: GO_Elite.remoteAnalysis(variables,'non-UI')
except Exception: 'GO-Elite failed for:',subdir
def identifyCommonGenes(resultsDirectory):
""" Compares results from parallel statistical analyses for unique and non-unique genetic donor workflows """
uniqueDonorGenes = {}
nonUniqueDonorGenes={}
folders = getFiles(resultsDirectory)
for folder in folders:
files = getFiles(resultsDirectory+'/'+folder,directories=False)
for file in files:
if '.txt' in file and 'GE.'== file[:3]:
filepath = resultsDirectory+'/'+folder+'/'+file
comparison = folder+':'+string.replace(file,'-UniqueDonors.txt','.txt')
if 'UniqueDonors.txt' in filepath:
uniqueDonorGenes = importResultsSummary(filepath,comparison,uniqueDonorGenes)
else:
nonUniqueDonorGenes = importResultsSummary(filepath,comparison,nonUniqueDonorGenes)
#nonUniqueDonorGenes = uniqueDonorGenes
from build_scripts import EnsemblImport
try: gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,platform,'key_by_array')
except Exception: gene_location_db={}
includeGlobalAdjustedPvals = False
if len(global_adjp_db)>0: ### When all comparisons are run together
#global_adjp_db[CovariateQuery,uniqueDonors,groups,geneID] = gs
if 'moderated' in probability_statistic:
try: statistics.moderateTestStats(global_adjp_db,probability_statistic) ### Moderates the original reported test p-value prior to adjusting
except Exception: print 'Moderated test failed... using student t-test instead'
statistics.adjustPermuteStats(global_adjp_db) ### sets the adjusted p-values for objects
includeGlobalAdjustedPvals = True
output_dir = resultsDirectory+'/CombinedResults/allTopGenes.txt'
eo = export.ExportFile(output_dir)
header = 'Comparison\tGeneID\tSymbol\tUniqueDonor-rawp\tNonUnique-adjp\tUniqueDonor-LogFold\tNonUnique-LogFold\tNonUnique-Avg1\tNonUnique-Avg2'
if includeGlobalAdjustedPvals:
header+='\tGlobalAdjustedP'
eo.write(header+'\n')
topComparisonAssociations={}
for comparison in uniqueDonorGenes:
if comparison in nonUniqueDonorGenes:
CovariateQuery,groups = string.split(comparison[:-4],':')
groups = tuple(string.split(groups[3:],'_vs_'))
comparison_dir = string.replace(comparison,':','/')[:-4]
do = export.ExportFile(resultsDirectory+'/Downregulated/'+comparison_dir+'-Combined.txt')
uo = export.ExportFile(resultsDirectory+'/Upregulated/'+comparison_dir+'-Combined.txt')
header = 'GeneID\tSy\tFoldChange\trawp\n'
uo.write(header)
do.write(header)
unique_gene_db = uniqueDonorGenes[comparison]
nonunique_gene_db = nonUniqueDonorGenes[comparison]
for gene in unique_gene_db: ### loop through the gene dictionary
if gene in nonunique_gene_db: ### common genes between unique and non-unique donors
ug = unique_gene_db[gene]
ng = nonunique_gene_db[gene]
values = [comparison,gene, ug.Symbol(),ug.Rawp(),ng.Adjp(),ug.LogFold(),ng.LogFold(),ng.Avg1(),ng.Avg2()]
if includeGlobalAdjustedPvals:
try:
gs = global_adjp_db[CovariateQuery,groups,gene]
ng_adjp = float(gs.AdjP())
values+=[str(ng_adjp)]
if platform == 'miRSeq' or platform == 'exon' and use_adjusted_p == False:
ng_adjp = float(ug.Rawp())
except Exception:
if platform == 'miRSeq' or platform == 'exon' and use_adjusted_p == False:
ng_adjp = float(ug.Rawp())
else:
ng_adjp = float(ug.Rawp())
values = string.join(values,'\t')+'\n'
eo.write(values)
if ng_adjp<pval_threshold:
try: topComparisonAssociations[gene].append((float(ug.Rawp()),values))
except Exception: topComparisonAssociations[gene] = [(float(ug.Rawp()),values)]
values = [ug.GeneID(), ug.SystemCode(), ug.LogFold(), ug.Rawp()]
values = string.join(values,'\t')+'\n'
try: chr = gene_location_db[ug.GeneID()][0]
except Exception: chr = ''
proceed = True
if 'Gender' in comparison:
if 'Y' in chr: proceed = False
if proceed:
if float(ug.LogFold())>0:
uo.write(values)
else:
do.write(values)
do.close()
uo.close()
eo.close()
print 'Matching Unique-Donor and NonUnique Donor results written to:',output_dir
### Write out the comparison for each gene with the most significant result (best associations)
output_dir = resultsDirectory+'/CombinedResults/eliteTopGenes.txt'
eo = export.ExportFile(output_dir)
eo.write('Comparison\tGeneID\tSymbol\tUniqueDonor-rawp\tNonUnique-adjp\tUniqueDonor-LogFold\tNonUnique-LogFold\tNonUnique-Avg1\tNonUnique-Avg2\n')
for gene in topComparisonAssociations:
topComparisonAssociations[gene].sort()
eo.write(topComparisonAssociations[gene][0][1])
eo.close()
print 'The most significant comparisons for each gene reported to:',output_dir
def getCommonCellLines(cellLines1,cellLines2,exp_cellLines1,exp_cellLines2,uniqueDonor_db,uniqueDonors,donor_db):
common_lines = list(cellLines1.viewkeys() & cellLines2.viewkeys() & exp_cellLines1.viewkeys() & exp_cellLines2.viewkeys())
common_lines.sort()
exclude = ['SC11-009','SC11-008'] ### selected the line with the greatest XIST for this donor
if uniqueDonors:
common_lines2=[]; donor_added=[]
for donorID in donor_db:
donor_db[donorID].sort()
for (priority,cellLine) in donor_db[donorID]: ### Prioritize based on sample QC and donor preference
if cellLine in common_lines and cellLine not in exclude:
if donorID not in donor_added and 'H9' not in cellLine:
common_lines2.append(cellLine)
donor_added.append(donorID)
common_lines = common_lines2
print 'Common Approved Lines:',common_lines
return common_lines
def downloadSynapseFile(synid,output_dir):
import synapseclient
import os,sys,string,shutil
syn = synapseclient.Synapse()
syn.login()
matrix = syn.get(synid, downloadLocation=output_dir, ifcollision="keep.local")
return matrix.path
def buildAdditionalMirTargetGeneSets():
miR_association_file = 'AltDatabase/ensembl/Hs/Hs_microRNA-Ensembl.txt'
output_dir = 'AltDatabase/EnsMart72/goelite/Hs/gene-mapp/Ensembl-'
eo = export.ExportFile(output_dir+'microRNATargets_All.txt')
header = 'GeneID\tSystemCode\tmiRNA\n'
eo.write(header)
miRNA_source_db={}
for line in open(unique.filepath(miR_association_file),'rU').xreadlines():
data = line.rstrip()
miRNA,ensembl,source = string.split(data,'\t')
output = ensembl+'\t\t'+miRNA+'\n'
eo.write(output)
sources = string.split(source,'|')
for source in sources:
try: miRNA_source_db[source].append(output)
except KeyError: miRNA_source_db[source]=[output]
eo.close()
for source in miRNA_source_db:
eo = export.ExportFile(output_dir+source+'.txt')
eo.write(header)
for line in miRNA_source_db[source]:
eo.write(line)
eo.close()
def returnSynFileLocations(file1,file2,output_dir):
if 'syn' in file1:
try: file1 = downloadSynapseFile(file1,output_dir)
except Exception:
print 'Is the destination file %s already open?' % file1;sys.exit()
if 'syn' in file2:
try: file2 = downloadSynapseFile(file2,output_dir)
except Exception:
print 'Is the destination file %s already open?' % file2;sys.exit()
return file1,file2
def synapseStore(file_dirs,root,parent_syn,executed_urls,used):
for file in file_dirs:
file_dir = root+'/'+file
file = synapseclient.File(file_dir, parent=parent_syn)
file = syn.store(file,executed=executed_urls,used=used)
def synapseStoreFolder(dir_path,parent_syn):
data_folder = synapseclient.Folder(dir_path, parent=parent_syn)
data_folder = syn.store(data_folder)
sub_parent = data_folder.id
return sub_parent
def synapseDirectoryUpload(expressionDir, parent_syn, executed_urls, used):
root = string.split(expressionDir,'/')[-1]
sub_parent = synapseStoreFolder(root,parent_syn)
folders = getFiles(expressionDir)
for folder in folders:
### Create the folder in Synapse
dir_path_level1 = expressionDir+'/'+folder
sub_parent1 = synapseStoreFolder(folder,sub_parent)
f2 = getFiles(dir_path_level1)
files = getFiles(dir_path_level1,False)
synapseStore(files,dir_path_level1,sub_parent1,executed_urls,used)
for folder in f2:
dir_path_level2 = dir_path_level1+'/'+folder
sub_parent2 = synapseStoreFolder(folder,sub_parent1)
f3 = getFiles(dir_path_level2)
files = getFiles(dir_path_level2,False)
synapseStore(files,dir_path_level2,sub_parent2,executed_urls,used)
for folder in f3:
dir_path_level3 = dir_path_level2+'/'+folder
sub_parent3 = synapseStoreFolder(folder,sub_parent2)
files = getFiles(dir_path_level3,False)
### These are the GO-Elite result files (not folders)
synapseStore(files,dir_path_level3,sub_parent3,executed_urls,used)
def exportGeneSetsFromCombined(filename):
firstLine=True
synapse_format = True
simple_format = False
reverse = True
comparison_to_gene={}
rootdir = export.findParentDir(filename)
file = export.findFilename(filename)
for line in open(filename,'rU').xreadlines():
data = line.rstrip()
values = string.split(data,'\t')
if firstLine:
firstLine=False
else:
comparison, gene, symbol, up_rawp, ng_adjp, up_logfold,ng_logofold, ng_avg1, ng_avg2 = values[:9]
comparison = string.replace(comparison,'GE.','')
prefix = string.split(comparison,':')[0]
state = string.split(prefix,'-')[0]
if state=='NA': state = 'All'
comparison = file[:-4]+'-'+string.split(comparison,':')[1]
comparison = string.replace(comparison,'allTopGenes-','')[:-4]
c1,c2 = string.split(comparison,'_vs_')
c1 = re.sub('[^0-9a-zA-Z]+', '', c1)
c2 = re.sub('[^0-9a-zA-Z]+', '', c2)
comparison = c1+'_vs_'+c2
#comparison = string.replace(comparison,':','-')
log_fold = float(up_logfold)
#print c1,c2,reverse,log_fold
if c1>c2:
comparison = c2+'_vs_'+c1
log_fold = log_fold *-1
if log_fold<0:
if synapse_format:
comparison+='__down'
else:
comparison = c2+'_vs_'+c1 ### reverse the regulation direction
else:
if synapse_format:
comparison+='__up'
if synapse_format:
if len(symbol) == 0: symbol = gene
gene = symbol
#comparison = string.replace(comparison,'_vs_','_')
comparison = string.replace(comparison,'NA','NotApplicable')
#comparison = string.replace(comparison,'MESO-5','MESO-EARLY')
comparison = string.replace(comparison,' ','_')
#if state == 'MESO': state = 'MESOEARLY'
if 'ENS' in gene:
SystemCode = 'En'
if ':' in gene:
gene = string.split(gene,':')[0]
genes = [gene]
elif 'cg' in gene:
SystemCode = 'En'
genes = []
for i in string.split(symbol,'|'):
if 'ENS' in i:
genes.append(i)
else:
SystemCode = 'Sy'
genes = [gene]
for g in genes:
if synapse_format:
if simple_format:
try: comparison_to_gene[comparison].append([g,log_fold])
except Exception: comparison_to_gene[comparison] = [[g,log_fold]]
else:
try: comparison_to_gene[state+'__'+comparison].append([g,log_fold])
except Exception: comparison_to_gene[state+'__'+comparison] = [[g,log_fold]]
elif simple_format:
try: comparison_to_gene[comparison].append([g,log_fold])
except Exception: comparison_to_gene[comparison] = [[g,log_fold]]
else:
try: comparison_to_gene[state+'-'+comparison].append([g,log_fold])
except Exception: comparison_to_gene[state+'-'+comparison] = [[g,log_fold]]
aro = export.ExportFile(rootdir+'/Regulated/combined.txt')
aro.write('Gene\tLogFold\tComparison\n')
for comparison in comparison_to_gene:
ro = export.ExportFile(rootdir+'/Regulated/'+comparison+'.txt')
ro.write('Gene\tSystemCode\n')
for (gene,logfold) in comparison_to_gene[comparison]:
ro.write(gene+'\t'+SystemCode+'\n')
aro.write(gene+'\t'+str(logfold)+'\t'+string.replace(comparison,'.txt','')+'\n')
ro.close()
aro.close()
if __name__ == '__main__':
################ Comand-line arguments ################
#buildAdditionalMirTargetGeneSets();sys.exit()
filename = '/Users/saljh8/Desktop/PCBC_MetaData_Comparisons/eXpress/CombinedResults/allTopGenes.txt' #DiffStateComps Reprogramming
#exportGeneSetsFromCombined(filename);sys.exit()
platform='RNASeq'
species='Hs'
probability_statistic = 'moderated t-test'
#probability_statistic = 'unpaired t-test'
minRPKM=-1000
logfold_threshold=math.log(1.3,2)
pval_threshold=0.05
use_adjusted_p = False
expression_files=[]
platforms=[]
metadata_files=[]
gender_restricted = None
runGOElite=False
compareEnrichmentProfiles=False
restrictCovariateTerm=None
compDiffState=None
runAgain=False
output_dir=None
include_only=''
diffStateQuery = 'NA'
used=[]
executed_urls=[]
restricted_gene_denominator={}
global_adjp_db={}
covariate_set = ['Cell Type of Origin Combined', 'Cell Type of Origin', 'Cell Line Type','Reprogramming Vector Type']
covariate_set+= ['Reprogramming Gene Combination','Gender','originating lab','Donor Life Stage','Culture_Conditions','C4 Karyotype Result','Small Molecules','Other Conditions During Reprogramming','XIST Level']
#covariate_set = ['Donor Life Stage','Other Conditions During Reprogramming','C4 Karyotype Result','Small Molecules','Other Conditions During Reprogramming']
#covariate_set = ['Cell Line Type']
diffState_set = ['SC','ECTO','DE','MESO-5','EB','MESO-15','MESO-30']
import getopt
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print 'Supply the argument --i location'
### python metaDataAnalysis.py --i /Volumes/SEQ-DATA\ 1/PCBC/RNASeq/July2014/MetaData/RNASeq_MetaData_July2014.txt --key "CellType" --value "Cell Type of Origin"
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['m=','i=','d=','c=','u=','p=','s=','g=','e=','ce=','rc=','cd=','o=','md=','in=','target=','parent=','urls=','used='])
for opt, arg in options:
if opt == '--m': metadata_files.append(arg)
if opt == '--o':
if output_dir==None:
output_dir = arg
else:
output_dir = [output_dir,arg]
if opt == '--i': expression_files.append(arg)
if opt == '--e': runGOElite=True
if opt == '--ce': compareEnrichmentProfiles = True
if opt == '--d': diffStateQuery=arg
if opt == '--c': CovariateQuery=arg
if opt == '--p': platforms.append(arg)
if opt == '--g': gender_restricted=arg
if opt == '--s': species=arg
if opt == '--rc': restrictCovariateTerm=arg
if opt == '--cd': compDiffState=arg
if opt == '--md': mirDataDir=arg
if opt == '--in': include_only=arg
if opt == '--target': target_dir=arg
if opt == '--parent': parent_syn=arg
if opt == '--urls': executed_urls.append(arg) ### options are: all, junction, exon, reference
if opt == '--used': used.append(arg)
if opt == '--u':
if string.lower(arg) == 'yes' or string.lower(arg) == 'true':
uniqueDonors=True
use_adjusted_p = False
else:
uniqueDonors = False
if string.lower(arg) == 'both':
runAgain = True
if len(used)>0:
###Upload existing results folder to Synapse
import synapseclient
import os,sys,string,shutil,getopt
syn = synapseclient.Synapse()
syn.login()
synapseDirectoryUpload(target_dir, parent_syn, executed_urls, used)
sys.exit()
elif compareEnrichmentProfiles:
#print expression_files
compareGOEliteEnrichmentProfiles(expression_files[0],expression_files[1])
elif runGOElite:
runGOEliteAnalysis(species,expression_files[0])
elif (len(expression_files)==1 and '.txt' in expression_files[0]) or (len(expression_files)==1 and 'syn' in expression_files[0]):
### Perform a covariate based analysis on the lone input expression file
metadata_file = metadata_files[0]
if 'syn' in metadata_file:
try: metadata_file = downloadSynapseFile(metadata_file,output_dir)
except Exception:
print 'Is the destination file %s already open?' % metadata_file;sys.exit()
expression_file = expression_files[0]
if 'syn' in expression_file:
try:expression_file = downloadSynapseFile(expression_file,output_dir)
except Exception:
print 'Is the destination file %s already open?' % expression_file;sys.exit()
if 'syn' in include_only:
try:include_only = downloadSynapseFile(include_only,output_dir)
except Exception:
print 'Is the destination file %s already open?' % include_only;sys.exit()
if len(platforms)>0: platform = platforms[0]
if platform == 'exon' or platform == 'methylation':
logfold_threshold=math.log(1.1892,2) ### equivalent to a 0.25 dPSI or 0.25 beta differences
if platform == 'exon':
logfold_threshold=math.log(1,2)
use_adjusted_p = False ### Too many drop-outs with lowdepth seq that adjusting will inherently exclude any significant changes
if platform == 'methylation':
use_adjusted_p = True
if platform == 'miRSeq':
use_adjusted_p = False
logfold_threshold=math.log(1,2)
if CovariateQuery != 'all': covariate_set = [CovariateQuery]
if diffStateQuery != 'all': diffState_set = [diffStateQuery]
print 'Filtering on adjusted p-value:',use_adjusted_p
from build_scripts import EnsemblImport
try: gene_location_db = EnsemblImport.getEnsemblGeneLocations(species,platform,'key_by_array')
except Exception: gene_location_db={}
if len(include_only)>0:
restricted_gene_denominator = importRestrictedSetOfGenesToQuery(include_only)
for CovariateQuery in covariate_set:
for diffStateQuery in diffState_set:
print 'Analyzing the covariate:',CovariateQuery, 'and diffState:',diffStateQuery, 'unique donor analysis:',uniqueDonors
if 'XIST' in CovariateQuery: gender_restricted='female'
genderRestricted = gender_restricted
try:
sample_metadata,groups_db,comps_db = prepareComparisonData(metadata_file,diffStateQuery,CovariateQuery,uniqueDonors,genderRestricted,platform=platform,compDiffState=compDiffState,restrictCovariateTerm=restrictCovariateTerm)
performDifferentialExpressionAnalysis(species,platform,expression_file,sample_metadata,groups_db,comps_db,diffStateQuery+'-'+CovariateQuery,uniqueDonors)
except Exception:
print traceback.format_exc()
if runAgain:
uniqueDonors=True
use_adjusted_p = False
print 'Analyzing the covariate:',CovariateQuery, 'and diffState:',diffStateQuery, 'unique donor analysis:',uniqueDonors
try:
sample_metadata,groups_db,comps_db = prepareComparisonData(metadata_file,diffStateQuery,CovariateQuery,uniqueDonors,genderRestricted,platform=platform,compDiffState=compDiffState,restrictCovariateTerm=restrictCovariateTerm)
performDifferentialExpressionAnalysis(species,platform,expression_file,sample_metadata,groups_db,comps_db,diffStateQuery+'-'+CovariateQuery,uniqueDonors)
except Exception: pass
uniqueDonors=False; use_adjusted_p = True
if platform == 'miRSeq' or platform == 'exon': use_adjusted_p = False
if runAgain:
root_exp_dir=export.findParentDir(expression_file)
identifyCommonGenes(root_exp_dir)
runGOEliteAnalysis(species,root_exp_dir)
try: compareGOEliteEnrichmentProfiles(root_exp_dir,mirDataDir)
except Exception: pass
elif '.txt' not in expression_files[0] and 'syn' not in expression_files[0]:
### Compare unique and non-unique results to get overlapps (single high-confidence result file)
### The parent directory that contain results from the above analysis serve as input
resultsDirectory = expression_files[0]
identifyCommonGenes(resultsDirectory)
else:
### Perform a correlation analysis between two omics technologies
expression_file1,expression_file2 = expression_files
platform1,platform2 = platforms
metadata_file1,metadata_file2 = metadata_files
output_dir1, output_dir2 = output_dir
print expression_files, output_dir1
print metadata_files, output_dir2
expression_file1,metadata_file1 = returnSynFileLocations(expression_file1,metadata_file1,output_dir1)
metadata_file2,expression_file2 = returnSynFileLocations(metadata_file2,expression_file2,output_dir2)
print expression_file1, expression_file2
print metadata_file1, metadata_file2
cellLines1,sample_metadata1,uniqueDonor_db,donor_db = prepareComparisonData(metadata_file1,diffStateQuery,None,uniqueDonors,gender_restricted,platform=platform1)
cellLines2,sample_metadata2,uniqueDonor_db,donor_db = prepareComparisonData(metadata_file2,diffStateQuery,None,uniqueDonors,gender_restricted,platform=platform2)
exp_cellLines1 = getDatasetSamples(expression_file1,sample_metadata1,cellLines1)
exp_cellLines2 = getDatasetSamples(expression_file2,sample_metadata2,cellLines2)
common_lines = getCommonCellLines(cellLines1,cellLines2,exp_cellLines1,exp_cellLines2,uniqueDonor_db,uniqueDonors,donor_db)
filtered_exp_data1, samplesToEvaluate = importExpressionData(species,platform1,expression_file1,cellLines1,common_lines)
filtered_exp_data2, samplesToEvaluate = importExpressionData(species,platform2,expression_file2,cellLines2,common_lines)
combineAndCompareMatrices(expression_file1,filtered_exp_data1,filtered_exp_data2,platform1,platform2,samplesToEvaluate) ### export results to two different files and a combined file
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/metaDataAnalysis_PCBC.py
|
metaDataAnalysis_PCBC.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy as np
import pylab as pl
import os.path
from collections import defaultdict
from sklearn.cluster import KMeans
try:from stats_scripts import statistics
except Exception: import statistics
import random
import UI
import export; reload(export)
import re
from stats_scripts import fishers_exact_test
import traceback
import warnings
import math
import export
def FishersExactTest(r,n,R,N):
z=0.0
"""
N is the total number of genes measured (Ensembl linked from denom) (total number of ) (number of exons evaluated)
R is the total number of genes meeting the criterion (Ensembl linked from input) (number of exonic/intronic regions overlaping with any CLIP peeks)
n is the total number of genes in this specific MAPP (Ensembl denom in MAPP) (number of exonic/intronic regions associated with the SF)
r is the number of genes meeting the criterion in this MAPP (Ensembl input in MAPP) (number of exonic/intronic regions with peeks overlapping with the SF)
With these values, we must create a 2x2 contingency table for a Fisher's Exact Test
that reports:
+---+---+ a is the # of IDs in the term regulated
| a | b | b is the # of IDs in the term not-regulated
+---+---+ c is the # of IDs not-in-term and regulated
| c | d | d is the # of IDs not-in-term and not-regulated
+---+---+
If we know r=20, R=80, n=437 and N=14480
+----+-----+
| 20 | 417 | 437
+----+-----+
| 65 |13978| 14043
+----+-----+
85 14395 14480
"""
if (R-N) == 0: return 0
elif r==0 and n == 0: return 0
else:
try:
#try:
z = (r - n*(R/N))/math.sqrt(n*(R/N)*(1-(R/N))*(1-((n-1)/(N-1))))
#except ZeroDivisionError: print 'r,_n,R,N: ', r,_n,R,N;kill
except Exception: print (r - n*(R/N)), n*(R/N),(1-(R/N)),(1-((n-1)/(N-1))),r,n,N,R;kill
a = r; b = n-r; c=R-r; d=N-R-b
table = [[int(a),int(b)], [int(c),int(d)]]
"""
print a,b; print c,d
import fishers_exact_test; table = [[a,b], [c,d]]
ft = fishers_exact_test.FishersExactTest(table)
print ft.probability_of_table(table); print ft.two_tail_p()
print ft.right_tail_p(); print ft.left_tail_p()
"""
try: ### Scipy version - cuts down rutime by ~1/3rd the time
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=RuntimeWarning) ### hides import warnings
oddsratio, pvalue = stats.fisher_exact(table)
# print pvalue
return pvalue,z
except Exception:
ft = fishers_exact_test.FishersExactTest(table)
# print ft.two_tail_p()
return ft.two_tail_p(),z
def header_file(fname, delimiter=None,Expand="no"):
head=0
header=[]
newheader=[]
with open(fname, 'rU') as fin:
for line in fin:
#print line
line = line.rstrip(os.linesep)
header=string.split(line,'\t')
if Expand=="yes":
if head==0:
for i in range(1,len(header)):
iq=header[i]
#iq=string.split(header[i],".")[0]
newheader.append(iq)
head=1
else:break
else:
if len(header)<3 or Expand=="no":
if header[0] not in newheader:
newheader.append(header[0])
#print len(newheader)
return newheader
def Enrichment(Inputfile,mutdict,mutfile,Expand,header):
import collections
import mappfinder
X=defaultdict(list)
prev=""
head=0
group=defaultdict(list)
enrichdict=defaultdict(float)
mut=export.findFilename(mutfile)
dire=export.findParentDir(Inputfile)
output_dir = dire+'MutationEnrichment'
export.createExportFolder(output_dir)
exportnam=output_dir+'/Enrichment_Results.txt'
export_enrich=open(exportnam,"w")
exportnam=output_dir+'/Enrichment_tophits.txt'
export_hit=open(exportnam,"w")
export_enrich.write("Mutations"+"\t"+"Cluster"+"\t"+"r"+"\t"+"R"+"\t"+"n"+"\t"+"Sensitivity"+"\t"+"Specificity"+"\t"+"z-score"+"\t"+"Fisher exact test"+"\t"+"adjp value"+"\n")
if Expand=="yes":
header2=header_file(Inputfile,Expand="yes")
for line in open(Inputfile,'rU').xreadlines():
if head >0:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
for i in range(1,len(q)):
if q[i]==str(1):
#group[q[0]].append(header2[i-1])
group[header2[i-1]].append(q[0])
else:
head+=1
continue
else:
for line in open(Inputfile,'rU').xreadlines():
line=line.rstrip('\r\n')
line=string.split(line,'\t')
#for i in range(1,len(line)):
group[line[2]].append(line[0])
total_Scores={}
for kiy in mutdict:
if kiy =="MDP":
print mutdict[kiy]
groupdict={}
remaining=[]
remaining=list(set(header) - set(mutdict[kiy]))
groupdict[1]=mutdict[kiy]
groupdict[2]=remaining
# export_enrich1.write(kiy)
for key2 in group:
r=float(len(list(set(group[key2])))-len(list(set(group[key2]) - set(mutdict[kiy]))))
n=float(len(group[key2]))
R=float(len(set(mutdict[kiy])))
N=float(len(header))
if r==0 or R==1.0:
print kiy,key2,r,n,R,N
pval=float(1)
z=float(0)
null_z = 0.000
zsd = mappfinder.ZScoreData(key2,r,R,z,null_z,n)
zsd.SetP(pval)
else:
try: z = Zscore(r,n,N,R)
except : z = 0.0000
### Calculate a Z-score assuming zero matching entries
try: null_z = Zscore(0,n,N,R)
except Exception: null_z = 0.000
try:
pval = mappfinder.FishersExactTest(r,n,R,N)
zsd = mappfinder.ZScoreData(key2,r,R,z,null_z,n)
zsd.SetP(pval)
except Exception:
pval=1.0
zsd = mappfinder.ZScoreData(key2,r,R,z,null_z,n)
zsd.SetP(pval)
#pass
if kiy in total_Scores:
signature_db = total_Scores[kiy]
signature_db[key2]=zsd ### Necessary format for the permutation function
else:
signature_db={key2:zsd}
total_Scores[kiy] = signature_db
sorted_results=[]
mutlabels={}
for kiy in total_Scores:
signature_db = total_Scores[kiy]
### Updates the adjusted p-value instances
mappfinder.adjustPermuteStats(signature_db)
for signature in signature_db:
zsd = signature_db[signature]
results = [kiy,signature,zsd.Changed(),zsd.Measured(),zsd.InPathway(),str(float(zsd.PercentChanged())/100.0),str(float(float(zsd.Changed())/float(zsd.InPathway()))), zsd.ZScore(), zsd.PermuteP(), zsd.AdjP()] #string.join(zsd.AssociatedIDs(),'|')
sorted_results.append([signature,float(zsd.PermuteP()),results])
sorted_results.sort() ### Sort by p-value
prev=""
for (sig,p,values) in sorted_results:
if sig!=prev:
flag=True
export_hit.write(string.join(values,'\t')+'\n')
if flag:
if (float(values[5])>=0.5 and float(values[6])>=0.5) or float(values[5])>=0.6 :
mutlabels[values[1]]=values[0]
flag=False
export_hit.write(string.join(values,'\t')+'\n')
export_enrich.write(string.join(values,'\t')+'\n')
prev=sig
if len(sorted_results)==0:
export_enrich.write(string.join([splicing_factor,'NONE','NONE','NONE','NONE','NONE','NONE'],'\t')+'\n')
export_enrich.close()
#print mutlabels
return mutlabels
def findsiggenepermut(mutfile):
samplelist=[]
mutdict=defaultdict(list)
head=0
#File with all the sample names
for exp1 in open(mutfile,"rU").xreadlines():
#print exp1
lin=exp1.rstrip('\r\n')
lin=string.split(lin,"\t")
if len(lin)>3:
if head==0:
for i in lin[1:]:
samplelist.append(i)
head=1
continue
else:
for j in range(1,len(lin)):
if lin[j]==str(1):
mutdict[lin[0]].append(samplelist[j-1])
else:
mutdict[lin[2]].append(lin[0])
return mutdict
def Zscore(r,n,N,R):
"""where N is the total number of events measured:
R is the total number of events meeting the criterion:
n is the total number of events in this specific reference gene-set:
r is the number of events meeting the criterion in the examined reference gene-set: """
N=float(N) ### This bring all other values into float space
z = (r - n*(R/N))/math.sqrt(n*(R/N)*(1-(R/N))*(1-((n-1)/(N-1))))
return z
if __name__ == '__main__':
import getopt
mutdict=defaultdict(list)
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['Inputfile=','Reference=','Expand='])
for opt, arg in options:
if opt == '--Inputfile': Inputfile=arg
elif opt == '--Reference':Reference=arg
elif opt =='--Expand': Expand=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
mutfile=Reference
header=header_file(mutfile)
mutdict=findsiggenepermut(mutfile)
Enrichment(Inputfile,mutdict,mutfile,Expand,header)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/MutationEnrichment_adj.py
|
MutationEnrichment_adj.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import warnings
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
import numpy as np
from sklearn.cluster import KMeans
import nimfa
from sklearn.decomposition import NMF
import os.path
from collections import defaultdict
import traceback
import export
from visualization_scripts import Orderedheatmap
#import statistics
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def filterRows(input_file,output_file,filterDB=None,logData=False):
orderlst={}
counter=[]
export_object = open(output_file,'w')
firstLine = True
Flag=0
for line in open(input_file,'rU').xreadlines(): ### Original expression file (source IDs)
#for i in filterDB:
flag1=0
data = cleanUpLine(line)
values = string.split(data,'\t')
if firstLine:
firstLine = False
if Flag==0:
export_object.write(line)
else:
#print values[0], filterDB
#sys.exit()
uid = values[0]
if uid in filterDB:
counter=[index for index, value in enumerate(filterDB) if value == uid]
for it in range(0,len(counter)):
orderlst[counter[it]]=line
for i in range(0,len(orderlst)):
try:
export_object.write(orderlst[i])
except Exception:
print i,filterDB[i]
continue
export_object.close()
print 'Filtered rows printed to:',output_file
def FilterGuideGeneFile(Guidefile,Guidefile_block,expressionInputFile,iteration,platform,uniqueIDs,symbolIDs):
""" Filters the original input expression file for Guide3 genes/events. Needed
Since NMF only can deal with positive values [Guide3 has negative values]"""
root_dir = export.findParentDir(expressionInputFile)[:-1]
if 'ExpressionInput' in root_dir:
root_dir = export.findParentDir(root_dir)
if 'Clustering' in Guidefile:
count=1
flag=True
rank_Count=0
prev=0
else:
count=0
val=[]
head=0
for line in open(Guidefile_block,'rU').xreadlines():
if head >count:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
#val.append(q[0])
if flag:
if int(q[1])==prev:
continue
else:
rank_Count+=1
prev=int(q[1])
else:
head+=1
continue
head=0
for line in open(Guidefile,'rU').xreadlines():
line=line.rstrip('\r\n')
q= string.split(line,'\t')
n=len(q)
if head >count:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
uid = q[0]
if uid not in uniqueIDs:
if uid in symbolIDs:
uid = symbolIDs[uid]
val.append(uid)
else:
continue
val.append(uid)
if platform != "PSI" and head==2:
rank_Count=rank_Count+int(q[1])
print rank_Count
head=head+1
else:
head+=1
if platform != "PSI" and q[0]=="column_clusters-flat":
rank_Count=int(q[n-1])
continue
output_dir = root_dir+'/NMF-SVM'
if os.path.exists(output_dir)==False:
export.createExportFolder(output_dir)
output_file = output_dir+'/NMFInput-Round'+str(iteration)+'.txt'
filterRows(expressionInputFile,output_file,filterDB=val)
return output_file,rank_Count
def NMFAnalysis(expressionInputFile,NMFinputDir,Rank,platform,iteration=0,strategy="conservative"):
root_dir = export.findParentDir(NMFinputDir)[:-1]
if 'ExpressionInput' in root_dir:
root_dir = export.findParentDir(root_dir)
if 'NMF-SVM' in root_dir:
root_dir = export.findParentDir(root_dir)
export.findFilename(NMFinputDir)
X=[]
header=[]
head=0
exportnam=root_dir+'/NMF-SVM/NMF/round'+str(iteration)+'NMFsnmf_versionr'+str(Rank)+'.txt'
export_res=export.ExportFile(exportnam)
exportnam_bin=root_dir+'/NMF-SVM/NMF/round'+str(iteration)+'NMFsnmf_binary'+str(Rank)+'.txt'
export_res1=export.ExportFile(exportnam_bin)
exportnam_bint=root_dir+'/NMF-SVM/NMF/round'+str(iteration)+'NMFsnmf_binary_t_'+str(Rank)+'.txt'
export_res5=export.ExportFile(exportnam_bint)
MF_input = root_dir+'/NMF-SVM/ExpressionInput/exp.NMF-MarkerFinder.txt'
export.customFileCopy(expressionInputFile,root_dir+'/NMF-SVM/ExpressionInput/exp.NMF-MarkerFinder.txt')
export_res4=open(string.replace(MF_input,'exp.','groups.'),"w")
export_res7=open(string.replace(MF_input,'exp.','comps.'),"w")
exportnam2=root_dir+'/NMF-SVM/SubtypeAnalyses/round'+str(iteration)+'Metadata'+str(Rank)+'.txt'
export_res2=export.ExportFile(exportnam2)
exportnam3=root_dir+'/NMF-SVM/SubtypeAnalyses/round'+str(iteration)+'Annotation'+str(Rank)+'.txt'
export_res3=export.ExportFile(exportnam3)
#if 'Clustering' in NMFinputDir:
# count=1
# start=2
#else:
count=0
start=1
#print Rank
for line in open(NMFinputDir,'rU').xreadlines():
line=line.rstrip('\r\n')
q= string.split(line,'\t')
if head >count:
val=[]
val2=[]
me=0.0
for i in range(start,len(q)):
try:
val2.append(float(q[i]))
except Exception:
continue
me=np.median(val2)
for i in range(start,len(q)):
try:
val.append(float(q[i]))
except Exception:
val.append(float(me))
#if q[1]==prev:
X.append(val)
else:
export_res1.write(line)
export_res.write(line)
export_res1.write("\n")
#export_res4.write(line)
#export_res4.write("\n")
export_res.write("\n")
header=q
head+=1
continue
group=defaultdict(list)
sh=[]
X=np.array(X)
#print X.shape
mat=[]
#mat=X
mat=zip(*X)
mat=np.array(mat)
#print mat.shape
#model = NMF(n_components=15, init='random', random_state=0)
#W = model.fit_transform(mat)
nmf = nimfa.Snmf(mat,seed="nndsvd", rank=int(Rank), max_iter=20,n_run=1,track_factor=False,theta=0.95)
nmf_fit = nmf()
W = nmf_fit.basis()
W=np.array(W)
#np.savetxt("basismatrix2.txt",W,delimiter="\t")
H=nmf_fit.coef()
H=np.array(H)
# np.savetxt("coefficientmatrix2.txt",H,delimiter="\t")
#print W.shape
sh=W.shape
export_res3.write("uid\tUID\tUID\n")
if int(Rank)==2:
par=1
else:
par=2
#for i in range(sh[1]):
# val=W[:,i]
# me=np.mean(val)
# st=np.std(val)
# export_res2.write(header[i+1])
# for j in range(sh[0]):
# if float(W[i][j])>=float(me+(par*st)):
#
# export_res2.write("\t"+str(1))
# else:
# export_res2.write("\t"+str(0))
#
# export_res2.write("\n")
if platform != 'PSI':
sh=W.shape
Z=[]
export_res5.write("uid")
export_res2.write("uid")
for i in range(sh[1]):
export_res5.write("\t"+'V'+str(i))
export_res2.write("\t"+'V'+str(i))
export_res3.write('V'+str(i)+"\t"+"Covariate"+"\t"+str(1)+"\n")
export_res5.write("\n")
export_res2.write("\n")
export_res3.write("\n")
for i in range(sh[0]):
new_val=[]
val=W[i,:]
export_res2.write(header[i+1])
export_res5.write(header[i+1])
export_res4.write(header[i+1])
flag=True
for j in range(sh[1]):
if W[i][j]==max(val) and flag:
export_res5.write("\t"+str(1))
export_res2.write("\t"+str(1))
new_val.append(1)
export_res4.write("\t"+str(j+1)+"\t"+'V'+str(j))
flag=False
else:
export_res5.write("\t"+str(0))
export_res2.write("\t"+str(0))
new_val.append(0)
Z.append(new_val)
export_res5.write("\n")
export_res2.write("\n")
export_res4.write("\n")
W=zip(*W)
W=np.array(W)
sh=W.shape
Z=zip(*Z)
Z=np.array(Z)
for i in range(sh[0]):
export_res.write('V'+str(i))
export_res1.write('V'+str(i))
for j in range(sh[1]):
export_res.write("\t"+str(W[i][j]))
export_res1.write("\t"+str(Z[i][j]))
export_res.write("\n")
export_res1.write("\n")
export_res.close()
export_res1.close()
export_res2.close()
export_res5.close()
Orderedheatmap.Classify(exportnam_bint)
return exportnam,exportnam_bin,exportnam2,exportnam3
else:
W=zip(*W)
W=np.array(W)
sh=W.shape
Z=[]
for i in range(sh[0]):
new_val=[]
val=W[i,:]
num=sum(i > 0.10 for i in val)
if num >40 or num <3:
compstd=True
else:
compstd=False
me=np.mean(val)
st=np.std(val)
#print 'V'+str(i)
export_res.write('V'+str(i))
export_res1.write('V'+str(i))
for j in range(sh[1]):
if compstd:
if float(W[i][j])>=float(me+(par*st)):
export_res1.write("\t"+str(1))
new_val.append(1)
else:
export_res1.write("\t"+str(0))
new_val.append(0)
else:
if float(W[i][j])>0.1:
export_res1.write("\t"+str(1))
new_val.append(1)
else:
export_res1.write("\t"+str(0))
new_val.append(0)
export_res.write("\t"+str(W[i][j]))
Z.append(new_val)
export_res.write("\n")
export_res1.write("\n")
# Z=zip(*Z)
Z=np.array(Z)
sh=Z.shape
Z_new=[]
val1=[]
Z1=[]
dellst=[]
export_res2.write("uid")
export_res5.write("uid")
for i in range(sh[0]):
indices=[]
val1=Z[i,:]
sum1=sum(val1)
flag=False
indices=[index for index, value in enumerate(val1) if value == 1]
for j in range(sh[0]):
val2=[]
if i!=j:
val2=Z[j,:]
sum2=sum([val2[x] for x in indices])
summ2=sum(val2)
try:
if float(sum2)/float(sum1)>0.5:
if summ2>sum1:
flag=True
#print str(i)
except Exception:
continue
if flag==False:
Z1.append(val1)
export_res2.write("\t"+'V'+str(i))
export_res5.write("\t"+'V'+str(i))
export_res3.write('V'+str(i)+"\t"+"Covariate"+"\t"+str(1)+"\n")
export_res2.write("\n")
export_res5.write("\n")
Z1=np.array(Z1)
Z=Z1
Z=zip(*Z)
Z=np.array(Z)
sh=Z.shape
for i in range(sh[0]):
val1=Z[i,:]
#print sum(val1)
#if sum(val)>2:
if sum(val1)>2:
val=[0 if x==1 else x for x in val1]
else:
val=val1
me=np.mean(val)
st=np.std(val)
export_res2.write(header[i+1])
export_res5.write(header[i+1])
for j in range(sh[1]):
if strategy=="conservative":
export_res2.write("\t"+str(val1[j]))
export_res5.write("\t"+str(val1[j]))
else:
export_res2.write("\t"+str(val[j]))
export_res5.write("\t"+str(val[j]))
export_res2.write("\n")
export_res5.write("\n")
Z_new.append(val)
Z_new=zip(*Z_new)
Z_new=np.array(Z_new)
sh=Z_new.shape
export_res5.close()
Orderedheatmap.Classify(exportnam_bint)
if strategy=="conservative":
return exportnam,exportnam_bin,exportnam2,exportnam3
else:
return exportnam,exportnam_bin,exportnam2,exportnam3
if __name__ == '__main__':
import getopt
mutdict=defaultdict(list)
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['Guidefile=','Rank=','PSI='])
for opt, arg in options:
if opt == '--Guidefile': Guidefile=arg
elif opt == '--Rank':Rank=arg
elif opt == '--PSI':PSI=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
inputfile=Guidefile
Rank=Rank
if Rank>1:
NMFAnalysis(inputfile,Rank,platform="RNASeq")
else:
pass
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/NMF_Analysis.py
|
NMF_Analysis.py
|
#Copyright 2017 Cincinnati Children's Hospital Medical Center, Research Foundation
#Author Meenakshi Venkatasubramanian - [email protected]
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
""" ICGS-NMF Module (Combatible with ICGS2 and splice-ICGS)
https://github.com/venkatmi/oncosplice
Steps applied in this workflow:
1 - Run splice-ICGS (Feature Selection)
2 - Block identification (Rank analysis)
3 - NMF Analysis (Initial subtype identification)
4 - Filter Event Annotation
5 - Meta data analysis (differential expression)
6 - Expand clusters (SVM sample classification)
7 - Mutation enrichment (MAF or VCF - optional)
8 - Correlation depletion (excluded biological confounding signatures)
"""
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import traceback
import sys, string, os
import RNASeq
import numpy as np
from stats_scripts import RNASeq_blockIdentification
from stats_scripts import NMF_Analysis; reload(NMF_Analysis)
from stats_scripts import filterEventAnnotation
from stats_scripts import metaDataAnalysis
from stats_scripts import ExpandSampleClusters; reload(ExpandSampleClusters)
from import_scripts import sampleIndexSelection
from stats_scripts import Correlationdepletion
import UI
import multiprocessing as mlp
import export
upd_guides=[]
import operator
from collections import OrderedDict
from collections import defaultdict
from stats_scripts import Kmeans
from stats_scripts import MutationEnrichment_adj as ME
from visualization_scripts import Orderedheatmap
from visualization_scripts import clustering; reload(clustering)
from sklearn.metrics.pairwise import euclidean_distances
from sklearn.metrics.pairwise import pairwise_distances
from sklearn.neighbors import KDTree
import community
import collections
from scipy.stats import variation
import networkx as nx
from sklearn.preprocessing import scale
from numpy import linalg as LA
import scipy
import warnings
warnings.filterwarnings('ignore')
def estimateK(inputfile):
header=[]
X=[]
head=0
counter=0
hgv={}
hgvgenes=[]
diclst={}
for line in open(inputfile,'rU').xreadlines():
if head==0:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
header=q
head=1
continue
else:
val=[]
line=line.rstrip('\r\n')
q= string.split(line,'\t')
#header.append(q[0])
for i in range(1,len(q)):
try:
val.append(float(q[i]))
except Exception:
continue
counter+=1
# break
X.append(val)
#X=zip(*X)
X=np.array(X)
try:
n=float(X.shape[0])
p=float(X.shape[1])
except: ### dimension error - assume k=30
return 15
X=scale(X)
Xt=np.transpose(X)
muTW=float((np.sqrt(n-1))+float(np.sqrt(p)))**2.0
sigmaTW=(float(np.sqrt(n - 1.0)) + float(np.sqrt(p))) * (1.0/float(np.sqrt(n - 1)) + 1.0/float(np.sqrt(p)))**(1.0/3.0)
sigmaHat=np.dot(Xt,X)
bd = 3.273 * sigmaTW + muTW
w,v = LA.eig(sigmaHat)
w=w.tolist()
k=0
for i in range(len(w)):
try:
if w[i]>bd:
k=k+1
except Exception:
if w[i].real>bd:
k=k+1
return k
def caldist(X,i,keys,keylist):
D=[]
Xxd=[]
newlist=[]
#for i in range(len(visited)):
#Xd=np.array(X[i])
#Xd=Xd.reshape(1, -1)
for ii in keys:
if ii==i: continue
newlist.append(ii)
Xxd.append(X[ii].tolist())
Xxd=np.array(Xxd)
Xd=X[i]
#Xd=Xxd
#Xxd=Xxd.tolist()
Xd=Xd.reshape(1, -1)
D=pairwise_distances(Xd,Xxd,metric='euclidean').tolist()
for q in range(len(np.argsort(D)[0])):
if newlist[q] in keylist:
continue
else:
key1=newlist[q]
break
return key1
def hgvfinder(inputfile,numVarGenes=500):
""" Find the highly variable genes by dispersion """
print 'Number of variable genes for dispersion:',numVarGenes
header=[]
X=[]
head=0
counter=0
hgv={}
hgvgenes=[]
for line in open(inputfile,'rU').xreadlines():
if head==0:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
count=len(q)-1
"""
if count >20000:
community=True
else:
community=False
"""
header=q
head=1
continue
else:
val=[]
line=line.rstrip('\r\n')
q= string.split(line,'\t')
#header.append(q[0])
for i in range(1,len(q)):
try:
val.append(float(q[i]))
except Exception:
continue
coun=len(set(val))
qq=q[0].lower()
if (qq.startswith("rpl") or qq.startswith("rps") or qq.startswith("mt-") or qq.startswith("ig")) and community:
continue
else:
if coun >5:
disp=float(np.var(val))/float(np.mean(val))
#me=float(np.mean(val))
hgv[q[0]]=disp
counter+=1
#if counter%500==0: print counter,
# break
#with open('hgv_0.1.txt', 'w') as f:
# for item in hgv:
# f.write(str(item)+"\t"+str(hgv[item]))
# f.write("\n")
#
hgv= sorted(hgv.items(), key=operator.itemgetter(1),reverse=True)
counter=0
for item,item2 in hgv:
if counter<numVarGenes: ### Number of highly variable genes for dispersion
hgvgenes.append(item)
counter+=1
output_file=inputfile[:-4]+'-filtered.txt'
#copy sample index selection file-mv
sampleIndexSelection.filterRows(inputfile,output_file,hgvgenes)
return output_file,count
def community_sampling(inputfile,downsample_cutoff):
""" This function performs downsampling of the input data using networkx to identify
initial distribution of cells, then Louvain clustering using the minimum resolution to
identify discrete initial clusters. """
header=[]
X=[]
head=0
counter=0
hgv={}
hgvgenes=[]
for line in open(inputfile,'rU').xreadlines():
if head==0:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
header=q
head=1
continue
else:
val=[]
line=line.rstrip('\r\n')
q= string.split(line,'\t')
#header.append(q[0])
for i in range(1,len(q)):
try:
val.append(float(q[i]))
except Exception:
continue
counter+=1
# break
X.append(val)
X=zip(*X)
X=np.array(X)
n=X.shape[0]
sampmark=[]
nn=X.shape[0]
nm=X.shape[1]
from annoy import AnnoyIndex
t=AnnoyIndex(nm,metric="euclidean")
for i in range(nn):
try: t.add_item(i,X[i])
except Exception: print i
t.build(100)
### t.save('ICGS.ann')
### u=AnnoyIndex(nm,metric="euclidean")
diclst={}
#### u.load('ICGS.ann')
#n1=25
print "creating graphs"
for i in range(nn):
#ind = tree.query([Xtemp[i]],k=10,return_distance=False,dualtree=True)
ind=t.get_nns_by_item(i,10)
diclst[i]=ind
G=nx.from_dict_of_lists(diclst)
# nx.write_adjlist(G,"test.adjlist")
#G=nx.read_adjlist("test.adjlist")
dendrogram= community.generate_dendrogram(G)
#for level in range(len(dendrogram) - 1):
level=0
pr= community.partition_at_level(dendrogram,level)
commun={}
comval={}
for key1 in pr:
try: commun[pr[key1]].append(key1)
except Exception: commun[pr[key1]]=[key1,]
try: comval[pr[key1]].append(X[int(key1)])
except Exception: comval[pr[key1]]=[X[int(key1)],]
print "Finding medians"
comindices=[]
downsamp_lim=downsample_cutoff*4
for key1 in comval:
k=downsamp_lim/len(comval)
if k<1: k=1
k2=len(comval[key1])
matri=np.array(comval[key1])
matri=np.array(matri)
#n=matri.shape[0]
D=pairwise_distances(matri,metric='euclidean').tolist()
D=np.array(D)
dist=np.mean(D,0)
if k2<k:
k=k2
count=0
for i in np.argsort(dist):
if count<k:
comindices.append(commun[key1][i])
count=count+1
sampmark=[]
for key1 in comindices:
#if count<2500:
#print key1
key=int(key1)
sampmark.append(header[key+1])
return sampmark
def PageRankSampling(inputfile,downsample_cutoff):
""" Google PageRank algorithm from networkX for graph-based link analysis """
header=[]
X=[]
head=0
counter=0
hgv={}
hgvgenes=[]
for line in open(inputfile,'rU').xreadlines():
if head==0:
line=line.rstrip('\r\n')
q= string.split(line,'\t')
header=q
head=1
continue
else:
val=[]
line=line.rstrip('\r\n')
q= string.split(line,'\t')
#header.append(q[0])
for i in range(1,len(q)):
try:
val.append(float(q[i]))
except Exception:
continue
counter+=1
X.append(val)
X=zip(*X)
X=np.array(X)
n=X.shape[0]
sampmark1=[]
downsamp_lim=downsample_cutoff*4
for iq in range(0,n,downsamp_lim):
jj=downsample_cutoff
if iq+downsamp_lim>n:
j=n-iq
else:
j=downsamp_lim
jj=int(float(j+1)/4.0)
jj=downsample_cutoff
#if jj<downsample_cutoff and n<3000:
#jj=n
Xtemp=X[iq:iq+j,]
nn=Xtemp.shape[0]
nm=Xtemp.shape[1]
diclst={}
from annoy import AnnoyIndex
t=AnnoyIndex(nm)
for i in range(nn):
t.add_item(i,Xtemp[i])
t.build(100)
t.save('ICGS.ann')
u=AnnoyIndex(nm)
u.load('ICGS.ann')
#tree = KDTree(X, leaf_size=10, metric='euclidean')
#n1=25
for i in range(nn):
#ind = tree.query([Xtemp[i]],k=10,return_distance=False,dualtree=True)
ind=u.get_nns_by_item(i,10)
diclst[i]=ind
# diclst[i]=ind.tolist()[0]
print "creating graphs"
G=nx.from_dict_of_lists(diclst)
#nx.write_adjlist(G,"test.adjlist")
#G=nx.read_adjlist("test.adjlist")
print "computing page rank"
pr=nx.pagerank(G)
pr= sorted(pr.items(), key=operator.itemgetter(1),reverse=True)
count=0
pr1=OrderedDict()
for (key1,key2) in pr:
if count<jj:
#print key1
key1=iq+int(key1)
pr1[key1,key2]=[]
#print header[key1-1]
sampmark1.append(key1)
count+=1
#with open('pangranresults_0.1.txt', 'w') as f:
#for (key1,key2) in pr:
#f.write(str(key1)+"\t"+str(key2)+"\n")
#f.write("\n")
samp=[]
sampdict={}
sampmark=[]
for (key1,key2) in pr1:
if len(samp)<len(pr1):
if key1 not in samp:
sampdict[key1]=[]
neighbours=list(G.adj[key1])
samp.append(key1)
for ii in range(len(neighbours)):
if neighbours[ii] not in samp and neighbours[ii] in sampmark1:
sampdict[key1].append(neighbours[ii])
samp.append(neighbours[ii])
else:
dup=[]
for key in sampdict:
if key1 in sampdict[key]:
neighbours=list(G.adj[key1])
for ii in range(len(neighbours)):
if neighbours[ii] not in samp and neighbours[ii] in sampmark1:
sampdict[key].append(neighbours[ii])
samp.append(neighbours[ii])
key=pr[0][0]
keylist=[]
keylist.append(key)
while len(keylist) <len(sampdict):
key=caldist(X,key,sampdict,keylist)
keylist.append(key)
for keys in range(len(keylist)):
sampmark.append(header[keylist[keys]+1])
for i in range(len(sampdict[keylist[keys]])):
sampmark.append(header[sampdict[keylist[keys]][i]+1])
#with open('pangranresults_0.1.txt', 'w') as f:
#for item in range(len(sampmark)):
#f.write(str(sampmark[item])+"\n")
#f.write("\n")
samptemp=[]
for i in range(len(header)):
if header[i] in sampmark:
samptemp.append(header[i])
sampmark=samptemp
if len(sampmark)>downsample_cutoff:
output_file=inputfile[:-4]+'-filtered.txt'
sampleIndexSelection.filterFile(inputfile,output_file,sampmark)
sampmark=PageRankSampling(output_file,downsample_cutoff)
return sampmark
else:
return sampmark
def filterPSIValues(filename):
fn = filepath(filename)
firstRow=True
header = True
rows=0
filtered=0
new_file = filename[:-4]+'-75p.txt'
ea = export.ExportFile(new_file)
for line in open(fn,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
header = False
eventindex=t.index('EventAnnotation')
t = [t[1]]+t[eventindex+1:]
header_length = len(t)-1
minimum_values_present = int(header_length)-1
not_detected = header_length-minimum_values_present
new_line = string.join(t,'\t')+'\n'
ea.write(new_line)
else:
t = [t[1]]+t[eventindex+1:]
missing_values_at_the_end = (header_length+1)-len(t)
missing = missing_values_at_the_end+t.count('')
if missing<not_detected:
new_line = string.join(t,'\t')+'\n'
ea.write(new_line)
filtered+=1
rows+=1
ea.close()
return newfile
def header_list(EventAnnot):
head=0
header=[]
with open(EventAnnot, 'rU') as fin:
for line in fin:
if head==0:
line = line.rstrip(os.linesep)
line=string.split(line,'\t')
startpos=line.index('EventAnnotation')
header.append('UID')
for i in range(startpos+1,len(line)):
header.append(line[i])
head=1
else:break
return header
def grpDict(grplst):
head=0
header={}
with open(grplst, 'rU') as fin:
for line in fin:
line = line.rstrip(os.linesep)
line=string.split(line,'\t')
#for i in range(len(line)):
try:header[line[2]].append(line[0])
except Exception: header[line[2]]=[line[0],]
return header
def FindTopUniqueEvents(Guidefile,psi,Guidedir):
head=0
guidekeys=[]
exportnam=os.path.join(Guidedir,"SplicingeventCount1.txt")
export_class=open(exportnam,"a")
tempkeys={}
global upd_guides
global train
omitcluster=0
unique_clusters={}
for line in open(Guidefile,'rU').xreadlines():
if head==0:
line1=line.rstrip('\r\n')
q= string.split(line1,'\t')
head=1
try:
uid=q.index('UID')
adjp=q.index('rawp')
dpsi=q.index('dPSI')
Clusterid=q.index('UpdatedClusterID')
cutoff=0.1
continue
except Exception:
uid=q.index('Symbol')
adjp=q.index('rawp')
dpsi=q.index('LogFold')
Clusterid=q.index('Symbol')
cutoff=0.58
else:
line1=line.rstrip('\r\n')
q= string.split(line1,'\t')
if abs(float(q[dpsi]))>cutoff and float(q[adjp])<0.01:
try:
tempkeys[q[Clusterid]].append([q[uid],float(q[adjp]),q[adjp+1]])
except KeyError:
tempkeys[q[Clusterid]]=[[q[uid],float(q[adjp]),q[adjp+1]],]
for i in tempkeys:
if len(tempkeys[i])>1:
tempkeys[i].sort(key=operator.itemgetter(1),reverse=False)
try:
unique_clusters[0].append(tempkeys[i][0])
except KeyError:
unique_clusters[0]=[tempkeys[i][0],]
else:
try:
unique_clusters[0].append(tempkeys[i][0])
except KeyError:
unique_clusters[0]=[tempkeys[i][0],]
try:
if len(unique_clusters[0])>1:
unique_clusters[0].sort(key=operator.itemgetter(1))
if len(unique_clusters[0])>10:
guidekeys=unique_clusters[0][0:150]
for i in range(0,len(guidekeys)):
upd_guides.append(guidekeys[i][0])
else:
omitcluster=1
else:
omitcluster=1
export_class.write(psi+"\t"+str(len(unique_clusters[0]))+"\n")
except Exception:
omitcluster=1
return omitcluster
def cleanUpLine(line):
line = string.replace(line,'\n','')
line = string.replace(line,'\c','')
data = string.replace(line,'\r','')
data = string.replace(data,'"','')
return data
def MergeResults(dire):
file_index={}
count=0
for filename in os.listdir(dire):
if ("Results_max" in filename or "Kmeans" in filename) and "._" not in filename and "ordered" not in filename and "max_t" not in filename:
file_index[filename]=count
count+=1
keylist={}
heads={}
for filename in os.listdir(dire):
if "Results_max" in filename or "Kmeans" in filename:
if "._" not in filename and "ordered" not in filename and "max_t" not in filename:
Guidefile=os.path.join(dire, filename)
head=0
for line in open(Guidefile,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
header=[]
if head==0:
head=1
for i in range(1,len(t)):
header.append(t[i])
heads[filename]=header
continue
else:
val=[]
key=t[0]
for i in range(1,len(t)):
val.append(t[i])
if key not in keylist:
keylist[key]=[[file_index[filename],val],]
else:
keylist[key].append([file_index[filename],val])
exportnam=os.path.join(dire,"MergedResult.txt")
export_class=open(exportnam,"w")
export_class.write("uid")
for filename in file_index:
export_class.write("\t")
export_class.write(string.join(heads[filename],"\t"))
export_class.write("\n")
for key in keylist:
export_class.write(key)
for filename in file_index:
for val1,val2 in keylist[key]:
if file_index[filename]==val1:
export_class.write("\t")
export_class.write(string.join(val2,"\t"))
break
export_class.write("\n")
return exportnam
def DetermineClusterFitness(allgenesfile,markerfile,filterfile,BinarizedOutput,rho_cutoff):
""" Determines whether a cluster has mutiple unique genes and hence should be used for SVM (AKA cluster fitness) """
header=True
genes=[]
nametemp=[]
for line in open(BinarizedOutput,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
header = False
else:
val=[]
for i in range(1,len(t)):
val.append(float(t[i]))
if sum(val)>2:
nametemp.append(t[0])
header=False
genes=[]
for line in open(filterfile,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
header = False
else:
genes.append(t[0])
allgenes={}
header=True
name=[]
for line in open(allgenesfile,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
uid = t[0] ### source ID not converted symbol
rho = t[2]
cluster = t[4]
if header:
header = False
else:
if float(rho)>0.3:
allgenes[uid]=cluster
header=True
markerdict={}
counter=1
group=[]
name=[]
common_geneIDs=0
marker_count=0
for line in open(markerfile,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
uid = t[0]
rho = t[2]
cluster = t[4]
marker_count+=1
if header:
header = False
else:
if uid in genes:
common_geneIDs+=1
#if rho_cutoff>0.4:rho_cutoff=0.4
rho_cutoff=0.3
#print rho_cutoff
#rho_cutoff=0.2
if float(rho)>rho_cutoff and cluster == allgenes[uid]:
try: markerdict[cluster].append([uid,float(rho)])
except Exception: markerdict[cluster]=[[uid,float(rho)]]
if (common_geneIDs+2)<marker_count:
print 'WARNING... only',common_geneIDs, 'out of', marker_count, 'gene IDs matched after conversion.'
for key in markerdict:
countr=1
if len(markerdict[key])>=2 and key in nametemp:
name.append(key+"_vs_Others.txt")
group.append(counter)
for i,j in markerdict[key] :
#if countr<30:
upd_guides.append(i)
countr+=1
counter+=1
return upd_guides,name,group
def sortFile(allgenesfile,rho_cutoff,name):
markergenes={}
val=[]
header=True
namelst=[]
for i in range(len(name)):
s=string.split(name[i],"_")[0]
namelst.append(s)
for line in open(allgenesfile,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
header = False
else:
values=[]
for i in range(len(t)):
if i ==0:
values.append(t[i])
if i ==2:
values.append(float(t[2]))
if i==4:
if "V" in t[4] and t[4] in namelst:
t[4]=string.replace(t[4],"V","")
values.append(t[4])
else:
values.append(t[4])
val.append(values)
val = sorted(val, key = operator.itemgetter(1),reverse=True)
val = sorted(val, key = operator.itemgetter(2))
count=0
prev="NA"
markerlst=[]
markergrps={}
for i in range(len(val)):
if val[i][2]==prev:
if count<60 and val[i][1]>=0.1: #rho_cutoff
try:markergrps[val[i][2]].append(val[i][0])
except Exception:markergrps[val[i][2]]=[val[i][0],]
markerlst.append(val[i][0])
count=count+1
prev=val[i][2]
else:
prev=val[i][2]
continue
else:
count=0
if val[i][1]>=0.1:
try:markergrps[val[i][2]].append(val[i][0])
except Exception:markergrps[val[i][2]]=[val[i][0],]
markerlst.append(val[i][0])
count=count+1
prev=val[i][2]
return markergrps,markerlst
def generateMarkerheatmap(processedInputExpFile,output_file,NMFSVM_centroid_cluster_dir,groupsdict,markergrps,header1,outputDir,root_dir,species,uniqueIDs):
""" Produces a final MarkerFinder result from ICGS-NMF """
matrix={}
header=True
samples=[]
samples2=[]
samples3=[]
samples_all=[]
samples2_all=[]
groups_list=[]
groups_list_all=[]
genes=[]
genes2=[]
exportnam2=root_dir+'/ICGS-NMF/FinalGroups.txt'
export_class2=open(exportnam2,"w")
for line in open(NMFSVM_centroid_cluster_dir,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
sampleOrder=[]
if header:
for i in range(len(t)):
if ":" in t[i]:
val=string.split(t[i],":")[1]
gr=string.split(val,"-")[1]
gr=string.split(gr,"_")[0]
gr=gr.replace("V","")
#sampleOrder.append(string.split(val,"-")[1])
sampleOrder.append(gr)
break
header=True
samp=[]
for line in open(processedInputExpFile,'rU').xreadlines():
data = line.rstrip()
t = string.split(data,'\t')
if header:
for i in range(1,len(t)):
samp.append(t[i])
header=False
continue
else:
for i in range(1,len(t)):
matrix[t[0],samp[i-1]]=t[i]
for i in range(len(sampleOrder)):
for j in range(len(groupsdict[sampleOrder[i]])):
export_class2.write(groupsdict[sampleOrder[i]][j]+"\t"+str(i+1)+"\t"+sampleOrder[i]+"\n")
if groupsdict[sampleOrder[i]][j] in header1:
samples.append(groupsdict[sampleOrder[i]][j])
groups_list.append(sampleOrder[i])
samples2.append(groupsdict[sampleOrder[i]][j])
samples3.append(sampleOrder[i]+':'+groupsdict[sampleOrder[i]][j])
for i in range(len(sampleOrder)):
for j in range(len(markergrps[sampleOrder[i]])):
uid = markergrps[sampleOrder[i]][j]
genes.append(uid)
if uid in uniqueIDs:
symbol = uniqueIDs[uid]
else:
symbol = uid
genes2.append((sampleOrder[i],uid))
MF_subsampled_export = outputDir+'/'+'MarkerFinder-subsampled-ordered.txt'
exportnam=open(MF_subsampled_export,"w")
exportnam.write(string.join(['UID','row_clusters-flat']+samples3,'\t')+'\n')
exportnam.write(string.join(['column_clusters-flat','']+groups_list,'\t')+'\n')
i=0
for i in range(len(genes)):
exportnam.write(genes2[i][1]+"\t"+genes2[i][0])
for j in range(len(samples)):
exportnam.write("\t"+matrix[genes[i],samples2[j]])
exportnam.write("\n")
exportnam.close()
export_class2.close()
graphic_links=[]
row_method=None
column_method=None
column_metric='euclidean'
row_metric='correlation'
color_gradient = 'yellow_black_blue'
transpose=False
import UI
Species=species
platform="RNASeq"
Vendor=""
gsp = UI.GeneSelectionParameters(Species,platform,Vendor)
gsp.setPathwaySelect('None Selected')
gsp.setGeneSelection('')
gsp.setOntologyID('')
gsp.setGeneSet('None Selected')
gsp.setJustShowTheseIDs('')
gsp.setTranspose(False)
gsp.setNormalize('median')
gsp.setGeneSelection('')
#gsp.setClusterGOElite('GeneOntology')
gsp.setClusterGOElite('BioMarkers')
graphic_links = clustering.runHCexplicit(MF_subsampled_export,graphic_links, row_method, row_metric, column_method,column_metric,color_gradient, gsp, display=False, Normalize=True,contrast=5)
graphic_links[-1][0] = MF_subsampled_export
if len(samp)>len(header1):
MF_all_export = outputDir+'/'+'MarkerFinder-Allsamples-ordered.txt'
all_cells_export=open(MF_all_export,"w")
for i in range(len(sampleOrder)):
for j in range(len(groupsdict[sampleOrder[i]])):
samples_all.append(sampleOrder[i]+":"+groupsdict[sampleOrder[i]][j])
groups_list_all.append(sampleOrder[i])
samples2_all.append(groupsdict[sampleOrder[i]][j])
all_cells_export.write(string.join(['UID','row_clusters-flat']+samples_all,'\t')+'\n')
all_cells_export.write(string.join(['column_clusters-flat','']+groups_list_all,'\t')+'\n')
for i in range(len(genes)):
all_cells_export.write(genes2[i][1]+"\t"+genes2[i][0])
for j in range(len(samples_all)):
all_cells_export.write("\t"+matrix[genes[i],samples2_all[j]])
all_cells_export.write("\n")
all_cells_export.close()
graphic_links = clustering.runHCexplicit(MF_all_export,graphic_links, row_method, row_metric, column_method,column_metric,color_gradient, gsp, display=False, Normalize=True,contrast=5)
graphic_links[-1][0] = MF_all_export
status = 'subsampled'
else:
status = 'not-subsampled'
return status, graphic_links
def callICGS(processedInputExpFile,species,rho_cutoff,dynamicCorrelation,platform,gsp):
#Run ICGS recursively to dynamically identify the best rho cutoff
graphic_links3,n = RNASeq.singleCellRNASeqWorkflow(species,platform,processedInputExpFile,mlp,dynamicCorrelation, rpkm_threshold=0, parameters=gsp)
if n>5000 and dynamicCorrelation:
rho_cutoff=rho_cutoff+0.1
gsp.setRhoCutoff(rho_cutoff)
print 'Increasing the Pearson rho threshold to:',rho_cutoff
graphic_links3,n,rho_cutoff=callICGS(processedInputExpFile,species,rho_cutoff,dynamicCorrelation,platform,gsp)
return graphic_links3,n,rho_cutoff
def getAllSourceIDs(fileame,species):
unique_ids={}
symbol_ids={}
IDtype='Symbol'
count=0
typeCount = 0
for line in open(fileame,'rU').xreadlines():
data = cleanUpLine(line)
uid = string.split(data,'\t')[0]
unique_ids[uid]=''
if count<100:
if 'ENS' in uid:
typeCount+=1
IDtype='Ensembl'
else:
try:
int(uid)
typeCount+=1
IDtype='EntrezGene'
except Exception:
pass
count+=1
### Check to see if these IDs are Ensembl IDs or EntrezGene
if typeCount>50: ### If over half of the IDs are EntrezGene or Ensembl
count=0
try:
import gene_associations
gene_annotations = gene_associations.importGeneData(species,IDtype)
except:
gene_annotations={}
for uid in gene_annotations:
if uid in unique_ids:
unique_ids[uid]=gene_annotations[uid].Symbol() #### Convert to Symbol
if 'LRG_' not in uid:
symbol_ids[gene_annotations[uid].Symbol()]=uid
count+=1
print count, IDtype, 'IDs with corresponding gene symbols out of', len(unique_ids)
return unique_ids, symbol_ids
def CompleteICGSWorkflow(root_dir,processedInputExpFile,EventAnnot,iteration,rho_cutoff,dynamicCorrelation,platform,species,scaling,gsp):
""" Run the entire ICGS-NMF workflow, recursively """
originalExpFile = EventAnnot
### Store a list of all valid original IDs (for ID conversions)
uniqueIDs, symbolIDs = getAllSourceIDs(processedInputExpFile,species)
if platform=='PSI':
### For splice-ICGS, the method performs signature depletion (removes correlated events from the prior round) on the Event Annotation file
FilteredEventAnnot=filterEventAnnotation.FilterFile(processedInputExpFile,EventAnnot,iteration)
graphic_links3 = RNASeq.singleCellRNASeqWorkflow(species, 'exons', processedInputExpFile,mlp, rpkm_threshold=0, parameters=gsp)
else:
### For single-cell RNA-Seq - run ICGS recursively to dynamically identify the best rho cutoff
graphic_links3,n,rho_cutoff=callICGS(processedInputExpFile,species,rho_cutoff,dynamicCorrelation,platform,gsp)
Guidefile=graphic_links3[-1][-1]
Guidefile=Guidefile[:-4]+'.txt'
#Guidefile="/Volumes/Pass/ICGS2_testrun/ExpressionInput/amplify/DataPlots/Clustering-exp.input-Guide3 AREG GZMA BTG1 CCL5 TMSB4X ITGA2B UBE2C IRF-hierarchical_euclidean_correlation.txt"
#rho_cutoff=0.2
try:
print "Running block identification for rank analyses - Round"+str(iteration)
try:
RNASeq_blockIdentification.correlateClusteredGenesParameters(Guidefile,rho_cutoff=0.4,hits_cutoff=4,hits_to_report=50,ReDefinedClusterBlocks=True,filter=True)
Guidefile_block=Guidefile[:-4]+'-BlockIDs.txt'
except Exception:
Guidefile_block=Guidefile
### Filters the original expression file for the guide3 genes [returns a filename similar to NMFInput-Round1.txt]
NMFinput,Rank=NMF_Analysis.FilterGuideGeneFile(Guidefile,Guidefile_block,processedInputExpFile,iteration,platform,uniqueIDs,symbolIDs)
#NMFinput="/Volumes/Pass/ICGS2_testrun/ExpressionInput/ICGS-interim/NMFInput-Round1.txt"
try: k = int(gsp.K())
except: k = None; #print traceback.format_exc()
if k==None:
k=estimateK(NMFinput)
Rank=k*2
if Rank>2 and platform=='PSI':
Rank=30
if Rank<5 and platform!='PSI':
Rank=10
### This function prepares files for differential expression analsyis (MetaDataAnalysis), MarkerFinder
filteredInputExpFile = string.replace(processedInputExpFile,'exp.','filteredExp.')
if '-OutliersRemoved' in Guidefile:
filteredInputExpFile = string.replace(filteredInputExpFile,'.txt','-OutliersRemoved.txt')
try: NMFResult,BinarizedOutput,Metadata,Annotation=NMF_Analysis.NMFAnalysis(filteredInputExpFile,NMFinput,Rank,platform,iteration)
except:
try:
Rank=k*1.5
NMFResult,BinarizedOutput,Metadata,Annotation=NMF_Analysis.NMFAnalysis(filteredInputExpFile,NMFinput,Rank,platform,iteration)
except:
Rank=k
NMFResult,BinarizedOutput,Metadata,Annotation=NMF_Analysis.NMFAnalysis(filteredInputExpFile,NMFinput,Rank,platform,iteration)
else:
Rank=k
print "Running NMF analyses for dimension reduction using "+str(Rank)+" ranks - Round"+str(iteration)
print "The number target number of clusters (k/rank) is:",k
filteredInputExpFile = string.replace(processedInputExpFile,'exp.','filteredExp.')
if '-OutliersRemoved' in Guidefile:
filteredInputExpFile = string.replace(filteredInputExpFile,'.txt','-OutliersRemoved.txt')
try:
NMFResult,BinarizedOutput,Metadata,Annotation=NMF_Analysis.NMFAnalysis(filteredInputExpFile,NMFinput,Rank,platform,iteration)
except Exception:
"Exception, choose a lower k value."
if Rank>1:
if platform == 'PSI':
print "Identifying cluster-specific differential splicing events"
findmarkers=False
else:
print 'Identifying cell-population specific genes'
findmarkers=True
if findmarkers:
import markerFinder
### Default path for the NMF clustered groups for MarkerFinder analysis
input_exp_file=root_dir+'/NMF-SVM/ExpressionInput/exp.NMF-MarkerFinder.txt'
logTransform = False
### Work around when performing this analysis on an alternative exon input cluster file
group_exp_file = input_exp_file
fl = UI.ExpressionFileLocationData(input_exp_file,'','',''); fl.setOutputDir(root_dir)
fl.setSpecies(species); fl.setVendor("3'array")
rpkm_threshold = 0.00
fl.setRPKMThreshold(rpkm_threshold)
fl.setCorrelationDirection('up')
compendiumType = 'protein_coding'
genesToReport = 60
correlateAll = True
markerFinder.analyzeData(input_exp_file,species,platform,compendiumType,geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl,logTransform=logTransform)
print 'MarkerFinder analysis complete'
#markerfile="/Volumes/Pass/Final_scicgs/ExpressionOutput/MarkerFinder/MarkerGenes_correlations-ReplicateBased.txt"
allgenesfile = root_dir+'/NMF-SVM/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
markerfile = root_dir+'/NMF-SVM/ExpressionOutput/MarkerFinder/MarkerGenes_correlations-ReplicateBased.txt'
guides=[]
### See if any unique genes are found in a cluster before using it for SVM
guides,name,group=DetermineClusterFitness(allgenesfile,markerfile,input_exp_file,BinarizedOutput,rho_cutoff)
counter=len(group)
else:
if platform=="PSI":
rootdir,CovariateQuery=metaDataAnalysis.remoteAnalysis(species,FilteredEventAnnot,Metadata,'PSI',0.1,use_adjusted_p,0.05,Annotation)
else:
rootdir,CovariateQuery=metaDataAnalysis.remoteAnalysis(species,processedInputExpFile,Metadata,'RNASeq',0.58,use_adjusted_p,0.05,Annotation)
counter=1
Guidedir=rootdir+CovariateQuery
PSIdir=rootdir+'ExpressionProfiles'
global upd_guides
upd_guides=[]
name=[]
group=[]
for filename in os.listdir(Guidedir):
if filename.startswith("PSI."):
Guidefile=os.path.join(Guidedir, filename)
psi=string.replace(filename,"PSI.","")
if filename.startswith("GE."):
Guidefile=os.path.join(Guidedir, filename)
psi=string.replace(filename,"GE.","")
PSIfile=os.path.join(PSIdir, psi)
omitcluster=FindTopUniqueEvents(Guidefile,psi,Guidedir)
if omitcluster==0:
group.append(counter)
name.append(psi)
counter+=1
upd_guides=[x for x in upd_guides if x != ""]
upd_guides=guides
upd_guides=list(set(upd_guides))
scaling=True
grplst=[]
############ Perform SVM classification to assign individual cells to valid-NMF clusters #############
### The below analysis is performed on the down-sampled expression file
if counter>2:
output_dir = root_dir+'/NMF-SVM'
if os.path.exists(output_dir)==False:
export.createExportFolder(output_dir)
#output_file = output_dir+'/SVMInput-Round'+str(iteration)+'.txt'
#ExpandSampleClusters.filterRows(processedInputExpFile,output_file,filterDB=upd_guides,logData=False)
if scaling:
output_fil=EventAnnot
output_file=output_dir+'/SVMInput-Round'+str(iteration)+'.txt'
#output_file1 = "/Users/meenakshi/Documents/Singlecellbest/exp.exp.CD34+.v5-log2_filtered.txt"
ExpandSampleClusters.filterRows(EventAnnot,output_file,filterDB=upd_guides,logData=False)
else:
output_file = output_dir+'/SVMInput-Round'+str(iteration)+'.txt'
ExpandSampleClusters.filterRows(processedInputExpFile,output_file,filterDB=upd_guides,logData=False)
header=ExpandSampleClusters.header_file(output_file)
print "Running SVM prediction for improved subtypes - Round"+str(iteration)
### Create teh training data for SVM
train,null=ExpandSampleClusters.TrainDataGeneration(output_file,BinarizedOutput,name,scaling,exports=False,rootDir=root_dir)
### Determine the medoids (use medoids for SVM but centroids for clustering)
grplst.append(group)
Expand=False ### If Expand == True, use all down-sampled cells for classification rather than medoids (similar cellHarmony)
if Expand==True:
grplst=[]
group=ExpandSampleClusters.Findgroups(BinarizedOutput,name)
grplst.append(group)
### Perform SVM
ExpandSampleClusters.Classify(header,train,output_file,grplst,name,iteration,platform,output_dir,root_dir)
### Create a groups file for the downsampled (or original) file
groupsfile = string.replace(originalExpFile,'exp.','groups.')
groupsfile_downsampled = string.replace(processedInputExpFile,'exp.','groups.')
finalgrpfile=root_dir+"/ICGS-NMF/FinalGroups.txt"
if groupsfile_downsampled == groupsfile:
pass
else:
export.customFileCopy(finalgrpfile,groupsfile_downsampled)
export.customFileCopy(finalgrpfile,groupsfile[:-4]+'-ICGS.txt')
export.customFileCopy(finalgrpfile,groupsfile[:-4]+'-markers.txt')
from shutil import copyfile
### Don't overwrite the original groups
updated_expfile = originalExpFile[:-4]+'-ICGS.txt'
copyfile(originalExpFile, updated_expfile)
if groupsfile_downsampled == groupsfile:
processedInputExpFile = updated_expfile
groupsfile=groupsfile[:-4]+'-ICGS.txt'
### Identify markers for the our final un-ordered clusters (clustering will need to be run after this)
markerFinder.analyzeData(processedInputExpFile,species,platform,compendiumType,geneToReport=genesToReport,correlateAll=correlateAll,AdditionalParameters=fl,logTransform=logTransform)
allgenesfile=root_dir+"/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt"
markergrps,markerlst=sortFile(allgenesfile,rho_cutoff,name)
if len(markergrps)!=len(name):
allgenesfile1 = root_dir+'/NMF-SVM/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
markergrps,markerlst=sortFile(allgenesfile1,rho_cutoff,name)
### To plot the heatmap, use the MarkerFinder genes (function pulls those genes out)
ExpandSampleClusters.filterRows(EventAnnot,processedInputExpFile[:-4]+'-markers.txt',filterDB=markerlst,logData=False) ### the processedInputExpFile is overwritten
groupsdict=grpDict(groupsfile)
SVMBinOutput=root_dir+"/NMF-SVM/SVMOutputs/round1SVC_Results_max.txt"
SVMBinOutput_t=root_dir+"/NMF-SVM/SVMOutputs/round1SVC_Results_max_t.txt"
import csv
from itertools import izip
a = izip(*csv.reader(open(SVMBinOutput,"rb"),delimiter='\t'))
csv.writer(open(SVMBinOutput_t, "wb"),delimiter='\t').writerows(a)
scaling=False ### will calculate centroids rather than medoids
centroids,centroid_heatmap_input=ExpandSampleClusters.TrainDataGeneration(processedInputExpFile[:-4]+'-markers.txt',SVMBinOutput_t,name,scaling,exports=True,rootDir=root_dir)
scaling=True
graphic_links=[]
row_method = "hopach"
column_method="hopach"
column_metric='cosine'
row_metric='correlation'
color_gradient = 'yellow_black_blue'
transpose=False
graphic_links = clustering.runHCexplicit(centroid_heatmap_input,graphic_links, row_method, row_metric, column_method,column_metric,color_gradient, transpose, display=False, Normalize=True)
NMFSVM_centroid_cluster_dir=graphic_links[0][1][:-4]+'.txt'
outputDir = root_dir+"/NMF-SVM/SVMOutputs"
header=ExpandSampleClusters.header_file(NMFinput)
status,graphic_links2=generateMarkerheatmap(processedInputExpFile[:-4]+'-markers.txt',output_file,NMFSVM_centroid_cluster_dir,groupsdict,markergrps,header,outputDir,root_dir,species,uniqueIDs)
import shutil
if status=='not-subsampled':
NMFSVM_centroid_cluster_graphics_dir=graphic_links2[0][1][:-4]
NMFSVM_centroid_cluster_dir=graphic_links2[0][0][:-4]
shutil.copy(NMFSVM_centroid_cluster_dir+'.txt',root_dir+"/ICGS-NMF/FinalMarkerHeatmap.txt")
shutil.copy(NMFSVM_centroid_cluster_graphics_dir+'.png',root_dir+"/ICGS-NMF/FinalMarkerHeatmap.png")
shutil.copy(NMFSVM_centroid_cluster_graphics_dir+'.pdf',root_dir+"/ICGS-NMF/FinalMarkerHeatmap.pdf")
shutil.copy(allgenesfile,root_dir+"/ICGS-NMF/MarkerGenes.txt")
final_exp_file = root_dir+"/ICGS-NMF/FinalMarkerHeatmap.txt"
else:
NMFSVM_centroid_cluster_graphics_dir=graphic_links2[0][1][:-4]
NMFSVM_centroid_cluster_dir=graphic_links2[0][0][:-4]
NMFSVM_centroid_cluster_graphics_dir2=graphic_links2[1][1][:-4]
NMFSVM_centroid_cluster_dir2=graphic_links2[1][0][:-4]
NMFSVM_centroid_cluster_dir=graphic_links2[0][0][:-4]
NMFSVM_centroid_cluster_dir1=graphic_links2[1][0][:-4]
shutil.copy(NMFSVM_centroid_cluster_dir+'.txt',root_dir+"/ICGS-NMF/FinalMarkerHeatmap_sampled.txt")
shutil.copy(NMFSVM_centroid_cluster_graphics_dir+'.png',root_dir+"/ICGS-NMF/FinalMarkerHeatmap_sampled.png")
shutil.copy(NMFSVM_centroid_cluster_graphics_dir+'.pdf',root_dir+"/ICGS-NMF/FinalMarkerHeatmap_sampled.pdf")
shutil.copy(NMFSVM_centroid_cluster_dir2+'.txt',root_dir+"/ICGS-NMF/FinalMarkerHeatmap_all.txt")
shutil.copy(NMFSVM_centroid_cluster_graphics_dir2+'.png',root_dir+"/ICGS-NMF/FinalMarkerHeatmap_all.png")
shutil.copy(NMFSVM_centroid_cluster_graphics_dir2+'.pdf',root_dir+"/ICGS-NMF/FinalMarkerHeatmap_all.pdf")
shutil.copy(allgenesfile,root_dir+"/ICGS-NMF/MarkerGenes.txt")
final_exp_file = root_dir+"/ICGS-NMF/FinalMarkerHeatmap_all.txt"
try:
### Build cell-type annotation FinalGroups file
goelite_path = export.findParentDir(NMFSVM_centroid_cluster_dir)[:-1]+'/GO-Elite/clustering/'+export.findFilename(NMFSVM_centroid_cluster_dir)+'/GO-Elite_results/pruned-results_z-score_elite.txt'
annotatedGroupsFile = RNASeq.predictCellTypesFromClusters(finalgrpfile, goelite_path)
group_alt = clustering.remoteAssignGroupColors(annotatedGroupsFile)
except:
print traceback.format_exc()
print 'Unable to export annotated groups file with predicted cell type names.'
group_alt=None
### Moved UMAP generation to the end (so the coordinates are the final coordinates and the user can review results earlier)
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(final_exp_file,geneFilter=markerlst)
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
#matrix = map(np.array, zip(*matrix)) ### coverts these to tuples
#column_header, row_header = row_header, column_header
finalOutputDir=root_dir+"/ICGS-NMF/"
#clustering.tSNE(np.array(matrix),column_header,dataset_name,group_db,display=False,showLabels=False,species=species,reimportModelScores=False)
try:
clustering.runUMAP(np.array(matrix),column_header,dataset_name,group_db,display=False, group_alt=group_alt,
showLabels=False,species=species,reimportModelScores=False,rootDir=root_dir,finalOutputDir=finalOutputDir)
except:
print traceback.format_exc()
"""
clustering.tSNE(processedInputExpFile,group_db=groupsdict,display=True,showLabels=False,row_header=None,colorByGene=None,species=None,reimportModelScores=False)
### MV need to do
Orderedfile,groupsdict=FindcentroidGroups(filtered,groupfile)
"""
### write final groups ordered
#exportGroups(root_dir+"/ICGS-NMF/FinalMarkerHeatmap.txt",root_dir+"/ICGS-NMF/FinalGroups.txt",platform)
if scaling:
flag=False
return flag,processedInputExpFile,EventAnnot,graphic_links3+graphic_links2
header=Correlationdepletion.header_file(NMFResult)
output_file=output_dir+'/DepletionInput-Round'+str(iteration)+".txt"
sampleIndexSelection.filterFile(processedInputExpFile[:-4]+'-markers.txt',output_file,header)
print "Running Correlation Depletion - Round"+str(iteration)
commonkeys,count=Correlationdepletion.FindCorrelations(NMFResult,output_file,name)
Depleted=Correlationdepletion.DepleteSplicingevents(commonkeys,output_file,count,processedInputExpFile)
processedInputExpFile=Deplete
flag=True
else:
print "No groups found!!! Re-analyze the data with a small k"
"""### Commented out in version 2.1.14
if iteration<2:
gsp.setK(2)
iteration=1
flag,processedInputExpFile,inputExpFile,graphic_links3=CompleteICGSWorkflow(root_dir,processedInputExpFile,originalExpFile,iteration,gsp.RhoCutoff(),dynamicCorrelation,platform,species,scaling,gsp)
try:
print "Running K-means analyses instead of NMF - Round"+str(iteration)
print "Extremely sparse data!! choose a small k"
header=[]
header=Kmeans.header_file(Guidefile_block)
Kmeans.KmeansAnalysis(Guidefile_block,header,processedInputExpFile,iteration)
flag=False
except Exception:
flag=False
"""
flag=False
else:
if Rank==1:
try:
print "Running K-means analyses instead of NMF - Round"+str(iteration)
print "Extremely sparse data!! choose a small k"
header=[]
header=Kmeans.header_file(Guidefile_block)
Kmeans.KmeansAnalysis(Guidefile_block,header,processedInputExpFile,iteration)
flag=False
except Exception:
flag=False
else:
flag=False
return flag,processedInputExpFile,EventAnnot,graphic_links3
except:
print traceback.format_exc()
print 'WARNING!!!! Error encountered in the NMF ICGS analysis... See the above report.'
flag=False
return flag,processedInputExpFile,EventAnnot,graphic_links3
def exportGroups(cluster_file,outdir,platform):
lineNum=1
for line in open(cluster_file,'rU').xreadlines():
data = cleanUpLine(line)
t = string.split(data,'\t')
if lineNum==1: names = t[2:]; lineNum+=1
elif lineNum==2: clusters = t[2:]; lineNum+=1
else: break
out_obj = export.ExportFile(outdir)
for name in names:
cluster = clusters[names.index(name)]
if platform == 'RNASeq':
if 'junction_quantification' not in name and '.bed' not in name:
name = name+'.bed'
elif 'junction_quantification.txt' not in name and '.txt' not in name and '.bed' not in name:
name = name+'.txt'
if ':' in name:
group,name = string.split(name,':')
if cluster=='NA': cluster = group
out_obj.write(name+'\t'+cluster+'\t'+cluster+'\n')
out_obj.close()
def runICGS_NMF(inputExpFile,scaling,platform,species,gsp,enrichmentInput='',dynamicCorrelation=True):
""" Export the filtered expression file then run downsampling analysis and prepares files for ICGS. After running ICGS, peform enrichment analyses """
try: downsample_cutoff = gsp.DownSample()
except: downsample_cutoff = 2500
try: numVarGenes = gsp.NumVarGenes()
except: numVarGenes = 500
print 'DownSample threshold =',downsample_cutoff, 'cells'
try: data_format = string.lower(gsp.CountsNormalization())
except: data_format = 'scaled'
### Scale and log2 normalize a counts expression file
if 'count' in data_format:
print 'Scaling counts as column normalized log2 values.',
from import_scripts import CountsNormalize
inputExpFile = CountsNormalize.normalizeDropSeqCountsMemoryEfficient(inputExpFile)
print 'Filtering the expression dataset (be patient).',
print_out, inputExpFile = RNASeq.singleCellRNASeqWorkflow(species,platform,inputExpFile,mlp,rpkm_threshold=0,parameters=gsp,reportOnly=True)
print 'Running ICGS-NMF'
### Find the parent dir of the output directory (expression file from the GUI will be stored in the output dir [ExpressionInput])
root_dir = export.findParentDir(inputExpFile)[:-1]
if 'ExpressionInput' in inputExpFile:
root_dir = export.findParentDir(root_dir)
exp_file_name = export.findFilename(inputExpFile)
### Assign the expression filename (for single-cell RNA-Seq rather than splicing)
if 'exp.' not in exp_file_name:
exp_file_name = 'exp.' + exp_file_name
########## Perform Downsampling for large datasets ##########
### Use dispersion (variance by mean) to define initial variable genes
inputExpFileVariableGenesDir,n=hgvfinder(inputExpFile,numVarGenes=numVarGenes) ### returns filtered expression file with 500 variable genes
if n>downsample_cutoff and scaling:
if n>15000: ### For extreemly large datasets, Louvain is used as a preliminary downsampling before pagerank
print 'Performing Community Clustering...'
inputExpFileScaled=inputExpFile[:-4]+'-Louvain-downsampled.txt'
### Louvain clustering for down-sampling from >25,000 to 10,000 cells
sampmark=community_sampling(inputExpFileVariableGenesDir,downsample_cutoff) ### returns list of Louvain downsampled cells
### Filer the original expression file using these downsampled cells
sampleIndexSelection.filterFile(inputExpFile,inputExpFileScaled,sampmark)
### Use dispersion (variance by mean) to define post-Louvain selected cell variable genes
inputExpFileVariableGenesDir,n=hgvfinder(inputExpFileScaled,numVarGenes=numVarGenes) ### returns filtered expression file with 500 variable genes
### Run PageRank on the Louvain/dispersion downsampled dataset
sampmark=PageRankSampling(inputExpFileVariableGenesDir,downsample_cutoff)
else:
### Directly run PageRank on the initial dispersion based dataset
sampmark=PageRankSampling(inputExpFileVariableGenesDir,downsample_cutoff)
### Write out final downsampled results to a new file
output_dir = root_dir+'/ExpressionInput'
try: export.createExportFolder(output_dir)
except: pass ### Already exists
processedInputExpFile = root_dir+'/ExpressionInput/'+exp_file_name[:-4]+'-PageRank-downsampled.txt' ### down-sampled file
sampleIndexSelection.filterFile(inputExpFile,processedInputExpFile,sampmark)
else:
output_dir = root_dir+'/ExpressionInput'
try: export.createExportFolder(output_dir)
except: pass ### Already exists
if platform == 'PSI':
### The PSI file name by default is not informative
processedInputExpFile=output_dir+"/exp.spliceICGS-input.txt"
export.customFileCopy(inputExpFile,processedInputExpFile)
elif 'ExpressionInput' not in inputExpFile:
processedInputExpFile = root_dir+'/'+exp_file_name
export.customFileCopy(inputExpFile,processedInputExpFile)
else: processedInputExpFile = inputExpFile
flag=True
iteration=1 ### Always equal to 1 for scRNA-Seq but can increment for splice-ICGS
### Recursively run ICGS with NMF
flag,processedInputExpFile,inputExpFile,graphic_links3=CompleteICGSWorkflow(root_dir,processedInputExpFile,
inputExpFile,iteration,gsp.RhoCutoff(),dynamicCorrelation,platform,species,scaling,gsp)
if platform == 'PSI':
output_dir = root_dir+'/SVMOutputs'
Combinedres=MergeResults(output_dir)
mutlabels={}
if enrichmentInput!='':
print "Running Mutation Enrichment Analyses"
Expand="yes"
mutdict=defaultdict(list)
header=ME.header_file(enrichmentInput)
mutdict=ME.findsiggenepermut(enrichmentInput)
mutlabels=ME.Enrichment(Combinedres,mutdict,enrichmentInput,Expand,header)
if platform == 'PSI':
print "Generating the final consolidated results"
Orderedheatmap.Classify(Combinedres,mutlabels,dire)
Orderedheatmap.Classify(Combinedres,mutlabels,dire,False)
print "successfully completed"
return graphic_links3
if __name__ == '__main__':
"""
processedInputExpFile="/Volumes/Pass/ICGS2_testrun/ExpressionInput/exp.input.txt"
matrix, column_header, row_header, dataset_name, group_db =clustering.importData(processedInputExpFile)
with warnings.catch_warnings():
warnings.filterwarnings("ignore",category=UserWarning) ### hides import warnings
#matrix = map(np.array, zip(*matrix)) ### coverts these to tuples
#column_header, row_header = row_header, column_header
directory=export.findParentDir(export.findParentDir(processedInputExpFile)[:-1])+"ICGS-NMF/"
#clustering.tSNE(np.array(matrix),column_header,dataset_name,group_db,display=False,showLabels=False,species="Hs",reimportModelScores=False)
clustering.umap(np.array(matrix),column_header,dataset_name,group_db,display=False,showLabels=False,species="Hs",reimportModelScores=False,directory=directory)
sys.exit()
"""
import getopt
rho_cutoff=0.2
dynamicCorrelation="optimize"
Mutationref=""
platform="RNASeq"
scaling=True
species="Hs"
row_method = 'hopach'
column_method = 'hopach'
row_metric = 'correlation'
column_metric = 'euclidean'
color_gradient = 'yellow_black_blue'
contrast=3
vendor = "RNASeq"
GeneSelection = ''
PathwaySelection = ''
GeneSetSelection = 'None Selected'
excludeCellCycle = False
restrictBy = 'protein_coding'
#restrictBy = 'None'
featurestoEvaluate = 'Genes'
ExpressionCutoff = 0
CountsCutoff = 0
if platform=="PSI":
FoldDiff = 1.2
else:
FoldDiff=4.0
ExpressionCutoff = 1
SamplesDiffering = 4
JustShowTheseIDs=''
removeOutliers = False
PathwaySelection=[]
array_type="RNASeq"
rho_cutoff=rho_cutoff
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['Input=','rho=','dynamicCorrelation=','Mutationref=','platform=','scaling=','species=','ExpressionCutoff=','CountsCutoff=','FoldDiff=','SamplesDiffering=','removeOutliers=','featurestoEvaluate=','restrictBy=','excludeCellCycle=','column_metric=','column_method=','row_method=','row_metric'])
for opt, arg in options:
#if opt == '--processedInputExpFile': processedInputExpFile=arg
if opt=='--Input':EventAnnot=arg # input file
if opt=='--rho':rho_cutoff=arg # rho cutoff
if opt=='--dynamicCorrelation':
if string.lower(dynamicCorrelation) == 'true' or string.lower(dynamicCorrelation) == 'optimize':
dynamicCorrelation=True #constant using the provided correlation,iteratively optimize correlation cutoff"
else:
dynamicCorrelation=False
if opt=='--Mutationref':Mutationref=arg #reference file provided for enrichment (format groups file)
if opt=='--platform':platform=arg
if opt=='--scaling':scaling=arg # True to scale for large datasets, False run with all samples
if opt=='--species':species=arg
if opt=='--ExpressionCutoff':ExpressionCutoff=arg
if opt=='--CountsCutoff':CountsCutoff=arg
if opt=='--FoldDiff':FoldDiff=arg
if opt=='--SamplesDiffering':SamplesDiffering=arg
if opt=='--removeOutliers':removeOutliers=arg
if opt=='--featurestoEvaluate':featurestoEvaluate=arg
if opt=='--restrictBy':restrictBy=arg
if opt=='--column_metric':column_metric=arg
if opt=='--column_method':column_method=arg
if opt=='--row_method':row_method=arg
if opt=='--row_metric':row_metric=arg
gsp = UI.GeneSelectionParameters(species,array_type,vendor)
gsp.setGeneSet(GeneSetSelection)
gsp.setPathwaySelect(PathwaySelection)
gsp.setGeneSelection(GeneSelection)
gsp.setJustShowTheseIDs(JustShowTheseIDs)
gsp.setNormalize('median')
gsp.setSampleDiscoveryParameters(ExpressionCutoff,CountsCutoff,FoldDiff,SamplesDiffering,removeOutliers,featurestoEvaluate,restrictBy,excludeCellCycle,column_metric,column_method,rho_cutoff)
runICGS_NMF(EventAnnot,scaling,dynamicCorrelation,platform,species,Mutationref,gsp)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/ICGS_NMF.py
|
ICGS_NMF.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import unique
import math
import random
import copy
try: from stats_scripts import salstat_stats
except Exception: null=[]; #print 'WARNING! The library file "salstat_stats" is not installed'
import traceback
try: ### Added for AltAnalyze - Nathan Salomonis, 1-24-2012
from math import *
import cmath as cm
from stats_scripts import mpmath as mpmath
from mpmath import *
from mpmath import functions as for_py2app
except Exception: null=[] #print 'WARNING! The library file "mpmath" is not installed'
def testMPMath():
print psi(0, 1), -euler
print psi(1, '1/4'), pi**2+8*catala
print psi(2, '1/2'), -14*apery
def LinearRegression_lm(ls1,ls2,return_rsqrd):
intercept = 0 ### when forced through the origin
from rpy import r
d = r.data_frame(x=ls1, y=ls2)
model = r("y ~ x - 1") ### when not forced through the origin it is r("y ~ x")
fitted_model = r.lm(model, data = d)
slope = fitted_model['coefficients']['x']
#intercept = fitted_model['coefficients']['(Intercept)']
if return_rsqrd == 'yes':
from scipy import stats
rsqrd = math.pow(stats.linregress(ls1,ls2)[2],2)
return slope,rsqrd
else: return slope
def LinearRegression(ls1,ls2,return_rsqrd):
intercept = 0 ### when forced through the origin
from rpy import r
r.library('MASS')
k = r.options(warn=-1) ### suppress all warning messages from R
#print ls1; print ls2
d = r.data_frame(x=ls1, y=ls2)
model = r("y ~ x - 1") ### when not forced through the origin it is r("y ~ x")
fitted_model = r.rlm(model, data = d) ###errors: rlm failed to converge in 20 steps - maxit=21
slope = fitted_model['coefficients']['x']
#intercept = fitted_model['coefficients']['(Intercept)']
if return_rsqrd == 'yes':
from scipy import stats
rsqrd = math.pow(stats.linregress(ls1,ls2)[2],2)
return slope,rsqrd
else:
return slope
def adjustPermuteStats(pval_db):
#1. Sort ascending the original input p value vector. Call this spval. Keep the original indecies so you can sort back.
#2. Define a new vector called tmp. tmp= spval. tmp will contain the BH p values.
#3. m is the length of tmp (also spval)
#4. i=m-1
#5 tmp[ i ]=min(tmp[i+1], min((m/i)*spval[ i ],1)) - second to last, last, last/second to last
#6. i=m-2
#7 tmp[ i ]=min(tmp[i+1], min((m/i)*spval[ i ],1))
#8 repeat step 7 for m-3, m-4,... until i=1
#9. sort tmp back to the original order of the input p values.
global spval; spval=[]
for element in pval_db:
zsd = pval_db[element]
try:
try: p = float(zsd.PermuteP())
except AttributeError: p = float(zsd[0]) ### When values are indeces rather than objects
except Exception: p = 1
spval.append([p,element])
spval.sort(); tmp = spval; m = len(spval); i=m-2; x=0 ###Step 1-4
#spval.sort(); tmp = spval; m = len(spval)-1; i=m-1; x=0 ###Step 1-4
while i > -1:
tmp[i]=min(tmp[i+1][0], min((float(m)/(i+1))*spval[i][0],1)),tmp[i][1]; i -= 1
for (adjp,element) in tmp:
zsd = pval_db[element]
try: zsd.SetAdjP(adjp)
except AttributeError: zsd[1] = adjp ### When values are indeces rather than objects
class GroupStats:
def __init__(self,log_fold,fold,p):
self.log_fold = log_fold; self.fold = fold; self.p = p
def LogFold(self): return self.log_fold
def Fold(self): return self.fold
def Pval(self): return self.p
def PermuteP(self): return self.p ### This is not a permute p, but the object name in the function is PermuteP
def SetAdjPIndex(self,index): self.adj_index = index
def SetPvalIndex(self,index): self.pval_index = index
def AdjIndex(self): return self.adj_index
def RawIndex(self): return self.pval_index
def SetAdjP(self,adjp): self.adj_p = adjp
def AdjP(self): return str(self.adj_p)
def setPval(self,p): self.p = p ### Typically re-set when a moderated statistic is calculated (e.g., emperical Bayesian - eBayes)
def SetMod(self,adjp): self.adj_p = adjp
def setMaxCount(self,max_count): self.max_count = max_count
def MaxCount(self): return self.max_count
def setAdditionalStats(self,data_list1,data_list2):
""" Obtain the statistics for a moderated t-test and store as object variables """
try:
sg,n1,n2,avg1,avg2 = FeatureVariance(data_list1,data_list2)
self.sg = sg; self.n1 = n1; self.n2 = n2; self.avg1 = avg1; self.avg2 = avg2
except Exception: pass
del data_list1; del data_list2 ### probably unnecessary
def setAdditionalWelchStats(self,data_list1,data_list2):
svar1,svar2,n1,n2,avg1,avg2,df = WelchTestFeatureVariance(data_list1,data_list2)
sg = svar1+svar2 ### gene-level variance - this is actually s sub g squared or s2g - take square root to get sg
self.sg = sg; self.n1 = n1; self.n2 = n2; self.avg1 = avg1; self.avg2 = avg2; self.df = df
self.svar1 = svar1; self.svar2 = svar2
del data_list1; del data_list2 ### probably unnecessary
def Avg1(self): return self.avg1
def Avg2(self): return self.avg2
def N1(self): return float(self.n1)
def N2(self): return float(self.n2)
def DF(self): return self.df
def Svar1(self): return self.svar1
def Svar2(self): return self.svar2
def setFeatureVariance(self,sg): self.sg = sg
def FeatureVariance(self): return self.sg
def Report(self):
output = str(self.Pval())+'|'+str(self.Fold())
return output
def __repr__(self): return self.Report()
class moderatedStatData(GroupStats):
def __init__(self,data_list1,data_list2):
""" Obtain the statistics for a moderated t-test and store as object variables """
sg,n1,n2,avg1,avg2 = FeatureVariance(data_list1,data_list2)
self.sg = sg; self.n1 = n1; self.n2 = n2; self.avg1 = avg1; self.avg2 = avg2
del data_list1; del data_list2 ### probably unnecessary
def moderateTestStats(pval_db,probability_statistic):
""" Calculate a moderated variance for each biological comparison based, based on the average variance of all genes or molecules.
This calculation should be identical for moderated student t-test p-values from the R package limma. Small variances might arrise
from differences in the precision float values stored by the different languages and threshold from the Newton Iteration step. This
implementation currently relies on first, second and third derivitive calculations (e.g., polygamma aka psi functions) from mpmath."""
#tst = salstat_stats.TwoSampleTests([],[]) ### Create object with two empty lists - will analyze in object database afterwards
#d0, s0_squared = tst.getModeratedStandardDeviation(pval_db)
d0, s0_squared = getModeratedStandardDeviation(pval_db,probability_statistic)
#print 'Prior degrees of freedom:',d0, 'and Prior s0 squared:',s0_squared
#d0 = 2.054191
#s0_squared = 0.01090202
for uid in pval_db:
gs = pval_db[uid]
if 'Welch' in probability_statistic:
ModeratedWelchTest(gs,d0, s0_squared)
else:
#tst.ModeratedTTestUnpaired(gs,d0, s0_squared)
ModeratedTTestUnpaired(gs,d0,s0_squared)
"""
if uid == '10367120':
print gs.Avg1(), gs.Avg2(), gs.FeatureVariance(), math.sqrt(gs.FeatureVariance()), gs.AdjP()
#gs.setFeatureVariance(math.sqrt(gs.FeatureVariance()))
#tst.ModeratedTTestUnpaired(gs,d0, s0_squared)
#print gs.Avg1(), gs.Avg2(), gs.FeatureVariance(), math.sqrt(gs.FeatureVariance()), gs.AdjP()
"""
def zscore(associated_in_group,in_static_interval,total,in_flexible_interval):
r = float(associated_in_group) #number of genes with this domain regulated (in one direction) (# genes regulated in pathway) (#peeks for the SF)
_n = float(in_static_interval) #measured genes in genomic interval - !!from chr_info!! (# genes regulated) (# of peaks for the CLIP)
N = float(total) #measured genes in the genome - total_count (#all genes evaluated on pathways) (# of peaks for CLIP that overlap )
R = float(in_flexible_interval) #genes in the hopach interval(not measured) - !!subtract max-min or from hopach_order (#genes in pathway)
if (R-N) == 0: return 0
elif r==0 and _n == 0: return 0
else:
try:
#try:
z = (r - _n*(R/N))/math.sqrt(_n*(R/N)*(1-(R/N))*(1-((_n-1)/(N-1))))
return z
#except ZeroDivisionError: print 'r,_n,R,N: ', r,_n,R,N;kill
except ValueError: print (r - _n*(R/N)), _n*(R/N)*(1-(R/N))*(1-((_n-1)/(N-1))),r,_n,N,R;kill
def factorial(n):
### Code from http://docs.python.org/lib/module-doctest.html
if not n >= 0:
raise ValueError("n must be >= 0")
if math.floor(n) != n:
raise ValueError("n must be exact integer")
if n+1 == n: # catch a value like 1e300
raise OverflowError("n too large")
result = 1
factor = 2
while factor <= n:
result *= factor
factor += 1
return result
def choose(n,x):
"""Equation represents the number of ways in which x objects can be selected from a total of n objects without regard to order."""
#(n x) = n!/(x!(n-x)!)
f = factorial
result = f(n)/(f(x)*f(n-x))
return result
def pearson(array1,array2):
item = 0
list_len = len(array1)
sum_a = 0
sum_b = 0
sum_c = 0
while item < list_len:
a = (array1[item] - avg(array1))*(array2[item] - avg(array2))
b = math.pow((array1[item] - avg(array1)),2)
c = math.pow((array2[item] - avg(array2)),2)
sum_a = sum_a + a
sum_b = sum_b + b
sum_c = sum_c + c
item = item + 1
try:
r = sum_a/math.sqrt(sum_b*sum_c)
except ZeroDivisionError:
print "ZeroDivisionError encountered: likely due to all control and experimental folds equal to zero. Results from Excel and Access data truncation.",quit
return r
def maxval(array):
### Same as _max_ but processes string values as floats
array2=[]
for i in array: array2.append(float(i))
return max(array2)
def permute(list):
if not list: #shuffle any sequence
return [list] #empty sequence
else:
try:
res = []
for i in range(len(list)):
rest = list[:i] + list[i+1:] #delete current node
for x in permute(rest): #permute the others
res.append(list[i:i+1] + x) #add node at front
return res
except TypeError:
print list,dog
def permute_arrays(a):
a.sort()
b = []; indices = []; permute_num = 10000
y = 0; x = 0; iter = 0; c = []
for ls in a:
x = x + len(ls)
indices.append(x)
tls = tuple(ls)
c.append(tls)
if iter == 0:
y = len(ls) ###number of elements in the first array
iter = 1
for index in ls:
b.append(index)
c = tuple(c) ### Original group organization, in tuple form (required for dictionsry keys)
#z = len(b) ###number of elements in the full array set
#c = choose(z,y) ###all possible unique permutations(only works for two arrays)
unique_array_permute_list = permute_at_random(b,indices,permute_num,c) ### send all indexes, len(array1), len(array2), possible permutations
###Below code also works but is VERY memory intensive (substitute out above line)
"""pz = permute(b); array_permute_list = []
for p in pz: ###for each permuted list
ls_pair = p[0:y]; ls_pair2 = p[y:]l ls_pair.sort();ls_pair2.sort();ls_pair = ls_pair;ls_pair2
array_permute_list.append(ls_pair)"""
return unique_array_permute_list
def permute_at_random(a,indices,permute_times,original_groups):
"""much more efficient method of generating all possible permuted lists"""
### Randomize original list, divide into two arrays, sort and store in a database
permute_db = {}; permute_ls = []; x = 0; null_hits = 0; done = 0
while done == 0:
b = copy.deepcopy(a); random.shuffle(b); y = 0
bx_set=[]
for index in indices:
if y == 0: bx = b[0:index]; bx.sort(); bx_set.append(bx)
else: bx = b[prev_index:index]; bx.sort(); bx_set.append(bx)
y += 1
prev_index = index
bn=[]
for new_list in bx_set:
bn.append(tuple(new_list))
bn = tuple(bn)
if bn in permute_db: null_hits += 1
else: permute_db[bn] = ''; null_hits = 0
x += 1
if (x>permute_times) or (null_hits>500):done = 1
#print len(permute_db), x
try: del permute_db[original_groups] ###Ensures that independent of the organization of the orginal arrays, the orginal is listed first
except KeyError: null = ''; ###Occurs when the max number of allowed permuations is greater than all possible
permute_ls.append(original_groups)
for entry in permute_db:
permute_ls.append(entry)
permute_ls.sort()
return permute_ls
def aspire_stringent(b1,e1,b2,e2):
baseline_ratio1 = b1; experimental_ratio1 = e1
baseline_ratio2 = b2; experimental_ratio2 = e2
Rin = baseline_ratio1/experimental_ratio1 # Rin=A/C
Rex = baseline_ratio2/experimental_ratio2 # Rin=B/D
I1=baseline_ratio1/(baseline_ratio1+baseline_ratio2)
I2=experimental_ratio1/(experimental_ratio1+experimental_ratio2)
#if (Rin>1 and Rex<1) or (Rin<1 and Rex>1):
in1=((Rex-1.0)*Rin)/(Rex-Rin)
in2=(Rex-1.0)/(Rex-Rin)
### dI = ((in1-in2)+(I1-I2))/2.0 #original equation
dI = ((in2-in1)+(I2-I1))/2.0 #modified to give propper exon inclusion
#dI_str = str(abs(dI)) #remove small decimal changes that would effect removal of duplicates
#numerator,decimal = string.split(dI_str,".")
#dI_str = numerator + '.' + decimal[0:5]
return dI
#else:return 'null'
def permute_p(null_list,true_value,n):
y = 0; z = 0
x = n # Can differ from len(null_list), since ASPIRE excludes non-significant entries
for value in null_list:
if value >= true_value:
y += 1
if value > true_value:
z += 1
return float(y)/float(x), y,x,z
def avg(array):
total = sum(map(float, array))
average = total/len(array)
return average
def stdev(array):
sum_dev = 0
x_bar = avg(array)
n = float(len(array))
for x in array:
x = float(x)
sq_deviation = math.pow((x-x_bar),2)
sum_dev += sq_deviation
try:
s_sqr = (1.0/(n-1.0))*sum_dev #s squared is the variance
s = math.sqrt(s_sqr)
except ZeroDivisionError:
s = 'null'
return s
def FeatureVariance(data_list1,data_list2):
"""Calculates the variance for a standard t-statistic to use for calculation of a moderated t-test"""
N1 = len(data_list1)
N2 = len(data_list2)
df = float((N1 + N2) - 2)
svar1 = math.pow(stdev(data_list1),2)
svar2 = math.pow(stdev(data_list2),2)
avg1 = avg(data_list1)
avg2 = avg(data_list2)
sg_squared = (svar1*(N1-1)+svar2*(N2-1))/df ### gene-level variance - this is actually s sub g squared or s2g - take square root to get sg
return sg_squared,N1,N2,avg1,avg2
def WelchTestFeatureVariance(data_list1,data_list2):
"""Calculates the variance for a standard t-statistic to use for calculation of a moderated t-test"""
n1 = len(data_list1)
n2 = len(data_list2)
svar1 = math.pow(stdev(data_list1),2)/n1
svar2 = math.pow(stdev(data_list2),2)/n2
avg1 = avg(data_list1)
avg2 = avg(data_list2)
try: df = math.pow((svar1+svar2),2)/((math.pow(svar1,2)/(n1-1)) + (math.pow(svar2,2)/(n2-1)))
except Exception: df = 1
return svar1,svar2,n1,n2,avg1,avg2,df
def getModeratedStandardDeviation(comparison_db,probability_statistic):
variance_ls=[]; e_sum=0; d0_2nd_moment_gene_sum = 0
for uid in comparison_db:
gs = comparison_db[uid] ### Object containing summary statistics needed for each uid (aka feature)
if 'Welch' in probability_statistic:
df = gs.DF()
else:
try: df = (gs.N1() + gs.N2()) - 2
except Exception,e: print e, gs, [gs.N1(), gs.N2()];kill
sg_squared = gs.FeatureVariance()
#print uid, df, sg_squared;kill
###calculate s0 and d0
if sg_squared > 1e-11:
zg = math.log(sg_squared)
eg = zg - psi(0,df/2.0) + math.log(df/2.0)
variance_ls.append((eg,df))
n = len(variance_ls) ### number of uids analyzed
### Get the mean eg for all IDs
for (eg,df) in variance_ls:
e_sum+=eg
e_avg = e_sum/len(variance_ls)
### Calculate the d0 2nd derivitive that will later need to be solved for d0
for (eg,df) in variance_ls:
d0_2nd_moment_gene_sum += ((math.pow(eg-e_avg,2)*n)/(n-1)) - psi(1,df/2)
d0_2nd_moment_solve = d0_2nd_moment_gene_sum/len(variance_ls)
#print [d0_2nd_moment_solve]
d0 = NewtonInteration(d0_2nd_moment_solve)*2
#print [d0]
d0 = float(d0)
e = cm.e
s0_squared = math.pow(e,e_avg+psi(0,d0/2) - math.log(d0/2))
return d0, s0_squared
def NewtonInteration(x):
""" Method used to emperically identify the best estimate when you can't solve for the variable of interest (in this case, d0 aka y)"""
y = 0.5 + (1/x)
proceed = 1
while proceed == 1:
if x>1e7: y = 1/math.sqrt(x); proceed = 0
elif x<1e-6: y = 1/x; proceed = 0
else:
d = (psi(1,y)*(1-(psi(1,y)/x)))/psi(2,y)
y = y + d
if (-d/y)< 1e-8:
proceed = 0
break
return y
def ModeratedWelchTest(gs,d0,s0_squared):
df = gs.DF()
### use the s0_squared for the pairwise comparison calculated in the getModeratedStandardDeviation
svar1 = (d0*s0_squared+df*gs.Svar1())/(d0+df)
svar2 = (d0*s0_squared+df*gs.Svar2())/(d0+df)
#svar = sg ### Use this to test and see if this gives the same result as a non-moderated t-test
if svar1 != 0 and svar2 != 0:
t = (gs.Avg1()-gs.Avg2())/math.sqrt(svar1+svar2)
prob = salstat_stats.betai(0.5*df,0.5,float(df)/(df+t*t))
else: prob = 1
#gs.SetAdjP(prob)
gs.setPval(prob)
#print [t, df, prob], 'ModeratedWelchTest'
def ModeratedTTestUnpaired(gs,d0,s0_squared):
""" This function was validated using output data from limma """
df = (gs.N1() + gs.N2()) - 2
sg_squared = gs.FeatureVariance()
### use the s0_squared for the pairwise comparison calculated in the getModeratedStandardDeviation
svar = (d0*s0_squared+df*sg_squared)/(d0+df) ### square root
#svar = sg ### Use this to test and see if this gives the same result as a non-moderated t-test
if svar != 0:
df = df+d0
t = (gs.Avg1()-gs.Avg2())/math.sqrt(svar*(1.0/gs.N1() + 1.0/gs.N2()))
prob = betai(0.5*df,0.5,float(df)/(df+t*t))
else: prob = 1
#print [t, df, prob], 'ModeratedTTestUnpaired'
#gs.SetAdjP(prob)
gs.setPval(prob)
def log_fold_conversion_fraction(array):
try:
new_array = []
for log_fold in array:
log_fold = float(log_fold)
real_fold = math.pow(2,log_fold); new_array.append(real_fold)
except TypeError:
log_fold = float(array)
new_array = math.pow(2,log_fold)
return new_array
def log_fold_conversion(array):
try:
new_array = []
for log_fold in array:
log_fold = float(log_fold)
if log_fold > 0 or log_fold == 0:
real_fold = math.pow(2,log_fold); new_array.append(real_fold)
else: real_fold = -1/(math.pow(2,log_fold)); new_array.append(real_fold)
except TypeError:
log_fold = float(array)
try:
if log_fold > 0 or log_fold == 0: new_array = math.pow(2,log_fold)
else: new_array = -1/(math.pow(2,log_fold))
except Exception:
print 'Error with fold transformation for the log fold:',log_fold
forceError
return new_array
def convert_to_log_fold(array):
list_status = 'yes'
try:
if len(array)>1: array = array
except TypeError: array2 = []; array2.append(array); array = array2; list_status = 'no'
new_array = []
for fold in array:
fold = float(fold)
if fold < -1: fold = -1/fold
#elif fold >-1 and fold <1: fold = 1
log_fold = math.log(fold,2)
new_array.append(log_fold)
if list_status == 'no': return new_array[0]
else: return new_array
def neg_folds_to_fractions(array):
try:
new_array = []
for fold in array:
try:
fold = float(fold)
except ValueError:
print fold, dog
if fold > 0:
fold = fold
new_array.append(fold)
else:
fold = -1/fold
new_array.append(fold)
except TypeError:
fold = float(array)
if fold > 0:
new_array = fold
else:
new_array = -1/fold
return new_array
def median(array):
array = list(array) ### If we don't do this we can modify the distribution of the original list object with sort!!!
array.sort()
len_float = float(len(array))
len_int = int(len(array))
if (len_float/2) == (len_int/2):
try: median_val = avg([array[(len_int/2)-1],array[(len_int/2)]])
except IndexError: median_val = ''
else:
try: median_val = array[len_int/2]
except IndexError: median_val = ''
return median_val
def int_check(value):
val_float = float(value)
val_int = int(value)
if val_float == val_int:
integer_check = 'yes'
if val_float != val_int:
integer_check = 'no'
return integer_check
def iqr(array, k1=75, k2=25):
array.sort()
n = len(array)
value1 = float((n*k1)/100)
value2 = float((n*k2)/100)
if int_check(value1) == 'no':
k1_val = int(value1) + 1
if int_check(value1) == 'yes':
k1_val = int(value1)
if int_check(value2) == 'no':
k2_val = int(value2) + 1
if int_check(value2) == 'yes':
k2_val = int(value2)
median_val = median(array)
upper75th = array[k1_val]
lower25th = array[k2_val]
int_qrt_range = upper75th - lower25th
return lower25th,median_val,upper75th,int_qrt_range
def paired_ttest(list1,list2,tails,variance):
i=0; dx_list=[]
for x in list1:
dx = float(list2[i])-float(list1[i]); dx_list.append(dx)
i+=1
avg_x = avg(dx_list)
sx = stdev(dx_list)
def ttest(list1,list2,tails,variance):
""" Although issues with this function and the incomplete beta were present in the past, evaluating these on 2-1-12
confirmed that these methods produce accurate p-values for various equal variance and unequal variance examples for equal and unequal sample sizes"""
val_list1=[]
val_list2=[]
n1 = float(len(list1))
n2 = float(len(list2))
#make sure values are not strings
for entry in list1:
entry = float(entry)
val_list1.append(entry)
for entry in list2:
entry = float(entry)
val_list2.append(entry)
if variance == 3: ### Unequal variance
var1 = math.pow(stdev(val_list1),2)/n1
var2 = math.pow(stdev(val_list2),2)/n2
try:
t = (avg(val_list1) - avg(val_list2))/math.sqrt(var1+var2)
df = math.pow((var1+var2),2)/((math.pow(var1,2)/(n1-1)) + (math.pow(var2,2)/(n2-1)))
#print (avg(val_list1), avg(val_list2)), math.sqrt(var1+var2), math.pow(stdev(val_list1),2), math.pow(stdev(val_list2),2)
"""
# Equivalent to the above - shows that the above df calculation is accurate
u = math.pow(stdev(val_list2),2)/math.pow(stdev(val_list1),2)
df2 = math.pow((1/n1)+(u/n2),2)/((1/(math.pow(n1,2)*(n1-1))) + (math.pow(u,2)/(math.pow(n2,2)*(n2-1))))
print df, df2;sys.exit()"""
except Exception: t=1; df=1; tails=2
#calculate the degree's of freedom
if variance == 2:
if n1 == n2: ### assuming equal size
var1 = math.pow(stdev(val_list1),2)
var2 = math.pow(stdev(val_list2),2)
sx = math.sqrt((var1+var2)/2)
#print sx, (avg(val_list1) - avg(val_list2));kill
try:
t = (avg(val_list1) - avg(val_list2))/(sx*math.sqrt(2/n1))
df = 2*n1-2
except Exception: t=1; df=1; tails=2
else:
var1 = math.pow(stdev(val_list1),2)
var2 = math.pow(stdev(val_list2),2)
a1 = 1.00/n1
a2 = 1.00/n2
sx = math.sqrt(((n1-1)*var1+(n2-1)*var2)/(n1+n2-2))
try:
t = (avg(val_list1) - avg(val_list2))/(sx*math.sqrt(a1+a2))
#t = (avg(val_list1) - avg(val_list2))/math.sqrt(sx*(a1+a2))
df = (n1 + n2 - 2)
except Exception: t=1; df=1; tails=2
return t,df,tails
def incompleteBeta(t,df):
p = salstat_stats.betai(0.5*df,0.5,df/(df+t*t))
return p
def t_probability(t,df):
### Works accurately for large df's when performing unequal variance tests - unlike t_probabilityOld
return incompleteBeta(t,df)
def t_probabilityOld(t,df):
"""P(abs(T)<t) is equivalent to the probability between -t and +t. So the two-sided p value for t is
1-P(abs(T)<t)."""
t = abs(t)
original_df = df
if df <0: df = df*-1
df=int(string.split(str(df),'.')[0]) ###alternatively could round down as - math.floor(number*10)/10
if original_df <0: df = df*-1
if df >100: df = 100
pi = 3.141592653589793238
if int(df)/2 == float(int(df))/2.0:
a = 'even'
else:
a = 'odd'
if a == 'even':
sdf1 = df - 2.0
x = 2; y = 1; z = 1; w = 1
while x < sdf1:
y = y*x; x = x + 2
sdf2 = df - 3.0
while z < sdf2:
w = w*z; z = z + 2.0
if a == 'odd':
sdf1 = df - 3.0
x = 2; y = 1; z = 1; w = 1
while x < sdf1:
y = y*x; x = x + 2.0
sdf2 = df - 2.0
while z < sdf2:
w = w*z; z = z + 2.0
theta = math.atan(t/math.sqrt(df))
if df == 1:
p = (2.0/pi)*theta
if df>1 and a =='odd':
store_var = 0
while sdf1 > 0:
var = (((y*(sdf1))/(w*(sdf2)))*math.pow(math.cos(theta),(sdf2)))
store_var = store_var + var
sdf1 = sdf1 - 2.0
sdf2 = sdf2 - 2.0
try:
w = w/sdf2
y = y/sdf1
except ZeroDivisionError:
continue
p = (2.0/pi)*(theta + math.sin(theta)*(math.cos(theta)+store_var))
#P(abs(T)<t) = (2/pi) * (theta + sin(theta) * (cos(theta)+ (2/3)*cos(theta)^3 + ... + ((2*4*...*(nu-3))/(1*3*...*(nu-2))) * cos(theta)^(nu-2) ))
if df>1 and a =='even':
store_var = 0
while sdf1 > 0:
var = (((w*(sdf2))/(y*(sdf1)))*math.pow(math.cos(theta),(sdf1)))
#print 'stats',w,y,sdf1
store_var = store_var + var
sdf1 = sdf1 - 2.0
sdf2 = sdf2 - 2.0
try:
w = w/sdf2
y = y/sdf1
except ZeroDivisionError:
continue
p = math.sin(theta)*(1.0 + store_var)
#p = math.sin(theta)*(1.0+(1.0/2.0)*math.pow(math.cos(theta),2.0)+((1.0*3.0)/(2.0*4.0))*math.pow(math.cos(theta),4.0) + ((w*(df-3.0))/(y*(df-2.0)))*math.pow(math.cos(theta),(df-2.0)))
#p= sin(theta)*(1 + 1/2*cos(theta)^2 + ((1*3)/(2*4))*cos(theta)^4 + ... + ((1*3*5*...*(nu-3))/(2*4*6*...*(nu-2))) * cos(theta)^(nu-2) )
#(1.0/2.0)*math.pow(math.cos(theta),2.0)+ ((1.0*3.0)/(2.0*4.0))*math.pow(math.cos(theta),4.0) + (1.0*3.0*5.0)/(2.0*4.0*6.0)*math.pow(math.cos(theta),(df-2.0))
p = 1-p
#print (2.0)/(3.0), ((w*(df-3.0))/(y*(df-2.0)))
return p
def rankExpectation(exp_db):
#exp_db[gene] = [53.4, 57.2]
# test both hypotheses separately (a>b) and (b>a)
# set a window for creating the normal distribution around all values to test - default is 50 genes on both sides - 100 total
#1) alength
# which ever is larger, max(mad(ax[r[x,1]:r[x,2]]),mm, max mad or minmad, take that
# can we emperically determine the min mad?
# pnorm is calculating a probability based on the z-score standard deviation
null=[]
def p_value(z):
"""A formula that is accurate to within 10^(-5) is the following:
P(z) = 1 - d(z)*(a1*t + a2*(t^2) + a3*(t^3)), where
z>=0,
P(z) is the standard normal cumulative,
d(z) is the standard normal density,
t = 1/(1+p*z),
p = 0.33267,
a1 = 0.4361836,
a2 = -0.1201676,
a3 = 0.9372980.
This is formula 26.2.16 from Abramowitz and Stegun. If z<0, use P(z) = 1 - P(-z).
If they need small tail probabilities with low relative error, the 10^(-5) possible error may be too large in some cases.
For large positive z, try
1-P(z) = d(z)*(1/(z+1/(z+2/(z+3/(z+4/(z+5/z)))))).
Check this in R to make sure relative errors are OK for large z. If not, extend to 6, 7, etc. (it's a continued fractions expansion).
d(z) = (1/(sqrt(2*pi))) * exp (-(z**2) / 2)"""
p = 0.33267
a1 = 0.4361836
a2 = -0.1201676
a3 = 0.9372980
t = 1/(1+(p*z))
pi = 3.141592653589793238
y = (1/(math.sqrt(2*pi)))* math.exp(-(z**2)/2)
if z >= 0:
p_val = 1-(y*((a1*t) + a2*(math.pow(t,2)) + a3*(math.pow(t,3))))
else:
z = z*(-1)
p_val = (y*((a1*t) + a2*(math.pow(t,2)) + a3*(math.pow(t,3))))
p_val = 2*(1-p_val)
return p_val
def bonferroni_p(z,correction):
p_val = p_value(z)
p_val = p_val*correction
return p_val
def GrandMean(arrays):
den = 0; num = 0; gn=0
for array in arrays:
x = avg(array); n = len(array); den += n; num += n*x; gn += n
gm = num/den
return gm,gn
def OneWayANOVA(arrays):
f,df1,df2 = Ftest(arrays)
p = fprob(df1,df2,f)
return p
def Ftest(arrays):
k = len(arrays); swsq_num=0; swsq_den=(-1)*k; sbsq_num=0; sbsq_den=(k-1); xg,ng = GrandMean(arrays)
for array in arrays:
try:
n=len(array); x=avg(array); s=stdev(array)
var1=(n-1)*(s**2); var2=n*((x-xg)**2)
swsq_num += var1; swsq_den += n; sbsq_num += var2
except Exception: null=[] ### Occurs when no variance - one sample for that group
swsq = swsq_num/swsq_den; sbsq = sbsq_num/sbsq_den
try: f = sbsq/swsq
except ZeroDivisionError: f = 0
df1=k-1; df2=ng-k
return f,df1,df2
def runComparisonStatistic(data_list1,data_list2,probability_statistic):
### This function uses the salstat_stats module from the SalStat statistics package http://salstat.sourceforge.net/
### This module is pure python and does not require other external libraries
if len(data_list1) == 1 or len(data_list2)==1: ### will return a p-value if one has multiple, but shouldn't
return 1
else:
if probability_statistic == 'unpaired t-test' or 'moderated' in probability_statistic:
p = OneWayANOVA([data_list1,data_list2]) ### faster implementation of unpaired equal variance t-test
else:
tst = salstat_stats.TwoSampleTests(data_list1,data_list2)
# options = unpaired t-test|paired t-test|Kolmogorov Smirnov|Mann Whitney U|Rank Sums
if probability_statistic == 'paired t-test': p = tst.TTestPaired()
elif probability_statistic == 'Kolmogorov Smirnov': p = tst.KolmogorovSmirnov()
elif probability_statistic == 'Mann Whitney U': p = tst.MannWhitneyU()
elif probability_statistic == 'Rank Sums': p = tst.RankSums()
elif probability_statistic == 'unpaired t-test' or 'moderated' in probability_statistic:
### Typically not run except during testing
p = tst.TTestUnpaired()
return p
###########Below Code Curtosey of Distribution functions and probabilities module
"""
AUTHOR(S): Sergio J. Rey [email protected]
Copyright (c) 2000-2005 Sergio J. Rey
Comments and/or additions are welcome (send e-mail to:
[email protected]).
"""
def fprob(dfnum, dfden, F):
"""Returns the (1-tailed) significance level (p-value) of an F
statistic given the degrees of freedom for the numerator (dfR-dfF) and
the degrees of freedom for the denominator (dfF).
Usage: fprob(dfnum, dfden, F) where usually dfnum=dfbn, dfden=dfwn """
p = betai(0.5*dfden, 0.5*dfnum, dfden/float(dfden+dfnum*F))
return p
def betacf(a,b,x):
"""This function evaluates the continued fraction form of the incomplete
Beta function, betai. (Adapted from: Numerical Recipies in C.)
Usage: betacf(a,b,x) """
ITMAX = 200; EPS = 3.0e-7
bm = az = am = 1.0; qab = a+b; qap = a+1.0; qam = a-1.0
bz = 1.0-qab*x/qap
for i in range(ITMAX+1):
em = float(i+1); tem = em + em
d = em*(b-em)*x/((qam+tem)*(a+tem)); ap = az + d*am; bp = bz+d*bm
d = -(a+em)*(qab+em)*x/((qap+tem)*(a+tem)); app = ap+d*az; bpp = bp+d*bz
aold = az; am = ap/bpp; bm = bp/bpp; az = app/bpp; bz = 1.0
if (abs(az-aold)<(EPS*abs(az))):
return az
print 'a or b too big, or ITMAX too small in Betacf.'
def betai(a,b,x):
"""Returns the incomplete beta function:
I-sub-x(a,b) = 1/B(a,b)*(Integral(0,x) of t^(a-1)(1-t)^(b-1) dt)
where a,b>0 and B(a,b) = G(a)*G(b)/(G(a+b)) where G(a) is the gamma
function of a. The continued fraction formulation is implemented here,
using the betacf function. (Adapted from: Numerical Recipies in C.)
Usage: betai(a,b,x)"""
if (x<0.0 or x>1.0): raise ValueError, 'Bad x in lbetai'
if (x==0.0 or x==1.0): bt = 0.0
else: bt = math.exp(gammln(a+b)-gammln(a)-gammln(b)+a*math.log(x)+b*math.log(1.0-x))
if (x<(a+1.0)/(a+b+2.0)): return bt*betacf(a,b,x)/float(a)
else: return 1.0-bt*betacf(b,a,1.0-x)/float(b)
def gammln(xx):
"""Returns the gamma function of xx. Gamma(z) = Integral(0,infinity) of t^(z-1)exp(-t) dt.
(Adapted from: Numerical Recipies in C.) Usage: gammln(xx) """
coeff = [76.18009173, -86.50532033, 24.01409822, -1.231739516, 0.120858003e-2, -0.536382e-5]
x = xx - 1.0
tmp = x + 5.5
tmp = tmp - (x+0.5)*math.log(tmp)
ser = 1.0
for j in range(len(coeff)):
x = x + 1
ser = ser + coeff[j]/x
return -tmp + math.log(2.50662827465*ser)
###########END Distribution functions and probabilities module
def simpleLinRegress(x,y):
### Seems to approximate "LinearRegression_lm" which uses lm but not rlm. RLM doesn't give the same value though.
### This is the standard the least squares formula or m = Sum( x_i * y_i ) / Sum( x_i * x_i )
i = 0; sum_val_num=0; sum_val_denom=0
for v in x:
try: sum_val_num+=(y[i]*x[i])
except Exception: print y[i],x[i], y, x;kill
sum_val_denom+=math.pow(x[i],2)
i+=1
slope = sum_val_num/sum_val_denom
return slope
def testModeratedStatistics():
dgt_ko = [5.405,5.375,5.614]
wt = [5.952,5.952,6.007]
d0 = 2.054191
s0_squared = 0.01090202
import reorder_arrays
gs = reorder_arrays.GroupStats(0,1,0)
gs.setAdditionalStats(wt,dgt_ko) ### Assuming equal variance
ModeratedTTestUnpaired(gs,d0,s0_squared)
gs.setAdditionalWelchStats(wt,dgt_ko) ### Assuming unequal variance
ModeratedWelchTest(gs,d0,s0_squared)
def matrixImport(filename):
matrix={}
compared_groups={} ### track which values correspond to which groups for pairwise group comparisons
original_data={}
headerRow=True
for line in open(filename,'rU').xreadlines():
original_line = line
data = line.rstrip()
values = string.split(data,'\t')
#print len(values)
if headerRow:
group_db={}
groups=[]
if ':' in data:
group_sample_list = map(lambda x: string.split(x,':'),values[1:])
index=1
for (g,s) in group_sample_list:
try: group_db[g].append(index)
except Exception: group_db[g] = [index]
index+=1
if g not in groups: groups.append(g)
else:
import ExpressionBuilder
search_dir = string.split(filename,'AltResults')[0]+'ExpressionInput'
files = unique.read_directory(search_dir)
for file in files:
if 'groups.' in file and '.txt' in file:
#print file
sample_group_db = ExpressionBuilder.simplerGroupImport(search_dir+'/'+file)
index=0; count=0
for s in values[1:]:
if s in sample_group_db:
g = sample_group_db[s]
try: group_db[g].append(index)
except Exception: group_db[g] = [index]
count+=1
if g not in groups: groups.append(g)
#else: print [s]
index+=1
#print count
headerRow = False
grouped_values=[]
original_data['header'] = original_line
else:
key = values[0]
values=values[1:]
grouped_floats=[]
float_values = []
associated_groups=[]
for g in groups: ### string values
gvalues_list=[]
for i in group_db[g]:
try:
if values[i] != '0':
try:
gvalues_list.append(float(values[i]))
except Exception: pass
else:
#try: gvalues_list.append('') ### Thus are missing values
#except Exception: pass
pass
except Exception:
#try: gvalues_list.append('') ### Thus are missing values
#except Exception: pass
pass
grouped_floats.append(gvalues_list)
if len(gvalues_list)>1:
associated_groups.append(g)
matrix[key] = grouped_floats
compared_groups[key] = associated_groups
if '\n' not in original_line:
original_line+='\n'
original_data[key] = original_line
last_line = line
return matrix,compared_groups,original_data
def runANOVA(filename,matrix,compared_groups):
try:
from import_scripts import AugmentEventAnnotations
annotationFile = string.replace(filename,'-clust.txt','_EventAnnotation.txt')
eventAnnotations = AugmentEventAnnotations.importPSIAnnotations(annotationFile)
except Exception:
#print traceback.format_exc();sys.exit()
eventAnnotations={}
import export
matrix_pvalues={}
all_matrix_pvalues={}
matrix_pvalues_list=[]
pairwise_matrix = {}
useAdjusted=False
pvals=[]
eo = export.ExportFile(filename[:-4]+'-pairwise.txt')
eo.write(string.join(['UID','Symbol','Description','Coordinates','Examined-Junction','Background-Major-Junction','AltExons','ProteinPredictions','EventAnnotation','Group1','Group2','rawp','G1-PSI','G2-PSI'],'\t')+'\n')
for key in matrix:
filtered_groups = []
### Import and add annotations for each event
try:
ea = eventAnnotations[key]
Symbol = ea.Symbol()
Description = ea.Description()
Junc1 = ea.Junc1()
Junc2 = ea.Junc2()
AltExons = ea.AltExons()
Coordinates = ea.Coordinates()
ProteinPredictions = ea.ProteinPredictions()
EventAnnotation = ea.EventAnnotation()
except Exception:
#print traceback.format_exc(); sys.exit()
Symbol = ''
Description = ''
Junc1 = ''
Junc2 = ''
AltExons = ''
ProteinPredictions = ''
EventAnnotation = ''
Coordinates = ''
for group in matrix[key]:
if len(group)>1:
filtered_groups.append(group)
try:
p = OneWayANOVA(filtered_groups)
pvals.append(p)
if useAdjusted == False:
if p < 0.05:
try:
### Perform all possible pairwise group comparisons
gi1=-1
major_groups={}
comparisons={}
group_names = compared_groups[key]
added=[]
for g1 in filtered_groups:
gi1+=1; gi2=-1
for g2 in filtered_groups:
gi2+=1
if g1!=g2:
if abs(avg(g1)-avg(g2))>0.1:
pairwise_p = OneWayANOVA([g1,g2])
if pairwise_p<0.05:
group1 = group_names[gi1]
group2 = group_names[gi2]
sorted_groups=[group1,group2]
sorted_groups.sort()
group1, group2 = sorted_groups
if (group1,group2) not in added:
#if key == 'Tnfaip8:ENSMUSG00000062210:E3.4-E8.1 ENSMUSG00000062210:E1.4-E8.1':
#print group1,'\t',group2,'\t',pairwise_p
added.append((group1,group2))
try: major_groups[group1]+=1
except Exception: major_groups[group1]=1
try: major_groups[group2]+=1
except Exception: major_groups[group2]=1
try: comparisons[group1].append(group2)
except Exception: comparisons[group1] = [group2]
try: comparisons[group2].append(group1)
except Exception: comparisons[group2] = [group1]
if g2<g1:
instance_proteinPredictions = string.replace(ProteinPredictions,'+','^^')
instance_proteinPredictions = string.replace(instance_proteinPredictions,'-','+')
instance_proteinPredictions = string.replace(instance_proteinPredictions,'^^','-')
else:
instance_proteinPredictions = ProteinPredictions
values = string.join([key,Symbol,Description,Coordinates,Junc1,Junc2,AltExons,
instance_proteinPredictions,EventAnnotation,group_names[gi1],group_names[gi2],
str(pairwise_p),str(avg(g1)),str(avg(g2))],'\t')+'\n'
eo.write(values)
#pairwise_matrix[key,group_names[gi1],group_names[gi2]] = pairwise_p,avg(g1),avg(g2)
major_group_list=[]
for group in major_groups: major_group_list.append([major_groups[group],group])
major_group_list.sort()
major_group_list.reverse()
top_group = major_group_list[0][1]
hits = top_group+'_vs_'+string.join(comparisons[top_group],'|')
"""
if major_group_list[0][0] == major_group_list[1][0]:
hits = major_group_list[0][1]+'|'+major_group_list[1][1]
else:
hits = major_group_list[0][1]
"""
except Exception:
#print traceback.format_exc();sys.exit()
hits=''
if len(added)>0:
matrix_pvalues[key]=[p,p]
matrix_pvalues_list.append((p,'',key,hits))
else:
matrix_pvalues[key]=[p,p]
matrix_pvalues_list.append((p,'',key,''))
all_matrix_pvalues[key]=[p,p]
#if 'CAMK2D' in key: print filtered_groups; print key, p
except Exception:
#print traceback.format_exc();sys.exit()
pass ### not enough values present or groups
pvals.sort()
#print pvals[:20]
adjustPermuteStats(all_matrix_pvalues)
adj_matrix_pvalues = copy.deepcopy(all_matrix_pvalues)
if useAdjusted: matrix_pvalues={}
matrix_pvalues_list.sort()
matrix_pvalues_list2=[]
for (p,null,key,hits) in matrix_pvalues_list:
p,adjp = adj_matrix_pvalues[key]
if useAdjusted:
if adj_matrix_pvalues[key][1] < 0.05:
matrix_pvalues_list2.append([key,str(p),str(adjp),hits])
matrix_pvalues[key] = adjp
else:
matrix_pvalues_list2.append([key,str(p),str(adjp),hits])
eo.close()
exportANOVAStats(filename,matrix_pvalues_list2)
print len(matrix_pvalues), 'ANOVA significant reciprocal PSI-junctions...'
return matrix_pvalues
def exportANOVAStats(filename,matrix_pvalues_list):
import export
export_name = filename[:-4]+'-stats.txt'
ee=export.ExportFile(export_name)
ee.write('SplicingEvent\tANOVA rawp\tANOVA adjp\tDriving Group(s)\n')
for ls in matrix_pvalues_list:
ee.write(string.join(ls,'\t')+'\n')
ee.close()
def returnANOVAFiltered(filename,original_data,matrix_pvalues):
import export
altExonFile = filename[:-4]+'-ANOVA.txt'
eo = export.ExportFile(filename[:-4]+'-ANOVA.txt')
eo.write(original_data['header'])
for key in matrix_pvalues:
eo.write(original_data[key])
last_line = original_data[key]
eo.close()
return altExonFile
if __name__ == '__main__':
dirfile = unique
filename = '/Users/saljh8/Desktop/top_alt_junctions-clust-Grimes_relativePE.txt'
filename = '/Volumes/SEQ-DATA/Jared/AltResults/AlternativeOutput/Hs_RNASeq_top_alt_junctions-PSI-clust.txt'
filename = '/Users/saljh8/Desktop/dataAnalysis/Mm_Simulation_AltAnalyze/AltResults/AlternativeOutput/Mm_RNASeq_top_alt_junctions-PSI-clust.txt'
#filename = '/Volumes/salomonis2/Grimes/tophat SKI KO/bams/AltResults/AlternativeOutput/Mm_RNASeq_top_alt_junctions-PSI-clust.txt'
matrix,compared_groups,original_data = matrixImport(filename)
matrix_pvalues=runANOVA(filename,matrix,compared_groups)
returnANOVAFiltered(filename,original_data,matrix_pvalues); sys.exit()
a = range(3, 18)
k=[]
for i in a:
y = choose(17,i)
k.append(y)
print sum(k)
#print choose(17,12)
#sys.exit()
r=589
n=1019
R=6605
N=10000
z = zscore(r,n,N,R)
print z, p_value(z);sys.exit()
testModeratedStatistics(); sys.exit()
high =[134, 146, 104, 119, 124, 161, 107, 83, 113, 129, 97, 123]
low = [70, 118, 101, 85, 107, 132, 94]
high=[0.71, 0.82, 0.82, 0.76, 0.76, 0.71, 0.71, 0.82]
low=[0.65, 0.53, 0.88, 0.59, 0.76, 0.59, 0.65]
#high = [102, 99, 90, 121, 114]
#low = [107, 125, 111, 117, 122]
#x,y = wt, dgt_ko
x,y = high[:7], low[:6]
x,y = high, low
p = OneWayANOVA([x,y])
print p
t,df,tails = ttest(x,y,2,3)
p = t_probability(t,df)
print p, t, df
sys.exit()
""" behaves well for
1) equal sample number, equal variance
"""
sys.exit()
testMPMath();sys.exit()
#r = pearson([0.0, -0.58999999999999997], [0.0, 0.000000])
#print rdf=7
#a = median([1,2,3.212,4]); print a; kill
a = [[-2.5157100000000003],[-2.3405800000000001, -1.6614700000000004], [-1.5, -1.7, -1.8]]
b = [[1, 2],[3, 4, 5]]
#f=OneWayANOVA(a)
f2=OneWayANOVA(b)
#print f2;sys.exit()
gamma = [[0,1,2,3],[4,5,6,7]]
delta = [[0.2,0.1,0.5,0.2],[1.2,1.4,1.3,1.0],[8,9,10,11],[12,13,14,15]]
f=OneWayANOVA(delta[:2])
#print f
t,df,tails = ttest(delta[0],delta[1],2,2); p1 = t_probability(t,df); print [t,df]
t,df,tails = ttest(delta[0],delta[1],2,3); p2 = t_probability(t,df); print [t,df]
print f, p1, p2
sys.exit()
r=1749
n=2536
R=9858
N=16595
z = zscore(r,n,N,R)
#print z;kill
x = choose(4,4)
#print x;kill
t=2.365
df = 3
t=2.28978775037
df = 12
x = [1,2,3,4,5]
y = [2,3,4,5,6]
b1 = 1.32773390271
e1= 1.05145574703
b2= 0.325021196935
e2= 0.267354914733
score1 = aspire_stringent(b1,e1,b2,e2)
e1= 1.051486632393623; e2= 0.2678618770638278
score2 = aspire_stringent(b1,e1,b2,e2)
print score1, score2
kill
x = [3,1,2,3,4,5]
y = [0.100,0.401,0.204,0.300,0.398,0.502]
x = [5.05, 6.75, 3.21, 2.66]
y = [1.65, 26.5, -5.93, 7.96]
s = LinearRegression(x,y,'no')
print [s]
s2 = simpleLinRegress(x,y)
s = LinearRegression(x,y,'no')
print [s], [s2]
kill
#t = 1.848
#df = 5
#p = t_probability(t,df)
#print p
"""
a = [[0,1,2],[3,4,5]]
a = [[0,1,2,3,4,5,6],[7,8,9,10,11,12,13]]
x = permute_arrays(a)
print len(x)
"""
#x= t_probability(9,9000)
#print x
#beta = permute_arrays(delta)
#print len(beta)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/statistics.py
|
statistics.py
|
#Permission is hereby granted, free of charge, to any person obtaining a copy
#of this software and associated documentation files (the "Software"), to deal
#in the Software without restriction, including without limitation the rights
#to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
#copies of the Software, and to permit persons to whom the Software is furnished
#to do so, subject to the following conditions:
#THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
#INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
#PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
#HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION
#OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE
#SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
import sys,string,os,copy, numpy, math
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
command_args = string.join(sys.argv,' ')
if len(sys.argv[1:])>0 and '--' in command_args: commandLine=True
else: commandLine=False
display_label_names = True
import traceback
from visualization_scripts import clustering
def removeMarkerFinderDoublets(heatmap_file,diff=1):
matrix, column_header, row_header, dataset_name, group_db, priorColumnClusters, priorRowClusters = clustering.remoteImportData(heatmap_file)
priorRowClusters.reverse()
if len(priorColumnClusters)==0:
for c in column_header:
cluster = string.split(c,':')[0]
priorColumnClusters.append(cluster)
for r in row_header:
cluster = string.split(r,':')[0]
priorRowClusters.append(cluster)
import collections
cluster_db = collections.OrderedDict()
i=0
for cluster in priorRowClusters:
try: cluster_db[cluster].append(matrix[i])
except: cluster_db[cluster] = [matrix[i]]
i+=1
transposed_data_matrix=[]
clusters=[]
for cluster in cluster_db:
cluster_cell_means = numpy.mean(cluster_db[cluster],axis=0)
cluster_db[cluster] = cluster_cell_means
transposed_data_matrix.append(cluster_cell_means)
if cluster not in clusters:
clusters.append(cluster)
transposed_data_matrix = zip(*transposed_data_matrix)
i=0
cell_max_scores=[]
cell_max_score_db = collections.OrderedDict()
for cell_scores in transposed_data_matrix:
cluster = priorColumnClusters[i]
cell = column_header[i]
ci = clusters.index(cluster)
#print ci, cell, cluster, cell_scores;sys.exit()
cell_state_score = cell_scores[ci] ### This is the score for that cell for it's assigned MarkerFinder cluster
alternate_state_scores=[]
for score in cell_scores:
if score != cell_state_score:
alternate_state_scores.append(score)
alt_max_score = max(alternate_state_scores)
alt_sum_score = sum(alternate_state_scores)
cell_max_scores.append([cell_state_score,alt_max_score,alt_sum_score]) ### max and secondary max score - max for the cell-state should be greater than secondary max
try: cell_max_score_db[cluster].append(([cell_state_score,alt_max_score,alt_sum_score]))
except: cell_max_score_db[cluster] = [[cell_state_score,alt_max_score,alt_sum_score]]
i+=1
for cluster in cell_max_score_db:
cluster_cell_means = numpy.median(cell_max_score_db[cluster],axis=0)
cell_max_score_db[cluster] = cluster_cell_means ### This is the cell-state mean score for all cells in that cluster and the alternative max mean score (difference gives you the threshold for detecting double)
i=0
#print len(cell_max_scores)
keep=['row_clusters-flat']
keep_alt=['row_clusters-flat']
remove = ['row_clusters-flat']
remove_alt = ['row_clusters-flat']
min_val = 1000
for (cell_score,alt_score,alt_sum) in cell_max_scores:
cluster = priorColumnClusters[i]
cell = column_header[i]
ref_max, ref_alt, ref_sum = cell_max_score_db[cluster]
ci = clusters.index(cluster)
ref_diff= math.pow(2,(ref_max-ref_alt))*diff #1.1
ref_alt = math.pow(2,(ref_alt))
cell_diff = math.pow(2,(cell_score-alt_score))
cell_score = math.pow(2,cell_score)
if cell_diff<min_val: min_val = cell_diff
if cell_diff>ref_diff and cell_diff>diff: #cell_score cutoff removes some, but cell_diff is more crucial
#if alt_sum<cell_score:
assignment=0 #1.2
keep.append(cell)
try: keep_alt.append(string.split(cell,':')[1]) ### if prefix added
except Exception:
keep_alt.append(cell)
else:
remove.append(cell)
try: remove_alt.append(string.split(cell,':')[1])
except Exception: remove_alt.append(cell)
assignment=1
#print assignment
i+=1
#print min_val
print 'Number of cells to keep:',len(keep), 'out of', len(column_header)
from import_scripts import sampleIndexSelection
input_file=heatmap_file
output_file = heatmap_file[:-4]+'-Singlets.txt'
try: sampleIndexSelection.filterFile(input_file,output_file,keep)
except: sampleIndexSelection.filterFile(input_file,output_file,keep_alt)
output_file = heatmap_file[:-4]+'-Multiplets.txt'
try: sampleIndexSelection.filterFile(input_file,output_file,remove)
except: sampleIndexSelection.filterFile(input_file,output_file,remove_alt)
if __name__ == '__main__':
import getopt
threshold=1
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Insufficient options provided";sys.exit()
else:
options, remainder = getopt.getopt(sys.argv[1:],'', ['i=','t=',])
#print sys.argv[1:]
for opt, arg in options:
if opt == '--i': inputFile=arg
elif opt == '--t': threshold=float(arg)
removeMarkerFinderDoublets(inputFile,diff=threshold)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/removeDoublets.py
|
removeDoublets.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import numpy as np
import pylab as pl
import os.path
import scipy
from collections import defaultdict
from sklearn.cluster import KMeans
from import_scripts import sampleIndexSelection
import export
def strip_first_col(fname, delimiter=None):
with open(fname, 'r') as fin:
for line in fin:
try:
yield line.split(delimiter, 1)[1]
except IndexError:
continue
def header_file(fname, delimiter=None):
head=0
header=[]
with open(fname, 'rU') as fin:
for line in fin:
if head==0:
line = line.rstrip(os.linesep)
line=string.split(line,'\t')
for i in line:
if ":" in i:
i=string.split(i,":")
header.append(i[1])
else:
header.append(i)
del header[:1]
head=1
else:break
return header
def DepleteSplicingevents(commonkeys,keylabel,count,InputFile):
eventlist=[]
#exportname=keylabel[:-4]+'correlationSelected_0.3.txt'
#export_res=open(exportname,"w")
exportdep=InputFile[:-4]+'cor_depleted.txt'
export_res1=open(exportdep,"w")
#export_res.write("splicingevent"+"\t"+"key"+"\n")
for event in commonkeys:
if commonkeys[event]==count:
eventlist.append(event)
# export_res.write(event+"\t"+str(1)+"\n")
head=0
for line in open(keylabel,'rU').xreadlines():
if head==0:
export_res1.write(line)
head=1
continue
else:
line1=line.rstrip('\r\n')
q= string.split(line1,'\t')
if q[0] in eventlist:
export_res1.write(line)
return exportdep
def FindCorrelations(filename,PSIfile,name):
X=defaultdict(list)
prev=""
head=0
for line in open(filename,'rU').xreadlines():
if head >0:
val=[]
line=line.rstrip('\r\n')
q= string.split(line,'\t')
for i in range(1,len(q)):
val.append(float(q[i]))
flag=0
for i in range(len(name)):
key1=q[0]+"_vs"
key2="vs_"+q[0]+".txt"
if key1 in name[i] or key2 in name[i]:
flag=1
if flag==1:
if q[0]==prev:
X[prev].append(val)
else:
prev=q[0]
X[prev].append(val)
else:
#print line
head+=1
continue
head=0
matrix=[]
eventnames=[]
Y={}
eventkeys=defaultdict(list)
commonkeys=defaultdict(int)
count=len(X)
for key in X:
#print key
X[key]=np.array(X[key])
#print X[key].shape
mat=[]
mat=zip(*X[key])
mat=np.array(mat)
#print mat.shape
mat=np.mean(mat,axis=1)
Y[key]=np.array(mat)
counter=defaultdict(int)
for line in open(PSIfile,'rU').xreadlines():
if head >0:
for key in Y:
list1=[]
list2=[]
mean=0.0
line=line.rstrip('\r\n')
#print line
q= string.split(line,'\t')
eventnames.append(q[0])
#print len(Y[key])
for i in range(1,len(q)):
try:
list1.append(float(q[i]))
list2.append(float(Y[key][i-1]))
except Exception:
continue
#print len(list1),len(list2)
rho=scipy.stats.pearsonr(list1,list2)
if abs(rho[0])<0.3:
commonkeys[q[0]]+=1
counter[key]+=1
else:
eventkeys[key].append([q[0],rho[0]])
else:
# print line
head+=1
continue
#for key in counter:
#print counter[key]
# export_key=open(PSIfile[:-4]+str(key)+'.txt',"w")
# for i,j in eventkeys[key]:
# export_key.write(i+"\t"+str(j)+"\n")
return commonkeys,count
if __name__ == '__main__':
import getopt
################ Comand-line arguments ################
if len(sys.argv[1:])<=1: ### Indicates that there are insufficient number of command-line arguments
print "Warning! Insufficient command line flags supplied."
sys.exit()
else:
analysisType = []
options, remainder = getopt.getopt(sys.argv[1:],'', ['Guidefile=','PSIfile='])
for opt, arg in options:
if opt == '--Guidefile': Guidefile=arg
elif opt =='--PSIfile':PSIfile=arg
else:
print "Warning! Command-line argument: %s not recognized. Exiting..." % opt; sys.exit()
#filename="/Users/meenakshi/Documents/leucegene/ICGS/Clustering-exp.Hs_RNASeq_top_alt_junctions367-Leucegene-75p_no149-Guide1 TRAK1&ENSG00000182606&I1.1_42075542-E2.1__E-hierarchical_cosine_correlation.txt"
#PSIfile="/Users/meenakshi/Documents/leucegene/ExpressionInput/exp.Hs_RNASeq_top_alt_junctions-PSI_EventAnnotation-367-Leucegene-75p-unique-filtered-filtered.txt"
#keylabel="/Users/meenakshi/Documents/leucegene/ExpressionInput/exp.round2_glmfilteredKmeans_label.txt"
header=header_file(Guidefile)
output_file=PSIfile[:-4]+"-filtered.txt"
sampleIndexSelection.filterFile(PSIfile,output_file,header)
commonkeys,count=FindCorrelations(Guidefile,output_file)
DepleteSplicingevents(commonkeys,output_file,count)
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/Correlationdepletion.py
|
Correlationdepletion.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import scipy, numpy
import statistics
from visualization_scripts import clustering
def evaluateMultiLinRegulatoryStructure(all_genes_TPM,MarkerFinder,SignatureGenes,state=None,query=None):
all_indexes, group_index, expressionData = loopThroughEachState(all_genes_TPM)
if state!=None:
states = [state] ### For example, we only want to look in annotated Multi-Lin's
else:
states = group_index
state_scores=[]
for state in states:
print '\n',state, 'running now.'
score = evaluateStateRegulatoryStructure(expressionData,all_indexes,group_index,MarkerFinder,SignatureGenes,state,query=query)
state_scores.append([score,state])
print state, score
state_scores.sort()
state_scores.reverse()
print state_scores
def loopThroughEachState(all_genes_TPM):
### Import all genes with TPM values for all cells
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(all_genes_TPM)
group_index={}
all_indexes=[]
for sampleName in group_db:
ICGS_state = group_db[sampleName][0]
try: group_index[ICGS_state].append(column_header.index(sampleName))
except Exception: group_index[ICGS_state] = [column_header.index(sampleName)]
all_indexes.append(column_header.index(sampleName))
for ICGS_state in group_index:
group_index[ICGS_state].sort()
all_indexes.sort()
expressionData = matrix, column_header, row_header, dataset_name, group_db
return all_indexes, group_index, expressionData
def evaluateStateRegulatoryStructure(expressionData, all_indexes,group_index,MarkerFinder,SignatureGenes,state,query=None):
"""Predict multi-lineage cells and their associated coincident lineage-defining TFs"""
useProbablityOfExpression=False
ICGS_State_as_Row = False
matrix, column_header, row_header, dataset_name, group_db = expressionData
def importGeneLists(fn):
genes={}
for line in open(fn,'rU').xreadlines():
data = clustering.cleanUpLine(line)
gene,cluster = string.split(data,'\t')[0:2]
genes[gene]=cluster
return genes
def importMarkerFinderHits(fn):
genes={}
genes_to_symbol={}
ICGS_State_ranked={}
skip=True
for line in open(fn,'rU').xreadlines():
data = clustering.cleanUpLine(line)
if skip: skip=False
else:
try:
gene,symbol,rho,ICGS_State = string.split(data,'\t')
except Exception:
gene,symbol,rho,rho_p,ICGS_State = string.split(data,'\t')
genes_to_symbol[gene]=symbol
#if ICGS_State!=state and float(rho)>0.0:
if float(rho)>0.3:
try: ICGS_State_ranked[ICGS_State].append([float(rho),gene,symbol])
except Exception: ICGS_State_ranked[ICGS_State] = [[float(rho),gene,symbol]]
for ICGS_State in ICGS_State_ranked:
ICGS_State_ranked[ICGS_State].sort()
ICGS_State_ranked[ICGS_State].reverse()
#print ICGS_State, ICGS_State_ranked[ICGS_State][:50]
for (rho,gene,symbol) in ICGS_State_ranked[ICGS_State][:50]:
genes[gene]=rho,ICGS_State ### Retain all population specific genes (lax)
genes[symbol]=rho,ICGS_State
return genes, genes_to_symbol
def importQueryDataset(fn):
matrix, column_header, row_header, dataset_name, group_db = clustering.importData(fn)
return matrix, column_header, row_header, dataset_name, group_db
signatureGenes = importGeneLists(SignatureGenes)
markerFinderGenes, genes_to_symbol = importMarkerFinderHits(MarkerFinder)
#print len(signatureGenes),len(markerFinderGenes)
### Determine for each gene, its population frequency per cell state
index=0
expressedGenesPerState={}
stateAssociatedMarkers={}
def freqCutoff(x,cutoff):
if x>cutoff: return 1 ### minimum expression cutoff
else: return 0
for row in matrix:
ICGS_state_gene_frq={}
gene = row_header[index]
for ICGS_state in group_index:
state_values = map(lambda i: row[i],group_index[ICGS_state])
def freqCheck(x):
if x>1: return 1 ### minimum expression cutoff
else: return 0
expStateCells = sum(map(lambda x: freqCheck(x),state_values))
statePercentage = (float(expStateCells)/len(group_index[ICGS_state]))
ICGS_state_gene_frq[ICGS_state] = statePercentage
datasets_values = map(lambda i: row[i],all_indexes)
all_cells_frq = sum(map(lambda x: freqCheck(x),datasets_values))/(len(datasets_values)*1.0)
all_states_frq = map(lambda x: ICGS_state_gene_frq[x],ICGS_state_gene_frq)
all_states_frq.sort() ### frequencies of all non-multilin states
states_expressed = sum(map(lambda x: freqCutoff(x,0.5),all_states_frq))/(len(all_states_frq)*1.0)
for State in ICGS_state_gene_frq:
state_frq = ICGS_state_gene_frq[State]
rank = all_states_frq.index(state_frq)
if state_frq > 0.25 and rank>0: #and states_expressed<0.75 #and all_cells_frq>0.75
if 'Rik' not in gene and 'Gm' not in gene and '-' not in gene:
if gene in markerFinderGenes:# and gene in markerFinderGenes:
if ICGS_State_as_Row:
ICGS_State = signatureGenes[gene]
if gene in markerFinderGenes:
if ICGS_State_as_Row == False:
rho, ICGS_State = markerFinderGenes[gene]
else:
rho, ICGS_Cell_State = markerFinderGenes[gene] #ICGS_Cell_State
#try: gene = genes_to_symbol[gene]
#except: gene = gene
score = int(rho*100*state_frq)*(float(rank)/len(all_states_frq))
try: expressedGenesPerState[ICGS_State].append((score,gene))
except Exception: expressedGenesPerState[ICGS_State]=[(score,gene)] #(rank*multilin_frq)
try: stateAssociatedMarkers[gene,ICGS_State].append(State)
except Exception: stateAssociatedMarkers[gene,ICGS_State] = [State]
index+=1
if query!=None:
matrix, column_header, row_header, dataset_name, group_db = importQueryDataset(query)
markers_to_exclude=[]
expressedGenesPerState2={}
for (gene,ICGS_State) in stateAssociatedMarkers:
if len(stateAssociatedMarkers[(gene,ICGS_State)])<2: # or len(stateAssociatedMarkers[(gene,ICGS_State)])>len(ICGS_state_gene_frq)/2.0:
markers_to_exclude.append(gene)
else:
print ICGS_State, gene, stateAssociatedMarkers[(gene,ICGS_State)]
for ICGS_State in expressedGenesPerState:
for (score,gene) in expressedGenesPerState[ICGS_State]:
if gene not in markers_to_exclude:
try: expressedGenesPerState2[ICGS_State].append((score,gene))
except Exception: expressedGenesPerState2[ICGS_State] = [(score,gene)]
expressedGenesPerState = expressedGenesPerState2
createPseudoCell=True
### The expressedGenesPerState defines genes and modules co-expressed in the multi-Lin
### Next, find the cells that are most frequent in mulitple states
representativeMarkers={}
for ICGS_State in expressedGenesPerState:
expressedGenesPerState[ICGS_State].sort()
expressedGenesPerState[ICGS_State].reverse()
if '1Multi' not in ICGS_State:
markers = expressedGenesPerState[ICGS_State]#[:5]
markers_unique = list(set(map(lambda x: x[1],list(markers))))
print ICGS_State,":",string.join(markers_unique,', ')
if createPseudoCell:
for gene in markers:
def getBinary(x):
if x>1: return 1
else: return 0
if gene[1] in row_header: ### Only for query datasets
row_index = row_header.index(gene[1])
if useProbablityOfExpression:
pvalues = calculateGeneExpressProbilities(matrix[row_index]) ### probability of expression
values = pvalues
else:
binaryValues = map(lambda x: getBinary(x), matrix[row_index])
values = binaryValues
#values = matrix[row_index]
#if gene[1]=='S100a8': print binaryValues;sys.exit()
try: representativeMarkers[ICGS_State].append(values)
except Exception: representativeMarkers[ICGS_State] = [values]
else:
representativeMarkers[ICGS_State]=markers[0][-1]
#int(len(markers)*.25)>5:
#print ICGS_State, markers
#sys.exit()
for ICGS_State in representativeMarkers:
if createPseudoCell:
signature_values = representativeMarkers[ICGS_State]
if useProbablityOfExpression:
signature_values = [numpy.sum(value) for value in zip(*signature_values)]
else:
signature_values = [float(numpy.mean(value)) for value in zip(*signature_values)]
representativeMarkers[ICGS_State] = signature_values
else:
gene = representativeMarkers[ICGS_State]
row_index = row_header.index(gene)
gene_values = matrix[row_index]
representativeMarkers[ICGS_State] = gene_values
### Determine for each gene, its population frequency per cell state
expressedStatesPerCell={}
multilin_probability={}
import export
print 'Writing results matrix to:',MarkerFinder[:-4]+'-cellStateScores.txt'
eo = export.ExportFile(MarkerFinder[:-4]+'-cellStateScores.txt')
eo.write(string.join(['UID']+column_header,'\t')+'\n')
print 'a'
print len(representativeMarkers)
for ICGS_State in representativeMarkers:
gene_values = representativeMarkers[ICGS_State]
index=0
scoreMatrix=[]
HitsCount=0
for cell in column_header:
value = gene_values[index]
"""
expressedLiklihood = '0'
if (value<0.05 and useProbablityOfExpression==True) or (value==1 and useProbablityOfExpression==False):
try: expressedStatesPerCell[cell].append(ICGS_State)
except Exception: expressedStatesPerCell[cell] = [ICGS_State]
expressedLiklihood = '1'
HitsCount+=1
if useProbablityOfExpression:
try: multilin_probability[cell].append(value)
except Exception: multilin_probability[cell] = [value]
"""
index+=1
HitsCount+=1
scoreMatrix.append(str(value))
if HitsCount>1:
#print ICGS_State,HitsCount
eo.write(string.join([ICGS_State]+scoreMatrix,'\t')+'\n')
eo.close()
sys.exit()
def multiply(values):
p = 1
for i in values:
if i>0:
p = p*i
else:
p = p*1.e-16
return p
cell_mutlilin_ranking=[]
for cell in expressedStatesPerCell:
#if 'Multi-Lin:Gmp.R3.10' in cell: sys.exit()
if useProbablityOfExpression:
p = numpy.mean(multilin_probability[cell]) ### mean state probability
lineageCount = expressedStatesPerCell[cell]
if useProbablityOfExpression:
cell_mutlilin_ranking.append((p,len(lineageCount),cell))
else:
cell_mutlilin_ranking.append((len(lineageCount),cell))
cell_mutlilin_ranking.sort()
if useProbablityOfExpression == False:
cell_mutlilin_ranking.reverse()
scores = []
state_scores={}
cellsPerState={} ### Denominator for z-score analysis
for cell in cell_mutlilin_ranking:
score = cell[0]
scores.append(score)
cell_state = string.split(cell[-1],':')[0]
try: cellsPerState[cell_state]+=1
except Exception: cellsPerState[cell_state]=1
try: state_scores[cell_state].append(float(score))
except Exception: state_scores[cell_state] = [float(score)]
scoreMean = numpy.mean(scores)
scoreSD = numpy.std(scores)
oneSD = scoreMean+scoreSD
twoSD = scoreMean+scoreSD+scoreSD
oneStandDeviationAway={}
twoStandDeviationsAway={}
oneStandDeviationAwayTotal=0
twoStandDeviationsAwayTotal=0
print 'Mean:',scoreMean
print 'STDev:',scoreSD
state_scores2=[]
for cell_state in state_scores:
state_scores2.append((numpy.mean(state_scores[cell_state]),cell_state))
i=0
for cell in cell_mutlilin_ranking:
score,cellName = cell
CellState,CellName = string.split(cellName,':')
if score>=oneSD:
try: oneStandDeviationAway[CellState]+=1
except Exception: oneStandDeviationAway[CellState]=1
oneStandDeviationAwayTotal+=1
if score>=twoSD:
try: twoStandDeviationsAway[CellState]+=1
except Exception: twoStandDeviationsAway[CellState]=1
twoStandDeviationsAwayTotal+=1
print cell, string.join(expressedStatesPerCell[cell[-1]],'|')
i+=1
state_scores2
state_scores2.sort()
state_scores2.reverse()
twoStandDeviationsAway = oneStandDeviationAway
twoStandDeviationsAwayTotal = oneStandDeviationAwayTotal
print '\n\n'
import statistics
zscores = []
for CellState in twoStandDeviationsAway:
#print CellState
highMetaScoreCells = twoStandDeviationsAway[CellState]
totalCellsPerState = cellsPerState[CellState]
r = highMetaScoreCells
n = twoStandDeviationsAwayTotal
R = totalCellsPerState
N = len(column_header)
z = statistics.zscore(r,n,N,R)
scores = [z, CellState,statistics.p_value(z)]
zscores.append(scores)
zscores.sort()
zscores.reverse()
for scores in zscores:
scores = string.join(map(str,scores),'\t')
print scores
"""
for i in state_scores2:
print str(i[0])+'\t'+str(i[1])"""
sys.exit()
return numpy.mean(state_scores)
def calculateGeneExpressProbilities(values, useZ=False):
### First calculate z-scores - scipy.stats.mstats.zscore for the entire matrix
avg = numpy.mean(values)
std = numpy.std(values)
if std ==0:
std = 0.1
if useZ:
values = map(lambda x: (x-avg)/std,values)
else:
values = map(lambda x: x*2,values)
p_values = 1 - scipy.special.ndtr(values)
return p_values
if __name__ == '__main__':
#query_dataset = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/exp.GSE81682_HTSeq-cellHarmony-filtered.txt'
all_tpm = '/Users/saljh8/Downloads/test1/exp.cellHarmony.txt'
markerfinder = '/Users/saljh8/Downloads/test1/AllGenes_correlations-ReplicateBased.txt'
signature_genes = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-10x/CITE-Seq_mLSK-60ADT/Merged/ExpressionInput/MF.txt'
state = 'DC'
#all_tpm = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/ExpressionInput/exp.Guide3-cellHarmony-revised.txt'
#markerfinder = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/updated.8.29.17/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
#signature_genes = '/Users/saljh8/Desktop/Old Mac/Desktop/Grimes/KashishNormalization/test/Panorama.txt'
query_dataset = None
query_dataset = '/Users/saljh8/Desktop/dataAnalysis/Collaborative/Grimes/All-Fluidigm/exp.NaturePan-PreGM-CD150-.txt'
query_dataset = None
"""
#all_tpm = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionInput/MultiLin/exp.Gottgens_HarmonizeReference.txt'
all_tpm = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/MultiLin/ExpressionInput/exp.Gottgens_HarmonizeReference.txt'
#signature_genes = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/MultiLin/ExpressionInput/Gottgens_HarmonizeReference.txt'
signature_genes = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/MultiLin/Gottgens_HarmonizeReference.txt'
#markerfinder = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/ExpressionOutput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
markerfinder = '/Users/saljh8/Desktop/Old Mac/Desktop/demo/Mm_Gottgens_3k-scRNASeq/MultiLin/ExpressionInput/MarkerFinder/AllGenes_correlations-ReplicateBased.txt'
state = 'Eryth_Multi-Lin'
"""
state = None
import getopt
options, remainder = getopt.getopt(sys.argv[1:],'', ['q=','expdir=','m=','ICGS=','state='])
#print sys.argv[1:]
for opt, arg in options:
if opt == '--q': query_dataset=arg
elif opt == '--expdir': all_tpm=arg
elif opt == '--m': markerfinder=arg
elif opt == '--ICGS': signature_genes=arg
elif opt == '--state': state=arg
#state = None
#evaluateMultiLinRegulatoryStructure(all_tpm,markerfinder,signature_genes,state);sys.exit()
evaluateMultiLinRegulatoryStructure(all_tpm,markerfinder,signature_genes,state,query = query_dataset);sys.exit()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/multiLineageScore.py
|
multiLineageScore.py
|
import sys,string,os
sys.path.insert(1, os.path.join(sys.path[0], '..')) ### import parent dir dependencies
import math
import copy
try: ### Added for AltAnalyze - Nathan Salomonis, 1-24-2012
from math import *
import cmath as cm
from stats_scripts import mpmath as mpmath
from mpmath import *
from stats_scripts import statistics
except Exception: null=[] #print 'WARNING! The library file "mpmath" is not installed'
# Short routines used in the functional constructs to reduce analysis time
def add(a,b): return a+b
def squared(a): return math.pow(a, 2)
def cubed(a): return math.pow(a, 3)
def quaded(a): return math.pow(a, 4)
def multiply(a,b): return a*b
def obsMinusExp(a,b): return (a-b)**2/b
def diffsquared(a,b): return (a-b)**2
def higher(a,b):
if a>b:
return 1
else:
return 0
def lower(a,b):
if a<b:
return 1
else:
return 0
def shellsort(inlist):
""" Shellsort algorithm. Sorts a 1D-list.
Usage: shellsort(inlist)
Returns: sorted-inlist, sorting-index-vector (for original list)
"""
n = len(inlist)
svec = copy.deepcopy(inlist)
ivec = range(n)
gap = n/2 # integer division needed
while gap >0:
for i in range(gap,n):
for j in range(i-gap,-1,-gap):
while j>=0 and svec[j]>svec[j+gap]:
temp = svec[j]
svec[j] = svec[j+gap]
svec[j+gap] = temp
itemp = ivec[j]
ivec[j] = ivec[j+gap]
ivec[j+gap] = itemp
gap = gap / 2 # integer division needed
# svec is now sorted inlist, and ivec has the order svec[i] = vec[ivec[i]]
return svec, ivec
def rankdata(inlist):
"""
Ranks the data in inlist, dealing with ties appropritely. Assumes
a 1D inlist. Adapted from Gary Perlman's |Stat ranksort.
Usage: rankdata(inlist)
Returns: a list of length equal to inlist, containing rank scores
"""
n = len(inlist)
svec, ivec = shellsort(inlist)
sumranks = 0
dupcount = 0
newlist = [0]*n
for i in range(n):
sumranks = sumranks + i
dupcount = dupcount + 1
if i==n-1 or svec[i] <> svec[i+1]:
averank = sumranks / float(dupcount) + 1
for j in range(i-dupcount+1,i+1):
newlist[ivec[j]] = averank
sumranks = 0
dupcount = 0
return newlist
def tiecorrect(rankvals):
"""
Corrects for ties in Mann Whitney U and Kruskal Wallis H tests. See
Siegel, S. (1956) Nonparametric Statistics for the Behavioral Sciences.
New York: McGraw-Hill. Code adapted from |Stat rankind.c code.
Usage: tiecorrect(rankvals)
Returns: T correction factor for U or H
"""
sorted = copy.copy(rankvals)
sorted.sort()
n = len(sorted)
T = 0.0
i = 0
while (i<n-1):
if sorted[i] == sorted[i+1]:
nties = 1
while (i<n-1) and (sorted[i] == sorted[i+1]):
nties = nties +1
i = i +1
T = T + nties**3 - nties
i = i+1
T = T / float(n**3-n)
return 1.0 - T
def sum (inlist):
"""
Returns the sum of the items in the passed list.
Usage: sum(inlist)
"""
s = 0
for item in inlist:
s = s + item
return s
# this is used by the single factor anova routines (only I think) & the SS
# value may not actually be needed!
def minimaldescriptives(inlist):
"""this function takes a clean list of data and returns the N, sum, mean
and sum of squares. """
N = 0
sum = 0.0
SS = 0.0
for i in range(len(inlist)):
N = N + 1
sum = sum + inlist[i]
SS = SS + (inlist[i] ** 2)
mean = sum / float(N)
return N, sum, mean, SS
###########################
## Probability functions ##
###########################
def chisqprob(chisq,df):
"""
Returns the (1-tailed) probability value associated with the provided
chi-square value and df. Adapted from chisq.c in Gary Perlman's |Stat.
Usage: chisqprob(chisq,df)
"""
BIG = 20.0
def ex(x):
BIG = 20.0
if x < -BIG:
return 0.0
else:
return math.exp(x)
if chisq <=0 or df < 1:
return 1.0
a = 0.5 * chisq
if df%2 == 0:
even = 1
else:
even = 0
if df > 1:
y = ex(-a)
if even:
s = y
else:
s = 2.0 * zprob(-math.sqrt(chisq))
if (df > 2):
chisq = 0.5 * (df - 1.0)
if even:
z = 1.0
else:
z = 0.5
if a > BIG:
if even:
e = 0.0
else:
e = math.log(math.sqrt(math.pi))
c = math.log(a)
while (z <= chisq):
e = math.log(z) + e
s = s + ex(c*z-a-e)
z = z + 1.0
return s
else:
if even:
e = 1.0
else:
e = 1.0 / math.sqrt(math.pi) / math.sqrt(a)
c = 0.0
while (z <= chisq):
e = e * (a/float(z))
c = c + e
z = z + 1.0
return (c*y+s)
else:
return s
def inversechi(prob, df):
"""This function calculates the inverse of the chi square function. Given
a p-value and a df, it should approximate the critical value needed to
achieve these functions. Adapted from Gary Perlmans critchi function in
C. Apologies if this breaks copyright, but no copyright notice was
attached to the relevant file."""
minchisq = 0.0
maxchisq = 99999.0
chi_epsilon = 0.000001
if (prob <= 0.0):
return maxchisq
elif (prob >= 1.0):
return 0.0
chisqval = df / math.sqrt(prob)
while ((maxchisq - minchisq) > chi_epsilon):
if (chisqprob(chisqval, df) < prob):
maxchisq = chisqval
else:
minchisq = chisqval
chisqval = (maxchisq + minchisq) * 0.5
return chisqval
def erfcc(x):
"""
Returns the complementary error function erfc(x) with fractional
error everywhere less than 1.2e-7. Adapted from Numerical Recipies.
Usage: erfcc(x)
"""
z = abs(x)
t = 1.0 / (1.0+0.5*z)
ans = t * math.exp(-z*z-1.26551223 + t*(1.00002368+t*(0.37409196+t* \
(0.09678418+t*(-0.18628806+t* \
(0.27886807+t*(-1.13520398+t* \
(1.48851587+t*(-0.82215223+t* \
0.17087277)))))))))
if x >= 0:
return ans
else:
return 2.0 - ans
def zprob(z):
"""
Returns the area under the normal curve 'to the left of' the given z value.
Thus,
for z<0, zprob(z) = 1-tail probability
for z>0, 1.0-zprob(z) = 1-tail probability
for any z, 2.0*(1.0-zprob(abs(z))) = 2-tail probability
Adapted from z.c in Gary Perlman's |Stat.
Usage: zprob(z)
"""
Z_MAX = 6.0 # maximum meaningful z-value
if z == 0.0:
x = 0.0
else:
y = 0.5 * math.fabs(z)
if y >= (Z_MAX*0.5):
x = 1.0
elif (y < 1.0):
w = y*y
x = ((((((((0.000124818987 * w
-0.001075204047) * w +0.005198775019) * w
-0.019198292004) * w +0.059054035642) * w
-0.151968751364) * w +0.319152932694) * w
-0.531923007300) * w +0.797884560593) * y * 2.0
else:
y = y - 2.0
x = (((((((((((((-0.000045255659 * y
+0.000152529290) * y -0.000019538132) * y
-0.000676904986) * y +0.001390604284) * y
-0.000794620820) * y -0.002034254874) * y
+0.006549791214) * y -0.010557625006) * y
+0.011630447319) * y -0.009279453341) * y
+0.005353579108) * y -0.002141268741) * y
+0.000535310849) * y +0.999936657524
if z > 0.0:
prob = ((x+1.0)*0.5)
else:
prob = ((1.0-x)*0.5)
return prob
def ksprob(alam):
"""
Computes a Kolmolgorov-Smirnov t-test significance level. Adapted from
Numerical Recipies.
Usage: ksprob(alam)
"""
fac = 2.0
sum = 0.0
termbf = 0.0
a2 = -2.0*alam*alam
for j in range(1,201):
term = fac*math.exp(a2*j*j)
sum = sum + term
if math.fabs(term)<=(0.001*termbf) or math.fabs(term)<(1.0e-8*sum):
return sum
fac = -fac
termbf = math.fabs(term)
return 1.0 # Get here only if fails to converge; was 0.0!!
def fprob (dfnum, dfden, F):
"""
Returns the (1-tailed) significance level (p-value) of an F
statistic given the degrees of freedom for the numerator (dfR-dfF) and
the degrees of freedom for the denominator (dfF).
Usage: fprob(dfnum, dfden, F) where usually dfnum=dfbn, dfden=dfwn
"""
p = betai(0.5*dfden, 0.5*dfnum, dfden/float(dfden+dfnum*F))
return p
def tprob(df, t):
return betai(0.5*df,0.5,float(df)/(df+t*t))
def inversef(prob, df1, df2):
"""This function returns the f value for a given probability and 2 given
degrees of freedom. It is an approximation using the fprob function.
Adapted from Gary Perlmans critf function - apologies if copyright is
broken, but no copyright notice was attached """
f_epsilon = 0.000001
maxf = 9999.0
minf = 0.0
if (prob <= 0.0) or (prob >= 1.0):
return 0.0
fval = 1.0 / prob
while (abs(maxf - minf) > f_epsilon):
if fprob(fval, df1, df2) < prob:
maxf = fval
else:
minf = fval
fval = (maxf + minf) * 0.5
return fval
def betacf(a,b,x):
"""
This function evaluates the continued fraction form of the incomplete
Beta function, betai. (Adapted from: Numerical Recipies in C.)
Usage: betacf(a,b,x)
"""
ITMAX = 200
EPS = 3.0e-7
bm = az = am = 1.0
qab = a+b
qap = a+1.0
qam = a-1.0
bz = 1.0-qab*x/qap
for i in range(ITMAX+1):
em = float(i+1)
tem = em + em
d = em*(b-em)*x/((qam+tem)*(a+tem))
ap = az + d*am
bp = bz+d*bm
d = -(a+em)*(qab+em)*x/((qap+tem)*(a+tem))
app = ap+d*az
bpp = bp+d*bz
aold = az
am = ap/bpp
bm = bp/bpp
az = app/bpp
bz = 1.0
if (abs(az-aold)<(EPS*abs(az))):
return az
#print 'a or b too big, or ITMAX too small in Betacf.'
def gammln(xx):
"""
Returns the gamma function of xx.
Gamma(z) = Integral(0,infinity) of t^(z-1)exp(-t) dt.
(Adapted from: Numerical Recipies in C.)
Usage: gammln(xx)
"""
coeff = [76.18009173, -86.50532033, 24.01409822, -1.231739516,
0.120858003e-2, -0.536382e-5]
x = xx - 1.0
tmp = x + 5.5
tmp = tmp - (x+0.5)*math.log(tmp)
ser = 1.0
for j in range(len(coeff)):
x = x + 1
ser = ser + coeff[j]/x
return -tmp + math.log(2.50662827465*ser)
def betai(a,b,x):
"""
Returns the incomplete beta function:
I-sub-x(a,b) = 1/B(a,b)*(Integral(0,x) of t^(a-1)(1-t)^(b-1) dt)
where a,b>0 and B(a,b) = G(a)*G(b)/(G(a+b)) where G(a) is the gamma
function of a. The continued fraction formulation is implemented here,
using the betacf function. (Adapted from: Numerical Recipies in C.)
Usage: betai(a,b,x)
"""
if (x<0.0 or x>1.0):
raise ValueError, 'Bad x in lbetai'
if (x==0.0 or x==1.0):
bt = 0.0
else:
bt = math.exp(gammln(a+b)-gammln(a)-gammln(b)+a*math.log(x)+b*
math.log(1.0-x))
if (x<(a+1.0)/(a+b+2.0)):
return bt*betacf(a,b,x)/float(a)
else:
return 1.0-bt*betacf(b,a,1.0-x)/float(b)
class Probabilities:
def __init__(self):
pass
# is this necessary?
def betai(self, a, b, x):
"""
returns the incomplete beta function
"""
if (x<0.0 or x>1.0):
raise ValueError, 'Bad x in lbetai'
if (x==0.0 or x==1.0):
bt = 0.0
else:
bt = math.exp(gammln(a+b)-gammln(a)-gammln(b)+a*math.log(x)+b*
math.log(1.0-x))
if (x<(a+1.0)/(a+b+2.0)):
self.prob = bt*betacf(a,b,x)/float(a)
else:
self.prob = 1.0-bt*betacf(b,a,1.0-x)/float(b)
def gammln(self, xx):
"""
returns the gamma function of xx.
"""
coeff = [76.18009173, -86.50532033, 24.01409822, -1.231739516,
0.120858003e-2, -0.536382e-5]
x = xx - 1.0
tmp = x + 5.5
tmp = tmp - (x+0.5)*math.log(tmp)
ser = 1.0
for j in range(len(coeff)):
x = x + 1
ser = ser + coeff[j]/x
return -tmp + math.log(2.50662827465*ser)
def betacf(self, a, b, x):
ITMAX = 200
EPS = 3.0e-7
bm = az = am = 1.0
qab = a+b
qap = a+1.0
qam = a-1.0
bz = 1.0-qab*x/qap
for i in range(ITMAX+1):
em = float(i+1)
tem = em + em
d = em*(b-em)*x/((qam+tem)*(a+tem))
ap = az + d*am
bp = bz+d*bm
d = -(a+em)*(qab+em)*x/((qap+tem)*(a+tem))
app = ap+d*az
bpp = bp+d*bz
aold = az
am = ap/bpp
bm = bp/bpp
az = app/bpp
bz = 1.0
if (abs(az-aold)<(EPS*abs(az))):
return az
###########################
## Test Classes ##
###########################
class FullDescriptives:
"""
class for producing a series of continuous descriptive statistics. The
variable "inlist" must have been cleaned of missing data. Statistics
available by method are: N, sum, mean, sample variance (samplevar),
variance, standard deviation (stddev), standard error (stderr), sum of
squares (sumsquares), sum of squared deviations (ssdevs), coefficient of
variation (coeffvar), skewness, kurtosis, median, mode, median absolute
deviation (mad), number of unique values (numberuniques).
"""
def __init__(self, inlist, name = '', missing = 0):
self.Name = name
if len(inlist) == 0:
self.N = 0
self.sum = self.mean = self.sumsquares = self.minimum = self.maximum = self.median = 0
self.mad = self.numberuniques = self.harmmean = self.ssdevs = self.samplevar = 0
self.geomean = self.variance = self.coeffvar = self.skewness = self.kurtosis = self.mode = 0
elif len(inlist) == 1:
self.N = self.numberuniques = 1
self.sum = self.mean = self.minimum = self.maximum = self.median = inlist[0]
self.mad = self.harmmean = self.geomean = inlist[0]
self.samplevar = self.variance = self.coeffvar = self.skewness = self.mode = 0
self.kurtosis = self.sumsquares = ssdevs = 0
elif len(inlist) > 1:
self.missing = missing
self.N = len(inlist)
self.sum = reduce(add, inlist)
try:
self.mean = self.sum / float(self.N)
except ZeroDivisionError:
self.mean = 0.0
self.sumsquares = reduce(add, map(squared, inlist))
difflist = []
self.sortlist = copy.copy(inlist)
self.sortlist.sort()
self.minimum = self.sortlist[0]
self.maximum = self.sortlist[len(self.sortlist)-1]
self.range = self.maximum - self.minimum
self.harmmean=0.0
medianindex = self.N / 2
if (self.N % 2):
self.median = self.sortlist[medianindex]
self.firstquartile = self.sortlist[((self.N + 1) / 4) - 1]
#print 'md ' + str(inlist[:medianindex])
else:
self.median = (self.sortlist[medianindex] + self.sortlist[medianindex-1]) / 2.0
self.firstquartile = self.sortlist[(self.N / 4) - 1]
#print 'md ' + str(self.firstquartile)
# median of ranks - useful in comparisons for KW & Friedmans
ranklist = rankdata(self.sortlist)
if (self.N % 2):
self.medianranks = ranklist[(self.N + 1) / 2]
else:
self.medianranks = ranklist[self.N / 2]
self.mad = 0.0
self.numberuniques = 0
for i in range(self.N):
difflist.append(inlist[i] - self.mean)
self.mad = self.mad + (inlist[i] - self.median)
uniques = 1
for j in range(self.N):
if (i != j):
if (inlist[i] == inlist[j]):
uniques = 0
if uniques:
self.numberuniques = self.numberuniques + 1
if (inlist[i] != 0.0):
self.harmmean = self.harmmean + (1.0/inlist[i])
if (self.harmmean != 0.0):
self.harmmean = self.N / self.harmmean
self.ssdevs = reduce(add, map(squared, difflist))
self.geomean = reduce(multiply, difflist)
try:
self.samplevar = self.ssdevs / float(self.N - 1)
except ZeroDivisionError:
self.samplevar = 0.0
try:
moment2 = self.ssdevs / float(self.N)
moment3 = reduce(add, map(cubed, difflist)) / float(self.N)
moment4 = reduce(add, map(quaded, difflist)) / float(self.N)
self.variance = self.ssdevs / float(self.N)
self.stddev = math.sqrt(self.samplevar)
self.coeffvar = self.stddev / self.mean
self.skewness = moment3 / (moment2 * math.sqrt(moment2))
self.kurtosis = (moment4 / math.pow(moment2, 2)) - 3.0
except ZeroDivisionError:
moment2 = 0.0
moment3 = 0.0
moment4 = 0.0
self.variance = 0.0
self.stderr = 0.0
self.coeffvar = 0.0
self.skewness = 0.0
self.kurtosis = 0.0
self.stderr = self.stddev / math.sqrt(self.N)
h = {}
for n in inlist:
try: h[n] = h[n]+1
except KeyError: h[n] = 1
a = map(lambda x: (x[1], x[0]), h.items())
self.mode = max(a)[1]
class OneSampleTests:
"""
This class produces single factor statistical tests.
"""
def __init__(self, data1, name = '', missing = 0):
"""
Pass the data to the init function.
"""
self.d1 = FullDescriptives(data1, name, missing)
def OneSampleTTest(self, usermean):
"""
This performs a single factor t test for a set of data and a user
hypothesised mean value.
Usage: OneSampleTTest(self, usermean)
Returns: t, df (degrees of freedom), prob (probability)
"""
if self.d1.N < 2:
self.t = 1.0
self.prob = -1.0
else:
self.df = self.d1.N - 1
svar = (self.df * self.d1.samplevar) / float(self.df)
self.t = (self.d1.mean - usermean) / math.sqrt(svar*(1.0/self.d1.N))
self.prob = betai(0.5*self.df,0.5,float(self.df)/(self.df+ \
self.t*self.t))
def OneSampleSignTest(self, data1, usermean):
"""
This method performs a single factor sign test. The data must be
supplied to this method along with a user hypothesised mean value.
Usage: OneSampleSignTest(self, data1, usermean)
Returns: nplus, nminus, z, prob.
"""
self.nplus=0
self.nminus=0
for i in range(len(data1)):
if (data1[i] < usermean):
self.nplus=self.nplus+1
if (data1[i] > usermean):
self.nminus=self.nminus+1
self.ntotal = add(self.nplus, self.nminus)
try:
self.z=(self.nplus-(self.ntotal/2)/math.sqrt(self.ntotal/2))
except ZeroDivisionError:
self.z=0
self.prob=-1.0
else:
self.prob=erfcc(abs(self.z) / 1.4142136)
def ChiSquareVariance(self, usermean):
"""
This method performs a Chi Square test for the variance ratio.
Usage: ChiSquareVariance(self, usermean)
Returns: df, chisquare, prob
"""
self.df = self.d1.N - 1
try:
self.chisquare = (self.d1.stderr / usermean) * self.df
except ZeroDivisionError:
self.chisquare = 0.0
self.prob = chisqprob(self.chisquare, self.df)
# class for two sample tests - instantiates descriptives class for both
# data sets, then has each test as a method
class TwoSampleTests:
"""This class performs a series of 2 sample statistical tests upon two
sets of data.
"""
# Note: some of the below has been slightly modified from the original code (AltAnalyze developers) to simply
# allow for more straight forward variable returns and eliminate variable passing redundancy
def __init__(self, data1, data2, name1 = '', name2 = '', \
missing1=0,missing2=0):
"""
The __init__ method retrieves a full set of descriptive statistics
for the two supplied data vectors.
"""
self.d1 = FullDescriptives(data1, name1, missing1)
self.d2 = FullDescriptives(data2, name2, missing2)
self.data1 = data1; self.data2 = data2
def TTestUnpaired(self):
"""
This performs an unpaired t-test.
Usage: TTestUnpaired()
Returns: t, df, prob
"""
self.df = (self.d1.N + self.d2.N) - 2
#self.d1.samplevar is stdev squared and N is the length of the list
svar = ((self.d1.N-1)*self.d1.samplevar+(self.d2.N-1)* \
self.d2.samplevar)/float(self.df) ### Equivalent to sg2
self.t = (self.d1.mean-self.d2.mean)/math.sqrt(svar* \
(1.0/self.d1.N + 1.0/self.d2.N))
self.prob = betai(0.5*self.df,0.5,float(self.df)/(self.df+self.t* \
self.t))
return self.prob
### Added for AltAnalyze - Nathan Salomonis, 1-24-2012
def getModeratedStandardDeviation(self,comparison_db):
""" Added for AltAnalyze - Nathan Salomonis, 1-24-2012
Adapted from TTestUnpaired and "Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments" by Gordon K. Smyth 2004
Analyzes all pairwise comparise comparisons for two groups to return a moderated sample variance calculated from two hyperparameters, d0 and s0
Returns: d0 and the square of s0
"""
variance_ls=[]; e_sum=0; d0_2nd_moment_gene_sum = []; median_var=[]
"""
for uid in comparison_db:
gs = comparison_db[uid] ### Object containing summary statistics needed for each uid (aka feature)
sg_squared = gs.FeatureVariance()
median_var.append(sg_squared)
m = statistics.median(median_var)
#print 'median_var',m
"""
for uid in comparison_db:
gs = comparison_db[uid]
try: df = (gs.N1() + gs.N2()) - 2
except Exception,e: print e, gs, [gs.N1(), gs.N2()];kill
sg_squared = gs.FeatureVariance()
#if sg_squared==0: sg_squared = 1e-8 #* m
if sg_squared > 1e-11: ### remove extreemly low variance genes from the analysis (log of sg_squared must be non-zero)
zg = math.log(sg_squared)
eg = float(zg - psi(0,df/2) + math.log(df/2))
variance_ls.append(eg)
#n = len(comparison_db) ### number of uids analyzed
n = len(variance_ls) ### number of uids analyzed
### Get the mean eg for all IDs
#e_avg = statistics.avg(variance_ls)
eg_sum=0
for eg in variance_ls: eg_sum += eg
e_avg = float(eg_sum/n) ### this is close but not exactly like limma - like do to the squared log error above
#print eg_sum; sys.exit()
### Calculate the d0 2nd derivitive that will later need to be solved for d0
for eg in variance_ls:
#d0_2nd_moment_gene_sum += (math.pow(eg-e_avg,2)*n)/((n-1) - psi(1,df/2))
d0_2nd_moment_gene_sum.append(math.pow(eg-e_avg,2))
"""
d0_sum = []
for y in d0_2nd_moment_gene_sum:
self.x = y
d0 = self.NewtonInteration(); d0_sum.append(d0)
d0 = statistics.avg(d0_sum)
"""
#print sum(d0_2nd_moment_gene_sum)
#print psi(1,df/2)
d0_2nd_moment_solve = (sum(d0_2nd_moment_gene_sum)/(n-1)) - psi(1,df/2)
self.x = float(d0_2nd_moment_solve)
#print [d0_2nd_moment_solve]
d0 = self.NewtonInteration() #"""
#print [d0]
d0 = float(d0)
#d0 = 2.054191
#s0_squared = 0.01090202
#s0_squared = 0.010905
e = cm.e
s0_squared = math.pow(e,e_avg+psi(0,d0/2) - math.log(d0/2))
return d0, s0_squared
def testNewtonInteration(self,x):
self.x = x
y = self.NewtonInteration()
print y, x, psi(1,y) ### x should equal psi(1,y)
def NewtonInteration(self):
""" Method used to emperically identify the best estimate when you can't solve for the variable of interest (in this case, d0 aka y).
This function was validated using limma"""
y = 0.5 + (1/self.x)
proceed = 1
while proceed == 1:
if self.x>1e7: y = 1/math.sqrt(x); proceed = 0
elif self.x<1e-6: y = 1/self.x; proceed = 0
else:
d = (psi(1,y)*(1-(psi(1,y)/self.x)))/psi(2,y) ### check 1- syntax with Kirsten
#print y, self.x, d
y = y + d
try:
if (-d/y)< 1e-8:
proceed = 0
break
except Exception: null=[]
#y = 1.0270955
return y*2
def ModeratedTTestUnpaired(self,gs,d0,s0_squared):
df = (gs.N1() + gs.N2()) - 2
sg_squared = gs.FeatureVariance()
### use the s0_squared for the pairwise comparison calculated in the getModeratedStandardDeviation
svar = (d0*s0_squared+df*sg_squared)/(d0+df) ### square root
#svar = sg ### Use this to test and see if this gives the same result as a non-moderated t-test
if svar != 0:
df = d0+df
self.t = (gs.Avg1()-gs.Avg2())/math.sqrt(svar*(1.0/gs.N1() + 1.0/gs.N2()))
prob = betai(0.5*df,0.5,float(df)/(df+self.t*self.t))
else: prob = 1
gs.SetAdjP(prob)
def TTestPaired(self):
"""
This method performs a paired t-test on two data sets. Both sets (as
vectors) need to be supplied.
Usage: TTestPaired(data1, data2)
Returns: t, df, prob
"""
if (self.d1.N != self.d2.N):
self.prob = -1.0
self.df = 0
self.t = 0.0
else:
cov = 0.0
self.df = self.d1.N - 1
for i in range(self.d1.N):
cov = cov + ((self.data1[i] - self.d1.mean) * (self.data2[i] - \
self.d2.mean))
cov = cov / float(self.df)
sd = math.sqrt((self.d1.samplevar + self.d2.samplevar - 2.0 * \
cov) / float(self.d1.N))
try:
self.t = (self.d1.mean - self.d2.mean) / sd
self.prob = betai(0.5*self.df,0.5,float(self.df)/(self.df+ \
self.t*self.t))
except ZeroDivisionError:
self.t = -1.0
self.prob = 0.0
return self.prob
def PearsonsCorrelation(self):
"""
This method performs a Pearsons correlation upon two sets of
data which are passed as vectors.
Usage: PearsonsCorrelation(data1, data2)
Returns: r, t, df, prob
"""
TINY = 1.0e-60
if (self.d1.N != self.d2.N):
self.prob = -1.0
else:
summult = reduce(add, map(multiply, self.data1, self.data2))
r_num = self.d1.N * summult - self.d1.sum * self.d2.sum
r_left = self.d1.N*self.d1.sumsquares-(self.d1.sum**2)
r_right= self.d2.N*self.d2.sumsquares-(self.d2.sum**2)
r_den = math.sqrt(r_left*r_right)
self.r = r_num / r_den
self.df = self.d1.N - 2
self.t = self.r*math.sqrt(self.df/((1.0-self.r+TINY)* \
(1.0+self.r+TINY)))
self.prob = betai(0.5*self.df,0.5,self.df/float \
(self.df+self.t*self.t))
return self.prob, self.r
def FTest(self, uservar):
"""
This method performs a F test for variance ratio and needs a user
hypothesised variance to be supplied.
Usage: FTest(uservar)
Returns: f, df1, df2, prob
"""
try:
self.f = (self.d1.samplevar / self.d2.samplevar) / uservar
except ZeroDivisionError:
self.f = 1.0
self.df1 = self.d1.N - 1
self.df2 = self.d2.N - 1
self.prob=fprob(self.df1, self.df2, self.f)
return self.prob
def TwoSampleSignTest(self):
"""
This method performs a 2 sample sign test for matched samples on 2
supplied data vectors.
Usage: TwoSampleSignTest(data1, data2)
Returns: nplus, nminus, ntotal, z, prob
"""
if (self.d1.N != self.d2.N):
self.prob=-1.0
else:
nplus=map(higher,self.data1,self.data2).count(1)
nminus=map(lower,self.data1,self.data2).count(1)
self.ntotal=nplus-nminus
mean=self.d1.N / 2
sd = math.sqrt(mean)
self.z = (nplus-mean)/sd
self.prob = erfcc(abs(self.z)/1.4142136)
return self.prob
def KendallsTau(self):
"""
This method performs a Kendalls tau correlation upon 2 data vectors.
Usage: KendallsTau(data1, data2)
Returns: tau, z, prob
"""
n1 = 0
n2 = 0
iss = 0
for j in range(self.d1.N-1):
for k in range(j,self.d2.N):
a1 = self.data1[j] - self.data1[k]
a2 = self.data2[j] - self.data2[k]
aa = a1 * a2
if (aa): # neither list has a tie
n1 = n1 + 1
n2 = n2 + 1
if aa > 0:
iss = iss + 1
else:
iss = iss -1
else:
if (a1):
n1 = n1 + 1
else:
n2 = n2 + 1
self.tau = iss / math.sqrt(n1*n2)
svar = (4.0*self.d1.N+10.0) / (9.0*self.d1.N*(self.d1.N-1))
self.z = self.tau / math.sqrt(svar)
self.prob = erfcc(abs(self.z)/1.4142136)
return self.prob
def KolmogorovSmirnov(self):
"""
This method performs a Kolmogorov-Smirnov test for unmatched samples
upon 2 data vectors.
Usage: KolmogorovSmirnov()
Returns: d, prob
"""
j1 = 0
j2 = 0
fn1 = 0.0
fn2 = 0.0
self.d = 0.0
data3 = self.d1.sortlist
data4 = self.d2.sortlist
while j1 < self.d1.N and j2 < self.d2.N:
d1=data3[j1]
d2=data4[j2]
if d1 <= d2:
fn1 = (j1)/float(self.d1.N)
j1 = j1 + 1
if d2 <= d1:
fn2 = (j2)/float(self.d2.N)
j2 = j2 + 1
dt = (fn2-fn1)
if math.fabs(dt) > math.fabs(self.d):
self.d = dt
try:
en = math.sqrt(self.d1.N*self.d2.N/float(self.d1.N+self.d2.N))
self.prob = ksprob((en+0.12+0.11/en)*abs(self.d))
except:
self.prob = 1.0
return self.prob
def SpearmansCorrelation(self):
"""
This method performs a Spearmans correlation upon 2 data sets as
vectors.
Usage: SpearmansCorrelation(data1, data2)
Returns: t, df, prob
"""
TINY = 1e-30
if self.d1.N <> self.d2.N:
self.prob= -1.0
else:
rankx = rankdata(self.data1)
ranky = rankdata(self.data2)
dsq = reduce(add, map(diffsquared, rankx, ranky))
self.rho = 1 - 6*dsq / float(self.d1.N*(self.d1.N**2-1))
self.t = self.rho * math.sqrt((self.d1.N-2) / \
((self.rho+1.0+TINY)*(1.0-self.rho+TINY)))
self.df = self.d1.N-2
self.prob = betai(0.5*self.df,0.5,self.df/(self.df+self.t*self.t))
return self.prob,self.rho
def RankSums(self):
"""
This method performs a Wilcoxon rank sums test for unpaired designs
upon 2 data vectors.
Usage: RankSums(data1, data2)
Returns: z, prob
"""
x = copy.copy(self.data1)
y = copy.copy(self.data2)
alldata = x + y
ranked = rankdata(alldata)
x = ranked[:self.d1.N]
y = ranked[self.d1.N:]
s = reduce(add, x)
expected = self.d1.N*(self.d1.N+self.d2.N+1) / 2.0
self.z = (s - expected) / math.sqrt(self.d1.N*self.d2.N* \
(self.d2.N+self.d2.N+1)/12.0)
self.prob = 2*(1.0 -zprob(abs(self.z)))
return self.prob
def SignedRanks(self):
"""
This method performs a Wilcoxon Signed Ranks test for matched samples
upon 2 data vectors.
Usage: SignedRanks(data1, data2)
Returns: wt, z, prob
"""
if self.d1.N <> self.d2.N:
self.prob = -1.0
else:
d=[]
for i in range(self.d1.N):
diff = self.data1[i] - self.data2[i]
if diff <> 0:
d.append(diff)
count = len(d)
absd = map(abs,d)
absranked = rankdata(absd)
r_plus = 0.0
r_minus = 0.0
for i in range(len(absd)):
if d[i] < 0:
r_minus = r_minus + absranked[i]
else:
r_plus = r_plus + absranked[i]
self.wt = min(r_plus, r_minus)
mn = count * (count+1) * 0.25
se = math.sqrt(count*(count+1)*(2.0*count+1.0)/24.0)
self.z = math.fabs(self.wt-mn) / se
self.prob = 2*(1.0 -zprob(abs(self.z)))
return self.prob
def MannWhitneyU(self):
"""
This method performs a Mann Whitney U test for unmatched samples on
2 data vectors.
Usage: MannWhitneyU(data1, data2)
Returns: bigu, smallu, z, prob
"""
ranked = rankdata(self.data1+self.data2)
rankx = ranked[0:self.d1.N]
u1 = self.d1.N*self.d2.N+(self.d1.N*(self.d1.N+1))/2.0-reduce\
(add, rankx)
u2 = self.d1.N*self.d2.N - u1
self.bigu = max(u1,u2)
self.smallu = min(u1,u2)
T = math.sqrt(tiecorrect(ranked))
if T == 0:
self.prob = -.10
self.z = -1.0
else:
sd = math.sqrt(T*self.d1.N*self.d2.N*(self.d1.N+self.d2.N+1)/12.0)
self.z = abs((self.bigu-self.d1.N*self.d2.N/2.0) / sd)
self.prob = 1.0-zprob(self.z)
return self.prob
def LinearRegression(self, x, y):
"""
This method performs a linear regression upon 2 data vectors.
Usage(LinearRegression(x,y)
Returns: r, df, t, prob, slope, intercept, sterrest
"""
TINY = 1.0e-20
if (self.d1.N != self.d2.N):
self.prob = -1.0
else:
summult = reduce(add, map(multiply, x, y))
r_num = float(self.d1.N*summult - self.d1.sum*self.d2.sum)
r_den = math.sqrt((self.d1.N*self.d1.sumsquares - \
(self.d1.sum**2))*(self.d2.N* \
self.d2.sumsquares - (self.d2.sum**2)))
try:
self.r = r_num / r_den
except ZeroDivisionError:
self.r = 0.0
#[] warning - z not used - is there a line missing here?
z = 0.5*math.log((1.0+self.r+TINY)/(1.0-self.r+TINY))
self.df = self.d1.N - 2
self.t = self.r*math.sqrt(self.df/((1.0-self.r+TINY)*(1.0+ \
self.r+TINY)))
self.prob = betai(0.5*self.df,0.5,self.df/(self.df+self.t*self.t))
self.slope = r_num / float(self.d1.N*self.d1.sumsquares - \
(self.d1.sum**2))
self.intercept = self.d2.mean - self.slope*self.d1.mean
self.sterrest = math.sqrt(1-self.r*self.r)*math.sqrt \
(self.d2.variance)
return self.prob
def PairedPermutation(self, x, y):
"""
This method performs a permutation test for matched samples upon 2
data vectors. This code was modified from Segal.
Usage: PairedPermutation(x,y)
Returns: utail, nperm, crit, prob
"""
self.utail = 0
self.nperm = 0
self.crit = 0.0
d = []
d.append(copy(x))
d.append(copy(x))
d.append(copy(y))
index = [1]*self.d1.N
for i in range(self.d1.N):
d[1][i] = x[i]-y[i]
d[2][i] = y[i]-x[i]
self.crit = self.crit + d[1][i]
#for j in range((self.d1.N-1), 0, -1):
while 1:
sum = 0
for i in range(self.d1.N):
sum = sum + d[index[i]][i]
self.nperm = self.nperm + 1
if (sum >= self.crit):
self.utail = self.utail + 1
for i in range((self.d1.N-1), 0, -1):
if (index[i] == 1):
index[i] = 2
continue
index[i] = 1
break
self.prob = float(self.utail / self.nperm)
return self.prob
def PointBiserialr(self, x, y):
TINY = 1e-30
if len(x) <> len(y):
return -1.0, -1.0
data = pstat.abut(x,y) # [] pstat module not available!
categories = pstat.unique(x)
if len(categories) <> 2:
return -1.0, -2.0
else: # [] there are 2 categories, continue
codemap = pstat.abut(categories,range(2))
recoded = pstat.recode(data,codemap,0) # [] can prob delete this line
x = pstat.linexand(data,0,categories[0])
y = pstat.linexand(data,0,categories[1])
xmean = mean(pstat.colex(x,1)) # [] use descriptives!
ymean = mean(pstat.colex(y,1)) # [] use descriptives!
n = len(data)
adjust = math.sqrt((len(x)/float(n))*(len(y)/float(n)))
rpb = (ymean - xmean)/samplestdev(pstat.colex(data,1))*adjust
df = n-2
t = rpb*math.sqrt(df/((1.0-rpb+TINY)*(1.0+rpb+TINY)))
prob = betai(0.5*df,0.5,df/(df+t*t)) # t already a float
return rpb, prob
def ChiSquare(self, x, y):
"""
This method performs a chi square on 2 data vectors.
Usage: ChiSquare(x,y)
Returns: chisq, df, prob
"""
self.df = len(x) - 1
if (self.df+1) != len(y):
self.chi = 0.0
self.prob = -1.0
else:
self.chisq = 0.0
for i in range(self.df+1):
self.chisq = self.chisq+((x[i]-y[i])**2)/float(y[i])
self.prob = chisqprob(self.chisq, self.df)
return self.prob
class ThreeSampleTests:
"""
This instantiates a class for three or more (actually 2 or more) sample
tests such as anova, Kruskal Wallis and Friedmans.
"""
def __init__(self):
self.prob = -1.0
def anovaWithin(self, inlist, ns, sums, means):
"""
This method is specialised for SalStat, and is best left alone.
For the brave:
Usage: anovaWithin(inlist, ns, sums, means). ns is a list of the N's,
sums is a list of the sums of each condition, and the same for means
being a list of means
Returns: SSint, SSres, SSbet, SStot, dfbet, dfwit, dfres, dftot, MSbet,
MSwit, MSres, F, prob.
"""
GN = 0
GS = 0.0
GM = 0.0
k = len(inlist)
meanlist = []
Nlist = []
for i in range(k):
GN = GN + ns[i]
GS = GS + sums[i]
Nlist.append(ns[i])
meanlist.append(means[i])
GM = GS / float(GN)
self.SSwit = 0.0
self.SSbet = 0.0
self.SStot = 0.0
for i in range(k):
for j in range(Nlist[i]):
diff = inlist[i][j] - meanlist[i]
self.SSwit = self.SSwit + (diff ** 2)
diff = inlist[i][j] - GM
self.SStot = self.SStot + (diff ** 2)
diff = meanlist[i] - GM
self.SSbet = self.SSbet + (diff ** 2)
self.SSbet = self.SSbet * float(GN / k)
self.SSint = 0.0
for j in range(ns[0]):
rowlist = []
for i in range(k):
rowlist.append(inlist[i][j])
n, sum, mean, SS = minimaldescriptives(rowlist)
self.SSint = self.SSint + ((mean - GM) ** 2)
self.SSint = self.SSint * k
self.SSres = self.SSwit - self.SSint
self.dfbet = k - 1
self.dfwit = GN - k
self.dfres = (ns[0] - 1) * (k - 1)
self.dftot = self.dfbet + self.dfwit + self.dfres
self.MSbet = self.SSbet / float(self.dfbet)
self.MSwit = self.SSwit / float(self.dfwit)
self.MSres = self.SSres / float(self.dfres)
self.F = self.MSbet / self.MSres
self.prob = fprob(self.dfbet, self.dfres, self.F)
def anovaBetween(self, descs):
"""
This method performs a univariate single factor between-subjects
analysis of variance on a list of lists (or a Numeric matrix). It is
specialised for SalStat and best left alone.
Usage: anovaBetween(descs). descs are a list of descriptives for each
condition.
Returns: SSbet, SSwit, SStot, dfbet, dferr, dftot, MSbet, MSerr, F, prob.
"""
GN = 0
GM = 0.0
self.SSwit = 0.0
self.SSbet = 0.0
self.SStot = 0.0
k = len(descs)
for i in range(k):
self.SSwit = self.SSwit + descs[i].ssdevs
GN = GN + descs[i].N
GM = GM + descs[i].mean
GM = GM / k
for i in range(k):
self.SSbet = self.SSbet + ((descs[i].mean - GM) ** 2)
self.SSbet = self.SSbet * descs[0].N
self.SStot = self.SSwit + self.SSbet
self.dfbet = k - 1
self.dferr = GN - k
self.dftot = self.dfbet + self.dferr
self.MSbet = self.SSbet / float(self.dfbet)
self.MSerr = self.SSwit / float(self.dferr)
try:
self.F = self.MSbet / self.MSerr
except ZeroDivisionError:
self.F = 1.0
self.prob = fprob(self.dfbet, self.dferr, self.F)
def KruskalWallisH(self, args):
"""
This method performs a Kruskal Wallis test (like a nonparametric
between subjects anova) on a list of lists.
Usage: KruskalWallisH(args).
Returns: h, prob.
"""
args = list(args)
n = [0]*len(args)
all = []
n = map(len,args)
for i in range(len(args)):
all = all + args[i]
ranked = rankdata(all)
T = tiecorrect(ranked)
for i in range(len(args)):
args[i] = ranked[0:n[i]]
del ranked[0:n[i]]
rsums = []
for i in range(len(args)):
rsums.append(sum(args[i])**2)
rsums[i] = rsums[i] / float(n[i])
ssbn = sum(rsums)
totaln = sum(n)
self.h = 12.0 / (totaln*(totaln+1)) * ssbn - 3*(totaln+1)
self.df = len(args) - 1
if T == 0:
self.h = 0.0
self.prob = 1.0
else:
self.h = self.h / float(T)
self.prob = chisqprob(self.h,self.df)
def FriedmanChiSquare(self, args):
"""
This method performs a Friedman chi square (like a nonparametric
within subjects anova) on a list of lists.
Usage: FriedmanChiSqure(args).
Returns: sumranks, chisq, df, prob.
"""
k = len(args)
n = len(args[0])
data=[]
for j in range(len(args[0])):
line=[]
for i in range(len(args)):
line.append(args[i][j])
data.append(line)
for i in range(len(data)):
data[i] = rankdata(data[i])
data2 = []
for j in range(len(data[0])):
line = []
for i in range(len(data)):
line.append(data[i][j])
data2.append(line)
self.sumranks = []
for i in range(k):
x = FullDescriptives(data2[i])
self.sumranks.append(x.sum)
ssbn = 0
sums = []
for i in range(k):
tmp = sum(data2[i])
ssbn = ssbn + (tmp ** 2)
sums.append(tmp/len(data2[i]))
self.chisq = (12.0 / (k*n*(k+1))) * ssbn - 3*n*(k+1)
self.df = k-1
self.prob = chisqprob(self.chisq,self.df)
def CochranesQ(self, inlist):
"""
This method performs a Cochrances Q test upon a list of lists.
Usage: CochranesQ(inlist)
Returns: q, df, prob
"""
k = len(inlist)
n = len(inlist[0])
self.df = k - 1
gtot = 0
for i in range(k):
g = 0
for j in range(n):
g = g + inlist[i][j]
gtot = gtot + (g ** 2)
l = lsq = 0
for i in range(n):
rowsum = 0
for j in range(k):
rowsum = rowsum + inlist[j][i]
l = l + rowsum
lsq = lsq + (rowsum ** 2)
self.q = ((k-1)*((k*gtot)-(l**2)))/((k*l)-lsq)
self.prob = chisqprob(self.q, self.df)
class FriedmanComp:
"""This class performs multiple comparisons on a Freidmans
test. Passed values are the medians, k (# conditions), n
(# samples), and the alpha value. Currently, all comparisons
are performed regardless. Assumes a balanced design.
ALSO: does not work yet!
"""
def __init__(self, medians, k, n, p):
crit = inversechi(p, k-1)
value = crit * math.sqrt((k * (k + 1)) / (6 * n * k))
self.outstr = '<p>Multiple Comparisons for Friedmans test:</p>'
self.outstr=self.outstr+'<br>Critical Value (>= for sig) = '+str(crit)
for i in range(len(medians)):
for j in range(i+1, len(medians)):
if (i != j):
self.outstr = self.outstr+'<br>'+str(i+1)+' against '+str(j+1)
diff = abs(medians[i] - medians[j])
self.outstr = self.outstr+' = '+str(diff)
class KWComp:
"""This class performs multiple comparisons on a Kruskal Wallis
test. Passed values are the medians, k (# conditions), n
(# samples), and the alpha value. Currently, all comparisons
are performed regardless. Assumes a balanced design.
Further note - not completed by any means! DO NO USE THIS YET!"""
def __init__(self, medians, k, n, p):
crit = inversechi(p, k-1)
value = crit * math.sqrt((k * (k + 1)) / (6 * n * k))
self.outstr = '<p>Multiple Comparisons for Friedmans test:</p>'
self.outstr=self.outstr+'<br>Critical Value (>= for sig) = '+str(crit)
for i in range(len(medians)):
for j in range(i+1, len(medians)):
if (i != j):
self.outstr = self.outstr+'<br>'+str(i+1)+' against '+str(j+1)
diff = abs(medians[i] - medians[j])
self.outstr = self.outstr+' = '+str(diff)
if __name__ == '__main__':
import sys
df = 4
eg = 0 - psi(0,df/2) + math.log(df/2)
#print eg; sys.exit()
#Below is modified from the original code for testing
#x = [1,2,3,4,5,6,7,8,9,10,11,12]
#y = [1,8,3,4,5,6,7,7,9,10,11,12]
x = [8.75204607,8.597121429,8.851280558,8.969386521]
y = [9.990671284,9.47208155,9.636987061,9.716990894,9.314016704,9.535469797,9.557272223,9.537994878,9.156588753,9.445428759,9.456354415,9.707186551,9.404502961]
x = [2.46282,2.58916,2.04632]
y = [3.64911,3.30122,3.52227]
x = [12.6898888,12.27990072]
y = [11.24108938,11.76942464]
x = [12.68979456,12.27987933]
y = [11.24024485,11.76516488]
x = [6606.06836,4971.92627]
y = [2418.0835,3479.70752]
x = [1.023376519,1.234599261]
y = [0.766677318,0.387252366]
from stats_scripts import statistics
k = statistics.stdev(x); l = statistics.stdev(y)
#print l*l
a = TwoSampleTests(x,y)
#print a.testNewtonInteration(1.5820968679895);sys.exit()
print a.TTestUnpaired(),'a.TTestUnpaired()'
print a.TTestPaired(),'a.TTestPaired()'
#print a.KendallsTau(),'a.KendallsTau()'
print a.KolmogorovSmirnov(),'a.KolmogorovSmirnov()'
print a.MannWhitneyU(),'a.MannWhitneyU()'
#print a.LinearRegression(),'a.LinearRegression()'
print a.SignedRanks(),'a.SignedRanks()'
#print a.PearsonsCorrelation(),'a.PearsonsCorrelation()'
print a.RankSums(),'a.RankSums()'
#print a.SpearmansCorrelation(),'a.SpearmansCorrelation()'
print a.TwoSampleSignTest(),'a.TwoSampleSignTest()'
#print a.PointBiserialr(),'a.PointBiserialr()'
#print a.ChiSquare(),'a.ChiSquare()'
#""" Testing code - ignore
# use - TTestUnpaired,TTestPaired,KolmogorovSmirnov,MannWhitneyU,RankSums
#"""
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/salstat_stats.py
|
salstat_stats.py
|
def monitor(f, input='print', output='print'):
"""
Returns a wrapped copy of *f* that monitors evaluation by calling
*input* with every input (*args*, *kwargs*) passed to *f* and
*output* with every value returned from *f*. The default action
(specify using the special string value ``'print'``) is to print
inputs and outputs to stdout, along with the total evaluation
count::
>>> from mpmath import *
>>> mp.dps = 5; mp.pretty = False
>>> diff(monitor(exp), 1) # diff will eval f(x-h) and f(x+h)
in 0 (mpf('0.99999999906867742538452148'),) {}
out 0 mpf('2.7182818259274480055282064')
in 1 (mpf('1.0000000009313225746154785'),) {}
out 1 mpf('2.7182818309906424675501024')
mpf('2.7182808')
To disable either the input or the output handler, you may
pass *None* as argument.
Custom input and output handlers may be used e.g. to store
results for later analysis::
>>> mp.dps = 15
>>> input = []
>>> output = []
>>> findroot(monitor(sin, input.append, output.append), 3.0)
mpf('3.1415926535897932')
>>> len(input) # Count number of evaluations
9
>>> print(input[3]); print(output[3])
((mpf('3.1415076583334066'),), {})
8.49952562843408e-5
>>> print(input[4]); print(output[4])
((mpf('3.1415928201669122'),), {})
-1.66577118985331e-7
"""
if not input:
input = lambda v: None
elif input == 'print':
incount = [0]
def input(value):
args, kwargs = value
print("in %s %r %r" % (incount[0], args, kwargs))
incount[0] += 1
if not output:
output = lambda v: None
elif output == 'print':
outcount = [0]
def output(value):
print("out %s %r" % (outcount[0], value))
outcount[0] += 1
def f_monitored(*args, **kwargs):
input((args, kwargs))
v = f(*args, **kwargs)
output(v)
return v
return f_monitored
def timing(f, *args, **kwargs):
"""
Returns time elapsed for evaluating ``f()``. Optionally arguments
may be passed to time the execution of ``f(*args, **kwargs)``.
If the first call is very quick, ``f`` is called
repeatedly and the best time is returned.
"""
once = kwargs.get('once')
if 'once' in kwargs:
del kwargs['once']
if args or kwargs:
if len(args) == 1 and not kwargs:
arg = args[0]
g = lambda: f(arg)
else:
g = lambda: f(*args, **kwargs)
else:
g = f
from timeit import default_timer as clock
t1=clock(); v=g(); t2=clock(); t=t2-t1
if t > 0.05 or once:
return t
for i in range(3):
t1=clock();
# Evaluate multiple times because the timer function
# has a significant overhead
g();g();g();g();g();g();g();g();g();g()
t2=clock()
t=min(t,(t2-t1)/10)
return t
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/usertools.py
|
usertools.py
|
from .libmp.backend import basestring, exec_
from .libmp import (MPZ, MPZ_ZERO, MPZ_ONE, int_types, repr_dps,
round_floor, round_ceiling, dps_to_prec, round_nearest, prec_to_dps,
ComplexResult, to_pickable, from_pickable, normalize,
from_int, from_float, from_str, to_int, to_float, to_str,
from_rational, from_man_exp,
fone, fzero, finf, fninf, fnan,
mpf_abs, mpf_pos, mpf_neg, mpf_add, mpf_sub, mpf_mul, mpf_mul_int,
mpf_div, mpf_rdiv_int, mpf_pow_int, mpf_mod,
mpf_eq, mpf_cmp, mpf_lt, mpf_gt, mpf_le, mpf_ge,
mpf_hash, mpf_rand,
mpf_sum,
bitcount, to_fixed,
mpc_to_str,
mpc_to_complex, mpc_hash, mpc_pos, mpc_is_nonzero, mpc_neg, mpc_conjugate,
mpc_abs, mpc_add, mpc_add_mpf, mpc_sub, mpc_sub_mpf, mpc_mul, mpc_mul_mpf,
mpc_mul_int, mpc_div, mpc_div_mpf, mpc_pow, mpc_pow_mpf, mpc_pow_int,
mpc_mpf_div,
mpf_pow,
mpf_pi, mpf_degree, mpf_e, mpf_phi, mpf_ln2, mpf_ln10,
mpf_euler, mpf_catalan, mpf_apery, mpf_khinchin,
mpf_glaisher, mpf_twinprime, mpf_mertens,
int_types)
import rational
import function_docs
new = object.__new__
class mpnumeric(object):
"""Base class for mpf and mpc."""
__slots__ = []
def __new__(cls, val):
raise NotImplementedError
class _mpf(mpnumeric):
"""
An mpf instance holds a real-valued floating-point number. mpf:s
work analogously to Python floats, but support arbitrary-precision
arithmetic.
"""
__slots__ = ['_mpf_']
def __new__(cls, val=fzero, **kwargs):
"""A new mpf can be created from a Python float, an int, a
or a decimal string representing a number in floating-point
format."""
prec, rounding = cls.context._prec_rounding
if kwargs:
prec = kwargs.get('prec', prec)
if 'dps' in kwargs:
prec = dps_to_prec(kwargs['dps'])
rounding = kwargs.get('rounding', rounding)
if type(val) is cls:
sign, man, exp, bc = val._mpf_
if (not man) and exp:
return val
v = new(cls)
v._mpf_ = normalize(sign, man, exp, bc, prec, rounding)
return v
elif type(val) is tuple:
if len(val) == 2:
v = new(cls)
v._mpf_ = from_man_exp(val[0], val[1], prec, rounding)
return v
if len(val) == 4:
sign, man, exp, bc = val
v = new(cls)
v._mpf_ = normalize(sign, MPZ(man), exp, bc, prec, rounding)
return v
raise ValueError
else:
v = new(cls)
v._mpf_ = mpf_pos(cls.mpf_convert_arg(val, prec, rounding), prec, rounding)
return v
@classmethod
def mpf_convert_arg(cls, x, prec, rounding):
if isinstance(x, int_types): return from_int(x)
if isinstance(x, float): return from_float(x)
if isinstance(x, basestring): return from_str(x, prec, rounding)
if isinstance(x, cls.context.constant): return x.func(prec, rounding)
if hasattr(x, '_mpf_'): return x._mpf_
if hasattr(x, '_mpmath_'):
t = cls.context.convert(x._mpmath_(prec, rounding))
if hasattr(t, '_mpf_'):
return t._mpf_
if hasattr(x, '_mpi_'):
a, b = x._mpi_
if a == b:
return a
raise ValueError("can only create mpf from zero-width interval")
raise TypeError("cannot create mpf from " + repr(x))
@classmethod
def mpf_convert_rhs(cls, x):
if isinstance(x, int_types): return from_int(x)
if isinstance(x, float): return from_float(x)
if isinstance(x, complex_types): return cls.context.mpc(x)
if isinstance(x, rational.mpq):
p, q = x._mpq_
return from_rational(p, q, cls.context.prec)
if hasattr(x, '_mpf_'): return x._mpf_
if hasattr(x, '_mpmath_'):
t = cls.context.convert(x._mpmath_(*cls.context._prec_rounding))
if hasattr(t, '_mpf_'):
return t._mpf_
return t
return NotImplemented
@classmethod
def mpf_convert_lhs(cls, x):
x = cls.mpf_convert_rhs(x)
if type(x) is tuple:
return cls.context.make_mpf(x)
return x
man_exp = property(lambda self: self._mpf_[1:3])
man = property(lambda self: self._mpf_[1])
exp = property(lambda self: self._mpf_[2])
bc = property(lambda self: self._mpf_[3])
real = property(lambda self: self)
imag = property(lambda self: self.context.zero)
conjugate = lambda self: self
def __getstate__(self): return to_pickable(self._mpf_)
def __setstate__(self, val): self._mpf_ = from_pickable(val)
def __repr__(s):
if s.context.pretty:
return str(s)
return "mpf('%s')" % to_str(s._mpf_, s.context._repr_digits)
def __str__(s): return to_str(s._mpf_, s.context._str_digits)
def __hash__(s): return mpf_hash(s._mpf_)
def __int__(s): return int(to_int(s._mpf_))
def __long__(s): return long(to_int(s._mpf_))
def __float__(s): return to_float(s._mpf_)
def __complex__(s): return complex(float(s))
def __nonzero__(s): return s._mpf_ != fzero
__bool__ = __nonzero__
def __abs__(s):
cls, new, (prec, rounding) = s._ctxdata
v = new(cls)
v._mpf_ = mpf_abs(s._mpf_, prec, rounding)
return v
def __pos__(s):
cls, new, (prec, rounding) = s._ctxdata
v = new(cls)
v._mpf_ = mpf_pos(s._mpf_, prec, rounding)
return v
def __neg__(s):
cls, new, (prec, rounding) = s._ctxdata
v = new(cls)
v._mpf_ = mpf_neg(s._mpf_, prec, rounding)
return v
def _cmp(s, t, func):
if hasattr(t, '_mpf_'):
t = t._mpf_
else:
t = s.mpf_convert_rhs(t)
if t is NotImplemented:
return t
return func(s._mpf_, t)
def __cmp__(s, t): return s._cmp(t, mpf_cmp)
def __lt__(s, t): return s._cmp(t, mpf_lt)
def __gt__(s, t): return s._cmp(t, mpf_gt)
def __le__(s, t): return s._cmp(t, mpf_le)
def __ge__(s, t): return s._cmp(t, mpf_ge)
def __ne__(s, t):
v = s.__eq__(t)
if v is NotImplemented:
return v
return not v
def __rsub__(s, t):
cls, new, (prec, rounding) = s._ctxdata
if type(t) in int_types:
v = new(cls)
v._mpf_ = mpf_sub(from_int(t), s._mpf_, prec, rounding)
return v
t = s.mpf_convert_lhs(t)
if t is NotImplemented:
return t
return t - s
def __rdiv__(s, t):
cls, new, (prec, rounding) = s._ctxdata
if isinstance(t, int_types):
v = new(cls)
v._mpf_ = mpf_rdiv_int(t, s._mpf_, prec, rounding)
return v
t = s.mpf_convert_lhs(t)
if t is NotImplemented:
return t
return t / s
def __rpow__(s, t):
t = s.mpf_convert_lhs(t)
if t is NotImplemented:
return t
return t ** s
def __rmod__(s, t):
t = s.mpf_convert_lhs(t)
if t is NotImplemented:
return t
return t % s
def sqrt(s):
return s.context.sqrt(s)
def ae(s, t, rel_eps=None, abs_eps=None):
return s.context.almosteq(s, t, rel_eps, abs_eps)
def to_fixed(self, prec):
return to_fixed(self._mpf_, prec)
def __round__(self, *args):
return round(float(self), *args)
mpf_binary_op = """
def %NAME%(self, other):
mpf, new, (prec, rounding) = self._ctxdata
sval = self._mpf_
if hasattr(other, '_mpf_'):
tval = other._mpf_
%WITH_MPF%
ttype = type(other)
if ttype in int_types:
%WITH_INT%
elif ttype is float:
tval = from_float(other)
%WITH_MPF%
elif hasattr(other, '_mpc_'):
tval = other._mpc_
mpc = type(other)
%WITH_MPC%
elif ttype is complex:
tval = from_float(other.real), from_float(other.imag)
mpc = self.context.mpc
%WITH_MPC%
if isinstance(other, mpnumeric):
return NotImplemented
try:
other = mpf.context.convert(other, strings=False)
except TypeError:
return NotImplemented
return self.%NAME%(other)
"""
return_mpf = "; obj = new(mpf); obj._mpf_ = val; return obj"
return_mpc = "; obj = new(mpc); obj._mpc_ = val; return obj"
mpf_pow_same = """
try:
val = mpf_pow(sval, tval, prec, rounding) %s
except ComplexResult:
if mpf.context.trap_complex:
raise
mpc = mpf.context.mpc
val = mpc_pow((sval, fzero), (tval, fzero), prec, rounding) %s
""" % (return_mpf, return_mpc)
def binary_op(name, with_mpf='', with_int='', with_mpc=''):
code = mpf_binary_op
code = code.replace("%WITH_INT%", with_int)
code = code.replace("%WITH_MPC%", with_mpc)
code = code.replace("%WITH_MPF%", with_mpf)
code = code.replace("%NAME%", name)
np = {}
exec_(code, globals(), np)
return np[name]
_mpf.__eq__ = binary_op('__eq__',
'return mpf_eq(sval, tval)',
'return mpf_eq(sval, from_int(other))',
'return (tval[1] == fzero) and mpf_eq(tval[0], sval)')
_mpf.__add__ = binary_op('__add__',
'val = mpf_add(sval, tval, prec, rounding)' + return_mpf,
'val = mpf_add(sval, from_int(other), prec, rounding)' + return_mpf,
'val = mpc_add_mpf(tval, sval, prec, rounding)' + return_mpc)
_mpf.__sub__ = binary_op('__sub__',
'val = mpf_sub(sval, tval, prec, rounding)' + return_mpf,
'val = mpf_sub(sval, from_int(other), prec, rounding)' + return_mpf,
'val = mpc_sub((sval, fzero), tval, prec, rounding)' + return_mpc)
_mpf.__mul__ = binary_op('__mul__',
'val = mpf_mul(sval, tval, prec, rounding)' + return_mpf,
'val = mpf_mul_int(sval, other, prec, rounding)' + return_mpf,
'val = mpc_mul_mpf(tval, sval, prec, rounding)' + return_mpc)
_mpf.__div__ = binary_op('__div__',
'val = mpf_div(sval, tval, prec, rounding)' + return_mpf,
'val = mpf_div(sval, from_int(other), prec, rounding)' + return_mpf,
'val = mpc_mpf_div(sval, tval, prec, rounding)' + return_mpc)
_mpf.__mod__ = binary_op('__mod__',
'val = mpf_mod(sval, tval, prec, rounding)' + return_mpf,
'val = mpf_mod(sval, from_int(other), prec, rounding)' + return_mpf,
'raise NotImplementedError("complex modulo")')
_mpf.__pow__ = binary_op('__pow__',
mpf_pow_same,
'val = mpf_pow_int(sval, other, prec, rounding)' + return_mpf,
'val = mpc_pow((sval, fzero), tval, prec, rounding)' + return_mpc)
_mpf.__radd__ = _mpf.__add__
_mpf.__rmul__ = _mpf.__mul__
_mpf.__truediv__ = _mpf.__div__
_mpf.__rtruediv__ = _mpf.__rdiv__
class _constant(_mpf):
"""Represents a mathematical constant with dynamic precision.
When printed or used in an arithmetic operation, a constant
is converted to a regular mpf at the working precision. A
regular mpf can also be obtained using the operation +x."""
def __new__(cls, func, name, docname=''):
a = object.__new__(cls)
a.name = name
a.func = func
a.__doc__ = getattr(function_docs, docname, '')
return a
def __call__(self, prec=None, dps=None, rounding=None):
prec2, rounding2 = self.context._prec_rounding
if not prec: prec = prec2
if not rounding: rounding = rounding2
if dps: prec = dps_to_prec(dps)
return self.context.make_mpf(self.func(prec, rounding))
@property
def _mpf_(self):
prec, rounding = self.context._prec_rounding
return self.func(prec, rounding)
def __repr__(self):
return "<%s: %s~>" % (self.name, self.context.nstr(self))
class _mpc(mpnumeric):
"""
An mpc represents a complex number using a pair of mpf:s (one
for the real part and another for the imaginary part.) The mpc
class behaves fairly similarly to Python's complex type.
"""
__slots__ = ['_mpc_']
def __new__(cls, real=0, imag=0):
s = object.__new__(cls)
if isinstance(real, complex_types):
real, imag = real.real, real.imag
elif hasattr(real, '_mpc_'):
s._mpc_ = real._mpc_
return s
real = cls.context.mpf(real)
imag = cls.context.mpf(imag)
s._mpc_ = (real._mpf_, imag._mpf_)
return s
real = property(lambda self: self.context.make_mpf(self._mpc_[0]))
imag = property(lambda self: self.context.make_mpf(self._mpc_[1]))
def __getstate__(self):
return to_pickable(self._mpc_[0]), to_pickable(self._mpc_[1])
def __setstate__(self, val):
self._mpc_ = from_pickable(val[0]), from_pickable(val[1])
def __repr__(s):
if s.context.pretty:
return str(s)
r = repr(s.real)[4:-1]
i = repr(s.imag)[4:-1]
return "%s(real=%s, imag=%s)" % (type(s).__name__, r, i)
def __str__(s):
return "(%s)" % mpc_to_str(s._mpc_, s.context._str_digits)
def __complex__(s):
return mpc_to_complex(s._mpc_)
def __pos__(s):
cls, new, (prec, rounding) = s._ctxdata
v = new(cls)
v._mpc_ = mpc_pos(s._mpc_, prec, rounding)
return v
def __abs__(s):
prec, rounding = s.context._prec_rounding
v = new(s.context.mpf)
v._mpf_ = mpc_abs(s._mpc_, prec, rounding)
return v
def __neg__(s):
cls, new, (prec, rounding) = s._ctxdata
v = new(cls)
v._mpc_ = mpc_neg(s._mpc_, prec, rounding)
return v
def conjugate(s):
cls, new, (prec, rounding) = s._ctxdata
v = new(cls)
v._mpc_ = mpc_conjugate(s._mpc_, prec, rounding)
return v
def __nonzero__(s):
return mpc_is_nonzero(s._mpc_)
__bool__ = __nonzero__
def __hash__(s):
return mpc_hash(s._mpc_)
@classmethod
def mpc_convert_lhs(cls, x):
try:
y = cls.context.convert(x)
return y
except TypeError:
return NotImplemented
def __eq__(s, t):
if not hasattr(t, '_mpc_'):
if isinstance(t, str):
return False
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
return s.real == t.real and s.imag == t.imag
def __ne__(s, t):
b = s.__eq__(t)
if b is NotImplemented:
return b
return not b
def _compare(*args):
raise TypeError("no ordering relation is defined for complex numbers")
__gt__ = _compare
__le__ = _compare
__gt__ = _compare
__ge__ = _compare
def __add__(s, t):
cls, new, (prec, rounding) = s._ctxdata
if not hasattr(t, '_mpc_'):
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
if hasattr(t, '_mpf_'):
v = new(cls)
v._mpc_ = mpc_add_mpf(s._mpc_, t._mpf_, prec, rounding)
return v
v = new(cls)
v._mpc_ = mpc_add(s._mpc_, t._mpc_, prec, rounding)
return v
def __sub__(s, t):
cls, new, (prec, rounding) = s._ctxdata
if not hasattr(t, '_mpc_'):
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
if hasattr(t, '_mpf_'):
v = new(cls)
v._mpc_ = mpc_sub_mpf(s._mpc_, t._mpf_, prec, rounding)
return v
v = new(cls)
v._mpc_ = mpc_sub(s._mpc_, t._mpc_, prec, rounding)
return v
def __mul__(s, t):
cls, new, (prec, rounding) = s._ctxdata
if not hasattr(t, '_mpc_'):
if isinstance(t, int_types):
v = new(cls)
v._mpc_ = mpc_mul_int(s._mpc_, t, prec, rounding)
return v
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
if hasattr(t, '_mpf_'):
v = new(cls)
v._mpc_ = mpc_mul_mpf(s._mpc_, t._mpf_, prec, rounding)
return v
t = s.mpc_convert_lhs(t)
v = new(cls)
v._mpc_ = mpc_mul(s._mpc_, t._mpc_, prec, rounding)
return v
def __div__(s, t):
cls, new, (prec, rounding) = s._ctxdata
if not hasattr(t, '_mpc_'):
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
if hasattr(t, '_mpf_'):
v = new(cls)
v._mpc_ = mpc_div_mpf(s._mpc_, t._mpf_, prec, rounding)
return v
v = new(cls)
v._mpc_ = mpc_div(s._mpc_, t._mpc_, prec, rounding)
return v
def __pow__(s, t):
cls, new, (prec, rounding) = s._ctxdata
if isinstance(t, int_types):
v = new(cls)
v._mpc_ = mpc_pow_int(s._mpc_, t, prec, rounding)
return v
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
v = new(cls)
if hasattr(t, '_mpf_'):
v._mpc_ = mpc_pow_mpf(s._mpc_, t._mpf_, prec, rounding)
else:
v._mpc_ = mpc_pow(s._mpc_, t._mpc_, prec, rounding)
return v
__radd__ = __add__
def __rsub__(s, t):
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
return t - s
def __rmul__(s, t):
cls, new, (prec, rounding) = s._ctxdata
if isinstance(t, int_types):
v = new(cls)
v._mpc_ = mpc_mul_int(s._mpc_, t, prec, rounding)
return v
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
return t * s
def __rdiv__(s, t):
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
return t / s
def __rpow__(s, t):
t = s.mpc_convert_lhs(t)
if t is NotImplemented:
return t
return t ** s
__truediv__ = __div__
__rtruediv__ = __rdiv__
def ae(s, t, rel_eps=None, abs_eps=None):
return s.context.almosteq(s, t, rel_eps, abs_eps)
complex_types = (complex, _mpc)
class PythonMPContext:
def __init__(ctx):
ctx._prec_rounding = [53, round_nearest]
ctx.mpf = type('mpf', (_mpf,), {})
ctx.mpc = type('mpc', (_mpc,), {})
ctx.mpf._ctxdata = [ctx.mpf, new, ctx._prec_rounding]
ctx.mpc._ctxdata = [ctx.mpc, new, ctx._prec_rounding]
ctx.mpf.context = ctx
ctx.mpc.context = ctx
ctx.constant = type('constant', (_constant,), {})
ctx.constant._ctxdata = [ctx.mpf, new, ctx._prec_rounding]
ctx.constant.context = ctx
def make_mpf(ctx, v):
a = new(ctx.mpf)
a._mpf_ = v
return a
def make_mpc(ctx, v):
a = new(ctx.mpc)
a._mpc_ = v
return a
def default(ctx):
ctx._prec = ctx._prec_rounding[0] = 53
ctx._dps = 15
ctx.trap_complex = False
def _set_prec(ctx, n):
ctx._prec = ctx._prec_rounding[0] = max(1, int(n))
ctx._dps = prec_to_dps(n)
def _set_dps(ctx, n):
ctx._prec = ctx._prec_rounding[0] = dps_to_prec(n)
ctx._dps = max(1, int(n))
prec = property(lambda ctx: ctx._prec, _set_prec)
dps = property(lambda ctx: ctx._dps, _set_dps)
def convert(ctx, x, strings=True):
"""
Converts *x* to an ``mpf`` or ``mpc``. If *x* is of type ``mpf``,
``mpc``, ``int``, ``float``, ``complex``, the conversion
will be performed losslessly.
If *x* is a string, the result will be rounded to the present
working precision. Strings representing fractions or complex
numbers are permitted.
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> mpmathify(3.5)
mpf('3.5')
>>> mpmathify('2.1')
mpf('2.1000000000000001')
>>> mpmathify('3/4')
mpf('0.75')
>>> mpmathify('2+3j')
mpc(real='2.0', imag='3.0')
"""
if type(x) in ctx.types: return x
if isinstance(x, int_types): return ctx.make_mpf(from_int(x))
if isinstance(x, float): return ctx.make_mpf(from_float(x))
if isinstance(x, complex):
return ctx.make_mpc((from_float(x.real), from_float(x.imag)))
prec, rounding = ctx._prec_rounding
if isinstance(x, rational.mpq):
p, q = x._mpq_
return ctx.make_mpf(from_rational(p, q, prec))
if strings and isinstance(x, basestring):
try:
_mpf_ = from_str(x, prec, rounding)
return ctx.make_mpf(_mpf_)
except ValueError:
pass
if hasattr(x, '_mpf_'): return ctx.make_mpf(x._mpf_)
if hasattr(x, '_mpc_'): return ctx.make_mpc(x._mpc_)
if hasattr(x, '_mpmath_'):
return ctx.convert(x._mpmath_(prec, rounding))
return ctx._convert_fallback(x, strings)
def isnan(ctx, x):
"""
Return *True* if *x* is a NaN (not-a-number), or for a complex
number, whether either the real or complex part is NaN;
otherwise return *False*::
>>> from mpmath import *
>>> isnan(3.14)
False
>>> isnan(nan)
True
>>> isnan(mpc(3.14,2.72))
False
>>> isnan(mpc(3.14,nan))
True
"""
if hasattr(x, "_mpf_"):
return x._mpf_ == fnan
if hasattr(x, "_mpc_"):
return fnan in x._mpc_
if isinstance(x, int_types) or isinstance(x, rational.mpq):
return False
x = ctx.convert(x)
if hasattr(x, '_mpf_') or hasattr(x, '_mpc_'):
return ctx.isnan(x)
raise TypeError("isnan() needs a number as input")
def isinf(ctx, x):
"""
Return *True* if the absolute value of *x* is infinite;
otherwise return *False*::
>>> from mpmath import *
>>> isinf(inf)
True
>>> isinf(-inf)
True
>>> isinf(3)
False
>>> isinf(3+4j)
False
>>> isinf(mpc(3,inf))
True
>>> isinf(mpc(inf,3))
True
"""
if hasattr(x, "_mpf_"):
return x._mpf_ in (finf, fninf)
if hasattr(x, "_mpc_"):
re, im = x._mpc_
return re in (finf, fninf) or im in (finf, fninf)
if isinstance(x, int_types) or isinstance(x, rational.mpq):
return False
x = ctx.convert(x)
if hasattr(x, '_mpf_') or hasattr(x, '_mpc_'):
return ctx.isinf(x)
raise TypeError("isinf() needs a number as input")
def isnormal(ctx, x):
"""
Determine whether *x* is "normal" in the sense of floating-point
representation; that is, return *False* if *x* is zero, an
infinity or NaN; otherwise return *True*. By extension, a
complex number *x* is considered "normal" if its magnitude is
normal::
>>> from mpmath import *
>>> isnormal(3)
True
>>> isnormal(0)
False
>>> isnormal(inf); isnormal(-inf); isnormal(nan)
False
False
False
>>> isnormal(0+0j)
False
>>> isnormal(0+3j)
True
>>> isnormal(mpc(2,nan))
False
"""
if hasattr(x, "_mpf_"):
return bool(x._mpf_[1])
if hasattr(x, "_mpc_"):
re, im = x._mpc_
re_normal = bool(re[1])
im_normal = bool(im[1])
if re == fzero: return im_normal
if im == fzero: return re_normal
return re_normal and im_normal
if isinstance(x, int_types) or isinstance(x, rational.mpq):
return bool(x)
x = ctx.convert(x)
if hasattr(x, '_mpf_') or hasattr(x, '_mpc_'):
return ctx.isnormal(x)
raise TypeError("isnormal() needs a number as input")
def isint(ctx, x, gaussian=False):
"""
Return *True* if *x* is integer-valued; otherwise return
*False*::
>>> from mpmath import *
>>> isint(3)
True
>>> isint(mpf(3))
True
>>> isint(3.2)
False
>>> isint(inf)
False
Optionally, Gaussian integers can be checked for::
>>> isint(3+0j)
True
>>> isint(3+2j)
False
>>> isint(3+2j, gaussian=True)
True
"""
if isinstance(x, int_types):
return True
if hasattr(x, "_mpf_"):
sign, man, exp, bc = xval = x._mpf_
return bool((man and exp >= 0) or xval == fzero)
if hasattr(x, "_mpc_"):
re, im = x._mpc_
rsign, rman, rexp, rbc = re
isign, iman, iexp, ibc = im
re_isint = (rman and rexp >= 0) or re == fzero
if gaussian:
im_isint = (iman and iexp >= 0) or im == fzero
return re_isint and im_isint
return re_isint and im == fzero
if isinstance(x, rational.mpq):
p, q = x._mpq_
return p % q == 0
x = ctx.convert(x)
if hasattr(x, '_mpf_') or hasattr(x, '_mpc_'):
return ctx.isint(x, gaussian)
raise TypeError("isint() needs a number as input")
def fsum(ctx, terms, absolute=False, squared=False):
"""
Calculates a sum containing a finite number of terms (for infinite
series, see :func:`~mpmath.nsum`). The terms will be converted to
mpmath numbers. For len(terms) > 2, this function is generally
faster and produces more accurate results than the builtin
Python function :func:`sum`.
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> fsum([1, 2, 0.5, 7])
mpf('10.5')
With squared=True each term is squared, and with absolute=True
the absolute value of each term is used.
"""
prec, rnd = ctx._prec_rounding
real = []
imag = []
other = 0
for term in terms:
reval = imval = 0
if hasattr(term, "_mpf_"):
reval = term._mpf_
elif hasattr(term, "_mpc_"):
reval, imval = term._mpc_
else:
term = ctx.convert(term)
if hasattr(term, "_mpf_"):
reval = term._mpf_
elif hasattr(term, "_mpc_"):
reval, imval = term._mpc_
else:
if absolute: term = ctx.absmax(term)
if squared: term = term**2
other += term
continue
if imval:
if squared:
if absolute:
real.append(mpf_mul(reval,reval))
real.append(mpf_mul(imval,imval))
else:
reval, imval = mpc_pow_int((reval,imval),2,prec+10)
real.append(reval)
imag.append(imval)
elif absolute:
real.append(mpc_abs((reval,imval), prec))
else:
real.append(reval)
imag.append(imval)
else:
if squared:
reval = mpf_mul(reval, reval)
elif absolute:
reval = mpf_abs(reval)
real.append(reval)
s = mpf_sum(real, prec, rnd, absolute)
if imag:
s = ctx.make_mpc((s, mpf_sum(imag, prec, rnd)))
else:
s = ctx.make_mpf(s)
if other is 0:
return s
else:
return s + other
def fdot(ctx, A, B=None, conjugate=False):
r"""
Computes the dot product of the iterables `A` and `B`,
.. math ::
\sum_{k=0} A_k B_k.
Alternatively, :func:`~mpmath.fdot` accepts a single iterable of pairs.
In other words, ``fdot(A,B)`` and ``fdot(zip(A,B))`` are equivalent.
The elements are automatically converted to mpmath numbers.
With ``conjugate=True``, the elements in the second vector
will be conjugated:
.. math ::
\sum_{k=0} A_k \overline{B_k}
**Examples**
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> A = [2, 1.5, 3]
>>> B = [1, -1, 2]
>>> fdot(A, B)
mpf('6.5')
>>> list(zip(A, B))
[(2, 1), (1.5, -1), (3, 2)]
>>> fdot(_)
mpf('6.5')
>>> A = [2, 1.5, 3j]
>>> B = [1+j, 3, -1-j]
>>> fdot(A, B)
mpc(real='9.5', imag='-1.0')
>>> fdot(A, B, conjugate=True)
mpc(real='3.5', imag='-5.0')
"""
if B:
A = zip(A, B)
prec, rnd = ctx._prec_rounding
real = []
imag = []
other = 0
hasattr_ = hasattr
types = (ctx.mpf, ctx.mpc)
for a, b in A:
if type(a) not in types: a = ctx.convert(a)
if type(b) not in types: b = ctx.convert(b)
a_real = hasattr_(a, "_mpf_")
b_real = hasattr_(b, "_mpf_")
if a_real and b_real:
real.append(mpf_mul(a._mpf_, b._mpf_))
continue
a_complex = hasattr_(a, "_mpc_")
b_complex = hasattr_(b, "_mpc_")
if a_real and b_complex:
aval = a._mpf_
bre, bim = b._mpc_
if conjugate:
bim = mpf_neg(bim)
real.append(mpf_mul(aval, bre))
imag.append(mpf_mul(aval, bim))
elif b_real and a_complex:
are, aim = a._mpc_
bval = b._mpf_
real.append(mpf_mul(are, bval))
imag.append(mpf_mul(aim, bval))
elif a_complex and b_complex:
#re, im = mpc_mul(a._mpc_, b._mpc_, prec+20)
are, aim = a._mpc_
bre, bim = b._mpc_
if conjugate:
bim = mpf_neg(bim)
real.append(mpf_mul(are, bre))
real.append(mpf_neg(mpf_mul(aim, bim)))
imag.append(mpf_mul(are, bim))
imag.append(mpf_mul(aim, bre))
else:
if conjugate:
other += a*ctx.conj(b)
else:
other += a*b
s = mpf_sum(real, prec, rnd)
if imag:
s = ctx.make_mpc((s, mpf_sum(imag, prec, rnd)))
else:
s = ctx.make_mpf(s)
if other is 0:
return s
else:
return s + other
def _wrap_libmp_function(ctx, mpf_f, mpc_f=None, mpi_f=None, doc="<no doc>"):
"""
Given a low-level mpf_ function, and optionally similar functions
for mpc_ and mpi_, defines the function as a context method.
It is assumed that the return type is the same as that of
the input; the exception is that propagation from mpf to mpc is possible
by raising ComplexResult.
"""
def f(x, **kwargs):
if type(x) not in ctx.types:
x = ctx.convert(x)
prec, rounding = ctx._prec_rounding
if kwargs:
prec = kwargs.get('prec', prec)
if 'dps' in kwargs:
prec = dps_to_prec(kwargs['dps'])
rounding = kwargs.get('rounding', rounding)
if hasattr(x, '_mpf_'):
try:
return ctx.make_mpf(mpf_f(x._mpf_, prec, rounding))
except ComplexResult:
# Handle propagation to complex
if ctx.trap_complex:
raise
return ctx.make_mpc(mpc_f((x._mpf_, fzero), prec, rounding))
elif hasattr(x, '_mpc_'):
return ctx.make_mpc(mpc_f(x._mpc_, prec, rounding))
raise NotImplementedError("%s of a %s" % (name, type(x)))
name = mpf_f.__name__[4:]
f.__doc__ = function_docs.__dict__.get(name, "Computes the %s of x" % doc)
return f
# Called by SpecialFunctions.__init__()
@classmethod
def _wrap_specfun(cls, name, f, wrap):
if wrap:
def f_wrapped(ctx, *args, **kwargs):
convert = ctx.convert
args = [convert(a) for a in args]
prec = ctx.prec
try:
ctx.prec += 10
retval = f(ctx, *args, **kwargs)
finally:
ctx.prec = prec
return +retval
else:
f_wrapped = f
f_wrapped.__doc__ = function_docs.__dict__.get(name, f.__doc__)
setattr(cls, name, f_wrapped)
def _convert_param(ctx, x):
if hasattr(x, "_mpc_"):
v, im = x._mpc_
if im != fzero:
return x, 'C'
elif hasattr(x, "_mpf_"):
v = x._mpf_
else:
if type(x) in int_types:
return int(x), 'Z'
p = None
if isinstance(x, tuple):
p, q = x
elif hasattr(x, '_mpq_'):
p, q = x._mpq_
elif isinstance(x, basestring) and '/' in x:
p, q = x.split('/')
p = int(p)
q = int(q)
if p is not None:
if not p % q:
return p // q, 'Z'
return ctx.mpq(p,q), 'Q'
x = ctx.convert(x)
if hasattr(x, "_mpc_"):
v, im = x._mpc_
if im != fzero:
return x, 'C'
elif hasattr(x, "_mpf_"):
v = x._mpf_
else:
return x, 'U'
sign, man, exp, bc = v
if man:
if exp >= -4:
if sign:
man = -man
if exp >= 0:
return int(man) << exp, 'Z'
if exp >= -4:
p, q = int(man), (1<<(-exp))
return ctx.mpq(p,q), 'Q'
x = ctx.make_mpf(v)
return x, 'R'
elif not exp:
return 0, 'Z'
else:
return x, 'U'
def _mpf_mag(ctx, x):
sign, man, exp, bc = x
if man:
return exp+bc
if x == fzero:
return ctx.ninf
if x == finf or x == fninf:
return ctx.inf
return ctx.nan
def mag(ctx, x):
"""
Quick logarithmic magnitude estimate of a number. Returns an
integer or infinity `m` such that `|x| <= 2^m`. It is not
guaranteed that `m` is an optimal bound, but it will never
be too large by more than 2 (and probably not more than 1).
**Examples**
>>> from mpmath import *
>>> mp.pretty = True
>>> mag(10), mag(10.0), mag(mpf(10)), int(ceil(log(10,2)))
(4, 4, 4, 4)
>>> mag(10j), mag(10+10j)
(4, 5)
>>> mag(0.01), int(ceil(log(0.01,2)))
(-6, -6)
>>> mag(0), mag(inf), mag(-inf), mag(nan)
(-inf, +inf, +inf, nan)
"""
if hasattr(x, "_mpf_"):
return ctx._mpf_mag(x._mpf_)
elif hasattr(x, "_mpc_"):
r, i = x._mpc_
if r == fzero:
return ctx._mpf_mag(i)
if i == fzero:
return ctx._mpf_mag(r)
return 1+max(ctx._mpf_mag(r), ctx._mpf_mag(i))
elif isinstance(x, int_types):
if x:
return bitcount(abs(x))
return ctx.ninf
elif isinstance(x, rational.mpq):
p, q = x._mpq_
if p:
return 1 + bitcount(abs(p)) - bitcount(q)
return ctx.ninf
else:
x = ctx.convert(x)
if hasattr(x, "_mpf_") or hasattr(x, "_mpc_"):
return ctx.mag(x)
else:
raise TypeError("requires an mpf/mpc")
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/ctx_mp_python.py
|
ctx_mp_python.py
|
from .libmp.backend import xrange
from .libmp import int_types, sqrt_fixed
# round to nearest integer (can be done more elegantly...)
def round_fixed(x, prec):
return ((x + (1<<(prec-1))) >> prec) << prec
class IdentificationMethods(object):
pass
def pslq(ctx, x, tol=None, maxcoeff=1000, maxsteps=100, verbose=False):
r"""
Given a vector of real numbers `x = [x_0, x_1, ..., x_n]`, ``pslq(x)``
uses the PSLQ algorithm to find a list of integers
`[c_0, c_1, ..., c_n]` such that
.. math ::
|c_1 x_1 + c_2 x_2 + ... + c_n x_n| < \mathrm{tol}
and such that `\max |c_k| < \mathrm{maxcoeff}`. If no such vector
exists, :func:`~mpmath.pslq` returns ``None``. The tolerance defaults to
3/4 of the working precision.
**Examples**
Find rational approximations for `\pi`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> pslq([-1, pi], tol=0.01)
[22, 7]
>>> pslq([-1, pi], tol=0.001)
[355, 113]
>>> mpf(22)/7; mpf(355)/113; +pi
3.14285714285714
3.14159292035398
3.14159265358979
Pi is not a rational number with denominator less than 1000::
>>> pslq([-1, pi])
>>>
To within the standard precision, it can however be approximated
by at least one rational number with denominator less than `10^{12}`::
>>> p, q = pslq([-1, pi], maxcoeff=10**12)
>>> print(p); print(q)
238410049439
75888275702
>>> mpf(p)/q
3.14159265358979
The PSLQ algorithm can be applied to long vectors. For example,
we can investigate the rational (in)dependence of integer square
roots::
>>> mp.dps = 30
>>> pslq([sqrt(n) for n in range(2, 5+1)])
>>>
>>> pslq([sqrt(n) for n in range(2, 6+1)])
>>>
>>> pslq([sqrt(n) for n in range(2, 8+1)])
[2, 0, 0, 0, 0, 0, -1]
**Machin formulas**
A famous formula for `\pi` is Machin's,
.. math ::
\frac{\pi}{4} = 4 \operatorname{acot} 5 - \operatorname{acot} 239
There are actually infinitely many formulas of this type. Two
others are
.. math ::
\frac{\pi}{4} = \operatorname{acot} 1
\frac{\pi}{4} = 12 \operatorname{acot} 49 + 32 \operatorname{acot} 57
+ 5 \operatorname{acot} 239 + 12 \operatorname{acot} 110443
We can easily verify the formulas using the PSLQ algorithm::
>>> mp.dps = 30
>>> pslq([pi/4, acot(1)])
[1, -1]
>>> pslq([pi/4, acot(5), acot(239)])
[1, -4, 1]
>>> pslq([pi/4, acot(49), acot(57), acot(239), acot(110443)])
[1, -12, -32, 5, -12]
We could try to generate a custom Machin-like formula by running
the PSLQ algorithm with a few inverse cotangent values, for example
acot(2), acot(3) ... acot(10). Unfortunately, there is a linear
dependence among these values, resulting in only that dependence
being detected, with a zero coefficient for `\pi`::
>>> pslq([pi] + [acot(n) for n in range(2,11)])
[0, 1, -1, 0, 0, 0, -1, 0, 0, 0]
We get better luck by removing linearly dependent terms::
>>> pslq([pi] + [acot(n) for n in range(2,11) if n not in (3, 5)])
[1, -8, 0, 0, 4, 0, 0, 0]
In other words, we found the following formula::
>>> 8*acot(2) - 4*acot(7)
3.14159265358979323846264338328
>>> +pi
3.14159265358979323846264338328
**Algorithm**
This is a fairly direct translation to Python of the pseudocode given by
David Bailey, "The PSLQ Integer Relation Algorithm":
http://www.cecm.sfu.ca/organics/papers/bailey/paper/html/node3.html
The present implementation uses fixed-point instead of floating-point
arithmetic, since this is significantly (about 7x) faster.
"""
n = len(x)
assert n >= 2
# At too low precision, the algorithm becomes meaningless
prec = ctx.prec
assert prec >= 53
if verbose and prec // max(2,n) < 5:
print("Warning: precision for PSLQ may be too low")
target = int(prec * 0.75)
if tol is None:
tol = ctx.mpf(2)**(-target)
else:
tol = ctx.convert(tol)
extra = 60
prec += extra
if verbose:
print("PSLQ using prec %i and tol %s" % (prec, ctx.nstr(tol)))
tol = ctx.to_fixed(tol, prec)
assert tol
# Convert to fixed-point numbers. The dummy None is added so we can
# use 1-based indexing. (This just allows us to be consistent with
# Bailey's indexing. The algorithm is 100 lines long, so debugging
# a single wrong index can be painful.)
x = [None] + [ctx.to_fixed(ctx.mpf(xk), prec) for xk in x]
# Sanity check on magnitudes
minx = min(abs(xx) for xx in x[1:])
if not minx:
raise ValueError("PSLQ requires a vector of nonzero numbers")
if minx < tol//100:
if verbose:
print("STOPPING: (one number is too small)")
return None
g = sqrt_fixed((4<<prec)//3, prec)
A = {}
B = {}
H = {}
# Initialization
# step 1
for i in xrange(1, n+1):
for j in xrange(1, n+1):
A[i,j] = B[i,j] = (i==j) << prec
H[i,j] = 0
# step 2
s = [None] + [0] * n
for k in xrange(1, n+1):
t = 0
for j in xrange(k, n+1):
t += (x[j]**2 >> prec)
s[k] = sqrt_fixed(t, prec)
t = s[1]
y = x[:]
for k in xrange(1, n+1):
y[k] = (x[k] << prec) // t
s[k] = (s[k] << prec) // t
# step 3
for i in xrange(1, n+1):
for j in xrange(i+1, n):
H[i,j] = 0
if i <= n-1:
if s[i]:
H[i,i] = (s[i+1] << prec) // s[i]
else:
H[i,i] = 0
for j in range(1, i):
sjj1 = s[j]*s[j+1]
if sjj1:
H[i,j] = ((-y[i]*y[j])<<prec)//sjj1
else:
H[i,j] = 0
# step 4
for i in xrange(2, n+1):
for j in xrange(i-1, 0, -1):
#t = floor(H[i,j]/H[j,j] + 0.5)
if H[j,j]:
t = round_fixed((H[i,j] << prec)//H[j,j], prec)
else:
#t = 0
continue
y[j] = y[j] + (t*y[i] >> prec)
for k in xrange(1, j+1):
H[i,k] = H[i,k] - (t*H[j,k] >> prec)
for k in xrange(1, n+1):
A[i,k] = A[i,k] - (t*A[j,k] >> prec)
B[k,j] = B[k,j] + (t*B[k,i] >> prec)
# Main algorithm
for REP in range(maxsteps):
# Step 1
m = -1
szmax = -1
for i in range(1, n):
h = H[i,i]
sz = (g**i * abs(h)) >> (prec*(i-1))
if sz > szmax:
m = i
szmax = sz
# Step 2
y[m], y[m+1] = y[m+1], y[m]
tmp = {}
for i in xrange(1,n+1): H[m,i], H[m+1,i] = H[m+1,i], H[m,i]
for i in xrange(1,n+1): A[m,i], A[m+1,i] = A[m+1,i], A[m,i]
for i in xrange(1,n+1): B[i,m], B[i,m+1] = B[i,m+1], B[i,m]
# Step 3
if m <= n - 2:
t0 = sqrt_fixed((H[m,m]**2 + H[m,m+1]**2)>>prec, prec)
# A zero element probably indicates that the precision has
# been exhausted. XXX: this could be spurious, due to
# using fixed-point arithmetic
if not t0:
break
t1 = (H[m,m] << prec) // t0
t2 = (H[m,m+1] << prec) // t0
for i in xrange(m, n+1):
t3 = H[i,m]
t4 = H[i,m+1]
H[i,m] = (t1*t3+t2*t4) >> prec
H[i,m+1] = (-t2*t3+t1*t4) >> prec
# Step 4
for i in xrange(m+1, n+1):
for j in xrange(min(i-1, m+1), 0, -1):
try:
t = round_fixed((H[i,j] << prec)//H[j,j], prec)
# Precision probably exhausted
except ZeroDivisionError:
break
y[j] = y[j] + ((t*y[i]) >> prec)
for k in xrange(1, j+1):
H[i,k] = H[i,k] - (t*H[j,k] >> prec)
for k in xrange(1, n+1):
A[i,k] = A[i,k] - (t*A[j,k] >> prec)
B[k,j] = B[k,j] + (t*B[k,i] >> prec)
# Until a relation is found, the error typically decreases
# slowly (e.g. a factor 1-10) with each step TODO: we could
# compare err from two successive iterations. If there is a
# large drop (several orders of magnitude), that indicates a
# "high quality" relation was detected. Reporting this to
# the user somehow might be useful.
best_err = maxcoeff<<prec
for i in xrange(1, n+1):
err = abs(y[i])
# Maybe we are done?
if err < tol:
# We are done if the coefficients are acceptable
vec = [int(round_fixed(B[j,i], prec) >> prec) for j in \
range(1,n+1)]
if max(abs(v) for v in vec) < maxcoeff:
if verbose:
print("FOUND relation at iter %i/%i, error: %s" % \
(REP, maxsteps, ctx.nstr(err / ctx.mpf(2)**prec, 1)))
return vec
best_err = min(err, best_err)
# Calculate a lower bound for the norm. We could do this
# more exactly (using the Euclidean norm) but there is probably
# no practical benefit.
recnorm = max(abs(h) for h in H.values())
if recnorm:
norm = ((1 << (2*prec)) // recnorm) >> prec
norm //= 100
else:
norm = ctx.inf
if verbose:
print("%i/%i: Error: %8s Norm: %s" % \
(REP, maxsteps, ctx.nstr(best_err / ctx.mpf(2)**prec, 1), norm))
if norm >= maxcoeff:
break
if verbose:
print("CANCELLING after step %i/%i." % (REP, maxsteps))
print("Could not find an integer relation. Norm bound: %s" % norm)
return None
def findpoly(ctx, x, n=1, **kwargs):
r"""
``findpoly(x, n)`` returns the coefficients of an integer
polynomial `P` of degree at most `n` such that `P(x) \approx 0`.
If no polynomial having `x` as a root can be found,
:func:`~mpmath.findpoly` returns ``None``.
:func:`~mpmath.findpoly` works by successively calling :func:`~mpmath.pslq` with
the vectors `[1, x]`, `[1, x, x^2]`, `[1, x, x^2, x^3]`, ...,
`[1, x, x^2, .., x^n]` as input. Keyword arguments given to
:func:`~mpmath.findpoly` are forwarded verbatim to :func:`~mpmath.pslq`. In
particular, you can specify a tolerance for `P(x)` with ``tol``
and a maximum permitted coefficient size with ``maxcoeff``.
For large values of `n`, it is recommended to run :func:`~mpmath.findpoly`
at high precision; preferably 50 digits or more.
**Examples**
By default (degree `n = 1`), :func:`~mpmath.findpoly` simply finds a linear
polynomial with a rational root::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> findpoly(0.7)
[-10, 7]
The generated coefficient list is valid input to ``polyval`` and
``polyroots``::
>>> nprint(polyval(findpoly(phi, 2), phi), 1)
-2.0e-16
>>> for r in polyroots(findpoly(phi, 2)):
... print(r)
...
-0.618033988749895
1.61803398874989
Numbers of the form `m + n \sqrt p` for integers `(m, n, p)` are
solutions to quadratic equations. As we find here, `1+\sqrt 2`
is a root of the polynomial `x^2 - 2x - 1`::
>>> findpoly(1+sqrt(2), 2)
[1, -2, -1]
>>> findroot(lambda x: x**2 - 2*x - 1, 1)
2.4142135623731
Despite only containing square roots, the following number results
in a polynomial of degree 4::
>>> findpoly(sqrt(2)+sqrt(3), 4)
[1, 0, -10, 0, 1]
In fact, `x^4 - 10x^2 + 1` is the *minimal polynomial* of
`r = \sqrt 2 + \sqrt 3`, meaning that a rational polynomial of
lower degree having `r` as a root does not exist. Given sufficient
precision, :func:`~mpmath.findpoly` will usually find the correct
minimal polynomial of a given algebraic number.
**Non-algebraic numbers**
If :func:`~mpmath.findpoly` fails to find a polynomial with given
coefficient size and tolerance constraints, that means no such
polynomial exists.
We can verify that `\pi` is not an algebraic number of degree 3 with
coefficients less than 1000::
>>> mp.dps = 15
>>> findpoly(pi, 3)
>>>
It is always possible to find an algebraic approximation of a number
using one (or several) of the following methods:
1. Increasing the permitted degree
2. Allowing larger coefficients
3. Reducing the tolerance
One example of each method is shown below::
>>> mp.dps = 15
>>> findpoly(pi, 4)
[95, -545, 863, -183, -298]
>>> findpoly(pi, 3, maxcoeff=10000)
[836, -1734, -2658, -457]
>>> findpoly(pi, 3, tol=1e-7)
[-4, 22, -29, -2]
It is unknown whether Euler's constant is transcendental (or even
irrational). We can use :func:`~mpmath.findpoly` to check that if is
an algebraic number, its minimal polynomial must have degree
at least 7 and a coefficient of magnitude at least 1000000::
>>> mp.dps = 200
>>> findpoly(euler, 6, maxcoeff=10**6, tol=1e-100, maxsteps=1000)
>>>
Note that the high precision and strict tolerance is necessary
for such high-degree runs, since otherwise unwanted low-accuracy
approximations will be detected. It may also be necessary to set
maxsteps high to prevent a premature exit (before the coefficient
bound has been reached). Running with ``verbose=True`` to get an
idea what is happening can be useful.
"""
x = ctx.mpf(x)
assert n >= 1
if x == 0:
return [1, 0]
xs = [ctx.mpf(1)]
for i in range(1,n+1):
xs.append(x**i)
a = ctx.pslq(xs, **kwargs)
if a is not None:
return a[::-1]
def fracgcd(p, q):
x, y = p, q
while y:
x, y = y, x % y
if x != 1:
p //= x
q //= x
if q == 1:
return p
return p, q
def pslqstring(r, constants):
q = r[0]
r = r[1:]
s = []
for i in range(len(r)):
p = r[i]
if p:
z = fracgcd(-p,q)
cs = constants[i][1]
if cs == '1':
cs = ''
else:
cs = '*' + cs
if isinstance(z, int_types):
if z > 0: term = str(z) + cs
else: term = ("(%s)" % z) + cs
else:
term = ("(%s/%s)" % z) + cs
s.append(term)
s = ' + '.join(s)
if '+' in s or '*' in s:
s = '(' + s + ')'
return s or '0'
def prodstring(r, constants):
q = r[0]
r = r[1:]
num = []
den = []
for i in range(len(r)):
p = r[i]
if p:
z = fracgcd(-p,q)
cs = constants[i][1]
if isinstance(z, int_types):
if abs(z) == 1: t = cs
else: t = '%s**%s' % (cs, abs(z))
([num,den][z<0]).append(t)
else:
t = '%s**(%s/%s)' % (cs, abs(z[0]), z[1])
([num,den][z[0]<0]).append(t)
num = '*'.join(num)
den = '*'.join(den)
if num and den: return "(%s)/(%s)" % (num, den)
if num: return num
if den: return "1/(%s)" % den
def quadraticstring(ctx,t,a,b,c):
if c < 0:
a,b,c = -a,-b,-c
u1 = (-b+ctx.sqrt(b**2-4*a*c))/(2*c)
u2 = (-b-ctx.sqrt(b**2-4*a*c))/(2*c)
if abs(u1-t) < abs(u2-t):
if b: s = '((%s+sqrt(%s))/%s)' % (-b,b**2-4*a*c,2*c)
else: s = '(sqrt(%s)/%s)' % (-4*a*c,2*c)
else:
if b: s = '((%s-sqrt(%s))/%s)' % (-b,b**2-4*a*c,2*c)
else: s = '(-sqrt(%s)/%s)' % (-4*a*c,2*c)
return s
# Transformation y = f(x,c), with inverse function x = f(y,c)
# The third entry indicates whether the transformation is
# redundant when c = 1
transforms = [
(lambda ctx,x,c: x*c, '$y/$c', 0),
(lambda ctx,x,c: x/c, '$c*$y', 1),
(lambda ctx,x,c: c/x, '$c/$y', 0),
(lambda ctx,x,c: (x*c)**2, 'sqrt($y)/$c', 0),
(lambda ctx,x,c: (x/c)**2, '$c*sqrt($y)', 1),
(lambda ctx,x,c: (c/x)**2, '$c/sqrt($y)', 0),
(lambda ctx,x,c: c*x**2, 'sqrt($y)/sqrt($c)', 1),
(lambda ctx,x,c: x**2/c, 'sqrt($c)*sqrt($y)', 1),
(lambda ctx,x,c: c/x**2, 'sqrt($c)/sqrt($y)', 1),
(lambda ctx,x,c: ctx.sqrt(x*c), '$y**2/$c', 0),
(lambda ctx,x,c: ctx.sqrt(x/c), '$c*$y**2', 1),
(lambda ctx,x,c: ctx.sqrt(c/x), '$c/$y**2', 0),
(lambda ctx,x,c: c*ctx.sqrt(x), '$y**2/$c**2', 1),
(lambda ctx,x,c: ctx.sqrt(x)/c, '$c**2*$y**2', 1),
(lambda ctx,x,c: c/ctx.sqrt(x), '$c**2/$y**2', 1),
(lambda ctx,x,c: ctx.exp(x*c), 'log($y)/$c', 0),
(lambda ctx,x,c: ctx.exp(x/c), '$c*log($y)', 1),
(lambda ctx,x,c: ctx.exp(c/x), '$c/log($y)', 0),
(lambda ctx,x,c: c*ctx.exp(x), 'log($y/$c)', 1),
(lambda ctx,x,c: ctx.exp(x)/c, 'log($c*$y)', 1),
(lambda ctx,x,c: c/ctx.exp(x), 'log($c/$y)', 0),
(lambda ctx,x,c: ctx.ln(x*c), 'exp($y)/$c', 0),
(lambda ctx,x,c: ctx.ln(x/c), '$c*exp($y)', 1),
(lambda ctx,x,c: ctx.ln(c/x), '$c/exp($y)', 0),
(lambda ctx,x,c: c*ctx.ln(x), 'exp($y/$c)', 1),
(lambda ctx,x,c: ctx.ln(x)/c, 'exp($c*$y)', 1),
(lambda ctx,x,c: c/ctx.ln(x), 'exp($c/$y)', 0),
]
def identify(ctx, x, constants=[], tol=None, maxcoeff=1000, full=False,
verbose=False):
"""
Given a real number `x`, ``identify(x)`` attempts to find an exact
formula for `x`. This formula is returned as a string. If no match
is found, ``None`` is returned. With ``full=True``, a list of
matching formulas is returned.
As a simple example, :func:`~mpmath.identify` will find an algebraic
formula for the golden ratio::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> identify(phi)
'((1+sqrt(5))/2)'
:func:`~mpmath.identify` can identify simple algebraic numbers and simple
combinations of given base constants, as well as certain basic
transformations thereof. More specifically, :func:`~mpmath.identify`
looks for the following:
1. Fractions
2. Quadratic algebraic numbers
3. Rational linear combinations of the base constants
4. Any of the above after first transforming `x` into `f(x)` where
`f(x)` is `1/x`, `\sqrt x`, `x^2`, `\log x` or `\exp x`, either
directly or with `x` or `f(x)` multiplied or divided by one of
the base constants
5. Products of fractional powers of the base constants and
small integers
Base constants can be given as a list of strings representing mpmath
expressions (:func:`~mpmath.identify` will ``eval`` the strings to numerical
values and use the original strings for the output), or as a dict of
formula:value pairs.
In order not to produce spurious results, :func:`~mpmath.identify` should
be used with high precision; preferably 50 digits or more.
**Examples**
Simple identifications can be performed safely at standard
precision. Here the default recognition of rational, algebraic,
and exp/log of algebraic numbers is demonstrated::
>>> mp.dps = 15
>>> identify(0.22222222222222222)
'(2/9)'
>>> identify(1.9662210973805663)
'sqrt(((24+sqrt(48))/8))'
>>> identify(4.1132503787829275)
'exp((sqrt(8)/2))'
>>> identify(0.881373587019543)
'log(((2+sqrt(8))/2))'
By default, :func:`~mpmath.identify` does not recognize `\pi`. At standard
precision it finds a not too useful approximation. At slightly
increased precision, this approximation is no longer accurate
enough and :func:`~mpmath.identify` more correctly returns ``None``::
>>> identify(pi)
'(2**(176/117)*3**(20/117)*5**(35/39))/(7**(92/117))'
>>> mp.dps = 30
>>> identify(pi)
>>>
Numbers such as `\pi`, and simple combinations of user-defined
constants, can be identified if they are provided explicitly::
>>> identify(3*pi-2*e, ['pi', 'e'])
'(3*pi + (-2)*e)'
Here is an example using a dict of constants. Note that the
constants need not be "atomic"; :func:`~mpmath.identify` can just
as well express the given number in terms of expressions
given by formulas::
>>> identify(pi+e, {'a':pi+2, 'b':2*e})
'((-2) + 1*a + (1/2)*b)'
Next, we attempt some identifications with a set of base constants.
It is necessary to increase the precision a bit.
>>> mp.dps = 50
>>> base = ['sqrt(2)','pi','log(2)']
>>> identify(0.25, base)
'(1/4)'
>>> identify(3*pi + 2*sqrt(2) + 5*log(2)/7, base)
'(2*sqrt(2) + 3*pi + (5/7)*log(2))'
>>> identify(exp(pi+2), base)
'exp((2 + 1*pi))'
>>> identify(1/(3+sqrt(2)), base)
'((3/7) + (-1/7)*sqrt(2))'
>>> identify(sqrt(2)/(3*pi+4), base)
'sqrt(2)/(4 + 3*pi)'
>>> identify(5**(mpf(1)/3)*pi*log(2)**2, base)
'5**(1/3)*pi*log(2)**2'
An example of an erroneous solution being found when too low
precision is used::
>>> mp.dps = 15
>>> identify(1/(3*pi-4*e+sqrt(8)), ['pi', 'e', 'sqrt(2)'])
'((11/25) + (-158/75)*pi + (76/75)*e + (44/15)*sqrt(2))'
>>> mp.dps = 50
>>> identify(1/(3*pi-4*e+sqrt(8)), ['pi', 'e', 'sqrt(2)'])
'1/(3*pi + (-4)*e + 2*sqrt(2))'
**Finding approximate solutions**
The tolerance ``tol`` defaults to 3/4 of the working precision.
Lowering the tolerance is useful for finding approximate matches.
We can for example try to generate approximations for pi::
>>> mp.dps = 15
>>> identify(pi, tol=1e-2)
'(22/7)'
>>> identify(pi, tol=1e-3)
'(355/113)'
>>> identify(pi, tol=1e-10)
'(5**(339/269))/(2**(64/269)*3**(13/269)*7**(92/269))'
With ``full=True``, and by supplying a few base constants,
``identify`` can generate almost endless lists of approximations
for any number (the output below has been truncated to show only
the first few)::
>>> for p in identify(pi, ['e', 'catalan'], tol=1e-5, full=True):
... print(p)
... # doctest: +ELLIPSIS
e/log((6 + (-4/3)*e))
(3**3*5*e*catalan**2)/(2*7**2)
sqrt(((-13) + 1*e + 22*catalan))
log(((-6) + 24*e + 4*catalan)/e)
exp(catalan*((-1/5) + (8/15)*e))
catalan*(6 + (-6)*e + 15*catalan)
sqrt((5 + 26*e + (-3)*catalan))/e
e*sqrt(((-27) + 2*e + 25*catalan))
log(((-1) + (-11)*e + 59*catalan))
((3/20) + (21/20)*e + (3/20)*catalan)
...
The numerical values are roughly as close to `\pi` as permitted by the
specified tolerance:
>>> e/log(6-4*e/3)
3.14157719846001
>>> 135*e*catalan**2/98
3.14166950419369
>>> sqrt(e-13+22*catalan)
3.14158000062992
>>> log(24*e-6+4*catalan)-1
3.14158791577159
**Symbolic processing**
The output formula can be evaluated as a Python expression.
Note however that if fractions (like '2/3') are present in
the formula, Python's :func:`~mpmath.eval()` may erroneously perform
integer division. Note also that the output is not necessarily
in the algebraically simplest form::
>>> identify(sqrt(2))
'(sqrt(8)/2)'
As a solution to both problems, consider using SymPy's
:func:`~mpmath.sympify` to convert the formula into a symbolic expression.
SymPy can be used to pretty-print or further simplify the formula
symbolically::
>>> from sympy import sympify
>>> sympify(identify(sqrt(2)))
2**(1/2)
Sometimes :func:`~mpmath.identify` can simplify an expression further than
a symbolic algorithm::
>>> from sympy import simplify
>>> x = sympify('-1/(-3/2+(1/2)*5**(1/2))*(3/2-1/2*5**(1/2))**(1/2)')
>>> x
(3/2 - 5**(1/2)/2)**(-1/2)
>>> x = simplify(x)
>>> x
2/(6 - 2*5**(1/2))**(1/2)
>>> mp.dps = 30
>>> x = sympify(identify(x.evalf(30)))
>>> x
1/2 + 5**(1/2)/2
(In fact, this functionality is available directly in SymPy as the
function :func:`~mpmath.nsimplify`, which is essentially a wrapper for
:func:`~mpmath.identify`.)
**Miscellaneous issues and limitations**
The input `x` must be a real number. All base constants must be
positive real numbers and must not be rationals or rational linear
combinations of each other.
The worst-case computation time grows quickly with the number of
base constants. Already with 3 or 4 base constants,
:func:`~mpmath.identify` may require several seconds to finish. To search
for relations among a large number of constants, you should
consider using :func:`~mpmath.pslq` directly.
The extended transformations are applied to x, not the constants
separately. As a result, ``identify`` will for example be able to
recognize ``exp(2*pi+3)`` with ``pi`` given as a base constant, but
not ``2*exp(pi)+3``. It will be able to recognize the latter if
``exp(pi)`` is given explicitly as a base constant.
"""
solutions = []
def addsolution(s):
if verbose: print("Found: ", s)
solutions.append(s)
x = ctx.mpf(x)
# Further along, x will be assumed positive
if x == 0:
if full: return ['0']
else: return '0'
if x < 0:
sol = ctx.identify(-x, constants, tol, maxcoeff, full, verbose)
if sol is None:
return sol
if full:
return ["-(%s)"%s for s in sol]
else:
return "-(%s)" % sol
if tol:
tol = ctx.mpf(tol)
else:
tol = ctx.eps**0.7
M = maxcoeff
if constants:
if isinstance(constants, dict):
constants = [(ctx.mpf(v), name) for (name, v) in constants.items()]
else:
namespace = dict((name, getattr(ctx,name)) for name in dir(ctx))
constants = [(eval(p, namespace), p) for p in constants]
else:
constants = []
# We always want to find at least rational terms
if 1 not in [value for (name, value) in constants]:
constants = [(ctx.mpf(1), '1')] + constants
# PSLQ with simple algebraic and functional transformations
for ft, ftn, red in transforms:
for c, cn in constants:
if red and cn == '1':
continue
t = ft(ctx,x,c)
# Prevent exponential transforms from wreaking havoc
if abs(t) > M**2 or abs(t) < tol:
continue
# Linear combination of base constants
r = ctx.pslq([t] + [a[0] for a in constants], tol, M)
s = None
if r is not None and max(abs(uw) for uw in r) <= M and r[0]:
s = pslqstring(r, constants)
# Quadratic algebraic numbers
else:
q = ctx.pslq([ctx.one, t, t**2], tol, M)
if q is not None and len(q) == 3 and q[2]:
aa, bb, cc = q
if max(abs(aa),abs(bb),abs(cc)) <= M:
s = quadraticstring(ctx,t,aa,bb,cc)
if s:
if cn == '1' and ('/$c' in ftn):
s = ftn.replace('$y', s).replace('/$c', '')
else:
s = ftn.replace('$y', s).replace('$c', cn)
addsolution(s)
if not full: return solutions[0]
if verbose:
print(".")
# Check for a direct multiplicative formula
if x != 1:
# Allow fractional powers of fractions
ilogs = [2,3,5,7]
# Watch out for existing fractional powers of fractions
logs = []
for a, s in constants:
if not sum(bool(ctx.findpoly(ctx.ln(a)/ctx.ln(i),1)) for i in ilogs):
logs.append((ctx.ln(a), s))
logs = [(ctx.ln(i),str(i)) for i in ilogs] + logs
r = ctx.pslq([ctx.ln(x)] + [a[0] for a in logs], tol, M)
if r is not None and max(abs(uw) for uw in r) <= M and r[0]:
addsolution(prodstring(r, logs))
if not full: return solutions[0]
if full:
return sorted(solutions, key=len)
else:
return None
IdentificationMethods.pslq = pslq
IdentificationMethods.findpoly = findpoly
IdentificationMethods.identify = identify
if __name__ == '__main__':
import doctest
doctest.testmod()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/identification.py
|
identification.py
|
from colorsys import hsv_to_rgb, hls_to_rgb
from .libmp import NoConvergence
from .libmp.backend import xrange
class VisualizationMethods(object):
plot_ignore = (ValueError, ArithmeticError, ZeroDivisionError, NoConvergence)
def plot(ctx, f, xlim=[-5,5], ylim=None, points=200, file=None, dpi=None,
singularities=[], axes=None):
r"""
Shows a simple 2D plot of a function `f(x)` or list of functions
`[f_0(x), f_1(x), \ldots, f_n(x)]` over a given interval
specified by *xlim*. Some examples::
plot(lambda x: exp(x)*li(x), [1, 4])
plot([cos, sin], [-4, 4])
plot([fresnels, fresnelc], [-4, 4])
plot([sqrt, cbrt], [-4, 4])
plot(lambda t: zeta(0.5+t*j), [-20, 20])
plot([floor, ceil, abs, sign], [-5, 5])
Points where the function raises a numerical exception or
returns an infinite value are removed from the graph.
Singularities can also be excluded explicitly
as follows (useful for removing erroneous vertical lines)::
plot(cot, ylim=[-5, 5]) # bad
plot(cot, ylim=[-5, 5], singularities=[-pi, 0, pi]) # good
For parts where the function assumes complex values, the
real part is plotted with dashes and the imaginary part
is plotted with dots.
.. note :: This function requires matplotlib (pylab).
"""
if file:
axes = None
fig = None
if not axes:
import pylab
fig = pylab.figure()
axes = fig.add_subplot(111)
if not isinstance(f, (tuple, list)):
f = [f]
a, b = xlim
colors = ['b', 'r', 'g', 'm', 'k']
for n, func in enumerate(f):
x = ctx.arange(a, b, (b-a)/float(points))
segments = []
segment = []
in_complex = False
for i in xrange(len(x)):
try:
if i != 0:
for sing in singularities:
if x[i-1] <= sing and x[i] >= sing:
raise ValueError
v = func(x[i])
if ctx.isnan(v) or abs(v) > 1e300:
raise ValueError
if hasattr(v, "imag") and v.imag:
re = float(v.real)
im = float(v.imag)
if not in_complex:
in_complex = True
segments.append(segment)
segment = []
segment.append((float(x[i]), re, im))
else:
if in_complex:
in_complex = False
segments.append(segment)
segment = []
if hasattr(v, "real"):
v = v.real
segment.append((float(x[i]), v))
except ctx.plot_ignore:
if segment:
segments.append(segment)
segment = []
if segment:
segments.append(segment)
for segment in segments:
x = [s[0] for s in segment]
y = [s[1] for s in segment]
if not x:
continue
c = colors[n % len(colors)]
if len(segment[0]) == 3:
z = [s[2] for s in segment]
axes.plot(x, y, '--'+c, linewidth=3)
axes.plot(x, z, ':'+c, linewidth=3)
else:
axes.plot(x, y, c, linewidth=3)
axes.set_xlim([float(_) for _ in xlim])
if ylim:
axes.set_ylim([float(_) for _ in ylim])
axes.set_xlabel('x')
axes.set_ylabel('f(x)')
axes.grid(True)
if fig:
if file:
pylab.savefig(file, dpi=dpi)
else:
pylab.show()
def default_color_function(ctx, z):
if ctx.isinf(z):
return (1.0, 1.0, 1.0)
if ctx.isnan(z):
return (0.5, 0.5, 0.5)
pi = 3.1415926535898
a = (float(ctx.arg(z)) + ctx.pi) / (2*ctx.pi)
a = (a + 0.5) % 1.0
b = 1.0 - float(1/(1.0+abs(z)**0.3))
return hls_to_rgb(a, b, 0.8)
def cplot(ctx, f, re=[-5,5], im=[-5,5], points=2000, color=None,
verbose=False, file=None, dpi=None, axes=None):
"""
Plots the given complex-valued function *f* over a rectangular part
of the complex plane specified by the pairs of intervals *re* and *im*.
For example::
cplot(lambda z: z, [-2, 2], [-10, 10])
cplot(exp)
cplot(zeta, [0, 1], [0, 50])
By default, the complex argument (phase) is shown as color (hue) and
the magnitude is show as brightness. You can also supply a
custom color function (*color*). This function should take a
complex number as input and return an RGB 3-tuple containing
floats in the range 0.0-1.0.
To obtain a sharp image, the number of points may need to be
increased to 100,000 or thereabout. Since evaluating the
function that many times is likely to be slow, the 'verbose'
option is useful to display progress.
.. note :: This function requires matplotlib (pylab).
"""
if color is None:
color = ctx.default_color_function
import pylab
if file:
axes = None
fig = None
if not axes:
fig = pylab.figure()
axes = fig.add_subplot(111)
rea, reb = re
ima, imb = im
dre = reb - rea
dim = imb - ima
M = int(ctx.sqrt(points*dre/dim)+1)
N = int(ctx.sqrt(points*dim/dre)+1)
x = pylab.linspace(rea, reb, M)
y = pylab.linspace(ima, imb, N)
# Note: we have to be careful to get the right rotation.
# Test with these plots:
# cplot(lambda z: z if z.real < 0 else 0)
# cplot(lambda z: z if z.imag < 0 else 0)
w = pylab.zeros((N, M, 3))
for n in xrange(N):
for m in xrange(M):
z = ctx.mpc(x[m], y[n])
try:
v = color(f(z))
except ctx.plot_ignore:
v = (0.5, 0.5, 0.5)
w[n,m] = v
if verbose:
print(n, "of", N)
rea, reb, ima, imb = [float(_) for _ in [rea, reb, ima, imb]]
axes.imshow(w, extent=(rea, reb, ima, imb), origin='lower')
axes.set_xlabel('Re(z)')
axes.set_ylabel('Im(z)')
if fig:
if file:
pylab.savefig(file, dpi=dpi)
else:
pylab.show()
def splot(ctx, f, u=[-5,5], v=[-5,5], points=100, keep_aspect=True, \
wireframe=False, file=None, dpi=None, axes=None):
"""
Plots the surface defined by `f`.
If `f` returns a single component, then this plots the surface
defined by `z = f(x,y)` over the rectangular domain with
`x = u` and `y = v`.
If `f` returns three components, then this plots the parametric
surface `x, y, z = f(u,v)` over the pairs of intervals `u` and `v`.
For example, to plot a simple function::
>>> from mpmath import *
>>> f = lambda x, y: sin(x+y)*cos(y)
>>> splot(f, [-pi,pi], [-pi,pi]) # doctest: +SKIP
Plotting a donut::
>>> r, R = 1, 2.5
>>> f = lambda u, v: [r*cos(u), (R+r*sin(u))*cos(v), (R+r*sin(u))*sin(v)]
>>> splot(f, [0, 2*pi], [0, 2*pi]) # doctest: +SKIP
.. note :: This function requires matplotlib (pylab) 0.98.5.3 or higher.
"""
import pylab
import mpl_toolkits.mplot3d as mplot3d
if file:
axes = None
fig = None
if not axes:
fig = pylab.figure()
axes = mplot3d.axes3d.Axes3D(fig)
ua, ub = u
va, vb = v
du = ub - ua
dv = vb - va
if not isinstance(points, (list, tuple)):
points = [points, points]
M, N = points
u = pylab.linspace(ua, ub, M)
v = pylab.linspace(va, vb, N)
x, y, z = [pylab.zeros((M, N)) for i in xrange(3)]
xab, yab, zab = [[0, 0] for i in xrange(3)]
for n in xrange(N):
for m in xrange(M):
fdata = f(ctx.convert(u[m]), ctx.convert(v[n]))
try:
x[m,n], y[m,n], z[m,n] = fdata
except TypeError:
x[m,n], y[m,n], z[m,n] = u[m], v[n], fdata
for c, cab in [(x[m,n], xab), (y[m,n], yab), (z[m,n], zab)]:
if c < cab[0]:
cab[0] = c
if c > cab[1]:
cab[1] = c
if wireframe:
axes.plot_wireframe(x, y, z, rstride=4, cstride=4)
else:
axes.plot_surface(x, y, z, rstride=4, cstride=4)
axes.set_xlabel('x')
axes.set_ylabel('y')
axes.set_zlabel('z')
if keep_aspect:
dx, dy, dz = [cab[1] - cab[0] for cab in [xab, yab, zab]]
maxd = max(dx, dy, dz)
if dx < maxd:
delta = maxd - dx
axes.set_xlim3d(xab[0] - delta / 2.0, xab[1] + delta / 2.0)
if dy < maxd:
delta = maxd - dy
axes.set_ylim3d(yab[0] - delta / 2.0, yab[1] + delta / 2.0)
if dz < maxd:
delta = maxd - dz
axes.set_zlim3d(zab[0] - delta / 2.0, zab[1] + delta / 2.0)
if fig:
if file:
pylab.savefig(file, dpi=dpi)
else:
pylab.show()
VisualizationMethods.plot = plot
VisualizationMethods.default_color_function = default_color_function
VisualizationMethods.cplot = cplot
VisualizationMethods.splot = splot
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/visualization.py
|
visualization.py
|
pi = r"""
`\pi`, roughly equal to 3.141592654, represents the area of the unit
circle, the half-period of trigonometric functions, and many other
things in mathematics.
Mpmath can evaluate `\pi` to arbitrary precision::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +pi
3.1415926535897932384626433832795028841971693993751
This shows digits 99991-100000 of `\pi`::
>>> mp.dps = 100000
>>> str(pi)[-10:]
'5549362464'
**Possible issues**
:data:`pi` always rounds to the nearest floating-point
number when used. This means that exact mathematical identities
involving `\pi` will generally not be preserved in floating-point
arithmetic. In particular, multiples of :data:`pi` (except for
the trivial case ``0*pi``) are *not* the exact roots of
:func:`~mpmath.sin`, but differ roughly by the current epsilon::
>>> mp.dps = 15
>>> sin(pi)
1.22464679914735e-16
One solution is to use the :func:`~mpmath.sinpi` function instead::
>>> sinpi(1)
0.0
See the documentation of trigonometric functions for additional
details.
"""
degree = r"""
Represents one degree of angle, `1^{\circ} = \pi/180`, or
about 0.01745329. This constant may be evaluated to arbitrary
precision::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +degree
0.017453292519943295769236907684886127134428718885417
The :data:`degree` object is convenient for conversion
to radians::
>>> sin(30 * degree)
0.5
>>> asin(0.5) / degree
30.0
"""
e = r"""
The transcendental number `e` = 2.718281828... is the base of the
natural logarithm (:func:`~mpmath.ln`) and of the exponential function
(:func:`~mpmath.exp`).
Mpmath can be evaluate `e` to arbitrary precision::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +e
2.7182818284590452353602874713526624977572470937
This shows digits 99991-100000 of `e`::
>>> mp.dps = 100000
>>> str(e)[-10:]
'2100427165'
**Possible issues**
:data:`e` always rounds to the nearest floating-point number
when used, and mathematical identities involving `e` may not
hold in floating-point arithmetic. For example, ``ln(e)``
might not evaluate exactly to 1.
In particular, don't use ``e**x`` to compute the exponential
function. Use ``exp(x)`` instead; this is both faster and more
accurate.
"""
phi = r"""
Represents the golden ratio `\phi = (1+\sqrt 5)/2`,
approximately equal to 1.6180339887. To high precision,
its value is::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +phi
1.6180339887498948482045868343656381177203091798058
Formulas for the golden ratio include the following::
>>> (1+sqrt(5))/2
1.6180339887498948482045868343656381177203091798058
>>> findroot(lambda x: x**2-x-1, 1)
1.6180339887498948482045868343656381177203091798058
>>> limit(lambda n: fib(n+1)/fib(n), inf)
1.6180339887498948482045868343656381177203091798058
"""
euler = r"""
Euler's constant or the Euler-Mascheroni constant `\gamma`
= 0.57721566... is a number of central importance to
number theory and special functions. It is defined as the limit
.. math ::
\gamma = \lim_{n\to\infty} H_n - \log n
where `H_n = 1 + \frac{1}{2} + \ldots + \frac{1}{n}` is a harmonic
number (see :func:`~mpmath.harmonic`).
Evaluation of `\gamma` is supported at arbitrary precision::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +euler
0.57721566490153286060651209008240243104215933593992
We can also compute `\gamma` directly from the definition,
although this is less efficient::
>>> limit(lambda n: harmonic(n)-log(n), inf)
0.57721566490153286060651209008240243104215933593992
This shows digits 9991-10000 of `\gamma`::
>>> mp.dps = 10000
>>> str(euler)[-10:]
'4679858165'
Integrals, series, and representations for `\gamma` in terms of
special functions include the following (there are many others)::
>>> mp.dps = 25
>>> -quad(lambda x: exp(-x)*log(x), [0,inf])
0.5772156649015328606065121
>>> quad(lambda x,y: (x-1)/(1-x*y)/log(x*y), [0,1], [0,1])
0.5772156649015328606065121
>>> nsum(lambda k: 1/k-log(1+1/k), [1,inf])
0.5772156649015328606065121
>>> nsum(lambda k: (-1)**k*zeta(k)/k, [2,inf])
0.5772156649015328606065121
>>> -diff(gamma, 1)
0.5772156649015328606065121
>>> limit(lambda x: 1/x-gamma(x), 0)
0.5772156649015328606065121
>>> limit(lambda x: zeta(x)-1/(x-1), 1)
0.5772156649015328606065121
>>> (log(2*pi*nprod(lambda n:
... exp(-2+2/n)*(1+2/n)**n, [1,inf]))-3)/2
0.5772156649015328606065121
For generalizations of the identities `\gamma = -\Gamma'(1)`
and `\gamma = \lim_{x\to1} \zeta(x)-1/(x-1)`, see
:func:`~mpmath.psi` and :func:`~mpmath.stieltjes` respectively.
"""
catalan = r"""
Catalan's constant `K` = 0.91596559... is given by the infinite
series
.. math ::
K = \sum_{k=0}^{\infty} \frac{(-1)^k}{(2k+1)^2}.
Mpmath can evaluate it to arbitrary precision::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +catalan
0.91596559417721901505460351493238411077414937428167
One can also compute `K` directly from the definition, although
this is significantly less efficient::
>>> nsum(lambda k: (-1)**k/(2*k+1)**2, [0, inf])
0.91596559417721901505460351493238411077414937428167
This shows digits 9991-10000 of `K`::
>>> mp.dps = 10000
>>> str(catalan)[-10:]
'9537871503'
Catalan's constant has numerous integral representations::
>>> mp.dps = 50
>>> quad(lambda x: -log(x)/(1+x**2), [0, 1])
0.91596559417721901505460351493238411077414937428167
>>> quad(lambda x: atan(x)/x, [0, 1])
0.91596559417721901505460351493238411077414937428167
>>> quad(lambda x: ellipk(x**2)/2, [0, 1])
0.91596559417721901505460351493238411077414937428167
>>> quad(lambda x,y: 1/(1+(x*y)**2), [0, 1], [0, 1])
0.91596559417721901505460351493238411077414937428167
As well as series representations::
>>> pi*log(sqrt(3)+2)/8 + 3*nsum(lambda n:
... (fac(n)/(2*n+1))**2/fac(2*n), [0, inf])/8
0.91596559417721901505460351493238411077414937428167
>>> 1-nsum(lambda n: n*zeta(2*n+1)/16**n, [1,inf])
0.91596559417721901505460351493238411077414937428167
"""
khinchin = r"""
Khinchin's constant `K` = 2.68542... is a number that
appears in the theory of continued fractions. Mpmath can evaluate
it to arbitrary precision::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +khinchin
2.6854520010653064453097148354817956938203822939945
An integral representation is::
>>> I = quad(lambda x: log((1-x**2)/sincpi(x))/x/(1+x), [0, 1])
>>> 2*exp(1/log(2)*I)
2.6854520010653064453097148354817956938203822939945
The computation of ``khinchin`` is based on an efficient
implementation of the following series::
>>> f = lambda n: (zeta(2*n)-1)/n*sum((-1)**(k+1)/mpf(k)
... for k in range(1,2*int(n)))
>>> exp(nsum(f, [1,inf])/log(2))
2.6854520010653064453097148354817956938203822939945
"""
glaisher = r"""
Glaisher's constant `A`, also known as the Glaisher-Kinkelin
constant, is a number approximately equal to 1.282427129 that
sometimes appears in formulas related to gamma and zeta functions.
It is also related to the Barnes G-function (see :func:`~mpmath.barnesg`).
The constant is defined as `A = \exp(1/12-\zeta'(-1))` where
`\zeta'(s)` denotes the derivative of the Riemann zeta function
(see :func:`~mpmath.zeta`).
Mpmath can evaluate Glaisher's constant to arbitrary precision:
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +glaisher
1.282427129100622636875342568869791727767688927325
We can verify that the value computed by :data:`glaisher` is
correct using mpmath's facilities for numerical
differentiation and arbitrary evaluation of the zeta function:
>>> exp(mpf(1)/12 - diff(zeta, -1))
1.282427129100622636875342568869791727767688927325
Here is an example of an integral that can be evaluated in
terms of Glaisher's constant:
>>> mp.dps = 15
>>> quad(lambda x: log(gamma(x)), [1, 1.5])
-0.0428537406502909
>>> -0.5 - 7*log(2)/24 + log(pi)/4 + 3*log(glaisher)/2
-0.042853740650291
Mpmath computes Glaisher's constant by applying Euler-Maclaurin
summation to a slowly convergent series. The implementation is
reasonably efficient up to about 10,000 digits. See the source
code for additional details.
References:
http://mathworld.wolfram.com/Glaisher-KinkelinConstant.html
"""
apery = r"""
Represents Apery's constant, which is the irrational number
approximately equal to 1.2020569 given by
.. math ::
\zeta(3) = \sum_{k=1}^\infty\frac{1}{k^3}.
The calculation is based on an efficient hypergeometric
series. To 50 decimal places, the value is given by::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +apery
1.2020569031595942853997381615114499907649862923405
Other ways to evaluate Apery's constant using mpmath
include::
>>> zeta(3)
1.2020569031595942853997381615114499907649862923405
>>> -psi(2,1)/2
1.2020569031595942853997381615114499907649862923405
>>> 8*nsum(lambda k: 1/(2*k+1)**3, [0,inf])/7
1.2020569031595942853997381615114499907649862923405
>>> f = lambda k: 2/k**3/(exp(2*pi*k)-1)
>>> 7*pi**3/180 - nsum(f, [1,inf])
1.2020569031595942853997381615114499907649862923405
This shows digits 9991-10000 of Apery's constant::
>>> mp.dps = 10000
>>> str(apery)[-10:]
'3189504235'
"""
mertens = r"""
Represents the Mertens or Meissel-Mertens constant, which is the
prime number analog of Euler's constant:
.. math ::
B_1 = \lim_{N\to\infty}
\left(\sum_{p_k \le N} \frac{1}{p_k} - \log \log N \right)
Here `p_k` denotes the `k`-th prime number. Other names for this
constant include the Hadamard-de la Vallee-Poussin constant or
the prime reciprocal constant.
The following gives the Mertens constant to 50 digits::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +mertens
0.2614972128476427837554268386086958590515666482612
References:
http://mathworld.wolfram.com/MertensConstant.html
"""
twinprime = r"""
Represents the twin prime constant, which is the factor `C_2`
featuring in the Hardy-Littlewood conjecture for the growth of the
twin prime counting function,
.. math ::
\pi_2(n) \sim 2 C_2 \frac{n}{\log^2 n}.
It is given by the product over primes
.. math ::
C_2 = \prod_{p\ge3} \frac{p(p-2)}{(p-1)^2} \approx 0.66016
Computing `C_2` to 50 digits::
>>> from mpmath import *
>>> mp.dps = 50; mp.pretty = True
>>> +twinprime
0.66016181584686957392781211001455577843262336028473
References:
http://mathworld.wolfram.com/TwinPrimesConstant.html
"""
ln = r"""
Computes the natural logarithm of `x`, `\ln x`.
See :func:`~mpmath.log` for additional documentation."""
sqrt = r"""
``sqrt(x)`` gives the principal square root of `x`, `\sqrt x`.
For positive real numbers, the principal root is simply the
positive square root. For arbitrary complex numbers, the principal
square root is defined to satisfy `\sqrt x = \exp(\log(x)/2)`.
The function thus has a branch cut along the negative half real axis.
For all mpmath numbers ``x``, calling ``sqrt(x)`` is equivalent to
performing ``x**0.5``.
**Examples**
Basic examples and limits::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> sqrt(10)
3.16227766016838
>>> sqrt(100)
10.0
>>> sqrt(-4)
(0.0 + 2.0j)
>>> sqrt(1+1j)
(1.09868411346781 + 0.455089860562227j)
>>> sqrt(inf)
+inf
Square root evaluation is fast at huge precision::
>>> mp.dps = 50000
>>> a = sqrt(3)
>>> str(a)[-10:]
'9329332814'
:func:`mpmath.iv.sqrt` supports interval arguments::
>>> iv.dps = 15; iv.pretty = True
>>> iv.sqrt([16,100])
[4.0, 10.0]
>>> iv.sqrt(2)
[1.4142135623730949234, 1.4142135623730951455]
>>> iv.sqrt(2) ** 2
[1.9999999999999995559, 2.0000000000000004441]
"""
cbrt = r"""
``cbrt(x)`` computes the cube root of `x`, `x^{1/3}`. This
function is faster and more accurate than raising to a floating-point
fraction::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> 125**(mpf(1)/3)
mpf('4.9999999999999991')
>>> cbrt(125)
mpf('5.0')
Every nonzero complex number has three cube roots. This function
returns the cube root defined by `\exp(\log(x)/3)` where the
principal branch of the natural logarithm is used. Note that this
does not give a real cube root for negative real numbers::
>>> mp.pretty = True
>>> cbrt(-1)
(0.5 + 0.866025403784439j)
"""
exp = r"""
Computes the exponential function,
.. math ::
\exp(x) = e^x = \sum_{k=0}^{\infty} \frac{x^k}{k!}.
For complex numbers, the exponential function also satisfies
.. math ::
\exp(x+yi) = e^x (\cos y + i \sin y).
**Basic examples**
Some values of the exponential function::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> exp(0)
1.0
>>> exp(1)
2.718281828459045235360287
>>> exp(-1)
0.3678794411714423215955238
>>> exp(inf)
+inf
>>> exp(-inf)
0.0
Arguments can be arbitrarily large::
>>> exp(10000)
8.806818225662921587261496e+4342
>>> exp(-10000)
1.135483865314736098540939e-4343
Evaluation is supported for interval arguments via
:func:`mpmath.iv.exp`::
>>> iv.dps = 25; iv.pretty = True
>>> iv.exp([-inf,0])
[0.0, 1.0]
>>> iv.exp([0,1])
[1.0, 2.71828182845904523536028749558]
The exponential function can be evaluated efficiently to arbitrary
precision::
>>> mp.dps = 10000
>>> exp(pi) #doctest: +ELLIPSIS
23.140692632779269005729...8984304016040616
**Functional properties**
Numerical verification of Euler's identity for the complex
exponential function::
>>> mp.dps = 15
>>> exp(j*pi)+1
(0.0 + 1.22464679914735e-16j)
>>> chop(exp(j*pi)+1)
0.0
This recovers the coefficients (reciprocal factorials) in the
Maclaurin series expansion of exp::
>>> nprint(taylor(exp, 0, 5))
[1.0, 1.0, 0.5, 0.166667, 0.0416667, 0.00833333]
The exponential function is its own derivative and antiderivative::
>>> exp(pi)
23.1406926327793
>>> diff(exp, pi)
23.1406926327793
>>> quad(exp, [-inf, pi])
23.1406926327793
The exponential function can be evaluated using various methods,
including direct summation of the series, limits, and solving
the defining differential equation::
>>> nsum(lambda k: pi**k/fac(k), [0,inf])
23.1406926327793
>>> limit(lambda k: (1+pi/k)**k, inf)
23.1406926327793
>>> odefun(lambda t, x: x, 0, 1)(pi)
23.1406926327793
"""
cosh = r"""
Computes the hyperbolic cosine of `x`,
`\cosh(x) = (e^x + e^{-x})/2`. Values and limits include::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> cosh(0)
1.0
>>> cosh(1)
1.543080634815243778477906
>>> cosh(-inf), cosh(+inf)
(+inf, +inf)
The hyperbolic cosine is an even, convex function with
a global minimum at `x = 0`, having a Maclaurin series
that starts::
>>> nprint(chop(taylor(cosh, 0, 5)))
[1.0, 0.0, 0.5, 0.0, 0.0416667, 0.0]
Generalized to complex numbers, the hyperbolic cosine is
equivalent to a cosine with the argument rotated
in the imaginary direction, or `\cosh x = \cos ix`::
>>> cosh(2+3j)
(-3.724545504915322565473971 + 0.5118225699873846088344638j)
>>> cos(3-2j)
(-3.724545504915322565473971 + 0.5118225699873846088344638j)
"""
sinh = r"""
Computes the hyperbolic sine of `x`,
`\sinh(x) = (e^x - e^{-x})/2`. Values and limits include::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> sinh(0)
0.0
>>> sinh(1)
1.175201193643801456882382
>>> sinh(-inf), sinh(+inf)
(-inf, +inf)
The hyperbolic sine is an odd function, with a Maclaurin
series that starts::
>>> nprint(chop(taylor(sinh, 0, 5)))
[0.0, 1.0, 0.0, 0.166667, 0.0, 0.00833333]
Generalized to complex numbers, the hyperbolic sine is
essentially a sine with a rotation `i` applied to
the argument; more precisely, `\sinh x = -i \sin ix`::
>>> sinh(2+3j)
(-3.590564589985779952012565 + 0.5309210862485198052670401j)
>>> j*sin(3-2j)
(-3.590564589985779952012565 + 0.5309210862485198052670401j)
"""
tanh = r"""
Computes the hyperbolic tangent of `x`,
`\tanh(x) = \sinh(x)/\cosh(x)`. Values and limits include::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> tanh(0)
0.0
>>> tanh(1)
0.7615941559557648881194583
>>> tanh(-inf), tanh(inf)
(-1.0, 1.0)
The hyperbolic tangent is an odd, sigmoidal function, similar
to the inverse tangent and error function. Its Maclaurin
series is::
>>> nprint(chop(taylor(tanh, 0, 5)))
[0.0, 1.0, 0.0, -0.333333, 0.0, 0.133333]
Generalized to complex numbers, the hyperbolic tangent is
essentially a tangent with a rotation `i` applied to
the argument; more precisely, `\tanh x = -i \tan ix`::
>>> tanh(2+3j)
(0.9653858790221331242784803 - 0.009884375038322493720314034j)
>>> j*tan(3-2j)
(0.9653858790221331242784803 - 0.009884375038322493720314034j)
"""
cos = r"""
Computes the cosine of `x`, `\cos(x)`.
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> cos(pi/3)
0.5
>>> cos(100000001)
-0.9802850113244713353133243
>>> cos(2+3j)
(-4.189625690968807230132555 - 9.109227893755336597979197j)
>>> cos(inf)
nan
>>> nprint(chop(taylor(cos, 0, 6)))
[1.0, 0.0, -0.5, 0.0, 0.0416667, 0.0, -0.00138889]
Intervals are supported via :func:`mpmath.iv.cos`::
>>> iv.dps = 25; iv.pretty = True
>>> iv.cos([0,1])
[0.540302305868139717400936602301, 1.0]
>>> iv.cos([0,2])
[-0.41614683654714238699756823214, 1.0]
"""
sin = r"""
Computes the sine of `x`, `\sin(x)`.
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> sin(pi/3)
0.8660254037844386467637232
>>> sin(100000001)
0.1975887055794968911438743
>>> sin(2+3j)
(9.1544991469114295734673 - 4.168906959966564350754813j)
>>> sin(inf)
nan
>>> nprint(chop(taylor(sin, 0, 6)))
[0.0, 1.0, 0.0, -0.166667, 0.0, 0.00833333, 0.0]
Intervals are supported via :func:`mpmath.iv.sin`::
>>> iv.dps = 25; iv.pretty = True
>>> iv.sin([0,1])
[0.0, 0.841470984807896506652502331201]
>>> iv.sin([0,2])
[0.0, 1.0]
"""
tan = r"""
Computes the tangent of `x`, `\tan(x) = \frac{\sin(x)}{\cos(x)}`.
The tangent function is singular at `x = (n+1/2)\pi`, but
``tan(x)`` always returns a finite result since `(n+1/2)\pi`
cannot be represented exactly using floating-point arithmetic.
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> tan(pi/3)
1.732050807568877293527446
>>> tan(100000001)
-0.2015625081449864533091058
>>> tan(2+3j)
(-0.003764025641504248292751221 + 1.003238627353609801446359j)
>>> tan(inf)
nan
>>> nprint(chop(taylor(tan, 0, 6)))
[0.0, 1.0, 0.0, 0.333333, 0.0, 0.133333, 0.0]
Intervals are supported via :func:`mpmath.iv.tan`::
>>> iv.dps = 25; iv.pretty = True
>>> iv.tan([0,1])
[0.0, 1.55740772465490223050697482944]
>>> iv.tan([0,2]) # Interval includes a singularity
[-inf, +inf]
"""
sec = r"""
Computes the secant of `x`, `\mathrm{sec}(x) = \frac{1}{\cos(x)}`.
The secant function is singular at `x = (n+1/2)\pi`, but
``sec(x)`` always returns a finite result since `(n+1/2)\pi`
cannot be represented exactly using floating-point arithmetic.
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> sec(pi/3)
2.0
>>> sec(10000001)
-1.184723164360392819100265
>>> sec(2+3j)
(-0.04167496441114427004834991 + 0.0906111371962375965296612j)
>>> sec(inf)
nan
>>> nprint(chop(taylor(sec, 0, 6)))
[1.0, 0.0, 0.5, 0.0, 0.208333, 0.0, 0.0847222]
Intervals are supported via :func:`mpmath.iv.sec`::
>>> iv.dps = 25; iv.pretty = True
>>> iv.sec([0,1])
[1.0, 1.85081571768092561791175326276]
>>> iv.sec([0,2]) # Interval includes a singularity
[-inf, +inf]
"""
csc = r"""
Computes the cosecant of `x`, `\mathrm{csc}(x) = \frac{1}{\sin(x)}`.
This cosecant function is singular at `x = n \pi`, but with the
exception of the point `x = 0`, ``csc(x)`` returns a finite result
since `n \pi` cannot be represented exactly using floating-point
arithmetic.
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> csc(pi/3)
1.154700538379251529018298
>>> csc(10000001)
-1.864910497503629858938891
>>> csc(2+3j)
(0.09047320975320743980579048 + 0.04120098628857412646300981j)
>>> csc(inf)
nan
Intervals are supported via :func:`mpmath.iv.csc`::
>>> iv.dps = 25; iv.pretty = True
>>> iv.csc([0,1]) # Interval includes a singularity
[1.18839510577812121626159943988, +inf]
>>> iv.csc([0,2])
[1.0, +inf]
"""
cot = r"""
Computes the cotangent of `x`,
`\mathrm{cot}(x) = \frac{1}{\tan(x)} = \frac{\cos(x)}{\sin(x)}`.
This cotangent function is singular at `x = n \pi`, but with the
exception of the point `x = 0`, ``cot(x)`` returns a finite result
since `n \pi` cannot be represented exactly using floating-point
arithmetic.
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> cot(pi/3)
0.5773502691896257645091488
>>> cot(10000001)
1.574131876209625656003562
>>> cot(2+3j)
(-0.003739710376336956660117409 - 0.9967577965693583104609688j)
>>> cot(inf)
nan
Intervals are supported via :func:`mpmath.iv.cot`::
>>> iv.dps = 25; iv.pretty = True
>>> iv.cot([0,1]) # Interval includes a singularity
[0.642092615934330703006419974862, +inf]
>>> iv.cot([1,2])
[-inf, +inf]
"""
acos = r"""
Computes the inverse cosine or arccosine of `x`, `\cos^{-1}(x)`.
Since `-1 \le \cos(x) \le 1` for real `x`, the inverse
cosine is real-valued only for `-1 \le x \le 1`. On this interval,
:func:`~mpmath.acos` is defined to be a monotonically decreasing
function assuming values between `+\pi` and `0`.
Basic values are::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> acos(-1)
3.141592653589793238462643
>>> acos(0)
1.570796326794896619231322
>>> acos(1)
0.0
>>> nprint(chop(taylor(acos, 0, 6)))
[1.5708, -1.0, 0.0, -0.166667, 0.0, -0.075, 0.0]
:func:`~mpmath.acos` is defined so as to be a proper inverse function of
`\cos(\theta)` for `0 \le \theta < \pi`.
We have `\cos(\cos^{-1}(x)) = x` for all `x`, but
`\cos^{-1}(\cos(x)) = x` only for `0 \le \Re[x] < \pi`::
>>> for x in [1, 10, -1, 2+3j, 10+3j]:
... print("%s %s" % (cos(acos(x)), acos(cos(x))))
...
1.0 1.0
(10.0 + 0.0j) 2.566370614359172953850574
-1.0 1.0
(2.0 + 3.0j) (2.0 + 3.0j)
(10.0 + 3.0j) (2.566370614359172953850574 - 3.0j)
The inverse cosine has two branch points: `x = \pm 1`. :func:`~mpmath.acos`
places the branch cuts along the line segments `(-\infty, -1)` and
`(+1, +\infty)`. In general,
.. math ::
\cos^{-1}(x) = \frac{\pi}{2} + i \log\left(ix + \sqrt{1-x^2} \right)
where the principal-branch log and square root are implied.
"""
asin = r"""
Computes the inverse sine or arcsine of `x`, `\sin^{-1}(x)`.
Since `-1 \le \sin(x) \le 1` for real `x`, the inverse
sine is real-valued only for `-1 \le x \le 1`.
On this interval, it is defined to be a monotonically increasing
function assuming values between `-\pi/2` and `\pi/2`.
Basic values are::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> asin(-1)
-1.570796326794896619231322
>>> asin(0)
0.0
>>> asin(1)
1.570796326794896619231322
>>> nprint(chop(taylor(asin, 0, 6)))
[0.0, 1.0, 0.0, 0.166667, 0.0, 0.075, 0.0]
:func:`~mpmath.asin` is defined so as to be a proper inverse function of
`\sin(\theta)` for `-\pi/2 < \theta < \pi/2`.
We have `\sin(\sin^{-1}(x)) = x` for all `x`, but
`\sin^{-1}(\sin(x)) = x` only for `-\pi/2 < \Re[x] < \pi/2`::
>>> for x in [1, 10, -1, 1+3j, -2+3j]:
... print("%s %s" % (chop(sin(asin(x))), asin(sin(x))))
...
1.0 1.0
10.0 -0.5752220392306202846120698
-1.0 -1.0
(1.0 + 3.0j) (1.0 + 3.0j)
(-2.0 + 3.0j) (-1.141592653589793238462643 - 3.0j)
The inverse sine has two branch points: `x = \pm 1`. :func:`~mpmath.asin`
places the branch cuts along the line segments `(-\infty, -1)` and
`(+1, +\infty)`. In general,
.. math ::
\sin^{-1}(x) = -i \log\left(ix + \sqrt{1-x^2} \right)
where the principal-branch log and square root are implied.
"""
atan = r"""
Computes the inverse tangent or arctangent of `x`, `\tan^{-1}(x)`.
This is a real-valued function for all real `x`, with range
`(-\pi/2, \pi/2)`.
Basic values are::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> atan(-inf)
-1.570796326794896619231322
>>> atan(-1)
-0.7853981633974483096156609
>>> atan(0)
0.0
>>> atan(1)
0.7853981633974483096156609
>>> atan(inf)
1.570796326794896619231322
>>> nprint(chop(taylor(atan, 0, 6)))
[0.0, 1.0, 0.0, -0.333333, 0.0, 0.2, 0.0]
The inverse tangent is often used to compute angles. However,
the atan2 function is often better for this as it preserves sign
(see :func:`~mpmath.atan2`).
:func:`~mpmath.atan` is defined so as to be a proper inverse function of
`\tan(\theta)` for `-\pi/2 < \theta < \pi/2`.
We have `\tan(\tan^{-1}(x)) = x` for all `x`, but
`\tan^{-1}(\tan(x)) = x` only for `-\pi/2 < \Re[x] < \pi/2`::
>>> mp.dps = 25
>>> for x in [1, 10, -1, 1+3j, -2+3j]:
... print("%s %s" % (tan(atan(x)), atan(tan(x))))
...
1.0 1.0
10.0 0.5752220392306202846120698
-1.0 -1.0
(1.0 + 3.0j) (1.000000000000000000000001 + 3.0j)
(-2.0 + 3.0j) (1.141592653589793238462644 + 3.0j)
The inverse tangent has two branch points: `x = \pm i`. :func:`~mpmath.atan`
places the branch cuts along the line segments `(-i \infty, -i)` and
`(+i, +i \infty)`. In general,
.. math ::
\tan^{-1}(x) = \frac{i}{2}\left(\log(1-ix)-\log(1+ix)\right)
where the principal-branch log is implied.
"""
acot = r"""Computes the inverse cotangent of `x`,
`\mathrm{cot}^{-1}(x) = \tan^{-1}(1/x)`."""
asec = r"""Computes the inverse secant of `x`,
`\mathrm{sec}^{-1}(x) = \cos^{-1}(1/x)`."""
acsc = r"""Computes the inverse cosecant of `x`,
`\mathrm{csc}^{-1}(x) = \sin^{-1}(1/x)`."""
coth = r"""Computes the hyperbolic cotangent of `x`,
`\mathrm{coth}(x) = \frac{\cosh(x)}{\sinh(x)}`.
"""
sech = r"""Computes the hyperbolic secant of `x`,
`\mathrm{sech}(x) = \frac{1}{\cosh(x)}`.
"""
csch = r"""Computes the hyperbolic cosecant of `x`,
`\mathrm{csch}(x) = \frac{1}{\sinh(x)}`.
"""
acosh = r"""Computes the inverse hyperbolic cosine of `x`,
`\mathrm{cosh}^{-1}(x) = \log(x+\sqrt{x+1}\sqrt{x-1})`.
"""
asinh = r"""Computes the inverse hyperbolic sine of `x`,
`\mathrm{sinh}^{-1}(x) = \log(x+\sqrt{1+x^2})`.
"""
atanh = r"""Computes the inverse hyperbolic tangent of `x`,
`\mathrm{tanh}^{-1}(x) = \frac{1}{2}\left(\log(1+x)-\log(1-x)\right)`.
"""
acoth = r"""Computes the inverse hyperbolic cotangent of `x`,
`\mathrm{coth}^{-1}(x) = \tanh^{-1}(1/x)`."""
asech = r"""Computes the inverse hyperbolic secant of `x`,
`\mathrm{sech}^{-1}(x) = \cosh^{-1}(1/x)`."""
acsch = r"""Computes the inverse hyperbolic cosecant of `x`,
`\mathrm{csch}^{-1}(x) = \sinh^{-1}(1/x)`."""
sinpi = r"""
Computes `\sin(\pi x)`, more accurately than the expression
``sin(pi*x)``::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> sinpi(10**10), sin(pi*(10**10))
(0.0, -2.23936276195592e-6)
>>> sinpi(10**10+0.5), sin(pi*(10**10+0.5))
(1.0, 0.999999999998721)
"""
cospi = r"""
Computes `\cos(\pi x)`, more accurately than the expression
``cos(pi*x)``::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> cospi(10**10), cos(pi*(10**10))
(1.0, 0.999999999997493)
>>> cospi(10**10+0.5), cos(pi*(10**10+0.5))
(0.0, 1.59960492420134e-6)
"""
sinc = r"""
``sinc(x)`` computes the unnormalized sinc function, defined as
.. math ::
\mathrm{sinc}(x) = \begin{cases}
\sin(x)/x, & \mbox{if } x \ne 0 \\
1, & \mbox{if } x = 0.
\end{cases}
See :func:`~mpmath.sincpi` for the normalized sinc function.
Simple values and limits include::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> sinc(0)
1.0
>>> sinc(1)
0.841470984807897
>>> sinc(inf)
0.0
The integral of the sinc function is the sine integral Si::
>>> quad(sinc, [0, 1])
0.946083070367183
>>> si(1)
0.946083070367183
"""
sincpi = r"""
``sincpi(x)`` computes the normalized sinc function, defined as
.. math ::
\mathrm{sinc}_{\pi}(x) = \begin{cases}
\sin(\pi x)/(\pi x), & \mbox{if } x \ne 0 \\
1, & \mbox{if } x = 0.
\end{cases}
Equivalently, we have
`\mathrm{sinc}_{\pi}(x) = \mathrm{sinc}(\pi x)`.
The normalization entails that the function integrates
to unity over the entire real line::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> quadosc(sincpi, [-inf, inf], period=2.0)
1.0
Like, :func:`~mpmath.sinpi`, :func:`~mpmath.sincpi` is evaluated accurately
at its roots::
>>> sincpi(10)
0.0
"""
expj = r"""
Convenience function for computing `e^{ix}`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> expj(0)
(1.0 + 0.0j)
>>> expj(-1)
(0.5403023058681397174009366 - 0.8414709848078965066525023j)
>>> expj(j)
(0.3678794411714423215955238 + 0.0j)
>>> expj(1+j)
(0.1987661103464129406288032 + 0.3095598756531121984439128j)
"""
expjpi = r"""
Convenience function for computing `e^{i \pi x}`.
Evaluation is accurate near zeros (see also :func:`~mpmath.cospi`,
:func:`~mpmath.sinpi`)::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> expjpi(0)
(1.0 + 0.0j)
>>> expjpi(1)
(-1.0 + 0.0j)
>>> expjpi(0.5)
(0.0 + 1.0j)
>>> expjpi(-1)
(-1.0 + 0.0j)
>>> expjpi(j)
(0.04321391826377224977441774 + 0.0j)
>>> expjpi(1+j)
(-0.04321391826377224977441774 + 0.0j)
"""
floor = r"""
Computes the floor of `x`, `\lfloor x \rfloor`, defined as
the largest integer less than or equal to `x`::
>>> from mpmath import *
>>> mp.pretty = False
>>> floor(3.5)
mpf('3.0')
.. note ::
:func:`~mpmath.floor`, :func:`~mpmath.ceil` and :func:`~mpmath.nint` return a
floating-point number, not a Python ``int``. If `\lfloor x \rfloor` is
too large to be represented exactly at the present working precision,
the result will be rounded, not necessarily in the direction
implied by the mathematical definition of the function.
To avoid rounding, use *prec=0*::
>>> mp.dps = 15
>>> print(int(floor(10**30+1)))
1000000000000000019884624838656
>>> print(int(floor(10**30+1, prec=0)))
1000000000000000000000000000001
The floor function is defined for complex numbers and
acts on the real and imaginary parts separately::
>>> floor(3.25+4.75j)
mpc(real='3.0', imag='4.0')
"""
ceil = r"""
Computes the ceiling of `x`, `\lceil x \rceil`, defined as
the smallest integer greater than or equal to `x`::
>>> from mpmath import *
>>> mp.pretty = False
>>> ceil(3.5)
mpf('4.0')
The ceiling function is defined for complex numbers and
acts on the real and imaginary parts separately::
>>> ceil(3.25+4.75j)
mpc(real='4.0', imag='5.0')
See notes about rounding for :func:`~mpmath.floor`.
"""
nint = r"""
Evaluates the nearest integer function, `\mathrm{nint}(x)`.
This gives the nearest integer to `x`; on a tie, it
gives the nearest even integer::
>>> from mpmath import *
>>> mp.pretty = False
>>> nint(3.2)
mpf('3.0')
>>> nint(3.8)
mpf('4.0')
>>> nint(3.5)
mpf('4.0')
>>> nint(4.5)
mpf('4.0')
The nearest integer function is defined for complex numbers and
acts on the real and imaginary parts separately::
>>> nint(3.25+4.75j)
mpc(real='3.0', imag='5.0')
See notes about rounding for :func:`~mpmath.floor`.
"""
frac = r"""
Gives the fractional part of `x`, defined as
`\mathrm{frac}(x) = x - \lfloor x \rfloor` (see :func:`~mpmath.floor`).
In effect, this computes `x` modulo 1, or `x+n` where
`n \in \mathbb{Z}` is such that `x+n \in [0,1)`::
>>> from mpmath import *
>>> mp.pretty = False
>>> frac(1.25)
mpf('0.25')
>>> frac(3)
mpf('0.0')
>>> frac(-1.25)
mpf('0.75')
For a complex number, the fractional part function applies to
the real and imaginary parts separately::
>>> frac(2.25+3.75j)
mpc(real='0.25', imag='0.75')
Plotted, the fractional part function gives a sawtooth
wave. The Fourier series coefficients have a simple
form::
>>> mp.dps = 15
>>> nprint(fourier(lambda x: frac(x)-0.5, [0,1], 4))
([0.0, 0.0, 0.0, 0.0, 0.0], [0.0, -0.31831, -0.159155, -0.106103, -0.0795775])
>>> nprint([-1/(pi*k) for k in range(1,5)])
[-0.31831, -0.159155, -0.106103, -0.0795775]
.. note::
The fractional part is sometimes defined as a symmetric
function, i.e. returning `-\mathrm{frac}(-x)` if `x < 0`.
This convention is used, for instance, by Mathematica's
``FractionalPart``.
"""
sign = r"""
Returns the sign of `x`, defined as `\mathrm{sign}(x) = x / |x|`
(with the special case `\mathrm{sign}(0) = 0`)::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> sign(10)
mpf('1.0')
>>> sign(-10)
mpf('-1.0')
>>> sign(0)
mpf('0.0')
Note that the sign function is also defined for complex numbers,
for which it gives the projection onto the unit circle::
>>> mp.dps = 15; mp.pretty = True
>>> sign(1+j)
(0.707106781186547 + 0.707106781186547j)
"""
arg = r"""
Computes the complex argument (phase) of `x`, defined as the
signed angle between the positive real axis and `x` in the
complex plane::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> arg(3)
0.0
>>> arg(3+3j)
0.785398163397448
>>> arg(3j)
1.5707963267949
>>> arg(-3)
3.14159265358979
>>> arg(-3j)
-1.5707963267949
The angle is defined to satisfy `-\pi < \arg(x) \le \pi` and
with the sign convention that a nonnegative imaginary part
results in a nonnegative argument.
The value returned by :func:`~mpmath.arg` is an ``mpf`` instance.
"""
fabs = r"""
Returns the absolute value of `x`, `|x|`. Unlike :func:`abs`,
:func:`~mpmath.fabs` converts non-mpmath numbers (such as ``int``)
into mpmath numbers::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> fabs(3)
mpf('3.0')
>>> fabs(-3)
mpf('3.0')
>>> fabs(3+4j)
mpf('5.0')
"""
re = r"""
Returns the real part of `x`, `\Re(x)`. Unlike ``x.real``,
:func:`~mpmath.re` converts `x` to a mpmath number::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> re(3)
mpf('3.0')
>>> re(-1+4j)
mpf('-1.0')
"""
im = r"""
Returns the imaginary part of `x`, `\Im(x)`. Unlike ``x.imag``,
:func:`~mpmath.im` converts `x` to a mpmath number::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> im(3)
mpf('0.0')
>>> im(-1+4j)
mpf('4.0')
"""
conj = r"""
Returns the complex conjugate of `x`, `\overline{x}`. Unlike
``x.conjugate()``, :func:`~mpmath.im` converts `x` to a mpmath number::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> conj(3)
mpf('3.0')
>>> conj(-1+4j)
mpc(real='-1.0', imag='-4.0')
"""
polar = r"""
Returns the polar representation of the complex number `z`
as a pair `(r, \phi)` such that `z = r e^{i \phi}`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> polar(-2)
(2.0, 3.14159265358979)
>>> polar(3-4j)
(5.0, -0.927295218001612)
"""
rect = r"""
Returns the complex number represented by polar
coordinates `(r, \phi)`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> chop(rect(2, pi))
-2.0
>>> rect(sqrt(2), -pi/4)
(1.0 - 1.0j)
"""
expm1 = r"""
Computes `e^x - 1`, accurately for small `x`.
Unlike the expression ``exp(x) - 1``, ``expm1(x)`` does not suffer from
potentially catastrophic cancellation::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> exp(1e-10)-1; print(expm1(1e-10))
1.00000008274037e-10
1.00000000005e-10
>>> exp(1e-20)-1; print(expm1(1e-20))
0.0
1.0e-20
>>> 1/(exp(1e-20)-1)
Traceback (most recent call last):
...
ZeroDivisionError
>>> 1/expm1(1e-20)
1.0e+20
Evaluation works for extremely tiny values::
>>> expm1(0)
0.0
>>> expm1('1e-10000000')
1.0e-10000000
"""
powm1 = r"""
Computes `x^y - 1`, accurately when `x^y` is very close to 1.
This avoids potentially catastrophic cancellation::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> power(0.99999995, 1e-10) - 1
0.0
>>> powm1(0.99999995, 1e-10)
-5.00000012791934e-18
Powers exactly equal to 1, and only those powers, yield 0 exactly::
>>> powm1(-j, 4)
(0.0 + 0.0j)
>>> powm1(3, 0)
0.0
>>> powm1(fadd(-1, 1e-100, exact=True), 4)
-4.0e-100
Evaluation works for extremely tiny `y`::
>>> powm1(2, '1e-100000')
6.93147180559945e-100001
>>> powm1(j, '1e-1000')
(-1.23370055013617e-2000 + 1.5707963267949e-1000j)
"""
root = r"""
``root(z, n, k=0)`` computes an `n`-th root of `z`, i.e. returns a number
`r` that (up to possible approximation error) satisfies `r^n = z`.
(``nthroot`` is available as an alias for ``root``.)
Every complex number `z \ne 0` has `n` distinct `n`-th roots, which are
equidistant points on a circle with radius `|z|^{1/n}`, centered around the
origin. A specific root may be selected using the optional index
`k`. The roots are indexed counterclockwise, starting with `k = 0` for the root
closest to the positive real half-axis.
The `k = 0` root is the so-called principal `n`-th root, often denoted by
`\sqrt[n]{z}` or `z^{1/n}`, and also given by `\exp(\log(z) / n)`. If `z` is
a positive real number, the principal root is just the unique positive
`n`-th root of `z`. Under some circumstances, non-principal real roots exist:
for positive real `z`, `n` even, there is a negative root given by `k = n/2`;
for negative real `z`, `n` odd, there is a negative root given by `k = (n-1)/2`.
To obtain all roots with a simple expression, use
``[root(z,n,k) for k in range(n)]``.
An important special case, ``root(1, n, k)`` returns the `k`-th `n`-th root of
unity, `\zeta_k = e^{2 \pi i k / n}`. Alternatively, :func:`~mpmath.unitroots`
provides a slightly more convenient way to obtain the roots of unity,
including the option to compute only the primitive roots of unity.
Both `k` and `n` should be integers; `k` outside of ``range(n)`` will be
reduced modulo `n`. If `n` is negative, `x^{-1/n} = 1/x^{1/n}` (or
the equivalent reciprocal for a non-principal root with `k \ne 0`) is computed.
:func:`~mpmath.root` is implemented to use Newton's method for small
`n`. At high precision, this makes `x^{1/n}` not much more
expensive than the regular exponentiation, `x^n`. For very large
`n`, :func:`~mpmath.nthroot` falls back to use the exponential function.
**Examples**
:func:`~mpmath.nthroot`/:func:`~mpmath.root` is faster and more accurate than raising to a
floating-point fraction::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = False
>>> 16807 ** (mpf(1)/5)
mpf('7.0000000000000009')
>>> root(16807, 5)
mpf('7.0')
>>> nthroot(16807, 5) # Alias
mpf('7.0')
A high-precision root::
>>> mp.dps = 50; mp.pretty = True
>>> nthroot(10, 5)
1.584893192461113485202101373391507013269442133825
>>> nthroot(10, 5) ** 5
10.0
Computing principal and non-principal square and cube roots::
>>> mp.dps = 15
>>> root(10, 2)
3.16227766016838
>>> root(10, 2, 1)
-3.16227766016838
>>> root(-10, 3)
(1.07721734501594 + 1.86579517236206j)
>>> root(-10, 3, 1)
-2.15443469003188
>>> root(-10, 3, 2)
(1.07721734501594 - 1.86579517236206j)
All the 7th roots of a complex number::
>>> for r in [root(3+4j, 7, k) for k in range(7)]:
... print("%s %s" % (r, r**7))
...
(1.24747270589553 + 0.166227124177353j) (3.0 + 4.0j)
(0.647824911301003 + 1.07895435170559j) (3.0 + 4.0j)
(-0.439648254723098 + 1.17920694574172j) (3.0 + 4.0j)
(-1.19605731775069 + 0.391492658196305j) (3.0 + 4.0j)
(-1.05181082538903 - 0.691023585965793j) (3.0 + 4.0j)
(-0.115529328478668 - 1.25318497558335j) (3.0 + 4.0j)
(0.907748109144957 - 0.871672518271819j) (3.0 + 4.0j)
Cube roots of unity::
>>> for k in range(3): print(root(1, 3, k))
...
1.0
(-0.5 + 0.866025403784439j)
(-0.5 - 0.866025403784439j)
Some exact high order roots::
>>> root(75**210, 105)
5625.0
>>> root(1, 128, 96)
(0.0 - 1.0j)
>>> root(4**128, 128, 96)
(0.0 - 4.0j)
"""
unitroots = r"""
``unitroots(n)`` returns `\zeta_0, \zeta_1, \ldots, \zeta_{n-1}`,
all the distinct `n`-th roots of unity, as a list. If the option
*primitive=True* is passed, only the primitive roots are returned.
Every `n`-th root of unity satisfies `(\zeta_k)^n = 1`. There are `n` distinct
roots for each `n` (`\zeta_k` and `\zeta_j` are the same when
`k = j \pmod n`), which form a regular polygon with vertices on the unit
circle. They are ordered counterclockwise with increasing `k`, starting
with `\zeta_0 = 1`.
**Examples**
The roots of unity up to `n = 4`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> nprint(unitroots(1))
[1.0]
>>> nprint(unitroots(2))
[1.0, -1.0]
>>> nprint(unitroots(3))
[1.0, (-0.5 + 0.866025j), (-0.5 - 0.866025j)]
>>> nprint(unitroots(4))
[1.0, (0.0 + 1.0j), -1.0, (0.0 - 1.0j)]
Roots of unity form a geometric series that sums to 0::
>>> mp.dps = 50
>>> chop(fsum(unitroots(25)))
0.0
Primitive roots up to `n = 4`::
>>> mp.dps = 15
>>> nprint(unitroots(1, primitive=True))
[1.0]
>>> nprint(unitroots(2, primitive=True))
[-1.0]
>>> nprint(unitroots(3, primitive=True))
[(-0.5 + 0.866025j), (-0.5 - 0.866025j)]
>>> nprint(unitroots(4, primitive=True))
[(0.0 + 1.0j), (0.0 - 1.0j)]
There are only four primitive 12th roots::
>>> nprint(unitroots(12, primitive=True))
[(0.866025 + 0.5j), (-0.866025 + 0.5j), (-0.866025 - 0.5j), (0.866025 - 0.5j)]
The `n`-th roots of unity form a group, the cyclic group of order `n`.
Any primitive root `r` is a generator for this group, meaning that
`r^0, r^1, \ldots, r^{n-1}` gives the whole set of unit roots (in
some permuted order)::
>>> for r in unitroots(6): print(r)
...
1.0
(0.5 + 0.866025403784439j)
(-0.5 + 0.866025403784439j)
-1.0
(-0.5 - 0.866025403784439j)
(0.5 - 0.866025403784439j)
>>> r = unitroots(6, primitive=True)[1]
>>> for k in range(6): print(chop(r**k))
...
1.0
(0.5 - 0.866025403784439j)
(-0.5 - 0.866025403784439j)
-1.0
(-0.5 + 0.866025403784438j)
(0.5 + 0.866025403784438j)
The number of primitive roots equals the Euler totient function `\phi(n)`::
>>> [len(unitroots(n, primitive=True)) for n in range(1,20)]
[1, 1, 2, 2, 4, 2, 6, 4, 6, 4, 10, 4, 12, 6, 8, 8, 16, 6, 18]
"""
log = r"""
Computes the base-`b` logarithm of `x`, `\log_b(x)`. If `b` is
unspecified, :func:`~mpmath.log` computes the natural (base `e`) logarithm
and is equivalent to :func:`~mpmath.ln`. In general, the base `b` logarithm
is defined in terms of the natural logarithm as
`\log_b(x) = \ln(x)/\ln(b)`.
By convention, we take `\log(0) = -\infty`.
The natural logarithm is real if `x > 0` and complex if `x < 0` or if
`x` is complex. The principal branch of the complex logarithm is
used, meaning that `\Im(\ln(x)) = -\pi < \arg(x) \le \pi`.
**Examples**
Some basic values and limits::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> log(1)
0.0
>>> log(2)
0.693147180559945
>>> log(1000,10)
3.0
>>> log(4, 16)
0.5
>>> log(j)
(0.0 + 1.5707963267949j)
>>> log(-1)
(0.0 + 3.14159265358979j)
>>> log(0)
-inf
>>> log(inf)
+inf
The natural logarithm is the antiderivative of `1/x`::
>>> quad(lambda x: 1/x, [1, 5])
1.6094379124341
>>> log(5)
1.6094379124341
>>> diff(log, 10)
0.1
The Taylor series expansion of the natural logarithm around
`x = 1` has coefficients `(-1)^{n+1}/n`::
>>> nprint(taylor(log, 1, 7))
[0.0, 1.0, -0.5, 0.333333, -0.25, 0.2, -0.166667, 0.142857]
:func:`~mpmath.log` supports arbitrary precision evaluation::
>>> mp.dps = 50
>>> log(pi)
1.1447298858494001741434273513530587116472948129153
>>> log(pi, pi**3)
0.33333333333333333333333333333333333333333333333333
>>> mp.dps = 25
>>> log(3+4j)
(1.609437912434100374600759 + 0.9272952180016122324285125j)
"""
log10 = r"""
Computes the base-10 logarithm of `x`, `\log_{10}(x)`. ``log10(x)``
is equivalent to ``log(x, 10)``.
"""
fmod = r"""
Converts `x` and `y` to mpmath numbers and returns `x \mod y`.
For mpmath numbers, this is equivalent to ``x % y``.
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> fmod(100, pi)
2.61062773871641
You can use :func:`~mpmath.fmod` to compute fractional parts of numbers::
>>> fmod(10.25, 1)
0.25
"""
radians = r"""
Converts the degree angle `x` to radians::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> radians(60)
1.0471975511966
"""
degrees = r"""
Converts the radian angle `x` to a degree angle::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> degrees(pi/3)
60.0
"""
atan2 = r"""
Computes the two-argument arctangent, `\mathrm{atan2}(y, x)`,
giving the signed angle between the positive `x`-axis and the
point `(x, y)` in the 2D plane. This function is defined for
real `x` and `y` only.
The two-argument arctangent essentially computes
`\mathrm{atan}(y/x)`, but accounts for the signs of both
`x` and `y` to give the angle for the correct quadrant. The
following examples illustrate the difference::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> atan2(1,1), atan(1/1.)
(0.785398163397448, 0.785398163397448)
>>> atan2(1,-1), atan(1/-1.)
(2.35619449019234, -0.785398163397448)
>>> atan2(-1,1), atan(-1/1.)
(-0.785398163397448, -0.785398163397448)
>>> atan2(-1,-1), atan(-1/-1.)
(-2.35619449019234, 0.785398163397448)
The angle convention is the same as that used for the complex
argument; see :func:`~mpmath.arg`.
"""
fibonacci = r"""
``fibonacci(n)`` computes the `n`-th Fibonacci number, `F(n)`. The
Fibonacci numbers are defined by the recurrence `F(n) = F(n-1) + F(n-2)`
with the initial values `F(0) = 0`, `F(1) = 1`. :func:`~mpmath.fibonacci`
extends this definition to arbitrary real and complex arguments
using the formula
.. math ::
F(z) = \frac{\phi^z - \cos(\pi z) \phi^{-z}}{\sqrt 5}
where `\phi` is the golden ratio. :func:`~mpmath.fibonacci` also uses this
continuous formula to compute `F(n)` for extremely large `n`, where
calculating the exact integer would be wasteful.
For convenience, :func:`~mpmath.fib` is available as an alias for
:func:`~mpmath.fibonacci`.
**Basic examples**
Some small Fibonacci numbers are::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for i in range(10):
... print(fibonacci(i))
...
0.0
1.0
1.0
2.0
3.0
5.0
8.0
13.0
21.0
34.0
>>> fibonacci(50)
12586269025.0
The recurrence for `F(n)` extends backwards to negative `n`::
>>> for i in range(10):
... print(fibonacci(-i))
...
0.0
1.0
-1.0
2.0
-3.0
5.0
-8.0
13.0
-21.0
34.0
Large Fibonacci numbers will be computed approximately unless
the precision is set high enough::
>>> fib(200)
2.8057117299251e+41
>>> mp.dps = 45
>>> fib(200)
280571172992510140037611932413038677189525.0
:func:`~mpmath.fibonacci` can compute approximate Fibonacci numbers
of stupendous size::
>>> mp.dps = 15
>>> fibonacci(10**25)
3.49052338550226e+2089876402499787337692720
**Real and complex arguments**
The extended Fibonacci function is an analytic function. The
property `F(z) = F(z-1) + F(z-2)` holds for arbitrary `z`::
>>> mp.dps = 15
>>> fib(pi)
2.1170270579161
>>> fib(pi-1) + fib(pi-2)
2.1170270579161
>>> fib(3+4j)
(-5248.51130728372 - 14195.962288353j)
>>> fib(2+4j) + fib(1+4j)
(-5248.51130728372 - 14195.962288353j)
The Fibonacci function has infinitely many roots on the
negative half-real axis. The first root is at 0, the second is
close to -0.18, and then there are infinitely many roots that
asymptotically approach `-n+1/2`::
>>> findroot(fib, -0.2)
-0.183802359692956
>>> findroot(fib, -2)
-1.57077646820395
>>> findroot(fib, -17)
-16.4999999596115
>>> findroot(fib, -24)
-23.5000000000479
**Mathematical relationships**
For large `n`, `F(n+1)/F(n)` approaches the golden ratio::
>>> mp.dps = 50
>>> fibonacci(101)/fibonacci(100)
1.6180339887498948482045868343656381177203127439638
>>> +phi
1.6180339887498948482045868343656381177203091798058
The sum of reciprocal Fibonacci numbers converges to an irrational
number for which no closed form expression is known::
>>> mp.dps = 15
>>> nsum(lambda n: 1/fib(n), [1, inf])
3.35988566624318
Amazingly, however, the sum of odd-index reciprocal Fibonacci
numbers can be expressed in terms of a Jacobi theta function::
>>> nsum(lambda n: 1/fib(2*n+1), [0, inf])
1.82451515740692
>>> sqrt(5)*jtheta(2,0,(3-sqrt(5))/2)**2/4
1.82451515740692
Some related sums can be done in closed form::
>>> nsum(lambda k: 1/(1+fib(2*k+1)), [0, inf])
1.11803398874989
>>> phi - 0.5
1.11803398874989
>>> f = lambda k:(-1)**(k+1) / sum(fib(n)**2 for n in range(1,int(k+1)))
>>> nsum(f, [1, inf])
0.618033988749895
>>> phi-1
0.618033988749895
**References**
1. http://mathworld.wolfram.com/FibonacciNumber.html
"""
altzeta = r"""
Gives the Dirichlet eta function, `\eta(s)`, also known as the
alternating zeta function. This function is defined in analogy
with the Riemann zeta function as providing the sum of the
alternating series
.. math ::
\eta(s) = \sum_{k=0}^{\infty} \frac{(-1)^k}{k^s}
= 1-\frac{1}{2^s}+\frac{1}{3^s}-\frac{1}{4^s}+\ldots
The eta function, unlike the Riemann zeta function, is an entire
function, having a finite value for all complex `s`. The special case
`\eta(1) = \log(2)` gives the value of the alternating harmonic series.
The alternating zeta function may expressed using the Riemann zeta function
as `\eta(s) = (1 - 2^{1-s}) \zeta(s)`. It can also be expressed
in terms of the Hurwitz zeta function, for example using
:func:`~mpmath.dirichlet` (see documentation for that function).
**Examples**
Some special values are::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> altzeta(1)
0.693147180559945
>>> altzeta(0)
0.5
>>> altzeta(-1)
0.25
>>> altzeta(-2)
0.0
An example of a sum that can be computed more accurately and
efficiently via :func:`~mpmath.altzeta` than via numerical summation::
>>> sum(-(-1)**n / n**2.5 for n in range(1, 100))
0.86720495150398402
>>> altzeta(2.5)
0.867199889012184
At positive even integers, the Dirichlet eta function
evaluates to a rational multiple of a power of `\pi`::
>>> altzeta(2)
0.822467033424113
>>> pi**2/12
0.822467033424113
Like the Riemann zeta function, `\eta(s)`, approaches 1
as `s` approaches positive infinity, although it does
so from below rather than from above::
>>> altzeta(30)
0.999999999068682
>>> altzeta(inf)
1.0
>>> mp.pretty = False
>>> altzeta(1000, rounding='d')
mpf('0.99999999999999989')
>>> altzeta(1000, rounding='u')
mpf('1.0')
**References**
1. http://mathworld.wolfram.com/DirichletEtaFunction.html
2. http://en.wikipedia.org/wiki/Dirichlet_eta_function
"""
factorial = r"""
Computes the factorial, `x!`. For integers `n \ge 0`, we have
`n! = 1 \cdot 2 \cdots (n-1) \cdot n` and more generally the factorial
is defined for real or complex `x` by `x! = \Gamma(x+1)`.
**Examples**
Basic values and limits::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for k in range(6):
... print("%s %s" % (k, fac(k)))
...
0 1.0
1 1.0
2 2.0
3 6.0
4 24.0
5 120.0
>>> fac(inf)
+inf
>>> fac(0.5), sqrt(pi)/2
(0.886226925452758, 0.886226925452758)
For large positive `x`, `x!` can be approximated by
Stirling's formula::
>>> x = 10**10
>>> fac(x)
2.32579620567308e+95657055186
>>> sqrt(2*pi*x)*(x/e)**x
2.32579597597705e+95657055186
:func:`~mpmath.fac` supports evaluation for astronomically large values::
>>> fac(10**30)
6.22311232304258e+29565705518096748172348871081098
Reciprocal factorials appear in the Taylor series of the
exponential function (among many other contexts)::
>>> nsum(lambda k: 1/fac(k), [0, inf]), exp(1)
(2.71828182845905, 2.71828182845905)
>>> nsum(lambda k: pi**k/fac(k), [0, inf]), exp(pi)
(23.1406926327793, 23.1406926327793)
"""
gamma = r"""
Computes the gamma function, `\Gamma(x)`. The gamma function is a
shifted version of the ordinary factorial, satisfying
`\Gamma(n) = (n-1)!` for integers `n > 0`. More generally, it
is defined by
.. math ::
\Gamma(x) = \int_0^{\infty} t^{x-1} e^{-t}\, dt
for any real or complex `x` with `\Re(x) > 0` and for `\Re(x) < 0`
by analytic continuation.
**Examples**
Basic values and limits::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for k in range(1, 6):
... print("%s %s" % (k, gamma(k)))
...
1 1.0
2 1.0
3 2.0
4 6.0
5 24.0
>>> gamma(inf)
+inf
>>> gamma(0)
Traceback (most recent call last):
...
ValueError: gamma function pole
The gamma function of a half-integer is a rational multiple of
`\sqrt{\pi}`::
>>> gamma(0.5), sqrt(pi)
(1.77245385090552, 1.77245385090552)
>>> gamma(1.5), sqrt(pi)/2
(0.886226925452758, 0.886226925452758)
We can check the integral definition::
>>> gamma(3.5)
3.32335097044784
>>> quad(lambda t: t**2.5*exp(-t), [0,inf])
3.32335097044784
:func:`~mpmath.gamma` supports arbitrary-precision evaluation and
complex arguments::
>>> mp.dps = 50
>>> gamma(sqrt(3))
0.91510229697308632046045539308226554038315280564184
>>> mp.dps = 25
>>> gamma(2j)
(0.009902440080927490985955066 - 0.07595200133501806872408048j)
Arguments can also be large. Note that the gamma function grows
very quickly::
>>> mp.dps = 15
>>> gamma(10**20)
1.9328495143101e+1956570551809674817225
"""
psi = r"""
Gives the polygamma function of order `m` of `z`, `\psi^{(m)}(z)`.
Special cases are known as the *digamma function* (`\psi^{(0)}(z)`),
the *trigamma function* (`\psi^{(1)}(z)`), etc. The polygamma
functions are defined as the logarithmic derivatives of the gamma
function:
.. math ::
\psi^{(m)}(z) = \left(\frac{d}{dz}\right)^{m+1} \log \Gamma(z)
In particular, `\psi^{(0)}(z) = \Gamma'(z)/\Gamma(z)`. In the
present implementation of :func:`~mpmath.psi`, the order `m` must be a
nonnegative integer, while the argument `z` may be an arbitrary
complex number (with exception for the polygamma function's poles
at `z = 0, -1, -2, \ldots`).
**Examples**
For various rational arguments, the polygamma function reduces to
a combination of standard mathematical constants::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> psi(0, 1), -euler
(-0.5772156649015328606065121, -0.5772156649015328606065121)
>>> psi(1, '1/4'), pi**2+8*catalan
(17.19732915450711073927132, 17.19732915450711073927132)
>>> psi(2, '1/2'), -14*apery
(-16.82879664423431999559633, -16.82879664423431999559633)
The polygamma functions are derivatives of each other::
>>> diff(lambda x: psi(3, x), pi), psi(4, pi)
(-0.1105749312578862734526952, -0.1105749312578862734526952)
>>> quad(lambda x: psi(4, x), [2, 3]), psi(3,3)-psi(3,2)
(-0.375, -0.375)
The digamma function diverges logarithmically as `z \to \infty`,
while higher orders tend to zero::
>>> psi(0,inf), psi(1,inf), psi(2,inf)
(+inf, 0.0, 0.0)
Evaluation for a complex argument::
>>> psi(2, -1-2j)
(0.03902435405364952654838445 + 0.1574325240413029954685366j)
Evaluation is supported for large orders `m` and/or large
arguments `z`::
>>> psi(3, 10**100)
2.0e-300
>>> psi(250, 10**30+10**20*j)
(-1.293142504363642687204865e-7010 + 3.232856260909107391513108e-7018j)
**Application to infinite series**
Any infinite series where the summand is a rational function of
the index `k` can be evaluated in closed form in terms of polygamma
functions of the roots and poles of the summand::
>>> a = sqrt(2)
>>> b = sqrt(3)
>>> nsum(lambda k: 1/((k+a)**2*(k+b)), [0, inf])
0.4049668927517857061917531
>>> (psi(0,a)-psi(0,b)-a*psi(1,a)+b*psi(1,a))/(a-b)**2
0.4049668927517857061917531
This follows from the series representation (`m > 0`)
.. math ::
\psi^{(m)}(z) = (-1)^{m+1} m! \sum_{k=0}^{\infty}
\frac{1}{(z+k)^{m+1}}.
Since the roots of a polynomial may be complex, it is sometimes
necessary to use the complex polygamma function to evaluate
an entirely real-valued sum::
>>> nsum(lambda k: 1/(k**2-2*k+3), [0, inf])
1.694361433907061256154665
>>> nprint(polyroots([1,-2,3]))
[(1.0 - 1.41421j), (1.0 + 1.41421j)]
>>> r1 = 1-sqrt(2)*j
>>> r2 = r1.conjugate()
>>> (psi(0,-r2)-psi(0,-r1))/(r1-r2)
(1.694361433907061256154665 + 0.0j)
"""
digamma = r"""
Shortcut for ``psi(0,z)``.
"""
harmonic = r"""
If `n` is an integer, ``harmonic(n)`` gives a floating-point
approximation of the `n`-th harmonic number `H(n)`, defined as
.. math ::
H(n) = 1 + \frac{1}{2} + \frac{1}{3} + \ldots + \frac{1}{n}
The first few harmonic numbers are::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(8):
... print("%s %s" % (n, harmonic(n)))
...
0 0.0
1 1.0
2 1.5
3 1.83333333333333
4 2.08333333333333
5 2.28333333333333
6 2.45
7 2.59285714285714
The infinite harmonic series `1 + 1/2 + 1/3 + \ldots` diverges::
>>> harmonic(inf)
+inf
:func:`~mpmath.harmonic` is evaluated using the digamma function rather
than by summing the harmonic series term by term. It can therefore
be computed quickly for arbitrarily large `n`, and even for
nonintegral arguments::
>>> harmonic(10**100)
230.835724964306
>>> harmonic(0.5)
0.613705638880109
>>> harmonic(3+4j)
(2.24757548223494 + 0.850502209186044j)
:func:`~mpmath.harmonic` supports arbitrary precision evaluation::
>>> mp.dps = 50
>>> harmonic(11)
3.0198773448773448773448773448773448773448773448773
>>> harmonic(pi)
1.8727388590273302654363491032336134987519132374152
The harmonic series diverges, but at a glacial pace. It is possible
to calculate the exact number of terms required before the sum
exceeds a given amount, say 100::
>>> mp.dps = 50
>>> v = 10**findroot(lambda x: harmonic(10**x) - 100, 10)
>>> v
15092688622113788323693563264538101449859496.864101
>>> v = int(ceil(v))
>>> print(v)
15092688622113788323693563264538101449859497
>>> harmonic(v-1)
99.999999999999999999999999999999999999999999942747
>>> harmonic(v)
100.000000000000000000000000000000000000000000009
"""
bernoulli = r"""
Computes the nth Bernoulli number, `B_n`, for any integer `n \ge 0`.
The Bernoulli numbers are rational numbers, but this function
returns a floating-point approximation. To obtain an exact
fraction, use :func:`~mpmath.bernfrac` instead.
**Examples**
Numerical values of the first few Bernoulli numbers::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(15):
... print("%s %s" % (n, bernoulli(n)))
...
0 1.0
1 -0.5
2 0.166666666666667
3 0.0
4 -0.0333333333333333
5 0.0
6 0.0238095238095238
7 0.0
8 -0.0333333333333333
9 0.0
10 0.0757575757575758
11 0.0
12 -0.253113553113553
13 0.0
14 1.16666666666667
Bernoulli numbers can be approximated with arbitrary precision::
>>> mp.dps = 50
>>> bernoulli(100)
-2.8382249570693706959264156336481764738284680928013e+78
Arbitrarily large `n` are supported::
>>> mp.dps = 15
>>> bernoulli(10**20 + 2)
3.09136296657021e+1876752564973863312327
The Bernoulli numbers are related to the Riemann zeta function
at integer arguments::
>>> -bernoulli(8) * (2*pi)**8 / (2*fac(8))
1.00407735619794
>>> zeta(8)
1.00407735619794
**Algorithm**
For small `n` (`n < 3000`) :func:`~mpmath.bernoulli` uses a recurrence
formula due to Ramanujan. All results in this range are cached,
so sequential computation of small Bernoulli numbers is
guaranteed to be fast.
For larger `n`, `B_n` is evaluated in terms of the Riemann zeta
function.
"""
stieltjes = r"""
For a nonnegative integer `n`, ``stieltjes(n)`` computes the
`n`-th Stieltjes constant `\gamma_n`, defined as the
`n`-th coefficient in the Laurent series expansion of the
Riemann zeta function around the pole at `s = 1`. That is,
we have:
.. math ::
\zeta(s) = \frac{1}{s-1} \sum_{n=0}^{\infty}
\frac{(-1)^n}{n!} \gamma_n (s-1)^n
More generally, ``stieltjes(n, a)`` gives the corresponding
coefficient `\gamma_n(a)` for the Hurwitz zeta function
`\zeta(s,a)` (with `\gamma_n = \gamma_n(1)`).
**Examples**
The zeroth Stieltjes constant is just Euler's constant `\gamma`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> stieltjes(0)
0.577215664901533
Some more values are::
>>> stieltjes(1)
-0.0728158454836767
>>> stieltjes(10)
0.000205332814909065
>>> stieltjes(30)
0.00355772885557316
>>> stieltjes(1000)
-1.57095384420474e+486
>>> stieltjes(2000)
2.680424678918e+1109
>>> stieltjes(1, 2.5)
-0.23747539175716
An alternative way to compute `\gamma_1`::
>>> diff(extradps(15)(lambda x: 1/(x-1) - zeta(x)), 1)
-0.0728158454836767
:func:`~mpmath.stieltjes` supports arbitrary precision evaluation::
>>> mp.dps = 50
>>> stieltjes(2)
-0.0096903631928723184845303860352125293590658061013408
**Algorithm**
:func:`~mpmath.stieltjes` numerically evaluates the integral in
the following representation due to Ainsworth, Howell and
Coffey [1], [2]:
.. math ::
\gamma_n(a) = \frac{\log^n a}{2a} - \frac{\log^{n+1}(a)}{n+1} +
\frac{2}{a} \Re \int_0^{\infty}
\frac{(x/a-i)\log^n(a-ix)}{(1+x^2/a^2)(e^{2\pi x}-1)} dx.
For some reference values with `a = 1`, see e.g. [4].
**References**
1. O. R. Ainsworth & L. W. Howell, "An integral representation of
the generalized Euler-Mascheroni constants", NASA Technical
Paper 2456 (1985),
http://ntrs.nasa.gov/archive/nasa/casi.ntrs.nasa.gov/19850014994_1985014994.pdf
2. M. W. Coffey, "The Stieltjes constants, their relation to the
`\eta_j` coefficients, and representation of the Hurwitz
zeta function", arXiv:0706.0343v1 http://arxiv.org/abs/0706.0343
3. http://mathworld.wolfram.com/StieltjesConstants.html
4. http://pi.lacim.uqam.ca/piDATA/stieltjesgamma.txt
"""
gammaprod = r"""
Given iterables `a` and `b`, ``gammaprod(a, b)`` computes the
product / quotient of gamma functions:
.. math ::
\frac{\Gamma(a_0) \Gamma(a_1) \cdots \Gamma(a_p)}
{\Gamma(b_0) \Gamma(b_1) \cdots \Gamma(b_q)}
Unlike direct calls to :func:`~mpmath.gamma`, :func:`~mpmath.gammaprod` considers
the entire product as a limit and evaluates this limit properly if
any of the numerator or denominator arguments are nonpositive
integers such that poles of the gamma function are encountered.
That is, :func:`~mpmath.gammaprod` evaluates
.. math ::
\lim_{\epsilon \to 0}
\frac{\Gamma(a_0+\epsilon) \Gamma(a_1+\epsilon) \cdots
\Gamma(a_p+\epsilon)}
{\Gamma(b_0+\epsilon) \Gamma(b_1+\epsilon) \cdots
\Gamma(b_q+\epsilon)}
In particular:
* If there are equally many poles in the numerator and the
denominator, the limit is a rational number times the remaining,
regular part of the product.
* If there are more poles in the numerator, :func:`~mpmath.gammaprod`
returns ``+inf``.
* If there are more poles in the denominator, :func:`~mpmath.gammaprod`
returns 0.
**Examples**
The reciprocal gamma function `1/\Gamma(x)` evaluated at `x = 0`::
>>> from mpmath import *
>>> mp.dps = 15
>>> gammaprod([], [0])
0.0
A limit::
>>> gammaprod([-4], [-3])
-0.25
>>> limit(lambda x: gamma(x-1)/gamma(x), -3, direction=1)
-0.25
>>> limit(lambda x: gamma(x-1)/gamma(x), -3, direction=-1)
-0.25
"""
beta = r"""
Computes the beta function,
`B(x,y) = \Gamma(x) \Gamma(y) / \Gamma(x+y)`.
The beta function is also commonly defined by the integral
representation
.. math ::
B(x,y) = \int_0^1 t^{x-1} (1-t)^{y-1} \, dt
**Examples**
For integer and half-integer arguments where all three gamma
functions are finite, the beta function becomes either rational
number or a rational multiple of `\pi`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> beta(5, 2)
0.0333333333333333
>>> beta(1.5, 2)
0.266666666666667
>>> 16*beta(2.5, 1.5)
3.14159265358979
Where appropriate, :func:`~mpmath.beta` evaluates limits. A pole
of the beta function is taken to result in ``+inf``::
>>> beta(-0.5, 0.5)
0.0
>>> beta(-3, 3)
-0.333333333333333
>>> beta(-2, 3)
+inf
>>> beta(inf, 1)
0.0
>>> beta(inf, 0)
nan
:func:`~mpmath.beta` supports complex numbers and arbitrary precision
evaluation::
>>> beta(1, 2+j)
(0.4 - 0.2j)
>>> mp.dps = 25
>>> beta(j,0.5)
(1.079424249270925780135675 - 1.410032405664160838288752j)
>>> mp.dps = 50
>>> beta(pi, e)
0.037890298781212201348153837138927165984170287886464
Various integrals can be computed by means of the
beta function::
>>> mp.dps = 15
>>> quad(lambda t: t**2.5*(1-t)**2, [0, 1])
0.0230880230880231
>>> beta(3.5, 3)
0.0230880230880231
>>> quad(lambda t: sin(t)**4 * sqrt(cos(t)), [0, pi/2])
0.319504062596158
>>> beta(2.5, 0.75)/2
0.319504062596158
"""
betainc = r"""
``betainc(a, b, x1=0, x2=1, regularized=False)`` gives the generalized
incomplete beta function,
.. math ::
I_{x_1}^{x_2}(a,b) = \int_{x_1}^{x_2} t^{a-1} (1-t)^{b-1} dt.
When `x_1 = 0, x_2 = 1`, this reduces to the ordinary (complete)
beta function `B(a,b)`; see :func:`~mpmath.beta`.
With the keyword argument ``regularized=True``, :func:`~mpmath.betainc`
computes the regularized incomplete beta function
`I_{x_1}^{x_2}(a,b) / B(a,b)`. This is the cumulative distribution of the
beta distribution with parameters `a`, `b`.
.. note :
Implementations of the incomplete beta function in some other
software uses a different argument order. For example, Mathematica uses the
reversed argument order ``Beta[x1,x2,a,b]``. For the equivalent of SciPy's
three-argument incomplete beta integral (implicitly with `x1 = 0`), use
``betainc(a,b,0,x2,regularized=True)``.
**Examples**
Verifying that :func:`~mpmath.betainc` computes the integral in the
definition::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> x,y,a,b = 3, 4, 0, 6
>>> betainc(x, y, a, b)
-4010.4
>>> quad(lambda t: t**(x-1) * (1-t)**(y-1), [a, b])
-4010.4
The arguments may be arbitrary complex numbers::
>>> betainc(0.75, 1-4j, 0, 2+3j)
(0.2241657956955709603655887 + 0.3619619242700451992411724j)
With regularization::
>>> betainc(1, 2, 0, 0.25, regularized=True)
0.4375
>>> betainc(pi, e, 0, 1, regularized=True) # Complete
1.0
The beta integral satisfies some simple argument transformation
symmetries::
>>> mp.dps = 15
>>> betainc(2,3,4,5), -betainc(2,3,5,4), betainc(3,2,1-5,1-4)
(56.0833333333333, 56.0833333333333, 56.0833333333333)
The beta integral can often be evaluated analytically. For integer and
rational arguments, the incomplete beta function typically reduces to a
simple algebraic-logarithmic expression::
>>> mp.dps = 25
>>> identify(chop(betainc(0, 0, 3, 4)))
'-(log((9/8)))'
>>> identify(betainc(2, 3, 4, 5))
'(673/12)'
>>> identify(betainc(1.5, 1, 1, 2))
'((-12+sqrt(1152))/18)'
"""
binomial = r"""
Computes the binomial coefficient
.. math ::
{n \choose k} = \frac{n!}{k!(n-k)!}.
The binomial coefficient gives the number of ways that `k` items
can be chosen from a set of `n` items. More generally, the binomial
coefficient is a well-defined function of arbitrary real or
complex `n` and `k`, via the gamma function.
**Examples**
Generate Pascal's triangle::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(5):
... nprint([binomial(n,k) for k in range(n+1)])
...
[1.0]
[1.0, 1.0]
[1.0, 2.0, 1.0]
[1.0, 3.0, 3.0, 1.0]
[1.0, 4.0, 6.0, 4.0, 1.0]
There is 1 way to select 0 items from the empty set, and 0 ways to
select 1 item from the empty set::
>>> binomial(0, 0)
1.0
>>> binomial(0, 1)
0.0
:func:`~mpmath.binomial` supports large arguments::
>>> binomial(10**20, 10**20-5)
8.33333333333333e+97
>>> binomial(10**20, 10**10)
2.60784095465201e+104342944813
Nonintegral binomial coefficients find use in series
expansions::
>>> nprint(taylor(lambda x: (1+x)**0.25, 0, 4))
[1.0, 0.25, -0.09375, 0.0546875, -0.0375977]
>>> nprint([binomial(0.25, k) for k in range(5)])
[1.0, 0.25, -0.09375, 0.0546875, -0.0375977]
An integral representation::
>>> n, k = 5, 3
>>> f = lambda t: exp(-j*k*t)*(1+exp(j*t))**n
>>> chop(quad(f, [-pi,pi])/(2*pi))
10.0
>>> binomial(n,k)
10.0
"""
rf = r"""
Computes the rising factorial or Pochhammer symbol,
.. math ::
x^{(n)} = x (x+1) \cdots (x+n-1) = \frac{\Gamma(x+n)}{\Gamma(x)}
where the rightmost expression is valid for nonintegral `n`.
**Examples**
For integral `n`, the rising factorial is a polynomial::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(5):
... nprint(taylor(lambda x: rf(x,n), 0, n))
...
[1.0]
[0.0, 1.0]
[0.0, 1.0, 1.0]
[0.0, 2.0, 3.0, 1.0]
[0.0, 6.0, 11.0, 6.0, 1.0]
Evaluation is supported for arbitrary arguments::
>>> rf(2+3j, 5.5)
(-7202.03920483347 - 3777.58810701527j)
"""
ff = r"""
Computes the falling factorial,
.. math ::
(x)_n = x (x-1) \cdots (x-n+1) = \frac{\Gamma(x+1)}{\Gamma(x-n+1)}
where the rightmost expression is valid for nonintegral `n`.
**Examples**
For integral `n`, the falling factorial is a polynomial::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(5):
... nprint(taylor(lambda x: ff(x,n), 0, n))
...
[1.0]
[0.0, 1.0]
[0.0, -1.0, 1.0]
[0.0, 2.0, -3.0, 1.0]
[0.0, -6.0, 11.0, -6.0, 1.0]
Evaluation is supported for arbitrary arguments::
>>> ff(2+3j, 5.5)
(-720.41085888203 + 316.101124983878j)
"""
fac2 = r"""
Computes the double factorial `x!!`, defined for integers
`x > 0` by
.. math ::
x!! = \begin{cases}
1 \cdot 3 \cdots (x-2) \cdot x & x \;\mathrm{odd} \\
2 \cdot 4 \cdots (x-2) \cdot x & x \;\mathrm{even}
\end{cases}
and more generally by [1]
.. math ::
x!! = 2^{x/2} \left(\frac{\pi}{2}\right)^{(\cos(\pi x)-1)/4}
\Gamma\left(\frac{x}{2}+1\right).
**Examples**
The integer sequence of double factorials begins::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> nprint([fac2(n) for n in range(10)])
[1.0, 1.0, 2.0, 3.0, 8.0, 15.0, 48.0, 105.0, 384.0, 945.0]
For large `x`, double factorials follow a Stirling-like asymptotic
approximation::
>>> x = mpf(10000)
>>> fac2(x)
5.97272691416282e+17830
>>> sqrt(pi)*x**((x+1)/2)*exp(-x/2)
5.97262736954392e+17830
The recurrence formula `x!! = x (x-2)!!` can be reversed to
define the double factorial of negative odd integers (but
not negative even integers)::
>>> fac2(-1), fac2(-3), fac2(-5), fac2(-7)
(1.0, -1.0, 0.333333333333333, -0.0666666666666667)
>>> fac2(-2)
Traceback (most recent call last):
...
ValueError: gamma function pole
With the exception of the poles at negative even integers,
:func:`~mpmath.fac2` supports evaluation for arbitrary complex arguments.
The recurrence formula is valid generally::
>>> fac2(pi+2j)
(-1.3697207890154e-12 + 3.93665300979176e-12j)
>>> (pi+2j)*fac2(pi-2+2j)
(-1.3697207890154e-12 + 3.93665300979176e-12j)
Double factorials should not be confused with nested factorials,
which are immensely larger::
>>> fac(fac(20))
5.13805976125208e+43675043585825292774
>>> fac2(20)
3715891200.0
Double factorials appear, among other things, in series expansions
of Gaussian functions and the error function. Infinite series
include::
>>> nsum(lambda k: 1/fac2(k), [0, inf])
3.05940740534258
>>> sqrt(e)*(1+sqrt(pi/2)*erf(sqrt(2)/2))
3.05940740534258
>>> nsum(lambda k: 2**k/fac2(2*k-1), [1, inf])
4.06015693855741
>>> e * erf(1) * sqrt(pi)
4.06015693855741
A beautiful Ramanujan sum::
>>> nsum(lambda k: (-1)**k*(fac2(2*k-1)/fac2(2*k))**3, [0,inf])
0.90917279454693
>>> (gamma('9/8')/gamma('5/4')/gamma('7/8'))**2
0.90917279454693
**References**
1. http://functions.wolfram.com/GammaBetaErf/Factorial2/27/01/0002/
2. http://mathworld.wolfram.com/DoubleFactorial.html
"""
hyper = r"""
Evaluates the generalized hypergeometric function
.. math ::
\,_pF_q(a_1,\ldots,a_p; b_1,\ldots,b_q; z) =
\sum_{n=0}^\infty \frac{(a_1)_n (a_2)_n \ldots (a_p)_n}
{(b_1)_n(b_2)_n\ldots(b_q)_n} \frac{z^n}{n!}
where `(x)_n` denotes the rising factorial (see :func:`~mpmath.rf`).
The parameters lists ``a_s`` and ``b_s`` may contain integers,
real numbers, complex numbers, as well as exact fractions given in
the form of tuples `(p, q)`. :func:`~mpmath.hyper` is optimized to handle
integers and fractions more efficiently than arbitrary
floating-point parameters (since rational parameters are by
far the most common).
**Examples**
Verifying that :func:`~mpmath.hyper` gives the sum in the definition, by
comparison with :func:`~mpmath.nsum`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> a,b,c,d = 2,3,4,5
>>> x = 0.25
>>> hyper([a,b],[c,d],x)
1.078903941164934876086237
>>> fn = lambda n: rf(a,n)*rf(b,n)/rf(c,n)/rf(d,n)*x**n/fac(n)
>>> nsum(fn, [0, inf])
1.078903941164934876086237
The parameters can be any combination of integers, fractions,
floats and complex numbers::
>>> a, b, c, d, e = 1, (-1,2), pi, 3+4j, (2,3)
>>> x = 0.2j
>>> hyper([a,b],[c,d,e],x)
(0.9923571616434024810831887 - 0.005753848733883879742993122j)
>>> b, e = -0.5, mpf(2)/3
>>> fn = lambda n: rf(a,n)*rf(b,n)/rf(c,n)/rf(d,n)/rf(e,n)*x**n/fac(n)
>>> nsum(fn, [0, inf])
(0.9923571616434024810831887 - 0.005753848733883879742993122j)
The `\,_0F_0` and `\,_1F_0` series are just elementary functions::
>>> a, z = sqrt(2), +pi
>>> hyper([],[],z)
23.14069263277926900572909
>>> exp(z)
23.14069263277926900572909
>>> hyper([a],[],z)
(-0.09069132879922920160334114 + 0.3283224323946162083579656j)
>>> (1-z)**(-a)
(-0.09069132879922920160334114 + 0.3283224323946162083579656j)
If any `a_k` coefficient is a nonpositive integer, the series terminates
into a finite polynomial::
>>> hyper([1,1,1,-3],[2,5],1)
0.7904761904761904761904762
>>> identify(_)
'(83/105)'
If any `b_k` is a nonpositive integer, the function is undefined (unless the
series terminates before the division by zero occurs)::
>>> hyper([1,1,1,-3],[-2,5],1)
Traceback (most recent call last):
...
ZeroDivisionError: pole in hypergeometric series
>>> hyper([1,1,1,-1],[-2,5],1)
1.1
Except for polynomial cases, the radius of convergence `R` of the hypergeometric
series is either `R = \infty` (if `p \le q`), `R = 1` (if `p = q+1`), or
`R = 0` (if `p > q+1`).
The analytic continuations of the functions with `p = q+1`, i.e. `\,_2F_1`,
`\,_3F_2`, `\,_4F_3`, etc, are all implemented and therefore these functions
can be evaluated for `|z| \ge 1`. The shortcuts :func:`~mpmath.hyp2f1`, :func:`~mpmath.hyp3f2`
are available to handle the most common cases (see their documentation),
but functions of higher degree are also supported via :func:`~mpmath.hyper`::
>>> hyper([1,2,3,4], [5,6,7], 1) # 4F3 at finite-valued branch point
1.141783505526870731311423
>>> hyper([4,5,6,7], [1,2,3], 1) # 4F3 at pole
+inf
>>> hyper([1,2,3,4,5], [6,7,8,9], 10) # 5F4
(1.543998916527972259717257 - 0.5876309929580408028816365j)
>>> hyper([1,2,3,4,5,6], [7,8,9,10,11], 1j) # 6F5
(0.9996565821853579063502466 + 0.0129721075905630604445669j)
Near `z = 1` with noninteger parameters::
>>> hyper(['1/3',1,'3/2',2], ['1/5','11/6','41/8'], 1)
2.219433352235586121250027
>>> hyper(['1/3',1,'3/2',2], ['1/5','11/6','5/4'], 1)
+inf
>>> eps1 = extradps(6)(lambda: 1 - mpf('1e-6'))()
>>> hyper(['1/3',1,'3/2',2], ['1/5','11/6','5/4'], eps1)
2923978034.412973409330956
Please note that, as currently implemented, evaluation of `\,_pF_{p-1}`
with `p \ge 3` may be slow or inaccurate when `|z-1|` is small,
for some parameter values.
When `p > q+1`, ``hyper`` computes the (iterated) Borel sum of the divergent
series. For `\,_2F_0` the Borel sum has an analytic solution and can be
computed efficiently (see :func:`~mpmath.hyp2f0`). For higher degrees, the functions
is evaluated first by attempting to sum it directly as an asymptotic
series (this only works for tiny `|z|`), and then by evaluating the Borel
regularized sum using numerical integration. Except for
special parameter combinations, this can be extremely slow.
>>> hyper([1,1], [], 0.5) # regularization of 2F0
(1.340965419580146562086448 + 0.8503366631752726568782447j)
>>> hyper([1,1,1,1], [1], 0.5) # regularization of 4F1
(1.108287213689475145830699 + 0.5327107430640678181200491j)
With the following magnitude of argument, the asymptotic series for `\,_3F_1`
gives only a few digits. Using Borel summation, ``hyper`` can produce
a value with full accuracy::
>>> mp.dps = 15
>>> hyper([2,0.5,4], [5.25], '0.08', force_series=True)
Traceback (most recent call last):
...
NoConvergence: Hypergeometric series converges too slowly. Try increasing maxterms.
>>> hyper([2,0.5,4], [5.25], '0.08', asymp_tol=1e-4)
1.0725535790737
>>> hyper([2,0.5,4], [5.25], '0.08')
(1.07269542893559 + 5.54668863216891e-5j)
>>> hyper([2,0.5,4], [5.25], '-0.08', asymp_tol=1e-4)
0.946344925484879
>>> hyper([2,0.5,4], [5.25], '-0.08')
0.946312503737771
>>> mp.dps = 25
>>> hyper([2,0.5,4], [5.25], '-0.08')
0.9463125037377662296700858
Note that with the positive `z` value, there is a complex part in the
correct result, which falls below the tolerance of the asymptotic series.
"""
hypercomb = r"""
Computes a weighted combination of hypergeometric functions
.. math ::
\sum_{r=1}^N \left[ \prod_{k=1}^{l_r} {w_{r,k}}^{c_{r,k}}
\frac{\prod_{k=1}^{m_r} \Gamma(\alpha_{r,k})}{\prod_{k=1}^{n_r}
\Gamma(\beta_{r,k})}
\,_{p_r}F_{q_r}(a_{r,1},\ldots,a_{r,p}; b_{r,1},
\ldots, b_{r,q}; z_r)\right].
Typically the parameters are linear combinations of a small set of base
parameters; :func:`~mpmath.hypercomb` permits computing a correct value in
the case that some of the `\alpha`, `\beta`, `b` turn out to be
nonpositive integers, or if division by zero occurs for some `w^c`,
assuming that there are opposing singularities that cancel out.
The limit is computed by evaluating the function with the base
parameters perturbed, at a higher working precision.
The first argument should be a function that takes the perturbable
base parameters ``params`` as input and returns `N` tuples
``(w, c, alpha, beta, a, b, z)``, where the coefficients ``w``, ``c``,
gamma factors ``alpha``, ``beta``, and hypergeometric coefficients
``a``, ``b`` each should be lists of numbers, and ``z`` should be a single
number.
**Examples**
The following evaluates
.. math ::
(a-1) \frac{\Gamma(a-3)}{\Gamma(a-4)} \,_1F_1(a,a-1,z) = e^z(a-4)(a+z-1)
with `a=1, z=3`. There is a zero factor, two gamma function poles, and
the 1F1 function is singular; all singularities cancel out to give a finite
value::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> hypercomb(lambda a: [([a-1],[1],[a-3],[a-4],[a],[a-1],3)], [1])
-180.769832308689
>>> -9*exp(3)
-180.769832308689
"""
hyp0f1 = r"""
Gives the hypergeometric function `\,_0F_1`, sometimes known as the
confluent limit function, defined as
.. math ::
\,_0F_1(a,z) = \sum_{k=0}^{\infty} \frac{1}{(a)_k} \frac{z^k}{k!}.
This function satisfies the differential equation `z f''(z) + a f'(z) = f(z)`,
and is related to the Bessel function of the first kind (see :func:`~mpmath.besselj`).
``hyp0f1(a,z)`` is equivalent to ``hyper([],[a],z)``; see documentation for
:func:`~mpmath.hyper` for more information.
**Examples**
Evaluation for arbitrary arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hyp0f1(2, 0.25)
1.130318207984970054415392
>>> hyp0f1((1,2), 1234567)
6.27287187546220705604627e+964
>>> hyp0f1(3+4j, 1000000j)
(3.905169561300910030267132e+606 + 3.807708544441684513934213e+606j)
Evaluation is supported for arbitrarily large values of `z`,
using asymptotic expansions::
>>> hyp0f1(1, 10**50)
2.131705322874965310390701e+8685889638065036553022565
>>> hyp0f1(1, -10**50)
1.115945364792025420300208e-13
Verifying the differential equation::
>>> a = 2.5
>>> f = lambda z: hyp0f1(a,z)
>>> for z in [0, 10, 3+4j]:
... chop(z*diff(f,z,2) + a*diff(f,z) - f(z))
...
0.0
0.0
0.0
"""
hyp1f1 = r"""
Gives the confluent hypergeometric function of the first kind,
.. math ::
\,_1F_1(a,b,z) = \sum_{k=0}^{\infty} \frac{(a)_k}{(b)_k} \frac{z^k}{k!},
also known as Kummer's function and sometimes denoted by `M(a,b,z)`. This
function gives one solution to the confluent (Kummer's) differential equation
.. math ::
z f''(z) + (b-z) f'(z) - af(z) = 0.
A second solution is given by the `U` function; see :func:`~mpmath.hyperu`.
Solutions are also given in an alternate form by the Whittaker
functions (:func:`~mpmath.whitm`, :func:`~mpmath.whitw`).
``hyp1f1(a,b,z)`` is equivalent
to ``hyper([a],[b],z)``; see documentation for :func:`~mpmath.hyper` for more
information.
**Examples**
Evaluation for real and complex values of the argument `z`, with
fixed parameters `a = 2, b = -1/3`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hyp1f1(2, (-1,3), 3.25)
-2815.956856924817275640248
>>> hyp1f1(2, (-1,3), -3.25)
-1.145036502407444445553107
>>> hyp1f1(2, (-1,3), 1000)
-8.021799872770764149793693e+441
>>> hyp1f1(2, (-1,3), -1000)
0.000003131987633006813594535331
>>> hyp1f1(2, (-1,3), 100+100j)
(-3.189190365227034385898282e+48 - 1.106169926814270418999315e+49j)
Parameters may be complex::
>>> hyp1f1(2+3j, -1+j, 10j)
(261.8977905181045142673351 + 160.8930312845682213562172j)
Arbitrarily large values of `z` are supported::
>>> hyp1f1(3, 4, 10**20)
3.890569218254486878220752e+43429448190325182745
>>> hyp1f1(3, 4, -10**20)
6.0e-60
>>> hyp1f1(3, 4, 10**20*j)
(-1.935753855797342532571597e-20 - 2.291911213325184901239155e-20j)
Verifying the differential equation::
>>> a, b = 1.5, 2
>>> f = lambda z: hyp1f1(a,b,z)
>>> for z in [0, -10, 3, 3+4j]:
... chop(z*diff(f,z,2) + (b-z)*diff(f,z) - a*f(z))
...
0.0
0.0
0.0
0.0
An integral representation::
>>> a, b = 1.5, 3
>>> z = 1.5
>>> hyp1f1(a,b,z)
2.269381460919952778587441
>>> g = lambda t: exp(z*t)*t**(a-1)*(1-t)**(b-a-1)
>>> gammaprod([b],[a,b-a])*quad(g, [0,1])
2.269381460919952778587441
"""
hyp1f2 = r"""
Gives the hypergeometric function `\,_1F_2(a_1,a_2;b_1,b_2; z)`.
The call ``hyp1f2(a1,b1,b2,z)`` is equivalent to
``hyper([a1],[b1,b2],z)``.
Evaluation works for complex and arbitrarily large arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> a, b, c = 1.5, (-1,3), 2.25
>>> hyp1f2(a, b, c, 10**20)
-1.159388148811981535941434e+8685889639
>>> hyp1f2(a, b, c, -10**20)
-12.60262607892655945795907
>>> hyp1f2(a, b, c, 10**20*j)
(4.237220401382240876065501e+6141851464 - 2.950930337531768015892987e+6141851464j)
>>> hyp1f2(2+3j, -2j, 0.5j, 10-20j)
(135881.9905586966432662004 - 86681.95885418079535738828j)
"""
hyp2f2 = r"""
Gives the hypergeometric function `\,_2F_2(a_1,a_2;b_1,b_2; z)`.
The call ``hyp2f2(a1,a2,b1,b2,z)`` is equivalent to
``hyper([a1,a2],[b1,b2],z)``.
Evaluation works for complex and arbitrarily large arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> a, b, c, d = 1.5, (-1,3), 2.25, 4
>>> hyp2f2(a, b, c, d, 10**20)
-5.275758229007902299823821e+43429448190325182663
>>> hyp2f2(a, b, c, d, -10**20)
2561445.079983207701073448
>>> hyp2f2(a, b, c, d, 10**20*j)
(2218276.509664121194836667 - 1280722.539991603850462856j)
>>> hyp2f2(2+3j, -2j, 0.5j, 4j, 10-20j)
(80500.68321405666957342788 - 20346.82752982813540993502j)
"""
hyp2f3 = r"""
Gives the hypergeometric function `\,_2F_3(a_1,a_2;b_1,b_2,b_3; z)`.
The call ``hyp2f3(a1,a2,b1,b2,b3,z)`` is equivalent to
``hyper([a1,a2],[b1,b2,b3],z)``.
Evaluation works for arbitrarily large arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> a1,a2,b1,b2,b3 = 1.5, (-1,3), 2.25, 4, (1,5)
>>> hyp2f3(a1,a2,b1,b2,b3,10**20)
-4.169178177065714963568963e+8685889590
>>> hyp2f3(a1,a2,b1,b2,b3,-10**20)
7064472.587757755088178629
>>> hyp2f3(a1,a2,b1,b2,b3,10**20*j)
(-5.163368465314934589818543e+6141851415 + 1.783578125755972803440364e+6141851416j)
>>> hyp2f3(2+3j, -2j, 0.5j, 4j, -1-j, 10-20j)
(-2280.938956687033150740228 + 13620.97336609573659199632j)
>>> hyp2f3(2+3j, -2j, 0.5j, 4j, -1-j, 10000000-20000000j)
(4.849835186175096516193e+3504 - 3.365981529122220091353633e+3504j)
"""
hyp2f1 = r"""
Gives the Gauss hypergeometric function `\,_2F_1` (often simply referred to as
*the* hypergeometric function), defined for `|z| < 1` as
.. math ::
\,_2F_1(a,b,c,z) = \sum_{k=0}^{\infty}
\frac{(a)_k (b)_k}{(c)_k} \frac{z^k}{k!}.
and for `|z| \ge 1` by analytic continuation, with a branch cut on `(1, \infty)`
when necessary.
Special cases of this function include many of the orthogonal polynomials as
well as the incomplete beta function and other functions. Properties of the
Gauss hypergeometric function are documented comprehensively in many references,
for example Abramowitz & Stegun, section 15.
The implementation supports the analytic continuation as well as evaluation
close to the unit circle where `|z| \approx 1`. The syntax ``hyp2f1(a,b,c,z)``
is equivalent to ``hyper([a,b],[c],z)``.
**Examples**
Evaluation with `z` inside, outside and on the unit circle, for
fixed parameters::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hyp2f1(2, (1,2), 4, 0.75)
1.303703703703703703703704
>>> hyp2f1(2, (1,2), 4, -1.75)
0.7431290566046919177853916
>>> hyp2f1(2, (1,2), 4, 1.75)
(1.418075801749271137026239 - 1.114976146679907015775102j)
>>> hyp2f1(2, (1,2), 4, 1)
1.6
>>> hyp2f1(2, (1,2), 4, -1)
0.8235498012182875315037882
>>> hyp2f1(2, (1,2), 4, j)
(0.9144026291433065674259078 + 0.2050415770437884900574923j)
>>> hyp2f1(2, (1,2), 4, 2+j)
(0.9274013540258103029011549 + 0.7455257875808100868984496j)
>>> hyp2f1(2, (1,2), 4, 0.25j)
(0.9931169055799728251931672 + 0.06154836525312066938147793j)
Evaluation with complex parameter values::
>>> hyp2f1(1+j, 0.75, 10j, 1+5j)
(0.8834833319713479923389638 + 0.7053886880648105068343509j)
Evaluation with `z = 1`::
>>> hyp2f1(-2.5, 3.5, 1.5, 1)
0.0
>>> hyp2f1(-2.5, 3, 4, 1)
0.06926406926406926406926407
>>> hyp2f1(2, 3, 4, 1)
+inf
Evaluation for huge arguments::
>>> hyp2f1((-1,3), 1.75, 4, '1e100')
(7.883714220959876246415651e+32 + 1.365499358305579597618785e+33j)
>>> hyp2f1((-1,3), 1.75, 4, '1e1000000')
(7.883714220959876246415651e+333332 + 1.365499358305579597618785e+333333j)
>>> hyp2f1((-1,3), 1.75, 4, '1e1000000j')
(1.365499358305579597618785e+333333 - 7.883714220959876246415651e+333332j)
An integral representation::
>>> a,b,c,z = -0.5, 1, 2.5, 0.25
>>> g = lambda t: t**(b-1) * (1-t)**(c-b-1) * (1-t*z)**(-a)
>>> gammaprod([c],[b,c-b]) * quad(g, [0,1])
0.9480458814362824478852618
>>> hyp2f1(a,b,c,z)
0.9480458814362824478852618
Verifying the hypergeometric differential equation::
>>> f = lambda z: hyp2f1(a,b,c,z)
>>> chop(z*(1-z)*diff(f,z,2) + (c-(a+b+1)*z)*diff(f,z) - a*b*f(z))
0.0
"""
hyp3f2 = r"""
Gives the generalized hypergeometric function `\,_3F_2`, defined for `|z| < 1`
as
.. math ::
\,_3F_2(a_1,a_2,a_3,b_1,b_2,z) = \sum_{k=0}^{\infty}
\frac{(a_1)_k (a_2)_k (a_3)_k}{(b_1)_k (b_2)_k} \frac{z^k}{k!}.
and for `|z| \ge 1` by analytic continuation. The analytic structure of this
function is similar to that of `\,_2F_1`, generally with a singularity at
`z = 1` and a branch cut on `(1, \infty)`.
Evaluation is supported inside, on, and outside
the circle of convergence `|z| = 1`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hyp3f2(1,2,3,4,5,0.25)
1.083533123380934241548707
>>> hyp3f2(1,2+2j,3,4,5,-10+10j)
(0.1574651066006004632914361 - 0.03194209021885226400892963j)
>>> hyp3f2(1,2,3,4,5,-10)
0.3071141169208772603266489
>>> hyp3f2(1,2,3,4,5,10)
(-0.4857045320523947050581423 - 0.5988311440454888436888028j)
>>> hyp3f2(0.25,1,1,2,1.5,1)
1.157370995096772047567631
>>> (8-pi-2*ln2)/3
1.157370995096772047567631
>>> hyp3f2(1+j,0.5j,2,1,-2j,-1)
(1.74518490615029486475959 + 0.1454701525056682297614029j)
>>> hyp3f2(1+j,0.5j,2,1,-2j,sqrt(j))
(0.9829816481834277511138055 - 0.4059040020276937085081127j)
>>> hyp3f2(-3,2,1,-5,4,1)
1.41
>>> hyp3f2(-3,2,1,-5,4,2)
2.12
Evaluation very close to the unit circle::
>>> hyp3f2(1,2,3,4,5,'1.0001')
(1.564877796743282766872279 - 3.76821518787438186031973e-11j)
>>> hyp3f2(1,2,3,4,5,'1+0.0001j')
(1.564747153061671573212831 + 0.0001305757570366084557648482j)
>>> hyp3f2(1,2,3,4,5,'0.9999')
1.564616644881686134983664
>>> hyp3f2(1,2,3,4,5,'-0.9999')
0.7823896253461678060196207
.. note ::
Evaluation for `|z-1|` small can currently be inaccurate or slow
for some parameter combinations.
For various parameter combinations, `\,_3F_2` admits representation in terms
of hypergeometric functions of lower degree, or in terms of
simpler functions::
>>> for a, b, z in [(1,2,-1), (2,0.5,1)]:
... hyp2f1(a,b,a+b+0.5,z)**2
... hyp3f2(2*a,a+b,2*b,a+b+0.5,2*a+2*b,z)
...
0.4246104461966439006086308
0.4246104461966439006086308
7.111111111111111111111111
7.111111111111111111111111
>>> z = 2+3j
>>> hyp3f2(0.5,1,1.5,2,2,z)
(0.7621440939243342419729144 + 0.4249117735058037649915723j)
>>> 4*(pi-2*ellipe(z))/(pi*z)
(0.7621440939243342419729144 + 0.4249117735058037649915723j)
"""
hyperu = r"""
Gives the Tricomi confluent hypergeometric function `U`, also known as
the Kummer or confluent hypergeometric function of the second kind. This
function gives a second linearly independent solution to the confluent
hypergeometric differential equation (the first is provided by `\,_1F_1` --
see :func:`~mpmath.hyp1f1`).
**Examples**
Evaluation for arbitrary complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hyperu(2,3,4)
0.0625
>>> hyperu(0.25, 5, 1000)
0.1779949416140579573763523
>>> hyperu(0.25, 5, -1000)
(0.1256256609322773150118907 - 0.1256256609322773150118907j)
The `U` function may be singular at `z = 0`::
>>> hyperu(1.5, 2, 0)
+inf
>>> hyperu(1.5, -2, 0)
0.1719434921288400112603671
Verifying the differential equation::
>>> a, b = 1.5, 2
>>> f = lambda z: hyperu(a,b,z)
>>> for z in [-10, 3, 3+4j]:
... chop(z*diff(f,z,2) + (b-z)*diff(f,z) - a*f(z))
...
0.0
0.0
0.0
An integral representation::
>>> a,b,z = 2, 3.5, 4.25
>>> hyperu(a,b,z)
0.06674960718150520648014567
>>> quad(lambda t: exp(-z*t)*t**(a-1)*(1+t)**(b-a-1),[0,inf]) / gamma(a)
0.06674960718150520648014567
[1] http://www.math.ucla.edu/~cbm/aands/page_504.htm
"""
hyp2f0 = r"""
Gives the hypergeometric function `\,_2F_0`, defined formally by the
series
.. math ::
\,_2F_0(a,b;;z) = \sum_{n=0}^{\infty} (a)_n (b)_n \frac{z^n}{n!}.
This series usually does not converge. For small enough `z`, it can be viewed
as an asymptotic series that may be summed directly with an appropriate
truncation. When this is not the case, :func:`~mpmath.hyp2f0` gives a regularized sum,
or equivalently, it uses a representation in terms of the
hypergeometric U function [1]. The series also converges when either `a` or `b`
is a nonpositive integer, as it then terminates into a polynomial
after `-a` or `-b` terms.
**Examples**
Evaluation is supported for arbitrary complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hyp2f0((2,3), 1.25, -100)
0.07095851870980052763312791
>>> hyp2f0((2,3), 1.25, 100)
(-0.03254379032170590665041131 + 0.07269254613282301012735797j)
>>> hyp2f0(-0.75, 1-j, 4j)
(-0.3579987031082732264862155 - 3.052951783922142735255881j)
Even with real arguments, the regularized value of 2F0 is often complex-valued,
but the imaginary part decreases exponentially as `z \to 0`. In the following
example, the first call uses complex evaluation while the second has a small
enough `z` to evaluate using the direct series and thus the returned value
is strictly real (this should be taken to indicate that the imaginary
part is less than ``eps``)::
>>> mp.dps = 15
>>> hyp2f0(1.5, 0.5, 0.05)
(1.04166637647907 + 8.34584913683906e-8j)
>>> hyp2f0(1.5, 0.5, 0.0005)
1.00037535207621
The imaginary part can be retrieved by increasing the working precision::
>>> mp.dps = 80
>>> nprint(hyp2f0(1.5, 0.5, 0.009).imag)
1.23828e-46
In the polynomial case (the series terminating), 2F0 can evaluate exactly::
>>> mp.dps = 15
>>> hyp2f0(-6,-6,2)
291793.0
>>> identify(hyp2f0(-2,1,0.25))
'(5/8)'
The coefficients of the polynomials can be recovered using Taylor expansion::
>>> nprint(taylor(lambda x: hyp2f0(-3,0.5,x), 0, 10))
[1.0, -1.5, 2.25, -1.875, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0]
>>> nprint(taylor(lambda x: hyp2f0(-4,0.5,x), 0, 10))
[1.0, -2.0, 4.5, -7.5, 6.5625, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0]
[1] http://www.math.ucla.edu/~cbm/aands/page_504.htm
"""
gammainc = r"""
``gammainc(z, a=0, b=inf)`` computes the (generalized) incomplete
gamma function with integration limits `[a, b]`:
.. math ::
\Gamma(z,a,b) = \int_a^b t^{z-1} e^{-t} \, dt
The generalized incomplete gamma function reduces to the
following special cases when one or both endpoints are fixed:
* `\Gamma(z,0,\infty)` is the standard ("complete")
gamma function, `\Gamma(z)` (available directly
as the mpmath function :func:`~mpmath.gamma`)
* `\Gamma(z,a,\infty)` is the "upper" incomplete gamma
function, `\Gamma(z,a)`
* `\Gamma(z,0,b)` is the "lower" incomplete gamma
function, `\gamma(z,b)`.
Of course, we have
`\Gamma(z,0,x) + \Gamma(z,x,\infty) = \Gamma(z)`
for all `z` and `x`.
Note however that some authors reverse the order of the
arguments when defining the lower and upper incomplete
gamma function, so one should be careful to get the correct
definition.
If also given the keyword argument ``regularized=True``,
:func:`~mpmath.gammainc` computes the "regularized" incomplete gamma
function
.. math ::
P(z,a,b) = \frac{\Gamma(z,a,b)}{\Gamma(z)}.
**Examples**
We can compare with numerical quadrature to verify that
:func:`~mpmath.gammainc` computes the integral in the definition::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> gammainc(2+3j, 4, 10)
(0.00977212668627705160602312 - 0.0770637306312989892451977j)
>>> quad(lambda t: t**(2+3j-1) * exp(-t), [4, 10])
(0.00977212668627705160602312 - 0.0770637306312989892451977j)
Argument symmetries follow directly from the integral definition::
>>> gammainc(3, 4, 5) + gammainc(3, 5, 4)
0.0
>>> gammainc(3,0,2) + gammainc(3,2,4); gammainc(3,0,4)
1.523793388892911312363331
1.523793388892911312363331
>>> findroot(lambda z: gammainc(2,z,3), 1)
3.0
Evaluation for arbitrarily large arguments::
>>> gammainc(10, 100)
4.083660630910611272288592e-26
>>> gammainc(10, 10000000000000000)
5.290402449901174752972486e-4342944819032375
>>> gammainc(3+4j, 1000000+1000000j)
(-1.257913707524362408877881e-434284 + 2.556691003883483531962095e-434284j)
Evaluation of a generalized incomplete gamma function automatically chooses
the representation that gives a more accurate result, depending on which
parameter is larger::
>>> gammainc(10000000, 3) - gammainc(10000000, 2) # Bad
0.0
>>> gammainc(10000000, 2, 3) # Good
1.755146243738946045873491e+4771204
>>> gammainc(2, 0, 100000001) - gammainc(2, 0, 100000000) # Bad
0.0
>>> gammainc(2, 100000000, 100000001) # Good
4.078258353474186729184421e-43429441
The incomplete gamma functions satisfy simple recurrence
relations::
>>> mp.dps = 25
>>> z, a = mpf(3.5), mpf(2)
>>> gammainc(z+1, a); z*gammainc(z,a) + a**z*exp(-a)
10.60130296933533459267329
10.60130296933533459267329
>>> gammainc(z+1,0,a); z*gammainc(z,0,a) - a**z*exp(-a)
1.030425427232114336470932
1.030425427232114336470932
Evaluation at integers and poles::
>>> gammainc(-3, -4, -5)
(-0.2214577048967798566234192 + 0.0j)
>>> gammainc(-3, 0, 5)
+inf
If `z` is an integer, the recurrence reduces the incomplete gamma
function to `P(a) \exp(-a) + Q(b) \exp(-b)` where `P` and
`Q` are polynomials::
>>> gammainc(1, 2); exp(-2)
0.1353352832366126918939995
0.1353352832366126918939995
>>> mp.dps = 50
>>> identify(gammainc(6, 1, 2), ['exp(-1)', 'exp(-2)'])
'(326*exp(-1) + (-872)*exp(-2))'
The incomplete gamma functions reduce to functions such as
the exponential integral Ei and the error function for special
arguments::
>>> mp.dps = 25
>>> gammainc(0, 4); -ei(-4)
0.00377935240984890647887486
0.00377935240984890647887486
>>> gammainc(0.5, 0, 2); sqrt(pi)*erf(sqrt(2))
1.691806732945198336509541
1.691806732945198336509541
"""
erf = r"""
Computes the error function, `\mathrm{erf}(x)`. The error
function is the normalized antiderivative of the Gaussian function
`\exp(-t^2)`. More precisely,
.. math::
\mathrm{erf}(x) = \frac{2}{\sqrt \pi} \int_0^x \exp(-t^2) \,dt
**Basic examples**
Simple values and limits include::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> erf(0)
0.0
>>> erf(1)
0.842700792949715
>>> erf(-1)
-0.842700792949715
>>> erf(inf)
1.0
>>> erf(-inf)
-1.0
For large real `x`, `\mathrm{erf}(x)` approaches 1 very
rapidly::
>>> erf(3)
0.999977909503001
>>> erf(5)
0.999999999998463
The error function is an odd function::
>>> nprint(chop(taylor(erf, 0, 5)))
[0.0, 1.12838, 0.0, -0.376126, 0.0, 0.112838]
:func:`~mpmath.erf` implements arbitrary-precision evaluation and
supports complex numbers::
>>> mp.dps = 50
>>> erf(0.5)
0.52049987781304653768274665389196452873645157575796
>>> mp.dps = 25
>>> erf(1+j)
(1.316151281697947644880271 + 0.1904534692378346862841089j)
Evaluation is supported for large arguments::
>>> mp.dps = 25
>>> erf('1e1000')
1.0
>>> erf('-1e1000')
-1.0
>>> erf('1e-1000')
1.128379167095512573896159e-1000
>>> erf('1e7j')
(0.0 + 8.593897639029319267398803e+43429448190317j)
>>> erf('1e7+1e7j')
(0.9999999858172446172631323 + 3.728805278735270407053139e-8j)
**Related functions**
See also :func:`~mpmath.erfc`, which is more accurate for large `x`,
and :func:`~mpmath.erfi` which gives the antiderivative of
`\exp(t^2)`.
The Fresnel integrals :func:`~mpmath.fresnels` and :func:`~mpmath.fresnelc`
are also related to the error function.
"""
erfc = r"""
Computes the complementary error function,
`\mathrm{erfc}(x) = 1-\mathrm{erf}(x)`.
This function avoids cancellation that occurs when naively
computing the complementary error function as ``1-erf(x)``::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> 1 - erf(10)
0.0
>>> erfc(10)
2.08848758376254e-45
:func:`~mpmath.erfc` works accurately even for ludicrously large
arguments::
>>> erfc(10**10)
4.3504398860243e-43429448190325182776
Complex arguments are supported::
>>> erfc(500+50j)
(1.19739830969552e-107492 + 1.46072418957528e-107491j)
"""
erfi = r"""
Computes the imaginary error function, `\mathrm{erfi}(x)`.
The imaginary error function is defined in analogy with the
error function, but with a positive sign in the integrand:
.. math ::
\mathrm{erfi}(x) = \frac{2}{\sqrt \pi} \int_0^x \exp(t^2) \,dt
Whereas the error function rapidly converges to 1 as `x` grows,
the imaginary error function rapidly diverges to infinity.
The functions are related as
`\mathrm{erfi}(x) = -i\,\mathrm{erf}(ix)` for all complex
numbers `x`.
**Examples**
Basic values and limits::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> erfi(0)
0.0
>>> erfi(1)
1.65042575879754
>>> erfi(-1)
-1.65042575879754
>>> erfi(inf)
+inf
>>> erfi(-inf)
-inf
Note the symmetry between erf and erfi::
>>> erfi(3j)
(0.0 + 0.999977909503001j)
>>> erf(3)
0.999977909503001
>>> erf(1+2j)
(-0.536643565778565 - 5.04914370344703j)
>>> erfi(2+1j)
(-5.04914370344703 - 0.536643565778565j)
Large arguments are supported::
>>> erfi(1000)
1.71130938718796e+434291
>>> erfi(10**10)
7.3167287567024e+43429448190325182754
>>> erfi(-10**10)
-7.3167287567024e+43429448190325182754
>>> erfi(1000-500j)
(2.49895233563961e+325717 + 2.6846779342253e+325717j)
>>> erfi(100000j)
(0.0 + 1.0j)
>>> erfi(-100000j)
(0.0 - 1.0j)
"""
erfinv = r"""
Computes the inverse error function, satisfying
.. math ::
\mathrm{erf}(\mathrm{erfinv}(x)) =
\mathrm{erfinv}(\mathrm{erf}(x)) = x.
This function is defined only for `-1 \le x \le 1`.
**Examples**
Special values include::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> erfinv(0)
0.0
>>> erfinv(1)
+inf
>>> erfinv(-1)
-inf
The domain is limited to the standard interval::
>>> erfinv(2)
Traceback (most recent call last):
...
ValueError: erfinv(x) is defined only for -1 <= x <= 1
It is simple to check that :func:`~mpmath.erfinv` computes inverse values of
:func:`~mpmath.erf` as promised::
>>> erf(erfinv(0.75))
0.75
>>> erf(erfinv(-0.995))
-0.995
:func:`~mpmath.erfinv` supports arbitrary-precision evaluation::
>>> mp.dps = 50
>>> x = erf(2)
>>> x
0.99532226501895273416206925636725292861089179704006
>>> erfinv(x)
2.0
A definite integral involving the inverse error function::
>>> mp.dps = 15
>>> quad(erfinv, [0, 1])
0.564189583547756
>>> 1/sqrt(pi)
0.564189583547756
The inverse error function can be used to generate random numbers
with a Gaussian distribution (although this is a relatively
inefficient algorithm)::
>>> nprint([erfinv(2*rand()-1) for n in range(6)]) # doctest: +SKIP
[-0.586747, 1.10233, -0.376796, 0.926037, -0.708142, -0.732012]
"""
npdf = r"""
``npdf(x, mu=0, sigma=1)`` evaluates the probability density
function of a normal distribution with mean value `\mu`
and variance `\sigma^2`.
Elementary properties of the probability distribution can
be verified using numerical integration::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> quad(npdf, [-inf, inf])
1.0
>>> quad(lambda x: npdf(x, 3), [3, inf])
0.5
>>> quad(lambda x: npdf(x, 3, 2), [3, inf])
0.5
See also :func:`~mpmath.ncdf`, which gives the cumulative
distribution.
"""
ncdf = r"""
``ncdf(x, mu=0, sigma=1)`` evaluates the cumulative distribution
function of a normal distribution with mean value `\mu`
and variance `\sigma^2`.
See also :func:`~mpmath.npdf`, which gives the probability density.
Elementary properties include::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> ncdf(pi, mu=pi)
0.5
>>> ncdf(-inf)
0.0
>>> ncdf(+inf)
1.0
The cumulative distribution is the integral of the density
function having identical mu and sigma::
>>> mp.dps = 15
>>> diff(ncdf, 2)
0.053990966513188
>>> npdf(2)
0.053990966513188
>>> diff(lambda x: ncdf(x, 1, 0.5), 0)
0.107981933026376
>>> npdf(0, 1, 0.5)
0.107981933026376
"""
expint = r"""
:func:`~mpmath.expint(n,z)` gives the generalized exponential integral
or En-function,
.. math ::
\mathrm{E}_n(z) = \int_1^{\infty} \frac{e^{-zt}}{t^n} dt,
where `n` and `z` may both be complex numbers. The case with `n = 1` is
also given by :func:`~mpmath.e1`.
**Examples**
Evaluation at real and complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> expint(1, 6.25)
0.0002704758872637179088496194
>>> expint(-3, 2+3j)
(0.00299658467335472929656159 + 0.06100816202125885450319632j)
>>> expint(2+3j, 4-5j)
(0.001803529474663565056945248 - 0.002235061547756185403349091j)
At negative integer values of `n`, `E_n(z)` reduces to a
rational-exponential function::
>>> f = lambda n, z: fac(n)*sum(z**k/fac(k-1) for k in range(1,n+2))/\
... exp(z)/z**(n+2)
>>> n = 3
>>> z = 1/pi
>>> expint(-n,z)
584.2604820613019908668219
>>> f(n,z)
584.2604820613019908668219
>>> n = 5
>>> expint(-n,z)
115366.5762594725451811138
>>> f(n,z)
115366.5762594725451811138
"""
e1 = r"""
Computes the exponential integral `\mathrm{E}_1(z)`, given by
.. math ::
\mathrm{E}_1(z) = \int_z^{\infty} \frac{e^{-t}}{t} dt.
This is equivalent to :func:`~mpmath.expint` with `n = 1`.
**Examples**
Two ways to evaluate this function::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> e1(6.25)
0.0002704758872637179088496194
>>> expint(1,6.25)
0.0002704758872637179088496194
The E1-function is essentially the same as the Ei-function (:func:`~mpmath.ei`)
with negated argument, except for an imaginary branch cut term::
>>> e1(2.5)
0.02491491787026973549562801
>>> -ei(-2.5)
0.02491491787026973549562801
>>> e1(-2.5)
(-7.073765894578600711923552 - 3.141592653589793238462643j)
>>> -ei(2.5)
-7.073765894578600711923552
"""
ei = r"""
Computes the exponential integral or Ei-function, `\mathrm{Ei}(x)`.
The exponential integral is defined as
.. math ::
\mathrm{Ei}(x) = \int_{-\infty\,}^x \frac{e^t}{t} \, dt.
When the integration range includes `t = 0`, the exponential
integral is interpreted as providing the Cauchy principal value.
For real `x`, the Ei-function behaves roughly like
`\mathrm{Ei}(x) \approx \exp(x) + \log(|x|)`.
The Ei-function is related to the more general family of exponential
integral functions denoted by `E_n`, which are available as :func:`~mpmath.expint`.
**Basic examples**
Some basic values and limits are::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> ei(0)
-inf
>>> ei(1)
1.89511781635594
>>> ei(inf)
+inf
>>> ei(-inf)
0.0
For `x < 0`, the defining integral can be evaluated
numerically as a reference::
>>> ei(-4)
-0.00377935240984891
>>> quad(lambda t: exp(t)/t, [-inf, -4])
-0.00377935240984891
:func:`~mpmath.ei` supports complex arguments and arbitrary
precision evaluation::
>>> mp.dps = 50
>>> ei(pi)
10.928374389331410348638445906907535171566338835056
>>> mp.dps = 25
>>> ei(3+4j)
(-4.154091651642689822535359 + 4.294418620024357476985535j)
**Related functions**
The exponential integral is closely related to the logarithmic
integral. See :func:`~mpmath.li` for additional information.
The exponential integral is related to the hyperbolic
and trigonometric integrals (see :func:`~mpmath.chi`, :func:`~mpmath.shi`,
:func:`~mpmath.ci`, :func:`~mpmath.si`) similarly to how the ordinary
exponential function is related to the hyperbolic and
trigonometric functions::
>>> mp.dps = 15
>>> ei(3)
9.93383257062542
>>> chi(3) + shi(3)
9.93383257062542
>>> chop(ci(3j) - j*si(3j) - pi*j/2)
9.93383257062542
Beware that logarithmic corrections, as in the last example
above, are required to obtain the correct branch in general.
For details, see [1].
The exponential integral is also a special case of the
hypergeometric function `\,_2F_2`::
>>> z = 0.6
>>> z*hyper([1,1],[2,2],z) + (ln(z)-ln(1/z))/2 + euler
0.769881289937359
>>> ei(z)
0.769881289937359
**References**
1. Relations between Ei and other functions:
http://functions.wolfram.com/GammaBetaErf/ExpIntegralEi/27/01/
2. Abramowitz & Stegun, section 5:
http://www.math.sfu.ca/~cbm/aands/page_228.htm
3. Asymptotic expansion for Ei:
http://mathworld.wolfram.com/En-Function.html
"""
li = r"""
Computes the logarithmic integral or li-function
`\mathrm{li}(x)`, defined by
.. math ::
\mathrm{li}(x) = \int_0^x \frac{1}{\log t} \, dt
The logarithmic integral has a singularity at `x = 1`.
Alternatively, ``li(x, offset=True)`` computes the offset
logarithmic integral (used in number theory)
.. math ::
\mathrm{Li}(x) = \int_2^x \frac{1}{\log t} \, dt.
These two functions are related via the simple identity
`\mathrm{Li}(x) = \mathrm{li}(x) - \mathrm{li}(2)`.
The logarithmic integral should also not be confused with
the polylogarithm (also denoted by Li), which is implemented
as :func:`~mpmath.polylog`.
**Examples**
Some basic values and limits::
>>> from mpmath import *
>>> mp.dps = 30; mp.pretty = True
>>> li(0)
0.0
>>> li(1)
-inf
>>> li(1)
-inf
>>> li(2)
1.04516378011749278484458888919
>>> findroot(li, 2)
1.45136923488338105028396848589
>>> li(inf)
+inf
>>> li(2, offset=True)
0.0
>>> li(1, offset=True)
-inf
>>> li(0, offset=True)
-1.04516378011749278484458888919
>>> li(10, offset=True)
5.12043572466980515267839286347
The logarithmic integral can be evaluated for arbitrary
complex arguments::
>>> mp.dps = 20
>>> li(3+4j)
(3.1343755504645775265 + 2.6769247817778742392j)
The logarithmic integral is related to the exponential integral::
>>> ei(log(3))
2.1635885946671919729
>>> li(3)
2.1635885946671919729
The logarithmic integral grows like `O(x/\log(x))`::
>>> mp.dps = 15
>>> x = 10**100
>>> x/log(x)
4.34294481903252e+97
>>> li(x)
4.3619719871407e+97
The prime number theorem states that the number of primes less
than `x` is asymptotic to `\mathrm{Li}(x)` (equivalently
`\mathrm{li}(x)`). For example, it is known that there are
exactly 1,925,320,391,606,803,968,923 prime numbers less than
`10^{23}` [1]. The logarithmic integral provides a very
accurate estimate::
>>> li(10**23, offset=True)
1.92532039161405e+21
A definite integral is::
>>> quad(li, [0, 1])
-0.693147180559945
>>> -ln(2)
-0.693147180559945
**References**
1. http://mathworld.wolfram.com/PrimeCountingFunction.html
2. http://mathworld.wolfram.com/LogarithmicIntegral.html
"""
ci = r"""
Computes the cosine integral,
.. math ::
\mathrm{Ci}(x) = -\int_x^{\infty} \frac{\cos t}{t}\,dt
= \gamma + \log x + \int_0^x \frac{\cos t - 1}{t}\,dt
**Examples**
Some values and limits::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> ci(0)
-inf
>>> ci(1)
0.3374039229009681346626462
>>> ci(pi)
0.07366791204642548599010096
>>> ci(inf)
0.0
>>> ci(-inf)
(0.0 + 3.141592653589793238462643j)
>>> ci(2+3j)
(1.408292501520849518759125 - 2.983617742029605093121118j)
The cosine integral behaves roughly like the sinc function
(see :func:`~mpmath.sinc`) for large real `x`::
>>> ci(10**10)
-4.875060251748226537857298e-11
>>> sinc(10**10)
-4.875060250875106915277943e-11
>>> chop(limit(ci, inf))
0.0
It has infinitely many roots on the positive real axis::
>>> findroot(ci, 1)
0.6165054856207162337971104
>>> findroot(ci, 2)
3.384180422551186426397851
Evaluation is supported for `z` anywhere in the complex plane::
>>> ci(10**6*(1+j))
(4.449410587611035724984376e+434287 + 9.75744874290013526417059e+434287j)
We can evaluate the defining integral as a reference::
>>> mp.dps = 15
>>> -quadosc(lambda t: cos(t)/t, [5, inf], omega=1)
-0.190029749656644
>>> ci(5)
-0.190029749656644
Some infinite series can be evaluated using the
cosine integral::
>>> nsum(lambda k: (-1)**k/(fac(2*k)*(2*k)), [1,inf])
-0.239811742000565
>>> ci(1) - euler
-0.239811742000565
"""
si = r"""
Computes the sine integral,
.. math ::
\mathrm{Si}(x) = \int_0^x \frac{\sin t}{t}\,dt.
The sine integral is thus the antiderivative of the sinc
function (see :func:`~mpmath.sinc`).
**Examples**
Some values and limits::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> si(0)
0.0
>>> si(1)
0.9460830703671830149413533
>>> si(-1)
-0.9460830703671830149413533
>>> si(pi)
1.851937051982466170361053
>>> si(inf)
1.570796326794896619231322
>>> si(-inf)
-1.570796326794896619231322
>>> si(2+3j)
(4.547513889562289219853204 + 1.399196580646054789459839j)
The sine integral approaches `\pi/2` for large real `x`::
>>> si(10**10)
1.570796326707584656968511
>>> pi/2
1.570796326794896619231322
Evaluation is supported for `z` anywhere in the complex plane::
>>> si(10**6*(1+j))
(-9.75744874290013526417059e+434287 + 4.449410587611035724984376e+434287j)
We can evaluate the defining integral as a reference::
>>> mp.dps = 15
>>> quad(sinc, [0, 5])
1.54993124494467
>>> si(5)
1.54993124494467
Some infinite series can be evaluated using the
sine integral::
>>> nsum(lambda k: (-1)**k/(fac(2*k+1)*(2*k+1)), [0,inf])
0.946083070367183
>>> si(1)
0.946083070367183
"""
chi = r"""
Computes the hyperbolic cosine integral, defined
in analogy with the cosine integral (see :func:`~mpmath.ci`) as
.. math ::
\mathrm{Chi}(x) = -\int_x^{\infty} \frac{\cosh t}{t}\,dt
= \gamma + \log x + \int_0^x \frac{\cosh t - 1}{t}\,dt
Some values and limits::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> chi(0)
-inf
>>> chi(1)
0.8378669409802082408946786
>>> chi(inf)
+inf
>>> findroot(chi, 0.5)
0.5238225713898644064509583
>>> chi(2+3j)
(-0.1683628683277204662429321 + 2.625115880451325002151688j)
Evaluation is supported for `z` anywhere in the complex plane::
>>> chi(10**6*(1+j))
(4.449410587611035724984376e+434287 - 9.75744874290013526417059e+434287j)
"""
shi = r"""
Computes the hyperbolic sine integral, defined
in analogy with the sine integral (see :func:`~mpmath.si`) as
.. math ::
\mathrm{Shi}(x) = \int_0^x \frac{\sinh t}{t}\,dt.
Some values and limits::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> shi(0)
0.0
>>> shi(1)
1.057250875375728514571842
>>> shi(-1)
-1.057250875375728514571842
>>> shi(inf)
+inf
>>> shi(2+3j)
(-0.1931890762719198291678095 + 2.645432555362369624818525j)
Evaluation is supported for `z` anywhere in the complex plane::
>>> shi(10**6*(1+j))
(4.449410587611035724984376e+434287 - 9.75744874290013526417059e+434287j)
"""
fresnels = r"""
Computes the Fresnel sine integral
.. math ::
S(x) = \int_0^x \sin\left(\frac{\pi t^2}{2}\right) \,dt
Note that some sources define this function
without the normalization factor `\pi/2`.
**Examples**
Some basic values and limits::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> fresnels(0)
0.0
>>> fresnels(inf)
0.5
>>> fresnels(-inf)
-0.5
>>> fresnels(1)
0.4382591473903547660767567
>>> fresnels(1+2j)
(36.72546488399143842838788 + 15.58775110440458732748279j)
Comparing with the definition::
>>> fresnels(3)
0.4963129989673750360976123
>>> quad(lambda t: sin(pi*t**2/2), [0,3])
0.4963129989673750360976123
"""
fresnelc = r"""
Computes the Fresnel cosine integral
.. math ::
C(x) = \int_0^x \cos\left(\frac{\pi t^2}{2}\right) \,dt
Note that some sources define this function
without the normalization factor `\pi/2`.
**Examples**
Some basic values and limits::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> fresnelc(0)
0.0
>>> fresnelc(inf)
0.5
>>> fresnelc(-inf)
-0.5
>>> fresnelc(1)
0.7798934003768228294742064
>>> fresnelc(1+2j)
(16.08787137412548041729489 - 36.22568799288165021578758j)
Comparing with the definition::
>>> fresnelc(3)
0.6057207892976856295561611
>>> quad(lambda t: cos(pi*t**2/2), [0,3])
0.6057207892976856295561611
"""
airyai = r"""
Computes the Airy function `\operatorname{Ai}(z)`, which is
the solution of the Airy differential equation `f''(z) - z f(z) = 0`
with initial conditions
.. math ::
\operatorname{Ai}(0) =
\frac{1}{3^{2/3}\Gamma\left(\frac{2}{3}\right)}
\operatorname{Ai}'(0) =
-\frac{1}{3^{1/3}\Gamma\left(\frac{1}{3}\right)}.
Other common ways of defining the Ai-function include
integrals such as
.. math ::
\operatorname{Ai}(x) = \frac{1}{\pi}
\int_0^{\infty} \cos\left(\frac{1}{3}t^3+xt\right) dt
\qquad x \in \mathbb{R}
\operatorname{Ai}(z) = \frac{\sqrt{3}}{2\pi}
\int_0^{\infty}
\exp\left(-\frac{t^3}{3}-\frac{z^3}{3t^3}\right) dt.
The Ai-function is an entire function with a turning point,
behaving roughly like a slowly decaying sine wave for `z < 0` and
like a rapidly decreasing exponential for `z > 0`.
A second solution of the Airy differential equation
is given by `\operatorname{Bi}(z)` (see :func:`~mpmath.airybi`).
Optionally, with *derivative=alpha*, :func:`airyai` can compute the
`\alpha`-th order fractional derivative with respect to `z`.
For `\alpha = n = 1,2,3,\ldots` this gives the derivative
`\operatorname{Ai}^{(n)}(z)`, and for `\alpha = -n = -1,-2,-3,\ldots`
this gives the `n`-fold iterated integral
.. math ::
f_0(z) = \operatorname{Ai}(z)
f_n(z) = \int_0^z f_{n-1}(t) dt.
The Ai-function has infinitely many zeros, all located along the
negative half of the real axis. They can be computed with
:func:`~mpmath.airyaizero`.
**Plots**
.. literalinclude :: /plots/ai.py
.. image :: /plots/ai.png
.. literalinclude :: /plots/ai_c.py
.. image :: /plots/ai_c.png
**Basic examples**
Limits and values include::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> airyai(0); 1/(power(3,'2/3')*gamma('2/3'))
0.3550280538878172392600632
0.3550280538878172392600632
>>> airyai(1)
0.1352924163128814155241474
>>> airyai(-1)
0.5355608832923521187995166
>>> airyai(inf); airyai(-inf)
0.0
0.0
Evaluation is supported for large magnitudes of the argument::
>>> airyai(-100)
0.1767533932395528780908311
>>> airyai(100)
2.634482152088184489550553e-291
>>> airyai(50+50j)
(-5.31790195707456404099817e-68 - 1.163588003770709748720107e-67j)
>>> airyai(-50+50j)
(1.041242537363167632587245e+158 + 3.347525544923600321838281e+157j)
Huge arguments are also fine::
>>> airyai(10**10)
1.162235978298741779953693e-289529654602171
>>> airyai(-10**10)
0.0001736206448152818510510181
>>> w = airyai(10**10*(1+j))
>>> w.real
5.711508683721355528322567e-186339621747698
>>> w.imag
1.867245506962312577848166e-186339621747697
The first root of the Ai-function is::
>>> findroot(airyai, -2)
-2.338107410459767038489197
>>> airyaizero(1)
-2.338107410459767038489197
**Properties and relations**
Verifying the Airy differential equation::
>>> for z in [-3.4, 0, 2.5, 1+2j]:
... chop(airyai(z,2) - z*airyai(z))
...
0.0
0.0
0.0
0.0
The first few terms of the Taylor series expansion around `z = 0`
(every third term is zero)::
>>> nprint(taylor(airyai, 0, 5))
[0.355028, -0.258819, 0.0, 0.0591713, -0.0215683, 0.0]
The Airy functions satisfy the Wronskian relation
`\operatorname{Ai}(z) \operatorname{Bi}'(z) -
\operatorname{Ai}'(z) \operatorname{Bi}(z) = 1/\pi`::
>>> z = -0.5
>>> airyai(z)*airybi(z,1) - airyai(z,1)*airybi(z)
0.3183098861837906715377675
>>> 1/pi
0.3183098861837906715377675
The Airy functions can be expressed in terms of Bessel
functions of order `\pm 1/3`. For `\Re[z] \le 0`, we have::
>>> z = -3
>>> airyai(z)
-0.3788142936776580743472439
>>> y = 2*power(-z,'3/2')/3
>>> (sqrt(-z) * (besselj('1/3',y) + besselj('-1/3',y)))/3
-0.3788142936776580743472439
**Derivatives and integrals**
Derivatives of the Ai-function (directly and using :func:`~mpmath.diff`)::
>>> airyai(-3,1); diff(airyai,-3)
0.3145837692165988136507873
0.3145837692165988136507873
>>> airyai(-3,2); diff(airyai,-3,2)
1.136442881032974223041732
1.136442881032974223041732
>>> airyai(1000,1); diff(airyai,1000)
-2.943133917910336090459748e-9156
-2.943133917910336090459748e-9156
Several derivatives at `z = 0`::
>>> airyai(0,0); airyai(0,1); airyai(0,2)
0.3550280538878172392600632
-0.2588194037928067984051836
0.0
>>> airyai(0,3); airyai(0,4); airyai(0,5)
0.3550280538878172392600632
-0.5176388075856135968103671
0.0
>>> airyai(0,15); airyai(0,16); airyai(0,17)
1292.30211615165475090663
-3188.655054727379756351861
0.0
The integral of the Ai-function::
>>> airyai(3,-1); quad(airyai, [0,3])
0.3299203760070217725002701
0.3299203760070217725002701
>>> airyai(-10,-1); quad(airyai, [0,-10])
-0.765698403134212917425148
-0.765698403134212917425148
Integrals of high or fractional order::
>>> airyai(-2,0.5); differint(airyai,-2,0.5,0)
(0.0 + 0.2453596101351438273844725j)
(0.0 + 0.2453596101351438273844725j)
>>> airyai(-2,-4); differint(airyai,-2,-4,0)
0.2939176441636809580339365
0.2939176441636809580339365
>>> airyai(0,-1); airyai(0,-2); airyai(0,-3)
0.0
0.0
0.0
Integrals of the Ai-function can be evaluated at limit points::
>>> airyai(-1000000,-1); airyai(-inf,-1)
-0.6666843728311539978751512
-0.6666666666666666666666667
>>> airyai(10,-1); airyai(+inf,-1)
0.3333333332991690159427932
0.3333333333333333333333333
>>> airyai(+inf,-2); airyai(+inf,-3)
+inf
+inf
>>> airyai(-1000000,-2); airyai(-inf,-2)
666666.4078472650651209742
+inf
>>> airyai(-1000000,-3); airyai(-inf,-3)
-333333074513.7520264995733
-inf
**References**
1. [DLMF]_ Chapter 9: Airy and Related Functions
2. [WolframFunctions]_ section: Bessel-Type Functions
"""
airybi = r"""
Computes the Airy function `\operatorname{Bi}(z)`, which is
the solution of the Airy differential equation `f''(z) - z f(z) = 0`
with initial conditions
.. math ::
\operatorname{Bi}(0) =
\frac{1}{3^{1/6}\Gamma\left(\frac{2}{3}\right)}
\operatorname{Bi}'(0) =
\frac{3^{1/6}}{\Gamma\left(\frac{1}{3}\right)}.
Like the Ai-function (see :func:`~mpmath.airyai`), the Bi-function
is oscillatory for `z < 0`, but it grows rather than decreases
for `z > 0`.
Optionally, as for :func:`~mpmath.airyai`, derivatives, integrals
and fractional derivatives can be computed with the *derivative*
parameter.
The Bi-function has infinitely many zeros along the negative
half-axis, as well as complex zeros, which can all be computed
with :func:`~mpmath.airybizero`.
**Plots**
.. literalinclude :: /plots/bi.py
.. image :: /plots/bi.png
.. literalinclude :: /plots/bi_c.py
.. image :: /plots/bi_c.png
**Basic examples**
Limits and values include::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> airybi(0); 1/(power(3,'1/6')*gamma('2/3'))
0.6149266274460007351509224
0.6149266274460007351509224
>>> airybi(1)
1.207423594952871259436379
>>> airybi(-1)
0.10399738949694461188869
>>> airybi(inf); airybi(-inf)
+inf
0.0
Evaluation is supported for large magnitudes of the argument::
>>> airybi(-100)
0.02427388768016013160566747
>>> airybi(100)
6.041223996670201399005265e+288
>>> airybi(50+50j)
(-5.322076267321435669290334e+63 + 1.478450291165243789749427e+65j)
>>> airybi(-50+50j)
(-3.347525544923600321838281e+157 + 1.041242537363167632587245e+158j)
Huge arguments::
>>> airybi(10**10)
1.369385787943539818688433e+289529654602165
>>> airybi(-10**10)
0.001775656141692932747610973
>>> w = airybi(10**10*(1+j))
>>> w.real
-6.559955931096196875845858e+186339621747689
>>> w.imag
-6.822462726981357180929024e+186339621747690
The first real root of the Bi-function is::
>>> findroot(airybi, -1); airybizero(1)
-1.17371322270912792491998
-1.17371322270912792491998
**Properties and relations**
Verifying the Airy differential equation::
>>> for z in [-3.4, 0, 2.5, 1+2j]:
... chop(airybi(z,2) - z*airybi(z))
...
0.0
0.0
0.0
0.0
The first few terms of the Taylor series expansion around `z = 0`
(every third term is zero)::
>>> nprint(taylor(airybi, 0, 5))
[0.614927, 0.448288, 0.0, 0.102488, 0.0373574, 0.0]
The Airy functions can be expressed in terms of Bessel
functions of order `\pm 1/3`. For `\Re[z] \le 0`, we have::
>>> z = -3
>>> airybi(z)
-0.1982896263749265432206449
>>> p = 2*power(-z,'3/2')/3
>>> sqrt(-mpf(z)/3)*(besselj('-1/3',p) - besselj('1/3',p))
-0.1982896263749265432206449
**Derivatives and integrals**
Derivatives of the Bi-function (directly and using :func:`~mpmath.diff`)::
>>> airybi(-3,1); diff(airybi,-3)
-0.675611222685258537668032
-0.675611222685258537668032
>>> airybi(-3,2); diff(airybi,-3,2)
0.5948688791247796296619346
0.5948688791247796296619346
>>> airybi(1000,1); diff(airybi,1000)
1.710055114624614989262335e+9156
1.710055114624614989262335e+9156
Several derivatives at `z = 0`::
>>> airybi(0,0); airybi(0,1); airybi(0,2)
0.6149266274460007351509224
0.4482883573538263579148237
0.0
>>> airybi(0,3); airybi(0,4); airybi(0,5)
0.6149266274460007351509224
0.8965767147076527158296474
0.0
>>> airybi(0,15); airybi(0,16); airybi(0,17)
2238.332923903442675949357
5522.912562599140729510628
0.0
The integral of the Bi-function::
>>> airybi(3,-1); quad(airybi, [0,3])
10.06200303130620056316655
10.06200303130620056316655
>>> airybi(-10,-1); quad(airybi, [0,-10])
-0.01504042480614002045135483
-0.01504042480614002045135483
Integrals of high or fractional order::
>>> airybi(-2,0.5); differint(airybi, -2, 0.5, 0)
(0.0 + 0.5019859055341699223453257j)
(0.0 + 0.5019859055341699223453257j)
>>> airybi(-2,-4); differint(airybi,-2,-4,0)
0.2809314599922447252139092
0.2809314599922447252139092
>>> airybi(0,-1); airybi(0,-2); airybi(0,-3)
0.0
0.0
0.0
Integrals of the Bi-function can be evaluated at limit points::
>>> airybi(-1000000,-1); airybi(-inf,-1)
0.000002191261128063434047966873
0.0
>>> airybi(10,-1); airybi(+inf,-1)
147809803.1074067161675853
+inf
>>> airybi(+inf,-2); airybi(+inf,-3)
+inf
+inf
>>> airybi(-1000000,-2); airybi(-inf,-2)
0.4482883750599908479851085
0.4482883573538263579148237
>>> gamma('2/3')*power(3,'2/3')/(2*pi)
0.4482883573538263579148237
>>> airybi(-100000,-3); airybi(-inf,-3)
-44828.52827206932872493133
-inf
>>> airybi(-100000,-4); airybi(-inf,-4)
2241411040.437759489540248
+inf
"""
airyaizero = r"""
Gives the `k`-th zero of the Airy Ai-function,
i.e. the `k`-th number `a_k` ordered by magnitude for which
`\operatorname{Ai}(a_k) = 0`.
Optionally, with *derivative=1*, the corresponding
zero `a'_k` of the derivative function, i.e.
`\operatorname{Ai}'(a'_k) = 0`, is computed.
**Examples**
Some values of `a_k`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> airyaizero(1)
-2.338107410459767038489197
>>> airyaizero(2)
-4.087949444130970616636989
>>> airyaizero(3)
-5.520559828095551059129856
>>> airyaizero(1000)
-281.0315196125215528353364
Some values of `a'_k`::
>>> airyaizero(1,1)
-1.018792971647471089017325
>>> airyaizero(2,1)
-3.248197582179836537875424
>>> airyaizero(3,1)
-4.820099211178735639400616
>>> airyaizero(1000,1)
-280.9378080358935070607097
Verification::
>>> chop(airyai(airyaizero(1)))
0.0
>>> chop(airyai(airyaizero(1,1),1))
0.0
"""
airybizero = r"""
With *complex=False*, gives the `k`-th real zero of the Airy Bi-function,
i.e. the `k`-th number `b_k` ordered by magnitude for which
`\operatorname{Bi}(b_k) = 0`.
With *complex=True*, gives the `k`-th complex zero in the upper
half plane `\beta_k`. Also the conjugate `\overline{\beta_k}`
is a zero.
Optionally, with *derivative=1*, the corresponding
zero `b'_k` or `\beta'_k` of the derivative function, i.e.
`\operatorname{Bi}'(b'_k) = 0` or `\operatorname{Bi}'(\beta'_k) = 0`,
is computed.
**Examples**
Some values of `b_k`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> airybizero(1)
-1.17371322270912792491998
>>> airybizero(2)
-3.271093302836352715680228
>>> airybizero(3)
-4.830737841662015932667709
>>> airybizero(1000)
-280.9378112034152401578834
Some values of `b_k`::
>>> airybizero(1,1)
-2.294439682614123246622459
>>> airybizero(2,1)
-4.073155089071828215552369
>>> airybizero(3,1)
-5.512395729663599496259593
>>> airybizero(1000,1)
-281.0315164471118527161362
Some values of `\beta_k`::
>>> airybizero(1,complex=True)
(0.9775448867316206859469927 + 2.141290706038744575749139j)
>>> airybizero(2,complex=True)
(1.896775013895336346627217 + 3.627291764358919410440499j)
>>> airybizero(3,complex=True)
(2.633157739354946595708019 + 4.855468179979844983174628j)
>>> airybizero(1000,complex=True)
(140.4978560578493018899793 + 243.3907724215792121244867j)
Some values of `\beta'_k`::
>>> airybizero(1,1,complex=True)
(0.2149470745374305676088329 + 1.100600143302797880647194j)
>>> airybizero(2,1,complex=True)
(1.458168309223507392028211 + 2.912249367458445419235083j)
>>> airybizero(3,1,complex=True)
(2.273760763013482299792362 + 4.254528549217097862167015j)
>>> airybizero(1000,1,complex=True)
(140.4509972835270559730423 + 243.3096175398562811896208j)
Verification::
>>> chop(airybi(airybizero(1)))
0.0
>>> chop(airybi(airybizero(1,1),1))
0.0
>>> u = airybizero(1,complex=True)
>>> chop(airybi(u))
0.0
>>> chop(airybi(conj(u)))
0.0
The complex zeros (in the upper and lower half-planes respectively)
asymptotically approach the rays `z = R \exp(\pm i \pi /3)`::
>>> arg(airybizero(1,complex=True))
1.142532510286334022305364
>>> arg(airybizero(1000,complex=True))
1.047271114786212061583917
>>> arg(airybizero(1000000,complex=True))
1.047197624741816183341355
>>> pi/3
1.047197551196597746154214
"""
ellipk = r"""
Evaluates the complete elliptic integral of the first kind,
`K(m)`, defined by
.. math ::
K(m) = \int_0^{\pi/2} \frac{dt}{\sqrt{1-m \sin^2 t}} \, = \,
\frac{\pi}{2} \,_2F_1\left(\frac{1}{2}, \frac{1}{2}, 1, m\right).
Note that the argument is the parameter `m = k^2`,
not the modulus `k` which is sometimes used.
**Plots**
.. literalinclude :: /plots/ellipk.py
.. image :: /plots/ellipk.png
**Examples**
Values and limits include::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> ellipk(0)
1.570796326794896619231322
>>> ellipk(inf)
(0.0 + 0.0j)
>>> ellipk(-inf)
0.0
>>> ellipk(1)
+inf
>>> ellipk(-1)
1.31102877714605990523242
>>> ellipk(2)
(1.31102877714605990523242 - 1.31102877714605990523242j)
Verifying the defining integral and hypergeometric
representation::
>>> ellipk(0.5)
1.85407467730137191843385
>>> quad(lambda t: (1-0.5*sin(t)**2)**-0.5, [0, pi/2])
1.85407467730137191843385
>>> pi/2*hyp2f1(0.5,0.5,1,0.5)
1.85407467730137191843385
Evaluation is supported for arbitrary complex `m`::
>>> ellipk(3+4j)
(0.9111955638049650086562171 + 0.6313342832413452438845091j)
A definite integral::
>>> quad(ellipk, [0, 1])
2.0
"""
agm = r"""
``agm(a, b)`` computes the arithmetic-geometric mean of `a` and
`b`, defined as the limit of the following iteration:
.. math ::
a_0 = a
b_0 = b
a_{n+1} = \frac{a_n+b_n}{2}
b_{n+1} = \sqrt{a_n b_n}
This function can be called with a single argument, computing
`\mathrm{agm}(a,1) = \mathrm{agm}(1,a)`.
**Examples**
It is a well-known theorem that the geometric mean of
two distinct positive numbers is less than the arithmetic
mean. It follows that the arithmetic-geometric mean lies
between the two means::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> a = mpf(3)
>>> b = mpf(4)
>>> sqrt(a*b)
3.46410161513775
>>> agm(a,b)
3.48202767635957
>>> (a+b)/2
3.5
The arithmetic-geometric mean is scale-invariant::
>>> agm(10*e, 10*pi)
29.261085515723
>>> 10*agm(e, pi)
29.261085515723
As an order-of-magnitude estimate, `\mathrm{agm}(1,x) \approx x`
for large `x`::
>>> agm(10**10)
643448704.760133
>>> agm(10**50)
1.34814309345871e+48
For tiny `x`, `\mathrm{agm}(1,x) \approx -\pi/(2 \log(x/4))`::
>>> agm('0.01')
0.262166887202249
>>> -pi/2/log('0.0025')
0.262172347753122
The arithmetic-geometric mean can also be computed for complex
numbers::
>>> agm(3, 2+j)
(2.51055133276184 + 0.547394054060638j)
The AGM iteration converges very quickly (each step doubles
the number of correct digits), so :func:`~mpmath.agm` supports efficient
high-precision evaluation::
>>> mp.dps = 10000
>>> a = agm(1,2)
>>> str(a)[-10:]
'1679581912'
**Mathematical relations**
The arithmetic-geometric mean may be used to evaluate the
following two parametric definite integrals:
.. math ::
I_1 = \int_0^{\infty}
\frac{1}{\sqrt{(x^2+a^2)(x^2+b^2)}} \,dx
I_2 = \int_0^{\pi/2}
\frac{1}{\sqrt{a^2 \cos^2(x) + b^2 \sin^2(x)}} \,dx
We have::
>>> mp.dps = 15
>>> a = 3
>>> b = 4
>>> f1 = lambda x: ((x**2+a**2)*(x**2+b**2))**-0.5
>>> f2 = lambda x: ((a*cos(x))**2 + (b*sin(x))**2)**-0.5
>>> quad(f1, [0, inf])
0.451115405388492
>>> quad(f2, [0, pi/2])
0.451115405388492
>>> pi/(2*agm(a,b))
0.451115405388492
A formula for `\Gamma(1/4)`::
>>> gamma(0.25)
3.62560990822191
>>> sqrt(2*sqrt(2*pi**3)/agm(1,sqrt(2)))
3.62560990822191
**Possible issues**
The branch cut chosen for complex `a` and `b` is somewhat
arbitrary.
"""
gegenbauer = r"""
Evaluates the Gegenbauer polynomial, or ultraspherical polynomial,
.. math ::
C_n^{(a)}(z) = {n+2a-1 \choose n} \,_2F_1\left(-n, n+2a;
a+\frac{1}{2}; \frac{1}{2}(1-z)\right).
When `n` is a nonnegative integer, this formula gives a polynomial
in `z` of degree `n`, but all parameters are permitted to be
complex numbers. With `a = 1/2`, the Gegenbauer polynomial
reduces to a Legendre polynomial.
**Examples**
Evaluation for arbitrary arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> gegenbauer(3, 0.5, -10)
-2485.0
>>> gegenbauer(1000, 10, 100)
3.012757178975667428359374e+2322
>>> gegenbauer(2+3j, -0.75, -1000j)
(-5038991.358609026523401901 + 9414549.285447104177860806j)
Evaluation at negative integer orders::
>>> gegenbauer(-4, 2, 1.75)
-1.0
>>> gegenbauer(-4, 3, 1.75)
0.0
>>> gegenbauer(-4, 2j, 1.75)
0.0
>>> gegenbauer(-7, 0.5, 3)
8989.0
The Gegenbauer polynomials solve the differential equation::
>>> n, a = 4.5, 1+2j
>>> f = lambda z: gegenbauer(n, a, z)
>>> for z in [0, 0.75, -0.5j]:
... chop((1-z**2)*diff(f,z,2) - (2*a+1)*z*diff(f,z) + n*(n+2*a)*f(z))
...
0.0
0.0
0.0
The Gegenbauer polynomials have generating function
`(1-2zt+t^2)^{-a}`::
>>> a, z = 2.5, 1
>>> taylor(lambda t: (1-2*z*t+t**2)**(-a), 0, 3)
[1.0, 5.0, 15.0, 35.0]
>>> [gegenbauer(n,a,z) for n in range(4)]
[1.0, 5.0, 15.0, 35.0]
The Gegenbauer polynomials are orthogonal on `[-1, 1]` with respect
to the weight `(1-z^2)^{a-\frac{1}{2}}`::
>>> a, n, m = 2.5, 4, 5
>>> Cn = lambda z: gegenbauer(n, a, z, zeroprec=1000)
>>> Cm = lambda z: gegenbauer(m, a, z, zeroprec=1000)
>>> chop(quad(lambda z: Cn(z)*Cm(z)*(1-z**2)*(a-0.5), [-1, 1]))
0.0
"""
laguerre = r"""
Gives the generalized (associated) Laguerre polynomial, defined by
.. math ::
L_n^a(z) = \frac{\Gamma(n+b+1)}{\Gamma(b+1) \Gamma(n+1)}
\,_1F_1(-n, a+1, z).
With `a = 0` and `n` a nonnegative integer, this reduces to an ordinary
Laguerre polynomial, the sequence of which begins
`L_0(z) = 1, L_1(z) = 1-z, L_2(z) = z^2-2z+1, \ldots`.
The Laguerre polynomials are orthogonal with respect to the weight
`z^a e^{-z}` on `[0, \infty)`.
**Plots**
.. literalinclude :: /plots/laguerre.py
.. image :: /plots/laguerre.png
**Examples**
Evaluation for arbitrary arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> laguerre(5, 0, 0.25)
0.03726399739583333333333333
>>> laguerre(1+j, 0.5, 2+3j)
(4.474921610704496808379097 - 11.02058050372068958069241j)
>>> laguerre(2, 0, 10000)
49980001.0
>>> laguerre(2.5, 0, 10000)
-9.327764910194842158583189e+4328
The first few Laguerre polynomials, normalized to have integer
coefficients::
>>> for n in range(7):
... chop(taylor(lambda z: fac(n)*laguerre(n, 0, z), 0, n))
...
[1.0]
[1.0, -1.0]
[2.0, -4.0, 1.0]
[6.0, -18.0, 9.0, -1.0]
[24.0, -96.0, 72.0, -16.0, 1.0]
[120.0, -600.0, 600.0, -200.0, 25.0, -1.0]
[720.0, -4320.0, 5400.0, -2400.0, 450.0, -36.0, 1.0]
Verifying orthogonality::
>>> Lm = lambda t: laguerre(m,a,t)
>>> Ln = lambda t: laguerre(n,a,t)
>>> a, n, m = 2.5, 2, 3
>>> chop(quad(lambda t: exp(-t)*t**a*Lm(t)*Ln(t), [0,inf]))
0.0
"""
hermite = r"""
Evaluates the Hermite polynomial `H_n(z)`, which may be defined using
the recurrence
.. math ::
H_0(z) = 1
H_1(z) = 2z
H_{n+1} = 2z H_n(z) - 2n H_{n-1}(z).
The Hermite polynomials are orthogonal on `(-\infty, \infty)` with
respect to the weight `e^{-z^2}`. More generally, allowing arbitrary complex
values of `n`, the Hermite function `H_n(z)` is defined as
.. math ::
H_n(z) = (2z)^n \,_2F_0\left(-\frac{n}{2}, \frac{1-n}{2},
-\frac{1}{z^2}\right)
for `\Re{z} > 0`, or generally
.. math ::
H_n(z) = 2^n \sqrt{\pi} \left(
\frac{1}{\Gamma\left(\frac{1-n}{2}\right)}
\,_1F_1\left(-\frac{n}{2}, \frac{1}{2}, z^2\right) -
\frac{2z}{\Gamma\left(-\frac{n}{2}\right)}
\,_1F_1\left(\frac{1-n}{2}, \frac{3}{2}, z^2\right)
\right).
**Plots**
.. literalinclude :: /plots/hermite.py
.. image :: /plots/hermite.png
**Examples**
Evaluation for arbitrary arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hermite(0, 10)
1.0
>>> hermite(1, 10); hermite(2, 10)
20.0
398.0
>>> hermite(10000, 2)
4.950440066552087387515653e+19334
>>> hermite(3, -10**8)
-7999999999999998800000000.0
>>> hermite(-3, -10**8)
1.675159751729877682920301e+4342944819032534
>>> hermite(2+3j, -1+2j)
(-0.07652130602993513389421901 - 0.1084662449961914580276007j)
Coefficients of the first few Hermite polynomials are::
>>> for n in range(7):
... chop(taylor(lambda z: hermite(n, z), 0, n))
...
[1.0]
[0.0, 2.0]
[-2.0, 0.0, 4.0]
[0.0, -12.0, 0.0, 8.0]
[12.0, 0.0, -48.0, 0.0, 16.0]
[0.0, 120.0, 0.0, -160.0, 0.0, 32.0]
[-120.0, 0.0, 720.0, 0.0, -480.0, 0.0, 64.0]
Values at `z = 0`::
>>> for n in range(-5, 9):
... hermite(n, 0)
...
0.02769459142039868792653387
0.08333333333333333333333333
0.2215567313631895034122709
0.5
0.8862269254527580136490837
1.0
0.0
-2.0
0.0
12.0
0.0
-120.0
0.0
1680.0
Hermite functions satisfy the differential equation::
>>> n = 4
>>> f = lambda z: hermite(n, z)
>>> z = 1.5
>>> chop(diff(f,z,2) - 2*z*diff(f,z) + 2*n*f(z))
0.0
Verifying orthogonality::
>>> chop(quad(lambda t: hermite(2,t)*hermite(4,t)*exp(-t**2), [-inf,inf]))
0.0
"""
jacobi = r"""
``jacobi(n, a, b, x)`` evaluates the Jacobi polynomial
`P_n^{(a,b)}(x)`. The Jacobi polynomials are a special
case of the hypergeometric function `\,_2F_1` given by:
.. math ::
P_n^{(a,b)}(x) = {n+a \choose n}
\,_2F_1\left(-n,1+a+b+n,a+1,\frac{1-x}{2}\right).
Note that this definition generalizes to nonintegral values
of `n`. When `n` is an integer, the hypergeometric series
terminates after a finite number of terms, giving
a polynomial in `x`.
**Evaluation of Jacobi polynomials**
A special evaluation is `P_n^{(a,b)}(1) = {n+a \choose n}`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> jacobi(4, 0.5, 0.25, 1)
2.4609375
>>> binomial(4+0.5, 4)
2.4609375
A Jacobi polynomial of degree `n` is equal to its
Taylor polynomial of degree `n`. The explicit
coefficients of Jacobi polynomials can therefore
be recovered easily using :func:`~mpmath.taylor`::
>>> for n in range(5):
... nprint(taylor(lambda x: jacobi(n,1,2,x), 0, n))
...
[1.0]
[-0.5, 2.5]
[-0.75, -1.5, 5.25]
[0.5, -3.5, -3.5, 10.5]
[0.625, 2.5, -11.25, -7.5, 20.625]
For nonintegral `n`, the Jacobi "polynomial" is no longer
a polynomial::
>>> nprint(taylor(lambda x: jacobi(0.5,1,2,x), 0, 4))
[0.309983, 1.84119, -1.26933, 1.26699, -1.34808]
**Orthogonality**
The Jacobi polynomials are orthogonal on the interval
`[-1, 1]` with respect to the weight function
`w(x) = (1-x)^a (1+x)^b`. That is,
`w(x) P_n^{(a,b)}(x) P_m^{(a,b)}(x)` integrates to
zero if `m \ne n` and to a nonzero number if `m = n`.
The orthogonality is easy to verify using numerical
quadrature::
>>> P = jacobi
>>> f = lambda x: (1-x)**a * (1+x)**b * P(m,a,b,x) * P(n,a,b,x)
>>> a = 2
>>> b = 3
>>> m, n = 3, 4
>>> chop(quad(f, [-1, 1]), 1)
0.0
>>> m, n = 4, 4
>>> quad(f, [-1, 1])
1.9047619047619
**Differential equation**
The Jacobi polynomials are solutions of the differential
equation
.. math ::
(1-x^2) y'' + (b-a-(a+b+2)x) y' + n (n+a+b+1) y = 0.
We can verify that :func:`~mpmath.jacobi` approximately satisfies
this equation::
>>> from mpmath import *
>>> mp.dps = 15
>>> a = 2.5
>>> b = 4
>>> n = 3
>>> y = lambda x: jacobi(n,a,b,x)
>>> x = pi
>>> A0 = n*(n+a+b+1)*y(x)
>>> A1 = (b-a-(a+b+2)*x)*diff(y,x)
>>> A2 = (1-x**2)*diff(y,x,2)
>>> nprint(A2 + A1 + A0, 1)
4.0e-12
The difference of order `10^{-12}` is as close to zero as
it could be at 15-digit working precision, since the terms
are large::
>>> A0, A1, A2
(26560.2328981879, -21503.7641037294, -5056.46879445852)
"""
legendre = r"""
``legendre(n, x)`` evaluates the Legendre polynomial `P_n(x)`.
The Legendre polynomials are given by the formula
.. math ::
P_n(x) = \frac{1}{2^n n!} \frac{d^n}{dx^n} (x^2 -1)^n.
Alternatively, they can be computed recursively using
.. math ::
P_0(x) = 1
P_1(x) = x
(n+1) P_{n+1}(x) = (2n+1) x P_n(x) - n P_{n-1}(x).
A third definition is in terms of the hypergeometric function
`\,_2F_1`, whereby they can be generalized to arbitrary `n`:
.. math ::
P_n(x) = \,_2F_1\left(-n, n+1, 1, \frac{1-x}{2}\right)
**Plots**
.. literalinclude :: /plots/legendre.py
.. image :: /plots/legendre.png
**Basic evaluation**
The Legendre polynomials assume fixed values at the points
`x = -1` and `x = 1`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> nprint([legendre(n, 1) for n in range(6)])
[1.0, 1.0, 1.0, 1.0, 1.0, 1.0]
>>> nprint([legendre(n, -1) for n in range(6)])
[1.0, -1.0, 1.0, -1.0, 1.0, -1.0]
The coefficients of Legendre polynomials can be recovered
using degree-`n` Taylor expansion::
>>> for n in range(5):
... nprint(chop(taylor(lambda x: legendre(n, x), 0, n)))
...
[1.0]
[0.0, 1.0]
[-0.5, 0.0, 1.5]
[0.0, -1.5, 0.0, 2.5]
[0.375, 0.0, -3.75, 0.0, 4.375]
The roots of Legendre polynomials are located symmetrically
on the interval `[-1, 1]`::
>>> for n in range(5):
... nprint(polyroots(taylor(lambda x: legendre(n, x), 0, n)[::-1]))
...
[]
[0.0]
[-0.57735, 0.57735]
[-0.774597, 0.0, 0.774597]
[-0.861136, -0.339981, 0.339981, 0.861136]
An example of an evaluation for arbitrary `n`::
>>> legendre(0.75, 2+4j)
(1.94952805264875 + 2.1071073099422j)
**Orthogonality**
The Legendre polynomials are orthogonal on `[-1, 1]` with respect
to the trivial weight `w(x) = 1`. That is, `P_m(x) P_n(x)`
integrates to zero if `m \ne n` and to `2/(2n+1)` if `m = n`::
>>> m, n = 3, 4
>>> quad(lambda x: legendre(m,x)*legendre(n,x), [-1, 1])
0.0
>>> m, n = 4, 4
>>> quad(lambda x: legendre(m,x)*legendre(n,x), [-1, 1])
0.222222222222222
**Differential equation**
The Legendre polynomials satisfy the differential equation
.. math ::
((1-x^2) y')' + n(n+1) y' = 0.
We can verify this numerically::
>>> n = 3.6
>>> x = 0.73
>>> P = legendre
>>> A = diff(lambda t: (1-t**2)*diff(lambda u: P(n,u), t), x)
>>> B = n*(n+1)*P(n,x)
>>> nprint(A+B,1)
9.0e-16
"""
legenp = r"""
Calculates the (associated) Legendre function of the first kind of
degree *n* and order *m*, `P_n^m(z)`. Taking `m = 0` gives the ordinary
Legendre function of the first kind, `P_n(z)`. The parameters may be
complex numbers.
In terms of the Gauss hypergeometric function, the (associated) Legendre
function is defined as
.. math ::
P_n^m(z) = \frac{1}{\Gamma(1-m)} \frac{(1+z)^{m/2}}{(1-z)^{m/2}}
\,_2F_1\left(-n, n+1, 1-m, \frac{1-z}{2}\right).
With *type=3* instead of *type=2*, the alternative
definition
.. math ::
\hat{P}_n^m(z) = \frac{1}{\Gamma(1-m)} \frac{(z+1)^{m/2}}{(z-1)^{m/2}}
\,_2F_1\left(-n, n+1, 1-m, \frac{1-z}{2}\right).
is used. These functions correspond respectively to ``LegendreP[n,m,2,z]``
and ``LegendreP[n,m,3,z]`` in Mathematica.
The general solution of the (associated) Legendre differential equation
.. math ::
(1-z^2) f''(z) - 2zf'(z) + \left(n(n+1)-\frac{m^2}{1-z^2}\right)f(z) = 0
is given by `C_1 P_n^m(z) + C_2 Q_n^m(z)` for arbitrary constants
`C_1`, `C_2`, where `Q_n^m(z)` is a Legendre function of the
second kind as implemented by :func:`~mpmath.legenq`.
**Examples**
Evaluation for arbitrary parameters and arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> legenp(2, 0, 10); legendre(2, 10)
149.5
149.5
>>> legenp(-2, 0.5, 2.5)
(1.972260393822275434196053 - 1.972260393822275434196053j)
>>> legenp(2+3j, 1-j, -0.5+4j)
(-3.335677248386698208736542 - 5.663270217461022307645625j)
>>> chop(legenp(3, 2, -1.5, type=2))
28.125
>>> chop(legenp(3, 2, -1.5, type=3))
-28.125
Verifying the associated Legendre differential equation::
>>> n, m = 2, -0.5
>>> C1, C2 = 1, -3
>>> f = lambda z: C1*legenp(n,m,z) + C2*legenq(n,m,z)
>>> deq = lambda z: (1-z**2)*diff(f,z,2) - 2*z*diff(f,z) + \
... (n*(n+1)-m**2/(1-z**2))*f(z)
>>> for z in [0, 2, -1.5, 0.5+2j]:
... chop(deq(mpmathify(z)))
...
0.0
0.0
0.0
0.0
"""
legenq = r"""
Calculates the (associated) Legendre function of the second kind of
degree *n* and order *m*, `Q_n^m(z)`. Taking `m = 0` gives the ordinary
Legendre function of the second kind, `Q_n(z)`. The parameters may
complex numbers.
The Legendre functions of the second kind give a second set of
solutions to the (associated) Legendre differential equation.
(See :func:`~mpmath.legenp`.)
Unlike the Legendre functions of the first kind, they are not
polynomials of `z` for integer `n`, `m` but rational or logarithmic
functions with poles at `z = \pm 1`.
There are various ways to define Legendre functions of
the second kind, giving rise to different complex structure.
A version can be selected using the *type* keyword argument.
The *type=2* and *type=3* functions are given respectively by
.. math ::
Q_n^m(z) = \frac{\pi}{2 \sin(\pi m)}
\left( \cos(\pi m) P_n^m(z) -
\frac{\Gamma(1+m+n)}{\Gamma(1-m+n)} P_n^{-m}(z)\right)
\hat{Q}_n^m(z) = \frac{\pi}{2 \sin(\pi m)} e^{\pi i m}
\left( \hat{P}_n^m(z) -
\frac{\Gamma(1+m+n)}{\Gamma(1-m+n)} \hat{P}_n^{-m}(z)\right)
where `P` and `\hat{P}` are the *type=2* and *type=3* Legendre functions
of the first kind. The formulas above should be understood as limits
when `m` is an integer.
These functions correspond to ``LegendreQ[n,m,2,z]`` (or ``LegendreQ[n,m,z]``)
and ``LegendreQ[n,m,3,z]`` in Mathematica. The *type=3* function
is essentially the same as the function defined in
Abramowitz & Stegun (eq. 8.1.3) but with `(z+1)^{m/2}(z-1)^{m/2}` instead
of `(z^2-1)^{m/2}`, giving slightly different branches.
**Examples**
Evaluation for arbitrary parameters and arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> legenq(2, 0, 0.5)
-0.8186632680417568557122028
>>> legenq(-1.5, -2, 2.5)
(0.6655964618250228714288277 + 0.3937692045497259717762649j)
>>> legenq(2-j, 3+4j, -6+5j)
(-10001.95256487468541686564 - 6011.691337610097577791134j)
Different versions of the function::
>>> legenq(2, 1, 0.5)
0.7298060598018049369381857
>>> legenq(2, 1, 1.5)
(-7.902916572420817192300921 + 0.1998650072605976600724502j)
>>> legenq(2, 1, 0.5, type=3)
(2.040524284763495081918338 - 0.7298060598018049369381857j)
>>> chop(legenq(2, 1, 1.5, type=3))
-0.1998650072605976600724502
"""
chebyt = r"""
``chebyt(n, x)`` evaluates the Chebyshev polynomial of the first
kind `T_n(x)`, defined by the identity
.. math ::
T_n(\cos x) = \cos(n x).
The Chebyshev polynomials of the first kind are a special
case of the Jacobi polynomials, and by extension of the
hypergeometric function `\,_2F_1`. They can thus also be
evaluated for nonintegral `n`.
**Plots**
.. literalinclude :: /plots/chebyt.py
.. image :: /plots/chebyt.png
**Basic evaluation**
The coefficients of the `n`-th polynomial can be recovered
using using degree-`n` Taylor expansion::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(5):
... nprint(chop(taylor(lambda x: chebyt(n, x), 0, n)))
...
[1.0]
[0.0, 1.0]
[-1.0, 0.0, 2.0]
[0.0, -3.0, 0.0, 4.0]
[1.0, 0.0, -8.0, 0.0, 8.0]
**Orthogonality**
The Chebyshev polynomials of the first kind are orthogonal
on the interval `[-1, 1]` with respect to the weight
function `w(x) = 1/\sqrt{1-x^2}`::
>>> f = lambda x: chebyt(m,x)*chebyt(n,x)/sqrt(1-x**2)
>>> m, n = 3, 4
>>> nprint(quad(f, [-1, 1]),1)
0.0
>>> m, n = 4, 4
>>> quad(f, [-1, 1])
1.57079632596448
"""
chebyu = r"""
``chebyu(n, x)`` evaluates the Chebyshev polynomial of the second
kind `U_n(x)`, defined by the identity
.. math ::
U_n(\cos x) = \frac{\sin((n+1)x)}{\sin(x)}.
The Chebyshev polynomials of the second kind are a special
case of the Jacobi polynomials, and by extension of the
hypergeometric function `\,_2F_1`. They can thus also be
evaluated for nonintegral `n`.
**Plots**
.. literalinclude :: /plots/chebyu.py
.. image :: /plots/chebyu.png
**Basic evaluation**
The coefficients of the `n`-th polynomial can be recovered
using using degree-`n` Taylor expansion::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(5):
... nprint(chop(taylor(lambda x: chebyu(n, x), 0, n)))
...
[1.0]
[0.0, 2.0]
[-1.0, 0.0, 4.0]
[0.0, -4.0, 0.0, 8.0]
[1.0, 0.0, -12.0, 0.0, 16.0]
**Orthogonality**
The Chebyshev polynomials of the second kind are orthogonal
on the interval `[-1, 1]` with respect to the weight
function `w(x) = \sqrt{1-x^2}`::
>>> f = lambda x: chebyu(m,x)*chebyu(n,x)*sqrt(1-x**2)
>>> m, n = 3, 4
>>> quad(f, [-1, 1])
0.0
>>> m, n = 4, 4
>>> quad(f, [-1, 1])
1.5707963267949
"""
besselj = r"""
``besselj(n, x, derivative=0)`` gives the Bessel function of the first kind
`J_n(x)`. Bessel functions of the first kind are defined as
solutions of the differential equation
.. math ::
x^2 y'' + x y' + (x^2 - n^2) y = 0
which appears, among other things, when solving the radial
part of Laplace's equation in cylindrical coordinates. This
equation has two solutions for given `n`, where the
`J_n`-function is the solution that is nonsingular at `x = 0`.
For positive integer `n`, `J_n(x)` behaves roughly like a sine
(odd `n`) or cosine (even `n`) multiplied by a magnitude factor
that decays slowly as `x \to \pm\infty`.
Generally, `J_n` is a special case of the hypergeometric
function `\,_0F_1`:
.. math ::
J_n(x) = \frac{x^n}{2^n \Gamma(n+1)}
\,_0F_1\left(n+1,-\frac{x^2}{4}\right)
With *derivative* = `m \ne 0`, the `m`-th derivative
.. math ::
\frac{d^m}{dx^m} J_n(x)
is computed.
**Plots**
.. literalinclude :: /plots/besselj.py
.. image :: /plots/besselj.png
.. literalinclude :: /plots/besselj_c.py
.. image :: /plots/besselj_c.png
**Examples**
Evaluation is supported for arbitrary arguments, and at
arbitrary precision::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> besselj(2, 1000)
-0.024777229528606
>>> besselj(4, 0.75)
0.000801070086542314
>>> besselj(2, 1000j)
(-2.48071721019185e+432 + 6.41567059811949e-437j)
>>> mp.dps = 25
>>> besselj(0.75j, 3+4j)
(-2.778118364828153309919653 - 1.5863603889018621585533j)
>>> mp.dps = 50
>>> besselj(1, pi)
0.28461534317975275734531059968613140570981118184947
Arguments may be large::
>>> mp.dps = 25
>>> besselj(0, 10000)
-0.007096160353388801477265164
>>> besselj(0, 10**10)
0.000002175591750246891726859055
>>> besselj(2, 10**100)
7.337048736538615712436929e-51
>>> besselj(2, 10**5*j)
(-3.540725411970948860173735e+43426 + 4.4949812409615803110051e-43433j)
The Bessel functions of the first kind satisfy simple
symmetries around `x = 0`::
>>> mp.dps = 15
>>> nprint([besselj(n,0) for n in range(5)])
[1.0, 0.0, 0.0, 0.0, 0.0]
>>> nprint([besselj(n,pi) for n in range(5)])
[-0.304242, 0.284615, 0.485434, 0.333458, 0.151425]
>>> nprint([besselj(n,-pi) for n in range(5)])
[-0.304242, -0.284615, 0.485434, -0.333458, 0.151425]
Roots of Bessel functions are often used::
>>> nprint([findroot(j0, k) for k in [2, 5, 8, 11, 14]])
[2.40483, 5.52008, 8.65373, 11.7915, 14.9309]
>>> nprint([findroot(j1, k) for k in [3, 7, 10, 13, 16]])
[3.83171, 7.01559, 10.1735, 13.3237, 16.4706]
The roots are not periodic, but the distance between successive
roots asymptotically approaches `2 \pi`. Bessel functions of
the first kind have the following normalization::
>>> quadosc(j0, [0, inf], period=2*pi)
1.0
>>> quadosc(j1, [0, inf], period=2*pi)
1.0
For `n = 1/2` or `n = -1/2`, the Bessel function reduces to a
trigonometric function::
>>> x = 10
>>> besselj(0.5, x), sqrt(2/(pi*x))*sin(x)
(-0.13726373575505, -0.13726373575505)
>>> besselj(-0.5, x), sqrt(2/(pi*x))*cos(x)
(-0.211708866331398, -0.211708866331398)
Derivatives of any order can be computed (negative orders
correspond to integration)::
>>> mp.dps = 25
>>> besselj(0, 7.5, 1)
-0.1352484275797055051822405
>>> diff(lambda x: besselj(0,x), 7.5)
-0.1352484275797055051822405
>>> besselj(0, 7.5, 10)
-0.1377811164763244890135677
>>> diff(lambda x: besselj(0,x), 7.5, 10)
-0.1377811164763244890135677
>>> besselj(0,7.5,-1) - besselj(0,3.5,-1)
-0.1241343240399987693521378
>>> quad(j0, [3.5, 7.5])
-0.1241343240399987693521378
Differentiation with a noninteger order gives the fractional derivative
in the sense of the Riemann-Liouville differintegral, as computed by
:func:`~mpmath.differint`::
>>> mp.dps = 15
>>> besselj(1, 3.5, 0.75)
-0.385977722939384
>>> differint(lambda x: besselj(1, x), 3.5, 0.75)
-0.385977722939384
"""
besseli = r"""
``besseli(n, x, derivative=0)`` gives the modified Bessel function of the
first kind,
.. math ::
I_n(x) = i^{-n} J_n(ix).
With *derivative* = `m \ne 0`, the `m`-th derivative
.. math ::
\frac{d^m}{dx^m} I_n(x)
is computed.
**Plots**
.. literalinclude :: /plots/besseli.py
.. image :: /plots/besseli.png
.. literalinclude :: /plots/besseli_c.py
.. image :: /plots/besseli_c.png
**Examples**
Some values of `I_n(x)`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> besseli(0,0)
1.0
>>> besseli(1,0)
0.0
>>> besseli(0,1)
1.266065877752008335598245
>>> besseli(3.5, 2+3j)
(-0.2904369752642538144289025 - 0.4469098397654815837307006j)
Arguments may be large::
>>> besseli(2, 1000)
2.480717210191852440616782e+432
>>> besseli(2, 10**10)
4.299602851624027900335391e+4342944813
>>> besseli(2, 6000+10000j)
(-2.114650753239580827144204e+2603 + 4.385040221241629041351886e+2602j)
For integers `n`, the following integral representation holds::
>>> mp.dps = 15
>>> n = 3
>>> x = 2.3
>>> quad(lambda t: exp(x*cos(t))*cos(n*t), [0,pi])/pi
0.349223221159309
>>> besseli(n,x)
0.349223221159309
Derivatives and antiderivatives of any order can be computed::
>>> mp.dps = 25
>>> besseli(2, 7.5, 1)
195.8229038931399062565883
>>> diff(lambda x: besseli(2,x), 7.5)
195.8229038931399062565883
>>> besseli(2, 7.5, 10)
153.3296508971734525525176
>>> diff(lambda x: besseli(2,x), 7.5, 10)
153.3296508971734525525176
>>> besseli(2,7.5,-1) - besseli(2,3.5,-1)
202.5043900051930141956876
>>> quad(lambda x: besseli(2,x), [3.5, 7.5])
202.5043900051930141956876
"""
bessely = r"""
``bessely(n, x, derivative=0)`` gives the Bessel function of the second kind,
.. math ::
Y_n(x) = \frac{J_n(x) \cos(\pi n) - J_{-n}(x)}{\sin(\pi n)}.
For `n` an integer, this formula should be understood as a
limit. With *derivative* = `m \ne 0`, the `m`-th derivative
.. math ::
\frac{d^m}{dx^m} Y_n(x)
is computed.
**Plots**
.. literalinclude :: /plots/bessely.py
.. image :: /plots/bessely.png
.. literalinclude :: /plots/bessely_c.py
.. image :: /plots/bessely_c.png
**Examples**
Some values of `Y_n(x)`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> bessely(0,0), bessely(1,0), bessely(2,0)
(-inf, -inf, -inf)
>>> bessely(1, pi)
0.3588729167767189594679827
>>> bessely(0.5, 3+4j)
(9.242861436961450520325216 - 3.085042824915332562522402j)
Arguments may be large::
>>> bessely(0, 10000)
0.00364780555898660588668872
>>> bessely(2.5, 10**50)
-4.8952500412050989295774e-26
>>> bessely(2.5, -10**50)
(0.0 + 4.8952500412050989295774e-26j)
Derivatives and antiderivatives of any order can be computed::
>>> bessely(2, 3.5, 1)
0.3842618820422660066089231
>>> diff(lambda x: bessely(2, x), 3.5)
0.3842618820422660066089231
>>> bessely(0.5, 3.5, 1)
-0.2066598304156764337900417
>>> diff(lambda x: bessely(0.5, x), 3.5)
-0.2066598304156764337900417
>>> diff(lambda x: bessely(2, x), 0.5, 10)
-208173867409.5547350101511
>>> bessely(2, 0.5, 10)
-208173867409.5547350101511
>>> bessely(2, 100.5, 100)
0.02668487547301372334849043
>>> quad(lambda x: bessely(2,x), [1,3])
-1.377046859093181969213262
>>> bessely(2,3,-1) - bessely(2,1,-1)
-1.377046859093181969213262
"""
besselk = r"""
``besselk(n, x)`` gives the modified Bessel function of the
second kind,
.. math ::
K_n(x) = \frac{\pi}{2} \frac{I_{-n}(x)-I_{n}(x)}{\sin(\pi n)}
For `n` an integer, this formula should be understood as a
limit.
**Plots**
.. literalinclude :: /plots/besselk.py
.. image :: /plots/besselk.png
.. literalinclude :: /plots/besselk_c.py
.. image :: /plots/besselk_c.png
**Examples**
Evaluation is supported for arbitrary complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> besselk(0,1)
0.4210244382407083333356274
>>> besselk(0, -1)
(0.4210244382407083333356274 - 3.97746326050642263725661j)
>>> besselk(3.5, 2+3j)
(-0.02090732889633760668464128 + 0.2464022641351420167819697j)
>>> besselk(2+3j, 0.5)
(0.9615816021726349402626083 + 0.1918250181801757416908224j)
Arguments may be large::
>>> besselk(0, 100)
4.656628229175902018939005e-45
>>> besselk(1, 10**6)
4.131967049321725588398296e-434298
>>> besselk(1, 10**6*j)
(0.001140348428252385844876706 - 0.0005200017201681152909000961j)
>>> besselk(4.5, fmul(10**50, j, exact=True))
(1.561034538142413947789221e-26 + 1.243554598118700063281496e-25j)
The point `x = 0` is a singularity (logarithmic if `n = 0`)::
>>> besselk(0,0)
+inf
>>> besselk(1,0)
+inf
>>> for n in range(-4, 5):
... print(besselk(n, '1e-1000'))
...
4.8e+4001
8.0e+3000
2.0e+2000
1.0e+1000
2302.701024509704096466802
1.0e+1000
2.0e+2000
8.0e+3000
4.8e+4001
"""
hankel1 = r"""
``hankel1(n,x)`` computes the Hankel function of the first kind,
which is the complex combination of Bessel functions given by
.. math ::
H_n^{(1)}(x) = J_n(x) + i Y_n(x).
**Plots**
.. literalinclude :: /plots/hankel1.py
.. image :: /plots/hankel1.png
.. literalinclude :: /plots/hankel1_c.py
.. image :: /plots/hankel1_c.png
**Examples**
The Hankel function is generally complex-valued::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hankel1(2, pi)
(0.4854339326315091097054957 - 0.0999007139290278787734903j)
>>> hankel1(3.5, pi)
(0.2340002029630507922628888 - 0.6419643823412927142424049j)
"""
hankel2 = r"""
``hankel2(n,x)`` computes the Hankel function of the second kind,
which is the complex combination of Bessel functions given by
.. math ::
H_n^{(2)}(x) = J_n(x) - i Y_n(x).
**Plots**
.. literalinclude :: /plots/hankel2.py
.. image :: /plots/hankel2.png
.. literalinclude :: /plots/hankel2_c.py
.. image :: /plots/hankel2_c.png
**Examples**
The Hankel function is generally complex-valued::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> hankel2(2, pi)
(0.4854339326315091097054957 + 0.0999007139290278787734903j)
>>> hankel2(3.5, pi)
(0.2340002029630507922628888 + 0.6419643823412927142424049j)
"""
lambertw = r"""
The Lambert W function `W(z)` is defined as the inverse function
of `w \exp(w)`. In other words, the value of `W(z)` is such that
`z = W(z) \exp(W(z))` for any complex number `z`.
The Lambert W function is a multivalued function with infinitely
many branches `W_k(z)`, indexed by `k \in \mathbb{Z}`. Each branch
gives a different solution `w` of the equation `z = w \exp(w)`.
All branches are supported by :func:`~mpmath.lambertw`:
* ``lambertw(z)`` gives the principal solution (branch 0)
* ``lambertw(z, k)`` gives the solution on branch `k`
The Lambert W function has two partially real branches: the
principal branch (`k = 0`) is real for real `z > -1/e`, and the
`k = -1` branch is real for `-1/e < z < 0`. All branches except
`k = 0` have a logarithmic singularity at `z = 0`.
The definition, implementation and choice of branches
is based on [Corless]_.
**Plots**
.. literalinclude :: /plots/lambertw.py
.. image :: /plots/lambertw.png
.. literalinclude :: /plots/lambertw_c.py
.. image :: /plots/lambertw_c.png
**Basic examples**
The Lambert W function is the inverse of `w \exp(w)`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> w = lambertw(1)
>>> w
0.5671432904097838729999687
>>> w*exp(w)
1.0
Any branch gives a valid inverse::
>>> w = lambertw(1, k=3)
>>> w
(-2.853581755409037807206819 + 17.11353553941214591260783j)
>>> w = lambertw(1, k=25)
>>> w
(-5.047020464221569709378686 + 155.4763860949415867162066j)
>>> chop(w*exp(w))
1.0
**Applications to equation-solving**
The Lambert W function may be used to solve various kinds of
equations, such as finding the value of the infinite power
tower `z^{z^{z^{\ldots}}}`::
>>> def tower(z, n):
... if n == 0:
... return z
... return z ** tower(z, n-1)
...
>>> tower(mpf(0.5), 100)
0.6411857445049859844862005
>>> -lambertw(-log(0.5))/log(0.5)
0.6411857445049859844862005
**Properties**
The Lambert W function grows roughly like the natural logarithm
for large arguments::
>>> lambertw(1000); log(1000)
5.249602852401596227126056
6.907755278982137052053974
>>> lambertw(10**100); log(10**100)
224.8431064451185015393731
230.2585092994045684017991
The principal branch of the Lambert W function has a rational
Taylor series expansion around `z = 0`::
>>> nprint(taylor(lambertw, 0, 6), 10)
[0.0, 1.0, -1.0, 1.5, -2.666666667, 5.208333333, -10.8]
Some special values and limits are::
>>> lambertw(0)
0.0
>>> lambertw(1)
0.5671432904097838729999687
>>> lambertw(e)
1.0
>>> lambertw(inf)
+inf
>>> lambertw(0, k=-1)
-inf
>>> lambertw(0, k=3)
-inf
>>> lambertw(inf, k=2)
(+inf + 12.56637061435917295385057j)
>>> lambertw(inf, k=3)
(+inf + 18.84955592153875943077586j)
>>> lambertw(-inf, k=3)
(+inf + 21.9911485751285526692385j)
The `k = 0` and `k = -1` branches join at `z = -1/e` where
`W(z) = -1` for both branches. Since `-1/e` can only be represented
approximately with binary floating-point numbers, evaluating the
Lambert W function at this point only gives `-1` approximately::
>>> lambertw(-1/e, 0)
-0.9999999999998371330228251
>>> lambertw(-1/e, -1)
-1.000000000000162866977175
If `-1/e` happens to round in the negative direction, there might be
a small imaginary part::
>>> mp.dps = 15
>>> lambertw(-1/e)
(-1.0 + 8.22007971483662e-9j)
>>> lambertw(-1/e+eps)
-0.999999966242188
**References**
1. [Corless]_
"""
barnesg = r"""
Evaluates the Barnes G-function, which generalizes the
superfactorial (:func:`~mpmath.superfac`) and by extension also the
hyperfactorial (:func:`~mpmath.hyperfac`) to the complex numbers
in an analogous way to how the gamma function generalizes
the ordinary factorial.
The Barnes G-function may be defined in terms of a Weierstrass
product:
.. math ::
G(z+1) = (2\pi)^{z/2} e^{-[z(z+1)+\gamma z^2]/2}
\prod_{n=1}^\infty
\left[\left(1+\frac{z}{n}\right)^ne^{-z+z^2/(2n)}\right]
For positive integers `n`, we have have relation to superfactorials
`G(n) = \mathrm{sf}(n-2) = 0! \cdot 1! \cdots (n-2)!`.
**Examples**
Some elementary values and limits of the Barnes G-function::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> barnesg(1), barnesg(2), barnesg(3)
(1.0, 1.0, 1.0)
>>> barnesg(4)
2.0
>>> barnesg(5)
12.0
>>> barnesg(6)
288.0
>>> barnesg(7)
34560.0
>>> barnesg(8)
24883200.0
>>> barnesg(inf)
+inf
>>> barnesg(0), barnesg(-1), barnesg(-2)
(0.0, 0.0, 0.0)
Closed-form values are known for some rational arguments::
>>> barnesg('1/2')
0.603244281209446
>>> sqrt(exp(0.25+log(2)/12)/sqrt(pi)/glaisher**3)
0.603244281209446
>>> barnesg('1/4')
0.29375596533861
>>> nthroot(exp('3/8')/exp(catalan/pi)/
... gamma(0.25)**3/sqrt(glaisher)**9, 4)
0.29375596533861
The Barnes G-function satisfies the functional equation
`G(z+1) = \Gamma(z) G(z)`::
>>> z = pi
>>> barnesg(z+1)
2.39292119327948
>>> gamma(z)*barnesg(z)
2.39292119327948
The asymptotic growth rate of the Barnes G-function is related to
the Glaisher-Kinkelin constant::
>>> limit(lambda n: barnesg(n+1)/(n**(n**2/2-mpf(1)/12)*
... (2*pi)**(n/2)*exp(-3*n**2/4)), inf)
0.847536694177301
>>> exp('1/12')/glaisher
0.847536694177301
The Barnes G-function can be differentiated in closed form::
>>> z = 3
>>> diff(barnesg, z)
0.264507203401607
>>> barnesg(z)*((z-1)*psi(0,z)-z+(log(2*pi)+1)/2)
0.264507203401607
Evaluation is supported for arbitrary arguments and at arbitrary
precision::
>>> barnesg(6.5)
2548.7457695685
>>> barnesg(-pi)
0.00535976768353037
>>> barnesg(3+4j)
(-0.000676375932234244 - 4.42236140124728e-5j)
>>> mp.dps = 50
>>> barnesg(1/sqrt(2))
0.81305501090451340843586085064413533788206204124732
>>> q = barnesg(10j)
>>> q.real
0.000000000021852360840356557241543036724799812371995850552234
>>> q.imag
-0.00000000000070035335320062304849020654215545839053210041457588
>>> mp.dps = 15
>>> barnesg(100)
3.10361006263698e+6626
>>> barnesg(-101)
0.0
>>> barnesg(-10.5)
5.94463017605008e+25
>>> barnesg(-10000.5)
-6.14322868174828e+167480422
>>> barnesg(1000j)
(5.21133054865546e-1173597 + 4.27461836811016e-1173597j)
>>> barnesg(-1000+1000j)
(2.43114569750291e+1026623 + 2.24851410674842e+1026623j)
**References**
1. Whittaker & Watson, *A Course of Modern Analysis*,
Cambridge University Press, 4th edition (1927), p.264
2. http://en.wikipedia.org/wiki/Barnes_G-function
3. http://mathworld.wolfram.com/BarnesG-Function.html
"""
superfac = r"""
Computes the superfactorial, defined as the product of
consecutive factorials
.. math ::
\mathrm{sf}(n) = \prod_{k=1}^n k!
For general complex `z`, `\mathrm{sf}(z)` is defined
in terms of the Barnes G-function (see :func:`~mpmath.barnesg`).
**Examples**
The first few superfactorials are (OEIS A000178)::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(10):
... print("%s %s" % (n, superfac(n)))
...
0 1.0
1 1.0
2 2.0
3 12.0
4 288.0
5 34560.0
6 24883200.0
7 125411328000.0
8 5.05658474496e+15
9 1.83493347225108e+21
Superfactorials grow very rapidly::
>>> superfac(1000)
3.24570818422368e+1177245
>>> superfac(10**10)
2.61398543581249e+467427913956904067453
Evaluation is supported for arbitrary arguments::
>>> mp.dps = 25
>>> superfac(pi)
17.20051550121297985285333
>>> superfac(2+3j)
(-0.005915485633199789627466468 + 0.008156449464604044948738263j)
>>> diff(superfac, 1)
0.2645072034016070205673056
**References**
1. http://www.research.att.com/~njas/sequences/A000178
"""
hyperfac = r"""
Computes the hyperfactorial, defined for integers as the product
.. math ::
H(n) = \prod_{k=1}^n k^k.
The hyperfactorial satisfies the recurrence formula `H(z) = z^z H(z-1)`.
It can be defined more generally in terms of the Barnes G-function (see
:func:`~mpmath.barnesg`) and the gamma function by the formula
.. math ::
H(z) = \frac{\Gamma(z+1)^z}{G(z)}.
The extension to complex numbers can also be done via
the integral representation
.. math ::
H(z) = (2\pi)^{-z/2} \exp \left[
{z+1 \choose 2} + \int_0^z \log(t!)\,dt
\right].
**Examples**
The rapidly-growing sequence of hyperfactorials begins
(OEIS A002109)::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(10):
... print("%s %s" % (n, hyperfac(n)))
...
0 1.0
1 1.0
2 4.0
3 108.0
4 27648.0
5 86400000.0
6 4031078400000.0
7 3.3197663987712e+18
8 5.56964379417266e+25
9 2.15779412229419e+34
Some even larger hyperfactorials are::
>>> hyperfac(1000)
5.46458120882585e+1392926
>>> hyperfac(10**10)
4.60408207642219e+489142638002418704309
The hyperfactorial can be evaluated for arbitrary arguments::
>>> hyperfac(0.5)
0.880449235173423
>>> diff(hyperfac, 1)
0.581061466795327
>>> hyperfac(pi)
205.211134637462
>>> hyperfac(-10+1j)
(3.01144471378225e+46 - 2.45285242480185e+46j)
The recurrence property of the hyperfactorial holds
generally::
>>> z = 3-4*j
>>> hyperfac(z)
(-4.49795891462086e-7 - 6.33262283196162e-7j)
>>> z**z * hyperfac(z-1)
(-4.49795891462086e-7 - 6.33262283196162e-7j)
>>> z = mpf(-0.6)
>>> chop(z**z * hyperfac(z-1))
1.28170142849352
>>> hyperfac(z)
1.28170142849352
The hyperfactorial may also be computed using the integral
definition::
>>> z = 2.5
>>> hyperfac(z)
15.9842119922237
>>> (2*pi)**(-z/2)*exp(binomial(z+1,2) +
... quad(lambda t: loggamma(t+1), [0, z]))
15.9842119922237
:func:`~mpmath.hyperfac` supports arbitrary-precision evaluation::
>>> mp.dps = 50
>>> hyperfac(10)
215779412229418562091680268288000000000000000.0
>>> hyperfac(1/sqrt(2))
0.89404818005227001975423476035729076375705084390942
**References**
1. http://www.research.att.com/~njas/sequences/A002109
2. http://mathworld.wolfram.com/Hyperfactorial.html
"""
rgamma = r"""
Computes the reciprocal of the gamma function, `1/\Gamma(z)`. This
function evaluates to zero at the poles
of the gamma function, `z = 0, -1, -2, \ldots`.
**Examples**
Basic examples::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> rgamma(1)
1.0
>>> rgamma(4)
0.1666666666666666666666667
>>> rgamma(0); rgamma(-1)
0.0
0.0
>>> rgamma(1000)
2.485168143266784862783596e-2565
>>> rgamma(inf)
0.0
A definite integral that can be evaluated in terms of elementary
integrals::
>>> quad(rgamma, [0,inf])
2.807770242028519365221501
>>> e + quad(lambda t: exp(-t)/(pi**2+log(t)**2), [0,inf])
2.807770242028519365221501
"""
loggamma = r"""
Computes the principal branch of the log-gamma function,
`\ln \Gamma(z)`. Unlike `\ln(\Gamma(z))`, which has infinitely many
complex branch cuts, the principal log-gamma function only has a single
branch cut along the negative half-axis. The principal branch
continuously matches the asymptotic Stirling expansion
.. math ::
\ln \Gamma(z) \sim \frac{\ln(2 \pi)}{2} +
\left(z-\frac{1}{2}\right) \ln(z) - z + O(z^{-1}).
The real parts of both functions agree, but their imaginary
parts generally differ by `2 n \pi` for some `n \in \mathbb{Z}`.
They coincide for `z \in \mathbb{R}, z > 0`.
Computationally, it is advantageous to use :func:`~mpmath.loggamma`
instead of :func:`~mpmath.gamma` for extremely large arguments.
**Examples**
Comparing with `\ln(\Gamma(z))`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> loggamma('13.2'); log(gamma('13.2'))
20.49400419456603678498394
20.49400419456603678498394
>>> loggamma(3+4j)
(-1.756626784603784110530604 + 4.742664438034657928194889j)
>>> log(gamma(3+4j))
(-1.756626784603784110530604 - 1.540520869144928548730397j)
>>> log(gamma(3+4j)) + 2*pi*j
(-1.756626784603784110530604 + 4.742664438034657928194889j)
Note the imaginary parts for negative arguments::
>>> loggamma(-0.5); loggamma(-1.5); loggamma(-2.5)
(1.265512123484645396488946 - 3.141592653589793238462643j)
(0.8600470153764810145109327 - 6.283185307179586476925287j)
(-0.05624371649767405067259453 - 9.42477796076937971538793j)
Some special values::
>>> loggamma(1); loggamma(2)
0.0
0.0
>>> loggamma(3); +ln2
0.6931471805599453094172321
0.6931471805599453094172321
>>> loggamma(3.5); log(15*sqrt(pi)/8)
1.200973602347074224816022
1.200973602347074224816022
>>> loggamma(inf)
+inf
Huge arguments are permitted::
>>> loggamma('1e30')
6.807755278982137052053974e+31
>>> loggamma('1e300')
6.897755278982137052053974e+302
>>> loggamma('1e3000')
6.906755278982137052053974e+3003
>>> loggamma('1e100000000000000000000')
2.302585092994045684007991e+100000000000000000020
>>> loggamma('1e30j')
(-1.570796326794896619231322e+30 + 6.807755278982137052053974e+31j)
>>> loggamma('1e300j')
(-1.570796326794896619231322e+300 + 6.897755278982137052053974e+302j)
>>> loggamma('1e3000j')
(-1.570796326794896619231322e+3000 + 6.906755278982137052053974e+3003j)
The log-gamma function can be integrated analytically
on any interval of unit length::
>>> z = 0
>>> quad(loggamma, [z,z+1]); log(2*pi)/2
0.9189385332046727417803297
0.9189385332046727417803297
>>> z = 3+4j
>>> quad(loggamma, [z,z+1]); (log(z)-1)*z + log(2*pi)/2
(-0.9619286014994750641314421 + 5.219637303741238195688575j)
(-0.9619286014994750641314421 + 5.219637303741238195688575j)
The derivatives of the log-gamma function are given by the
polygamma function (:func:`~mpmath.psi`)::
>>> diff(loggamma, -4+3j); psi(0, -4+3j)
(1.688493531222971393607153 + 2.554898911356806978892748j)
(1.688493531222971393607153 + 2.554898911356806978892748j)
>>> diff(loggamma, -4+3j, 2); psi(1, -4+3j)
(-0.1539414829219882371561038 - 0.1020485197430267719746479j)
(-0.1539414829219882371561038 - 0.1020485197430267719746479j)
The log-gamma function satisfies an additive form of the
recurrence relation for the ordinary gamma function::
>>> z = 2+3j
>>> loggamma(z); loggamma(z+1) - log(z)
(-2.092851753092733349564189 + 2.302396543466867626153708j)
(-2.092851753092733349564189 + 2.302396543466867626153708j)
"""
siegeltheta = r"""
Computes the Riemann-Siegel theta function,
.. math ::
\theta(t) = \frac{
\log\Gamma\left(\frac{1+2it}{4}\right) -
\log\Gamma\left(\frac{1-2it}{4}\right)
}{2i} - \frac{\log \pi}{2} t.
The Riemann-Siegel theta function is important in
providing the phase factor for the Z-function
(see :func:`~mpmath.siegelz`). Evaluation is supported for real and
complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> siegeltheta(0)
0.0
>>> siegeltheta(inf)
+inf
>>> siegeltheta(-inf)
-inf
>>> siegeltheta(1)
-1.767547952812290388302216
>>> siegeltheta(10+0.25j)
(-3.068638039426838572528867 + 0.05804937947429712998395177j)
Arbitrary derivatives may be computed with derivative = k
>>> siegeltheta(1234, derivative=2)
0.0004051864079114053109473741
>>> diff(siegeltheta, 1234, n=2)
0.0004051864079114053109473741
The Riemann-Siegel theta function has odd symmetry around `t = 0`,
two local extreme points and three real roots including 0 (located
symmetrically)::
>>> nprint(chop(taylor(siegeltheta, 0, 5)))
[0.0, -2.68609, 0.0, 2.69433, 0.0, -6.40218]
>>> findroot(diffun(siegeltheta), 7)
6.28983598883690277966509
>>> findroot(siegeltheta, 20)
17.84559954041086081682634
For large `t`, there is a famous asymptotic formula
for `\theta(t)`, to first order given by::
>>> t = mpf(10**6)
>>> siegeltheta(t)
5488816.353078403444882823
>>> -t*log(2*pi/t)/2-t/2
5488816.745777464310273645
"""
grampoint = r"""
Gives the `n`-th Gram point `g_n`, defined as the solution
to the equation `\theta(g_n) = \pi n` where `\theta(t)`
is the Riemann-Siegel theta function (:func:`~mpmath.siegeltheta`).
The first few Gram points are::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> grampoint(0)
17.84559954041086081682634
>>> grampoint(1)
23.17028270124630927899664
>>> grampoint(2)
27.67018221781633796093849
>>> grampoint(3)
31.71797995476405317955149
Checking the definition::
>>> siegeltheta(grampoint(3))
9.42477796076937971538793
>>> 3*pi
9.42477796076937971538793
A large Gram point::
>>> grampoint(10**10)
3293531632.728335454561153
Gram points are useful when studying the Z-function
(:func:`~mpmath.siegelz`). See the documentation of that function
for additional examples.
:func:`~mpmath.grampoint` can solve the defining equation for
nonintegral `n`. There is a fixed point where `g(x) = x`::
>>> findroot(lambda x: grampoint(x) - x, 10000)
9146.698193171459265866198
**References**
1. http://mathworld.wolfram.com/GramPoint.html
"""
siegelz = r"""
Computes the Z-function, also known as the Riemann-Siegel Z function,
.. math ::
Z(t) = e^{i \theta(t)} \zeta(1/2+it)
where `\zeta(s)` is the Riemann zeta function (:func:`~mpmath.zeta`)
and where `\theta(t)` denotes the Riemann-Siegel theta function
(see :func:`~mpmath.siegeltheta`).
Evaluation is supported for real and complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> siegelz(1)
-0.7363054628673177346778998
>>> siegelz(3+4j)
(-0.1852895764366314976003936 - 0.2773099198055652246992479j)
The first four derivatives are supported, using the
optional *derivative* keyword argument::
>>> siegelz(1234567, derivative=3)
56.89689348495089294249178
>>> diff(siegelz, 1234567, n=3)
56.89689348495089294249178
The Z-function has a Maclaurin expansion::
>>> nprint(chop(taylor(siegelz, 0, 4)))
[-1.46035, 0.0, 2.73588, 0.0, -8.39357]
The Z-function `Z(t)` is equal to `\pm |\zeta(s)|` on the
critical line `s = 1/2+it` (i.e. for real arguments `t`
to `Z`). Its zeros coincide with those of the Riemann zeta
function::
>>> findroot(siegelz, 14)
14.13472514173469379045725
>>> findroot(siegelz, 20)
21.02203963877155499262848
>>> findroot(zeta, 0.5+14j)
(0.5 + 14.13472514173469379045725j)
>>> findroot(zeta, 0.5+20j)
(0.5 + 21.02203963877155499262848j)
Since the Z-function is real-valued on the critical line
(and unlike `|\zeta(s)|` analytic), it is useful for
investigating the zeros of the Riemann zeta function.
For example, one can use a root-finding algorithm based
on sign changes::
>>> findroot(siegelz, [100, 200], solver='bisect')
176.4414342977104188888926
To locate roots, Gram points `g_n` which can be computed
by :func:`~mpmath.grampoint` are useful. If `(-1)^n Z(g_n)` is
positive for two consecutive `n`, then `Z(t)` must have
a zero between those points::
>>> g10 = grampoint(10)
>>> g11 = grampoint(11)
>>> (-1)**10 * siegelz(g10) > 0
True
>>> (-1)**11 * siegelz(g11) > 0
True
>>> findroot(siegelz, [g10, g11], solver='bisect')
56.44624769706339480436776
>>> g10, g11
(54.67523744685325626632663, 57.54516517954725443703014)
"""
riemannr = r"""
Evaluates the Riemann R function, a smooth approximation of the
prime counting function `\pi(x)` (see :func:`~mpmath.primepi`). The Riemann
R function gives a fast numerical approximation useful e.g. to
roughly estimate the number of primes in a given interval.
The Riemann R function is computed using the rapidly convergent Gram
series,
.. math ::
R(x) = 1 + \sum_{k=1}^{\infty}
\frac{\log^k x}{k k! \zeta(k+1)}.
From the Gram series, one sees that the Riemann R function is a
well-defined analytic function (except for a branch cut along
the negative real half-axis); it can be evaluated for arbitrary
real or complex arguments.
The Riemann R function gives a very accurate approximation
of the prime counting function. For example, it is wrong by at
most 2 for `x < 1000`, and for `x = 10^9` differs from the exact
value of `\pi(x)` by 79, or less than two parts in a million.
It is about 10 times more accurate than the logarithmic integral
estimate (see :func:`~mpmath.li`), which however is even faster to evaluate.
It is orders of magnitude more accurate than the extremely
fast `x/\log x` estimate.
**Examples**
For small arguments, the Riemann R function almost exactly
gives the prime counting function if rounded to the nearest
integer::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> primepi(50), riemannr(50)
(15, 14.9757023241462)
>>> max(abs(primepi(n)-int(round(riemannr(n)))) for n in range(100))
1
>>> max(abs(primepi(n)-int(round(riemannr(n)))) for n in range(300))
2
The Riemann R function can be evaluated for arguments far too large
for exact determination of `\pi(x)` to be computationally
feasible with any presently known algorithm::
>>> riemannr(10**30)
1.46923988977204e+28
>>> riemannr(10**100)
4.3619719871407e+97
>>> riemannr(10**1000)
4.3448325764012e+996
A comparison of the Riemann R function and logarithmic integral estimates
for `\pi(x)` using exact values of `\pi(10^n)` up to `n = 9`.
The fractional error is shown in parentheses::
>>> exact = [4,25,168,1229,9592,78498,664579,5761455,50847534]
>>> for n, p in enumerate(exact):
... n += 1
... r, l = riemannr(10**n), li(10**n)
... rerr, lerr = nstr((r-p)/p,3), nstr((l-p)/p,3)
... print("%i %i %s(%s) %s(%s)" % (n, p, r, rerr, l, lerr))
...
1 4 4.56458314100509(0.141) 6.1655995047873(0.541)
2 25 25.6616332669242(0.0265) 30.1261415840796(0.205)
3 168 168.359446281167(0.00214) 177.609657990152(0.0572)
4 1229 1226.93121834343(-0.00168) 1246.13721589939(0.0139)
5 9592 9587.43173884197(-0.000476) 9629.8090010508(0.00394)
6 78498 78527.3994291277(0.000375) 78627.5491594622(0.00165)
7 664579 664667.447564748(0.000133) 664918.405048569(0.000511)
8 5761455 5761551.86732017(1.68e-5) 5762209.37544803(0.000131)
9 50847534 50847455.4277214(-1.55e-6) 50849234.9570018(3.35e-5)
The derivative of the Riemann R function gives the approximate
probability for a number of magnitude `x` to be prime::
>>> diff(riemannr, 1000)
0.141903028110784
>>> mpf(primepi(1050) - primepi(950)) / 100
0.15
Evaluation is supported for arbitrary arguments and at arbitrary
precision::
>>> mp.dps = 30
>>> riemannr(7.5)
3.72934743264966261918857135136
>>> riemannr(-4+2j)
(-0.551002208155486427591793957644 + 2.16966398138119450043195899746j)
"""
primepi = r"""
Evaluates the prime counting function, `\pi(x)`, which gives
the number of primes less than or equal to `x`. The argument
`x` may be fractional.
The prime counting function is very expensive to evaluate
precisely for large `x`, and the present implementation is
not optimized in any way. For numerical approximation of the
prime counting function, it is better to use :func:`~mpmath.primepi2`
or :func:`~mpmath.riemannr`.
Some values of the prime counting function::
>>> from mpmath import *
>>> [primepi(k) for k in range(20)]
[0, 0, 1, 2, 2, 3, 3, 4, 4, 4, 4, 5, 5, 6, 6, 6, 6, 7, 7, 8]
>>> primepi(3.5)
2
>>> primepi(100000)
9592
"""
primepi2 = r"""
Returns an interval (as an ``mpi`` instance) providing bounds
for the value of the prime counting function `\pi(x)`. For small
`x`, :func:`~mpmath.primepi2` returns an exact interval based on
the output of :func:`~mpmath.primepi`. For `x > 2656`, a loose interval
based on Schoenfeld's inequality
.. math ::
|\pi(x) - \mathrm{li}(x)| < \frac{\sqrt x \log x}{8 \pi}
is returned. This estimate is rigorous assuming the truth of
the Riemann hypothesis, and can be computed very quickly.
**Examples**
Exact values of the prime counting function for small `x`::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> iv.dps = 15; iv.pretty = True
>>> primepi2(10)
[4.0, 4.0]
>>> primepi2(100)
[25.0, 25.0]
>>> primepi2(1000)
[168.0, 168.0]
Loose intervals are generated for moderately large `x`:
>>> primepi2(10000), primepi(10000)
([1209.0, 1283.0], 1229)
>>> primepi2(50000), primepi(50000)
([5070.0, 5263.0], 5133)
As `x` increases, the absolute error gets worse while the relative
error improves. The exact value of `\pi(10^{23})` is
1925320391606803968923, and :func:`~mpmath.primepi2` gives 9 significant
digits::
>>> p = primepi2(10**23)
>>> p
[1.9253203909477020467e+21, 1.925320392280406229e+21]
>>> mpf(p.delta) / mpf(p.a)
6.9219865355293e-10
A more precise, nonrigorous estimate for `\pi(x)` can be
obtained using the Riemann R function (:func:`~mpmath.riemannr`).
For large enough `x`, the value returned by :func:`~mpmath.primepi2`
essentially amounts to a small perturbation of the value returned by
:func:`~mpmath.riemannr`::
>>> primepi2(10**100)
[4.3619719871407024816e+97, 4.3619719871407032404e+97]
>>> riemannr(10**100)
4.3619719871407e+97
"""
primezeta = r"""
Computes the prime zeta function, which is defined
in analogy with the Riemann zeta function (:func:`~mpmath.zeta`)
as
.. math ::
P(s) = \sum_p \frac{1}{p^s}
where the sum is taken over all prime numbers `p`. Although
this sum only converges for `\mathrm{Re}(s) > 1`, the
function is defined by analytic continuation in the
half-plane `\mathrm{Re}(s) > 0`.
**Examples**
Arbitrary-precision evaluation for real and complex arguments is
supported::
>>> from mpmath import *
>>> mp.dps = 30; mp.pretty = True
>>> primezeta(2)
0.452247420041065498506543364832
>>> primezeta(pi)
0.15483752698840284272036497397
>>> mp.dps = 50
>>> primezeta(3)
0.17476263929944353642311331466570670097541212192615
>>> mp.dps = 20
>>> primezeta(3+4j)
(-0.12085382601645763295 - 0.013370403397787023602j)
The prime zeta function has a logarithmic pole at `s = 1`,
with residue equal to the difference of the Mertens and
Euler constants::
>>> primezeta(1)
+inf
>>> extradps(25)(lambda x: primezeta(1+x)+log(x))(+eps)
-0.31571845205389007685
>>> mertens-euler
-0.31571845205389007685
The analytic continuation to `0 < \mathrm{Re}(s) \le 1`
is implemented. In this strip the function exhibits
very complex behavior; on the unit interval, it has poles at
`1/n` for every squarefree integer `n`::
>>> primezeta(0.5) # Pole at s = 1/2
(-inf + 3.1415926535897932385j)
>>> primezeta(0.25)
(-1.0416106801757269036 + 0.52359877559829887308j)
>>> primezeta(0.5+10j)
(0.54892423556409790529 + 0.45626803423487934264j)
Although evaluation works in principle for any `\mathrm{Re}(s) > 0`,
it should be noted that the evaluation time increases exponentially
as `s` approaches the imaginary axis.
For large `\mathrm{Re}(s)`, `P(s)` is asymptotic to `2^{-s}`::
>>> primezeta(inf)
0.0
>>> primezeta(10), mpf(2)**-10
(0.00099360357443698021786, 0.0009765625)
>>> primezeta(1000)
9.3326361850321887899e-302
>>> primezeta(1000+1000j)
(-3.8565440833654995949e-302 - 8.4985390447553234305e-302j)
**References**
Carl-Erik Froberg, "On the prime zeta function",
BIT 8 (1968), pp. 187-202.
"""
bernpoly = r"""
Evaluates the Bernoulli polynomial `B_n(z)`.
The first few Bernoulli polynomials are::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(6):
... nprint(chop(taylor(lambda x: bernpoly(n,x), 0, n)))
...
[1.0]
[-0.5, 1.0]
[0.166667, -1.0, 1.0]
[0.0, 0.5, -1.5, 1.0]
[-0.0333333, 0.0, 1.0, -2.0, 1.0]
[0.0, -0.166667, 0.0, 1.66667, -2.5, 1.0]
At `z = 0`, the Bernoulli polynomial evaluates to a
Bernoulli number (see :func:`~mpmath.bernoulli`)::
>>> bernpoly(12, 0), bernoulli(12)
(-0.253113553113553, -0.253113553113553)
>>> bernpoly(13, 0), bernoulli(13)
(0.0, 0.0)
Evaluation is accurate for large `n` and small `z`::
>>> mp.dps = 25
>>> bernpoly(100, 0.5)
2.838224957069370695926416e+78
>>> bernpoly(1000, 10.5)
5.318704469415522036482914e+1769
"""
polylog = r"""
Computes the polylogarithm, defined by the sum
.. math ::
\mathrm{Li}_s(z) = \sum_{k=1}^{\infty} \frac{z^k}{k^s}.
This series is convergent only for `|z| < 1`, so elsewhere
the analytic continuation is implied.
The polylogarithm should not be confused with the logarithmic
integral (also denoted by Li or li), which is implemented
as :func:`~mpmath.li`.
**Examples**
The polylogarithm satisfies a huge number of functional identities.
A sample of polylogarithm evaluations is shown below::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> polylog(1,0.5), log(2)
(0.693147180559945, 0.693147180559945)
>>> polylog(2,0.5), (pi**2-6*log(2)**2)/12
(0.582240526465012, 0.582240526465012)
>>> polylog(2,-phi), -log(phi)**2-pi**2/10
(-1.21852526068613, -1.21852526068613)
>>> polylog(3,0.5), 7*zeta(3)/8-pi**2*log(2)/12+log(2)**3/6
(0.53721319360804, 0.53721319360804)
:func:`~mpmath.polylog` can evaluate the analytic continuation of the
polylogarithm when `s` is an integer::
>>> polylog(2, 10)
(0.536301287357863 - 7.23378441241546j)
>>> polylog(2, -10)
-4.1982778868581
>>> polylog(2, 10j)
(-3.05968879432873 + 3.71678149306807j)
>>> polylog(-2, 10)
-0.150891632373114
>>> polylog(-2, -10)
0.067618332081142
>>> polylog(-2, 10j)
(0.0384353698579347 + 0.0912451798066779j)
Some more examples, with arguments on the unit circle (note that
the series definition cannot be used for computation here)::
>>> polylog(2,j)
(-0.205616758356028 + 0.915965594177219j)
>>> j*catalan-pi**2/48
(-0.205616758356028 + 0.915965594177219j)
>>> polylog(3,exp(2*pi*j/3))
(-0.534247512515375 + 0.765587078525922j)
>>> -4*zeta(3)/9 + 2*j*pi**3/81
(-0.534247512515375 + 0.765587078525921j)
Polylogarithms of different order are related by integration
and differentiation::
>>> s, z = 3, 0.5
>>> polylog(s+1, z)
0.517479061673899
>>> quad(lambda t: polylog(s,t)/t, [0, z])
0.517479061673899
>>> z*diff(lambda t: polylog(s+2,t), z)
0.517479061673899
Taylor series expansions around `z = 0` are::
>>> for n in range(-3, 4):
... nprint(taylor(lambda x: polylog(n,x), 0, 5))
...
[0.0, 1.0, 8.0, 27.0, 64.0, 125.0]
[0.0, 1.0, 4.0, 9.0, 16.0, 25.0]
[0.0, 1.0, 2.0, 3.0, 4.0, 5.0]
[0.0, 1.0, 1.0, 1.0, 1.0, 1.0]
[0.0, 1.0, 0.5, 0.333333, 0.25, 0.2]
[0.0, 1.0, 0.25, 0.111111, 0.0625, 0.04]
[0.0, 1.0, 0.125, 0.037037, 0.015625, 0.008]
The series defining the polylogarithm is simultaneously
a Taylor series and an L-series. For certain values of `z`, the
polylogarithm reduces to a pure zeta function::
>>> polylog(pi, 1), zeta(pi)
(1.17624173838258, 1.17624173838258)
>>> polylog(pi, -1), -altzeta(pi)
(-0.909670702980385, -0.909670702980385)
Evaluation for arbitrary, nonintegral `s` is supported
for `z` within the unit circle:
>>> polylog(3+4j, 0.25)
(0.24258605789446 - 0.00222938275488344j)
>>> nsum(lambda k: 0.25**k / k**(3+4j), [1,inf])
(0.24258605789446 - 0.00222938275488344j)
It is also currently supported outside of the unit circle for `z`
not too large in magnitude::
>>> polylog(1+j, 20+40j)
(-7.1421172179728 - 3.92726697721369j)
>>> polylog(1+j, 200+400j)
Traceback (most recent call last):
...
NotImplementedError: polylog for arbitrary s and z
**References**
1. Richard Crandall, "Note on fast polylogarithm computation"
http://people.reed.edu/~crandall/papers/Polylog.pdf
2. http://en.wikipedia.org/wiki/Polylogarithm
3. http://mathworld.wolfram.com/Polylogarithm.html
"""
bell = r"""
For `n` a nonnegative integer, ``bell(n,x)`` evaluates the Bell
polynomial `B_n(x)`, the first few of which are
.. math ::
B_0(x) = 1
B_1(x) = x
B_2(x) = x^2+x
B_3(x) = x^3+3x^2+x
If `x = 1` or :func:`~mpmath.bell` is called with only one argument, it
gives the `n`-th Bell number `B_n`, which is the number of
partitions of a set with `n` elements. By setting the precision to
at least `\log_{10} B_n` digits, :func:`~mpmath.bell` provides fast
calculation of exact Bell numbers.
In general, :func:`~mpmath.bell` computes
.. math ::
B_n(x) = e^{-x} \left(\mathrm{sinc}(\pi n) + E_n(x)\right)
where `E_n(x)` is the generalized exponential function implemented
by :func:`~mpmath.polyexp`. This is an extension of Dobinski's formula [1],
where the modification is the sinc term ensuring that `B_n(x)` is
continuous in `n`; :func:`~mpmath.bell` can thus be evaluated,
differentiated, etc for arbitrary complex arguments.
**Examples**
Simple evaluations::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> bell(0, 2.5)
1.0
>>> bell(1, 2.5)
2.5
>>> bell(2, 2.5)
8.75
Evaluation for arbitrary complex arguments::
>>> bell(5.75+1j, 2-3j)
(-10767.71345136587098445143 - 15449.55065599872579097221j)
The first few Bell polynomials::
>>> for k in range(7):
... nprint(taylor(lambda x: bell(k,x), 0, k))
...
[1.0]
[0.0, 1.0]
[0.0, 1.0, 1.0]
[0.0, 1.0, 3.0, 1.0]
[0.0, 1.0, 7.0, 6.0, 1.0]
[0.0, 1.0, 15.0, 25.0, 10.0, 1.0]
[0.0, 1.0, 31.0, 90.0, 65.0, 15.0, 1.0]
The first few Bell numbers and complementary Bell numbers::
>>> [int(bell(k)) for k in range(10)]
[1, 1, 2, 5, 15, 52, 203, 877, 4140, 21147]
>>> [int(bell(k,-1)) for k in range(10)]
[1, -1, 0, 1, 1, -2, -9, -9, 50, 267]
Large Bell numbers::
>>> mp.dps = 50
>>> bell(50)
185724268771078270438257767181908917499221852770.0
>>> bell(50,-1)
-29113173035759403920216141265491160286912.0
Some even larger values::
>>> mp.dps = 25
>>> bell(1000,-1)
-1.237132026969293954162816e+1869
>>> bell(1000)
2.989901335682408421480422e+1927
>>> bell(1000,2)
6.591553486811969380442171e+1987
>>> bell(1000,100.5)
9.101014101401543575679639e+2529
A determinant identity satisfied by Bell numbers::
>>> mp.dps = 15
>>> N = 8
>>> det([[bell(k+j) for j in range(N)] for k in range(N)])
125411328000.0
>>> superfac(N-1)
125411328000.0
**References**
1. http://mathworld.wolfram.com/DobinskisFormula.html
"""
polyexp = r"""
Evaluates the polyexponential function, defined for arbitrary
complex `s`, `z` by the series
.. math ::
E_s(z) = \sum_{k=1}^{\infty} \frac{k^s}{k!} z^k.
`E_s(z)` is constructed from the exponential function analogously
to how the polylogarithm is constructed from the ordinary
logarithm; as a function of `s` (with `z` fixed), `E_s` is an L-series
It is an entire function of both `s` and `z`.
The polyexponential function provides a generalization of the
Bell polynomials `B_n(x)` (see :func:`~mpmath.bell`) to noninteger orders `n`.
In terms of the Bell polynomials,
.. math ::
E_s(z) = e^z B_s(z) - \mathrm{sinc}(\pi s).
Note that `B_n(x)` and `e^{-x} E_n(x)` are identical if `n`
is a nonzero integer, but not otherwise. In particular, they differ
at `n = 0`.
**Examples**
Evaluating a series::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> nsum(lambda k: sqrt(k)/fac(k), [1,inf])
2.101755547733791780315904
>>> polyexp(0.5,1)
2.101755547733791780315904
Evaluation for arbitrary arguments::
>>> polyexp(-3-4j, 2.5+2j)
(2.351660261190434618268706 + 1.202966666673054671364215j)
Evaluation is accurate for tiny function values::
>>> polyexp(4, -100)
3.499471750566824369520223e-36
If `n` is a nonpositive integer, `E_n` reduces to a special
instance of the hypergeometric function `\,_pF_q`::
>>> n = 3
>>> x = pi
>>> polyexp(-n,x)
4.042192318847986561771779
>>> x*hyper([1]*(n+1), [2]*(n+1), x)
4.042192318847986561771779
"""
cyclotomic = r"""
Evaluates the cyclotomic polynomial `\Phi_n(x)`, defined by
.. math ::
\Phi_n(x) = \prod_{\zeta} (x - \zeta)
where `\zeta` ranges over all primitive `n`-th roots of unity
(see :func:`~mpmath.unitroots`). An equivalent representation, used
for computation, is
.. math ::
\Phi_n(x) = \prod_{d\mid n}(x^d-1)^{\mu(n/d)} = \Phi_n(x)
where `\mu(m)` denotes the Moebius function. The cyclotomic
polynomials are integer polynomials, the first of which can be
written explicitly as
.. math ::
\Phi_0(x) = 1
\Phi_1(x) = x - 1
\Phi_2(x) = x + 1
\Phi_3(x) = x^3 + x^2 + 1
\Phi_4(x) = x^2 + 1
\Phi_5(x) = x^4 + x^3 + x^2 + x + 1
\Phi_6(x) = x^2 - x + 1
**Examples**
The coefficients of low-order cyclotomic polynomials can be recovered
using Taylor expansion::
>>> from mpmath import *
>>> mp.dps = 15; mp.pretty = True
>>> for n in range(9):
... p = chop(taylor(lambda x: cyclotomic(n,x), 0, 10))
... print("%s %s" % (n, nstr(p[:10+1-p[::-1].index(1)])))
...
0 [1.0]
1 [-1.0, 1.0]
2 [1.0, 1.0]
3 [1.0, 1.0, 1.0]
4 [1.0, 0.0, 1.0]
5 [1.0, 1.0, 1.0, 1.0, 1.0]
6 [1.0, -1.0, 1.0]
7 [1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0]
8 [1.0, 0.0, 0.0, 0.0, 1.0]
The definition as a product over primitive roots may be checked
by computing the product explicitly (for a real argument, this
method will generally introduce numerical noise in the imaginary
part)::
>>> mp.dps = 25
>>> z = 3+4j
>>> cyclotomic(10, z)
(-419.0 - 360.0j)
>>> fprod(z-r for r in unitroots(10, primitive=True))
(-419.0 - 360.0j)
>>> z = 3
>>> cyclotomic(10, z)
61.0
>>> fprod(z-r for r in unitroots(10, primitive=True))
(61.0 - 3.146045605088568607055454e-25j)
Up to permutation, the roots of a given cyclotomic polynomial
can be checked to agree with the list of primitive roots::
>>> p = taylor(lambda x: cyclotomic(6,x), 0, 6)[:3]
>>> for r in polyroots(p[::-1]):
... print(r)
...
(0.5 - 0.8660254037844386467637232j)
(0.5 + 0.8660254037844386467637232j)
>>>
>>> for r in unitroots(6, primitive=True):
... print(r)
...
(0.5 + 0.8660254037844386467637232j)
(0.5 - 0.8660254037844386467637232j)
"""
meijerg = r"""
Evaluates the Meijer G-function, defined as
.. math ::
G^{m,n}_{p,q} \left( \left. \begin{matrix}
a_1, \dots, a_n ; a_{n+1} \dots a_p \\
b_1, \dots, b_m ; b_{m+1} \dots b_q
\end{matrix}\; \right| \; z ; r \right) =
\frac{1}{2 \pi i} \int_L
\frac{\prod_{j=1}^m \Gamma(b_j+s) \prod_{j=1}^n\Gamma(1-a_j-s)}
{\prod_{j=n+1}^{p}\Gamma(a_j+s) \prod_{j=m+1}^q \Gamma(1-b_j-s)}
z^{-s/r} ds
for an appropriate choice of the contour `L` (see references).
There are `p` elements `a_j`.
The argument *a_s* should be a pair of lists, the first containing the
`n` elements `a_1, \ldots, a_n` and the second containing
the `p-n` elements `a_{n+1}, \ldots a_p`.
There are `q` elements `b_j`.
The argument *b_s* should be a pair of lists, the first containing the
`m` elements `b_1, \ldots, b_m` and the second containing
the `q-m` elements `b_{m+1}, \ldots b_q`.
The implicit tuple `(m, n, p, q)` constitutes the order or degree of the
Meijer G-function, and is determined by the lengths of the coefficient
vectors. Confusingly, the indices in this tuple appear in a different order
from the coefficients, but this notation is standard. The many examples
given below should hopefully clear up any potential confusion.
**Algorithm**
The Meijer G-function is evaluated as a combination of hypergeometric series.
There are two versions of the function, which can be selected with
the optional *series* argument.
*series=1* uses a sum of `m` `\,_pF_{q-1}` functions of `z`
*series=2* uses a sum of `n` `\,_qF_{p-1}` functions of `1/z`
The default series is chosen based on the degree and `|z|` in order
to be consistent with Mathematica's. This definition of the Meijer G-function
has a discontinuity at `|z| = 1` for some orders, which can
be avoided by explicitly specifying a series.
Keyword arguments are forwarded to :func:`~mpmath.hypercomb`.
**Examples**
Many standard functions are special cases of the Meijer G-function
(possibly rescaled and/or with branch cut corrections). We define
some test parameters::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> a = mpf(0.75)
>>> b = mpf(1.5)
>>> z = mpf(2.25)
The exponential function:
`e^z = G^{1,0}_{0,1} \left( \left. \begin{matrix} - \\ 0 \end{matrix} \;
\right| \; -z \right)`
>>> meijerg([[],[]], [[0],[]], -z)
9.487735836358525720550369
>>> exp(z)
9.487735836358525720550369
The natural logarithm:
`\log(1+z) = G^{1,2}_{2,2} \left( \left. \begin{matrix} 1, 1 \\ 1, 0
\end{matrix} \; \right| \; -z \right)`
>>> meijerg([[1,1],[]], [[1],[0]], z)
1.178654996341646117219023
>>> log(1+z)
1.178654996341646117219023
A rational function:
`\frac{z}{z+1} = G^{1,2}_{2,2} \left( \left. \begin{matrix} 1, 1 \\ 1, 1
\end{matrix} \; \right| \; z \right)`
>>> meijerg([[1,1],[]], [[1],[1]], z)
0.6923076923076923076923077
>>> z/(z+1)
0.6923076923076923076923077
The sine and cosine functions:
`\frac{1}{\sqrt \pi} \sin(2 \sqrt z) = G^{1,0}_{0,2} \left( \left. \begin{matrix}
- \\ \frac{1}{2}, 0 \end{matrix} \; \right| \; z \right)`
`\frac{1}{\sqrt \pi} \cos(2 \sqrt z) = G^{1,0}_{0,2} \left( \left. \begin{matrix}
- \\ 0, \frac{1}{2} \end{matrix} \; \right| \; z \right)`
>>> meijerg([[],[]], [[0.5],[0]], (z/2)**2)
0.4389807929218676682296453
>>> sin(z)/sqrt(pi)
0.4389807929218676682296453
>>> meijerg([[],[]], [[0],[0.5]], (z/2)**2)
-0.3544090145996275423331762
>>> cos(z)/sqrt(pi)
-0.3544090145996275423331762
Bessel functions:
`J_a(2 \sqrt z) = G^{1,0}_{0,2} \left( \left.
\begin{matrix} - \\ \frac{a}{2}, -\frac{a}{2}
\end{matrix} \; \right| \; z \right)`
`Y_a(2 \sqrt z) = G^{2,0}_{1,3} \left( \left.
\begin{matrix} \frac{-a-1}{2} \\ \frac{a}{2}, -\frac{a}{2}, \frac{-a-1}{2}
\end{matrix} \; \right| \; z \right)`
`(-z)^{a/2} z^{-a/2} I_a(2 \sqrt z) = G^{1,0}_{0,2} \left( \left.
\begin{matrix} - \\ \frac{a}{2}, -\frac{a}{2}
\end{matrix} \; \right| \; -z \right)`
`2 K_a(2 \sqrt z) = G^{2,0}_{0,2} \left( \left.
\begin{matrix} - \\ \frac{a}{2}, -\frac{a}{2}
\end{matrix} \; \right| \; z \right)`
As the example with the Bessel *I* function shows, a branch
factor is required for some arguments when inverting the square root.
>>> meijerg([[],[]], [[a/2],[-a/2]], (z/2)**2)
0.5059425789597154858527264
>>> besselj(a,z)
0.5059425789597154858527264
>>> meijerg([[],[(-a-1)/2]], [[a/2,-a/2],[(-a-1)/2]], (z/2)**2)
0.1853868950066556941442559
>>> bessely(a, z)
0.1853868950066556941442559
>>> meijerg([[],[]], [[a/2],[-a/2]], -(z/2)**2)
(0.8685913322427653875717476 + 2.096964974460199200551738j)
>>> (-z)**(a/2) / z**(a/2) * besseli(a, z)
(0.8685913322427653875717476 + 2.096964974460199200551738j)
>>> 0.5*meijerg([[],[]], [[a/2,-a/2],[]], (z/2)**2)
0.09334163695597828403796071
>>> besselk(a,z)
0.09334163695597828403796071
Error functions:
`\sqrt{\pi} z^{2(a-1)} \mathrm{erfc}(z) = G^{2,0}_{1,2} \left( \left.
\begin{matrix} a \\ a-1, a-\frac{1}{2}
\end{matrix} \; \right| \; z, \frac{1}{2} \right)`
>>> meijerg([[],[a]], [[a-1,a-0.5],[]], z, 0.5)
0.00172839843123091957468712
>>> sqrt(pi) * z**(2*a-2) * erfc(z)
0.00172839843123091957468712
A Meijer G-function of higher degree, (1,1,2,3):
>>> meijerg([[a],[b]], [[a],[b,a-1]], z)
1.55984467443050210115617
>>> sin((b-a)*pi)/pi*(exp(z)-1)*z**(a-1)
1.55984467443050210115617
A Meijer G-function of still higher degree, (4,1,2,4), that can
be expanded as a messy combination of exponential integrals:
>>> meijerg([[a],[2*b-a]], [[b,a,b-0.5,-1-a+2*b],[]], z)
0.3323667133658557271898061
>>> chop(4**(a-b+1)*sqrt(pi)*gamma(2*b-2*a)*z**a*\
... expint(2*b-2*a, -2*sqrt(-z))*expint(2*b-2*a, 2*sqrt(-z)))
0.3323667133658557271898061
In the following case, different series give different values::
>>> chop(meijerg([[1],[0.25]],[[3],[0.5]],-2))
-0.06417628097442437076207337
>>> meijerg([[1],[0.25]],[[3],[0.5]],-2,series=1)
0.1428699426155117511873047
>>> chop(meijerg([[1],[0.25]],[[3],[0.5]],-2,series=2))
-0.06417628097442437076207337
**References**
1. http://en.wikipedia.org/wiki/Meijer_G-function
2. http://mathworld.wolfram.com/MeijerG-Function.html
3. http://functions.wolfram.com/HypergeometricFunctions/MeijerG/
4. http://functions.wolfram.com/HypergeometricFunctions/MeijerG1/
"""
clsin = r"""
Computes the Clausen sine function, defined formally by the series
.. math ::
\mathrm{Cl}_s(z) = \sum_{k=1}^{\infty} \frac{\sin(kz)}{k^s}.
The special case `\mathrm{Cl}_2(z)` (i.e. ``clsin(2,z)``) is the classical
"Clausen function". More generally, the Clausen function is defined for
complex `s` and `z`, even when the series does not converge. The
Clausen function is related to the polylogarithm (:func:`~mpmath.polylog`) as
.. math ::
\mathrm{Cl}_s(z) = \frac{1}{2i}\left(\mathrm{Li}_s\left(e^{iz}\right) -
\mathrm{Li}_s\left(e^{-iz}\right)\right)
= \mathrm{Im}\left[\mathrm{Li}_s(e^{iz})\right] \quad (s, z \in \mathbb{R}),
and this representation can be taken to provide the analytic continuation of the
series. The complementary function :func:`~mpmath.clcos` gives the corresponding
cosine sum.
**Examples**
Evaluation for arbitrarily chosen `s` and `z`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> s, z = 3, 4
>>> clsin(s, z); nsum(lambda k: sin(z*k)/k**s, [1,inf])
-0.6533010136329338746275795
-0.6533010136329338746275795
Using `z + \pi` instead of `z` gives an alternating series::
>>> clsin(s, z+pi)
0.8860032351260589402871624
>>> nsum(lambda k: (-1)**k*sin(z*k)/k**s, [1,inf])
0.8860032351260589402871624
With `s = 1`, the sum can be expressed in closed form
using elementary functions::
>>> z = 1 + sqrt(3)
>>> clsin(1, z)
0.2047709230104579724675985
>>> chop((log(1-exp(-j*z)) - log(1-exp(j*z)))/(2*j))
0.2047709230104579724675985
>>> nsum(lambda k: sin(k*z)/k, [1,inf])
0.2047709230104579724675985
The classical Clausen function `\mathrm{Cl}_2(\theta)` gives the
value of the integral `\int_0^{\theta} -\ln(2\sin(x/2)) dx` for
`0 < \theta < 2 \pi`::
>>> cl2 = lambda t: clsin(2, t)
>>> cl2(3.5)
-0.2465045302347694216534255
>>> -quad(lambda x: ln(2*sin(0.5*x)), [0, 3.5])
-0.2465045302347694216534255
This function is symmetric about `\theta = \pi` with zeros and extreme
points::
>>> cl2(0); cl2(pi/3); chop(cl2(pi)); cl2(5*pi/3); chop(cl2(2*pi))
0.0
1.014941606409653625021203
0.0
-1.014941606409653625021203
0.0
Catalan's constant is a special value::
>>> cl2(pi/2)
0.9159655941772190150546035
>>> +catalan
0.9159655941772190150546035
The Clausen sine function can be expressed in closed form when
`s` is an odd integer (becoming zero when `s` < 0)::
>>> z = 1 + sqrt(2)
>>> clsin(1, z); (pi-z)/2
0.3636895456083490948304773
0.3636895456083490948304773
>>> clsin(3, z); pi**2/6*z - pi*z**2/4 + z**3/12
0.5661751584451144991707161
0.5661751584451144991707161
>>> clsin(-1, z)
0.0
>>> clsin(-3, z)
0.0
It can also be expressed in closed form for even integer `s \le 0`,
providing a finite sum for series such as
`\sin(z) + \sin(2z) + \sin(3z) + \ldots`::
>>> z = 1 + sqrt(2)
>>> clsin(0, z)
0.1903105029507513881275865
>>> cot(z/2)/2
0.1903105029507513881275865
>>> clsin(-2, z)
-0.1089406163841548817581392
>>> -cot(z/2)*csc(z/2)**2/4
-0.1089406163841548817581392
Call with ``pi=True`` to multiply `z` by `\pi` exactly::
>>> clsin(3, 3*pi)
-8.892316224968072424732898e-26
>>> clsin(3, 3, pi=True)
0.0
Evaluation for complex `s`, `z` in a nonconvergent case::
>>> s, z = -1-j, 1+2j
>>> clsin(s, z)
(-0.593079480117379002516034 + 0.9038644233367868273362446j)
>>> extraprec(20)(nsum)(lambda k: sin(k*z)/k**s, [1,inf])
(-0.593079480117379002516034 + 0.9038644233367868273362446j)
"""
clcos = r"""
Computes the Clausen cosine function, defined formally by the series
.. math ::
\mathrm{\widetilde{Cl}}_s(z) = \sum_{k=1}^{\infty} \frac{\cos(kz)}{k^s}.
This function is complementary to the Clausen sine function
:func:`~mpmath.clsin`. In terms of the polylogarithm,
.. math ::
\mathrm{\widetilde{Cl}}_s(z) =
\frac{1}{2}\left(\mathrm{Li}_s\left(e^{iz}\right) +
\mathrm{Li}_s\left(e^{-iz}\right)\right)
= \mathrm{Re}\left[\mathrm{Li}_s(e^{iz})\right] \quad (s, z \in \mathbb{R}).
**Examples**
Evaluation for arbitrarily chosen `s` and `z`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> s, z = 3, 4
>>> clcos(s, z); nsum(lambda k: cos(z*k)/k**s, [1,inf])
-0.6518926267198991308332759
-0.6518926267198991308332759
Using `z + \pi` instead of `z` gives an alternating series::
>>> s, z = 3, 0.5
>>> clcos(s, z+pi)
-0.8155530586502260817855618
>>> nsum(lambda k: (-1)**k*cos(z*k)/k**s, [1,inf])
-0.8155530586502260817855618
With `s = 1`, the sum can be expressed in closed form
using elementary functions::
>>> z = 1 + sqrt(3)
>>> clcos(1, z)
-0.6720334373369714849797918
>>> chop(-0.5*(log(1-exp(j*z))+log(1-exp(-j*z))))
-0.6720334373369714849797918
>>> -log(abs(2*sin(0.5*z))) # Equivalent to above when z is real
-0.6720334373369714849797918
>>> nsum(lambda k: cos(k*z)/k, [1,inf])
-0.6720334373369714849797918
It can also be expressed in closed form when `s` is an even integer.
For example,
>>> clcos(2,z)
-0.7805359025135583118863007
>>> pi**2/6 - pi*z/2 + z**2/4
-0.7805359025135583118863007
The case `s = 0` gives the renormalized sum of
`\cos(z) + \cos(2z) + \cos(3z) + \ldots` (which happens to be the same for
any value of `z`)::
>>> clcos(0, z)
-0.5
>>> nsum(lambda k: cos(k*z), [1,inf])
-0.5
Also the sums
.. math ::
\cos(z) + 2\cos(2z) + 3\cos(3z) + \ldots
and
.. math ::
\cos(z) + 2^n \cos(2z) + 3^n \cos(3z) + \ldots
for higher integer powers `n = -s` can be done in closed form. They are zero
when `n` is positive and even (`s` negative and even)::
>>> clcos(-1, z); 1/(2*cos(z)-2)
-0.2607829375240542480694126
-0.2607829375240542480694126
>>> clcos(-3, z); (2+cos(z))*csc(z/2)**4/8
0.1472635054979944390848006
0.1472635054979944390848006
>>> clcos(-2, z); clcos(-4, z); clcos(-6, z)
0.0
0.0
0.0
With `z = \pi`, the series reduces to that of the Riemann zeta function
(more generally, if `z = p \pi/q`, it is a finite sum over Hurwitz zeta
function values)::
>>> clcos(2.5, 0); zeta(2.5)
1.34148725725091717975677
1.34148725725091717975677
>>> clcos(2.5, pi); -altzeta(2.5)
-0.8671998890121841381913472
-0.8671998890121841381913472
Call with ``pi=True`` to multiply `z` by `\pi` exactly::
>>> clcos(-3, 2*pi)
2.997921055881167659267063e+102
>>> clcos(-3, 2, pi=True)
0.008333333333333333333333333
Evaluation for complex `s`, `z` in a nonconvergent case::
>>> s, z = -1-j, 1+2j
>>> clcos(s, z)
(0.9407430121562251476136807 + 0.715826296033590204557054j)
>>> extraprec(20)(nsum)(lambda k: cos(k*z)/k**s, [1,inf])
(0.9407430121562251476136807 + 0.715826296033590204557054j)
"""
whitm = r"""
Evaluates the Whittaker function `M(k,m,z)`, which gives a solution
to the Whittaker differential equation
.. math ::
\frac{d^2f}{dz^2} + \left(-\frac{1}{4}+\frac{k}{z}+
\frac{(\frac{1}{4}-m^2)}{z^2}\right) f = 0.
A second solution is given by :func:`~mpmath.whitw`.
The Whittaker functions are defined in Abramowitz & Stegun, section 13.1.
They are alternate forms of the confluent hypergeometric functions
`\,_1F_1` and `U`:
.. math ::
M(k,m,z) = e^{-\frac{1}{2}z} z^{\frac{1}{2}+m}
\,_1F_1(\tfrac{1}{2}+m-k, 1+2m, z)
W(k,m,z) = e^{-\frac{1}{2}z} z^{\frac{1}{2}+m}
U(\tfrac{1}{2}+m-k, 1+2m, z).
**Examples**
Evaluation for arbitrary real and complex arguments is supported::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> whitm(1, 1, 1)
0.7302596799460411820509668
>>> whitm(1, 1, -1)
(0.0 - 1.417977827655098025684246j)
>>> whitm(j, j/2, 2+3j)
(3.245477713363581112736478 - 0.822879187542699127327782j)
>>> whitm(2, 3, 100000)
4.303985255686378497193063e+21707
Evaluation at zero::
>>> whitm(1,-1,0); whitm(1,-0.5,0); whitm(1,0,0)
+inf
nan
0.0
We can verify that :func:`~mpmath.whitm` numerically satisfies the
differential equation for arbitrarily chosen values::
>>> k = mpf(0.25)
>>> m = mpf(1.5)
>>> f = lambda z: whitm(k,m,z)
>>> for z in [-1, 2.5, 3, 1+2j]:
... chop(diff(f,z,2) + (-0.25 + k/z + (0.25-m**2)/z**2)*f(z))
...
0.0
0.0
0.0
0.0
An integral involving both :func:`~mpmath.whitm` and :func:`~mpmath.whitw`,
verifying evaluation along the real axis::
>>> quad(lambda x: exp(-x)*whitm(3,2,x)*whitw(1,-2,x), [0,inf])
3.438869842576800225207341
>>> 128/(21*sqrt(pi))
3.438869842576800225207341
"""
whitw = r"""
Evaluates the Whittaker function `W(k,m,z)`, which gives a second
solution to the Whittaker differential equation. (See :func:`~mpmath.whitm`.)
**Examples**
Evaluation for arbitrary real and complex arguments is supported::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> whitw(1, 1, 1)
1.19532063107581155661012
>>> whitw(1, 1, -1)
(-0.9424875979222187313924639 - 0.2607738054097702293308689j)
>>> whitw(j, j/2, 2+3j)
(0.1782899315111033879430369 - 0.01609578360403649340169406j)
>>> whitw(2, 3, 100000)
1.887705114889527446891274e-21705
>>> whitw(-1, -1, 100)
1.905250692824046162462058e-24
Evaluation at zero::
>>> for m in [-1, -0.5, 0, 0.5, 1]:
... whitw(1, m, 0)
...
+inf
nan
0.0
nan
+inf
We can verify that :func:`~mpmath.whitw` numerically satisfies the
differential equation for arbitrarily chosen values::
>>> k = mpf(0.25)
>>> m = mpf(1.5)
>>> f = lambda z: whitw(k,m,z)
>>> for z in [-1, 2.5, 3, 1+2j]:
... chop(diff(f,z,2) + (-0.25 + k/z + (0.25-m**2)/z**2)*f(z))
...
0.0
0.0
0.0
0.0
"""
ber = r"""
Computes the Kelvin function ber, which for real arguments gives the real part
of the Bessel J function of a rotated argument
.. math ::
J_n\left(x e^{3\pi i/4}\right) = \mathrm{ber}_n(x) + i \mathrm{bei}_n(x).
The imaginary part is given by :func:`~mpmath.bei`.
**Plots**
.. literalinclude :: /plots/ber.py
.. image :: /plots/ber.png
**Examples**
Verifying the defining relation::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> n, x = 2, 3.5
>>> ber(n,x)
1.442338852571888752631129
>>> bei(n,x)
-0.948359035324558320217678
>>> besselj(n, x*root(1,8,3))
(1.442338852571888752631129 - 0.948359035324558320217678j)
The ber and bei functions are also defined by analytic continuation
for complex arguments::
>>> ber(1+j, 2+3j)
(4.675445984756614424069563 - 15.84901771719130765656316j)
>>> bei(1+j, 2+3j)
(15.83886679193707699364398 + 4.684053288183046528703611j)
"""
bei = r"""
Computes the Kelvin function bei, which for real arguments gives the
imaginary part of the Bessel J function of a rotated argument.
See :func:`~mpmath.ber`.
"""
ker = r"""
Computes the Kelvin function ker, which for real arguments gives the real part
of the (rescaled) Bessel K function of a rotated argument
.. math ::
e^{-\pi i/2} K_n\left(x e^{3\pi i/4}\right) = \mathrm{ker}_n(x) + i \mathrm{kei}_n(x).
The imaginary part is given by :func:`~mpmath.kei`.
**Plots**
.. literalinclude :: /plots/ker.py
.. image :: /plots/ker.png
**Examples**
Verifying the defining relation::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> n, x = 2, 4.5
>>> ker(n,x)
0.02542895201906369640249801
>>> kei(n,x)
-0.02074960467222823237055351
>>> exp(-n*pi*j/2) * besselk(n, x*root(1,8,1))
(0.02542895201906369640249801 - 0.02074960467222823237055351j)
The ker and kei functions are also defined by analytic continuation
for complex arguments::
>>> ker(1+j, 3+4j)
(1.586084268115490421090533 - 2.939717517906339193598719j)
>>> kei(1+j, 3+4j)
(-2.940403256319453402690132 - 1.585621643835618941044855j)
"""
kei = r"""
Computes the Kelvin function kei, which for real arguments gives the
imaginary part of the (rescaled) Bessel K function of a rotated argument.
See :func:`~mpmath.ker`.
"""
struveh = r"""
Gives the Struve function
.. math ::
\,\mathbf{H}_n(z) =
\sum_{k=0}^\infty \frac{(-1)^k}{\Gamma(k+\frac{3}{2})
\Gamma(k+n+\frac{3}{2})} {\left({\frac{z}{2}}\right)}^{2k+n+1}
which is a solution to the Struve differential equation
.. math ::
z^2 f''(z) + z f'(z) + (z^2-n^2) f(z) = \frac{2 z^{n+1}}{\pi (2n-1)!!}.
**Examples**
Evaluation for arbitrary real and complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> struveh(0, 3.5)
0.3608207733778295024977797
>>> struveh(-1, 10)
-0.255212719726956768034732
>>> struveh(1, -100.5)
0.5819566816797362287502246
>>> struveh(2.5, 10000000000000)
3153915652525200060.308937
>>> struveh(2.5, -10000000000000)
(0.0 - 3153915652525200060.308937j)
>>> struveh(1+j, 1000000+4000000j)
(-3.066421087689197632388731e+1737173 - 1.596619701076529803290973e+1737173j)
A Struve function of half-integer order is elementary; for example:
>>> z = 3
>>> struveh(0.5, 3)
0.9167076867564138178671595
>>> sqrt(2/(pi*z))*(1-cos(z))
0.9167076867564138178671595
Numerically verifying the differential equation::
>>> z = mpf(4.5)
>>> n = 3
>>> f = lambda z: struveh(n,z)
>>> lhs = z**2*diff(f,z,2) + z*diff(f,z) + (z**2-n**2)*f(z)
>>> rhs = 2*z**(n+1)/fac2(2*n-1)/pi
>>> lhs
17.40359302709875496632744
>>> rhs
17.40359302709875496632744
"""
struvel = r"""
Gives the modified Struve function
.. math ::
\,\mathbf{L}_n(z) = -i e^{-n\pi i/2} \mathbf{H}_n(i z)
which solves to the modified Struve differential equation
.. math ::
z^2 f''(z) + z f'(z) - (z^2+n^2) f(z) = \frac{2 z^{n+1}}{\pi (2n-1)!!}.
**Examples**
Evaluation for arbitrary real and complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> struvel(0, 3.5)
7.180846515103737996249972
>>> struvel(-1, 10)
2670.994904980850550721511
>>> struvel(1, -100.5)
1.757089288053346261497686e+42
>>> struvel(2.5, 10000000000000)
4.160893281017115450519948e+4342944819025
>>> struvel(2.5, -10000000000000)
(0.0 - 4.160893281017115450519948e+4342944819025j)
>>> struvel(1+j, 700j)
(-0.1721150049480079451246076 + 0.1240770953126831093464055j)
>>> struvel(1+j, 1000000+4000000j)
(-2.973341637511505389128708e+434290 - 5.164633059729968297147448e+434290j)
Numerically verifying the differential equation::
>>> z = mpf(3.5)
>>> n = 3
>>> f = lambda z: struvel(n,z)
>>> lhs = z**2*diff(f,z,2) + z*diff(f,z) - (z**2+n**2)*f(z)
>>> rhs = 2*z**(n+1)/fac2(2*n-1)/pi
>>> lhs
6.368850306060678353018165
>>> rhs
6.368850306060678353018165
"""
appellf1 = r"""
Gives the Appell F1 hypergeometric function of two variables,
.. math ::
F_1(a,b_1,b_2,c,x,y) = \sum_{m=0}^{\infty} \sum_{n=0}^{\infty}
\frac{(a)_{m+n} (b_1)_m (b_2)_n}{(c)_{m+n}}
\frac{x^m y^n}{m! n!}.
This series is only generally convergent when `|x| < 1` and `|y| < 1`,
although :func:`~mpmath.appellf1` can evaluate an analytic continuation
with respecto to either variable, and sometimes both.
**Examples**
Evaluation is supported for real and complex parameters::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> appellf1(1,0,0.5,1,0.5,0.25)
1.154700538379251529018298
>>> appellf1(1,1+j,0.5,1,0.5,0.5j)
(1.138403860350148085179415 + 1.510544741058517621110615j)
For some integer parameters, the F1 series reduces to a polynomial::
>>> appellf1(2,-4,-3,1,2,5)
-816.0
>>> appellf1(-5,1,2,1,4,5)
-20528.0
The analytic continuation with respect to either `x` or `y`,
and sometimes with respect to both, can be evaluated::
>>> appellf1(2,3,4,5,100,0.5)
(0.0006231042714165329279738662 + 0.0000005769149277148425774499857j)
>>> appellf1('1.1', '0.3', '0.2+2j', '0.4', '0.2', 1.5+3j)
(-0.1782604566893954897128702 + 0.002472407104546216117161499j)
>>> appellf1(1,2,3,4,10,12)
-0.07122993830066776374929313
For certain arguments, F1 reduces to an ordinary hypergeometric function::
>>> appellf1(1,2,3,5,0.5,0.25)
1.547902270302684019335555
>>> 4*hyp2f1(1,2,5,'1/3')/3
1.547902270302684019335555
>>> appellf1(1,2,3,4,0,1.5)
(-1.717202506168937502740238 - 2.792526803190927323077905j)
>>> hyp2f1(1,3,4,1.5)
(-1.717202506168937502740238 - 2.792526803190927323077905j)
The F1 function satisfies a system of partial differential equations::
>>> a,b1,b2,c,x,y = map(mpf, [1,0.5,0.25,1.125,0.25,-0.25])
>>> F = lambda x,y: appellf1(a,b1,b2,c,x,y)
>>> chop(x*(1-x)*diff(F,(x,y),(2,0)) +
... y*(1-x)*diff(F,(x,y),(1,1)) +
... (c-(a+b1+1)*x)*diff(F,(x,y),(1,0)) -
... b1*y*diff(F,(x,y),(0,1)) -
... a*b1*F(x,y))
0.0
>>>
>>> chop(y*(1-y)*diff(F,(x,y),(0,2)) +
... x*(1-y)*diff(F,(x,y),(1,1)) +
... (c-(a+b2+1)*y)*diff(F,(x,y),(0,1)) -
... b2*x*diff(F,(x,y),(1,0)) -
... a*b2*F(x,y))
0.0
The Appell F1 function allows for closed-form evaluation of various
integrals, such as any integral of the form
`\int x^r (x+a)^p (x+b)^q dx`::
>>> def integral(a,b,p,q,r,x1,x2):
... a,b,p,q,r,x1,x2 = map(mpmathify, [a,b,p,q,r,x1,x2])
... f = lambda x: x**r * (x+a)**p * (x+b)**q
... def F(x):
... v = x**(r+1)/(r+1) * (a+x)**p * (b+x)**q
... v *= (1+x/a)**(-p)
... v *= (1+x/b)**(-q)
... v *= appellf1(r+1,-p,-q,2+r,-x/a,-x/b)
... return v
... print("Num. quad: %s" % quad(f, [x1,x2]))
... print("Appell F1: %s" % (F(x2)-F(x1)))
...
>>> integral('1/5','4/3','-2','3','1/2',0,1)
Num. quad: 9.073335358785776206576981
Appell F1: 9.073335358785776206576981
>>> integral('3/2','4/3','-2','3','1/2',0,1)
Num. quad: 1.092829171999626454344678
Appell F1: 1.092829171999626454344678
>>> integral('3/2','4/3','-2','3','1/2',12,25)
Num. quad: 1106.323225040235116498927
Appell F1: 1106.323225040235116498927
Also incomplete elliptic integrals fall into this category [1]::
>>> def E(z, m):
... if (pi/2).ae(z):
... return ellipe(m)
... return 2*round(re(z)/pi)*ellipe(m) + mpf(-1)**round(re(z)/pi)*\
... sin(z)*appellf1(0.5,0.5,-0.5,1.5,sin(z)**2,m*sin(z)**2)
...
>>> z, m = 1, 0.5
>>> E(z,m); quad(lambda t: sqrt(1-m*sin(t)**2), [0,pi/4,3*pi/4,z])
0.9273298836244400669659042
0.9273298836244400669659042
>>> z, m = 3, 2
>>> E(z,m); quad(lambda t: sqrt(1-m*sin(t)**2), [0,pi/4,3*pi/4,z])
(1.057495752337234229715836 + 1.198140234735592207439922j)
(1.057495752337234229715836 + 1.198140234735592207439922j)
**References**
1. [WolframFunctions]_ http://functions.wolfram.com/EllipticIntegrals/EllipticE2/26/01/
2. [SrivastavaKarlsson]_
3. [CabralRosetti]_
4. [Vidunas]_
5. [Slater]_
"""
angerj = r"""
Gives the Anger function
.. math ::
\mathbf{J}_{\nu}(z) = \frac{1}{\pi}
\int_0^{\pi} \cos(\nu t - z \sin t) dt
which is an entire function of both the parameter `\nu` and
the argument `z`. It solves the inhomogeneous Bessel differential
equation
.. math ::
f''(z) + \frac{1}{z}f'(z) + \left(1-\frac{\nu^2}{z^2}\right) f(z)
= \frac{(z-\nu)}{\pi z^2} \sin(\pi \nu).
**Examples**
Evaluation for real and complex parameter and argument::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> angerj(2,3)
0.4860912605858910769078311
>>> angerj(-3+4j, 2+5j)
(-5033.358320403384472395612 + 585.8011892476145118551756j)
>>> angerj(3.25, 1e6j)
(4.630743639715893346570743e+434290 - 1.117960409887505906848456e+434291j)
>>> angerj(-1.5, 1e6)
0.0002795719747073879393087011
The Anger function coincides with the Bessel J-function when `\nu`
is an integer::
>>> angerj(1,3); besselj(1,3)
0.3390589585259364589255146
0.3390589585259364589255146
>>> angerj(1.5,3); besselj(1.5,3)
0.4088969848691080859328847
0.4777182150870917715515015
Verifying the differential equation::
>>> v,z = mpf(2.25), 0.75
>>> f = lambda z: angerj(v,z)
>>> diff(f,z,2) + diff(f,z)/z + (1-(v/z)**2)*f(z)
-0.6002108774380707130367995
>>> (z-v)/(pi*z**2) * sinpi(v)
-0.6002108774380707130367995
Verifying the integral representation::
>>> angerj(v,z)
0.1145380759919333180900501
>>> quad(lambda t: cos(v*t-z*sin(t))/pi, [0,pi])
0.1145380759919333180900501
**References**
1. [DLMF]_ section 11.10: Anger-Weber Functions
"""
webere = r"""
Gives the Weber function
.. math ::
\mathbf{E}_{\nu}(z) = \frac{1}{\pi}
\int_0^{\pi} \sin(\nu t - z \sin t) dt
which is an entire function of both the parameter `\nu` and
the argument `z`. It solves the inhomogeneous Bessel differential
equation
.. math ::
f''(z) + \frac{1}{z}f'(z) + \left(1-\frac{\nu^2}{z^2}\right) f(z)
= -\frac{1}{\pi z^2} (z+\nu+(z-\nu)\cos(\pi \nu)).
**Examples**
Evaluation for real and complex parameter and argument::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> webere(2,3)
-0.1057668973099018425662646
>>> webere(-3+4j, 2+5j)
(-585.8081418209852019290498 - 5033.314488899926921597203j)
>>> webere(3.25, 1e6j)
(-1.117960409887505906848456e+434291 - 4.630743639715893346570743e+434290j)
>>> webere(3.25, 1e6)
-0.00002812518265894315604914453
Up to addition of a rational function of `z`, the Weber function coincides
with the Struve H-function when `\nu` is an integer::
>>> webere(1,3); 2/pi-struveh(1,3)
-0.3834897968188690177372881
-0.3834897968188690177372881
>>> webere(5,3); 26/(35*pi)-struveh(5,3)
0.2009680659308154011878075
0.2009680659308154011878075
Verifying the differential equation::
>>> v,z = mpf(2.25), 0.75
>>> f = lambda z: webere(v,z)
>>> diff(f,z,2) + diff(f,z)/z + (1-(v/z)**2)*f(z)
-1.097441848875479535164627
>>> -(z+v+(z-v)*cospi(v))/(pi*z**2)
-1.097441848875479535164627
Verifying the integral representation::
>>> webere(v,z)
0.1486507351534283744485421
>>> quad(lambda t: sin(v*t-z*sin(t))/pi, [0,pi])
0.1486507351534283744485421
**References**
1. [DLMF]_ section 11.10: Anger-Weber Functions
"""
lommels1 = r"""
Gives the Lommel function `s_{\mu,\nu}` or `s^{(1)}_{\mu,\nu}`
.. math ::
s_{\mu,\nu}(z) = \frac{z^{\mu+1}}{(\mu-\nu+1)(\mu+\nu+1)}
\,_1F_2\left(1; \frac{\mu-\nu+3}{2}, \frac{\mu+\nu+3}{2};
-\frac{z^2}{4} \right)
which solves the inhomogeneous Bessel equation
.. math ::
z^2 f''(z) + z f'(z) + (z^2-\nu^2) f(z) = z^{\mu+1}.
A second solution is given by :func:`~mpmath.lommels2`.
**Plots**
.. literalinclude :: /plots/lommels1.py
.. image :: /plots/lommels1.png
**Examples**
An integral representation::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> u,v,z = 0.25, 0.125, mpf(0.75)
>>> lommels1(u,v,z)
0.4276243877565150372999126
>>> (bessely(v,z)*quad(lambda t: t**u*besselj(v,t), [0,z]) - \
... besselj(v,z)*quad(lambda t: t**u*bessely(v,t), [0,z]))*(pi/2)
0.4276243877565150372999126
A special value::
>>> lommels1(v,v,z)
0.5461221367746048054932553
>>> gamma(v+0.5)*sqrt(pi)*power(2,v-1)*struveh(v,z)
0.5461221367746048054932553
Verifying the differential equation::
>>> f = lambda z: lommels1(u,v,z)
>>> z**2*diff(f,z,2) + z*diff(f,z) + (z**2-v**2)*f(z)
0.6979536443265746992059141
>>> z**(u+1)
0.6979536443265746992059141
**References**
1. [GradshteynRyzhik]_
2. [Weisstein]_ http://mathworld.wolfram.com/LommelFunction.html
"""
lommels2 = r"""
Gives the second Lommel function `S_{\mu,\nu}` or `s^{(2)}_{\mu,\nu}`
.. math ::
S_{\mu,\nu}(z) = s_{\mu,\nu}(z) + 2^{\mu-1}
\Gamma\left(\tfrac{1}{2}(\mu-\nu+1)\right)
\Gamma\left(\tfrac{1}{2}(\mu+\nu+1)\right) \times
\left[\sin(\tfrac{1}{2}(\mu-\nu)\pi) J_{\nu}(z) -
\cos(\tfrac{1}{2}(\mu-\nu)\pi) Y_{\nu}(z)
\right]
which solves the same differential equation as
:func:`~mpmath.lommels1`.
**Plots**
.. literalinclude :: /plots/lommels2.py
.. image :: /plots/lommels2.png
**Examples**
For large `|z|`, `S_{\mu,\nu} \sim z^{\mu-1}`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> lommels2(10,2,30000)
1.968299831601008419949804e+40
>>> power(30000,9)
1.9683e+40
A special value::
>>> u,v,z = 0.5, 0.125, mpf(0.75)
>>> lommels2(v,v,z)
0.9589683199624672099969765
>>> (struveh(v,z)-bessely(v,z))*power(2,v-1)*sqrt(pi)*gamma(v+0.5)
0.9589683199624672099969765
Verifying the differential equation::
>>> f = lambda z: lommels2(u,v,z)
>>> z**2*diff(f,z,2) + z*diff(f,z) + (z**2-v**2)*f(z)
0.6495190528383289850727924
>>> z**(u+1)
0.6495190528383289850727924
**References**
1. [GradshteynRyzhik]_
2. [Weisstein]_ http://mathworld.wolfram.com/LommelFunction.html
"""
appellf2 = r"""
Gives the Appell F2 hypergeometric function of two variables
.. math ::
F_2(a,b_1,b_2,c_1,c_2,x,y) = \sum_{m=0}^{\infty} \sum_{n=0}^{\infty}
\frac{(a)_{m+n} (b_1)_m (b_2)_n}{(c_1)_m (c_2)_n}
\frac{x^m y^n}{m! n!}.
The series is generally absolutely convergent for `|x| + |y| < 1`.
**Examples**
Evaluation for real and complex arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> appellf2(1,2,3,4,5,0.25,0.125)
1.257417193533135344785602
>>> appellf2(1,-3,-4,2,3,2,3)
-42.8
>>> appellf2(0.5,0.25,-0.25,2,3,0.25j,0.25)
(0.9880539519421899867041719 + 0.01497616165031102661476978j)
>>> chop(appellf2(1,1+j,1-j,3j,-3j,0.25,0.25))
1.201311219287411337955192
>>> appellf2(1,1,1,4,6,0.125,16)
(-0.09455532250274744282125152 - 0.7647282253046207836769297j)
A transformation formula::
>>> a,b1,b2,c1,c2,x,y = map(mpf, [1,2,0.5,0.25,1.625,-0.125,0.125])
>>> appellf2(a,b1,b2,c1,c2,x,y)
0.2299211717841180783309688
>>> (1-x)**(-a)*appellf2(a,c1-b1,b2,c1,c2,x/(x-1),y/(1-x))
0.2299211717841180783309688
A system of partial differential equations satisfied by F2::
>>> a,b1,b2,c1,c2,x,y = map(mpf, [1,0.5,0.25,1.125,1.5,0.0625,-0.0625])
>>> F = lambda x,y: appellf2(a,b1,b2,c1,c2,x,y)
>>> chop(x*(1-x)*diff(F,(x,y),(2,0)) -
... x*y*diff(F,(x,y),(1,1)) +
... (c1-(a+b1+1)*x)*diff(F,(x,y),(1,0)) -
... b1*y*diff(F,(x,y),(0,1)) -
... a*b1*F(x,y))
0.0
>>> chop(y*(1-y)*diff(F,(x,y),(0,2)) -
... x*y*diff(F,(x,y),(1,1)) +
... (c2-(a+b2+1)*y)*diff(F,(x,y),(0,1)) -
... b2*x*diff(F,(x,y),(1,0)) -
... a*b2*F(x,y))
0.0
**References**
See references for :func:`~mpmath.appellf1`.
"""
appellf3 = r"""
Gives the Appell F3 hypergeometric function of two variables
.. math ::
F_3(a_1,a_2,b_1,b_2,c,x,y) = \sum_{m=0}^{\infty} \sum_{n=0}^{\infty}
\frac{(a_1)_m (a_2)_n (b_1)_m (b_2)_n}{(c)_{m+n}}
\frac{x^m y^n}{m! n!}.
The series is generally absolutely convergent for `|x| < 1, |y| < 1`.
**Examples**
Evaluation for various parameters and variables::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> appellf3(1,2,3,4,5,0.5,0.25)
2.221557778107438938158705
>>> appellf3(1,2,3,4,5,6,0); hyp2f1(1,3,5,6)
(-0.5189554589089861284537389 - 0.1454441043328607980769742j)
(-0.5189554589089861284537389 - 0.1454441043328607980769742j)
>>> appellf3(1,-2,-3,1,1,4,6)
-17.4
>>> appellf3(1,2,-3,1,1,4,6)
(17.7876136773677356641825 + 19.54768762233649126154534j)
>>> appellf3(1,2,-3,1,1,6,4)
(85.02054175067929402953645 + 148.4402528821177305173599j)
>>> chop(appellf3(1+j,2,1-j,2,3,0.25,0.25))
1.719992169545200286696007
Many transformations and evaluations for special combinations
of the parameters are possible, e.g.:
>>> a,b,c,x,y = map(mpf, [0.5,0.25,0.125,0.125,-0.125])
>>> appellf3(a,c-a,b,c-b,c,x,y)
1.093432340896087107444363
>>> (1-y)**(a+b-c)*hyp2f1(a,b,c,x+y-x*y)
1.093432340896087107444363
>>> x**2*appellf3(1,1,1,1,3,x,-x)
0.01568646277445385390945083
>>> polylog(2,x**2)
0.01568646277445385390945083
>>> a1,a2,b1,b2,c,x = map(mpf, [0.5,0.25,0.125,0.5,4.25,0.125])
>>> appellf3(a1,a2,b1,b2,c,x,1)
1.03947361709111140096947
>>> gammaprod([c,c-a2-b2],[c-a2,c-b2])*hyp3f2(a1,b1,c-a2-b2,c-a2,c-b2,x)
1.03947361709111140096947
The Appell F3 function satisfies a pair of partial
differential equations::
>>> a1,a2,b1,b2,c,x,y = map(mpf, [0.5,0.25,0.125,0.5,0.625,0.0625,-0.0625])
>>> F = lambda x,y: appellf3(a1,a2,b1,b2,c,x,y)
>>> chop(x*(1-x)*diff(F,(x,y),(2,0)) +
... y*diff(F,(x,y),(1,1)) +
... (c-(a1+b1+1)*x)*diff(F,(x,y),(1,0)) -
... a1*b1*F(x,y))
0.0
>>> chop(y*(1-y)*diff(F,(x,y),(0,2)) +
... x*diff(F,(x,y),(1,1)) +
... (c-(a2+b2+1)*y)*diff(F,(x,y),(0,1)) -
... a2*b2*F(x,y))
0.0
**References**
See references for :func:`~mpmath.appellf1`.
"""
appellf4 = r"""
Gives the Appell F4 hypergeometric function of two variables
.. math ::
F_4(a,b,c_1,c_2,x,y) = \sum_{m=0}^{\infty} \sum_{n=0}^{\infty}
\frac{(a)_{m+n} (b)_{m+n}}{(c_1)_m (c_2)_n}
\frac{x^m y^n}{m! n!}.
The series is generally absolutely convergent for
`\sqrt{|x|} + \sqrt{|y|} < 1`.
**Examples**
Evaluation for various parameters and arguments::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> appellf4(1,1,2,2,0.25,0.125)
1.286182069079718313546608
>>> appellf4(-2,-3,4,5,4,5)
34.8
>>> appellf4(5,4,2,3,0.25j,-0.125j)
(-0.2585967215437846642163352 + 2.436102233553582711818743j)
Reduction to `\,_2F_1` in a special case::
>>> a,b,c,x,y = map(mpf, [0.5,0.25,0.125,0.125,-0.125])
>>> appellf4(a,b,c,a+b-c+1,x*(1-y),y*(1-x))
1.129143488466850868248364
>>> hyp2f1(a,b,c,x)*hyp2f1(a,b,a+b-c+1,y)
1.129143488466850868248364
A system of partial differential equations satisfied by F4::
>>> a,b,c1,c2,x,y = map(mpf, [1,0.5,0.25,1.125,0.0625,-0.0625])
>>> F = lambda x,y: appellf4(a,b,c1,c2,x,y)
>>> chop(x*(1-x)*diff(F,(x,y),(2,0)) -
... y**2*diff(F,(x,y),(0,2)) -
... 2*x*y*diff(F,(x,y),(1,1)) +
... (c1-(a+b+1)*x)*diff(F,(x,y),(1,0)) -
... ((a+b+1)*y)*diff(F,(x,y),(0,1)) -
... a*b*F(x,y))
0.0
>>> chop(y*(1-y)*diff(F,(x,y),(0,2)) -
... x**2*diff(F,(x,y),(2,0)) -
... 2*x*y*diff(F,(x,y),(1,1)) +
... (c2-(a+b+1)*y)*diff(F,(x,y),(0,1)) -
... ((a+b+1)*x)*diff(F,(x,y),(1,0)) -
... a*b*F(x,y))
0.0
**References**
See references for :func:`~mpmath.appellf1`.
"""
zeta = r"""
Computes the Riemann zeta function
.. math ::
\zeta(s) = 1+\frac{1}{2^s}+\frac{1}{3^s}+\frac{1}{4^s}+\ldots
or, with `a \ne 1`, the more general Hurwitz zeta function
.. math ::
\zeta(s,a) = \sum_{k=0}^\infty \frac{1}{(a+k)^s}.
Optionally, ``zeta(s, a, n)`` computes the `n`-th derivative with
respect to `s`,
.. math ::
\zeta^{(n)}(s,a) = (-1)^n \sum_{k=0}^\infty \frac{\log^n(a+k)}{(a+k)^s}.
Although these series only converge for `\Re(s) > 1`, the Riemann and Hurwitz
zeta functions are defined through analytic continuation for arbitrary
complex `s \ne 1` (`s = 1` is a pole).
The implementation uses three algorithms: the Borwein algorithm for
the Riemann zeta function when `s` is close to the real line;
the Riemann-Siegel formula for the Riemann zeta function when `s` is
large imaginary, and Euler-Maclaurin summation in all other cases.
The reflection formula for `\Re(s) < 0` is implemented in some cases.
The algorithm can be chosen with ``method = 'borwein'``,
``method='riemann-siegel'`` or ``method = 'euler-maclaurin'``.
The parameter `a` is usually a rational number `a = p/q`, and may be specified
as such by passing an integer tuple `(p, q)`. Evaluation is supported for
arbitrary complex `a`, but may be slow and/or inaccurate when `\Re(s) < 0` for
nonrational `a` or when computing derivatives.
**Examples**
Some values of the Riemann zeta function::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> zeta(2); pi**2 / 6
1.644934066848226436472415
1.644934066848226436472415
>>> zeta(0)
-0.5
>>> zeta(-1)
-0.08333333333333333333333333
>>> zeta(-2)
0.0
For large positive `s`, `\zeta(s)` rapidly approaches 1::
>>> zeta(50)
1.000000000000000888178421
>>> zeta(100)
1.0
>>> zeta(inf)
1.0
>>> 1-sum((zeta(k)-1)/k for k in range(2,85)); +euler
0.5772156649015328606065121
0.5772156649015328606065121
>>> nsum(lambda k: zeta(k)-1, [2, inf])
1.0
Evaluation is supported for complex `s` and `a`:
>>> zeta(-3+4j)
(-0.03373057338827757067584698 + 0.2774499251557093745297677j)
>>> zeta(2+3j, -1+j)
(389.6841230140842816370741 + 295.2674610150305334025962j)
The Riemann zeta function has so-called nontrivial zeros on
the critical line `s = 1/2 + it`::
>>> findroot(zeta, 0.5+14j); zetazero(1)
(0.5 + 14.13472514173469379045725j)
(0.5 + 14.13472514173469379045725j)
>>> findroot(zeta, 0.5+21j); zetazero(2)
(0.5 + 21.02203963877155499262848j)
(0.5 + 21.02203963877155499262848j)
>>> findroot(zeta, 0.5+25j); zetazero(3)
(0.5 + 25.01085758014568876321379j)
(0.5 + 25.01085758014568876321379j)
>>> chop(zeta(zetazero(10)))
0.0
Evaluation on and near the critical line is supported for large
heights `t` by means of the Riemann-Siegel formula (currently
for `a = 1`, `n \le 4`)::
>>> zeta(0.5+100000j)
(1.073032014857753132114076 + 5.780848544363503984261041j)
>>> zeta(0.75+1000000j)
(0.9535316058375145020351559 + 0.9525945894834273060175651j)
>>> zeta(0.5+10000000j)
(11.45804061057709254500227 - 8.643437226836021723818215j)
>>> zeta(0.5+100000000j, derivative=1)
(51.12433106710194942681869 + 43.87221167872304520599418j)
>>> zeta(0.5+100000000j, derivative=2)
(-444.2760822795430400549229 - 896.3789978119185981665403j)
>>> zeta(0.5+100000000j, derivative=3)
(3230.72682687670422215339 + 14374.36950073615897616781j)
>>> zeta(0.5+100000000j, derivative=4)
(-11967.35573095046402130602 - 218945.7817789262839266148j)
>>> zeta(1+10000000j) # off the line
(2.859846483332530337008882 + 0.491808047480981808903986j)
>>> zeta(1+10000000j, derivative=1)
(-4.333835494679647915673205 - 0.08405337962602933636096103j)
>>> zeta(1+10000000j, derivative=4)
(453.2764822702057701894278 - 581.963625832768189140995j)
For investigation of the zeta function zeros, the Riemann-Siegel
Z-function is often more convenient than working with the Riemann
zeta function directly (see :func:`~mpmath.siegelz`).
Some values of the Hurwitz zeta function::
>>> zeta(2, 3); -5./4 + pi**2/6
0.3949340668482264364724152
0.3949340668482264364724152
>>> zeta(2, (3,4)); pi**2 - 8*catalan
2.541879647671606498397663
2.541879647671606498397663
For positive integer values of `s`, the Hurwitz zeta function is
equivalent to a polygamma function (except for a normalizing factor)::
>>> zeta(4, (1,5)); psi(3, '1/5')/6
625.5408324774542966919938
625.5408324774542966919938
Evaluation of derivatives::
>>> zeta(0, 3+4j, 1); loggamma(3+4j) - ln(2*pi)/2
(-2.675565317808456852310934 + 4.742664438034657928194889j)
(-2.675565317808456852310934 + 4.742664438034657928194889j)
>>> zeta(2, 1, 20)
2432902008176640000.000242
>>> zeta(3+4j, 5.5+2j, 4)
(-0.140075548947797130681075 - 0.3109263360275413251313634j)
>>> zeta(0.5+100000j, 1, 4)
(-10407.16081931495861539236 + 13777.78669862804508537384j)
>>> zeta(-100+0.5j, (1,3), derivative=4)
(4.007180821099823942702249e+79 + 4.916117957092593868321778e+78j)
Generating a Taylor series at `s = 2` using derivatives::
>>> for k in range(11): print("%s * (s-2)^%i" % (zeta(2,1,k)/fac(k), k))
...
1.644934066848226436472415 * (s-2)^0
-0.9375482543158437537025741 * (s-2)^1
0.9946401171494505117104293 * (s-2)^2
-1.000024300473840810940657 * (s-2)^3
1.000061933072352565457512 * (s-2)^4
-1.000006869443931806408941 * (s-2)^5
1.000000173233769531820592 * (s-2)^6
-0.9999999569989868493432399 * (s-2)^7
0.9999999937218844508684206 * (s-2)^8
-0.9999999996355013916608284 * (s-2)^9
1.000000000004610645020747 * (s-2)^10
Evaluation at zero and for negative integer `s`::
>>> zeta(0, 10)
-9.5
>>> zeta(-2, (2,3)); mpf(1)/81
0.01234567901234567901234568
0.01234567901234567901234568
>>> zeta(-3+4j, (5,4))
(0.2899236037682695182085988 + 0.06561206166091757973112783j)
>>> zeta(-3.25, 1/pi)
-0.0005117269627574430494396877
>>> zeta(-3.5, pi, 1)
11.156360390440003294709
>>> zeta(-100.5, (8,3))
-4.68162300487989766727122e+77
>>> zeta(-10.5, (-8,3))
(-0.01521913704446246609237979 + 29907.72510874248161608216j)
>>> zeta(-1000.5, (-8,3))
(1.031911949062334538202567e+1770 + 1.519555750556794218804724e+426j)
>>> zeta(-1+j, 3+4j)
(-16.32988355630802510888631 - 22.17706465801374033261383j)
>>> zeta(-1+j, 3+4j, 2)
(32.48985276392056641594055 - 51.11604466157397267043655j)
>>> diff(lambda s: zeta(s, 3+4j), -1+j, 2)
(32.48985276392056641594055 - 51.11604466157397267043655j)
**References**
1. http://mathworld.wolfram.com/RiemannZetaFunction.html
2. http://mathworld.wolfram.com/HurwitzZetaFunction.html
3. http://www.cecm.sfu.ca/personal/pborwein/PAPERS/P155.pdf
"""
dirichlet = r"""
Evaluates the Dirichlet L-function
.. math ::
L(s,\chi) = \sum_{k=1}^\infty \frac{\chi(k)}{k^s}.
where `\chi` is a periodic sequence of length `q` which should be supplied
in the form of a list `[\chi(0), \chi(1), \ldots, \chi(q-1)]`.
Strictly, `\chi` should be a Dirichlet character, but any periodic
sequence will work.
For example, ``dirichlet(s, [1])`` gives the ordinary
Riemann zeta function and ``dirichlet(s, [-1,1])`` gives
the alternating zeta function (Dirichlet eta function).
Also the derivative with respect to `s` (currently only a first
derivative) can be evaluated.
**Examples**
The ordinary Riemann zeta function::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> dirichlet(3, [1]); zeta(3)
1.202056903159594285399738
1.202056903159594285399738
>>> dirichlet(1, [1])
+inf
The alternating zeta function::
>>> dirichlet(1, [-1,1]); ln(2)
0.6931471805599453094172321
0.6931471805599453094172321
The following defines the Dirichlet beta function
`\beta(s) = \sum_{k=0}^\infty \frac{(-1)^k}{(2k+1)^s}` and verifies
several values of this function::
>>> B = lambda s, d=0: dirichlet(s, [0, 1, 0, -1], d)
>>> B(0); 1./2
0.5
0.5
>>> B(1); pi/4
0.7853981633974483096156609
0.7853981633974483096156609
>>> B(2); +catalan
0.9159655941772190150546035
0.9159655941772190150546035
>>> B(2,1); diff(B, 2)
0.08158073611659279510291217
0.08158073611659279510291217
>>> B(-1,1); 2*catalan/pi
0.5831218080616375602767689
0.5831218080616375602767689
>>> B(0,1); log(gamma(0.25)**2/(2*pi*sqrt(2)))
0.3915943927068367764719453
0.3915943927068367764719454
>>> B(1,1); 0.25*pi*(euler+2*ln2+3*ln(pi)-4*ln(gamma(0.25)))
0.1929013167969124293631898
0.1929013167969124293631898
A custom L-series of period 3::
>>> dirichlet(2, [2,0,1])
0.7059715047839078092146831
>>> 2*nsum(lambda k: (3*k)**-2, [1,inf]) + \
... nsum(lambda k: (3*k+2)**-2, [0,inf])
0.7059715047839078092146831
"""
coulombf = r"""
Calculates the regular Coulomb wave function
.. math ::
F_l(\eta,z) = C_l(\eta) z^{l+1} e^{-iz} \,_1F_1(l+1-i\eta, 2l+2, 2iz)
where the normalization constant `C_l(\eta)` is as calculated by
:func:`~mpmath.coulombc`. This function solves the differential equation
.. math ::
f''(z) + \left(1-\frac{2\eta}{z}-\frac{l(l+1)}{z^2}\right) f(z) = 0.
A second linearly independent solution is given by the irregular
Coulomb wave function `G_l(\eta,z)` (see :func:`~mpmath.coulombg`)
and thus the general solution is
`f(z) = C_1 F_l(\eta,z) + C_2 G_l(\eta,z)` for arbitrary
constants `C_1`, `C_2`.
Physically, the Coulomb wave functions give the radial solution
to the Schrodinger equation for a point particle in a `1/z` potential; `z` is
then the radius and `l`, `\eta` are quantum numbers.
The Coulomb wave functions with real parameters are defined
in Abramowitz & Stegun, section 14. However, all parameters are permitted
to be complex in this implementation (see references).
**Plots**
.. literalinclude :: /plots/coulombf.py
.. image :: /plots/coulombf.png
.. literalinclude :: /plots/coulombf_c.py
.. image :: /plots/coulombf_c.png
**Examples**
Evaluation is supported for arbitrary magnitudes of `z`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> coulombf(2, 1.5, 3.5)
0.4080998961088761187426445
>>> coulombf(-2, 1.5, 3.5)
0.7103040849492536747533465
>>> coulombf(2, 1.5, '1e-10')
4.143324917492256448770769e-33
>>> coulombf(2, 1.5, 1000)
0.4482623140325567050716179
>>> coulombf(2, 1.5, 10**10)
-0.066804196437694360046619
Verifying the differential equation::
>>> l, eta, z = 2, 3, mpf(2.75)
>>> A, B = 1, 2
>>> f = lambda z: A*coulombf(l,eta,z) + B*coulombg(l,eta,z)
>>> chop(diff(f,z,2) + (1-2*eta/z - l*(l+1)/z**2)*f(z))
0.0
A Wronskian relation satisfied by the Coulomb wave functions::
>>> l = 2
>>> eta = 1.5
>>> F = lambda z: coulombf(l,eta,z)
>>> G = lambda z: coulombg(l,eta,z)
>>> for z in [3.5, -1, 2+3j]:
... chop(diff(F,z)*G(z) - F(z)*diff(G,z))
...
1.0
1.0
1.0
Another Wronskian relation::
>>> F = coulombf
>>> G = coulombg
>>> for z in [3.5, -1, 2+3j]:
... chop(F(l-1,eta,z)*G(l,eta,z)-F(l,eta,z)*G(l-1,eta,z) - l/sqrt(l**2+eta**2))
...
0.0
0.0
0.0
An integral identity connecting the regular and irregular wave functions::
>>> l, eta, z = 4+j, 2-j, 5+2j
>>> coulombf(l,eta,z) + j*coulombg(l,eta,z)
(0.7997977752284033239714479 + 0.9294486669502295512503127j)
>>> g = lambda t: exp(-t)*t**(l-j*eta)*(t+2*j*z)**(l+j*eta)
>>> j*exp(-j*z)*z**(-l)/fac(2*l+1)/coulombc(l,eta)*quad(g, [0,inf])
(0.7997977752284033239714479 + 0.9294486669502295512503127j)
Some test case with complex parameters, taken from Michel [2]::
>>> mp.dps = 15
>>> coulombf(1+0.1j, 50+50j, 100.156)
(-1.02107292320897e+15 - 2.83675545731519e+15j)
>>> coulombg(1+0.1j, 50+50j, 100.156)
(2.83675545731519e+15 - 1.02107292320897e+15j)
>>> coulombf(1e-5j, 10+1e-5j, 0.1+1e-6j)
(4.30566371247811e-14 - 9.03347835361657e-19j)
>>> coulombg(1e-5j, 10+1e-5j, 0.1+1e-6j)
(778709182061.134 + 18418936.2660553j)
The following reproduces a table in Abramowitz & Stegun, at twice
the precision::
>>> mp.dps = 10
>>> eta = 2; z = 5
>>> for l in [5, 4, 3, 2, 1, 0]:
... print("%s %s %s" % (l, coulombf(l,eta,z),
... diff(lambda z: coulombf(l,eta,z), z)))
...
5 0.09079533488 0.1042553261
4 0.2148205331 0.2029591779
3 0.4313159311 0.320534053
2 0.7212774133 0.3952408216
1 0.9935056752 0.3708676452
0 1.143337392 0.2937960375
**References**
1. I.J. Thompson & A.R. Barnett, "Coulomb and Bessel Functions of Complex
Arguments and Order", J. Comp. Phys., vol 64, no. 2, June 1986.
2. N. Michel, "Precise Coulomb wave functions for a wide range of
complex `l`, `\eta` and `z`", http://arxiv.org/abs/physics/0702051v1
"""
coulombg = r"""
Calculates the irregular Coulomb wave function
.. math ::
G_l(\eta,z) = \frac{F_l(\eta,z) \cos(\chi) - F_{-l-1}(\eta,z)}{\sin(\chi)}
where `\chi = \sigma_l - \sigma_{-l-1} - (l+1/2) \pi`
and `\sigma_l(\eta) = (\ln \Gamma(1+l+i\eta)-\ln \Gamma(1+l-i\eta))/(2i)`.
See :func:`~mpmath.coulombf` for additional information.
**Plots**
.. literalinclude :: /plots/coulombg.py
.. image :: /plots/coulombg.png
.. literalinclude :: /plots/coulombg_c.py
.. image :: /plots/coulombg_c.png
**Examples**
Evaluation is supported for arbitrary magnitudes of `z`::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> coulombg(-2, 1.5, 3.5)
1.380011900612186346255524
>>> coulombg(2, 1.5, 3.5)
1.919153700722748795245926
>>> coulombg(-2, 1.5, '1e-10')
201126715824.7329115106793
>>> coulombg(-2, 1.5, 1000)
0.1802071520691149410425512
>>> coulombg(-2, 1.5, 10**10)
0.652103020061678070929794
The following reproduces a table in Abramowitz & Stegun,
at twice the precision::
>>> mp.dps = 10
>>> eta = 2; z = 5
>>> for l in [1, 2, 3, 4, 5]:
... print("%s %s %s" % (l, coulombg(l,eta,z),
... -diff(lambda z: coulombg(l,eta,z), z)))
...
1 1.08148276 0.6028279961
2 1.496877075 0.5661803178
3 2.048694714 0.7959909551
4 3.09408669 1.731802374
5 5.629840456 4.549343289
Evaluation close to the singularity at `z = 0`::
>>> mp.dps = 15
>>> coulombg(0,10,1)
3088184933.67358
>>> coulombg(0,10,'1e-10')
5554866000719.8
>>> coulombg(0,10,'1e-100')
5554866221524.1
Evaluation with a half-integer value for `l`::
>>> coulombg(1.5, 1, 10)
0.852320038297334
"""
coulombc = r"""
Gives the normalizing Gamow constant for Coulomb wave functions,
.. math ::
C_l(\eta) = 2^l \exp\left(-\pi \eta/2 + [\ln \Gamma(1+l+i\eta) +
\ln \Gamma(1+l-i\eta)]/2 - \ln \Gamma(2l+2)\right),
where the log gamma function with continuous imaginary part
away from the negative half axis (see :func:`~mpmath.loggamma`) is implied.
This function is used internally for the calculation of
Coulomb wave functions, and automatically cached to make multiple
evaluations with fixed `l`, `\eta` fast.
"""
ellipfun = r"""
Computes any of the Jacobi elliptic functions, defined
in terms of Jacobi theta functions as
.. math ::
\mathrm{sn}(u,m) = \frac{\vartheta_3(0,q)}{\vartheta_2(0,q)}
\frac{\vartheta_1(t,q)}{\vartheta_4(t,q)}
\mathrm{cn}(u,m) = \frac{\vartheta_4(0,q)}{\vartheta_2(0,q)}
\frac{\vartheta_2(t,q)}{\vartheta_4(t,q)}
\mathrm{dn}(u,m) = \frac{\vartheta_4(0,q)}{\vartheta_3(0,q)}
\frac{\vartheta_3(t,q)}{\vartheta_4(t,q)},
or more generally computes a ratio of two such functions. Here
`t = u/\vartheta_3(0,q)^2`, and `q = q(m)` denotes the nome (see
:func:`~mpmath.nome`). Optionally, you can specify the nome directly
instead of `m` by passing ``q=<value>``, or you can directly
specify the elliptic parameter `k` with ``k=<value>``.
The first argument should be a two-character string specifying the
function using any combination of ``'s'``, ``'c'``, ``'d'``, ``'n'``. These
letters respectively denote the basic functions
`\mathrm{sn}(u,m)`, `\mathrm{cn}(u,m)`, `\mathrm{dn}(u,m)`, and `1`.
The identifier specifies the ratio of two such functions.
For example, ``'ns'`` identifies the function
.. math ::
\mathrm{ns}(u,m) = \frac{1}{\mathrm{sn}(u,m)}
and ``'cd'`` identifies the function
.. math ::
\mathrm{cd}(u,m) = \frac{\mathrm{cn}(u,m)}{\mathrm{dn}(u,m)}.
If called with only the first argument, a function object
evaluating the chosen function for given arguments is returned.
**Examples**
Basic evaluation::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> ellipfun('cd', 3.5, 0.5)
-0.9891101840595543931308394
>>> ellipfun('cd', 3.5, q=0.25)
0.07111979240214668158441418
The sn-function is doubly periodic in the complex plane with periods
`4 K(m)` and `2 i K(1-m)` (see :func:`~mpmath.ellipk`)::
>>> sn = ellipfun('sn')
>>> sn(2, 0.25)
0.9628981775982774425751399
>>> sn(2+4*ellipk(0.25), 0.25)
0.9628981775982774425751399
>>> chop(sn(2+2*j*ellipk(1-0.25), 0.25))
0.9628981775982774425751399
The cn-function is doubly periodic with periods `4 K(m)` and `4 i K(1-m)`::
>>> cn = ellipfun('cn')
>>> cn(2, 0.25)
-0.2698649654510865792581416
>>> cn(2+4*ellipk(0.25), 0.25)
-0.2698649654510865792581416
>>> chop(cn(2+4*j*ellipk(1-0.25), 0.25))
-0.2698649654510865792581416
The dn-function is doubly periodic with periods `2 K(m)` and `4 i K(1-m)`::
>>> dn = ellipfun('dn')
>>> dn(2, 0.25)
0.8764740583123262286931578
>>> dn(2+2*ellipk(0.25), 0.25)
0.8764740583123262286931578
>>> chop(dn(2+4*j*ellipk(1-0.25), 0.25))
0.8764740583123262286931578
"""
jtheta = r"""
Computes the Jacobi theta function `\vartheta_n(z, q)`, where
`n = 1, 2, 3, 4`, defined by the infinite series:
.. math ::
\vartheta_1(z,q) = 2 q^{1/4} \sum_{n=0}^{\infty}
(-1)^n q^{n^2+n\,} \sin((2n+1)z)
\vartheta_2(z,q) = 2 q^{1/4} \sum_{n=0}^{\infty}
q^{n^{2\,} + n} \cos((2n+1)z)
\vartheta_3(z,q) = 1 + 2 \sum_{n=1}^{\infty}
q^{n^2\,} \cos(2 n z)
\vartheta_4(z,q) = 1 + 2 \sum_{n=1}^{\infty}
(-q)^{n^2\,} \cos(2 n z)
The theta functions are functions of two variables:
* `z` is the *argument*, an arbitrary real or complex number
* `q` is the *nome*, which must be a real or complex number
in the unit disk (i.e. `|q| < 1`). For `|q| \ll 1`, the
series converge very quickly, so the Jacobi theta functions
can efficiently be evaluated to high precision.
The compact notations `\vartheta_n(q) = \vartheta_n(0,q)`
and `\vartheta_n = \vartheta_n(0,q)` are also frequently
encountered. Finally, Jacobi theta functions are frequently
considered as functions of the half-period ratio `\tau`
and then usually denoted by `\vartheta_n(z|\tau)`.
Optionally, ``jtheta(n, z, q, derivative=d)`` with `d > 0` computes
a `d`-th derivative with respect to `z`.
**Examples and basic properties**
Considered as functions of `z`, the Jacobi theta functions may be
viewed as generalizations of the ordinary trigonometric functions
cos and sin. They are periodic functions::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> jtheta(1, 0.25, '0.2')
0.2945120798627300045053104
>>> jtheta(1, 0.25 + 2*pi, '0.2')
0.2945120798627300045053104
Indeed, the series defining the theta functions are essentially
trigonometric Fourier series. The coefficients can be retrieved
using :func:`~mpmath.fourier`::
>>> mp.dps = 10
>>> nprint(fourier(lambda x: jtheta(2, x, 0.5), [-pi, pi], 4))
([0.0, 1.68179, 0.0, 0.420448, 0.0], [0.0, 0.0, 0.0, 0.0, 0.0])
The Jacobi theta functions are also so-called quasiperiodic
functions of `z` and `\tau`, meaning that for fixed `\tau`,
`\vartheta_n(z, q)` and `\vartheta_n(z+\pi \tau, q)` are the same
except for an exponential factor::
>>> mp.dps = 25
>>> tau = 3*j/10
>>> q = exp(pi*j*tau)
>>> z = 10
>>> jtheta(4, z+tau*pi, q)
(-0.682420280786034687520568 + 1.526683999721399103332021j)
>>> -exp(-2*j*z)/q * jtheta(4, z, q)
(-0.682420280786034687520568 + 1.526683999721399103332021j)
The Jacobi theta functions satisfy a huge number of other
functional equations, such as the following identity (valid for
any `q`)::
>>> q = mpf(3)/10
>>> jtheta(3,0,q)**4
6.823744089352763305137427
>>> jtheta(2,0,q)**4 + jtheta(4,0,q)**4
6.823744089352763305137427
Extensive listings of identities satisfied by the Jacobi theta
functions can be found in standard reference works.
The Jacobi theta functions are related to the gamma function
for special arguments::
>>> jtheta(3, 0, exp(-pi))
1.086434811213308014575316
>>> pi**(1/4.) / gamma(3/4.)
1.086434811213308014575316
:func:`~mpmath.jtheta` supports arbitrary precision evaluation and complex
arguments::
>>> mp.dps = 50
>>> jtheta(4, sqrt(2), 0.5)
2.0549510717571539127004115835148878097035750653737
>>> mp.dps = 25
>>> jtheta(4, 1+2j, (1+j)/5)
(7.180331760146805926356634 - 1.634292858119162417301683j)
Evaluation of derivatives::
>>> mp.dps = 25
>>> jtheta(1, 7, 0.25, 1); diff(lambda z: jtheta(1, z, 0.25), 7)
1.209857192844475388637236
1.209857192844475388637236
>>> jtheta(1, 7, 0.25, 2); diff(lambda z: jtheta(1, z, 0.25), 7, 2)
-0.2598718791650217206533052
-0.2598718791650217206533052
>>> jtheta(2, 7, 0.25, 1); diff(lambda z: jtheta(2, z, 0.25), 7)
-1.150231437070259644461474
-1.150231437070259644461474
>>> jtheta(2, 7, 0.25, 2); diff(lambda z: jtheta(2, z, 0.25), 7, 2)
-0.6226636990043777445898114
-0.6226636990043777445898114
>>> jtheta(3, 7, 0.25, 1); diff(lambda z: jtheta(3, z, 0.25), 7)
-0.9990312046096634316587882
-0.9990312046096634316587882
>>> jtheta(3, 7, 0.25, 2); diff(lambda z: jtheta(3, z, 0.25), 7, 2)
-0.1530388693066334936151174
-0.1530388693066334936151174
>>> jtheta(4, 7, 0.25, 1); diff(lambda z: jtheta(4, z, 0.25), 7)
0.9820995967262793943571139
0.9820995967262793943571139
>>> jtheta(4, 7, 0.25, 2); diff(lambda z: jtheta(4, z, 0.25), 7, 2)
0.3936902850291437081667755
0.3936902850291437081667755
**Possible issues**
For `|q| \ge 1` or `\Im(\tau) \le 0`, :func:`~mpmath.jtheta` raises
``ValueError``. This exception is also raised for `|q|` extremely
close to 1 (or equivalently `\tau` very close to 0), since the
series would converge too slowly::
>>> jtheta(1, 10, 0.99999999 * exp(0.5*j))
Traceback (most recent call last):
...
ValueError: abs(q) > THETA_Q_LIM = 1.000000
"""
eulernum = r"""
Gives the `n`-th Euler number, defined as the `n`-th derivative of
`\mathrm{sech}(t) = 1/\cosh(t)` evaluated at `t = 0`. Equivalently, the
Euler numbers give the coefficients of the Taylor series
.. math ::
\mathrm{sech}(t) = \sum_{n=0}^{\infty} \frac{E_n}{n!} t^n.
The Euler numbers are closely related to Bernoulli numbers
and Bernoulli polynomials. They can also be evaluated in terms of
Euler polynomials (see :func:`~mpmath.eulerpoly`) as `E_n = 2^n E_n(1/2)`.
**Examples**
Computing the first few Euler numbers and verifying that they
agree with the Taylor series::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> [eulernum(n) for n in range(11)]
[1.0, 0.0, -1.0, 0.0, 5.0, 0.0, -61.0, 0.0, 1385.0, 0.0, -50521.0]
>>> chop(diffs(sech, 0, 10))
[1.0, 0.0, -1.0, 0.0, 5.0, 0.0, -61.0, 0.0, 1385.0, 0.0, -50521.0]
Euler numbers grow very rapidly. :func:`~mpmath.eulernum` efficiently
computes numerical approximations for large indices::
>>> eulernum(50)
-6.053285248188621896314384e+54
>>> eulernum(1000)
3.887561841253070615257336e+2371
>>> eulernum(10**20)
4.346791453661149089338186e+1936958564106659551331
Comparing with an asymptotic formula for the Euler numbers::
>>> n = 10**5
>>> (-1)**(n//2) * 8 * sqrt(n/(2*pi)) * (2*n/(pi*e))**n
3.69919063017432362805663e+436961
>>> eulernum(n)
3.699193712834466537941283e+436961
Pass ``exact=True`` to obtain exact values of Euler numbers as integers::
>>> print(eulernum(50, exact=True))
-6053285248188621896314383785111649088103498225146815121
>>> print(eulernum(200, exact=True) % 10**10)
1925859625
>>> eulernum(1001, exact=True)
0
"""
eulerpoly = r"""
Evaluates the Euler polynomial `E_n(z)`, defined by the generating function
representation
.. math ::
\frac{2e^{zt}}{e^t+1} = \sum_{n=0}^\infty E_n(z) \frac{t^n}{n!}.
The Euler polynomials may also be represented in terms of
Bernoulli polynomials (see :func:`~mpmath.bernpoly`) using various formulas, for
example
.. math ::
E_n(z) = \frac{2}{n+1} \left(
B_n(z)-2^{n+1}B_n\left(\frac{z}{2}\right)
\right).
Special values include the Euler numbers `E_n = 2^n E_n(1/2)` (see
:func:`~mpmath.eulernum`).
**Examples**
Computing the coefficients of the first few Euler polynomials::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> for n in range(6):
... chop(taylor(lambda z: eulerpoly(n,z), 0, n))
...
[1.0]
[-0.5, 1.0]
[0.0, -1.0, 1.0]
[0.25, 0.0, -1.5, 1.0]
[0.0, 1.0, 0.0, -2.0, 1.0]
[-0.5, 0.0, 2.5, 0.0, -2.5, 1.0]
Evaluation for arbitrary `z`::
>>> eulerpoly(2,3)
6.0
>>> eulerpoly(5,4)
423.5
>>> eulerpoly(35, 11111111112)
3.994957561486776072734601e+351
>>> eulerpoly(4, 10+20j)
(-47990.0 - 235980.0j)
>>> eulerpoly(2, '-3.5e-5')
0.000035001225
>>> eulerpoly(3, 0.5)
0.0
>>> eulerpoly(55, -10**80)
-1.0e+4400
>>> eulerpoly(5, -inf)
-inf
>>> eulerpoly(6, -inf)
+inf
Computing Euler numbers::
>>> 2**26 * eulerpoly(26,0.5)
-4087072509293123892361.0
>>> eulernum(26)
-4087072509293123892361.0
Evaluation is accurate for large `n` and small `z`::
>>> eulerpoly(100, 0.5)
2.29047999988194114177943e+108
>>> eulerpoly(1000, 10.5)
3.628120031122876847764566e+2070
>>> eulerpoly(10000, 10.5)
1.149364285543783412210773e+30688
"""
spherharm = r"""
Evaluates the spherical harmonic `Y_l^m(\theta,\phi)`,
.. math ::
Y_l^m(\theta,\phi) = \sqrt{\frac{2l+1}{4\pi}\frac{(l-m)!}{(l+m)!}}
P_l^m(\cos \theta) e^{i m \phi}
where `P_l^m` is an associated Legendre function (see :func:`~mpmath.legenp`).
Here `\theta \in [0, \pi]` denotes the polar coordinate (ranging
from the north pole to the south pole) and `\phi \in [0, 2 \pi]` denotes the
azimuthal coordinate on a sphere. Care should be used since many different
conventions for spherical coordinate variables are used.
Usually spherical harmonics are considered for `l \in \mathbb{N}`,
`m \in \mathbb{Z}`, `|m| \le l`. More generally, `l,m,\theta,\phi`
are permitted to be complex numbers.
.. note ::
:func:`~mpmath.spherharm` returns a complex number, even the value is
purely real.
**Plots**
.. literalinclude :: /plots/spherharm40.py
`Y_{4,0}`:
.. image :: /plots/spherharm40.png
`Y_{4,1}`:
.. image :: /plots/spherharm41.png
`Y_{4,2}`:
.. image :: /plots/spherharm42.png
`Y_{4,3}`:
.. image :: /plots/spherharm43.png
`Y_{4,4}`:
.. image :: /plots/spherharm44.png
**Examples**
Some low-order spherical harmonics with reference values::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> theta = pi/4
>>> phi = pi/3
>>> spherharm(0,0,theta,phi); 0.5*sqrt(1/pi)*expj(0)
(0.2820947917738781434740397 + 0.0j)
(0.2820947917738781434740397 + 0.0j)
>>> spherharm(1,-1,theta,phi); 0.5*sqrt(3/(2*pi))*expj(-phi)*sin(theta)
(0.1221506279757299803965962 - 0.2115710938304086076055298j)
(0.1221506279757299803965962 - 0.2115710938304086076055298j)
>>> spherharm(1,0,theta,phi); 0.5*sqrt(3/pi)*cos(theta)*expj(0)
(0.3454941494713354792652446 + 0.0j)
(0.3454941494713354792652446 + 0.0j)
>>> spherharm(1,1,theta,phi); -0.5*sqrt(3/(2*pi))*expj(phi)*sin(theta)
(-0.1221506279757299803965962 - 0.2115710938304086076055298j)
(-0.1221506279757299803965962 - 0.2115710938304086076055298j)
With the normalization convention used, the spherical harmonics are orthonormal
on the unit sphere::
>>> sphere = [0,pi], [0,2*pi]
>>> dS = lambda t,p: fp.sin(t) # differential element
>>> Y1 = lambda t,p: fp.spherharm(l1,m1,t,p)
>>> Y2 = lambda t,p: fp.conj(fp.spherharm(l2,m2,t,p))
>>> l1 = l2 = 3; m1 = m2 = 2
>>> print(fp.quad(lambda t,p: Y1(t,p)*Y2(t,p)*dS(t,p), *sphere))
(1+0j)
>>> m2 = 1 # m1 != m2
>>> print(fp.chop(fp.quad(lambda t,p: Y1(t,p)*Y2(t,p)*dS(t,p), *sphere)))
0.0
Evaluation is accurate for large orders::
>>> spherharm(1000,750,0.5,0.25)
(3.776445785304252879026585e-102 - 5.82441278771834794493484e-102j)
Evaluation works with complex parameter values::
>>> spherharm(1+j, 2j, 2+3j, -0.5j)
(64.44922331113759992154992 + 1981.693919841408089681743j)
"""
scorergi = r"""
Evaluates the Scorer function
.. math ::
\operatorname{Gi}(z) =
\operatorname{Ai}(z) \int_0^z \operatorname{Bi}(t) dt +
\operatorname{Bi}(z) \int_z^{\infty} \operatorname{Ai}(t) dt
which gives a particular solution to the inhomogeneous Airy
differential equation `f''(z) - z f(z) = 1/\pi`. Another
particular solution is given by the Scorer Hi-function
(:func:`~mpmath.scorerhi`). The two functions are related as
`\operatorname{Gi}(z) + \operatorname{Hi}(z) = \operatorname{Bi}(z)`.
**Plots**
.. literalinclude :: /plots/gi.py
.. image :: /plots/gi.png
.. literalinclude :: /plots/gi_c.py
.. image :: /plots/gi_c.png
**Examples**
Some values and limits::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> scorergi(0); 1/(power(3,'7/6')*gamma('2/3'))
0.2049755424820002450503075
0.2049755424820002450503075
>>> diff(scorergi, 0); 1/(power(3,'5/6')*gamma('1/3'))
0.1494294524512754526382746
0.1494294524512754526382746
>>> scorergi(+inf); scorergi(-inf)
0.0
0.0
>>> scorergi(1)
0.2352184398104379375986902
>>> scorergi(-1)
-0.1166722172960152826494198
Evaluation for large arguments::
>>> scorergi(10)
0.03189600510067958798062034
>>> scorergi(100)
0.003183105228162961476590531
>>> scorergi(1000000)
0.0000003183098861837906721743873
>>> 1/(pi*1000000)
0.0000003183098861837906715377675
>>> scorergi(-1000)
-0.08358288400262780392338014
>>> scorergi(-100000)
0.02886866118619660226809581
>>> scorergi(50+10j)
(0.0061214102799778578790984 - 0.001224335676457532180747917j)
>>> scorergi(-50-10j)
(5.236047850352252236372551e+29 - 3.08254224233701381482228e+29j)
>>> scorergi(100000j)
(-8.806659285336231052679025e+6474077 + 8.684731303500835514850962e+6474077j)
Verifying the connection between Gi and Hi::
>>> z = 0.25
>>> scorergi(z) + scorerhi(z)
0.7287469039362150078694543
>>> airybi(z)
0.7287469039362150078694543
Verifying the differential equation::
>>> for z in [-3.4, 0, 2.5, 1+2j]:
... chop(diff(scorergi,z,2) - z*scorergi(z))
...
-0.3183098861837906715377675
-0.3183098861837906715377675
-0.3183098861837906715377675
-0.3183098861837906715377675
Verifying the integral representation::
>>> z = 0.5
>>> scorergi(z)
0.2447210432765581976910539
>>> Ai,Bi = airyai,airybi
>>> Bi(z)*(Ai(inf,-1)-Ai(z,-1)) + Ai(z)*(Bi(z,-1)-Bi(0,-1))
0.2447210432765581976910539
**References**
1. [DLMF]_ section 9.12: Scorer Functions
"""
scorerhi = r"""
Evaluates the second Scorer function
.. math ::
\operatorname{Hi}(z) =
\operatorname{Bi}(z) \int_{-\infty}^z \operatorname{Ai}(t) dt -
\operatorname{Ai}(z) \int_{-\infty}^z \operatorname{Bi}(t) dt
which gives a particular solution to the inhomogeneous Airy
differential equation `f''(z) - z f(z) = 1/\pi`. See also
:func:`~mpmath.scorergi`.
**Plots**
.. literalinclude :: /plots/hi.py
.. image :: /plots/hi.png
.. literalinclude :: /plots/hi_c.py
.. image :: /plots/hi_c.png
**Examples**
Some values and limits::
>>> from mpmath import *
>>> mp.dps = 25; mp.pretty = True
>>> scorerhi(0); 2/(power(3,'7/6')*gamma('2/3'))
0.4099510849640004901006149
0.4099510849640004901006149
>>> diff(scorerhi,0); 2/(power(3,'5/6')*gamma('1/3'))
0.2988589049025509052765491
0.2988589049025509052765491
>>> scorerhi(+inf); scorerhi(-inf)
+inf
0.0
>>> scorerhi(1)
0.9722051551424333218376886
>>> scorerhi(-1)
0.2206696067929598945381098
Evaluation for large arguments::
>>> scorerhi(10)
455641153.5163291358991077
>>> scorerhi(100)
6.041223996670201399005265e+288
>>> scorerhi(1000000)
7.138269638197858094311122e+289529652
>>> scorerhi(-10)
0.0317685352825022727415011
>>> scorerhi(-100)
0.003183092495767499864680483
>>> scorerhi(100j)
(-6.366197716545672122983857e-9 + 0.003183098861710582761688475j)
>>> scorerhi(50+50j)
(-5.322076267321435669290334e+63 + 1.478450291165243789749427e+65j)
>>> scorerhi(-1000-1000j)
(0.0001591549432510502796565538 - 0.000159154943091895334973109j)
Verifying the differential equation::
>>> for z in [-3.4, 0, 2, 1+2j]:
... chop(diff(scorerhi,z,2) - z*scorerhi(z))
...
0.3183098861837906715377675
0.3183098861837906715377675
0.3183098861837906715377675
0.3183098861837906715377675
Verifying the integral representation::
>>> z = 0.5
>>> scorerhi(z)
0.6095559998265972956089949
>>> Ai,Bi = airyai,airybi
>>> Bi(z)*(Ai(z,-1)-Ai(-inf,-1)) - Ai(z)*(Bi(z,-1)-Bi(-inf,-1))
0.6095559998265972956089949
"""
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/function_docs.py
|
function_docs.py
|
import operator
import libmp
from .libmp.backend import basestring
from .libmp import (
int_types, MPZ_ONE,
prec_to_dps, dps_to_prec, repr_dps,
round_floor, round_ceiling,
fzero, finf, fninf, fnan,
mpf_le, mpf_neg,
from_int, from_float, from_str, from_rational,
mpi_mid, mpi_delta, mpi_str,
mpi_abs, mpi_pos, mpi_neg, mpi_add, mpi_sub,
mpi_mul, mpi_div, mpi_pow_int, mpi_pow,
mpi_from_str,
mpci_pos, mpci_neg, mpci_add, mpci_sub, mpci_mul, mpci_div, mpci_pow,
mpci_abs, mpci_pow, mpci_exp, mpci_log,
ComplexResult)
mpi_zero = (fzero, fzero)
from .ctx_base import StandardBaseContext
new = object.__new__
def convert_mpf_(x, prec, rounding):
if hasattr(x, "_mpf_"): return x._mpf_
if isinstance(x, int_types): return from_int(x, prec, rounding)
if isinstance(x, float): return from_float(x, prec, rounding)
if isinstance(x, basestring): return from_str(x, prec, rounding)
class ivmpf(object):
"""
Interval arithmetic class. Precision is controlled by iv.prec.
"""
def __new__(cls, x=0):
return cls.ctx.convert(x)
def __int__(self):
a, b = self._mpi_
if a == b:
return int(libmp.to_int(a))
raise ValueError
@property
def real(self): return self
@property
def imag(self): return self.ctx.zero
def conjugate(self): return self
@property
def a(self):
a, b = self._mpi_
return self.ctx.make_mpf((a, a))
@property
def b(self):
a, b = self._mpi_
return self.ctx.make_mpf((b, b))
@property
def mid(self):
ctx = self.ctx
v = mpi_mid(self._mpi_, ctx.prec)
return ctx.make_mpf((v, v))
@property
def delta(self):
ctx = self.ctx
v = mpi_delta(self._mpi_, ctx.prec)
return ctx.make_mpf((v,v))
@property
def _mpci_(self):
return self._mpi_, mpi_zero
def _compare(*args):
raise TypeError("no ordering relation is defined for intervals")
__gt__ = _compare
__le__ = _compare
__gt__ = _compare
__ge__ = _compare
def __contains__(self, t):
t = self.ctx.mpf(t)
return (self.a <= t.a) and (t.b <= self.b)
def __str__(self):
return mpi_str(self._mpi_, self.ctx.prec)
def __repr__(self):
if self.ctx.pretty:
return str(self)
a, b = self._mpi_
n = repr_dps(self.ctx.prec)
a = libmp.to_str(a, n)
b = libmp.to_str(b, n)
return "mpi(%r, %r)" % (a, b)
def _compare(s, t, cmpfun):
if not hasattr(t, "_mpi_"):
try:
t = s.ctx.convert(t)
except:
return NotImplemented
return cmpfun(s._mpi_, t._mpi_)
def __eq__(s, t): return s._compare(t, libmp.mpi_eq)
def __ne__(s, t): return s._compare(t, libmp.mpi_ne)
def __lt__(s, t): return s._compare(t, libmp.mpi_lt)
def __le__(s, t): return s._compare(t, libmp.mpi_le)
def __gt__(s, t): return s._compare(t, libmp.mpi_gt)
def __ge__(s, t): return s._compare(t, libmp.mpi_ge)
def __abs__(self):
return self.ctx.make_mpf(mpi_abs(self._mpi_, self.ctx.prec))
def __pos__(self):
return self.ctx.make_mpf(mpi_pos(self._mpi_, self.ctx.prec))
def __neg__(self):
return self.ctx.make_mpf(mpi_neg(self._mpi_, self.ctx.prec))
def ae(s, t, rel_eps=None, abs_eps=None):
return s.ctx.almosteq(s, t, rel_eps, abs_eps)
class ivmpc(object):
def __new__(cls, re=0, im=0):
re = cls.ctx.convert(re)
im = cls.ctx.convert(im)
y = new(cls)
y._mpci_ = re._mpi_, im._mpi_
return y
def __repr__(s):
if s.ctx.pretty:
return str(s)
return "iv.mpc(%s, %s)" % (repr(s.real), repr(s.imag))
def __str__(s):
return "(%s + %s*j)" % (str(s.real), str(s.imag))
@property
def a(self):
(a, b), (c,d) = self._mpci_
return self.ctx.make_mpf((a, a))
@property
def b(self):
(a, b), (c,d) = self._mpci_
return self.ctx.make_mpf((b, b))
@property
def c(self):
(a, b), (c,d) = self._mpci_
return self.ctx.make_mpf((c, c))
@property
def d(self):
(a, b), (c,d) = self._mpci_
return self.ctx.make_mpf((d, d))
@property
def real(s):
return s.ctx.make_mpf(s._mpci_[0])
@property
def imag(s):
return s.ctx.make_mpf(s._mpci_[1])
def conjugate(s):
a, b = s._mpci_
return s.ctx.make_mpc((a, mpf_neg(b)))
def overlap(s, t):
t = s.ctx.convert(t)
real_overlap = (s.a <= t.a <= s.b) or (s.a <= t.b <= s.b) or (t.a <= s.a <= t.b) or (t.a <= s.b <= t.b)
imag_overlap = (s.c <= t.c <= s.d) or (s.c <= t.d <= s.d) or (t.c <= s.c <= t.d) or (t.c <= s.d <= t.d)
return real_overlap and imag_overlap
def __contains__(s, t):
t = s.ctx.convert(t)
return t.real in s.real and t.imag in s.imag
def _compare(s, t, ne=False):
if not isinstance(t, s.ctx._types):
try:
t = s.ctx.convert(t)
except:
return NotImplemented
if hasattr(t, '_mpi_'):
tval = t._mpi_, mpi_zero
elif hasattr(t, '_mpci_'):
tval = t._mpci_
if ne:
return s._mpci_ != tval
return s._mpci_ == tval
def __eq__(s, t): return s._compare(t)
def __ne__(s, t): return s._compare(t, True)
def __lt__(s, t): raise TypeError("complex intervals cannot be ordered")
__le__ = __gt__ = __ge__ = __lt__
def __neg__(s): return s.ctx.make_mpc(mpci_neg(s._mpci_, s.ctx.prec))
def __pos__(s): return s.ctx.make_mpc(mpci_pos(s._mpci_, s.ctx.prec))
def __abs__(s): return s.ctx.make_mpf(mpci_abs(s._mpci_, s.ctx.prec))
def ae(s, t, rel_eps=None, abs_eps=None):
return s.ctx.almosteq(s, t, rel_eps, abs_eps)
def _binary_op(f_real, f_complex):
def g_complex(ctx, sval, tval):
return ctx.make_mpc(f_complex(sval, tval, ctx.prec))
def g_real(ctx, sval, tval):
try:
return ctx.make_mpf(f_real(sval, tval, ctx.prec))
except ComplexResult:
sval = (sval, mpi_zero)
tval = (tval, mpi_zero)
return g_complex(ctx, sval, tval)
def lop_real(s, t):
ctx = s.ctx
if not isinstance(t, ctx._types): t = ctx.convert(t)
if hasattr(t, "_mpi_"): return g_real(ctx, s._mpi_, t._mpi_)
if hasattr(t, "_mpci_"): return g_complex(ctx, (s._mpi_, mpi_zero), t._mpci_)
return NotImplemented
def rop_real(s, t):
ctx = s.ctx
if not isinstance(t, ctx._types): t = ctx.convert(t)
if hasattr(t, "_mpi_"): return g_real(ctx, t._mpi_, s._mpi_)
if hasattr(t, "_mpci_"): return g_complex(ctx, t._mpci_, (s._mpi_, mpi_zero))
return NotImplemented
def lop_complex(s, t):
ctx = s.ctx
if not isinstance(t, s.ctx._types):
try:
t = s.ctx.convert(t)
except (ValueError, TypeError):
return NotImplemented
return g_complex(ctx, s._mpci_, t._mpci_)
def rop_complex(s, t):
ctx = s.ctx
if not isinstance(t, s.ctx._types):
t = s.ctx.convert(t)
return g_complex(ctx, t._mpci_, s._mpci_)
return lop_real, rop_real, lop_complex, rop_complex
ivmpf.__add__, ivmpf.__radd__, ivmpc.__add__, ivmpc.__radd__ = _binary_op(mpi_add, mpci_add)
ivmpf.__sub__, ivmpf.__rsub__, ivmpc.__sub__, ivmpc.__rsub__ = _binary_op(mpi_sub, mpci_sub)
ivmpf.__mul__, ivmpf.__rmul__, ivmpc.__mul__, ivmpc.__rmul__ = _binary_op(mpi_mul, mpci_mul)
ivmpf.__div__, ivmpf.__rdiv__, ivmpc.__div__, ivmpc.__rdiv__ = _binary_op(mpi_div, mpci_div)
ivmpf.__pow__, ivmpf.__rpow__, ivmpc.__pow__, ivmpc.__rpow__ = _binary_op(mpi_pow, mpci_pow)
ivmpf.__truediv__ = ivmpf.__div__; ivmpf.__rtruediv__ = ivmpf.__rdiv__
ivmpc.__truediv__ = ivmpc.__div__; ivmpc.__rtruediv__ = ivmpc.__rdiv__
class ivmpf_constant(ivmpf):
def __new__(cls, f):
self = new(cls)
self._f = f
return self
def _get_mpi_(self):
prec = self.ctx._prec[0]
a = self._f(prec, round_floor)
b = self._f(prec, round_ceiling)
return a, b
_mpi_ = property(_get_mpi_)
class MPIntervalContext(StandardBaseContext):
def __init__(ctx):
ctx.mpf = type('ivmpf', (ivmpf,), {})
ctx.mpc = type('ivmpc', (ivmpc,), {})
ctx._types = (ctx.mpf, ctx.mpc)
ctx._constant = type('ivmpf_constant', (ivmpf_constant,), {})
ctx._prec = [53]
ctx._set_prec(53)
ctx._constant._ctxdata = ctx.mpf._ctxdata = ctx.mpc._ctxdata = [ctx.mpf, new, ctx._prec]
ctx._constant.ctx = ctx.mpf.ctx = ctx.mpc.ctx = ctx
ctx.pretty = False
StandardBaseContext.__init__(ctx)
ctx._init_builtins()
def _mpi(ctx, a, b=None):
if b is None:
return ctx.mpf(a)
return ctx.mpf((a,b))
def _init_builtins(ctx):
ctx.one = ctx.mpf(1)
ctx.zero = ctx.mpf(0)
ctx.inf = ctx.mpf('inf')
ctx.ninf = -ctx.inf
ctx.nan = ctx.mpf('nan')
ctx.j = ctx.mpc(0,1)
ctx.exp = ctx._wrap_mpi_function(libmp.mpi_exp, libmp.mpci_exp)
ctx.sqrt = ctx._wrap_mpi_function(libmp.mpi_sqrt)
ctx.ln = ctx._wrap_mpi_function(libmp.mpi_log, libmp.mpci_log)
ctx.cos = ctx._wrap_mpi_function(libmp.mpi_cos, libmp.mpci_cos)
ctx.sin = ctx._wrap_mpi_function(libmp.mpi_sin, libmp.mpci_sin)
ctx.tan = ctx._wrap_mpi_function(libmp.mpi_tan)
ctx.gamma = ctx._wrap_mpi_function(libmp.mpi_gamma, libmp.mpci_gamma)
ctx.loggamma = ctx._wrap_mpi_function(libmp.mpi_loggamma, libmp.mpci_loggamma)
ctx.rgamma = ctx._wrap_mpi_function(libmp.mpi_rgamma, libmp.mpci_rgamma)
ctx.factorial = ctx._wrap_mpi_function(libmp.mpi_factorial, libmp.mpci_factorial)
ctx.fac = ctx.factorial
ctx.eps = ctx._constant(lambda prec, rnd: (0, MPZ_ONE, 1-prec, 1))
ctx.pi = ctx._constant(libmp.mpf_pi)
ctx.e = ctx._constant(libmp.mpf_e)
ctx.ln2 = ctx._constant(libmp.mpf_ln2)
ctx.ln10 = ctx._constant(libmp.mpf_ln10)
ctx.phi = ctx._constant(libmp.mpf_phi)
ctx.euler = ctx._constant(libmp.mpf_euler)
ctx.catalan = ctx._constant(libmp.mpf_catalan)
ctx.glaisher = ctx._constant(libmp.mpf_glaisher)
ctx.khinchin = ctx._constant(libmp.mpf_khinchin)
ctx.twinprime = ctx._constant(libmp.mpf_twinprime)
def _wrap_mpi_function(ctx, f_real, f_complex=None):
def g(x, **kwargs):
if kwargs:
prec = kwargs.get('prec', ctx._prec[0])
else:
prec = ctx._prec[0]
x = ctx.convert(x)
if hasattr(x, "_mpi_"):
return ctx.make_mpf(f_real(x._mpi_, prec))
if hasattr(x, "_mpci_"):
return ctx.make_mpc(f_complex(x._mpci_, prec))
raise ValueError
return g
@classmethod
def _wrap_specfun(cls, name, f, wrap):
if wrap:
def f_wrapped(ctx, *args, **kwargs):
convert = ctx.convert
args = [convert(a) for a in args]
prec = ctx.prec
try:
ctx.prec += 10
retval = f(ctx, *args, **kwargs)
finally:
ctx.prec = prec
return +retval
else:
f_wrapped = f
setattr(cls, name, f_wrapped)
def _set_prec(ctx, n):
ctx._prec[0] = max(1, int(n))
ctx._dps = prec_to_dps(n)
def _set_dps(ctx, n):
ctx._prec[0] = dps_to_prec(n)
ctx._dps = max(1, int(n))
prec = property(lambda ctx: ctx._prec[0], _set_prec)
dps = property(lambda ctx: ctx._dps, _set_dps)
def make_mpf(ctx, v):
a = new(ctx.mpf)
a._mpi_ = v
return a
def make_mpc(ctx, v):
a = new(ctx.mpc)
a._mpci_ = v
return a
def _mpq(ctx, pq):
p, q = pq
a = libmp.from_rational(p, q, ctx.prec, round_floor)
b = libmp.from_rational(p, q, ctx.prec, round_ceiling)
return ctx.make_mpf((a, b))
def convert(ctx, x):
if isinstance(x, (ctx.mpf, ctx.mpc)):
return x
if isinstance(x, ctx._constant):
return +x
if isinstance(x, complex) or hasattr(x, "_mpc_"):
re = ctx.convert(x.real)
im = ctx.convert(x.imag)
return ctx.mpc(re,im)
if isinstance(x, basestring):
v = mpi_from_str(x, ctx.prec)
return ctx.make_mpf(v)
if hasattr(x, "_mpi_"):
a, b = x._mpi_
else:
try:
a, b = x
except (TypeError, ValueError):
a = b = x
if hasattr(a, "_mpi_"):
a = a._mpi_[0]
else:
a = convert_mpf_(a, ctx.prec, round_floor)
if hasattr(b, "_mpi_"):
b = b._mpi_[1]
else:
b = convert_mpf_(b, ctx.prec, round_ceiling)
if a == fnan or b == fnan:
a = fninf
b = finf
assert mpf_le(a, b), "endpoints must be properly ordered"
return ctx.make_mpf((a, b))
def nstr(ctx, x, n=5, **kwargs):
x = ctx.convert(x)
if hasattr(x, "_mpi_"):
return libmp.mpi_to_str(x._mpi_, n, **kwargs)
if hasattr(x, "_mpci_"):
re = libmp.mpi_to_str(x._mpci_[0], n, **kwargs)
im = libmp.mpi_to_str(x._mpci_[1], n, **kwargs)
return "(%s + %s*j)" % (re, im)
def mag(ctx, x):
x = ctx.convert(x)
if isinstance(x, ctx.mpc):
return max(ctx.mag(x.real), ctx.mag(x.imag)) + 1
a, b = libmp.mpi_abs(x._mpi_)
sign, man, exp, bc = b
if man:
return exp+bc
if b == fzero:
return ctx.ninf
if b == fnan:
return ctx.nan
return ctx.inf
def isnan(ctx, x):
return False
def isinf(ctx, x):
return x == ctx.inf
def isint(ctx, x):
x = ctx.convert(x)
a, b = x._mpi_
if a == b:
sign, man, exp, bc = a
if man:
return exp >= 0
return a == fzero
return None
def ldexp(ctx, x, n):
a, b = ctx.convert(x)._mpi_
a = libmp.mpf_shift(a, n)
b = libmp.mpf_shift(b, n)
return ctx.make_mpf((a,b))
def absmin(ctx, x):
return abs(ctx.convert(x)).a
def absmax(ctx, x):
return abs(ctx.convert(x)).b
def atan2(ctx, y, x):
y = ctx.convert(y)._mpi_
x = ctx.convert(x)._mpi_
return ctx.make_mpf(libmp.mpi_atan2(y,x,ctx.prec))
def _convert_param(ctx, x):
if isinstance(x, libmp.int_types):
return x, 'Z'
if isinstance(x, tuple):
p, q = x
return (ctx.mpf(p) / ctx.mpf(q), 'R')
x = ctx.convert(x)
if isinstance(x, ctx.mpf):
return x, 'R'
if isinstance(x, ctx.mpc):
return x, 'C'
raise ValueError
def _is_real_type(ctx, z):
return isinstance(z, ctx.mpf) or isinstance(z, int_types)
def _is_complex_type(ctx, z):
return isinstance(z, ctx.mpc)
def hypsum(ctx, p, q, types, coeffs, z, maxterms=6000, **kwargs):
coeffs = list(coeffs)
num = range(p)
den = range(p,p+q)
#tol = ctx.eps
s = t = ctx.one
k = 0
while 1:
for i in num: t *= (coeffs[i]+k)
for i in den: t /= (coeffs[i]+k)
k += 1; t /= k; t *= z; s += t
if t == 0:
return s
#if abs(t) < tol:
# return s
if k > maxterms:
raise ctx.NoConvergence
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/ctx_iv.py
|
ctx_iv.py
|
import operator
import math
import cmath
# Irrational (?) constants
pi = 3.1415926535897932385
e = 2.7182818284590452354
sqrt2 = 1.4142135623730950488
sqrt5 = 2.2360679774997896964
phi = 1.6180339887498948482
ln2 = 0.69314718055994530942
ln10 = 2.302585092994045684
euler = 0.57721566490153286061
catalan = 0.91596559417721901505
khinchin = 2.6854520010653064453
apery = 1.2020569031595942854
logpi = 1.1447298858494001741
def _mathfun_real(f_real, f_complex):
def f(x, **kwargs):
if type(x) is float:
return f_real(x)
if type(x) is complex:
return f_complex(x)
try:
x = float(x)
return f_real(x)
except (TypeError, ValueError):
x = complex(x)
return f_complex(x)
f.__name__ = f_real.__name__
return f
def _mathfun(f_real, f_complex):
def f(x, **kwargs):
if type(x) is complex:
return f_complex(x)
try:
return f_real(float(x))
except (TypeError, ValueError):
return f_complex(complex(x))
f.__name__ = f_real.__name__
return f
def _mathfun_n(f_real, f_complex):
def f(*args, **kwargs):
try:
return f_real(*(float(x) for x in args))
except (TypeError, ValueError):
return f_complex(*(complex(x) for x in args))
f.__name__ = f_real.__name__
return f
# Workaround for non-raising log and sqrt in Python 2.5 and 2.4
# on Unix system
try:
math.log(-2.0)
def math_log(x):
if x <= 0.0:
raise ValueError("math domain error")
return math.log(x)
def math_sqrt(x):
if x < 0.0:
raise ValueError("math domain error")
return math.sqrt(x)
except (ValueError, TypeError):
math_log = math.log
math_sqrt = math.sqrt
pow = _mathfun_n(operator.pow, lambda x, y: complex(x)**y)
log = _mathfun_n(math_log, cmath.log)
sqrt = _mathfun(math_sqrt, cmath.sqrt)
exp = _mathfun_real(math.exp, cmath.exp)
cos = _mathfun_real(math.cos, cmath.cos)
sin = _mathfun_real(math.sin, cmath.sin)
tan = _mathfun_real(math.tan, cmath.tan)
acos = _mathfun(math.acos, cmath.acos)
asin = _mathfun(math.asin, cmath.asin)
atan = _mathfun_real(math.atan, cmath.atan)
cosh = _mathfun_real(math.cosh, cmath.cosh)
sinh = _mathfun_real(math.sinh, cmath.sinh)
tanh = _mathfun_real(math.tanh, cmath.tanh)
floor = _mathfun_real(math.floor,
lambda z: complex(math.floor(z.real), math.floor(z.imag)))
ceil = _mathfun_real(math.ceil,
lambda z: complex(math.ceil(z.real), math.ceil(z.imag)))
cos_sin = _mathfun_real(lambda x: (math.cos(x), math.sin(x)),
lambda z: (cmath.cos(z), cmath.sin(z)))
cbrt = _mathfun(lambda x: x**(1./3), lambda z: z**(1./3))
def nthroot(x, n):
r = 1./n
try:
return float(x) ** r
except (ValueError, TypeError):
return complex(x) ** r
def _sinpi_real(x):
if x < 0:
return -_sinpi_real(-x)
n, r = divmod(x, 0.5)
r *= pi
n %= 4
if n == 0: return math.sin(r)
if n == 1: return math.cos(r)
if n == 2: return -math.sin(r)
if n == 3: return -math.cos(r)
def _cospi_real(x):
if x < 0:
x = -x
n, r = divmod(x, 0.5)
r *= pi
n %= 4
if n == 0: return math.cos(r)
if n == 1: return -math.sin(r)
if n == 2: return -math.cos(r)
if n == 3: return math.sin(r)
def _sinpi_complex(z):
if z.real < 0:
return -_sinpi_complex(-z)
n, r = divmod(z.real, 0.5)
z = pi*complex(r, z.imag)
n %= 4
if n == 0: return cmath.sin(z)
if n == 1: return cmath.cos(z)
if n == 2: return -cmath.sin(z)
if n == 3: return -cmath.cos(z)
def _cospi_complex(z):
if z.real < 0:
z = -z
n, r = divmod(z.real, 0.5)
z = pi*complex(r, z.imag)
n %= 4
if n == 0: return cmath.cos(z)
if n == 1: return -cmath.sin(z)
if n == 2: return -cmath.cos(z)
if n == 3: return cmath.sin(z)
cospi = _mathfun_real(_cospi_real, _cospi_complex)
sinpi = _mathfun_real(_sinpi_real, _sinpi_complex)
def tanpi(x):
try:
return sinpi(x) / cospi(x)
except OverflowError:
if complex(x).imag > 10:
return 1j
if complex(x).imag < 10:
return -1j
raise
def cotpi(x):
try:
return cospi(x) / sinpi(x)
except OverflowError:
if complex(x).imag > 10:
return -1j
if complex(x).imag < 10:
return 1j
raise
INF = 1e300*1e300
NINF = -INF
NAN = INF-INF
EPS = 2.2204460492503131e-16
_exact_gamma = (INF, 1.0, 1.0, 2.0, 6.0, 24.0, 120.0, 720.0, 5040.0, 40320.0,
362880.0, 3628800.0, 39916800.0, 479001600.0, 6227020800.0, 87178291200.0,
1307674368000.0, 20922789888000.0, 355687428096000.0, 6402373705728000.0,
121645100408832000.0, 2432902008176640000.0)
_max_exact_gamma = len(_exact_gamma)-1
# Lanczos coefficients used by the GNU Scientific Library
_lanczos_g = 7
_lanczos_p = (0.99999999999980993, 676.5203681218851, -1259.1392167224028,
771.32342877765313, -176.61502916214059, 12.507343278686905,
-0.13857109526572012, 9.9843695780195716e-6, 1.5056327351493116e-7)
def _gamma_real(x):
_intx = int(x)
if _intx == x:
if _intx <= 0:
#return (-1)**_intx * INF
raise ZeroDivisionError("gamma function pole")
if _intx <= _max_exact_gamma:
return _exact_gamma[_intx]
if x < 0.5:
# TODO: sinpi
return pi / (_sinpi_real(x)*_gamma_real(1-x))
else:
x -= 1.0
r = _lanczos_p[0]
for i in range(1, _lanczos_g+2):
r += _lanczos_p[i]/(x+i)
t = x + _lanczos_g + 0.5
return 2.506628274631000502417 * t**(x+0.5) * math.exp(-t) * r
def _gamma_complex(x):
if not x.imag:
return complex(_gamma_real(x.real))
if x.real < 0.5:
# TODO: sinpi
return pi / (_sinpi_complex(x)*_gamma_complex(1-x))
else:
x -= 1.0
r = _lanczos_p[0]
for i in range(1, _lanczos_g+2):
r += _lanczos_p[i]/(x+i)
t = x + _lanczos_g + 0.5
return 2.506628274631000502417 * t**(x+0.5) * cmath.exp(-t) * r
gamma = _mathfun_real(_gamma_real, _gamma_complex)
def rgamma(x):
try:
return 1./gamma(x)
except ZeroDivisionError:
return x*0.0
def factorial(x):
return gamma(x+1.0)
def arg(x):
if type(x) is float:
return math.atan2(0.0,x)
return math.atan2(x.imag,x.real)
# XXX: broken for negatives
def loggamma(x):
if type(x) not in (float, complex):
try:
x = float(x)
except (ValueError, TypeError):
x = complex(x)
try:
xreal = x.real
ximag = x.imag
except AttributeError: # py2.5
xreal = x
ximag = 0.0
# Reflection formula
# http://functions.wolfram.com/GammaBetaErf/LogGamma/16/01/01/0003/
if xreal < 0.0:
if abs(x) < 0.5:
v = log(gamma(x))
if ximag == 0:
v = v.conjugate()
return v
z = 1-x
try:
re = z.real
im = z.imag
except AttributeError: # py2.5
re = z
im = 0.0
refloor = floor(re)
if im == 0.0:
imsign = 0
elif im < 0.0:
imsign = -1
else:
imsign = 1
return (-pi*1j)*abs(refloor)*(1-abs(imsign)) + logpi - \
log(sinpi(z-refloor)) - loggamma(z) + 1j*pi*refloor*imsign
if x == 1.0 or x == 2.0:
return x*0
p = 0.
while abs(x) < 11:
p -= log(x)
x += 1.0
s = 0.918938533204672742 + (x-0.5)*log(x) - x
r = 1./x
r2 = r*r
s += 0.083333333333333333333*r; r *= r2
s += -0.0027777777777777777778*r; r *= r2
s += 0.00079365079365079365079*r; r *= r2
s += -0.0005952380952380952381*r; r *= r2
s += 0.00084175084175084175084*r; r *= r2
s += -0.0019175269175269175269*r; r *= r2
s += 0.0064102564102564102564*r; r *= r2
s += -0.02955065359477124183*r
return s + p
_psi_coeff = [
0.083333333333333333333,
-0.0083333333333333333333,
0.003968253968253968254,
-0.0041666666666666666667,
0.0075757575757575757576,
-0.021092796092796092796,
0.083333333333333333333,
-0.44325980392156862745,
3.0539543302701197438,
-26.456212121212121212]
def _digamma_real(x):
_intx = int(x)
if _intx == x:
if _intx <= 0:
raise ZeroDivisionError("polygamma pole")
if x < 0.5:
x = 1.0-x
s = pi*cotpi(x)
else:
s = 0.0
while x < 10.0:
s -= 1.0/x
x += 1.0
x2 = x**-2
t = x2
for c in _psi_coeff:
s -= c*t
if t < 1e-20:
break
t *= x2
return s + math_log(x) - 0.5/x
def _digamma_complex(x):
if not x.imag:
return complex(_digamma_real(x.real))
if x.real < 0.5:
x = 1.0-x
s = pi*cotpi(x)
else:
s = 0.0
while abs(x) < 10.0:
s -= 1.0/x
x += 1.0
x2 = x**-2
t = x2
for c in _psi_coeff:
s -= c*t
if abs(t) < 1e-20:
break
t *= x2
return s + cmath.log(x) - 0.5/x
digamma = _mathfun_real(_digamma_real, _digamma_complex)
# TODO: could implement complex erf and erfc here. Need
# to find an accurate method (avoiding cancellation)
# for approx. 1 < abs(x) < 9.
_erfc_coeff_P = [
1.0000000161203922312,
2.1275306946297962644,
2.2280433377390253297,
1.4695509105618423961,
0.66275911699770787537,
0.20924776504163751585,
0.045459713768411264339,
0.0063065951710717791934,
0.00044560259661560421715][::-1]
_erfc_coeff_Q = [
1.0000000000000000000,
3.2559100272784894318,
4.9019435608903239131,
4.4971472894498014205,
2.7845640601891186528,
1.2146026030046904138,
0.37647108453729465912,
0.080970149639040548613,
0.011178148899483545902,
0.00078981003831980423513][::-1]
def _polyval(coeffs, x):
p = coeffs[0]
for c in coeffs[1:]:
p = c + x*p
return p
def _erf_taylor(x):
# Taylor series assuming 0 <= x <= 1
x2 = x*x
s = t = x
n = 1
while abs(t) > 1e-17:
t *= x2/n
s -= t/(n+n+1)
n += 1
t *= x2/n
s += t/(n+n+1)
n += 1
return 1.1283791670955125739*s
def _erfc_mid(x):
# Rational approximation assuming 0 <= x <= 9
return exp(-x*x)*_polyval(_erfc_coeff_P,x)/_polyval(_erfc_coeff_Q,x)
def _erfc_asymp(x):
# Asymptotic expansion assuming x >= 9
x2 = x*x
v = exp(-x2)/x*0.56418958354775628695
r = t = 0.5 / x2
s = 1.0
for n in range(1,22,4):
s -= t
t *= r * (n+2)
s += t
t *= r * (n+4)
if abs(t) < 1e-17:
break
return s * v
def erf(x):
"""
erf of a real number.
"""
x = float(x)
if x != x:
return x
if x < 0.0:
return -erf(-x)
if x >= 1.0:
if x >= 6.0:
return 1.0
return 1.0 - _erfc_mid(x)
return _erf_taylor(x)
def erfc(x):
"""
erfc of a real number.
"""
x = float(x)
if x != x:
return x
if x < 0.0:
if x < -6.0:
return 2.0
return 2.0-erfc(-x)
if x > 9.0:
return _erfc_asymp(x)
if x >= 1.0:
return _erfc_mid(x)
return 1.0 - _erf_taylor(x)
gauss42 = [\
(0.99839961899006235, 0.0041059986046490839),
(-0.99839961899006235, 0.0041059986046490839),
(0.9915772883408609, 0.009536220301748501),
(-0.9915772883408609,0.009536220301748501),
(0.97934250806374812, 0.014922443697357493),
(-0.97934250806374812, 0.014922443697357493),
(0.96175936533820439,0.020227869569052644),
(-0.96175936533820439, 0.020227869569052644),
(0.93892355735498811, 0.025422959526113047),
(-0.93892355735498811,0.025422959526113047),
(0.91095972490412735, 0.030479240699603467),
(-0.91095972490412735, 0.030479240699603467),
(0.87802056981217269,0.03536907109759211),
(-0.87802056981217269, 0.03536907109759211),
(0.8402859832618168, 0.040065735180692258),
(-0.8402859832618168,0.040065735180692258),
(0.7979620532554873, 0.044543577771965874),
(-0.7979620532554873, 0.044543577771965874),
(0.75127993568948048,0.048778140792803244),
(-0.75127993568948048, 0.048778140792803244),
(0.70049459055617114, 0.052746295699174064),
(-0.70049459055617114,0.052746295699174064),
(0.64588338886924779, 0.056426369358018376),
(-0.64588338886924779, 0.056426369358018376),
(0.58774459748510932, 0.059798262227586649),
(-0.58774459748510932, 0.059798262227586649),
(0.5263957499311922, 0.062843558045002565),
(-0.5263957499311922, 0.062843558045002565),
(0.46217191207042191, 0.065545624364908975),
(-0.46217191207042191, 0.065545624364908975),
(0.39542385204297503, 0.067889703376521934),
(-0.39542385204297503, 0.067889703376521934),
(0.32651612446541151, 0.069862992492594159),
(-0.32651612446541151, 0.069862992492594159),
(0.25582507934287907, 0.071454714265170971),
(-0.25582507934287907, 0.071454714265170971),
(0.18373680656485453, 0.072656175243804091),
(-0.18373680656485453, 0.072656175243804091),
(0.11064502720851986, 0.073460813453467527),
(-0.11064502720851986, 0.073460813453467527),
(0.036948943165351772, 0.073864234232172879),
(-0.036948943165351772, 0.073864234232172879)]
EI_ASYMP_CONVERGENCE_RADIUS = 40.0
def ei_asymp(z, _e1=False):
r = 1./z
s = t = 1.0
k = 1
while 1:
t *= k*r
s += t
if abs(t) < 1e-16:
break
k += 1
v = s*exp(z)/z
if _e1:
if type(z) is complex:
zreal = z.real
zimag = z.imag
else:
zreal = z
zimag = 0.0
if zimag == 0.0 and zreal > 0.0:
v += pi*1j
else:
if type(z) is complex:
if z.imag > 0:
v += pi*1j
if z.imag < 0:
v -= pi*1j
return v
def ei_taylor(z, _e1=False):
s = t = z
k = 2
while 1:
t = t*z/k
term = t/k
if abs(term) < 1e-17:
break
s += term
k += 1
s += euler
if _e1:
s += log(-z)
else:
if type(z) is float or z.imag == 0.0:
s += math_log(abs(z))
else:
s += cmath.log(z)
return s
def ei(z, _e1=False):
typez = type(z)
if typez not in (float, complex):
try:
z = float(z)
typez = float
except (TypeError, ValueError):
z = complex(z)
typez = complex
if not z:
return -INF
absz = abs(z)
if absz > EI_ASYMP_CONVERGENCE_RADIUS:
return ei_asymp(z, _e1)
elif absz <= 2.0 or (typez is float and z > 0.0):
return ei_taylor(z, _e1)
# Integrate, starting from whichever is smaller of a Taylor
# series value or an asymptotic series value
if typez is complex and z.real > 0.0:
zref = z / absz
ref = ei_taylor(zref, _e1)
else:
zref = EI_ASYMP_CONVERGENCE_RADIUS * z / absz
ref = ei_asymp(zref, _e1)
C = (zref-z)*0.5
D = (zref+z)*0.5
s = 0.0
if type(z) is complex:
_exp = cmath.exp
else:
_exp = math.exp
for x,w in gauss42:
t = C*x+D
s += w*_exp(t)/t
ref -= C*s
return ref
def e1(z):
# hack to get consistent signs if the imaginary part if 0
# and signed
typez = type(z)
if type(z) not in (float, complex):
try:
z = float(z)
typez = float
except (TypeError, ValueError):
z = complex(z)
typez = complex
if typez is complex and not z.imag:
z = complex(z.real, 0.0)
# end hack
return -ei(-z, _e1=True)
_zeta_int = [\
-0.5,
0.0,
1.6449340668482264365,1.2020569031595942854,1.0823232337111381915,
1.0369277551433699263,1.0173430619844491397,1.0083492773819228268,
1.0040773561979443394,1.0020083928260822144,1.0009945751278180853,
1.0004941886041194646,1.0002460865533080483,1.0001227133475784891,
1.0000612481350587048,1.0000305882363070205,1.0000152822594086519,
1.0000076371976378998,1.0000038172932649998,1.0000019082127165539,
1.0000009539620338728,1.0000004769329867878,1.0000002384505027277,
1.0000001192199259653,1.0000000596081890513,1.0000000298035035147,
1.0000000149015548284]
_zeta_P = [-3.50000000087575873, -0.701274355654678147,
-0.0672313458590012612, -0.00398731457954257841,
-0.000160948723019303141, -4.67633010038383371e-6,
-1.02078104417700585e-7, -1.68030037095896287e-9,
-1.85231868742346722e-11][::-1]
_zeta_Q = [1.00000000000000000, -0.936552848762465319,
-0.0588835413263763741, -0.00441498861482948666,
-0.000143416758067432622, -5.10691659585090782e-6,
-9.58813053268913799e-8, -1.72963791443181972e-9,
-1.83527919681474132e-11][::-1]
_zeta_1 = [3.03768838606128127e-10, -1.21924525236601262e-8,
2.01201845887608893e-7, -1.53917240683468381e-6,
-5.09890411005967954e-7, 0.000122464707271619326,
-0.000905721539353130232, -0.00239315326074843037,
0.084239750013159168, 0.418938517907442414, 0.500000001921884009]
_zeta_0 = [-3.46092485016748794e-10, -6.42610089468292485e-9,
1.76409071536679773e-7, -1.47141263991560698e-6, -6.38880222546167613e-7,
0.000122641099800668209, -0.000905894913516772796, -0.00239303348507992713,
0.0842396947501199816, 0.418938533204660256, 0.500000000000000052]
def zeta(s):
"""
Riemann zeta function, real argument
"""
if not isinstance(s, (float, int)):
try:
s = float(s)
except (ValueError, TypeError):
try:
s = complex(s)
if not s.imag:
return complex(zeta(s.real))
except (ValueError, TypeError):
pass
raise NotImplementedError
if s == 1:
raise ValueError("zeta(1) pole")
if s >= 27:
return 1.0 + 2.0**(-s) + 3.0**(-s)
n = int(s)
if n == s:
if n >= 0:
return _zeta_int[n]
if not (n % 2):
return 0.0
if s <= 0.0:
return 2.**s*pi**(s-1)*_sinpi_real(0.5*s)*_gamma_real(1-s)*zeta(1-s)
if s <= 2.0:
if s <= 1.0:
return _polyval(_zeta_0,s)/(s-1)
return _polyval(_zeta_1,s)/(s-1)
z = _polyval(_zeta_P,s) / _polyval(_zeta_Q,s)
return 1.0 + 2.0**(-s) + 3.0**(-s) + 4.0**(-s)*z
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/math2.py
|
math2.py
|
import operator
import sys
from .libmp import int_types, mpf_hash, bitcount, from_man_exp, HASH_MODULUS
new = object.__new__
def create_reduced(p, q, _cache={}):
key = p, q
if key in _cache:
return _cache[key]
x, y = p, q
while y:
x, y = y, x % y
if x != 1:
p //= x
q //= x
v = new(mpq)
v._mpq_ = p, q
# Speedup integers, half-integers and other small fractions
if q <= 4 and abs(key[0]) < 100:
_cache[key] = v
return v
class mpq(object):
"""
Exact rational type, currently only intended for internal use.
"""
__slots__ = ["_mpq_"]
def __new__(cls, p, q=1):
if type(p) is tuple:
p, q = p
elif hasattr(p, '_mpq_'):
p, q = p._mpq_
return create_reduced(p, q)
def __repr__(s):
return "mpq(%s,%s)" % s._mpq_
def __str__(s):
return "(%s/%s)" % s._mpq_
def __int__(s):
a, b = s._mpq_
return a // b
def __nonzero__(s):
return bool(s._mpq_[0])
__bool__ = __nonzero__
def __hash__(s):
a, b = s._mpq_
if sys.version >= "3.2":
inverse = pow(b, HASH_MODULUS-2, HASH_MODULUS)
if not inverse:
h = sys.hash_info.inf
else:
h = (abs(a) * inverse) % HASH_MODULUS
if a < 0: h = -h
if h == -1: h = -2
return h
else:
if b == 1:
return hash(a)
# Power of two: mpf compatible hash
if not (b & (b-1)):
return mpf_hash(from_man_exp(a, 1-bitcount(b)))
return hash((a,b))
def __eq__(s, t):
ttype = type(t)
if ttype is mpq:
return s._mpq_ == t._mpq_
if ttype in int_types:
a, b = s._mpq_
if b != 1:
return False
return a == t
return NotImplemented
def __ne__(s, t):
ttype = type(t)
if ttype is mpq:
return s._mpq_ != t._mpq_
if ttype in int_types:
a, b = s._mpq_
if b != 1:
return True
return a != t
return NotImplemented
def _cmp(s, t, op):
ttype = type(t)
if ttype in int_types:
a, b = s._mpq_
return op(a, t*b)
if ttype is mpq:
a, b = s._mpq_
c, d = t._mpq_
return op(a*d, b*c)
return NotImplementedError
def __lt__(s, t): return s._cmp(t, operator.lt)
def __le__(s, t): return s._cmp(t, operator.le)
def __gt__(s, t): return s._cmp(t, operator.gt)
def __ge__(s, t): return s._cmp(t, operator.ge)
def __abs__(s):
a, b = s._mpq_
if a >= 0:
return s
v = new(mpq)
v._mpq_ = -a, b
return v
def __neg__(s):
a, b = s._mpq_
v = new(mpq)
v._mpq_ = -a, b
return v
def __pos__(s):
return s
def __add__(s, t):
ttype = type(t)
if ttype is mpq:
a, b = s._mpq_
c, d = t._mpq_
return create_reduced(a*d+b*c, b*d)
if ttype in int_types:
a, b = s._mpq_
v = new(mpq)
v._mpq_ = a+b*t, b
return v
return NotImplemented
__radd__ = __add__
def __sub__(s, t):
ttype = type(t)
if ttype is mpq:
a, b = s._mpq_
c, d = t._mpq_
return create_reduced(a*d-b*c, b*d)
if ttype in int_types:
a, b = s._mpq_
v = new(mpq)
v._mpq_ = a-b*t, b
return v
return NotImplemented
def __rsub__(s, t):
ttype = type(t)
if ttype is mpq:
a, b = s._mpq_
c, d = t._mpq_
return create_reduced(b*c-a*d, b*d)
if ttype in int_types:
a, b = s._mpq_
v = new(mpq)
v._mpq_ = b*t-a, b
return v
return NotImplemented
def __mul__(s, t):
ttype = type(t)
if ttype is mpq:
a, b = s._mpq_
c, d = t._mpq_
return create_reduced(a*c, b*d)
if ttype in int_types:
a, b = s._mpq_
return create_reduced(a*t, b)
return NotImplemented
__rmul__ = __mul__
def __div__(s, t):
ttype = type(t)
if ttype is mpq:
a, b = s._mpq_
c, d = t._mpq_
return create_reduced(a*d, b*c)
if ttype in int_types:
a, b = s._mpq_
return create_reduced(a, b*t)
return NotImplemented
def __rdiv__(s, t):
ttype = type(t)
if ttype is mpq:
a, b = s._mpq_
c, d = t._mpq_
return create_reduced(b*c, a*d)
if ttype in int_types:
a, b = s._mpq_
return create_reduced(b*t, a)
return NotImplemented
def __pow__(s, t):
ttype = type(t)
if ttype in int_types:
a, b = s._mpq_
if t:
if t < 0:
a, b, t = b, a, -t
v = new(mpq)
v._mpq_ = a**t, b**t
return v
raise ZeroDivisionError
return NotImplemented
mpq_1 = mpq((1,1))
mpq_0 = mpq((0,1))
mpq_1_2 = mpq((1,2))
mpq_3_2 = mpq((3,2))
mpq_1_4 = mpq((1,4))
mpq_1_16 = mpq((1,16))
mpq_3_16 = mpq((3,16))
mpq_5_2 = mpq((5,2))
mpq_3_4 = mpq((3,4))
mpq_7_4 = mpq((7,4))
mpq_5_4 = mpq((5,4))
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/rational.py
|
rational.py
|
__version__ = '0.17'
from .usertools import monitor, timing
from .ctx_fp import FPContext
from .ctx_mp import MPContext
from .ctx_iv import MPIntervalContext
fp = FPContext()
mp = MPContext()
iv = MPIntervalContext()
fp._mp = mp
mp._mp = mp
iv._mp = mp
mp._fp = fp
fp._fp = fp
mp._iv = iv
fp._iv = iv
iv._iv = iv
# XXX: extremely bad pickle hack
import ctx_mp as _ctx_mp
_ctx_mp._mpf_module.mpf = mp.mpf
_ctx_mp._mpf_module.mpc = mp.mpc
make_mpf = mp.make_mpf
make_mpc = mp.make_mpc
extraprec = mp.extraprec
extradps = mp.extradps
workprec = mp.workprec
workdps = mp.workdps
autoprec = mp.autoprec
maxcalls = mp.maxcalls
memoize = mp.memoize
mag = mp.mag
bernfrac = mp.bernfrac
qfrom = mp.qfrom
mfrom = mp.mfrom
kfrom = mp.kfrom
taufrom = mp.taufrom
qbarfrom = mp.qbarfrom
ellipfun = mp.ellipfun
jtheta = mp.jtheta
kleinj = mp.kleinj
qp = mp.qp
qhyper = mp.qhyper
qgamma = mp.qgamma
qfac = mp.qfac
nint_distance = mp.nint_distance
plot = mp.plot
cplot = mp.cplot
splot = mp.splot
odefun = mp.odefun
jacobian = mp.jacobian
findroot = mp.findroot
multiplicity = mp.multiplicity
isinf = mp.isinf
isnan = mp.isnan
isnormal = mp.isnormal
isint = mp.isint
almosteq = mp.almosteq
nan = mp.nan
rand = mp.rand
absmin = mp.absmin
absmax = mp.absmax
fraction = mp.fraction
linspace = mp.linspace
arange = mp.arange
mpmathify = convert = mp.convert
mpc = mp.mpc
mpi = iv._mpi
nstr = mp.nstr
nprint = mp.nprint
chop = mp.chop
fneg = mp.fneg
fadd = mp.fadd
fsub = mp.fsub
fmul = mp.fmul
fdiv = mp.fdiv
fprod = mp.fprod
quad = mp.quad
quadgl = mp.quadgl
quadts = mp.quadts
quadosc = mp.quadosc
pslq = mp.pslq
identify = mp.identify
findpoly = mp.findpoly
richardson = mp.richardson
shanks = mp.shanks
nsum = mp.nsum
nprod = mp.nprod
difference = mp.difference
diff = mp.diff
diffs = mp.diffs
diffs_prod = mp.diffs_prod
diffs_exp = mp.diffs_exp
diffun = mp.diffun
differint = mp.differint
taylor = mp.taylor
pade = mp.pade
polyval = mp.polyval
polyroots = mp.polyroots
fourier = mp.fourier
fourierval = mp.fourierval
sumem = mp.sumem
sumap = mp.sumap
chebyfit = mp.chebyfit
limit = mp.limit
matrix = mp.matrix
eye = mp.eye
diag = mp.diag
zeros = mp.zeros
ones = mp.ones
hilbert = mp.hilbert
randmatrix = mp.randmatrix
swap_row = mp.swap_row
extend = mp.extend
norm = mp.norm
mnorm = mp.mnorm
lu_solve = mp.lu_solve
lu = mp.lu
unitvector = mp.unitvector
inverse = mp.inverse
residual = mp.residual
qr_solve = mp.qr_solve
cholesky = mp.cholesky
cholesky_solve = mp.cholesky_solve
det = mp.det
cond = mp.cond
expm = mp.expm
sqrtm = mp.sqrtm
powm = mp.powm
logm = mp.logm
sinm = mp.sinm
cosm = mp.cosm
mpf = mp.mpf
j = mp.j
exp = mp.exp
expj = mp.expj
expjpi = mp.expjpi
ln = mp.ln
im = mp.im
re = mp.re
inf = mp.inf
ninf = mp.ninf
sign = mp.sign
eps = mp.eps
pi = mp.pi
ln2 = mp.ln2
ln10 = mp.ln10
phi = mp.phi
e = mp.e
euler = mp.euler
catalan = mp.catalan
khinchin = mp.khinchin
glaisher = mp.glaisher
apery = mp.apery
degree = mp.degree
twinprime = mp.twinprime
mertens = mp.mertens
ldexp = mp.ldexp
frexp = mp.frexp
fsum = mp.fsum
fdot = mp.fdot
sqrt = mp.sqrt
cbrt = mp.cbrt
exp = mp.exp
ln = mp.ln
log = mp.log
log10 = mp.log10
power = mp.power
cos = mp.cos
sin = mp.sin
tan = mp.tan
cosh = mp.cosh
sinh = mp.sinh
tanh = mp.tanh
acos = mp.acos
asin = mp.asin
atan = mp.atan
asinh = mp.asinh
acosh = mp.acosh
atanh = mp.atanh
sec = mp.sec
csc = mp.csc
cot = mp.cot
sech = mp.sech
csch = mp.csch
coth = mp.coth
asec = mp.asec
acsc = mp.acsc
acot = mp.acot
asech = mp.asech
acsch = mp.acsch
acoth = mp.acoth
cospi = mp.cospi
sinpi = mp.sinpi
sinc = mp.sinc
sincpi = mp.sincpi
cos_sin = mp.cos_sin
cospi_sinpi = mp.cospi_sinpi
fabs = mp.fabs
re = mp.re
im = mp.im
conj = mp.conj
floor = mp.floor
ceil = mp.ceil
nint = mp.nint
frac = mp.frac
root = mp.root
nthroot = mp.nthroot
hypot = mp.hypot
fmod = mp.fmod
ldexp = mp.ldexp
frexp = mp.frexp
sign = mp.sign
arg = mp.arg
phase = mp.phase
polar = mp.polar
rect = mp.rect
degrees = mp.degrees
radians = mp.radians
atan2 = mp.atan2
fib = mp.fib
fibonacci = mp.fibonacci
lambertw = mp.lambertw
zeta = mp.zeta
altzeta = mp.altzeta
gamma = mp.gamma
rgamma = mp.rgamma
factorial = mp.factorial
fac = mp.fac
fac2 = mp.fac2
beta = mp.beta
betainc = mp.betainc
psi = mp.psi
#psi0 = mp.psi0
#psi1 = mp.psi1
#psi2 = mp.psi2
#psi3 = mp.psi3
polygamma = mp.polygamma
digamma = mp.digamma
#trigamma = mp.trigamma
#tetragamma = mp.tetragamma
#pentagamma = mp.pentagamma
harmonic = mp.harmonic
bernoulli = mp.bernoulli
bernfrac = mp.bernfrac
stieltjes = mp.stieltjes
hurwitz = mp.hurwitz
dirichlet = mp.dirichlet
bernpoly = mp.bernpoly
eulerpoly = mp.eulerpoly
eulernum = mp.eulernum
polylog = mp.polylog
clsin = mp.clsin
clcos = mp.clcos
gammainc = mp.gammainc
gammaprod = mp.gammaprod
binomial = mp.binomial
rf = mp.rf
ff = mp.ff
hyper = mp.hyper
hyp0f1 = mp.hyp0f1
hyp1f1 = mp.hyp1f1
hyp1f2 = mp.hyp1f2
hyp2f1 = mp.hyp2f1
hyp2f2 = mp.hyp2f2
hyp2f0 = mp.hyp2f0
hyp2f3 = mp.hyp2f3
hyp3f2 = mp.hyp3f2
hyperu = mp.hyperu
hypercomb = mp.hypercomb
meijerg = mp.meijerg
appellf1 = mp.appellf1
appellf2 = mp.appellf2
appellf3 = mp.appellf3
appellf4 = mp.appellf4
hyper2d = mp.hyper2d
bihyper = mp.bihyper
erf = mp.erf
erfc = mp.erfc
erfi = mp.erfi
erfinv = mp.erfinv
npdf = mp.npdf
ncdf = mp.ncdf
expint = mp.expint
e1 = mp.e1
ei = mp.ei
li = mp.li
ci = mp.ci
si = mp.si
chi = mp.chi
shi = mp.shi
fresnels = mp.fresnels
fresnelc = mp.fresnelc
airyai = mp.airyai
airybi = mp.airybi
airyaizero = mp.airyaizero
airybizero = mp.airybizero
scorergi = mp.scorergi
scorerhi = mp.scorerhi
ellipk = mp.ellipk
ellipe = mp.ellipe
ellipf = mp.ellipf
ellippi = mp.ellippi
elliprc = mp.elliprc
elliprj = mp.elliprj
elliprf = mp.elliprf
elliprd = mp.elliprd
elliprg = mp.elliprg
agm = mp.agm
jacobi = mp.jacobi
chebyt = mp.chebyt
chebyu = mp.chebyu
legendre = mp.legendre
legenp = mp.legenp
legenq = mp.legenq
hermite = mp.hermite
pcfd = mp.pcfd
pcfu = mp.pcfu
pcfv = mp.pcfv
pcfw = mp.pcfw
gegenbauer = mp.gegenbauer
laguerre = mp.laguerre
spherharm = mp.spherharm
besselj = mp.besselj
j0 = mp.j0
j1 = mp.j1
besseli = mp.besseli
bessely = mp.bessely
besselk = mp.besselk
besseljzero = mp.besseljzero
besselyzero = mp.besselyzero
hankel1 = mp.hankel1
hankel2 = mp.hankel2
struveh = mp.struveh
struvel = mp.struvel
angerj = mp.angerj
webere = mp.webere
lommels1 = mp.lommels1
lommels2 = mp.lommels2
whitm = mp.whitm
whitw = mp.whitw
ber = mp.ber
bei = mp.bei
ker = mp.ker
kei = mp.kei
coulombc = mp.coulombc
coulombf = mp.coulombf
coulombg = mp.coulombg
lambertw = mp.lambertw
barnesg = mp.barnesg
superfac = mp.superfac
hyperfac = mp.hyperfac
loggamma = mp.loggamma
siegeltheta = mp.siegeltheta
siegelz = mp.siegelz
grampoint = mp.grampoint
zetazero = mp.zetazero
riemannr = mp.riemannr
primepi = mp.primepi
primepi2 = mp.primepi2
primezeta = mp.primezeta
bell = mp.bell
polyexp = mp.polyexp
expm1 = mp.expm1
powm1 = mp.powm1
unitroots = mp.unitroots
cyclotomic = mp.cyclotomic
mangoldt = mp.mangoldt
secondzeta = mp.secondzeta
nzeros = mp.nzeros
backlunds = mp.backlunds
lerchphi = mp.lerchphi
# be careful when changing this name, don't use test*!
def runtests():
"""
Run all mpmath tests and print output.
"""
import os.path
from inspect import getsourcefile
from .tests import runtests as tests
testdir = os.path.dirname(os.path.abspath(getsourcefile(tests)))
importdir = os.path.abspath(testdir + '/../..')
tests.testit(importdir, testdir)
def doctests():
try:
import psyco; psyco.full()
except ImportError:
pass
import sys
from timeit import default_timer as clock
filter = []
for i, arg in enumerate(sys.argv):
if '__init__.py' in arg:
filter = [sn for sn in sys.argv[i+1:] if not sn.startswith("-")]
break
import doctest
globs = globals().copy()
for obj in globs: #sorted(globs.keys()):
if filter:
if not sum([pat in obj for pat in filter]):
continue
sys.stdout.write(str(obj) + " ")
sys.stdout.flush()
t1 = clock()
doctest.run_docstring_examples(globs[obj], {}, verbose=("-v" in sys.argv))
t2 = clock()
print(round(t2-t1, 3))
if __name__ == '__main__':
doctests()
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/__init__.py
|
__init__.py
|
from .ctx_base import StandardBaseContext
import math
import cmath
import math2
import function_docs
from .libmp import mpf_bernoulli, to_float, int_types
import libmp
class FPContext(StandardBaseContext):
"""
Context for fast low-precision arithmetic (53-bit precision, giving at most
about 15-digit accuracy), using Python's builtin float and complex.
"""
def __init__(ctx):
StandardBaseContext.__init__(ctx)
# Override SpecialFunctions implementation
ctx.loggamma = math2.loggamma
ctx._bernoulli_cache = {}
ctx.pretty = False
ctx._init_aliases()
_mpq = lambda cls, x: float(x[0])/x[1]
NoConvergence = libmp.NoConvergence
def _get_prec(ctx): return 53
def _set_prec(ctx, p): return
def _get_dps(ctx): return 15
def _set_dps(ctx, p): return
_fixed_precision = True
prec = property(_get_prec, _set_prec)
dps = property(_get_dps, _set_dps)
zero = 0.0
one = 1.0
eps = math2.EPS
inf = math2.INF
ninf = math2.NINF
nan = math2.NAN
j = 1j
# Called by SpecialFunctions.__init__()
@classmethod
def _wrap_specfun(cls, name, f, wrap):
if wrap:
def f_wrapped(ctx, *args, **kwargs):
convert = ctx.convert
args = [convert(a) for a in args]
return f(ctx, *args, **kwargs)
else:
f_wrapped = f
f_wrapped.__doc__ = function_docs.__dict__.get(name, f.__doc__)
setattr(cls, name, f_wrapped)
def bernoulli(ctx, n):
cache = ctx._bernoulli_cache
if n in cache:
return cache[n]
cache[n] = to_float(mpf_bernoulli(n, 53, 'n'), strict=True)
return cache[n]
pi = math2.pi
e = math2.e
euler = math2.euler
sqrt2 = 1.4142135623730950488
sqrt5 = 2.2360679774997896964
phi = 1.6180339887498948482
ln2 = 0.69314718055994530942
ln10 = 2.302585092994045684
euler = 0.57721566490153286061
catalan = 0.91596559417721901505
khinchin = 2.6854520010653064453
apery = 1.2020569031595942854
glaisher = 1.2824271291006226369
absmin = absmax = abs
def is_special(ctx, x):
return x - x != 0.0
def isnan(ctx, x):
return x != x
def isinf(ctx, x):
return abs(x) == math2.INF
def isnormal(ctx, x):
if x:
return x - x == 0.0
return False
def isnpint(ctx, x):
if type(x) is complex:
if x.imag:
return False
x = x.real
return x <= 0.0 and round(x) == x
mpf = float
mpc = complex
def convert(ctx, x):
try:
return float(x)
except:
return complex(x)
power = staticmethod(math2.pow)
sqrt = staticmethod(math2.sqrt)
exp = staticmethod(math2.exp)
ln = log = staticmethod(math2.log)
cos = staticmethod(math2.cos)
sin = staticmethod(math2.sin)
tan = staticmethod(math2.tan)
cos_sin = staticmethod(math2.cos_sin)
acos = staticmethod(math2.acos)
asin = staticmethod(math2.asin)
atan = staticmethod(math2.atan)
cosh = staticmethod(math2.cosh)
sinh = staticmethod(math2.sinh)
tanh = staticmethod(math2.tanh)
gamma = staticmethod(math2.gamma)
rgamma = staticmethod(math2.rgamma)
fac = factorial = staticmethod(math2.factorial)
floor = staticmethod(math2.floor)
ceil = staticmethod(math2.ceil)
cospi = staticmethod(math2.cospi)
sinpi = staticmethod(math2.sinpi)
cbrt = staticmethod(math2.cbrt)
_nthroot = staticmethod(math2.nthroot)
_ei = staticmethod(math2.ei)
_e1 = staticmethod(math2.e1)
_zeta = _zeta_int = staticmethod(math2.zeta)
# XXX: math2
def arg(ctx, z):
z = complex(z)
return math.atan2(z.imag, z.real)
def expj(ctx, x):
return ctx.exp(ctx.j*x)
def expjpi(ctx, x):
return ctx.exp(ctx.j*ctx.pi*x)
ldexp = math.ldexp
frexp = math.frexp
def mag(ctx, z):
if z:
return ctx.frexp(abs(z))[1]
return ctx.ninf
def isint(ctx, z):
if hasattr(z, "imag"): # float/int don't have .real/.imag in py2.5
if z.imag:
return False
z = z.real
try:
return z == int(z)
except:
return False
def nint_distance(ctx, z):
if hasattr(z, "imag"): # float/int don't have .real/.imag in py2.5
n = round(z.real)
else:
n = round(z)
if n == z:
return n, ctx.ninf
return n, ctx.mag(abs(z-n))
def _convert_param(ctx, z):
if type(z) is tuple:
p, q = z
return ctx.mpf(p) / q, 'R'
if hasattr(z, "imag"): # float/int don't have .real/.imag in py2.5
intz = int(z.real)
else:
intz = int(z)
if z == intz:
return intz, 'Z'
return z, 'R'
def _is_real_type(ctx, z):
return isinstance(z, float) or isinstance(z, int_types)
def _is_complex_type(ctx, z):
return isinstance(z, complex)
def hypsum(ctx, p, q, types, coeffs, z, maxterms=6000, **kwargs):
coeffs = list(coeffs)
num = range(p)
den = range(p,p+q)
tol = ctx.eps
s = t = 1.0
k = 0
while 1:
for i in num: t *= (coeffs[i]+k)
for i in den: t /= (coeffs[i]+k)
k += 1; t /= k; t *= z; s += t
if abs(t) < tol:
return s
if k > maxterms:
raise ctx.NoConvergence
def atan2(ctx, x, y):
return math.atan2(x, y)
def psi(ctx, m, z):
m = int(m)
if m == 0:
return ctx.digamma(z)
return (-1)**(m+1) * ctx.fac(m) * ctx.zeta(m+1, z)
digamma = staticmethod(math2.digamma)
def harmonic(ctx, x):
x = ctx.convert(x)
if x == 0 or x == 1:
return x
return ctx.digamma(x+1) + ctx.euler
nstr = str
def to_fixed(ctx, x, prec):
return int(math.ldexp(x, prec))
def rand(ctx):
import random
return random.random()
_erf = staticmethod(math2.erf)
_erfc = staticmethod(math2.erfc)
def sum_accurately(ctx, terms, check_step=1):
s = ctx.zero
k = 0
for term in terms():
s += term
if (not k % check_step) and term:
if abs(term) <= 1e-18*abs(s):
break
k += 1
return s
|
AltAnalyze
|
/AltAnalyze-2.1.3.15.tar.gz/AltAnalyze-2.1.3.15/altanalyze/stats_scripts/mpmath/ctx_fp.py
|
ctx_fp.py
|
Subsets and Splits
No community queries yet
The top public SQL queries from the community will appear here once available.